regulatory perspective on assuring ingredient quality -...
TRANSCRIPT

Regulatory Perspective on
Assuring Ingredient Quality
PQRI Conference
December 15, 2009
Rockville, MD
Steven M. Wolfgang, Ph.D.,
Acting Associate Director, Regulatory Science
US Food and Drug Administration
Division of Manufacturing & Product Quality
Center for Drug Evaluation & Research

Economically Motivated Adulteration?

Outline
• Pharmaceutical Ingredients are Drugs!
• Globalization begets complexity and risk
• CGMP Regulations and Guidance
• Quality Management Systems for Acceptance of Drug Components
• Component Supplier Quality Management
• Modernization of Detection Systems

Definition of Drug• FD&C Act (law) Section 201(g)(1)
– recognized in an official US compendium: United
States Pharmacopeia, Homoeopathic
Pharmacopoeia, or National Formulary
– intended to provide diagnosis, cure, mitigation,
treatment, or prevention of disease
– intended to affect the structure or any function of the
body
– intended for use as a COMPONENT of any article
meeting above criteria
EXCIPIENTS and APIs are DRUGS!
ADDITIVES REMOVED DURING PROCESSING are DRUGS!

All parties which manufacture (includes testing), process, pack, or hold an ingredient or drug product
are responsible for meeting CGMPs.
Adulterated Drug Component = Adulterated Drug Product
Regulatory Requirements for Supply Chain Members

Adulteration
• 501(b) – drugs that purport to be official
articles but do not meet the USP
monograph requirement (public standard)
• 501(c) – non-compendial drugs that do not
meet the purported specification
(proprietary standard)
• 501(d) – substitution (EMA)

Adulteration - GMP
• 501(a)(2)(B) – not manufactured (and held) in
accordance with CGMP, as appropriate for the
intended use
– 21 CFR 210/211 only apply to finished drugs
• Q9 (risk management) & Q10 (quality systems)
– Q7A is a nonbinding consensus standard (guidance)
for APIs, generally accepted by regulators WW
– IPEC/PQG Excipient GMP is a nonbinding standard
(guideline), aiming to achieve consensus standard
status

8
“
Globalization of FDA Regulated Drugs

If the Trends Continue…..
0
500
1000
1500
2000
2500
3000
3500
4000
1992 1994 1996 1998 2000 2002 2004 2006 2008
Calendar Year
Nu
mb
er
of
Reg
iste
red
Sit
es
Registered Domestic Sites
Registered Foreign Sites
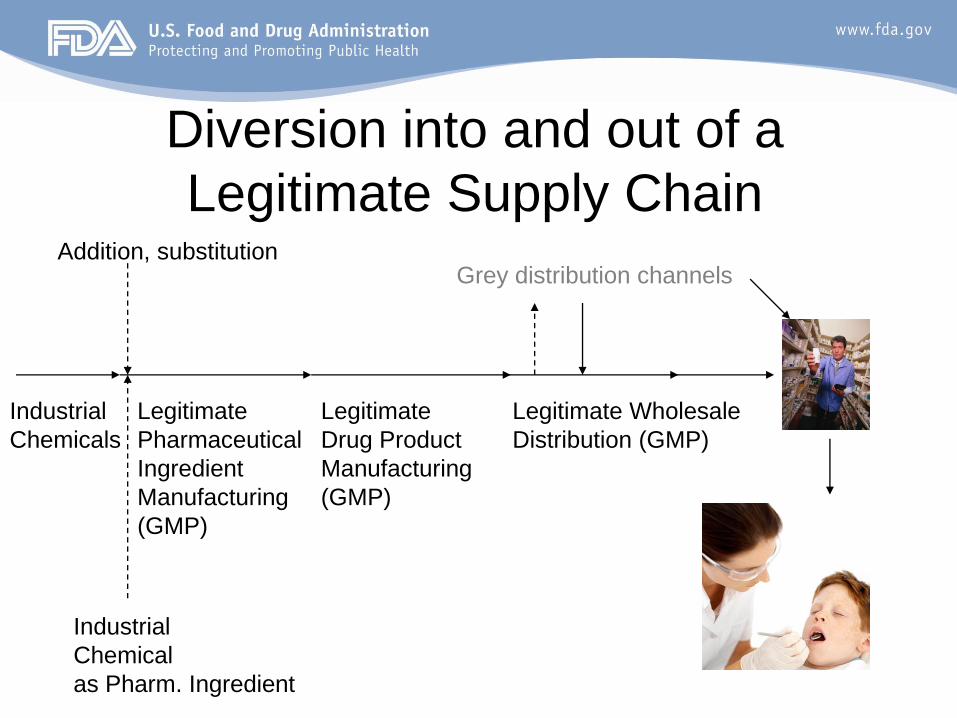
Diversion into and out of a
Legitimate Supply Chain
Legitimate
Drug Product
Manufacturing
(GMP)
Industrial
Chemical
as Pharm. Ingredient
Legitimate Wholesale
Distribution (GMP)
Legitimate
Pharmaceutical
Ingredient
Manufacturing
(GMP)
Addition, substitutionGrey distribution channels
Industrial
Chemicals

• Deliberate Acts of Deception
– Misrepresenting identity or source or an ingredient
– Fabricating data or falsely labeling an ingredient purporting it
to be of suitable quality for drug use
• Adulteration by Substitution
– Economically Motivated Adulteration (EMA) even
targets commodities:
• Drug ingredient raw material supply
– Melamine for crude protein (source of excipients)
– OSCS for heparin (source of API)
• Drug ingredient supply
– DEG for Glycerin or Polyol excipient
Risk of Illicit Activity within Distribution Network

Complexity of Holding and Distribution
Within Globalized Supply Chain
Ingredient manufacturers
primary manufacturing sites
ingredient finishing sites
Brokers and traders
Forwarding, warehousing and bulk storage
Distributors
Repackaging and relabeling
Certificates of analysis

Globalization Might Increase Exposure to Risk
• Lack of traceability
• Acceptance of information from supplier without
adequate verification
• Incomplete Supply Chain oversight
• Limited FDA import screening of ingredients
• Risk of illicit activity within distribution network

14
Lack of Traceability
and Acceptance of Information from Supplier
Without Adequate Verification
• Lack of knowledge about manufacturing sites and distribution routes
• Over-reliance on Certificates of Analysis
• CoA not from original manufacturer
• CoA not adequately verified for accuracy
• Use of non-specific ID test
• Testing based on composite samples
• Reliance on vendor questionnaire without auditing

Limited Supplier Quality Management System
Inadequate qualification programs
No written quality agreement
Limited quality monitoring
Limited Oversight of Remote Manufacturing Sites
No on-site audit by drug product manufacturer
Infrequent inspections
FDA
other regulatory authorities
regional regulatory authority
Incomplete Supply Chain Oversight

Limited FDA Import Screening
• Drug Import Volumes Far Exceed Human Resources
– Which shipments should be scrutinized?
• Importation Documentation
– Provides limited traceability back to original
manufacturer
– Excipient need not be declared as drug
• Excipient manufacturers are excluded from
registration requirement

CGMP Requirements for Drug
Manufacturers - 21 CFR Part 211• Control of Components (Subpart E)
– 211.84 places burden on drug manufacturers to prevent adulterated ingredients from being used
• Representative samples (so as not to mask variation)
• Appropriate testing or examination
• Appropriate written specifications
– Verification of identity
– Allows use of CoA data for conformance to other specifications provided data is verified periodically for accuracy

Caveats Re: Reliance Upon CoA
• ID testing of drug components shall be specific (or must
perform additional tests able to support unequivocal ID)
• Use of the vendor CoA applies only if there is ongoing
verification of reliability of data and the absence of
adulteration (not evident via specification)
– 501(a)(2)(B) requires that drug components are
manufactured in accordance with appropriate GMP
• suitable controls in SC pertaining to manufacturing,
processing, packing, holding, distribution
• change management system
– supported by periodic audits

Caveats: Sampling and Testing
• Homogeneity is a function of the drug component manufacturing process – lot size does not necessarily equate to a batch
– factors associated with production rate or equipment size
• Cannot assume homogeneity within a lot– among different containers
– within container
• Test results have no meaning unless sampling plan and degree of variation in each test attribute is known– especially important for functionality related characteristics
– affects how one interprets the significance of data on CoA

Glycerin Guidance http://www.fda.gov/downloads/Drugs/GuidanceComplian
ceRegulatoryInformation/Guidances/ucm070347.pdf
• Reiterates 211.84(d)(2) requirement for specific
ID testing when not performing full USP testing
– testing has to be capable of detecting DEG
– applies to all recipients of Glycerin USP, not
limited to those which formulate or compound
• Recommends intimate knowledge of the
members of the supply chain
– traceability

Supply Chain GMP/GDP/Security
• All members of a drug component SC should have an adequate Quality System
• Manufacturing controls should provide added assurance that drug componentts are not adulterated and consistently meet the agreed-upon user requirements
– includes in-process and finished product testing
• Documentation should allow traceability of an excipient to its starting materials
– starting materials can be a significant source of variation or a point at which adulteration occurs
• Documentation should provide forward traceability
• Packaging and holding practices (transport, storage) should prevent deterioration and tampering

Caveats Re: Intended Use of an Excipient
• Comprehensively assessed and qualified for intended use
– Appropriate excipient specifications
– Appropriate excipient GMP
• e.g. for use in a sterile drug
– Functionality
• dosage form
• manufacturing of drug product
– Safety relevant to drug product
• level of exposure via DP
• route of administration
• patient population

Functionality Testing
Building Quality into Drug Product
• Manufacturers should link quality of ingredient to performance as part of supplier qualification– measurable physical or chemical attribute
• predicts performance
– desired performance is in context of drug product
– determine suitable ingredient compositions (ranges) for mixtures or polymers
• Design space - base development work on establishing range of use or predictive model?
• Use of signature to identify and verify helps assure functionality

Use of a 3rd Party Audits• Identifying credible 3rd parties
– Impartiality
– Qualifications• training and expertise of auditors
– Audits apply appropriate GMP guideline• preferably developed via consensus among practitioners/users
• preferably based on principles of GMP and quality for drugs
• Supplier maintenance and monitoring, coverage– Frequency of audit
– Scope of audit• Quality management systems
• Systems applicable to preventing contamination and mixups
• Controls on supply chain security and integrity
• Corrective measures

USP’s Role
• USP Ingredient Monograph Revisions
– improve specificity of ID and assay tests focusing on APIs/excipients at risk to EMA
• non-specific ID testing
– qualitative wet chemistry (color changes, formation of precipitate)
– indicators of presence of functional group
• non-specific assays
– N assays
• specifications that allow undetected adulterants to be present
– DEG/EG in polyols

USP Activities of Interest to FDA• USP Ingredient Monograph Revisions
– specify modern instrumental methods to replace less sophisticated, less capable methods
• wet chemistry, acid-base titration
• TLC
– manufacturers and consortia can provide any assistance
• Outreach
– to improve global standards (e.g. MOU with China)
– to improve detection of adulteration in developing countries
• Overcoming challenges to develop reference standards for excipients that are not single molecular entity

Achieving Compendial Modernization
• Use of screening tools to flag suspect ingredients– for further testing to determine if adulterated
– for suspicion of having deviated from the legitimate supply chain
• ID testing via pattern recognition– comparison with spectral signature (library) for
verification of authenticity• only accept from approved manufacturing sites
– verification supports qualification of ingredient and its supplier
– verification supports acceptance of data on CoA

That’s all folks!