computer-aided molecular design using tabu search molecular design (camd) has the potential to...
TRANSCRIPT

Computer-Aided Molecular Design Using Tabu Search
B. Lin,1 S. Chavali,2 K. Camarda2 and D. C. Miller1,*
1Department of Chemical Engineering, Rose-Hulman Institute of Technology, 5500 Wabash Avenue, Terre Haute, IN 47803, USA 2Department of Chemical and Petroleum Engineering, University of Kansas, 1530 W. 15th, 4132 Learned Hall, Lawrence, KS 66045, USA
Abstract
A detailed implementation of the Tabu Search (TS) algorithm for computer-aided
molecular design (CAMD) of transition metal catalysts is presented in this paper.
Previous CAMD research has applied deterministic methods or genetic algorithms to the
solution of the optimization problems which arise from the search for a molecule
satisfying a set of property targets. In this work, properties are estimated using
correlations based on connectivity indices, which allows the TS algorithm to use several
novel operators to generate neighbors, such as swap and move, which would have no
effect with a traditional group contribution-based approach. In addition, the formulation
of the neighbor generation process guarantees that molecular valency and connectivity
constraints are met, resulting in a complete molecular structure. Results on two case
studies using TS are compared with a deterministic approach and show that TS is able to
provide a list of good candidate molecules while using a much smaller amount of
computation time.
Introduction

Computer-aided molecular design (CAMD) has the potential to greatly decrease the time
and effort required to develop new molecular entities by reducing the need for costly and
time-consuming trial-and-error experiments. Instead, a slate of candidate molecules
which are predicted to have the desired properties can be used as a starting point for more
focused experimental synthesis. This general method is applicable to such varied
products as catalysts, polymers, solvents, and detergents. Hairston (1998) recently
reported that a computational algorithm has been successfully used to design a new
cancer-fighting pharmaceutical.
The CAMD methodology consists of solving both a forward and a backward
problem (Venkatasubramanian et al., 1994). The forward problem predicts properties
based on molecule structure; the backward step identifies a structure to obtain a molecule
with a given set of target properties. Property prediction is usually based on either group
contribution methods or topological indices. The group contribution approach has been
most widely reported (Gani et al., 1991; Venkatasubramanian et al., 1994; Maranas,
1996; Vaidyanathan & El-Halwagi, 1996; Harper & Gani, 2000; Sahinidis &
Tawarmalani, 2000; Harper et al., 2003; Sahinidis et al., 2003). Constantinou & Gani
(1994) and Constantinou et al. (1995) described a two level group contribution method
which utilizes molecular structure information to estimate the physical and
thermodynamic properties of pure components. A three-level group contribution method
proposed by Marrero & Gani (2001) exhibits improved accuracy and applicability to deal
with bio-chemically and environmentally-related compounds. Because most group
contribution methods cannot adequately account for steric effects (Wang & Milne, 1994),
several researchers have begun using topological indices in an effort to obtain more

accurate property prediction (Kier & Hall, 1976; Raman & Maranas, 1998; Camarda &
Maranas, 1999; Siddhaye et al., 2000; Harper et al., 2003). Whichever approach is
chosen, existing structure-property information is regressed to form an empirical model
relating the molecule structure to the properties of interest. The inverse problem is
essentially a mixed integer optimization problem, whether it is solved explicitly and
deterministically as in Maranas (1996), stochastically via a genetic algorithm-based
approach as in Venkatasubramaniam et al. (1994), or via a generate and test approach
such as those of Gordeeva et al. (1990), Friedler et al. (1998), Harper & Gani (2000) and
Harper et al. (2003).
Many researchers have reported solutions to the backward problem to determine
the molecular structure. Combinatorial and heuristic-based enumeration approaches have
been reported (Gani & Brignole, 1983; Joback & Stephanopoulos, 1989). Kier et al.
(1993) used a graph reconstruction approach to determine feasible molecular structures
with bounded physical property values, while Vaidynathan & El-Halwagi (1996)
described an interval analysis approach for the computer-aided synthesis of polymers and
blends. CAMD problems have recently been formulated as mixed-integer linear/nonlinear
programming (MILP/MINLP) problems. Maranas (1996) transformed the nonconvex
MINLP formulation into a tractable MILP model by expressing integer variables as a
linear combination of binary variables and replacing the products of continuous and
binary variables with linear inequality constraints. Using connectivity indices, Camarda
& Maranas (1999) described a convex MINLP representation for solving several polymer
design problems. Churi & Achenie (1996) solved a refrigerant design problem with an
augmented penalty-outer approximation (AP/OA) algorithm. A reduced dimension

branch-and-bound algorithm was presented by Ostrovsky et al. (2003) to design optimal
solvents used as cleaning agents in the printing industry. Sahinidis et al. (2000, 2003)
reported a branch-and-reduce algorithm for identifying a replacement of Freon.
Stochastic optimization approaches have also been developed as alternate strategies for
rigorous deterministic methods. Venkatasubramaniam et al. (1994) employed a genetic
algorithm (GA) for polymer design in which properties are estimated via group
contribution methods. Marcoulaki & Kokossis (1998) described a simulated annealing
(SA) approach for the design of refrigerants and liquid-liquid extraction solvents. Wang
& Achenie (2002) presented a hybrid global optimization approach that combines the OA
and SA algorithms for several solvent design problems.
Tabu search (TS) is a heuristic approach for solving combinatorial optimization
problems by using a guided, local search procedure to explore the entire solution space
without becoming easily trapped in local optima. It differs from other stochastic
optimization techniques by maintaining lists of previous solutions (usually termed
“memory”) that help guide the search process. These lists are useful for CAMD since
they provide a direct method to track near-optimal solutions. The ability of TS to
efficiently find a set of near-optimal solutions is particularly useful since all property
prediction algorithms have limited accuracy and problem formulations normally do not
include all relevant properties. Thus, the determination of the global optimum is not as
critical as finding a set of near-optimal solutions. Deterministic approaches can generate
such a list by multiple MINLP solves and application of integer cuts, but for large
problems this becomes prohibitively computationally expensive. Other stochastic
approaches, as well as generate and test strategies, can also generate such a near-optimal

candidate list. By identifying a range of potential target molecules, TS avoids missing
potentially useful molecules and allows the use of other criteria (such as ease of
synthesis) to perform a final ranking of the candidates.
Transition metal catalysts are an important class of molecule for creating other
molecules efficiently with minimal impact on the environment; however, the catalysts
themselves are often extremely toxic and harmful to the environment. For example, most
propylene oxide is produced using homogenous catalysts containing molybdenum, which
are effective but also harmful to the environment. Since in many cases a significant
amount of such catalyst is lost to the environment (Allen & Shonnard, 2002), new
materials which show improved catalytic activity and less toxicity are highly desired.
This paper describes a framework for using TS to design transition-metal catalysts.
Property Prediction via Connectivity Indices
In order for a molecular design algorithm to be successful, physical properties of interest,
such as density, toxicity, solubility, or reactivity within a given system must be estimated
with a reasonable accuracy using only a very small computational effort. Once the
properties of a transition metal catalyst can be predicted from structure, the problem of
designing a new catalyst can be formulated as an optimization problem. We employ
connectivity indices, which are numerical values that describe the electronic structure of
a molecule, to characterize the molecule and to correlate its internal structure with
physical properties of interest. These indices connect the molecular structure to the
properties of interest by providing a mathematical description of the molecule as a whole.

Molecular connectivity indices were first introduced by Randić (1975). Kier &
Hall (1976) reported correlations between connectivity indices and many key properties
of organic compounds, such as density, solubility, and toxicity. Furthermore, these
indices can be computed with a minimum of computational effort. Many of the
applications of connectivity indices have been reviewed by Trinajstic (1983). Kier et al.
(1993) and Gordeeva et al. (1990) reported the use of connectivity indices within a
molecular design context, while Raman & Maranas (1998) first incorporated connectivity
indices into an optimization framework. Camarda and Maranas (1999) used connectivity
indices as property descriptors to design polymers that have pre-specified property
values. Siddhaye et al. (2000) employed connectivity indices to predict the physical
properties for the design of novel pharmaceutical products via combinatorial
optimization.
In earlier molecular design work, group contribution methods were used to
estimate the values of physical properties, as in Gani, et al.(1991), Venkatasubramanian
et al. (1994), and Maranas (1996). The works of Constantinou & Gani (1994) and
Marrero & Gani (2001) extend the basic group contribution approach by considering
combinations of functional groups and thus take into account second-order effects when
used to predict physical properties. Connectivity indices, however, take into account the
entire molecular structure of a compound. By using higher-order connectivity indices,
third-order and higher structural effects can be included in structure property relations.
Harper & Gani (2000) and Harper et al. (2003) have combined group contribution with
connectivity indices to include these higher-order effects. Raman & Maranas (1998) and
Harper et al. (2003) have shown that these higher order effects are able to give more

accurate property descriptions. Furthermore, when a molecular design problem is solved
using these indices, a complete molecular structure is obtained, and no secondary
problem must be solved to recover the final molecular structure. Harper & Gani (2000)
and Meniai et al. (1998) have also reported the design of complete molecular structures.
Connectivity indices also provide a method to compute the properties of mixtures, since
the connectivity indices of individual compounds can be combined to estimate mixture
properties in certain cases (Kim et al., 1992; Johnson et al., 2002).
Computational property estimation algorithms first decompose a molecule into
smaller units. Then, a set of basic groups that can potentially be part of the candidate
molecules are defined. A basic group is defined as a single non-hydrogen atom in a given
valence state, bonded to some number of hydrogen atoms. Atomic connectivity indices
are defined over each basic group.
Basic groups of the molecule CHCl2F are shown in Table 1. The values are the
simple atomic connectivity indices that refer to the number of bonds which can be formed
with other groups. The v values are atomic valence connectivity indices that describe
the electronic structure of each basic group, including lone-pair electrons and
electronegativity. For transition metals, which can assume multiple valence states, the
definition of v is based on the number of electrons participating in the bonding, instead
of those present in the outer shell. Once the atomic connectivity indices of basic groups
are defined, the molecular connectivity indices can be computed for the entire molecule.
Molecule structure is expressed with a hydrogen-suppressed graph (as shown in
Figure 1). The zero order molecular connectivity indices 0 and V0 are the sum of each

basic group (the sum over all vertices), which describe the identity of the groups in a
given molecule (see Table 2).
Higher order connectivity indices can also be defined and have been used to give
a more precise description of molecular structure (Kier & Hall, 1976). In this work, we
have used the second-order connectivity indices, 2 and V2 , to give added accuracy to
correlations. These two indices are sums over each of the triplets in the molecule, that is,
over each possible combination of three bonded groups (see Figure 1).
Once the equations defining the (molecular) connectivity indices are in place,
these indices can be used in empirical correlations to predict the physical properties of
novel transition-metal catalysts. Table 3 shows the basic groups employed in the design
of a molybdenum catalyst for an epoxidation reaction. (To limit the search space, we set
an upper bound on the maximum number of groups in a molecule.) The correlation
derived in this work for density is:
0 0 1 1 2 2
2 2 20 0 0 1 1
2 22 2 1 2
55351 75800 7663 40901 1784 72046 607
24695 649 12271 65.4
1793 8.9 72323
v v v
v v
v
(1)
This correlation was developed from regressing 23 Mo-centered complexes and resulted
in a correlation coefficient of 0.962 and a sum of squared error of 0.944. Using this
correlation, an optimization problem has been formulated whose optimal solutions are
molecules which most closely match a set of target property values.
Problem Formulation
When zeroth, first and second order connectivity indices are employed for
property estimation, a molecule is represented mathematically using sets of binary

variables. First, a vector of binary variables, iz , is defined. An element of this vector
equals one if the ith group exists in the molecule; otherwise, it equals zero. Second, a
partitioned adjacency matrix with elements kjif ,, is determined. When basic groups i and
j are bonded with a kth-multiplicity bond, 1,, kjif ; otherwise, 0,, kjif . These sets of
variables provide sufficient information to compute the connectivity indices and, thus,
estimate molecular properties. Along with these definitions, property correlations using
the connectivity indices are included in the overall formulation.
The objective is to determine the molecule that minimizes the difference between
the target property values. This can be written as:
targetscale
1min Obj m mm R m
P PP
(2)
where R is the set of all targeted properties, Pm is the estimated value of property m,
Pmscale is a scale factor used to weight the importance of one property relative to another,
and Pmtarget is the target value for property m.
Structural constraints are added to ensure the obtained molecules are fully
connected and satisfy valency by being connected with the appropriate types of bonds.
Other constraints include bounds on the variables and property values. The resulting
formulation is a large, nonconvex MINLP model for CAMD.
Table 3 shows the set of 8 basic group types used for designing a molybdenum
catalyst for an epoxidation reaction. The total number of available groups is
221
, i
iMaxN . Since only single bonds are involved, the size of the adjacency matrix f
is elements 48412222 . Each element, , ,i j kf , can be either 0 or 1, and the total

number of independent variables is 25322122
1
ii . In order to apply TS effectively,
the original MINLP model of Siddhaye et al. (2000) has been altered to handle basic
groups and the connectivity between them rather than dealing with the elements of the
adjacency matrix directly.
TS Implementation
TS begins by determining an initial solution. Additional solutions (termed
neighbors) are generated by modifying the existing solution through a sequence of
moves. The best new neighbor (x’) is used as the starting point for the next iteration
unless it is on a tabu list. Thus, even if no neighbor solutions are better than the initial
solution, one of these is still chosen as the starting point for the next iteration. A record of
the best solution ever found (x*) is separately maintained. In addition, the tabu lists
provide an adaptive memory that guides the search by taking advantage of historical
information. This memory enables TS to make strategic choices and achieve responsive
exploration.
The standard TS algorithm (Glover & Laguna, 1997; Lin et al., 2003; Lin &
Miller, 2004b) is adapted to represent the molecule efficiently. Each solution is a
molecule consisting of a series of fully connected groups. Therefore, the initial solution is
constructed by connecting basic groups together. In generating neighbor solutions,
operators are also designed to handle basic groups. The procedure of building an initial
solution is described using the basic groups listed in Table 3. The progression of the
building process is shown in Table 4. In the CAMD framework, a mainchain is defined as
the list within a molecule with the largest number of groups. It is determined during the

process of constructing a molecule. The length of sidechains (list of groups connecting to
the mainchain) is constrained to be less than that of the mainchain. For catalyst design
test cases, the number of Mo groups is constrained to be either 1 or 2.
Building an initial solution
Step 1. One of the basic groups is selected as a root group. For simplicity, a group with
only one bond, –NH2, is chosen.
Step 2. Another group is added to the root group. If the second group has only one bond,
i.e., –OH, there are no empty bonds in the molecule. Since the molecule is fully
connected, the initial solution, hydroxylamine (H2N–OH), would be obtained;
however, this violates the constraint requiring at least one Mo atom. Thus, another
group must be chosen. In this example, this step adds basic group #1 from Table
3, resulting in molecule fragment for step 2 shown in Table 4.
Step 3. Now, the molecule consists of two groups. Additional groups are needed to
connect to the empty bonds. Since the length of the mainchain is 2, the length of
the sidechains can be either 1 or 2. When the length is 1, another group with a
single bond, –OH, is attached as shown in Table 4. As sidechains are added to the
molecule, the mainchain identity and length are updated so that it is always the
longest chain in the molecule.
Step 4. The length of the second sidechain is 2, resulting in a branch that consists of two
groups. Thus, –O–Cl, are added.
Step 5. Another sidechain of length 1, –CH3, is added. One free bond is still existing in
the molecule.

Step 6. If a group with only one bond is selected, i.e. –OH, the molecule is fully
connected, and the initial solution is obtained as shown in Table 4. If the selected
group has more than one bond, steps 2-5 are repeated until the molecule is fully
connected.
A set of candidate solutions will be generated and evaluated at each iteration. These
candidates (or neighbor solutions) will be constructed by modifying the current molecule.
Neighbor generation operators
Two sets of operators are defined: local and global. Local operators maintain the
main backbone of the current molecule, and are implemented for the purpose of localized
search; global operators generate neighbor solutions that are significantly different from
the current one in order diversify the solutions at each iteration. There are six local
operators:
1. Replace
This operator replaces a group in the current molecule with another available basic
group. If –OH is chosen, it can be replaced by –Cl, –OH, or –CH3. If the group –
O– is replaced by–CH2–, a new molecule will be obtained (see Table 5)
2. Insert
A basic group that is available will be inserted in front of the selected group in the
current molecule. Suppose the group, –CH3, is chosen, a group with two single
bonds can be inserted on the sidechain. The new molecule by inserting –CH2– is
shown in Table 5.
3. Delete

This operator deletes the selected group in the current solution. Therefore, the
operator cannot be applied to a group with only a single bond, since it will results in
an infeasible molecule. In this example, only the molybdenum or the oxygen group
can be deleted. The molecule in Table 5 shows the result of the deletion of –O–.
4. Swap
Two groups of the current molecule are exchanged by applying the swap operator.
Suppose the group –CH3 is selected, another group with only a single bond will be
chosen. The molecule after swapping –Cl with –CH3 is shown in Table 5. Since
molecules obtained from swapping two groups connecting to the same “parent
group” are equivalent, groups to be swapped are required to be different and not
connected to the same “parent”.
5. Move
This operator moves the selected group within the existing molecule. Suppose the
group –O– is selected to be moved after the group –NH2. A new molecule is
obtained as shown in Table 5.
6. Combination
This operator combines the 5 operators to form a new one. A random number
between 1 and 5 is first generated. For example, 3, then 3 of the 5 operators, such as
Move, Insert, Swap, will be applied sequentially to the current molecule and
generate a neighbor solution.
The following three global operators help TS perform a diversified search to investigate
the whole solution space sufficiently:
7. SideChain_Rebuild

If the selected group is located on a sidechain of the molecule, the sidechain will be
replaced by a newly-generated one.
8. MainChain_Rebuild
If a mainchain group is selected, all groups that are located after it will be deleted
and the rest part of the molecule is reconstructed.
9. Total_Rebuild
If the first or the last group of the molecule is selected, the whole molecule will be
discarded and replaced by a new molecule that is built following the steps of
generating an initial solution.
The two sets of operators are selected based on the stage of locating a solution. Global
operators are more frequently selected at the starting stage to favor a diversified search
and local operators are selected more frequently at the end of the search process to locate
the final solution precisely.
After obtaining a molecule, all elements of the adjacency matrix can be determined
according to the connectivity among the groups. Then, with the provided simple atomic
indices and valence indices, the molecular connectivity indices can be calculated, and the
properties of this molecule are estimated with the property-structure correlations. At each
iteration, certain solutions are classified as tabu (forbidden) and added to tabu lists. At the
same time, the tabu property of other solutions will expire, and they will be removed
from the tabu lists. In this way, tabu lists are updated continuously and adapt to the
current state of the search. The use of adaptive memory enables TS to exhibit learning
and creates a more flexible and effective search.

Tabu lists
In the TS algorithm, each molecule consists of a set of fully connected groups,
which are classified into mainchain and sidechain groups. Figure 2 shows a solution
located by TS based on basic groups listed in Table 3; the mainchain is identified as:
MoCl CH2 Cl
with 3 sidechains that are all of length 1, the group –OH. If both the mainchain and
sidechain groups, as well as connectivity relations are included in tabu lists, the update
and maintenance of the lists will be time-consuming. Therefore, both recency-based and
frequency-based tabu lists only keep track of mainchain groups.
The recency-based tabu list records recently visited solutions, and is called short-
term memory. At each iteration, if the best neighbor is not better than the current
solution, it is classified as tabu and added to the recency-based tabu list. The tabu
property remains active throughout the tabu tenure, which is empirically set equal to the
total number of basic groups available for building the molecule (22 for this case).
A segment of the recency-based tabu list is shown is Table 6. The first column
shows the number of iterations during which a solution will be kept on the recency-based
tabu list. The first solution on the recency-based tabu list with an objective function
0.0036 has the following mainchain:
MoNH2 CH2 CH3
It will be released from the list after 22 iterations, unless a best neighbor with the same
mainchain is located within this period. In this case, the tabu property is overridden by
the improved-best aspiration criterion (Glover & Laguna, 1997).

The frequency-based tabu list provides long-term memory and keeps track of the
solutions that have been visited most frequently. The frequency tabu property is denoted
with an index. When a previous solution is revisited, its frequency index will be
incremented; otherwise, the frequency index decreases. This allows TS to determine
whether the search process has become trapped in a specific area for a long time.
Table 7 shows six solutions on the frequency-based tabu list. The 1st solution with
an objective function value of 0.4848 has the lowest frequency index. This solution will
be replaced by new solutions if the frequency-based tabu list is full. The 4th solution with
the following mainchain structure:
MoOH O CH3
has been visited most frequently, since its frequency index (12.75) is much larger than
that of the other solutions.
Because the tabu lists track the occurrence mainchain groups, the danger of auto
isomorphism negatively impacting the algorithm is reduced. Even without considering
auto isomorphism, it is possible that some neighbor solutions will be the same as others.
To avoid becoming stuck with a best solution always being a rearranged version of the
same molecule, the tabu list will recognize the mainchain and force the selection of a new
starting point for the next generation of neighbors.
Intensification and diversification
Based on knowledge of the current search status as provided through the tabu
lists, intensification and diversification strategies are used to control the search area.
Intensification is carried out by generating neighbor solutions employing the local

operators. Global operators increase diversification and are implemented based on
frequency-based tabu lists. If the maximum frequency index of solutions on the
frequency-based tabu list is larger than a predefined threshold (in this case, the threshold
is empirically set to 22), TS is deemed to have been searching around a specific solution
too often. Thus, the current solution will be replaced by a new randomly generated
solution by applying the global operators, and the search process will be restarted. This
diversification strategy is similar to the restart mechanism used in other stochastic
optimization approaches; however, the restart is not purely random. Instead, it is guided
by historical information.
In some cases, TS cannot locate improved solutions after the restart operation and
additional procedures will be required. For example, if the current best solution has the
objective function of 0.0094 and TS initiates a restart, if the best neighbor after several
iterations is not less than 0.0094, another intensification strategy will pull the search back
to the formerly promising area, by assigning the current solution to the best neighbor
recorded before the restart operation.
Aspiration Criterion
An aspiration criterion based on the a sigmoid function is employed to invalidate
tabu property in certain cases and helps to maintain an appropriate balance between
diversification and intensification (Lin & Miller, 2004a,b). Since an intensified search is
favored at the end, and a broadly diversified search, at the beginning, the aspiration
criterion allows the tabu property to be overridden more towards the end of the search.
This helps to ensure that the tabu lists properly encourage a broad search early on.

Constraint Handling
In this application, constraints are handled in a way that guarantees the feasibility
of the molecule. Since TS operates on basic groups directly, both valence and
connectivity constraints are handled during the process of neighbor generation as
previously described. For example, both local and global operators ensure the proper type
of bond (single, double or triple) to be connected. In addition, the number of empty bonds
is checked to guarantee that valency will be satisfied in the current solution. Groups that
would cause a violation of either connectivity or valency are not allowed. Otherwise all
groups can interconnect.
Case Studies
The effectiveness of the TS algorithm for CAMD is demonstrated with the
following two case studies. The parameters for each case are as follows:
1. The number of neighbor solutions generated at each iteration is equal to the
product of the number of basic groups and the maximal number of groups in a
molecule;
2. The number of iterations is 200;
3. The length of the tabu list is defined to be the same as the maximal number of
groups in a molecule;
The TS algorithm is compiled using the gcc compiler on a Pentium III 1.0 Ghz CPU,
1024 MB memory PC running Redhat Linux 7.1.

Case 1
The first case is the design of a molybdenum catalyst for an epoxidation reaction. Eight
basic groups (see Table 3) are used to define the search space. The purpose of this case is
to evaluate the effectiveness of the formulation and to compare TS with a deterministic
solution method. Thus, only a single target property, density, is used. While density is an
important property of a homogenous catalyst, it does not define the catalytic activity or
any other critical properties and, thus, cannot alone define an effective catalyst. However,
density is closely related to solubility for these systems, and solubility defines the amount
of potential uptake by humans. Therefore, a combination of density and toxicity would be
needed to assess exposure risk. The target density value is 4172 kg/m3, which is the
density of a commonly used homogenous transition-metal catalyst. The correlation for
density this property is given by eq. (1).
The number of neighbor solutions is 8 * 22 = 176. The 5 best molecules and the
probability that a molecule is found are obtained from 100 trials. Suboptimal solutions
are either the final output of a test or an intermediate result of certain iteration. If the
suboptimal solution is the final result of a trial, the probability is incremented by 1%.
Since intermediate solutions of some trials may be even better than the final result of
other trials, the probability of such suboptimal solutions is denoted as < 1% (see Table 8)
to indicate that it was located, but was never a final solution. The best solution, with the
objective function value of 0.000111, corresponds to a density of 4172.46, which is only
found once in 100 trials. Solution 3 (0.000238) has 7 basic groups and is found 80% of
the time. The corresponding density value of 4172.99 kg/m3 is very close to the best
solution. Since the difference between the best solution and the 5th best solution is less

than 0.2%, this shows that TS can successfully determine several promising catalyst
molecules for further experimental verification.
It is especially useful to identify and record near-optimal solutions since the
density correlation has an error of approximately 4%. Thus, near-optimal solutions are
almost as likely to be strong candidates for synthesis as the optimal one. Furthermore,
many other factors, such as ease of synthesis, have not been taken into account within the
optimization formulation. Thus, a user would like to have multiple options to choose
from.
TS provided the results in Table 8 in 90 seconds on a PIII personal PC. In
comparison, using Outer Approximation via the DICOPT solver in GAMS, only an
integer feasible solution was found after 20 minutes on a Sun Ultra 10 workstation. The
structure (see Figure 3) resulted in an objective function of 5.67.
Case 2
The second case study involves 10 basic groups (see Table 9). The properties to be
optimized are electronegativity, oxidation state and toxicity. The electronegativity is
calculated as follows:
, ,1 1
* ( ) ( )Mo Tot BondN N N
i j li j i l
Elec f Elec i Elec j
(3)
where )(iElec is the electronegativity of group i, f is an element of the adjacency matrix,
MoN is the number of rows allotted to all the molybdenum groups, TotN is the total
number of basic groups (20 in this example), BondN is type of bonds of the basic groups

(2 in this example, single and double). The lower bound is 0 and the upper bound is 3.84
eV. Oxidation state is the sum of the oxidation states of all the molybdenum groups:
1
* ( )MoN
ii
Oxid z Oxid i
(4)
where )(iOxid is the oxidation state of the ith group and z(i) is an element of the existence
vector. The target value of the oxidation state is the set 8,7,6 . Toxicity is
determined with the following correlation for the LC50:
0 0 1 1 2 250
0 2 1 2 1 2
0 1 0 1 0 2
log( ) 27.8 5.49 10.6 6.65 2.93 4.40 0.368
0.710( ) 5.59( ) 0.0761( )2.10 0.516 0.0295
v v v v
v v
v v v v v
LC
(5)
The LC50 was developed using 34 data points obtained from Syracuse Research
Corporation (2000). The resulting correlation has a correlation coefficient of 0.953 and a
sum of squared error of 2.083.
In this example, the objective function is the weighted summation of these
properties:
500.6 0.3 0.1Obj LC Elec Oxid (6)
The weights on the properties are freely adjustable and could be based on the preference
of the designer. For this case study, these weights are simply examples and do not
directly impact the conclusions of the paper.
The number of neighbor solutions is 10 * 20 = 200. The length of the tabu list is
20. The 5 best molecules obtained from 100 TS trials are shown in Table 10. The optimal
solution with an objective function of 1.286401 is obtained in 40 trials and the second
optimal solution is located the remaining 60% of the time. The difference between the
optimal solution and the 10th sub-optimal solution (shown below):

Cl ONH ClMo MoO CH2NH
is smaller than 1%. (The value of its objective function is 1.297251.) In addition, more
than 60 near optimal solutions are obtained for this case study, which shows that TS can
locate a large number of near-optimal solutions for further experimental verification.
In comparison, employing Outer Approximation via the DICOPT solver in
GAMS, only an integer feasible solution was found after 20 minutes of CPU time on a
Sun Ultra 10 workstation. The structure (see Figure 4) resulted in an objective function of
5.55. While the deterministic algorithm may be able to find and guarantee the global
optimum to this problem after expending a large amount of CPU time, we see that TS can
generate a list of near-optimal solutions within a short amount of time. This is very useful
to a researcher searching for novel alternatives to current catalysts, since the list of near-
optimal solutions provides options which can be narrowed down by employing other
factors such as cost, ease of synthesis, and estimated values of physical and chemical
properties not included in the original design.
Conclusions
A detailed implementation of the TS algorithm to CAMD problems is presented
in this paper. Although other optimization approaches have been applied to CAMD with
properties predicted using group contribution techniques, the TS algorithm implemented
with novel neighbor-generating operators and combined with property prediction via
connectivity index-based correlations provides a powerful technique for generating lists

of near-optimal molecular candidates for a given application. In addition, the tabu lists
help TS search the solution space both in a diversified way, to cover the entire search
space, and in an intensified manner, to locate the final solution precisely. Moreover, TS is
able to locate a large number of near optimal solutions within a short time as shown in
the two case studies.
Acknowledgements
This project was supported by the National Science Foundation through grant CTS-
0224887. B. Lin acknowledges additional support from the Department of Chemical
Engineering of Rose-Hulman Institute of Technology.
Nomenclature
simple atomic connectivity indices that refer to the number of bonds which
can be formed with other groups
v atomic valence connectivity indices that describe the electronic structure of
each basic group
0 zero order molecular connectivity indices
V0 zero order molecular valence connectivity indices
2 second-order molecular connectivity indices
V2 second-order molecular valence connectivity indices
iz An existence vector showing wither the ith group exists in the molecule

kjif ,, A partitioned binary adjacency matrix showing when basic groups i and j are
bonded with a kth-multiplicity bond
R set of all targeted properties
Pm the estimated value of property m
Pmscale a scale factor used to weight the importance of one property relative to
another
Pmtarget the target value for property m
x’ best new neighbor solution
x* the best solution ever found
)(iElec electronegativity of group i in electronvolts (eV)
MoN the number of rows allotted to all the molybdenum groups
TotN total number of basic groups
BondN type of bonds of the basic groups (1 = single, 2 = double)
Oxid sum of the oxidation states of all the Mo groups
)(iOxid the oxidation state of the of the ith group
LC50 Lethal concentration killing 50% of the test population, a measure of toxicity
References
Allen, D. & D. Shonnard (2002). Green Engineering: Environmentally Conscious Design
of Chemical Processes. Upper Saddle River, New Jersey: Prentice-Hall.
Camarda, K.V. & C.D. Maranas (1999). Optimization in Polymer Design using
Connectivity Indices. Industrial & Engineering Chemistry Research 38: 1884-
1892.

Churi, N. & L.E.K. Achenie (1996). A Novel Mathematical Programming Model for
Computer Aided Molecular Design. Ind. Eng. Chem. Res. 35(10): 3788-3794.
Constaninou, L. & R. Gani (1994). New Group Contribution Method for Estimating
Properties of Pure Components. AIChE Journal 40(10): 1697-1710.
Constantinou, L., R. Gani & J.P. O'Connell (1995). Estimation of the Acentric Factor and
the Liquid Molar Volume AT 298K Through a New Group Contribution Method.
Fluid Phase Equilibria 103: 11-22.
Friedler, F., L.T. Fan, L. Katotai & A. Dallos (1998). A Combinatorial Approach for
Generating Candidate Molecules with Desired Properties Based on Group
Contribution. Computers & Chemical Engineering 22(6): 809-817.
Gani, R. & E.A. Brignole (1983). Molecular Design of Solvents for Liquid Extraction
based on UNIFAC. Fluid Phase Equilibria 13: 331-340.
Gani, R., B. Nielsen & A. Fredenslund (1991). A Group Contribution Approach to
Computer-Aided Molecular Design. AIChE Journal 37(9): 1318-1332.
Glover, F. & M. Laguna (1997). Tabu Search. Boston: Kluwer Academic Publishers.
Gordeeva, E.V., M.S. Molchanova & N.S. Zefirov (1990). General Methodology and
Computer Program for the Exhaustive Restoring of Chemical Structures by
Molecular Connectivity Indexes. Solution of the Inverse Problem in
QSAR/QSPR. Tetrahedron Computer Methodology, 3, 389.
Hairston, D.W. (1998). New Molecules Get on the Fast Track. Chemical Engineering:
30-33.
Harper, P.M. & R. Gani (2000). A Multi-Step and Multi-Level Approach for Computer
Aided Molecular Design. Computers & Chemical Engineering 24(2-7): 677-683.

Harper, P.M., M. Hostrup & R. Gani (2003). A Hybrid CAMD Method. in Computer
Aided Melecular Design: Theory and Practice. L. E. K. Achenie, R. Gani & V.
Venkatasubramanian Eds. Amsterdam: Elsevier. 12: 122-169.
Joback, K. G., & G. Stephanopoulos(1989). Designing Molecules Possessing Desired
Physical Property Values. in Proceedings of the Foundations of Computer-Aided
Process Design (FOCAPD), Snowmass, CO(July 12-14). Eds. J.J. Siirola, I.
Grossmann and Geo. Stephanopoulos, CACHE-Elsevier, 363-387.
Johnson, K., B. Lin, D.C. Miller & K.V. Camarda (2002). Molecular Design of Polymer
Coatings using Optimization Techniques. Poster 246b, AIChE Annual Meeting,
Indianapolis, Indiana.
Kier, L.B. & L.H. Hall (1976). Molecular Connectivity in Chemistry and Drug Research.
New York: Academic Press.
Kier, L. B., H. H. Lowell & J. F. Frazer (1993). Design of molecules from Quantitative
Structure-Activity Relationship Model. 1. Information Transfer between Path and
Vertex Degree Counts, J. Chem. Inf. Comput. Sci., 33, 142
Kim, U.-R., K.-S. Min, M.-J. Lee, S.-H. Kim & B.-J. Jeong (1992). The Study of
Physical Properties for the Organic Compounds and their Binary Mixture
according to Molecular Connectivity Method. Journal of the Korean Chemical
Society 36(4): 485-495.
Lin, B., S. Chavali, K. Camarda & D.C. Miller (2003) Using Tabu Search to Solve
MINLP Problems for PSE, In Process Systems Engineering 2003 (Proceeding of
8th International Symposium on Process Systems Engineering, KunMing, China)
B. Chen & A.W. Westerberg, Eds. Elsevier, 541-546.

Lin, B. & D.C. Miller (2004a). Solving Heat Exchanger Network Synthesis Problems
with Tabu Search. Computers & Chemical Engineering 28(8): 1451-1464.
Lin, B. & D.C. Miller (2004b). Tabu search algorithm for chemical process optimization.
Computers & Chemical Engineering, In press.
Maranas, C.D. (1996). Optimal Computer-aided Molecular Design: A Polymer Design
Case Study. Ind. Eng. Chem. Res. 35: 3403-3414.
Marcoulaki, E.C. & A.C. Kokossis (1998). Molecular Design Synthesis Using Stochastic
Optimization as a Tool for Scoping and Screening. Computers & Chemical
Engineering 22(Suppl.): S11-18.
Marrero, J. & R. Gani (2001). Group Contribution Based Estimation of Pure Component
Properties. Fluid Phase Equilibria 183: 183-208.
Meniai, A. H., D. M. T. Newsham & B. Khalfaoui, (1998), Solvent Design for Liquid
Extraction Using Calculated Molecular Interaction Paramters, Trans. IchemE,
76(A11), 942-950.
Ostrovsky, G.M., L.E.K. Achenie & M. Sinha (2003). A Reduced Dimension Branch-
and-Bound Algorithm for Molecular Design. Computers & Chemical Engineering
27(4): 551-567.
Raman, V.S. & C.D. Maranas (1998). Optimization in Product Design with Properties
Correlated with Topological Indices. Computers & Chemical Engineering 22(6):
747-763.
Randic, M. (1975). On Characterization of Molecular Branching. J.Am.Chem.Soc. 97:
6609-6615.

Sahinidis, N.V. & M. Tawarmalani (2000). Applications of Global Optimization to
Process and Molecular Design. Computers & Chemical Engineering 24: 2157-
2169.
Sahinidis, N.V., M. Tawarmalani & M. Yu (2003). Design of Alternative Refrigerants via
Global Optimization. AIChE Journal 49(7): 1761-1774.
Siddhaye, S., K.V. Camarda, E. Topp & M. Southard (2000). Design of Novel
Pharmaceutical Product via Combinatorial Optimization. Computers & Chemical
Engineering 24: 701-704.
Syracuse Research Corporation, (2000) User’s Guide for the Physprop Database.
Syracuse Research Corp., North Syracuse, New York.
Trinajstic, N. (1983). Chemical Graph Theory, CRC Press.
Vaidyanathan, R. & M.M. El-Halwagi (1996). Computer-Aided Synthesis of Polymers
and Blends with Target Properties. Ind. Eng. Chem. Res. 35(2): 627-634.
Venkatasubramanian, V., K. Chan & J.M. Caruthers (1994). Computer-aided Molecular
Design Using Genetic Algorithm. Computers & Chemical Engineering 18(9):
833-844.
Wang, S. & G.W.A. Milne (1994). Graph Theory and Group Contributions in the
Estimation of Boiling Points. J. Chem. Inf. Comput. Sci 34: 1242-1250.
Wang, Y. & L.E.K. Achenie (2002). A Hybrid Global Optimization Approach for
Solvent Design. Computers & Chemical Engineering 26: 1415-1425.
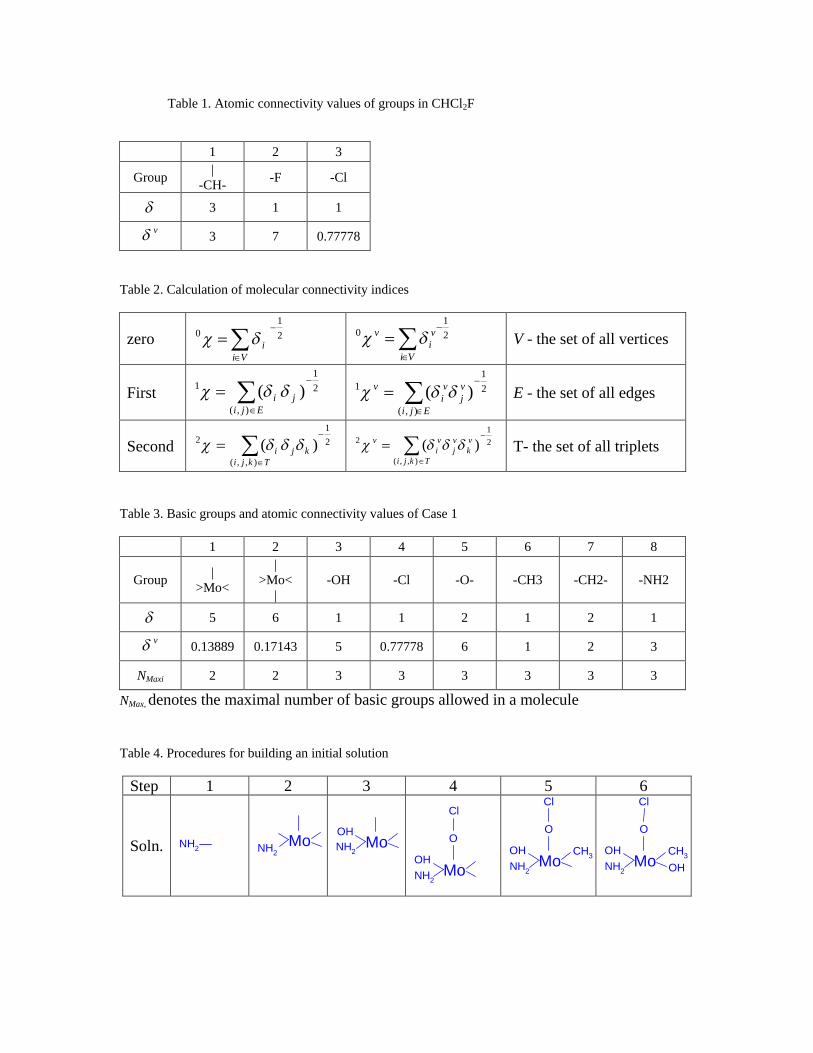
Table 1. Atomic connectivity values of groups in CHCl2F
1 2 3
Group | -CH- -F -Cl
3 1 1 v 3 7 0.77778
Table 2. Calculation of molecular connectivity indices
zero
Vii
21
0
Vi
vi
v 21
0 V - the set of all vertices
First
Ejiji
),(
21
1 )(
Eji
vj
vi
v
),(
21
1 )( E - the set of all edges
Second
Tkjikji
),,(
21
2 )(
Tkji
vk
vj
vi
v
),,(
21
2 )( T- the set of all triplets
Table 3. Basic groups and atomic connectivity values of Case 1
1 2 3 4 5 6 7 8
Group | >Mo<
| >Mo<
| -OH -Cl -O- -CH3 -CH2- -NH2
5 6 1 1 2 1 2 1 v 0.13889 0.17143 5 0.77778 6 1 2 3
NMaxi 2 2 3 3 3 3 3 3
NMax, denotes the maximal number of basic groups allowed in a molecule Table 4. Procedures for building an initial solution
Step 1 2 3 4 5 6
Soln. NH2 MoNH2
MoNH2
OH
MoNH2
OH
O
Cl
MoNH2
OH
O
Cl
CH3
MoNH2
OH
O
Cl
CH3
OH
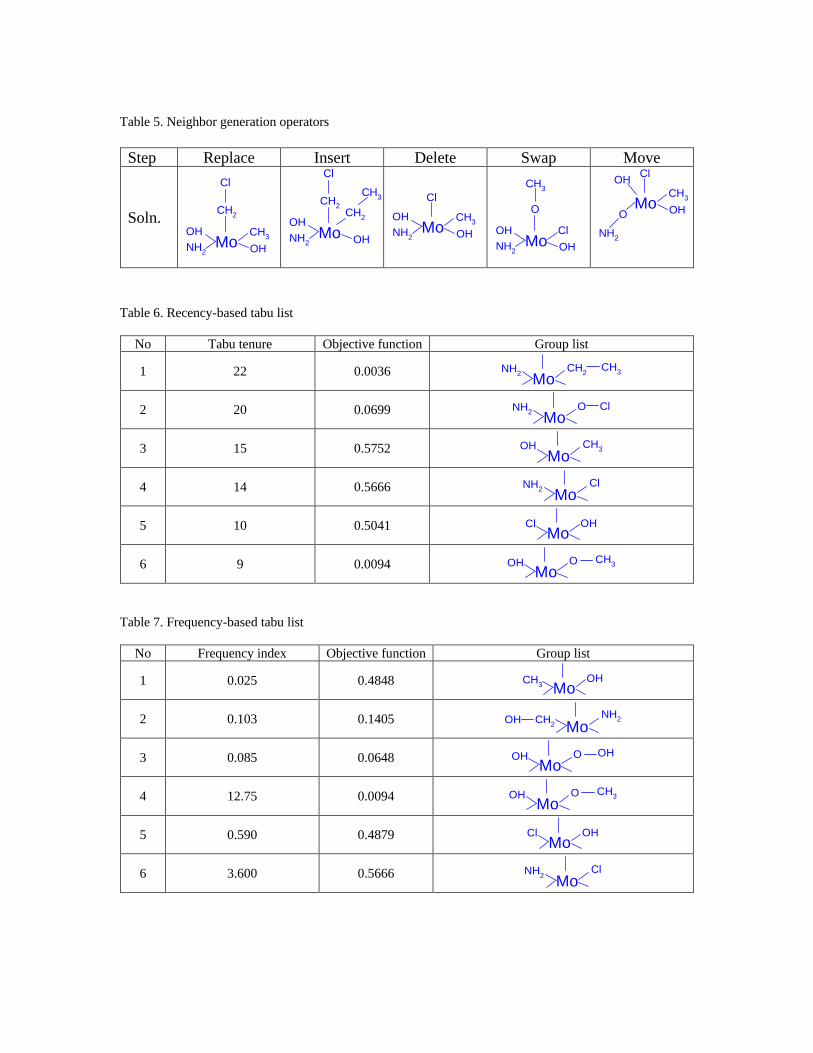
Table 5. Neighbor generation operators
Step Replace Insert Delete Swap Move
Soln. MoNH2
OH
CH2
Cl
CH3
OH MoNH2
OH
CH2
Cl
CH3
OH
CH2
MoNH2
OH
Cl
CH3
OH MoNH2
OH
O
Cl
CH3
OH
Mo
NH2
OH
O
Cl
CH3
OH
Table 6. Recency-based tabu list
No Tabu tenure Objective function Group list
1 22 0.0036 MoNH2 CH2 CH3
2 20 0.0699 MoNH2 O Cl
3 15 0.5752 MoOH CH3
4 14 0.5666 MoNH2
Cl
5 10 0.5041 MoOHCl
6 9 0.0094 MoOH O CH3
Table 7. Frequency-based tabu list
No Frequency index Objective function Group list
1 0.025 0.4848 MoOHCH3
2 0.103 0.1405 MoOH CH2NH2
3 0.085 0.0648 MoOH O OH
4 12.75 0.0094 MoOH O CH3
5 0.590 0.4879 MoOHCl
6 3.600 0.5666 MoNH2
Cl
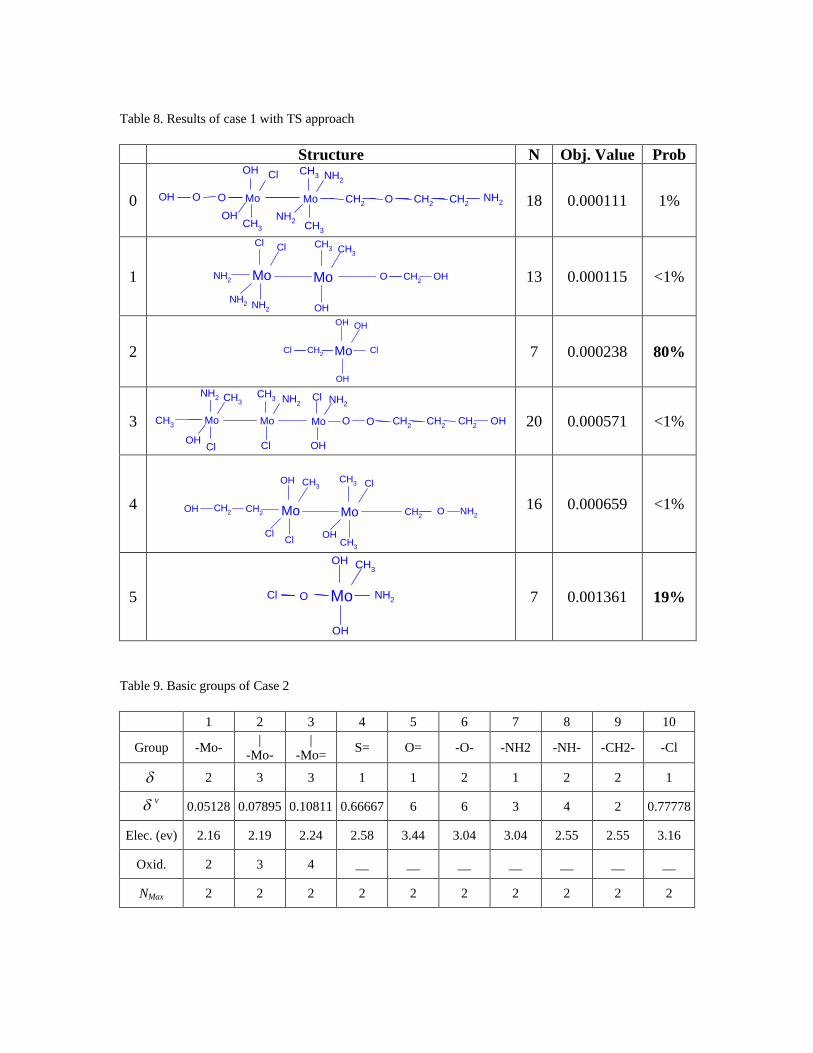
Table 8. Results of case 1 with TS approach
Structure N Obj. Value Prob
0 O OOH
OH Cl
CH3OH
CH3
CH3
NH2
NH2
CH2 O CH2 CH2NH2MoMo
18 0.000111 1%
1
CH3 CH3
MoMo
OH
Cl
NH2
NH2NH2
O CH2 OH
Cl
13 0.000115 <1%
2 Mo
OH
Cl
OH
CH2
OH
Cl
7 0.000238 80%
3 O O OH
Cl
CH3
OH
CH3
CH3NH2 NH2
CH2 CH2Mo Mo
Cl
Cl
OH
NH2
Mo CH2
20 0.000571 <1%
4
CH3
CH3
MoMo
OH
Cl
NH2OCH2OH
Cl
CH2 CH2
OH
Cl
CH3
16 0.000659 <1%
5 Mo
OH
Cl
CH3
O
OH
NH2
7 0.001361 19%
Table 9. Basic groups of Case 2
1 2 3 4 5 6 7 8 9 10
Group -Mo- | -Mo-
| -Mo= S= O= -O- -NH2 -NH- -CH2- -Cl
2 3 3 1 1 2 1 2 2 1 v 0.05128 0.07895 0.10811 0.66667 6 6 3 4 2 0.77778
Elec. (ev) 2.16 2.19 2.24 2.58 3.44 3.04 3.04 2.55 2.55 3.16
Oxid. 2 3 4 __ __ __ __ __ __ __
NMax 2 2 2 2 2 2 2 2 2 2
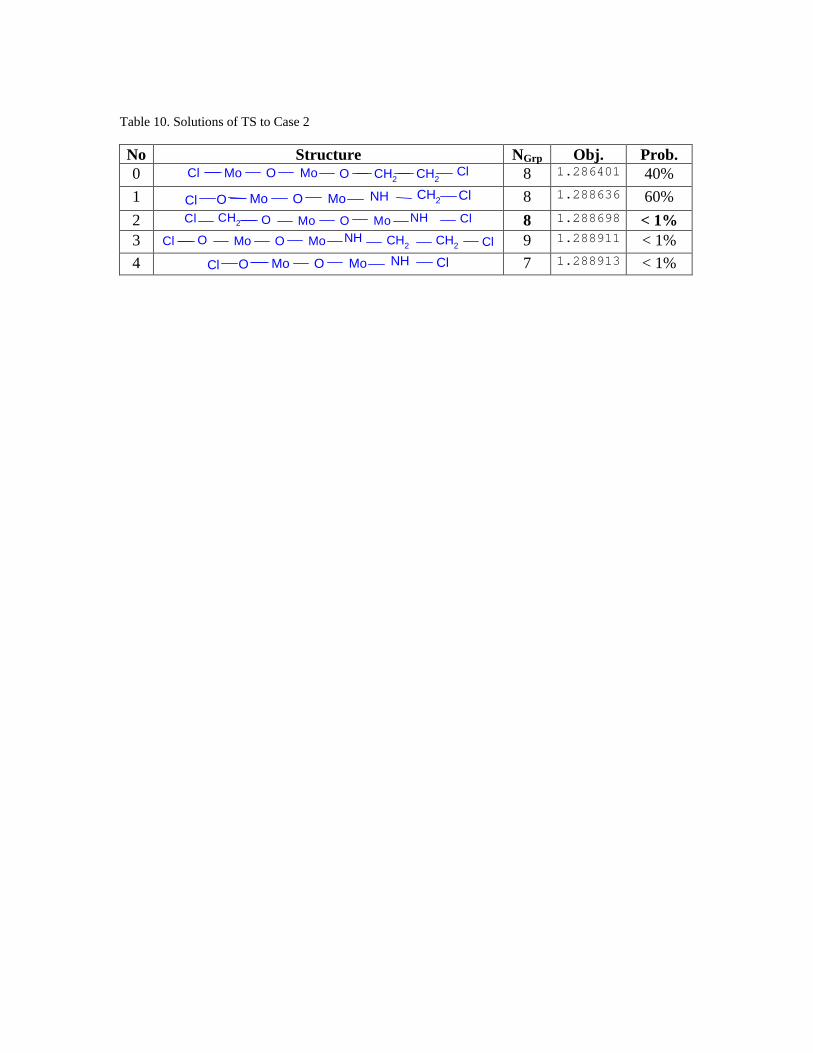
Table 10. Solutions of TS to Case 2 No Structure NGrp Obj. Prob. 0 Cl O CH2 CH2
ClMo MoO 8 1.286401 40% 1 ClO NH CH2Cl Mo MoO 8 1.288636 60% 2 Cl O NHCH2 ClMo MoO 8 1.288698 < 1% 3 Cl O NH CH2 ClMo MoO CH2 9 1.288911 < 1% 4 ClO NHCl Mo MoO 7 1.288913 < 1%

Figure 1. Hydrogen-suppressed graph of CHCl2F
Mo
OH
Cl
OH
CH2
OH
Cl
Figure 2. A sample molecule for case 1 located by TS
Mo
OH
Cl
O
NH2
Mo
OHCH3
CH3
OH
NH2
Cl Figure 3. Integer solution for case 1 found with OA algorithm
222 NHCHMoMoNH Figure 4. Integer solution for case 2 found with OA algorithm
Cl
CH
Cl F
Vertex
Edge Triplet