universit`a degli studi di parma - fis.unipr.itgigi/tirocini/tesi_carmen_mandalari.pdf · 1...
TRANSCRIPT

1
UNIVERSIT`A DEGLI STUDI DI PARMA Facoltà di Scienze Matematiche, Fisiche e Naturali Corso di Laurea in Fisica
Microspettroscopia Raman applicata a campioni di interesse artistico
Relatore: Chiar.mo Prof. MARCO P. FONTANA Correlatore: Dott. Bersani Danilo Laureanda: Carmen Mandalari
ANNO ACCADEMICO 2005-2006

2
INDICE
Nota introduttiva ........................................................................................ 4
1 INTRODUZIONE ...................................................................... 5
1.1 Lavoro compiuto........................................................................................ 5
1.2 Problemi incontrati ................................................................................... 6
1.3 Risultati ...................................................................................................... 9
2 SPETTROSCOPIA RAMAN ................................................... 10
2.1 Informazioni preliminari........................................................................ 10
2.2 Classicamente........................................................................................... 13
2.3 Quantisticamente..................................................................................... 15
2.4 Effetto Raman risonante......................................................................... 16
3 DESCRIZIONE APPARATO STRUMENTALE.................... 18
3.1 Laser ......................................................................................................... 18
3.2 T64000 ...................................................................................................... 18 3.2.1 Microscopio .....................................................................................................18 3.2.2 Premonocromatore ...........................................................................................19 3.2.3 Monocromatore e CCD....................................................................................21 3.2.4 Acquisizione dati..............................................................................................22
4 DESCRIZIONE CAMPIONI UTILIZZATI ........................... 23
4.1 Lapislazzuli .............................................................................................. 23
4.2 Cromato di piombo ................................................................................. 28
4.3 Campione 1 .............................................................................................. 30
4.4 Campione 3 .............................................................................................. 31
4.5 Campione 6 .............................................................................................. 32
4.6 Campione 11 ............................................................................................ 32
5 ANALISI DATI ........................................................................ 34
5.1 Lapislazzuli ............................................................................................... 35 5.1.1 Zona blu ..........................................................................................................35 5.1.2 Zona azzurra frastagliata.................................................................................36 5.1.3 Zona argentata nella zona bianca ....................................................................37

3
5.1.4 Zona bianca .....................................................................................................38 5.1.5 Zona azzurra......................................................................................................40 5.1.6 Zona dorata nel blu ...........................................................................................41 5.1.7 Zona dorata nel bianco......................................................................................42 5.1.8 Zona azzurra vicino al dorato............................................................................43 5.1.9 Zona bianca vicino all’azzurro..........................................................................45 5.1.10 Zona blu scuro...................................................................................................46 5.1.11 Zona blu cobalto................................................................................................47
5.2 Cromato di piombo.................................................................................. 48
5.3 Campione 11 ............................................................................................. 52 5.3.1 Zona bianca frammento dimensioni maggiori ..................................................52 5.3.2 Zona bianca frammento dimensioni minori ......................................................53 5.3.3 Zona grigia scura frammento dimensioni minori..............................................54
5.4 Campione 1 ............................................................................................... 54 5.4.1 Zona scura vicino al bordo.................................................................................54
5.5 Campione 6 .............................................................................................. 55 5.5.1 Zona bianca frammento dimensioni maggiori ...................................................55 5.5.2 Zona scura di confine frammento dimensioni maggiori ....................................56 5.5.3 Zona bianca frammento dimensioni minori .......................................................56
5.6 Campione 3 .............................................................................................. 57
6 Conclusioni........................................................................................... 58
Bibliografia .............................................................................................. 65
Ringraziamenti ......................................................................................... 66

4
Nota introduttiva
L’arte e la fisica due materie completamente diverse, la prima prettamente umanistica la
seconda completamente scientifica, che punti d’incontro potrebbero mai avere? A detta di tutti
nessuno. Quando ho parlato del mio tirocinio tutti mi hanno chiesto ma come è possibile che una
laureanda in fisica possa trattare di storia dell’arte?
Ebbene io posso dirvi che non è proprio così, l’arte e la fisica si possono incontrare e la
seconda è molto utile alla prima. Il lavoro del restauratore è molto spesso supportato da quello del
fisico che ha gli strumenti e le conoscenze adatte per poter analizzare le opere d’arte e dare
informazioni per quanto riguarda l’esecutore, la data di esecuzione, i pigmenti utilizzati, la tecnica
usata e la sua “salute” in modo da decidere quale sia il miglior modo di intervenire per
salvaguardare e conservare l’opera. Il lavoro di restauro è infatti volto alla conservazione del bene
culturale e l’analisi fisica è atta a investigare i materiali che lo compongono, i restauri precedenti e
tutte le sostanze esterne che hanno contribuito alla rovina o all’offuscamento dell’opera.
Il mio lavoro è consistito, prima di tutto, nell’imparare a utilizzare la microspettroscopia
Raman e, in particolar modo, il T64000 della Jobin-Yvon per poi analizzare un campione di
lapislazzuli, minerale utilizzato dagli artisti per il blu cobalto dei loro bellissimi cieli, di cromato di
piombo da cui si ricava il giallo della terra, e frammenti provenienti dalla seconda cappella di San
Giovanni Evangelista in Parma di cui non si conoscevano i costituenti. Ho fatto degli spettri Raman
di ogni campione a mia disposizione e da questi sono risalita al materiale costituente.
La fisica e l’arte si possono quindi incontrare e una non esclude l’altra.

5
1 INTRODUZIONE
Ora scendo nei particolari e spiego meglio in cosa è consistito il mio lavoro, quali sono stati
problemi che ho affrontato anche dovuti alla tecnica utilizzata e in linea di massima quali sono stati
i risultati ottenuti.
1.1 Lavoro compiuto
Il primo passo compiuto durante il mio tirocinio è stato quello di prendere familiarità con la
spettroscopia Raman, capire la sua utilità, il suo funzionamento e, in particolar modo imparare ad
utilizzare lo strumento grazie al quale ho potuto compiere le misure sui campioni a me affidati, il
T64000 della Jobin-Yvon. Questo è un lavoro già fatto da molti infatti la spettroscopia Raman è
una delle tecniche più utilizzate per sondare la struttura e la dinamica dei materiali, come vedremo.
Ad esempio nello stesso dipartimento di fisica dell’Università di Parma sono state effettuate
delle investigazioni sugli affreschi dell’abbazia di San Giovanni Evangelista in Parma, la stessa
oggetto della mia analisi. Il mio lavoro può essere inserito in questa serie di indagini. Grazie alle
ricerche precedenti si sono potuti confrontare i dipinti attribuiti al Parmigianino (Girolamo
Francesco Maria Mazzola, 1503-1540) e quelli attribuiti a Michelangelo Anselmi (1492?-1556?) e
constatare le diverse tecniche pittoriche e le influenze reciproche. L’analisi micro-raman, in questo
caso, sono state effettuate con un apparato DILOR (Jobin-Yvon) Labram e sono state utilizzate le
righe laser a 632.8 nm e 784.8 nm. Anche in questo caso i pigmenti sono stati identificati grazie
agli spettri Raman già conosciuti. Dalle osservazioni fatte si è potuto risalire alle differenze di
realizzazione degli affreschi: Parmigianino dipingeva larghe aree senza seguire i contorni dei
disegni preparatori mentre Anselmi preferiva seguire i suoi disegni e dipingere piccole aree.
Dall’analisi di frammenti sono stati poi identificati i vari pigmenti e i vari materiali presenti negli
affreschi rivelando una presenza massiccia, in entrambi gli artisti, di calcite e di gesso, quest’ultimo
ipotizzando derivi dalla solforizzazione del carbonato di calcio. I risultati sono stati poi riassunti in
una tabella e da essa si è potuto constatare che i due pittori utilizzavano più o meno gli stessi
pigmenti ad eccezione di quello blu: Parmigianino prediligeva l’azurite, la lazurite e raramente lo
smalt(?) mentre Anselmi usava quest’ultimo e l’azurite. Il carbonato di calcio è l’unico pigmento
bianco utilizzato. Un’analisi più profonda è riuscita a spiegare anche le differenti tecniche pittoriche
utilizzate di due artisti: Parmigianino usava oltre all’affresco la tempera che gli permetteva di
lavorare su zone più larghe mentre Anselmi prediligeva la tecnica a fresco e quindi il suo lavoro si
limitava su piccole zone.
Questo è un procedimento simile a quello che ho compiuto. L’obiettivo del mio tirocinio è
stato, infatti, quello di “mappare” i campioni cioè di assegnare ad ogni zona in cui essi potevano
essere divisi in base al colore e alla morfologia visibile ad occhio nudo, il materiale costituente e i

6
relativi modi di vibrazione a partire dagli spettri Raman ottenuti. Per fare ciò ho utilizzato il T64000
con righe laser a 488 nm, a 676 nm e, solo per alcuni campioni, a 647 nm. Nel mio caso
particolare ho sfruttato l’effetto Raman risonante, che ci permette di dare informazioni sulla
struttura dei materiali osservando i diversi picchi e le loro differenti intensità a seconda che eccito il
campione con riga blu o riga rossa del laser.
1.2 Problemi incontrati
Sembrerebbe tutto semplice tutto facile; si tratta in parole povere di fare degli spettri e da
essi risalire a elementi e composti di cui si conoscono già gli spettri ma non è così in realtà. Per
fare questo lavoro ci sono abbastanza difficoltà.
Prima fra tutte è il non danneggiamento del campione quindi bisogna stare attenti alla
potenza del laser di modo da non rovinare il reperto. Possiamo notare che a una determinata
potenza del laser, i.e 100 mW, si vede solo luminescenza o addirittura il rilevatore va in
saturazione cioè non riesce più a misurare l’intensità dello spettro Raman. Abbiamo perciò deciso
di diminuire la potenza fino ad arrivare a quella di 25 mW, grazie alla quale non si presenta più
saturazione dello strumento e le zone molto luminose non danno molta luminescenza come prima.
Per sapere se il campione è stato danneggiato o meno dall’intensità della potenza possiamo
osservare che lo spettro non è più riproducibile nel primo caso. Bisogna osservare uno spettro che
non presenta cambiamenti nel tempo e che sia riproducibile, indice di non danneggiamento del
campione altrimenti vuol dire che abbiamo falsato le nostre misure.
La focalizzazione sulla zona del campione è molto difficile in quanto esso non ha una
superficie piana ma spesso molto frastagliata, ruvida al tatto e inclinata; per questo la luce laser,
colpendolo non ortogonalmente, produce della luce diffusa che a volte “nasconde” il segnale
Raman vero e proprio che è molto debole, in generale, e quindi bisogna stare molto attenti al punto
in cui il laser arriva. Ci sono molti tentativi da fare prima di vedere un bello spettro Raman. Nelle
figure 1-1, 1-2, 1-3 si può vedere uno spettro ottenuto con molta luminescenza rappresentata da
quella riga quasi obliqua, uno spettro ottenuto con la giusta focalizzazione e uno spettro con molta
luminescenza ma che comunque presenta un picco Raman riconoscibile:

7
Figura 1-1 si tratta della zona blu scuro del campione numero 6. E’ una multiacquisizione da 50 a 1250 cm-1 con una potenza del laser di 25 mW. Non si osserva alcun picco. Riga laser 488 nm.
Figura 1-2 è lo spettro preso nella zona blu del lapislazzuli che si presenta piatta al tatto. Si possono osservare i picchi caratteristici del materiale. Multiacquisizione da 100 a 1250 cm-1 Riga laser 488 nm.
8
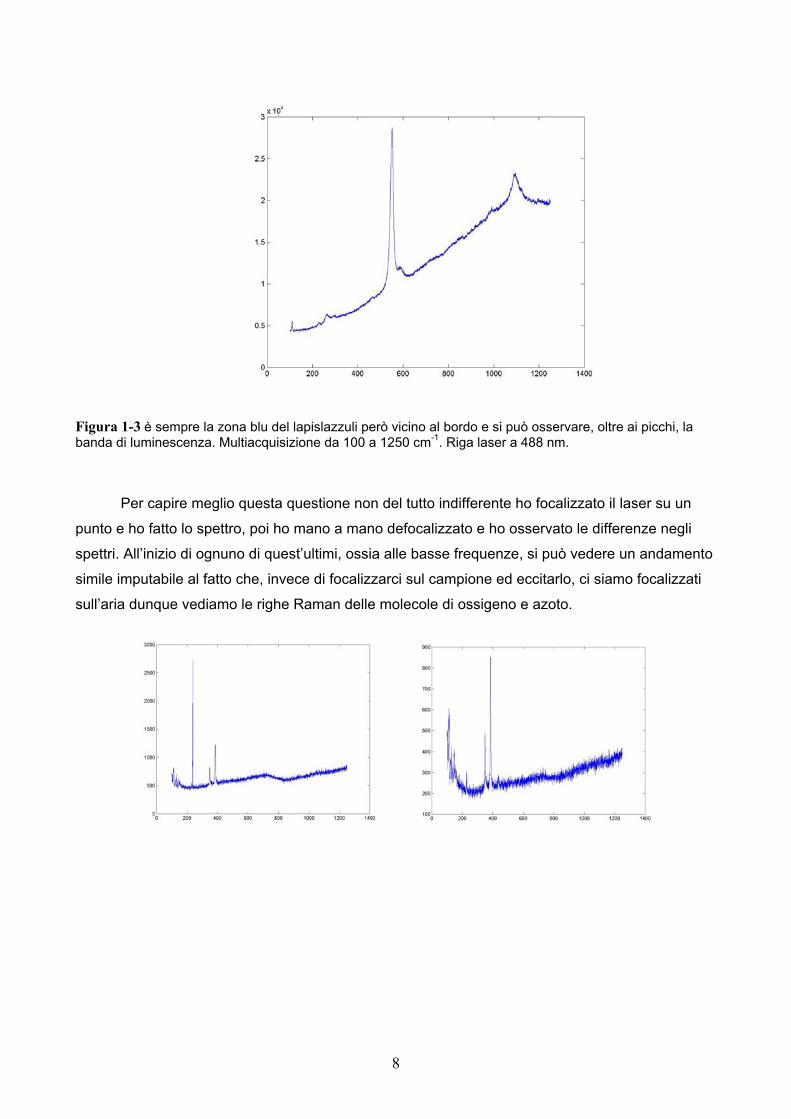
Figura 1-3 è sempre la zona blu del lapislazzuli però vicino al bordo e si può osservare, oltre ai picchi, la banda di luminescenza. Multiacquisizione da 100 a 1250 cm-1. Riga laser a 488 nm.
Per capire meglio questa questione non del tutto indifferente ho focalizzato il laser su un
punto e ho fatto lo spettro, poi ho mano a mano defocalizzato e ho osservato le differenze negli
spettri. All’inizio di ognuno di quest’ultimi, ossia alle basse frequenze, si può vedere un andamento
simile imputabile al fatto che, invece di focalizzarci sul campione ed eccitarlo, ci siamo focalizzati
sull’aria dunque vediamo le righe Raman delle molecole di ossigeno e azoto.

9
Figura 1-4 la prima è la focalizzazione sulla zona argentata del lapislazzuli che è molto inclinata e quindi presenta una forte luminescenza; la seconda e la terza rappresentano una defocalizzazione e si può osservare un andamento simile alle basse frequenze dovuto probabilmente all’eccitazione dell’aria.
Un altro problema non banale è stato quello di riconoscere le varie zone di colore che, a
prima vista sembravano tutte uguali, ma in realtà avevano sfumature diverse indice di miscelanze
di elementi e composti. Nei lapislazzuli vedremo che ci sono almeno tre tipi di blu diverso e ognuno
di essi presenta uno spettro Raman differente.
Inoltre gli artisti non sempre hanno utilizzato un solo pigmento per realizzare il colore poi
usato nell’opera, quindi, nei nostri campioni, non sarà presente un solo pigmento o comunque una
sola sostanza ma ci sarà una mescolanza e tutti contribuiranno allo spettro Raman con i loro
picchi. A volte, perciò, lo spettro non è riscontrabile così com’è ottenuto, nella letteratura ma
bisogna cercare quello più vicino al dato sperimentale.
Il lavoro del fisico al servizio del restauro e della conservazione dei beni culturali non è
quindi semplice e breve ma è un lavoro piuttosto lungo che deve tenere conto di varie eventualità.
1.3 Risultati
Dopo aver ovviato e cercato di risolvere o minimizzare i problemi che mano a mano
venivano riscontrati ho analizzato gli spettri ottenuti ricavando le frequenze a cui si riscontravano i
picchi caratteristici, ho confrontato queste con quelle della letteratura e ho racchiuso tutto in una
tabella grazie alla quale dai dati spettroscopici si può risalire a materiale costituente la zona di
analisi e infine ai modi vibrazionali attivati dall’eccitazione della luce laser.

10
2 SPETTROSCOPIA RAMAN
2.1 Informazioni preliminari
In generale, l’indagine su un qualunque sistema fisico avviene studiando la sua risposta B
ad un dato stimolo A che ci permette di sondare la sua struttura e la sua dinamica su una
determinata scala spazio-temporale. Questo è quello che avviene per una qualunque
investigazione fisica su un sistema: la spettroscopia Raman rientra in questo tipo di indagine e può
essere spiegata in questo modo purchè si tenga presente che la quantità misurata è una densità
spettrale, che si riferisce, come vedremo, alle eccitazioni vibrazionali.
Quando lo stimolo e il sistema interagiscono con energia piccola rispetto a quelle che
caratterizzano la dinamica interna di quest’ultimo si può supporre che la risposta sia proporzionale
allo stimolo: AB ⋅= ξ . Si parla perciò di risposta lineare. ξ è funzione del tempo e dei parametri
che caratterizzano il sistema e non dipende da A. Se la perturbazione è forte non si potranno più
trascurare i contributi non lineari e ξ sarà funzione anche dello stimolo.
Lo stimolo usato nel Raman per avere la risposta è la radiazione elettromagnetica visibile:
la si fa incidere sul campione e si osserva come viene modificato lo spettro dopo aver attraversato
il materiale. In esso infatti, oltre alla frequenza della radiazione incidente, se ne possono osservare
altre dovute all’interazione della sonda con i moti vibrazionali a livello atomico e molecolare che noi
stiamo andando a studiare. Da qui si ha una descrizione a livello locale, dell’ordine di grandezza
della distanza interatomica della struttura del materiale studiato.
Quando una radiazione incide su un determinato campione una parte di essa viene diffusa
elasticamente in tutte le direzioni senza perdita di energia, cioè alla stessa frequenza dell’onda
incidente: questo è dovuto agli urti elastici tra i quanti e le molecole e si parla di diffusione elastica
o Rayleigh. Una parte minore di questa, però, viene diffusa anelasticamente cedendo (diffusione
Raman Stokes) o acquisendo (diffusione Raman anti-Stokes) energia nell’interazione con la
molecola: si vedono perciò frequenze che differiscono per quanti d’energia vibrazionali. La
diffusione elastica è più probabile, la diffusione Stokes è più favorita di quella anti-Stokes come si
può vedere nella 2.1. La differenza tra i due picchi sembrerebbe una contraddizione della
conservazione dell’energia ma la spiegherò più avanti quando parleremo dell’effetto Raman dal
punto di vista quantistico: le righe Stokes e quelle anti-Stokes sono un fenomeno tipicamente
quantistico.
Ogni materiale ha dei propri moti vibrazionali quindi l’interazione tra questi e la radiazione
incidente è specifica per ognuno: ogni sostanza ha il suo spettro Raman caratteristico perciò da
esso possiamo risalire al composto. È quello che ci proponiamo di fare mappando i nostri campioni
in questa esperienza.

11
Figura 2-1 come si può osservare l’intensità del picco Rayleigh è molto elevata rispetto agli altri due, indice di probabilità maggiore. Anche il picco Stokes è più grande di quello anti-Stokes. Sembrerebbe una contraddizione della conservazione dell’energia ma non è così: la differenza è spiegabile solo con la meccanica quantistica.
L’onda elettromagnetica che va a incidere sul campione ha un vettore d’onda ki invece
quello che noi studiamo è l’onda diffusa avente un determinato vettore kd. Nel caso dello scattering
Raman ω, cioè la frequenza dell’onda diffusa, è indipendente da q= kd – ki , vettore di scattering.
In linea di principio, variando q misuriamo la dipendenza delle frequenze ωi dal vettore
d’onda k, dato che in un cristallo deve valere q=k. Si può dire, in generale, che la legge di
dispersione che lega le frequenze di vibrazione dei moti vibrazionali che avvengono all’interno
della sostanza al vettore di scattering q è caratterizzata da rami acustici per cui la frequenza ω
tende a 0 linearmente nel limite di 0→k e da rami ottici caratterizzati dal fatto che ω tende a
valori finiti nel limite di 0→k . Dato che la lunghezza d’onda della luce 0a>>λ si ha che 0≈= kq .
Questo spiega perché in un cristallo solo i modi vibrazionali al centro della zona di Brillouin
contribuiscono allo spettro Raman e, dunque, anche la sostanziale indipendenza di ω da k.
Si sa dalla meccanica quantistica che ogni atomo è descritto da più livelli energetici
descrivibili da funzioni d’onda soluzioni dell’equazione di Schrodinger stazionaria. Essi
costituiscono una base completa e ogni stato eccitato dell’atomo può essere descritto da una loro
combinazione lineare. I livelli sono infiniti e stazionari e l’atomo, all’equilibrio, si trova nello stato di
energia inferiore. Ora, facendo arrivare una radiazione elettromagnetica descrivibile tramite un
potenziale vettore A essa interagisce con gli stati dell’elettrone descritto con quantità di moto p .
Se l’energia degli elettroni interessati dall’interazione con la radiazione sonda è molto
piccola rispetto a quella portata da essa allora ci troviamo in regime non relativistico e possiamo
considerare solo l’interazione elettrica come perturbazione.

12
Infatti, dal punto di vista quantistico, per risolvere l’equazione di Schrodinger e trovare così
le funzioni d’onda degli stati, dobbiamo utilizzare la teoria perturbativa il cui termine di
perturbazione che si aggiunge all’hamiltoniana è: pAmce
⋅ , dove A è il potenziale vettore del
campo magnetico che oscilla nel tempo perciò la perturbazione dipende dal tempo.
Quando faccio interagire la radiazione visibile con una qualunque sostanza,
quantisticamente, faccio passare un elettrone che si trova in un determinato stato elettronico
caratterizzato da un’energia Ei in un altro individuato da energia Ef: ci può essere transizione
dall’uno all’altro se e solo se l’energia dell’onda incidente è pari alla differenza di energia tra i 2
livelli, if EE −=ω . Se l’energia è maggiore o minore non ci può essere transizione.
In realtà noi dobbiamo considerare anche gli stati vibrazionali che appartengono alla sostanza
perché gli atomi non sono fermi all’interno di un materiale ma si muovono continuamente rispetto
alla loro posizione d’equilibrio e bisogna considerare anche questo.
La funzione d’onda che descrive lo stato in cui si trova un elettrone della sostanza è
descritta da entrambi i contributi mescolati tra loro in modo da dare lo stato effettivo dell’elettrone:
per l’approssimazione adiabatica possiamo rappresentare lo stato quantico con il prodotto tra la
funzione elettronica e quella vibrazionale. Perché? Semplicemente perché l’ordine di grandezza
della massa dell’elettrone è 3 volte più piccolo di quello della massa del nucleo perciò si può
pensare che esso, quando avviene una transizione descritta in precedenza, rimane fermo. In realtà
la divisione non è così bene netta perché la funzione elettronica dipende parametricamente da R,
lo spostamento rispetto alla posizione d’equilibrio iniziale, e quella vibrazionale dal numero
quantico che compete allo stato elettronico. Per questo in ogni stato elettronico ci sono tanti
sottolivelli vibrazionali e si può affermare che le transizioni avvengono nello stesso stato elettronico
tra i sottolivelli vibrazionali.
Per la spettroscopia Raman il discorso è solo in parte vero: essa studia le transizioni tra i
sottolivelli vibrazionali che però avvengono grazie a una transizione virtuale tra due stati elettronici.
Nella figura 2.3 possiamo vedere due stati elettronici caratterizzati dai numeri quantici n, m che
contengono al loro interno vari sottolivelli vibrazionali. Se la radiazione incidente ha energia pari
alla differenza tra le energie di due livelli vibrazionali allora c’è una transizione che può avvenire
sia direttamente (assorbimento infrarosso) sia indirettamente passando attraverso una transizione
di grande energia che dura un tempo molto piccolo rispetto alla durata del processo che andiamo a
studiare e che non conserva l’energia, una transizione, perciò, virtuale che alla fine riporta allo
stato elettronico di partenza ma in un sottolivello vibrazionale diverso. Questo ultimo passaggio è

13
studiato grazie alla spettroscopia Raman. In particolar modo si studia l’azione della transizione
facendo una media temporale.
Figura 2-2 come si può osservare nella figura per ogni stato elettronico caratterizzato dagli stati quantici n,
m ci sono vari sotolivelli vibrazionali. La transizione verticale azzurra rappresenta il passaggio diretto tra i sottolivelli vibrazionali mentre la riga blu è la transizione virtuale di cui abbiamo parlato nel testo.
Si dice, così, che l’effetto Raman è un effetto del secondo ordine: c’è la simultanea distruzione o
creazione di 2 quanti.
2.2 Classicamente
Dal punto di vista classico per spiegare la spettroscopia Raman bisogna introdurre il vettore
polarizzabilità elettrica che descrive la deformazione della distribuzione di carica in un atomo
eccitato da un’onda, descrivibile mediante un dipolo elettrico oscillante.
L’effetto Raman è legato alle fluttuazioni di questa grandezza a livello atomico e molecolare
perciò si dice che esso è un effetto locale, che sonda la dinamica del moto degli atomi e delle
molecole, che causa le fluttuazioni. I moti sono quindi le vibrazioni, le rotazioni e le traslazioni
rispetto a una determinata posizione d’equilibrio. La scala spaziale è dell’ordine dell’Angstrom
mentre quella temporale corrispondente è del picosecondo.
La densità dei dipoli indotti è esprimibile come ),()( tEtP ωα= , dove α(t) è il tensore di
polarizzabilità. Nel caso della diffusione elastica (Rayleigh) la dipendenza temporale è dovuta solo
al campo elettrico oscillante nel tempo ma, nel caso del Raman, bisogna considerare anche la
dipendenza temporale di α. Quello che ci interessa per la dinamica è la media temporale del
quadrato della derivata della polarizzabilità rispetto allo spostamento u degli atomi e delle

14
molecole dalla posizione d’equilibrio R0, che è proporzionale all’intensità 2
0
∂∂
≈Ru
I α. Grazie a
questo valore calcolato in R0 possiamo conoscere la simmetria della sostanza che stiamo
analizzando. Alla posizione d’equilibrio rispetto alla quale gli atomi e le molecole si muovono la
derivata è diversa da zero. Se infatti la derivata all’equilibrio è nulla il modo vibrazionale, istante
per istante, modula la polarizzabilità ma la sua media temporale, l’intensità che la spettroscopia
misura, è nulla.
Se la polarizzabilità in funzione della distanza fra atomi delle molecole rispetto all’equilibrio
è una funzione monotona crescente, la media temporale non sarà nulla: si dice che il modo di
vibrazione corrispondente è attivo in Raman perché la derivata all’equilibrio è sempre diversa da
zero. Se, invece, la vibrazione non è simmetrica allora la funzione aumenta sia all’allontanamento
sia all’avvicinamento degli atomi costituenti la molecola rispetto all’equilibrio. In questo caso se la
derivata è uguale a zero il modo non è attivo in Raman; se è diversa da zero allora è attiva in
Raman. Da questo fatto discendono le regole di simmetria che, sono caratteristiche della
spettroscopia Raman, e, formalmente, per ogni simmetria molecolare o cristallina si ricavano dalla
teoria dei gruppi di simmetria.
Abbiamo già descritto un’altra peculiarità dell’effetto Raman, che limita alle vibrazioni delle
branche ottiche delle curve di dispersione, nella zona 0≈k , la possibilità di contribuire allo spettro.
Un cristallo è un sistema molto particolare perché le particelle costituenti sono distribuite
spazialmente in un reticolo ordinato quindi godono di simmetria traslazionale discreta. Vale, in
questo caso, la regola di conservazione del vettor d’onda purchè q sia un vettore del reticolo
reciproco, cioè il reticolo di nodi immerso nello spazio dei vettori d’onda: qkk si =− . La
funzione di distribuzione delle frequenze dei modi vibrazionali, cioè la densità degli stati
vibrazionali, è continua quindi lo spettro risultante dell’effetto Raman dovrebbe avere una
distribuzione continua rispecchiante questa e dipendente dalle leggi di dispersione. Ma in realtà
non è così, proprio per il fatto che 0≈= kq : solo i fononi al centro della zona di Brillouin sono
interessati e quindi è come se le curve di dispersione non ci fossero. Lo spettro Raman è perciò
costituito solo da una serie di picchi “molecolari” attribuibili al limitato numero di frequenze
associate ai gradi di libertà della cella unitaria del cristallo.
Se il solido è invece amorfo l’uguaglianza tra Q e k non deve più valere perché non è più
possibile assegnare un preciso vettore d’onda ai modi vibrazionali come accade per un cristallo,
non c’è più un reticolo che si ripete nello spazio, perciò lo spettro Raman diventa continuo e riflette
più o meno la densità di stati vibrazionali.
In ogni caso da questo tipo di variazione spettrale si hanno informazioni sull’eventuale
disordine nel materiale.

15
2.3 Quantisticamente
La teoria classica però non riesce a spiegare un effetto peculiare dell’effetto Raman: la
presenza del picco Stokes e di quello anti-Stokes aventi intensità differenti anche se relativi a
frequenze is ωωω ±= 0 ove ω0 è la frequenza dell’onda incidente e ωi è la frequenza minore o
maggiore che costituiscono la banda di intensità. Questa discrepanza si risolve solo dal punto di
vista quantistico considerando il fattore statistico di Bose-Einstein:
−
=
1
1),(TkBe
Tnω
ωh
, che tiene
conto della quantizzazione dei modi vibrazionali e della diversità dei vari processi.
Infatti possiamo sviluppare la polarizzabilità attorno alla posizione d’equilibrio di ogni
particella R0j j=1,…,N: ...21),(
2
+
∂∂
+
∂∂
= kjkj
jj
uuuuPu
uPtrPδ , considerando che il termine
lineare corrisponde ad eventi in cui c’è lo scambio solo di un quanto mentre quello quadratico si
riferisce alla creazione o alla distruzione di due quanti. Da u, che ci indica lo spostamento
dell’atomo o della molecola rispetto alla posizione d’equilibrio, passiamo alle coordinate normali ξ e
per avere informazioni sull’intensità Raman calcoliamo 2ξ . Classicamente TkB→2ξ
,
quantisticamente
[ ]ω
ωξ 1),(2 +=
Tn per gli eventi Stokes e
[ ]ωωξ ),(2 Tn
= per quelli anti-
Stokes. Come si può vedere il rapporto tra le intensità è diverso da 1: la fisica classica non
prevede questo fenomeno anzi per essa questo va contro la conservazione dell’energia che deve,
invece, sempre valere (come abbiamo già detto precedentemente).
Nel caso quantistico, però, non si usa la polarizzabilità classica. Quantisticamente quello
che ci importa conoscere è la matrice di transizione tra i due stati quantici in cui si trova all’inizio
l’elettrone e in cui va a finire dopo la perturbazione.
L’effetto Raman è spiegabile con la teoria delle perturbazioni dipendenti dal tempo al
secondo ordine perché è un effetto a due quanti: c’è la contemporanea creazione o distruzione di
due fotoni conservando l’energia dello stato iniziale e di quello finale. L’hamiltoniana perturbante è:
pAmeH ⋅
−= dove A è il potenziale vettore del campo elettrico incidente e p la quantità di moto
dell’elettrone.

16
L’intensità Raman è descrivibile tramite la polarizzabilità di transizione dallo stato iniziale
⟩0 a quello finale ⟩f : 20 fPI αβ≈ dove ∑
+
+
−
=i
i
ifi
i
ifif MMMMP
)()()(
)()()(
00
0
00
00
ωωωωβααβ
αβ (2.3.1) e
∑=j jerM α, β=x, y,z è l’operatore dipolare, ifM )( α ecc sono i corrispondenti elementi di
matrice.
L’effetto Raman, dal punto di vista quantistico, è quindi spiegabile grazie a una sommatoria
i∑ degli elementi di matrice con cui si descrivono gli stati quantici in cui si trovano gli elettroni.
Siccome vale l’approssimazione adiabatica e quindi possiamo separare le funzioni che
descrivono, rispettivamente, gli stati elettronici e quelli vibrazionali possiamo dire che la transizione
avviene tra due stati vibrazionali all’interno dello stesso stato elettronico, però, come abbiamo già
detto, questa avviene tramite una transizione virtuale tra gli stati elettronici per il Raman. Le
funzioni elettroniche dipendono parametricamente da R (per questo non è possibile la separazione
totale tra le due funzioni), gli elementi di matrice M dipendono solo da esse per l’approssimazione
adiabatica perciò essi dipendono da u come la polarizzabilità classica. Da qui in poi si può usare la
stessa teoria descritta precedentemente riguardante la simmetria delle molecole.
2.4 Effetto Raman risonante
L’effetto Raman è dovuto alle fluttuazioni vibrazionali che, quantisticamente, si descrivono
con la dipendenza della polarizzabilità di transizione e delle frequenze di transizione dagli
spostamenti u rispetto alla posizione di equilibrio. Se quindi eccitiamo il campione con una
radiazione di frequenza vicina a quelle di transizione elettronica del sistema ω0i dalla formula
(2.3.1) osserviamo che c’è un termine con un denominatore divergente: il contributo di questo sarà
determinante nella sommatoria. Questo, sperimentalmente, si traduce in un aumento significativo
dell’intensità di un picco relativo a una determinata frequenza o nella “comparsa” di un picco che,
ad una diversa frequenza di incidenza non era visibile, data la bassa intensità. Il denominatore
dominerà addirittura il numeratore quindi a questo punto possiamo semplificare l’espressione della
polarizzabilità di transizione considerando solo lo stato elettronico risonante e aggiungendo ad
esso un termine di smorzamento: ( ) ( )[ ]( ) f
f
iMM
P f νωω
ν αβνναβ
Γ+−=
010
101, , dove ora possiamo
considerare gli elementi di matrice M come indipendenti dagli spostamenti u.
Vediamo dunque come nel caso dell’effetto Raman risonante siamo sensibili alla
modulazione del contributo alla polarizzabilità totale connesso con l’interazione delle vibrazioni con

17
il singolo stato elettronico risonante: saranno dunque solo quelle che interagiscono con questo
stato che avranno un aumento dell’intensità che può arrivare anche a 4 ordini di grandezza.
Questo è l’effetto caratteristico del Raman che andiamo ad analizzare studiando questi tipi
di campioni artistici ed eccitandoli con luce laser blu (488 nm) e rossa (647 e 676 nm).

18
3 DESCRIZIONE APPARATO STRUMENTALE
Per la mappatura a livello microscopico dei campioni ci siamo serviti di un laser ad Argon e
Kripton e del T64000, un triplo monocromatore che permette l’illuminazione da microscopio,
grazie al quale è possibile studiare un effetto debole come quello Raman. Importante è il rilevatore
connesso a questo strumento: un CCD (charge-coupled device) che, oltre ad avere elevatissima
sensitività, ci permette di studiare simultaneamente una larga banda spettrale, invece di una
lunghezza d’onda per volta.
Parliamo di queste componenti descrivendole separatamente.
3.1 Laser
La sorgente delle righe è Argon per 488 nm e Kripton per le altre due righe usate nel rosso
e vicino infrarosso. Al suo interno c’è un prisma che possiamo ruotare per scegliere la riga del
laser da utilizzare per le nostre analisi. Dopo essere uscito attraversa un filtro interferenziale
diverso a seconda della riga laser selezionata, che serve per eliminare le righe di emissione
spontanea della scarica nel gas (plasmi).
3.2 T64000
3.2.2 Microscopio
Dopo aver attraversato una lamina semiriflettente che lascia passare solo metà
dell’intensità, la luce laser arriva all’obiettivo di un microscopio grazie al quale incide sul campione
posto sul suo piano focale. Gli obiettivi a disposizione sono 10x, 50x e 100x. Ho utilizzato quello
10x e ho focalizzato il laser sull’oggetto utilizzando un televisore collegato a una telecamera.
Da qui poi la luce retrodiffusa viene raccolta, ripassa attraverso la lamina semiriflettente e
poi viene filtrata spazialmente da un “pinhole”, che ha la funzione di chiudere la geometria
confocale e infine viene inviata nel premonocromatore come vedremo.
Bisogna stare molto attenti alla focalizzazione perché una sbagliata potrebbe pregiudicare
la raccolta dei dati.

19
Figura 3-1 questo è il microscopio dove si può vedere la telecamera collegata grazie alla quale possiamo vedere su uno schermo lo spot del laser e la struttura del campione in modo da focalizzare al meglio il fascio
e gli obiettivi puntati sul piano focale.
3.2.2 Premonocromatore
La luce retrodiffusa attraversa poi una serie di lenti,che servono a collimare il fascio sulla
fenditura d’entrata del premonocromatore e attraversa una lamina λ/2 che ha il compito di
cambiare la sua polarizzazione poiché essa oscilla sul piano focale xy mentre il premonocromatore
accetta principalmente luce di polarizzazione verticale. Prima della fenditura però c’è una ruota con
cui si può scegliere la configurazione micro e quella macro, che evita il passaggio dal microscopio.
Noi scegliamo la prima per il nostro scopo di mappatura.

20
Figura 3-2 in questa foto si può vedere la ruota che permette la selezione del cammino della luce fino all’apertura del premonocromatore e l’apertura della fenditura del terzo monocromatore manipolabile
manualmente.
Una volta entrato si trova davanti un doppio monocromatore. C’è, difatti, un doppio reticolo
che può essere usato in forma additiva o sottrattiva a seconda della somma o differenza della
dispersione angolare data dai reticoli.
Noi lo usiamo nella configurazione sottrativa: infatti noi vogliamo eliminare la luce spuria del
laser e avere solamente lo spettro Raman. In figura 3.3 è rappresentata la configurazione
sottrattiva del triplo premonocromatore fino al rilevatore:
Figura 3-3 qui si può vedere che il primo reticolo disperde la luce, il secondo ricompone lo spettro dopo che ha attraversato una finestra spettrale che l’ha ridotto eliminando la luce laser e il terzo disperde quest’ultimo
prima di arrivare al CCD.

21
La luce entra nel premonocromatore all’interno del quale trova il primo reticolo che la
disperde. Qui la luce trova davanti a sé una fenditura la cui apertura può essere controllata dal
software e in grado di “tagliare” la luce laser e far passare solo lo spettro Raman. Essa però deve
essere abbastanza larga da far passare più spettro Raman possibile e non solo una parte. Per
decidere l’apertura di questa c’è un comando nel software che indica l’apertura relativa alla riga
laser che stiamo utilizzando. A seconda di questa la fenditura permette la raccolta dati in un range
di cm-1 differenti: se si usa la riga blu l’apertura massima a cui si può arrivare è 800 cm-1 mentre
con la riga a 676 nm essa è 200 cm--- 111. Questo succede perché la finestra spettrale dipende dalla
lunghezza d’onda del laser, rispetto a cui si misura la sua larghezza; siccome l’energia di un’onda
è legata alla lunghezza d’onda tramite una relazione di proporzionalità inversa, λ
=E , a parità di
lunghezza d’onda l’intervallo di frequenze da analizzare è maggiore per il blu e minore per il rosso.
Si può selezionare così solo un intervallo dello spettro che vogliamo analizzare. Bisogna stare
molto attenti a non selezionare un intervallo che comprenda la luce laser altrimenti rischiamo di
bruciare il CCD.
Il secondo reticolo fa l’opposto di quello che faceva il primo: lo spettro disperso viene
compattato di nuovo in modo da avere il segnale entrante all’inizio senza la luce del laser. Da qui,
grazie a uno specchio concavo, viene inviato alla fenditura di ingresso del terzo monocromatore, il
vero e proprio spettrografo.
Noi possiamo aggiustare manualmente le larghezze della fenditura d’ingresso nel
premonocromatore e quella d’entrata nel monocromatore vero e proprio.
3.2.3 Monocromatore e CCD
Arrivato a questo il fascio viene disperso tramite un reticolo e mandato sul CCD, il sensore
al silicio grazie al quale i dati vengono digitalizzati. Il rivelatore è un array bidimensionale di
2048X512 e ogni nodo del reticolo è un pixel. Ognuno di essi è sensibile al singolo fotone e ci dice
la posizione dello spettro e l’intensità del segnale.
Di solito per la rilevazione si usava un fotomoltiplicatore ma il CCD è molto più conveniente
perché ci consente di lavorare simultaneamente su uno spettro di frequenze ben più ampio e ci
consente di diminuire di molto la potenza del laser utilizzato in modo da evitare il danneggiamento
del campione analizzato data la sua maggiore efficienza.
C’è anche un sistema di raffreddamento dato da azoto liquido per eliminare il cosiddetto
rumore di buio, particolarmente elevato per il silicio usato per il CCD.

22
3.2.4 Acquisizione dati
C’è un software in dotazione grazie al quale è possibile vedere gli spettri Raman e
l’illuminazione del CCD. Possiamo decidere l’intervallo di frequenze da analizzare modificando
l’apertura della finestra spettrale di cui abbiamo precedentemente parlato e, a volte, siccome
abbiamo bisogno di una misura di un ampio spettro di frequenze e non è possibile farlo, data la
massima apertura consentita a seconda della luce laser, è possibile fare delle multiacquisizioni su
vari intervalli. È possibile anche trovare l’intensità e le frequenze dei picchi Raman osservabili e
fare così analisi dati.

23
4 DESCRIZIONE CAMPIONI UTILIZZATI
I campioni a nostra disposizione sono stati di due tipi. Abbiamo avuto dei minerali di cui
conoscevamo la composizione e di cui abbiamo fatto una mappatura per conoscere
qualitativamente l’abbondanza di alcuni composti nelle varie zone che si potevano riconoscere alla
vista. Abbiamo poi avuto dei frammenti della seconda cappella della chiesa di San Giovanni
Evangelista in Parma utilizzati nel lavoro di cui ho parlato in precedenza per quanto riguarda lo
studio delle opere del Parmigianino. In questo ultimo caso si è trattato non solo di mappare il
campione ma di risalire dallo spettro sperimentale ai suoi costituenti in modo da conoscere i
pigmenti utilizzati dall’artista per le sue opere.
Cercherò di spiegare cosa vuol dire mappatura del campione visto che è uno degli obiettivi
del mio lavoro. Entrambi i tipi presentano alla vista delle diverse zone di colore attribuibili ai diversi
composti presenti in quella determinata regione; in più, essi presentano delle superfici non lisce e
piane quindi oltre alla differenziazione di colore c’è n’è una dovuta alla loro morfologia: essa
contribuisce o meno alla troppa o poca luminescenza presente nei dati sperimentali e alla
focalizzazione.
Entriamo nei particolari delle zone dei diversi campioni partendo dal lapislazzuli e dal
cromato di piombo, i minerali di cui abbiamo fatto solo la mappatura, e poi descriviamo i frammenti
mostrando la loro provenienza dalla cappella.
4.1 Lapislazzuli
Il lapislazzuli è il nome comune che si utilizza per indicare una roccia costituita
principalmente da lazurite 24648 ),,()(),( ClSSOAlSiOCaNa , minerale che ha delle impurezze di
zolfo che lo rendono di colore azzurro. È il tipico pigmento utilizzato per il blu dei dipinti ed è
caratteristico dell’arte. Il nostro campione non è solo di colore blu ma presenta quattro zone ben
diverse a prima vista: una zona blu scuro, una azzurra, una bianca e una dorata.

24
Figura 4-1 in questa foto in cui il lapislazzuli è ripreso dall’alto possiamo osservare tutte le 4 zone: la bianca, la blu, l’azzurra (sulla sinistra) e le macchie di marroni che non siamo riusciti a focalizzare.
Lo zolfo è il responsabile dell’effetto Raman risonante che noi andiamo a studiare infatti alle
frequenze del laser blu esso va in risonanza.
All’interno di queste zone grandi di colore possiamo osservare delle diverse sfumature che
diverranno l’oggetto della nostra analisi fine del campione.
La zona blu scuro presenta, in realtà, una colorazione che assomiglia al blu cobalto e l’altra
più scura; nella zona bianca ci sono dei punti argentati e delle zone quasi marroni (che però non
siamo riusciti ad analizzare per la loro morfologia che non ha permesso la corretta focalizzazione
della regione); la zona azzurra ha molte sfumature dovute alla vicinanza o meno alla regione blu o
bianca.
Figura 4-2 in questa foto si possono osservare le diverse tonalità del blu e la parte argentata dovuta probabilmente alla presenza di pirite. Si vede anche il puntino argentato nel bianco e le diverse tonalità di
azzurro nella parte superiore più scuro perché in mezzo alla zona blu e in quella inferiore più chiaro.

25
Il minerale ha una forma a cubottaedro con il vertice superiore troncato, che presenta,
perciò, una superficie piana; ci sono, però, anche, delle superfici inclinate di difficile focalizzazione.
La zona bianca, in più, è molto ruvida al tatto perché molto “frastagliata”. La zona dorata che si
estende sia in quella blu sia in quella bianca è inclinata su un lato della piramide.
Figura 4-3 nella foto possiamo vedere, dall’alto, la tipica forma a cuboottaedro e sono riconoscibili le due grandi zone di colore bianca e blu. Nella figura successiva vediamo il cubottaedro, il solido a cui si avvicina il nostro lapislazzuli.
In base a queste informazioni sul colore e sulla morfologia abbiamo individuato 11 zone da
analizzare nel lapislazzuli:
• Blu piatta (vedi figura 4.3)
• Azzurra frastagliata su un lato del cuboottaedro;

26
Figura 4-4 in questa foto si può notare la parte azzurra che si trova tra quella blu e quella bianca ed è la zona che abbiamo analizzato e di cui stiamo parlando.
• Punto argentato nella zona bianca posizionato su un vertice del cuboottaedro (vedi
figura 4.2);
• Bianca;
• Azzurra;
• Dorata nella zona blu;
• Dorata nella zona bianca;
Figura 4-5 si può osservare la zona dorata sia in quella blu sia in quella bianca.
• Azzurra vicino alla zona dorata precedente;
• Bianca vicino alla zona azzurra;
• Blu scuro;
• Blu cobalto.
27
Come si può notare abbiamo incluso anche delle zone di colore uguale però le abbiamo
differenziate in base alla vicinanza o meno alla zona bianca che supponiamo essere calcite.
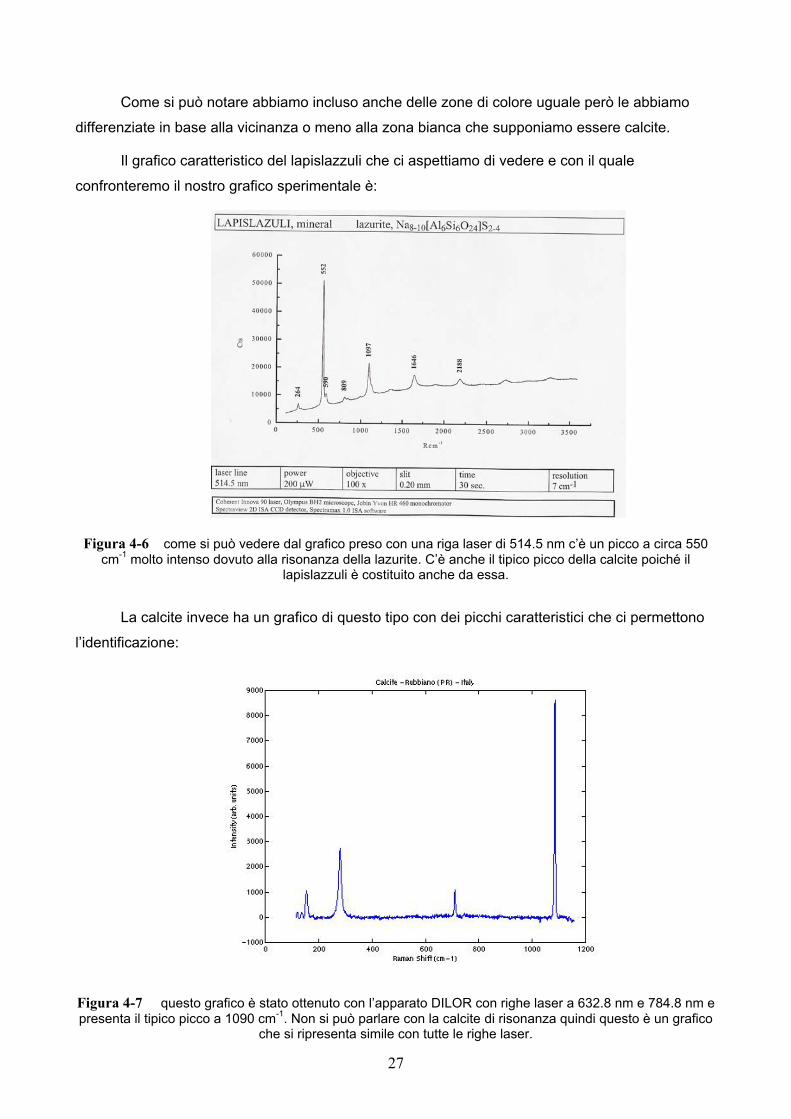
Il grafico caratteristico del lapislazzuli che ci aspettiamo di vedere e con il quale
confronteremo il nostro grafico sperimentale è:
Figura 4-6 come si può vedere dal grafico preso con una riga laser di 514.5 nm c’è un picco a circa 550 cm-1 molto intenso dovuto alla risonanza della lazurite. C’è anche il tipico picco della calcite poiché il
lapislazzuli è costituito anche da essa.
La calcite invece ha un grafico di questo tipo con dei picchi caratteristici che ci permettono
l’identificazione:
Figura 4-7 questo grafico è stato ottenuto con l’apparato DILOR con righe laser a 632.8 nm e 784.8 nm e presenta il tipico picco a 1090 cm-1. Non si può parlare con la calcite di risonanza quindi questo è un grafico
che si ripresenta simile con tutte le righe laser.

28
Supponiamo che la zona dorata presente in questo minerale siano tracce di pirite e quindi ci
aspettiamo di vedere un grafico così:
Figura 4-8 anche questo grafico è stato ottenuto con l’apparato DILOR con righe a 632.8 nm e 784.8 nm.
4.2 Cromato di piombo
Questo è un altro tipico minerale utilizzato per il giallo dei dipinti. Il suo nome comune è
giallo cromo. La sua formula chimica è 4PbCrO . A seconda della maggiore o minore presenza del
cromo questo minerale si presenta con diverse sfumature del rosso; nel nostro caso abbiamo
visibile 4 zone:
• Rosa;
• Rosso;
• Giallo;
• Grigio.

29
Figura 4-9 in questa foto sono riconoscibili tutte e 4 le zone di colore del nostro campione e ci mostra anche la sua forma.
Per quanto riguarda la morfologia possiamo osservare che nelle zone rosso e rosa sono
presenti dei cristalli prismatici a contorno poligonale, che sembrano dei “cilindretti” regolari a
causa delle numerose facce e che indicano un certo ordine all’interno di questo materiale mentre
la zona grigia si presenta amorfa e in più all’interno di essa sono presenti le parti gialle. Ho cercato
di focalizzarmi sempre sui cilindri colorati per quanto mi è stato possibile ma alcuni presentavano
molta fluorescenza soprattutto con il laser blu.
Figura 4-10 in questa foto presa dalla letteratura possiamo vedere i cristalli prismatici a facce poligonali che non siamo riusciti a fotografare nel nostro campione ma che vi sono simili.

30
Il cromato di piombo, a differenza del lapislazzuli, va in risonanza con il laser rosso e il
grafico che si può osservare presenta dei picchi ben marcati e modulati che indicano anche il
grado di ordine/disordine della zona che stiamo analizzando:
Figura 4-11 tipico grafico di risonanza infatti la riga laser con cui è stato ottenuto è 647,1 nm, riga rossa. Si può osservare la tipica serie di picchi a circa 300 cm-1.
4.3 Campione 1
È costituito da due frammenti di dimensioni differenti: uno è abbastanza grande rispetto
all’altro e si presenta di colore marrone scuro da un lato e bianco dall’altro mentre quello di
dimensioni minori è bianco con chiazze marroni scure.
È stato molto difficile fare misure con questo campione a causa delle sue dimensioni molto
piccole che portavano a molta luminescenza e a difficile focalizzazione.
Siamo riusciti ad analizzare solo 1 zona riferita al frammento di dimensioni maggiori:
• Bianca;
Le altre zone visibili a occhio nudo sono state difficili da focalizzare e presentavano molta
luminescenza senza alcuno spettro Raman.
Questi due frammenti sono stati prelevati dalla zona della cappella osservabile nella
seguente foto:

31
Figura 4-12 si può vedere la colorazione scura che presenta il frammento.
Ipotizziamo che ci sia la presenza di calcite, la zona bianca e di qualche pigmento usato per
la colorazione scura di cui non conosciamo nulla a priori.
4.4 Campione 3
È composto da un frammento di dimensioni abbastanza grandi rispetto ad agli altri due;
principalmente abbiamo analizzato questo che si presenta di colore bianco con delle macchie
grigie da un lato e dall’altro una regione grigia centrale molto inclinata e difficile da focalizzare.
Le zone facilmente visibili e analizzabili sono:
• Grigia all’interno di quella bianca;
• Bianca;
• Grigia.
Per quanto riguarda il frammento di dimensioni maggiori, mentre per quello minore:
• Bianca.
La regione della cappella da cui sono stati prelevati è:
Figura 4-13 sono il bianco e il grigio dei frammenti.

32
Supponiamo che la zona bianca sia calcite o gesso e che la colorazione scura sia un
pigmento di cui non conosciamo nulla.
4.5 Campione 6
Sono 3 frammenti di cui analizzeremo solo quello di dimensioni maggiori. Esso si presenta
di colore prevalentemente bianco con alcune macchie blu, che all’analisi precedente risultano
essere lapislazzuli. Le zone analizzate sono state per il frammento di dimensioni maggiori:
• Bianca;
• Scura di confine.
Per quello di dimensioni minori invece:
• Bianca di confine.
La zona di provenienza è rappresentata nella seguente foto:
Figura 4-14 questa è la colorazione dei frammenti.
Sappiamo già che il grafico che dovremmo vedere è quello del lapislazzuli quindi
verificheremo questo dato noto, in più, supponiamo la presenza della calcite o del gesso per
quanto riguarda la regione bianca.
4.6 Campione 11
Si presenta come due frammento di dimensioni abbastanza grandi di colore bianco a
chiazze marrone scuro. La provenienza è:

33
Figura 4-15 ecco il marrone e il bianco del frammento analizzato.
Anche in questo caso ipotizziamo la presenza di calcite data la colorazione.
Le zone analizzate sono state per il frammento maggiore:
• Bianca.
Per il frammento minore:
• Bianca;
• Grigio scuro.

34
5 ANALISI DATI Tutti i grafici sono stati ottenuti con potenza laser di 25 mW, quella che all’inizio abbiamo visto non
danneggia il campione. Il tempo di acquisizione è minimo 30 secondi per poter osservare un
segnale al di là della luminescenza. Le aperture delle fenditure del premonocromatore e del
monocromatore sono le stesse: 300 µm.
Se ci sono dei cambiamenti per quanto riguarda la finestra spettrale, la potenza laser, la
durata dell’acquisizione e le aperture delle fenditure sono scritti nelle didascalie dei grafici. Una
potenza maggiore di 25 mW, soprattutto per quanto riguarda la riga rossa, sottintende il lavoro
descritto nel primo paragrafo per risolvere il problema del danneggiamento. I grafici dei lapislazzuli
con la riga rossa sono stati ottenuti con potenza di 107 mW misurata prima del filtro interferenziale
che sappiamo quasi dimezzare l’intensità. I grafici del rosso per i frammenti sono invece ottenuti
con potenza laser di 106 mW. Per il cromato di piombo è stata utilizzata una potenza di 99 mW
con la riga a 676 nm e sono state fatte delle multiacquisizioni da 100 a 1200 cm-1 della durata di 60
secondi ognuna.
L’intensità dei grafici è espressa in numero di conteggi al secondo. Le ascisse dei picchi
sono state ottenute utilizzando MatLab.

35
5.1 Lapislazzuli
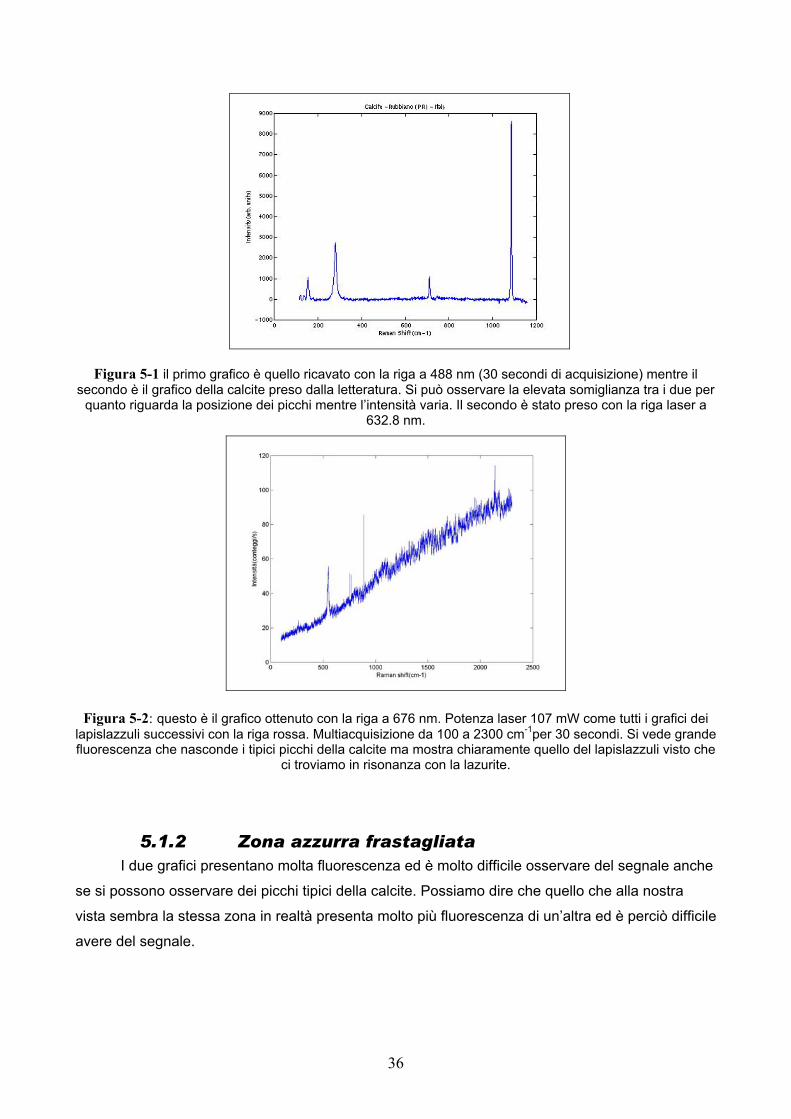
5.1.1 Zona blu Con la linea a 488 nm si può osservare uno spettro molto simile a quello della calcite che
presenta infatti il picco caratteristico a 1090 cm-1 corrispondente allo stiramento simmetrico del
carbonato mentre con la riga a 676 nm si vede solo il picco a circa 500 cm-1 peculiare del
lapislazzuli. Si tratta di effetto Raman risonante infatti come si può evincere dalla figura alcuni
picchi presenti con la riga blu non ci sono con quella rossa mentre l’unico picco in comune tra le
due presenta un’intensità maggiore rispetto all’altra. Come avevo detto in precedenza quando ci
troviamo in risonanza è possibile che ci sia un termine che contribuisce maggiormente e che abbia
un’intensità così elevata da poter trascurare tutti gli altri . Con la riga a 676 nm lo spettro si vede
abbastanza male e c’è molta fluorescenza che nasconde molto segnale infatti si può dire che
siamo in risonanza per quanto riguarda la lazurite infatti si vede ben marcato e facilmente il tipico
picco a circa 500 cm-1 anche se alcuni picchi della calcite come quello a circa 200 e a 1090 cm-1
sono ancora visibili essendo quelli che danno più segnale anche con la riga blu.
36
Figura 5-1 il primo grafico è quello ricavato con la riga a 488 nm (30 secondi di acquisizione) mentre il secondo è il grafico della calcite preso dalla letteratura. Si può osservare la elevata somiglianza tra i due per
quanto riguarda la posizione dei picchi mentre l’intensità varia. Il secondo è stato preso con la riga laser a 632.8 nm.
Figura 5-2: questo è il grafico ottenuto con la riga a 676 nm. Potenza laser 107 mW come tutti i grafici dei lapislazzuli successivi con la riga rossa. Multiacquisizione da 100 a 2300 cm-1per 30 secondi. Si vede grande fluorescenza che nasconde i tipici picchi della calcite ma mostra chiaramente quello del lapislazzuli visto che
ci troviamo in risonanza con la lazurite.
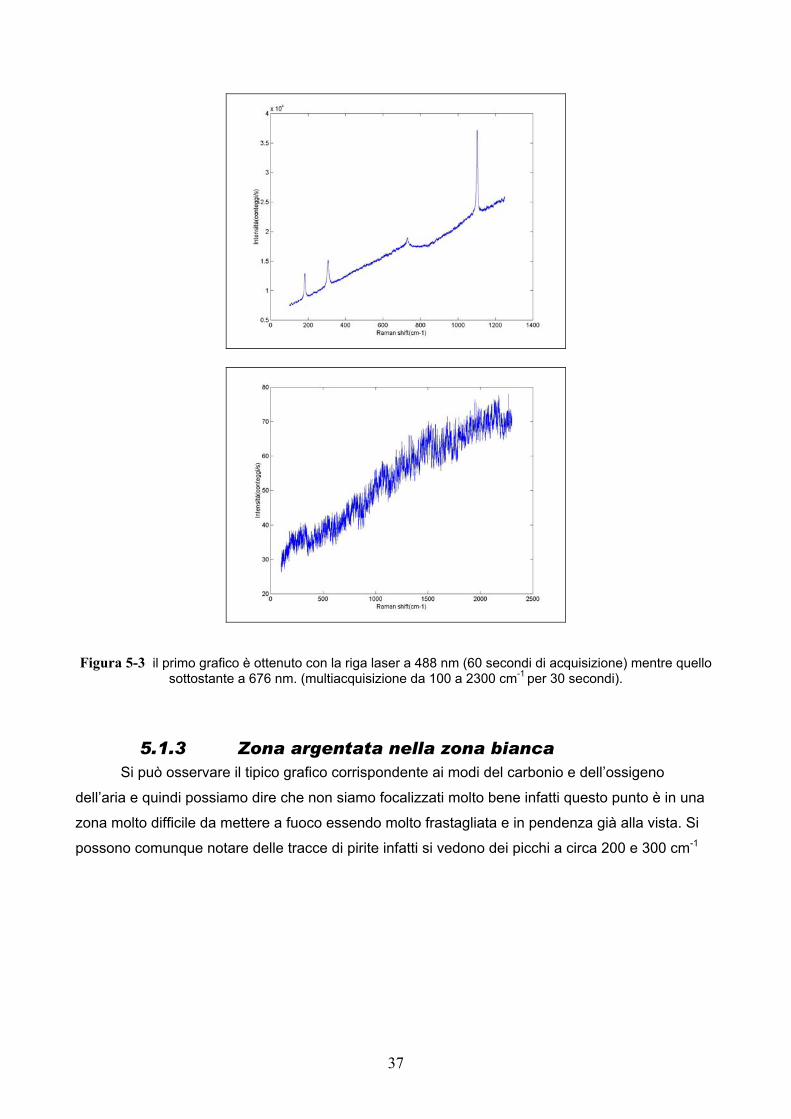
5.1.2 Zona azzurra frastagliata I due grafici presentano molta fluorescenza ed è molto difficile osservare del segnale anche
se si possono osservare dei picchi tipici della calcite. Possiamo dire che quello che alla nostra
vista sembra la stessa zona in realtà presenta molto più fluorescenza di un’altra ed è perciò difficile
avere del segnale.
37
Figura 5-3 il primo grafico è ottenuto con la riga laser a 488 nm (60 secondi di acquisizione) mentre quello sottostante a 676 nm. (multiacquisizione da 100 a 2300 cm-1 per 30 secondi).
5.1.3 Zona argentata nella zona bianca Si può osservare il tipico grafico corrispondente ai modi del carbonio e dell’ossigeno
dell’aria e quindi possiamo dire che non siamo focalizzati molto bene infatti questo punto è in una
zona molto difficile da mettere a fuoco essendo molto frastagliata e in pendenza già alla vista. Si
possono comunque notare delle tracce di pirite infatti si vedono dei picchi a circa 200 e 300 cm-1
38
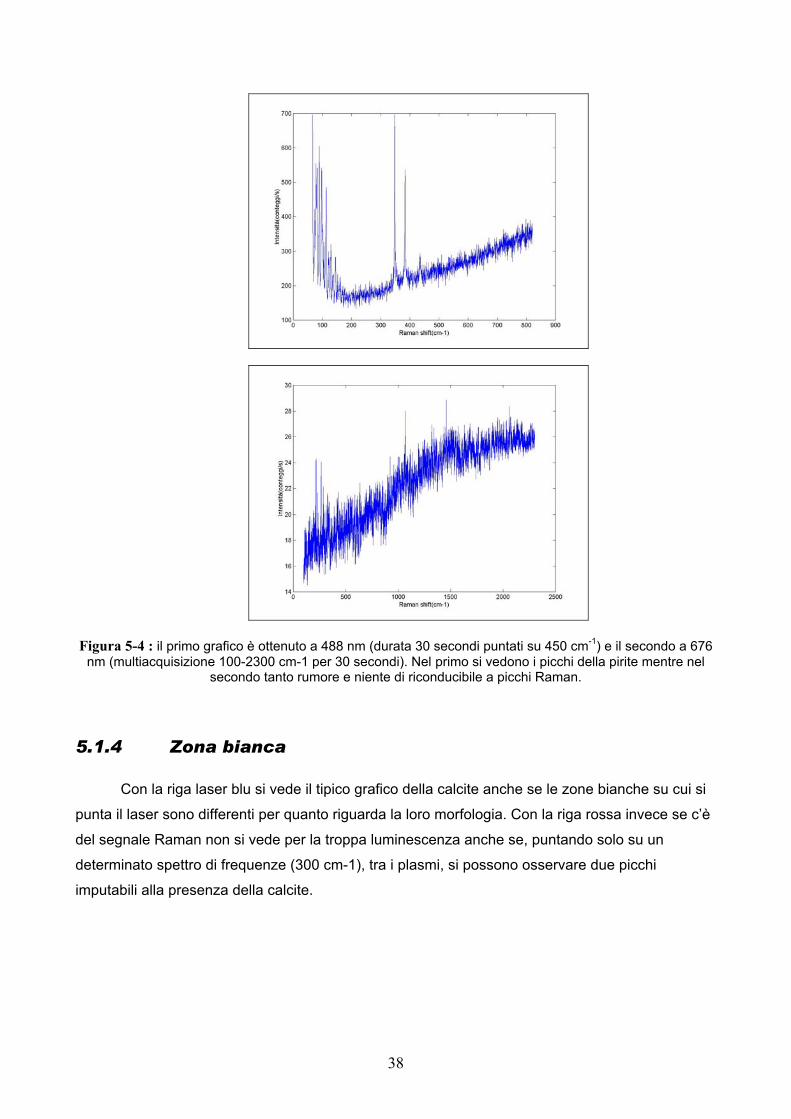
Figura 5-4 : il primo grafico è ottenuto a 488 nm (durata 30 secondi puntati su 450 cm-1) e il secondo a 676 nm (multiacquisizione 100-2300 cm-1 per 30 secondi). Nel primo si vedono i picchi della pirite mentre nel
secondo tanto rumore e niente di riconducibile a picchi Raman.
5.1.4 Zona bianca
Con la riga laser blu si vede il tipico grafico della calcite anche se le zone bianche su cui si
punta il laser sono differenti per quanto riguarda la loro morfologia. Con la riga rossa invece se c’è
del segnale Raman non si vede per la troppa luminescenza anche se, puntando solo su un
determinato spettro di frequenze (300 cm-1), tra i plasmi, si possono osservare due picchi
imputabili alla presenza della calcite.

39
Figura 5-5 il primo grafico è ottenuto con la riga a 488nm (30 secondi); il secondo e il terzo con
quella a 676 nm. Il secondo è una multiacquisizione da 100 a 2300 cm-1(30 secondi); il terzo è puntato su
300 cm-1 (60 secondi).
40
5.1.5 Zona azzurra
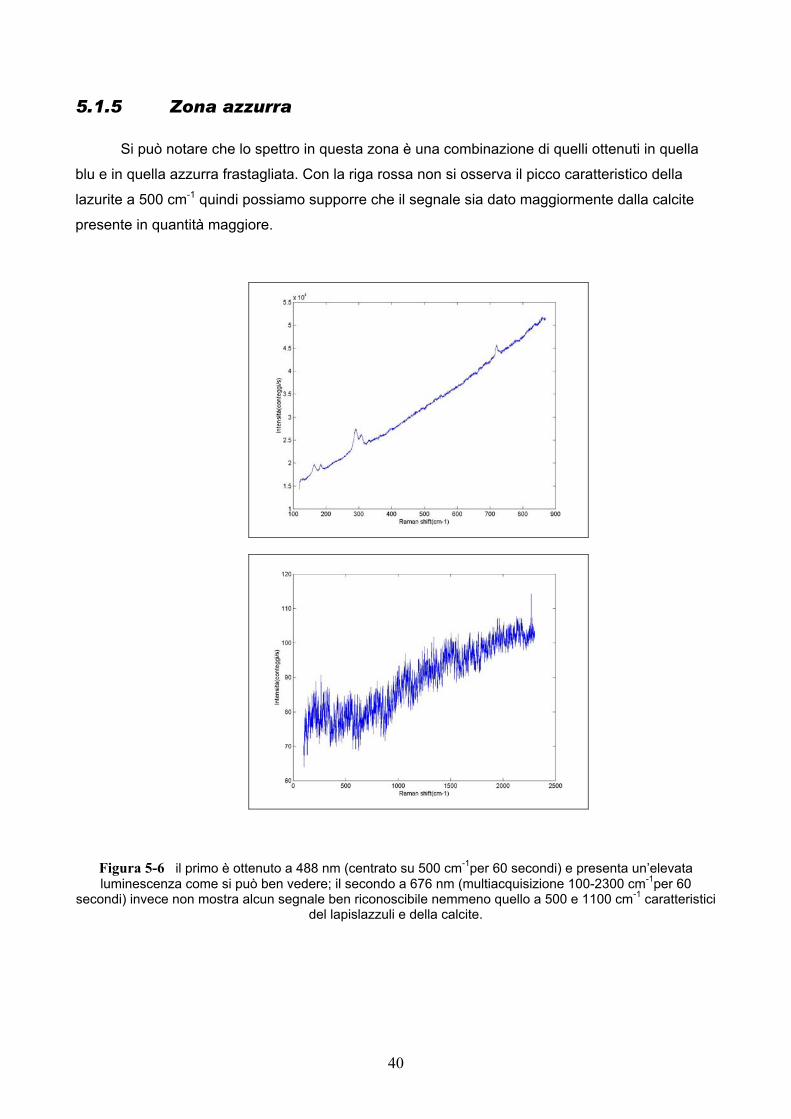
Si può notare che lo spettro in questa zona è una combinazione di quelli ottenuti in quella
blu e in quella azzurra frastagliata. Con la riga rossa non si osserva il picco caratteristico della
lazurite a 500 cm-1 quindi possiamo supporre che il segnale sia dato maggiormente dalla calcite
presente in quantità maggiore.
Figura 5-6 il primo è ottenuto a 488 nm (centrato su 500 cm-1per 60 secondi) e presenta un’elevata luminescenza come si può ben vedere; il secondo a 676 nm (multiacquisizione 100-2300 cm-1per 60
secondi) invece non mostra alcun segnale ben riconoscibile nemmeno quello a 500 e 1100 cm-1 caratteristici del lapislazzuli e della calcite.

41
5.1.6 Zona dorata nel blu
Anche in questo caso la focalizzazione su questa zona è stata ardua; tanto è vero ci sono
dei picchi a basse frequenze imputabili ai modi dell’ossigeno e dell’azoto. Nonostante questo,
però, si possono riconoscere con la luce laser a 488 nm i picchi della pirite mentre con quella a
676 nm c’è troppa luminescenza e troppi plasmi.
Figura 5-7 il primo a 488 nm è molto disturbato (60 secondi) mentre il secondo a 676 nm(puntati su 300 per 60 secondi) presenta dei picchi imputabili a dei plasmi.

42
5.1.7 Zona dorata nel bianco Si osserva un segnale di fluorescenza troppo forte e non si può vedere alcun picco a parte
quello della lazurite che però non si osserva con la riga rossa a differenza delle altre zone che
presentano il picco a 500 cm-1 solo con la riga a 676 nm. Nel grafico con la riga blu si osserva un
cambiamento significativo nell’andamento probabilmente dovuto a problemi della strumentazione;
questo salto che si può vedere non è fluorescenza e non può essere spiegato in nessun modo se
non in un cambiamento di qualche parametro dello strumento.
Figura 5-8 il primo è ottenuto con la riga a 488 nm (30 secondi) e presenta molta fluorescenza oltre al picco poco intenso della lazurite; il secondo è ottenuto con la riga a 676 nm (puntati su 600 cm-1 per 60
secondi) e c’è solo del rumore.

43
5.1.8 Zona azzurra vicino al dorato Si riesce a vedere il picco della lazurite a circa 500 cm-1. Considerando questo spettro e
quello precedente si può osservare che quando c’è un forte segnale di fluorescenza si riesce ad
avere il picco a 500 cm-1 anche con la riga blu. Anche in questo caso si osserva quel salto
imputabile alla strumentazione. Nel rosso si vedono molti picchi dovuti alla risonanza della lazurite
però il picco a 300 cm-1 è molto più forte rispetto a quello a 500 cm-1 a differenza di quello che
accade normalmente. Si può vedere che con la riga a 676 nm il picco a 1090 cm-1 che potrebbe
essere attributo alla presenza di calcite è in realtà molto più simile a quello della lazurite infatti si
può osservare una gobba peculiare.
Figura 5-9 è ottenuto con la riga a 488 nm (30 secondi) e presenta molta fluorescenza anche se si riconoscono dei picchi intorno a 600 cm-1.

44
Figura 5-10 sono ottenuti con la riga a 676 nm (60 secondi) intorno alla zona in cui con la riga blu si vedevano i picchi per notare delle somiglianze o meno. Si vedono i picchi caratteristici della lazurite a 500
cm-1 e della calcite a 1090 cm-1.

45
5.1.9 Zona bianca vicino all’azzurro E’ il tipico grafico della calcite con i picchi caratteristici.
Figura 5-11 è ottenuto con la riga blu (30 secondi) ed è uguale a meno delle intensità al grafico della calcite.

46
Figura 5-12 sono entrambi ottenuti con la riga a 676 nm intorno alle posizioni in cui si notavano i picchi con il blu; il primo è puntato su 250 cm-1, il secondo su 700 cm-1, per entrambi il tempo di acquisizione è di 60
secondi.
5.1.10 Zona blu scuro Come si può osservare dai grafici con la riga blu del laser c’è un picco molto pronunciato a
circa 500 cm-1, tipico del lapislazzuli, e uno a 1090 cm-1, caratteristico della calcite. A differenza,
però, delle altre zone del campione in cui compaiono entrambi questi picchi, il picco del lapislazzuli
è molto più intenso di quello della calcite: si evince, perciò, una maggiore quantità di questa
sostanza in questa particolare zona.
Con la riga rossa si osserva solo il picco della lazurite molto meno intenso rispetto a quello
di prima.

47
Figura 5-13 primo è ottenuto con la riga blu (30 secondi) c’è molta fluorescenza ma si possono riconoscere i picchi della lazurite e della calcite; il secondo è ottenuto con la riga rossa (60 secondi) e oltre
alla luminescenza c’è il solo picco della lazurite.
5.1.11 Zona blu cobalto
Si osserva il picco caratteristico della lazurite con il laser blu e si vede molto pronunciata
una “piccola” gobbetta che negli altri grafici è solo accennata. C’è sempre il picco a 1090 cm-1 e
“compare” un picco a circa 1000 cm-1, di cui non si conosce la provenienza.
Con il laser rosso, invece, c’è molto luminescenza, come si può osservare, e intorno a 500
cm-1, dove dovrebbe esserci il picco del lapislazzuli, c’è una serie di picchi di difficile
assegnazione; c’è, invece, un picco intorno ai 250 cm-1.

48
Figura 5-14 il primo ottenuto con la riga blu e si vedono tutti i picchi caratteristici; il secondo con la riga rossa presenta un picco ben accentuato mai visto e altri di incerta origine.
5.2 Cromato di piombo
Nelle zone colorate si può osservare uno spettro molto strutturato e intenso con la riga a
676 nm e a 647 nm perché con essa si assiste al fenomeno del Raman risonante. Con il blu si
possono vedere dei picchi intorno a 300 cm-1 e a 800 cm-1 come con il rosso e questo ci dice che il
sistema è disordinato. Difatti se ci mettiamo su uno dei prismi a facce poligonali caratteristici della
struttura del giallo cromo vediamo uno spettro poco intenso ma avente tutti i picchi tipici; se,
invece, siamo su una zona “amorfa” non si nota nulla di tutto ciò. È quello che accade nei grafici a
seguire: la zona rosa è senza “cilindretti” e l’andamento dello spettro è abbastanza continuo
mentre la zona rossa è uno di questi quindi si ha un accenno dello spettro di risonananza. In
alcune zone abbiamo del cromato di piombo puro mentre nelle altre c’è una certa mescolanza.
Quando la zona di colore (dal rosso al giallo) cambia, il sistema diventa più disordinato e c’è più
mescolanza di sostanze.

49
Figura 5-15 questa serie di tre grafici si riferisce alla zona rosa che possiamo osservare: il primo è ottenuto con la riga blu e si può notare la difficile focalizzazione, il secondo con la riga a 676 nm (10 secondi) e il
terzo a 647 nm (60 secondi).

50
Figura 5-16 sono tre grafici della zona rossa: il primo è ottenuto con la riga a 488 nm (60 secondi) e non si
vede nessun picco mentre con le righe a 647 nm (secondo 60 secondi) e a 676 nm (terzo 10 secondi) si vede la risonanza del cromato di piombo.

51
Figura 5-17 zona gialla: con la riga laser a 488 nm (30 secondi) si riesce a vedere il picco più intenso a circa 800 cm-1 anche se molto meno intenso rispetto agli altri mentre con le altre due righe nel rosso si vede
il grafico di risonanza. Il secondo è con la 647 (60 secondi) e il terzo con 676 (10 secondi)

52
Figura 5-18 zona grigia: con la riga a 488 nm (60 secondi) non si vede alcun picco mentre con la riga a 676 nm (10 secondi) si può osservare solo del rumore; questa può essere una zona fortemente disordinata.
5.3 Campione 11
5.3.1 Zona bianca frammento dimensioni maggiori
Con il blu si vedono i due picchi caratteristici della calcite (290 e 1094 cm-1) mentre con il
rosso (647 nm) solo quello a 1090cm-1 nonostante la fluorescenza elevata. Con la riga a 676 nm
si vedono dei picchi riconducibili ai plasmi: abbiamo fatto la stessa misura con le stesse condizioni
utilizzando della carta; il grafico risultante è stato lo stesso, quindi, i picchi osservabili non sono utili
alla nostra analisi.

53
Figura 5-19 primi due grafici ottenuti con il blu con diverso intervallo di frequenze considerato attorno ai picchi caratteristici che ci aspetteremmo( potenza laser 10 mW acquisizione di 60 secondi; il primo puntato su 500 cm-1 il secondo su 1100 cm-1); il terzo con la riga a 676 nm (puntati su 1050 cm-1 per 180 secondi)
ma oltre a dei plasmi non si vede nulla.
5.3.2 Zona bianca frammento dimensioni minori
Ci sono anche qui i picchi caratteristici della calcite oltre a molta luminescenza con la riga
blu e rumore con quella rossa (676 nm) che si presenterà molto simile in altre zone di questi
frammenti perciò non metterò più questo tipo di grafici vista la loro somiglianza.

54
Figura 5-20 grafici ottenuti con luce blu: il primo su un ampio spettro di frequenze il secondo attorno solo a 1090 cm-1 per osservare meglio la struttura di questo picco (potenza laser 10 mW per 60 secondi di
acquisizione).
5.3.3 Zona grigia scura frammento dimensioni minori
Con la riga blu non si riesce a vedere alcun picco Raman a causa dell’elevata
luminescenza. Con la riga rossa a 676 nm si vede solo il rumore.
5.4 Campione 1
5.4.1 Zona scura vicino al bordo
Per il rosso si vede il picco a circa 1300 cm-1 anche se c’è troppa fluorescenza anche con il
blu infatti non si vede nulla neanche nell’altra zona colorata.

55
Figura 5-21 sono ottenuti con la riga rossa a 647 nm (potenza laser 15 mW per 60 secondi); in particolare il secondo è centrato attorno a 1100 cm-1.
5.5 Campione 6
5.5.1 Zona bianca frammento dimensioni maggiori Nelle zone bianche c’è troppa luminescenza anche se si riesce ad osservare il picco attorno
a 1100 cm-1 tipico della calcite e non riusciamo a dire nulla sugli altri picchi.
Figura 5-22 questo è il grafico della zone bianca con la luce blu (potenza laser 35 mW acquisizione per 90
secondi); quello con la riga rossa presenta solo del rumore come quelli precedenti.
56
5.5.2 Zona scura frammento dimensioni maggiori
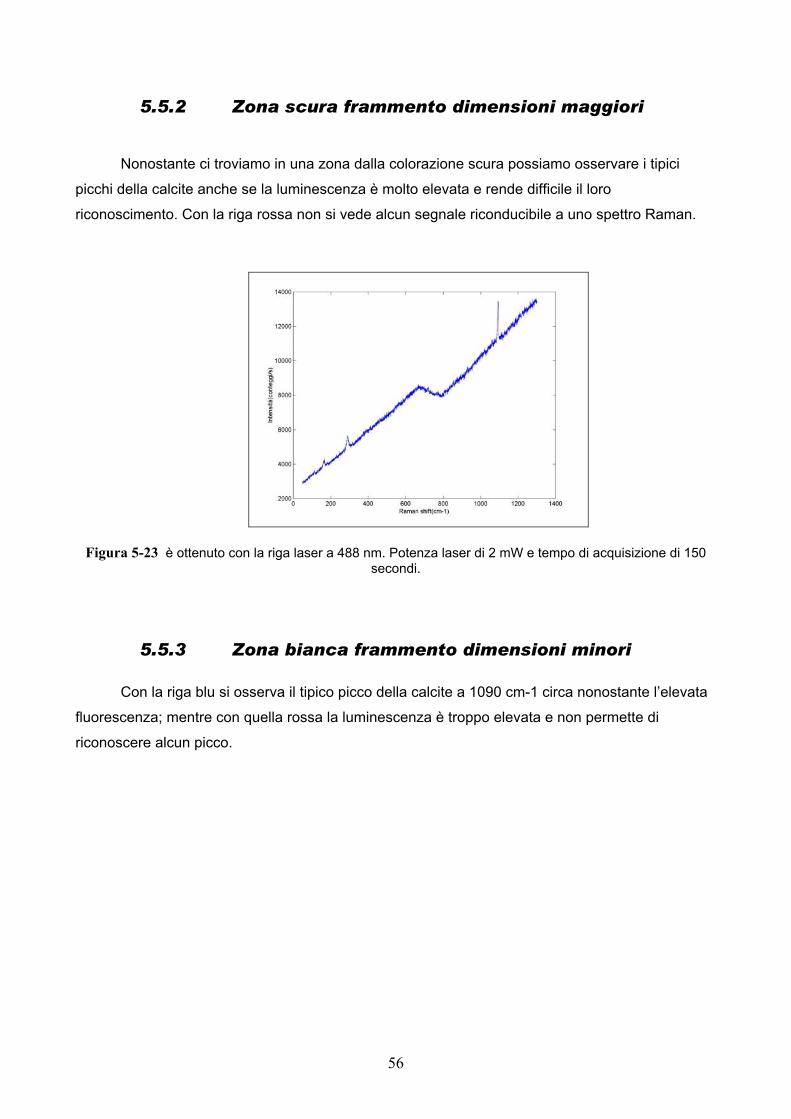
Nonostante ci troviamo in una zona dalla colorazione scura possiamo osservare i tipici
picchi della calcite anche se la luminescenza è molto elevata e rende difficile il loro
riconoscimento. Con la riga rossa non si vede alcun segnale riconducibile a uno spettro Raman.
Figura 5-23 è ottenuto con la riga laser a 488 nm. Potenza laser di 2 mW e tempo di acquisizione di 150 secondi.
5.5.3 Zona bianca frammento dimensioni minori
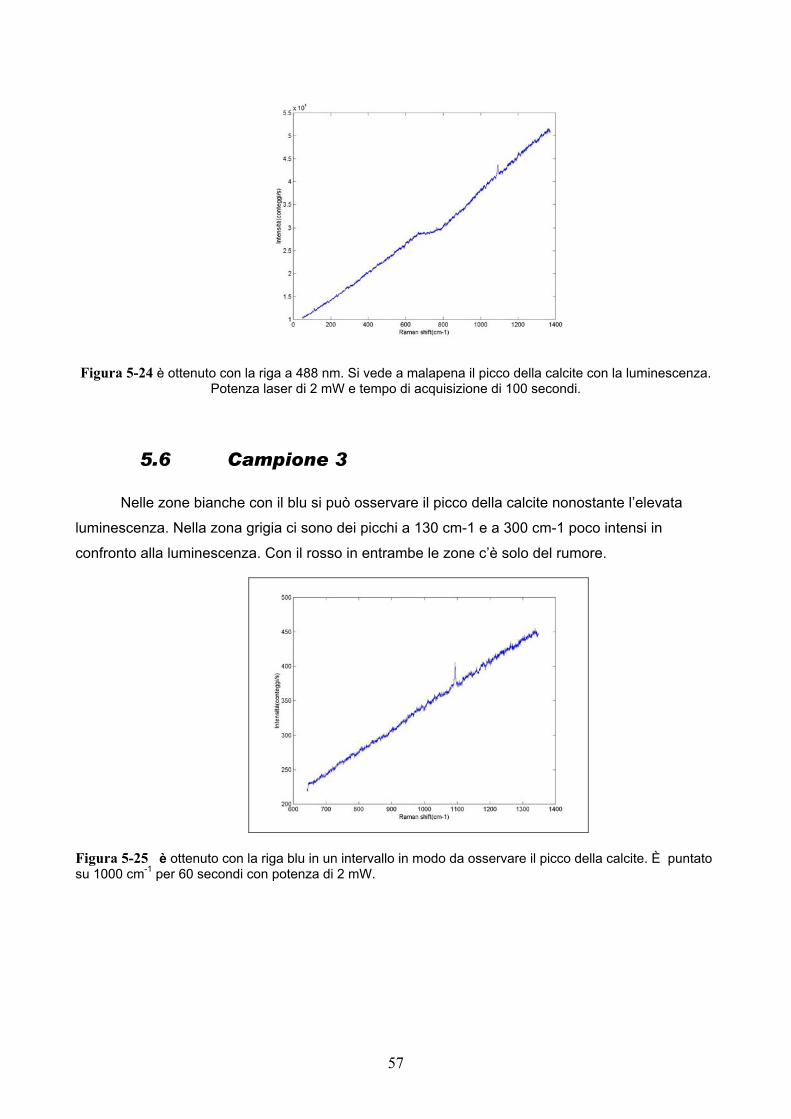
Con la riga blu si osserva il tipico picco della calcite a 1090 cm-1 circa nonostante l’elevata
fluorescenza; mentre con quella rossa la luminescenza è troppo elevata e non permette di
riconoscere alcun picco.
57
Figura 5-24 è ottenuto con la riga a 488 nm. Si vede a malapena il picco della calcite con la luminescenza. Potenza laser di 2 mW e tempo di acquisizione di 100 secondi.
5.6 Campione 3
Nelle zone bianche con il blu si può osservare il picco della calcite nonostante l’elevata
luminescenza. Nella zona grigia ci sono dei picchi a 130 cm-1 e a 300 cm-1 poco intensi in
confronto alla luminescenza. Con il rosso in entrambe le zone c’è solo del rumore.
Figura 5-25 è ottenuto con la riga blu in un intervallo in modo da osservare il picco della calcite. È puntato su 1000 cm-1 per 60 secondi con potenza di 2 mW.

58
6 Conclusioni
Di seguito possiamo vedere una tabella che riassume tutti i dati acquisiti e, grazie a questa, possiamo risalire al composto in una
determinata regione dei nostri campioni e al modo di vibrazione con cui interagisce la radiazione incidente:
Zone di colore visibili Picchi exc 488nm (cm-1) Materiali Modi vibrazionali
Picchi exc
676nm (cm-1)
Materiali Modi vibrazionali
Lapislazzuli
blu piatta 165 Calcite Modi del reticolo 547,2 Lapislazzuli 1υ di −3S
287 Calcite Modi del reticolo Tracce di calcite
716 Calcite 4υ del carbonato
1087 Calcite stiramento simmetrico CO3
azzurra frastagliata 184 Calcite Modi del reticolo 553 Lapislazzuli 1υ di −3S
306 Calcite Modi del reticolo Tracce di calcite
729 Calcite 4υ del carbonato
1100 Calcite stiramento simmetrico CO3 punto argentato zona bianca 348,4 Pirite 383,6 Pirite 433,2 Pirite
bianca 181 Calcite Modi del reticolo 179,3 Calcite Modi del reticolo
303 Calcite Modi del reticolo 301,8 Calcite Modi del reticolo
726 Calcite 4υ del carbonato
1100 Calcite stiramento simmetrico CO3
azzurra 165 Miscuglio blu piatta Azzurra frastagliata Modi del reticolo
181 Modi del reticolo (290-306) Modi del reticolo

59
719 4υ del carbonato
dorata nella zona blu 383,9 Pirite 432,3 dorata nella zona bianca 348,4 Pirite 383,9
551,6 Lazurite 1υ di −3S
azzurra vicino alla zona dorata 348,4 Pirite
380,6
548,4 Lapislazzuli 1υ di −3S
1090,3 Calcite,Lapislazzuli Stiramento simmetrico carbonato
bianca vicino alla zona azzurra 181 Calcite Modi del reticolo
306 Calcite Modi del reticolo 729 Calcite 4υ del carbonato
1100 Calcite Stiramento simmetrico del carbonato
blu scuro 258 Lapislazzuli
(548-587) Lapislazzuli 1υ di −3S
1090 Lapislazzuli Stiramento simmetrico del carbonato
blu cobalto 264,5 Lapislazzuli 300 471
(551,6-587,1) Lapislazzuli 1υ di −3S
987,1 1093,2

60
Zone di colore Picchi exc 488nm(cm-1) Picchi exc 676nm (cm-1) Riga a 647 Modi vibrazionali
Cromato di piombo
grigia luminescenza rumore luminescenza rosa luminescenza (118,1-137,5) (125-141) Vibrazione legame Pb-Cr 181,6 187
(327,9-340,3-359,7-377,3-402) (330-345-365-383-405) Vibrazione legame Cr-O
841 844 Vibrazione legame Pb-O rossa 306,5 135,8 141,1 Vibrazione legame Pb-Cr
345,1 (328-340,3-361,5-377,3-402) (334,5-342,6-364,7-382,9-405) Vibrazione legame Cr-O
541,9 839,3 846,3 Vibrazione legame Pb-O 832,2 1269,5 1303,8 1418,6 gialla 341,9 137,5 (123-141) Vibrazione legame Pb-Cr 835,5 (328-338,5-361,4-379-402) 187 841 (332-345-365-383-405) Vibrazione legame Cr-O 844 Vibrazione legame Pb-O 1272
Zone di colore
Picchi exc 488nm (cm-1) Materiali Picchi exc 676nm (cm-1) Materiali Modi vibrazionali Riga a 647
Campione 11 Frammento dimensioni
maggiori
bianca 290 Calcite Modi di reticolo
1093 Calcite stiramento simmetrico carbonato
1091,9
Frammento dimensioni minori bianca 165 Calcite 925,3 Modi di reticolo 290 Calcite 1070 calcite Modi di reticolo

61
719 Calcite 4υ del carbonato
1093 Calcite Stiramento simmetrico carbonato
grigio scuro 1334 Non rintracciabile
Zone di colore Picchi exc 488nm (cm-1) Picchi exc 676nm (cm-1) Modi vibrazionali Riga a
647 Materiali
Campione 1 Frammento dimensioni
maggiori
Scura vicino al bordo Luminescenza Luminescenza 1271,9 Non
rintracciato 1306,9
Zone di colore Picchi (exc 488nm) (cm-1) Materiali Modi vibrazionali Campione 6
Frammento dimensioni maggiori Scura di confine 165 Modi del reticolo 290 Modi del reticolo 1094 Stiramento simmetrico carbonato bianca 1094 Calcite Stiramento simmetrico carbonato
Frammento dimensioni minori
bianca di confine 1087 Calcite Stiramento simmetrico carbonato

62
Zone di colore Picchi (exc 488nm) (cm-
1) Materiali Modi vibrazionali
Campione 3 Frammento dimensioni
maggiori
bianca all'interno di quella grigia 165 Calcite Modi di
reticolo
292 Modi di reticolo
bianca 1092 Calcite Stiramento simmetrico carbonato
grigia 2025,3 Frammento dimensioni minori
bianca 1092 calcite Stiramento simmetrico carbonato

63
Come possiamo osservare non tutti i picchi sono stati riconosciuti e non a tutti sono stati
attribuiti i modi di vibrazione corrispondenti.
Per il lapislazzuli possiamo confermare che esso è costituito principalmente da lazurite ma
presenta anche degli altri composti quali il carbonato di calcio e la pirite. Lo zolfo, uno dei
costituenti principali, è il responsabile della risonanza che osserviamo utilizzando la riga laser blu
infatti il picco caratteristico a circa 500 cm-1 corrisponde al modo vibrazionale dello ione −3S ed è
molto più intenso eccitando il campione con la riga a 488 nm. C’è anche della calcite visibile grazie
al picco intorno a 1090 cm-1 corrispondente allo stiramento simmetrico del carbonato. I picchi a
bassa frequenza sono riferibili a modi del reticolo mentre il picco a circa 700 cm-1 è dovuto a 4υ del
carbonato.
Gli spettri della calcite presente non sono influenzati dell’effetto risonante infatti quasi tutti i
grafici con qualunque riga laser presentano il suo picco caratteristico se la luminescenza non è
troppo elevata.
Per il cromato di piombo possiamo osservare l’effetto Raman risonante con la riga rossa del
laser: si riconoscono così, nel grafico, tre bande centrate più o meno attorno a 130 cm-1, 350 cm-1
e 850 cm-1. Dalla letteratura si evince che i modi a bassa frequenza corrispondono alle vibrazioni
del legame Pb-Cr, che coinvolgono gli atomi con numero atomico maggiore; possiamo supporre
che le bande centrate su 350 cm-1, più articolate e strutturate, corrispondano alla vibrazione Cr-O
mentre l’ultima banda ad alta frequenza è dovuta al legame Pb-O.
Anche con la riga blu si riescono a riconoscere dei picchi intorno alle bande riconoscibili con
il rosso, però sono solo degli accenni. Nella zona grigia non si riconoscono dei picchi ma solo della
luminescenza.
Possiamo osservare anche che se siamo in una zona amorfa con la riga blu allora il grafico
ha un andamento piuttosto continuo a parte dei picchi poco intensi come nel caso della zone gialla
mentre se siamo su un prisma a facce poligonali (zona rossa) allora i picchi sono molto più intensi.
Con la riga rossa si vede lo stesso grafico in ogni zona colorata però la banda a 350 cm-1 è più o
meno articolata strutturata a seconda dell’ordine o del disordine della regione che stiamo
analizzando. La zona gialla è più disordinata di quella rossa e si può supporre che nella prima ci
sia più mescolanza.
Caso particolare da menzionare è il picco a circa 600 cm-1 osservabile nella zona rossa con
la luce blu: abbiamo cercato di attribuirlo ma è stato molto difficile anzi impossibile date le poche
informazioni in nostro possesso. Nell’analisi delle rocce bisognerebbe considerare anche la loro

64
provenienza, la loro genesi in modo da sapere quali disomogeneità si potrebbero vedere e quali
elementi si possono trovare. Per la mancanza di queste informazioni è stato molto difficile
l’attribuzione.
In tutti e 4 i frammenti si può notare la presenza del carbonato di calcio con il suo picco
caratteristico a 1090 cm -1. Nel campione 6 un’analisi precedente aveva ipotizzato la presenza di
lapislazzuli ma noi non siamo riusciti a riscontrarli sia usando la luce rossa sia usando quella blu.
Nel campione 1 si può osservare un picco a circa 1300 cm-1 che non siamo riusciti ad
identificare infatti molti composti organici hanno questo tipo di picco e quello che li caratterizza è
l’andamento a basse frequenze: nel nostro caso questo è molto disturbato e non siamo riusciti a
riconoscere alcun picco Raman perciò l’identificazione è stata impossibile.
L’analisi attraverso la microspettroscopia Raman di questi pigmenti utilizzati dagli artisti mi
ha permesso di mostrare la grande utilità dell’effetto Raman risonante accoppiato alla possibilità di
sondare zone microscopiche del campione nei casi, tipicamente come quelli da noi osservati, dove
ci sono vari tipi di eterogeneità (chimiche, strutturali, morfologiche…) che rendono molto difficile la
loro analisi quantitativa.

65
Bibliografia
1) D. Bersani, G.Antonioli, P.P. Lottici, A. Casoli Raman microspectrometric
investigation of wall paintings in S.Giovanni Evangelista Abbey in Parma: a comparison
between two artists of the 16th century
2) dispense fornite dal prof. Fontana per il corso di Spettroscopia
3) dispense fornite dal prof. Ghezzi per il corso di Introduzione allo stato solido
4) a cura di Lucia Fornari Scianchi Parmigianino e il manierismo europeo Atti del
convegno internazionale di studi- Parma 13-15 giugno 2002 Silvana Editoriale
5) N.Cavalca Corso di diploma universitario in metodologie fisiche: Tecniche fisiche di
indagine applicate al restauro degli affreschi in San Giovanni Evangelista e nella
Cattedrale di Parma
6) Manuale d’istruzioni T64000
7) M. Cardona Light scattering in solids: R.M Martin L.M. Falicov Resonant Raman
Scattering
8) C. Biellmann P. Gillet High-pressure and high-temperature behaviour of calcite,
aragonite and dolomite: a Raman spetroscopy study
9) N. Gobeltz-Hautecoeur Occupancy of the Solidate Cages in the Blue Ultramarine
Pigments

66
Ringraziamenti
Ringrazio, innanzitutto, il mio relatore, il prof. Marco Fontana, che mi ha permesso di svolgere il mio lavoro di tesi accomunando la fisica e la storia
dell’arte, mia grande passione, e per aver corretto pazientemente i miei numerosi errori. Grazie per la disponibilità mostratami.
Un ringraziamento al mio correlatore, il dott. Danilo Bersani, per avermi
fornito i campioni da analizzare con la spettroscopia Raman e avermi aiutato nella loro identificazione.
Ringrazio Paolo Camorani e il prof. Luigi Cristofolini per avermi aiutato a
capire il funzionamento del T64000 e tutto quello che riguardava il laboratorio dove ho svolto il mio tirocinio.
Un ringraziamento particolare va ai miei compagni di studio insieme ai quali sono arrivata a questo traguardo. Ognuno di loro ha un motivo specifico per
essere ringraziato ma lo spazio è ridotto e non posso, purtroppo, dedicarmi a ognuno di voi: grazie a tutti per aver rallegrato questi tre anni. Grazie a voi
per il viaggio in Olanda e per le cene a casa della Marti e per tutto.
Non vogliatemi male ma tra voi rivolgo un ringraziamento speciale a Luca e alla Marti per aver condiviso con me le esperienze di laboratorio. Come avrei
fatto senza di voi? Un ringraziamento particolare a Davide, che ha spartito con me il tirocinio e mi ha fatto compagnia nel primo periodo in comune di apprendimento del funzionamento del T64000 durante le, a volte, lunghe acquisizioni dei dati.
Grazie ai miei amici per volermi bene e per essermi sempre accanto,
soprattutto nell’ultimo periodo in cui mi sono data per.. dispersa…
Grazie a Filippo per avermi prestato il computer portatile senza il quale non sarei riuscita nel mio lavoro.
Ringrazio tantissimo i miei genitori, a cui dedico il mio lavoro, per tutto quello che hanno fatto per me e per avermi insegnato a non mollare mai. Grazie per avermi sopportato e supportato. Grazie a tutta la mia famiglia perché mi ama
e crede in me nonostante tutto. Grazie!
Grazie a tutte le persone che mi amano, mi sopportano e credono in me.
Grazie!!

67

68

69

70