immunotherapy reengineering chimeric antigen receptor t ... · immunotherapy reengineering chimeric...
TRANSCRIPT

IMMUNOTHERAPY
Reengineering chimeric antigenreceptor T cells for targeted therapyof autoimmune diseaseChristoph T. Ellebrecht,1 Vijay G. Bhoj,2 Arben Nace,1 Eun Jung Choi,1 Xuming Mao,1
Michael Jeffrey Cho,1 Giovanni Di Zenzo,3 Antonio Lanzavecchia,4 John T. Seykora,1
George Cotsarelis,1 Michael C. Milone,2*† Aimee S. Payne1*†
Ideally, therapy for autoimmune diseases should eliminate pathogenic autoimmune cellswhile sparing protective immunity, but feasible strategies for such an approach have beenelusive. Here, we show that in the antibody-mediated autoimmune disease pemphigusvulgaris (PV), autoantigen-based chimeric immunoreceptors can direct T cells to killautoreactive B lymphocytes through the specificity of the B cell receptor (BCR). Weengineered human T cells to express a chimeric autoantibody receptor (CAAR), consistingof the PV autoantigen, desmoglein (Dsg) 3, fused to CD137-CD3z signaling domains.Dsg3 CAAR-T cells exhibit specific cytotoxicity against cells expressing anti-Dsg3 BCRsin vitro and expand, persist, and specifically eliminate Dsg3-specific B cells in vivo.CAAR-T cells may provide an effective and universal strategy for specific targeting ofautoreactive B cells in antibody-mediated autoimmune disease.
Pemphigus vulgaris (PV) is a life-threateningautoimmune blistering disease caused byautoantibodies to the keratinocyte adhe-sion protein Dsg3 (1). CD20-targeted B celldepletion results in short-term disease re-
mission in 95% of pemphigus patients, but 81%relapse and fatal infection may occur (2). AfterCD20-targeted depletion, serum autoantibodytiters drop, indicating that short-lived plasma-blasts are the source of autoantibodies in PVand targeting of CD20+memoryB cell precursorsindirectly depletes autoantibody-secreting CD20–
plasmablasts (3, 4). Relapsing pemphigus demon-strates the same anti-Dsg3 B cell clones observedduring active disease, whereas disease remissionis associated with disappearance of circulatinganti-Dsg3B cells (5). Thus, targeted elimination ofanti-Dsg3 memory B cells should cure PV with-out the risks of general immunosuppression.Recently, chimeric antigen receptor (CAR) tech-
nology has revolutionized cancer therapy. ACD19-specific CAR, consisting of an extracellularsingle-chain variable fragment (scFv) antibodyto CD19 fused to T cell cytoplasmic signalingdomains, activates T cell cytotoxicity upon con-tact with CD19+ B cells, causing specific andpermanent elimination of B cells and durableremission of leukemia (6–14). In PV, pathogenicmemoryB cells express anti-Dsg3 B cell receptors(BCRs). We reasoned that by expressing Dsg3 asthe extracellular domain of a chimeric immuno-
receptor, cytotoxicity would become specific foronly those B cells bearing anti-Dsg3 BCRs, pro-viding targeted therapy for PV without generalimmunosuppression. Such a strategy would di-rectly eliminate surface immunoglobulin (sIg)+
anti-Dsg3 memory B cells and indirectly elimi-nate sIg− Dsg3-specific short-lived plasma cellsthat produce the disease-causing antibodies. Wethus created a chimeric autoantibody receptor(CAAR) (fig. S1A), with the autoantigen Dsg3 asthe CAAR extracellular domain, in order to en-gineer T cells to kill autoimmune B cells in PV.Dsg3 consists of five extracellular cadherin
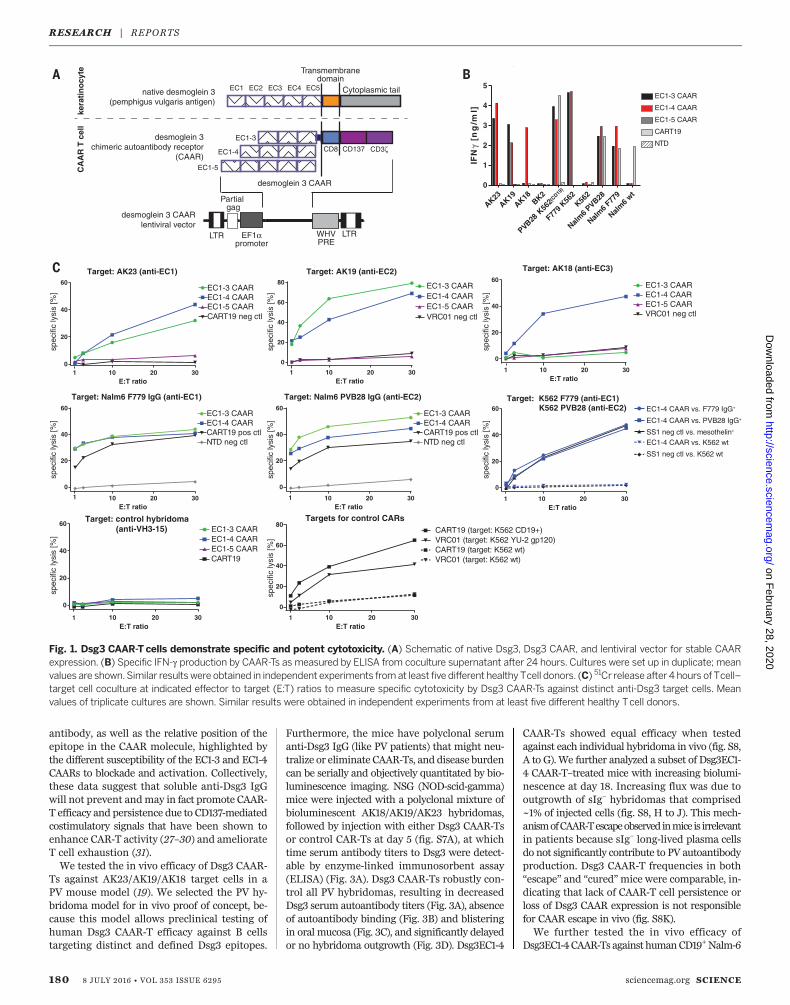
(EC) domains, with residues important for celladhesion residing in EC1 and EC2 (15). Auto-antibodies to EC1, EC2, EC3, EC4, and EC5 occurin 91, 71, 51, 19, and 12% of PV sera; no PV seratarget only the EC4 and/or EC5 domains (16).Since T cell activation depends on the intermem-brane distance of the immunologic synapse (17),we reasoned that shorter conformational frag-ments of Dsg3 should enhance CAAR efficacy.We therefore designed a panel of Dsg3 CAARsfor expression in primary humanT cells (Fig. 1A),using Dsg3 EC1-3/EC1-4/EC1-5 as the extracellu-lar domain, fused to a dimerization-competentCD8a transmembrane (18) and CD137-CD3z cy-toplasmic signaling domains, which were usedsuccessfully in CD19 CAR clinical trials (6, 7).EC1-3/EC1-4 CAARs demonstrate robust surfaceexpression of mature, conformational Dsg3 inprimary human T cells, whereas EC1-5 CAARexpression is more variable (fig. S1, B to D).We first evaluated the ability of Dsg3 CAAR-T
cells (CAAR-Ts) to kill anti-Dsg3 B cells in vitro,using antibody-secreting hybridomas that tar-get EC1 (AK23), EC2 (AK19), and EC3-4 (AK18)(19), or K562/Nalm-6 cells expressing pathogenicF779/anti-EC1 or PVB28/anti-EC2 IgG cloned fromPV patients (20, 21) (fig. S2). All BCRs were ex-
pressed at a density comparable to humanB cells(fig. S3). Dsg3EC1-3/EC1-4 CAAR-Ts demonstrateinterferon-g (IFN-g) secretion and specific cytol-ysis against anti-EC1/EC2 but not control tar-gets (Fig. 1, B and C, and fig. S4). Dsg3EC1-5CAAR-Ts exhibitminimal cytolysis, andDsg3EC1-3CAAR-Ts do not lyse anti-EC3/4 targets. Thus,the Dsg3EC1-4 CAAR demonstrates the best com-bination of potency and breadth, with specificcytolysis of cells expressing anti-Dsg3 BCRs.To investigate the mechanism of CAAR-T ac-
tivation, we examined the molecular organiza-tion within CAAR-Ts upon binding sIg target bytotal internal reflection fluorescence microscopy.CAAR-Ts form immunologic synapses analogousto T cell receptor (TCR)–peptide/major histocom-patibility complex (MHC) interaction (22, 23),with actin reorganization and centripetal move-ment of CAAR-IgG clusters resulting in a centralsupramolecular activation complex (SMAC)–likestructure (fig. S5A and movie S1). The proteintyrosine phosphatase CD45 is excluded from earlyCAAR-IgG microclusters (fig. S5B and movie S2),similar to findings with the anti-CD19 CAR (24)and the native TCR-MHC synapse (25), sugges-tive of a kinetic segregation model for CAAR ac-tivation (26).PV patients have serum anti-Dsg3 IgG that
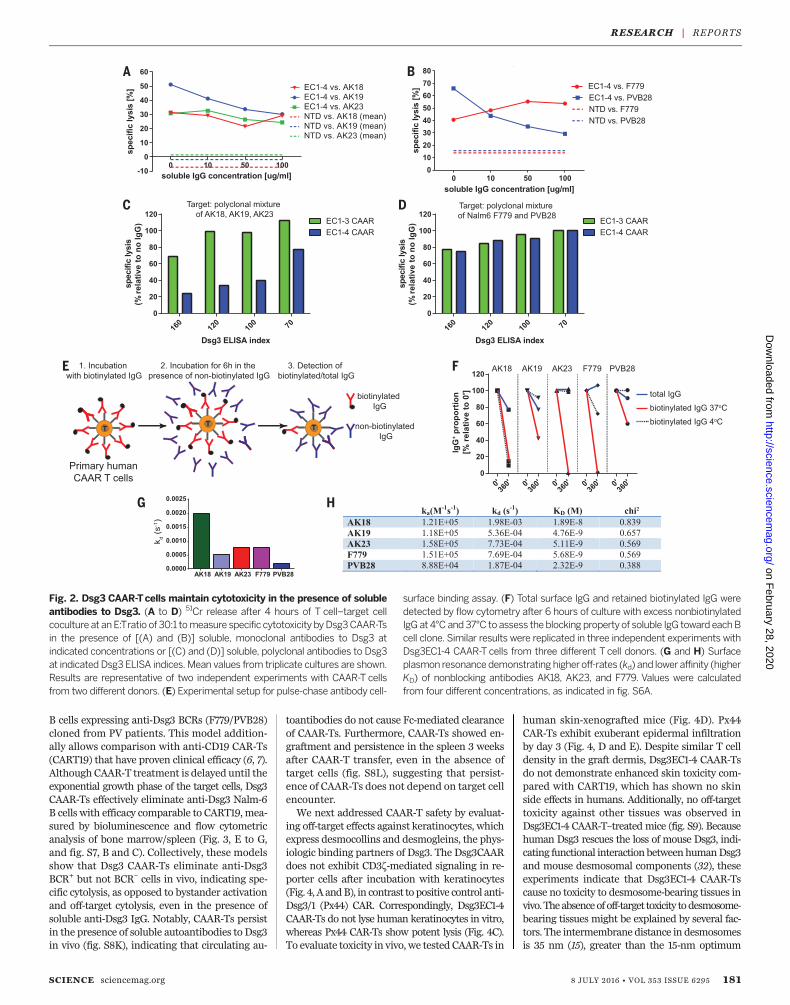
might neutralize, or alternatively could help stim-ulate, Dsg3 CAAR-Ts. We therefore evaluatedDsg3 CAAR-T cytotoxicity in the presence of sol-uble monoclonal IgGmatching the targeted BCRto maximize the neutralization capacity of thesoluble IgG. Soluble anti-Dsg3 IgG decreases butstill preserves compelling CAAR-T cytotoxicityagainst AK19/PVB28, minimally affects AK23/AK18, and potentiates cytotoxicity against F779targets (Fig. 2, A and B). Because CAAR-Ts willencounter polyclonal anti-Dsg3 IgG inPVpatients,we next tested Dsg3 CAAR-T cytotoxicity againstpolyclonal targets in the presence of polyclonalPV serum IgG. Dsg3EC1-3, and to a lesser extentDsg3EC1-4, CAAR-Ts retained efficient levels ofcytolysis against AK hybridomas (Fig. 2C), de-spite the presence of PV serum IgG as well assecreted anti-Dsg3 IgG by these hybridomas.Both Dsg3EC1-3 and EC1-4 CAAR-Ts effectivelykilled Nalm6 cells expressing human anti-Dsg3IgG in the presence of PV serum IgG (Fig. 2D).Surface binding competition assays indicate thatantibodies that inhibit cytotoxicity (AK19/PVB28)persist on the CAAR-T surface, which reduces thenumber of accessible CAAR molecules to bindtarget cells, while noninhibitory F779/AK23/AK18aremore rapidly replacedby competing antibodies,with AK18 replacement occurring even at 4°C,suggesting low affinity (Fig. 2, E and F). Indeed,surface plasmon resonance analysis indicates thatnoninhibitory antibodies tend to have fasteroff-rates and lower affinity (Fig. 2, G and H,and fig. S6A). Furthermore, F779 and PV serumIgG stimulate Dsg3EC1-4 CAAR-Ts, more so thanthe Dsg3EC1-3 CAAR-Ts, to secrete low levels ofIFN-g and proliferate (fig. S6, B and C). Thus,the effect of soluble antibodies to Dsg3 on CAAR-Tfunction and activation is a complex process thatis affected by affinity and binding kinetics of the
RESEARCH | REPORTS
SCIENCE sciencemag.org 8 JULY 2016 • VOL 353 ISSUE 6295 179
1Department of Dermatology, University of Pennsylvania,Philadelphia, PA 19104, USA. 2Department of Pathology andLaboratory Medicine, University of Pennsylvania,Philadelphia, PA 19104, USA. 3Laboratory of Molecular andCellular Biology, Istituto Dermopatico dell’Immacolata(IDI-IRCCS), 00167 Rome, Italy. 4Institute for Research inBiomedicine, 6500 Bellinzona, Switzerland.*Corresponding author. Email: [email protected](A.S.P.); [email protected] (M.C.M.) †These authorscontributed equally to this work.
on February 28, 2020
http://science.sciencem
ag.org/D
ownloaded from
antibody, as well as the relative position of theepitope in the CAAR molecule, highlighted bythe different susceptibility of the EC1-3 and EC1-4CAARs to blockade and activation. Collectively,these data suggest that soluble anti-Dsg3 IgGwill not prevent andmay in fact promote CAAR-T efficacy and persistence due to CD137-mediatedcostimulatory signals that have been shown toenhance CAR-T activity (27–30) and ameliorateT cell exhaustion (31).We tested the in vivo efficacy of Dsg3 CAAR-
Ts against AK23/AK19/AK18 target cells in aPV mouse model (19). We selected the PV hy-bridoma model for in vivo proof of concept, be-cause this model allows preclinical testing ofhuman Dsg3 CAAR-T efficacy against B cellstargeting distinct and defined Dsg3 epitopes.
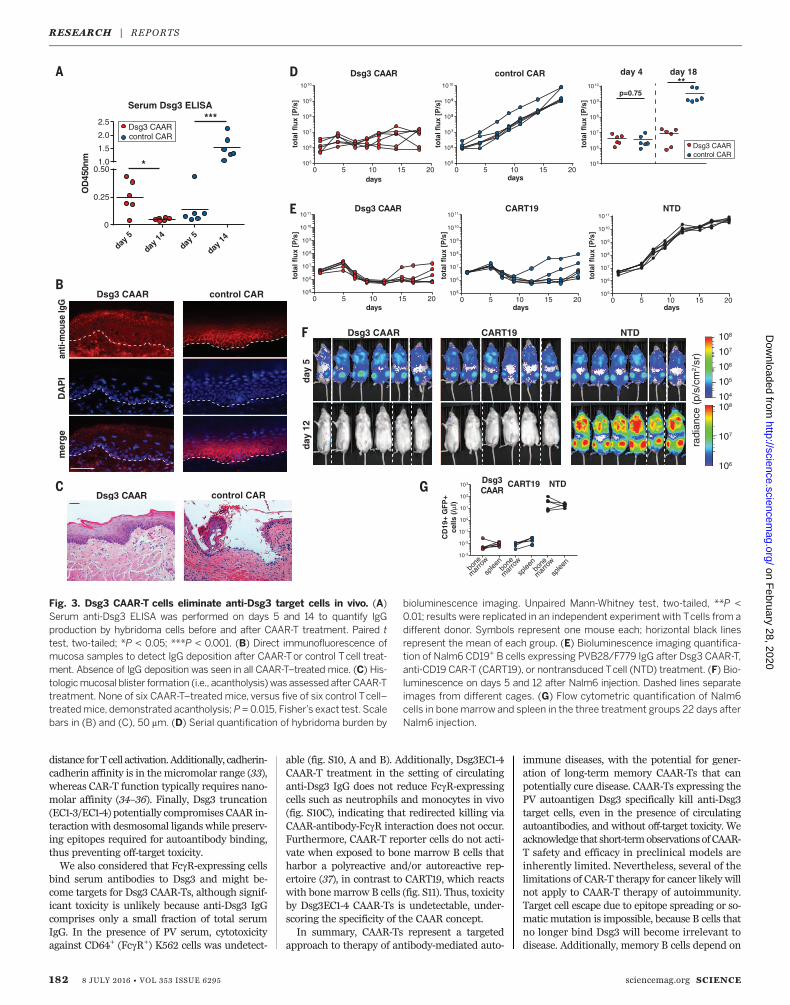
Furthermore, the mice have polyclonal serumanti-Dsg3 IgG (like PV patients) that might neu-tralize or eliminate CAAR-Ts, and disease burdencan be serially and objectively quantitated by bio-luminescence imaging. NSG (NOD-scid-gamma)mice were injected with a polyclonal mixture ofbioluminescent AK18/AK19/AK23 hybridomas,followed by injection with either Dsg3 CAAR-Tsor control CAR-Ts at day 5 (fig. S7A), at whichtime serum antibody titers to Dsg3 were detect-able by enzyme-linked immunosorbent assay(ELISA) (Fig. 3A). Dsg3 CAAR-Ts robustly con-trol all PV hybridomas, resulting in decreasedDsg3 serum autoantibody titers (Fig. 3A), absenceof autoantibody binding (Fig. 3B) and blisteringin oral mucosa (Fig. 3C), and significantly delayedor no hybridoma outgrowth (Fig. 3D). Dsg3EC1-4
CAAR-Ts showed equal efficacy when testedagainst each individual hybridoma in vivo (fig. S8,A to G). We further analyzed a subset of Dsg3EC1-4 CAAR-T–treated mice with increasing biolumi-nescence at day 18. Increasing flux was due tooutgrowth of sIg– hybridomas that comprised~1% of injected cells (fig. S8, H to J). This mech-anismofCAAR-Tescapeobservedinmiceis irrelevantin patients because sIg– long-lived plasma cellsdo not significantly contribute to PV autoantibodyproduction. Dsg3 CAAR-T frequencies in both“escape” and “cured”mice were comparable, in-dicating that lack of CAAR-T cell persistence orloss of Dsg3 CAAR expression is not responsiblefor CAAR escape in vivo (fig. S8K).We further tested the in vivo efficacy of
Dsg3EC1-4CAAR-Ts against humanCD19+Nalm-6
180 8 JULY 2016 • VOL 353 ISSUE 6295 sciencemag.org SCIENCE
Target: AK18 (anti-EC3)
EC1-3 CAAREC1-4 CAAREC1-5 CAARVRC01 neg ctl
10 20 30
0
20
40
60
E:T ratio
spec
ific
lysi
s [%
]
spec
ific
lysi
s [%
]sp
ecifi
c ly
sis
[%]
spec
ific
lysi
s [%
]
spec
ific
lysi
s [%
]sp
ecifi
c ly
sis
[%]
spec
ific
lysi
s [%
]
spec
ific
lysi
s [%
]
10 20 30
0
20
40
60
80
Target: AK19 (anti-EC2)
E:T ratio
EC1-3 CAAREC1-4 CAAR
VRC01 neg ctlEC1-5 CAAR
10 20 30
0
20
40
60
E:T ratio
EC1-4 CAAR vs. F779 IgG+
EC1-4 CAAR vs. PVB28 IgG+
SS1 neg ctl vs. mesothelin+
EC1-4 CAAR vs. K562 wt
SS1 neg ctl vs. K562 wt
Target: K562 F779 (anti-EC1)
LTR LTR
Partial gag
EF1αpromoter
WHVPRE
Transmembranedomain
native desmoglein 3(pemphigus vulgaris antigen)
desmoglein 3chimeric autoantibody receptor
(CAAR)
desmoglein 3 CAAR
EC1-3
EC1-4
EC1-5
CD137 CD3ζCD8
desmoglein 3 CAARlentiviral vector
EC1 EC4EC2 EC3 EC5 Cytoplasmic tail k
erat
ino
cyte
CA
AR
T c
ell
10 20 30
0
20
40
60
Target: Nalm6 F779 IgG (anti-EC1)
E:T ratio
EC1-3 CAAREC1-4 CAARCART19 pos ctl NTD neg ctl
10 20 30
0
20
40
60
Target: Nalm6 PVB28 IgG (anti-EC2)
E:T ratio
EC1-3 CAAREC1-4 CAARCART19 pos ctlNTD neg ctl
Target: AK23 (anti-EC1)
10 20 300
20
40
60
E:T ratio
EC1-3 CAAREC1-4 CAAR
CART19 neg ctlEC1-5 CAAR
1
11 1
1 1
11
10 20 30
0
20
40
60
80
E:T ratio
CART19 (target: K562 CD19+)VRC01 (target: K562 YU-2 gp120)
VRC01 (target: K562 wt)CART19 (target: K562 wt)
Targets for control CARs
10 20 30
0
20
40
60Target: control hybridoma
(anti-VH3-15)
E:T ratio
EC1-3 CAAREC1-4 CAAREC1-5 CAARCART19
AK23AK19
AK18BK2
PVB28
K562
F779 K
562
K562
Nalm6 P
VB28
Nalm6 F
779
Nalm6 w
t0
1
2
3
4
5
IFN
γ [n
g/m
l]
EC1-3 CAAR
EC1-4 CAAR
EC1-5 CAAR
CART19
NTD
(CD19
)
K562 PVB28 (anti-EC2)
Fig. 1. Dsg3 CAAR-T cells demonstrate specific and potent cytotoxicity. (A) Schematic of native Dsg3, Dsg3 CAAR, and lentiviral vector for stable CAARexpression. (B) Specific IFN-g production by CAAR-Ts as measured by ELISA from coculture supernatant after 24 hours. Cultures were set up in duplicate; meanvalues are shown.Similar resultswere obtained in independent experiments fromat least five different healthy Tcell donors. (C) 51Cr release after 4 hours of Tcell–target cell coculture at indicated effector to target (E:T) ratios to measure specific cytotoxicity by Dsg3 CAAR-Ts against distinct anti-Dsg3 target cells. Meanvalues of triplicate cultures are shown. Similar results were obtained in independent experiments from at least five different healthy Tcell donors.
RESEARCH | REPORTSon F
ebruary 28, 2020
http://science.sciencemag.org/
Dow
nloaded from
B cells expressing anti-Dsg3 BCRs (F779/PVB28)cloned from PV patients. This model addition-ally allows comparison with anti-CD19 CAR-Ts(CART19) that have proven clinical efficacy (6, 7).Although CAAR-T treatment is delayed until theexponential growth phase of the target cells, Dsg3CAAR-Ts effectively eliminate anti-Dsg3 Nalm-6B cells with efficacy comparable to CART19, mea-sured by bioluminescence and flow cytometricanalysis of bone marrow/spleen (Fig. 3, E to G,and fig. S7, B and C). Collectively, these modelsshow that Dsg3 CAAR-Ts eliminate anti-Dsg3BCR+ but not BCR– cells in vivo, indicating spe-cific cytolysis, as opposed to bystander activationand off-target cytolysis, even in the presence ofsoluble anti-Dsg3 IgG. Notably, CAAR-Ts persistin the presence of soluble autoantibodies to Dsg3in vivo (fig. S8K), indicating that circulating au-
toantibodies do not cause Fc-mediated clearanceof CAAR-Ts. Furthermore, CAAR-Ts showed en-graftment and persistence in the spleen 3 weeksafter CAAR-T transfer, even in the absence oftarget cells (fig. S8L), suggesting that persist-ence of CAAR-Ts does not depend on target cellencounter.We next addressed CAAR-T safety by evaluat-
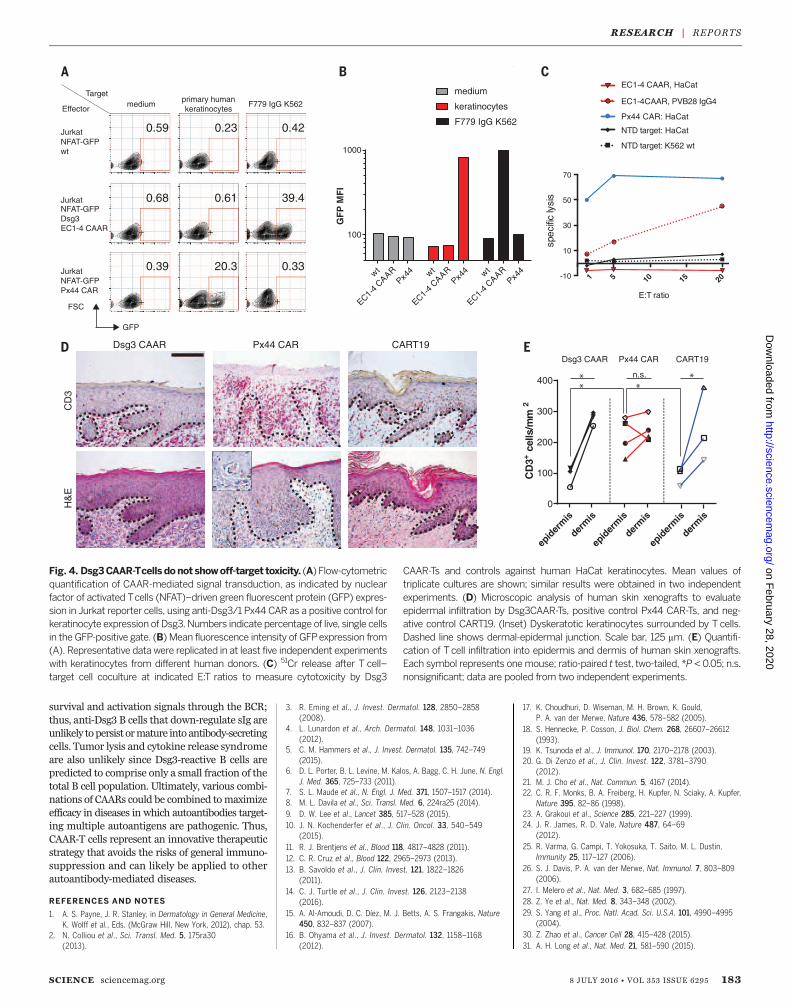
ing off-target effects against keratinocytes, whichexpress desmocollins and desmogleins, the phys-iologic binding partners of Dsg3. The Dsg3CAARdoes not exhibit CD3z-mediated signaling in re-porter cells after incubation with keratinocytes(Fig. 4, A andB), in contrast to positive control anti-Dsg3/1 (Px44) CAR. Correspondingly, Dsg3EC1-4CAAR-Ts do not lyse human keratinocytes in vitro,whereas Px44 CAR-Ts show potent lysis (Fig. 4C).To evaluate toxicity in vivo, we tested CAAR-Ts in
human skin-xenografted mice (Fig. 4D). Px44CAR-Ts exhibit exuberant epidermal infiltrationby day 3 (Fig. 4, D and E). Despite similar T celldensity in the graft dermis, Dsg3EC1-4 CAAR-Tsdo not demonstrate enhanced skin toxicity com-pared with CART19, which has shown no skinside effects in humans. Additionally, no off-targettoxicity against other tissues was observed inDsg3EC1-4 CAAR-T–treated mice (fig. S9). Becausehuman Dsg3 rescues the loss of mouse Dsg3, indi-cating functional interactionbetweenhumanDsg3and mouse desmosomal components (32), theseexperiments indicate that Dsg3EC1-4 CAAR-Tscause no toxicity to desmosome-bearing tissues invivo. Theabsenceof off-target toxicity todesmosome-bearing tissues might be explained by several fac-tors. The intermembrane distance in desmosomesis 35 nm (15), greater than the 15-nm optimum
SCIENCE sciencemag.org 8 JULY 2016 • VOL 353 ISSUE 6295 181
Fig. 2. Dsg3 CAAR-Tcells maintain cytotoxicity in the presence of solubleantibodies to Dsg3. (A to D) 51Cr release after 4 hours of T cell–target cellcoculture at anE:Tratio of 30:1 tomeasure specific cytotoxicity byDsg3CAAR-Tsin the presence of [(A) and (B)] soluble, monoclonal antibodies to Dsg3 atindicated concentrations or [(C) and (D)] soluble, polyclonal antibodies to Dsg3at indicated Dsg3 ELISA indices. Mean values from triplicate cultures are shown.Results are representative of two independent experiments with CAAR-T cellsfrom two different donors. (E) Experimental setup for pulse-chase antibody cell-
surface binding assay. (F) Total surface IgG and retained biotinylated IgG weredetected by flow cytometry after 6 hours of culture with excess nonbiotinylatedIgG at 4°C and37°C to assess theblocking propertyof soluble IgG toward eachBcell clone. Similar results were replicated in three independent experiments withDsg3EC1-4 CAAR-T cells from three different T cell donors. (G and H) Surfaceplasmon resonancedemonstrating higheroff-rates (kd) and lower affinity (higherKD) of nonblocking antibodies AK18, AK23, and F779. Values were calculatedfrom four different concentrations, as indicated in fig. S6A.
RESEARCH | REPORTSon F
ebruary 28, 2020
http://science.sciencemag.org/
Dow
nloaded from
distance forT cell activation.Additionally, cadherin-cadherin affinity is in the micromolar range (33),whereas CAR-T function typically requires nano-molar affinity (34–36). Finally, Dsg3 truncation(EC1-3/EC1-4) potentially compromises CAAR in-teraction with desmosomal ligands while preserv-ing epitopes required for autoantibody binding,thus preventing off-target toxicity.We also considered that FcgR-expressing cells
bind serum antibodies to Dsg3 and might be-come targets for Dsg3 CAAR-Ts, although signif-icant toxicity is unlikely because anti-Dsg3 IgGcomprises only a small fraction of total serumIgG. In the presence of PV serum, cytotoxicityagainst CD64+ (FcgR+) K562 cells was undetect-
able (fig. S10, A and B). Additionally, Dsg3EC1-4CAAR-T treatment in the setting of circulatinganti-Dsg3 IgG does not reduce FcgR-expressingcells such as neutrophils and monocytes in vivo(fig. S10C), indicating that redirected killing viaCAAR-antibody-FcgR interaction does not occur.Furthermore, CAAR-T reporter cells do not acti-vate when exposed to bone marrow B cells thatharbor a polyreactive and/or autoreactive rep-ertoire (37), in contrast to CART19, which reactswith bone marrow B cells (fig. S11). Thus, toxicityby Dsg3EC1-4 CAAR-Ts is undetectable, under-scoring the specificity of the CAAR concept.In summary, CAAR-Ts represent a targeted
approach to therapy of antibody-mediated auto-
immune diseases, with the potential for gener-ation of long-term memory CAAR-Ts that canpotentially cure disease. CAAR-Ts expressing thePV autoantigen Dsg3 specifically kill anti-Dsg3target cells, even in the presence of circulatingautoantibodies, and without off-target toxicity. Weacknowledge that short-termobservationsofCAAR-T safety and efficacy in preclinical models areinherently limited. Nevertheless, several of thelimitations of CAR-T therapy for cancer likely willnot apply to CAAR-T therapy of autoimmunity.Target cell escape due to epitope spreading or so-matic mutation is impossible, because B cells thatno longer bind Dsg3 will become irrelevant todisease. Additionally, memory B cells depend on
182 8 JULY 2016 • VOL 353 ISSUE 6295 sciencemag.org SCIENCE
106
107
108
control CARDsg3 CAARd
ay 5
day
12
CART19Dsg3 CAAR NTD
104
105
106
107
108
radi
ance
(p/
s/cm
2 /sr
)
0 5 10 15 20105
106
107
108
109
1010
1011Dsg3 CAAR
days
tota
l flu
x [P
/s]
tota
l flu
x [P
/s]
tota
l flu
x [P
/s]
tota
l flu
x [P
/s]
tota
l flu
x [P
/s]
tota
l flu
x [P
/s]
0 5 10 15 20105
106
107
108
109
1010
1011CART19
days0 5 10 15 20
105
106
107
108
109
1010
1011NTD
days
10-3
10-2
10-1
100
101
102
103 CART19Dsg3CAAR
NTD
bone
mar
row
splee
n
0 5 10 15 20105
106
107
108
109
1010
control CAR
days0 5 10 15 20
105
106
107
108
109
1010
Dsg3 CAAR
days
Dsg3 CAAR control CAR
105
106
107
108
109
1010
day 4 day 18
p=0.75**
anti-
mou
se Ig
GD
AP
Im
erg
e
day5
day14
day5
day14
0
0.25
0.501.0
1.5
2.0
2.5
OD
450n
m
Serum Dsg3 ELISA
*
***
Dsg3 CAARcontrol CAR
bone
mar
row
splee
nbo
ne
mar
row
splee
n
CD
19+
GF
P+
cells
(/µ
l)
Dsg3 CAARcontrol CAR
Fig. 3. Dsg3 CAAR-T cells eliminate anti-Dsg3 target cells in vivo. (A)Serum anti-Dsg3 ELISA was performed on days 5 and 14 to quantify IgGproduction by hybridoma cells before and after CAAR-T treatment. Paired ttest, two-tailed; *P < 0.05; ***P < 0.001. (B) Direct immunofluorescence ofmucosa samples to detect IgG deposition after CAAR-Tor control Tcell treat-ment. Absence of IgG deposition was seen in all CAAR-T–treated mice. (C) His-tologicmucosal blister formation (i.e., acantholysis) was assessed after CAAR-Ttreatment. None of six CAAR-T–treated mice, versus five of six control Tcell–treatedmice, demonstrated acantholysis;P =0.015, Fisher’s exact test. Scalebars in (B) and (C), 50 mm. (D) Serial quantification of hybridoma burden by
bioluminescence imaging. Unpaired Mann-Whitney test, two-tailed, **P <0.01; results were replicated in an independent experiment with Tcells from adifferent donor. Symbols represent one mouse each; horizontal black linesrepresent the mean of each group. (E) Bioluminescence imaging quantifica-tion of Nalm6 CD19+ B cells expressing PVB28/F779 IgG after Dsg3 CAAR-T,anti-CD19 CAR-T (CART19), or nontransduced Tcell (NTD) treatment. (F) Bio-luminescence on days 5 and 12 after Nalm6 injection. Dashed lines separateimages from different cages. (G) Flow cytometric quantification of Nalm6cells in bone marrow and spleen in the three treatment groups 22 days afterNalm6 injection.
RESEARCH | REPORTSon F
ebruary 28, 2020
http://science.sciencemag.org/
Dow
nloaded from
survival and activation signals through the BCR;thus, anti-Dsg3 B cells that down-regulate sIg areunlikely topersist ormature intoantibody-secretingcells. Tumor lysis and cytokine release syndromeare also unlikely since Dsg3-reactive B cells arepredicted to comprise only a small fraction of thetotal B cell population. Ultimately, various combi-nations of CAARs could be combined tomaximizeefficacy in diseases in which autoantibodies target-ing multiple autoantigens are pathogenic. Thus,CAAR-T cells represent an innovative therapeuticstrategy that avoids the risks of general immuno-suppression and can likely be applied to otherautoantibody-mediated diseases.
REFERENCES AND NOTES
1. A. S. Payne, J. R. Stanley, in Dermatology in General Medicine,K. Wolff et al., Eds. (McGraw Hill, New York, 2012), chap. 53.
2. N. Colliou et al., Sci. Transl. Med. 5, 175ra30(2013).
3. R. Eming et al., J. Invest. Dermatol. 128, 2850–2858(2008).
4. L. Lunardon et al., Arch. Dermatol. 148, 1031–1036(2012).
5. C. M. Hammers et al., J. Invest. Dermatol. 135, 742–749(2015).
6. D. L. Porter, B. L. Levine, M. Kalos, A. Bagg, C. H. June, N. Engl.J. Med. 365, 725–733 (2011).
7. S. L. Maude et al., N. Engl. J. Med. 371, 1507–1517 (2014).8. M. L. Davila et al., Sci. Transl. Med. 6, 224ra25 (2014).9. D. W. Lee et al., Lancet 385, 517–528 (2015).10. J. N. Kochenderfer et al., J. Clin. Oncol. 33, 540–549
(2015).11. R. J. Brentjens et al., Blood 118, 4817–4828 (2011).12. C. R. Cruz et al., Blood 122, 2965–2973 (2013).13. B. Savoldo et al., J. Clin. Invest. 121, 1822–1826
(2011).14. C. J. Turtle et al., J. Clin. Invest. 126, 2123–2138
(2016).15. A. Al-Amoudi, D. C. Díez, M. J. Betts, A. S. Frangakis, Nature
450, 832–837 (2007).16. B. Ohyama et al., J. Invest. Dermatol. 132, 1158–1168
(2012).
17. K. Choudhuri, D. Wiseman, M. H. Brown, K. Gould,P. A. van der Merwe, Nature 436, 578–582 (2005).
18. S. Hennecke, P. Cosson, J. Biol. Chem. 268, 26607–26612(1993).
19. K. Tsunoda et al., J. Immunol. 170, 2170–2178 (2003).20. G. Di Zenzo et al., J. Clin. Invest. 122, 3781–3790
(2012).21. M. J. Cho et al., Nat. Commun. 5, 4167 (2014).22. C. R. F. Monks, B. A. Freiberg, H. Kupfer, N. Sciaky, A. Kupfer,
Nature 395, 82–86 (1998).23. A. Grakoui et al., Science 285, 221–227 (1999).24. J. R. James, R. D. Vale, Nature 487, 64–69
(2012).25. R. Varma, G. Campi, T. Yokosuka, T. Saito, M. L. Dustin,
Immunity 25, 117–127 (2006).26. S. J. Davis, P. A. van der Merwe, Nat. Immunol. 7, 803–809
(2006).27. I. Melero et al., Nat. Med. 3, 682–685 (1997).28. Z. Ye et al., Nat. Med. 8, 343–348 (2002).29. S. Yang et al., Proc. Natl. Acad. Sci. U.S.A. 101, 4990–4995
(2004).30. Z. Zhao et al., Cancer Cell 28, 415–428 (2015).31. A. H. Long et al., Nat. Med. 21, 581–590 (2015).
SCIENCE sciencemag.org 8 JULY 2016 • VOL 353 ISSUE 6295 183
0.23
0.61
20.3
0.59
0.68 39.4
0.33
0.42
0.39
Effector
Targetmedium
primary humankeratinocytes
F779 IgG K562
Jurkat
Jurkat
NFAT-GFPwt
NFAT-GFPDsg3EC1-4 CAAR
JurkatNFAT-GFPPx44 CAR
GFP
FSC
wt
EC1-4
CAARPx4
4 wt
EC1-4
CAARPx4
4 wt
EC1-4
CAARPx4
4
100
1000
GF
P M
FI
keratinocytes
medium
F779 IgG K562
Px44 CAR
H&
E
Dsg3 CAAR
CD
3
CART19
5 10 15 20-10
10
30
50
70
E:T ratio
spec
ific
lysi
s
EC1-4CAAR, PVB28 IgG4
NTD target: K562 wt
EC1-4 CAAR, HaCat
NTD target: HaCat
Px44 CAR: HaCat
1
epid
erm
is
derm
is
epid
erm
is
derm
is
epid
erm
is
derm
is0
100
200
300
400
CD
3+ce
lls/m
m2
Dsg3 CAAR Px44 CAR CART19
Fig. 4. Dsg3CAAR-Tcells donot showoff-target toxicity. (A) Flow-cytometricquantification of CAAR-mediated signal transduction, as indicated by nuclearfactor of activated Tcells (NFAT)–driven green fluorescent protein (GFP) expres-sion in Jurkat reporter cells, using anti-Dsg3/1 Px44 CAR as a positive control forkeratinocyte expression of Dsg3.Numbers indicate percentage of live, single cellsin the GFP-positive gate. (B) Mean fluorescence intensity of GFPexpression from(A). Representative data were replicated in at least five independent experimentswith keratinocytes from different human donors. (C) 51Cr release after T cell–target cell coculture at indicated E:T ratios to measure cytotoxicity by Dsg3
CAAR-Ts and controls against human HaCat keratinocytes. Mean values oftriplicate cultures are shown; similar results were obtained in two independentexperiments. (D) Microscopic analysis of human skin xenografts to evaluateepidermal infiltration by Dsg3CAAR-Ts, positive control Px44 CAR-Ts, and neg-ative control CART19. (Inset) Dyskeratotic keratinocytes surrounded by T cells.Dashed line shows dermal-epidermal junction. Scale bar, 125 mm. (E) Quantifi-cation of Tcell infiltration into epidermis and dermis of human skin xenografts.Each symbol represents onemouse; ratio-paired t test, two-tailed, *P < 0.05; n.s.nonsignificant; data are pooled from two independent experiments.
RESEARCH | REPORTSon F
ebruary 28, 2020
http://science.sciencemag.org/
Dow
nloaded from

32. D. A. Culton et al., J. Invest. Dermatol. 135, 1590–1597(2015).
33. P. Katsamba et al., Proc. Natl. Acad. Sci. U.S.A. 106,11594–11599 (2009).
34. M. Hudecek et al., Clin. Cancer Res. 19, 3153–3164(2013).
35. K. Watanabe et al., Blood 124, 4799 (2014).36. S. Srivastava, S. R. Riddell, Trends Immunol. 36, 494–502
(2015).37. H. Wardemann et al., Science 301, 1374–1377
(2003).
ACKNOWLEDGMENTS
We thank D. Margolis, S. Prouty, T. Dentchev, C.-Y. Tsai, andS. Nunez-Cruz for technical assistance and consultation on thestudies and J. R. Stanley for helpful discussions. The data reportedin this manuscript are tabulated in the main paper and in the
supplementary materials. Constructs and cell lines are availablefrom the corresponding authors under a Material TransferAgreement with the University of Pennsylvania. C.T.E., V.G.B.,M.C.M, and A.S.P. have filed patent PCT/US15/28872, 2015, whichrelates to compositions and methods of chimeric autoantibodyreceptor T cells. C.T.E. and A.S.P. have filed Provisional PatentApplication 62/222,132, 2015, which relates to the VRC01 chimericantigen receptor. Research reported in this publication wassupported in part by the Penn Institute for Immunology (A.S.P. andM.C.M.); the Dermatology Foundation Charles and Daneen StiefelScholar Award (A.S.P.); the National Institute of Arthritis andMusculoskeletal and Skin Diseases of NIH (A.S.P., R01-AR057001and R01-AR068288; M.J.C., T32-AR007465 and F31-AR066456; G.C.,R01-AR055309; and Skin Diseases Research Core grant P30-AR057217); Deutsche Forschungsgemeinschaft (C.T.E., EL711/1-1);the National Cancer Institute of NIH (V.G.B., T32-CA009140); theNational Heart, Lung, and Blood Institute of NIH (V.G.B., K12-
HL087064); and the Italian Ministry of Health (G.D.Z., RicercaFinalizzata RF10-2309790). The content is solely the responsibility ofthe authors and does not necessarily represent the official views ofthe National Institutes of Health.
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/353/6295/179/suppl/DC1Materials and MethodsFigs. S1 to S11Table S1Movies S1 and S2References (38–55)
17 March 2016; accepted 9 June 2016Published online 30 June 201610.1126/science.aaf6756
184 8 JULY 2016 • VOL 353 ISSUE 6295 sciencemag.org SCIENCE
RESEARCH | REPORTSon F
ebruary 28, 2020
http://science.sciencemag.org/
Dow
nloaded from

Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease
Antonio Lanzavecchia, John T. Seykora, George Cotsarelis, Michael C. Milone and Aimee S. PayneChristoph T. Ellebrecht, Vijay G. Bhoj, Arben Nace, Eun Jung Choi, Xuming Mao, Michael Jeffrey Cho, Giovanni Di Zenzo,
originally published online June 30, 2016DOI: 10.1126/science.aaf6756 (6295), 179-184.353Science
, this issue p. 179Scienceeffective way to treat antibody-driven autoimmune diseases.targeted and killed B cells that express antibodies targeting desmoglein 3, hinting that such a strategy may be an desmoglein 3 fused to signaling domains that activate T cells. When given to diseased mice, the engineered T cellsdiseases. They engineered T cells to express chimeric receptors consisting of the disease-causing autoantigen
asked whether a similar approach might also work against antibody-driven autoimmuneet al.cancers, Ellebrecht disease-causing cells. Inspired by the clinical success of using chimeric antigen receptor T cells to treat certain types of
Autoimmune diseases such as lupus and rheumatoid arthritis lack therapies that specifically target only theEngineering T cells to treat autoimmunity
ARTICLE TOOLS http://science.sciencemag.org/content/353/6295/179
MATERIALSSUPPLEMENTARY http://science.sciencemag.org/content/suppl/2016/06/29/science.aaf6756.DC1
CONTENTRELATED
http://stke.sciencemag.org/content/sigtrans/9/447/ra95.fullhttp://stm.sciencemag.org/content/scitransmed/7/303/303ra139.fullhttp://stm.sciencemag.org/content/scitransmed/6/261/261ra151.fullhttp://stm.sciencemag.org/content/scitransmed/7/275/275ra22.fullhttp://stm.sciencemag.org/content/scitransmed/7/307/307ra156.fullhttp://science.sciencemag.org/content/sci/353/6294/14.full
REFERENCES
http://science.sciencemag.org/content/353/6295/179#BIBLThis article cites 54 articles, 20 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.ScienceScience, 1200 New York Avenue NW, Washington, DC 20005. The title (print ISSN 0036-8075; online ISSN 1095-9203) is published by the American Association for the Advancement ofScience
Copyright © 2016, American Association for the Advancement of Science
on February 28, 2020
http://science.sciencem
ag.org/D
ownloaded from