diversas teorias del daño cerebral en la enfermedad de alzheimer
TRANSCRIPT

MODELOS DE DAÑO CEREBRAL EN ENFERMEDAD DE ALZHEIMER: UNA BREVE REVISIÓN
Fabian A. Pavez Reyes & Luis A. Ramírez Sepúlveda
Introducción. En 1906 el Dr. Alois Alzheimer describió el caso de una paciente de 51 años correlacionando el cuadro clínico neuropsiquiátrico con lesiones características a la AP: los ovillos neurofibrilares. En 1910, Kraepelin denominaría este cuadro en su Manual de Psiquiatría como “Enfermedad de Alzheimer” (EA).
Se concibe como una enfermedad neurodegenerativa correspondiente a la más común de las demencias (50-60% de éstas) que cursa con pérdida neuronal selectiva a nivel hipocampal, corteza entorrinal y núcleos de Meynert y tiene lesiones características: placas seniles y ovillos neurofibrilares.
La EA afecta a 15 millones de personas en el mundo. Con un claro perfil etáreo, 5% en mayores de 65 años y 20% en mayores de 80 años. A los 90 años, un 50% desarrollará la enfermedad. Existe una forma familiar, de herencia autosómica dominante con distintos grados de penetrancia (<5% de las EA), y una forma esporádica (mayoritaria). El objetivo de la presente revisión es describir de manera breve los principales mecanismos de daño cerebrales vinculados a la EA en la actual. La relevancia del hallazgo de un modelo de daño cerebral que dé sustento a la enfermedad se encuentra en la posibilidad de generar un blanco terapéutico que optimice el tratamiento de la enfermedad.
Existen numerosas teorías: Cascada de B-Amiloide, Hiperfosforilación de proteína Tau, Mecanismos inflamatorios-inmunológicos, Estrés oxidativo, Cerebrovascular-hiperhomocisteinemia; entre otras.
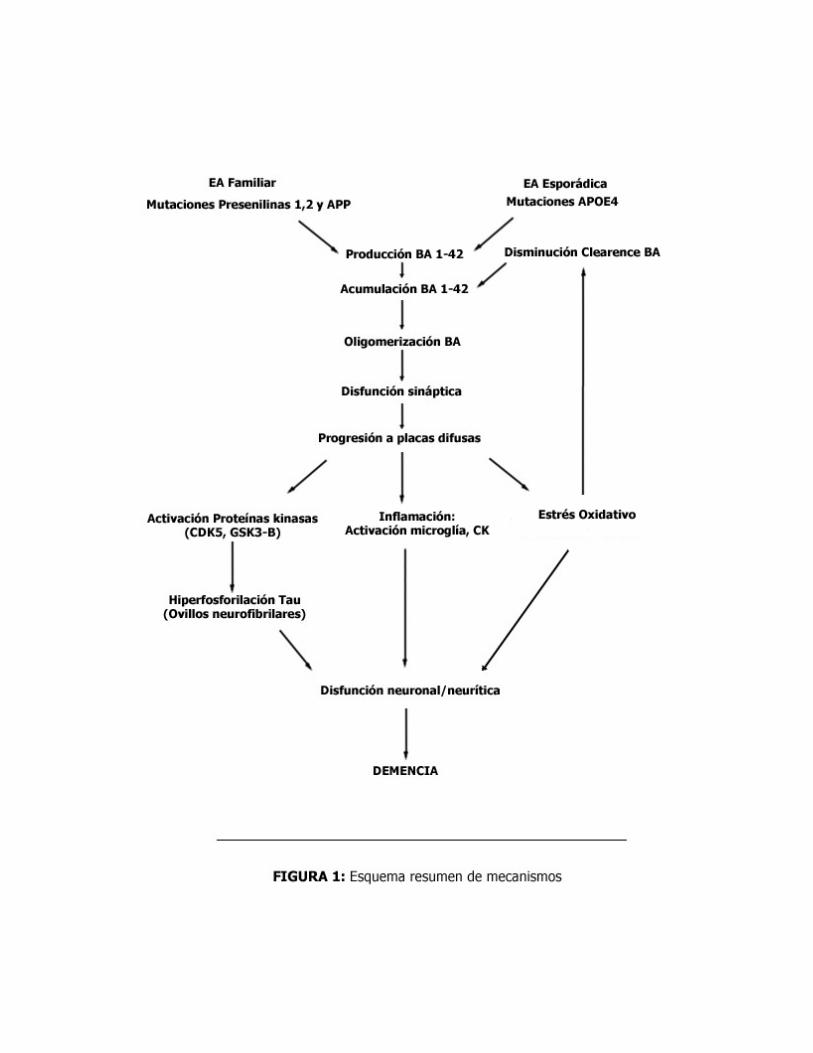
En la presente revisión pondremos énfasis en las cuatro primeras, sugiriendo la posibilidad de que no sólo una de las teorías pueda dar cuenta de la patogénesis de la EA; sino más bien pensando en que pueden vincularse en distinto grado dentro de una red de fenómenos no necesariamente secuenciales que finalmente redundarán en las manifestaciones clínicas de esta enfermedad.

Teoría de la Cascada de Beta-Amiloide (BA) El origen de esta teoría radica en el hallazgo de que la EA familiar y esporádica tiene en
común la formación de depósitos de BA. El mecanismo propuesto plantea que las placas seniles contienen productos del procesamiento de la Proteína precursora del amiloide (APP, del inglés Amiloyd precursor protein) la cual es una proteína de membrana cuya función no está completamente clara, pero se cree podría ser factor de crecimiento. APP bajo la acción de secretasas, moduladas por presenilinas, se cliva formando -entre otras proteínas de menor peso molecular- el BA, el cual agrega en placas y contribuiría al daño celular.
Las secretasas son alpha, beta y gamma secretasas. Las dos últimas estarían mayormente
ligadas a la génesis del BA; particularmente la gamma secretasa, cuya estructura pareciera corresponder a una presenilina. Si bien no está claro, al menos se acepta que es presenilina-dependiente. Por su parte, las presenilinas son el foco en la EA familiar, puesto que mutaciones a este nivel llevan en última instancia a la acumulación de BA; por otra parte, también se han descrito mutaciones a nivel de APP en las regiones de corte de las secretasas.
En las formas esporádicas, las alteraciones se encuentran principalmente a nivel del alelo
APOE4, lo cual se traduce en reducción de los niveles de APOE, facilitando el depósito de BA. Los mecanismos propuestos para esto son que podría funcionar como chaperona patológica, además del daño generado al disminuir la eficiencia en la reparación neuronal y el rol como transportador de colesterol a nivel cerebral.
Cabe destacar que las placas seniles pueden encontrarse en pacientes ancianos que no
presentan EA, por lo cual son marcadores de senilidad. Se puede depositar BA en el cerebro de los ancianos sin desarrollar la enfermedad, ya que ésta se encuentra determinada por el daño citotóxico que produce un tipo específico de BA (Beta Amiloide 1-42) el cual a su vez depende de los factores mencionados previamente. En la molécula de BA 1-42 existe metionina en la posición 35 la cual es clave en las reacciones de estrés oxidativo. Puede haber manifestación de enfermedad de Alzheimer en ausencia de placas seniles; además, su densidad no se correlaciona a severidad de la enfermedad.
Los mecanismos de citotoxicidad por los que actúa el BA son la activación inmune (microglía) lo
cual se traduce en inflamación, liberación de citoquinas, neurotoxicidad directa y estrés oxidativo. Además, puede inducir apoptosis a través de una enzima, la Aβ-binding alcohol dehydrogenase
(ABAD), uniéndose a la mitocondria. Esto se asocia a muerte cerebral. Se han encontrado altos niveles de ABAD en pacientes con enfermedad de Alzheimer.
Por otra parte, el BA puede dificultar la perfusión mediante su acumulación en arteriolas y
capilares, y afectar los contactos sinápticos. Este último mecanismo de daño sería producto de la

acumulación de oligómeros pequeños de BA, involucrando disfunción de las sinapsis colinérgicas y glutamatérgicas. Por excitotoxicidad, se produce gran liberación de Glutamato desde las células gliales lo que se traduce en activación exagerada de R-NMDA, esto genera aumento del calcio intracelular, lo cual a su vez activa la Oxído Nítrico sintetasa llevando a daño oxidativo. Proteína Tau y EA.
Se trata de una proteína asociada a microtúbulos, esencial en la estabilidad de éstos y en transporte axonal. La agregación de Tau genera filamentos helicoidales pareados conocidos como ovillos neurofibrilares, esto disminuye la estabilidad de los microtúbulos y desemboca en muerte neuronal.
Relación Tau-BA: El Beta Amiloide activa proteínas kinasa (Glicógeno sintasa kinasa 3β,
GSK3β, y kinasa dependiente de ciclina 5, CDK5); las cuales a su vez dependen de caseína kinasa, calmodulina y PK dependiente de calcio, entre otras, para llevar a la hiperfosforilación de Tau. Esto redunda en oligomerización y nucleación de la proteína, seguido de un proceso de glicolización no enzimática mediante el cual se generan enlaces covalentes que estabilizan la estructura llevando a la formación de los ovillos neurofibrilares.
Datos de investigación básica sugieren que la inhibición farmacológica de CDK5 disminuye la
neurotoxicidad de BA. Por otra parte, en ratones que no expresan proteína Tau no se genera neurotoxicidad, sugiriendo que la hiperfosforilación de ésta secundaria a la acción de kinasas dependientes de la activación por BA constituye uno de los mecanismos fundamentales de daño cerebral y afirmando la idea de un modelo integrado en el que diversas teorías confluyen para explicar cómo se determinan las bases de la enfermedad. Daño Oxidativo.
La respiración mitocondrial es un proceso bioquímico característico de las células con metabolismo aeróbico. Este proceso lleva, inevitablemente, a la producción de radicales libres. Dicho fenómeno se encuentra aumentado en los procesos inflamatorios, las alteraciones vasculares y en el envejecimiento, debido a la esperable falla en los mecanismos de reparación antioxidante.
El estrés oxidativo es aún más importante a nivel del SNC. Esto, debido al gran consumo de
oxígeno que demanda y la alta concentración de lípidos (en la forma de ácidos grasos poli-insaturados) susceptibles de ser oxidados. La peroxidación lipídica se vincularía a daño cerebral, considerando las funciones de componente estructural, moduladores celulares y segundos mensajeros que los lípidos poseen.

Las primeras evidencias que ponen al estrés oxidativo como unos de los responsables en la etiopatogenia de la Enfermedad de Alzheimer fueron aportadas por Stadtman, quien postuló la presencia de un aumento de proteínas oxidadas (como carbonilos) en EA. Sin embargo, hasta ahora no ha sido posible aclarar si tales procesos son un mecanismo causal o sólo forman parte de la propagación del daño celular.
Este modelo propone que el óxido nítrico (NO) en grandes cantidades, facilitaría su unión a
anión superóxido (O2-) para formar peroxinitrito (ONOO-), siendo este último el causante del estrés oxidativo y nitrosatrivo que llevaría, en último término, al daño mitocondrial. A su vez, el BA bloquea la cadena respiratoria I, disminuye ATP, lleva a disfunción mitocondrial y estrés oxidativo. En suma, se produce disminución de ATP y disminución de la remoción de agregados de BA.
Por otra parte, el estrés oxidativo sería secundario al daño mitocondrial previo (en etapas
tempranas de la EA). Estudios de análisis mitocondriales por hibridación in situ e inmunohistoquímica darían cuenta de polimorfismos en el DNA mitocondrial en neuronas de pacientes con EA. En dichas neuronas, la cantidad de DNA mitocondrial y proteínas mitocondriales sería 3 a 4 veces superior comparado con mitocondrias de neuronas de pacientes sin EA, siendo el número de mitocondrias similar en ambos casos. Análisis ultraestructurales (por microscopía electrónica) mostraron presencia de DNA mitocondrial en lisosomas de células de pacientes con EA. Mitocondrias y retículo endoplásmico eran fagocitados, liberando metales (Fe, Cu) al citoplasma, lo que aumentaría los procesos de óxido-reducción. Además, metales en combinación con ácidos nucleicos formarían ácidos nucleicos de una hebra, sensibles a nucleasas. Por último, se plantea que NADPH oxidasa u otra xantina oxidasa (aumentada en pacientes con EA) contribuiría a incrementar el aporte de oxígeno reactivo de otras especies generadoras, como el peróxido de hidrógeno (H2O2) producido por la mitocondria. Inflamación.
Desde fines de los ’80 ha aparecido cierta evidencia epidemiológica respecto de que el tratamiento crónico con AINEs disminuiría la incidencia de EA en la población.
Aunque las neuronas secretan citoquinas en forma constitutiva, serían la microglía
(macrófagos del SNC encargados de comandar la muerte celular programada o apoptosis de neuronas durante el desarrollo cerebral) y los astrocitos los generadores principales de factores proinflamatorios (IL-1β, IL-6, TNFα, etc.). El Aβ activaría la microglía, produciendo activación de la fagocitosis y daño sobre neuronas vecinas (mecanismo directo) y liberación de citoquinas, NO y neurotoxinas (mecanismo indirecto). En este sentido, se especula que los AINEs contribuirían a frenar a esta microglía activada por Aβ.

Cabe mencionar que, en relación a la edad, se ha demostrado que IL-6 y TNFα aumentarían su producción con el paso de los años. Asimismo, personas portadoras de Síndrome de Down presentarían un riesgo aumentado de desarrollar EA hacia la senectud, debido en parte a que en estos pacientes hay aumento de IL-1 y de niveles de microglía activada; aunque no se debe dejar de mencionar en este subgrupo la influencia de la alteración genética por sí misma, pues existe un locus ligado a la EA en el cromosoma 21.
Existen ciertas evidencias a favor de mecanismos inmunológicos. Por ejemplo, el año 2001 se
inició el uso de inmunoterapia en EA con inmunización pasiva (anticuerpos anti BA) y activa. Debió suspenderse el año 2002 por la alta tasa de encefalitis en los pacientes tratados. El año 2003 se publicó el resultado de la autopsia de una paciente fallecida por TEP, ésta arrojó disminución de placas de BA en corteza cerebral en relación a 7 pacientes controles de características similares no inmunizados, siendo relevante el hecho de que las regiones sin placas contenían gran cantidad de microglías que co-localizaban con la inmunodetección para BA, lo que sugiere que la inmunización contra BA promovió la fagocitosis de la microglía.
También se ha demostrado mejoría cognitiva en modelos animales inmunizados, lo que permite
plantear que en fases tempranas de la enfermedad existirían mecanismos reversibles que anteceden a la muerte neuronal, ya sean éstos a nivel de sinapsis, de traducción de señales o de conducción neural.
Finalmente, existe un reporte de Canadian Study of Health and Aging del año 2001, en el cual
mediante análisis estadístico de pacientes mayores de 65 años con deterioro cognitivo se encontró que aquellos vacunados contra difteria, tétanos, polio e influenza presentaron menor riesgo de desarrollar EA.
Otros mecanismos propuestos. La hipótesis cerebrovascular plantea daño definido por disminución del clearence de BA cerebral y disminución en el flujo de nutrientes por disfunción de vasos sanguíneos cerebrales. Existe evidencia de que los pacientes con antecedentes de AVE isquémico tienen aumentada la expresión de APP seguido de aumento en depósito de BA. Se ha vinculado el daño vascular a hiperhomocisteinemia, sin que quede claro si ésta es factor de riesgo o marcador de enfermedad, pues el aumento de homocisteína en el plasma precede temporalmente las manifestaciones de alteraciones cognitivas.

Lecturas Recomendadas.
1. Von Bernhardi R. Mecanismos neurobiológicos de la enfermedad de Alzheimer. Rev. Chil. Neuro-psiquiatr. 2005; 43(2):123-132.
2. Maccioni C., Arzola M., Mujica L., Maccioni R. Nuevos paradigmas en el estudio de la
patogénesis de la enfermedad de Alzheimer. Rev. Chil. Neuro-psiquiatr. 2003; 41(2):33-46. 3. Perry G., Ávila J., Casadesus G., Nunomura A., Tabaton M., Cash A., Aliev G., Wataya T.,
Shimohama S., Drew K., Atwood C., Smith M. La función del estrés oxidativo en la patogénesis de la enfermedad de Alzheimer. Rev. Chil. Neuro-psiquiatr. 2003; 41(2):47-52.
4. Blennow K., De Leon M., Zetterberg H. Alzheimer’s disease. Lancet. 2006; 368:387-403. 5. Flajolet M., He G., Heiman M., Lin A., Nairn A., Greengard P. Regulation of Alzheimer’s
disease amyloid-beta formation by casin kinase I. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104(10):4159-4164.
6. Bradford F., Sanjay G. A review of antioxidants and Alzheimer’s disease. Annals of Clinical
Psychiatry. 2005; 14(4):269-286. 7. Butterfield A. Amyloid b-peptide (1-42)-induced oxidative stress and neurotoxicity:
implications for neurodegeneration in Alzheimer’s disease brain. Free Radical Research. 2002; 36(12):1307-1313.
8. Rosenberg P. Clinical aspects of inflammation in Alzheimer’s disease. International Review
of Psychiatry. 2005; 17(6):503–514. 9. Nielke M., Lyketsos C. Lipids and the pathogenesis of Alzheimer’s disease: is there a link?
International Review of Psychiatry. 2006; 18(2):173–186. 10. Eikelenbooma P., Hoogendijka W., Jonkera C., van Tilburga W. Inmunological mechanisms
and the spectrum of psychiatric syndromes in Alzheimer’s disease. Journal of Psychiatric Research. 2002; 36:269-280.
11. Alkont D., Sun M., Nelson T. PKC signaling deficits : a mechanistic hypothesis for the origins
of Alzheimer’s disease. Trends in Pharmacological Sciences. 2006; 28(2):51-60.

12. Tolnay M., Probst A. The neuropathological spectrum of neurodegenerative tauopathies. Life. 2003; 55(6):299-305.
13. Ruppin E. Neural modelling of psychiatric disorders. Network Computation in Neural
Systems. 1995;6:635-656.