analytical biochemistry comparative analysis of inducible expression systems in transient...
TRANSCRIPT

ANALYTICALBIOCHEMISTRY
Analytical Biochemistry 334 (2004) 9–19
www.elsevier.com/locate/yabio
Comparative analysis of inducible expression systems in transient transfection studies
Mirella L. Meyer-Ficca1, Ralph G. Meyer¤,1, Heike Kaiser, Alexandra R. Brack, Reinhard Kandolf, Jan-Heiner Küpper2
Department of Molecular Pathology, University Hospital of Tübingen, Liebermeisterstr. 8, D-72076 Tübingen, Germany
Received 25 February 2004Available online 25 August 2004
Abstract
Ectopic protein expression in mammalian cells is a valuable tool to analyze protein functions. Increasingly, inducible promotersare being used for regulated gene expression. Here, we compare expression maxima, induction rates, and “leakiness” of the followingpromoter systems: (I) two tetracycline-responsive Tet systems (Tet-On, Tet-OV), (II) the glucocorticoid-responsive mouse mammarytumor virus promoter (MMTVprom), (III) the ecdysone-inducible promoter (EcP), and (IV) the T7 promoter/T7 RNA polymerasesystem (T7P). The systems were analyzed by expressing an enhanced green Xuorescent protein (EGFP) luciferase fusion reporter pro-tein in transiently transfected cells. Expression was assessed qualitatively by Xuorescence microscopy of the EGFP component andquantitatively by measuring the enzymatic activity of the luciferase component of the fusion protein. Basal expression levels (“leaki-ness”) were ranked Tet-On > Tet-OV > MMTVprom > EcP > T7P. Induction rates were EcP > MMTVprom > T7P > Tet-OV > Tet-On. Expression maxima were ranked. Tet-On > Tet-OV > MMTVprom > EcP > T7P. To increase T7-promoter-mediated expressionwe inserted an internal ribosomal entry site element into the T7 expression cassette. In presence of T7 RNA polymerase this modiWedT7 promoter achieved expression levels of 42% of a Rous Sarcoma virus promoter, while keeping basal expression extremely low. 2004 Elsevier Inc. All rights reserved.
Keywords: Inducible promoter systems; Transient transfection; Stable transfection; GFP; Luciferase; Cytotoxic proteins; Tetracycline; Glucocorti-coid receptor; Dexamethasone; Ecdysone; IRES
Expression of bacterial or viral proteins or overex-pression of cellular genes often poses problems related totoxic side eVects caused by the target proteins. Depend-ing on the cell type used for investigation of the(over)expressed protein, low transfection rates and sideeVects of the transfection method often limit the successof expression experiments. Stable transfection proce-dures using selectable markers are often used to over-
* Corresponding author. Fax: +520 626 5990/+1 520 626 8567.E-mail address: [email protected] (R.G. Meyer).
1 Present address: Department of Pharmacology and Toxicology,University of Arizona College of Pharmacy, Arizona Cancer Center,Tucson, AZ 85724, USA.
2 Present address: Heart BioSystems GmbH., Im Neuenheimer Feld583, D-69120 Heidelberg, Germany.
0003-2697/$ - see front matter 2004 Elsevier Inc. All rights reserved.doi:10.1016/j.ab.2004.07.011
come these problems. Classic approaches using stablytransfected constitutive promoters have been plagued bycounterselection problems and by the statistical neces-sity to investigate a number of individual clones to testthe proposed scientiWc hypothesis.
The available gene expression systems have thereforebeen complemented by a number of inducible promotersystems in the past 10 years. Researchers are at sometime challenged to make a decision with respect to whichof the available systems is the most suitable one for theirenvisaged investigations. Unfortunately, so far there islimited information available on systematic and compar-ative analyses of known inducible promoter systems.
In particular, the expression of toxic proteins oftenproves to be challenging. For example, the regulated

10 M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19
expression of toxic proteins is desirable in the context ofbasic research on cytotoxic or pro-apoptotic mecha-nisms of some bacterial [1] or viral proteins. If expres-sion of toxic genes is attempted in primary cells,transfection often is not the method of choice, andinstead the generation of viral vectors may be consid-ered. Problems with regard to the generation of thesevectors can arise because the expressed toxic proteins ofinterest may damage viral helper cells and thus preventformation of recombinant viral particles. Here again,tightly regulated gene expression using inducible pro-moters may be a possible way to circumvent this prob-lem. For instance, tetracycline-dependent promotersystems have already been made commercially availablefor use in adenoviral and retroviral vectors.
In this study, four diVerent expression systems werecomparatively tested in transient transfection experi-ments to evaluate their versatility for regulated expres-sion. While this work can not provide a complete surveyover the variety of available systems, it is focused on theanalyses of three more commonly used inducible pro-moters, namely the glucocorticoid-inducible mousemammary tumor virus (MMTV)3 promoter [2] (MMTVsystem), the ecdysone-inducible Drosophila-analog pro-moter/receptor (EcP) [3] (EcP system), and the tetracy-cline-dependent Tet system [4]. In addition, the T7 RNApolymerase-dependent T7 promoter (T7P) [5] (T7 sys-tem) was tested as an alternative system that is widelyutilized in bacterial expression and that may hold somepotential for eukaryotic expression also.
The MMTV promoter is regulated by the presence ofactivated glucocorticoid receptor molecules. Binding ofglucocorticoids, such as dexamethasone, as ligands tothe receptor molecules releases receptor molecules froma cytoplasmatic complex with heat shock protein 70.This allows for their translocalization into the nucleus,where they bind to speciWc glucocorticoid-responsiveDNA elements (GRE) to subsequently enhance tran-scription of the promoter. For use as an inducibleexpression system, both an expression cassette allowingfor the glucocorticoid-regulated expression of the geneof interest and a second cassette for constitutive expres-sion of the glucocorticoid receptor are applied. The lat-ter cassette is necessary only for those cell typesexpressing limiting amounts of endogenous glucocorti-coid receptor.
The ecdysone-inducible system [3], too, depends onthe binding of a hormone—in this case the insect hor-mone ecdysone—to an insect receptor protein. Bindingof ecdysone or synthetic analogs (muristerone A, ponas-
3 Abbreviations used: MMTV, mouse mammary tumor virus; ECP,ecdysone promoter; Tet, tetracyclin; T7P, T7 promoter; GRE, gluco-corticoid-response elements; ECR, ecdysone receptor; RXR, retinoidX receptor; ORF, open reading frame; EGFP, enhanced green Xuores-cent protein; GFL, green Xuorescent luciferase; IRES, internal ribo-somal entry.
terone A) leads to the formation of a heterodimer of theengineered ecdysone receptor (EcR), which is a fusionprotein consisting of the EcR DNA binding domainfused to a herpes simplex virus VP16 transactivationdomain and the retinoid X receptor (RXR), a mamma-lian homolog to the Drosophila protein ultraspinacle.The heterodimer binds to the ecdysone/glucocorticoid-responsive elements in the synthetic ecdysone-responsiveIND promoter and thereby activates transcription of anengineered downstream minimal heat shock promoter.Biological eVects of insect steroid hormones on mamma-lian cells are unlikely to occur and thus pleiotropiceVects can be excluded. In analogy to the glucocorticoid-regulated system, this system relies on the presence of amammalian nuclear receptor protein (RXR) in the cellbut here in combination with a transactivating recombi-nant receptor (EcR) which both must be expressed tofacilitate expression of the desired transgene.
The third expression system tested in this study, thetetracycline-regulated Tet system, is based on the tetra-cycline resistance operon of Escherichia coli transposonT10. In the Tet-On system, the receptor molecule is thechimeric synthetic heterodimer rtTA, which is a fusionprotein of the DNA binding domain of the bacterial tet-racycline repressor and the herpes simplex virus VP16transactivation domain. Upon binding of tetracycline, acomplex is formed that activates transcription of a targetpromoter consisting of multiple operators upstream of acore promoter [4]. In the Tet-OV system, the receptormolecule is the chimeric heterodimer tTA, which uponbinding of tetracycline represses expression of tetracy-cline-responsive promoter. In this case, maximal expres-sion is achieved in the absence of tetracycline.
All systems described above require the formation ofcomplexes of transactivating receptor molecules withchemical inducer substances. These promoter systemsare dependent on the availability of speciWc receptormolecules and therefore each requires constitutiveexpression of the appropriate receptor gene(s) to facili-tate expression of the target gene.
In addition to the systems described above, the T7-pro-moter-regulated expression system was evaluated inmammalian cells. The T7 promoter is less commonly usedfor eukaryotic cells although its utilization as a mamma-lian expression system has been proposed before [6,7]. Inthis expression system, only the T7-phage-derived RNApolymerase has the ability to initiate transcription fromthe phage T7 promoter. Transcription is terminated bystem loops formed in the so-called T7 terminator regiondownstream of the gene ORF. In contrast to RNA-poly-merase-II-derived RNA molecules, transcripts producedby T7 RNA polymerase only rarely have 5�-cap structures[8] and polyadenylated 3�-RNA ends. While this system isnot truly inducible, but rather works in an “On/OV”mode, it was included in this study for its potential valuein the expression of very toxic genes.

M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19 11
All these inducible promoters were operationallylinked with a reporter gene, i.e., a fusion gene (green Xuo-rescent luciferase; GFL) consisting of the gene forenhanced green Xuoresecence protein (EGFP) and theWreXy luciferase gene. The respective inducible expres-sion systems were qualitatively and quantitatively evalu-ated by Xuorescence microscopy and luminometricluciferase assays after transient transfection experiments.Expression rates achieved with the T7 promoter couldbe increased 30-fold by the insertion of a DNA elementcorresponding to the coxsackievius B3 5� nontranslatedregion, which contains an internal ribosomal entry site(IRES).
Material and methods
Expression plasmids
Reporter plasmids containing a reporter gene encod-ing an EGFP/WreXy luciferase fusion protein wereconstructed for all inducible expression systems tested.N-terminal fusion of the EGFP cDNA to the luciferasegene in vector pGL3 (Promega, Madison, WI, USA)resulted in the dual-function reporter protein designatedgreen Xuorescent luciferase; the resulting promoterlessplasmid was therefore named pGFL [9]. Insertion of aRous sarcoma virus (RSV, strain Schmidt–Ruppin,EMBL Database Accession No. L29198) promoterresulted in the control plasmid pRSV-GFL [9]. Isolationof the GFL cDNA from the plasmid pGFL by restric-tion digestion with BglII and cloning of this fragmentinto the BamHI restriction site of vector pIND (Invitro-gen, Carlsbad, CA, USA) led to the ecdysone-induciblereporter construct pIND-GFL. Expression of the recep-tor molecules was achieved by using the plasmidpVgRXR (Invitrogen).
The mouse mammary tumor virus promoter reporterplasmid pMMTV-GFL was synthesized by inserting theGFL fragment into the EcoRI-generated vector frag-ment of plasmid pPARP93 [10]. Overexpression of thehuman glucocorticoide receptor was achieved by utiliza-tion of plasmid pHG0 [2,11].
The tetracycline-responsive reporter plasmid pTet-GFL was generated by inserting the NheI/XbaI fragmentfrom pGFL into the XbaI restriction site of pUHD10-3[4]. For analysis of the Tet-On system the receptorexpression plasmid pUHD172-neo [4] was used toexpress the transactivating rtTA receptor, while for theTet-OV system the commercially available plasmidpTetOV (Clontech) was used to express the repressingtTA receptor.
Generation of the T7 promoter reporter gene vectorwas performed in several steps. First a BglII/EcoRV T7promoter/T7 terminator fragment from pET3a (Nova-gen, Merck, Darmstadt, Germany) was ligated into
pGL3 (Promega), which was cut with BamHI and thenblunted and cut with BglII. The resulting plasmid wasnamed pGT7PT7T. The residual pGL3-derived multiplecloning site was removed by digestion with BglII andKpnI and subsequent religation; a diVerent multiplecloning site (PmeI fragment from vector pIND (Invitro-gen)) was inserted between the T7 promoter and the T7terminator using a NdeI site, resulting in pT7-MCS. AGFL fragment was generated by cutting pGFL withBglII with a subsequent T4 DNA polymerase reaction toWll in the 5� overhang, followed by a HindIII digestion.The resulting fragment was ligated into pT7-MCS cutwith HindIII/EcoRV leading to the reporter vector pT7-GFL.
Plasmid pCMV-T7Pol was utilized as an expressionvector for a nuclear T7 RNA polymerase [7,12]. Thisplasmid was generated by ligating the ORF encodingT7-RNA polymerase from plasmid pAR3132 [7] cutwith HindIII/EcoRV fragment into plasmid pshuttle-CMV [13].
A modiWed T7 expression plasmid pT7-IRES-GFLand the control plasmid pIRES-GFL were synthesizedby inserting the complete 765-bp nontranslatedupstream sequence of coxsackievirus B3 [14] containingan IRES element into vectors pT7GFL and pGFL,respectively [15].
Cell culture
911 Cells [16] (Invitrogen) were grown under stan-dard cell culture conditions (37 °C, 5% CO2) in Dul-becco’s modiWed Eagle’s medium (DMEM)supplemented with 10% fetal bovine serum (FBS),100 U/ml penicillin, and 100 �g/ml streptomycin (allGibco/BRL, Invitrogen) or in DMEM supplementedwith 10% Tet-approved fetal bovine serum (Clontech),where applicable.
Lipofection
911 cells were seeded in 24-well plates at a density of5 £ 104 and allowed to attach overnight. Three hoursprior to adding the transfection mixture the medium wasremoved and 500 �l of fresh culture medium was addedto each 24-well plate. The transfection mixture used foreach well consisted of 0.8�l FuGene 5 reagent (Roche,Mannheim, Germany) and 280 ng of plasmid DNA mix-ture in 25 �l of serum-free DMEM. Each DNA mixturecontained 28 fmol receptor plasmid and 14 fmol expres-sion plasmid and, where applicable, appropriateamounts of carrier plasmid DNA (pshuttle; [13]). A totalof 25 �l of transfection solution was added to each 24-well plate and lipofection was allowed to proceed for12 h. After 12 h, the supernatant medium was replacedby fresh medium and expression of the inducible pro-moter systems was induced by adding the required

12 M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19
inducing chemical agent to a Wnal concentration of 1 �M(ponasterone A, dexamethasone, and doxycycline for theecdysone-, the glucocorticoid-, and the tetracycline-responsive promoters, respectively). As an internal con-trol and to monitor transfection eYciency, plasmidpCMV-� Gal (BD Biosciences Clontech, Palo Alto, CA,USA) was cotransfected in all samples.
Reporter gene assays
Green Xuorescent reporter protein (GFL) expressionand �-galactosidase staining were each documentedusing inverse light and Xuorescence microscopy (Axio-vert 35, Zeiss, Oberkochen, Germany).
Luciferase activity was determined using a lumino-meter (Berthold, Bad Wildbad, Germany) and the lucif-erase assay system with reporter lysis buVer (Promega)according to the manufacturer’s guidelines. Values werenormalized to total cell protein content.
�-Galactosidase activity was determined qualitativelyby performing an in situ staining procedure [17] andquantitatively by photometrically determining theamount of cleaved ortho-nitrophenyl-�-galactopyrano-side using the �-galactosidase enzyme assay system(Invitrogen) with reporter lysis buVer (Promega) accord-ing to the manufacturers’ instructions. All assays werecarried out in triplicates and experimental sets wererepeated three times.
In vitro transcription and translation
Coupled in vitro transcription and translation analy-ses were carried out using a rabbit-reticulocyte-based T7expression kit (Promega) in accordance to the protocolsupplied by the manufacturer. As templates, 2 �g of line-arized pT7GFL and pT7IRESGFL, pIRESGFL, orpT7luc control DNA (Promega) were used per reaction.Samples were subjected to luciferase activity assays asdescribed above.
Results
As a model system for characterization of the fourdiVerent inducible promoter systems (Tet-On, EcP,MMTV, and T7 systems) we performed transient trans-fection experiments in the human embryonic retino-blast cell line 911 which was chosen because it can betransfected extremely well. Plasmids that induciblyexpress the reporter gene green Xuorescent luciferaseunder control of the described inducible promoterswere cotransfected with the corresponding receptorplasmids or the T7 RNA polymerase expression plas-mid and adequately induced, if required. Fig. 1 providesan overview of the diVerent expression cassettes used inthis study.
Microscopic evaluation of induced and basal expressionlevels of the tested promoter systems
Expression of the GFL reporter protein obtainedwith the control promoter construct pRSV-GFL (Figs.2A and B) was comparable to the expression achievedunder control of the induced EcP system (Fig. 2C), bothin number of Xuorescent cells and in intensity of theobserved Xuorescence. In the absence of ponasterone Ano Xuorescence was observed (data not shown). Simi-larly, expression in the induced MMTV system appearedcomparable to that in the RSV promoter (Fig. 2D). As
Fig. 1. Schematic overview of the promoter and receptor constructsused in this study. On the left side, cassettes for the expression ofreceptors are shown. On the right side, the respective promoter cas-settes driving expression of the EGFP-luciferase reporter (GFL) aredepicted. (A) Tet-On and Tet-OV system; (B) MMTV promoter sys-tem; (C) ecdysone-inducible system; (D) T7 promoter and T7 RNApolymerase system; (E) modiWed T7 promoter cassette containing theIRES sequence of the CVB3 5�NTR region; (F) promoterless controlplasmid containing the IRES sequence identical to that in (E). BGH,bovine growth hormone; CMVi.e., human cytomegalovirus immediateearly promoter; GFL, enhanced green Xuorescent protein/WreXy lucif-erase fusion gene; hGR, human glucocorticoid receptor; pA, polyade-nylation signal; SV40, simian virus 40; CVB3 5�UTR, 5� untranslatedregion of coxsackievirus B3; TK, thymidine kinase of human herpessimplex virus type 1; MMTV, mouse mammary tumor virus; RXR,retinoid X receptor; VgEcR, modiWed ecdysone receptor; E/GRE,ecdysone/glucocorticoid-responsive element; HSP70, minimal heatshock promoter; RSV, rous sarcoma virus promoter; TetO, tetracy-cline-responsive element; rtTA, recombinant tetracycline-responsivetransactivator; tTA, tetracycline-responsive repressor: T7Pro, phageT7 promoter; T7 RNA Pol, phage T7 RNA polymerase; T7Ter, phageT7 terminator.

M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19 13
observed in the EcP system, the MMTV promoteryielded no visible Xuorescence without hormone induc-tion. Transfection of plasmid pT7-GFL produced hardlyany visible Xuorescence with coexpression of the T7RNA polymerase (Fig. 2E) and no Xuorescence at allwithout coexpression of the T7 RNA polymerase (notshown). However, insertion of an IRES-containingCVB3 DNA element between T7 promoter and transla-tional start of the reporter gene results in clearly visibleXuorescence signals with the T7P system (see below, Fig.2F). Transfection and induction of the Tet-On promoterreporter plasmid pTet-GFL with doxycycline resulted innumerous bright Xuorescent cells (Fig. 2H), but even inthe absence of doxycycline Xuorescing cells could beobserved (Fig. 2G), indicating high background activityof this expression system.
Quantitative evaluation of the tested promoter systems
To compare the expression levels obtained by thediVerent promoter systems at the quantitative level,transfected cells were lysed and luciferase activity inthe lysates was determined. Lysates of cells transfectedeither with the promoterless plasmid pGFL or withplasmid pRSV-GFL using the constitutive RSV pro-moter were used as negative and positive controls,respectively. Luciferase activities obtained from thetested promoter systems were calculated as percentageof RSV promoter activity for better comparison. In thediVerent systems we obtained the following results(Fig. 3).
Fig. 2. Qualitative analysis of the promoter systems. Equimolaramounts of plasmid mix were transiently transfected into 911 cells foreach system. Twelve hours after transfection, the respective inducingreagent was added (see Material and methods). Forty-eight hours afterinduction Xuorescence signals were analyzed by immunoXuorescencemicroscopy using an Axiovert microscope and microphotographswere taken. (A) pRSV-GFL (positive control); (B) phase contrastimage of (A); (C) EcP system (pIND-GFL/pVgRXR/PonA); (D)MMTV promoter (pMMTV-GFL/pHG0/dex); (E) T7 promoter sys-tem (pT7P-GFL/pCMVT7-Pol); (F) modiWed T7 promoter system(pT7-IRES-GFL/pCMV-T7Pol) (see below for details); (G) Tet-Onsystem (pTet-On-GFL/pUHD172-1neo) without induction; (H) Tet-On system, as in G but after doxycyline induction. A promoterlessplasmid pGFL used as a negative control gave no visible Xuorescencesignals (not shown).
Tet-On systemGFL expression in the induced Tet-On system was
more than 5-fold higher than RSV promoter expressionlevels. In absence of doxycycline induction, luciferaseactivity still reached more than 1.5-fold of RSV promoteractivity. Therefore, the basal expression level (leakiness)of this promoter system was even higher than those of thepositive control (Fig. 3, Tet-On). In the absence ofthe receptor plasmid, luciferase activity reached values inthe range of 5% of RSV promoter activity, irrespectiveof the presence or absence of doxycycline. This intrinsicexpression corresponds to »1% of the induced and »3%of the noninduced Tet-On promoter activity.
MMTV promoterInduction of the MMTV promoter resulted in lucifer-
ase activities of about 50% of RSV promoter activity. Inabsence of dexamethasone, promoter activities were onlyabout 0.4 and 0.2% of RSV promoter activity, in thepresence and in the absence of receptor plasmid pHG0,respectively. Leakiness and intrinsic promoter activityare therefore less than 1% of the induced MMTV pro-moter. Addition of dexamethasone in the absence of thereceptor plasmid led to an expression rate of 3.2%related to RSV promoter activity, which is a 10-foldinduction factor and equivalent to »7% of induced levelin the presence of receptor plasmid (Fig. 3, MMTV).
Ecdysone promoterThe maximal expression value achieved after ponas-
terone A induction of the EcP system was about 80% of
Fig. 3. Quantitative analysis of the tested promoter systems. Tran-siently transfected 911 cells (see Fig. 2) were lysed in reporter lysisbuVer (Promega) and protein content and luciferase activity weredetermined. The promoterless plasmid pGFL was transfected as a neg-ative control and pRSV-GFL as a positive control (right). The averagevalues of three independent experiments, each performed in quintu-plets, is shown. The highest expression level was obtained with the Tet-On system but with a relatively high basal expression level. Theecdysone- and the dexamethasone-inducible systems, EcP andMMTV, respectively, had expression levels comparable to RSV pro-moter activity after induction, i.e., 100-fold higher than basal expres-sion levels. Expression of the T7 promoter was only about 1% of RSVactivity in the presence of T7 RNA polymerase. Basal expression with-out T7 RNA polymerase was less than 0.01% of RSV promoterexpression levels.

14 M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19
RSV promoter expression. In the absence of ponaster-one A, the basal expression (leakiness) was in the rangeof 1% of RSV promoter activity in the presence of thereceptor plasmid pVgRXR. This corresponds to »1.4%of the induced promoter. In the absence of the receptorplasmid pVgRXR, the intrinsic expression rate wasreduced to »0.2% of RSV promoter activity, with orwithout ponasterone A induction, respectively (Fig. 3,EcP).
T7 promoterCotransfection of the T7-promoter-dependent
reporter plasmid pT7-GFL with the T7 RNA polymer-ase expression plasmid pCMV-T7Pol yielded only 1.7%of the enzymatic activity observed with the RSV pro-moter. The basal expression of the transfected pT7-GFL reporter plasmid without coexpression of T7RNA polymerase resulted in a luciferase activity of0.005% of RSV promoter achieved luciferase activity,i.e., ca. 1% of the expression in the presence of T7 RNApolymerase (Fig. 3, T7P). The basal expression of pT7-GFL is therefore even below the basal expression of thepromoterless control plasmid pGFL. The basal activityof pGFL might be due to a slight promoter activity ofthe SmaI site in the multiple cloning site sequence [18].In pT7-GFL the SmaI site was removed during thecloning procedure (see Material and methods). Conclu-sively, the T7 promoter system had the lowest basalactivity, but even with coexpression of the T7 RNApolymerase this system achieved only relatively lowmaximal expression levels.
ModiWcation of the T7 promoter system
We assumed that the relatively low expression levelsobtained by T7 promoter plasmid transfections were dueto ineYcient translation of the transcribed mRNA,because T7 RNA polymerase transcripts mostly do notcontain a 5�-cap structure to facilitate translation initia-tion [8]. To increase expression levels, the 5� nontrans-lated region of a coxsackievirus B3 (CVB3) cDNA wasinserted between the T7 promoter and the translationstart of the reporter gene (Fig. 1E). This DNA fragmentencodes a RNA that forms a hairpin structure calledIRES, to which translation initiation factors bind andfacilitate 5�-cap, independent-translation initiation [19–21]. In the modiWed expression system, the principle ofT7 RNA polymerase transcription regulation remainedunaltered except for the presence of the CVB3 IRESsequence in the 5� end of the transcribed RNA mole-cules.
Qualitative evaluation of the modiWed T7 promoter system
Cotransfection experiments were performed asdescribed above. When compared to the original
pT7-GFL/pCMV-T7-pol system, modiWcation of theT7 reporter plasmid increased the GFL expression lev-els enough to allow for microscopic observation ofXuorescence in pT7IRESGFL and pCMV-T7-Polcotransfected cells (Fig. 2F). In contrast, cells trans-fected with the reporter plasmid pT7IRESGFL alonedid not show any microscopically visible Xuorescence(not shown).
Quantitative evaluation of the modiWed T7 promotersystem
While expression achieved by the original T7 pro-moter system reached only levels of less than 2% of RSVpromoter expression (Fig. 4a), the IRES-containingplasmid pT7-IRES-GFL (Fig. 1) yielded luciferaseactivities of »40% of RSV promoter activities (Fig. 4b).In the absence of T7 RNA polymerase the basal expres-sion level of the modiWed T7-IRES-GFL plasmid isabout 1.5-fold higher than that of the original plasmidpT7-GFL but with only 0.02% of RSV promoter expres-sion. Transfection of a promoterless control plasmidpIRES-GFL (Fig. 1F) ruled out that the IRES sequenceof the CVB3 5� nontranslated region per se contains anypromoter activity. Expression levels obtained by trans-fecting this plasmid either in the presence or in theabsence of T7 RNA polymerase were »0.01% of RSVpromoter activity and this was in the range of the basalexpression of the pT7-IRES-GFL plasmid (Fig. 4c). Theinsertion of the CVB3 IRES therefore increased theexpression rate achievable by the T7 promoter by a fac-tor of 30.
Fig. 4. Improvement of T7 expression level by insertion of a CVB3IRES. One hundred thousand 911 cells were transiently transfected asdescribed in Fig. 2. Cell lysates were prepared and protein contents andluciferase activity were determined in the sample. Transfection of thepromoterless plasmid pGFL was used as a negative control (f) andpRSV-GFL as a positive control (e). Insertion of the CVB3 5�NTRcontaining an IRES sequence increased expression levels to 41.86% ofRSV promoter levels (b), while the basal expression level was onlyslightly raised to 0.02% of RSV promoter activity compared to 0.01%of RSV promoter activity with original T7 promoter (a). The IRES-containing element without any promoter had 0.01% of RSV promoteractivity. It therefore did not have any signiWcant promoter activity,independent of presence or absence of T7 RNA polymerase (c).
M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19 15
In vitro transcription/translation of T7 promoterconstructs
Coupled in vitro transcription and translation assayswere performed to analyze whether enhanced transcrip-tion or translation takes place in the modiWed constructwhich contains the CVB3 5� nontranslated region, pT7-IRES-GFL. In contrast to the luciferase activity resultsobtained after transfection of cells, the results of the invitro transcription and translation assays indicate that in
Fig. 5. In vitro transcription and translation of pT7-GFL, pT7-IRES-GFL, and pIRES-GFL. Two micrograms of plasmid DNA were usedin a coupled in vitro transcription and translation reaction. The lucif-erase activity of 5 �l reaction sample was determined. The average oftwo independent experiments carried out in triplicates is shown. Inser-tion of the IRES-containing element between the T7 promoter and theGFL reporter gene led to 95% decreased in vitro expression of thereporter. In the absence of the T7 promoter, GFL expression was notdetectable in the in vitro system.
this experimental setup the insertion of the IRES mark-edly reduced luciferase activity (Fig. 5) by about 95%.
Comparison of Tet-On and Tet-OV systems
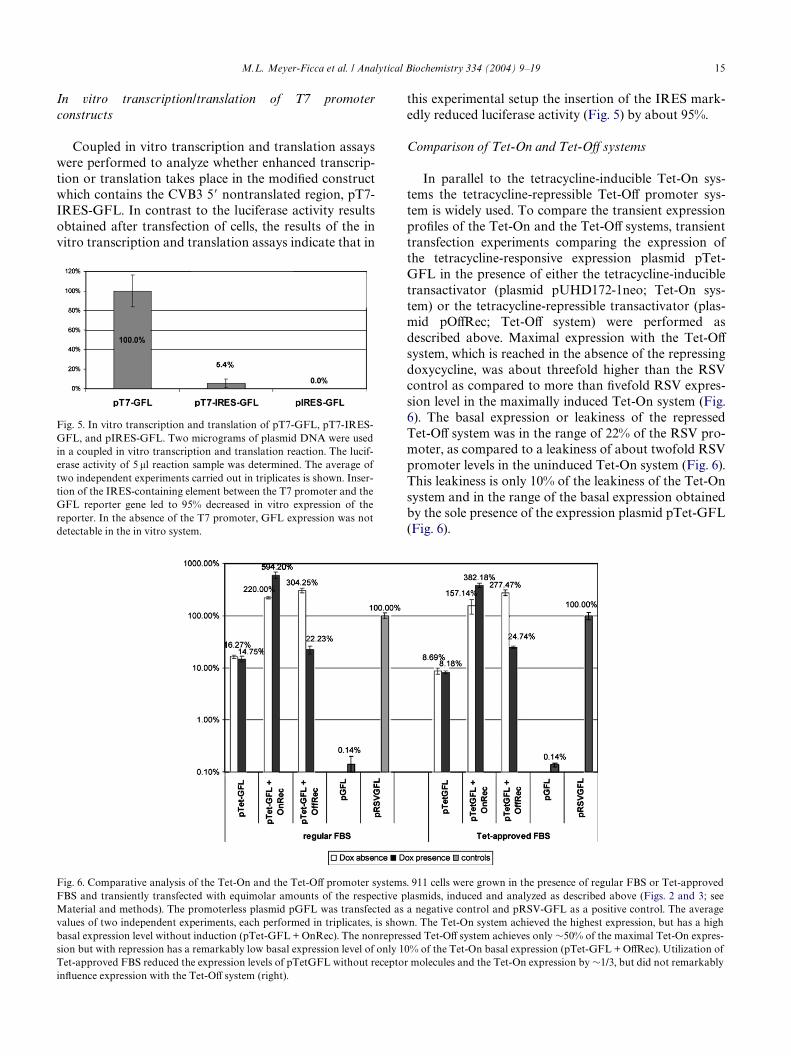
In parallel to the tetracycline-inducible Tet-On sys-tems the tetracycline-repressible Tet-OV promoter sys-tem is widely used. To compare the transient expressionproWles of the Tet-On and the Tet-OV systems, transienttransfection experiments comparing the expression ofthe tetracycline-responsive expression plasmid pTet-GFL in the presence of either the tetracycline-inducibletransactivator (plasmid pUHD172-1neo; Tet-On sys-tem) or the tetracycline-repressible transactivator (plas-mid pOVRec; Tet-OV system) were performed asdescribed above. Maximal expression with the Tet-OV
system, which is reached in the absence of the repressingdoxycycline, was about threefold higher than the RSVcontrol as compared to more than Wvefold RSV expres-sion level in the maximally induced Tet-On system (Fig.6). The basal expression or leakiness of the repressedTet-OV system was in the range of 22% of the RSV pro-moter, as compared to a leakiness of about twofold RSVpromoter levels in the uninduced Tet-On system (Fig. 6).This leakiness is only 10% of the leakiness of the Tet-Onsystem and in the range of the basal expression obtainedby the sole presence of the expression plasmid pTet-GFL(Fig. 6).
Fig. 6. Comparative analysis of the Tet-On and the Tet-OV promoter systems. 911 cells were grown in the presence of regular FBS or Tet-approvedFBS and transiently transfected with equimolar amounts of the respective plasmids, induced and analyzed as described above (Figs. 2 and 3; seeMaterial and methods). The promoterless plasmid pGFL was transfected as a negative control and pRSV-GFL as a positive control. The averagevalues of two independent experiments, each performed in triplicates, is shown. The Tet-On system achieved the highest expression, but has a highbasal expression level without induction (pTet-GFL + OnRec). The nonrepressed Tet-OV system achieves only »50% of the maximal Tet-On expres-sion but with repression has a remarkably low basal expression level of only 10% of the Tet-On basal expression (pTet-GFL + OVRec). Utilization ofTet-approved FBS reduced the expression levels of pTetGFL without receptor molecules and the Tet-On expression by »1/3, but did not remarkably
inXuence expression with the Tet-OV system (right).
16 M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19
To be able to directly compare the results obtainedfor the diVerent systems all transfection experimentswere performed in the presence of regular fetal bovineserum. To further test the inXuence of possible antibio-tica traces in the regular FBS, the transfection experi-ments testing the Tet-On and the Tet-OV system wererepeated in cells cultured and transfected in commer-cially available “Tet system approved” FBS. In the Tet-On system, utilization of the approved FBS reduced theleakiness in both absence and presence of Tet-On trans-activators, but the maximally achievable Tet-On expres-sion was reduced by »1/3 also. Expression levels in theTet-OV system were not markedly altered (Fig. 6).
Discussion
The aim of this study was to compare the suitabilityof diVerent commonly used inducible promoter systemsfor regulated protein expression in transiently trans-fected cells.
The tetracycline-inducible Tet system (Tet-On andTet-OV), the ecdysone-inducible promoter (EcP), theglucocorticoid-responsive MMTV promoter (MMTV),and the T7 promoter (T7) in combination with expres-sion of T7 RNA polymerase were analyzed (see summa-rized data in Table 1).
Analyses of the inducible expression systems werefocused on four main criteria: (1) maximum expressionperformance (strength) of the promoters in theirinduced states, (2) “tightness” of expression in theuninduced state, (3) amplitude of expression increaseas a marker of inducibility, and (4) feasibility in han-dling and side eVects of the induction or the inducingagent used.
The Tet-On system yielded very high induced expres-sion levels (more than 5-fold of RSV promoter activity).However, the induction rate was only 3.7-fold, becausethe basal expression level was relatively high (approx.1.5-fold of RSV promoter activity). The tetracycline-repressible Tet-OV system, which was tested additionallyas an alternative to the Tet-On system, also obtained
high maximal expression levels with 3-fold RSVpromoter activity but reached only 50–70% of the Tet-On activity. The Tet-OV induction rate was 13-fold andtherefore signiWcantly higher than the Tet-On inductionrate. Although the repressed expression was only »10%of the Tet-On leakiness, it still reached 22% of RSV pro-moter activity.
The high basal expression levels in the Tet systems aremost probably due to the fact that, under conditions oftransient transfections, plasmids are usually not chroma-tinized, which has been described to be an important fac-tor in tight regulation of the Tet promoter [22–24]. Itshould be mentioned at this point that the Tet systemhas been utilized successfully in the context of retroviralvectors [25], as retroviral infection of cells results in theintegration and chromatization of the viral ORF, i.e., thetransduced expression cassette.
Tetracyclines are used widely in veterinarian medicineand are routinely applied to a wide variety of livestock.The presence of tetracycline traces in our media cannotbe ruled out, as commercially available fetal bovineserum was used to supplement the media. To estimatethe amount of unintended induction by residual tracesof tetracycline in the FBS, the inXuence of regular serumon the leakiness of the Tet-On and the Tet-OV systemswas analyzed by experimental comparison with commer-cially available, speciWcally tested tetracycline-freeserum. While no diVerence between the sera wasobserved in the Tet-OV system, both leakiness and maxi-mal expression were reduced in the Tet-On system whenthe Tet-system-approved serum was used.
Using Tet-approved FBS therefore improves the leak-iness of the Tet-On system but will reduce the maximalexpression level. As an alternative to using tetracycline-tested serum with the Tet-On system, traces of tetracy-cline in the medium can be removed by puriWcation withactivated charcoal, which will add some cost and/oreVort to the utilization of this inducible system.
Additional versions of Tet-On promoters have alsobeen developed and are available today. These promot-ers combine activation and silencing elements andthereby achieve a signiWcant reduction of the basal
Table 1Comparison of the analyzed promoter systems
Maximal expression rates, leakiness (expression in the absence of inducing reagent or T7 RNA polymerase), induction rates, and induction sub-stances are shown.
a Ratio (nonrepressed/repressed) in the Tet-OV system.b Doxycyline is repressing agent in the Tet-OV system.
Promoter system
Maximal expression (% of RSV prom. expr.)
Leakiness (% of RSV prom. expr.)
Induction ratea (ratio ind./non induced)
Inducing agentb
T7P 1.7 § 0.3 0.005 § 0.0002 58.8 pCMV-T7-RNApolEcP 77.5 § 15 1.1 § 0.15 73.7 Ponasterone [1 �M]MMTV 48.4 § 10 0.4 § 0.05 120.0 Dexamethasone [1 �M]Tet-On 585.4 § 15 155 § 24 3.7 Doxycycline [1 �M]Tet-OV
a 304.3 § 28 22.2 § 3.7 13.6 Doxycycline [1 �M]T7IRES 41.9 § 6 0.02 § 0.004 1761.0 pCMV-T7-RNApol

M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19 17
activity [26,27] as compared to the Wrst-generation Tet-On system used in the present study. While earlier stud-ies had shown that the Tet-On system was inferior foruse in recombinant adenoviral vectors compared to theTet-OV systems [28,29], the combination of tetracy-cline-responsive transactivators with tetracycline-con-trolled transcriptional silencers allowed for thedevelopment of a recently published Tet-On systemcontaining adenoviral vector with very good inductionproperties [30].
In conclusion, both the Tet-On and the Tet-OV sys-tems are valuable tools to achieve high levels of overex-pression of nontoxic proteins, i.e., abundant cellularproteins. While the Tet-OV system allows for a higherdegree of regulation, the Tet-On system can achieve thehighest expression levels of all systems tested in thisstudy.
The MMTV promoter is activated by glucocorticoidssuch as the synthetic dexamethasone. Fetal bovine serumused for cell culture is predicted to contain traces of nat-ural glucocorticoid hormones that can bind to the recep-tor. The puriWcation steps in removing the residualendogenous hormones from sera (stripping) are similarto those used to remove tetracyclines from commerciallyavailable fetal bovine serum. Moreover, endogenoushuman growth receptor (hGR) molecules, which occurnaturally in many cell types, are activated upon additionof dexamethasone to the medium. As a result, cellularpromoters that are naturally regulated by this transcrip-tion factor are inXuenced by the addition of dexametha-sone. Therefore, a hormone response and possibly achange of cellular transcription patterns cannot beexcluded as a potential side eVect of this promoter sys-tem. Overexpression of hGR may potentiate these eVectsby changing cellular transcription in cells otherwiseimpervious to hormone stimulation.
We hypothesize that in the 911 cells tested in the pres-ent study there may be some endogenous glucocorticoidreceptor present as dexamethasone induction of theMMTV promoter was possible even in the absence ofthe receptor plasmid pHG0. If this was the case, theamount of endogenously expressed glucocorticoid recep-tor seemed to be limiting the expression rate, as expres-sion increases only 14-fold from »0.2% of RSV activityto »3% of RSV activity compared to 120-fold from 0.4%to »50% of RSV promoter activity with additionalreceptor overexpression.
If controllable low-level overexpression is desired, theMMTV promoter can be a valuable tool in cells whichendogenously express human growth receptor. Here,inducible expression can be achieved using the MMTVpromoter, eliminating the need to express additionalreceptor molecules.
In contrast to the MMTV promoter, the ecdysone-inducible promoter is entirely dependent on coexpres-sion of adequate transactivator molecules and shows
hardly any increase in expression level without coexpres-sion of the transactivator. Ecdysone is an insecthormone that has no known pleiotropic eVects on mam-malian cells, which is an advantage of the ecdysone-responsive system. The ecdysone-inducible promoter(EcP) proved to be quite suitable for inducible expres-sion under transient transfection conditions with aninduction rate of »70-fold. Maximal expression rates ofthe EcP were 80% of RSV promoter activity with lowobserved leakiness of the system (basal expression wasonly in the range of 1% of RSV promoter activity). Theecdysone system is a very versatile system allowingsimultaneously for a high degree of expression controland relatively high expression levels.
The T7 promoter system is conceptually diVerentfrom the other systems which require a small moleculeas an inducing agent to start transcription. In the case ofthe T7 promoter, transcription is performed by addi-tionally expressed T7 RNA polymerase. This system hadby far the lowest basal expression level. Unfortunately,this was combined with very low maximal expressionlevels in the presence of T7 RNA polymerase of only1.7% of RSV promoter activity and is therefore in therange of the basal expression levels of the ecdysone-responsive system or the MMTV promoter system. Thisvery low expression maximum could be raised 30-foldby inserting the coxsackievirus B3 5� nontranslatedregion, which contains an internal ribosomal entry site(IRES) and facilitates 5�-cap-independent translationinitiation. The beneWt of using an IRES-containing ele-ment to enhance T7-promoter-dependent expressionwas also reported earlier [6,31–33]. The expression maxi-mum of the modiWed system was »40% of RSV pro-moter level, while the basal expression level of themodiWed T7-promoter-containing construct was only0.02% of RSV promoter activity and therefore stillextremely low. Unexpectedly, the increased expressionlevels achieved with the IRES-containing plasmid werenot found in in vitro transcription and translationassays. In contrast to the luciferase activity resultsobtained after transfection of cells, the results of the invitro transcription and translation assays indicate thatin this experimental setup the insertion of the IRESmarkedly reduced luciferase activity (Fig. 5), namely by»95%. This indicates that the IRES sequence most likelydoes not enhance transcription of the reporter gene. Inthe context of the in vitro expression system, which isoptimized for 5�-cap-independent translation of T7RNA polymerase transcripts, the IRES most likely doesnot provide any advantageous translation initiation alsocompared to the simple T7 promoter construct pT7-GFL. It can be hypothesized that the IRES structurepresent in the 5� nontranslated region increases cap-independent translation initiation only in the context ofintact cells. As a possible reason, it may be argued thatthe majority of RNA molecules synthesized by T7 RNA

18 M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19
polymerase are not capped and are therefore translatedvery ineYciently in living cells but not in an in vitrotranslation experiment. Moreover, the uncapped T7RNA polymerase transcripts are very rapidly degraded[8] in living cells. In the in vitro transcription and trans-lation reaction degradation of the T7-synthesizedmRNA molecules is prevented by the presence of RNaseinhibitors. There is a marked preference for translationof mRNA molecules with 5�-cap structures, which formthe majority of cellular mRNA molecules. In the cell, inwhich uncapped and capped transcripts compete forbinding to the ribosomes, the addition of a 5�-IRESsequence to the mRNA probably provides an alterna-tive to a 5�-cap. Hairpin-rich IRES structures bindtranslation initiation factors and improve translationinitiation in vivo [21,34,35] and might protect RNAfrom rapid degradation, as was shown for E. coli [36]. Inthe noncompetitive situation during an in vitro transla-tion the presence of an IRES sequence reduces theexpression rate; here it probably hampers the transla-tion process.
Despite the improved expression levels, the T7 pro-moter/T7 RNA polymerase system turned out to have amajor disadvantage: Expression of T7 RNA polymer-ase seemed to inXuence the overall protein expression inmammalian cells, as could be seen by reduced expres-sion levels of the �-galactosidase when T7-RNA poly-merase was coexpressed (not shown). Taken together,these Wndings suggest limited applications for the cur-rent IRES-improved T7 promoter/T7 RNA polymerasesystem in mammalian cells. However, it is well conceiv-able to modify this system by introducing recombinantpuriWed T7 RNA polymerase into target cells by usingone of the commercially available protein transfectionreagents. Thus, expression could be induced in targetcells that have been stably or transiently transfectedwith the gene of interest under the control of the T7/IRES promoter as long as T7 RNA polymerase isapplied. Furthermore, recombinant adenovirus [37] andvaccinia virus [38] have been published and can be usedto trigger expression in transfected target cells, also. Inconclusion, although the T7 promoter system is not atrue inducible system, it appears suitable for the expres-sion of very toxic proteins, because the basal expressionlevel in the absence of T7 RNA polymerase is neglecta-bly small, as was also observed by others [37]. Expres-sion of the T7 RNA polymerase will allow for areasonable expression of the gene of interest but at theexpense of inXuencing cellular metabolism by T7 RNApolymerase itself.
Other studies report induction behavior of, e.g., theTet system to be dependent on the cell line utilized [39].This means that the 911 cell study presented here gives ageneral guideline, but speciWc conditions should betested for every cell line or primary cell culture usingsuch an assay system as reported here.
Acknowledgement
This work was supported in part by grants of theIZKF Tübingen and of Bundesministerium fuer Bildungund Forschung (BMBF, 01GE9903). Heike Kaiser wassupported by the fortüne program of the UniversityHospital of Tübingen (No. 951-0-0).
References
[1] K. Keyvani, I. Baur, W. Paulus, Tetracycline-controlled expressionbut not toxicity of an attenuated diphtheria toxin mutant, Life Sci.64 (1999) 1719–1724.
[2] V. Kumar, S. Green, G. Stack, M. Berry, J.R. Jin, P. Chambon,Functional domains of the human estrogen receptor, Cell 51(1987) 941–951.
[3] D. No, T.P. Yao, R.M. Evans, Ecdysone-inducible gene expressionin mammalian cells and transgenic mice, Proc. Natl. Acad. Sci.USA 93 (1996) 3346–3351.
[4] M. Gossen, H. Bujard, EYcacy of tetracycline-controlled geneexpression is inXuenced by cell type: commentary, Biotechniques19 (1995) 213–216 discussion 216–217.
[5] A. Lieber, U. Kiessling, M. Strauss, High level gene expression inmammalian cells by a nuclear T7-phase RNA polymerase, NucleicAcids Res. 17 (1989) 8485–8493.
[6] O. Elroy-Stein, T.R. Fuerst, B. Moss, Cap-independent translationof mRNA conferred by encephalomyocarditis virus 5� sequenceimproves the performance of the vaccinia virus/bacteriophage T7hybrid expression system, Proc. Natl. Acad. Sci. USA 86 (1989)6126–6130.
[7] F.W. Studier, A.H. Rosenberg, J.J. Dunn, J.W. DubendorV, Use ofT7 RNA polymerase to direct expression of cloned genes, Meth-ods Enzymol. 185 (1990) 60–89.
[8] T.R. Fuerst, B. Moss, Structure and stability of mRNA synthe-sized by vaccinia virus-encoded bacteriophage T7 RNA polymer-ase in mammalian cells. Importance of the 5� untranslated leader,J. Mol. Biol. 206 (1989) 333–348.
[9] R.G. Meyer, J.H. Kupper, R. Kandolf, H.P. Rodemann, Earlygrowth response-1 gene (Egr-1) promoter induction by ionizingradiation in U87 malignant glioma cells in vitro, Eur. J. Biochem.269 (2002) 337–346.
[10] L. Van Gool, R. Meyer, E. Tobiasch, C. Cziepluch, J.C. Jauniaux,A. Mincheva, P. Lichter, G.G. Poirier, A. Burkle, J.H. Kupper,Overexpression of human poly(ADP-ribose) polymerase in trans-fected hamster cells leads to increased poly(ADP-ribosyl)ationand cellular sensitization to gamma irradiation, Eur. J. Biochem.244 (1997) 15–20.
[11] J.H. Kupper, M. Muller, M.K. Jacobson, J. Tatsumi-Miyajima,D.L. Coyle, E.L. Jacobson, A. Burkle, trans-dominant inhibitionof poly(ADP-ribosyl)ation sensitizes cells against gamma-irradia-tion and N-methyl-N�-nitro-N-nitrosoguanidine but does not limitDNA replication of a polyomavirus replicon, Mol. Cell. Biol. 15(1995) 3154–3163.
[12] J.J. Dunn, B. Krippl, K.E. Bernstein, H. Westphal, F.W. Studier,Targeting bacteriophage T7 RNA polymerase to the mammaliancell nucleus, Gene 68 (1988) 259–266.
[13] T.C. He, S. Zhou, L.T. da Costa, J. Yu, K.W. Kinzler, B. Vogel-stein, A simpliWed system for generating recombinant adenovi-ruses, Proc. Natl. Acad. Sci. USA 95 (1998) 2509–2514.
[14] W.M. Klump, I. Bergmann, B.C. Muller, D. Ameis, R. Kandolf,Complete nucleotide sequence of infectious Coxsackievirus B3cDNA: two initial 5� uridine residues are regained during plus-strand RNA synthesis, J. Virol. 64 (1990) 1573–1583.

M.L. Meyer-Ficca et al. / Analytical Biochemistry 334 (2004) 9–19 19
[15] R.G. Meyer, M.L. Meyer-Ficca, H. Kaiser, H.C. Selinka, R. Kan-dolf, J.H. Küpper, Plasmid-based generation of recombinant cox-sackievirus B3 particles carrying capsid gene replacementreplicons, Virus Res. 104 (2004) 17–26.
[16] F.J. Fallaux, O. Kranenburg, S.J. Cramer, A. Houweling, H. VanOrmondt, R.C. Hoeben, A.J. Van Der Eb, Characterization of 911:a new helper cell line for the titration and propagation of earlyregion 1-deleted adenoviral vectors, Hum. Gene Ther. 7 (1996)215–222.
[17] D. Panicali, A. Grzelecki, C. Huang, Vaccinia virus vectors utiliz-ing the beta-galactosidase assay for rapid selection of recombinantviruses and measurement of gene expression, Gene 47 (1986) 193–199.
[18] A.L. Yang, M.F. Festing, A promoter function of the CCCGGGSma I recognition sequence and its speciWc role in determining p53status and identifying DNA damaging agents, Biochem. Biophys.Res. Commun. 281 (2001) 506–510.
[19] D. Yang, J.E. Wilson, D.R. Anderson, L. Bohunek, C. Cordeiro, R.Kandolf, B.M. McManus, In vitro mutational and inhibitory anal-ysis of the cis-acting translational elements within the 5� untrans-lated region of coxsackievirus B3: potential targets for antiviralaction of antisense oligomers, Virology 228 (1997) 63–73.
[20] Z. Liu, C.M. Carthy, P. Cheung, L. Bohunek, J.E. Wilson, B.M.McManus, D. Yang, Structural and functional analysis of the 5�
untranslated region of coxsackievirus B3 RNA: in vivo transla-tional and infectivity studies of full-length mutants, Virology 265(1999) 206–217.
[21] D. Yang, P. Cheung, Y. Sun, J. Yuan, H. Zhang, C.M. Carthy,D.R. Anderson, L. Bohunek, J.E. Wilson, B.M. McManus, Ashine-dalgarno-like sequence mediates in vitro ribosomal internalentry and subsequent scanning for translation initiation of coxsac-kievirus B3 RNA, Virology 305 (2003) 31–43.
[22] S. Freundlieb, U. Baron, A.L. Bonin, M. Gossen, H. Bujard, Use oftetracycline-controlled gene expression systems to study mamma-lian cell cycle, Methods Enzymol. 283 (1997) 159–173.
[23] M. Gossen, A.L. Bonin, H. Bujard, Control of gene activity inhigher eukaryotic cells by prokaryotic regulatory elements, TrendsBiochem. Sci. 18 (1993) 471–475.
[24] M. Gossen, H. Bujard, Tight control of gene expression in mam-malian cells by tetracycline-responsive promoters, Proc. Natl.Acad. Sci. USA 89 (1992) 5547–5551.
[25] W. Paulus, I. Baur, F.M. Boyce, X.O. BreakeWeld, S.A. Reeves,Self-contained, tetracycline-regulated retroviral vector system forgene delivery to mammalian cells, J. Virol. 70 (1996) 62–67.
[26] S. Freundlieb, C. Schirra-Muller, H. Bujard, A tetracycline con-trolled activation/repression system with increased potential forgene transfer into mammalian cells, J. Gene Med. 1 (1999) 4–12.
[27] K. Forster, V. Helbl, T. Lederer, S. Urlinger, N. Wittenburg, W.Hillen, Tetracycline-inducible expression systems with reducedbasal activity in mammalian cells, Nucleic Acids Res. 27 (1999)708–710.
[28] H. Mizuguchi, T. Hayakawa, Characteristics of adenovirus-medi-ated tetracycline-controllable expression system, Biochim. Bio-phys. Acta 1568 (2001) 21–29.
[29] H. Mizuguchi, T. Hayakawa, The tet-oV system is more eVectivethan the tet-on system for regulating transgene expression in a sin-gle adenovirus vector, J. Gene Med. 4 (2002) 240–247.
[30] H. Mizuguchi, Z.L. Xu, F. Sakurai, T. Mayumi, T. Hayakawa,Tight positive regulation of transgene expression by a single ade-novirus vector containing the rtTA and tTS expression cassettesin separate genome regions, Hum. Gene Ther. 14 (2003) 1265–1277.
[31] O. Elroy-Stein, B. Moss, Cytoplasmic expression system basedon constitutive synthesis of bacteriophage T7 RNA polymerasein mammalian cells, Proc. Natl. Acad. Sci. USA 87 (1990) 6743–6747.
[32] T. Wilk, H. Mierswa, H.G. Krausslich, J.J. Dunn, V. Bosch,Expression of biologically active HIV glycoproteins using a T7RNA polymerase-based eucaryotic vector system, Virus Genes 6(1992) 229–246.
[33] A. Kohl, A. Billecocq, C. Prehaud, F.Z. Yadani, M. Bouloy, Tran-sient gene expression in mammalian and mosquito cells using arecombinant Semliki Forest virus expressing T7 RNA polymer-ase, Appl. Microbiol. Biotechnol. 53 (1999) 51–56.
[34] S.K. Jang, T.V. Pestova, C.U. Hellen, G.W. Witherell, E. Wimmer,Cap-independent translation of picornavirus RNAs: structure andfunction of the internal ribosomal entry site, Enzyme 44 (1990)292–309.
[35] M. Schmid, E. Wimmer, IRES-controlled protein synthesis andgenome replication of poliovirus, Arch. Virol. Suppl. 9 (1994) 279–289.
[36] K.E. Baker, G.A. Mackie, Ectopic RNase E sites promote bypassof 5�-end-dependent mRNA decay in Escherichia coli, Mol.Microbiol. 47 (2003) 75–88.
[37] J. Myung, N. Khalap, G. Kalkeri, R. Garry, S. Dash, Induciblemodel to study negative strand RNA synthesis and assembly ofhepatitis C virus from a full-length cDNA clone, J. Virol. Methods94 (2001) 55–67.
[38] M.R. Mohamed, E.G. Niles, Transient and inducible expression ofvaccinia/t7 recombinant viruses, Methods Mol. Biol. 269 (2004)41–50.
[39] C.E. Ackland-Berglund, D.A. Leib, EYcacy of tetracycline-con-trolled gene expression is inXuenced by cell type, Biotechniques 18(1995) 196–200.