whole exome sequencing identifies crb1 defect in an unusual maculopathy phenotype
TRANSCRIPT

Whole Exome Sequencing Identifies CRB1Defect in an Unusual Maculopathy Phenotype
Stephen H Tsang MD PhD12 Tomas Burke MD13 Maris Oll MD14 Suzanne Yzer MD PhD15
Winston Lee MA1 Yajing (Angela) Xie MA1 Rando Allikmets PhD12
Objective To report a new phenotype caused by mutations in the CRB1 gene in a family with 2 affectedsiblings
Design Molecular genetics and observational case studiesParticipants Two affected siblings and 3 unaffected family membersMethods Each subject received a complete ophthalmic examination together with color fundus photog-
raphy fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) Micro-perimetry 1 (MP-1) mapping and electroretinogram (ERG) analysis were performed on the proband Screening fordisease-causing mutations was performed by whole exome sequencing in 3 family members followed bysegregation analyses in the entire family
Main Outcome Measures Appearance of the macula as examined by clinical examination fundusphotography FAF imaging SD-OCT and visual function by MP-1 and ERG
Results The proband and her affected brother exhibited unusual previously unreported findings of a maculardystrophy with relative sparing of the retinal periphery beyond the vascular arcades The FAF imaging showedseverely affected areas of hypoautofluorescence that extended nasally beyond the optic disc in both eyes Acentral macular patch of retinal pigment epithelium (RPE) sparing was evident in both eyes on FAF whereasphotoreceptor sparing was documented in the right eye only using SD-OCT The affected brother presented withirregular patterns of autofluorescence in both eyes characterized by concentric rings of alternating hyper- andhypoautofluorescence and foveal sparing of photoreceptors and RPE as seen on SD-OCT bilaterally Afternegative results in screening for mutations in candidate genes including ABCA4 and PRPH2 DNA from 3membersof the family including both affected siblings and their mother was screened by whole exome sequencingresulting in identification of 2 CRB1 missense mutations cC3991TpR1331C and cC4142TpP1381L whichsegregated with the disease in the family Of the 2 the pR1331C CRB1 mutation has not been described beforeand the pP1381L variant has been described in 1 patient with Leber congenital amaurosis
Conclusions This report illustrates a novel presentation of a macular dystrophy caused by CRB1 mutationsBoth affected siblings exhibited a relatively well-developed retinal structure and preservation of generalized retinalfunction An unusual 5-year progression of macular atrophy alone was observed that has not been described inany other CRB1-associated phenotypes Ophthalmology 2014-1e10 ordf 2014 by the American Academy ofOphthalmology
Supplemental material is available at wwwaaojournalorg
Mutations in the CRB1 gene (Mendelian Inheritance in Man604210) have been associated with a variety of generalizedretinal dystrophies ranging from retinitis pigmentosa (RP) toLeber congenital amaurosis (LCA)1e4 Retinitis pigmentosarefers to a group of clinically and genetically heterogeneousdisorders affecting 15 million people worldwide Reportedcases of RP associated with mutations in CRB1 (RP12phenotype) present with an early disease onset includingnystagmus hyperopia optic nerve head drusen relativeattenuation of the vessels a maculopathy and nummulartype of pigmentation in the periphery256 CRB1 mutationsalso have been correlated to retinal vascular sheathingpreserved para-arteriolar retinal pigment epithelium (RPE)and the development of Coats-like exudative vasculopathya condition of abnormally permeable blood vessels leading
2014 by the American Academy of OphthalmologyPublished by Elsevier Inc
to exudation and retinal detachment457 Mutations in theCRB1 gene have been detected in 10 to 13 of patientswith LCA one of the most severe forms of retinal dystrophycharacterized by onset in the first year of life nystagmussluggish pupillary and oculodigital reflexes and an extin-guished electroretinogram (ERG)48e10 Phenotypicallypatients with LCA with CRB1 mutations usually show thedescribed RP12 characteristics including the early-onsetmaculopathy (macular dysplasia)
CRB1 is the human homologue of the gene encoding thecrumbs (Crb) protein in Drosophila melanogaster and isexpressed in the fetal brain and the inner segments ofphotoreceptors in humans211 CRB1 also is expressed in thebrain kidney colon stomach lung and testis1213 CRB1maps to chromosome 1q313 and is composed of 12 exons
1ISSN 0161-642014$ - see front matterhttpdxdoiorg101016jophtha201403010
Ophthalmology Volume - Number - Month 2014
that are translated into 2 protein isoforms the larger ofwhich possesses a cytoplasmic domain containing FERM-and PDZ-binding motifs that enable adherin junction for-mation and actin skeleton association14 CRB1 is crucial forthe assembly of the zonula adherens in Drosophila and hasbeen found to be localized at the apical membrane1516 Asimilar distribution in the outer limiting membranes ofepithelial cells Muumlller cells and photoreceptor inner seg-ments has been observed in mouse and human ret-inas111718 Developmentally CRB1 has been shown todetermine embryonic epithelium and peripheral neurons inDrosophila1516 In addition both human and mouse CRB1proteins are involved in photoreceptor morphogen-esis111719 Mouse models of CRB1 have been extensivelystudied and used to show developmental defects anddisorganization of the retina in mutants particularly dis-ruptions of the outer limiting membrane and the formationof retinal folds or pseudorosettes1719 These findingscorrelate with the appearance of the developmentallyimmature retinas in patients with CRB1 mutations Theretinas of such patients appear thickened and often exhibitaltered laminar organization through the disruption ofdevelopmental apoptosis19e22
More than 150 disease-associated variants have beendescribed to date in the CRB1 gene the most common ofwhich is the pC948Y variant in exon 9125112023e26 Wedescribe the clinical appearance of a combination of novelCRB1 variants that were associated with an unusual andpreviously not described phenotype in 2 affected siblings ofIrish descent
Methods
Patients and Clinical Evaluation
Two patients the proband and her affected brother along with anunaffected sister mother and father were enrolled in the studyunder the protocol AAAB6560 after obtaining full consent Theprotocol was approved by the institutional review board atColumbia University and adhered to tenets set out in the Decla-ration of Helsinki
Each patient underwent a complete ophthalmic examination bya retinal physician (SHT) which included color fundus photog-raphy with an FF 450plus Fundus Camera (Carl Zeiss Meditec AGJena Germany) Fundus autofluorescence (FAF) images wereobtained using a confocal scanning-laser ophthalmoscope (Hei-delberg Retina Angiograph 2 Heidelberg Engineering Dos-senheim Germany) by illuminating the fundus with argon laserlight (488 nm) and viewing the resultant fluorescence through aband pass filter with a short wavelength cutoff at 495 nm Simul-taneous FAF and spectral-domain optical coherence tomography(SD-OCT) images were acquired using a Spectralis HRAthornOCT(Heidelberg Engineering Heidelberg Germany) Color-codedretinal thickness maps were exported from Heidelberg Explorerv 5460 (Heidelberg Engineering Heidelberg Germany) soft-ware that had automatically calculated the thickness of the retina(from internal limiting membrane to Bruchrsquos membrane) usingraster SD-OCT scans acquired on the Heidelberg Spectralis Thesegmentations performed by the software were manually adjustedwhere necessary The retinal thickness measurements are auto-matically mapped onto an infrared fundus image and color-codedaccording to thickness (micrograms) Electroretinography was
2
carried out using the Diagnosys Espion Electrophysiology System(Diagnosys LLC Littleton MA) For all recordings the pupilswere maximally dilated before full-field ERG testing using guttatetropicamide (1) and phenylephrine hydrochloride (25) and thecorneas were anesthetized with guttate proparacaine 05 Silver-impregnated fiber electrodes (DTL Diagnosys LLC) were usedwith a ground electrode on the forehead Full-field ERGs to testgeneralized retinal function were performed using extended testingprotocols incorporating the International Society for ClinicalElectrophysiology of Vision standard27 Microperimetry (NidekInstruments Inc Padova Italy NAVIS software version 173)mapping was carried out in proband using the 10-2 pattern afterpupil dilation with 1 tropicamide and after a 15-minute adapta-tion period to the background luminance
Genetic Analyses
The proband was initially screened for variants in the ABCA4 andPRPH2 genes by direct Sanger sequencing revealing no disease-associated variants Because the phenotype did not suggesttesting any other candidate genes the family was subjected to thewhole exome sequencing and analysis
Exome sequencing was performed for the 2 affected siblingsand their unaffected mother Three to 5 mm of genomic DNAextracted from peripheral blood were exome captured andsequenced at Axeq Technologies (Rockville MD available atwwwaxeqcom accessed September 10 2013) In-solutionsequence capture was performed using Nimblegen capture array(SeqCap EZ Exome Library v30) with 64 Mb target regionMassively parallel sequencing of the enriched library was per-formed on Illumina HiSeq platform with 100 base pair paired-endreads Sequencing reads were generated in the fastq format afternucleotide calling and quality score assessment was performedusing instrument-specific Real Time Analysis software (IlluminaSan Diego CA) Read pairs were aligned to the human referencegenome (hg19) using Burrows-Wheeler Aligner (available athttpbio-bwasourceforgenet) and duplicate reads were removedwith PICARD tools (available at httppicardsourceforgenet)Uniquely mapped on-target reads were extracted and singlenucleotide polymorphism (SNP) and indel calling were performedwith Samtools (httpsamtoolssourceforgenet) Variants wereannotated using ANNOVAR (httpwwwopenbioinformaticsorgannovar) All variants of interest were confirmed by Sangersequencing and segregation analyses were performed in the entirefamily
Results
Clinical Examination
The proband a 45-year-old woman of Irish descent had noticed adecrease in her vision starting around her mid-20s Her brotheraged 41 years at the time of examination had reported similaronset of visual symptoms in his early 30s Neither the proband northe affected sibling had undergone ophthalmic examination beforethe onset of symptoms therefore there are no data on the pre-symptomatic retinal state in either case
The third sibling and parents reported no major issues withvision (Fig 1) The family members recalled a significant vision lossin their maternal grandmother and a paternal aunt beginning in the6th and 7th decades of life However in both cases the causeremained unknown and both of these individuals were deceasedwith no available clinical records Both the proband and heraffected sibling did not have any contributory ophthalmic orsystemic illnesses and both denied a history of smoking
Figure 1 Pedigree of the family Open circles and squares represent theunaffected female and male family members respectively closed circles andsquares represent the affected female and male patients The mother andfather in this pedigree are heterozygous for the pP1381L and p R1331Cmutations respectively The affected siblings are compound heterozygousfor both mutations
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
Table 1 summarizes the demographic and clinical findings fromthe initial evaluation of both affected siblings Best-corrected vi-sual acuity in the proband was 2040 in the right eye and 20400 inthe left eye at presentation A slit-lamp examination of the anteriorsegment showed unremarkable results Dilated fundoscopyrevealed a clear vitreous and healthy vascular arcades There wasno optic disc swelling however temporal pallor was present inboth eyes (Fig 2A F) There were no signs of macular edema in theright eye although both maculae exhibited a mottled granularlyspeckled appearance with a continuous annulus of atrophycircumscribing an island of preserved RPE in the foveal regionA more generalized ldquowipe-outrdquo of RPE in the left macula wasobserved No peripapillary sparing was noted in either eye andmost notable atrophy of the retina and RPE was observed nasalto the disc in both eyes Central fixation was spared according tothe patientrsquos ability to follow a fixation target (Fig 2A F)
The younger brother of the proband presented with similarocular findings His best-corrected visual acuity was 2040 in theright eye and 2070 in the left eye Cystoid macular edema (CME)was present in the left eye that responded to treatment with oralacetazolamide (500 mg) and guttate nepafenac resulting in theimprovement of visual acuity to 2030 with an associated reductionin the size of intraretinal cysts The anterior segment examinationresults were unremarkable No vitreous opacities were found andboth the optic nerves and the fundus vasculature appeared healthyBoth maculae exhibited similar granular characteristics observed in
Table 1 Summary of Demographi
Family Age (yrs) BCVA Snellen (logMAR) OD BCVA Snelle
Proband 45 2040 20Brother 41 2040 20Sister 33 dMother 69 2050 20Father 85 2020 20
e frac14 not applicable BCVA frac14 best-corrected visual acuity logMAR frac14 logarithNot clinically examined underwent only genetic testing
the proband however clear parafoveal atrophy was seen in themaculae (right eye gt left eye) along with a few foci of hyper-pigmentation (Fig 2L Q)
Both parents of the affected siblings were examined withdilated fundus examination There was no vascular attenuation orintraretinal pigment migration in either parent However peri-papillary atrophy was present in both parents Choroidal thinningdue to myopia was observed in the motherrsquos left eye A nonsig-nificant epiretinal membrane was observed in the temporal maculaof her left eye Normal retinal layers and intact photoreceptors wereapparent in both parents (Fig 3B D F H)
Fundus Autofluorescence
Fundus autofluorescence imaging in the proband showed hyper-autofluorescence suggestive of preservation of a central island ofRPE in the right eye and parafoveal preservation of an RPE islandin the left eye The macula in both eyes appeared to be enshroudedwith a large cloud of hypoautofluorescence extending temporallypast the optic discs Peripheral areas of relatively normal FAFpattern were marked with dark punctate changes in areasapproaching the hypoautofluorescent cloud (Fig 2B C G H)
In the affected brother the central maculae of both eyesappeared hypoautofluorescent more so in the left eye There wasassociated hypoautofluorescence along the peripapillary area andthe proximal superotemporal arcades in both eyes However themore peripheral maculae appeared relatively hyperautofluorescentcompared with the surrounding extramacular retina There weremultiple discrete punctate hyperautofluorescent foci in themaculae predominating nasally and temporally (Fig 2N S) Apetaloid pattern of alternating hyper- and hypoautofluorescencewas seen in both eyes but was again more obvious in the lefteye (Fig 2S)
Spectral-Domain Optical Coherence Tomography
Horizontal SD-OCT line scans through the central maculae of botheyes of the proband showed an absence of the outer nuclear layerinner segment ellipsoid band or innerouter segment junction ofthe photoreceptors and RPE except in the foveal region of the righteye where the structure of both the photoreceptors and RPEappeared anomalous but present suggestive of relative sparing(Fig 2D E) This sparing co-localized with the region of hyper-autofluorescence Of note similar sparing of the photoreceptorswas not seen in the foveal region of the left eye despite a hyper-autofluorescent signal (Fig 2I J) The SD-OCT scans in the righteye of the affected sibling resembled that of the proband Howeverthe photoreceptor sparing on SD-OCT was less obvious None-theless in the foveal region of the siblingrsquos right eye there wasevidence of a residual inner segment ellipsoid band in the fovealregion and a disorganized atrophic outer nuclear layer On closeinspection of the outer nuclear layer in the temporal parafoveal
c Clinical and Genetic Data
n (logMAR) OS Condition Age of Onset CRB1 Mutation
400 Affected Mid-20s R1331C P1381L70 Affected Early 30s R1331C P1381Ld Unaffected d wt wt40 Unaffected d wt P1381L20 Unaffected d R1331C wt
m of the minimum angle of resolution OD frac14 right eye OS frac14 left eye
3
Figure 2 Fundus photographs fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans of the right andleft eyes of a 45-year-old woman (proband) and her 41-year-old affected brother with the same CRB1 mutations Fundus photography in the probandexhibited stable fixation (needle position) temporal pallor of the optic discs attenuated retinal vasculature and extensive retinal pigment epithelium (RPE)changes (A F) The FAF imaging reveals predominant hypoautofluorescence across the macula consistent with widespread RPE atrophy although relativehyperautofluorescence was documented in the foveal and parafoveal regions of the right and left eyes and respectively consistent with sparing of the RPE (BC G H) The SD-OCT scans reveal relative sparing of foveal photoreceptors (outer nuclear layer inner segment ellipsoid band) and RPE in the right eye(D E) and an absence of all except RPE in the left eye (I J) Fundus photographs in the affected brother also showed extensive loss of macular RPE (L Q)The FAF revealed concentric rings of alternating hyper- and hypoautofluorescence encircling the macula (M R) and there were petaloid patterns centrallygreatest in the left eye (N S) The SD-OCT scans through the fovea in the right eye revealed an area of relative foveal sparing (O P) and cystoid macularedema (CME) was detected in the left eye (T U) There was possible early cystoid change in the temporal parafoveal macula of the right eye (O) Relativelynormal laminar architecture is preserved in the maculae of both affected siblings Electroretinography in the proband reveals overall preservation ofgeneralized cone and rod function without any obvious implicit time shifts (K) ISe frac14 inner segment ellipsoid OD frac14 right eye ONL frac14 outer nuclear layerOS frac14 left eye
Ophthalmology Volume - Number - Month 2014
macula of the right eye there was possible early cystoid change(Fig 2O) There was definite CME in the left eye visible on SD-OCT (Fig 2T U) Despite the presence of CME in the left eyethe outer nuclear layer and inner segment ellipsoid bandappeared well preserved in the central macula A 3-year follow-up scan of the affected siblingrsquos left eye was acquired after treat-ment (Fig 4 available at wwwaaojournalorg) and showed thatthere was persistence of CME although it had reduced Evenwith progressive photoreceptor and RPE layer disruption retinalthickness appeared to be within normal limits in both affectedsiblings (excluding the siblingrsquos left eye with CME) withreasonably well-preserved retinal lamination
Retinal thickness in the proband and affected brother wasassessed using color-coded thickness maps over the macula of each
4
eye (Fig 5) Compared with an age-matched control both eyes ofthe proband and the right eye of her affected sibling exhibited areduction in retinal thickness in the central macula consistent withatrophy There was a trend toward increased thickness relative tothe control in the more peripheral macula The left eye of theprobandrsquos sibling had grossly increased macular thickness centrallydue to CME
Electroretinogram Analysis
Scotopic responses in the proband showed a generalized preser-vation of retinal rod function A marginal implicit time delaywas detected in the photopic responses suggestive of cone
Figure 3 Fundus photographs 488 nm fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans in theparental carriers of the CRB1 mutation No significant retinal pigment epithelium (RPE) and abnormal AF patterns were observed Both parents exhibitedperipapillary atrophy in both eyes greatest in the mother and related to her high myopia Minor choroidal thinning in the mother also was attributed to thismyopia (B D) The SD-OCT of her left eye revealed a nonsignificant epiretinal membrane Otherwise normal retinal layers and intact photoreceptors werepresent in both parents FAF frac14 fundus autofluorescence OD frac14 right eye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
involvement however both the waveform and amplitudes werewithin normal limits compared with age-matched controls(Fig 2K)
Progression and Visual Function
Microperimetry 1 (MP-1) mapping results (in decibels) wererecorded in the proband at the initial visit and registered to acorresponding FAF image with the built-in Navis software Pre-served visual function was documented within the region ofhyperautofluorescence in both eyes However across the atrophichypoautofluorescent macula retinal sensitivities of 0 decibels wererecorded The test documented steady foveal fixation as assessedby the fixation tracker on the mapping instrument A subsequentrecording after 5 years showed a notable reduction in sensitivity in
the right eye and an almost complete loss of function in the left eye(Fig 6)
Genetic Analyses
Whole exome sequencing was performed in the 2 affected siblingsand their unaffected mother An average of 113 million total readswere generated for each sample with an average of 65 million(58) nonredundant unique reads mapped to the 64 MB exomeregion (Table 2) (available at wwwaaojournalorg) More than90 of the target regions have gt10 coverage with an averagemean depth of coverage of 80 On average there were a totalof 99 041 variants identified for each sample of which 20 730(21) were in protein coding sequences Some 98 of allcoding variants were SNPs and 2 were insertions and deletions
5
Figure 5 Color-coded macular thickness (mm) maps of the CRB1-affected proband and her brother compared with an age-matched normal subject Themaps show a reduced (darker colors) thickness in the central macula in both eyes of the proband and in the right eye of her sibling compared with thecontrol with a trend toward increased (brighter colors) thickness relative to the control in the more peripheral macula The left eye of the probandrsquos siblinghad grossly increased macular thickness due to cystoid macular edema (CME)
Ophthalmology Volume - Number - Month 2014
(indels) The ratio of nonsynonymous to synonymous variants wasclose to 11 in all samples For initial filtering of variants theminimum SNP quality value was set at 20 and the minimumtotal read depth was set at 5
Considering the autosomal recessive inheritance pattern of thedisease and that both affected siblings had to have the same causalmutations we focused on genes that had at least 2 shared variantsin the 2 affected siblings (Table 3) We further assumed that thedisease-causing variants have to be rare thereby filtering thesequence data for new variants and those found in lt05 indbSNP135 and in the 1000 Genomes databases After excluding
6
synonymous variants and all coding and intronic variants that didnot affect splicing the number of candidate genes was narroweddown to 19 After assessing the phase of remaining variants usingthe sequences of the mother only 3 possible candidate genesremained CRB1 NADK and DNAH12 The segregation of vari-ants in these genes with the disease was tested in the entire familyincluding the unaffected sibling and the father
Dynein axonemal heavy chain 12 protein is a large proteinwith multiple rare variants involved in microtubule-associatedmotor protein complexes composed of several heavy light andintermediate chains NADK catalyzes the synthesis of NADP from
Figure 6 Functional and macular progression assessment through fundus autofluorescence (FAF) imaging and microperimetry (MP)-1 mapping Over thecourse of 5 years preservation of the central island of hyperautofluorescent retinal pigment epithelium (RPE) was maintained however the spatial functionhad become more constricted A comparative overlay of MP-1 and FAF showed that the scotomata (0 decibels) recorded on the MP-1 corresponded toatrophic (hypoautofluorescent) regions at the initial examination in the right (A) and left (B) eyes and in the same area of both eyes respectively (C D)after 5 years OD frac14 righteye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
NAD and ATP and exists in several isoforms Neither of these 2genes has been associated with eye disease phenotypes and there isno direct mechanism how these would be involved in retinaldystrophies
This left CRB1 as the only plausible candidate gene for thedisease phenotype segregating with the 2 variants because it is awell-known gene in retinal dystrophies (RetNet httpssphutheduretnet accessed September 10 2013) The 2 CRB1 var-iants shared by the affected siblings (cC3991TpR1331C andC4142TpP1381L) are not present in ESP6500 dbSNP135 and1000 Genomes database although the pP1381L variant wasrecently identified as disease associated in 1 patient with LCA28
Both variants are predicted to be deleterious or damaging bySorting Intolerant from Tolerant (SIFT httpsiftjcviorg)Polyphen-2 and MutationTaster programs Nucleotide positionsof the 2 mutations are highly conserved as predicted by phyloPThe novel cC3991T variant that results in the pR1331C mutationis 1 nucleotide before a previously reported benign variantcA3992G which results in the pR1331H amino acid change
Table 3 Variants Identified in the 2 Affected Individuals
Variant Filters No SNVs No Genes
Total variants shared between bothaffected siblings
67 951 20 181
Not present in dbSNP135 commonand lt05 in 1000 genomes
5733 4499
Missense nonsense indels and splicing 392 364Recessive 47 19Phase-assessed 6 3
SNV frac14 single nucleotide variation
The latter is considered benign however unlike arginine to his-tidine change (Grantham distance 29) there is large physico-chemical difference between arginine and cysteine (Granthamdistance 180) Creation of a cysteine residue at this position alsoresults in 2 consecutive cysteines in the amino acid chainBecause EGF-like domain is characterized by 6 conserved cys-teines forming 3 disulfide bonds and modification from arginineto histidine at this position is considered benign pR1331C alsocould be a relatively milder mutation which could explain themilder phenotype compared with other CRB1-associatedphenotypes
Discussion
The clinical and genetic findings were summarized in afamily in whom 2 rare likely disease-associated CRB1missense variants segregated with an unusual maculardystrophy phenotype Previously reported phenotypesassociated with mutations in CRB1 include those consistentwith an early-onset RP associated with preserved para-arteriolar RPE or Coats-like exudative vasculopathy Thepatients described in this study exhibited unusual pheno-typic characteristics that have not been described beforeand therefore eluded clinical diagnosis by several retinalspecialists Both affected siblings presented with decreasedvisual acuity but without nyctalopia Furthermore onfundoscopy there was a relatively normal-appearing pe-riphery without any of the RP12 characteristics such asnummular hyperpigmentation and some degree of vascularattenuation The disease was largely confined to the mac-ula In both cases the reported age of onset was between
7
Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu

Ophthalmology Volume - Number - Month 2014
that are translated into 2 protein isoforms the larger ofwhich possesses a cytoplasmic domain containing FERM-and PDZ-binding motifs that enable adherin junction for-mation and actin skeleton association14 CRB1 is crucial forthe assembly of the zonula adherens in Drosophila and hasbeen found to be localized at the apical membrane1516 Asimilar distribution in the outer limiting membranes ofepithelial cells Muumlller cells and photoreceptor inner seg-ments has been observed in mouse and human ret-inas111718 Developmentally CRB1 has been shown todetermine embryonic epithelium and peripheral neurons inDrosophila1516 In addition both human and mouse CRB1proteins are involved in photoreceptor morphogen-esis111719 Mouse models of CRB1 have been extensivelystudied and used to show developmental defects anddisorganization of the retina in mutants particularly dis-ruptions of the outer limiting membrane and the formationof retinal folds or pseudorosettes1719 These findingscorrelate with the appearance of the developmentallyimmature retinas in patients with CRB1 mutations Theretinas of such patients appear thickened and often exhibitaltered laminar organization through the disruption ofdevelopmental apoptosis19e22
More than 150 disease-associated variants have beendescribed to date in the CRB1 gene the most common ofwhich is the pC948Y variant in exon 9125112023e26 Wedescribe the clinical appearance of a combination of novelCRB1 variants that were associated with an unusual andpreviously not described phenotype in 2 affected siblings ofIrish descent
Methods
Patients and Clinical Evaluation
Two patients the proband and her affected brother along with anunaffected sister mother and father were enrolled in the studyunder the protocol AAAB6560 after obtaining full consent Theprotocol was approved by the institutional review board atColumbia University and adhered to tenets set out in the Decla-ration of Helsinki
Each patient underwent a complete ophthalmic examination bya retinal physician (SHT) which included color fundus photog-raphy with an FF 450plus Fundus Camera (Carl Zeiss Meditec AGJena Germany) Fundus autofluorescence (FAF) images wereobtained using a confocal scanning-laser ophthalmoscope (Hei-delberg Retina Angiograph 2 Heidelberg Engineering Dos-senheim Germany) by illuminating the fundus with argon laserlight (488 nm) and viewing the resultant fluorescence through aband pass filter with a short wavelength cutoff at 495 nm Simul-taneous FAF and spectral-domain optical coherence tomography(SD-OCT) images were acquired using a Spectralis HRAthornOCT(Heidelberg Engineering Heidelberg Germany) Color-codedretinal thickness maps were exported from Heidelberg Explorerv 5460 (Heidelberg Engineering Heidelberg Germany) soft-ware that had automatically calculated the thickness of the retina(from internal limiting membrane to Bruchrsquos membrane) usingraster SD-OCT scans acquired on the Heidelberg Spectralis Thesegmentations performed by the software were manually adjustedwhere necessary The retinal thickness measurements are auto-matically mapped onto an infrared fundus image and color-codedaccording to thickness (micrograms) Electroretinography was
2
carried out using the Diagnosys Espion Electrophysiology System(Diagnosys LLC Littleton MA) For all recordings the pupilswere maximally dilated before full-field ERG testing using guttatetropicamide (1) and phenylephrine hydrochloride (25) and thecorneas were anesthetized with guttate proparacaine 05 Silver-impregnated fiber electrodes (DTL Diagnosys LLC) were usedwith a ground electrode on the forehead Full-field ERGs to testgeneralized retinal function were performed using extended testingprotocols incorporating the International Society for ClinicalElectrophysiology of Vision standard27 Microperimetry (NidekInstruments Inc Padova Italy NAVIS software version 173)mapping was carried out in proband using the 10-2 pattern afterpupil dilation with 1 tropicamide and after a 15-minute adapta-tion period to the background luminance
Genetic Analyses
The proband was initially screened for variants in the ABCA4 andPRPH2 genes by direct Sanger sequencing revealing no disease-associated variants Because the phenotype did not suggesttesting any other candidate genes the family was subjected to thewhole exome sequencing and analysis
Exome sequencing was performed for the 2 affected siblingsand their unaffected mother Three to 5 mm of genomic DNAextracted from peripheral blood were exome captured andsequenced at Axeq Technologies (Rockville MD available atwwwaxeqcom accessed September 10 2013) In-solutionsequence capture was performed using Nimblegen capture array(SeqCap EZ Exome Library v30) with 64 Mb target regionMassively parallel sequencing of the enriched library was per-formed on Illumina HiSeq platform with 100 base pair paired-endreads Sequencing reads were generated in the fastq format afternucleotide calling and quality score assessment was performedusing instrument-specific Real Time Analysis software (IlluminaSan Diego CA) Read pairs were aligned to the human referencegenome (hg19) using Burrows-Wheeler Aligner (available athttpbio-bwasourceforgenet) and duplicate reads were removedwith PICARD tools (available at httppicardsourceforgenet)Uniquely mapped on-target reads were extracted and singlenucleotide polymorphism (SNP) and indel calling were performedwith Samtools (httpsamtoolssourceforgenet) Variants wereannotated using ANNOVAR (httpwwwopenbioinformaticsorgannovar) All variants of interest were confirmed by Sangersequencing and segregation analyses were performed in the entirefamily
Results
Clinical Examination
The proband a 45-year-old woman of Irish descent had noticed adecrease in her vision starting around her mid-20s Her brotheraged 41 years at the time of examination had reported similaronset of visual symptoms in his early 30s Neither the proband northe affected sibling had undergone ophthalmic examination beforethe onset of symptoms therefore there are no data on the pre-symptomatic retinal state in either case
The third sibling and parents reported no major issues withvision (Fig 1) The family members recalled a significant vision lossin their maternal grandmother and a paternal aunt beginning in the6th and 7th decades of life However in both cases the causeremained unknown and both of these individuals were deceasedwith no available clinical records Both the proband and heraffected sibling did not have any contributory ophthalmic orsystemic illnesses and both denied a history of smoking
Figure 1 Pedigree of the family Open circles and squares represent theunaffected female and male family members respectively closed circles andsquares represent the affected female and male patients The mother andfather in this pedigree are heterozygous for the pP1381L and p R1331Cmutations respectively The affected siblings are compound heterozygousfor both mutations
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
Table 1 summarizes the demographic and clinical findings fromthe initial evaluation of both affected siblings Best-corrected vi-sual acuity in the proband was 2040 in the right eye and 20400 inthe left eye at presentation A slit-lamp examination of the anteriorsegment showed unremarkable results Dilated fundoscopyrevealed a clear vitreous and healthy vascular arcades There wasno optic disc swelling however temporal pallor was present inboth eyes (Fig 2A F) There were no signs of macular edema in theright eye although both maculae exhibited a mottled granularlyspeckled appearance with a continuous annulus of atrophycircumscribing an island of preserved RPE in the foveal regionA more generalized ldquowipe-outrdquo of RPE in the left macula wasobserved No peripapillary sparing was noted in either eye andmost notable atrophy of the retina and RPE was observed nasalto the disc in both eyes Central fixation was spared according tothe patientrsquos ability to follow a fixation target (Fig 2A F)
The younger brother of the proband presented with similarocular findings His best-corrected visual acuity was 2040 in theright eye and 2070 in the left eye Cystoid macular edema (CME)was present in the left eye that responded to treatment with oralacetazolamide (500 mg) and guttate nepafenac resulting in theimprovement of visual acuity to 2030 with an associated reductionin the size of intraretinal cysts The anterior segment examinationresults were unremarkable No vitreous opacities were found andboth the optic nerves and the fundus vasculature appeared healthyBoth maculae exhibited similar granular characteristics observed in
Table 1 Summary of Demographi
Family Age (yrs) BCVA Snellen (logMAR) OD BCVA Snelle
Proband 45 2040 20Brother 41 2040 20Sister 33 dMother 69 2050 20Father 85 2020 20
e frac14 not applicable BCVA frac14 best-corrected visual acuity logMAR frac14 logarithNot clinically examined underwent only genetic testing
the proband however clear parafoveal atrophy was seen in themaculae (right eye gt left eye) along with a few foci of hyper-pigmentation (Fig 2L Q)
Both parents of the affected siblings were examined withdilated fundus examination There was no vascular attenuation orintraretinal pigment migration in either parent However peri-papillary atrophy was present in both parents Choroidal thinningdue to myopia was observed in the motherrsquos left eye A nonsig-nificant epiretinal membrane was observed in the temporal maculaof her left eye Normal retinal layers and intact photoreceptors wereapparent in both parents (Fig 3B D F H)
Fundus Autofluorescence
Fundus autofluorescence imaging in the proband showed hyper-autofluorescence suggestive of preservation of a central island ofRPE in the right eye and parafoveal preservation of an RPE islandin the left eye The macula in both eyes appeared to be enshroudedwith a large cloud of hypoautofluorescence extending temporallypast the optic discs Peripheral areas of relatively normal FAFpattern were marked with dark punctate changes in areasapproaching the hypoautofluorescent cloud (Fig 2B C G H)
In the affected brother the central maculae of both eyesappeared hypoautofluorescent more so in the left eye There wasassociated hypoautofluorescence along the peripapillary area andthe proximal superotemporal arcades in both eyes However themore peripheral maculae appeared relatively hyperautofluorescentcompared with the surrounding extramacular retina There weremultiple discrete punctate hyperautofluorescent foci in themaculae predominating nasally and temporally (Fig 2N S) Apetaloid pattern of alternating hyper- and hypoautofluorescencewas seen in both eyes but was again more obvious in the lefteye (Fig 2S)
Spectral-Domain Optical Coherence Tomography
Horizontal SD-OCT line scans through the central maculae of botheyes of the proband showed an absence of the outer nuclear layerinner segment ellipsoid band or innerouter segment junction ofthe photoreceptors and RPE except in the foveal region of the righteye where the structure of both the photoreceptors and RPEappeared anomalous but present suggestive of relative sparing(Fig 2D E) This sparing co-localized with the region of hyper-autofluorescence Of note similar sparing of the photoreceptorswas not seen in the foveal region of the left eye despite a hyper-autofluorescent signal (Fig 2I J) The SD-OCT scans in the righteye of the affected sibling resembled that of the proband Howeverthe photoreceptor sparing on SD-OCT was less obvious None-theless in the foveal region of the siblingrsquos right eye there wasevidence of a residual inner segment ellipsoid band in the fovealregion and a disorganized atrophic outer nuclear layer On closeinspection of the outer nuclear layer in the temporal parafoveal
c Clinical and Genetic Data
n (logMAR) OS Condition Age of Onset CRB1 Mutation
400 Affected Mid-20s R1331C P1381L70 Affected Early 30s R1331C P1381Ld Unaffected d wt wt40 Unaffected d wt P1381L20 Unaffected d R1331C wt
m of the minimum angle of resolution OD frac14 right eye OS frac14 left eye
3
Figure 2 Fundus photographs fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans of the right andleft eyes of a 45-year-old woman (proband) and her 41-year-old affected brother with the same CRB1 mutations Fundus photography in the probandexhibited stable fixation (needle position) temporal pallor of the optic discs attenuated retinal vasculature and extensive retinal pigment epithelium (RPE)changes (A F) The FAF imaging reveals predominant hypoautofluorescence across the macula consistent with widespread RPE atrophy although relativehyperautofluorescence was documented in the foveal and parafoveal regions of the right and left eyes and respectively consistent with sparing of the RPE (BC G H) The SD-OCT scans reveal relative sparing of foveal photoreceptors (outer nuclear layer inner segment ellipsoid band) and RPE in the right eye(D E) and an absence of all except RPE in the left eye (I J) Fundus photographs in the affected brother also showed extensive loss of macular RPE (L Q)The FAF revealed concentric rings of alternating hyper- and hypoautofluorescence encircling the macula (M R) and there were petaloid patterns centrallygreatest in the left eye (N S) The SD-OCT scans through the fovea in the right eye revealed an area of relative foveal sparing (O P) and cystoid macularedema (CME) was detected in the left eye (T U) There was possible early cystoid change in the temporal parafoveal macula of the right eye (O) Relativelynormal laminar architecture is preserved in the maculae of both affected siblings Electroretinography in the proband reveals overall preservation ofgeneralized cone and rod function without any obvious implicit time shifts (K) ISe frac14 inner segment ellipsoid OD frac14 right eye ONL frac14 outer nuclear layerOS frac14 left eye
Ophthalmology Volume - Number - Month 2014
macula of the right eye there was possible early cystoid change(Fig 2O) There was definite CME in the left eye visible on SD-OCT (Fig 2T U) Despite the presence of CME in the left eyethe outer nuclear layer and inner segment ellipsoid bandappeared well preserved in the central macula A 3-year follow-up scan of the affected siblingrsquos left eye was acquired after treat-ment (Fig 4 available at wwwaaojournalorg) and showed thatthere was persistence of CME although it had reduced Evenwith progressive photoreceptor and RPE layer disruption retinalthickness appeared to be within normal limits in both affectedsiblings (excluding the siblingrsquos left eye with CME) withreasonably well-preserved retinal lamination
Retinal thickness in the proband and affected brother wasassessed using color-coded thickness maps over the macula of each
4
eye (Fig 5) Compared with an age-matched control both eyes ofthe proband and the right eye of her affected sibling exhibited areduction in retinal thickness in the central macula consistent withatrophy There was a trend toward increased thickness relative tothe control in the more peripheral macula The left eye of theprobandrsquos sibling had grossly increased macular thickness centrallydue to CME
Electroretinogram Analysis
Scotopic responses in the proband showed a generalized preser-vation of retinal rod function A marginal implicit time delaywas detected in the photopic responses suggestive of cone
Figure 3 Fundus photographs 488 nm fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans in theparental carriers of the CRB1 mutation No significant retinal pigment epithelium (RPE) and abnormal AF patterns were observed Both parents exhibitedperipapillary atrophy in both eyes greatest in the mother and related to her high myopia Minor choroidal thinning in the mother also was attributed to thismyopia (B D) The SD-OCT of her left eye revealed a nonsignificant epiretinal membrane Otherwise normal retinal layers and intact photoreceptors werepresent in both parents FAF frac14 fundus autofluorescence OD frac14 right eye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
involvement however both the waveform and amplitudes werewithin normal limits compared with age-matched controls(Fig 2K)
Progression and Visual Function
Microperimetry 1 (MP-1) mapping results (in decibels) wererecorded in the proband at the initial visit and registered to acorresponding FAF image with the built-in Navis software Pre-served visual function was documented within the region ofhyperautofluorescence in both eyes However across the atrophichypoautofluorescent macula retinal sensitivities of 0 decibels wererecorded The test documented steady foveal fixation as assessedby the fixation tracker on the mapping instrument A subsequentrecording after 5 years showed a notable reduction in sensitivity in
the right eye and an almost complete loss of function in the left eye(Fig 6)
Genetic Analyses
Whole exome sequencing was performed in the 2 affected siblingsand their unaffected mother An average of 113 million total readswere generated for each sample with an average of 65 million(58) nonredundant unique reads mapped to the 64 MB exomeregion (Table 2) (available at wwwaaojournalorg) More than90 of the target regions have gt10 coverage with an averagemean depth of coverage of 80 On average there were a totalof 99 041 variants identified for each sample of which 20 730(21) were in protein coding sequences Some 98 of allcoding variants were SNPs and 2 were insertions and deletions
5
Figure 5 Color-coded macular thickness (mm) maps of the CRB1-affected proband and her brother compared with an age-matched normal subject Themaps show a reduced (darker colors) thickness in the central macula in both eyes of the proband and in the right eye of her sibling compared with thecontrol with a trend toward increased (brighter colors) thickness relative to the control in the more peripheral macula The left eye of the probandrsquos siblinghad grossly increased macular thickness due to cystoid macular edema (CME)
Ophthalmology Volume - Number - Month 2014
(indels) The ratio of nonsynonymous to synonymous variants wasclose to 11 in all samples For initial filtering of variants theminimum SNP quality value was set at 20 and the minimumtotal read depth was set at 5
Considering the autosomal recessive inheritance pattern of thedisease and that both affected siblings had to have the same causalmutations we focused on genes that had at least 2 shared variantsin the 2 affected siblings (Table 3) We further assumed that thedisease-causing variants have to be rare thereby filtering thesequence data for new variants and those found in lt05 indbSNP135 and in the 1000 Genomes databases After excluding
6
synonymous variants and all coding and intronic variants that didnot affect splicing the number of candidate genes was narroweddown to 19 After assessing the phase of remaining variants usingthe sequences of the mother only 3 possible candidate genesremained CRB1 NADK and DNAH12 The segregation of vari-ants in these genes with the disease was tested in the entire familyincluding the unaffected sibling and the father
Dynein axonemal heavy chain 12 protein is a large proteinwith multiple rare variants involved in microtubule-associatedmotor protein complexes composed of several heavy light andintermediate chains NADK catalyzes the synthesis of NADP from
Figure 6 Functional and macular progression assessment through fundus autofluorescence (FAF) imaging and microperimetry (MP)-1 mapping Over thecourse of 5 years preservation of the central island of hyperautofluorescent retinal pigment epithelium (RPE) was maintained however the spatial functionhad become more constricted A comparative overlay of MP-1 and FAF showed that the scotomata (0 decibels) recorded on the MP-1 corresponded toatrophic (hypoautofluorescent) regions at the initial examination in the right (A) and left (B) eyes and in the same area of both eyes respectively (C D)after 5 years OD frac14 righteye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
NAD and ATP and exists in several isoforms Neither of these 2genes has been associated with eye disease phenotypes and there isno direct mechanism how these would be involved in retinaldystrophies
This left CRB1 as the only plausible candidate gene for thedisease phenotype segregating with the 2 variants because it is awell-known gene in retinal dystrophies (RetNet httpssphutheduretnet accessed September 10 2013) The 2 CRB1 var-iants shared by the affected siblings (cC3991TpR1331C andC4142TpP1381L) are not present in ESP6500 dbSNP135 and1000 Genomes database although the pP1381L variant wasrecently identified as disease associated in 1 patient with LCA28
Both variants are predicted to be deleterious or damaging bySorting Intolerant from Tolerant (SIFT httpsiftjcviorg)Polyphen-2 and MutationTaster programs Nucleotide positionsof the 2 mutations are highly conserved as predicted by phyloPThe novel cC3991T variant that results in the pR1331C mutationis 1 nucleotide before a previously reported benign variantcA3992G which results in the pR1331H amino acid change
Table 3 Variants Identified in the 2 Affected Individuals
Variant Filters No SNVs No Genes
Total variants shared between bothaffected siblings
67 951 20 181
Not present in dbSNP135 commonand lt05 in 1000 genomes
5733 4499
Missense nonsense indels and splicing 392 364Recessive 47 19Phase-assessed 6 3
SNV frac14 single nucleotide variation
The latter is considered benign however unlike arginine to his-tidine change (Grantham distance 29) there is large physico-chemical difference between arginine and cysteine (Granthamdistance 180) Creation of a cysteine residue at this position alsoresults in 2 consecutive cysteines in the amino acid chainBecause EGF-like domain is characterized by 6 conserved cys-teines forming 3 disulfide bonds and modification from arginineto histidine at this position is considered benign pR1331C alsocould be a relatively milder mutation which could explain themilder phenotype compared with other CRB1-associatedphenotypes
Discussion
The clinical and genetic findings were summarized in afamily in whom 2 rare likely disease-associated CRB1missense variants segregated with an unusual maculardystrophy phenotype Previously reported phenotypesassociated with mutations in CRB1 include those consistentwith an early-onset RP associated with preserved para-arteriolar RPE or Coats-like exudative vasculopathy Thepatients described in this study exhibited unusual pheno-typic characteristics that have not been described beforeand therefore eluded clinical diagnosis by several retinalspecialists Both affected siblings presented with decreasedvisual acuity but without nyctalopia Furthermore onfundoscopy there was a relatively normal-appearing pe-riphery without any of the RP12 characteristics such asnummular hyperpigmentation and some degree of vascularattenuation The disease was largely confined to the mac-ula In both cases the reported age of onset was between
7
Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu

Figure 1 Pedigree of the family Open circles and squares represent theunaffected female and male family members respectively closed circles andsquares represent the affected female and male patients The mother andfather in this pedigree are heterozygous for the pP1381L and p R1331Cmutations respectively The affected siblings are compound heterozygousfor both mutations
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
Table 1 summarizes the demographic and clinical findings fromthe initial evaluation of both affected siblings Best-corrected vi-sual acuity in the proband was 2040 in the right eye and 20400 inthe left eye at presentation A slit-lamp examination of the anteriorsegment showed unremarkable results Dilated fundoscopyrevealed a clear vitreous and healthy vascular arcades There wasno optic disc swelling however temporal pallor was present inboth eyes (Fig 2A F) There were no signs of macular edema in theright eye although both maculae exhibited a mottled granularlyspeckled appearance with a continuous annulus of atrophycircumscribing an island of preserved RPE in the foveal regionA more generalized ldquowipe-outrdquo of RPE in the left macula wasobserved No peripapillary sparing was noted in either eye andmost notable atrophy of the retina and RPE was observed nasalto the disc in both eyes Central fixation was spared according tothe patientrsquos ability to follow a fixation target (Fig 2A F)
The younger brother of the proband presented with similarocular findings His best-corrected visual acuity was 2040 in theright eye and 2070 in the left eye Cystoid macular edema (CME)was present in the left eye that responded to treatment with oralacetazolamide (500 mg) and guttate nepafenac resulting in theimprovement of visual acuity to 2030 with an associated reductionin the size of intraretinal cysts The anterior segment examinationresults were unremarkable No vitreous opacities were found andboth the optic nerves and the fundus vasculature appeared healthyBoth maculae exhibited similar granular characteristics observed in
Table 1 Summary of Demographi
Family Age (yrs) BCVA Snellen (logMAR) OD BCVA Snelle
Proband 45 2040 20Brother 41 2040 20Sister 33 dMother 69 2050 20Father 85 2020 20
e frac14 not applicable BCVA frac14 best-corrected visual acuity logMAR frac14 logarithNot clinically examined underwent only genetic testing
the proband however clear parafoveal atrophy was seen in themaculae (right eye gt left eye) along with a few foci of hyper-pigmentation (Fig 2L Q)
Both parents of the affected siblings were examined withdilated fundus examination There was no vascular attenuation orintraretinal pigment migration in either parent However peri-papillary atrophy was present in both parents Choroidal thinningdue to myopia was observed in the motherrsquos left eye A nonsig-nificant epiretinal membrane was observed in the temporal maculaof her left eye Normal retinal layers and intact photoreceptors wereapparent in both parents (Fig 3B D F H)
Fundus Autofluorescence
Fundus autofluorescence imaging in the proband showed hyper-autofluorescence suggestive of preservation of a central island ofRPE in the right eye and parafoveal preservation of an RPE islandin the left eye The macula in both eyes appeared to be enshroudedwith a large cloud of hypoautofluorescence extending temporallypast the optic discs Peripheral areas of relatively normal FAFpattern were marked with dark punctate changes in areasapproaching the hypoautofluorescent cloud (Fig 2B C G H)
In the affected brother the central maculae of both eyesappeared hypoautofluorescent more so in the left eye There wasassociated hypoautofluorescence along the peripapillary area andthe proximal superotemporal arcades in both eyes However themore peripheral maculae appeared relatively hyperautofluorescentcompared with the surrounding extramacular retina There weremultiple discrete punctate hyperautofluorescent foci in themaculae predominating nasally and temporally (Fig 2N S) Apetaloid pattern of alternating hyper- and hypoautofluorescencewas seen in both eyes but was again more obvious in the lefteye (Fig 2S)
Spectral-Domain Optical Coherence Tomography
Horizontal SD-OCT line scans through the central maculae of botheyes of the proband showed an absence of the outer nuclear layerinner segment ellipsoid band or innerouter segment junction ofthe photoreceptors and RPE except in the foveal region of the righteye where the structure of both the photoreceptors and RPEappeared anomalous but present suggestive of relative sparing(Fig 2D E) This sparing co-localized with the region of hyper-autofluorescence Of note similar sparing of the photoreceptorswas not seen in the foveal region of the left eye despite a hyper-autofluorescent signal (Fig 2I J) The SD-OCT scans in the righteye of the affected sibling resembled that of the proband Howeverthe photoreceptor sparing on SD-OCT was less obvious None-theless in the foveal region of the siblingrsquos right eye there wasevidence of a residual inner segment ellipsoid band in the fovealregion and a disorganized atrophic outer nuclear layer On closeinspection of the outer nuclear layer in the temporal parafoveal
c Clinical and Genetic Data
n (logMAR) OS Condition Age of Onset CRB1 Mutation
400 Affected Mid-20s R1331C P1381L70 Affected Early 30s R1331C P1381Ld Unaffected d wt wt40 Unaffected d wt P1381L20 Unaffected d R1331C wt
m of the minimum angle of resolution OD frac14 right eye OS frac14 left eye
3
Figure 2 Fundus photographs fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans of the right andleft eyes of a 45-year-old woman (proband) and her 41-year-old affected brother with the same CRB1 mutations Fundus photography in the probandexhibited stable fixation (needle position) temporal pallor of the optic discs attenuated retinal vasculature and extensive retinal pigment epithelium (RPE)changes (A F) The FAF imaging reveals predominant hypoautofluorescence across the macula consistent with widespread RPE atrophy although relativehyperautofluorescence was documented in the foveal and parafoveal regions of the right and left eyes and respectively consistent with sparing of the RPE (BC G H) The SD-OCT scans reveal relative sparing of foveal photoreceptors (outer nuclear layer inner segment ellipsoid band) and RPE in the right eye(D E) and an absence of all except RPE in the left eye (I J) Fundus photographs in the affected brother also showed extensive loss of macular RPE (L Q)The FAF revealed concentric rings of alternating hyper- and hypoautofluorescence encircling the macula (M R) and there were petaloid patterns centrallygreatest in the left eye (N S) The SD-OCT scans through the fovea in the right eye revealed an area of relative foveal sparing (O P) and cystoid macularedema (CME) was detected in the left eye (T U) There was possible early cystoid change in the temporal parafoveal macula of the right eye (O) Relativelynormal laminar architecture is preserved in the maculae of both affected siblings Electroretinography in the proband reveals overall preservation ofgeneralized cone and rod function without any obvious implicit time shifts (K) ISe frac14 inner segment ellipsoid OD frac14 right eye ONL frac14 outer nuclear layerOS frac14 left eye
Ophthalmology Volume - Number - Month 2014
macula of the right eye there was possible early cystoid change(Fig 2O) There was definite CME in the left eye visible on SD-OCT (Fig 2T U) Despite the presence of CME in the left eyethe outer nuclear layer and inner segment ellipsoid bandappeared well preserved in the central macula A 3-year follow-up scan of the affected siblingrsquos left eye was acquired after treat-ment (Fig 4 available at wwwaaojournalorg) and showed thatthere was persistence of CME although it had reduced Evenwith progressive photoreceptor and RPE layer disruption retinalthickness appeared to be within normal limits in both affectedsiblings (excluding the siblingrsquos left eye with CME) withreasonably well-preserved retinal lamination
Retinal thickness in the proband and affected brother wasassessed using color-coded thickness maps over the macula of each
4
eye (Fig 5) Compared with an age-matched control both eyes ofthe proband and the right eye of her affected sibling exhibited areduction in retinal thickness in the central macula consistent withatrophy There was a trend toward increased thickness relative tothe control in the more peripheral macula The left eye of theprobandrsquos sibling had grossly increased macular thickness centrallydue to CME
Electroretinogram Analysis
Scotopic responses in the proband showed a generalized preser-vation of retinal rod function A marginal implicit time delaywas detected in the photopic responses suggestive of cone
Figure 3 Fundus photographs 488 nm fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans in theparental carriers of the CRB1 mutation No significant retinal pigment epithelium (RPE) and abnormal AF patterns were observed Both parents exhibitedperipapillary atrophy in both eyes greatest in the mother and related to her high myopia Minor choroidal thinning in the mother also was attributed to thismyopia (B D) The SD-OCT of her left eye revealed a nonsignificant epiretinal membrane Otherwise normal retinal layers and intact photoreceptors werepresent in both parents FAF frac14 fundus autofluorescence OD frac14 right eye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
involvement however both the waveform and amplitudes werewithin normal limits compared with age-matched controls(Fig 2K)
Progression and Visual Function
Microperimetry 1 (MP-1) mapping results (in decibels) wererecorded in the proband at the initial visit and registered to acorresponding FAF image with the built-in Navis software Pre-served visual function was documented within the region ofhyperautofluorescence in both eyes However across the atrophichypoautofluorescent macula retinal sensitivities of 0 decibels wererecorded The test documented steady foveal fixation as assessedby the fixation tracker on the mapping instrument A subsequentrecording after 5 years showed a notable reduction in sensitivity in
the right eye and an almost complete loss of function in the left eye(Fig 6)
Genetic Analyses
Whole exome sequencing was performed in the 2 affected siblingsand their unaffected mother An average of 113 million total readswere generated for each sample with an average of 65 million(58) nonredundant unique reads mapped to the 64 MB exomeregion (Table 2) (available at wwwaaojournalorg) More than90 of the target regions have gt10 coverage with an averagemean depth of coverage of 80 On average there were a totalof 99 041 variants identified for each sample of which 20 730(21) were in protein coding sequences Some 98 of allcoding variants were SNPs and 2 were insertions and deletions
5
Figure 5 Color-coded macular thickness (mm) maps of the CRB1-affected proband and her brother compared with an age-matched normal subject Themaps show a reduced (darker colors) thickness in the central macula in both eyes of the proband and in the right eye of her sibling compared with thecontrol with a trend toward increased (brighter colors) thickness relative to the control in the more peripheral macula The left eye of the probandrsquos siblinghad grossly increased macular thickness due to cystoid macular edema (CME)
Ophthalmology Volume - Number - Month 2014
(indels) The ratio of nonsynonymous to synonymous variants wasclose to 11 in all samples For initial filtering of variants theminimum SNP quality value was set at 20 and the minimumtotal read depth was set at 5
Considering the autosomal recessive inheritance pattern of thedisease and that both affected siblings had to have the same causalmutations we focused on genes that had at least 2 shared variantsin the 2 affected siblings (Table 3) We further assumed that thedisease-causing variants have to be rare thereby filtering thesequence data for new variants and those found in lt05 indbSNP135 and in the 1000 Genomes databases After excluding
6
synonymous variants and all coding and intronic variants that didnot affect splicing the number of candidate genes was narroweddown to 19 After assessing the phase of remaining variants usingthe sequences of the mother only 3 possible candidate genesremained CRB1 NADK and DNAH12 The segregation of vari-ants in these genes with the disease was tested in the entire familyincluding the unaffected sibling and the father
Dynein axonemal heavy chain 12 protein is a large proteinwith multiple rare variants involved in microtubule-associatedmotor protein complexes composed of several heavy light andintermediate chains NADK catalyzes the synthesis of NADP from
Figure 6 Functional and macular progression assessment through fundus autofluorescence (FAF) imaging and microperimetry (MP)-1 mapping Over thecourse of 5 years preservation of the central island of hyperautofluorescent retinal pigment epithelium (RPE) was maintained however the spatial functionhad become more constricted A comparative overlay of MP-1 and FAF showed that the scotomata (0 decibels) recorded on the MP-1 corresponded toatrophic (hypoautofluorescent) regions at the initial examination in the right (A) and left (B) eyes and in the same area of both eyes respectively (C D)after 5 years OD frac14 righteye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
NAD and ATP and exists in several isoforms Neither of these 2genes has been associated with eye disease phenotypes and there isno direct mechanism how these would be involved in retinaldystrophies
This left CRB1 as the only plausible candidate gene for thedisease phenotype segregating with the 2 variants because it is awell-known gene in retinal dystrophies (RetNet httpssphutheduretnet accessed September 10 2013) The 2 CRB1 var-iants shared by the affected siblings (cC3991TpR1331C andC4142TpP1381L) are not present in ESP6500 dbSNP135 and1000 Genomes database although the pP1381L variant wasrecently identified as disease associated in 1 patient with LCA28
Both variants are predicted to be deleterious or damaging bySorting Intolerant from Tolerant (SIFT httpsiftjcviorg)Polyphen-2 and MutationTaster programs Nucleotide positionsof the 2 mutations are highly conserved as predicted by phyloPThe novel cC3991T variant that results in the pR1331C mutationis 1 nucleotide before a previously reported benign variantcA3992G which results in the pR1331H amino acid change
Table 3 Variants Identified in the 2 Affected Individuals
Variant Filters No SNVs No Genes
Total variants shared between bothaffected siblings
67 951 20 181
Not present in dbSNP135 commonand lt05 in 1000 genomes
5733 4499
Missense nonsense indels and splicing 392 364Recessive 47 19Phase-assessed 6 3
SNV frac14 single nucleotide variation
The latter is considered benign however unlike arginine to his-tidine change (Grantham distance 29) there is large physico-chemical difference between arginine and cysteine (Granthamdistance 180) Creation of a cysteine residue at this position alsoresults in 2 consecutive cysteines in the amino acid chainBecause EGF-like domain is characterized by 6 conserved cys-teines forming 3 disulfide bonds and modification from arginineto histidine at this position is considered benign pR1331C alsocould be a relatively milder mutation which could explain themilder phenotype compared with other CRB1-associatedphenotypes
Discussion
The clinical and genetic findings were summarized in afamily in whom 2 rare likely disease-associated CRB1missense variants segregated with an unusual maculardystrophy phenotype Previously reported phenotypesassociated with mutations in CRB1 include those consistentwith an early-onset RP associated with preserved para-arteriolar RPE or Coats-like exudative vasculopathy Thepatients described in this study exhibited unusual pheno-typic characteristics that have not been described beforeand therefore eluded clinical diagnosis by several retinalspecialists Both affected siblings presented with decreasedvisual acuity but without nyctalopia Furthermore onfundoscopy there was a relatively normal-appearing pe-riphery without any of the RP12 characteristics such asnummular hyperpigmentation and some degree of vascularattenuation The disease was largely confined to the mac-ula In both cases the reported age of onset was between
7
Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu
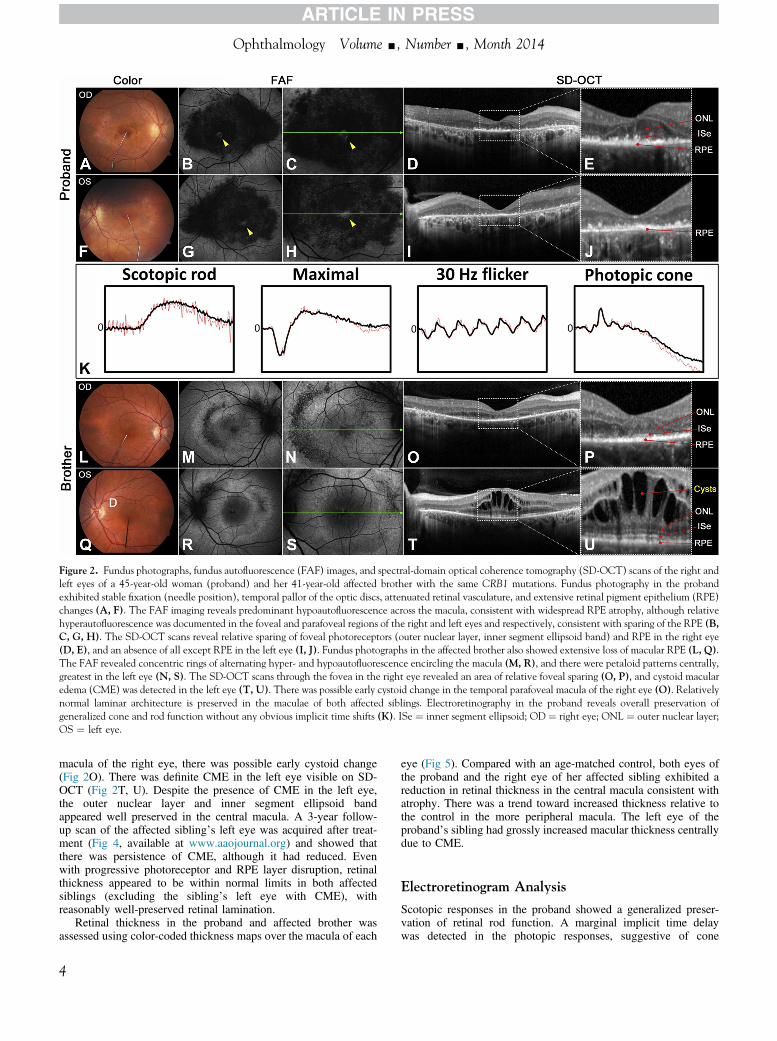
Figure 2 Fundus photographs fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans of the right andleft eyes of a 45-year-old woman (proband) and her 41-year-old affected brother with the same CRB1 mutations Fundus photography in the probandexhibited stable fixation (needle position) temporal pallor of the optic discs attenuated retinal vasculature and extensive retinal pigment epithelium (RPE)changes (A F) The FAF imaging reveals predominant hypoautofluorescence across the macula consistent with widespread RPE atrophy although relativehyperautofluorescence was documented in the foveal and parafoveal regions of the right and left eyes and respectively consistent with sparing of the RPE (BC G H) The SD-OCT scans reveal relative sparing of foveal photoreceptors (outer nuclear layer inner segment ellipsoid band) and RPE in the right eye(D E) and an absence of all except RPE in the left eye (I J) Fundus photographs in the affected brother also showed extensive loss of macular RPE (L Q)The FAF revealed concentric rings of alternating hyper- and hypoautofluorescence encircling the macula (M R) and there were petaloid patterns centrallygreatest in the left eye (N S) The SD-OCT scans through the fovea in the right eye revealed an area of relative foveal sparing (O P) and cystoid macularedema (CME) was detected in the left eye (T U) There was possible early cystoid change in the temporal parafoveal macula of the right eye (O) Relativelynormal laminar architecture is preserved in the maculae of both affected siblings Electroretinography in the proband reveals overall preservation ofgeneralized cone and rod function without any obvious implicit time shifts (K) ISe frac14 inner segment ellipsoid OD frac14 right eye ONL frac14 outer nuclear layerOS frac14 left eye
Ophthalmology Volume - Number - Month 2014
macula of the right eye there was possible early cystoid change(Fig 2O) There was definite CME in the left eye visible on SD-OCT (Fig 2T U) Despite the presence of CME in the left eyethe outer nuclear layer and inner segment ellipsoid bandappeared well preserved in the central macula A 3-year follow-up scan of the affected siblingrsquos left eye was acquired after treat-ment (Fig 4 available at wwwaaojournalorg) and showed thatthere was persistence of CME although it had reduced Evenwith progressive photoreceptor and RPE layer disruption retinalthickness appeared to be within normal limits in both affectedsiblings (excluding the siblingrsquos left eye with CME) withreasonably well-preserved retinal lamination
Retinal thickness in the proband and affected brother wasassessed using color-coded thickness maps over the macula of each
4
eye (Fig 5) Compared with an age-matched control both eyes ofthe proband and the right eye of her affected sibling exhibited areduction in retinal thickness in the central macula consistent withatrophy There was a trend toward increased thickness relative tothe control in the more peripheral macula The left eye of theprobandrsquos sibling had grossly increased macular thickness centrallydue to CME
Electroretinogram Analysis
Scotopic responses in the proband showed a generalized preser-vation of retinal rod function A marginal implicit time delaywas detected in the photopic responses suggestive of cone
Figure 3 Fundus photographs 488 nm fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans in theparental carriers of the CRB1 mutation No significant retinal pigment epithelium (RPE) and abnormal AF patterns were observed Both parents exhibitedperipapillary atrophy in both eyes greatest in the mother and related to her high myopia Minor choroidal thinning in the mother also was attributed to thismyopia (B D) The SD-OCT of her left eye revealed a nonsignificant epiretinal membrane Otherwise normal retinal layers and intact photoreceptors werepresent in both parents FAF frac14 fundus autofluorescence OD frac14 right eye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
involvement however both the waveform and amplitudes werewithin normal limits compared with age-matched controls(Fig 2K)
Progression and Visual Function
Microperimetry 1 (MP-1) mapping results (in decibels) wererecorded in the proband at the initial visit and registered to acorresponding FAF image with the built-in Navis software Pre-served visual function was documented within the region ofhyperautofluorescence in both eyes However across the atrophichypoautofluorescent macula retinal sensitivities of 0 decibels wererecorded The test documented steady foveal fixation as assessedby the fixation tracker on the mapping instrument A subsequentrecording after 5 years showed a notable reduction in sensitivity in
the right eye and an almost complete loss of function in the left eye(Fig 6)
Genetic Analyses
Whole exome sequencing was performed in the 2 affected siblingsand their unaffected mother An average of 113 million total readswere generated for each sample with an average of 65 million(58) nonredundant unique reads mapped to the 64 MB exomeregion (Table 2) (available at wwwaaojournalorg) More than90 of the target regions have gt10 coverage with an averagemean depth of coverage of 80 On average there were a totalof 99 041 variants identified for each sample of which 20 730(21) were in protein coding sequences Some 98 of allcoding variants were SNPs and 2 were insertions and deletions
5
Figure 5 Color-coded macular thickness (mm) maps of the CRB1-affected proband and her brother compared with an age-matched normal subject Themaps show a reduced (darker colors) thickness in the central macula in both eyes of the proband and in the right eye of her sibling compared with thecontrol with a trend toward increased (brighter colors) thickness relative to the control in the more peripheral macula The left eye of the probandrsquos siblinghad grossly increased macular thickness due to cystoid macular edema (CME)
Ophthalmology Volume - Number - Month 2014
(indels) The ratio of nonsynonymous to synonymous variants wasclose to 11 in all samples For initial filtering of variants theminimum SNP quality value was set at 20 and the minimumtotal read depth was set at 5
Considering the autosomal recessive inheritance pattern of thedisease and that both affected siblings had to have the same causalmutations we focused on genes that had at least 2 shared variantsin the 2 affected siblings (Table 3) We further assumed that thedisease-causing variants have to be rare thereby filtering thesequence data for new variants and those found in lt05 indbSNP135 and in the 1000 Genomes databases After excluding
6
synonymous variants and all coding and intronic variants that didnot affect splicing the number of candidate genes was narroweddown to 19 After assessing the phase of remaining variants usingthe sequences of the mother only 3 possible candidate genesremained CRB1 NADK and DNAH12 The segregation of vari-ants in these genes with the disease was tested in the entire familyincluding the unaffected sibling and the father
Dynein axonemal heavy chain 12 protein is a large proteinwith multiple rare variants involved in microtubule-associatedmotor protein complexes composed of several heavy light andintermediate chains NADK catalyzes the synthesis of NADP from
Figure 6 Functional and macular progression assessment through fundus autofluorescence (FAF) imaging and microperimetry (MP)-1 mapping Over thecourse of 5 years preservation of the central island of hyperautofluorescent retinal pigment epithelium (RPE) was maintained however the spatial functionhad become more constricted A comparative overlay of MP-1 and FAF showed that the scotomata (0 decibels) recorded on the MP-1 corresponded toatrophic (hypoautofluorescent) regions at the initial examination in the right (A) and left (B) eyes and in the same area of both eyes respectively (C D)after 5 years OD frac14 righteye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
NAD and ATP and exists in several isoforms Neither of these 2genes has been associated with eye disease phenotypes and there isno direct mechanism how these would be involved in retinaldystrophies
This left CRB1 as the only plausible candidate gene for thedisease phenotype segregating with the 2 variants because it is awell-known gene in retinal dystrophies (RetNet httpssphutheduretnet accessed September 10 2013) The 2 CRB1 var-iants shared by the affected siblings (cC3991TpR1331C andC4142TpP1381L) are not present in ESP6500 dbSNP135 and1000 Genomes database although the pP1381L variant wasrecently identified as disease associated in 1 patient with LCA28
Both variants are predicted to be deleterious or damaging bySorting Intolerant from Tolerant (SIFT httpsiftjcviorg)Polyphen-2 and MutationTaster programs Nucleotide positionsof the 2 mutations are highly conserved as predicted by phyloPThe novel cC3991T variant that results in the pR1331C mutationis 1 nucleotide before a previously reported benign variantcA3992G which results in the pR1331H amino acid change
Table 3 Variants Identified in the 2 Affected Individuals
Variant Filters No SNVs No Genes
Total variants shared between bothaffected siblings
67 951 20 181
Not present in dbSNP135 commonand lt05 in 1000 genomes
5733 4499
Missense nonsense indels and splicing 392 364Recessive 47 19Phase-assessed 6 3
SNV frac14 single nucleotide variation
The latter is considered benign however unlike arginine to his-tidine change (Grantham distance 29) there is large physico-chemical difference between arginine and cysteine (Granthamdistance 180) Creation of a cysteine residue at this position alsoresults in 2 consecutive cysteines in the amino acid chainBecause EGF-like domain is characterized by 6 conserved cys-teines forming 3 disulfide bonds and modification from arginineto histidine at this position is considered benign pR1331C alsocould be a relatively milder mutation which could explain themilder phenotype compared with other CRB1-associatedphenotypes
Discussion
The clinical and genetic findings were summarized in afamily in whom 2 rare likely disease-associated CRB1missense variants segregated with an unusual maculardystrophy phenotype Previously reported phenotypesassociated with mutations in CRB1 include those consistentwith an early-onset RP associated with preserved para-arteriolar RPE or Coats-like exudative vasculopathy Thepatients described in this study exhibited unusual pheno-typic characteristics that have not been described beforeand therefore eluded clinical diagnosis by several retinalspecialists Both affected siblings presented with decreasedvisual acuity but without nyctalopia Furthermore onfundoscopy there was a relatively normal-appearing pe-riphery without any of the RP12 characteristics such asnummular hyperpigmentation and some degree of vascularattenuation The disease was largely confined to the mac-ula In both cases the reported age of onset was between
7
Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu

Figure 3 Fundus photographs 488 nm fundus autofluorescence (FAF) images and spectral-domain optical coherence tomography (SD-OCT) scans in theparental carriers of the CRB1 mutation No significant retinal pigment epithelium (RPE) and abnormal AF patterns were observed Both parents exhibitedperipapillary atrophy in both eyes greatest in the mother and related to her high myopia Minor choroidal thinning in the mother also was attributed to thismyopia (B D) The SD-OCT of her left eye revealed a nonsignificant epiretinal membrane Otherwise normal retinal layers and intact photoreceptors werepresent in both parents FAF frac14 fundus autofluorescence OD frac14 right eye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
involvement however both the waveform and amplitudes werewithin normal limits compared with age-matched controls(Fig 2K)
Progression and Visual Function
Microperimetry 1 (MP-1) mapping results (in decibels) wererecorded in the proband at the initial visit and registered to acorresponding FAF image with the built-in Navis software Pre-served visual function was documented within the region ofhyperautofluorescence in both eyes However across the atrophichypoautofluorescent macula retinal sensitivities of 0 decibels wererecorded The test documented steady foveal fixation as assessedby the fixation tracker on the mapping instrument A subsequentrecording after 5 years showed a notable reduction in sensitivity in
the right eye and an almost complete loss of function in the left eye(Fig 6)
Genetic Analyses
Whole exome sequencing was performed in the 2 affected siblingsand their unaffected mother An average of 113 million total readswere generated for each sample with an average of 65 million(58) nonredundant unique reads mapped to the 64 MB exomeregion (Table 2) (available at wwwaaojournalorg) More than90 of the target regions have gt10 coverage with an averagemean depth of coverage of 80 On average there were a totalof 99 041 variants identified for each sample of which 20 730(21) were in protein coding sequences Some 98 of allcoding variants were SNPs and 2 were insertions and deletions
5
Figure 5 Color-coded macular thickness (mm) maps of the CRB1-affected proband and her brother compared with an age-matched normal subject Themaps show a reduced (darker colors) thickness in the central macula in both eyes of the proband and in the right eye of her sibling compared with thecontrol with a trend toward increased (brighter colors) thickness relative to the control in the more peripheral macula The left eye of the probandrsquos siblinghad grossly increased macular thickness due to cystoid macular edema (CME)
Ophthalmology Volume - Number - Month 2014
(indels) The ratio of nonsynonymous to synonymous variants wasclose to 11 in all samples For initial filtering of variants theminimum SNP quality value was set at 20 and the minimumtotal read depth was set at 5
Considering the autosomal recessive inheritance pattern of thedisease and that both affected siblings had to have the same causalmutations we focused on genes that had at least 2 shared variantsin the 2 affected siblings (Table 3) We further assumed that thedisease-causing variants have to be rare thereby filtering thesequence data for new variants and those found in lt05 indbSNP135 and in the 1000 Genomes databases After excluding
6
synonymous variants and all coding and intronic variants that didnot affect splicing the number of candidate genes was narroweddown to 19 After assessing the phase of remaining variants usingthe sequences of the mother only 3 possible candidate genesremained CRB1 NADK and DNAH12 The segregation of vari-ants in these genes with the disease was tested in the entire familyincluding the unaffected sibling and the father
Dynein axonemal heavy chain 12 protein is a large proteinwith multiple rare variants involved in microtubule-associatedmotor protein complexes composed of several heavy light andintermediate chains NADK catalyzes the synthesis of NADP from
Figure 6 Functional and macular progression assessment through fundus autofluorescence (FAF) imaging and microperimetry (MP)-1 mapping Over thecourse of 5 years preservation of the central island of hyperautofluorescent retinal pigment epithelium (RPE) was maintained however the spatial functionhad become more constricted A comparative overlay of MP-1 and FAF showed that the scotomata (0 decibels) recorded on the MP-1 corresponded toatrophic (hypoautofluorescent) regions at the initial examination in the right (A) and left (B) eyes and in the same area of both eyes respectively (C D)after 5 years OD frac14 righteye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
NAD and ATP and exists in several isoforms Neither of these 2genes has been associated with eye disease phenotypes and there isno direct mechanism how these would be involved in retinaldystrophies
This left CRB1 as the only plausible candidate gene for thedisease phenotype segregating with the 2 variants because it is awell-known gene in retinal dystrophies (RetNet httpssphutheduretnet accessed September 10 2013) The 2 CRB1 var-iants shared by the affected siblings (cC3991TpR1331C andC4142TpP1381L) are not present in ESP6500 dbSNP135 and1000 Genomes database although the pP1381L variant wasrecently identified as disease associated in 1 patient with LCA28
Both variants are predicted to be deleterious or damaging bySorting Intolerant from Tolerant (SIFT httpsiftjcviorg)Polyphen-2 and MutationTaster programs Nucleotide positionsof the 2 mutations are highly conserved as predicted by phyloPThe novel cC3991T variant that results in the pR1331C mutationis 1 nucleotide before a previously reported benign variantcA3992G which results in the pR1331H amino acid change
Table 3 Variants Identified in the 2 Affected Individuals
Variant Filters No SNVs No Genes
Total variants shared between bothaffected siblings
67 951 20 181
Not present in dbSNP135 commonand lt05 in 1000 genomes
5733 4499
Missense nonsense indels and splicing 392 364Recessive 47 19Phase-assessed 6 3
SNV frac14 single nucleotide variation
The latter is considered benign however unlike arginine to his-tidine change (Grantham distance 29) there is large physico-chemical difference between arginine and cysteine (Granthamdistance 180) Creation of a cysteine residue at this position alsoresults in 2 consecutive cysteines in the amino acid chainBecause EGF-like domain is characterized by 6 conserved cys-teines forming 3 disulfide bonds and modification from arginineto histidine at this position is considered benign pR1331C alsocould be a relatively milder mutation which could explain themilder phenotype compared with other CRB1-associatedphenotypes
Discussion
The clinical and genetic findings were summarized in afamily in whom 2 rare likely disease-associated CRB1missense variants segregated with an unusual maculardystrophy phenotype Previously reported phenotypesassociated with mutations in CRB1 include those consistentwith an early-onset RP associated with preserved para-arteriolar RPE or Coats-like exudative vasculopathy Thepatients described in this study exhibited unusual pheno-typic characteristics that have not been described beforeand therefore eluded clinical diagnosis by several retinalspecialists Both affected siblings presented with decreasedvisual acuity but without nyctalopia Furthermore onfundoscopy there was a relatively normal-appearing pe-riphery without any of the RP12 characteristics such asnummular hyperpigmentation and some degree of vascularattenuation The disease was largely confined to the mac-ula In both cases the reported age of onset was between
7
Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu
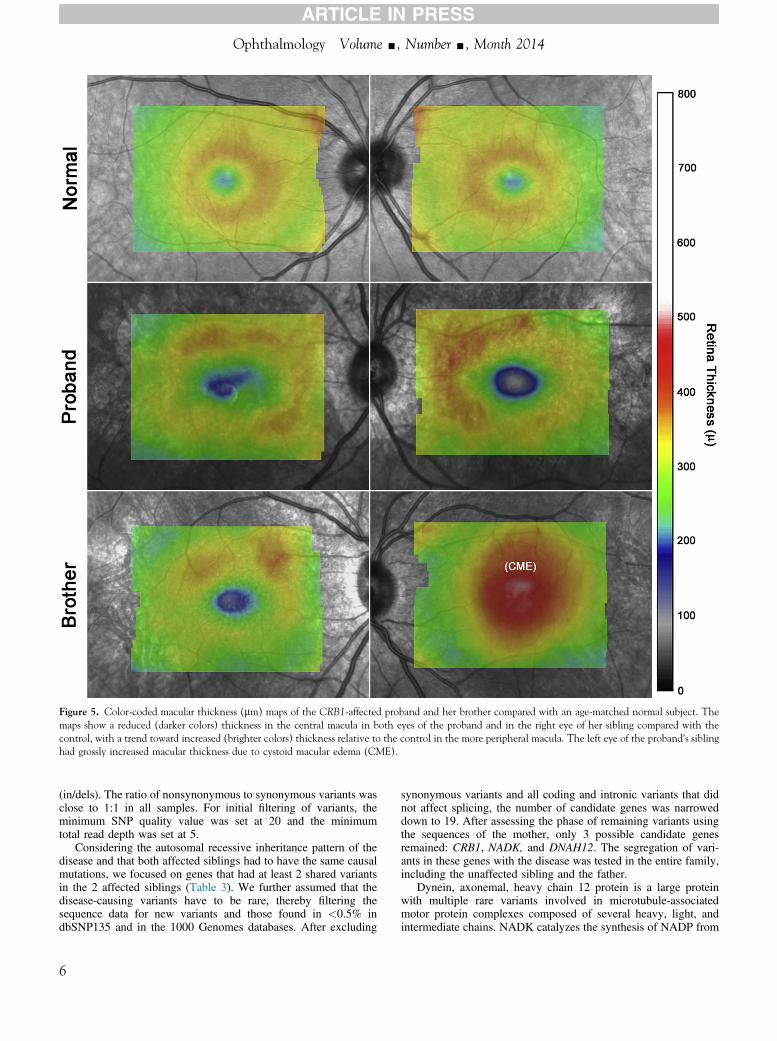
Figure 5 Color-coded macular thickness (mm) maps of the CRB1-affected proband and her brother compared with an age-matched normal subject Themaps show a reduced (darker colors) thickness in the central macula in both eyes of the proband and in the right eye of her sibling compared with thecontrol with a trend toward increased (brighter colors) thickness relative to the control in the more peripheral macula The left eye of the probandrsquos siblinghad grossly increased macular thickness due to cystoid macular edema (CME)
Ophthalmology Volume - Number - Month 2014
(indels) The ratio of nonsynonymous to synonymous variants wasclose to 11 in all samples For initial filtering of variants theminimum SNP quality value was set at 20 and the minimumtotal read depth was set at 5
Considering the autosomal recessive inheritance pattern of thedisease and that both affected siblings had to have the same causalmutations we focused on genes that had at least 2 shared variantsin the 2 affected siblings (Table 3) We further assumed that thedisease-causing variants have to be rare thereby filtering thesequence data for new variants and those found in lt05 indbSNP135 and in the 1000 Genomes databases After excluding
6
synonymous variants and all coding and intronic variants that didnot affect splicing the number of candidate genes was narroweddown to 19 After assessing the phase of remaining variants usingthe sequences of the mother only 3 possible candidate genesremained CRB1 NADK and DNAH12 The segregation of vari-ants in these genes with the disease was tested in the entire familyincluding the unaffected sibling and the father
Dynein axonemal heavy chain 12 protein is a large proteinwith multiple rare variants involved in microtubule-associatedmotor protein complexes composed of several heavy light andintermediate chains NADK catalyzes the synthesis of NADP from
Figure 6 Functional and macular progression assessment through fundus autofluorescence (FAF) imaging and microperimetry (MP)-1 mapping Over thecourse of 5 years preservation of the central island of hyperautofluorescent retinal pigment epithelium (RPE) was maintained however the spatial functionhad become more constricted A comparative overlay of MP-1 and FAF showed that the scotomata (0 decibels) recorded on the MP-1 corresponded toatrophic (hypoautofluorescent) regions at the initial examination in the right (A) and left (B) eyes and in the same area of both eyes respectively (C D)after 5 years OD frac14 righteye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
NAD and ATP and exists in several isoforms Neither of these 2genes has been associated with eye disease phenotypes and there isno direct mechanism how these would be involved in retinaldystrophies
This left CRB1 as the only plausible candidate gene for thedisease phenotype segregating with the 2 variants because it is awell-known gene in retinal dystrophies (RetNet httpssphutheduretnet accessed September 10 2013) The 2 CRB1 var-iants shared by the affected siblings (cC3991TpR1331C andC4142TpP1381L) are not present in ESP6500 dbSNP135 and1000 Genomes database although the pP1381L variant wasrecently identified as disease associated in 1 patient with LCA28
Both variants are predicted to be deleterious or damaging bySorting Intolerant from Tolerant (SIFT httpsiftjcviorg)Polyphen-2 and MutationTaster programs Nucleotide positionsof the 2 mutations are highly conserved as predicted by phyloPThe novel cC3991T variant that results in the pR1331C mutationis 1 nucleotide before a previously reported benign variantcA3992G which results in the pR1331H amino acid change
Table 3 Variants Identified in the 2 Affected Individuals
Variant Filters No SNVs No Genes
Total variants shared between bothaffected siblings
67 951 20 181
Not present in dbSNP135 commonand lt05 in 1000 genomes
5733 4499
Missense nonsense indels and splicing 392 364Recessive 47 19Phase-assessed 6 3
SNV frac14 single nucleotide variation
The latter is considered benign however unlike arginine to his-tidine change (Grantham distance 29) there is large physico-chemical difference between arginine and cysteine (Granthamdistance 180) Creation of a cysteine residue at this position alsoresults in 2 consecutive cysteines in the amino acid chainBecause EGF-like domain is characterized by 6 conserved cys-teines forming 3 disulfide bonds and modification from arginineto histidine at this position is considered benign pR1331C alsocould be a relatively milder mutation which could explain themilder phenotype compared with other CRB1-associatedphenotypes
Discussion
The clinical and genetic findings were summarized in afamily in whom 2 rare likely disease-associated CRB1missense variants segregated with an unusual maculardystrophy phenotype Previously reported phenotypesassociated with mutations in CRB1 include those consistentwith an early-onset RP associated with preserved para-arteriolar RPE or Coats-like exudative vasculopathy Thepatients described in this study exhibited unusual pheno-typic characteristics that have not been described beforeand therefore eluded clinical diagnosis by several retinalspecialists Both affected siblings presented with decreasedvisual acuity but without nyctalopia Furthermore onfundoscopy there was a relatively normal-appearing pe-riphery without any of the RP12 characteristics such asnummular hyperpigmentation and some degree of vascularattenuation The disease was largely confined to the mac-ula In both cases the reported age of onset was between
7
Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu
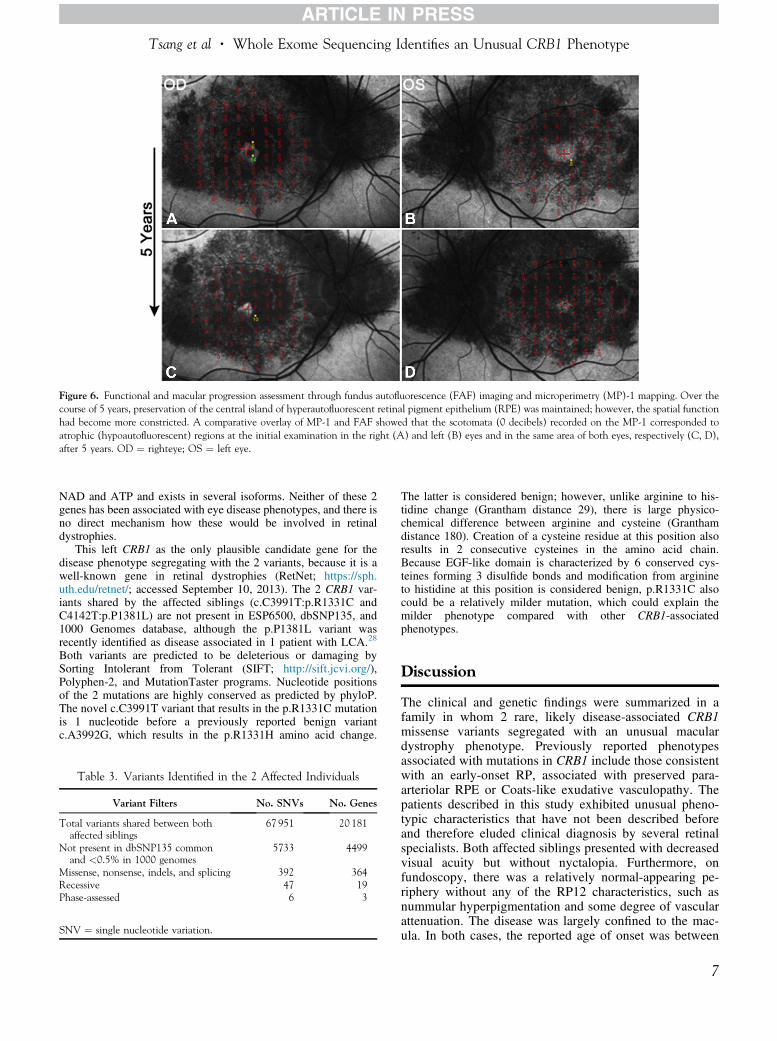
Figure 6 Functional and macular progression assessment through fundus autofluorescence (FAF) imaging and microperimetry (MP)-1 mapping Over thecourse of 5 years preservation of the central island of hyperautofluorescent retinal pigment epithelium (RPE) was maintained however the spatial functionhad become more constricted A comparative overlay of MP-1 and FAF showed that the scotomata (0 decibels) recorded on the MP-1 corresponded toatrophic (hypoautofluorescent) regions at the initial examination in the right (A) and left (B) eyes and in the same area of both eyes respectively (C D)after 5 years OD frac14 righteye OS frac14 left eye
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
NAD and ATP and exists in several isoforms Neither of these 2genes has been associated with eye disease phenotypes and there isno direct mechanism how these would be involved in retinaldystrophies
This left CRB1 as the only plausible candidate gene for thedisease phenotype segregating with the 2 variants because it is awell-known gene in retinal dystrophies (RetNet httpssphutheduretnet accessed September 10 2013) The 2 CRB1 var-iants shared by the affected siblings (cC3991TpR1331C andC4142TpP1381L) are not present in ESP6500 dbSNP135 and1000 Genomes database although the pP1381L variant wasrecently identified as disease associated in 1 patient with LCA28
Both variants are predicted to be deleterious or damaging bySorting Intolerant from Tolerant (SIFT httpsiftjcviorg)Polyphen-2 and MutationTaster programs Nucleotide positionsof the 2 mutations are highly conserved as predicted by phyloPThe novel cC3991T variant that results in the pR1331C mutationis 1 nucleotide before a previously reported benign variantcA3992G which results in the pR1331H amino acid change
Table 3 Variants Identified in the 2 Affected Individuals
Variant Filters No SNVs No Genes
Total variants shared between bothaffected siblings
67 951 20 181
Not present in dbSNP135 commonand lt05 in 1000 genomes
5733 4499
Missense nonsense indels and splicing 392 364Recessive 47 19Phase-assessed 6 3
SNV frac14 single nucleotide variation
The latter is considered benign however unlike arginine to his-tidine change (Grantham distance 29) there is large physico-chemical difference between arginine and cysteine (Granthamdistance 180) Creation of a cysteine residue at this position alsoresults in 2 consecutive cysteines in the amino acid chainBecause EGF-like domain is characterized by 6 conserved cys-teines forming 3 disulfide bonds and modification from arginineto histidine at this position is considered benign pR1331C alsocould be a relatively milder mutation which could explain themilder phenotype compared with other CRB1-associatedphenotypes
Discussion
The clinical and genetic findings were summarized in afamily in whom 2 rare likely disease-associated CRB1missense variants segregated with an unusual maculardystrophy phenotype Previously reported phenotypesassociated with mutations in CRB1 include those consistentwith an early-onset RP associated with preserved para-arteriolar RPE or Coats-like exudative vasculopathy Thepatients described in this study exhibited unusual pheno-typic characteristics that have not been described beforeand therefore eluded clinical diagnosis by several retinalspecialists Both affected siblings presented with decreasedvisual acuity but without nyctalopia Furthermore onfundoscopy there was a relatively normal-appearing pe-riphery without any of the RP12 characteristics such asnummular hyperpigmentation and some degree of vascularattenuation The disease was largely confined to the mac-ula In both cases the reported age of onset was between
7
Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu

Table 5 Potential Modifier Genes for CRB1-Associated Phenotype
Gene DNA Change Protein Change PP-2 SIFT Mutation Taster PhyloP In ProbandBrother
MPDZ C1970T P657L Benign Deleterious Disease causing Weakly conserved ProbandUSH2A A10999C T3667P NA Deleterious Polymorphism Not conserved BothRPGRIP1 C2794G P932A Probably damaging Tolerated Disease causing Moderately conserved BothTOPORS G2138A R713K Benign Tolerated Polymorphism Moderately conserved BothCDHR1 A1868G N623S Benign Tolerated Polymorphism Highly conserved BothCEP290 G4237C D1413H Benign Deleterious Disease causing Moderately conserved ProbandSNRNP200 A3315G A1105A Synonymous Synonymous Synonymous Synonymous Probandc2orf71 G3789A L1263L Synonymous Synonymous Synonymous Synonymous Brother
NA frac14 not applicable PP-2frac14Polymorphism Phenotyping v2 (PolyPhen-2 httpgeneticsbwhharvardedupph2) PhyloP frac14 Phylogenetic p-values (httpcompgenbscbcornelleduphastphyloP) SIFT frac14 Sorting Intolerant from Tolerant (httpsiftjcviorg)
Ophthalmology Volume - Number - Month 2014
the mid-20s and early 30s however the proband hadprogressed to a more advanced disease stage as evidencedby her comparatively lower visual acuity and the extent ofmacular atrophy on FAF imaging and SD-OCT Of note asmall patch of RPE was preserved in the central macularregions and the overlying retina was found to be func-tionally intact through microperimetry testing in the pro-band Spectral-domain OCT documented relative fovealsparing of the photoreceptors in the right eye of the pro-band and in both eyes of the affected sibling although thelatter also had CME in his left eye Although the probanddid not exhibit any CME at the time of examination retinallaminar changes seen in her foveal SD-OCT scans may bedue to macular edema from an earlier disease stageHyperautofluorescence in the nasal retina and fovealsparing are rare phenomena observed in some cases ofABCA4- and PRPH2-associated maculopathies Clinicalfindings in the affected brother exhibited unusual alter-nating patterns of autofluorescent rings marked withpunctate changes in the central macula resembling abullrsquos-eye lesion reported in other related retinalconditions
Patients with CRB1-associated phenotypes typicallypresent with a thick underdeveloped retina characteristi-cally exhibiting loss of laminar layering A study of micecarrying the rd8 mutation in Crb1 has shown an analogousphenotype of a thickened retina and loss of distinct retinallayering22 This developmental disorganization has beenattributed to the role of CRB1 in embryonic retinaldevelopment in mice and humans more specificallyphotoreceptor morphogenesis Abnormal thickening andloss of laminar layering were not seen in the presentedcases Although some laminar disruption is evident thedistinct layers can in general be distinguished and retinalthickness appears normal Preserved electrophysiologicfunction was observed in full-field ERG measurementsThese findings are consistent with the presence of periph-eral retinal sparing but distinct from previous reports ofCRB1-assciated phenotypes in which ERG was extin-guished and undetectable because of abnormal developmentof the embryonic retina
The molecular genetic reasons for the observed distinctphenotype may be attributed to a specific combination ofCRB1 alleles to modifier genes or both Furthermore the
8
modifying effect of nongenetic factors (eg environ-mental) has been suggested as a reason for phenotypevariation in CRB1 retinopathy24 As discussed previouslythe combination of the alleles specifically the newpR1331C variant may have a different effect on theprotein function than other CRB1 alleles The exomesequences also were analyzed for variants in possiblemodifier genes especially those that have been shown tointeract with the CRB1 protein or belong to the samepathway(s) (Table 4 available at wwwaaojournalorg)Specifically genes that encode for proteins involved inthe CRUMBS network those that have been co-immunoprecipitated with CRB1 or retinal ciliopathy pro-teins in the ciliary compartment were analyzed for possiblemodifier variants (Table 5)
Crb1 in mice localizes to the outer limiting membranebetween the subapical surface or region and adherensjunction of Muumlller glia cells19 In the outer limitingmembrane Crb1 interacts with MPP5 via its PDZ bindingdomain and EPB41L5 with its FERM binding site At thislocation MPP5 organizes a protein scaffold that includesthe MAGUK family members MPP3 and MPP4 Inaddition MPP5 also has been found to interact with LIN-7 PAR6 PATJ MUPP1 EZRIN and the neuronalGABA transporter GAT114 No mutations that were sharedbetween both affected siblings were found except for a rareheterozygous missense mutation in MPDZ a gene thatcodes for multi-PDZ domain protein-1 (MUPP1) in theproband (Table 5) MUPP1 interacts with the intracellulardomain of CRB1 via association with the PDZ domain ofMPP5
Some rare heterozygous variants in genes known tocause retinal dystrophies such as RP LCA and CRDwere shared by both affected siblings The specific variantsand in silico prediction of their pathogenicity are shown inTable 5 One study has suggested that the USHERINprotein network has physical connection to the CRUMBSprotein complex via interaction of MPP5 with MPP1 andthe multi-PDZ protein whirlin at the outer limiting mem-brane14 WHRN and USH2A co-localize at the outerlimiting membrane and the connecting cilium of photore-ceptors29 Mutations in USH2A are associated withrecessive Usher syndrome type 2a and recessive RP Astudy of Spanish families with LCA has suggested that
Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu

Tsang et al Whole Exome Sequencing Identifies an Unusual CRB1 Phenotype
variants in RPGRIP1 along with GUCY2D and AIPL1could be modifier alleles of CRB130 Several studies haveidentified mutations in the TOPORS gene that causedominant RP3132 TOPORS is a cilia-centrosomal proteinthat localizes to the basal bodies of connecting cilium andto the centrosomes of cultured cells Morpholino-mediatedsilencing of topors in zebrafish embryos demonstrateddefective retinal development and failure to form outersegments33 CDHR1 (PCDH21) encodes a photoreceptor-specific cadherin that co-localizes at the base of outersegment with Prominin 1 It is involved in disc morpho-genesis and causes cone-rod dystrophy in a mutatedform34 In addition sequence changes in CEP290 andSNRNP200 were detected in the proband and 1 variant inthe c2orf71 gene in the affected brother Recessivemutations in all these genes have been associated withLCA RP or CRD However because both patientsexhibited similar disease phenotype and the combinationof CRB1 variants was unique to this family we were notable to assign modifier role to any of the identifiedvariants or to nongenetic modifiers
In conclusion manifestation of only focal disease inCRB1-associated degeneration in this family was distinctfrom all previously described retinal dysfunction caused bymutations in CRB1 Instead of a generalized retinal degen-eration with dysplastic retinae seen in other CRB1-associ-ated cases patients in this study exhibited a slowlyprogressive focal disease Patients also were lacking allwell-known phenotypic features of CRB1-associated dis-ease such as peripheral nummular pigmentation preservedpara-arteriolar RPE or Coats-like vasculopathy Althoughno unequivocal evidence was found for any modifier allelesthat could explain the clinical findings variants in severalgenes were identified that could modulate the phenotype inthis family Identification of gene- and especially mutation-specific phenotypes will aid in directing future DNA testingand selecting treatment options for patients with maculardystrophies
References
1 Clark GR Crowe P Muszynska D et al Development of adiagnostic genetic test for simplex and autosomal recessiveretinitis pigmentosa Ophthalmology 20101172169ndash77
2 den Hollander AI ten Brink JB de Kok YJ et al Mutations ina human homologue of Drosophila crumbs cause retinitispigmentosa (RP12) Nat Genet 199923217ndash21
3 Lotery AJ Jacobson SG Fishman GA et al Mutations in theCRB1 gene cause Leber congenital amaurosis Arch Oph-thalmol 2001119415ndash20
4 Lotery AJ Malik A Shami SA et al CRB1 mutations mayresult in retinitis pigmentosa without para-arteriolar RPEpreservation Ophthalmic Genet 200122163ndash9
5 Bernal S Calaf M Garcia-Hoyos M et al Study of theinvolvement of the RGR CRPB1 and CRB1 genes in thepathogenesis of autosomal recessive retinitis pigmentosa[report online] J Med Genet 200340e89 Available athttpjmgbmjcomcontent407e89long Accessed October14 2013
6 Heckenlively JR Preserved para-arteriole retinal pigmentepithelium (PPRPE) in retinitis pigmentosa Birth Defects OrigArtic Ser 198218193ndash6
7 den Hollander AI Heckenlively JR van den Born LI et alLeber congenital amaurosis and retinitis pigmentosa withCoats-like exudative vasculopathy are associated with muta-tions in the crumbs homologue 1 (CRB1) gene Am J HumGenet 200169198ndash203
8 den Hollander AI Johnson K de Kok YJ et al CRB1 hasa cytoplasmic domain that is functionally conserved be-tween human and Drosophila Hum Mol Genet 2001102767ndash73
9 den Hollander AI Roepman R Koenekoop RK Cremers FPLeber congenital amaurosis genes proteins and diseasemechanisms Prog Retin Eye Res 200827391ndash419
10 Franceschetti A Dieterle P Diagnostic and prognosticimportance of the electroretinogram in tapetoretinal degener-ation with reduction of the visual field and hemeralopia [inFrench] Confin Neurol 195414184ndash6
11 den Hollander AI Ghiani M de Kok YJ et al Isolation ofCrb1 a mouse homologue of Drosophila crumbs and analysisof its expression pattern in eye and brain Mech Dev 2002110203ndash7
12 Roh MH Makarova O Liu CJ et al The Maguk proteinPals1 functions as an adapter linking mammalian homo-logues of Crumbs and Discs Lost J Cell Biol 2002157161ndash72
13 Watanabe T Miyatani S Katoh I et al Expression of a novelsecretory form (Crb1s) of mouse Crumbs homologue Crb1 inskin development Biochem Biophys Res Commun 2004313263ndash70
14 Gosens I den Hollander AI Cremers FP Roepman RComposition and function of the Crumbs protein complex inthe mammalian retina Exp Eye Res 200886713ndash26
15 Tepass U Crumbs a component of the apical membrane isrequired for zonula adherens formation in primary epithelia ofDrosophila Dev Biol 1996177217ndash25
16 Tepass U Theres C Knust E crumbs encodes an EGF-likeprotein expressed on apical membranes of Drosophila epithe-lial cells and required for organization of epithelia Cell199061787ndash99
17 Mehalow AK Kameya S Smith RS et al CRB1 is essentialfor external limiting membrane integrity and photoreceptormorphogenesis in the mammalian retina Hum Mol Genet2003122179ndash89
18 Pellikka M Tanentzapf G Pinto M et al Crumbs theDrosophila homologue of human CRB1RP12 is essential forphotoreceptor morphogenesis Nature 2002416143ndash9
19 van de Pavert SA Kantardzhieva A Malysheva A et alCrumbs homologue 1 is required for maintenance of photo-receptor cell polarization and adhesion during light exposureJ Cell Sci 20041174169ndash77
20 Jacobson SG Cideciyan AV Aleman TS et al Crumbshomolog 1 (CRB1) mutations result in a thick human retinawith abnormal lamination Hum Mol Genet 2003121073ndash8
21 van de Pavert SA Meuleman J Malysheva A et alA single amino acid substitution (Cys249Trp) in Crb1causes retinal degeneration and deregulates expression ofpituitary tumor transforming gene Pttg1 J Neurosci200727564ndash73
22 Aleman TS Cideciyan AV Aguirre GK et al Human CRB1-associated retinal degeneration comparison with the rd8 Crb1-mutant mouse model Invest Ophthalmol Vis Sci 2011526898ndash910
9
Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu

Ophthalmology Volume - Number - Month 2014
23 Booij JC Florijn RJ ten Brink JB et al Identification ofmutations in the AIPL1 CRB1 GUCY2D RPE65 andRPGRIP1 genes in patients with juvenile retinitis pigmen-tosa [report online] J Med Genet 200542e67 Available athttpjmgbmjcomcontent4211e67long Accessed October14 2013
24 Bujakowska K Audo I Mohand-Said S et al CRB1 mutationsin inherited retinal dystrophies Hum Mutat 201233306ndash15
25 Tosi J Tsui I Lima LH et al Case report autofluorescenceimaging and phenotypic variance in a sibling pair with early-onset retinal dystrophy due to defective CRB1 function CurrEye Res 200934395ndash400
26 Zernant J Kulm M Dharmaraj S et al Genotyping micro-array (disease chip) for Leber congenital amaurosis detectionof modifier alleles Invest Ophthalmol Vis Sci 2005463052ndash9
27 Marmor MF Fulton AB Holder GE et al International So-ciety for Clinical Electrophysiology of Vision ISCEV Stan-dard for full-field clinical electroretinography (2008 update)Doc Ophthalmol 200911869ndash77
28 Henderson RH Mackay DS Li Z et al Phenotypic variabilityin patients with retinal dystrophies due to mutations in CRB1Br J Ophthalmol 201195811ndash7
10
29 van Wijk E van der Zwaag B Peters T et al The DFNB31gene product whirlin connects to the Usher protein network inthe cochlea and retina by direct association with USH2A andVLGR1 Hum Mol Genet 200615751ndash65
30 Vallespin E Cantalapiedra D Riveiro-Alvarez R et alMutation screening of 299 Spanish families with retinal dys-trophies by Leber congenital amaurosis genotyping micro-array Invest Ophthalmol Vis Sci 2007485653ndash61
31 Chakarova CF Papaioannou MG Khanna H et al Mutationsin TOPORS cause autosomal dominant retinitis pigmentosawith perivascular retinal pigment epithelium atrophy Am JHum Genet 2007811098ndash103
32 Papaioannou M Chakarova CF Prescott DC et al A newlocus (RP31) for autosomal dominant retinitis pigmentosamaps to chromosome 9p Hum Genet 2005118501ndash3
33 Chakarova CF Khanna H Shah AZ et al TOPORS impli-cated in retinal degeneration is a cilia-centrosomal proteinHum Mol Genet 201120975ndash87
34 Henderson RH Li Z Abd El Aziz MM et al Biallelic mu-tation of protocadherin-21 (PCDH21) causes retinal degener-ation in humans Mol Vis [serial online] 20101646ndash52Available at httpwwwmolvisorgmolvisv16a6 AccessedOctober 14 2013
Footnotes and Financial Disclosures
Originally received June 18 2013Final revision March 7 2014Accepted March 7 2014Available online --- Manuscript no 2013-9751 Department of Ophthalmology Columbia University New York NewYork2 Department of Pathology amp Cell Biology Columbia University NewYork New York3 Department of Ophthalmology Stoke Mandeville Hospital AylesburyBuckinghamshire United Kingdom4 University Eye Clinic Tartu University Tartu Estonia5 Rotterdam Eye Hospital Rotterdam The Netherlands
Financial Disclosure(s)The author(s) have no proprietary or commercial interest in any materialsdiscussed in this article
Supported in part by grants from the National Eye InstituteNational In-stitutes of Health EY021163 EY019861 EY018213 and EY019007 (CoreSupport for Vision Research) Stichting Wetenschappelijk OnderzoekOogziekenhuis Rotterdam Rotterdamse Blindenbelangen StichtingBlindenhulp Gelderse Blinden Stichting Landelijke Stichting voor Blindenen Slechtzienden Foundation Fighting Blindness (Owings Mills MD) andunrestricted funds from Research to Prevent Blindness (New York NY) tothe Department of Ophthalmology Columbia University
Abbreviations and AcronymsCME frac14 cystoid macular edema ERG frac14 electroretinogram FAF frac14 fundusautofluorescenceLCAfrac14Leber congenital amaurosisMP-1frac14microperimetry1 MUPP1 frac14 multi-PDZ domain protein-1 RP frac14 retinitis pigmentosaRPE frac14 retinal pigment epithelium SD-OCT frac14 spectral-domain opticalcoherence tomography SNPfrac14 single nucleotide polymorphism
CorrespondenceRando Allikmets PhD Eye Institute Research Room 202 160 FortWashington Avenue New York NY 10032 E-mail rla22columbiaedu