carbon supported pt–pd alloy as an ethanol tolerant oxygen reduction electrocatalyst for direct...
TRANSCRIPT

i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 0
Avai lab le a t www.sc iencedi rec t .com
j ourna l homepage : www.e lsev ier . com/ loca te /he
Carbon supported Pt–Pd alloy as an ethanol tolerant oxygenreduction electrocatalyst for direct ethanol fuel cells
T. Lopesa, E. Antolinia,b,*, E.R. Gonzaleza
aInstituto de Quımica de Sao Carlos, USP, C.P. 780, Sao Carlos, SP 13560-970, BrazilbScuola di Scienza dei Materiali, Chemistry, Via 25 aprile 22, 16016, Cogoleto, Genova, Italy
a r t i c l e i n f o
Article history:
Received 5 December 2007
Received in revised form
22 March 2008
Accepted 2 May 2008
Available online 16 September 2008
Keywords:
Direct ethanol fuel cells (DEFC)
Electrocatalyst
Platinum
Palladium
Oxygen reduction
* Corresponding author. Scuola di Scienza deE-mail address: [email protected] (E. An
0360-3199/$ – see front matter ª 2008 Interndoi:10.1016/j.ijhydene.2008.05.030
a b s t r a c t
A carbon supported Pt–Pd catalyst with a Pt:Pd atomic ratio 77:23 was prepared by
reduction of metal precursors with formic acid and characterized by EDX, XRD and XPS
techniques. A decrease of the lattice parameter compared with that of pure Pt was
observed, indicating the formation of a Pt–Pd alloy. Tests in H2SO4 solution in the absence
of ethanol showed that the Pd-containing is slightly more active than pure Pt for the
oxygen reduction reaction (ORR). In the presence of ethanol a larger increase in over-
potential of the ORR on pure Pt than that on Pt–Pd was found, indicating a higher ethanol
tolerance of the binary catalyst. The enhanced performance at 90 �C of the direct ethanol
fuel cell with Pt–Pd/C as cathode material confirmed the results of half cell tests, and was
essentially ascribed to a reduced ethanol adsorption on Pt–Pd.
ª 2008 International Association for Hydrogen Energy. Published by Elsevier Ltd. All rights
reserved.
1. Introduction electrochemical oxidation kinetics on the Pt/C cathode. The
Ethanol is an attractive liquid fuel for direct alcohol fuelled
systems. It is the major renewable biofuel obtained from the
fermentation of biomass, and ethanol is less toxic than
methanol. Wang et al. [1] compared the performance of fuel
cells operating on various methanol-alternative fuels. They
found that ethanol is a promising alternative fuel with an
electrochemical activity comparable to that of methanol.
In the direct ethanol fuel cell (DEFC), the ethanol fed to
the anode compartment can permeate through the electro-
lyte to the cathode, similar to what happens in the direct
methanol fuel cell (DMFC), i.e. methanol crossover. Song
et al. [2] found that the ethanol permeated to the cathode
exhibited a less serious effect on the cell performance
compared to methanol because of both its smaller perme-
ability through the Nafion� membrane and its slower
i Materiali, Via 25 apriletolini).ational Association for H
influence of ethanol crossover on the DEFC performance,
however, is not negligible [3,4]. The effect of the ethanol
concentration and the operating temperature on the ethanol
crossover rate was investigated by Andreadis and Tsiakaras
[3]. They found that the ethanol crossover rate dependence
on the ethanol feed concentration is an almost linear func-
tion presenting a maximum at about [CH3CH2OH]¼ 10 M.
They observed also that the ethanol crossover rate increases
as the temperature increases and the apparent activation
energy is about 4 kcal/mol indicating the physical nature of
the crossover process. As reported by Song et al. [4], the
method of preparation of the membrane electrode assembly
(MEA) also affects the ethanol crossover. Rousseau et al. [5]
evaluated the crossover of ethanol during the DEFC opera-
tion from the amount of reaction products of ethanol
oxidation. For Nafion� 115 and 112 membranes, the evaluated
22, 16016, Cogoleto, Genova, Italy.
ydrogen Energy. Published by Elsevier Ltd. All rights reserved.

i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 05564
crossover rates at 20 �C and [CH3CH2OH]¼ 1 M were
8.66� 10�8 and 13� 10�8 mol cm�2 s�1, respectively.
Another main objective in the development of DEFCs is the
achievement of anode catalysts with high activity for ethanol
oxidation [6] and, due to the low activity of Pt for the oxygen
reduction reaction (ORR), research on cathode catalysts
alternative to pure Pt are also in progress. The requirements of
a suitable cathode material for the DEFC are an improved ORR
activity and an ethanol tolerance higher than pure Pt. The
alloys of transition metals, such as V, Cr, Co, Ti and Ni, with
platinum have been found to exhibit significantly higher
electrocatalytic activities towards the oxygen reduction reac-
tion than platinum alone in low temperature fuel cells [7–16].
These Pt–M alloy electrocatalysts improve both the perfor-
mance and the resistance to sintering and coalescence of the
nanoparticles under the operating conditions of the phos-
phoric acid and proton exchange membrane fuel cells.
Many papers were devoted to the research of platinum
based methanol tolerant cathodes for DMFCs, as reported by
Antolini et al. in a recent review [17]. The negative effect of
methanol crossover on cell performance is mitigated by using
Pt-based binary catalysts [17]. Higher methanol tolerance is
reported in the literature for non-noble metal electrocatalysts
based on chalcogenides [18–20] and macrocycles of transition
metals [21,22]. These electrocatalysts have shown nearly
the same activity for the ORR in the absence as well as in
the presence of methanol. However in methanol free elec-
trolytes, these materials did not reach the catalytic activity of
dispersed platinum.
Conversely, few works were addressed to the development
of ethanol tolerant oxygen reduction catalysts for DEFCs.
Savadogo and Rodriguez-Varela studied the catalytic activity
of carbon supported Ru [23] and unsupported Pd and Pd–Co
[24] catalysts for the ORR in an acid medium with and without
ethanol. They found that these catalysts have a high tolerance
to ethanol; their ORR activity in the absence of ethanol,
however, was considerably lower than that of Pt. In our
previous work [25], a single direct ethanol fuel cell with Pt–Co/
C as cathode catalyst performed better than the cell with Pt/C.
Recent works showed that the addition of Pd to Pt increases
the ORR activity of platinum [26–28] and that the depen-
dence of the ORR activity on the Pd content goes through
a maximum. Li et al. [26] prepared Pt–Pd/C (Pt:Pd¼ 3:1 and 1:1)
and Pt/C catalysts by a modified polyol method. They found
that the catalytic activity for the ORR of Pt–Pd/C in the Pt:Pd
atomic ratio 3:1 is improved compared with that of Pt/C or Pt–
Pd/C (1:1). They found that O2 is more readily adsorbed and
easily dissociated on the Pd-modified Pt surface. According to
the authors, this result mainly originates from the weakening
of the O–O bond on Pd-modified Pt clusters. Ye and Crooks
[27] prepared Pt–Pd bimetallic nanoparticles containing an
average of 180 atoms and composed of seven different
Pt:Pd ratios within sixth-generation, hydroxyl-terminated,
poly(amidoamine) dendrimers. Cyclic voltammetry and
rotating disk voltammetry measurements showed that the
Pt:Pd ratio of the nanoparticles determines their efficiency for
the oxygen reduction reaction (ORR). The maximum activity
for the ORR occurs at a Pt:Pd ratio of 5:1, which corresponds to
a relative mass activity enhancement of 2.4 compared to
otherwise identical monometallic Pt nanoparticles. Finally, Xu
and Lin [28] found that an electrodeposited Pt–Pd (9:1) catalyst
presents significantly higher stability and catalytic activity for
both the methanol oxidation reaction (MOR) and the ORR than
the corresponding electrodeposited Pt.
On this basis, a carbon supported Pt–Pd catalyst was
prepared by reduction of metal precursors with formic acid.
This synthesis method was successfully used to prepare
carbon supported Pt–Sn alloy catalysts [29]. In a previous work
carried out in our laboratory on a Pt–Pd/C catalyst with a Pt:Pd
atomic ratio 9:1 [30], it was found that this Pd-containing
catalyst has higher ethanol tolerance than Pt/C under ORR
operation. Moreover, a single DEFC with Pt–Pd (9:1) catalyst as
cathode material presented better performance than that
with Pt. Preliminary results indicated that a DEFC with a
Pt–Pd/C catalyst with Pt:Pd atomic ratio 3:1 as cathode mate-
rial performs better than that with Pt–Pd/C (9:1). On this basis
we selected to focus this work on the properties of Pt–Pd/C
(3:1) as oxygen reduction, ethanol tolerant catalyst for DEFCs.
2. Experimental
2.1. Catalyst preparation
A carbon supported Pt–Pd catalyst with nominal Pt:Pd atomic
ratio 75:25 was prepared by reduction of metal precursors
with formic acid. An appropriate amount of carbon powder
(Vulcan XC-72, Cabot, 240 m2 g�1) was suspended in 2 M for-
mic acid solution and the suspension heated to 80 �C. Chlor-
oplatinic acid (H2PtCl6$6H2O, Johnson Matthey) solution and
a palladium chloride (PdCl2$2H2O, MERCK) solution were
slowly added to the carbon suspension. The suspension was
left to cool at room temperature and the solid filtered and
dried in an oven at 80 �C for 1 h. The material was 20 wt.%
metal (Ptþ Pd) on carbon.
2.2. Characterization of the catalyst
The atomic ratio of the Pt–Pd/C catalyst was determined by
the EDX technique coupled to a scanning electron microscope
LEO Mod. 440 with a silicon detector with Be window and
applying 20 keV.
X-ray diffractograms of the catalysts were obtained at the
D12A-XRD1 beam line of the Brazilian Synchrotron Light
Laboratory. Scans were done for 2q values between 20 and
100�. The lattice parameters were obtained by refining the unit
cell dimensions by the least squares method [31].
X-ray photoelectron spectroscopy (XPS) experiments were
carried out using a conventional Al-Ka radiation (1486.6 eV),
in an ultrahigh vacuum system at a base pressure of
1� 10�10 mbar, using a VSW HA 100 analyzer operated in the
fixed analyzer transmission mode with a pass energy of
58.7 eV.
2.3. Electrochemical measurements
In order to test the electrochemical behavior in sulphuric acid
solution (with and without ethanol), the electrocatalysts were
used to make gas diffusion electrodes (GDE). A diffusion layer
was made with carbon powder (Vulcan XC-72) and 15 wt.%
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 0 5565
polytetrafluoroethylene (PTFE) and applied over a carbon cloth
(PWB-3, Stackpole). On top of this layer, the electrocatalyst
was applied in the form of a homogeneous dispersion of
Pt–Pd/C or Pt/C, Nafion� solution (5 wt.%, Aldrich) and iso-
propanol (Merck). All electrodes were made to contain 1 mg
Pt cm�2.
The oxidation of ethanol on Pt–Pd/C and Pt/C was tested in
a direct ethanol fuel cell system fed with a 1 M ethanol solu-
tion at the anode. Hydrogen was supplied to the cathode,
which operated simultaneously as auxiliary and reference
electrode. The experiments were done at room temperature,
40 and 90 �C with a 1285 A Solartron Potentiostat connected to
a personal computer and using the software CorrWare for
Windows (Scribner).
For the DEFC studies, the electrodes were hot pressed on
both sides of a Nafion� 115 membrane at 125 �C and
50 kg cm�2 for 2 min. The Nafion� membranes were pre-
treated with a 3 wt.% solution of H2O2, washed and then
treated with a 0.5 M solution of H2SO4. The geometric area of
the electrodes was 4.6 cm2, and the anode materials were
20 wt.% Pt/C and PtRu/C (1:1) from E-TEK. The cell polarization
data at 60 �C/1 atm and 90 �C/3 atm O2 pressure were obtained
by circulating a 1 M aqueous ethanol solution at the anode.
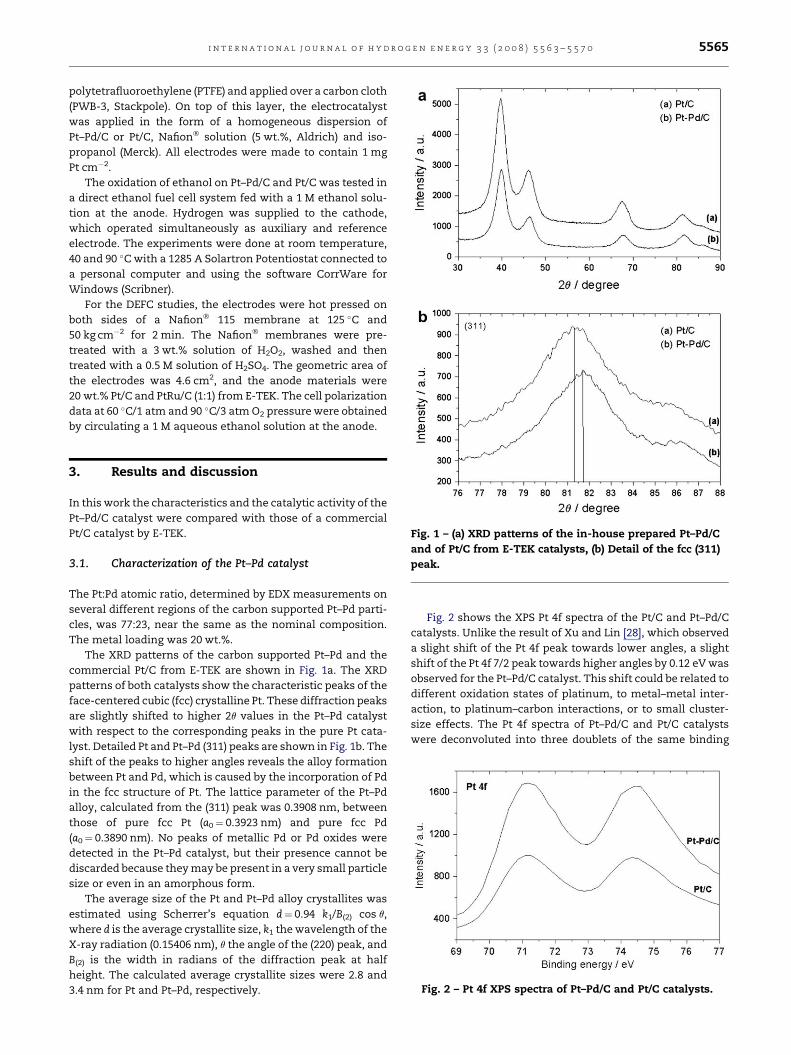
Fig. 1 – (a) XRD patterns of the in-house prepared Pt–Pd/C
and of Pt/C from E-TEK catalysts, (b) Detail of the fcc (311)
peak.
Fig. 2 – Pt 4f XPS spectra of Pt–Pd/C and Pt/C catalysts.
3. Results and discussion
In this work the characteristics and the catalytic activity of the
Pt–Pd/C catalyst were compared with those of a commercial
Pt/C catalyst by E-TEK.
3.1. Characterization of the Pt–Pd catalyst
The Pt:Pd atomic ratio, determined by EDX measurements on
several different regions of the carbon supported Pt–Pd parti-
cles, was 77:23, near the same as the nominal composition.
The metal loading was 20 wt.%.
The XRD patterns of the carbon supported Pt–Pd and the
commercial Pt/C from E-TEK are shown in Fig. 1a. The XRD
patterns of both catalysts show the characteristic peaks of the
face-centered cubic (fcc) crystalline Pt. These diffraction peaks
are slightly shifted to higher 2q values in the Pt–Pd catalyst
with respect to the corresponding peaks in the pure Pt cata-
lyst. Detailed Pt and Pt–Pd (311) peaks are shown in Fig. 1b. The
shift of the peaks to higher angles reveals the alloy formation
between Pt and Pd, which is caused by the incorporation of Pd
in the fcc structure of Pt. The lattice parameter of the Pt–Pd
alloy, calculated from the (311) peak was 0.3908 nm, between
those of pure fcc Pt (a0¼ 0.3923 nm) and pure fcc Pd
(a0¼ 0.3890 nm). No peaks of metallic Pd or Pd oxides were
detected in the Pt–Pd catalyst, but their presence cannot be
discarded because they may be present in a very small particle
size or even in an amorphous form.
The average size of the Pt and Pt–Pd alloy crystallites was
estimated using Scherrer’s equation d¼ 0.94 k1/B(2) cos q,
where d is the average crystallite size, k1 the wavelength of the
X-ray radiation (0.15406 nm), q the angle of the (220) peak, and
B(2) is the width in radians of the diffraction peak at half
height. The calculated average crystallite sizes were 2.8 and
3.4 nm for Pt and Pt–Pd, respectively.
Fig. 2 shows the XPS Pt 4f spectra of the Pt/C and Pt–Pd/C
catalysts. Unlike the result of Xu and Lin [28], which observed
a slight shift of the Pt 4f peak towards lower angles, a slight
shift of the Pt 4f 7/2 peak towards higher angles by 0.12 eV was
observed for the Pt–Pd/C catalyst. This shift could be related to
different oxidation states of platinum, to metal–metal inter-
action, to platinum–carbon interactions, or to small cluster-
size effects. The Pt 4f spectra of Pt–Pd/C and Pt/C catalysts
were deconvoluted into three doublets of the same binding
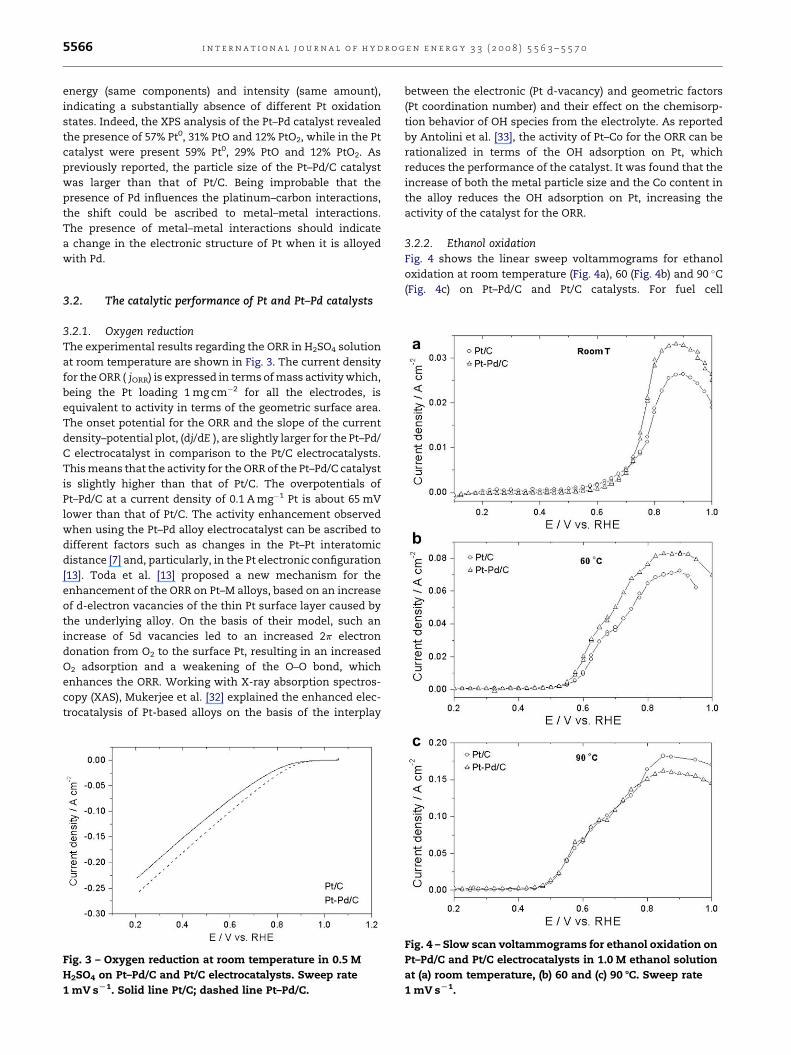
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 05566
energy (same components) and intensity (same amount),
indicating a substantially absence of different Pt oxidation
states. Indeed, the XPS analysis of the Pt–Pd catalyst revealed
the presence of 57% Pt0, 31% PtO and 12% PtO2, while in the Pt
catalyst were present 59% Pt0, 29% PtO and 12% PtO2. As
previously reported, the particle size of the Pt–Pd/C catalyst
was larger than that of Pt/C. Being improbable that the
presence of Pd influences the platinum–carbon interactions,
the shift could be ascribed to metal–metal interactions.
The presence of metal–metal interactions should indicate
a change in the electronic structure of Pt when it is alloyed
with Pd.
3.2. The catalytic performance of Pt and Pt–Pd catalysts
3.2.1. Oxygen reductionThe experimental results regarding the ORR in H2SO4 solution
at room temperature are shown in Fig. 3. The current density
for the ORR ( jORR) is expressed in terms of mass activity which,
being the Pt loading 1 mg cm�2 for all the electrodes, is
equivalent to activity in terms of the geometric surface area.
The onset potential for the ORR and the slope of the current
density–potential plot, (dj/dE ), are slightly larger for the Pt–Pd/
C electrocatalyst in comparison to the Pt/C electrocatalysts.
This means that the activity for the ORR of the Pt–Pd/C catalyst
is slightly higher than that of Pt/C. The overpotentials of
Pt–Pd/C at a current density of 0.1 A mg�1 Pt is about 65 mV
lower than that of Pt/C. The activity enhancement observed
when using the Pt–Pd alloy electrocatalyst can be ascribed to
different factors such as changes in the Pt–Pt interatomic
distance [7] and, particularly, in the Pt electronic configuration
[13]. Toda et al. [13] proposed a new mechanism for the
enhancement of the ORR on Pt–M alloys, based on an increase
of d-electron vacancies of the thin Pt surface layer caused by
the underlying alloy. On the basis of their model, such an
increase of 5d vacancies led to an increased 2p electron
donation from O2 to the surface Pt, resulting in an increased
O2 adsorption and a weakening of the O–O bond, which
enhances the ORR. Working with X-ray absorption spectros-
copy (XAS), Mukerjee et al. [32] explained the enhanced elec-
trocatalysis of Pt-based alloys on the basis of the interplay
Fig. 3 – Oxygen reduction at room temperature in 0.5 M
H2SO4 on Pt–Pd/C and Pt/C electrocatalysts. Sweep rate
1 mV sL1. Solid line Pt/C; dashed line Pt–Pd/C.
between the electronic (Pt d-vacancy) and geometric factors
(Pt coordination number) and their effect on the chemisorp-
tion behavior of OH species from the electrolyte. As reported
by Antolini et al. [33], the activity of Pt–Co for the ORR can be
rationalized in terms of the OH adsorption on Pt, which
reduces the performance of the catalyst. It was found that the
increase of both the metal particle size and the Co content in
the alloy reduces the OH adsorption on Pt, increasing the
activity of the catalyst for the ORR.
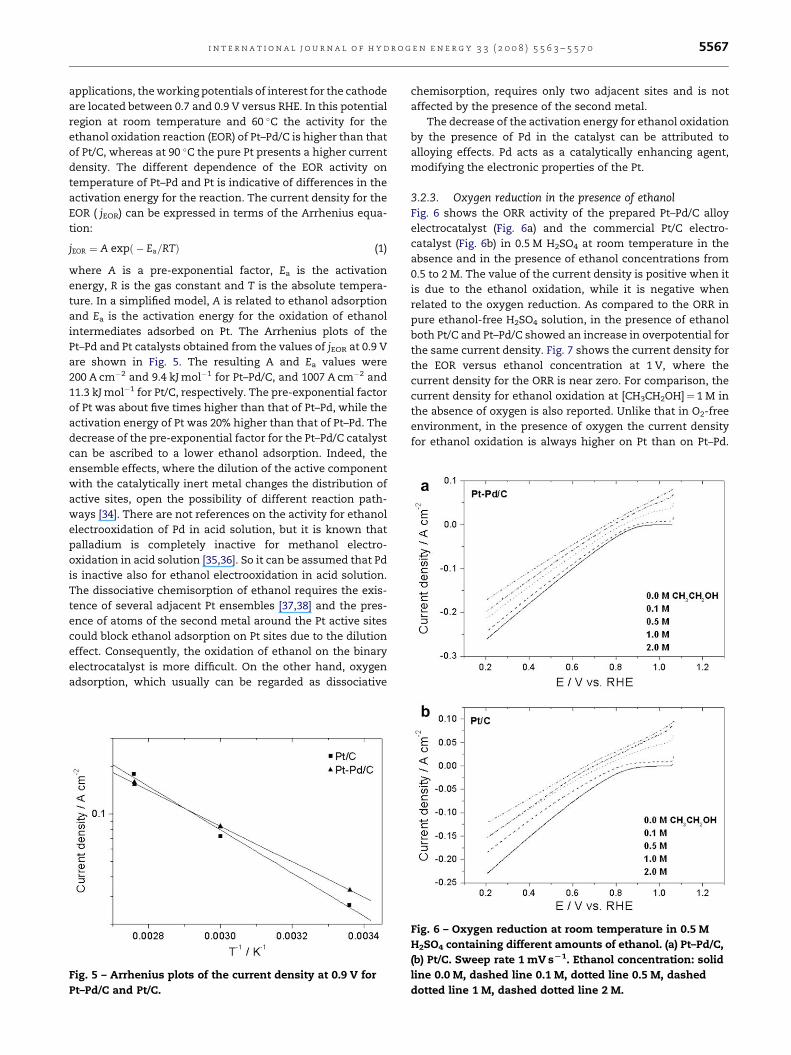
3.2.2. Ethanol oxidationFig. 4 shows the linear sweep voltammograms for ethanol
oxidation at room temperature (Fig. 4a), 60 (Fig. 4b) and 90 �C
(Fig. 4c) on Pt–Pd/C and Pt/C catalysts. For fuel cell
Fig. 4 – Slow scan voltammograms for ethanol oxidation on
Pt–Pd/C and Pt/C electrocatalysts in 1.0 M ethanol solution
at (a) room temperature, (b) 60 and (c) 90 8C. Sweep rate
1 mV sL1.
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 0 5567
applications, the working potentials of interest for the cathode
are located between 0.7 and 0.9 V versus RHE. In this potential
region at room temperature and 60 �C the activity for the
ethanol oxidation reaction (EOR) of Pt–Pd/C is higher than that
of Pt/C, whereas at 90 �C the pure Pt presents a higher current
density. The different dependence of the EOR activity on
temperature of Pt–Pd and Pt is indicative of differences in the
activation energy for the reaction. The current density for the
EOR ( jEOR) can be expressed in terms of the Arrhenius equa-
tion:
jEOR ¼ A expð � Ea=RTÞ (1)
where A is a pre-exponential factor, Ea is the activation
energy, R is the gas constant and T is the absolute tempera-
ture. In a simplified model, A is related to ethanol adsorption
and Ea is the activation energy for the oxidation of ethanol
intermediates adsorbed on Pt. The Arrhenius plots of the
Pt–Pd and Pt catalysts obtained from the values of jEOR at 0.9 V
are shown in Fig. 5. The resulting A and Ea values were
200 A cm�2 and 9.4 kJ mol�1 for Pt–Pd/C, and 1007 A cm�2 and
11.3 kJ mol�1 for Pt/C, respectively. The pre-exponential factor
of Pt was about five times higher than that of Pt–Pd, while the
activation energy of Pt was 20% higher than that of Pt–Pd. The
decrease of the pre-exponential factor for the Pt–Pd/C catalyst
can be ascribed to a lower ethanol adsorption. Indeed, the
ensemble effects, where the dilution of the active component
with the catalytically inert metal changes the distribution of
active sites, open the possibility of different reaction path-
ways [34]. There are not references on the activity for ethanol
electrooxidation of Pd in acid solution, but it is known that
palladium is completely inactive for methanol electro-
oxidation in acid solution [35,36]. So it can be assumed that Pd
is inactive also for ethanol electrooxidation in acid solution.
The dissociative chemisorption of ethanol requires the exis-
tence of several adjacent Pt ensembles [37,38] and the pres-
ence of atoms of the second metal around the Pt active sites
could block ethanol adsorption on Pt sites due to the dilution
effect. Consequently, the oxidation of ethanol on the binary
electrocatalyst is more difficult. On the other hand, oxygen
adsorption, which usually can be regarded as dissociative
Fig. 5 – Arrhenius plots of the current density at 0.9 V for
Pt–Pd/C and Pt/C.
chemisorption, requires only two adjacent sites and is not
affected by the presence of the second metal.
The decrease of the activation energy for ethanol oxidation
by the presence of Pd in the catalyst can be attributed to
alloying effects. Pd acts as a catalytically enhancing agent,
modifying the electronic properties of the Pt.
3.2.3. Oxygen reduction in the presence of ethanolFig. 6 shows the ORR activity of the prepared Pt–Pd/C alloy
electrocatalyst (Fig. 6a) and the commercial Pt/C electro-
catalyst (Fig. 6b) in 0.5 M H2SO4 at room temperature in the
absence and in the presence of ethanol concentrations from
0.5 to 2 M. The value of the current density is positive when it
is due to the ethanol oxidation, while it is negative when
related to the oxygen reduction. As compared to the ORR in
pure ethanol-free H2SO4 solution, in the presence of ethanol
both Pt/C and Pt–Pd/C showed an increase in overpotential for
the same current density. Fig. 7 shows the current density for
the EOR versus ethanol concentration at 1 V, where the
current density for the ORR is near zero. For comparison, the
current density for ethanol oxidation at [CH3CH2OH]¼ 1 M in
the absence of oxygen is also reported. Unlike that in O2-free
environment, in the presence of oxygen the current density
for ethanol oxidation is always higher on Pt than on Pt–Pd.
Fig. 6 – Oxygen reduction at room temperature in 0.5 M
H2SO4 containing different amounts of ethanol. (a) Pt–Pd/C,
(b) Pt/C. Sweep rate 1 mV sL1. Ethanol concentration: solid
line 0.0 M, dashed line 0.1 M, dotted line 0.5 M, dashed
dotted line 1 M, dashed dotted line 2 M.
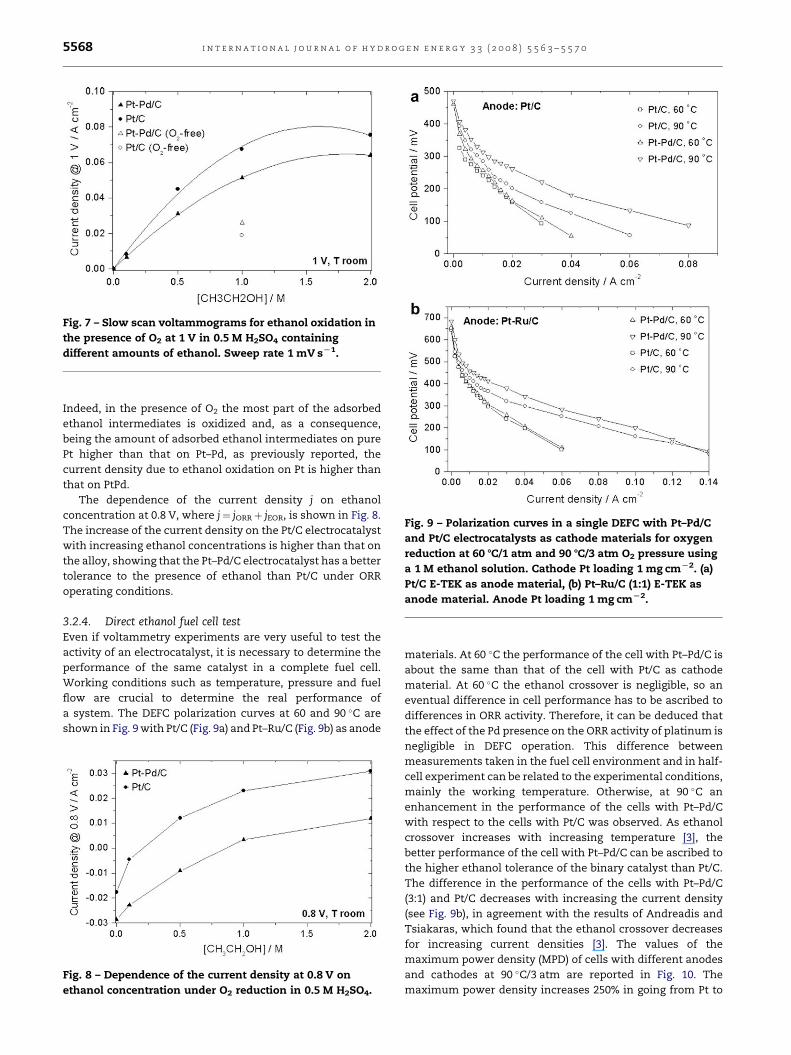
Fig. 7 – Slow scan voltammograms for ethanol oxidation in
the presence of O2 at 1 V in 0.5 M H2SO4 containing
different amounts of ethanol. Sweep rate 1 mV sL1.
Fig. 9 – Polarization curves in a single DEFC with Pt–Pd/C
and Pt/C electrocatalysts as cathode materials for oxygen
reduction at 60 8C/1 atm and 90 8C/3 atm O2 pressure using
a 1 M ethanol solution. Cathode Pt loading 1 mg cmL2. (a)
Pt/C E-TEK as anode material, (b) Pt–Ru/C (1:1) E-TEK as
anode material. Anode Pt loading 1 mg cmL2.
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 05568
Indeed, in the presence of O2 the most part of the adsorbed
ethanol intermediates is oxidized and, as a consequence,
being the amount of adsorbed ethanol intermediates on pure
Pt higher than that on Pt–Pd, as previously reported, the
current density due to ethanol oxidation on Pt is higher than
that on PtPd.
The dependence of the current density j on ethanol
concentration at 0.8 V, where j¼ jORRþ jEOR, is shown in Fig. 8.
The increase of the current density on the Pt/C electrocatalyst
with increasing ethanol concentrations is higher than that on
the alloy, showing that the Pt–Pd/C electrocatalyst has a better
tolerance to the presence of ethanol than Pt/C under ORR
operating conditions.
3.2.4. Direct ethanol fuel cell testEven if voltammetry experiments are very useful to test the
activity of an electrocatalyst, it is necessary to determine the
performance of the same catalyst in a complete fuel cell.
Working conditions such as temperature, pressure and fuel
flow are crucial to determine the real performance of
a system. The DEFC polarization curves at 60 and 90 �C are
shown in Fig. 9 with Pt/C (Fig. 9a) and Pt–Ru/C (Fig. 9b) as anode
Fig. 8 – Dependence of the current density at 0.8 V on
ethanol concentration under O2 reduction in 0.5 M H2SO4.
materials. At 60 �C the performance of the cell with Pt–Pd/C is
about the same than that of the cell with Pt/C as cathode
material. At 60 �C the ethanol crossover is negligible, so an
eventual difference in cell performance has to be ascribed to
differences in ORR activity. Therefore, it can be deduced that
the effect of the Pd presence on the ORR activity of platinum is
negligible in DEFC operation. This difference between
measurements taken in the fuel cell environment and in half-
cell experiment can be related to the experimental conditions,
mainly the working temperature. Otherwise, at 90 �C an
enhancement in the performance of the cells with Pt–Pd/C
with respect to the cells with Pt/C was observed. As ethanol
crossover increases with increasing temperature [3], the
better performance of the cell with Pt–Pd/C can be ascribed to
the higher ethanol tolerance of the binary catalyst than Pt/C.
The difference in the performance of the cells with Pt–Pd/C
(3:1) and Pt/C decreases with increasing the current density
(see Fig. 9b), in agreement with the results of Andreadis and
Tsiakaras, which found that the ethanol crossover decreases
for increasing current densities [3]. The values of the
maximum power density (MPD) of cells with different anodes
and cathodes at 90 �C/3 atm are reported in Fig. 10. The
maximum power density increases 250% in going from Pt to

Fig. 10 – Histograms of the maximum power density of
direct ethanol fuel cells operating at 90 8C with Pt/C and
Pt–Pd/C as cathode materials, and Pt/C and Pt–Ru/C as
anode materials.
i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 0 5569
Pt–Ru as anode material, and 70% in going from Pt to Pt–Pd as
cathode material. The gain in cell performance in going from
a cell using pure Pt both as anode and cathode material to
a cell with Pt–Ru and Pt–Pd as anode and cathode materials,
respectively (DMPDTot¼ 15.3 mW cm�2) is about the sum of
the gains in the performance of the cell with Pt–Ru as
anode and Pt as cathode (DMPD¼ 10.8 mW cm�2), and the
cell with Pt as anode and Pt–Pd as cathode material
(DMPD¼ 3.3 mW cm�2). As reported by Lopes et al. [25], tests
in a DEFC at various temperatures showed an enhancement of
the cell performance when Pt–Co/C (3:1) was used as cathode
material with respect to the cell with Pt/C, both in terms of
mass activity and in terms of specific activity. Considering
that Pt–Co/C and Pt/C have the same activity for the EOR in the
cathodic potential region, the improvement was ascribed to
the higher ORR activity of the binary alloy catalyst. The gain in
the performance of the cell operating at 90 �C/3 atm with
Pt as anode and Pt–Co/C as cathode material with respect to
the cell with Pt/C both as anode and cathode material
(DMPD¼ 2.8 mW cm�2) was slightly lower than that observed
in this work comparing the performance of DEFCs with Pt/C
and Pt–Pd/C as cathode materials. In the former case,
however, the enhanced performance of the cell with Pt–Co/C
as cathode material with respect to that with Pt/C was
ascribed to the higher ORR activity of the Co-containing
catalyst.
On the basis of the experiments in H2SO4 solution
described above, the poorer performance of the cell operating
at 90 �C with Pt/C catalyst with respect to that with Pt–Pd/C as
cathode material can be essentially ascribed to Pt poisoning
owing to ethanol crossover. As shown in this work, Pt
poisoning and, as a consequence, the decrease in DEFC
performance due to ethanol crossover can be reduced using
an ethanol tolerant catalyst.
4. Conclusions
The activity for the oxygen reduction reaction on carbon
supported Pt–Pd electrocatalysts prepared by reduction of
metal precursors with formic acid was investigated in
sulphuric acid both in the absence and in the presence of
ethanol and compared with a commercial Pt/C catalyst. In
ethanol-free sulphuric acid the Pt–Pd/C alloy catalyst showed
a slightly higher activity towards the oxygen reduction
compared to pure platinum. In the presence of ethanol
a higher increase in overpotential of the ORR on pure Pt than
that on Pt–Pd was found, indicating a higher ethanol tolerance
of the binary catalyst. Tests in DEFC at 60 �C indicated that the
performance of the cell with Pt–Pd/C was about the same than
that of the cell with Pt/C as cathode material, while at 90 �C an
enhancement of the cell performance when Pt–Pd/C was used
as cathode material was observed with respect to the cell with
Pt/C. Considering that at 60 �C the ethanol crossover is negli-
gible, the improvement of DEFC performance at 90 �C was
ascribed to the higher ethanol tolerance of Pt–Pd/C.
Acknowledgements
The authors thank CAPES/Brazil, Progr. PVE 2007, and the
Conselho Nacional de Desenvolvimento Cientıfico e Tecnolo-
gico (CNPq, Proc. 142097/2005-5), for financial assistance to the
project. Thanks are also due to the Brazilian Synchrotron Light
Laboratory, LNLS, for helping with the physical characteriza-
tion of the catalysts.
r e f e r e n c e s
[1] Wang J, Wasmus S, Savinell RF. Evaluation of ethanol,1-propanol, and 2-propanol in a direct oxidation polymer-electrolyte fuel cell. J Electrochem Soc 1995;142:4218.
[2] Song S, Zhou W, Liang Z, Cai R, Sun G, Xin Q, et al. The effectof methanol and ethanol cross-over on the performance ofPtRu/C-based anode DAFCs. Appl Catal B Environ 2005;55:65.
[3] Andreadis G, Tsiakaras P. Ethanol crossover and directethanol PEM fuel cell performance modeling andexperimental validation. Chem Eng Sci 2006;61:7497.
[4] Song S, Wang G, Zhou W, Zhao X, Sun G, Xin Q, et al. Theeffect of the MEA preparation procedure on both ethanolcrossover and DEFC performance. J Power Sources 2005;140:103.
[5] Rousseau S, Coutanceau C, Lamy C, Leger J-M. Direct ethanolfuel cell (DEFC) electrical performances and reaction productsdistribution under operating conditions with differentplatinum-based anodes. J Power Sources 2006;158:18.
[6] Antolini E. Catalysts for direct ethanol fuel cells. J PowerSources 2007;170:1.
[7] Jalan V, Taylor EJ. Importance of interatomic spacing incatalytic reduction of oxygen in phosphoric acid. JElectrochem Soc 1983;130:2299.
[8] Beard BC, Ross PN. Characterization of a titanium-promotedsupported platinum electrocatalyst. J Electrochem Soc 1986;133:1839.
[9] Paffett MT, Berry GJ, Gottesfeld S. Oxygen reduction atPt0.65Cr0.35, Pt0.2Cr0.8 and roughened platinum. J ElectrochemSoc 1988;135:1431.
[10] Beard BC, Ross PN. The structure and activity of Pt–Co alloysas oxygen reduction electrocatalysts. J Electrochem Soc 1990;137:3368.
[11] Mukerjee S, Srinivasan S. Enhanced electrocatalysis ofoxygen reduction on platinum alloys in proton exchangemembrane fuel cells. J Electroanal Chem 1993;357:201.

i n t e r n a t i o n a l j o u r n a l o f h y d r o g e n e n e r g y 3 3 ( 2 0 0 8 ) 5 5 6 3 – 5 5 7 05570
[12] Watanabe M, Tsurumi K, Mizukami T, Nakamura T,Stonehart P. Activity and stability of ordered and disorderedCo–Pt alloys for phosphoric acid fuel cells. J Electrochem Soc1994;141:2659.
[13] Toda T, Igarashi H, Uchida H, Watanabe M. Enhancement ofelectroreduction of oxygen on Pt alloys with Fe, Ni and Co. JElectrochem Soc 1999;146:3750.
[14] Min M, Cho J, Cho K, Kim H. Particle size and alloying effectsof Pt-based alloy catalysts for fuel cell applications.Electrochim Acta 2000;45:4211.
[15] Paulus UA, Scherer GG, Wokaun A, Schmidt TJ,Stamenkovic V, Radmilovic V, et al. Oxygen reduction oncarbon-supported Pt–Ni and Pt–Co alloy catalysts. J PhysChem B 2002;106:4181.
[16] Antolini E, Passos RR, Ticianelli EA. Electrocatalysis ofoxygen reduction on a carbon supported platinum–vanadium alloy in polymer electrolyte fuel cells. ElectrochimActa 2002;48:263.
[17] Antolini E, Lopes T, Gonzalez ER. An overview of platinum-based catalysts as methanol-resistant oxygen reductionmaterials for direct methanol fuel cells. J Alloys Comp 2008;461:253.
[18] Urban PM, Funke A, Muller JT, Himmen M, Docter A.Catalytic processes in solid polymer electrolyte fuel cellsystems. Appl Catal A 2001;221:459.
[19] Schubert B, Tributsch H, Alonso-Vante N, Perrin A. Influenceof d-state density and chemistry of transition metal clusterselenides on electrocatalysis. J Catal 1988;12:384.
[20] Alonso-Vante N, Tributsch H. Energy conversion catalysisusing semiconducting transition metal cluster compounds.Nature 1986;323:431.
[21] Jiang R, Chu D. Remarkably active catalysts for theelectroreduction of O2 to H2O for use in acidic electrolytecontaining concentrated methanol. J Electrochem Soc 2000;147:4605.
[22] Convert P, Coutanceau C, Crouigneau P, Gloaguen F, Lamy C.Electrodes modified by electrodeposition of CoTAAcomplexes as selective oxygen cathodes in a direct methanolfuel cel. J Appl Electrochem 2001;31:945.
[23] Savadogo O, Rodriguez-Varela FJ. Ethanol-tolerant oxygenreduction reaction (ORR) cathodes for direct ethanol fuel cellapplications. ECS Trans 2006;1:331.
[24] Savadogo O, Rodriguez-Varela FJ. Palladium-alloy catalystsas ethanol tolerant cathodes for direct alcohol fuel cell(DEFC) applications. ECS Trans 2006;1:247.
[25] Lopes T, Antolini E, Gonzalez ER. Carbon supported Pt–Co(3:1) alloy as improved cathode electrocatalyst for directethanol fuel cells. J Power Sources 2007;164:111.
[26] Li H, Sun G, Li N, Sun S, Su D, Xin Q. Design and preparationof highly active Pt–Pd/C catalyst for the oxygen reductionreaction. J Phys Chem C 2007;111:5605.
[27] Ye H, Crooks RM. Effect of elemental composition of PtPdbimetallic nanoparticles containing an average of 180 atomson the kinetics of the electrochemical oxygen reductionreaction. J Am Chem Soc 2007;129:3627.
[28] Xu Y, Lin X. Facile fabrication and electrocatalytic activity ofPt0.9Pd0.1 alloy film catalysts. J Power Sources 2007;170:13.
[29] Colmati F, Antolini E, Gonzalez ER. Ethanol oxidation oncarbon supported Pt–Sn electrocatalysts prepared byreduction with formic acid. J Electrochem Soc 2007;154:B39.
[30] Lopes T, Antolini E, Gonzalez ER. Nanostructured PtPd alloysas cathode catalysts for direct ethanol fuel cells. In: 211thECS Meeting, Chicago, Illinois; May 6–10 2007.
[31] Mascarenhas YP, Pinheiro JMV. Programa para Calculo deParametro de Rede pelo Metodo de Minimos Quadrados.SBPC; 1985.
[32] Mukerjee S, Srinivasan S, Soriaga MP, McBreen J. role ofstructural and electronic properties of Pt and Pt alloys onelectrocatalysis of oxygen reduction. J Electrochem Soc 1995;142:1409.
[33] Antolini E, Salgado JRC, Giz MJ, Gonzalez ER. Effects ofgeometric and electronic factors on ORR activity of carbonsupported Pt–Co electrocatalysts in PEM fuel cells. Int JHydrogen Energy 2005;30:1213.
[34] Markovic NM, Ross PN. Surface science studies of model fuelcell electrocatalysts. Surf Sci Rep 2002;45:121.
[35] Li HQ, Xin Q, Li WZ, Zhou ZZ, Jiang LH, Yang SH, et al. Animproved palladium-based DMFCs cathode catalyst. ChemCommun 2004:2776.
[36] Creus AH, Gimeno Y, Diaz P, Vazquez L, Gonzalez S,Salvarezza RC, et al. Influence of the nanostructure ofpalladium mesoparticles on the kinetics of molecular oxygenelectroreduction. J Phys Chem B 2004;108:10785.
[37] Lamy C, Lima A, Le Rhun V, Coutanceau C, Leger J-M. Recentadvances in the development of direct alcohol fuel cells(DAFC). J Power Sources 2002;105:283.
[38] Gasteiger HA, Markovic NM, Ross PN, Cairns EJ. Electro-oxidation of small organic molecules on well-characterizedPt–Ru alloys. Electrochim Acta 1994;39:1825.