calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo
TRANSCRIPT

Calcineurin-Mediated Hypertrophy Protects CardiomyocytesFrom Apoptosis In Vitro and In Vivo
An Apoptosis-Independent Model of Dilated Heart Failure
Leon J. De Windt, Hae W. Lim, Tyler Taigen, Detlef Wencker, Gianluigi Condorelli,Gerald W. Dorn II, Richard N. Kitsis, Jeffery D. Molkentin
Abstract—We have previously shown that the calcium-calmodulin–regulated phosphatase calcineurin (PP2B) is sufficientto induce cardiac hypertrophy that transitions to heart failure in transgenic mice. Given the rapid onset of heart failurein these mice, we hypothesized that calcineurin signaling would stimulate myocardial cell apoptosis. However, utilizingmultiple approaches, we determined that calcineurin-mediated hypertrophy protected cardiac myocytes from apoptosis,suggesting a model of heart failure that is independent of apoptosis. Adenovirally mediated gene transfer of aconstitutively active calcineurin cDNA (AdCnA) was performed in cultured neonatal rat cardiomyocytes to elucidate themechanism whereby calcineurin affected myocardial cell viability. AdCnA infection, which induced myocytehypertrophy and atrial natriuretic factor expression, protected against apoptosis induced by 2-deoxyglucose orstaurosporine, as assessed by terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL)labeling, caspase-3 activation, DNA laddering, and cellular morphology. The level of protection conferred by AdCnAwas similar to that of adenoviral Bcl-xL gene transfer or hypertrophy induced by phenylephrine. In vivo, failing heartsfrom calcineurin-transgenic mice did not demonstrate increased TUNEL labeling and, in fact, demonstrated a resistanceto ischemia/reperfusion–induced apoptosis. We determined that the mechanism whereby calcineurin afforded protectionfrom apoptosis was partially mediated by nuclear factor of activated T cells (NFAT3) signaling and partially byAkt/protein kinase B (PKB) signaling. Although calcineurin activation protected myocytes from apoptosis, inhibition ofcalcineurin with cyclosporine was not sufficient to induce TUNEL labeling in Gqa-transgenic mice or in culturedcardiomyocytes. Collectively, these data identify a calcineurin-dependent mouse model of dilated heart failure that isindependent of apoptosis.(Circ Res. 2000;86:255-263.)
Key Words: calcineurinn apoptosisn cardiac hypertrophyn phenylephrinen caspase-3
Heart failure can result from a wide range of pathologicalconditions such as infarction, chronic left ventricular
hypertrophy, ischemic cardiomyopathy, idiopathic dilatedcardiomyopathy, hypertrophic cardiomyopathy, valvularheart disease, and viral infection. In response to thesepathologic conditions, cardiomyocytes may undergo apopto-sis, or programmed cell death, which has been postulated tocontribute to the progressive pathology associated with heartfailure.1–4 Apoptosis is an energy-requiring process thatresults in cellular shrinkage, nuclear condensation, and DNAfragmentation in response to developmental cues or to acti-vation/inhibition of specific reactive signaling cascades (re-viewed in References 5 and 6).
Increased hypertrophic signaling via either of the hetero-trimeric G proteins, Gqa or Gsa, in transgenic mouse heartswas shown to promote cardiomyocyte apoptosis and cardio-
myopathy, suggesting that overstimulation of G protein–coupled receptors might promote cell death.7,8 Consistentwith this interpretation, pharmacological stimulation ofb-adrenergic receptors in vivo caused apoptosis independentof left ventricular hypertrophy or tachycardia.9 In vitrostudies in cultured primary cardiomyocytes also implicatecertain signaling pathways in apoptosis. Stretch-mediatedrelease of angiotensin II is associated with p53 activation andcardiomyocyte apoptosis.10,11Similarly, atrial natriuretic fac-tor (ANF) and tumor necrosis factor-a likely promote car-diomyocyte apoptosis.12,13 Mitogen-activated protein kinase(MAPK) activation is also thought to be a regulator ofapoptosis. Specifically, activation of p38a may promotecardiomyocyte apoptosis.14,15
In contrast, numerous studies have demonstrated protectionfrom apoptosis through activation of certain intracellular
Received July 6, 1999; accepted November 17, 1999.From the Departments of Pediatrics (L.J.D.W., H.W.L., T.T., J.D.M.) and Cardiology (G.W.D.), University of Cincinnati, and Division of Molecular
Cardiovascular Biology (L.J.D.W., H.W.L., T.T., J.D.M.), Children’s Hospital Medical Center, Cincinnati, Ohio; Departments of Medicine and CellBiology (D.W., R.N.K.), Albert Einstein College of Medicine, The Bronx, NY; and Kimmel Cancer Center and Department of Microbiology andImmunology (G.C.), Thomas Jefferson University, Philadelphia, Pa.
Correspondence to Jeffery D. Molkentin, Department of Pediatrics, Division of Molecular Cardiovascular Biology, Children’s Hospital Medical Center,3333 Burnet Ave, Cincinnati, OH 45229-3039. E-mail [email protected]
© 2000 American Heart Association, Inc.
Circulation Researchis available at http://www.circresaha.org
255
Integrative Physiology
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from

signaling pathways. Cardiotrophin-1 signaling through thegp130 receptor was shown to protect cardiomyocytes fromapoptosis.16 Induction of cardiac hypertrophy through MKK6and P38b has also been associated with protection fromapoptosis.14,15 Insulin-like growth factor-1 (IGF-1) stimula-tion protects cardiomyocytes from apoptosis through phos-phatidylinositol 3-kinase and MAPK-dependent signalingpathways.17–19 Hypertrophic agonists such as phenylephrine(PE) and endothelin-1 are also associated with protectionfrom apoptosis, suggesting that hypertrophy itself may conferprotection.20
We recently described a novel mediator of cardiac hyper-trophy through the calcium-calmodulin–regulated intracellu-lar phosphatase calcineurin (PP2B) and the transcriptionalregulator nuclear factor of activated T cells (NFAT3).21
Expression of a constitutively active form of calcineurin intransgenic mouse hearts resulted in profound cardiac hyper-trophy that progressed to dilated heart failure within 8 to 12weeks.21 It was of interest to determine whether hypertrophicsignaling mediated by calcineurin induced apoptosis, whichmight explain the rapid progression to heart failure in thesemice. However, we determined that cardiomyocyte terminaldeoxynucleotidyltransferase–mediated dUTP nick end label-ing (TUNEL) in calcineurin-transgenic hearts is not signifi-cantly different from that in wild-type hearts. In fact,calcineurin-transgenic hearts were significantly less suscep-tible to ischemia/reperfusion–induced apoptosis than werewild-type hearts. To define the mechanism whereby cal-cineurin signaling might protect cardiac myocytes fromapoptosis, we generated a calcineurin adenovirus (AdCnA) toinfect cultured rat neonatal cardiomyocytes. AdCnA infectioninduced a hypertrophic response that was protective againststaurosporine- and 2-deoxyglucose–induced apoptosis. Thelikely mechanism whereby calcineurin protects myocytesfrom apoptosis is associated with NFAT3 and Akt/proteinkinase B (PKB) activation.
Materials and MethodsThe mouse model of ischemia/reperfusion by ligation of the leftdescending coronary artery was performed as described previously.22
Primary cultures of neonatal rat cardiomyocytes were obtained asdescribed previously.23 E1a-deleted, replication-deficient adenovirusexpressing a constitutively activated form of mouse calcineurin Aaamino acids 1 to 398 (AdCnA), full-length human Bcl-xL (AdBcl-xL), or a constitutively nuclear form of NFAT3 (AdNFAT3) wasinitially constructed in pACCMV-pLpA and cotransfected intoHEK293 cells with pJM17 as described before.24 For the generationof the adenovirus expressing a calcineurin-inhibitory peptide (Ad-cain), a 582-bp fragment corresponding to amino acids 1989 to 2182of cain25,26 was generated by polymerase chain reaction and sub-cloned with a flag epitope as aHindIII fragment into pACCMV-pLpA. All initial recombinants were plaque-purified, expanded, andtitered by duplicate plaque assays in monolayers of HEK293 usingthe agarose gel overlay method.27 Adenoviral infection of cardio-myocytes was performed at a multiplicity of infection of 100plaque-forming units in 2 mL (6-cm culture dishes) of DMEMsupplemented with 2% FBS for 2 hours at 37°C in a humidified, 5%CO2 incubator. Under these infection conditions,'99% of the cellswere positive for protein expression by immunocytochemistry orstainedb-galactosidase positive after 24 hours. TUNEL labeling ofcultured cardiomyocytes or tissue sections was performed with theCardioTACS kit (Trevigen) as recommended by the manufacturer.Cardiomyocytes were prepared for immunocytochemistry as de-
scribed previously.21 Data are expressed as mean6SEM, and differ-ences were evaluated for significance using the Studentt test forunpaired data or 1-way ANOVA followed by the Bonferroni post testwhen appropriate.
An expanded Materials and Methods section is available online athttp://www.circresaha.org.
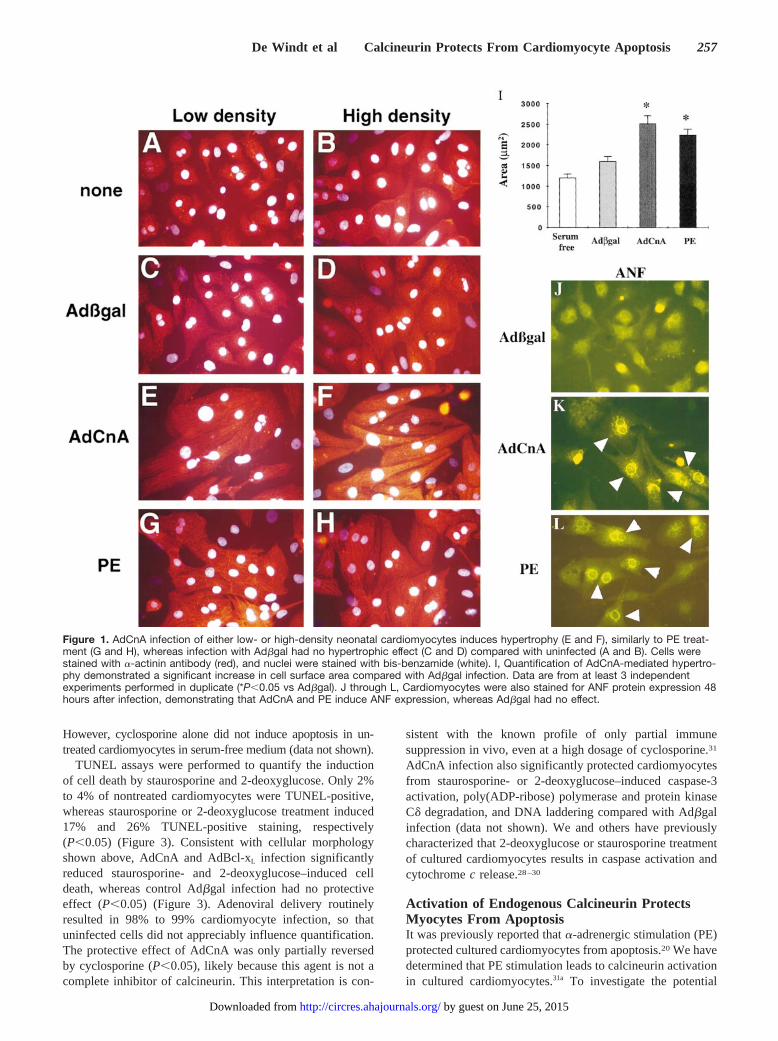
ResultsCalcineurin Adenovirus Drives NeonatalCardiomyocyte Hypertrophy In VitroTo investigate the importance of calcineurin signaling in vitro,we generated a replication-deficient adenovirus expressing theconstitutively active form of mouse calcineurin Aa (AdCnA)(amino acids 1 to 398) to perform gene transfer in culturedcardiac myocytes. Consistent with the effect of activated cal-cineurin expression in vivo, AdCnA induced morphologicalhypertrophy, whereas control infection with ab-galactosidaseadenovirus (Adbgal) had no effect (Figure 1A through 1H). Thehypertrophic response was independent of cellular density andproduced a quantitative increase in cell surface area 96 hoursafter infection (Figure 1I). AdCnA-infected cardiomyocytes alsodemonstrated greater sarcomeric organization (Figure 1E and1F), rhythmic beating 24 hours after infection, and ANF proteinexpression (Figure 1K). Quantification of ANF expression in arepresentative experiment revealed that among cardiomyocytes,9.2% (62.4%) of those infected with Adbgal, 39% (62.5%) ofthose infected with AdCnA, and 54% (62.7%) of those treatedwith PE were positive (P,0.05). These data indicate thatexpression of activated calcineurin is sufficient to drive thehypertrophic program in cultured neonatal cardiomyocytes.
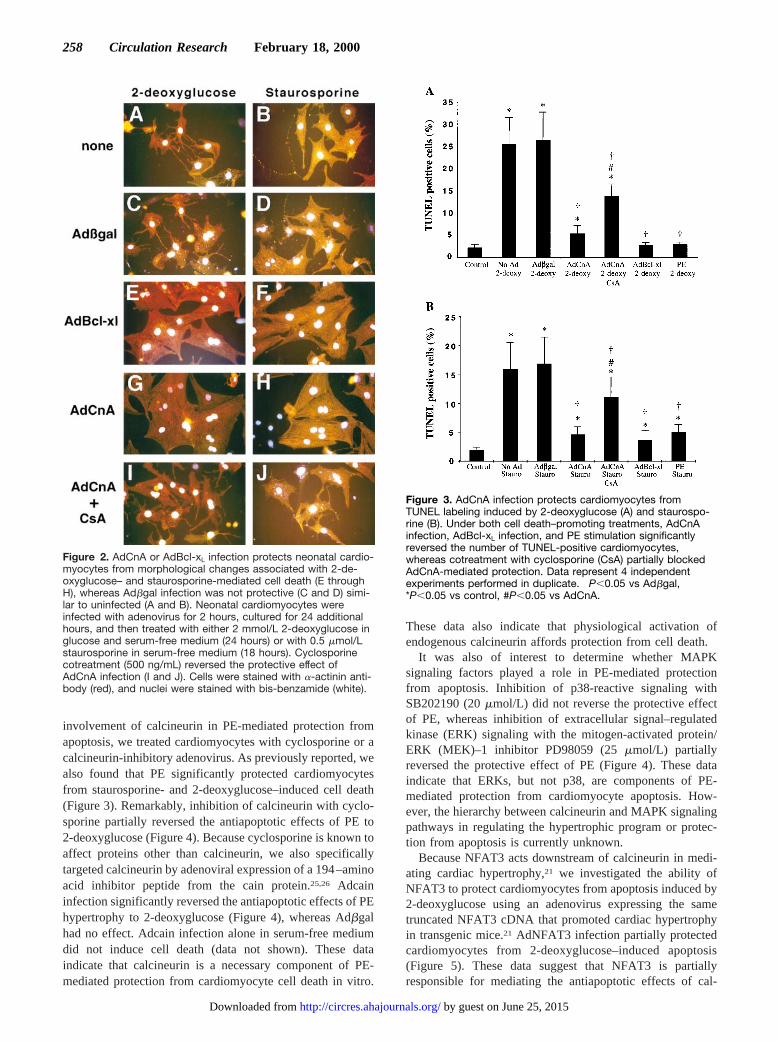
Calcineurin Adenoviral Infection ProtectsCardiomyocytes From ApoptosisActivation of certain intracellular signaling pathways in cardio-myocytes has been shown to profoundly affect myocardial cellviability. It was of interest to determine whether constitutivecalcineurin activation would promote or protect cardiomyocytesfrom cell death. To this end, we determined that AdCnAinfection of cultured neonatal cardiomyocytes did not induceapoptosis over any time period compared with Adbgal infectionor uninfected cells (data not shown). In contrast, AdCnAinfection was found to protect cultured cardiomyocytes fromapoptosis induced by 2 different pharmacological agents. Cellswere cultured for 24 hours after adenoviral infection and treatedwith either staurosporine (1.0mmol/L) or 2-deoxyglucose(2 mmol/L) in glucose- and serum-free medium. Staurosporinewas previously shown to induce cardiomyocyte apoptosis in acaspase-dependent manner.28 Similarly, metabolic inhibitionwith 2-deoxyglucose was also shown to induce apoptosis incultured cardiomyocytes.29,30The data show that AdCnA infec-tion protected cardiomyocytes from morphological featuresassociated with cell death, whereas Adbgal infection had noprotective effect (Figure 2C, 2D, 2G, and 2H). As a positivecontrol for these assays, we generated an adenovirus expressingthe antiapoptotic gene Bcl-xL. AdBcl-xL infection also protectedcardiomyocytes from both staurosporine- and 2-deoxyglucose–mediated effects (Figure 2E and 2F). As a further control, thecalcineurin-inhibitory drug cyclosporine was added to AdCnA-infected cardiomyocytes, resulting in a reversal of protection.
256 Circulation Research February 18, 2000
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from
However, cyclosporine alone did not induce apoptosis in un-treated cardiomyocytes in serum-free medium (data not shown).
TUNEL assays were performed to quantify the inductionof cell death by staurosporine and 2-deoxyglucose. Only 2%to 4% of nontreated cardiomyocytes were TUNEL-positive,whereas staurosporine or 2-deoxyglucose treatment induced17% and 26% TUNEL-positive staining, respectively(P,0.05) (Figure 3). Consistent with cellular morphologyshown above, AdCnA and AdBcl-xL infection significantlyreduced staurosporine- and 2-deoxyglucose–induced celldeath, whereas control Adbgal infection had no protectiveeffect (P,0.05) (Figure 3). Adenoviral delivery routinelyresulted in 98% to 99% cardiomyocyte infection, so thatuninfected cells did not appreciably influence quantification.The protective effect of AdCnA was only partially reversedby cyclosporine (P,0.05), likely because this agent is not acomplete inhibitor of calcineurin. This interpretation is con-
sistent with the known profile of only partial immunesuppression in vivo, even at a high dosage of cyclosporine.31
AdCnA infection also significantly protected cardiomyocytesfrom staurosporine- or 2-deoxyglucose–induced caspase-3activation, poly(ADP-ribose) polymerase and protein kinaseCd degradation, and DNA laddering compared with Adbgalinfection (data not shown). We and others have previouslycharacterized that 2-deoxyglucose or staurosporine treatmentof cultured cardiomyocytes results in caspase activation andcytochromec release.28–30
Activation of Endogenous Calcineurin ProtectsMyocytes From ApoptosisIt was previously reported thata-adrenergic stimulation (PE)protected cultured cardiomyocytes from apoptosis.20 We havedetermined that PE stimulation leads to calcineurin activationin cultured cardiomyocytes.31a To investigate the potential
Figure 1. AdCnA infection of either low- or high-density neonatal cardiomyocytes induces hypertrophy (E and F), similarly to PE treat-ment (G and H), whereas infection with Adbgal had no hypertrophic effect (C and D) compared with uninfected (A and B). Cells werestained with a-actinin antibody (red), and nuclei were stained with bis-benzamide (white). I, Quantification of AdCnA-mediated hypertro-phy demonstrated a significant increase in cell surface area compared with Adbgal infection. Data are from at least 3 independentexperiments performed in duplicate (*P,0.05 vs Adbgal). J through L, Cardiomyocytes were also stained for ANF protein expression 48hours after infection, demonstrating that AdCnA and PE induce ANF expression, whereas Adbgal had no effect.
De Windt et al Calcineurin Protects From Cardiomyocyte Apoptosis 257
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from
involvement of calcineurin in PE-mediated protection fromapoptosis, we treated cardiomyocytes with cyclosporine or acalcineurin-inhibitory adenovirus. As previously reported, wealso found that PE significantly protected cardiomyocytesfrom staurosporine- and 2-deoxyglucose–induced cell death(Figure 3). Remarkably, inhibition of calcineurin with cyclo-sporine partially reversed the antiapoptotic effects of PE to2-deoxyglucose (Figure 4). Because cyclosporine is known toaffect proteins other than calcineurin, we also specificallytargeted calcineurin by adenoviral expression of a 194–aminoacid inhibitor peptide from the cain protein.25,26 Adcaininfection significantly reversed the antiapoptotic effects of PEhypertrophy to 2-deoxyglucose (Figure 4), whereas Adbgalhad no effect. Adcain infection alone in serum-free mediumdid not induce cell death (data not shown). These dataindicate that calcineurin is a necessary component of PE-mediated protection from cardiomyocyte cell death in vitro.
These data also indicate that physiological activation ofendogenous calcineurin affords protection from cell death.
It was also of interest to determine whether MAPKsignaling factors played a role in PE-mediated protectionfrom apoptosis. Inhibition of p38-reactive signaling withSB202190 (20mmol/L) did not reverse the protective effectof PE, whereas inhibition of extracellular signal–regulatedkinase (ERK) signaling with the mitogen-activated protein/ERK (MEK)–1 inhibitor PD98059 (25mmol/L) partiallyreversed the protective effect of PE (Figure 4). These dataindicate that ERKs, but not p38, are components of PE-mediated protection from cardiomyocyte apoptosis. How-ever, the hierarchy between calcineurin and MAPK signalingpathways in regulating the hypertrophic program or protec-tion from apoptosis is currently unknown.
Because NFAT3 acts downstream of calcineurin in medi-ating cardiac hypertrophy,21 we investigated the ability ofNFAT3 to protect cardiomyocytes from apoptosis induced by2-deoxyglucose using an adenovirus expressing the sametruncated NFAT3 cDNA that promoted cardiac hypertrophyin transgenic mice.21 AdNFAT3 infection partially protectedcardiomyocytes from 2-deoxyglucose–induced apoptosis(Figure 5). These data suggest that NFAT3 is partiallyresponsible for mediating the antiapoptotic effects of cal-
Figure 2. AdCnA or AdBcl-xL infection protects neonatal cardio-myocytes from morphological changes associated with 2-de-oxyglucose– and staurosporine-mediated cell death (E throughH), whereas Adbgal infection was not protective (C and D) simi-lar to uninfected (A and B). Neonatal cardiomyocytes wereinfected with adenovirus for 2 hours, cultured for 24 additionalhours, and then treated with either 2 mmol/L 2-deoxyglucose inglucose and serum-free medium (24 hours) or with 0.5 mmol/Lstaurosporine in serum-free medium (18 hours). Cyclosporinecotreatment (500 ng/mL) reversed the protective effect ofAdCnA infection (I and J). Cells were stained with a-actinin anti-body (red), and nuclei were stained with bis-benzamide (white).
Figure 3. AdCnA infection protects cardiomyocytes fromTUNEL labeling induced by 2-deoxyglucose (A) and staurospo-rine (B). Under both cell death–promoting treatments, AdCnAinfection, AdBcl-xL infection, and PE stimulation significantlyreversed the number of TUNEL-positive cardiomyocytes,whereas cotreatment with cyclosporine (CsA) partially blockedAdCnA-mediated protection. Data represent 4 independentexperiments performed in duplicate. †P,0.05 vs Adbgal,*P,0.05 vs control, #P,0.05 vs AdCnA.
258 Circulation Research February 18, 2000
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from
cineurin activation in cardiomyocytes. However, AdNFAT3infection was not as protective as AdCnA infection, suggest-ing that calcineurin provides protection by additionalmechanisms.
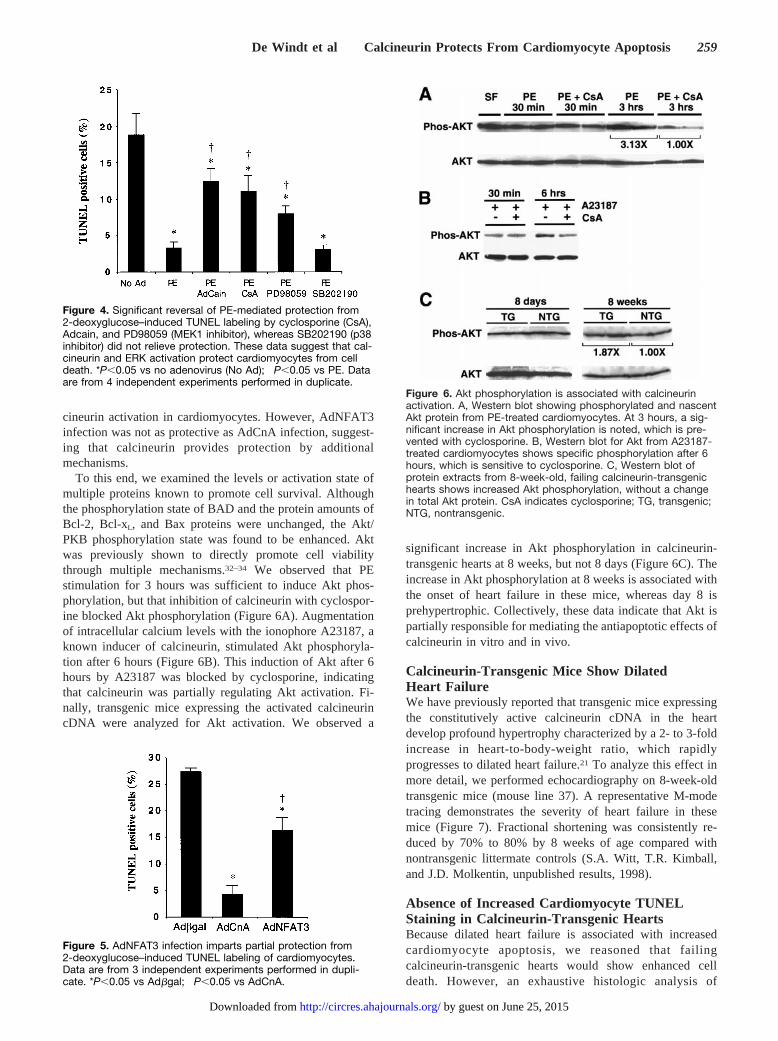
To this end, we examined the levels or activation state ofmultiple proteins known to promote cell survival. Althoughthe phosphorylation state of BAD and the protein amounts ofBcl-2, Bcl-xL, and Bax proteins were unchanged, the Akt/PKB phosphorylation state was found to be enhanced. Aktwas previously shown to directly promote cell viabilitythrough multiple mechanisms.32–34 We observed that PEstimulation for 3 hours was sufficient to induce Akt phos-phorylation, but that inhibition of calcineurin with cyclospor-ine blocked Akt phosphorylation (Figure 6A). Augmentationof intracellular calcium levels with the ionophore A23187, aknown inducer of calcineurin, stimulated Akt phosphoryla-tion after 6 hours (Figure 6B). This induction of Akt after 6hours by A23187 was blocked by cyclosporine, indicatingthat calcineurin was partially regulating Akt activation. Fi-nally, transgenic mice expressing the activated calcineurincDNA were analyzed for Akt activation. We observed a
significant increase in Akt phosphorylation in calcineurin-transgenic hearts at 8 weeks, but not 8 days (Figure 6C). Theincrease in Akt phosphorylation at 8 weeks is associated withthe onset of heart failure in these mice, whereas day 8 isprehypertrophic. Collectively, these data indicate that Akt ispartially responsible for mediating the antiapoptotic effects ofcalcineurin in vitro and in vivo.
Calcineurin-Transgenic Mice Show DilatedHeart FailureWe have previously reported that transgenic mice expressingthe constitutively active calcineurin cDNA in the heartdevelop profound hypertrophy characterized by a 2- to 3-foldincrease in heart-to-body-weight ratio, which rapidlyprogresses to dilated heart failure.21 To analyze this effect inmore detail, we performed echocardiography on 8-week-oldtransgenic mice (mouse line 37). A representative M-modetracing demonstrates the severity of heart failure in thesemice (Figure 7). Fractional shortening was consistently re-duced by 70% to 80% by 8 weeks of age compared withnontransgenic littermate controls (S.A. Witt, T.R. Kimball,and J.D. Molkentin, unpublished results, 1998).
Absence of Increased Cardiomyocyte TUNELStaining in Calcineurin-Transgenic HeartsBecause dilated heart failure is associated with increasedcardiomyocyte apoptosis, we reasoned that failingcalcineurin-transgenic hearts would show enhanced celldeath. However, an exhaustive histologic analysis of
Figure 4. Significant reversal of PE-mediated protection from2-deoxyglucose–induced TUNEL labeling by cyclosporine (CsA),Adcain, and PD98059 (MEK1 inhibitor), whereas SB202190 (p38inhibitor) did not relieve protection. These data suggest that cal-cineurin and ERK activation protect cardiomyocytes from celldeath. *P,0.05 vs no adenovirus (No Ad); †P,0.05 vs PE. Dataare from 4 independent experiments performed in duplicate.
Figure 5. AdNFAT3 infection imparts partial protection from2-deoxyglucose–induced TUNEL labeling of cardiomyocytes.Data are from 3 independent experiments performed in dupli-cate. *P,0.05 vs Adbgal; †P,0.05 vs AdCnA.
Figure 6. Akt phosphorylation is associated with calcineurinactivation. A, Western blot showing phosphorylated and nascentAkt protein from PE-treated cardiomyocytes. At 3 hours, a sig-nificant increase in Akt phosphorylation is noted, which is pre-vented with cyclosporine. B, Western blot for Akt from A23187-treated cardiomyocytes shows specific phosphorylation after 6hours, which is sensitive to cyclosporine. C, Western blot ofprotein extracts from 8-week-old, failing calcineurin-transgenichearts shows increased Akt phosphorylation, without a changein total Akt protein. CsA indicates cyclosporine; TG, transgenic;NTG, nontransgenic.
De Windt et al Calcineurin Protects From Cardiomyocyte Apoptosis 259
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from
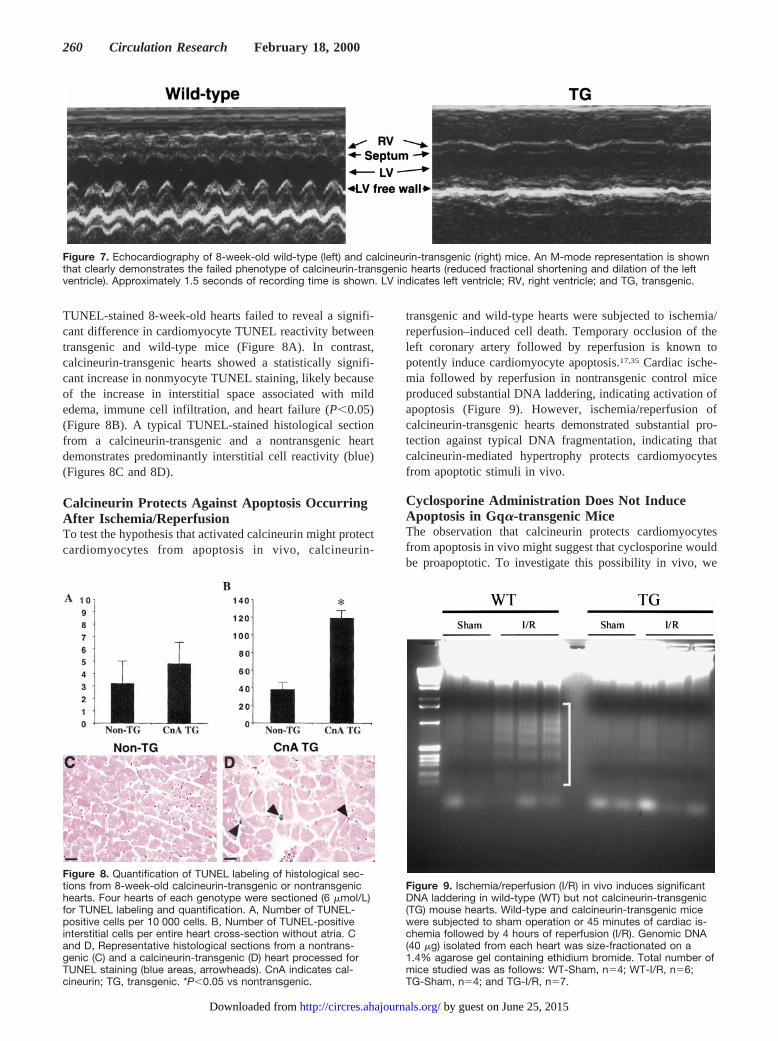
TUNEL-stained 8-week-old hearts failed to reveal a signifi-cant difference in cardiomyocyte TUNEL reactivity betweentransgenic and wild-type mice (Figure 8A). In contrast,calcineurin-transgenic hearts showed a statistically signifi-cant increase in nonmyocyte TUNEL staining, likely becauseof the increase in interstitial space associated with mildedema, immune cell infiltration, and heart failure (P,0.05)(Figure 8B). A typical TUNEL-stained histological sectionfrom a calcineurin-transgenic and a nontransgenic heartdemonstrates predominantly interstitial cell reactivity (blue)(Figures 8C and 8D).
Calcineurin Protects Against Apoptosis OccurringAfter Ischemia/ReperfusionTo test the hypothesis that activated calcineurin might protectcardiomyocytes from apoptosis in vivo, calcineurin-
transgenic and wild-type hearts were subjected to ischemia/reperfusion–induced cell death. Temporary occlusion of theleft coronary artery followed by reperfusion is known topotently induce cardiomyocyte apoptosis.17,35 Cardiac ische-mia followed by reperfusion in nontransgenic control miceproduced substantial DNA laddering, indicating activation ofapoptosis (Figure 9). However, ischemia/reperfusion ofcalcineurin-transgenic hearts demonstrated substantial pro-tection against typical DNA fragmentation, indicating thatcalcineurin-mediated hypertrophy protects cardiomyocytesfrom apoptotic stimuli in vivo.
Cyclosporine Administration Does Not InduceApoptosis in Gqa-transgenic MiceThe observation that calcineurin protects cardiomyocytesfrom apoptosis in vivo might suggest that cyclosporine wouldbe proapoptotic. To investigate this possibility in vivo, we
Figure 7. Echocardiography of 8-week-old wild-type (left) and calcineurin-transgenic (right) mice. An M-mode representation is shownthat clearly demonstrates the failed phenotype of calcineurin-transgenic hearts (reduced fractional shortening and dilation of the leftventricle). Approximately 1.5 seconds of recording time is shown. LV indicates left ventricle; RV, right ventricle; and TG, transgenic.
Figure 8. Quantification of TUNEL labeling of histological sec-tions from 8-week-old calcineurin-transgenic or nontransgenichearts. Four hearts of each genotype were sectioned (6 mmol/L)for TUNEL labeling and quantification. A, Number of TUNEL-positive cells per 10 000 cells. B, Number of TUNEL-positiveinterstitial cells per entire heart cross-section without atria. Cand D, Representative histological sections from a nontrans-genic (C) and a calcineurin-transgenic (D) heart processed forTUNEL staining (blue areas, arrowheads). CnA indicates cal-cineurin; TG, transgenic. *P,0.05 vs nontransgenic.
Figure 9. Ischemia/reperfusion (I/R) in vivo induces significantDNA laddering in wild-type (WT) but not calcineurin-transgenic(TG) mouse hearts. Wild-type and calcineurin-transgenic micewere subjected to sham operation or 45 minutes of cardiac is-chemia followed by 4 hours of reperfusion (I/R). Genomic DNA(40 mg) isolated from each heart was size-fractionated on a1.4% agarose gel containing ethidium bromide. Total number ofmice studied was as follows: WT-Sham, n54; WT-I/R, n56;TG-Sham, n54; and TG-I/R, n57.
260 Circulation Research February 18, 2000
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from
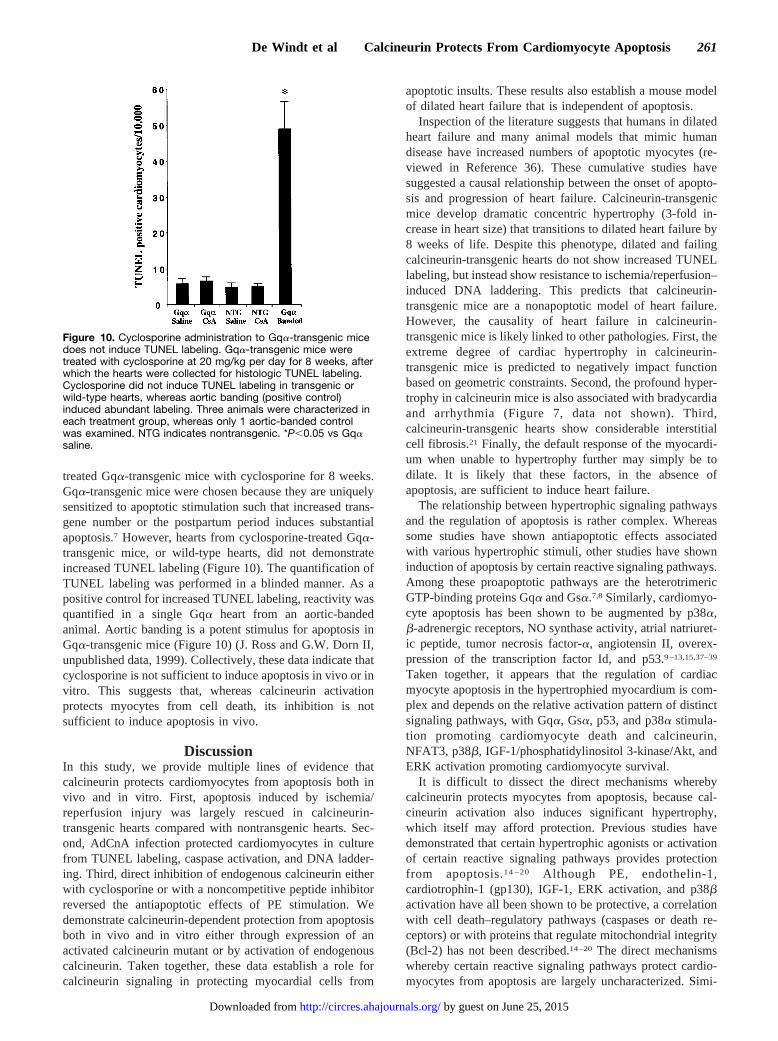
treated Gqa-transgenic mice with cyclosporine for 8 weeks.Gqa-transgenic mice were chosen because they are uniquelysensitized to apoptotic stimulation such that increased trans-gene number or the postpartum period induces substantialapoptosis.7 However, hearts from cyclosporine-treated Gqa-transgenic mice, or wild-type hearts, did not demonstrateincreased TUNEL labeling (Figure 10). The quantification ofTUNEL labeling was performed in a blinded manner. As apositive control for increased TUNEL labeling, reactivity wasquantified in a single Gqa heart from an aortic-bandedanimal. Aortic banding is a potent stimulus for apoptosis inGqa-transgenic mice (Figure 10) (J. Ross and G.W. Dorn II,unpublished data, 1999). Collectively, these data indicate thatcyclosporine is not sufficient to induce apoptosis in vivo or invitro. This suggests that, whereas calcineurin activationprotects myocytes from cell death, its inhibition is notsufficient to induce apoptosis in vivo.
DiscussionIn this study, we provide multiple lines of evidence thatcalcineurin protects cardiomyocytes from apoptosis both invivo and in vitro. First, apoptosis induced by ischemia/reperfusion injury was largely rescued in calcineurin-transgenic hearts compared with nontransgenic hearts. Sec-ond, AdCnA infection protected cardiomyocytes in culturefrom TUNEL labeling, caspase activation, and DNA ladder-ing. Third, direct inhibition of endogenous calcineurin eitherwith cyclosporine or with a noncompetitive peptide inhibitorreversed the antiapoptotic effects of PE stimulation. Wedemonstrate calcineurin-dependent protection from apoptosisboth in vivo and in vitro either through expression of anactivated calcineurin mutant or by activation of endogenouscalcineurin. Taken together, these data establish a role forcalcineurin signaling in protecting myocardial cells from
apoptotic insults. These results also establish a mouse modelof dilated heart failure that is independent of apoptosis.
Inspection of the literature suggests that humans in dilatedheart failure and many animal models that mimic humandisease have increased numbers of apoptotic myocytes (re-viewed in Reference 36). These cumulative studies havesuggested a causal relationship between the onset of apopto-sis and progression of heart failure. Calcineurin-transgenicmice develop dramatic concentric hypertrophy (3-fold in-crease in heart size) that transitions to dilated heart failure by8 weeks of life. Despite this phenotype, dilated and failingcalcineurin-transgenic hearts do not show increased TUNELlabeling, but instead show resistance to ischemia/reperfusion–induced DNA laddering. This predicts that calcineurin-transgenic mice are a nonapoptotic model of heart failure.However, the causality of heart failure in calcineurin-transgenic mice is likely linked to other pathologies. First, theextreme degree of cardiac hypertrophy in calcineurin-transgenic mice is predicted to negatively impact functionbased on geometric constraints. Second, the profound hyper-trophy in calcineurin mice is also associated with bradycardiaand arrhythmia (Figure 7, data not shown). Third,calcineurin-transgenic hearts show considerable interstitialcell fibrosis.21 Finally, the default response of the myocardi-um when unable to hypertrophy further may simply be todilate. It is likely that these factors, in the absence ofapoptosis, are sufficient to induce heart failure.
The relationship between hypertrophic signaling pathwaysand the regulation of apoptosis is rather complex. Whereassome studies have shown antiapoptotic effects associatedwith various hypertrophic stimuli, other studies have showninduction of apoptosis by certain reactive signaling pathways.Among these proapoptotic pathways are the heterotrimericGTP-binding proteins Gqa and Gsa.7,8 Similarly, cardiomyo-cyte apoptosis has been shown to be augmented by p38a,b-adrenergic receptors, NO synthase activity, atrial natriuret-ic peptide, tumor necrosis factor-a, angiotensin II, overex-pression of the transcription factor Id, and p53.9–13,15,37–39
Taken together, it appears that the regulation of cardiacmyocyte apoptosis in the hypertrophied myocardium is com-plex and depends on the relative activation pattern of distinctsignaling pathways, with Gqa, Gsa, p53, and p38a stimula-tion promoting cardiomyocyte death and calcineurin,NFAT3, p38b, IGF-1/phosphatidylinositol 3-kinase/Akt, andERK activation promoting cardiomyocyte survival.
It is difficult to dissect the direct mechanisms wherebycalcineurin protects myocytes from apoptosis, because cal-cineurin activation also induces significant hypertrophy,which itself may afford protection. Previous studies havedemonstrated that certain hypertrophic agonists or activationof certain reactive signaling pathways provides protectionfrom apoptosis.14 –20 Although PE, endothelin-1,cardiotrophin-1 (gp130), IGF-1, ERK activation, and p38bactivation have all been shown to be protective, a correlationwith cell death–regulatory pathways (caspases or death re-ceptors) or with proteins that regulate mitochondrial integrity(Bcl-2) has not been described.14–20 The direct mechanismswhereby certain reactive signaling pathways protect cardio-myocytes from apoptosis are largely uncharacterized. Simi-
Figure 10. Cyclosporine administration to Gqa-transgenic micedoes not induce TUNEL labeling. Gqa-transgenic mice weretreated with cyclosporine at 20 mg/kg per day for 8 weeks, afterwhich the hearts were collected for histologic TUNEL labeling.Cyclosporine did not induce TUNEL labeling in transgenic orwild-type hearts, whereas aortic banding (positive control)induced abundant labeling. Three animals were characterized ineach treatment group, whereas only 1 aortic-banded controlwas examined. NTG indicates nontransgenic. *P,0.05 vs Gqasaline.
De Windt et al Calcineurin Protects From Cardiomyocyte Apoptosis 261
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from

larly, calcineurin-transgenic hearts and AdCnA-infected car-diomyocytes did not demonstrate a perturbation in the Bcl-2–to–Bax ratio or the Bcl-xL–to–Bax ratio. However, onemechanism whereby calcineurin signaling directly protectscardiac myocytes is through activation of PKB/Akt. Akt isknown to downregulate caspase-9 activation, phosphorylateBAD, and directly activate nuclear factorkB to promote cellsurvival.32–34 Although we have demonstrated that Akt acti-vation is associated with calcineurin-induced hypertrophy, itis not clear how this activation occurs. However, it is likelythat calcineurin signaling first activates the hypertrophicprogram, which then indirectly leads to Akt activation,providing protection from cell death. NFAT3 adenoviral genetransfer also provided partial protection from 2-deoxyglu-cose–induced TUNEL labeling, suggesting an additionalmechanism whereby calcineurin provides protection. How-ever, the direct mechanism whereby NFAT3 provides protec-tion from cell death is unknown, but it can be speculated thatprotection is linked to the hypertrophic response itself.
The observation that calcineurin protects cardiomyocytesfrom apoptosis might also suggest that cyclosporine treatmentcould promote myocardial cell apoptosis in vivo. However,patients on chronic cyclosporine therapy have not beenreported to be at increased risk for heart failure, although anincreased propensity toward hypertrophy has been de-scribed.40 We directly addressed this issue experimentally bytreating a mouse model that is prone to apoptosis (Gqa) withcyclosporine at 20 mg/kg per day for 8 weeks.7 Character-ization of TUNEL labeling did not reveal an induction of celldeath in wild-type or Gqa-transgenic mice with cyclosporine,yet aortic banding induced a profound increase (Figure 10, J.Ross and G.W. Dorn, unpublished data, 1999). In other celltypes, cyclosporine has been shown to inhibit apoptosis bypreventing mitochondrial pore transition, which is regulatedby cyclophilin-D (reviewed in Reference 41). These datasuggests that cyclosporine has diverse intracellular effects,some of which protect cells and others of which may beproapoptotic. Indeed, calcineurin was recently shown toeither protect cells from apoptosis or to directly induceapoptosis depending on cross talk between other intracellularsignaling pathways.42
Calcineurin has been shown to promote viability in multi-ple cell types by suppressing apoptosis,43–46 whereas otherstudies have shown that calcineurin can activate apoptosis indisparate cell types.47–50These differing accounts suggest thatthe role of calcineurin in controlling cell viability is complexand integrated with other signaling pathways. In cardiomyo-cytes, it is likely that calcineurin functions in concert withother reactive signaling pathways to achieve a balancedactivation of cellular hypertrophy, which is beneficial tocellular viability.
AcknowledgmentsThis work was supported by NIH Grants HL-69562 and HL-62927,a Scholar Award from the Pew Foundation, a local American HeartAssociation affiliate grant-in-aid (to J.D.M.), and NIH GrantsHL60665 and HL61550 (to R.N.K.). H.W.L. was supported by NIHTraining Grant T32 ES07051. We thank the veterinary staff atChildren’s Hospital Medical Center for excellenttechnical assistance.
References1. Itoh G, Tamura J, Suzuki M, Suzuki Y, Ikeda H, Koike M, Nomura M,
Jie T, Ito K. DNA fragmentation of human infarcted myocardial cellsdemonstrated by the nick end labeling method and DNA agarose gelelectrophoresis.Am J Pathol. 1995;146:1325–1331.
2. Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen KM, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction.Circulation.1997;95:320–323.
3. Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ,Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes inend-stage heart failure.N Engl J Med. 1996;335:1182–1189.
4. Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, QuainiE, Di Loreto C, Beltrami CA, Krajewski S, Reed JC, Anversa P. Apo-ptosis in the failing human heart.N Engl J Med. 1997;336:1131–1141.
5. MacLellan WR, Schneider MD. Death by design: programmed cell deathin cardiovascular biology and disease.Circ Res. 1997;81:137–144.
6. Haunstetter A, Izumo S. Apoptosis: basic mechanisms and implicationsfor cardiovascular disease.Circ Res. 1998;82:1111–1129.
7. Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, ChienKR, Brown JH, Dorn GW II. Enhanced Gaq signaling: a commonpathway mediates cardiac hypertrophy and apoptotic heart failure.ProcNatl Acad Sci U S A.1998;95:10140–10145.
8. Geng YJ, Ishikawa Y, Vatner DE, Wagner TE, Bishop SP, Vatner SF,Homcy CJ. Apoptosis of cardiac myocytes in Gsa transgenic mice.CircRes. 1999;84:34–42.
9. Shizukida Y, Buttrick PM, Geenen DL, Borczuk AC, Kitsis RN,Sonnenblick EH.b-Adrenergic stimulation causes cardiocyte apoptosis:influence of tachycardia and hypertrophy.Am J Physiol. 1998;275:H961–H968.
10. Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, Malhotra A,Kajstura J, Anversa P. Stretch-mediated release of angiotensin II inducesmyocytes apoptosis by activating p53 that enhances the local renin-an-giotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell.J Clin Invest. 1998;101:1326–1342.
11. Leri A, Liu Y, Claudio PP, Kajstura J, Wang X, Wang S, Kang P,Malhotra A, Anversa P. Insulin-like growth factor-1 induces Mdm2 anddown-regulates p53, attenuating the myocyte renin-angiotensin systemand stretch-mediated apoptosis.Am J Pathol. 1999;154:567–580.
12. Wu CF, Bishopric NH, Pratt RE. Atrial natriuretic peptide induces apo-ptosis in neonatal rat cardiac myocytes.J Biol Chem. 1997;272:14860–14866.
13. Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, KoretskyAP, Demetris AJ, Feldman AM. Dilated cardiomyopathy in transgenicmice with cardiac-specific overexpression of tumor necrosis factor-alpha.Circ Res. 1997;81:627–635.
14. Zechner D, Craig R, Hanford DS, McDonough PM, Sabbadini RA,Glembotski CC. MKK6 activates myocardial cell NF-kB and inhibitsapoptosis in a p38 mitogen-activated protein kinase-dependent manner.J Biol Chem. 1998;273:8232–8239.
15. Wang Y, Huang S, Sah VP, Ross J Jr, Heller Brown J, Han J, Chien KR.Cardiac muscle cell hypertrophy and apoptosis induced by distinctmembers of the p38 mitogen-activated protein kinase family.J BiolChem. 1998;273:2161–2168.
16. Sheng Z, Knowlton K, Chen J, Hoshijima M, Brown JH, Chien KR.Cardiotrophin 1 (CT-1) inhibition of cardiac myocyte apoptosis via amitogen-activated protein kinase-dependent pathway: divergence fromdownstream CT-1 signals for myocardial cell hypertrophy.J Biol Chem.1997;272:5783–5791.
17. Li Q, Li B, Wang X, Leri A, Jana KP, Liu Y, Kajstura J, Baserga R,Anversa P. Overexpression of insulin-like growth factor-1 in miceprotects from myocyte death after infarction, attenuating ventricular dila-tation, wall stress, and cardiac hypertrophy.J Clin Invest. 1997;100:1991–1999.
18. Parrizas M, Saltiel AR, LeRoith D. Insulin-like growth factor 1 inhibitsapoptosis using the phosphatidylinositol 3-kinase and mitogen-activatedprotein kinase pathways.J Biol Chem. 1997;272:154–161.
19. Wang L, Ma W, Markovich R, Chen J-W, Wang PH. Regulation ofcardiomyocyte apoptotic signaling by insulin-like growth factor-1.CircRes. 1998;83:516–522.
20. Zhu H, Qi M, McElwee-Witmer SA, Merkel-Jordan L, Perrone MH,Clark KL, Zilberstein A. The relationship between hypertrophy andapoptosis in cultured neonatal ventricular cardiomyocytes.Circulation.1998;96(suppl I):I-347. Abstract.
262 Circulation Research February 18, 2000
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from

21. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J,Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway forcardiac hypertrophy.Cell. 1998;93:215–228.
22. Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, LordEM, Koch CJ, Kitsis RN. Myocyte apoptosis during acute myocardialinfarction in the mouse localizes to hypoxic regions but occurs indepen-dently of p53.J Clin Invest. 1997;100:1363–1372.
23. De Windt LJ, Willemsen PHM, Popping S, Van der Vusse GJ, RenemanRS, Van Bilsen M. Cloning and cellular distribution of a group IIphospholipase A2 expressed in the heart.J Mol Cell Cardiol. 1997;8:2095–2106.
24. Gomez-Foix AM, Coats WS, Baque S, Alam T, Gerard RD, NewgardCB. Adenovirus-mediated transfer of the muscle glycogen phosphorylasegene into hepatocytes confers altered regulation of glycogen metabolism.J Biol Chem. 1992;267:25129–25134.
25. Sun L, Youn HD., Loh C, Stolow M, He W, Liu JO. Cabin 1, a negativeregulator for calcineurin signaling in T lymphocytes.Immunity. 1998;8:703–711.
26. Lai MM, Burnett PE, Wolosker H, Blackshaw S, Snyder SH. Cain, anovel physiologic protein inhibitor of calcineurin.J Biol Chem. 1998;273:18325–18331.
27. Mittereder N, March KL, Trapnell BC. Evaluation of the concentrationand bioactivity of adenovirus vectors for gene therapy.J Virol. 1996;70:7498–7509.
28. Yue TL, Wang C, Romanic AM, Kikly K, Keller P, DeWolf WE Jr, HartTK, Thomas HC, Storer B, Gu JL, Wang X, Feuerstein GZ. Staurospo-rine-induced apoptosis in cardiomyocytes: a potential role of caspase-3.JMol Cell Cardiol. 1998;30:495–507.
29. Malhotra R, Brosius FC III. Glucose uptake and glycolysis reduce hyp-oxia-induced apoptosis in cultured neonatal rat cardiac myocytes.J BiolChem. 1999;274:12567–12575.
30. Bialik S, Cryns VL, Drincic A, Miyata S, Wollowick AL, Srinivasan A,Kitsis RN. The mitochondrial apoptotic pathway is activated by serumand glucose deprivation in cardiac myocytes.Circ Res. 1999;85:403–414.
31. Batiuk TD, Urmson J, Vincent D, Yatscoff RW, Halloran PF. Quanti-tating immunosuppression.Transplantation. 1996;61:1618–1624.
31a.Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition ofcalcineurin prevents agonist-induced cardiomyocyte hypertrophy.ProcNatl Acad Sci U S A.In press.
32. del Peso L, Gonzalez-Garcia M, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt.Science.1997;278:687–689.
33. Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, StanbridgeE, Frisch S, Reed JC. Regulation of cell death protease caspase-9 byphosphorylation.Science. 1998;282:1381–1321.
34. Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signaling.Nature. 1999;401:86–90.
35. Reiss K, Kajstura J, Zhang X, Li P, Olivetti G, Anversa P. Acutemyocardial infarction leads to upregulation of the IGF-1 autocrine
system, DNA replication, and nuclear mitotic division in the remainingviable cardiac myocytes.Exp Cell Res. 1994;213:463–472.
36. Haunstetter A, Izumo S. Apoptosis: basic mechanisms and implicationsfor cardiovascular disease.Circ Res. 1998;82:1111–1129.
37. Ing DJ, Dzau VJ, Webster KA, Bishopric NH. Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bax, and Bcl-x.CircRes. 1999;84:21–33.
38. Koglin J, Granville DJ, Glysing-Jensen T, Mudgett JS, Carthy CM,McManus BM, Russell ME. Attenuated acute cardiac rejection inNOS2–/– recipients correlates with reduced apoptosis.Circulation. 1999;99:836–842.
39. Bryant D, Becker L, Richardson J, Shelton J, Franco F, Peshock R,Thompson M, Giroir B. Cardiac failure in transgenic mice with myo-cardial expression of tumor necrosis factor-alpha.Circulation. 1998;97:1375–1381.
40. Ventura HO, Malik FS, Mehra MR, Stapelton DD, Smart FW. Mech-anisms of hypertension in cardiac transplantation and the role of cyclo-sporine.Curr Opin Cardiol. 1997;12:375–381.
41. Crompton M. The mitochondrial permeability pore and its role in celldeath.Biochem J. 1999;341:233–249.
42. Lotem J, Kama R, Sachs L. Suppression or induction of apoptosis byopposing pathways downstream from calcium-activated calcineurin.ProcNatl Acad Sci U S A.1999;96:12016–12020.
43. Asada A, Zhao Y, Kondo S, Iwata M. Induction of thymocyte apoptosisby Ca21-independent protein kinase C (nPKC) activation and its regu-lation by calcineurin activation.J Biol Chem. 1998;273:28392–28398.
44. Lotem J, Sachs L. Different mechanisms for suppression of apoptosis bycytokines and calcium mobilizing compounds.Proc Natl Acad Sci U S A.1998;95:4601–4606.
45. Davis PK, Dudek SM, Johnson GV. Select alterations in protein kinasesand phosphatases during apoptosis of differentiated PC12 cells.J Neu-rochem. 1997;68:2338–2347.
46. Zhao Y, Tozawa Y, Iseki R, Mukai M, Iwata M. Calcineurin activationprotects T cells from glucocorticoid-induced apoptosis.J Immunol. 1995;154:6346–6354.
47. Shibasaki F, McKeon F. Calcineurin functions in Ca(21)-activated celldeath in mammalian cells.J Cell Biol. 1995;131:735–743.
48. Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin andmitochondrial function in glutamate-induced neuronal cell death.FEBSLett. 1996;394:321–324.
49. Toth R, Szegezdi E, Molnar G, Lord JM, Fesus L, Szondy Z. Regulationof cell surface expression of Fas (CD95) ligand and susceptibility to Fas(CD95)-mediated apoptosis in activation-induced T cell death involvescalcineurin and protein kinase C, respectively.Eur J Immunol. 1999;29:383–393.
50. Denecker G, Vandenabeele P, Grooten J, Penning LC, Declercq W,Beyaert R, Buurman WA, Fiers W. Differential role of calcium in tumournecrosis factor-mediated apoptosis and secretion of granulocyte-macro-phage colony-stimulating factor in a T cell hybridoma.Cytokine. 1997;9:631–638.
De Windt et al Calcineurin Protects From Cardiomyocyte Apoptosis 263
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from

Dorn II, Richard N. Kitsis and Jeffery D. MolkentinLeon J. De Windt, Hae W. Lim, Tyler Taigen, Detlef Wencker, Gianluigi Condorelli, Gerald W.
and In Vivo: An Apoptosis-Independent Model of Dilated Heart FailureCalcineurin-Mediated Hypertrophy Protects Cardiomyocytes From Apoptosis In Vitro
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2000 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/01.RES.86.3.2552000;86:255-263Circ Res.
http://circres.ahajournals.org/content/86/3/255World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2000/02/15/86.3.255.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 25, 2015http://circres.ahajournals.org/Downloaded from