aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release...
TRANSCRIPT

Racha Estephan, Randi B. Silver, Daria Mochly-Rosen and Roberto LeviKenichiro Koda, Mariselis Salazar-Rodriguez, Federico Corti, Noel Yan-Ki Chan,
Inhibiting Local Renin Release From Cardiac Mast CellsAldehyde Dehydrogenase Activation Prevents Reperfusion Arrhythmias by
ISSN: 1524-4539 Copyright © 2010 American Heart Association. All rights reserved. Print ISSN: 0009-7322. Online
72514Circulation is published by the American Heart Association. 7272 Greenville Avenue, Dallas, TX
published online August 9, 2010Circulation
1.citationhttp://circ.ahajournals.org/content/early/2010/08/09/CIRCULATIONAHA.110.95248
located on the World Wide Web at: The online version of this article, along with updated information and services, is
http://circ.ahajournals.org/content/suppl/2010/08/05/CIRCULATIONAHA.110.952481.DC1.htmlData Supplement (unedited) at:
http://www.lww.com/reprintsReprints: Information about reprints can be found online at
[email protected]. E-mail:
Fax:Kluwer Health, 351 West Camden Street, Baltimore, MD 21202-2436. Phone: 410-528-4050. Permissions: Permissions & Rights Desk, Lippincott Williams & Wilkins, a division of Wolters
http://circ.ahajournals.org//subscriptions/Subscriptions: Information about subscribing to Circulation is online at
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

Aldehyde Dehydrogenase Activation Prevents ReperfusionArrhythmias by Inhibiting Local Renin Release From
Cardiac Mast CellsKenichiro Koda, MD*; Mariselis Salazar-Rodriguez, MPharm*; Federico Corti, MPharm;
Noel Yan-Ki Chan, BS; Racha Estephan, PhD; Randi B. Silver, PhD;Daria Mochly-Rosen, PhD; Roberto Levi, MD, DSc
Background—Renin released by ischemia/reperfusion from cardiac mast cells activates a local renin-angiotensin system(RAS). This exacerbates norepinephrine release and reperfusion arrhythmias (ventricular tachycardia and fibrillation),making RAS a new therapeutic target in myocardial ischemia.
Methods and Results—We investigated whether ischemic preconditioning (IPC) prevents cardiac RAS activation in guineapig hearts ex vivo. When ischemia/reperfusion (20 minutes of ischemia/30 minutes of reperfusion) was preceded by IPC(two 5-minute ischemia/reperfusion cycles), renin and norepinephrine release and ventricular tachycardia and fibrillationduration were markedly decreased, a cardioprotective anti-RAS effect. Activation and blockade of adenosine A2b/A3
receptors and activation and inhibition of protein kinase C� (PKC�) mimicked and prevented, respectively, the anti-RASeffects of IPC. Moreover, activation of A2b/A3 receptors or activation of PKC� prevented degranulation and reninrelease elicited by peroxide in cultured mast cells (HMC-1). Activation and inhibition of mitochondrial aldehydedehydrogenase type-2 (ALDH2) also mimicked and prevented, respectively, the cardioprotective anti-RAS effects ofIPC. Furthermore, ALDH2 activation inhibited degranulation and renin release by reactive aldehydes in HMC-1.Notably, PKC� and ALDH2 were both activated by A2b/A3 receptor stimulation in HMC-1, and PKC� inhibitionprevented ALDH2 activation.
Conclusions—The results uncover a signaling cascade initiated by A2b/A3 receptors, which triggers PKC�-mediatedALDH2 activation in cardiac mast cells, contributing to IPC-induced cardioprotection by preventing mast cell reninrelease and the dysfunctional consequences of local RAS activation. Thus, unlike classic IPC in which cardiacmyocytes are the main target, cardiac mast cells are the critical site at which the cardioprotective anti-RAS effectsof IPC develop. (Circulation. 2010;122:771-781.)
Key Words: arrhythmia � ischemia � norepinephrine � renin � reperfusion
How critical mast cells are in cardiac pathophysiology isnot well understood. Numerous mast cells are present in
the mammalian heart (�50 000 mast cells per 1 g humanheart tissue) in close proximity to vessels and nerves, andtheir density markedly increases in heart failure, ischemiccardiomyopathy, and experimental infarct models.1 Mastcells synthesize, store, and release a variety of mediators. Werecently reported that cardiac mast cells are also an importantsource of the aspartyl protease renin.2 When released byischemia/reperfusion (I/R), this renin initiates the activationof a local renin-angiotensin system (RAS); the locally formedangiotensin II (Ang II) exacerbates norepinephrine releasefrom cardiac sympathetic nerves and elicits reperfusion ar-rhythmias.3 When mast cells are depleted or pharmacologi-
cally stabilized, renin and norepinephrine release and reper-fusion arrhythmias are markedly reduced.3 Thus, the releaseof mast cell–derived renin represents a new target in theprevention and treatment of ischemic cardiac dysfunction.
Editorial see p 761Clinical Perspective on p 781
Brief ischemic exposures have repeatedly been shown toprotect the heart both structurally and functionally from asubsequent prolonged exposure to I/R. Ischemic precondi-tioning (IPC), as this phenomenon is known, has been theobject of wide attention since its discovery.4–6 Adenosinerelease and activation/translocation of protein kinase C(PKC) have been identified as necessary steps in the cardio-
Received March 9, 2010; accepted June 14, 2010.From the Departments of Pharmacology (K.K., M.S.-R., F.C., N.Y.-K.C., R.L.) and Physiology and Biophysics (R.E., R.B.S.), Weill Cornell Medical
College, New York, NY, and Department of Chemical and Systems Biology (D.M.-R.), Stanford University School of Medicine, Stanford, Calif.*The first 2 authors contributed equally to this work.The online-only Data Supplement is available with this article at http://circ.ahajournals.org/cgi/content/full/CIRCULATIONAHA.110.952481/DC1.Correspondence to Roberto Levi, MD, DSc, Department of Pharmacology, Weill Cornell Medical College, 1300 York Ave, New York, NY 10065.
E-mail [email protected]© 2010 American Heart Association, Inc.
Circulation is available at http://circ.ahajournals.org DOI: 10.1161/CIRCULATIONAHA.110.952481
771 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

protection afforded by classic IPC (eg, infarct size reduc-tion).6–8 Of the PKC family of serine/threonine kinases, thePKC� isoform has been shown to play a key cardioprotectiverole against I/R9,10 and to exhibit antisecretory activity inmast cells.11
Accordingly, we set out to determine whether IPC mightalso prevent the activation of a local cardiac RAS initiated bythe release of mast cell renin and, if so, whether this novelIPC paradigm involves the activation of PKC� in mast cells.Given that phosphorylation of mitochondrial aldehyde dehy-drogenase type-2 (ALDH2) by PKC� correlates with cardio-protection,12 we hypothesized that IPC could promote PKC�-induced activation of ALDH2, which would then remove thetoxic aldehydes known to degranulate mast cells such asacetaldehyde and 4-hydroxynonenal (4-HNE), which areformed by lipid peroxidation.13–15 Our study outlines a novelprotective anti-RAS effect of IPC; we find that the sequentialactivation of adenosine A2b and A3 receptors, PKC�, andALDH2 in cardiac mast cells diminishes the release of reninelicited by I/R and thus curtails Ang II– and norepinephrine-induced arrhythmias.
MethodsSee the online-only Data Supplement for the completeMethods section.
Ex Vivo HeartsIn total, 132 male Hartley guinea pigs (weight, 300 to 350 g; CharlesRiver Laboratories, Kingston, NY) were anesthetized with CO2 andeuthanized by stunning with approval from the Institutional AnimalCare and Use Committee. Isolated hearts were perfused at constantpressure with oxygenated Ringer at 37°C in a Langendorff apparatus(Radnoti Glass Technology, Monrovia, Calif).
Ischemia/ReperfusionAfter equilibration, all hearts were subjected to 20 minutes of globalischemia followed by 30 minutes of reperfusion. For IPC, two5-minute cycles of ischemia were each followed by 5 minutes ofreperfusion. For pharmacological prevention of IPC, antagonistswere perfused for 20 minutes (glyceryl trinitrate [GTN] for 30minutes) before and during IPC and then washed out for 15 minutesbefore I/R. For pharmacological preconditioning, given agents wereperfused for two 5-minute cycles before I/R except for �V1-1 (PKC�inhibitor), which was administered during the entire 30-minutereperfusion following the 20-minute ischemia. For prevention ofpharmacological preconditioning, antagonists were perfused for 20minutes (GTN for 30 minutes) before and during pharmacologicalpreconditioning and then washed out for 15 minutes before I/R.
Coronary flow was measured every 2 minutes; samples wereassayed for renin, norepinephrine, �-hexosaminidase, and creatinephosphokinase (CPK). Surface ECG was obtained from the leftventricle and right atrium, recorded in digital format, and analyzedwith Power Laboratory/8SP (AdInstrument, Colorado Springs,Colo).
Cell CultureThe human mastocytoma cell line (HMC-1) was a gift of from Dr I.Biaggioni (Vanderbilt University, Nashville, Tenn). Cells weremaintained in suspension culture as previously described.2
�-Hexosaminidase and Renin Assay�-Hexosaminidase and renin coronary overflow was measured aspreviously described.3 HMC-1 cells were suspended in Ringerbuffer, and equal volumes were divided into aliquots in Eppendorftubes and incubated at 37°C with a given agent (ie, Alda-1,
��RACK, or LUF5835 plus IBMECA) for 10 minutes (preceded ornot by a 30-minute incubation with GTN). Acetaldehyde, H2O2, or4-HNE was subsequently added for 20 minutes. All results werenormalized and expressed as percent above control.
Norepinephrine AssayCoronary effluent was assayed for norepinephrine by high-performance liquid chromatography with electrochemical detectionas previously described.3
CPK AssayCoronary effluent was assayed for CPK release with a CPK assay kit(Genzyme Diagnostics, Charlottetown, Prince Edward Island,Canada).
Polymerase Chain Reaction and ImmunostainingFor reverse-transcription polymerase chain reaction (RT-PCR), totalRNA was extracted from HMC-1 cells with TRIzol reagent (Invitro-gen, Carlsbad, Calif), 1 �g total RNA from each sample was reversetranscribed, and complementary DNA was amplified by RT-PCRwith a Qiagen (Valencia, Calif) 1-step RT-PCR kit. PCR productswere analyzed by agarose gel electrophoresis and ethidium bromidestaining. For immunostaining, HMC-1 cells were fixed and perme-abilized on glass slides and stained with the goat anti–A2b receptorantibody (Santa Cruz Biotechnology, Santa Cruz, Calif) conjugatedto Alexa Fluor 488 donkey anti-goat IgG and with rabbit anti–A3
receptor antibody (Santa Cruz) conjugated to Alexa Fluor 488donkey anti-rabbit IgG. Nuclei were stained with DAPI. For immu-nofluorescence, cells were examined with an inverted fluorescencemicroscope (Nikon Eclipse TE 2000-U, Morrell Instruments,Melville, NY) interfaced to an electron multiplying charge-coupleddevice (Hamamatsu Photonics, Bridgewater, NJ) and processedwith Metamorph software (version 6.2, Universal Imaging Corp,Downington, Pa).
Translocation of PKC�Cytosolic and membrane fractions of HMC-1 cells were separated,and Western blot analysis was performed with a PKC�-specificantibody (Santa Cruz).
ALDH2 Enzymatic Activity AssayALDH2 activity in HMC-1 cells was determined spectrophotometri-cally by monitoring the reductive reaction of NAD� to NADH at 340nm as previously described.16
Drugs and ChemicalsAcetaldehyde, H2O2, IBMECA, MRS1754, MRS1523, DPCPX,chelerythrine, 5-hydroxydecanoate, and cyanamide were purchasedfrom Sigma-Aldrich (St Louis, Mo); 4-HNE in ethanol solution waspurchased from Cayman Chemical (Ann Arbor, Mich). LUF5835was a gift from Dr M.W. Beukers (University of Leiden, Leiden, theNetherlands); EXP3174 was a gift from Merck Sharp & Dohme Ltd(Whitehouse Station, NJ); ��RACK, �V1-1, and Alda-1 weresynthesized in Dr Mochly-Rosen’s laboratory (Stanford UniversitySchool of Medicine, Palo Alto, Calif). Phorbol 12-myristate 13-acetate was purchased from LC Laboratories (Woburn, Mass). GTNwas purchased from Hospira Inc (Lake Forest, Ill). Human plasmaangiotensinogen was purchased from Calbiochem (San Diego,Calif).
StatisticsData are presented as mean�SEM. Nonparametric tests were usedthroughout the study. For 2-group comparisons, the Mann–Whitneytest was used (Figures 1 and 2). For comparisons among �2 groups,the Kruskal-Wallis test followed by the posthoc Dunn test was used(Figures 1 through 3, 4D, 4F, and 5 through 7). GraphPad Prismversion 4.03 for Windows (GraphPad Software, San Diego, Calif)was used. Values of P�0.05 were considered statistically significant.
772 Circulation August 24, 2010
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from
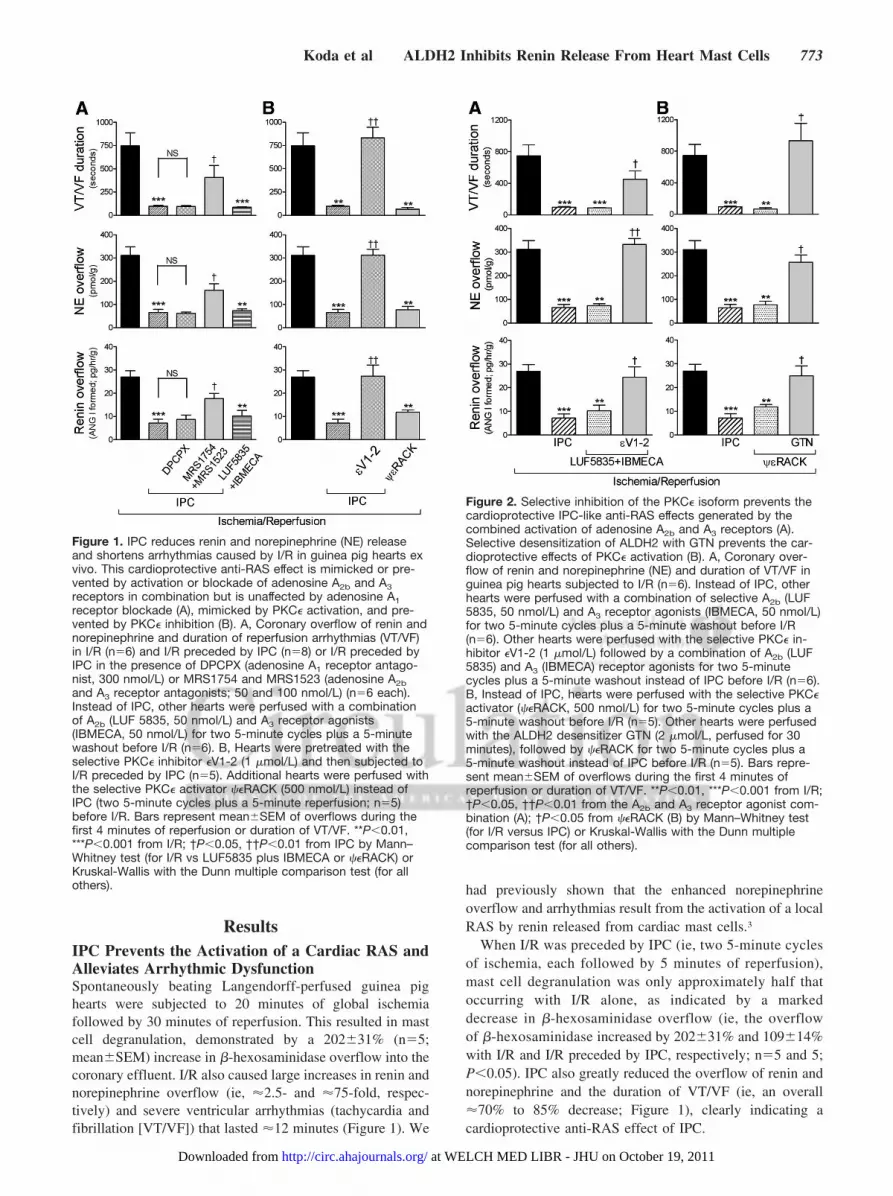
ResultsIPC Prevents the Activation of a Cardiac RAS andAlleviates Arrhythmic DysfunctionSpontaneously beating Langendorff-perfused guinea pighearts were subjected to 20 minutes of global ischemiafollowed by 30 minutes of reperfusion. This resulted in mastcell degranulation, demonstrated by a 202�31% (n�5;mean�SEM) increase in �-hexosaminidase overflow into thecoronary effluent. I/R also caused large increases in renin andnorepinephrine overflow (ie, �2.5- and �75-fold, respec-tively) and severe ventricular arrhythmias (tachycardia andfibrillation [VT/VF]) that lasted �12 minutes (Figure 1). We
had previously shown that the enhanced norepinephrineoverflow and arrhythmias result from the activation of a localRAS by renin released from cardiac mast cells.3
When I/R was preceded by IPC (ie, two 5-minute cyclesof ischemia, each followed by 5 minutes of reperfusion),mast cell degranulation was only approximately half thatoccurring with I/R alone, as indicated by a markeddecrease in �-hexosaminidase overflow (ie, the overflowof �-hexosaminidase increased by 202�31% and 109�14%with I/R and I/R preceded by IPC, respectively; n�5 and 5;P�0.05). IPC also greatly reduced the overflow of renin andnorepinephrine and the duration of VT/VF (ie, an overall�70% to 85% decrease; Figure 1), clearly indicating acardioprotective anti-RAS effect of IPC.
Figure 1. IPC reduces renin and norepinephrine (NE) releaseand shortens arrhythmias caused by I/R in guinea pig hearts exvivo. This cardioprotective anti-RAS effect is mimicked or pre-vented by activation or blockade of adenosine A2b and A3receptors in combination but is unaffected by adenosine A1receptor blockade (A), mimicked by PKC� activation, and pre-vented by PKC� inhibition (B). A, Coronary overflow of renin andnorepinephrine and duration of reperfusion arrhythmias (VT/VF)in I/R (n�6) and I/R preceded by IPC (n�8) or I/R preceded byIPC in the presence of DPCPX (adenosine A1 receptor antago-nist, 300 nmol/L) or MRS1754 and MRS1523 (adenosine A2band A3 receptor antagonists; 50 and 100 nmol/L) (n�6 each).Instead of IPC, other hearts were perfused with a combinationof A2b (LUF 5835, 50 nmol/L) and A3 receptor agonists(IBMECA, 50 nmol/L) for two 5-minute cycles plus a 5-minutewashout before I/R (n�6). B, Hearts were pretreated with theselective PKC� inhibitor �V1-2 (1 �mol/L) and then subjected toI/R preceded by IPC (n�5). Additional hearts were perfused withthe selective PKC� activator ��RACK (500 nmol/L) instead ofIPC (two 5-minute cycles plus a 5-minute reperfusion; n�5)before I/R. Bars represent mean�SEM of overflows during thefirst 4 minutes of reperfusion or duration of VT/VF. **P�0.01,***P�0.001 from I/R; †P�0.05, ††P�0.01 from IPC by Mann–Whitney test (for I/R vs LUF5835 plus IBMECA or ��RACK) orKruskal-Wallis with the Dunn multiple comparison test (for allothers).
Figure 2. Selective inhibition of the PKC� isoform prevents thecardioprotective IPC-like anti-RAS effects generated by thecombined activation of adenosine A2b and A3 receptors (A).Selective desensitization of ALDH2 with GTN prevents the car-dioprotective effects of PKC� activation (B). A, Coronary over-flow of renin and norepinephrine (NE) and duration of VT/VF inguinea pig hearts subjected to I/R (n�6). Instead of IPC, otherhearts were perfused with a combination of selective A2b (LUF5835, 50 nmol/L) and A3 receptor agonists (IBMECA, 50 nmol/L)for two 5-minute cycles plus a 5-minute washout before I/R(n�6). Other hearts were perfused with the selective PKC� in-hibitor �V1-2 (1 �mol/L) followed by a combination of A2b (LUF5835) and A3 (IBMECA) receptor agonists for two 5-minutecycles plus a 5-minute washout instead of IPC before I/R (n�6).B, Instead of IPC, hearts were perfused with the selective PKC�activator (��RACK, 500 nmol/L) for two 5-minute cycles plus a5-minute washout before I/R (n�5). Other hearts were perfusedwith the ALDH2 desensitizer GTN (2 �mol/L, perfused for 30minutes), followed by ��RACK for two 5-minute cycles plus a5-minute washout instead of IPC before I/R (n�5). Bars repre-sent mean�SEM of overflows during the first 4 minutes ofreperfusion or duration of VT/VF. **P�0.01, ***P�0.001 from I/R;†P�0.05, ††P�0.01 from the A2b and A3 receptor agonist com-bination (A); †P�0.05 from ��RACK (B) by Mann–Whitney test(for I/R versus IPC) or Kruskal-Wallis with the Dunn multiplecomparison test (for all others).
Koda et al ALDH2 Inhibits Renin Release From Heart Mast Cells 773
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

Notably, the overflow of renin and norepinephrine duringpreconditioning was only slightly and not significantly higherthan that in nonconditioned hearts (renin overflow, 7.10�0.99pg � h�1 � g�1 Ang I formed during IPC and 6.81�2.12pg � h�1 � g�1 Ang I in control conditions; norepinephrine over-flow, 3.82�1.17 pmol/g during IPC versus 4.02�0.77 pmol/g incontrol conditions; mean�SEM; n�8�6; P�0.95 and P�0.75for renin and norepinephrine, respectively). Thus, the findingthat IPC markedly attenuated the I/R-induced release of reninand norepinephrine was not due to depletion of renin andnorepinephrine pools during preconditioning.
Combined Activation of Adenosine A2b and A3
Receptors Mimics the Cardioprotective Anti-RASEffects of IPCAdenosine, PKC, and mitochondrial ATP-sensitive potassiumchannels (mKATP) have been identified as necessary steps in the
cardioprotection afforded by classic IPC (eg, infarct size reduc-tion).6–8 Furthermore, although Ang II is widely perceived as adeleterious agent, it has also been found to mimic the cardio-protective effects of IPC.17–19 Thus, we investigated whetheradenosine, mKATP channels, Ang II, and PKC play a role in thecardioprotective anti-RAS IPC paradigm.
First, we assessed whether inhibition or activation of adeno-sine receptors prevents or mimics, respectively, the IPC-mediated attenuation of renin release in hearts subjected to I/R.We found that the combined blockade of adenosine A2b and A3
receptors with the MRS1754 (50 nmol/L)20 and MRS1523 (100nmol/L)21 compounds prevented the IPC-induced attenuation ofrenin and norepinephrine release and the alleviation of reperfu-sion arrhythmias (Figure 1A). Conversely, the combined activa-tion of A2b and A3 receptors with the LUF5835 (50 nmol/L)22
and IBMECA (50 nmol/L)23 compounds mimicked the cardio-protective anti-RAS effects of IPC (Figure 1A). In contrast,activation of A2b or A3 receptors alone failed to mimic theeffects of IPC (renin overflow, 26.94�2.80, 19.77�3.9, and24.37�2.58 pg � h�1 � g�1 Ang I formed for I/R, I/R plusLUF5835, and I/R plus IBMECA, respectively; n�6�5�5,P�0.20). Blockade of adenosine A1 receptors with DPCPX(300 nmol/L; IC50�18 nmol/L)24 was also ineffective (Figure1A). Thus, only the combined activation of A2b and A3 receptorsappears to induce the cardioprotective anti-RAS effects of IPC.
Because the opening of mKATP channels is known to beinvolved in the mediation of classic IPC-induced cardioprotec-tion,7,25,26 we questioned whether these channels may also playa role in the anti-RAS effects of IPC. Thus, we induced IPC inthe presence of the mKATP antagonist 5-hydroxydecanoate(100 �mol/L; IC50�30 �mol/L).27 5-Hydroxydecanoate failedto affect the cardioprotective anti-RAS effects of IPC. In fact, theIPC-induced attenuation of renin and norepinephrine release andthe abbreviation of reperfusion arrhythmias were the same in thepresence and absence of 5-hydroxydecanoate (Figure IA in theonline-only Data Supplement). Hence, the cardioprotective anti-RAS effects of IPC do not appear to depend on the opening ofmKATP channels.
According to early reports in rabbits and rats, Ang II mim-icked the cardioprotective effects of classic IPC.17–19 Thus, wedetermined whether Ang II, which is locally produced in theheart subjected to I/R,3 contributes to the anti-RAS effects ofIPC. For this, we induced IPC in hearts perfused with the AT1
receptor antagonist EXP3174. EXP3174 (300 nmol/L; IC50�6nmol/L)28 did not prevent the cardioprotective anti-RAS effectsof IPC (Figure IA in the online-only Data Supplement). In fact,the IPC-induced attenuation of renin and norepinephrine releaseand the abbreviation of reperfusion arrhythmias were the same inthe presence and absence of EXP3174 (Figure IA in theonline-only Data Supplement). Therefore, AT1 receptors areprobably not involved in the mediation of the cardioprotectiveanti-RAS effects of IPC.
Translocation of PKC� Mediates theCardioprotective Anti-RAS Effects of AdenosineA2b and A3 Receptor ActivationBecause PKC activation/translocation is likely to be involvedin the cardioprotective effects of classic IPC,8,29 we nextinvestigated the role of PKC in the cardioprotective anti-RAS
Figure 3. Selective activation of ALDH2 with Alda-1 mimics thecardioprotective anti-RAS effects of IPC. ALDH inhibition withcyanamide or selective ALDH2 desensitization with GTN eachprevents the effects of IPC and Alda-1. A, Coronary overflow ofrenin and norepinephrine (NE) and duration of VT/VF in guineapigs hearts subjected to I/R (n�6) or I/R preceded by IPC eitherin the absence (n�8) or presence (n�5) of the ALDH inhibitorcyanamide (CYA; 5 mmol/L). Other hearts were perfused withthe selective ALDH-2 activator Alda-1 (20 �mol/L) instead ofIPC (two 5-minute cycles plus a 5-minute washout; n�5) beforeI/R. Other hearts were perfused with cyanamide, followed byAlda-1 (two 5-minute cycles plus a 5-minute reperfusion)instead of IPC before I/R (n�5). B, Hearts were pretreated withthe ALDH2 desensitizer GTN (2 �mol/L for 30 minutes) and thensubjected to I/R preceded by IPC (n�5). Some hearts were per-fused with Alda-1 instead of IPC before I/R (n�5). Other heartswere perfused with GTN (2 �mol/L for 30 minutes), followedby Alda-1 instead of IPC before I/R (n�5). Bars representmean�SEM of overflows collected during the first 4 minutes ofreperfusion or duration of VT/VF. *P�0.05, **P�0.01, ***P�0.001from I/R; #P�0.05, ##P�0.01, ###P�0.001 from IPC; †P�0.05,††P�0.01 from Alda-1 by Kruskal-Wallis with the Dunn multiplecomparison test.
774 Circulation August 24, 2010
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

effects of IPC. Treatment of hearts with the general, non–isoform-selective PKC activator phorbol 12-myristate 13-acetate (PMA; 0.05 nmol/L, two 5-minute cycles before I/R)mimicked the protective effects of IPC on renin and norepi-nephrine release and reperfusion arrhythmia duration (FigureIB in the online-only Data Supplement). Moreover, inhibitionof PKC with the specific but non–isoform-selective chel-erythrine (2.8 �mol/L) prevented the effects of IPC on thesame parameters (Figure IB in the online-only Data Supple-ment). Thus, general PKC activation appears to mediate thecardioprotective anti-RAS effects of IPC.
Of the PKC family of serine/threonine kinases, the PKC�isoform has been shown to play a key cardioprotective roleagainst I/R.9,10,30,31 Thus, we tested whether ��RACK, aselective activator peptide of PKC�,32 mimics the anti-RASeffects of IPC. ��RACK (500 nmol/L), perfused for two5-minute cycles followed by a 5-minute washout before I/R,mimicked the protective anti-RAS effects of IPC. Indeed, theoverflow of renin and norepinephrine and the duration ofVT/VF were reduced by �55% to 90% compared with I/Rhearts (Figure 1B). Moreover, selective inhibition of PKC�with �V1-2 (1 �mol/L)33 prevented the effects of IPC on thesame parameters (Figure 1B). Thus, PKC� activation appearsto be required and sufficient for the genesis of the cardiopro-tective anti-RAS effects of IPC.
Stimulation of adenosine A2b and A3 receptors mimickedthe anti-RAS effects of IPC (see Figure 1A); selectiveactivation of the PKC� isoform also had anti-RAS effectssimilar to IPC (see Figure 1B). Given that adenosine is knownto activate PKC, thus initiating the traditional preconditioningcascade,6–8 we determined whether the anti-RAS IPC-likeeffects of A2b and A3 receptors rely on the consequentactivation of PKC�. To verify this notion, we assessedwhether PKC� blockade would prevent the IPC-like effects ofA2b and A3 receptor agonists. We found that selectiveinhibition of the PKC� isozyme with �V1-2 (1 �mol/L)33
prevented the anti-RAS IPC-like effects resulting from thecombined activation of A2b and A3 receptors (Figure 2A).Thus, A2b and A3 receptor–mediated activation of PKC�appears to be the first significant step in the anti-RASpreconditioning pathway.
Activation of ALDH2 Is Pivotal for theCardioprotective Anti-RAS Effects of PKC�Because the cardioprotective infarct-sparing effects of PKC�activation30 have been found to depend on phosphorylation ofmitochondrial ALDH2,12,34 we next assessed whether theanti-RAS effects of IPC are also determined by ALDH2activation. For this, we assessed whether inhibition/inactiva-tion of ALDH2 would abolish the anti-RAS effects of IPCand whether activation of ALDH2 would mimic them. Wefound that GTN, perfused for 30 minutes at a concentrationknown to inactivate ALDH2 (2 �mol/L),12 prevented thecardioprotective anti-RAS effects of ��RACK (ie, GTNabolished the ��RACK-induced inhibition of renin and nor-epinephrine release and the alleviation of VT/VF; Figure 2B).We also found that the general ALDH inhibitors cyanamide(5 mmol/L)12 and GTN prevented the anti-RAS effects of IPC(Figure 3A and 3B). Conversely, selective activation of
ALDH2 with Alda-112,34 (20 �mol/L, two 5-minute cycles)reproduced all of the anti-RAS effects of IPC, an action thatwas also prevented by cyanamide (Figure 3A) and bypretreatment with GTN (Figure 3B). Collectively, thesefindings suggest that ALDH2 activation by PKC� is a crucialmechanistic step in the development of the anti-RAS effectsof IPC.
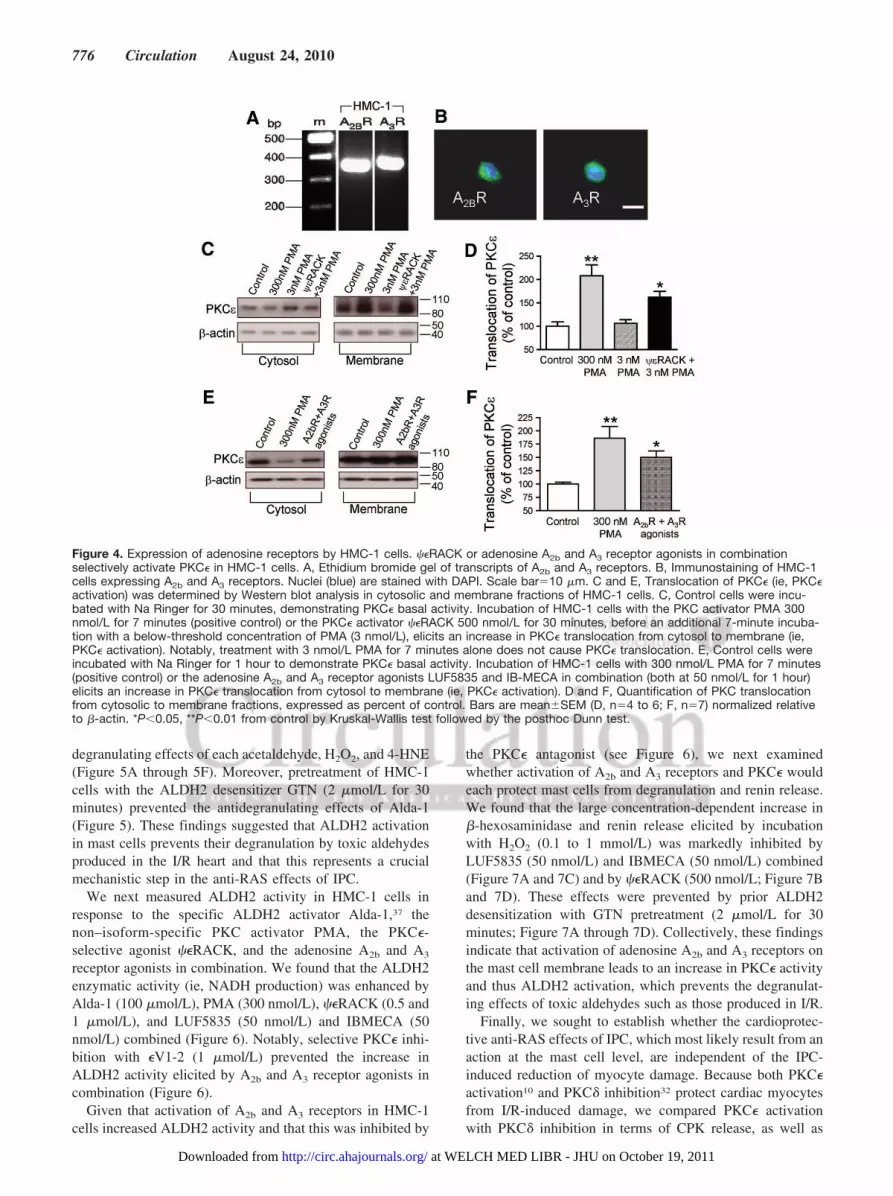
Mast Cells Are the Site of the CardioprotectiveAnti-RAS Action of IPCGiven the pivotal role that mast cells play in the activation ofRAS in the heart,3 cardiac mast cells are likely to be the siteat which the anti-RAS effects of IPC develop. Becausecombined activation of adenosine A2b and A3 receptorsmimics the cardioprotective anti-RAS effects of IPC, whereasthe combined blockade of the same receptors prevents theanti-RAS effects (see Figure 1A), we first ascertained thepresence of A2b and A3 receptors on mast cells. For this, weused human mast cells in culture (HMC-1 cells). Total RNA(1 �g) was extracted from HMC-1 cells, reverse transcribed,and amplified by PCR using sense and antisense primersspecific for human A2b and A3 receptor genes. Figure 4Adepicts an ethidium bromide–stained gel showing that theHMC-1 PCR products for these adenosine receptor subtypesare consistent with those reported by others.35 HMC-1 cellswere also immunopositive for the 2 adenosine receptorsubtypes (Figure 4B).
We next made certain that mast cell PKC� can be activated.Using Western analysis in cytosolic and membrane fractionsof HMC-1 cells, we found that the phorbol ester PMA(positive control) markedly increased the translocation ofPKC� from cytosol to membrane (ie, a hallmark of PKC�activation; Figure 4C through 4F). Incubation of HMC-1 cellswith the PKC� activator ��RACK (500 nmol/L for 30minutes) before a 7-minute incubation with a below-thresholdconcentration of PMA (3 nmol/L)36 also significantly trans-located PKC� from cytosol to membrane (Figure 4C and 4D).Moreover, incubating HMC-1 cells with the adenosine A2b
and A3 receptor agonists in combination (LUF5835 andIBMECA, 50 nmol/L each for 1 hour) also translocated PKC�(Figure 4E and 4F).
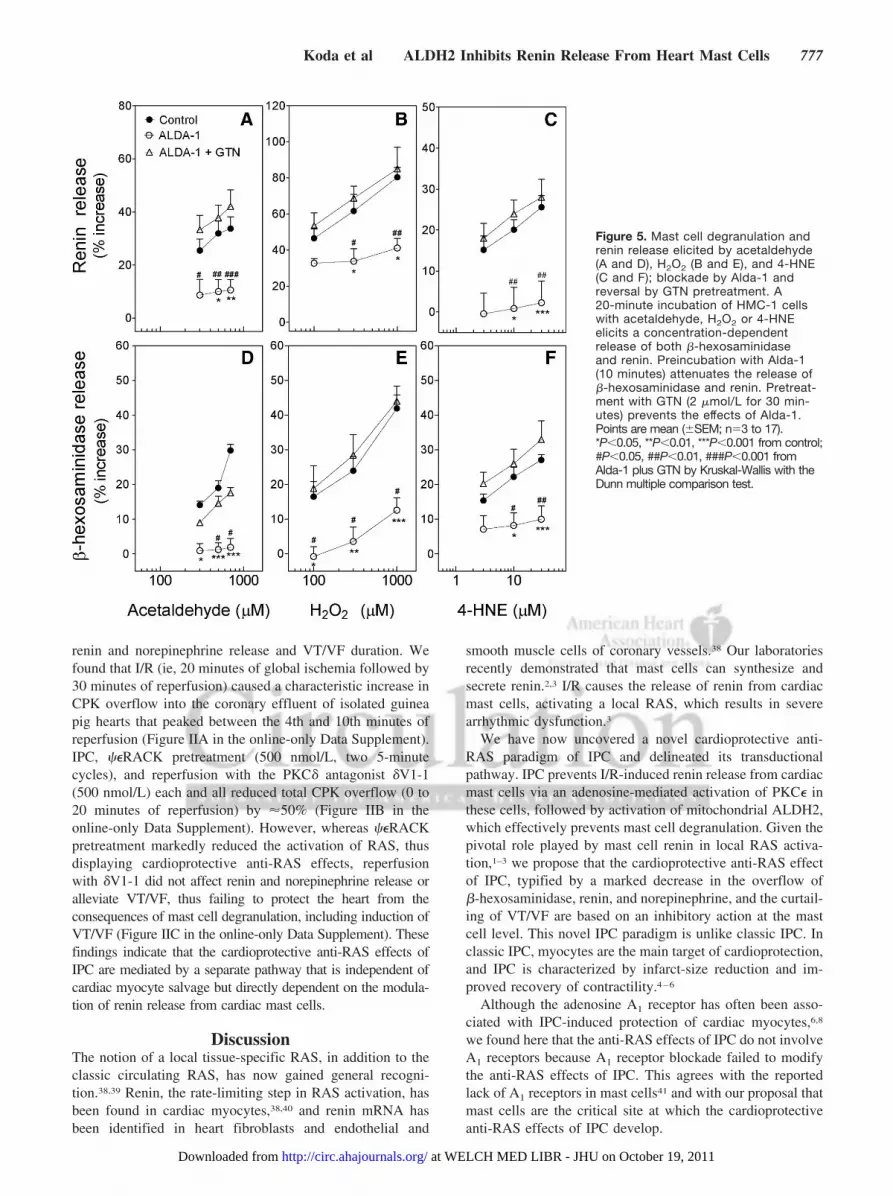
Our findings in isolated guinea pig hearts and culturedmast cells suggested that IPC may result from the activationof adenosine A2b and A3 receptors expressed by cardiac mastcells and consequent PKC�-dependent activation of mito-chondrial ALDH2. Thus, we next investigated the role ofALDH2 in mast cell degranulation and renin release elicitedby prototypic toxic compounds formed in I/R. For this, wemeasured mast cell degranulation in response to acetalde-hyde, 4-HNE, another toxic aldehyde that accumulates duringcardiac ischemia, and hydrogen peroxide (H2O2), whichtriggers toxic aldehydes formation by membrane lipid per-oxidation.13,15 Incubation of HMC-1 cells with acetaldehyde(300 to 700 �mol/L), H2O2 (0.1 to 1 mmol/L), or 4-HNE (3to 30 �mol/L) elicited a concentration-dependent increase inthe release of �-hexosaminidase (�15% to 42%, an indica-tion of mast cell degranulation) and renin (�15% to 80%;Figure 5). Notably, preincubation of HMC-1 cells with theALDH2 activator Alda-1 (20 �mol/L)12,34 prevented the
Koda et al ALDH2 Inhibits Renin Release From Heart Mast Cells 775
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from
degranulating effects of each acetaldehyde, H2O2, and 4-HNE(Figure 5A through 5F). Moreover, pretreatment of HMC-1cells with the ALDH2 desensitizer GTN (2 �mol/L for 30minutes) prevented the antidegranulating effects of Alda-1(Figure 5). These findings suggested that ALDH2 activationin mast cells prevents their degranulation by toxic aldehydesproduced in the I/R heart and that this represents a crucialmechanistic step in the anti-RAS effects of IPC.
We next measured ALDH2 activity in HMC-1 cells inresponse to the specific ALDH2 activator Alda-1,37 thenon–isoform-specific PKC activator PMA, the PKC�-selective agonist ��RACK, and the adenosine A2b and A3
receptor agonists in combination. We found that the ALDH2enzymatic activity (ie, NADH production) was enhanced byAlda-1 (100 �mol/L), PMA (300 nmol/L), ��RACK (0.5 and1 �mol/L), and LUF5835 (50 nmol/L) and IBMECA (50nmol/L) combined (Figure 6). Notably, selective PKC� inhi-bition with �V1-2 (1 �mol/L) prevented the increase inALDH2 activity elicited by A2b and A3 receptor agonists incombination (Figure 6).
Given that activation of A2b and A3 receptors in HMC-1cells increased ALDH2 activity and that this was inhibited by
the PKC� antagonist (see Figure 6), we next examinedwhether activation of A2b and A3 receptors and PKC� wouldeach protect mast cells from degranulation and renin release.We found that the large concentration-dependent increase in�-hexosaminidase and renin release elicited by incubationwith H2O2 (0.1 to 1 mmol/L) was markedly inhibited byLUF5835 (50 nmol/L) and IBMECA (50 nmol/L) combined(Figure 7A and 7C) and by ��RACK (500 nmol/L; Figure 7Band 7D). These effects were prevented by prior ALDH2desensitization with GTN pretreatment (2 �mol/L for 30minutes; Figure 7A through 7D). Collectively, these findingsindicate that activation of adenosine A2b and A3 receptors onthe mast cell membrane leads to an increase in PKC� activityand thus ALDH2 activation, which prevents the degranulat-ing effects of toxic aldehydes such as those produced in I/R.
Finally, we sought to establish whether the cardioprotec-tive anti-RAS effects of IPC, which most likely result from anaction at the mast cell level, are independent of the IPC-induced reduction of myocyte damage. Because both PKC�activation10 and PKC� inhibition32 protect cardiac myocytesfrom I/R-induced damage, we compared PKC� activationwith PKC� inhibition in terms of CPK release, as well as
Figure 4. Expression of adenosine receptors by HMC-1 cells. ��RACK or adenosine A2b and A3 receptor agonists in combinationselectively activate PKC� in HMC-1 cells. A, Ethidium bromide gel of transcripts of A2b and A3 receptors. B, Immunostaining of HMC-1cells expressing A2b and A3 receptors. Nuclei (blue) are stained with DAPI. Scale bar�10 �m. C and E, Translocation of PKC� (ie, PKC�activation) was determined by Western blot analysis in cytosolic and membrane fractions of HMC-1 cells. C, Control cells were incu-bated with Na Ringer for 30 minutes, demonstrating PKC� basal activity. Incubation of HMC-1 cells with the PKC activator PMA 300nmol/L for 7 minutes (positive control) or the PKC� activator ��RACK 500 nmol/L for 30 minutes, before an additional 7-minute incuba-tion with a below-threshold concentration of PMA (3 nmol/L), elicits an increase in PKC� translocation from cytosol to membrane (ie,PKC� activation). Notably, treatment with 3 nmol/L PMA for 7 minutes alone does not cause PKC� translocation. E, Control cells wereincubated with Na Ringer for 1 hour to demonstrate PKC� basal activity. Incubation of HMC-1 cells with 300 nmol/L PMA for 7 minutes(positive control) or the adenosine A2b and A3 receptor agonists LUF5835 and IB-MECA in combination (both at 50 nmol/L for 1 hour)elicits an increase in PKC� translocation from cytosol to membrane (ie, PKC� activation). D and F, Quantification of PKC translocationfrom cytosolic to membrane fractions, expressed as percent of control. Bars are mean�SEM (D, n�4 to 6; F, n�7) normalized relativeto �-actin. *P�0.05, **P�0.01 from control by Kruskal-Wallis test followed by the posthoc Dunn test.
776 Circulation August 24, 2010
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from
renin and norepinephrine release and VT/VF duration. Wefound that I/R (ie, 20 minutes of global ischemia followed by30 minutes of reperfusion) caused a characteristic increase inCPK overflow into the coronary effluent of isolated guineapig hearts that peaked between the 4th and 10th minutes ofreperfusion (Figure IIA in the online-only Data Supplement).IPC, ��RACK pretreatment (500 nmol/L, two 5-minutecycles), and reperfusion with the PKC� antagonist �V1-1(500 nmol/L) each and all reduced total CPK overflow (0 to20 minutes of reperfusion) by �50% (Figure IIB in theonline-only Data Supplement). However, whereas ��RACKpretreatment markedly reduced the activation of RAS, thusdisplaying cardioprotective anti-RAS effects, reperfusionwith �V1-1 did not affect renin and norepinephrine release oralleviate VT/VF, thus failing to protect the heart from theconsequences of mast cell degranulation, including induction ofVT/VF (Figure IIC in the online-only Data Supplement). Thesefindings indicate that the cardioprotective anti-RAS effects ofIPC are mediated by a separate pathway that is independent ofcardiac myocyte salvage but directly dependent on the modula-tion of renin release from cardiac mast cells.
DiscussionThe notion of a local tissue-specific RAS, in addition to theclassic circulating RAS, has now gained general recogni-tion.38,39 Renin, the rate-limiting step in RAS activation, hasbeen found in cardiac myocytes,38,40 and renin mRNA hasbeen identified in heart fibroblasts and endothelial and
smooth muscle cells of coronary vessels.38 Our laboratoriesrecently demonstrated that mast cells can synthesize andsecrete renin.2,3 I/R causes the release of renin from cardiacmast cells, activating a local RAS, which results in severearrhythmic dysfunction.3
We have now uncovered a novel cardioprotective anti-RAS paradigm of IPC and delineated its transductionalpathway. IPC prevents I/R-induced renin release from cardiacmast cells via an adenosine-mediated activation of PKC� inthese cells, followed by activation of mitochondrial ALDH2,which effectively prevents mast cell degranulation. Given thepivotal role played by mast cell renin in local RAS activa-tion,1–3 we propose that the cardioprotective anti-RAS effectof IPC, typified by a marked decrease in the overflow of�-hexosaminidase, renin, and norepinephrine, and the curtail-ing of VT/VF are based on an inhibitory action at the mastcell level. This novel IPC paradigm is unlike classic IPC. Inclassic IPC, myocytes are the main target of cardioprotection,and IPC is characterized by infarct-size reduction and im-proved recovery of contractility.4–6
Although the adenosine A1 receptor has often been asso-ciated with IPC-induced protection of cardiac myocytes,6,8
we found here that the anti-RAS effects of IPC do not involveA1 receptors because A1 receptor blockade failed to modifythe anti-RAS effects of IPC. This agrees with the reportedlack of A1 receptors in mast cells41 and with our proposal thatmast cells are the critical site at which the cardioprotectiveanti-RAS effects of IPC develop.
Figure 5. Mast cell degranulation andrenin release elicited by acetaldehyde(A and D), H2O2 (B and E), and 4-HNE(C and F); blockade by Alda-1 andreversal by GTN pretreatment. A20-minute incubation of HMC-1 cellswith acetaldehyde, H2O2 or 4-HNEelicits a concentration-dependentrelease of both �-hexosaminidaseand renin. Preincubation with Alda-1(10 minutes) attenuates the release of�-hexosaminidase and renin. Pretreat-ment with GTN (2 �mol/L for 30 min-utes) prevents the effects of Alda-1.Points are mean (�SEM; n�3 to 17).*P�0.05, **P�0.01, ***P�0.001 from control;#P�0.05, ##P�0.01, ###P�0.001 fromAlda-1 plus GTN by Kruskal-Wallis with theDunn multiple comparison test.
Koda et al ALDH2 Inhibits Renin Release From Heart Mast Cells 777
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

Mast cells are known to express both adenosine A2b and A3
receptors42,43 (see also Figure 4). We found that the combinedactivation of A2b and A3 receptors displayed IPC-like effects:It attenuated the release of renin and norepinephrine andalleviated reperfusion arrhythmias associated with I/R. Thesecardioprotective anti-RAS effects were similar to those af-forded by IPC, which in fact was abolished by blockade ofadenosine A2b and A3 receptors in combination. AlthoughA2b and A3 receptors are known as low-affinity receptors (Ki
�5 and 1 �mol/L for A2b and A3, respectively),44 both werelikely activated by endogenous adenosine during IPC. Indeed,interstitial adenosine was shown to reach a 7-�mol/L levelafter 6 minutes of IPC in the isolated heart.45
Significantly, the combined activation of A2b and A3
receptors in HMC-1 cells in culture also prevented peroxide-induced degranulation and renin release, supporting ourconclusion that these mast cell receptors play a major role inthe anti-RAS effects of IPC. Activation of either A2b or A3
receptor alone failed to mimic the cardioprotective effects ofIPC, demonstrating the necessity that both receptors beactivated for the initiation of IPC. Other actions of adenosinesuch as coronary dilatation have also been shown to require aconcomitant activation of both A2b and A3 receptors.46
That the activation of A2b and A3 receptors in HMC-1 cellsprevents degranulation and renin release concurs with the
protective antisecretory effect of A2b receptors, demonstratedby enhanced mast cell activation when A2b receptors aredeleted in mice.47 Yet, other investigators have shown thatactivation of A2b and A3 receptors promotes the release ofmediators and cytokines from human lung fragments, ratRBL-2H3 cells, HMC-1 cells, and macrophages from A2b
receptor–deleted mice.42,43,48 The discrepancy between thesefindings and ours most likely depends on differences in cellsand animal species and on the different stimuli used todegranulate mast cells.
Opening of KATP channels in the inner membrane ofmitochondria in cardiac myocytes has been found to contrib-ute to the protective effects of classic IPC.6–8Yet, the mKATP
channel antagonist 5-hydroxydecanoate27 failed to modify thecardioprotective anti-RAS effects of IPC, indicating thatthese mitochondrial channels are not involved in the mastcell–dependent anti-RAS effects of IPC.
Having established that activation of A2b and A3 receptorscontributes to the anti-RAS effects of IPC, we asked whether
Figure 6. Activation of adenosine A2b and A3 receptors in combi-nation or PKC� increases ALDH2 activity in HMC-1 cells. Selectiveinhibition of the PKC� isoform prevents the increase in ALDH2activity by the combined activation of adenosine A2b and A3 recep-tors. Incubation of HMC-1 cells with the selective ALDH2 activatorAlda-1 (100 �mol/L for 10 minutes), the PKC activator PMA (300nmol/L for 10 minutes), the PKC�-selective activator ��RACK (0.5and 1 �mol/L for 30 minutes), or the adenosine A2b and A3 recep-tor agonists LUF5835 and IB-MECA in combination (both at 50nmol/L for 1 hour) increases ALDH2 activity (measured by the rateof NADH production at 340 nm). Pretreatment of HMC-1 cells withthe selective PKC� antagonist ��V1-2 (1 �mol/L for 20 minutes)prevents the effects of A2b and A3 receptor activation. Bars aremean percent increases from control (�SEM; n�4 to 8). BasalNADH production was 3.63�0.25 �mol � min�1 � /mg�1 protein.*P�0.05, **P�0.01, ***P�0.001 from control; #P�0.05 from thecombination of A2b and A3 receptor agonists by Kruskal-Wallis testfollowed by the posthoc Dunn test.
Figure 7. Activation of A2b and A3 receptors (A and C) or PKC�(B and D) inhibits mast cell degranulation and renin release elic-ited by H2O2; prevention by GTN pretreatment. Incubation ofHMC-1 cells with H2O2 for 20 minutes elicits a concentration-dependent release of both �-hexosaminidase and renin. Prein-cubation of HMC-1 cells with the A2b and A3 receptor agonistsLUF5835 and IB-MECA (10 minutes) in combination or with��RACK (10 minutes) attenuates the H2O2-induced release of�-hexosaminidase and renin. Pretreatment with GTN (2 �mol/Lfor 30 minutes) prevents the effects of A2b and A3 receptors andPKC�. Points are mean (�SEM; n�3 to 19). *P�0.05, **P�0.01,***P�0.001 from control; #P�0.05, ##P�0.01, ###P�0.001from LUF5835 plus IB-MECA plus GTN or ��RACK plus GTN,respectively, by Kruskal-Wallis with the Dunn multiple compari-son test.
778 Circulation August 24, 2010
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

A2b and A3 receptors might signal via PKC�, given that thisisoform has both cardioprotective9,10 and antisecretory prop-erties in mast cell–like RBL-H3 cells.11 Indeed, we found thatselective blockade of PKC� with �V1-233 abolished theanti-RAS effects of IPC, whereas selective activation ofPKC� with ��RACK10 mimicked them. Notably, the IPC-likeanti-RAS effects of A2b and A3 receptor agonists in combi-nation were prevented by selective PKC� inhibition. Collec-tively, at this point, our findings suggested that the initial stepin the anti-RAS signaling sequence of IPC could be anadenosine-induced stimulation of A2b and A3 receptors re-sulting in PKC� translocation in cardiac mast cells. In fact,A2b and A3 receptor activation in HMC-1 cells caused thetranslocation of PKC� from cytosol to membrane, the hall-mark of PKC� activation.
Acetaldehyde and 4-HNE are formed during I/R,14,15 inpart by lipid peroxidation caused by reactive oxygen speciessuch as hydrogen peroxide.13 These toxic aldehydes, knownto elicit mast cell degranulation,49 can be removed bymitochondrial ALDH2, a phosphorylation target of PKC�.12
Indeed, we found that Alda-1, a selective ALDH2-activatingdrug,12,34,37 not only mimicked the cardioprotective anti-RASeffects of IPC in the guinea pig heart ex vivo but alsoprevented the degranulating effects of acetaldehyde, H2O2,and 4-HNE in HMC-1 cells in culture. These effects wereabolished by selective inactivation of ALDH2 with GTN.Similarly, pretreatment of HMC-1 cells with GTN preventedthe antidegranulating effects caused by activation of A2b andA3 receptors or PKC�. Hence, these findings add furthersupport to the proposal that activation of ALDH2 in cardiacmast cells is the final crucial step of the protective anti-RASpathway.
Because A2b and A3 receptors, PKC�, and ALDH2 are alsopresent in cardiac myocytes,37,50 it is conceivable that inaddition to mast cells, activation of ALDH2 in cardiacmyocytes mediates cardioprotection from I/R. To rule out thispossibility, we used the PKC� inhibitor �V1-1, which pro-tects cardiac myocytes from damage by I/R51 but does notaffect mast cell degranulation (Palandiyani SS and Mochly-Rosen D, unpublished results). When we compared thecardioprotective effects resulting from PKC� activation withthose caused by PKC� inhibition,32 we found that the mastcell–dependent anti-RAS effects of IPC were clearly separa-ble from the protective effects on cardiac myocytes. Onewould have expected that the cardioprotective effects ofPKC� inhibition might reduce reperfusion arrhythmias, ifthey were to originate mostly from myocytes. On the con-trary, PKC� inhibition, which reduced myocyte damage byI/R to a similar extent as PKC� activation, did not abolishreperfusion arrhythmias. Thus, mast cell renin release andRAS activation are the predominant cause of arrhythmicdysfunction in I/R, and activation of ALDH2 in mast cells isan important cardioprotective mechanism in ischemicconditions.
Cardiac myocytes have been reported to express all of theRAS components, including renin.38,40 Renin mRNA has alsobeen found in heart fibroblasts and in endothelial and smoothmuscle cells of coronary vessels.38 Thus, it is conceivable thatin addition to cardiac mast cells, myocytes and other cells in
the heart may contribute to renin production and RASactivation in I/R. In fact, the incidence of reperfusion arrhyth-mias was markedly reduced, but not completely abolished, inhearts isolated from mice lacking mast cells or in guinea pighearts perfused with mast cell–stabilizing agents.3 This sug-gests that other cells in the heart could release renin inresponse to I/R.
ConclusionsWe propose that adenosine released from various cells duringI/R and IPC activates A2b and A3 receptors on the surface ofcardiac mast cells and that this is followed by activation/translocation of PKC�, which then increases the catalyticactivity of mitochondrial ALDH2 (see Figure 8). By elimi-nating reactive aldehydes and their mast cell–degranulatingeffects, ALDH2 prevents renin release and RAS activation.This reduces Ang II formation, inhibits excessive norepineph-rine release, and prevents the generation of reperfusionarrhythmias. Notably, the relevance of our findings extendsbeyond the disclosure of a new IPC mechanism. Indeed,although the discovery of IPC has generated a wealth ofstudies, its clinical translation has yet to come to fruition.52
As the search for cardioprotective drugs continues unabated,our findings elucidate novel basic mechanisms of pharmaco-logical cardioprotection (ie, the detoxification of reactivealdehydes and ROS in I/R by increasing the catalytic activityof mitochondrial ALDH2, thus alleviating the dysfunctionalconsequences of RAS activation in the heart). This newfinding suggests that in addition to protecting cardiac myo-cytes from I/R-induced injury, drugs that prevent cardiacmast cell degranulation may prevent the activation of a localRAS, thus providing an additional benefit to patients withmyocardial infarction and perhaps in other cardiac oxidativestress conditions such as those occurring in heart failure.
Sources of FundingThis work was supported by National Institutes of Health grantsHL34215, HL73400 (Dr Koda, Dr Estephan, Dr Silver, Dr Levi, M.Salazar-Rodriguez, F. Corti), HL47073, HL46403 (Dr Levi), andAA11147 (Dr Mochly-Rosen); by CDCH B-09-11-3999-2005 ofCentral University of Venezuela (M. Salazar-Rodriguez); and by aPharmaceutical Research and Manufacturers of America Foundation
Figure 8. Proposed mechanisms of the cardioprotective anti-RAS effects of IPC.
Koda et al ALDH2 Inhibits Renin Release From Heart Mast Cells 779
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

predoctoral fellowship (N. Yan-Ki Chan). We thank Paul J. Christos,DrPH, for statistical help, partially supported by the Clinical Trans-lational Science Center grant UL1-RR024996.
DisclosuresDr Mochly-Rosen is the founder of KAI Pharmaceuticals Inc, acompany that plans to bring PKC regulators to the clinic. However,none of the work in her laboratory is in collaboration with orsupported by the company. The other authors report no disclosures.
References1. Reid AC, Silver RB, Levi R. Renin: at the heart of the mast cell. Immunol
Rev. 2007;217:123–140.2. Silver RB, Reid AC, Mackins CJ, Askwith T, Schaefer U, Herzlinger D,
Levi R. Mast cells: a unique source of renin. Proc Natl Acad Sci U S A.2004;101:13607–13612.
3. Mackins CJ, Kano S, Seyedi N, Schafer U, Reid AC, Machida T, SilverRB, Levi R. Cardiac mast cell-derived renin promotes local angiotensinformation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J Clin Invest. 2006;116:1063–1070.
4. Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: adelay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136.
5. Murphy E, Steenbergen C. Mechanisms underlying acute protection fromcardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609.
6. Cohen MV, Downey JM. Adenosine: trigger and mediator of cardiopro-tection. Basic Res Cardiol. 2008;103:203–215.
7. Gross GJ, Auchampach JA. Blockade of ATP-sensitive potassiumchannels prevents myocardial preconditioning in dogs. Circ Res. 1992;70:223–233.
8. Peart JN, Headrick JP. Adenosinergic cardioprotection: multiplereceptors, multiple pathways. Pharmacol Ther. 2007;114:208–221.
9. Liu GS, Cohen MV, Mochly-Rosen D, Downey JM. Protein kinase C-� isresponsible for the protection of preconditioning in rabbit cardiomyo-cytes. J Mol Cell Cardiol. 1999;31:1937–1948.
10. Inagaki K, Begley R, Ikeno F, Mochly-Rosen D. Cardioprotection byepsilon-protein kinase C activation from ischemia: continuous deliveryand antiarrhythmic effect of an epsilon-protein kinase C-activatingpeptide. Circulation. 2005;111:44–50.
11. Ozawa K, Yamada K, Kazanietz MG, Blumberg PM, Beaven MA. Dif-ferent isozymes of protein kinase C mediate feedback inhibition of phos-pholipase C and stimulatory signals for exocytosis in rat RBL-2H3 cells.J Biol Chem. 1993;268:2280–2283.
12. Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD,Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces is-chemic damage to the heart. Science. 2008;321:1493–1495.
13. Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of4-hydroxynonenal, malonaldehyde and related aldehydes. Free RadicBiol Med. 1991;11:81–128.
14. Cordis GA, Maulik N, Bagchi D, Engelman RM, Das DK. Estimation ofthe extent of lipid peroxidation in the ischemic and reperfused heart bymonitoring lipid metabolic products with the aid of high-performanceliquid chromatography. J Chromatogr. 1993;632:97–103.
15. Eaton P, Li JM, Hearse DJ, Shattock MJ. Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am J Physiol. 1999;276:H935–H943.
16. Churchill EN, Disatnik MH, Mochly-Rosen D. Time-dependent andethanol-induced cardiac protection from ischemia mediated by mito-chondrial translocation of varepsilonPKC and activation of aldehydedehydrogenase 2. J Mol Cell Cardiol. 2009;46:278–284.
17. Liu Y, Tsuchida A, Cohen MV, Downey JM. Pretreatment with angio-tensin II activates protein kinase C and limits myocardial infarction inisolated rabbit hearts. J Mol Cell Cardiol. 1995;27:883–892.
18. Diaz RJ, Wilson GJ. Selective blockade of AT1 angiotensin II receptorsabolishes ischemic preconditioning in isolated rabbit hearts. J Mol CellCardiol. 1997;29:129–139.
19. Das S, Otani H, Maulik N, Das DK. Redox regulation of angiotensin IIpreconditioning of the myocardium requires MAP kinase signaling. J MolCell Cardiol. 2006;41:248–2455.
20. Kim YC, Ji X, Melman N, Linden J, Jacobson KA. Anilide derivatives ofan 8-phenylxanthine carboxylic congener are highly potent and selectiveantagonists at human A2B adenosine receptors. J Med Chem. 2000;43:1165–1172.
21. Li AH, Moro S, Melman N, Ji XD, Jacobson KA. Structure-activityrelationships and molecular modeling of 3, 5-diacyl-2,4-dialkylpyridinederivatives as selective A3 adenosine receptor antagonists. J Med Chem.1998;41:3186–3201.
22. Baraldi PG, Tabrizi MA, Fruttarolo F, Romagnoli R, Preti D. Recentimprovements in the development of A2B adenosine receptor agonists.Purinergic Signal. 2008;4:287–303.
23. Giannella E, Mochmann HC, Levi R. Ischemic preconditioning preventsthe impairment of hypoxic coronary vasodilatation caused by ischemia/reperfusion: role of adenosine A1/A3 and bradykinin B2 receptor acti-vation. Circ Res. 1997;81:415–422.
24. Kobayashi T, Ikeda K, Kumanishi T. Functional characterization of anendogenous Xenopus oocyte adenosine receptor. Br J Pharmacol. 2002;135:313–322.
25. Grover GJ, Garlid KD. ATP-sensitive potassium channels: a review oftheir cardioprotective pharmacology. J Mol Cell Cardiol. 2000;32:677–695.
26. Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM. Acetylcholine,bradykinin, opioids, and phenylephrine, but not adenosine, trigger pre-conditioning by generating free radicals and opening mitochondrialK(ATP) channels. Circ Res. 2001;89:273–278.
27. Li X, Rapedius M, Baukrowitz T, Liu GX, Srivastava DK, Daut J, HanleyPJ. 5-Hydroxydecanoate and coenzyme A are inhibitors of native sar-colemmal KATP channels in inside-out patches. Biochim Biophysic Acta.2010;1800:385–391.
28. Chang RS, Siegl PK, Clineschmidt BV, Mantlo NB, Chakravarty PK,Greenlee WJ, Patchett AA, Lotti VJ. In vitro pharmacology of L-158,809,a new highly potent and selective angiotensin II receptor antagonist.J Pharmacol Exp Ther. 1992;262:133–138.
29. Downey JM, Davis AM, Cohen MV. Signaling pathways in ischemicpreconditioning. Heart Fail Rev. 2007;12:181–188.
30. Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X, Bolli R.Ischemic preconditioning induces selective translocation of protein kinaseC isoforms epsilon and eta in the heart of conscious rabbits withoutsubcellular redistribution of total protein kinase C activity. Circ Res.1997;81:404–414.
31. Jaburek M, Costa AD, Burton JR, Costa CL, Garlid KD. MitochondrialPKC epsilon and mitochondrial ATP-sensitive K� channel copurify andcoreconstitute to form a functioning signaling module in proteoliposomes.Circ Res. 2006;99:878–883.
32. Inagaki K, Hahn HS, Dorn GW, Mochly-Rosen D. Additive protection ofthe ischemic heart ex vivo by combined treatment with delta-proteinkinase C inhibitor and epsilon-protein kinase C activator. Circulation.2003;108:869–875.
33. Johnson JA, Gray MO, Chen CH, Mochly-Rosen D. A protein kinase Ctranslocation inhibitor as an isozyme-selective antagonist of cardiacfunction. J Biol Chem. 1996;271:24962–24966.
34. Ping P. Getting to the heart of proteomics. N Engl J Med. 2009;360:532–534.
35. Feoktistov I, Ryzhov S, Goldstein AE, Biaggioni I. Mast cell-mediatedstimulation of angiogenesis: cooperative interaction between A2B and A3
adenosine receptors. Circ Res. 2003;92:485–492.36. Dorn GW, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ,
Csukai M, Wu G, Lorenz JN, Mochly-Rosen D. Sustained in vivo cardiacprotection by a rationally designed peptide that causes epsilon proteinkinase C translocation. Proc Natl Acad Sci U S A. 1999;96:12798–12803.
37. Budas GR, Disatnik MH, Mochly-Rosen D. Aldehyde dehydrogenase 2 incardiac protection: a new therapeutic target? Trends Cardiovasc Med.2009;19:158–164.
38. Dostal DE, Baker KM. The cardiac renin-angiotensin system: conceptual,or a regulator of cardiac function? Circ Res. 1999;85:643–650.
39. Campbell DJ. Critical review of prorenin and (pro)renin receptorresearch. Hypertension. 2008;51:1259–1264.
40. Barlucchi L, Leri A, Dostal DE, Fiordaliso F, Tada H, Hintze TH,Kajstura J, Nadal-Ginard B, Anversa P. Canine ventricular myocytespossess a renin-angiotensin system that is upregulated with heart failure.Circ Res. 2001;88:298–304.
41. Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3 adenosine receptoris the unique adenosine receptor which facilitates release of allergicmediators in mast cells. J Biol Chem. 1993;268:16887–16890.
42. Linden J. Cloned adenosine A3 receptors: pharmacological properties,species differences and receptor functions. Trends Pharmacol Sci. 1994;15:298–306.
43. Feoktistov I, Biaggioni I. Adenosine A2B receptors. Pharmacol Rev.1997;49:381–402.
780 Circulation August 24, 2010
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

44. Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK,Jacobson KA, Leff P, Williams M. Nomenclature and classification ofpurinoceptors. Pharmacol Rev. 1994;46:143–156.
45. Headrick JP. Ischemic preconditioning: bioenergetic and metabolicchanges and the role of endogenous adenosine. J Mol Cell Cardiol.1996;28:1227–1240.
46. Hinschen AK, Rose’Meyer RB, Headrick JP. Adenosine receptorsubtypes mediating coronary vasodilation in rat hearts. J CardiovascPharmacol. 2003;41:73–80.
47. Hua X, Kovarova M, Chason KD, Nguyen M, Koller BH, Tilley SL.Enhanced mast cell activation in mice deficient in the A2b adenosinereceptor. J Exp Med. 2007;204:117–128.
48. Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, BlackburnMR, Biaggioni I, Feoktistov I. Effect of A2B adenosine receptor gene
ablation on adenosine-dependent regulation of proinflammatory cyto-kines. J Pharmacol Exp Ther. 2008;324:694–700.
49. Kawano T, Matsuse H, Kondo Y, Machida I, Saeki S, Tomari S, MitsutaK, Obase Y, Fukushima C, Shimoda T, Kohno S. Acetaldehyde induceshistamine release from human airway mast cells to cause bronchocon-striction. Int Arch Allergy Immunol. 2004;134:233–239.
50. Auchampach JA, Bolli R. Adenosine receptor subtypes in the heart:therapeutic opportunities and challenges. Am J Physiol. 1999;276:H1113–H1116.
51. Wilson GJ, Diaz RJ. The myocardial no-reflow phenomenon: role ofdeltaPKC. Cardiovasc Res. 2007;73:623–625.
52. Downey JM, Cohen MV. Why do we still not have cardioprotectivedrugs? Circ J. 2009;73:1171–1177.
CLINICAL PERSPECTIVEIschemia/reperfusion is a known cause of cardiac dysfunction that is often accompanied by infarction, contractile failure,and severe arrhythmias. The production of reactive oxygen species and reactive aldehydes is considered a prominent causebecause aldehyde detoxification by mitochondrial aldehyde dehydrogenase type-2 displays cardioprotective effects. Werecently reported that ischemia/reperfusion elicits the release of renin from cardiac mast cells, which in turn activates alocal renin-angiotensin system, causing severe arrhythmic dysfunction. Reactive oxygen species and reactive aldehydes arelikely responsible for mast cell degranulation and renin release; hence, means to prevent such effects could be clinicallybeneficial. We describe here a novel pathway that is independent of cardiomyocyte salvage and relies instead on aldehydedehydrogenase type-2 activation in mast cell mitochondria. Aldehyde dehydrogenase type-2 detoxifies reactive aldehydesproduced in ischemia/reperfusion, thus preventing mast cell degranulation and renin release. This avoids the activation ofa local renin-angiotensin system and reperfusion arrhythmias. This new finding suggests that in addition to protectingcardiac myocytes from ischemia/reperfusion-induced injury, drugs that prevent cardiac mast cell degranulation mayprevent the activation of a local renin-angiotensin system, thus providing an additional benefit to patients with myocardialinfarction and perhaps in other cardiac oxidative stress conditions such as those occurring in heart failure.
Koda et al ALDH2 Inhibits Renin Release From Heart Mast Cells 781
at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

Supplemental Materials MATERIALS AND METHODS
Animals. 132 male Hartley guinea pigs weighing 300-350 grams (Charles River
Laboratories, Kingston, NY) were used for the study. All experiments were
approved by the Institutional Animal Care and Use Committee of Weill Cornell
Medical College.
Perfusion of guinea-pig hearts ex vivo. Guinea pigs were anesthetized with
CO2 and euthanized by stunning. Hearts were rapidly isolated and perfused at
constant pressure (55 cm H2O) with oxygenated Ringer’s solution (NaCl 154
mmol/L, KCl 5.61 mmol/L, CaCl2 2.16 mmol/L, NaHCO3 5.95 mmol/L and
dextrose 5.55 mmol/L) at 37oC in a Langendorff apparatus (Radnoti Glass
Technology Inc., Monrovia, CA).
Ischemia/reperfusion (I/R): following an equilibration period all hearts were
subjected to 20-min global ischemia induced by complete cessation of coronary
perfusion, followed by 30-min reperfusion (I/R). Ischemic preconditioning (IPC):
hearts were subjected to 2 x 5-min cycles of ischemia, each followed by 5-min
reperfusion. Pharmacological prevention of IPC: hearts were perfused with a
given antagonist for 20 min (30 min for GTN) before and during IPC, and then
washed out for 15 min before I/R. Pharmacological preconditioning: hearts were
perfused for 2 x 5-min cycles with a given agent, except for the selective PKCδ
inhibitor δV1-1 which was administered during the entire 30-min reperfusion
following the 20-min ischemia. Prevention of pharmacological preconditioning:
hearts were perfused with a given antagonist for 20 min (30 min for GTN) before
1 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

and during pharmacological preconditioning. All agents were then washed out for
15 min before I/R.
Coronary flow was measured by timed collections of the effluent every 2-
min; all samples were assayed for renin and norepinephrine, some for β-
hexosaminidase and creatine phosphokinase (CPK). Surface ECG was obtained
from leads attached to the left ventricle and the right atrium, recorded in digital
format through each experiment, and analyzed using Power Lab/8SP
(AdInstrument, Colorado Springs, CO). Onset and duration of reperfusion
arrhythmias were recorded and evaluated according to the Lambeth
conventions.1
Cell culture. The human mastocytoma cell line (HMC-1) was kindly provided to
us by Dr. I. Biaggioni (Vanderbilt University, Nashville, TN) and J. H. Butterfield
(Mayo Clinic, Rochester, MN). Cells were maintained in suspension culture at
high density in Iscove’s modified Dulbecco’s medium supplemented with 10%
FBS and 1.2 mmol/L monothioglycerol and kept at 37°C, 5% CO2.
β-Hexosaminidase and renin assay. Coronary effluent from guinea-pig hearts
ex vivo was assayed for β-hexosaminidase using the method of Schwartz et al.2
Samples of coronary effluent (20 µL) were placed in the well of a 96-well plate
and total β-hexosaminidase content was determined. For the measurement of
renin overflow, coronary effluent was immediately concentrated 8-fold by
centrifugal filtration (Millipore, Billerica, MA). Concentrated samples were
incubated overnight with human angiotensinogen and then renin activity
(angiotensin I formed) was determined by GammaCoat Plasma Renin Activity 125I
2 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

Radioimmunoassay (DiaSorin, Stillwater, MN). The detection limit was
approximately 0.01 pmol.
For HMC-1 cells, 4 to 6 pooled confluent flasks were pelleted and cells
washed twice with Ringer buffer (pH 7.4). Pelleted HMC-1 were then
resuspended in Ringer buffer and equal volumes of cell suspension were
aliquoted in Eppendorf tubes and incubated at 37°C with gentle agitation. Cell
suspensions were then incubated with a given agent (i.e., Alda-1, ψεRACK or
LUF5835 + IBMECA) for 10 min (preceded or not by a 30-min incubation with
GTN). Acetaldehyde, H2O2 or 4-HNE was subsequently added to the incubation
mixture for 20 minutes. At the end of protocol, samples were transferred to ice
and then centrifuged at 1,000 g for 10 min. Supernatants were collected and kept
in ice until β-hexosaminidase and renin release was evaluated. Cell pellets were
lysed with 0.5% triton X-100 and centrifuged at 13,000 g. Supernatants were
then used for protein and total β-hexosaminidase content determination. All
results were normalized and expressed as percent above control.
NE assay. Coronary effluent was assayed for norepinephrine by HPLC with
electrochemical detection as previously described.3 The detection limit was
approximately 0.2 pmol.
CPK assay. Coronary effluent was assayed for creatine phosphokinase release
using a creatine kinase assay kit (Genzyme Diagnostics, Charlottetown, PE,
Canada). Substrate and buffer reagent were added to the collected coronary
effluent and then incubated at 37oC for 2 min. The change in absorbance at 340
3 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

nm at one- min intervals was recorded in the spectrometer until the change in
absorbance was constant.
PCR and immunostaining. RT-PCR: total RNA was extracted from HMC-1 cells
using TRIzol reagent (Invitrogen, Carlsbad, CA), 1 µg of total RNA from each
sample was reverse-transcribed and cDNA amplified by RT-PCR using a
QIAGEN One-step RT-PCR kit. The sense primers specific for adenosine A2b-
and A3-receptors were: 5’-TAAGATCTTCCTGGTGGCCT-3’, and 5-
AGATGCCCAACAACAGCACT-3’, respectively. Antisense primers specific for
adenosine A2b- and A3-receptors were: 5’-GCTTGGCAGAGAAGATAC-3’, and 5’-
ATCTGACGGTAAGCTTGACC-3’, respectively. The amplification profile used
was: 50°C for 30 min, 95°C for 15 min, then 94°C for 30 sec, 55°C for 30 sec,
72°C for 1.5 min (40 cycles), and finally 72°C for 10 min. PCR products
generated were, ~330 and ~354 bp for adenosine A2b- and A3-receptor,
respectively. PCR products were analyzed by agarose gel electrophoresis and
ethidium bromide staining.
Immunostaining: HMC-1 cells were fixed and permeabilized on glass
slides and stained with the goat anti-A2b-receptor Ab (Santa Cruz Biotechnology,
Santa Cruz, CA)(1:200) conjugated to Alexa Fluor 488 donkey anti-goat IgG
(1:500) and with rabbit anti-A3-receptor Ab (Santa Cruz) (1:200) conjugated to
Alexa Fluor 488 donkey anti-rabbit IgG (1:500). Nuclei were stained with DAPI.
For viewing immunofluorescence, cells were examined with an inverted
fluorescence microscope (Nikon Eclipse TE 2000-U) interfaced to an electron
4 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

multiplying charge coupled device (Hamamatsu) and processed with Metamorph
software (version 6.2; Universal Imaging Corp.).
Translocation of PKCε. Following incubation of HMC-1 cells with PMA, the
PKCε-selective agonist ψεRACK, or the adenosine A2b- and A3-receptor agonists
LUF5835 and IB-MECA in combination, cells were homogenized in buffer (200
µL) (20 mmol/L Tris-HCl, 2 mmol/L EDTA, 10 mmol/L EGTA, 0.25 M sucrose, β-
mercaptoethanol and 1X protease inhibitors cocktail). Cell homogenates were
then passed through syringes with needle (30 Gauge) 10 times, and cell lysates
were centrifuged at 100,000 g for 30 min to collect cytosolic fractions
(supernatant). The pellets were resuspended in homogenization buffer (50 µL)
with 1% Triton X-100, and then centrifuged at 100,000 g for 30 min to collect
membrane fractions (supernatant). Translocation of PKCε was assessed by
using a PKCε-specific antibody (Santa Cruz; 1:1000 dilution) in Western blot
analysis. Methods for Western blot analysis were as previously described.3 The
ratio of PKCε in membrane to that in cytosol was expressed as PKCε
translocation.
ALDH2 enzymatic activity assay. Enzymatic activity of ALDH2 in HMC-1 cells
was determined spectrophotometrically by monitoring the reductive reaction of
NAD+ to NADH at 340 nm as previously described.4 The assays were carried out
in 50 mmol/L sodium pyrophosphate buffer, pH = 9.0, at 25oC. 300 µg of cell
lysates and 2.5 mmol/L NAD were added to the buffer. To start the reaction, 10
mmol/L acetaldehyde was added and the accumulation of NADH was recorded
for 3 min with measurements being taken every 15 s. ALDH2 reaction rates were
5 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

calculated as µmol NADH/min/mg proteins, and compared to cells that were
treated with Na Ringer (control) and expressed as % increase from control.
Drugs and chemicals. Acetaldehyde (17.8 M) and H2O2 (30% w/w, 9.8 M) were
purchased from Sigma-Aldrich (St. Louis, MO); 4-hydroxy Nonenal (4-HNE) in
ethanol solution was purchased from Cayman Chemical and dissolved in DMSO
to 300 mmol/L stock concentration after solvent evaporation. DMSO
concentration during experiments was always below 0.1% and did not affect the
cell response. LUF5825 was a gift from Dr. M.W. Beukers (University of Leiden,
Leiden, Netherlands); EXP3174 was a gift from Merck Sharp & Dohme Ltd
(Whitehouse Station, NJ); ψεRACK, δV1-1 and Alda-1 were synthesized in the
Mochly-Rosen lab (Stanford University School of Medicine, Palo Alto, CA).
IBMECA, MRS1754, MRS1523, DPCPX, chelerythrine, 5-hydroxydecanoate and
cyanamide were purchased from Sigma-Aldrich. Phorbol 12-myristate 13-acetate
was purchased from LC Laboratories (Woburn, MA). Glyceryl trinitrate was
purchased from Hospira Inc. (Lake Forest, IL). Human plasma angiotensinogen
was purchased from Calbiochem (San Diego, CA). Cyanamide and chelerythrine
were dissolved in water; EXP3174, DPCPX and 5-hydroxydecanoate were
dissolved in ethanol; IBMECA, MRS1754, MRS1523, phorbol 12-myristate 13-
acetate, and Alda-1 were dissolved in DMSO.
Figure Legends
Figure 1. IPC reduces renin and NE release, and shortens arrhythmias
caused by I/R in guinea-pig hearts ex vivo. This cardioprotective anti-RAS
6 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

effect is mimicked or prevented by activation or blockade of adenosine A2b-
and A3-receptors in combination, but unaffected by adenosine A1-receptor
blockade (Panel A), and mimicked by PKCε activation and prevented by
PKCε inhibition (Panel B). Panel A: Coronary overflow of renin and NE, and
duration of reperfusion arrhythmias (VT/VF) in isolated hearts subjected to 20-
min global ischemia and 30-min reperfusion (n=6). In other hearts, I/R was
preceded by IPC (2 x 5-min ischemia cycles each followed by 5-min reperfusion;
n=8). Other hearts were subjected to I/R preceded by IPC in the presence of
DPCPX (300 nmol/L, adenosine A1-receptor antagonist) or MRS1754 and
MRS1523 (50 and 100 nmol/L, adenosine A2b- and A3-receptor antagonists,
respectively) (n=6 each). Instead of IPC, other hearts were perfused with a
combination of A2b- (LUF 5835; 50 nmol/L) and A3-receptor agonists (IBMECA;
50 nmol/L) for 2 x 5-min cycles each followed by 5-min washout before I/R (n=6).
Panel B: Hearts underwent I/R preceded by IPC either in the absence (n=8) or
presence (n=5) of the selective PKCε inhibitor εV1-2 (1 µmol/L). Additional hearts
were perfused with the selective PKCε activator ψεRACK instead of IPC (500
nmol/L, 2 x 5-min cycles each followed by 5-min reperfusion; n=5) before I/R.
Basal, pre-ischemic overflows of active renin (i.e., ANG I formed) and NE were
6.81 ± 2.12 pg/hr/g and 4.02 ± 0.77 pmol/g, respectively. Bars represent means ±
SEM of overflows during the first 4 min of reperfusion or duration of VT/VF. **
And ***, P<0.01 and P<0.001, respectively, from I/R; † and ††, P<0.05 and P<0.01
from IPC, respectively, by Mann-Whitney test (for I/R vs LUF5835 + IBMECA or
ψεRACK) or Kruskal-Wallis with Dunn’s multiple comparison test (for all others).
7 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

Figure 2. Selective inhibition of the PKCε isoform prevents the
cardioprotective IPC-like anti-RAS effects generated by the combined
activation of adenosine A2b- and A3-receptors (Panel A). Selective
desensitization of ALDH2 with glyceryl trinitrate (GTN) prevents the
cardioprotective IPC-like anti-RAS effects generated by PKCε activation
(Panel B). Panel A: Coronary overflow of renin and NE, and duration of VT/VF in
ex vivo guinea-pig hearts subjected to I/R (n=6). Instead of IPC, other hearts
were perfused with a combination of selective A2b- (LUF 5835; 50 nmol/L) and
A3-receptor agonists (IBMECA; 50 nmol/L) for 2 x 5-min cycles each followed by
5-min washout before I/R (n=5). Other hearts were perfused with the selective
PKCε inhibitor εV1-2 (1 µmol/L), followed by a combination of A2b- (LUF 5835; 50
nmol/L) and A3-receptor agonists (IBMECA; 50 nmol/L) for 2 x 5-min cycles each
followed by 5-min washout instead of IPC before I/R (n=6). Panel B: Instead of
IPC, hearts were perfused with the selective PKCε activator (ψεRACK; 500
nmol/L) for 2 x 5-min cycles each followed by 5-min washout before I/R (n=5).
Other hearts were perfused with the ALDH2 desensitizer glyceryl trinitrate (GTN;
2µmol/L, perfused for 30 min), followed by ψεRACK (500 nmol/L) for 2 x 5-min
cycles each followed by 5-min washout instead of IPC before I/R (n=5). Bars
represent means ± SEM of overflows during the first 4 min of reperfusion or
duration of VT/VF. ** And ***, P<0.01 and P<0.001 from I/R, respectively; † and ††,
P<0.05 and P<0.01, respectively from the A2bR and A3R agonist combination
8 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

(Panel A), and †, P<0.05 from ψεRACK (Panel B), by Mann-Whitney test (for I/R
vs IPC) or Kruskal-Wallis test with Dunn’s multiple comparison test (for all others).
Figure 3. Selective activation of ALDH2 with Alda-1 mimics the
cardioprotective anti-RAS effects of IPC. General ALDH inhibition with
cyanamide or selective desensitization of ALDH2 with glyceryl trinitrate
(GTN), each prevents the cardioprotective anti-RAS effects of IPC and Alda-
1. Panel A: Coronary overflow of renin and NE, and duration of VT/VF in guinea-
pigs hearts subjected to I/R (n=6) or I/R preceded by IPC either in the absence
(n=8) or presence (n=5) of the ALDH inhibitor cyanamide (CYA; 5 mmol/L). Other
hearts were perfused with the selective ALDH-2 activator Alda-1 instead of IPC
(20 µmol/L, 2 x 5-min cycles each followed by 5-min washout; n=5) before I/R.
Other hearts were perfused with CYA (5 mmol/L), followed by Alda-1 (20 µmol/L,
2 x 5 min cycles each followed by 5-min reperfusion) instead of IPC before I/R
(n=5). Panel B: Hearts were pretreated with the ALDH2 desensitizer glyceryl
trinitrate (nitroglycerin, GTN; 2 µmol/L, perfused for 30 min) and then subjected
to I/R preceded by IPC (n=5). Other hearts were perfused with the selective
ALDH-2 activator Alda-1 instead of IPC (20 µmol/L, 2 x 5-min cycles each
followed by 5-min reperfusion; n=5) before I/R. Other hearts were perfused with
GTN (2 µmol/L) for 30 min, followed by Alda-1 (20 µmol/L, 2 x 5 min cycles each
followed by 5-min reperfusion) instead of IPC before I/R (n=5). Bars represent
means ± SEM of overflows collected during the first 4 min of reperfusion or
duration of VT/VF. *, ** And ***, P<0.05, P<0.01 and P<0.001 from I/R,
9 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

respectively; #, ## and ###, P<0.05, P<0.01 and <0.001 from IPC, respectively; †
and ††, P<0.05 and P<0.01 from Alda-1, respectively, by Kruskal-Wallis test with
Dunn’s multiple comparison test.
Figure 4. Expression of adenosine receptors by HMC-1 cells. ψεRACK or
adenosine A2b- and A3-receptor agonists in combination induces selective
activation of PKCε in HMC-1 cells. A, Ethidium bromide gel of transcripts of the
adenosine A2b- and A3-receptor sub-types in HMC-1 cells. B, Immuno-staining of
HMC-1 cells expressing the adenosine A2b- and A3-receptor sub-types. Nuclei
(blue) are stained with DAPI. Scale bar = 10 μm. C and E, Translocation of PKCε
(i.e., activity of PKCε) was determined by Western blot analysis in cytosolic and
membrane fractions of HMC-1 cells. C, Control cells were incubated with Na
Ringer for 30 min demonstrating the basal activity of PKCε. Incubation of HMC-1
cells with the PKC activator phorbol ester myristate (PMA) 300 nmol/L for 7 min
(positive control) or the PKCε activator ψεRACK 500 nmol/L for 30 min, prior to
an additional 7-min incubation with a below-threshold concentration of PMA (3
nmol/L), elicits an increase in PKCε translocation from cytosol to membrane (i.e.,
PKCε activation). Notably, treatment with 3 nmol/L PMA for 7 min alone does not
cause PKCε translocation. E, Control cells were incubated with Na Ringer for 1
hr demonstrating the basal activity of PKCε. Incubation of HMC-1 cells with 300
nmol/L PMA for 7 min (positive control) or the adenosine A2b- and A3-receptor
agonists LUF5835 and IB-MECA in combination (both at 50 nmol/L; 1 hour)
elicits an increase in PKCε translocation from cytosol to membrane (i.e., PKCε
10 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

activation). D and F, quantification of PKCε translocation from cytosolic to
membrane fractions, expressed as % of control. Bars are means ± SEM (D, n=4-
6; F, n=7) normalized relative to β-actin. * And **, P<0.05 and P<0.01, from
control respectively, by Kruskal-Wallis test followed by post-hoc Dunn’s test.
Figure 5. Mast cell degranulation and renin release elicited by acetaldehyde
(A and D), H2O2 (B and E) and 4-HNE (C and F): blockade by Alda-1 and its
reversal by pretreatment with GTN. Incubation of HMC-1 cells with
acetaldehyde (300-700 µmol/L, 20 min), H2O2 (0.1-1 mmol/L, 20 min) or 4-HNE
(3-30 µmol/L, 20 min) elicits a concentration-dependent release of both β-HEX
and renin. Pre-incubation of HMC-1 cells with the ALDH2 activator Alda-1 (20
µmol/L, 10 min) attenuates the acetaldehyde- , H2O2- and 4-HNE-induced
release of β-HEX and renin. Pre-treatment of HMC-1 cells with the ALDH2-
desensitizer glyceryl trinitrate (GTN, 2 µmol/L, 30 min) prevents the effects of
Alda-1. Points are means (± SEM; n=3-17). *, ** and ***, P<0.05, P<0.01 and
P<0.001, from control, respectively; #, ## and ###, P<0.05 and P<0.01, from Alda-1
+ GTN respectively, by Kruskal-Wallis with Dunn’s multiple comparison test.
Figure 6. Activation of adenosine A2b- and A3-receptors in combination or
PKCε increases ALDH2 activity in HMC-1 cells. Selective inhibition of the
PKCε isoform prevents the increase in ALDH2 activity by the combined
activation of adenosine A2b- and A3-receptors. Incubation of HMC-1 cells with
the selective ALDH2 activator Alda-1 (100 µmol/L; 10 min), the PKC activator
11 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

phorbol ester myristate (PMA) (300 nmol/L; 10 min), the PKCε-selective agonist
ψεRACK (0.5 and 1 µmol/L; 30 min), or the adenosine A2b- and A3-receptor
agonists LUF5835 and IB-MECA in combination (both at 50 nmol/L; 1 hour)
increases ALDH2 activity (measured by the rate of NADH production at 340 nm).
Pre-treatment of HMC-1 cells with the PKCε antagonist εV1-2 (1 µmol/L; 20 min)
prevents the effects of adenosine A2b- and A3-receptor activation. Bars are mean
percent increases from control (±SEM; n=4-8). Basal NADH production was 3.63
± 0.25 µmol/min/mg protein. *, ** and ***, P<0.05, P<0.01 and P<0.001 from
control, respectively, by Kruskal-Wallis test followed by post-hoc Dunn’s test. #,
P<0.05 from the A2b- and A3-receptor agonists combination by Kruskal-Wallis test
followed by post-hoc Dunn’s test.
Figure 7. Activation of adenosine A2b- and A3-receptors (A and C), or PKCε
(B and D), inhibits mast cell degranulation and renin release elicited by
H2O2: prevention by GTN pretreatment. Incubation of HMC-1 cells with H2O2
for 20 min elicits a concentration-dependent release of both β-HEX and renin.
Pre-incubation of HMC-1 cells with the adenosine A2b- and A3-receptor agonists
LUF5835 and IB-MECA (both at 50 nmol/L, 10 min) in combination, or with the
PKCε activator (ψεRACK, 500 nmol/L, 10 min) attenuates the H2O2-induced
release of β-HEX and renin. Pre-treatment of HMC-1 cells with the ALDH2
desensitizer glyceryl trinitrate (GTN, 2 µmol/L, 30 min) prevents the effects
resulting from the activation of A2b- and A3-receptors and PKCε. Points are
means (± SEM; n=3-19). *, ** and ***, P<0.05, P<0.01 and P<0.001, from control,
12 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

respectively; #, ## and ###, P<0.05, P<0.01 and P<0.001 from LUF5835 + IB-
MECA + GTN or ψεRACK + GTN, respectively, by Kruskal-Wallis with Dunn’s
multiple comparison test.
Figure 8. Proposed mechanisms of the cardioprotective anti-RAS effects of
ischemic preconditioning.
Legend to Supplemental Figure I. The cardioprotective anti-RAS effects of
IPC are unaffected by mitochondrial KATP channel blockade or angiotensin
AT1-receptor blockade (Panel A), and mimicked by general PKC activation
and prevented by general PKC inhibition (Panel B). Panel A: Coronary
overflow of renin and NE, and duration of VT/VF in ex vivo guinea-pig hearts
subjected to I/R (as in Fig.1; n=6). Other hearts underwent I/R preceded by IPC
in the presence of 5-hydroxydecanoate, a mitochondrial KATP channel blocker (5-
HD; 100 µmol/L; n=6), or EXP3174, an angiotensin AT1-receptor antagonist (300
nmol/L; n=5). Panel B: Hearts were pretreated with the general PKC inhibitor
chelerythrine (2.8 µmol/L; Chel) and then subjected to I/R preceded by IPC (n=6).
Additional hearts were perfused with the general PKC activator phorbol ester
myristate (0.05 nmol/L; PMA) instead of IPC (2 x 5-min cycles + 5-min
reperfusion; n=6) before I/R. Bars are means ± SEM of overflows during the first
4 min of reperfusion or duration of VT/VF. ** And ***, P<0.01 and <0.001 from I/R,
respectively; †† and †††, P<0.01 and P<0.001 from IPC, respectively, by Mann-
13 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

Whitney test (for I/R vs PMA) or Kruskal-Wallis with Dunn’s multiple comparison
test (for all others).
Legend to Supplemental Figure II. Selective inhibition of PKCδ during
reperfusion following ischemia prevents CPK release from isolated guinea-
pig hearts (Panel A and B), but fails to affect the cardioprotective anti-RAS
effect of IPC (Panel C). Time course of CPK overflow (Panel A) and total CPK
release during 20 min of reperfusion (Panel B) in ex vivo guinea-pig hearts
subjected to I/R preceded or not by IPC or PKCε activation (with ψεRACK, 500
nmol/L) (n=5-7). Other hearts underwent I/R and received the selective PKCδ
inhibitor δV1-1 (500 nmol/L, n=4) during the 30-min reperfusion. Panel C:
Coronary overflow of renin, NE, and duration of VT/VF, in ex vivo guinea-pig
hearts subjected to I/R, preceded or not by IPC (n=8 and 6, respectively) or the
selective PKCε activator ψεRACK (500 nmol/L) for 2 x 5-min cycles each
followed by 5-min washout (n=5). Other hearts underwent I/R and received the
selective PKCδ inhibitor δV1-1 (500 nmol/L) during the 30-min reperfusion (n=4).
In Panel A and B, bars represent means ± SEM of overflows collected during the
first 20 min of reperfusion. In Panel C, bars represent means ± SEM of overflows
collected during the first 4 min of reperfusion or duration of VT/VF. *, ** And ***,
P<0.05, P<0.01 and P<0.001 from I/R, respectively, by Mann-Whitney test.
Reference List 1. Walker MJ, Curtis MJ, Hearse DJ, Campbell RW, Janse MJ, Yellon DM,
Cobbe SM, Coker SJ, Harness JB, Harron DW, Higgins AJ, Julian DG, Lab
14 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from

MJ, Manning AS, Northover BJ, Parratt JR, Riemersma RA, Riva E, Russell DC, Sheridan DJ, Winslow E, Woodward B. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res. 1988;22:447-455.
2. Schwartz LB, Austen KF, Wasserman SI. Immunologic release of beta-hexosaminidase and beta-glucuronidase from purified rat serosal mast cells. J Immunol. 1979;123:1445-1450.
3. Seyedi N, Mackins CJ, Machida T, Reid AC, Silver RB, Levi R. Histamine H3-receptor-induced attenuation of norepinephrine exocytosis: a decreased PKA activity mediates a reduction in intracellular calcium. J Pharmacol Exp Ther. 2005;312:272-280.
4. Churchill EN, Disatnik MH, Mochly-Rosen D. Time-dependent and ethanol-induced cardiac protection from ischemia mediated by mitochondrial translocation of varepsilonPKC and activation of aldehyde dehydrogenase 2. J Mol Cell Cardiol. 2009;46:278-284.
15 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from
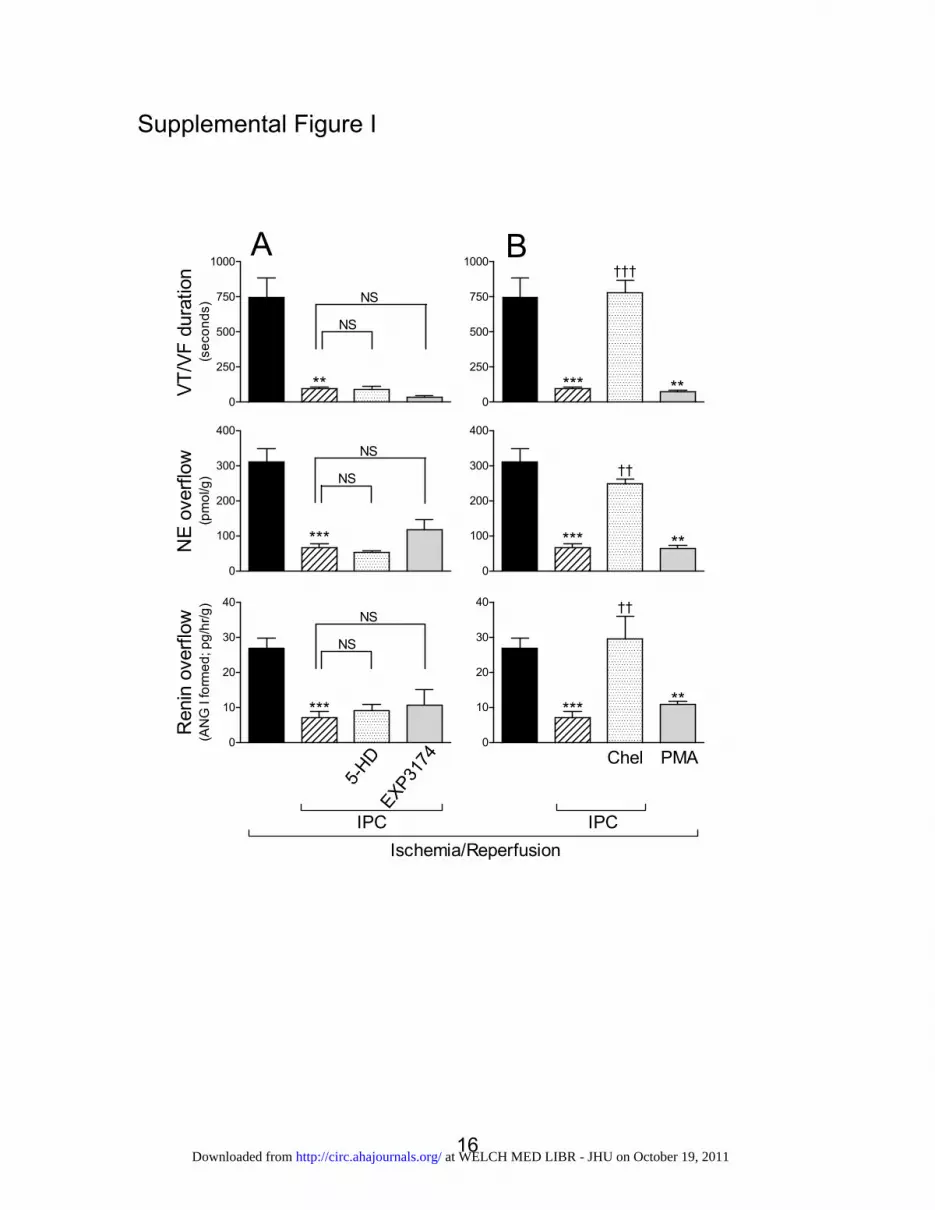
Supplemental Figure I
0
250
500
750
1000
**
NS
NS
VT/V
F du
ratio
n(s
econ
ds)
0
250
500
750
1000
*** **
†††
0
100
200
300
400
***
NS
NS
NE
over
flow
(pm
ol/g
)
0
100
200
300
400
*** **
††
0
10
20
30
40
***
NS
NS
Ren
in o
verfl
ow(A
NG
I fo
rmed
; pg/
hr/g
)
0
10
20
30
40
*****
††
Ischemia/ReperfusionIPC
Chel PMA
IPC
A B
5-HD
EXP3174
16 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from
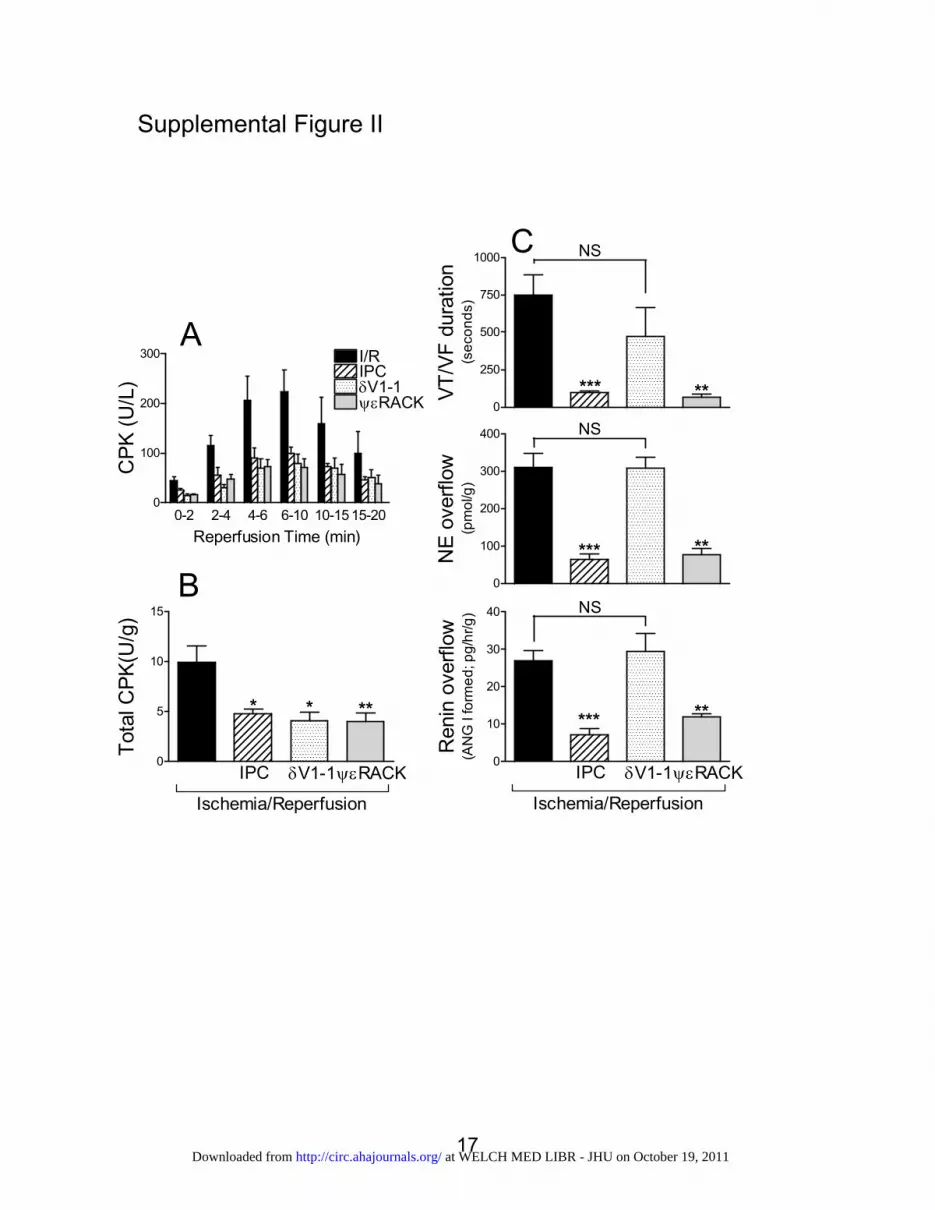
Supplemental Figure II
0-2 2-4 4-6 6-10 10-15 15-200
100
200
300
δV1-1
I/R
ψεRACK
IPC
Reperfusion Time (min)
CPK
(U/L
)
0
250
500
750
1000
*** **
NS
VT/V
F du
ratio
n(s
econ
ds)
0
5
10
15
* * **
Tota
l CPK
(U/g
)
0
100
200
300
400
*** **
NSN
E ov
erflo
w(p
mol
/g)
0
10
20
30
40
*** **
NS
Ren
in o
verfl
ow(A
NG
I fo
rmed
; pg/
hr/g
)
A
B
C
Ischemia/Reperfusion
δV1-1ψεRACKIPC
Ischemia/Reperfusion
δV1-1ψεRACKIPC
17 at WELCH MED LIBR - JHU on October 19, 2011http://circ.ahajournals.org/Downloaded from