okko t. pyykkÖ - uef electronic...
TRANSCRIPT

DIS
SE
RT
AT
ION
S | O
KK
O T
. PY
YK
KÖ
| IDIO
PA
TH
IC N
OR
MA
L P
RE
SS
UR
E H
YD
RO
CE
PH
AL
US
: A S
TU
DY
... | No
336
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
Dissertations in Health Sciences
Dissertations in Health Sciences
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
OKKO T. PYYKKÖ
IDIOPATHIC NORMAL PRESSURE HYDROCEPHALUSA study of epidemiology, genetics, and cerebrospinal �uid
OKKO T. PYYKKÖ
ISBN 978-952-61-2059-1ISSN 1798-5706
Idiopathic normal pressure hydrocephalus isa slowly progressive syndrome in the elderly
characterized by gait disorder, cognitivedeterioration, and urinary incontinence.
Compared to earlier studies, a higher annualincidence of the syndrome was noted in a
Finnish population with an increasing trend.High vascular comorbidity and mortality wasobserved without a major neuroin�ammatorycomponent. Apolipoprotein E genotypes did
not differentiate patients with idiopathicnormal pressure hydrocephalus from healthy
age- and gender-matched controls.


Idiopathic normal pressurehydrocephalus: a study of
epidemiology, genetics, andcerebrospinal fluid


I
OKKO T. PYYKKÖ
Idiopathic normal pressurehydrocephalus: a study of
epidemiology, genetics, andcerebrospinal fluid
To be presented by permission of the Faculty of Health Sciences, University of Eastern Finlandfor public examination at Kuopio University Hospital Auditorium 2, Kuopio,
on Saturday 2nd April at 12 noon
Publications of the University of Eastern FinlandDissertations in Health Sciences
Number 336
Institute of Clinical Medicine – Neurology and NeurosurgerySchool of Medicine, Faculty of Health Sciences, University of Eastern Finland
and Neurosurgery of Neuro Center, Kuopio University HospitalKuopio
2016

II
Grano OyJyväskylä, 2016
Series Editors: Professor Veli-Matti Kosma, M.D., Ph.D.Institute of Clinical Medicine, Pathology
Faculty of Health Sciences
Professor Hannele Turunen, Ph.D.Department of Nursing Science
Faculty of Health Sciences
Professor Olli Gröhn, Ph.D.A.I. Virtanen Institute for Molecular Sciences
Faculty of Health Sciences
Professor Kai Kaarniranta, M.D., Ph.D.Institute of Clinical Medicine, Ophthalmology
Faculty of Health Sciences
Lecturer Veli-Pekka Ranta, Ph.D. (pharmacy)School of Pharmacy
Faculty of Health Sciences
Distributor: University of Eastern Finland
Kuopio Campus LibraryP.O.Box 1627
FI-70211 Kuopio, Finlandhttp://www.uef.fi/kirjasto
ISBN (print): 978-952-61-2059-1ISBN (pdf): 978-952-61-2060-7
ISSN (print): 1798-5706ISSN (pdf): 1798-5714
ISSN-L: 1798-5706

III
Author’s address: Department of Neurosurgery Institute of Clinical Medicine, School of Medicine University of Eastern Finland KUOPIO FINLAND
Supervisors: Docent Ville Leinonen, M.D., Ph.D. Department of Neurosurgery Institute of Clinical Medicine, School of Medicine University of Eastern Finland KUOPIO FINLAND
Docent Anne M. Koivisto, M.D., Ph.D. Department of Neurology Institute of Clinical Medicine, School of Medicine University of Eastern Finland KUOPIO FINLAND
Professor Mikko Hiltunen., Ph.D. Institute of Biomedicine University of Eastern Finland KUOPIO FINLAND
Reviewers: Docent Leena Kivipelto, M.D., Ph.D. Department of Neurosurgery University of Helsinki and Helsinki University Hospital HELSINKI FINLAND
Docent Pauli Helén, M.D., Ph.D. Department of Neurosurgery University of Tampere and Tampere University Hospital TAMPERE FINLAND
Opponent: Professor emeritus Carsten Wikkelsø, M.D., Ph.D. Institute of Neuroscience and Physiology
Sahlgrenska Academy at the University of Gothenburg GOTHENBURG SWEDEN

IV

V
Pyykkö, Okko T.Idiopathic normal pressure hydrocephalus: a study of epidemiology, genetics, and cerebrospinal fluidUniversity of Eastern Finland, Faculty of Health SciencesPublications of the University of Eastern Finland. Dissertations in Health Sciences 336. 2016. 83 p.
ISBN (print): 978-952-61-2059-1ISBN (pdf): 978-952-61-2060-7ISSN (print): 1798-5706ISSN (pdf): 1798-5714ISSN-L: 1798-5706
ABSTRACT
Idiopathic normal pressure hydrocephalus (iNPH) is a slowly progressive syndrome in the elderly characterized by gait disorder, cognitive deterioration, and urinary incontinence. Symptoms of iNPH can be relieved and even reversed by ventriculoperitoneal shunt surgery in most patients. The aim of this thesis was to investigate incidence, comorbidities, mortality, and causes of death in iNPH. In addition, the potential effects of apolipoprotein E (APOE) genotypes in the diagnostics and prognostics in iNPH was explored. Another aim was to evaluate a large panel of cerebrospinal fluid (CSF) biomarkers in iNPH and how they relate to brain biopsy findings. Kuopio NPH registry consists of all evaluated possible iNPH patients from the Kuopio Univer-sity Hospital catchment population since 1993 and contains clinical baseline and follow-up data, other hospital diagnoses, and medications. In addition, all patients in the registry underwent a frontal cortical biopsy during a diagnostic intracranial pressure monitoring or shunt surgery. An-nual population of the catchment area and causes of death were obtained from the national regis-tries. Patients with a blood sample available for DNA extraction were genotyped for APOE. Fur-thermore, lumbar and ventricular CSF samples were analyzed for several biomarkers: beta amyloid (Aβ) isoforms, soluble amyloid precursor protein (sAPP) isoforms, proinflammatory cytokines, and biomarkers of neuronal damage. Compared to earlier studies, a higher annual incidence of iNPH was noted in Middle and East-ern Finnish populations with an increasing trend. While being relatively uncommon in the total population (1.8 / 100,000 / year), a much higher age-specific cumulative incidence of iNPH was seen in persons aged 70 or older (15 / 100,000 / year). The most common comorbidity in patients with iNPH was arterial hypertension. Additionally, type 2 diabetes mellitus was twice more common in patients with iNPH compared to controls (23% vs. 13%, p = 0.002). The most common causes of death in iNPH were ischaemic heart disease (22%) and cerebrovascular disease (19%). In contrast to previous small-scale studies, APOE genotypes did not differ between 113 iNPH patients and 687 healthy elderly controls. However, APOE ε4 allele was associated with Alzheimer’s disease (OR 4.3, 95% CI 2.0–9.3) and presence of Aβ in the brain biopsy (OR 8.7, 95% CI 3.8–20), which is in line with previous studies. Decreased levels of sAPP alpha in the CSF were identified as a potential di-agnostic biomarker for iNPH. No neuroinflammatory findings were identified in the CSF samples of patients with iNPH.
National Library of Medical Classifications: WL 300, WL 203, QU 450, WA 900Medical Subject Headings: Hydrocephalus, Normal Pressure; Incidence; Comorbidity; Survival; Cause of Death; Risk Factors, Genes; Genetics; Genotype; Polymorphism, Genetic; Apolipoproteins E; Cerebrospinal Fluid; Biomarkers; Amyloid beta-Peptides; Cohort Studies; Finland

VI

VII
Pyykkö, Okko T.Idiopaattinen normaalipaineinen hydrokefalus: epidemiologia, genetiikka ja aivo-selkäydinnesteItä-Suomen yliopisto, terveystieteiden tiedekuntaItä-Suomen yliopiston julkaisuja. Terveystieteiden tiedekunnan väitöskirjat 336. 2016. 83 s.
ISBN (nid.): 978-952-61-2059-1ISBN (pdf): 978-952-61-2060-7ISSN (nid.): 1798-5706ISSN (pdf): 1798-5714ISSN-L: 1798-5706
TIIVISTELMÄ
Idiopaattinen normaalipaineinen hydrokefalus (iNPH) on iäkkäillä esiintyvä hitaasti etenevä oireyhtymä, jonka tyyppioireita ovat kävelyvaikeudet, kognitiivinen heikentyminen ja virtsan-pidätysvaikeus. Useimmilla potilailla oireistoa voidaan helpottaa tai jopa parantaa kokonaan leik-kauksella, jossa sunttikatetri viedään aivokammiosta vatsaonteloon. Tämän väitöskirjan tavoit-teena oli tutkia iNPH:n esiintyvyyttä, liitännäissairauksia, kuolleisuutta ja kuolemansyitä. Lisäksi tavoitteena oli arvioida apolipoproteiini E (APOE) -genotyyppauksen mahdollista hyötyä iNPH:n diagnostiikassa ja ennusteessa. Päämääränä oli myös arvioida laajasti aivo-selkäydinnestemerkki-aineita iNPH-potilailla ja kuinka merkkiaineet liittyvät aivobiopsian löydöksiin.
Kuopion NPH-rekisteri koostuu potilaista, jotka on lähetetty arvioon Kuopion yliopistollisen sairaalan neurokirurgian klinikkaan väestövastuualueelta vuodesta 1993 alkaen. Rekisteri sisältää kliiniset lähtö- ja seurantatiedot, muut sairaaladiagnoosit sekä lääkitykset. Lisäksi kaikilta re-kisterin potilailta otettiin otsalohkon aivokuorinäyte diagnostisen aivopainemittauksen tai sunt-tileikkauksen aikana. Väestövastuualueen väkiluku ja potilaiden kuolemansyyt hankittiin Tilas-tokeskuksesta. Potilaat, joilla oli verinäyte DNA:n eristystä varten, genotyypattiin APOE:n suhteen. Lumbaali- ja aivokammiopunktiolla otetuista aivo-selkäydinnestenäytteistä arvioitiin lisäksi useita merkkiaineita: beeta-amyloidin (Aβ) isoformit, liukenevan amyloidiprekursoriproteiinin (sAPP) isoformit, proinflammatorisia sytokiineja ja neuraalisen vaurion merkkiaineita.
Aiempiin tutkimuksiin verraten iNPH:n ilmaantuvuus oli korkeampi keski- ja itäsuomalaises-sa väestössä lisääntyen tutkimuksen seuranta-aikana. Vaikka iNPH on koko väestössä harvinainen (1,8 / 100 000 / vuosi), nähtiin 70-vuotiailla ja vanhemmilla selvästi korkeampi ikäspesifinen ilmaan-tuvuus (15 / 100 000 / vuosi). Yleisin iNPH:n liitännäissairaus on verenpainetauti. Myös tyypin 2 diabetes oli kaksi kertaa yleisempi iNPH-potilailla verrattuna verrokkipotilaisiin (23 % vs. 13 %, p = 0,002). Iskeemiset sydäntaudit (22 %) ja aivoverisuonien sairaudet (19 %) olivat yleisin kuole-mansyy iNPH-potilailla. APOE-genotyyppijakauma ei eronnut 113 iNPH-potilaan ja 687 terveen verrokin välillä, vaikka aiempien pienten tutkimusten perusteella on ajateltu toisin. Kuitenkin APOE:n ε4-alleelin yhteys Alzheimerin tautiin (vetosuhde 4,3; 95 %:n luottamusväli 2,0–9,3) ja Aβ:n esiintymiseen aivobiopsiassa (vetosuhde 8,7; 95 %:n luottamusväli 3,8–20) oli vahva, mikä on aiem-missakin tutkimuksissa raportoitu. Aivo-selkäydinnestenäytteestä määritetyn sAPP-alfan matala pitoisuus tunnistettiin mahdolliseksi diagnostiseksi löydökseksi iNPH-potilailla. iNPH-potilaiden aivo-selkäydinnesteessä ei todettu neuroinflammatorisia löydöksiä.
Luokitus: WL 300, WL 203, QU 450, WA 900Yleinen Suomalainen Asiasanasto: hydrokefalia; epidemiologia; ilmaantuvuus; riskitekijät; geenit; perinnöl-lisyystiede; genotyyppi; aivo-selkäydinneste; merkkiaineet

VIII

IX
One might say science is the sum total of our knowledge of the universe, the great library of the known, but the practice of sci-ence happens at the border between the known and the unknown. Standing on the shoulders of giants, we peer into the darkness with eyes opened not in fear but in wonder.
Professor Brian Cox – in Wonders of the Universe

X

XI
Acknowledgements
This study was carried out in the Neurosurgery of Neuro Center, Kuopio University Hospital and the Institute of Clinical Medicine – Neurology and Neurosurgery, University of Eastern Finland during the years 2009–2016. I am grateful for the possibility to have studied and worked under the Doctoral Programme of Clinical Research at the Doctoral School of the University of Eastern Finland. Numerous people have helped me during this process, and I would like to thank them for their support.
I wish to express my deepest gratitude to my principal supervisor Docent Ville Leinonen. Your end-less enthusiasm and energy was crucial in all stages of this thesis, from my early years as a medical student to my current assignment as a resident physician. Without your endless dedication and support, completion of this thesis would have been impossible. The way you find time on top of all your responsibilities to help me in things big and small was indispensable. Your ability to connect clinical and scientific work is truly inspirational.
Furthermore, I wish to express my warmest gratitude to my co-supervisors Docent Anne Koivisto and Professor Mikko Hiltunen. In addition to the encouragement you gave me, I am grateful for the deep scientific understanding and knowledge that were irreplaceable during this process. It has been a privilege to make this thesis under your supervision.
I want to express my gratitude to Professor Juha E. Jääskeläinen. As well as teaching me the art of scientific writing, your dedication and interest to the whole research project has been inspiring. I am in awe of your ability of answering emails within five minutes – at any time of the day.
I greatly appreciate and express my sincere gratitude to the reviewers of this doctoral thesis Docent Leena Kivipelto and Docent Pauli Helén. Your valuable comments improved this thesis consider-ably. I wish to thank Mrs Lisa Angela Stango for the linguistic revision of the thesis.
I have been privileged to work with many talented people within the Kuopio NPH and Early AD group: Seppo Helisalmi, Juhani A. A. Mölsä, Jaana Rummukainen, Irina Alafuzoff, Sakari Savo-lainen, Hilkka Soininen, Jaakko Rinne, Miikka Lumela, Ossi Nerg, Toni T. Seppälä, Sanna-Kaisa Herukka, Lakshman Puli, Henrik Zetterberg, Hanna-Mari Niskasaari, Timo Niskasaari, Jussi Pihla-jamäki, Tuomas Rauramaa, Maria Kojouhkova, Antti Luikku and Antti Junkkari. This thesis would not exist without your massive contribution. I wish to thank Marita Voutilainen and Seija Kasanen for all the practical help during this project.
This thesis was written mainly on my spare time during medical studies, military service, and sub-sequent clinical work. I am thankful for all the support and encouragement I got from my fellow medical students, brothers-in-arms, and collegues.
Especially, I want to acknowledge my two comrades of our dynamic trio in medical school, Alise and Mikko. There are no words to describe the importance that our friendship has had in my life. From my time in the military service, I want to thank Varis (the gamer), Pyykkönen (the dentist), Pekonen (the player), Saarijärvi (the short one), and Tuohino (the one with many names). Although I have been busy with my life lately, some friendships have begun in Lahti the Business City even before this research project. I wish to thank Arttu from elementary school (who thought my name is Obo), Tuomas Helin from the same era (with whom I skied), and Jani from high school (whose best man I was honored to be). It has been a joy to share many good memories and moments in these years with all of you.
Foremost, I wish to thank my parents Sirpa and Arto as well as my sister Elli. Your kindness, sym-

XII
pathy, and intelligence have been most precious to me. The compassion, support, and everything you have done and given to me since my childhood is the reason why I was able to start and finish this project. Finally, I wish to express my gratitude to my significant other, Erika. Your help and the time I have been able to spend with you have been invaluable. You are the world to me.
14th of February 2016, in Lahti
Okko T. Pyykkö
This work was supported by grants from the Finnish Medical Foundation, Kuopio University Hos-pital, the A. A. Laaksonen Fund of The Northern Savo Regional Fund of the Finnish Cultural Foun-dation, Aino and Akseli Koskimies Fund of the Finnish Medical Society Duodecim, Maire Taponen Foundation, University of Eastern Finland, Academy of Finland (JPND-EADB), VTR grant V16001 of Kuopio University Hospital, Sigrid Jusélius Foundation, the Strategic Funding of the University of Eastern Finland (UEF-Brain), FP7, Grant Agreement no 601055, VPH Dementia Research En-abled by IT VPH-DARE@IT., and BIOMARKAPD project in the JPND programme.

XIII
List of the original publications
This dissertation is based on the following original publications:
I Pyykkö OT, Nerg O, Niskasaari H-M, Niskasaari T, Koivisto AM, Hiltunen M, Pihlajamäki J, Rauramaa T, Kojoukhova M, Alafuzoff I, Soininen S, Jääskeläinen JE, Leinonen V. Incidence, comorbidities, and mortality in idiopathic normal pressure hydrocephalus. Submitted manuscript.
II Pyykkö OT, Helisalmi S, Koivisto AM, Mölsä JA, Rummukainen J, Nerg O, Alafuzoff I, Savolainen S, Soininen H, Jääskeläinen JE, Rinne J, Leinonen V, Hiltunen M. APOE4 predicts amyloid-β in cortical brain biopsy but not idiopathic normal pressure hydrocephalus. Journal of Neurology, Neurosurgery, and Psychiatry 83: 1119–24, 2012.
III Pyykkö OT, Lumela M, Rummukainen J, Nerg O, Seppälä TT, Herukka SK, Koivisto AM, Alafuzoff I, Puli L, Savolainen S, Soininen H, Jääskeläinen JE, Hiltunen M, Zetterberg H, Leinonen V. Cerebrospinal fluid biomarker and brain biopsy findings in idiopathic normal pressure hydrocephalus. PLOS ONE 9:e91974, 2014.
The publications have been adapted with permission from the copyright owners.

XIV

XV
Contents
1 INTRODUCTION 1
2 REVIEW OF THE LITERATURE 32.1 Definition of idiopathic normal pressure hydrocephalus (iNPH) 32.2 Clinical features of iNPH 32.3 Epidemiology of iNPH 3
2.3.1 Incidence 32.3.2 Prevalence 4
2.4 Pathophysiology of iNPH 42.4.1 Cerebrospinal fluid circulation and hydrocephalus 42.4.2 Pathophysiological theories 4
2.5 Genetic background of iNPH 82.5.1 Familial occurence 82.5.2 Apolipoprotein E ε4 as a potential risk factor 102.5.3 Other potential genetic risk factors 12
2.6 Diagnostics of iNPH 122.6.1 Diagnostic criteria 122.6.2 Neuroradiology 152.6.3 Neuropsychology 162.6.4 Tests of CSF circulation dynamics 162.6.5 Diagnosis of iNPH post mortem 162.6.6 Differential diagnosis 172.6.7 CSF biomarkers as potential future diagnostic tools 18
2.7 Comorbidities in iNPH 182.7.1 Degenerative brain disease 182.7.2 Vascular disease and type 2 diabetes mellitus 192.7.3 Other comorbidities 19
2.8 Treatment of iNPH 202.9 Prognosis of iNPH 20
2.9.1 Natural course 202.9.2 Outcome of treatment 202.9.3 Mortality and causes of death 21
3 AIMS OF THE STUDY 25
4 INCIDENCE, COMORBIDITIES, AND MORTALITY IN IDIOPATHIC NORMAL PRESSURE HYDROCEPHALUS 27
4.1 Abstract 274.1.1 Objective 274.1.2 Methods 274.1.3 Results 274.1.4 Conclusions 27
4.2 Introduction 274.3 Methods 28
4.3.1 Kuopio NPH Registry and protocol 284.3.2 Participants 284.3.3 Shunt treatment and shunt response 294.3.4 Comorbidities and causes of death 294.3.5 Immunohistochemistry and histological evaluation of brain biopsy 294.3.6 Statistical analysis 294.3.7 Ethical issues 29

XVI
4.4 Results 294.4.1 Study population 294.4.2 Catchment population and the incidence of iNPH 314.4.3 Vascular disease and T2DM in iNPH 314.4.4 Brain biopsy findings 324.4.5 Mortality and causes of death in iNPH 32
4.5 Discussion 35
5 APOE4 PREDICTS AMYLOID-Β IN CORTICAL BRAIN BIOPSY BUT NOT IDIOPATHIC NORMAL PRESSURE HYDROCEPHALUS 37
5.1 Abstract 375.1.1 Objective 375.1.2 Methods 375.1.3 Results 375.1.4 Conclusions 37
5.2 Introduction 375.3 Methods 38
5.3.1 Catchment population of the Kuopio University Hospital 385.3.2 Kuopio NPH Registry 385.3.3 Participants 385.3.4 Shunt treatment and shunt response 405.3.5 Final clinical diagnosis 405.3.6 Immunohistochemical staining and histological evaluation of brain biopsy 415.3.7 APOE genotyping 415.3.8 Statistical analysis 415.3.9 Ethical issues 41
5.4 Results 415.4.1 APOE and shunt responsive iNPH 415.4.2 APOE and shunt nonresponsive iNPH 415.4.3 APOE and Aβ plaques in brain biopsy 425.4.4 APOE and final clinical diagnosis of AD 42
5.5 Discussion 44
6 CEREBROSPINAL FLUID BIOMARKER AND BRAIN BIOPSY FINDINGS IN IDIOPATHIC NORMAL PRESSURE HYDROCEPHALUS 47
6.1 Abstract 476.1.1 Objective 476.1.2 Methods 476.1.3 Results 476.1.4 Conclusions 47
6.2 Introduction 476.3 Methods 48
6.3.1 Ethics statement 486.3.2 Kuopio NPH Registry and protocol 496.3.3 Participants 496.3.4 Immunohistochemistry and histological evaluation 516.3.5 APOE genotyping 516.3.6 CSF samples and biomarker analyses 516.3.7 Statistical analysis 51
6.4 Results 516.4.1 Aβ and sAPP isoforms 516.4.2 Proinflammatory cytokines 576.4.3 Biomarkers of neuronal damage 57
6.5 Discussion 58

XVII
7 GENERAL DISCUSSION 61
8 CONCLUSIONS 63
9 REFERENCES 65

XVIII

XIX
AbbreviationsACE/ACE Angiotensin I converting enzyme gene/proteinACZ AcetazolamideAD Alzheimer’s diseaseAβ Amyloid betaAβ38/40/40 Amyloid beta isoprotein, length 38/40/42 amino acidsAPOE/apoE Apolipoprotein E gene/proteinAPP Amyloid precursor proteinBBB Blood–brain barrierCBF Cerebral blood flowCNS Central nervous systemCSF Cerebrospinal fluidCT Computed tomographyDESH Disproportionately enlarged subarachnoid space hydro- cephalusDLB Dementia with Lewy bodiesELD External lumbar drainageETINPH Essential tremor–idiopathic normal pressure hydrocepha- lus syndromeETV Endoscopic third ventriculos- tomyHbA1c Haemoglobin A1cHPτ Hyperphosphorylated tau proteinHSPG Heparan sulfate proteoglycanICP Intracranial pressureIL InterleukiniNPH Idiopathic normal pressure hydrocephalus
KUH Kuopio University HospitalLPS Lumboperitoneal shuntMBP Myelin basic proteinMCP-1 Monocyte chemoattractant protein-1MMSE Mini-Mental State Examina- tionMRI Magnetic resonance imagingNFL Neurofilament light proteinNPH Normal pressure hydrocepha- lusp-tau 181 Tau phosphorylated at threo- nine 181PD Parkinson’s diseasePDD Parkinson’s disease dementiaPET Positron emission tomographysAPPα/β Soluble amyloid precursor protein alpha/betaSFMBT1 Scm-like with four MBT do- mains 1 genesNPH Secondary normal pressure hydrocephalusSPECT Single-photon emission com- puted tomographyT2DM Type 2 diabetes mellitusTf-1/Tf-2 Transferrin-1/transferrin-2 isoform ratioTNF-α Tumor necrosis factor-alphaVAS Ventriculoatrial shuntVCI Vascular cognitive impairmentVLDL Very low-density lipoproteinVPS Ventriculoperitoneal shunt

XX

1
1 Introduction
In 1964 doctor Salomon Hakim described a syndrome of symptomatic hydrocephalus with normal cerebrospinal fluid (CSF) pressure in his thesis Some Observations on C.S.F. Pressure. Hydrocephalic Syndrome in Adults with “Normal” C.S.F. Pressure (1). The following year Hakim published his find-ings with Raymond D. Adams in the scientific papers (2, 3). Despite the absence of elevated pres-sure observed in the CSF system of the presented patients, a diversion of CSF with an implantable shunt device corrected this condition (2). While Hakim’s thesis was the first one to identify normal pressure hydrocephalus (NPH) as an independent treatable syndrome, case reports of patients with similar clinical features and findings had been reported by Foltz and Ward in 1956 (4) and McHugh in 1964 (5). NPH is characterized by a triad of symptoms: gait disorder, cognitive impairment, and urinary incontinence in adult patients with findings of communicating hydrocephalus in a neuroradiologi-cal study of the brain and CSF pressure within normal range (2). Secondary NPH occurs after head trauma, subarachnoid haemorrhage, or other insult to the brain (6). In contrast, when no such pre-disposing factors are identified, the syndrome is described as primary or idiopathic NPH (iNPH) (7). The differential diagnosis of iNPH includes Alzheimer’s disease (AD), Parkinson’s disease, and cerebrovascular disease (7). Arterial hypertension is the most common comorbidity in persons with iNPH and the burden of cardiovascular risk factors is higher compared to the general popu-lation (8, 9). Unlike the differential diagnostic disorders, iNPH is an uncommon syndrome with a reported incidence between 0.5–5.5 cases per 100 000 inhabitants a year (10–15). Although iNPH is considered to be sporadic in nature, a number of case reports of familial iNPH has emerged (16–21), supporting the potential genetic predisposition. Some previous studies have suggested a possibility of similar genetical background in iNPH and AD. An overrepresenta-tion of the ε4 allele of the apolipoprotein E (APOE) gene was noted in one small study (22) and a better response to shunt treatment in patients with the ε3/ε3 genotype compared to patients with other genotypes was reported in another paper (23). Diagnostic tests of CSF circulation dynamics are currently in the clinical practice of iNPH (7). In contrast, several potential CSF biomarkers have been proposed (24), but are not included in the cur-rent diagnostic criteria. In AD, determing CSF levels of amyloid beta 42 (Aβ42), hyperphosphoryl-ated tau (HPτ) 181, and total tau are used as supplemental diagnostic tests (25), and can be utilized to differentiate AD from iNPH (26). During the six decades after Hakim’s and Adams’ seminal publication, a significant number of papers studying iNPH have been published. While our understanding of the special clinical problem of iNPH has expanded, many questions remain unanswered. This doctoral thesis focuses on a cohort of patients from a defined geographical area refered to the Kuopio University Hospital (KUH) as suspected iNPH patients from Middle and Eastern Finland from 1993 to 2010. In addi-tion, association of APOE genotype and various CSF biomarkers with AD and cortical brain biopsy findings are explored.

2

3
2 Review of the literature
2.1 DEFINITION OF IDIOPATHIC NORMAL PRESSURE HYDROCEPHALUS (INPH)
Idiopathic normal pressure hydrocephalus (iNPH) is typically defined as a slowly progressive clinical syndrome of gait disturbance, cognitive deterioration, and urinary incontinence in the el-derly with a normotensive impairment of cerebrospinal fluid (CSF) circulation in conjunction with ventricular dilation in the absence of identified predisposing factors (27). While this is the standard definition of iNPH given in most papers, interestingly, there is currently no consensus statement on the definition in scientific literature.
2.2 CLINICAL FEATURES OF INPH
Principally, an impairment of gait, cognition, and urinary continence define the classic triad of symptoms of NPH (2) including the idiopathic type (28). Gait disturbance is the most frequent symptom in iNPH with a reported frequency between 91–100% in larger series (6, 29–32). Papers focusing on the gait analysis of iNPH describe the following frequent abnormalities: slowness, dis-equilibrium, decreased and variable stride length, decreased foot-to-floor clearance, broad-based, arrestments (freezing), and difficulties in turning around (dyspraxia) (33–35). Atypically, paratonic rigidity with brisk deep tendon reflexes and Babinski sign have sometimes been observed (33). Cognitive deterioration is present in 78–88% of the iNPH patients at the time of the diagnosis (6, 29–31). Studies of neuropsychological profile of iNPH have shown impairment in several ar-eas, notably in psychomotor speed, executive functions, attention and concentration, memory and learning, visuospatial functions, and dexterity (36–41). Clinically, the neuropsychological profile of iNPH bears a resemblance to dementia of the subcortical type (37). Urinary incontinence is the least common symptom of the triad with a frequency of 60–90% in iNPH patients (6, 29–31). In iNPH patients, lower urinary tract symptoms include storage symp-toms with urinary urgency, nocturia, and urgency incontinence being the most frequent ones; and voiding symptoms with retardation in initiating urination and prolongation/poor flow reported as the most common of the voiding symptoms (42). Urodynamic abnormalities seen in iNPH are indicative of detrusor overactivity of the bladder, and primarily explained by an exaggerated mic-turition reflex and frontal lobe hypoperfusion as the proposed mechanisms (42). Importantly, even though considered to be one of the defining features of iNPH, a complete triad of symptoms is observed only in half of the iNPH patients (6, 30, 31). Additional potential symptoms of iNPH, such as hypokinetic motor deficit of the upper extremities (43), headache (44), and psychiatric symptoms of apathy, anxiety, and depression (45, 46), have also been presented in the literature.
2.3 EPIDEMIOLOGY OF INPH
2.3.1 IncidenceGenerally regarded as an uncommon disease, iNPH has been in focus only in few epidemiological studies. Hospital-based studies of iNPH in the Netherlands (10), Sweden (12), Norway (14), and the United States of America (15) have reported an incidence of 0.5–1.2 shunted iNPH patients per 100,000 inhabitants per year, while in Germany an estimated annual incidence of 1.2 iNPH patients considered for surgical intervention per 100,000 inhabitants was reported in a survey-based study (11) (Table 1). An American study reported an age-specific incidence of 5.8 cases of noncongenital hydrocephalus without predisposing intracranial bleed in computed tomography (CT) scan per 100,000 inhabitants aged 65 or older (47), while a Japanese study presented an annual incidence

4
of 1.2 possible iNPH patients per 1000 inhabitants aged 70 or older (48) (Table 1). The diagnosis of iNPH is relatively rare in the total population, whereas it is markedly more common in the elderly. The only true population-based epidemiological study of iNPH was conducted in Norway (13). Derived from prevalence data, a minimum incidence of 5.5 probable iNPH patients per 100,000 in-habitants per year was estimated, which indicates iNPH to be an underdiagnosed or undertreated disease as most hospital-based studies reported considerably lower incidence figures (13) (Table 1). In contrast, a single paper reported no diagnosed iNPH patients in a population-based dementia study of five years in the United States of America (49) (Table 1).
2.3.2 PrevalenceLogically, most papers exploring the prevalence of iNPH study a defined population of elderly in a single community. Prevalence between 0.01 –2.9% of the elderly population has been established in studies carried out in San Marino (50), Germany (51), Singapore (52), Spain (53), Japan (48, 54–56), Turkey (57), and Sweden (58) (Table 1). In total population, probable iNPH has a prevalence of 21.9 per 100,000 inhabitants as reported in a Norwegian study (13) (Table 1).
2.4 PATHOPHYSIOLOGY OF INPH
2.4.1 Cerebrospinal fluid circulation and hydrocephalusCSF is formed in the choroid plexus, the ependyma, and the parenchyma of the ventricular system of the brain (59–61). Eighty percent of the CSF formation occurs in the choroid plexus, which com-prises 60% of the total internal surface area of the ventricles (60). CSF is formed at the rate of 20 ml/h or 500 ml/day (60) by means of passive filtration across the capillary endothelium and regulated secretion across the choroidal epithelium (61). The current classical concept of the CSF circulation, or the third circulation, was introduced by Harvey Cushing in 1926 (61, 62). CSF formed in the lateral ventricles flows through the paired in-terventricular foramina to the third ventricle and through the cerebral aqueduct to the fourth ven-tricle (59). The fluid exits the ventricular system through the median aperture and lateral apertures of the fourth ventricle into the subarachnoid space or drains through the obex to the spinal canal (59). From the subarachnoid space, CSF is absorbed into the blood circulation via the villi of the arachnoid granulations projecting into the venous sinuses (59–61). In recent years, this traditional view of CSF circulation has been challenged and more complex models of CSF turnover have been presented (61, 63). If the rate of CSF formation and absorption is in equilibrium, the total volume of CSF in adults is 90–330 ml (60, 61, 64) with 50–100 ml in the spinal CSF space (64, 65). In the pathological states of the CSF turnover, the formation and absorption of CSF is in a state of disequilibrium leading to a disproportionally large volume of CSF intracranially, or hydrocephalus. Potential causative fac-tors include an obstruction of CSF flow within the ventricular system or at the subarachnoid space (decreased absorption), or rarely a papilloma of the choroid plexus (increased formation) (59).
2.4.2 Pathophysiological theoriesThe cause of iNPH is unknown and the pathogenesis is poorly understood. Originally, Hakim and Adams hypothesized that an increase of intracranial pressure (ICP) precedes the normal pressure state dilating the ventricles and as the cause of the hydrocephalus is relieved, a new balance of CSF formation and absorption develops lowering the pressure but leaving the ventricles dilated (2). Despite the normal pressure conditions, more force is applied to the brain as the ventricular surface area is larger compared to the initial hydrocephalic phase (force = pressure × area), ultimately caus-ing the neurologic symptoms by means of tangential shearing forces affecting the paracentral fibers of the corona radiata (2, 66). A myriad number of pathophysiological theories for iNPH have been subsequently proposed. Currently most theories of the pathogenesis in iNPH stress the pathological CSF dynamics, the role of vascular changes, or metabolic changes related to CSF stagnation. As the name suggests, the mean CSF pressure in iNPH is within normal range of 5–15 mmHg (2). Nevertheless, several disturbances of CSF dynamics have been reported. Intermittent rises in the CSF pressure (B-waves) (67) can be observed in a prolonged CSF pressure monitoring in iNPH

5
(68) especially during sleep (69). In addition, abnormal ICP pulsatility has been associated with positive response to shunt treatment (70). Magnetic resonance imaging (MRI) studies have dem-ostrated CSF flow hyperdynamics, such as increased aqueductal stroke volume (71, 72). A reduced global cerebral blood flow (CBF) in iNPH was reported originally in 1969 (73, 74) and has been verified since in numerous studies (75). Additionally, a reduction in regional CBF and the presence of deep white matter ischaemia is well-documented in iNPH (75–80). Whether the abnormal cerebral circulation and vascular changes are a cause or effect of the ventricular dilation and iNPH is under debate. Some authors have suggested that dilated ventricles stretch the anterior cerebral arteries over the corpus callosum (81) and the pathologic periventricular CSF absorption and the increase of intraparenchymal pressure (82) leads to the reduced blood flow causing is-chaemia. In contrast, it has been postulated that periventricular ischaemic injury causes decreased tensile strength in brain tissue and subsequently leads to ventriculomegaly (75, 76). A more recent theory depicts arterial circulatory changes as an epiphenomena to the lowered intracranial compli-ance due to venous hypertension, which leads to decreased absorption of CSF via the arachnoid granulations in the superior sagittal sinus (71). Another theory suggests that iNPH is preceded by benign external hydrocephalus in childhood and followed by a loss of compensatory mechanisms after deep white matter ischaemia in adulthood, consequently leading to symptomatic hydroceph-alus (72). Frequent comorbid vascular diseases reported in iNPH patients can be seen as supportive features for the vascular theories of iNPH (see 2.7.2 Vascular risk factors and vascular disease). A single theory recognizes the presence of disturbed CSF dynamics and ischaemic changes, but hypothesizes that the stagnation and reduced turnover of CSF leads to the accumulation of amyloid beta (Aβ) and tau (τ) proteins and potentially other toxic metabolic products in the interstitial fluid of the brain parenchyma, subsequently causing dementia (83–86). In contrast to this theory, most patients with iNPH (with or without dementia) do not show Aβ or tau proteins in brain biopsy (87, 88). In another theory, metabolic disturbance was proposed to be decoupled from CSF dynamics at a certain ‘point of no return’ of the pathogenesis of iNPH (89).

6
Study Country Region Incidence Prevalence NotesCasmiro et al. 1989(50)
San Marino Not specified – 2/396 (0.5%) of 67–87 yo inhabitants
67, 72, 77, 82, and 87 yo inhabitants evaluated
Vanneste et al. 1992(10)
The Netherlands City of Amsterdam 0.5 / 100,000 / year – Shunted iNPH patients (n = 127)
Alexander et al. 1995(47)
United States of America
Seattle,Washington
5.8* / 100,000 / year – Noncongenital hydrocephalus without predisposing intracranial bleed in CT scan (n = 8)
Trenkwalder et al. 1995(51)
Germany Two Bavarian villages – 4/982 (0.4%) of >65 yo inhabitants
Only inhabitants with basal ganglia symptoms (45/982) evaluated as a part of parkinsonism study
Krauss and Halve 2004(11)
Germany Several 1.2 / 100,000 / year – Estimation. iNPH patients considered for surgical intervention
Tan et al. 2004(52)
Singapore Several – 1/14,906 (0.01%) of ≥50 yo inhabitants
Only inhabitants with positive Parkinson disease screening questionnaire (1246/14906) evaluated
Tisell et al. 2005(12)
Sweden Nationwide 0.9 / 100,000 / year – Shunted iNPH patients (n = 243)
Knopman et al. 2006(49)
United States of America
Rochester, Minnesota 0 – No NPH cases were reported during 1990–1994 in the study population
Gascon-Bayarri et al. 2007(53)
Spain El Prat de Llobregat, Catalonia
– 1/1754 (0.06%) of ≥70 yo inhabitants
Only inhabitants with dementia (165/1754) evaluated
Brean and Eide 2008(13)
Norway Vestfold County 5.5 / 100,000 / year 48/219,478 (21.9 / 100,000) Probable iNPH. Incidence calculated from the prevalence data.
Hiraoka et al. 2008(54)
Japan Town of Tajiri – 5/170 (2.9%) of ≥65 yo inhabitants
Possible iNPH
Arslantaş et al. 2009(57)
Turkey Middle Anatolia – 1/3200 (0.03%) of ≥55 yo inhabitants
Only inhabitants with dementia (262/3200) evaluated
Brean et al. 2009(14)
Norway Nationwide 1.1 / 100,000 / year – Shunted iNPH patients (n = 252)
Iseki et al. 2009(55)
Japan Town of Takahata & City of Sagae
– 4/790 (0.5%) of >61 yo inhabitants
Possible iNPH
Tanaka et al. 2009(56)
Japan Town of Tajiri – 7/497 (1.4%) of ≥65 yo inhabitants
Possible iNPH
Klassen and Ahlskog 2011(15)
United States of America
Olmsted County, Minnesota
1.2 / 100,000 / year – Shunted for iNPH (n = 13)(3.7 / 100 000 / year clinically suspected iNPH)
Iseki et al. 2014(48)
Japan Town of Takahata 1.2** / 1000 / year 3/211 (1.4%) of 80 yo inhabitants
Possible iNPH
Jaraj et al. 2014(58)
Sweden City of Gothenburg – 26/1,238 (2.1%) of ≥70 yo inhabitants
Probable iNPH
*Age-specific incidence (≥65 yo inhabitants)**Age-specific incidence (≥70 yo inhabitants)CT = computed tomography
Table 1. Incidence and prevalence of idiopathic normal pressure hydrocephalus (iNPH) in literature.

7
Study Country Region Incidence Prevalence NotesCasmiro et al. 1989(50)
San Marino Not specified – 2/396 (0.5%) of 67–87 yo inhabitants
67, 72, 77, 82, and 87 yo inhabitants evaluated
Vanneste et al. 1992(10)
The Netherlands City of Amsterdam 0.5 / 100,000 / year – Shunted iNPH patients (n = 127)
Alexander et al. 1995(47)
United States of America
Seattle,Washington
5.8* / 100,000 / year – Noncongenital hydrocephalus without predisposing intracranial bleed in CT scan (n = 8)
Trenkwalder et al. 1995(51)
Germany Two Bavarian villages – 4/982 (0.4%) of >65 yo inhabitants
Only inhabitants with basal ganglia symptoms (45/982) evaluated as a part of parkinsonism study
Krauss and Halve 2004(11)
Germany Several 1.2 / 100,000 / year – Estimation. iNPH patients considered for surgical intervention
Tan et al. 2004(52)
Singapore Several – 1/14,906 (0.01%) of ≥50 yo inhabitants
Only inhabitants with positive Parkinson disease screening questionnaire (1246/14906) evaluated
Tisell et al. 2005(12)
Sweden Nationwide 0.9 / 100,000 / year – Shunted iNPH patients (n = 243)
Knopman et al. 2006(49)
United States of America
Rochester, Minnesota 0 – No NPH cases were reported during 1990–1994 in the study population
Gascon-Bayarri et al. 2007(53)
Spain El Prat de Llobregat, Catalonia
– 1/1754 (0.06%) of ≥70 yo inhabitants
Only inhabitants with dementia (165/1754) evaluated
Brean and Eide 2008(13)
Norway Vestfold County 5.5 / 100,000 / year 48/219,478 (21.9 / 100,000) Probable iNPH. Incidence calculated from the prevalence data.
Hiraoka et al. 2008(54)
Japan Town of Tajiri – 5/170 (2.9%) of ≥65 yo inhabitants
Possible iNPH
Arslantaş et al. 2009(57)
Turkey Middle Anatolia – 1/3200 (0.03%) of ≥55 yo inhabitants
Only inhabitants with dementia (262/3200) evaluated
Brean et al. 2009(14)
Norway Nationwide 1.1 / 100,000 / year – Shunted iNPH patients (n = 252)
Iseki et al. 2009(55)
Japan Town of Takahata & City of Sagae
– 4/790 (0.5%) of >61 yo inhabitants
Possible iNPH
Tanaka et al. 2009(56)
Japan Town of Tajiri – 7/497 (1.4%) of ≥65 yo inhabitants
Possible iNPH
Klassen and Ahlskog 2011(15)
United States of America
Olmsted County, Minnesota
1.2 / 100,000 / year – Shunted for iNPH (n = 13)(3.7 / 100 000 / year clinically suspected iNPH)
Iseki et al. 2014(48)
Japan Town of Takahata 1.2** / 1000 / year 3/211 (1.4%) of 80 yo inhabitants
Possible iNPH
Jaraj et al. 2014(58)
Sweden City of Gothenburg – 26/1,238 (2.1%) of ≥70 yo inhabitants
Probable iNPH

8
2.5 GENETIC BACKGROUND OF INPH
2.5.1 Familial occurencePapers exploring the genetics of iNPH are scarce, even though genetic predisposition seems con-ceivable, as there are several reported cases of possible familial iNPH. Three case studies have reported siblings developing a shunt-responsive iNPH with no identified contributing environ-mental factors (16–18). In addition, an overrepresentation of iNPH symptoms (7.1%) in the relatives of iNPH patients compared to control relatives (0.7%) have been noted and an iNPH pedigree with four affected individuals was published by the same group (20). The most recent report of potential familial iNPH presents a total of four cases in two generations with atypical clinical features (21). The largest iNPH pedigree in literature was reported in Japan with four cases of diagnosed iNPH and four individuals with suspected NPH in three generations (Figure 1) (19). Author hypothesized that in the familial subgroup of iNPH, the syndrome is inherited in an autosomal-dominant fashion (19).
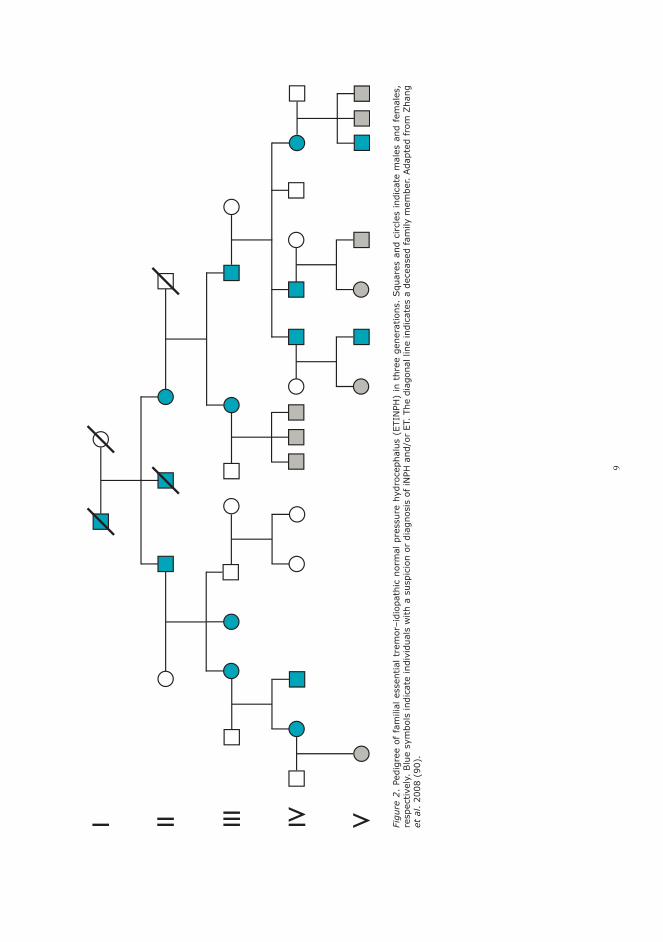
A novel heritable syndrome of essential tremor–idiopathic normal pressure hydrocephalus (ETINPH) was introduced in 2008 (90). In five generations, the affected members of the kindred developed essential tremor at the ages of 16–44 years and later iNPH at the onset age of >65 years in an autosomal-dominant pattern (Figure 2) (90). In the genetical analyses, neither copy number changes nor susceptible genetic defects linked to established loci associated with tremor and Par-kinson’s disease (PD) were identified (90). In the following genome-wide linkage scan, the locus of ETINPH gene was mapped to chromosome 19q12 –13.31, which comprises several potential neu-ronal genes (91).
Figure 1. Pedigree of familial idiopathic normal pressure hydrocephalus (iNPH) in three generations. Squares and circles indicate males and females, respectively. Blue symbols indicate individuals with a suspicion or diagnosis of iNPH. The diagonal line indicates a deceased family member. Adapted from Takahashi et al. 2011 (19).
9
Figu
re 2
. Pe
digr
ee o
f fa
mili
al e
ssen
tial t
rem
or–i
diop
athi
c no
rmal
pre
ssur
e hy
droc
epha
lus
(ETI
NPH
) in
thr
ee g
ener
atio
ns.
Squ
ares
and
circl
es in
dica
te m
ales
and
fem
ales
, re
spec
tivel
y. B
lue
sym
bols
indi
cate
indi
vidu
als
with
a s
uspi
cion
or
diag
nosi
s of
iNPH
and
/or
ET.
The
diag
onal
line
indi
cate
s a
dece
ased
fam
ily m
embe
r. Ada
pted
fro
m Z
hang
et
al.
2008
(90
).

10
2.5.2 Apolipoprotein E ε4 as a potential risk factorApolipoprotein E (apoE) is a 299 amino acid glycoprotein with a relative molecular weight of 34 kD (92). The main function of apoE in lipid metabolism is redistribution of lipids among cells of dif-ferent organs and within an organ or tissue (92, 93). At molecular level, the function of apoE can be derived from its structure: the globular N-terminus domain (residues 1–191) contains the binding site for the low-density lipoprotein (LDL) receptor and the helical C-terminus (residues 216–299) contains the major lipid binding site (94). At cellular level, in addition to the general cholesterol redistribution, apoE participates in several neurobiological processes (93, 95). The gene coding for apoE is located in the long arm of chromosome 19 (Figure 3) (96–98). The APOE gene consists of 3.6 kilobases with four exons and three introns (Figure 3) (95). Primarily, APOE is expressed in the liver and central nervous system (CNS) (cerebral cortex, hippocampus, cerebellum, medulla) in addition to the adrenal gland, testis, skin, kidney, spleen, adipose tissue, and various tissue macrophages (93, 95). The polymorphism of the APOE was first discovered in 1977 utilizing isoelectric focusing (99) and expanded further with two-dimensional electrophoresis (100). Altogether three different apoE isoproteins are coded by three different APOE alleles: ε2, ε3, and ε4 (101). In the diploid karyotope, six different genotypes are possible: three homozygous (ε2/ε2, ε3/ε3, and ε4/ε4) and three heterozy-gous (ε2/ε3, ε2/ε4, and ε3/ε4) leading to six corresponding phenotypes (E2/E2, E3/E3, E4/E4, E2/E3, E2/E4, and E3/E4) (101). ApoE3 is accepted as the common form and apoE2 and apoE4 as mutations differing from apoE3 at positions 112 or 158 by a single amino acid (Figure 3) (93, 102). Both sites of variation reside at the N-terminus domain of apoE (Figure 3). The structural dissimilarity of apoE isoproteins reflects the variations in the interactions be-tween the apoE and LDL receptor, very low-density lipoprotein (VLDL) receptor, the apoE re-ceptor-2, and megalin (93). Isoform-specific differences in the binding affinity to heparan sulfate proteoglycans (HSPG) have been noted (93). These differences in molecular interactions translate to cellular neurobiology and pathology. Fundamentally, apoE2 and apoE3 are more neuroprotective than apoE4 (93, 103). Described functional impairments of apoE4 include reduced Aβ clearance, increased accumulation of neurofibrillary tangles, phosphorylation of tau, inhibition of neuronal outgrowth, impairment of neuronal plasticity, impairment of blood–brain barrier (BBB), reduced protection against oxidative stress, and increased neurotoxicity and neuroinflammation (93, 95, 103–105) (Figure 4). Due to the inferior functionality of apoE4, several neurological diseases have been associated with the presence of ε4 allele. The connection between APOE genotypes and AD was described in 1993 (106). APOE ε4 allele is the major genetic risk factor for AD – it is overrepresented and advances the onset of the disease (102, 107). In other neurological disorders such as head trauma, stroke and PD, an association of ε4 allele and unfavourable outcome have been reported (105). In addition to neurological diseases, APOE ε4 has been associated with cardiovascular diseases (108). In iNPH, an overrepresentation of ε4 allele in NPH patients (23.0%) compared to controls (6.9%) was reported in a small Italian study (22). No differences were reported in the distribution of the alpha 1-antichymotrypsin or presenillin 1 gene types in the same study (22). In the earlier cited Canadian case study (2.5.1 Familial occurence), both affected individuals were homozygous for the ε3 allele (18). In an Icelandic preliminary report, an association with APOE ε3/ε3 genotype and a favourable gait response to shunt surgery in iNPH patients (n = 15) was noted (23), however, no follow-up study was published.

11
1 2 3 4 5 6 7 8 9
1510 11 12 13 14 16 17 18
19 20 21 22 XY
19q13
5’ 3’
EXON 1 EXON 2 EXON 3 EXON 4
rs42935 rs7412
NH2 COOH
Receptorbinding
Lipidbinding
Hinge
112 158CysCysArg
CysArgArg
apoE2apoE3apoE4
Figure 3. Locus of the apolipoprotein E (APOE) gene in chromosome 19 and two sites of variation responsible for the three different apoE isoproteins. Adapted from Alzheimer Research Forum (98) and Kim et al. 2014 (102).

12
2.5.3 Other potential genetic risk factorsIn a Spanish study comprising 112 NPH patients and 124 controls, the polymorphism of the angio-tensin I converting enzyme (ACE) was investigated (109). ACE codes a significant enzyme of the renin–angiotensin–aldosterone system, which is a major contributor to blood pressure regulation (110). The polymorphism of the ACE gene is based on insertion (I) and deletion (D) alleles, leading to three different genotypes: I/I, D/D, I/D, of which, in particular, the D/D genotype has been as-sociated with cardiovascular diseases (110). The study found no differences in the distribution of the alleles between patients with NPH and controls, however, the D/D and I/D genotypes showed a weaker response to shunting compared to the I/I genotype (109). There are no other publications in the literature regarding the polymorphism of ACE in NPH. In a small study in Japan, a segmental copy number loss of the Scm-like with four MBT do-mains 1 gene (SFMBT1) was observed in four of the eight cases with features of iNPH on MRI, while in ten control subjects, no such finding was reported (111). Due to the small number of sub-jects and unusual setting, the finding requires further study with a larger cohort with diagnosed iNPH patients and healthy controls. If the finding is replicated in a larger setting, it may shed light on the pathogenesis of iNPH.
2.6 DIAGNOSTICS OF INPH
2.6.1 Diagnostic criteriaEvidence-based international guidelines for the diagnosis of iNPH were published by an independ-ent study group in 2005 (7). Based on signs and symptoms (see 2.2 Clinical features), brain imaging, and physiological tests, iNPH is classified into unlikely, possible, and probable categories accord-ing to the certainty of the diagnosis (Table 2) (7).
Mechanisms of injury
Amyloid betaplaque & neuro-fibrillary tangleaccumulation
Excitotoxicamino acids
Neuro-inflammation
Oxidativestress
NEURONAL DAMAGE
apoE2 & apoE3 apoE4
EFFECTIVE REPAIR AND PROTECTION POOR REPAIR AND PROTECTION
Figure 4. Mechanisms of neuronal injury and the effect of the different apolipoprotein E (apoE) isoforms. Adapted from Mahley and Huang 1999 (104) and Kim et al. 2009 (103).

13
Table 2. Classification of idiopathic normal pressure hydrocephalus (iNPH) into probable, possible, and unlikely categories according to the international diagnostic guidelines. Adapted from Relkin et al. 2005 (7).
Probable iNPHThe diagnosis of Probable iNPH is based on clinical history, brain imaging, physical findings, and physiological criteria.
I. HistoryReported symptoms should be corroborated by an informant familiar with the patient’s premorbid and current condition, and must include
a. Insidious onset (versus acute)b. Origin after age 40 yrc. A minimum duration of at least 3 to 6 mod. No evidence of an antecedent event such as head trauma, intracerebral hemorrhage,
meningitis, or other known causes of secondary hydrocephaluse. Progression over timef. No other neurological, psychiatric, or general medical conditions that are sufficient to
explain the presenting symptoms
II. Brain imagingA brain imaging study (CT or MRI) performed after onset of symptoms must show evidence of
a. Ventricular enlargement not entirely attributable to cerebral atrophy or congenital enlargement (Evans’ index >0.3 or comparable measure)
b. No macroscopic obstruction to CSF flowc. At least one of the following supportive features
1. Enlargement of the temporal horns of the lateral ventricles not entirely attributable to hippocampus atrophy
2. Callosal angle of 40 degrees or more3. Evidence of altered brain water content, including periventricular signal changes on
CT and MRI not attributable to microvascular ischemic changes or demyelination4. An aqueductal or fourth ventricular flow void on MRI
Other brain imaging findings may be supportive of an iNPH diagnosis but are not required for a probable designation
1. A brain imaging study performed before onset of symptoms showing smaller ventricular size or without evidence of hydrocephalus
2. Radionuclide cisternogram showing delayed clearance of radiotracer over the cerebral convexities after 48–72h
3. Cine MRI study or other technique showing increased ventricular flow rate4. A SPECT-acetazolamide challenge showing decreased periventricular perfusion that is
not altered by acetazolamide
III. ClinicalBy classic definitions, findings of gait/balance disturbance must be present, plus at least one other area of impairment in cognition, urinary symptoms, or both.
With respect to gait/balance, at least two of the following should be present and not be entirely attributable to other conditionsa. Decreased step heightb. Decreased step lengthc. Decreased cadence (speed of walking)d. Increased trunk sway during walkinge. Widened standing basef. Toes turned outward on walkingg. Retropulsion (spontaneous or provoked)h. En bloc turning (turning requiring three or more steps for 180 degrees)i. Impaired walking balance, as evidenced by two or more corrections out of eight steps
on tandem gait testing

14
With respect to cognition, there must be documented impairment (adjusted for age and educational attainment) and/or decrease in performance on a cognitive screening instrument (such as the Minimental State examination), or evidence of at least two of the following on examination that is not fully attributable to other conditions
a. Psychomotor slowing (increased response latency)b. Decreased fine motor speedc. Decreased fine motor accuracyd. Difficulty dividing or maintaining attentione. Impaired recall, especially for recent eventsf. Executive dysfunction, such as impairment in multistep procedures, working memory,
formulation of abstractions/similarities, insightg. Behavioral or personality changes
To document symptoms in the domain of urinary continence, either one of the following should be present
a. Episodic or persistent urinary incontinence not attributable to primary urological disorders
b. Persistent urinary incontinencec. Urinary and fecal incontinence
Or any two of the following should be presenta. Urinary urgency as defined by frequent perception of a pressing need to voidb. Urinary frequency as defined by more than six voiding episodes in an average 12-hour
period despite normal fluid intakec. Nocturia as defined by the need to urinate more than two times in an average night
IV. PhysiologicalCSF opening pressure in the range of 5–18 mmHg (or 70–245 mmH2O) as determined by a lumbar puncture or a comparable procedure. Appropriately measured pressures that are significantly higher or lower than this range are not consistent with a probable iNPH diagnosis.
Possible iNPHA diagnosis of Possible iNPH is based on historical, brain imaging, and clinical and physiological criteria
I. HistoryReported symptoms may
a. Have a subacute or indeterminate mode of onsetb. Begin at any age after childhoodc. May have less than 3 mo or indeterminate durationd. May follow events such as mild head trauma, remote history of intracerebral
hemorrhage, or childhood and adolescent meningitis or other conditions that in the judgment of the clinician are not likely to be causally related
e. Coexist with other neurological, psychiatric, or general medical disorders but in the judgment of the clinician not be entirely attributable to these conditions
f. Be nonprogressive or not clearly progressive
II. Brain imagingVentricular enlargement consistent with hydrocephalus but associated with any of the following
a. Evidence of cerebral atrophy of sufficient severity to potentially explain ventricular sizeb. Structural lesions that may influence ventricular size
Table 2 Continued.

15
Preceding the international diagnostic criteria, a set of guidelines were introduced in Japan in 2004 (112, 113). Originally, the Japanese guidelines followed the same scheme of diagnosis and the categories of possible and probable iNPH, but added a category of “definite iNPH”, if a positive response to shunting was observed (113). However, in the revision of the guidelines in 2012, a novel subgroup of iNPH with disproportionately enlarged subarachnoid space hydrocephalus (DESH) findings in the MRI, which negates the need for additional physiogical tests in these patients, was included in the guidelines (27).
2.6.2 NeuroradiologyHistorically, hydrocephalus was visualized with ventriculography (114) and more commonly with pneumoencephalography (115), where air injected to the CSF space created a contrast between the air-filled ventricles and the solid brain parenchyma. This was the golden standard of ventricular imaging until cross-sectional imaging modalities were introduced (116). In the anatomical cross-sectional imaging studies of the brain, e.g. CT and MRI, the contrast between the CSF and brain parenchyma is high and the ventricular size and the potential cause of the CSF flow obstruction can be readily assessed noninvasively (116). The pivotal neuroradiological finding in iNPH is the enlargement of the brain ventricles with-out the presence of a causative factor such as cerebral atrophy, congenital enlargement, or mac-roscopic obstruction of the CSF flow (7). Other anatomical findings of iNPH seen in radiographic imaging include enlarged temporal horns (not attributable to hippocampus atrophy) and lateral sulci, callosal angle of 40 degrees or more, periventricular signal changes suggestive of altered water content of the brain, and tight subarachnoid space at high convexity (Table 2) (7, 27). A com-mon method of ventricular size quantification is the calculation of Evans’ index by dividing the maximal horizontal width of the frontal horns by the maximal width of the inner skull at the same level (Figure 5) (117).
III. ClinicalSymptoms of either
a. Incontinence and/or cognitive impairment in the absence of an observable gait or balance disturbance
b. Gait disturbance or dementia alone
IV. PhysiologicalOpening pressure measurement not available or pressure outside the range required for probable iNPH
Unlikely iNPH1. No evidence of ventriculomegaly2. Signs of increased intracranial pressure such as papilledema3. No component of the clinical triad of iNPH is present4. Symptoms explained by other causes (e.g. spinal stenosis)
CT = computed tomography; MRI = magnetic resonance imaging; CSF = cerebrospinal fluid; SPECT = single-photon emission computed tomography
Table 2 Continued.

16
2.6.3 NeuropsychologyAccording to the international diagnostic criteria, at least two of the following findings must be present in neuropsychological examination, if cognitive deterioration is attributed to iNPH: psy-chomotor slowing (increased response latency); decreased fine motor speed; decreased fine motor accuracy; difficulty dividing or maintaining attention; impaired recall, especially for recent events; executive dysfunction, such as impairment in multistep procedures, working memory, formulation of abstractions/similarities, insight; and behavioral or personality changes (Table 2) (7). Various cognitive tests, including Minimental State examination (118) for screening and more comprehensive neuropsychological test batteries, are utilized to differentiate iNPH-related cogni-tive deterioration from other diseases causing cognitive impairment (see 2.6.6 Differential diagno-sis and 2.7 Comorbidities). Wechsler Memory Scale logical memory subtest, Rey Auditory Verbal Learning Test, Trail Making B test, Rey-Osterrieth Complex Figure test, Line-Tracing test, Alzhei-mer’s Disease Assessment Scale, Wechsler Adult Intelligence Scale-Revised test, Stroop tests, and Grooved Pegboard test have been reported to be useful in the diagnostics and prognostication of iNPH (36, 38–41).
2.6.4 Tests of CSF circulation dynamicsIn probable iNPH the CSF opening pressure, as measured by a lumbar puncture or comparable method, must be within 5–18 mmHg (70–245 mmH2O) (Table 2) (7). Other supporting findings for iNPH in examinations of CSF physiology include sporadic rises in the CSF pressure in a direct ICP monitoring (67–69, 87, 119) and a low CSF outflow conductance in various infusion tests (119, 120). The predictive value of CSF removal via a lumbar puncture was first noted by Hakim (1, 2). Lumbar CSF bolus removal (tap test) and external lumbar drainage (ELD) reduce the CSF pressure and absorption (essentially creating a temporary shunt) by removing 40–50 ml and 300–720 ml of CSF, respectively (119, 121, 122). The specificity of the tap test is considered to be high (72–100%), but sensitivity low (26–62%) (119). In contrast, ELD is both sensitive (50–100%) and specific (60–100%), but requires hospitalization and the rate of complications is higher compared to the tap test (119).
2.6.5 Diagnosis of iNPH post mortemThere are no identified pathognomonic signs of iNPH in the autopsy. On gross examination of
A
B
Figure 5. Axial magnetic resonance imaging (MRI) scan demonstrating calculation of Evans’ index by dividing the maximal horizontal width of the frontal horns (a) by the maximal width of the inner skull (b). Evans’ index >0.3 is suggestive of hydrocepha-lus including idiopathic normal pressure hydroceph-alus (iNPH) (7).

17
the brain, dilated brain ventricles and possible fibrous thickening of the leptomeninges have been reported (143). Microscopic examination may reveal gaps in the ependymal lining, gliosis of the periventricular region, and ischaemic lesions in the deep white matter (143, 144).
2.6.6 Differential diagnosisAlzheimer’s disease (AD) was first described in 1907 by Alois Alzheimer (145). Characteristic neuro-pathological findings include cerebral Aβ plaques and neurofibrillary tangles composed of hyper-phosphorylated tau (HPτ) aggregates in elderly patients with cognitive deterioration (146, 147). Ac-cording to the amyloid cascade hypothesis, molecular and structural pathological changes precede the symptoms and clinical manifestation of the disease by decades (Figure 6) (148, 149). Common methods to evaluate the presence of accumulated amyloid in live persons include CSF sampling (see 2.6.7 CSF biomarkers as potential future diagnostic tools), positron emission tomography (PET) imaging (utilizing the 11C-labelled Pittsburgh compound B (150), [18F]flutemetamol (151), or [18F]florbetaben (152)) or rarely, brain biopsy (87, 153). Additionally, systemic inflammation and neuroinflammation may contribute to the pathogenesis of AD (154).
In a neuroradiological study, medial temporal lobe atrophy is the central finding in AD, and while AD patients may show ventricular enlargement, concommitant cortical atrophy is usually present in such cases (155–157). In AD, difficulties in learning and impairment of episodic memory or other cognitive domains such as visuospatial or linguistic skills are usually the early symptoms, whereas in iNPH, psychomotor slowing is typically present (7, 88, 158). In the progression of AD, extrapyramidal signs and symptoms may develop, however, typically in the more advanced stage of the disease, stance is usually narrower compared to iNPH (159). Vascular cognitive impairment (VCI) is an umbrella term, which includes a spectrum of cerebro-vascular diseases with cognitive symptoms from early mild cognitive impairment to late-stage de-mentia, and often presents in conjunction with AD in the elderly (160, 161). VCI can occur after stroke (cortical type) or due to ischaemia in the white matter (subcortical type), multiple small la-cunae infarcts, or infarct at a strategic site (160). Subcortical vascular degeneration with or without dementia is the most important subtype of VCI regarding the differential diagnosis of iNPH, as the
COGNITIVELY NORMAL MCI DEMENTIA
Normal
Abnormal
Biom
arke
r mag
nitu
de
Clinical disease stage
Amyloid betaTau-mediated neuronal injuryBrain structureMemoryClinical function
Figure 6. Biomarkers and progression of mild cognitive impairment (MCI) to Alzheimer’s disease (AD). AD-related pathology occurs years before manifestation of clinical symptoms. Adapted from Jack et al. 2010 (149).

18
gait, congnitive, and urinary symptoms bear a resemblance to iNPH (7, 160–162). In neuroimag-ing, ischaemic deep white matter lesions are the hallmark findings of subcortical VCI (160–162), however, such findings are also frequent in iNPH (9, 76, 82, 163–167). Gait disturbances and other iNPH-like symptoms may occur after stroke as well (7). Dementia with Lewy bodies (DLB) and Parkinson’s disease (PD) with (PDD) or without dementia share the disturbance of α-synuclein metabolism and formation of Lewy bodies, ultimately lead-ing to neurodegeneration (168). Clinically, DLB is characterized by neuropsychiatric symptoms, such as hallucinations and psychoses; adverse reaction to neuroleptic drugs; autonomic symptoms; REM-sleep disturbances; and cognitive impairment, which includes fluctuations in attention, exec-utive function, visuospatial function, language function, memory, and behavior (168). The majority of patients with DLB eventually develop extrapyramidal motor disorder defined by bradykinesia, gait disorder, and postural disturbances without major tremor (168). However, patients with PDD develop motor deficits and a unilateral tremor years before clear cognitive deterioration occurs (168). Differential diagnosis between Lewy body disorders (PD, PDD, DLB) and iNPH is based on differences between clinical features (2.2 Clinical features) and radiological findings of iNPH that are not associated with Lewy body disorders (2.6.2 Neuroradiology). Common diseases that may present with one or more of the iNPH symptoms (gait disorder, cognitive impairment, urinary incontinence) include frontotemporal dementia, traumatic brain in-jury, brain tumors, other hydrocephalic disorders, depression, spinal stenosis, and primary uro-logical disorders (7).
2.6.7 CSF biomarkers as potential future diagnostic toolsCSF biomarkers are not included in the international diagnostic guidelines of iNPH. However, low CSF levels of amyloid beta 42 (Aβ42) (123) and increased levels of HPτ 181 and total tau (124), are supportive findings in the diagnostics of AD (25) and are helpful in differential diagnostics between AD and iNPH (26). Additionally, elevation of CSF tau has been associated with several other neuro-degenerative disorders (125). Aβ is formed by the proteolytic cleavage of amyloid precursor protein (APP) (126). The cleavage of APP by the amyloidogenic β-secretase pathway produces additionally soluble APP beta (sAPPβ), while the α-secretase pathway precludes the formation of Aβ and results in the introduction of soluble APP alpha (sAPPα) (126). Lower CSF levels of both sAPP isoforms have been reported in iNPH compared to healthy individuals and AD (127–129), and subsequent increase in the ventricular CSF levels of sAPP isoforms after shunt surgery was noted in a single paper (129). In addition, sAPPα has shown a potential prognostic value in shunted iNPH patients (128). Several other CSF biomarkers have been studied in iNPH, but none of them have become a rou-tine diagnostic tool (130). Findings of proinflammatory cytokines in the CSF of iNPH patients have been rather contradictory (129, 131–136). In contrast, elevated levels of neurofilament light (NFL) protein in iNPH and secondary NPH have been reported consistently in several publications (129, 137–140). However, as NFL reflects axonal damage and neuronal loss, higher CSF NFL levels have been reported in various other dementing disorders including AD and VCI compared to healthy individuals (141). A single article reported abnormal CSF transferrin-1/transferrin-2 isoform (Tf-1/Tf-2) ratios in patients with iNPH compared to healthy controls and AD patients (142).
2.7 COMORBIDITIES IN INPH
2.7.1 Degenerative brain diseaseWhile AD is important in the differential diagnostics of iNPH (2.6.6.1 Alzheimer’s disease), these two diseases frequently coexist as iNPH-AD (9, 88). In a single study, iNPH-AD was reported in 12% of shunt-responding iNPH patients and iNPH-VCI in 5% (88). AD-related findings have been reported in 62% of NPH patients (pooled data from autopsy studies) on neuropathological exami-nation (144, 169). In addition, Aβ plaques together with neurofibrillary tangles have been present in 23% of the living NPH patients (pooled data) in studies utilizing brain biopsy (144, 170, 171). Moreover, similar findings have been reported in CSF and PET studies (26, 150, 151). Current un-derstanding indicates that AD pathology in iNPH patients is associated with a worse outcome after shunt surgery, but does not affect survival (9, 87, 88, 153, 170, 172–174). Nevertheless, a panel of

19
experts of the International Society for Hydrocephalus and Cerebrospinal Fluid Disorders recom-mends shunt treatment in patients with mixed iNPH-AD findings, however, the cognitive outcome in particular may be less satisfactory compared to typical iNPH patients (9). The common concur-rence of iNPH and AD has lead some researchers to suggest common pathological pathways for the two diseases (83) (see 2.4.2 Pathophysiological theories). Even if no AD-related pathological findings are observed in the brain biopsy and shunt surgery is successful, in some persons with iNPH a clinical ‘hydrocephalic’ dementia has been diagnosed (88).
2.7.2 Vascular disease and type 2 diabetes mellitusThe burden of vascular risk factors (Table 3) increases an individual’s risk of developing vascular diseases such as coronary heart disease and potentially life-threatening complications (e.g. myocar-dial infarction) (175). A few studies have reported increased frequency of various vascular risk fac-tors in iNPH patients (176–178). In particular, the prevalence of systemic arterial hypertension has been reported to be higher in iNPH (42–88%) compared to the age-matched hospital or population-based controls in several papers (8, 9, 176–180). Interestingly, one study reported a better cogni-tive response to shunt surgery in iNPH patients without a diagnosis of hypertension compared to hypertensive patients (180).
The prevalence of cardiovascular disease has been reported to be high in patients with iNPH (8, 176–178). In addition, deep white matter lesions in brain imaging and a diagnosed comorbid cerebrovascular disease are common in iNPH (8, 9, 76, 82, 163–166, 177, 181–183). Type 2 diabetes mellitus (T2DM) is frequently associated with vascular diseases (184) and the reported relative prevalence of T2DM in iNPH is high (16–52%) (8, 176–178, 185). Although the association of vascular disease and iNPH is well-established, the underlying pathophysiology is currently not understood (9) (see 2.4.2 Pathophysiological theories).
2.7.3 Other comorbiditiesA single paper reported an overrepresentation of glaucoma in 72 patients with iNPH compared to 72 age-matched non-iNPH hydrocephalic controls (18% vs. 5.6%, p = 0.02) (186). The authors hy-pothesized that similar mechanisms could be responsible for the normotensive glaucomatous optic neuropathy and axonal injury in iNPH (186). Interestingly, shunt treatment may cause progression of glaucoma due to a decrease of CSF pressure, which in turn increases translaminar pressure gradient of the optic nerve (187). Other potential comorbidities that can cause or worsen iNPH-
Male sexAge (men ≥55 years; women ≥65 years)SmokingHypertensionDyslipidaemia
Total cholesterol >4.9 mmol/L, and/orLow-density lipoprotein cholesterol >3.0 mmol/L, and/orHigh-density lipoprotein cholesterol: men <1.0 mmol/L, women <1.2 mmol/L, and/orTriglycerides >1.7 mmol/L
HyperglycaemiaAbnormal fasting plasma glucoseAbnormal glucose tolerance test
Abdominal obesity (waist circumference men ≥102 cm; women ≥88 cm) (in Caucasians)Family history of premature cardiovascular disease (men aged <55 years; women aged <65 years)
Table 3. List of cardiovascular risk factors. Adapted from Mancia et al. 2013 (175).

20
like symptoms include lumbar and cervical spinal stenosis, osteoarthrosis, and primary urological diseases (9).
2.8 TREATMENT OF INPH
The treatment of iNPH by CSF shunt surgery has been established since the first description of the disease (2, 3). A shunt is an implantable device that comprises of a proximal catheter, a one-way valve, and a distal catheter creating an alternative pathway for the CSF drainage. Shunt systems are categorized by the placement of the catheters: a ventriculoperitoneal (VPS) (188) and a ventricu-loatrial (VAS) (189) shunt drain CSF from a lateral ventricle of the brain to the peritoneal cavity or to the right atrium of the heart, respectively; while a lumboperitoneal shunt (LPS) (190) drains CSF from the lumbar CSF space to the peritoneal cavity. There are currently no randomized controlled trials comparing shunt surgery with a closed shunt or sham surgery (no shunt), and thus the level I evidence on the efficacy of shunt surgery in iNPH is lacking (191). The highest level of evidence to date is presented by a Japanese open-label randomized trial that compared immediate (<1 month after randomization) and postponed (3 months) LPS surgery and reported a 65% improvement in the first group and 5% in the latter at three months (p < 0.0001) concluding that LPS may be beneficial in the treatment of iNPH (192). While there is no level I evidence on shunt surgery, several studies have reported a beneficial out-come after shunt surgery in most selected patients (see 2.9.2 Outcome of treatment) and VPS sur-gery remains the recommended treatment for iNPH (193). An alternative management for hydrocephalus is the endoscopic third ventriculostomy (ETV) that is mainly utilized for the obstructive hydrocephalus (194, 195). In ETV, the floor of the third ventricle is perforated endoscopically creating an alternative tract from the ventricular CSF system to the skull base subarachnoid space (194, 195). The role of ETV in the nonobstructive hydrocepha-lus, such as iNPH, is controversial, since although success rates similar to shunting have been re-ported (196), an open-label, noncrossover, randomized trial found VPS to be superior compared to ETV in iNPH (197). A Cochrane Review found no high-quality evidence to determine the effective-ness of ETV in patients with iNPH (198). There is currently no established pharmacological treatment for iNPH. However, acetazola-mide (ACZ), a carbonic anhydrase inhibitor that reduces CSF production, is widely used in glauco-ma (199), idiopathic intracranial hypertension (200, 201), and for the prophylaxis of acute mountain sickness (202). In iNPH, ACZ has been subject to a single case report and two small studies that have demonstrated a potential to alleviate gait symptoms and to reduce white matter hyperintensi-ties (203–205). However, as these studies have a very small number of patients (n = 1–15) and no control group, there is currently no scientific evidence for the efficacy of ACZ in iNPH.
2.9 PROGNOSIS OF INPH
2.9.1 Natural courseThe natural course of iNPH in unshunted patients is unclear (206). The studies that have included unshunted iNPH patients, have shown most patients to deteriorate and some to remain stable (206–213). However, all the cited studies that included unshunted iNPH patients suffer from selec-tion bias, as no randomization was undertaken. A double-blind, randomized trial with a closed shunt or sham surgery as a control would give insight on the natural course of iNPH in unselected patients. A trial has been published at a small scale (n = 14) with iNPH patients with findings of comorbid subcortical vascular dementia and negative findings in CSF infusion studies, suggesting that shunt treatment is beneficial to these patients (183).
2.9.2 Outcome of treatmentA substantial number of papers studying the outcome of shunt treatment in iNPH have been pub-lished. A systematic review comprising 64 studies and 3,063 patients with iNPH reported an aver-age improvement of 71% in short-term (3–12 months after surgery) and 65% in long-term (at least more than 3 years after surgery) with an average of 1% mortality and complications rate of 10.4% (214). A better outcome (82%) and lower mortality (0.2%) were noted in the studies published in

21
2006 or later compared to earlier papers (214). The most common complications of shunt treatment include a subdural haemorrhage or effusion (6.3%), infections (3%), and a new onset of seizures (0.7%) (214). In ETV, higher mortality (3.2% vs. 0.5%) and short-term complication rates (17.9% vs. 11.8%) have been reported (215).
2.9.3 Mortality and causes of deathHigh frequency of vascular disease comorbidity seems to affect the mortality and causes of death in patients with iNPH – cerebrovascular and cardiovascular disease comprise 52% of the causes of death in iNPH (pooled data from eight studies including a total of 110 deceased iNPH patients) (177, 216–222). Following vascular diseases, the most common causes of death in patients with iNPH are injury (15%) and malignant neoplasms (11%) (177, 216–222). Interestingly, dementia or iNPH as a cause of death is rare (0.1%) (177, 216–222). For a full review of the literature regarding mortality and causes of death in iNPH, see Table 4. No causes of death were provided for control populations in the cited studies, however, ischaemic heart disease and stroke are the most common causes of death in high income countries (223).

22
Table 4. Mortality and causes of death in idiopathic normal pressure hydrocephalus (iNPH) in literature.
aRangebMeancMean±SDdMedian
Study Patients, n Follow-up time, mo Deaths, n (%) Cause of deathGreenberg et al. 1977(216)
73 iNPH 3–66a
9.7b (series 1)16.7b (series 2)
29 (39.7) 24 unrelated to shunt surgery, otherwise not specified. 5 patients died within one month of surgery: myocardial infarction (n = 1), stroke (n = 1), pneumonia (n = 1), perforated duodenal ulcer (n = 1), pulmonary embolism and uraemia (n = 1).
Black 1980(217)
62 iNPH 2.25–75a
36.5b5 (8.1) Myocardial infarction (n = 1), stroke (n = 1), pneumonia (n = 1), pulmonary embolism (n =
1), unknown (n = 1). All within 3 months of surgery.Raftopoulos et al. 1996(218)
23 iNPH 9–68a 13 (56.5) Stroke (n = 4), myocardial infarction (n = 3), digestive tract neoplasia (n = 1), ovarian neoplasia (n = 1), pneumonia (n = 1), septicemia (n = 1), struck by a car (n = 1), not specified (n = 1).
Boon et al. 1998(219)
101 iNPH <1–12a
10.9±3.1c (group 1)10.7±3.2c (group 2)
16 (15.8) Pneumonia (n = 4), cardiac failure (n = 3), stroke (n = 3), mesenteric arterial thrombosis (n = 1), peritonitis (n = 1), head injury (n = 1), unknown (n = 3).
Malm et al. 2000(220)
42 iNPH <3–36a 12 (28.6) Intracerebral hemorrhage (n = 4), myocardial infarction (n = 2), abdominal cancer (n = 2), pulmonary complications following shunt surgery (n = 1), ruptured aortic aneurysm (n = 1), dementia (n = 1), complications following a subdural hematoma (n = 1).
Aygok et al. 2005(258)
50 iNPH 36 4 (8.0) Unrelated to shunt surgery, otherwise not specified.
Spagnoli et al. 2006(221)
66 iNPH 4–96a
52±24.8c16 (24.2) Cardiac failure (n = 4), stroke (n = 3), pneumonia (n = 3), kidney cancer (n = 1), unknown
(n = 5).Tisell et al. 2006(259)
109 total38 iNPH
<3–74a
50.4d29 (28.7) total14 (36.8) iNPH
Heart disease (n = 7), malignancy (n = 7), infection (n = 7), stroke (n = 5), epileptic status (n = 1), choking (n = 1), unknown (n = 1). No separate data for the iNPH subgroup was provided.
Kahlon et al. 2007(260)
75 total58 iNPH
>6–91a
6.1±4.6c (short-term)66±16.2c (long-term)
28 (37.3) total Cardiovascular disease (n = 9), malignancy (n = 7), pneumonia (n = 3), septicaemia (n = 2), dementia (n = 2), pancreatitis (n = 1), unknown (n = 4). No separate data for the iNPH subgroup was provided.
Mirzayan et al. 2010(177)
51 iNPH 18.8±16.6c (short-term)80.9±51.6c (long-term)
29 (56.9) Cerebral infarction (n = 12), cardiac failure (n = 7), cancer (n = 2), pneumonia (n = 2), acute respiratory distress syndrome (n = 1), pulmonary embolism (n = 1), renal failure (n = 1), unknown (n = 3).
Sprung et al. 2010(261)
165 total69 iNPH
12 9 (5.5) total Unspecified internal disease (n = 3), myocardial infarction (n = 2), pneumonia (n = 2), aortofemoral bypass operation (n = 1), stroke (n = 1). No separate data for the iNPH subgroup was provided.
Lundkvist et al. 2011(262)
72 iNPH 3–7935d
7 (9.7) Unrelated to shunt surgery, otherwise not specified.
Leinonen et al. 2012(170)
468 NPH 0–204a 267 (57.1) Cardiovascular (n = 49), ischaemic stroke (n = 26), haemorrhagic stroke (n = 7), any dementia (n = 46), infection (n = 23), other (n = 77), data not available (n = 39). No separate data for the iNPH subgroup was provided.
Stranjalis et al. 2012(263)
238 iNPH Not specified 4 (1.7) Shunt-related death (n = 3), unrelated to shunt surgery (n = 1). Only hospital mortality reported.
Gölz et al. 2014(222)
147 iNPH 72 (for 61 patients, others died or lost to follow-up)
69 (46.9) Pneumonia (n = 7), cardiovascular diseases and heart attacks (n = 5), malignancy (n = 5), stroke (n = 3), multiple organ failure following liver dysfunction (n = 3), renal failure (n = 2), sepsis due to peritonitis (n = 1), acute paraplegia (n = 1), not specified (n = 42).

23
Study Patients, n Follow-up time, mo Deaths, n (%) Cause of deathGreenberg et al. 1977(216)
73 iNPH 3–66a
9.7b (series 1)16.7b (series 2)
29 (39.7) 24 unrelated to shunt surgery, otherwise not specified. 5 patients died within one month of surgery: myocardial infarction (n = 1), stroke (n = 1), pneumonia (n = 1), perforated duodenal ulcer (n = 1), pulmonary embolism and uraemia (n = 1).
Black 1980(217)
62 iNPH 2.25–75a
36.5b5 (8.1) Myocardial infarction (n = 1), stroke (n = 1), pneumonia (n = 1), pulmonary embolism (n =
1), unknown (n = 1). All within 3 months of surgery.Raftopoulos et al. 1996(218)
23 iNPH 9–68a 13 (56.5) Stroke (n = 4), myocardial infarction (n = 3), digestive tract neoplasia (n = 1), ovarian neoplasia (n = 1), pneumonia (n = 1), septicemia (n = 1), struck by a car (n = 1), not specified (n = 1).
Boon et al. 1998(219)
101 iNPH <1–12a
10.9±3.1c (group 1)10.7±3.2c (group 2)
16 (15.8) Pneumonia (n = 4), cardiac failure (n = 3), stroke (n = 3), mesenteric arterial thrombosis (n = 1), peritonitis (n = 1), head injury (n = 1), unknown (n = 3).
Malm et al. 2000(220)
42 iNPH <3–36a 12 (28.6) Intracerebral hemorrhage (n = 4), myocardial infarction (n = 2), abdominal cancer (n = 2), pulmonary complications following shunt surgery (n = 1), ruptured aortic aneurysm (n = 1), dementia (n = 1), complications following a subdural hematoma (n = 1).
Aygok et al. 2005(258)
50 iNPH 36 4 (8.0) Unrelated to shunt surgery, otherwise not specified.
Spagnoli et al. 2006(221)
66 iNPH 4–96a
52±24.8c16 (24.2) Cardiac failure (n = 4), stroke (n = 3), pneumonia (n = 3), kidney cancer (n = 1), unknown
(n = 5).Tisell et al. 2006(259)
109 total38 iNPH
<3–74a
50.4d29 (28.7) total14 (36.8) iNPH
Heart disease (n = 7), malignancy (n = 7), infection (n = 7), stroke (n = 5), epileptic status (n = 1), choking (n = 1), unknown (n = 1). No separate data for the iNPH subgroup was provided.
Kahlon et al. 2007(260)
75 total58 iNPH
>6–91a
6.1±4.6c (short-term)66±16.2c (long-term)
28 (37.3) total Cardiovascular disease (n = 9), malignancy (n = 7), pneumonia (n = 3), septicaemia (n = 2), dementia (n = 2), pancreatitis (n = 1), unknown (n = 4). No separate data for the iNPH subgroup was provided.
Mirzayan et al. 2010(177)
51 iNPH 18.8±16.6c (short-term)80.9±51.6c (long-term)
29 (56.9) Cerebral infarction (n = 12), cardiac failure (n = 7), cancer (n = 2), pneumonia (n = 2), acute respiratory distress syndrome (n = 1), pulmonary embolism (n = 1), renal failure (n = 1), unknown (n = 3).
Sprung et al. 2010(261)
165 total69 iNPH
12 9 (5.5) total Unspecified internal disease (n = 3), myocardial infarction (n = 2), pneumonia (n = 2), aortofemoral bypass operation (n = 1), stroke (n = 1). No separate data for the iNPH subgroup was provided.
Lundkvist et al. 2011(262)
72 iNPH 3–7935d
7 (9.7) Unrelated to shunt surgery, otherwise not specified.
Leinonen et al. 2012(170)
468 NPH 0–204a 267 (57.1) Cardiovascular (n = 49), ischaemic stroke (n = 26), haemorrhagic stroke (n = 7), any dementia (n = 46), infection (n = 23), other (n = 77), data not available (n = 39). No separate data for the iNPH subgroup was provided.
Stranjalis et al. 2012(263)
238 iNPH Not specified 4 (1.7) Shunt-related death (n = 3), unrelated to shunt surgery (n = 1). Only hospital mortality reported.
Gölz et al. 2014(222)
147 iNPH 72 (for 61 patients, others died or lost to follow-up)
69 (46.9) Pneumonia (n = 7), cardiovascular diseases and heart attacks (n = 5), malignancy (n = 5), stroke (n = 3), multiple organ failure following liver dysfunction (n = 3), renal failure (n = 2), sepsis due to peritonitis (n = 1), acute paraplegia (n = 1), not specified (n = 42).

24

25
3 Aims of the study
The aim of this thesis was to investigate the general characteristics of a large cohort of patients from a defined geographical area with possible iNPH and to evaluate potential biomarkers.
The specific aims were:
1.) To determine incidence of iNPH in Middle and East Finnish populations (i).
2.) To assess comorbidities, mortality, and causes of death in patients with iNPH (i).
3.) To study the effects of APOE genotypes in the diagnostics and prognostics in iNPH pa-tients (ii).
4.) To evaluate a large panel of CSF biomarkers in iNPH and how they relate to brain biopsy findings (iii).
Roman numerals refer to original publications.

26

27
4 Incidence, comorbidities, and mortality in idiopathic normal pressure hydrocephalus
4.1 ABSTRACT
4.1.1 ObjectiveTo investigate incidence, comorbidities, mortality, and causes of death in idiopathic normal pres-sure hydrocephalus (iNPH).
4.1.2 MethodsA cohort of 536 patients with possible NPH from a defined population with a median follow-up time of 5.1 years (range 0.04–19.9) was included in the study. Patients were evaluated by brain imaging and intraventricular pressure monitoring with a brain biopsy immunostained against amyloid-β and hyperphosphorylated tau. Hospital records were reviewed for vascular diseases and type 2 diabetes mellitus (T2DM). Death certificates and yearly population of the catchment area were obtained from national registries.
4.1.3 ResultsA total of 283 patients had a clinical diagnosis of iNPH leading to a median annual incidence of 1.58 iNPH patients per 100,000 inhabitants (range 0.8–4.5). Alzeimer’s disease-related brain biopsy findings were less frequent in iNPH compared to the non-iNPH patients (p<0.05). An overrepre-sentation of hypertension (52% vs. 33%, p<0.001) and T2DM (23% vs. 13%, p=0.002) was noted in iNPH patients. Age (hazards ratio (HR) 1.04/year, 95% confidence interval (CI) 1.03–1.06, p<0.001) and T2DM (HR 1.63, 95% CI 1.23–2.16, p<0.001) increased the risk of death in the iNPH patients and in the total population. iNPH was associated with decreased risk of death (HR 0.63, 95% CI 0.50–0.78, p<0.001). The most frequent causes of death were cardiovascular and cerebrovascular disease. Dementia as a cause of death was more common in non-iNPH patients compared to iNPH (27% vs. 10%, p<0.001).
4.1.4 ConclusionsHypertension and T2DM are common in iNPH and the latter causes excess mortality in the affected patients.
4.2 INTRODUCTION
Idiopathic normal pressure hydrocephalus (iNPH) is characterized by gait disturbance, cognitive decline, and urinary incontinence in the elderly with signs of communicating hydrocephalus in a neuroradiological study, while the cerebrospinal fluid (CSF) pressure is within normal range, and no predisposing factors such as subarachnoid hemorrhage are observed (2, 3). Despite the absence of elevated CSF pressure, the diversion of CSF with an implantable shunt device relieves and even reverses the symptoms in most patients (193). iNPH is generally regarded as a rare disease with a reported incidence of 0.5–1.2 shunted iNPH patients per 100,000 inhabitants per year in hospital-based studies conducted in the Netherlands (10), Germany (11), Sweden (12), Norway (14), and the United States of America (15) (Table 1). In Japan, a hundredfold incidence (1.2 / 1000 inhabitants / year) of possible iNPH was reported in inhabitants aged 70 or older (48) (Table 1). The only true epidemiological population-based study of iNPH reported an incidence of at least 5.5 per 100,000 inhabitants per year in Norway indicating iNPH to be an underdiagnosed or undertreated disease (13) (Table 1). Interestingly, a single study

28
reported no diagnosed iNPH patients in a population-based dementia study of five years in the United States of America among 560 patients with nondegenerative and nonvascular dementia (49) (Table 1). Arterial hypertension is the best documented concomitant disease with iNPH with a reported prevalence of 42–88% among iNPH patients (8, 9, 176–180). A single article noted a better cogni-tive outcome following shunt surgery in iNPH patients without hypertension compared to iNPH patients with diagnosed hypertension (180). Additionally, white matter vascular degeneration in brain imaging (8, 9, 76, 82, 163, 164, 177), cardiovascular disease (8, 176–178), vascular risk factors (176–178), and type 2 diabetes mellitus (T2DM) (8, 176–178) have been frequently associated with iNPH. It is currently unknown which mechanisms affect the concurrence of vascular disease and iNPH (9). A heavy burden of vascular disease reflects the mortality and causes of death in patients with iNPH: the most frequently reported causes of death are cerebrovascular disease and heart diseases (177, 216–222). For a summary of mortality and causes of death in iNPH, see Table 4. Alzheimer’s disease (AD) is considered to be significant in the differential diagnostics of iNPH with cognitive symptoms, however, the two diseases are not exclusive, and in fact, frequently coex-ist as iNPH-AD (9). AD-related neuropathological findings have been reported in 62% of the NPH cases on autopsy (144, 169) and 34% of the living NPH patients studied with brain biopsy (144, 170). While pathological findings of AD in iNPH patients predict a worse cognitive outcome of shunt surgery, current expert opinion (9) does not recommend against shunting in iNPH-AD patients, as some benefit is still usually seen (87, 153, 170, 172, 173). Thus, shunt surgery in iNPH patients with a comorbid neurodegenerative disease should be considered individually depending on the disease severity and estimated benefits.
Objectives of the current study:1. to determine the incidence of iNPH in Middle and Eastern Finland,2. study the association between vascular diseases, T2DM, and iNPH,3. assess the cumulative mortality and causes of death in iNPH.
4.3 METHODS
4.3.1 Kuopio NPH Registry and protocolThe patients of the current study were recruited from a defined geographical area in Middle and Eastern Finland, of which the sole serving acute and elective neurosurgical unit is the department of neurosurgery of the Kuopio University Hospital (KUH). There are four central hospitals (Central Finland Central Hospital, Mikkeli Central Hospital, Savonlinna Central Hospital, and North Kare-lia Central Hospital) in the KUH area with neurological units, which refer possible NPH patients to KUH Neurosurgery. The annual population of the KUH catchment area during the recruitment period was obtained from Statistics Finland (224). Patients fulfilling the following criteria were further assessed in KUH Neurosurgery as possible NPH patients: (1) primary evaluation and examination by a neurologist; (2) one to three symptoms suggestive of NPH (gait disorder, cognitive decline, urinary incontinence); and (3) NPH-related brain imaging findings. The diagnostic workup protocol of KUH Neurosurgery for possible NPH included a clinical examination, computed tomography (CT) or magnetic resonance imaging (MRI) scan of the brain, and a 24h intraventricular pressure monitoring together with a frontal cortical brain biopsy. During the final year of the recruitment period, 13 patients underwent a tap test and an infusion study instead of intracranial pressure (ICP) monitoring, and the cortical brain biopsy was obtained during the shunt operation (225). Kuopio NPH Registry (www.uef.fi/nph) consists of all evaluated possible NPH patients from the KUH catchment population since 1993 and contains clinical baseline and follow-up data, other hospital diagnoses, medications, causes of death from national registries, and neuropathological findings.
4.3.2 ParticipantsAll patients meeting the criteria above during the recruitment period (1993–2010) were initially en-rolled to the study. Patients outside the current KUH catchment area and patients diagnosed with secondary NPH were excluded from the current study. Included patients were followed-up until

29
death or the end of December 2013. All patients or their proxies gave a written, informed consent prior to participation in the study.
4.3.3 Shunt treatment and shunt responseIn iNPH patients, ICP criteria indicating shunt treatment were (1) a basal ICP pressure between 10 and 20 mmHg continuously during the 24h monitoring, or (2) the presence of A-waves or more than 30% B-waves, when basal pressure was between 5 and 10 mmHg (207). The shunt response was clinically evaluated at 2–3 months after surgery, and any subjective or objective improvement in the patient’s gait, memory, or urinary continence was graded as a positive shunt response (87).
4.3.4 Comorbidities and causes of deathPresence or absence of a diagnosed arterial hypertension, coronary artery disease, atrial fibrilla-tion, chronic heart failure, and T2DM were reviewed from hospital records. Causes of death were obtained from national clinical registries.
4.3.5 Immunohistochemistry and histological evaluation of brain biopsyA frontal cortex sample approximately 10 mm in length and 2 mm in diameter was obtained during the ICP monitoring or shunt operation. A hematoxylin-eosin staining revealed that in most sam-ples both grey and white matter were represented. Seven micrometre thick sections were stained applying immunohistochemistry to visualize β-amyloid (Aβ) (6 clone 6F/3D) and hyperphospho-rylated tau (HPτ) (clone AT8) as described earlier (87). Immunoreactivity for each antibody was assessed as present or absent.
4.3.6 Statistical analysisFor nominal variables (sex, leading symptom, immunoreactivity, comorbidities, causes of death), differences between groups were analyzed with the χ2 test or Fisher’s exact test, when applicable. The Mann–Whitney U test was utilized in continuous variables (age and follow-up time). In mul-tiple comparisons, Bonferroni correction was used. Correlation of incidence was analyzed with Spearman’s correlation and linear regression. In survival analyses, Kaplan-Meier with log rank test and Cox proportional hazards model were applied. IBM SPSS Statistics for Mac (version 22.0.0.0, IBM, Armonk, NY, USA) was used in the statistical analyses. The level of significance was set at p < 0.05.
4.3.7 Ethical issuesThis study was approved by the Research Ethics Committee of the Northern Savo Hospital District, The Finnish National Supervisory Authority for Welfare and Health, and The Finnish Ministry of Social Affairs and Health.
4.4 RESULTS
4.4.1 Study populationAltogether 635 patients met the initial inclusion criteria for the study. A total of 99 patients were then excluded (40 patients outside of the current KUH catchment area, 59 patients with secondary NPH), which lead to the final study population of 536 patients with possible iNPH. Of the final study population, 283 patients had a clinical diagnosis of iNPH and 253 patients did not have iNPH. Overall, 269 of the iNPH patients were shunted with 229 patients (85%) showing a positive response. Patient characteristics of iNPH and non-iNPH patients are summarized in Table 5. Both groups were similar in age (median age at the time of ICP monitoring 73.3 vs. 73.5 years), however, there were more women in the iNPH patient group (57% vs. 46%, p = 0.02). Gait disorder was the most common leading symptom in the iNPH group, while cognitive decline was more frequent in the other group (p < 0.001).

30
iNPH non-iNPH p post hoc p
n 283 253
Age (years) (median (range)) 73.28 (43.49–87.86)
73.51 (29.57–86.13)
0.82a
Women (n (%)) 161 (57) 117 (46) 0.015b
Follow-up time (years) (median (range))
5.56 (0.04–19.87)
4.83 (0.05–19.70)
0.019a
Leading symptom (n (%))c <0.001d
Gait impairment 127 (50) 50 (21) <0.001b,e
Cognitive decline 61 (24) 88 (37) 0.01b,e
Other 67 (26) 103 (43) <0.001b,e
Immunoreactivity (n (%))f <0.001d
Aβ – HPτ – 169 (60) 109 (43) <0.001b,e
Aβ + HPτ – 86 (30) 99 (39) 0.14b,e
Aβ + HPτ + 24 (8) 41 (16) 0.03b,e
Aβ – HPτ + 3 (1) 2 (1) 0.99b,e
Comorbidities (n (%))
Arterial hypertension 148(52) 84 (33) <0.001b
Cardiovascular disease 53 (18) 61 (24) 0.14b
Atrial fibrillation 20 (7) 9 (4) 0.09b
Chronic heart failure 18 (6) 25 (10) 0.15b
Type 2 diabetes mellitus 65 (23) 32 (13)g 0.002b
Mortality (deaths) (n (%)) 144 (51) 181 (72) <0.001b
Causes of death (n (%))h <0.001d
Heart disease 31 (22) 30 (17) 0.99b,e
Cerebrovascular disease 27 (19) 41 (23) 0.99b,e
Malignant neoplasm 18 (13) 16 (9) 0.99b,e
Infection 11 (8) 16 (9) 0.99b,e
Injury 4 (3) 8 (4) 0.99b,e
Dementia 14 (10) 48 (27) <0.001b,e
iNPH 13 (9.) 0 (0) <0.001b,e
Other 25 (17) 22 (12) 0.99b,e
Table 5. Characteristics, immunoreactivity (for amyloid-β (Aβ) and hyperphosphorylated tau (HPτ)), comorbidi-amyloid-β (Aβ) and hyperphosphorylated tau (HPτ)), comorbidi-β (Aβ) and hyperphosphorylated tau (HPτ)), comorbidi- (Aβ) and hyperphosphorylated tau (HPτ)), comorbidi-β) and hyperphosphorylated tau (HPτ)), comorbidi-and hyperphosphorylated tau (HPτ)), comorbidi-τ)), comorbidi-comorbidi-ties, mortality, and causes of death in 283 patients with idiopathic normal pressure hydrocephalus (iNPH) and 253 non-iNPH patients.
aMann-Whitney U testbFisher’s exact testcSufficient data for the determination of a leading symptom was unavailable in 28 iNPH patients and 12 non-iNPH patientsdχ2 testeBonferroni correctedfBiopsy data was unavailable from 2 non-iNPH patients and 1 iNPH patientgPresence/absence of T2DM was unknown in three patients from the non-iNPH grouphCause of death was not available in 1 iNPH patient

31
4.4.2 Catchment population and the incidence of iNPHCatchment population of KUH Neurosurgery decreased from 875,184 to 842,918 inhabitants dur-ing the recruitment period (1993–2010) of the current study (Figure 7). In contrast, the number of inhabitants aged 70 or older increased constantly from 87,112 to 120,423 during the study period. The cumulative incidence of iNPH ranged from 0.8 to 4.5 cases per 100,000 inhabitants per year with a mean incidence of 1.84 and standard deviation (SD) of 0.99 (Figure 7). Incidence correlated with the increasing proportion of inhabitants aged 70 or older in the population (Spearman’s ρ = 0.77, p < 0.001) and with the year of diagnosis (Spearman’s ρ = 0.77, p < 0.001). A linear regression analysis including the proportion of inhabitants aged 70 or older and the year of diagnosis resulted in correlation coefficient R = 0.82 and coefficient of determination R2 = 0.67, p < 0.001. In the inhabit-ants aged 70 or older, the cumulative age-specific incidence of iNPH ranged from 7.1 to 31.6 with a mean incidence of 14.65 (SD 6.51).
4.4.3 Vascular disease and T2DM in iNPHHypertension was significantly more frequent in patients with a diagnosis of iNPH compared to non-iNPH patients with a prevalence of 52% in iNPH and 33% in non-iNPH (p < 0.001) (Table 5). Similar overrepresentation was noted in the frequency of T2DM (23% vs. 12%, p = 0.002) (Table 5). No differences were observed in other vascular diseases (Table 5).
Inci
denc
e ra
tes
per 1
00,0
00 in
habi
tant
s
0
1
2
3
4
5
6
7
8
250,000
500,000
750,000
1,000,000
0
1993
1994
1995
1996
2010
1997
1998
1999
2000
2001
2002
2003
2004
2005
2006
2007
2008
2009
Year
Popu
latio
n
Possible NPH
iNPH
Inhabitants aged under 70 years
Inhabitants aged 70 or older
Figure 7. Annual incidence of possible idiopathic normal pressure hydrocephalus (iNPH) and iNPH in Middle and Eastern Finland from 1993 to 2010 with annual population of the catchment area shown in the background.

32
4.4.4 Brain biopsy findingsBiopsy findings are summarized in Table 5. iNPH patients lacked more frequently Aβ and HPτ immunoreactivity (p < 0.001), whereas non-iNPH patients displayed Aβ+ HPτ+ more commonly (p = 0.03) (Table 5).
4.4.5 Mortality and causes of death in iNPHDuring the follow-up 144 (51%) patients with iNPH and 181 (72%) patients with a final diagnosis other than iNPH died (p < 0.001) (Table 5). In iNPH, median estimated survival time was 9.0 years (95% confidence interval (CI) 7.6–10.5 years) and in non-iNPH patients 5.8 years (95% CI 4.9–6.7 years) (p < 0.001) (Figure 8).
0 5 10 15 200
0.2
0.4
0.6
0.8
1.0
Time (years)
Cum
ulat
ive
surv
ival
Log rank testp < 0.001
iNPH (n = 293)non-iNPH (n = 259)
293
259 123 40 8
161 50 9
Number at risk
iNPH
non-iNPH
Figure 8. Kaplan-Meier survival plot of 283 patients with idiopathic normal pressure hydrocephalus (iNPH) (blue line) and 253 non-iNPH patients (red line). Vertical tick-marks indicate censored cases.

33
The causes of death for both groups are summarized in Table 5 and Figure 9. The most common cause of death in patients with iNPH was heart disease (22%) and cerebrovascular disease (23%) for the non-iNPH patients. Dementia was a more frequent cause of death in non-iNPH patients compared to iNPH patients (27% vs. 10%, p < 0.001).
A Cox proportional hazards model was implemented to study the survival of the patients fur-ther (Figure 10). In the multivariable analysis, significant covariates were age (Hazards ratio (HR) 1.04/year, 95% CI 1.03–1.06, p < 0.001), chronic heart failure (HR 1.51, 95% CI 1.03–2.21, p = 0.04), T2DM (HR 1.63, 95% CI 1.23–2.16, p < 0.001), and iNPH (HR 0.63, 95% CI 0.50–0.78, p <0.001) in the total population (Figure 10). In patients with iNPH (within-group analysis), significant covariates were age (HR 1.08/year, 95% CI 1.04–1.11, p < 0.001), atrial fibrillation (HR 2.27, 95% CI 1.20–4.29, p = 0.01), and T2DM (HR 1.71, 95% CI 1.15–2.54, p = 0.008).
Heart disease
Cerebrovascular disease
Malignant neoplasm
Infection
Injury
Dementia
iNPH
Other
0 % 10 % 20 % 30 %
n = 16
n = 30
n = 31
n = 41
n = 27
n = 18
n = 16
n = 11
n = 48
n = 14
n = 13
n = 22
n = 25
n = 0
n = 8
n = 4
p < 0.001
p < 0.001
Caus
e of
dea
th
Figure 9. Causes of death in 143 deceased idiopathic normal pressure hydrocephalus (iNPH) patients (lighter shade lower bars) and 185 non-iNPH patients (darker shade upper bars). P-values were determined using a Mann–Whitney U test.

34
01.
02.
03.
04.
05.
0
Decr
ease
dris
k of
dea
thIn
crea
sed
risk
of d
eath
Adju
sted
haz
ard
ratio
(95%
CI)
Cova
riate
n
HR (9
5% C
I)
p
Sex
Fem
ale
275
1
M
ale
254
1.18
(0.9
3–1.
49)
0.18
Age
(yea
r)
1.
04 (1
.03–
1.06
) <0
.001
Imm
unor
eact
ivity
A
– HP
– 27
6 1
A+
HP–
183
0.96
(0.7
4–1.
23)
0.73
A+
HP+
65
1.26
(0.8
8–1.
81)
0.20
A–
HP+
5 1.
54 (0
.56–
4.26
) 0.
40
Coro
nary
hea
rt di
seas
e 11
1 1.
12 (0
.86–
1.46
) 0.
41
Atria
l fib
rilla
tion
29
1.33
(0.8
1–2.
18)
0.27
Oth
er a
rryt
hmia
25
1.
33 (0
.83–
2.12
) 0.
24
Chro
nic
hear
t fai
lure
42
1.
51 (1
.03–
2.21
) 0.
04
Arte
rial h
yper
tens
ion
232
1.00
(0.7
9–1.
27)
0.98
T2D
M
97
1.63
(1.2
3–2.
16)
<0.0
01
iNPH
28
2 0.
63 (0
.50–
0.78
) <0
.001
Figu
re 1
0. A
mul
tivar
iabl
e Cox
pro
port
iona
l haz
ards
mod
el o
f 529
pat
ient
s w
ith s
uspe
cted
nor
mal
pre
ssur
e hy
droc
epha
lus
(NPH
) an
d a
fore
st p
lot
of a
djus
ted
haza
rds
ratio
s (H
R)
with
95%
con
fiden
ce in
terv
als
(CI)
. 7
patie
nts
wer
e ex
clud
ed f
rom
the
ana
lysi
s du
e to
insu
ffici
ent
patie
nt r
ecor
ds.
Aβ
= a
myl
oid-
β; H
Pτ =
hyp
erph
osph
oryl
ated
tau
; T2
DM
= t
ype
2 di
abet
es m
ellit
us;
iNPH
= id
iopa
thic
nor
mal
pre
ssur
e hy
droc
epha
lus.

35
4.5 DISCUSSION
This is currently the largest study to evaluate incidence, comorbidities, mortality and causes of death in iNPH. The main finding was the demonstration of a major vascular and diabetic comor-bidity in patients with iNPH and how it reflects the mortality and causes of death in iNPH in a large cohort from a defined geographical area during a long-term follow-up. Mean incidence of iNPH in the study population was 1.84 / 100,000 / year (SD 0.99) with a mark-edly higher age-specific incidence in the elderly (70 or older) of 14.65 (SD 6.51). The incidence of iNPH increased during the recruitment period (1993–2010) both in the total population and in the elderly, possibly due to better recognition of the disease among general practitioners and in spe-cialist care. The incidence figures we found are higher than previously reported in hospital-based studies (10–12, 14, 15) but lower than in population-based studies (13, 48). It is unclear whether iNPH is more frequent in Finland compared to other countries or that the observed differences are due to dissimilarities in the study designs. As expected, arterial hypertension was frequent in iNPH patients with a prevalence of 52% among patients with a diagnosis of iNPH (Table 5), which is in line with the reported prevalences in previous studies (42–88%) (8, 9, 176–180). According to the multivariable model, a diagnosis of hypertension did not affect mortality (Figure 10). Additionally, T2DM was twice more frequent in patients with iNPH compared to non-iNPH patients of the cohort (23% vs. 13%, p = 0.002) (Table 5), which is a confirmatory finding for earlier studies (8, 176–178). Interestingly, although T2DM increased the risk of death in the total popula-tion (HR 1.63, 95% CI 1.23–2.16, p < 0.001) and in patients with iNPH (HR 1.71, 95% CI 1.15–2.54, p = 0.008), the overall mortality of iNPH patients was lower and thus iNPH was associated with a decreased risk of death compared to non-iNPH patients in the cohort (HR 0.63, 95% CI 0.50–0.78, p <0.001) (Figure 10). In future studies, the relationship of vascular disease and iNPH should be explored more: is the high vascular comorbidity in iNPH due to shared genetic or environmental factors, or are the diseases interlinked with the same pathological processes? As reported earlier by our group (87, 88, 170), AD-related brain biopsy findings (Aβ+ HPτ+) were reported in 8.5% of the patients with iNPH. A tendency towards higher mortality was noted in patients with Aβ+ HPτ+, possibly reflecting AD-associated mortality (Figure 10). In iNPH, leading causes of death were heart disease (22%) and cerebrovascular disease (18%), which is similar to pooled data from earlier reports (24% and 28%, respectively) (177, 216–222). Conversely, in our cohort, dementia was listed as a main cause of death in 10% of patients with iNPH, however, in previous studies (177, 216–222) this figure was only 0.1% (pooled data). Infec-tion and injury were more frequently listed as a cause of death in earlier studies (20% and 15%) compared to our findings (8% and 3%) (177, 216–222). The frequency of malignant neoplasm as a cause of death was similar in our cohort (10.9% vs. 12.8%). Importantly, none of the cited studies reported iNPH as a cause death, however, in our cohort, iNPH was listed as a main cause of death in 9%. The observed differences in the causes of death are probably explained by the differences in listing and determining causes of death in different countries rather than actual pathobiological processes. Of the thirteen patients with iNPH listed as a main cause of death, one patient died of complications following ICP measurement (in the autopsy, immediate cause of death was aspira-tion pneumonia), and the twelve other patients had a chain of events unrelated to ICP measure-ment or shunt surgery. However, as no autopsy was performed in these patients, the true final cause of death is not certain. Strengths of the current study included: a large cohort evaluated by cortical brain biopsy from a defined catchment area, a long-term follow-up, and systematically kept medical records for com-prehensive retrospective review. The retrospective nature of the study caused some limitations: blood pressure measurements or blood glucose tests (a fasting glucose test, a glucose tolerance test, or haemoglobin A1c (HbA1c)) were not systematically commenced to diagnose potentially undiagnosed cases of hypertension or T2DM. In addition, shunted patients were routinely reviewed only at 2–3 months after surgery, and no validated objective outcome measures were used. Lack of age- and gender-matched population-based controls is also a limitation to our study. More clinical studies are needed to explore the relationship between vascular disease and iNPH

36
with systematic evaluation of blood pressure and blood glucose levels. Does aggresive treatment of hypertension or T2DM affect the symptoms or progression of iNPH? Are there potential genetic or environmental factors that would explain the high vascular comorbidity in iNPH? To conclude, we present the incidence of iNPH in a defined population in Middle and East-ern Finland. We confirm a high comorbidity of arterial hypertension and T2DM in patients with iNPH. Although iNPH was associated with a decreased risk of death in the patient cohort, a high frequency of T2DM caused excess mortality in these patients. Additionally, dementia was rather common as a cause of death in patients with iNPH.

37
5 APOE4 predicts amyloid-β in cortical brain biopsy but not idiopathic normal pressure hydrocephalus
5.1 ABSTRACT
5.1.1 ObjectiveTo investigate the association of apolipoprotein E (APOE) genotype, especially the APOE4 allele, (1) to idiopathic normal pressure hydrocephalus (iNPH), and (2) to amyloid-β (Aβ) plaques in cortical brain biopsies of presumed NPH patients with and without a final clinical diagnosis of Alzheimer’s disease (AD).
5.1.2 MethodsAltogether 202 patients with presumed NPH were evaluated by intraventricular pressure moni-toring and frontal cortical biopsy immunostained against Aβ (134 semiquantified by Aβ plaques/mm2). The 202 patients and 687 cognitively healthy individuals were genotyped for APOE. The final clinical diagnoses in a median follow up of 3.9 years were: 113 iNPH (94 shunt-responsive, 16 shunt-non-responsive, 3 not shunted); 36 AD (12 mixed iNPH + AD); 53 others.
5.1.3 ResultsThe APOE genotypes distributed similarly in the 94 shunt-responsive and 16 non-responsive iNPH patients and healthy controls. In the multivariate analysis, the APOE4 allele correlated indepen-dently with Aβ plaques in the cortical biopsies (odds ratio 8.7, 95% confidence interval 3.6–20, p < 0.001). The APOE4 allele in presumed NPH predicted later AD as follows: sensitivity 61%; specific-ity 77%; positive predictive value 37%; negative predictive value 90%.
5.1.4 ConclusionsIn presumed NPH patients, APOE4 associates independently with the presence of Aβ plaques in the frontal cortical biopsy. APOE4 is not a risk factor for iNPH and does not predict the response to shunt. Our data further supports the view that the iNPH syndrome is a distinct dementing disease.
5.2 INTRODUCTION
Normal pressure hydrocephalus (NPH) is an uncommon dementing disorder in the elderly, pre-senting with enlarged ventricles but normal or slightly elevated CSF pressure. NPH is character-ised by three clinical symptoms: gait disorder, impaired cognition and urinary incontinence (2, 3). Symptoms are often relieved, or even reversed, by CSF shunt insertion, the treatment of choice at present (226). Idiopathic NPH (iNPH) is a form of NPH without any known predisposing factors, such as brain injury, subarachnoid haemorrhage, or meningitis (7). Various procedures to evalu-ate CSF dynamics in patients with presumed NPH are used to identify those who would benefit from CSF shunting. These include the CSF tap test, external lumbar drainage test (119), infusion tests (119, 120), and intraventricular (87) or intracranial (70) pressure (ICP) monitoring. The most frequent differential diagnoses of iNPH are Alzheimer ’s disease (AD) and vascular dementia (87). In NPH patients, ICP monitoring and shunt insertion, both requiring a burr hole, allow for a small brain biopsy to assess the histological signs of AD and other dementias (87). AD is character-ised by beta-amyloid (Aβ) plaques and neurofibrillary tangles composed of hyperphosphorylated tau (HPτ) in the brain of patients with amnesic cognitive decline (146, 147, 227). The amyloid cas-cade hypothesis suggests that Aβ starts to accumulate even decades before the clinical manifesta-

38
tions of AD (148, 228). During life, Aβ can be detected with brain biopsy (87, 153) and evaluated by positron emission tomography (PET) utilising the 11C-labelled Pittsburgh compound B (150). Apolipoprotein E (apoE) has a significant role in neurobiology (105). There are three different apoE isoproteins coded by three different APOE alleles (ε2, ε3, and ε4) with six different genotypes (101). The APOE genotypes correlate with Aβ accumulation in amyloid-targeted PET (229, 230) and in autopsy studies (231–235). To date, the APOE4 allele is the most important independent genetic risk factor for late onset AD (102, 106, 107, 236, 237) (http://www.alzgene.org). APOE4 is also as-sociated with other neurological disorders (105, 238) including iNPH (22, 23). To date, the possible association of APOE and iNPH has been explored in only two small cohorts.
The objectives of this study were to determine:1. the distribution of APOE genotypes in 94 shunt responsive iNPH patients and in 687 cogni-
tively healthy controls,2. the predictive value of APOE genotypes in the shunt response of 110 shunted iNPH patients,3. the predictive value of APOE4 allele for the presence of Aβ plaques in frontal cortical biopsies
of 199 patients with presumed NPH and for the final clinical diagnosis of AD.
5.3 METHODS
5.3.1 Catchment population of the Kuopio University HospitalNeurosurgery of Kuopio University Hospital (KUH) is the sole provider of full time acute and elec-tive neurosurgical services for the KUH catchment population in Middle and Eastern Finland (239). The KUH area contains four central hospitals with neurological units and catchment areas of their own. During the recruitment period of the present study, from 1993 to 2009, the geographic area remained the same, the population decreased from 874,228 to 842,496, and the proportion of men remained unchanged at 49%.
5.3.2 Kuopio NPH RegistryPatients fulfilling the following criteria were further assessed in KUH Neurosurgery as presumed NPH patients: (1) primary evaluation and examination by a neurologist; (2) one to three symp-toms suggestive of NPH (gait disorder, cognitive impairment, and urinary incontinence); and (3) NPH-related brain imaging findings. Since 1993, the diagnostic workup of KUH Neurosurgery for presumed NPH has included a clinical examination, computed tomography (CT) or magnetic resonance imaging (MRI) scan and 24h intraventricular ICP monitoring together with a right fron-tal cortical biopsy. Kuopio NPH Registry (http://www.uef.fi/nph) consists of all presumed NPH patients from the KUH catchment population since 1993 with (a) clinical baseline and follow-up data and (b) other hospital diagnoses, medications, causes of death from national registries, and neuropathological findings.
5.3.3 ParticipantsAll patients from 1993 to 2009 with presumed NPH, a frontal cortical brain biopsy, and a venous blood sample available until November 2009 (n = 202) from the Kuopio NPH Registry were in-cluded in this study (Table 6, Figure 11). The control population consisted of 687 individuals from Eastern Finland with no signs of cognitive decline on interview or neuropsychological testing (240, 241) (Table 6).

39
Var
iab
les
Pre
sum
ed N
PH
an
d c
orti
cal b
iop
sy s
amp
le a
vaila
ble
(n
= 2
02
)C
ontr
ols
Fin
al c
linic
al d
iag
nos
is o
f iN
PH
a
(n =
11
3)
Mix
ed iN
PH
an
d A
Dc
Oth
er fi
nal
clin
ical
d
iag
nos
is t
han
iNP
H(n
= 7
7)
Sh
un
t re
spon
der
Sh
un
t n
onre
spon
der
Not
sh
un
ted
bA
Dd
Oth
ere
n94
163
1224
5368
7M
edia
n ag
e, y
r (r
ange
, yr
)70
(43
–83)
71 (
64–8
4)78
(74
–85)
79 (
66–8
3)72
.5 (
53–8
2)65
(25
–83)
70 (
37–8
7)Fe
mal
es,
%56
8110
050
5432
60M
edia
n fo
llow
-up
time,
yr
(ran
ge,
yr)
3.3
(0.8
–17)
5.1
(1.2
–17)
7.6
(0.2
–9.4
)2.
9 (1
.1–7
.9)
3.3
(0.4
–13)
5.8
(1.0
–17)
Lead
ing
sym
ptom
, n
(%)
Gai
t di
sord
erM
emor
y im
pairm
ent
Oth
erU
ndefi
nedf
63 (
67)
13 (
14)
16 (
17)
2 (2
)
9 (5
6)3
(19)
2 (1
3)2
(13)
3 (1
00)
0 (0
)0
(0)
0 (0
)
5 (4
2)6
(50)
1 (8
)0
3 (1
3)17
(71
)2
(8)
2 (8
)
16 (
30)
11 (
21)
23 (
43)
3 (6
)Im
mun
orea
ctiv
ity,
n (%
)Aβ
– H
Pt –
Aβ
+ H
Pt –
Aβ
+ H
Pt +
Aβ
– H
Pt +
53 (
56)
25 (
27)
9 (1
0)7
(7)
10 (
63)
5 (3
1)1
(6)
0 (0
)
2 (6
7)1
(33)
0 (0
)0
(0)
1 (8
)10
(83
)1
(8)
0 (0
)
4 (1
7)9
(38)
11 (
46)
0
37 (
70)
13 (
25)
3 (6
)0
(0)
APO
E ge
noty
pe,
n (%
)ε2
/ε3
ε2/ε
4ε3
/ε3
ε3/ε
4ε4
/ε4
6 (6
)0
(0)
70 (
75)
17 (
18)
1 (1
)
2 (1
3)0
(0)
11 (
69)
3 (1
9)0
(0)
0 (0
)1
(33)
2 (6
7)0
(0)
0 (0
)
0 (0
)0
(0)
4 (3
3)7
(58)
1 (8
)
1 (4
)1
(4)
9 (3
8)11
(46
)2
(8)
3 (6
)1
(2)
34 (
64)
13 (
25)
2 (4
)
47 (
7)9
(1)
446
(65)
158
(23)
27 (
4)
Tabl
e 6.
Clin
ical
cha
ract
eris
tics,
am
yloi
d-β
(Aβ)
and
hyp
erph
osph
oryl
ated
tau
(H
Pt)
prot
eins
in r
ight
fro
ntal
cor
tical
bra
in b
iops
y, a
nd a
polip
opro
tein
E (
APO
E) g
enot
ypes
of
202
pres
umed
nor
mal
pre
ssur
e hy
droc
epha
lus
(NPH
) pa
tient
s. A
POE
geno
type
s of
687
cog
nitiv
ely
heal
thy
cont
rols
als
o pr
esen
ted.
a 112
pat
ient
s di
d no
t de
velo
p AD
in t
he fol
low
-up
and
1 pa
tient
had
insu
ffici
ent
patie
nt d
ata
avai
labl
e to
dia
gnos
e or
exc
lude
AD
b 1 p
atie
nt d
ecea
sed
befo
re s
hunt
ope
ratio
n, 1
ref
used
, an
d 1
was
not
shu
nted
due
to
heav
y co
mor
bidi
ties
c Pat
ient
s w
ith a
clin
ical
dia
gnos
is o
f iN
PH (
11 s
hunt
ed,
7 be
nefit
ed),
who
dev
elop
ed A
D in
the
fol
low
-up
d Of
the
199
pres
umed
NPH
pat
ient
s w
ith s
uffic
ient
fol
low
-up
data
for
a fi
nal c
linic
al d
iagn
osis
of AD
e Cas
es w
ith o
ther
neu
rode
gene
rativ
e di
agno
sis
than
iNPH
or
AD
, in
clud
ing
2 ca
ses
with
insu
ffici
ent
data
to
diag
nose
or
excl
ude
AD
f 9 c
ases
did
not
hav
e su
ffici
ent
patie
nt d
ata
to d
eter
min
e a
lead
ing
sym
ptom
.iN
PH =
idio
path
ic N
PH,
AD
= A
lzhe
imer
’s d
isea
se,
APO
E =
apo
lipop
rote
in E

40
5.3.4 Shunt treatment and shunt responseA ventriculoperitoneal shunt (with Codman–Hakim medium pressure valve or PS Medical me-dium pressure valve, after revision in a few cases to low or high pressure valve) was inserted in 137 of 202 presumed NPH patients. Indications for the shunt treatment were: (1) basal ICP >10 mmHg but <20 mmHg continuously or (2) the presence of any A waves or more than 30% B waves during the 24h ICP monitoring period when basal pressure was between 5 and 10 mmHg (207). The clinical response to shunt was evaluated at 2–3 months at the KUH Neurosurgery outpa-tient clinic. Thereafter, patients were re-examined only if they developed symptoms suggestive of shunt malfunction or progression of disease. Any subjective or objective improvement in patient gait, memory or urinary continence was graded as a positive shunt response (87).
5.3.5 Final clinical diagnosisPatients were followed-up until death or the end of November 2010, for a median follow-up time of 3.9 years (range 0.2–17.3 years). For the final clinical diagnosis, all available clinical data from the hospitals in the KUH catchment area were retrospectively reviewed by a neurologist. Possible
385 patients’ blood samplesunavailable at the time of the
study (deceased, refused,no contact)
25 patients with insufficientfollow-up data for a clinical
diagnosis of AD
587 consecutive presumed NPH patientswith a frontal cortical brain biopsy
STUDY POPULATION202 patients with availableDNA for APOE genotyping
177 patients with sufficientfollow-up data available
124 APOE4 noncarriers 53 APOE4 carriers
35 (28 %)patients Aβ +
89 (72 %)patients Aβ –
41 (77 %)patients Aβ +
12 (23 %)patients Aβ –
8 (23 %) patientswith a final
diagnosis of AD
3 (3.4 %) patientswith a final
diagnosis of AD
17 (41 %) patientswith a final
diagnosis of AD
1 (8.3 %) patientswith a final
diagnosis of AD
Figure 11. Flowchart of 587 consecutive patients evaluated with a right frontal cortical biopsy for presumed normal pressure hydrocephalus (NPH) between 1993–2009 from an Eastern Finnish catchment population. The present study population consists of 202 patients with DNA available for genotyping, with 199 patients with suf-ficient follow-up data for the determination of Alzheimer’s disease (AD). APOE4 = apolipoprotein E ε4 allele, Aβ + = immunoreactivity for amyloid-β protein.

41
or probable AD was diagnosed in 24 patients according to the criteria of the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disor-ders Association (NINCDS–ADRDA) and Diagnostic and Statistical Manual of Mental Disorders, fourth edition (DSM-IV) (87, 242, 243). iNPH was diagnosed in 113 patients according to the follow-ing criteria: abnormal ICP findings indicating shunt surgery; no factors causing secondary NPH; and no clinical AD at the end of follow-up. In addition, there were 12 mixed cases of iNPH and AD (Table 6).
5.3.6 Immunohistochemical staining and histological evaluation of brain biopsyThe 202 paraffin embedded biopsy samples were sectioned and stained with haematoxylin–eo-sin and immunostained with monoclonal antibodies directed at Aβ (6F/3D) and HPτ (AT8), as described previously (87). In the histological evaluation, immunoreactivity for Aβ and HPτ in all samples was graded as present or absent by a neuropathologist. In 134 samples, Aβ was semiquan-tified by counting fleecy, diffuse and dense plaques in the whole cortical grey matter seen in the biopsy under light microscopy (magnification X 100) and dividing the total number of Aβ plaques by the area of grey matter (mm2).
5.3.7 APOE genotypingGenomic DNA was extracted from 200µl of venous blood (Illustra Blood GenomicPrep Mini Spin Kit; GE Healthcare, Little Chalfont, UK) and stored at –20°C. A PCR method was used in the analy-sis, as described previously (244). APOE genotyping was carried out using the ABI3100 analyser (Applied Biosystems, Carlsbad, California, USA) and allele peaks were determined using GeneMa-pper software (Applied Biosystems).
5.3.8 Statistical analysisDifferences between groups were analysed with the χ2 test for nominal variables and with the Mann–Whitney U test for continuous variables. Univariate and multivariate logistic regression analysis was used to evaluate the association of APOE4 status and other covariates with the three end variables: final diagnosis of iNPH in presumed NPH patients; presence of amyloid in cortical biopsy; and final diagnosis of AD in presumed NPH patients. Patients with both iNPH and AD as the final clinical diagnoses were excluded from the analysis of APOE in shunt responsive iNPH patients and cognitively healthy controls, and from the analysis of shunt response, but were included in all other analyses. PASW Statistics for Mac (V.18.0.3, SPSS Inc.) was used and the level of significance was set at p < 0.05.
5.3.9 Ethical issuesThe study was accepted by KUH Research Ethical Committee, The Finnish National Supervisory Authority for Welfare and Health, and The Finnish Ministry of Social Affairs and Health. All sub-jects or their proxies gave a written, informed consent prior to participation in the study.
5.4 RESULTS
5.4.1 APOE and shunt responsive iNPHThe distribution of the APOE genotypes was similar in the 94 shunt responsive iNPH patients (12 developed AD during follow-up and were excluded) and in the 687 healthy elderly controls (p = 0.27) (Table 6). The proportion of APOE4 carriers was lower in the shunt responsive iNPH patients (19%) compared with the control population (28%) (p = 0.06). Both groups had a similar gender distribution (56% vs. 60% females) and median age (70 years vs. 70 years). There were fewer APOE4 carriers in patients with a final clinical diagnosis of iNPH (24%) compared with presumed NPH patients with a different final clinical diagnosis (44%; p = 0.008) (Table 6).
5.4.2 APOE and shunt nonresponsive iNPHThere were 16 iNPH patients who did not respond to shunt. They did not develop clinical AD in a median follow-up time of 5.1 years (Table 6). The 94 shunt responsive and 16 nonresponsive iNPH patients had a similar distribution of APOE genotypes (p = 0.47) and proportion of APOE4 carriers

42
(19% vs. 19%; p = 0.72).
5.4.3 APOE and Aβ plaques in brain biopsyThere were 199 patients with APOE status, immunohistochemical evaluation of the right frontal cortical biopsy and sufficient follow-up data available to diagnose or exclude AD (Figure 11). Aβ plaques were detected in 88 cases (44%) at a median age of 75 years and no Aβ plaques were seen in 111 cases at a median age of 71 years. APOE4 allele was present in 60 cases (30%) (Figure 11). In the multivariate logistic regression analysis (Table 7), independent predictors of Aβ plaques were APOE4 (odds ratio (OR) 8.7, 95% confidence interval (CI) 3.8–20), age at biopsy (OR/year 1.08, 95% CI 1.03–1.1) and final clinical diagnosis of AD (OR 7.9, 95% CI 2.3–27). Aβ load (plaques/mm2), semiquantified in 134 cases, was higher (p < 0.001) in APOE4 carriers (mean 12) than in non-APOE4 carriers (mean 2.5).
5.4.4 APOE and final clinical diagnosis of ADOf the 199 patients, 24 had a final clinical diagnosis of AD at a median follow-up time of 3.3 years and, in addition, 12 iNPH patients developed AD at a median follow-up time of 2.9 years. Of the combined 36 AD cases, 22 (61%) were APOE4 carriers. In the multivariate logistic regression analy-sis (Table 7), memory impairment at biopsy (OR 9.3, 95% CI 3.4–26), clinical diagnosis of iNPH (OR 0.19, 95% CI 0.06–0.55), both Aβ plaques and HPτ (OR 9.2, 95% CI 2.0–43), or Aβ plaques without HPτ (OR 5.8, 95% CI 1.6–22) in the cortical brain biopsy were independent predictors of AD. In uni-variate logistic regression, APOE4 associated with AD (OR 4.3, 95% CI 2.0–9.3, p < 0.001), but when adjusted for other covariates in a multivariate model, the OR halved (OR 1.8, 95% CI 0.61–5.5, p = 0.23), which was mainly explained by inclusion of cortical pathology to the analysis (Table 7). The APOE4 allele in presumed NPH predicts AD as follows: sensitivity 61%; specificity 77%; positive predictive value 37%; negative predictive value 90%.

43
Cov
aria
tes
iNP
H a
s en
d v
aria
ble
(n
= 1
70
)aA
β a
s en
d v
aria
ble
(n
= 1
99
)bA
D a
s en
d v
aria
ble
(n
= 1
92
)c
nO
dd
s ra
tio
(95
% C
I)p
nO
dd
s ra
tio
(95
% C
I)p
nO
dd
s ra
tio
(95
% C
I)p
Age
at
biop
sy/y
ear
1.02
(0.
98–1
.07)
0.35
1.08
(1.
03–1
.13)
0.00
21.
06 (
0.99
–1.1
4)0.
07
Mal
eFe
mal
e79 91
1 2.13
(0.
99–4
.61)
Ref
0.05
95 104
1 1.65
(0.
79–3
.44)
Ref
0.18
92 100
1 1.67
(0.
57–4
.84)
Ref
0.35
APO
E4 n
onca
rrie
rAPO
E4 c
arrier
116
541 0.
73 (
0.29
–1.8
2)Re
f0.
5013
960
1 8.67
(3.
75–2
0.05
)Re
f<
0.00
0113
260
1 1.83
(0.
61–5
.46)
Ref
0.28
Lead
ing
sym
ptom
Oth
er t
han
gait
diso
rder
mem
ory
impa
irm
ent
Gai
t di
sord
erM
emor
y im
pairm
ent
86 84
1 4.41
(1.
93–1
0.09
)
Ref
<0.
001
149
50
1 0.81
(0.
31–2
.11)
Ref
0.67
142
50
1 9.33
(3.
38–2
5.77
)
Ref
<0.
0001
Imm
unoh
isto
chem
istr
yAβ
– H
Pt –
Aβ
+ H
Pt –
Aβ
+ H
Pt +
Aβ
– H
Pt +
89 57 23
1 1.01
(0.
36–2
.80)
0.61
(0.
16–2
.30)
Excl
uded
Ref
0.99
0.47
105
63 24
1 5.84
(1.
56–2
1.83
)9.
21 (
1.99
–42.
73)
Excl
uded
Ref
0.00
90.
005
NPH N
ot N
PHiN
PHsN
PH
53 124
22
1 1.06
(0.
42–2
.64)
0.96
(0.
23–4
.07)
Ref
0.91
0.96
53 117
22
1 0.19
(0.
06–0
.55)
0.54
(0.
79–3
.71)
Ref
0.00
20.
53Alz
heim
er’s
dis
ease
Not
pre
sent
Pres
ent
137
331 0.
24 (
0.09
–0.6
5)Re
f0.
005
163
361 7.
91 (
2.30
–27.
17)
Ref
0.00
1
Tabl
e 7.
Mul
tivar
iate
logi
stic
reg
ress
ion
anal
yses
of 2
02 p
resu
med
nor
mal
pre
ssur
e hy
droc
epha
lus
(NPH
) pa
tient
s w
ith a
fron
tal c
ortic
al b
iops
y an
d ap
olip
opro
tein
E (
APO
E)
geno
type
ava
ilabl
e.
a 22
sNPH
pat
ient
s, 7
iNPH
pat
ient
s w
ith A
β –
HPt
+,
and
3 pa
tient
s w
ith in
suffi
cien
t fo
llow
-up
data
for
a fi
nal c
linic
al d
iagn
osis
of
AD
wer
e ex
clud
ed f
rom
the
ana
lysi
sb 3
pat
ient
s w
ith in
suffi
cien
t fo
llow
-up
data
for
a fi
nal c
linic
al d
iagn
osis
of AD
wer
e ex
clud
ed fro
m t
he a
naly
sis
c 7 n
on-A
D p
atie
nts
with
Aβ
– H
Pt +
, an
d 3
patie
nts
with
insu
ffici
ent
follo
w-u
p da
ta for
a fi
nal c
linic
al d
iagn
osis
of AD
wer
e ex
clud
ed f
rom
the
ana
lysi
s.iN
PH =
idio
path
ic N
PH,
Aβ
= a
myl
oid
beta
pro
tein
, AD
= A
lzhe
imer
’s d
isea
se,
CI
= c
onfid
ence
inte
rval
, APO
E4 =
apo
lipop
rote
in E
ε4
alle
le,
HPt
= h
yper
phos
phor
ylat
ed
tau
prot
ein,
sN
PH =
sec
onda
ry N
PH

44
5.5 DISCUSSION
This is the first sizeable cohort to assess the prognostic value of APOE alleles in iNPH patients. The APOE genotypes were equally distributed in shunt responsive iNPH patients and cognitively unimpaired controls, and did not predict the shunt response in iNPH patients. On the other hand, the APOE4 allele independently predicted Aβ plaques in the frontal cortical biopsy. The strengths of our study included: a large NPH cohort evaluated by 24h intraventricular ICP monitoring and frontal cortical biopsy; long term follow-up for possible development of AD or other forms of de-mentia; and a large cognitively unimpaired control population. Weaknesses included: a retrospec-tive study; only six APOE4 homozygotes; a limited number of patients with no shunt response; systematic assessment of shunt response only at 2–3 months; dichotomised shunt response that overlooked the symptom triad of iNPH; and lack of validated objective outcome measures. It is clear that at least in prospective clinical trials, shunted patients should be followed-up routinely for a significantly longer time and with symptom specific outcome scales. In contrast with our study, one small series suggested that APOE4 would be overrepresented in NPH patients (22). The study included 13 patients (mean age 72 years) with MRI and neuropsy-chological data suggestive of NPH and 108 apparently normal controls (mean age 81 years) with no neurological disorders. In our study, iNPH patients were evaluated by brain imaging, ICP monitor-ing and frontal cortical biopsy, and they were followed-up for a shunt response and for possible later clinical development of other forms of dementia. Thus with a notably larger iNPH patient cohort, more sound methodology and better differentiation of AD cases, our data is more reliable. In the diagnostic workup of presumed NPH, APOE genotyping would be beneficial as APOE4 allele carriership indicates an increased risk of AD – or iNPH with concomitant AD. Importantly, the APOE4 allele does not exclude iNPH. Furthermore, APOE genotypes did not predict shunt response in our 121 shunted iNPH patients. Instead, in a series of 15 iNPH patients evaluated by gait analysis before and after CSF drainage, the APOE ε3/ε3 genotype seemed to predict improved gait (23). It should be noted that the cited study bears significant weaknesses, including a very small number of patients and no detection of primary or comorbid AD patients by means of brain biopsy, potentially invalidating the shunt response. Taking these facts into account, we regard our findings as more solid. However, the value of APOE genotyping in the prediction of shunt response should be evaluated in further studies with a longer follow-up of shunt response and by detailed analysis of gait, cognition (e.g., CERAD (245)) and urinary incontinence (e.g., structured NPH-questionnaires). There is no consensus on the indications for shunt treatment in iNPH based on ICP diagnostics in literature. Some of our patients whose ICP monitoring did not support NPH might have benefit-ed from a shunt. In contrast, a significant number of patients did not respond to shunt despite posi-tive ICP findings. On the other hand, classification of true or definite iNPH often includes a positive response to shunt treatment. However, the response may be impeded by concomitant diseases or progression of iNPH beyond the possible point of no return. As the presumed NPH patients in our cohort were referred to KUH Neurosurgery presenting with NPH-related symptoms and history, in addition to CT and/or MRI scans with signs of enlarged brain ventricles disproportionate to the size of the sulci in the cerebral convexities, presumed NPH corresponds to possible iNPH accord-ing to the iNPH guidelines (7). Probable iNPH in the guidelines (7) translates to having a final clini-cal diagnosis of iNPH in our cohort as these patients had ICP abnormalities in further physiological diagnostic tests. In our study, the APOE4 allele was strongly associated (OR 8.7) with Aβ plaques in the frontal cortical brain biopsy. Semiquantification of the Aβ load in the biopsies further emphasised the as-sociation, which is in line with previous autopsy studies (231–235) and recent PET imaging studies of Aβ (229, 230). Along with previous studies, we also found a significant association of age to Aβ deposits in the brain. Our study is the first to evaluate the association of APOE4 with Aβ plaques in the living human brain. Importantly, in our study and in a PET study for amyloid (230), accumula-tion of Aβ was detected approximately three years earlier in APOE4 carriers than in non-carriers. Aβ accumulation in the brain is a potential preclinical indicator for the risk of future AD, exist-ing far earlier than any clinical signs of the disease (149, 246). Brain biopsy provides a unique re-search window to the living human brain and also allows the study of the relationship of APOE4 or

45
other biomarkers to molecular biology and the progress of AD. The current AlzGene meta-analysis (247) gives an OR of 3.68 for APOE4 versus APOE3 as a single biomarker in the prediction of AD. In our study, the OR was 4.3 in the univariate analysis, but addition of Aβ and HPτ pathology in the frontal cortical biopsy to the analysis, diluted the OR of APOE4 to 1.8. In a previous study of 198 patients with amnesic mild cognitive impairment and 98 AD patients, APOE4 dose correlated stronger with low CSF Aβ1–42 levels than with cognitive status (248). In the present study, there were six APOE4 homozygotes and three had a final clinical diagnosis of AD. The large variation in follow-up time of patients may have had an effect on the results concerning AD patients, as the principal risk factor for AD is age, and many more patients would have deteriorated if followed-up for longer. AD cannot be diagnosed solely on APOE genotype. However, APOE polymorphisms can predict Aβ pathology in the brain, and combined with other biomarkers, probably increase the diagnostic accuracy of AD. Further research is needed to elucidate the genetics and pathobiology of iNPH as well as the potential diagnostic and prognostic biomarkers. In addition, prospective follow-up studies are re-quired to clarify the predictive value of AD-related pathology and genetics to the long term re-sponse to CSF shunt treatment in iNPH. In conclusion, in presumed NPH patients, APOE4 associates with the Aβ plaques in the frontal cortical biopsy. APOE4 is not a risk factor for iNPH and does not predict the response to shunt.

46

47
6 Cerebrospinal fluid biomarker and brain biopsy findings in idiopathic normal pressure hydrocephalus
6.1 ABSTRACT
6.1.1 ObjectiveTo investigate the role of soluble amyloid precursor protein (sAPP) and amyloid beta (Aβ) iso-forms, proinflammatory cytokines, and biomarkers of neuronal damage in the cerebrospinal fluid (CSF) in relation to brain biopsy Aβ and hyperphosphorylated tau (HPτ) findings in patients with idiopathic normal pressure hydrocephalus (iNPH) and Alzheimer’s disease (AD).
6.1.2 MethodsThe study population comprised 102 patients with possible NPH with cortical brain biopsies, ven-tricular and lumbar CSF samples, and DNA available. The final clinical diagnoses were: 53 iNPH (91% shunt-responders), 26 AD (10 mixed iNPH-AD), and 23 others. Biopsy samples were immu-nostained against Aβ and HPτ. CSF levels of AD-related biomarkers (Aβ42, p-tau, total tau), non-AD-related Aβ isoforms (Aβ38, Aβ40), sAPP isoforms (sAPPα, sAPPβ), proinflammatory cytokines (several interleukins (IL), interferon-gamma, monocyte chemoattractant protein-1, tumor necrosis factor-alpha) and biomarkers of neuronal damage (neurofilament light and myelin basic protein) were measured. All patients were genotyped for APOE.
6.1.3 ResultsLumbar CSF levels of sAPPα were lower (p<0.05) in patients with shunt-responsive iNPH com-pared to non-iNPH patients. sAPPβ showed a similar trend (p=0.06). CSF sAPP isoform levels showed no association to Aβ or HPτ in the brain biopsy. Quantified Aβ load in the brain biopsy showed a negative correlation with CSF levels of Aβ42 in ventricular (r=–0.295, p=0.003) and lumbar (r=–0.356, p=0.01) samples, while the levels of Aβ38 and Aβ40 showed no correlation. CSF levels of proinflammatory cytokines and biomarkers of neuronal damage did not associate to the brain biopsy findings, diagnosis, or shunt response. Higher lumbar/ventricular CSF IL-8 ratios (p<0.001) were seen in lumbar samples collected after ventriculostomy compared to the samples collected before the procedure.
6.1.4 ConclusionsThe role of sAPP isoforms in iNPH seems to be independent from the amyloid cascade. No neuro-inflammatory background was observed in iNPH or AD.
6.2 INTRODUCTION
Idiopathic normal pressure hydrocephalus (iNPH) is a progressive neurodegenerative disorder of unknown etiology in the elderly presenting with gait disorder, cognitive impairment, and urinary incontinence, with enlarged ventricles of the brain but normal or slightly elevated cerebrospinal fluid (CSF) pressure (2, 3). Currently there is no pathological hallmark for iNPH (144). Studies sug-gesting some potential genetic background of iNPH have been published (18, 20). The present treat-ment of choice in iNPH is CSF diversion with an implanted shunt that relieves or even reverses the symptoms. Various procedures to evaluate CSF dynamics in patients with possible iNPH are used to identify those who could benefit from CSF shunting. These include the CSF tap test, external lumbar drainage test, infusion tests, and intraventricular or intracranial pressure (ICP) monitoring

48
(7, 70, 87, 120). The most frequent differential diagnoses of iNPH are atypical Alzheimer’s disease (AD) and vascular dementia (87, 88). AD is characterized by the hallmark lesions of amyloid-β (Aβ) plaques and neurofibrillary tan-gles composed of hyperphosphorylated tau (HPτ) in the brain of patients with amnestic cognitive decline (146, 147, 227). The amyloid cascade hypothesis states that Aβ starts to accumulate decades before the clinical manifestations of AD (148, 228). In vivo, Aβ can be detected directly with brain biopsy (87, 153), or indirectly by observing low levels of Aβ in CSF (26). Fibrillar Aβ can also be evaluated by positron emission tomography (PET) utilizing the 11C-labelled Pittsburgh compound B (150) or [18F]flutemetamol (151). Although common pathways for iNPH and AD have been proposed (83), the findings in genetic (249) and Aβ studies (87) suggest differences in etiologies of the two diseases. Aβ and HPτ in the CSF may help to differentiate iNPH and AD patient groups or detect comorbid AD in iNPH (26). In addition, these biomarkers have shown a potency to predict response to shunt in iNPH (174, 250). Aβ originates from a cell membrane-spanning protein, amyloid precursor protein (APP), which has diverse roles in normal neuronal function (126). Soluble APP alpha (sAPPα) and beta (sAPPβ) result from the cleavage of APP by α- and β-secretases, respectively. Low CSF levels of sAPP iso-forms have been reported in post-stroke and iNPH patients compared to AD and normal healthy controls (127–129, 251). In addition, sAPPα has shown a marked prognostic value for cognitive performance following shunt surgery (128), and subsequent increase of ventricular sAPP-levels has been noted in shunt-responders (129). Abnormal levels of proinflammatory cytokines, such as interleukins (IL), interferon-gamma (IFN-γ), monocyte chemoattractant protein-1 (MCP-1), and tumor necrosis factor-alpha (TNF-α), in CSF have been noted in various diseases of the nervous system, including AD (252). In iNPH, several proinflammatory cytokines have been studied, but none of them have proven to be useful in diagnostics (130). Lower levels of IL-1β in NPH compared to AD were reported in a single paper (131), while increased levels of IL-4 and IL-10 were reported in patients with NPH compared to healthy individuals in another study, but no significant difference was seen between NPH and oth-er dementias (133). No differences were found between NPH and AD or healthy controls in studies comparing the levels of IL-8, IL-10, IL-12 (p40 and p70), IFN-γ, and transforming growth factor-β1 (TGF- β1) (129, 134). However, iNPH patients did show increased levels of MCP-1 compared to healthy individuals (129). Prior to treatment, higher TNF-α concentrations (and subsequent nor-malization after shunting) in CSF were observed in NPH patients compared to healthy controls in a single study (132); however, these results did not replicate in a more recent study with solely idiopathic NPH patients (135). Elevated levels of neurofilament light protein (NFL) in the CSF, indicating neuronal death and axonal loss, have been found in iNPH and secondary NPH in several studies (129, 137–140). In-creased levels of myelin basic protein (MBP) in the CSF is a well-established biomarker for demy-elination and myelin damage in the central nervous system (253). Furthermore, elevated levels of MBP have been reported in NPH (254). To our knowledge, studies assessing the association between proinflammatory cytokines and biomarkers of neuronal damage in CSF, and cortical brain biopsy have not been published to date.
The objectives of the current study were:1. to determine the levels of AD-related biomarkers (Aβ42, p-tau, total tau), non-AD-related Aβ
isoforms (Aβ38, Aβ40), sAPP isoforms (sAPPα, sAPPβ), proinflammatory cytokines (IL 1β, 2, 4, 5, 8, 10, 12p70, and 13, IFN-γ, MCP-1, TNF-α) and biomarkers of neuronal damage (NFL, MBP) in lumbar and ventricular CSF, and how they correlate;
2. study the relationship between the CSF biomarkers and cortical brain biopsy;3. assess the diagnostic and prognostic value of the CSF biomarkers in iNPH and AD.
6.3 METHODS
6.3.1 Ethics statementThis study was approved by the Kuopio University Hospital (KUH) Research Ethical Committee, The Finnish National Supervisory Authority for Welfare and Health, and The Finnish Ministry of

49
Social Affairs and Health. All participants or their proxies gave a written, informed consent prior to participation in the study. If the clinician suspected dementia to significantly affect the capacity of the patient to consent, a next of kin, caretaker or guardian consented on behalf of the participant. When a consent was obtained from a participant’s proxy, the patient’s own opinion was inquired and considered, and no patient was recruited against their will.
6.3.2 Kuopio NPH Registry and protocolNeurosurgery of KUH solely provides full-time acute and elective neurosurgical services for the KUH catchment population in Middle and Eastern Finland. In addition, the KUH area contains four central hospitals with neurological units and catchment areas of their own. Patients fulfilling the following criteria were further assessed in KUH Neurosurgery as possible NPH patients: (1) primary evaluation and examination by a neurologist indicating NPH; (2) one to three symptoms suggestive of NPH (gait disorder, cognitive impairment, urinary incontinence); and (3) NPH related brain imaging findings (enlarged ventricles (Evans’ index > 0.3) together with obliterated cortical sulci). The diagnostic workup protocol of KUH Neurosurgery for possible NPH included a clinical examination, computed tomography (CT) or magnetic resonance imaging (MRI) scan, and 24h intraventricular ICP monitoring together with a frontal cortical brain biopsy. Kuopio NPH Registry (www.uef.fi/nph) consists of all evaluated possible NPH patients from the KUH catchment population since 1993. The ICP criteria for shunt treatment in iNPH patients were (1) a basal ICP pressure between 10 and 20 mmHg continuously, or (2) the presence of A-waves or more than 30% B-waves during the 24h monitoring, when basal pressure was between 5 and 10 mmHg (207).
6.3.3 ParticipantsAltogether 102 patients, 51 men and 51 women, with a median age of 74.6 years (range 47–87 years) from the Kuopio NPH Registry with cortical brain biopsy, APOE genotype, and a ventricular CSF sample available were included in the study (Table 8). 63 patients were diagnosed with iNPH according to the protocol above, and were shunted with ventriculoperitoneal shunt (PS Medical medium pressure or adjustable valve). The clinical response to shunt was evaluated at 2–3 months after surgery, and any subjective or objective improvement in patient gait, memory or urinary continence was graded as a positive shunt response. Clinical AD was diagnosed according to a pro-tocol described earlier (87, 88) in 26 patients (including 10 patients with initial/primary diagnosis of iNPH) in a median follow-up time of 2.3 years (range 0.2–6.2 years).

50
Pos
sib
le N
PH
(n
= 1
02
)
Fin
al c
linic
al d
iag
nos
is o
f iN
PH
(n =
53
)N
o d
iag
nos
is o
f iN
PH
(n =
39
)C
har
acte
rist
ics
Sh
un
t re
spon
der
Sh
un
t n
onre
spon
der
Mix
ed iN
PH
+ A
DA
DO
ther
n48
510
1623
Age
(ye
ars)
(m
edia
n (r
ange
))72
.7 (
63.8
–87.
3)82
.8 (
79.9
–86.
2)78
.4 (
69.8
–86.
7)78
.3 (
54.1
–85.
7)70
.6 (
47.1
–81.
7)
Wom
en (
n (%
))23
(48
)3
(60)
3 (3
0)10
(63
)23
(52
)Fo
llow
-up
time
(yea
rs)
(med
ian
(ran
ge))
2.51
(0.
82–6
.22)
2.16
(1.
27–3
.21)
1.95
(0.
99–3
.60)
1.88
(0.
35–3
.06)
2.35
(0.
19-4
.88)
Lead
ing
sym
ptom
, n
(%)*
Gai
t di
sord
er29
(73
)3
(100
)2
(29)
3 (2
1)8
(42)
Mem
ory
impa
irm
ent
9 (2
3)0
(0)
5 (7
1)10
(71
)5
(26)
Oth
er2
(5)
0 (0
)0
(0)
1 (7
)6
(31)
Imm
unor
eact
ivity
(n
(%))
Aβ
– H
Pτ –
24 (
50)
2 (4
0)3
(30)
4 (2
5)15
(65
)Aβ
+ H
Pτ –
17 (
35)
2 (4
0)2
(20)
6 (3
8)6
(26)
Aβ
+ H
Pτ +
4 (8
)1
(20)
5 (5
0)6
(38)
1 (4
)Aβ
– H
Pτ +
3 (6
)0
(0)
0 (0
)0
(0)
1 (4
)APO
E-ε4
car
rier
s (n
(%
))9
(19)
2 (4
0)7
(70)
7 (4
4)4
(17)
Tabl
e 8.
Cha
ract
eris
tics,
bra
in b
iops
y fin
ding
s, a
nd a
polip
opro
tein
E ε
4 (A
POE-
ε4)
stat
uses
of 10
2 pa
tient
s w
ith p
ossi
ble
norm
al p
ress
ure
hydr
ocep
halu
s (N
PH).
*Suf
ficie
nt d
ata
for
the
dete
rmin
atio
n of
a le
adin
g sy
mpt
om w
as u
nava
ilabl
e in
10
iNPH
pat
ient
s, 3
mix
ed iN
PH +
AD
pat
ient
s, 2
AD
pat
ient
s an
d 4
patie
nts
with
oth
er fi
nal
clin
ical
dia
gnos
isAPO
E =
apo
lipop
rote
in E
gen
e, C
SF
= c
ereb
rosp
inal
flui
d, N
PH =
nor
mal
pre
ssur
e hy
droc
epha
lus,
iN
PH =
idi
opat
hic
NPH
, AD
= A
lzhe
imer
’s d
isea
se,
Aβ
= a
myl
oid
beta
pr
otei
n, H
Pτ =
hyp
erph
osph
oryl
ated
tau
pro
tein

51
6.3.4 Immunohistochemistry and histological evaluationBiopsy samples representing frontal cortex and subcortical white matter were stained with hema-toxylin–eosin and immunostained with monoclonal antibodies directed to Aβ (6F/3D) and HPτ (AT8) as described earlier in detail (87). Positive Aβ immunostain was further quantified and re-ported as the ratio of area covered by Aβ to total area of cortex in the biopsy as described earlier (26).
6.3.5 APOE genotypingDNA was extracted from venous blood using commercial kit according to manufacturer’s protocol (Illustra Blood GenomicPrep Mini Spin Kit, GE Healthcare, Little Chalfont, UK). A standard PCR method was used in the APOE genotyping (244).
6.3.6 CSF samples and biomarker analysesThe ventricular CSF samples were collected immediately after the placement of the intraventricu-lar catheter (first 1 mL discarded) in the ICP measurement procedure. In addition, a lumbar CSF sample was available in 49 patients. The lumbar samples were obtained through a lumbar puncture prior to the ICP measurement protocol (12 patients) or 24 –48 hours after introducing the ventricular catheter (37 patients). Levels of AD biomarkers (Aβ42, p-tau, total tau) were measured from the CSF samples using commercial ELISA kits (Innotest β-amyloid1–42, Innotest Phosphotau(181P), Innotest Tau-Ag, In-nogenetics, Ghent, Belgium) according to the manufacturer’s protocol at a validated laboratory in Neurology (www.uef.fi/neuro), University of Eastern Finland, Kuopio, Finland as described earlier (26). Aβ isoforms (Aβ38, Aβ40), sAPP isoforms (sAPPα, sAPPβ), and the proinflammatory cytokines (IL 1β, 2, 4, 5, 8, 10, 12p70, and 13, IFN-γ, MCP-1, TNF-α) were analyzed utilizing commercially available multiplexed assays (Meso Scale Discovery, Gaithersburg, MD, USA) (129, 255), and NFL and MBP concentrations were measured using commercial ELISA kits (NF-Light, UmanDiag-nostics, Umeå, Sweden, and ACTIVE MBP, Diagnostic Systems Laboratories, Webster, TX, USA, respectively). All analyses were performed according to the manufacturers’ protocols by board-certified laboratory technicians at the Clinical Neurochemistry Laboratory, Sahlgrenska University Hospital, Mölndal. Each set of biomarker measurements was performed on one day, using one batch of reagents. All clinical, immunohistochemical, and laboratory analyses were performed blinded to the re-sult information of each other.
6.3.7 Statistical analysisNonparametric Kruskal–Wallis H and Mann–Whitney U tests were used for comparing CSF levels of measured biomarkers between different groups, and the Wilcoxon signed-rank test for com-parisons of two related samples. To define the correlation between different proinflammatory cy-tokines, Pearson correlation with Bonferroni correction (k = 55) was applied. Patients with tauopathy but no amyloid (n = 4) were excluded from the analyses of different biomarkers in relation to biopsy findings, and iNPH patients with comorbid AD (n = 10) and iNPH patients with negative shunt-response (n = 5) from the analyses comparing true iNPH patients to non-iNPH patients. Some cytokines were below the lower limit of detection of the assays and graded as zero concentration in statistical analyses. One patient had an insufficient CSF sample for the analysis of sAPPs, and another for the analysis of Aβ isoforms Aβ38 and Aβ40. IBM SPSS Statistics for Mac (version 19.0.0.2, IBM, Armonk, NY, USA) was used in the statistical analyses. The level of significance was set at p < 0.05.
6.4 RESULTS
6.4.1 Aβ and sAPP isoformsLumbar CSF levels of sAPPα were significantly lower (p < 0.05) in patients with iNPH and a posi-tive shunt reponse compared to non-iNPH patients, while sAPPβ showed a similar tendency (p = 0.06, Table 9, Figure 12B and 12D). However, no such association was seen for sAPP isoforms in

52
ventricular CSF samples (p = 0.37–0.47, Table 9, Figure 12A and 12C). In iNPH patients, a tendency towards lower levels of sAPPα and sAPPβ were observed (p = 0.23–0.49) in shunt-responders com-pared to nonresponders in ventricular and lumbar CSF (Table 9). Ventricular CSF levels of Aβ42 differed (p = 0.003) between different brain biopsy groups (Table 10, Figure 13C). Patients with positive Aβ and HPτ immunoreactivity in the cortical brain biopsy showed significantly lower CSF levels of Aβ42 compared to the Aβ positive and HPτ negative group (post hoc p = 0.008), and to the Aβ and HPτ negative group (post hoc p = 0.005, Table 10, Fig-ure 13C). Similar associations were seen in lumbar samples (data not shown). Quantified Aβ load showed a negative correlation with the levels of CSF Aβ42 in ventricular (Pearson’s r = –0.295, p = 0.003) and lumbar (Pearson’s r = –0.356, p = 0.01) samples (Figure 14). However, the CSF levels of other Aβ isoforms (Aβ38, Aβ40) and sAPP isoforms (sAPPα, sAPPβ) did not correlate (p = 0.59–0.95) with the presence of Aβ or HPτ in the biopsy (Table 10, Figure 13A–B). Furthermore, there was no correlation between the levels of sAPP isoforms in the CSF and Aβ load in cortical brain biopsy (p = 0.84–0.92). There were no statistically significant differences between the levels of CSF Aβ or tau biomarkers in shunt-responding and nonresponding iNPH patients.

53
Possible NPH (n = 102)Final clinical diagnosis of iNPH (n = 53)
No diagnosis of iNPH(n = 39)
CSF Biomarker*
Shunt responder
Shunt non-responder
Mixed iNPH + AD
AD Other
n 48 5 10 16 23Aβ42
Lumbar 587 (141) 563 (241) 487 (256) 584 (248) 667 (181)Ventricular 476 (203) 428 (250) 450 (134) 422 (259) 542 (271)
P-tau 181Lumbar 35.3 (15.5) 38.0 (14.8) 41.1 (22.1) 47.1 (13.6) 43.2 (15.5)Ventricular 77.1 (51.7) 50.4 (14.3) 93.1 (39.9) 81.3 (51.2) 91.8 (133)
Total tauLumbar 239 (156) 255 (121) 211 (135) 294 (164) 252 (98.9)Ventricular 1210 (1186) 562 (443) 1500 (1320) 1361 (1687) 1698 (3313)
sAPPαLumbar 217 (156) 472 (560) 350 (137) 325 (238) 424 (300)Ventricular 237 (241) 374 (484) 261 (193) 223 (216) 304 (284)
sAPPβLumbar 84.3 (63.4) 193 (200) 102 (44.9) 108 (67.9) 158 (101)Ventricular 88.3 (92.3) 169 (235) 110 (92.6) 83.6 (81.7) 123 (109)
IL-8Lumbar 1101 (2440) 318 (374) 85.7 (73.1) 461 (592) 466 (420)Ventricular 20.2 (20.8) 33.0 (51.3) 23.0 (5.76) 85.9 (286) 19.1 (11.0)
IL-8 ratioLumbar/ventricular 132 (446) 55.0 (84.1) 3.05 (2.01) 51.7 (105) 20.9 (14.5)
MCP-1Lumbar 3398 (2865) 1876 (766) 785 (515) 2618 (2881) 3262 (2061)Ventricular 748 (280) 1096 (688) 784 (187) 819 (436) 758 (264)
TNF-αLumbar 4.38 (7.80) 1.88 (1.40) 0.50 (0.86) 2.56 (3.45) 2.76 (2.58)Ventricular 0.31 (0.62) 0.28 (0.63) 0.25 (0.55) 0.57 (1.16) 0.00 (0.00)
NFLLumbar 2511 (1798) 6545 (6242) 2153 (830) 2007 (867) 1399 (538)Ventricular 886 (681) 6692 (9723) 1993 (1715) 1567 (2152) 1010 (864)
MBPLumbar 117 (170) 27.0 (9.93) 17.5 (–) 49.9 (57.7) 90.6 (61.7)Ventricular 8.22 (12.1) 12.9 (19.2) 50.5 (73.5) 9.03 (18.8) 21.2 (30.8)
Table 9. Cerebrospinal fluid (CSF) biomarker levels of 102 patients with possible normal pressure hydrocephalus (NPH).
*Mean (SD) CSF concentrations in ng/L.CSF = cerebrospinal fluid, Aβ42 = amyloid beta 1-42, p-tau 181 = tau phosphorylated at threonine 181, sAPP = soluable amyloid precursor protein, IL-8 = interleukin 8, MCP-1 = monocyte chemoattractant protein-1, TNF-α = tumor necrosis factor-alpha, NFL = neurofilament light protein, MBP = myelin basic protein

54
ImmunoreactivityVariable Aβ– HPτ– Aβ+ HPτ– Aβ– HPτ+ Aβ+ HPτ+ Totaln
Lumbar 23 17 1 8 49Ventricular 48 33 4 17 102
Aβ38*Lumbar 682 (370) 754 (365) 923 (–) 849 (352) 739 (360)Ventricular 583 (612) 533 (305) 564 (544) 615 (464) 572 (499)
Aβ40*Lumbar 6105 (3007) 6,170 (2157) 8143 (–) 6735 (1916) 6272 (2521)Ventricular 4544 (3162) 4,768 (2203) 4858 (3598) 5101 (3082) 4721 (2855)
Aβ42* Lumbar 641 (184) 596 (167) 758 (–) 393 (108) 588 (187)Ventricular 529 (240) 485 (188) 463 (327) 320 (162) 477 (225)
P-tau 181* Lumbar 37.8 (15.9) 37.0 (15.0) 45.6 (–) 48.9 (16.0) 39.5 (15.7)Ventricular 81.3 (97.3) 73.6 (47.3) 83.5 (23.3) 95.8 (57.1) 81.3 (75.5)
Total tau* Lumbar 238 (139) 246 (164) 287 (–) 296 (127) 251 (143)Ventricular 1423 (2386) 1054 (1104) 1387 (415) 1652 (1970) 1340 (1924)
sAPPα* Lumbar 300 (287) 288 (211) 178 (–) 323 (261) 298 (251)Ventricular 266 (283) 264 (263) 179 (190) 249 (184) 259 (256)
sAPPβ* Lumbar 112 (106) 110 (77.0) 92 (–) 106 (75.8) 110 (89.5)Ventricular 104 (116) 105 (107) 69.5 (65.2) 95.3 (76.6) 102 (105)
IL-8* Lumbar 852 (2299) 435 (732) 197 (–) 1218 (1901) 754 (1792)Ventricular 43.5 (165) 22.8 (24.3) 16.9 (8.04) 15.6 (10.0) 31.1 (114)
IL-8 ratio**
Lumbar/ventricular 154 (525) 36.9 (51.3) 9.12 (–) 154 (133) 112 (370)
MCP-1* Lumbar 2994 (2628) 2519 (2354) 3059 (–) 3637 (3323) 2935 (2602)Ventricular 767 (391) 820 (304) 704 (191) 771 (165) 782 (327)
TNF-α* Lumbar 2.77 (4.94) 2.55 (2.85) 1.27 (–) 6.88 (11.2) 3.34 (5.91)Ventricular 0.28 (0.74) 0.22 (0.59) 0.36 (0.72) 0.34 (0.67) 0.27 (0.67)
NFL* Lumbar 2216 (2044) 3135 (3073) 1180 (–) 2479 (1987) 2557 (2419)Ventricular 1127 (1385) 2082 (4153) 618 (448) 1213 (1307) 1430 (2615)
MBP* Lumbar 71.5 (77.6) 82.2 (71.6) 48.9 (–) 160 (275) 89.9 (131)Ventricular 12.7 (24.2) 17.1 (33.5) 30.5 (23.5) 9.65 (18.7) 14.4 (26.8)
Table 10. Lumbar and ventricular cerebrospinal fluid (CSF) biomarkers in different brain biopsy findings.
*Mean (SD) CSF concentrations in ng/L.**Mean (SD). Only cases with CSF sample obtained after 24h ICP monitoring included (n = 37).Aβ = amyloid beta protein, HPτ = hyperphosphorylated tau protein, Aβ38/40/42 = amyloid beta 1-38/40/42, p-tau 181 = tau phosphorylated at threonine 181, sAPP = soluable amyloid precursor protein, IL-8 = interleukin 8, MCP-1 = monocyte chemoattractant protein-1, TNF-α = tumor necrosis factor-alpha, NFL = neurofilament light protein, MBP = myelin basic protein
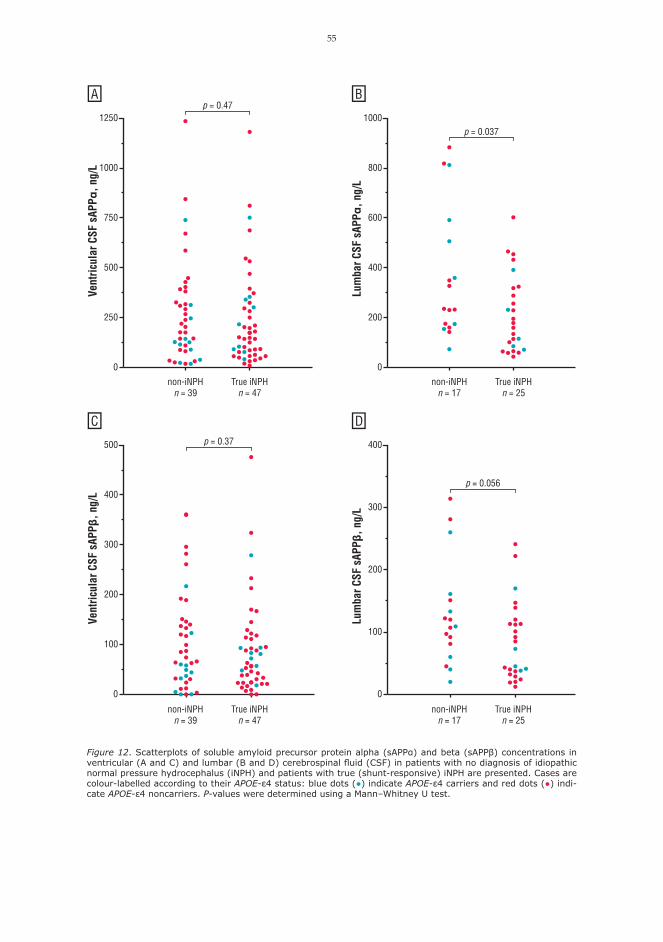
55
Vent
ricul
ar C
SF s
APPα
, ng/
L
Lum
bar C
SF s
APPα
, ng/
L
1250
1000
750
500
250
0
1000
800
600
400
200
0
non-iNPHn = 39
True iNPHn = 47
non-iNPHn = 17
True iNPHn = 25
p = 0.037
non-iNPHn = 39
True iNPHn = 47
non-iNPHn = 17
True iNPHn = 25
500
400
300
200
100
0
Vent
ricul
ar C
SF s
APPβ
, ng/
L
Lum
bar C
SF s
APPβ
, ng/
L
400
300
200
100
0
p = 0.056
A B
C D
p = 0.47
p = 0.37
Figure 12. Scatterplots of soluble amyloid precursor protein alpha (sAPPα) and beta (sAPPβ) concentrations in ventricular (A and C) and lumbar (B and D) cerebrospinal fluid (CSF) in patients with no diagnosis of idiopathic normal pressure hydrocephalus (iNPH) and patients with true (shunt-responsive) iNPH are presented. Cases are colour-labelled according to their APOE-ε4 status: blue dots (●) indicate APOE-ε4 carriers and red dots (●) indi-cate APOE-ε4 noncarriers. P-values were determined using a Mann–Whitney U test.

56
1600
1200 800
400 0
4000
1000
0
7500
5000
2500 0
2000
0
1750
0
Aβ +
HPτ
–n
= 32
Aβ –
HPτ
–n
= 48
Aβ +
HPτ
+n
= 17
Aβ +
HPτ
–n
= 32
Aβ –
HPτ
–n
= 48
Aβ +
HPτ
+n
= 17
Ventricular CSF Aβ1–38, ng/L
Ventricular CSF Aβ1–40, ng/L
AB
1200
1000 800
600
400
200 0
Aβ +
HPτ
–n
= 33
Aβ –
HPτ
–n
= 48
Aβ +
HPτ
+n
= 17
Ventricular CSF Aβ1–42, ng/L
p =
0.00
3
p =
0.00
5 p =
0.00
8
C
Figu
re 1
3. S
catt
erpl
ots
of a
myl
oid
beta
1-3
8 (A
β38)
(A),
Aβ4
0 (B
), a
nd A
β42
(C)
in g
roup
s of
pos
itive
/neg
ativ
e Aβ
and
hype
rpho
spho
ryla
ted
tau
(HPτ
) im
mun
orea
ctiv
ity in
br
ain
biop
sy a
re p
rese
nted
. Cas
es a
re c
olou
r-la
belle
d ac
cord
ing
to t
heir A
POE-
ε4 s
tatu
s: b
lue
dots
(●)
indi
cate
APO
E-ε4
car
rier
s an
d re
d do
ts (
●) in
dica
te A
POE-
ε4 n
onca
rri-
ers.
P-v
alue
s w
ere
dete
rmin
ed u
sing
a K
rusk
al–W
allis
H t
est
and
post
-hoc
Man
n–W
hitn
ey U
tes
t w
ith B
onfe
rron
i cor
rect
ion.
Onl
y st
atis
tical
ly s
igni
fican
t p-
valu
es a
re s
how
n.

57
Lum
bar C
SF Aβ4
2, n
g/L
Vent
ricul
ar C
SF Aβ4
2, n
g/L
1086420
1250
1000
750
500
250
0
1250
1000
750
500
250
01086420
r = –0.295p = 0.003
n = 98
r = –0.356p = 0.01
n = 48
Biopsy amyloid area, % Biopsy amyloid area, %
A B
Figure 14. Scatterplots of ventricular (A) and lumbar (B) cerebrospinal fluid (CSF) amyloid beta 1-42 (Aβ42) levels in relation to the percentage of Aβ area in frontal cortical brain biopsies are presented. Cases are colour-labelled according to their APOE-ε4 status: blue dots (●) indicate APOE-ε4 carriers and red dots (●) indicate APOE-ε4 noncarriers. Correlation coefficients and p-values were determined using Pearson correlation.
6.4.2 Proinflammatory cytokinesSeveral cytokines were present at concentrations below the lower limit of detection of the assay. All tested proinflammatory cytokines showed positive correlations between each other in ventricular and lumbar CSF samples. All ventricular and lumbar CSF IL-8 samples and all but one ventricular CSF MCP-1 sample were above the lower limit of detection, and the two cytokines and TNF-α were chosen for further analyses from the proinflammatory cytokines measured (Table 9 and 10). Lumbar CSF samples showed higher IL-8 levels compared to ventricular samples (p < 0.001, Table 9 and 10). Moreover, lumbar/ventricular CSF IL-8 ratios (p < 0.001) were significantly higher in samples collected after the ventriculostomy and ICP measurement compared to those collected before the procedure (Table 9 and 10, Figure 15). The levels of tested proinflammatory cytokines in the CSF showed no association to the pres-ence of Aβ or HPτ in brain biopsy, diagnosis of iNPH or AD, or shunt response in iNPH patients (Table 9).
6.4.3 Biomarkers of neuronal damageThere was no significant relation of CSF NFL or MBP levels to the brain biopsy findings (Table 10) or to the diagnosis of iNPH or AD (Table 9). In iNPH, a tendency (p = 0.05) towards lower ventricu-lar CSF NFL values was seen in shunt-responders (Table 9).

58
6.5 DISCUSSION
This is the first study to explore Aβ and sAPP isoforms, proinflammatory cytokines, and biomark-ers of neuronal damage in the CSF in conjunction with cortical brain biopsy. The major finding in the current study was the demonstration of the independent role of sAPPα in iNPH, which is not explained by cortical Aβ pathology. In iNPH, decreased levels of sAPP isoforms in lumbar CSF have been reported in previous studies (127–129). As predicted, the level of lumbar CSF sAPPα was lower, whereas sAPPβ showed a similar trend in patients with shunt-responsive iNPH compared to non-iNPH patients in our patient cohort. Interestingly, in ventricular samples no association with iNPH was noted, which could be explained by the ventriculostomy procedure as an invasive sample collection method or by concentration of proteins in lumbar CSF. However, the reason for unaffected ventricular levels of sAPP isoforms is uncertain, as the ventricular CSF could be expected to reflect the periventricular metabolism better than lumbar CSF. The pathobiological role of sAPP isoforms in iNPH seems to
0–500–1000–1500
400
300
200
100
0
2300
2200
Lum
bar/v
entri
cula
r CSF
IL-8
ratio
Time between lumbar and ventricular (0) CSF samples, h
Lumbar CSF sample obtained before ventriculostomy (n = 12)
Lumbar CSF sample obtained after ventriculostomy (n = 37)
Figure 15. Scatterplot of lumbar/ventricular cerebrospinal fluid (CSF) interleukin 8 (IL-8) ratio in individual cases in relation to the time difference between lumbar and ventricular samples is presented. Cases are colour-labelled according to whether lumbar CSF sample was collected before (yellow dot (●)) or after (red dot (●)) the ven-tricular sample.

59
be unconnected to the amyloidogenic pathway as there was no correlation between sAPP isoform levels in the CSF and Aβ load in the cortical brain biopsy. The reason for the lowering of sAPP iso-form levels in untreated iNPH remains unclear. However, as the levels are restored upon successful shunt treatment it has been hypothesized that the lowering may reflect metabolic impairment in brain tissue affected by iNPH (129). In any case, our data suggests that the observed APP changes are independent of Aβ pathology or are so early in the cascade that they do not reflect current tissue pathology. As expected, CSF levels of Aβ42 showed a negative association and correlation to Aβ load in the brain, as published earlier (26), and as suggested by amyloid-imaging studies (256). In contrast, no such association was seen between the CSF levels of Aβ38 or Aβ40 with positive Aβ or HPτ immu-noreactivity in the cortical brain biopsy. Our findings support the non-amyloidogenic role of Aβ38 and Aβ40 in the living human brain. As predicted, AD patients had lower Aβ42 and higher p-tau levels in the CSF compared to non-AD patients, although the differences did not reach statistical significance. One explanation for this is that non-AD patients show similar CSF Aβ42 and p-tau findings as biomarkers of comorbid AD tissue pathology without clinical dementia of Alzheimer’s type. As no pathological hallmark le-sions have been identified in iNPH (144), the role of Aβ and tau in iNPH remains elusive. However, there are patients with mixed pathologies i.e., patients with AD-related pathology and later demen-tia but still initial objective response for shunt surgery (88). Interestingly, patients with iNPH-AD had lowest levels of CSF Aβ42, and highest frequency of APOE-ε4 carriers. Eighty percent (8/10) of these patients showed a favourable response to shunt treatment. In contrast to two previous studies (174, 250), we found no prognostic potential in the levels of CSF Aβ42 or tau in shunted iNPH patients. The differences in the results may be explained by the different sample collection time (most the samples in the current study were collected after ven-triculostomy). It should also be noted that cited studies included fewer patients. In iNPH, reports of abnormal levels of proinflammatory cytokines (IL-1β, IL-4, IL-10, MCP-1, TNF-α) in the CSF have been published (129, 131–133), while contradictory findings in studies comparing proinflammatory cytokines (IL-8, IL-10, IL-12 (p40 and p70), IFN-γ, TNF-α, TGF- β1) have also been reported (129, 134, 135). In the current study,we attempted to measure a wide panel of different proinflammatory cytokines from ventricular and lumbar CSF, and positive correlations between cytokines were seen. However, we also noted that most of the cytokines are present in CSF at concentrations that are close to or below the lower limit of detection of the assay. In fact, these low concentrations, which are technically challenging to measure, may explain some of the varying results in the published literature. Here, we focused on the cytokines that could be robustly quantified in at least a subset of samples, i.e., IL-8. No association of proinflammatory cytokines in the CSF with the diagnosis of iNPH or AD or the presence of Aβ or HPτ in brain biopsy was seen. In patients with iNPH, proinflammatory cytokines did not show a prognostic value in shunt sur-gery. Consequently, our data suggests the role of neuroinflammation in iNPH and AD to be of little importance. Instead, an inflammatory response was seen in lumbar CSF samples collected after the ventriculostomy and ICP measurement. In addition, increased CSF tau and p-tau levels were observed in lumbar samples obtained after ventriculostomy as reported in earlier studies (26, 140). In consequence, levels of biomarkers in post-ventriculostomy lumbar CSF may not reflect the true values of the biomarkers in these patients. Previous studies have reported increased levels of CSF NFL in NPH (129, 137–140). In our cohort, NFL showed higher lumbar CSF levels in patients with shunt-responsive iNPH compared to non-iNPH patients (Table 9), but not to a significant degree. Interestingly, iNPH patients with positive shunt response showed a tendency towards lower NFL levels in ventricular CSF compared to shunt-nonresponsive iNPH patients. As NFL reflects subcortical axonal damage, perhaps high NFL could represent more severe and less recovering injury in the hydrocephalic brain. The strengths of the current study included: a large NPH cohort evaluated by cortical brain biopsy, utilization of ventricular and lumbar CSF samples in the analyses, a wide panel of tested CSF biomarkers, and evaluation of clinical outcome and other dementing disorders in the follow-up. The limitations of this study included: a limited number of patients with no shunt response, systematic assessment of shunt response only at 2–3 months, dichotomised shunt response scale, lack of validated objective outcome measures, and lumbar CSF sample available only in half of

60
the cases. It is obvious that at least in a prospective research setting, shunted patients should be followed-up for a significantly longer time and validated outcome measures should be utilized. Five patients with diagnostic findings suggesting iNPH did not respond to shunt, possibly due to comorbidities or misdiagnosis, and thus these patients were excluded from the comparisons of (‘true’) iNPH patients and non-iNPH patients, in addition to the iNPH patients who were diag-nosed with comorbid AD in the follow-up. More basic science and clinical studies evaluating the biology and potential role as diagnostic and prognostic biomarker of sAPPα and β are needed in the future. To conclude, the role of sAPP isoforms in iNPH seems to be unconnected to the Aβ cascade pathway, but rather may be explained by a metabolic failure or ischemia in the brain. No elevations in the levels of proinflammatory cytokines in the CSF were observed in the different diagnostic groups. Consequently, neuroinflammation in iNPH and AD require further study. None of the tested CSF biomarkers showed a tendency to discriminate between iNPH and non-iNPH patients or shunt-responders and nonresponders in iNPH in a clinical setting.

61
7 General discussion
This doctoral thesis studied currently the largest consecutive iNPH cohort through a unique win-dow of brain pathology with the implementation of cortical brain biopsy. Spanning over three decades, a major research opportunity to assess long-term morbidity and mortality in iNPH was identified. The most important findings of the thesis were: defining the incidence of iNPH in Mid-dle and Eastern Finland; confirmation of a high vascular comorbidity and how it affects the mortal-ity and the causes of death in iNPH; clarification of the genetical background of iNPH regarding the APOE genotypes; describing the independent role of APP in iNPH; and the evaluation of neu-roinflammation in iNPH. iNPH is an uncommon disease. Previous hospital-based studies performed in the Netherlands (10), Scandinavia (12, 14), and the United States of America (15) have presented incidence figures between 0.5–1.2 shunted iNPH patients per 100,000 inhabitants per year. In this thesis, a mean incidence of 1.8 iNPH patients / 100,000 / year was reported during 1993–2010 in Middle and East-ern Finland with a growing trend. It is not known whether these figures truly indicate a higher incidence of iNPH in Finland compared to other countries, or if the observed disparity is due to differences in the design of the study or variety in the detection and selection of the patients. How-ever, an even higher incidence of iNPH has been reported in population-based studies (13, 48). In persons aged 70 or older, the age-specific incidence of iNPH was notably higher at 15 cases / 100,000 / year. The possibility of iNPH should be considered in the differential diagnostics of gait and cog-nitive symptoms in the elderly, as most iNPH patients show a positive response to shunt surgery. The most common reported comorbidity in patients with iNPH is systemic arterial hyperten-sion (8, 9, 176–180). In accordance with previous studies, a high frequency of hypertension (52%) was observed in iNPH patients. In addition, T2DM was also frequent in persons with iNPH (23%). High vascular comorbidity reflected the causes of death in patients with iNPH: the most common causes of death were heart disease and cerebrovascular disease. Interestingly, despite the heavy burden of vascular disease, overall mortality was lower and estimated survival higher in patients with iNPH compared to non-iNPH patients in the patient cohort. Differential diagnostic options of iNPH such as AD and VCI carry worse prognosis compared to shunted iNPH patients. In future studies and in clinical practice, blood pressure monitoring and blood glucose tests (a fasting glu-cose test, a glucose tolerance test, or HbA1c) should be utilized to identify previously undiagnosed cases of hypertension and T2DM in iNPH patients, as T2DM increases the risk of death in the affected patients. The observed high vascular comorbidity supports the vascular theories of patho-genesis of iNPH (2.4.2 Pathophysiological theories). As there are several published studies of potential familial iNPH, a genetic predisposition to iNPH seems likely (16–18, 20). Identifying the genetic background of iNPH would be beneficial in the diagnostics of iNPH and would shed light on the pathogenesis of the disease. Previously, APOE ε4 allele, the most important genetic risk factor for AD, was suggested to be overrepresented in iNPH in a single study (22). Even though the difference was statistically significant (p = 0.02), the cited study shows major limitations: only a small number of NPH patients (n = 13), no differenta-tion between idiopathic and secondary NPH, incomplete representation of APOE genotypes, and remarkably lower frequency of APOE ε4 in the controls compared to other studies. However, with a superior number of patients and healthy controls and more rigorous differential diagnostics, no differences between iNPH and control subjects were observed in the APOE genotypes in this doc-toral thesis. As expected, APOE ε4 associated with the presence of Aβ plaques in the brain biopsy and the diagnosis of AD. APOE does not seem to have a major role in the pathogenesis of iNPH, however, APOE genotyping is often used in randomized clinical trials associated to neurodegener-ative diseases, and in the future it may be useful in planning individualized therapy for the patients (257). The effect of the amyloid cascade was further investigated in this thesis. Importantly, lower CSF

62
levels of sAPP isoforms were observed in shunt-responsive iNPH patients compared to non-iNPH subjects. This finding is in line with earlier studies of APP in iNPH (127–129). Although lumbar CSF sAPPα levels were lower (p < 0.05) in patients with shunt-responsive iNPH compared to non-iNPH patients, a significant overlap of concentrations were observed in both patient groups, which limits the clinical value of the finding. Importantly, the differences in sAPP levels in the lumbar or ventricular CSF were not reflective of cortical Aβ pathology, as demonstrated in this doctoral thesis. Instead, CSF levels of Aβ42 and HPτ show weak to moderate correlation with frontal cortical brain biopsy findings, as has been demonstrated in previous studies (26) and in this thesis. Brain biopsy is a useful tool in the differential diagnostics of iNPH at least in a research setting and for atypical cases, when specific diagnosis is needed but cannot be made with support of conventional nonin-vasive diagnostic tools, such as an MRI scan (87, 170). Neuroinflammation in iNPH has been studied by measuring the levels of proinflammatory cytokines in the CSF (129, 131–135). The results have been variable and none of the tested cytokines have proven to be useful in the diagnosis of iNPH (130). In this thesis, a wide panel of proinflam-matory cytokines were examined from the lumbar and ventricular CSF of iNPH patients to iden-tify possible novel biomarkers in iNPH, but no diagnostic or prognostic potential was observed. In consequence, neuroinflammation does not seem to have a measurable effect on iNPH at least from a clinical perspective. As predicted, a marked inflammatory response was seen in lumbar CSF samples collected after the ventriculostomy and ICP measurement procedure. As the CSF shunt treatment is associated with potentially lethal complications such as subdural haematoma and in-tracranial infections, there is critical need for future studies to identify noninvasive biomarkers to diagnose and to predict the response for shunt surgery in iNPH. There are limitations regarding the results presented in this thesis. Retrospective design of the studies meant that some clinical data was unavailable (e.g. systematic blood pressure measure-ments or blood glucose tests to find previously undiagnosed cases of hypertension or T2DM). For shunted iNPH patients there were no routine long-term follow-up visits, and no validated objective outcome measures were used to evaluate shunt response. There is an important selection bias as the patients in the cohort were referred to a tertiary university hospital, which may have limited the number of patients with only mild or early symptoms and patients at the other end of the spectrum with very poor general health due to other diseases and potentially old age. In addition, age- and gender-matched population-based controls were not utilized in all the studies. To conclude, while the understanding of the special clinical problem of iNPH has been ex-panded, the true pathogenesis of the disease remains a mystery. The most promising lines of future research explored in this doctoral thesis are the high vascular comorbidity and the role of sAPP isoforms. As a genetic predisposition to iNPH seems likely, a great potential lies in the genomic study of iNPH in the future.

63
8 Conclusions
In conclusion:
1.) The diagnosis of iNPH is uncommon in the total population (1.8 / 100,000 / year). The inci-dence of iNPH increases with ageing and the age-specific incidence in the inhabitants aged 70 or older is 15 / 100,000 / year in Middle and East Finnish populations (i).
2.) Systemic arterial hypertension and T2DM are frequent comorbidities in patients with iNPH, and the latter causes excess mortality in the affected patients. Heart disease and cerebrovas-cular disease are the most common causes of death in iNPH (i).
3.) APOE ε4 is not a genetic risk factor for iNPH and does not predict the response to shunt. Instead, the presence of APOE ε4 allele predisposes to concomitant AD pathology (ii).
4.) The role of sAPP isoforms in iNPH seems to be independent from the cerebral Aβ pathology. No neuroinflammatory findings were observed in patients with iNPH (iii).
Roman numerals refer to original publications.

64

65
9 References
1. Hakim S. Some observations on CSF pressure. Hydrocephalic syndrome in adults with ”normal” CSF pressure. Recognition of a new syndrome. Javeriana University School of Medicine, Bogotá, Colombia; 1964.
2. Hakim S, Adams RD. The special clinical problem of symptomatic hydrocephalus with normal cerebrospinal fluid pressure. Observations on cerebrospinal fluid hydrodynamics. J Neurol Sci 1965 Jul-Aug;2(4):307-327.
3. Adams RD, Fischer CM, Hakim S, Ojemann RG, Sweet WH. Symptomatic occult hydrocephalus with ”normal” cerebrospinal-fluid pressure. A treatable syndrome. N Engl J Med 1965 Jul 15;273:117-126.
4. Foltz EL, Ward AA Jr. Communicating hydrocephalus from subarachnoid bleeding. J Neurosurg 1956 Nov;13(6):546-566.
5. McHugh PR. Occult hydrocephalus. Q J Med 1964 Apr;33:297-308.
6. McGirt MJ, Woodworth G, Coon AL, Thomas G, Williams MA, Rigamonti D. Diagnosis, treatment, and analysis of long-term outcomes in idiopathic normal-pressure hydrocephalus. Neurosurgery 2005 Oct;57(4):699-705; discussion 699-705.
7. Relkin N, Marmarou A, Klinge P, Bergsneider M, Black PM. Diagnosing idiopathic normal-pressure hydrocephalus. Neurosurgery 2005 Sep;57(3 Suppl):S4-16; discussion ii-v.
8. Krauss JK, Regel JP, Vach W, Droste DW, Borremans JJ, Mergner T. Vascular risk factors and arteriosclerotic disease in idiopathic normal-pressure hydrocephalus of the elderly. Stroke 1996 Jan;27(1):24-29.
9. Malm J, Graff-Radford NR, Ishikawa M, Kristensen B, Leinonen V, Mori E, et al. Influence of comorbidities in idiopathic normal pressure hydrocephalus – research and clinical care. A report of the ISHCSF task force on comorbidities in INPH. Fluids Barriers CNS 2013 Jun 10;10(1):22-8118-10-22.
10. Vanneste J, Augustijn P, Dirven C, Tan WF, Goedhart ZD. Shunting normal-pressure hydrocephalus: do the benefits outweigh the risks? A multicenter study and literature review. Neurology 1992 Jan;42(1):54-59.
11. Krauss JK, Halve B. Normal pressure hydrocephalus: survey on contemporary diagnostic algorithms and therapeutic decision-making in clinical practice. Acta Neurochir (Wien) 2004 Apr;146(4):379-88; discussion 388.
12. Tisell M, Höglund M, Wikkelsø C. National and regional incidence of surgery for adult hydrocephalus in Sweden. Acta Neurol Scand 2005 Aug;112(2):72-75.
13. Brean A, Eide PK. Prevalence of probable idiopathic normal pressure hydrocephalus in a Norwegian population. Acta Neurol Scand 2008 Jul;118(1):48-53.

66
14. Brean A, Fredø HL, Sollid S, Müller T, Sundstrøm T, Eide PK. Five-year incidence of surgery for idiopathic normal pressure hydrocephalus in Norway. Acta Neurol Scand 2009 Nov;120(5):314-316.
15. Klassen BT, Ahlskog JE. Normal pressure hydrocephalus: how often does the diagnosis hold water? Neurology 2011 Sep 20;77(12):1119-1125.
16. Portenoy RK, Berger A, Gross E. Familial occurrence of idiopathic normal-pressure hydrocephalus. Arch Neurol 1984 Mar;41(3):335-337.
17. Lohmann F, Hofmann C, Kehler U. Normal pressure hydrocephalus in identical twins: a case report. German Medical Science Portal 2004 04/23/2004.
18. Cusimano MD, Rewilak D, Stuss DT, Barrera-Martinez JC, Salehi F, Freedman M. Normal-pressure hydrocephalus: is there a genetic predisposition? Can J Neurol Sci 2011 Mar;38(2):274-281.
19. Takahashi Y, Kawanami T, Nagasawa H, Iseki C, Hanyu H, Kato T. Familial normal pressure hydrocephalus (NPH) with an autosomal-dominant inheritance: a novel subgroup of NPH. J Neurol Sci 2011 Sep 15;308(1-2):149-151.
20. McGirr A, Cusimano MD. Familial aggregation of idiopathic normal pressure hydrocephalus: novel familial case and a family study of the NPH triad in an iNPH patient cohort. J Neurol Sci 2012 Oct 15;321(1-2):82-88.
21. Liouta E, Liakos F, Koutsarnakis C, Katsaros V, Stranjalis G. Novel case of familial normal pressure hydrocephalus. Psychiatry Clin Neurosci 2014 Jul;68(7):583-584.
22. Nacmias B, Tedde A, Guarnieri BM, Petruzzi C, Ortenzi L, Serio A, et al. Analysis of apolipoprotein E, alpha1-antichymotrypsin and presenilin-1 genes polymorphisms in dementia caused by normal pressure hydrocephalus in man. Neurosci Lett 1997 Jul 4;229(3):177-180.
23. Gudmundsson G, Kristjansdottir G, Cook E, Olafsson I. Association of ApoE genotype with clinical features and outcome in idiopathic normal pressure hydrocephalus (iNPH): a preliminary report. Acta Neurochir (Wien) 2009 Nov;151(11):1511-1512.
24. Tarnaris A, Watkins LD, Kitchen ND. Biomarkers in chronic adult hydrocephalus. Cerebrospinal Fluid Res 2006 Oct 4;3:11.
25. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011 May;7(3):263-269.
26. Seppälä TT, Nerg O, Koivisto AM, Rummukainen J, Puli L, Zetterberg H, et al. CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology 2012 May 15;78(20):1568-1575.
27. Mori E, Ishikawa M, Kato T, Kazui H, Miyake H, Miyajima M, et al. Guidelines for management of idiopathic normal pressure hydrocephalus: second edition. Neurol Med Chir (Tokyo) 2012;52(11):775-809.

67
28. Fisher CM. The clinical picture in occult hydrocephalus. Clin Neurosurg 1977;24:270-284.
29. Mori K. Management of idiopathic normal-pressure hydrocephalus: a multiinstitutional study conducted in Japan. J Neurosurg 2001 Dec;95(6):970-973.
30. Marmarou A, Young HF, Aygok GA, Sawauchi S, Tsuji O, Yamamoto T, et al. Diagnosis and management of idiopathic normal-pressure hydrocephalus: a prospective study in 151 patients. J Neurosurg 2005 Jun;102(6):987-997.
31. Hashimoto M, Ishikawa M, Mori E, Kuwana N, Study of INPH on neurological improvement (SINPHONI). Diagnosis of idiopathic normal pressure hydrocephalus is supported by MRI-based scheme: a prospective cohort study. Cerebrospinal Fluid Res 2010 Oct 31;7:18-8454-7-18.
32. Klinge P, Hellström P, Tans J, Wikkelsø C, European iNPH Multicentre Study Group. One-year outcome in the European multicentre study on iNPH. Acta Neurol Scand 2012 Sep;126(3):145-153.
33. Wikkelsø C, Andersson H, Blomstrand C, Lindqvist G, Svendsen P. Normal pressure hydrocephalus. Predictive value of the cerebrospinal fluid tap-test. Acta Neurol Scand 1986 Jun;73(6):566-573.
34. Stolze H, Kuhtz-Buschbeck JP, Drücke H, Jöhnk K, Diercks C, Palmie S, et al. Gait analysis in idiopathic normal pressure hydrocephalus – which parameters respond to the CSF tap test? Clin Neurophysiol 2000 Sep;111(9):1678-1686.
35. Bugalho P, Guimarães J. Gait disturbance in normal pressure hydrocephalus: a clinical study. Parkinsonism Relat Disord 2007 Oct;13(7):434-437.
36. Duinkerke A, Williams MA, Rigamonti D, Hillis AE. Cognitive recovery in idiopathic normal pressure hydrocephalus after shunt. Cogn Behav Neurol 2004 Sep;17(3):179-184.
37. Devito EE, Pickard JD, Salmond CH, Iddon JL, Loveday C, Sahakian BJ. The neuropsychology of normal pressure hydrocephalus (NPH). Br J Neurosurg 2005 Jun;19(3):217-224.
38. Thomas G, McGirt MJ, Woodworth G, Heidler J, Rigamonti D, Hillis AE, et al. Baseline neuropsychological profile and cognitive response to cerebrospinal fluid shunting for idiopathic normal pressure hydrocephalus. Dement Geriatr Cogn Disord 2005;20(2-3):163-168.
39. Ogino A, Kazui H, Miyoshi N, Hashimoto M, Ohkawa S, Tokunaga H, et al. Cognitive impairment in patients with idiopathic normal pressure hydrocephalus. Dement Geriatr Cogn Disord 2006;21(2):113-119.
40. Hellström P, Edsbagge M, Archer T, Tisell M, Tullberg M, Wikkelsø C. The neuropsychology of patients with clinically diagnosed idiopathic normal pressure hydrocephalus. Neurosurgery 2007 Dec;61(6):1219-26; discussion 1227-8.
41. Hellström P, Klinge P, Tans J, Wikkelsø C. The neuropsychology of iNPH: findings and evaluation of tests in the European multicentre study. Clin Neurol Neurosurg 2012 Feb;114(2):130-134.
42. Sakakibara R, Kanda T, Sekido T, Uchiyama T, Awa Y, Ito T, et al. Mechanism of bladder

68
dysfunction in idiopathic normal pressure hydrocephalus. Neurourol Urodyn 2008;27(6):507-510.
43. Nowak DA, Topka HR. Broadening a classic clinical triad: the hypokinetic motor disorder of normal pressure hydrocephalus also affects the hand. Exp Neurol 2006 Mar;198(1):81-87.
44. Pujari S, Kharkar S, Metellus P, Shuck J, Williams MA, Rigamonti D. Normal pressure hydrocephalus: long-term outcome after shunt surgery. J Neurol Neurosurg Psychiatry 2008 Nov;79(11):1282-1286.
45. Kito Y, Kazui H, Kubo Y, Yoshida T, Takaya M, Wada T, et al. Neuropsychiatric symptoms in patients with idiopathic normal pressure hydrocephalus. Behav Neurol 2009;21(3):165-174.
46. Oliveira MF, Oliveira JR, Rotta JM, Pinto FC. Psychiatric symptoms are present in most of the patients with idiopathic normal pressure hydrocephalus. Arq Neuropsiquiatr 2014 Jun;72(6):435-438.
47. Alexander EM, Wagner EH, Buchner DM, Cain KC, Larson EB. Do surgical brain lesions present as isolated dementia? A population-based study. J Am Geriatr Soc 1995 Feb;43(2):138-143.
48. Iseki C, Takahashi Y, Wada M, Kawanami T, Adachi M, Kato T. Incidence of idiopathic normal pressure hydrocephalus (iNPH): a 10-year follow-up study of a rural community in Japan. J Neurol Sci 2014 Apr 15;339(1-2):108-112.
49. Knopman DS, Petersen RC, Cha RH, Edland SD, Rocca WA. Incidence and causes of nondegenerative nonvascular dementia: a population-based study. Arch Neurol 2006 Feb;63(2):218-221.
50. Casmiro M, Benassi G, Cacciatore FM, D’Alessandro R. Frequency of idiopathic normal pressure hydrocephalus. Arch Neurol 1989 Jun;46(6):608.
51. Trenkwalder C, Schwarz J, Gebhard J, Ruland D, Trenkwalder P, Hense HW, et al. Starnberg trial on epidemiology of Parkinsonism and hypertension in the elderly. Prevalence of Parkinson’s disease and related disorders assessed by a door-to-door survey of inhabitants older than 65 years. Arch Neurol 1995 Oct;52(10):1017-1022.
52. Tan LC, Venketasubramanian N, Hong CY, Sahadevan S, Chin JJ, Krishnamoorthy ES, et al. Prevalence of Parkinson disease in Singapore: Chinese vs Malays vs Indians. Neurology 2004 Jun 8;62(11):1999-2004.
53. Gascón-Bayarri J, Reñé R, Del Barrio JL, De Pedro-Cuesta J, Ramón JM, Manubens JM, et al. Prevalence of dementia subtypes in El Prat de Llobregat, Catalonia, Spain: the PRATICON study. Neuroepidemiology 2007;28(4):224-234.
54. Hiraoka K, Meguro K, Mori E. Prevalence of idiopathic normal-pressure hydrocephalus in the elderly population of a Japanese rural community. Neurol Med Chir (Tokyo) 2008 May;48(5):197-99; discussion 199-200.
55. Iseki C, Kawanami T, Nagasawa H, Wada M, Koyama S, Kikuchi K, et al. Asymptomatic ventriculomegaly with features of idiopathic normal pressure hydrocephalus on MRI (AVIM) in the elderly: a prospective study in a Japanese population. J Neurol Sci 2009 Feb 15;277(1-2):54-57.

69
56. Tanaka N, Yamaguchi S, Ishikawa H, Ishii H, Meguro K. Prevalence of possible idiopathic normal-pressure hydrocephalus in Japan: the Osaki-Tajiri project. Neuroepidemiology 2009;32(3):171-175.
57. Arslantaş D, Özbabalık D, Metintaş S, Özkan S, Kalyoncu C, Özdemir G, et al. Prevalence of dementia and associated risk factors in Middle Anatolia, Turkey. J Clin Neurosci 2009 Nov;16(11):1455-1459.
58. Jaraj D, Rabiei K, Marlow T, Jensen C, Skoog I, Wikkelsø C. Prevalence of idiopathic normal-pressure hydrocephalus. Neurology 2014 Apr 22;82(16):1449-1454.
59. Milhorat TH. The third circulation revisited. J Neurosurg 1975 Jun;42(6):628-645.
60. McComb JG. Recent research into the nature of cerebrospinal fluid formation and absorption. J Neurosurg 1983 Sep;59(3):369-383.
61. Brinker T, Stopa E, Morrison J, Klinge P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 2014 May 1;11:10-8118-11-10. eCollection 2014.
62. Cushing H. Studies in intracranial physiology & surgery: the third circulation, the hypophysis, the gliomas: the Cameron prize lectures delivered at the University of Edinburgh, October 19–22, 1925. Oxford: 1926.
63. Bulat M, Klarica M. Recent insights into a new hydrodynamics of the cerebrospinal fluid. Brain Res Rev 2011 Jan 1;65(2):99-112.
64. Hodel J, Lebret A, Petit E, Leclerc X, Zins M, Vignaud A, et al. Imaging of the entire cerebrospinal fluid volume with a multistation 3D SPACE MR sequence: feasibility study in patients with hydrocephalus. Eur Radiol 2013 Jun;23(6):1450-1458.
65. Edsbagge M, Starck G, Zetterberg H, Ziegelitz D, Wikkelsø C. Spinal cerebrospinal fluid volume in healthy elderly individuals. Clin Anat 2011 Sep;24(6):733-740.
66. Hakim S, Venegas JG, Burton JD. The physics of the cranial cavity, hydrocephalus and normal pressure hydrocephalus: mechanical interpretation and mathematical model. Surg Neurol 1976 Mar;5(3):187-210.
67. Lundberg N. Continuous recording and control of ventricular fluid pressure in neurosurgical practice. Acta Psychiatr Scand Suppl 1960;36(149):1-193.
68. Crockard HA, Hanlon K, Duda EE, Mullan JF. Hydrocephalus as a cause of dementia: evaluation by computerised tomography and intracranial pressure monitoring. J Neurol Neurosurg Psychiatry 1977 Aug;40(8):736-740.
69. Stephensen H, Andersson N, Eklund A, Malm J, Tisell M, Wikkelsø C. Objective B wave analysis in 55 patients with non-communicating and communicating hydrocephalus. J Neurol Neurosurg Psychiatry 2005 Jul;76(7):965-970.
70. Eide PK, Sorteberg W. Diagnostic intracranial pressure monitoring and surgical management in idiopathic normal pressure hydrocephalus: a 6-year review of 214 patients. Neurosurgery 2010 Jan;66(1):80-91.
71. Bateman GA. The pathophysiology of idiopathic normal pressure hydrocephalus: cerebral

70
ischemia or altered venous hemodynamics? AJNR Am J Neuroradiol 2008 Jan;29(1):198-203.
72. Bradley WG Jr. CSF flow in the brain in the context of normal pressure hydrocephalus. AJNR Am J Neuroradiol 2015 May;36(5):831-838.
73. Greitz TV, Grepe AO, Kalmér MS, Lopez J. Pre- and postoperative evaluation of cerebral blood flow in low-pressure hydrocephalus. J Neurosurg 1969 Dec;31(6):644-651.
74. Greitz T. Cerebral blood flow in occult hydrocephalus studied with angiography and the 133Xenon clearance method. Acta Radiol Diagn (Stockh) 1969 Sep;8(5):376-384.
75. Owler BK, Pickard JD. Normal pressure hydrocephalus and cerebral blood flow: a review. Acta Neurol Scand 2001 Dec;104(6):325-342.
76. Bradley WG Jr, Whittemore AR, Watanabe AS, Davis SJ, Teresi LM, Homyak M. Association of deep white matter infarction with chronic communicating hydrocephalus: implications regarding the possible origin of normal-pressure hydrocephalus. AJNR Am J Neuroradiol 1991 Jan-Feb;12(1):31-39.
77. Momjian S, Owler BK, Czosnyka Z, Czosnyka M, Pena A, Pickard JD. Pattern of white matter regional cerebral blood flow and autoregulation in normal pressure hydrocephalus. Brain 2004 May;127(Pt 5):965-972.
78. Sasaki H, Ishii K, Kono AK, Miyamoto N, Fukuda T, Shimada K, et al. Cerebral perfusion pattern of idiopathic normal pressure hydrocephalus studied by SPECT and statistical brain mapping. Ann Nucl Med 2007 Jan;21(1):39-45.
79. Ziegelitz D, Starck G, Kristiansen D, Jakobsson M, Hultenmo M, Mikkelsen IK, et al. Cerebral perfusion measured by dynamic susceptibility contrast MRI is reduced in patients with idiopathic normal pressure hydrocephalus. J Magn Reson Imaging 2014 Jun;39(6):1533-1542.
80. Ziegelitz D, Arvidsson J, Hellström P, Tullberg M, Wikkelsø C, Starck G. In patients with idiopathic normal pressure hydrocephalus postoperative cerebral perfusion changes measured by dynamic susceptibility contrast magnetic resonance imaging correlate with clinical improvement. J Comput Assist Tomogr 2015 Jul-Aug;39(4):531-540.
81. Mathew NT, Meyer JS, Hartmann A, Ott EO. Abnormal cerebrospinal fluid-blood flow dynamics. Implications in diagnosis, treatment, and prognosis in normal pressure hydrocephalus. Arch Neurol 1975 Oct;32(10):657-664.
82. Kristensen B, Malm J, Fagerland M, Hietala SO, Johansson B, Ekstedt J, et al. Regional cerebral blood flow, white matter abnormalities, and cerebrospinal fluid hydrodynamics in patients with idiopathic adult hydrocephalus syndrome. J Neurol Neurosurg Psychiatry 1996 Mar;60(3):282-288.
83. Silverberg GD, Mayo M, Saul T, Rubenstein E, McGuire D. Alzheimer’s disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol 2003 Aug;2(8):506-511.
84. Silverberg GD. Normal pressure hydrocephalus (NPH): ischaemia, CSF stagnation or both. Brain 2004 May;127(Pt 5):947-948.
85. Silverberg GD, Miller MC, Machan JT, Johanson CE, Caralopoulos IN, Pascale CL, et al.

71
Amyloid and tau accumulate in the brains of aged hydrocephalic rats. Brain Res 2010 Mar 4;1317:286-296.
86. Silverberg GD, Miller MC, Pascale CL, Caralopoulos IN, Agca Y, Agca C, et al. Kaolin-induced chronic hydrocephalus accelerates amyloid deposition and vascular disease in transgenic rats expressing high levels of human APP. Fluids Barriers CNS 2015 Jan 24;12(1):2-8118-12-2. eCollection 2015.
87. Leinonen V, Koivisto AM, Savolainen S, Rummukainen J, Tamminen JN, Tillgren T, et al. Amyloid and tau proteins in cortical brain biopsy and Alzheimer’s disease. Ann Neurol 2010;68(4):446-453.
88. Koivisto AM, Alafuzoff I, Savolainen S, Sutela A, Rummukainen J, Kurki M, et al. Poor cognitive outcome in shunt-responsive idiopathic normal pressure hydrocephalus. Neurosurgery 2013 Jan;72(1):1-8.
89. Kondziella D, Sonnewald U, Tullberg M, Wikkelsø C. Brain metabolism in adult chronic hydrocephalus. J Neurochem 2008 Aug;106(4):1515-1524.
90. Zhang J, Williams MA, Rigamonti D. Heritable essential tremor-idiopathic normal pressure hydrocephalus (ETINPH). Am J Med Genet A 2008 Feb 15;146A(4):433-439.
91. Zhang J, Carr CW, Rigamonti D, Badr A. Genome-wide linkage scan maps ETINPH gene to chromosome 19q12-13.31. Hum Hered 2010;69(4):262-267.
92. Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 1988 Apr 29;240(4852):622-630.
93. Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis 2014 Dec;72 Pt A:3-12.
94. Weisgraber KH. Apolipoprotein E: structure-function relationships. Adv Protein Chem 1994;45:249-302.
95. Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 2000;1:507-537.
96. Olaisen B, Teisberg P, Gedde-Dahl T Jr. The locus for apolipoprotein E (apoE) is linked to the complement component C3 (C3) locus on chromosome 19 in man. Hum Genet 1982;62(3):233-236.
97. Lusis AJ, Heinzmann C, Sparkes RS, Scott J, Knott TJ, Geller R, et al. Regional mapping of human chromosome 19: organization of genes for plasma lipid transport (APOC1, -C2, and -E and LDLR) and the genes C3, PEPD, and GPI. Proc Natl Acad Sci U S A 1986 Jun;83(11):3929-3933.
98. Alzheimer Research Forum. Chromosome 19: location of published AD candidate genes. Available at: http://www.alzgene.org/chromo.asp?c=19. Accessed 14/02, 2016.
99. Utermann G, Hees M, Steinmetz A. Polymorphism of apolipoprotein E and occurrence of dysbetalipoproteinaemia in man. Nature 1977 Oct 13;269(5629):604-607.
100. Zannis VI, Just PW, Breslow JL. Human apolipoprotein E isoprotein subclasses are genetically

72
determined. Am J Hum Genet 1981 Jan;33(1):11-24.
101. Zannis VI, Breslow JL, Utermann G, Mahley RW, Weisgraber KH, Havel RJ, et al. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J Lipid Res 1982 Aug;23(6):911-914.
102. Kim DH, Yeo SH, Park JM, Choi JY, Lee TH, Park SY, et al. Genetic markers for diagnosis and pathogenesis of Alzheimer’s disease. Gene 2014 Jul 25;545(2):185-193.
103. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009 Aug 13;63(3):287-303.
104. Mahley RW, Huang Y. Apolipoprotein E: from atherosclerosis to Alzheimer’s disease and beyond. Curr Opin Lipidol 1999 Jun;10(3):207-217.
105. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A 2006 Apr 11;103(15):5644-5651.
106. Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993 Aug 13;261(5123):921-923.
107. Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci 2014;37:79-100.
108. Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 2007 Sep 19;298(11):1300-1311.
109. del Mar Matarín M, Poca MA, Bartrés-Faz D, Mataró M, Clemente IC, Solé-Padullés C, et al. Angiotensin I converting enzyme polymorphism effects in patients with normal pressure hydrocephalus syndrome before and after surgery. J Neurol 2005 Feb;252(2):191-196.
110. Rudnicki M, Mayer G. Significance of genetic polymorphisms of the renin-angiotensin-aldosterone system in cardiovascular and renal disease. Pharmacogenomics 2009 Mar;10(3):463-476.
111. Kato T, Sato H, Emi M, Seino T, Arawaka S, Iseki C, et al. Segmental copy number loss of SFMBT1 gene in elderly individuals with ventriculomegaly: a community-based study. Intern Med 2011;50(4):297-303.
112. Ishikawa M, Guideline Committe for Idiopathic Normal Pressure Hydrocephalus, Japanese Society of Normal Pressure Hydrocephalus. Clinical guidelines for idiopathic normal pressure hydrocephalus. Neurol Med Chir (Tokyo) 2004 Apr;44(4):222-223.
113. Ishikawa M, Hashimoto M, Kuwana N, Mori E, Miyake H, Wachi A, et al. Guidelines for management of idiopathic normal pressure hydrocephalus. Neurol Med Chir (Tokyo) 2008;48 Suppl:S1-23.
114. Dandy WE. Ventriculography following the injection of air into the cerebral ventricles. Ann Surg 1918 Jul;68(1):5-11.

73
115. Dandy WE. Röntgenography of the brain after the injection of air into the spinal canal. Ann Surg 1919 Oct;70(4):397-403.
116. Hoeffner EG, Mukherji SK, Srinivasan A, Quint DJ. Neuroradiology back to the future: brain imaging. AJNR Am J Neuroradiol 2012 Jan;33(1):5-11.
117. Evans WA Jr. An encephalographic ratio for estimating ventricular enlargement and cerebral atrophy. Arch Neurol Psychiatry 1942;47(6):931-937.
118. Folstein MF, Folstein SE, McHugh PR. ”Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975 Nov;12(3):189-198.
119. Marmarou A, Bergsneider M, Klinge P, Relkin N, Black PM. The value of supplemental prognostic tests for the preoperative assessment of idiopathic normal-pressure hydrocephalus. Neurosurgery 2005 Sep;57(3 Suppl):S17-28; discussion ii-v.
120. Sundström N, Andersson K, Marmarou A, Malm J, Eklund A. Comparison between 3 infusion methods to measure cerebrospinal fluid outflow conductance. J Neurosurg 2010 Dec;113(6):1294-1303.
121. Wikkelsø C, Andersson H, Blomstrand C, Lindqvist G. The clinical effect of lumbar puncture in normal pressure hydrocephalus. J Neurol Neurosurg Psychiatry 1982 Jan;45(1):64-69.
122. Haan J, Thomeer RT. Predictive value of temporary external lumbar drainage in normal pressure hydrocephalus. Neurosurgery 1988 Feb;22(2):388-391.
123. Motter R, Vigo-Pelfrey C, Kholodenko D, Barbour R, Johnson-Wood K, Galasko D, et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann Neurol 1995 Oct;38(4):643-648.
124. Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E. Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease? Mol Chem Neuropathol 1995 Dec;26(3):231-245.
125. van Harten AC, Kester MI, Visser PJ, Blankenstein MA, Pijnenburg YA, van der Flier WM, et al. Tau and p-tau as CSF biomarkers in dementia: a meta-analysis. Clin Chem Lab Med 2011 Mar;49(3):353-366.
126. Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 2003 May;70(1):1-32.
127. Ray B, Reyes PF, Lahiri DK. Biochemical studies in normal pressure hydrocephalus (NPH) patients: change in CSF levels of amyloid precursor protein (APP), amyloid-beta (Aβ) peptide and phospho-tau. J Psychiatr Res 2011 Apr;45(4):539-547.
128. Miyajima M, Nakajima M, Ogino I, Miyata H, Motoi Y, Arai H. Soluble amyloid precursor protein α in the cerebrospinal fluid as a diagnostic and prognostic biomarker for idiopathic normal pressure hydrocephalus. Eur J Neurol 2012 Jun 4;20(2):236-242.
129. Jeppsson A, Zetterberg H, Blennow K, Wikkelsø C. Idiopathic normal-pressure hydrocephalus: pathophysiology and diagnosis by CSF biomarkers. Neurology 2013 Apr 9;80(15):1385-1392.

74
130. Tarnaris A, Toma AK, Kitchen ND, Watkins LD. Ongoing search for diagnostic biomarkers in idiopathic normal pressure hydrocephalus. Biomark Med 2009 Dec;3(6):787-805.
131. Cacabelos R, Barquero M, García P, Alvarez XA, Varela de Seijas E. Cerebrospinal fluid interleukin-1 beta (IL-1 beta) in Alzheimer’s disease and neurological disorders. Methods Find Exp Clin Pharmacol 1991 Sep;13(7):455-458.
132. Tarkowski E, Tullberg M, Fredman P, Wikkelsø C. Normal pressure hydrocephalus triggers intrathecal production of TNF-alpha. Neurobiol Aging 2003 Sep;24(5):707-714.
133. Stoeck K, Bodemer M, Ciesielczyk B, Meissner B, Bartl M, Heinemann U, et al. Interleukin 4 and interleukin 10 levels are elevated in the cerebrospinal fluid of patients with Creutzfeldt-Jakob disease. Arch Neurol 2005 Oct;62(10):1591-1594.
134. Rota E, Bellone G, Rocca P, Bergamasco B, Emanuelli G, Ferrero P. Increased intrathecal TGF-beta1, but not IL-12, IFN-gamma and IL-10 levels in Alzheimer’s disease patients. Neurol Sci 2006 Apr;27(1):33-39.
135. Leinonen V, Menon LG, Carroll RS, Dello Iacono D, Grevet J, Jääskeläinen JE, et al. Cerebrospinal fluid biomarkers in idiopathic normal pressure hydrocephalus. Int J Alzheimers Dis 2011;2011:312526.
136. Sosvorova L, Vcelak J, Mohapl M, Vitku J, Bicikova M, Hampl R. Selected pro- and anti-inflammatory cytokines in cerebrospinal fluid in normal pressure hydrocephalus. Neuro Endocrinol Lett 2014;35(7):586-593.
137. Tullberg M, Rosengren L, Blomsterwall E, Karlsson JE, Wikkelsø C. CSF neurofilament and glial fibrillary acidic protein in normal pressure hydrocephalus. Neurology 1998 Apr;50(4):1122-1127.
138. Tullberg M, Blennow K, Månsson JE, Fredman P, Tisell M, Wikkelsø C. Ventricular cerebrospinal fluid neurofilament protein levels decrease in parallel with white matter pathology after shunt surgery in normal pressure hydrocephalus. Eur J Neurol 2007 Mar;14(3):248-254.
139. Ågren-Wilsson A, Lekman A, Sjöberg W, Rosengren L, Blennow K, Bergenheim AT, et al. CSF biomarkers in the evaluation of idiopathic normal pressure hydrocephalus. Acta Neurol Scand 2007 Nov;116(5):333-339.
140. Tullberg M, Blennow K, Månsson JE, Fredman P, Tisell M, Wikkelsø C. Cerebrospinal fluid markers before and after shunting in patients with secondary and idiopathic normal pressure hydrocephalus. Cerebrospinal Fluid Res 2008 Apr 25;5:9-8454-5-9.
141. Petzold A, Keir G, Warren J, Fox N, Rossor MN. A systematic review and meta-analysis of CSF neurofilament protein levels as biomarkers in dementia. Neurodegener Dis 2007;4(2-3):185-194.
142. Futakawa S, Nara K, Miyajima M, Kuno A, Ito H, Kaji H, et al. A unique N-glycan on human transferrin in CSF: a possible biomarker for iNPH. Neurobiol Aging 2012 Aug;33(8):1807-1815.
143. Love S. Neuropathological investigation of dementia: a guide for neurologists. J Neurol Neurosurg Psychiatry 2005 Dec;76 Suppl 5:v8-14.

75
144. Leinonen V, Koivisto AM, Savolainen S, Rummukainen J, Sutela A, Vanninen R, et al. Post-mortem findings in 10 patients with presumed normal-pressure hydrocephalus and review of the literature. Neuropathol Appl Neurobiol 2012 Feb;38(1):72-86.
145. Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie und psychisch-gerichtliche Medizin 1907;64:146-148.
146. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82(4):239-259.
147. Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 1997 Jul-Aug;18(4):351-357.
148. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002 Jul 19;297(5580):353-356.
149. Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010 Jan;9(1):119-128.
150. Leinonen V, Alafuzoff I, Aalto S, Suotunen T, Savolainen S, Nagren K, et al. Assessment of beta-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11-labeled Pittsburgh Compound B. Arch Neurol 2008 Oct;65(10):1304-1309.
151. Rinne JO, Wong DF, Wolk DA, Leinonen V, Arnold SE, Buckley C, et al. [18F]Flutemetamol PET imaging and cortical biopsy histopathology for fibrillar amyloid β detection in living subjects with normal pressure hydrocephalus: pooled analysis of four studies. Acta Neuropathol 2012 Dec;124(6):833-845.
152. Richards D, Sabbagh MN. Florbetaben for PET Imaging of beta-amyloid plaques in the brain. Neurol Ther 2014 Nov 27;3(2):79-88.
153. Hamilton R, Patel S, Lee EB, Jackson EM, Lopinto J, Arnold SE, et al. Lack of shunt response in suspected idiopathic normal pressure hydrocephalus with Alzheimer disease pathology. Ann Neurol 2010 Mar 8;68(4):535-540.
154. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 2015 Apr;14(4):388-405.
155. Scheltens P, Leys D, Barkhof F, Huglo D, Weinstein HC, Vermersch P, et al. Atrophy of medial temporal lobes on MRI in ”probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry 1992 Oct;55(10):967-972.
156. Scheltens P, Fox N, Barkhof F, De Carli C. Structural magnetic resonance imaging in the practical assessment of dementia: beyond exclusion. Lancet Neurol 2002 May;1(1):13-21.
157. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet 2011 Mar 19;377(9770):1019-1031.
158. Mendez MF. Early-onset Alzheimer’s disease: nonamnestic subtypes and type 2 AD. Arch Med Res 2012 Nov;43(8):677-685.

76
159. Morgan D, Funk M, Crossley M, Basran J, Kirk A, Dal Bello-Haas V. The potential of gait analysis to contribute to differential diagnosis of early stage dementia: current research and future directions. Can J Aging 2007 Spring;26(1):19-32.
160. O’Brien JT, Erkinjuntti T, Reisberg B, Roman G, Sawada T, Pantoni L, et al. Vascular cognitive impairment. Lancet Neurol 2003 Feb;2(2):89-98.
161. Moorhouse P, Rockwood K. Vascular cognitive impairment: current concepts and clinical developments. Lancet Neurol 2008 Mar;7(3):246-255.
162. Román GC, Erkinjuntti T, Wallin A, Pantoni L, Chui HC. Subcortical ischaemic vascular dementia. Lancet Neurol 2002 Nov;1(7):426-436.
163. Tullberg M, Jensen C, Ekholm S, Wikkelsø C. Normal pressure hydrocephalus: vascular white matter changes on MR images must not exclude patients from shunt surgery. AJNR Am J Neuroradiol 2001 Oct;22(9):1665-1673.
164. Tullberg M, Hultin L, Ekholm S, Mansson JE, Fredman P, Wikkelsø C. White matter changes in normal pressure hydrocephalus and Binswanger disease: specificity, predictive value and correlations to axonal degeneration and demyelination. Acta Neurol Scand 2002 Jun;105(6):417-426.
165. Bugalho P, Alves L. Normal-pressure hydrocephalus: white matter lesions correlate negatively with gait improvement after lumbar puncture. Clin Neurol Neurosurg 2007 Nov;109(9):774-778.
166. Lenfeldt N, Larsson A, Nyberg L, Birgander R, Eklund A, Malm J. Diffusion tensor imaging reveals supplementary lesions to frontal white matter in idiopathic normal pressure hydrocephalus. Neurosurgery 2011 Jun;68(6):1586-93; discussion 1593.
167. Kojoukhova M, Koivisto AM, Korhonen R, Remes AM, Vanninen R, Soininen H, et al. Feasibility of radiological markers in idiopathic normal pressure hydrocephalus. Acta Neurochir (Wien) 2015 Oct;157(10):1709-1719.
168. Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 2007 Mar 13;68(11):812-819.
169. Cabral D, Beach TG, Vedders L, Sue LI, Jacobson S, Myers K, et al. Frequency of Alzheimer’s disease pathology at autopsy in patients with clinical normal pressure hydrocephalus. Alzheimers Dement 2011 Sep;7(5):509-513.
170. Leinonen V, Koivisto AM, Alafuzoff I, Pyykkö OT, Rummukainen J, von und zu Fraunberg M, et al. Cortical brain biopsy in long-term prognostication of 468 patients with possible normal pressure hydrocephalus. Neurodegener Dis 2012;10(1-4):166-169.
171. Pomeraniec IJ, Bond AE, Lopes MB, Jane JA S. Concurrent Alzheimer’s pathology in patients with clinical normal pressure hydrocephalus: correlation of high-volume lumbar puncture results, cortical brain biopsies, and outcomes. J Neurosurg 2015 Sep 4:1-7.
172. Golomb J, Wisoff J, Miller DC, Boksay I, Kluger A, Weiner H, et al. Alzheimer’s disease comorbidity in normal pressure hydrocephalus: prevalence and shunt response. J Neurol Neurosurg Psychiatry 2000 Jun;68(6):778-781.

77
173. Bech-Azeddine R, Høgh P, Juhler M, Gjerris F, Waldemar G. Idiopathic normal-pressure hydrocephalus: clinical comorbidity correlated with cerebral biopsy findings and outcome of cerebrospinal fluid shunting. J Neurol Neurosurg Psychiatry 2007 Feb;78(2):157-161.
174. Patel S, Lee EB, Xie SX, Law A, Jackson EM, Arnold SE, et al. Phosphorylated tau/amyloid beta 1-42 ratio in ventricular cerebrospinal fluid reflects outcome in idiopathic normal pressure hydrocephalus. Fluids Barriers CNS 2012 Mar 23;9(1):7-8118-9-7.
175. Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Böhm M, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J 2013 Jul;34(28):2159-2219.
176. Casmiro M, D’Alessandro R, Cacciatore FM, Daidone R, Calbucci F, Lugaresi E. Risk factors for the syndrome of ventricular enlargement with gait apraxia (idiopathic normal pressure hydrocephalus): a case-control study. J Neurol Neurosurg Psychiatry 1989 Jul;52(7):847-852.
177. Mirzayan MJ, Luetjens G, Borremans JJ, Regel JP, Krauss JK. Extended long-term (> 5 years) outcome of cerebrospinal fluid shunting in idiopathic normal pressure hydrocephalus. Neurosurgery 2010 Aug;67(2):295-301.
178. Eide PK, Pripp AH. Increased prevalence of cardiovascular disease in idiopathic normal pressure hydrocephalus patients compared to a population-based cohort from the HUNT3 survey. Fluids Barriers CNS 2014 Aug 19;11:19-8118-11-19. eCollection 2014.
179. Graff-Radford NR, Godersky JC. Idiopathic normal pressure hydrocephalus and systemic hypertension. Neurology 1987 May;37(5):868-871.
180. Kazui H, Mori E, Ohkawa S, Okada T, Kondo T, Sakakibara R, et al. Predictors of the disappearance of triad symptoms in patients with idiopathic normal pressure hydrocephalus after shunt surgery. J Neurol Sci 2013 May 15;328(1-2):64-69.
181. Earnest MP, Fahn S, Karp JH, Rowland LP. Normal pressure hydrocephalus and hypertensive cerebrovascular disease. Arch Neurol 1974 Oct;31(4):262-266.
182. Koto A, Rosenberg G, Zingesser LH, Horoupian D, Katzman R. Syndrome of normal pressure hydrocephalus: possible relation to hypertensive and arteriosclerotic vasculopathy. J Neurol Neurosurg Psychiatry 1977 Jan;40(1):73-79.
183. Tisell M, Tullberg M, Hellström P, Edsbagge M, Högfeldt M, Wikkelsø C. Shunt surgery in patients with hydrocephalus and white matter changes. J Neurosurg 2011 May;114(5):1432-1438.
184. Beckman JA, Creager MA, Libby P. Diabetes and atherosclerosis: epidemiology, pathophysiology, and management. JAMA 2002 May 15;287(19):2570-2581.
185. Jacobs L. Diabetes mellitus in normal pressure hydrocephalus. J Neurol Neurosurg Psychiatry 1977 Apr;40(4):331-335.
186. Chang TC, Singh K. Glaucomatous disease in patients with normal pressure hydrocephalus. J Glaucoma 2009 Mar;18(3):243-246.
187. Chen BH, Drucker MD, Louis KM, Richards DW. Progression of normal-tension glaucoma

78
after ventriculoperitoneal shunt to decrease cerebrospinal fluid pressure. J Glaucoma 2014 Oct 27.
188. Ames RH. Ventriculo-peritoneal shunts in the management of hydrocephalus. J Neurosurg 1967 Dec;27(6):525-529.
189. Pudenz RH. The ventriculo-atrial shunt. J Neurosurg 1966 Nov;25(5):602-608.
190. Spetzler RF, Wilson CB, Grollmus JM. Percutaneous lumboperitoneal shunt. Technical note. J Neurosurg 1975 Dec;43(6):770-773.
191. Esmonde T, Cooke S. Shunting for normal pressure hydrocephalus (NPH). Cochrane Database Syst Rev 2002;(3)(3):CD003157.
192. Kazui H, Miyajima M, Mori E, Ishikawa M, SINPHONI-2 Investigators. Lumboperitoneal shunt surgery for idiopathic normal pressure hydrocephalus (SINPHONI-2): an open-label randomised trial. Lancet Neurol 2015 Jun;14(6):585-594.
193. Bergsneider M, Black PM, Klinge P, Marmarou A, Relkin N. Surgical management of idiopathic normal-pressure hydrocephalus. Neurosurgery 2005 Sep;57(3 Suppl):S29-39; discussion ii-v.
194. Mixter W. Ventriculoscopy and puncture of the floor of the third ventricle. Boston Med Surg J 1923(188):227-228.
195. Hellwig D, Grotenhuis JA, Tirakotai W, Riegel T, Schulte DM, Bauer BL, et al. Endoscopic third ventriculostomy for obstructive hydrocephalus. Neurosurg Rev 2005 Jan;28(1):1-34; discussion 35-8.
196. Gangemi M, Maiuri F, Naddeo M, Godano U, Mascari C, Broggi G, et al. Endoscopic third ventriculostomy in idiopathic normal pressure hydrocephalus: an Italian multicenter study. Neurosurgery 2008 Jul;63(1):62-7; discussion 67-9.
197. Pinto FC, Saad F, Oliveira MF, Pereira RM, Miranda FL, Tornai JB, et al. Role of endoscopic third ventriculostomy and ventriculoperitoneal shunt in idiopathic normal pressure hydrocephalus: preliminary results of a randomized clinical trial. Neurosurgery 2013 May;72(5):845-854.
198. Tudor KI, Tudor M, McCleery J, Car J. Endoscopic third ventriculostomy (ETV) for idiopathic normal pressure hydrocephalus (iNPH). Cochrane Database Syst Rev 2015 Jul 29;7:CD010033.
199. European Glaucoma Society. Terminology and guidelines for glaucoma. 2014.
200. Biousse V, Bruce BB, Newman NJ. Update on the pathophysiology and management of idiopathic intracranial hypertension. J Neurol Neurosurg Psychiatry 2012 May;83(5):488-494.
201. NORDIC Idiopathic Intracranial Hypertension Study Group Writing Committee, Wall M, McDermott MP, Kieburtz KD, Corbett JJ, Feldon SE, et al. Effect of acetazolamide on visual function in patients with idiopathic intracranial hypertension and mild visual loss: the idiopathic intracranial hypertension treatment trial. JAMA 2014 Apr 23-30;311(16):1641-1651.
202. Low EV, Avery AJ, Gupta V, Schedlbauer A, Grocott MP. Identifying the lowest effective dose of acetazolamide for the prophylaxis of acute mountain sickness: systematic review and

79
meta-analysis. BMJ 2012 Oct 18;345:e6779.
203. Aimard G, Vighetto A, Gabet JY, Bret P, Henry E. Acetazolamide: an alternative to shunting in normal pressure hydrocephalus? Preliminary results. Rev Neurol (Paris) 1990;146(6-7):437-439.
204. García-Gascó P, Salame Gamarra F, Tenllado Doblas P, Chazarra Talens C. Complete resolution of chronic hydrocephalus of adult with acetazolamide. Med Clin (Barc) 2005 Apr 9;124(13):516-517.
205. Alperin N, Oliu CJ, Bagci AM, Lee SH, Kovanlikaya I, Adams D, et al. Low-dose acetazolamide reverses periventricular white matter hyperintensities in iNPH. Neurology 2014 Apr 15;82(15):1347-1351.
206. Toma AK, Stapleton S, Papadopoulos MC, Kitchen ND, Watkins LD. Natural history of idiopathic normal-pressure hydrocephalus. Neurosurg Rev 2011 Oct;34(4):433-439.
207. Savolainen S, Hurskainen H, Paljarvi L, Alafuzoff I, Vapalahti M. Five-year outcome of normal pressure hydrocephalus with or without a shunt: predictive value of the clinical signs, neuropsychological evaluation and infusion test. Acta Neurochir (Wien) 2002 Jun;144(6):515-23; discussion 523.
208. Eide PK, Brean A. Intracranial pulse pressure amplitude levels determined during preoperative assessment of subjects with possible idiopathic normal pressure hydrocephalus. Acta Neurochir (Wien) 2006 Nov;148(11):1151-6; discussion 1156.
209. Pfisterer WK, Aboul-Enein F, Gebhart E, Graf M, Aichholzer M, Mühlbauer M. Continuous intraventricular pressure monitoring for diagnosis of normal-pressure hydrocephalus. Acta Neurochir (Wien) 2007 Oct;149(10):983-90; discussion 990.
210. Brean A, Eide PK. Assessment of idiopathic normal pressure patients in neurological practice: the role of lumbar infusion testing for referral of patients to neurosurgery. Eur J Neurol 2008 Jun;15(6):605-612.
211. Scollato A, Tenenbaum R, Bahl G, Celerini M, Salani B, Di Lorenzo N. Changes in aqueductal CSF stroke volume and progression of symptoms in patients with unshunted idiopathic normal pressure hydrocephalus. AJNR Am J Neuroradiol 2008 Jan;29(1):192-197.
212. Razay G, Vreugdenhil A, Liddell J. A prospective study of ventriculo-peritoneal shunting for idiopathic normal pressure hydrocephalus. J Clin Neurosci 2009 Sep;16(9):1180-1183.
213. Andrén K, Wikkelsø C, Tisell M, Hellström P. Natural course of idiopathic normal pressure hydrocephalus. J Neurol Neurosurg Psychiatry 2014 Jul;85(7):806-810.
214. Toma AK, Papadopoulos MC, Stapleton S, Kitchen ND, Watkins LD. Systematic review of the outcome of shunt surgery in idiopathic normal-pressure hydrocephalus. Acta Neurochir (Wien) 2013 Oct;155(10):1977-1980.
215. Chan AK, McGovern RA, Zacharia BE, Mikell CB, Bruce SS, Sheehy JP, et al. Inferior short-term safety profile of endoscopic third ventriculostomy compared with ventriculoperitoneal shunt placement for idiopathic normal-pressure hydrocephalus: a population-based study. Neurosurgery 2013 Dec;73(6):951-60; discussion 960-1.

80
216. Greenberg JO, Shenkin HA, Adam R. Idiopathic normal pressure hydrocephalus– a report of 73 patients. J Neurol Neurosurg Psychiatry 1977 Apr;40(4):336-341.
217. Black PM. Idiopathic normal-pressure hydrocephalus. Results of shunting in 62 patients. J Neurosurg 1980 Mar;52(3):371-377.
218. Raftopoulos C, Massager N, Baleriaux D, Deleval J, Clarysse S, Brotchi J. Prospective analysis by computed tomography and long-term outcome of 23 adult patients with chronic idiopathic hydrocephalus. Neurosurgery 1996 Jan;38(1):51-59.
219. Boon AJ, Tans JT, Delwel EJ, Egeler-Peerdeman SM, Hanlo PW, Wurzer HA, et al. Dutch Normal-Pressure Hydrocephalus Study: randomized comparison of low- and medium-pressure shunts. J Neurosurg 1998 Mar;88(3):490-495.
220. Malm J, Kristensen B, Stegmayr B, Fagerlund M, Koskinen LO. Three-year survival and functional outcome of patients with idiopathic adult hydrocephalus syndrome. Neurology 2000 Aug 22;55(4):576-578.
221. Spagnoli D, Innocenti L, Bello L, Pluderi M, Bacigaluppi S, Tomei G, et al. Impact of cerebrovascular disease on the surgical treatment of idiopathic normal pressure hydrocephalus. Neurosurgery 2006 Sep;59(3):545-52; discussion 545-52.
222. Gölz L, Ruppert FH, Meier U, Lemcke J. Outcome of modern shunt therapy in patients with idiopathic normal pressure hydrocephalus 6 years postoperatively. J Neurosurg 2014 Oct;121(4):771-775.
223. World Health Organization. The top 10 causes of death – Fact sheet N°310 – The 10 leading causes of death by country income group (2012). 2014; Available at: http://www.who.int/mediacentre/factsheets/fs310/en/. Accessed 14/02, 2016.
224. Statistics Finland. Tables on the subject area of: Population Structure. 2015; Available at: http://pxnet2.stat.fi/PXWeb/pxweb/en/StatFin/StatFin__vrm__vaerak/?tablelist=true. Accessed 14/02, 2016.
225. Laiterä T, Kurki MI, Pursiheimo JP, Zetterberg H, Helisalmi S, Rauramaa T, et al. The expression of transthyretin and amyloid-beta protein precursor is altered in the brain of idiopathic normal pressure hydrocephalus patients. J Alzheimers Dis 2015 Oct 27;48(4):959-968.
226. Bergsneider M, Miller C, Vespa PM, Hu X. Surgical management of adult hydrocephalus. Neurosurgery 2008 Feb;62 Suppl 2:643-59; discussion 659-60.
227. Thal DR, Rub U, Orantes M, Braak H. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 2002 Jun 25;58(12):1791-1800.
228. Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med 2010 Jan 28;362(4):329-344.
229. Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010 Jan;67(1):122-131.
230. Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol 2011

81
Jan;69(1):181-192.
231. Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A 1993 Oct 15;90(20):9649-9653.
232. Nagy Z, Esiri MM, Jobst KA, Johnston C, Litchfield S, Sim E, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience 1995 Dec;69(3):757-761.
233. Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, et al. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol 1996 Jan;39(1):62-70.
234. Kok E, Haikonen S, Luoto T, Huhtala H, Goebeler S, Haapasalo H, et al. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann Neurol 2009 Jun;65(6):650-657.
235. Nicoll JA, Savva GM, Stewart J, Matthews FE, Brayne C, Ince P, et al. Association between APOE genotype, neuropathology and dementia in the older population of England and Wales. Neuropathol Appl Neurobiol 2010 Sep 30.
236. Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 1993 Mar 1;90(5):1977-1981.
237. Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010 May 12;303(18):1832-1840.
238. Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol 2011 Mar;10(3):241-252.
239. Huttunen T, von und zu Fraunberg M, Frosen J, Lehecka M, Tromp G, Helin K, et al. Saccular intracranial aneurysm disease: distribution of site, size, and age suggests different etiologies for aneurysm formation and rupture in 316 familial and 1454 sporadic eastern Finnish patients. Neurosurgery 2010 Apr;66(4):631-8; discussion 638.
240. Lehtovirta M, Soininen H, Helisalmi S, Mannermaa A, Helkala EL, Hartikainen P, et al. Clinical and neuropsychological characteristics in familial and sporadic Alzheimer’s disease: relation to apolipoprotein E polymorphism. Neurology 1996 Feb;46(2):413-419.
241. Vepsäläinen S, Helisalmi S, Mannermaa A, Pirttilä T, Soininen H, Hiltunen M. Combined risk effects of IDE and NEP gene variants on Alzheimer disease. J Neurol Neurosurg Psychiatry 2009 Nov;80(11):1268-1270.
242. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984 Jul;34(7):939-944.

82
243. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington DC: American Psychiatric Association; 1994.
244. Tsukamoto K, Watanabe T, Matsushima T, Kinoshita M, Kato H, Hashimoto Y, et al. Determination by PCR-RFLP of apo E genotype in a Japanese population. J Lab Clin Med 1993 Apr;121(4):598-602.
245. Morris JC, Heyman A, Mohs RC, Hughes JP, van Belle G, Fillenbaum G, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 1989 Sep;39(9):1159-1165.
246. Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 2010 Jul 20;75(3):230-238.
247. Alzheimer Research Forum. Meta-analysis of all published AD association studies (case-control only) APOE_E2/3/4. 2010; Available at: http://www.alzgene.org/meta.asp?geneID=83. Accessed 14/02, 2016.
248. Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Shaw LM, Trojanowski JQ, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol 2010 Mar;67(3):308-316.
249. Pyykkö OT, Helisalmi S, Koivisto AM, Mölsä JA, Rummukainen J, Nerg O, et al. APOE4 predicts amyloid-β in cortical brain biopsy but not idiopathic normal pressure hydrocephalus. J Neurol Neurosurg Psychiatry 2012 Nov;83(11):1119-1124.
250. Tarnaris A, Toma AK, Chapman MD, Keir G, Kitchen ND, Watkins LD. Use of cerebrospinal fluid amyloid-beta and total tau protein to predict favorable surgical outcomes in patients with idiopathic normal pressure hydrocephalus. J Neurosurg 2011 Jul;115(1):145-150.
251. Selnes P, Blennow K, Zetterberg H, Grambaite R, Rosengren L, Johnsen L, et al. Effects of cerebrovascular disease on amyloid precursor protein metabolites in cerebrospinal fluid. Cerebrospinal Fluid Res 2010 Jul 30;7:10-8454-7-10.
252. Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res 2010 Jun;88(8):1615-1631.
253. Whitaker JN. Myelin basic protein in cerebrospinal fluid and other body fluids. Mult Scler 1998 Feb;4(1):16-21.
254. Sutton LN, Wood JH, Brooks BR, Barrer SJ, Kline M, Cohen SR. Cerebrospinal fluid myelin basic protein in hydrocephalus. J Neurosurg 1983 Sep;59(3):467-470.
255. Zetterberg H, Andreasson U, Hansson O, Wu G, Sankaranarayanan S, Andersson ME, et al. Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Arch Neurol 2008 Aug;65(8):1102-1107.
256. Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann Neurol 2006 Mar;59(3):512-519.
257. Hanson AJ, Craft S, Banks WA. The APOE genotype: modification of therapeutic responses in

83
Alzheimer’s disease. Curr Pharm Des 2015;21(1):114-120.
258. Aygok G, Marmarou A, Young HF. Three-year outcome of shunted idiopathic NPH patients. Acta Neurochir Suppl 2005;95:241-245.
259. Tisell M, Hellström P, Ahl-Börjesson G, Barrows G, Blomsterwall E, Tullberg M, et al. Long-term outcome in 109 adult patients operated on for hydrocephalus. Br J Neurosurg 2006 Aug;20(4):214-221.
260. Kahlon B, Sjunnesson J, Rehncrona S. Long-term outcome in patients with suspected normal pressure hydrocephalus. Neurosurgery 2007 Feb;60(2):327-32; discussion 332.
261. Sprung C, Schlosser HG, Lemcke J, Meier U, Messing-Jünger M, Trost HA, et al. The adjustable proGAV shunt: a prospective safety and reliability multicenter study. Neurosurgery 2010 Mar;66(3):465-474.
262. Lundkvist B, Koskinen LO, Birgander R, Eklund A, Malm J. Cerebrospinal fluid dynamics and long-term survival of the Strata valve in idiopathic normal pressure hydrocephalus. Acta Neurol Scand 2011 Aug;124(2):115-121.
263. Stranjalis G, Kalamatianos T, Koutsarnakis C, Loufardaki M, Stavrinou L, Sakas DE. Twelve-year hospital outcomes in patients with idiopathic hydrocephalus. Acta Neurochir Suppl 2012;113:115-117.

DIS
SE
RT
AT
ION
S | O
KK
O T
. PY
YK
KÖ
| IDIO
PA
TH
IC N
OR
MA
L P
RE
SS
UR
E H
YD
RO
CE
PH
AL
US
: A S
TU
DY
... | No
336
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
Dissertations in Health Sciences
Dissertations in Health Sciences
PUBLICATIONS OF THE UNIVERSITY OF EASTERN FINLAND
OKKO T. PYYKKÖ
IDIOPATHIC NORMAL PRESSURE HYDROCEPHALUSA study of epidemiology, genetics, and cerebrospinal �uid
OKKO T. PYYKKÖ
ISBN 978-952-61-2059-1ISSN 1798-5706
Idiopathic normal pressure hydrocephalus isa slowly progressive syndrome in the elderly
characterized by gait disorder, cognitivedeterioration, and urinary incontinence.
Compared to earlier studies, a higher annualincidence of the syndrome was noted in a
Finnish population with an increasing trend.High vascular comorbidity and mortality wasobserved without a major neuroin�ammatorycomponent. Apolipoprotein E genotypes did
not differentiate patients with idiopathicnormal pressure hydrocephalus from healthy
age- and gender-matched controls.