trace elements from combustion and gasification of coal—an equilibrium approach
TRANSCRIPT

Pergamon Prog. Energy Combust. ScL Vol. 20, pp. 115 138, 1994 Copyright © 1994 Elsevier Science Lid
Printed in Great Britain. All rights reserved 0360-1285/94 $26.00
0360-1285(94)E0004-M
TRACE ELEMENTS FROM COMBUSTION AND GASIFICATION OF COAL--AN EQUILIBRIUM APPROACH
FLEMMING FRANDSEN, KIM DAM-JOHANSEN a n d PETER RASMUSSEN
Department of Chemical Engineering, Technical University of Denmark, Building 229, DK-2800 Lyngby, Denmark
Received 4 March 1994
Abstract--The fate of several trace elements in the thermal conversion of coal has been investigated, assuming global equilibrium and using an in-house database and a Fortran-77 computer code for the calculations. The format and content of the database DGFDBASE, containing reduced data on AG~i(T) for approximately 800 chemical species of the elements AI, As, B, Be, Br, C, Ca, Cd, CI, Co, Cr, F, Fe, Ga, Ge, H, Hg, K, Mg, N, Na, Ni, O, P, Pb, S, Sb, Se, Si, Sn, Ti, V and Zn are described. Results of thermodynamic equilibrium calculations performed using 'the total Gibbs free energy minimization' program MINGTSYS on simple systems containing one of the trace elements As, B, Be, Cd, Co, Cr, Ga, Ge, Hg, Ni, P, Pb, Sb, Se, Sn, Ti, V and Zn are presented and compared with results from the literature. Combustion as well as gasification conditions have been considered.
At oxidizing conditions all the trace elements considered form at least one stable condensed phase in the temperature range from 300-2000 K. Regarding the condensed phase being stable at the lowest temperatures, the trace elements can be divided into two groups, the first of sulfate forming elements (this group includes the elements Be, Cd, Co, Cr, Hg, Ni, Pb, Sb, Sn, V, and Zn) and the latter of oxide- hydroxide forming elements (this group includes the elements: As, B, Ga, Ge, P, Se and Ti).
At reducing conditions, the behavior of the trace elements considered is complex, and no simple classification of the elements is possible.
CONTENTS
1. Introduction 116 2. Description of the Model 117
2.1. Model considerations 117 2.2. The program MINGTSYS 117 2.3. The database DGFDBASE 117
3. The Range of Parameters Used 118 3.1. The range of pressure, temperature and composition used I 18 3.2. The classification nomenclature 118 3.3. The basis of the calculations 119
4. An Example: the Equilibrium Distribution of Mercury 119 4.1. Chemical species encountered 119 4.2. Equilibrium distribution of Hg 119
4.2.1. The system Hg/O 119 4.2.2. The system Hg/O/CI 119 4.2.3. The equilibrium chemistry of Hg 119
5. Equilibrium Characterization of Coal Conversion Systems 120 5.1. A literature survey 120 5.2. Thermodynamic stable phases of trace elements 122
5.2.1. Arsenic 122 5.2.2. Boron 123 5.2.3. Beryllium 123 5.2.4. Cadmium 124 5.2.5. Cobalt 124 5.2.6. Chromium 125 5.2.7. Gallium 126 5.2.8. Germanium 128 5.2.9. Mercury 128 5.2.10. Nickel 128 5.2.11. Phosphorus 129 5.2.12. Lead 129 5.2.13. Antimony 131 5.2.14. Selenium 131 5.2.15. Tin 132 5.2.16. Titanium 133 5.2.17. Vanadium 133 5.2.18. Zinc 133
115

116 F. FRANDSEN et al.
6. Summary 7. Conclusion Acknowledgements References
134 136 137 137
I. INTRODUCTION
While the release of the main elements C, O, H, S, N building up the organic matrix of a coal and the behavior of the minor elements AI, Ca, Fe, K, Mg, Na and Si building up the inorganic mineral fraction of the coal, have been investigated over several years, the fate of trace elements in coal combustion is a relatively new subject.
A trace element is defined as an element, usually from one of the subgroups in the periodic table, occurring in coal in concentrations below 1000 ppmw. 1
Several of the trace elements are vaporized during pyrolysis and combustion or gasification and recondensed on the surface of fly ash particles, 2 ~3 during flue gas cooling. Full-scale measurements a4 21 have revealed significant amounts of certain trace elements (e.g. B, Hg and Se) in the flue gas leaving the stack, causing an undesired direct gaseous emis- sion of these trace elements, among which some are suspected of possessing toxicological effects toward the environment or to cause generic or biological changes in human beings. 1.21-23
In order to decrease the emission of trace element species, knowledge is needed about the behavior of trace elements in the combustion zone and the pen- etration of flue gas cleaning equipment by trace elements. Knowledge about the behavior of trace elements in the combustion zone is important when estimating the release of trace elements from a burning coal particle and the partitioning of trace elements between the gaseous and condensed phases generated during combustion. Regarding the penetra- tion of flue gas cleaning equipment, Clarke and Sioss 21 mention as an example that small amounts of trace elements in the vapor phase (such as Hg) can be removed by adsorption on ash collected in fabric filters.
A certain knowledge about the chemistry of trace elements in cooled flue gases is necessary to under- stand the phenomenon of transport of trace elements through modern coal-fired power plants. The aim of this review is to clarify the equilibrium chemistry of trace elements in oxidative as well as reductive flue gases.
Coal combustion is a very complex process involv- ing numerous homogeneous and heterogeneous reac- tions during the course of pyrolysis, char burnout and combustion of volatiles. Many of the reactions in coal combustion are known--or believed--to be kinetically controlled. Chemical kinetics deal with the rates of chemical reactions and the dependence of reaction rates on factors such as temperature and concentration.
Except for mercury, almost no kinetic data for the reactions of trace elements in hot flue gases are avail- able. Thus, as a first approach a global equilibrium analysis (GEA) is performed to pin-point thermo- dynamic stable chemical and physical forms of the trace elements as functions of temperature, pressure and total composition in a combustion system.
In GEA the composition of a system at a given pressure and temperature is calculated by minimizing the total Gibbs free energy of the system. The method used is outlined in Section 2. When the total Gibbs free energy is at a minimum, the system is in thermo- dynamic equilibrium, i.e., the system consists of ther- modynamic stable chemical species and phases. The word global is used, because only global parameters such as pressure, temperature and total composition are taken into account--no local conditions, e.g., pressure or temperature gradients, are considered. The system is at its final state, where all possible reactions--homogeneous and heterogeneous--have reached equilibrium.
When used on combustion or gasification systems GEA has several limitations. (1) In the furnace zone, where the temperature is
high (T > 1500 K in pulverized coal-fired sys- tems) the reaction rates may be high enough to reach equilibrium, provided that the residence time is long enough.
(2) In low-temperature (T < 800 K) zones of the com- bustion system, e.g., around an electrostatic pre- cipitator, the reaction rates may be too low to reach equilibrium, even for long residence times.
(3) In the furnace (flame) zone, mixing phenomena may introduce local conditions e.g., temperature and/or composition gradients not taken into account in the global equilibrium analysis.
(4) All relevant chemical species occurring in the real combustion system must be taken into account, otherwise the output from the GEA may be misleading.
Thus, when used on real combustion systems, care must be taken when interpreting the results of a GEA.
However, the equilibrium approach has been adopted by several investigators, but usually equilib- rium distributions for only a few trace elements at either reducing or oxidizing conditions are reported. In this study, equilibrium distributions are reported for the 18 trace elements: As, B, Be, Cd, Co, Cr, Ga, Ge, Hg, Ni, P, Pb, Sb, Se, Sn, Ti, V and Zn, which are among the elements of greatest concern with respect to coal utilization. 1~ Reducing and oxidizing conditions are considered and the results are com- pared qualitatively with results published in the literature.

Trace elements from coal combustion I 17
2. DE S CRIPTION OF THE M O D E L
2.1. Model Considerations
GEA has been applied for many years in order to understand the various processes in combustion sys- tems and numerous computer algorithms and thermo- dynamic databases have been developed for this pur- pose. The authors are familiar with the HSC Chemis- try Software 24 and ChemSage 25 minimization algo- rithms. A list of available metallurgical databases is given by Bale and Eriksson. a4 An introduction to this subject is given by Smith and Missen. a6
In the GEA model used in this study, temperature, pressure and total composition are specified. The gas is assumed ideal and all condensed phases are consid- ered pure (see Section 2.2). The presence of ash- forming elements, i.e., AI, Ca, Fe, K, Mg, Na and Si is not taken into account, because molten oxide systems, e.g., systems containing aluminosilicates are expected to behave highly nonideally and because only very few thermodynamic data have been found for chemical species containing both ash forming and trace elements. Babushkin et al. z7 give an intro- duction to the interaction between silicates (ceramics) and various metals, including AI, Co, Cr, Cu, Fe, Mn, Mo, Nb, Ni, Pb, Ta, Ti and V.
Comparison of the equilibrium distribution of a trace element in a system with and without the presence of other trace elements has showed almost similar results. The competition for oxygen is be- lieved to only be of importance in systems with air excess numbers close to 1. Thus, since the air excess number in this study has been set to 0.6 and 1.2 (see Section 3), the trace elements are considered individually.
2.2. The Program M I N G T S Y S
kth element, n~ is the number of moles of the ith chemical species and b~ is the number of atom masses of the kth element in the ith chemical species. A total mole-balance is given by:
N
nt = Z ni. (3) i = l
If G~ is set arbitrarily to zero for all elements in their standard states and if the standard states are chosen to be the pure components at 1 bar, then the equilibrium expression for component i is given by:
K
AG°yl + RTln(ai) + ~ 2kb~ k = 0, (4) k = l
where AG.~i is the change in Gibbs energy of forma- tion of component i and 2k is the Lagrange multiplica- tor for the kth element.
Equations (2), (3) and (4) make up a (N + K + 1) x (N + K + l)-system of nonlinear equations,
with N unknown n~ values, K unknown 2k values and one unknown n t value. An algorithm developed and described by Michelsen 29 is used to solve the non- linear equations.
For a system, the elemental composition, tempera- ture and pressure must be specified. The program enables the user to choose fuels with different com- positions and to specifiy a value of the air excess number,
For a fuel with the general main component molar composition C,HpSrNoO~.(H20)u the stoichiometric reaction between coal and oxygen from the air is given by: 3°
C~H~S~N~O.'(H 20)~(cr)
+ ( ~ + ~ + 7 + ~ - 2 ) O 2 ( g ) - - - ~ C O 2 ( g ' 4
The program MINGTSYS (a Fortran-77 compu- ter code) minimizes the total Gibbs free energy, Gt:
- n i \ R T + ln(ai) (1) R T i=1
of a mass-balance constrained system, using the method of undetermined Lagrangian multipliers. An introduction to this method is given by Smith and Missen 26 and by Greiner. 28
In Eq. (1) G symbolizes the Gibbs energy, super- script t denotes total, R is the universal gas constant, T is the absolute temperature, a i is the activity of component i, subscript f i denotes formation of com- ponent i and N is the total number of chemical species. In a system of N chemical species composed of K elements, a mass balance for the kth element is given by:
N nlblk -- Bk = 0, (2)
i=1
where Bk is the total number of atom masses of the
assuming all nitrogen in the coal is converted to nitric oxide. (g) denotes the gaseous state and (cr) denotes the crystalline state. The stoichiometric air requirement is given by:
4 2 2 Lm~, = , (6)
Y1(O2)
where Yl(O2) is the mole fraction of oxygen in air. The air excess number (2) used in MINGTSYS is now defined by:
L 2 - , (7)
Lmin
where L is the actual air supply.
2.3. The Database DGFDBASE
A database, DGFDBASE, has been developed in

118 F. FRANDSEN et al.
TABLE I. Composition (%(w/w)) of the subbituminous coal used in the equilibrium calculations performed in this work. C, O, H, S and N denotes the respective chemical elements; A denotes ash and W denotes water; 'dry' and 'wet' denote
bases of composition
Coal-rank Subbituminous
C(dry) 54.8 O(dry) 33.8 H(dry) 6.3 S(dry) 0.3 N(dry) 1.2 A(dry) 3.6 W(wet) 24.57
TABLE 2. Initial concentrations of trace elements and halo- gens considered in this study. The values for each element corresponds to a mean concentration of the element in
coal. Unit: ppmw unless otherwise stated--% (= %(w/w))
Element Concentration
Arsenic ! Boron 50 Beryllium 1 Cadmium 0.05 Cobalt 5 Chromium 30 Gallium 5 Germanium 5 Mercury 0.1 Nickel 25 Phosphorus 50 Lead 25 Antimony I Selenium 1 Tin 5 Titanium 0.05(%) Vanadium 25 Zinc 100 Bromine 25 Chlorine 300 Fluorine 100
ASCII codes for the program MINGTSYS. For the general reaction:
/)iS1 -1-/32S2 "~ . . . . . ~ V R SR-----~
I)R+ISR+ 1 ~- "'" "~ I)R+pSR+ P (8)
including a total of R + P reactants and products, the change in the Gibbs free energy caused by the reaction may be calculated from:
R+P AG~i = ~ vjG~, (9)
j+l
where v~ and Gfl are the stoichiometric coefficient and the Gibbs free energy for component j in Eq. (8), respectively, where vj is positive for products and negative for reactants.
If Eq. (8) is the reaction of formation of some chemical species X, Eq. (9) is the definition of the Gibbs free energy of formation of X, AG~x. The G~
values are generated by reducing values of the func- tion:
H o G F E j = - - ( - a J ° - - ~ 298"j ) (10)
(known as the Gibbs free energy (GFE) function in the literature 31) tabulated as a function of the temperature, using a conventional least-squares curve-fitting algorithm, and the fitting function:
G F E i ( T ) = A I + B i T + CIT 2 + DIT 3 + EIT a. ( l l )
The database DGFDBASE contains the polynomial coefficients for approximately 800 chemical species (sulfides, sulfates, carbonates, oxides, halides, and silicates) of 33 elements, calculated using the equa- tions outlined above. The thermochemical data 31-4° cover the temperature range 300-2000 K for both condensed and gaseous states (if data are available) and are cross-checked whenever possible.
3. THE RANGE OF PARAMETERS USED
In this section the range of parameters (pressure, temperature, composition and air excess) used in the calculations is described. The classification nomen- clature for trace element containing systems is de- scribed. Finally the chemical species considered in each specific system is outlined.
3. I. The Range of Pressure, Temperature and Composition Used
In this work equilibrium calculations have been performed with a total pressure of 1 atm and the temperature between 350 K and 2000 K, provided that thermodynamic data are available in the whole temperature range.
A subbituminous coal with the composition shown in Table 1 has been used as fuel and the air excess number (2) has been set to equal 0.6 (gasification) or 1.2 (combustion). The air is assumed to be composed of 78.5%(mol/mol) N2, 20.5%(mol/mol) 02 and l%(mol/mol) H20. Table 2 shows the initial concentration of the trace elements and halogens.
3.2. The Classification Nomenclature
The thermodynamic fate of the trace elements has been characterized in systems containing one trace element and the elements carbon (C), oxygen (O), hydrogen (H), sulfur (S) and nitrogen (N). In addi- tion, in some systems a halogen i.e., bromine (Br), chlorine (CI) or fluorine (F), is present.
The following nomenclature has been used to classify the systems:
X/CON/Y, (12)

Trace elements from coal combustion 119
TABLE 3. The content of Hg-containing chemical species in the database DGFDBASE. 'Upper temp.' denotes the upper limit of the temperature range where data are available for the actual chemical species: g is the gaseous state; cr is the
crystalline state and cr, l is the condensed state
Gaseous Condensed
Species Upper temp. Species Upper temp.
Hg(g) 2000 K Hg(cr, l) 2000 K HgBr(g) 2000 K HgBr2(cr, I ) 1500 K HgBr2(g) 2000 K Hg2Br2(cr ) 1500 K HgCl(g) 2000 K HgCl2(cr,l ) 1500 K HgCI2(g ) 2000 K Hg2Cl2(cr ) 1500 K HgF(g) 2000 K HgF2(cr,l ) 2000 K HgF2(g ) 2000 K Hg2F2(cr) 1500 K HgH(g) 2000 K HgO(cr) 1000 K HgO(g) 2000 K HgS(cr) 1100 K HgS(g) 2000 K HgSe(cr) 1000 K HgSe(g) 2000 K HgSO4(cr ) 800 K
where X is the chemical symbol of a trace element (e.g., As), CON is a redox indicator and Y is the chemical symbol of a halogen, Y ~ {Br,CI,F}. If CON = O the system is oxidizing, 2 = 1.2 (combustion), if CON = R the system is reducing, 2 = 0.6 (gasification).
As an example Hg/O symbolizes an oxidizing system with Hg present in addition to C, O, H, S and N, while As/R/C1 symbolizes a reducing system with As and CI present in addition to C, O, H, S and N.
3.3. The Basis of the Calculations
In oxidizing systems (2 = 1.2) the following gase- ous combustion products are taken into account: H2, N2, 02, CO, CO2, COS, H20, H2S, NH3, NO, NO 2, SO 2, SO3.
In the case of a reducing system (2 = 0.6), C(cr) is added to the list. If a halogen, Y, is present, the compounds Y2(g) and HY(g), Y e {Br, CI,F} are also taken into account.
4. AN EXAMPLE: THE EQUILIBRIUM DISTRIBUTION OF MERCURY
As an example this section contains a description of the equilibrium distribution of Hg in combustion flue gases. A list of the chemical species considered is given, in addition to plots showing the equilibrium distribution of mercury as a function of the tempera- ture. The results obtained are interpreted by suggest- ing a number of equilibrium reactions important in the system. Finally, two examples of the numerical accuracy encountered are given.
chemical species are considered in addition to the list of classical combustion products given in Section 3.3: Hg(g), HgH(g), HgO(g), HgS(g), Hg(cr,l), HgO(cr), HgS(cr) and HgSO4(cr), where (or,I) de- notes the condensed state.
If chlorine also is present, e.g., in the system Hg/O/CI, the following chemical species are included: HgCl(g), HgCI2(g), HgDl:(cr,1) and Hg2Cl~(cr ).
A chemical species is only considered within the temperature range where thermochemical data are available, e.g., HgSO4(cr) is only taken into account in the temperature range 350-800 K. No extrapola- tion of data has been made.
4.2. Equilibrium Distribution of rig
Below the different equilibrium distributions en- countered in the oxidative systems Hg/O (no access to chlorine) and Hg/O/CI (access to clorine) are outlined. At reducing conditions only elemental gase- ous mercury, Hg(g) is formed.
4.2.1. The system Hg/O
The equilibrium distribution of Hg in the system Hg/O, at standard oxidizing conditions, is shown in Fig. 1.
Crystalline mercury(II)sulfate, HgSO4(cr) is stable up to 540 K. Above 590 K an equilibrium between Hg(g) and HgO(g) exists, with Hg(g) carrying more than 90%(mol/mol) of the mercury present. The equi- librium between Hg(g) and HgO(g) is gradually shifted towards Hg(g) with increasing temperatures. If no HgSO4(cr) is allowed to form, HgO(cr) is the stable form of mercury at low temperatures (T < 440 K).
4.2.2. The system Hg/O/CI
If chlorine is present in addition to the elements C, O, H, S, N and Hg, at standard oxidizing conditions, the equilibrium distribution shown in Fig. 2 is obtained.
The crystalline mercury(II)sulfate, HgSO4(cr), is stable up to 380 K. Between 380 K and 700 K gase- ous mercury(II)chloride is stable. At 650 K formation of Hg(g) begins. Above 700 K small amounts of HgO(g) are formed, reaching a maximum occurrence around 800 K. Above 950 K an equilibrium between Hg(g) and HgO(g), with elemental gaseous mercury, Hg(g), as the major stable form, is shifted gradually towards Hg(g) with increasing temperatures. If HgSOa(cr) is not considered in the calculation, HgCI2(g ) is the stable form of mercury below 650 K.
4.1. Chemical Species Encountered
The Hg-carrying chemical species contained in the database DGFDBASE, are shown in Table 3.
In the systems Hg/O and Hg/R the following
4.2.3. The equilibrium chemistry of rig
At equilibrium, the reactions of mercury with com- bustion flue gases produce three gaseous forms: HgCi z, HgO and Hg. In addition, HgSO4 is stable at

120 F. FRANDSEN et al.
HgSO4(cr) Hg(g) oo
03 I
- ~ -
n
~ HgO(g) 0 , i , i , = , i , i , i ,
35o 4~o sso 6so 7so 85o 9so Temperature (K)
FIG. 1. Equilibrium distribution of Hg (%(moles Hg/moles Hg total)) at standard oxidizing conditions (in the Hg/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and cHg,o =
1).2 ppmw.
r , o
E L C , q
I
*6
HgCl2(g)
Q )
( 3 -
HgSO4(cr) . ~
3 5 0 ' , , i , i , 450 550 650 750 850
Temperature (K)
Hg(g)
_ (g) 9 5 0
FIG. 2. Equilibrium distribution of Hg (%(moles Hg/moles Hg total)) at standard oxidizing conditions (in the Hg/O/Cl system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2, c.g.o = 0.2
ppmw and CcLo = 300 ppmw.
low temperatures. The results of the equilibrium calculations performed are interpreted below, and important equilibrium reactions are suggested.
In the Hg/O system at standard oxidizing condi- tions, by lowering the temperature, HgSO,(cr) is formed around 590 K by the reaction:
HgO(g) + SO2(g ) + "~)2(g) ~ HgSO,(cr). (13)
If HgSO4(cr) is not considered in the calculation, crystalline mercury oxide will form by condensation of HgO(g):
HgO(g) ~=- HgO(cr). (14)
Above 590 K HgO(g) gradually decomposes with increasing temperatures, according to the reaction:
HgO(g) ~=- Hg(g) + ½02(g ). (15)
In the Hg/O/Cl system at oxidizing conditions, by lowering the temperature, HgSO4(cr) is formed by the reaction:
HgCl2(g) + SO2(g) + O2(g) -,~ HgSO#(cr) + Cl2(g ). (16)
At higher temperatures HgClE(g ) reacts with H20(g):
HgCl2(g ) + H20(g) ~- HgO(g) + 2HCl(g). (17)
Above 950 K the equilibrium reaction (Eq. (15)) is established and gradually shifts towards the right with increasing temperature.
5. EQUILIBRIUM CHARACTERIZATION OF COAL CONVERSION SYSTEMS
5.1. A L i tera ture Survey
The idea of pin-pointing the stable phases of minor and trace elements by thermodynamic calculations has been adopted by several authors. Table 4 gives a list of major thermodynamic studies in the last 15 years. The bases of some of the studies are described below and the results are compared with the authors' calculations in Section 5.2.
In order to study the release of trace elements in fluidized-bed combustion systems, Alvin et aL 42 per- formed thermodynamic calculations of the equilib- rium distribution of 14 elements, for operating condi- tions corresponding to atmospheric and pressurized fluidized-bed combustion systems. The thermody- namic projections were calculated at equilibrium at 10, 100 and 3009/o excess oxygen, with the total pressure varying between 1 and 10 atm. Each projec- tion was carried out for a 4 ~ sulfur content coal, with a steady-state SOx value in the effluent corre- sponding to 0, 90 and 99~o S removal. The tempera- ture range was 300-1200 K,
Kalfadelis and Magee 43 performed equilibrium cal- culations on trace element containing systems under reducing (gasification) conditions, and have com- pared the results with experimental results from the industrial COED-gasification process. 189 chemical species of 21 elements were considered.
Alvin 44 performed an extensive study on minor and trace element reactions in fluidized-bed combustion processes. The potential volatility and interactions of 26 minor and trace elements with clay (AI,Si) and carbonate (Ca,Mg) constituents in the coal and sorb- ent (for desulfurization) are parametrically investi- gated on the basis of thermodynamic equilibrium calculations. Operation ranges of different fluidized- bed combustor designs were used and several import- ant reactions involving trace elements were suggested.
Moberg et al. 45 made a basic study of the thermo- dynamic behavior of trace elements during the com- bustion of coal. Calculations were performed with a model coal and 20~ excess air. The total pressure

Trace elements from coal combustion 121
TABLE 4. Survey of thermodynamic studies of the behavior of minor and trace elements in combustion systems. In addition to the elements shown, the main elements (C, H, O, S, N) and the halogens (Br, CI, F) are implemented in
most of the studies
Elements considered: Reference:
AI,B,Be,Co,Cr,Fe,Hg, K,Mn,Mo,Na,Ni,Pb,Ti As,B,Ba,Be,Cd,Co,Cr, Cu,Ge,Hg, Mn,Mo,Ni,P, Pb,Sb,Se,Sn,U,V,Zn AI,As,B,Be,Cd,Co,Cr, Cu,Fe,Ga,Ge,Hg,K,Mn, Mo,Na,Ni,Pb,Sb,Se, Si,Sn,Ti,V,Zn,Zr AI,As,Ca,Cd,Fe,Hg,Pb, Se,Si,Zn Cd,Hg, Pb,Zn AI,B,Ca,Fe,Mg, Si AI,Ba,Be,Ca,Fe,Mg,Si, Sr AI,Ca,Fe,Mg,P,Pb,Si AI,As,B,Ba,Bi,Ca,Cd, Co,Cr,Cu,Fe,Ga,Ge,Hg,
Alvin e t al. (1977) 42
Kalfadelis and Magee (1977) 43
Alvin (1982) 44
Moberg et al. (1982) 45 Mojtahedi et al. (1987) 46 Malaykh et al. (I 988) 47
Malykh e t al. (1988) 4a Shpirt e t al. (1988) 49
K,Mg, Mn,Mo,Na,P,Pb,Sb, Se,Si,Sn,Ti,V,Zn,Zr METCR (1989) s° AI,As,Ca,Fe,Si,Se Malykh and Pertsikov (1990)5 t Hg Nordin e t al. (1990) 52 Hg, Se Von Fahlke (1992) ~a
was 1 atm and the temperature was varied between 400 and 2300 K. The gas phase was assumed to be ideal and the condensed phases were considered as pure phases. A total of 17 elements building up 500 chemical species, were involved.
Mojtahedi e t a l . 46 investigated thermodynamic equilibrium distributions of the trace elements Cd, Hg, Pb, and Zn, under the conditions of atmospheric and pressurized fluidized-bed combustion and gasifi- cation. Calculations were performed at temperatures from 500 K to 1600 K, at pressures between 1 and 10 atm, with the excess air ratio (2) 0.5 and 1.3 and with a high-sulfur coal (4.7~o(W/W)) and a low-sulfur coal (1.1~(w/w)). Altogether 100 chemical species (67 gaseous and 33 condensed) of the four trace elements were considered.
Malykh e t a l . 47 calculated the concentrations of fluorine, chlorine and boron compounds in waste gases from coal-processing operations. Combustion, gasification and coking processes were considered. Combustion was considered with the air excess number equal to 1.0 and i.2 for a pressure of 0.1 MPa and at temperatures between 700 K and 1700 K. Gasification was considered at a pressure P = 11 atm, and the temperature varying between 900 K and 1500 K. The initial composition for the gasifica- tion process was calculated assuming the formation of CO, H z and CH4 from the carbon and hydrogen of organic matter in the coal. Coking was regarded as a process of heating the coal without the access of air at P = 1 atm, and with the temperatures rising from 900 K to 1700 K.
Malykh e t a l . 4a performed thermodynamic equilib-
rium calculations of coal combustion products, with the aim of finding the stable chemical forms of the trace elements Ba, Be and Sr and their phase distribu- tion. The thermodynamic system consisted of 367 chemical species--85 in the condensed phase and 282 in the gas phase---of 15 elements. The computer code used provided the possibility of imposing definite conditions of the yield of final products, i.e., of modeling the combustion process not only with re- spect to its initial but also to its final parameters. Two models were considered: one in which the condensed compounds represent individual phases and the other in which the condensed phases form an ideal solution.
Shpirt e t al . 49 investigated the behavior of lead in the high-temperature oxidation of coals. Their calcu- lations were performed for temperatures of 473- 2273 K at a pressure of 0.1 MPa with 2 = 1.0 and 2 = i.2. Two model coals with different composi- tions were used. Initial concentrations of 20 ppmw lead coal and 200 ppmw lead coal, respectively, were used. The calculations took into account 96 com- pounds in the gas phase and 86 condensed phases of 13 elements. The condensed phases were treated both as individual phases and as ideal solutions.
At the Morgantown Energy Technology Center (METC), a project with the aim of determining the forms and relative abundances of trace inorganic vapor species during the combustion and gasification of coal was carried out in the late 1980s. The final report of this project was published in 1989 (see METCRS°). Experimental studies were performed using mass spectrometry on each coal sample at low temperature and pressure. Vapor species were identi- fied and their abundances were measured over the temperature range 300-1700 K. Coincident with the experimental investigations, trace element vaporiza- tion behavior was evaluated by Gibbs energy minimi- zation calculations. A database including thermo- chemical data for approximately 400 pure condensed, solution and gaseous components of 29 minor and trace elements was used. Calculations were per- formed with the pressure between 10 25 and 1 atm, the temperature between 300 K and 1800 K, and under the assumption that the possible chemical phases included a gas, in addition to single-compo- nent solid phases and a liquid-solution mixture.
Malykh and Pertsikov 51 performed thermodyanic studies of the fate of the trace elements Se and As, during combustion, coking and gasification. The ob- jective was to identify chemical compounds of sele- nium and arsenic in an equilibrium composition of a complex multicomponent system. Coal combustion was analyzed in the temperature range 700-1700 K, at a pressure of 1 atm and coking in the temperature range 700-1500 K, at a pressure of 1 atm. The coal gasification model assumed a mixture of air and water with transition of the organic coal components into CO, H 2 and CH4 at i I atm. The temperature was varied between 700 K and 1700 K. A total of 168 gaseous and 45 condensed phases were considered.

122 F. FR^NO~ et aL
J
As20s(cr)
40e(g)
800 9(30 Temperoture (K)
AsO(g)
Ck
U3 L
q)
(D cl
700 1000
FIG. 3. Equilibrium distribution of As (%(moles As/moles As total)) at standard oxidizing conditions (in the As/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: ~ = 1.2 and c^s,o = I
ppmw.
oo
q)
LD I b3o < , n
q) EL
As2S2(cr,I) AsO(g)
C350 ' 5~0 750 950 1150 Temperature (K)
FIe. 4. Equilibrium distribution of As (%(moles As/moles As total)) at standard reducing conditions (in the As/R system) as a function of the temperature, in a flue gas from combustion of a subbituminons coal: I = 0.6 and cA,.o = 1
ppmw.
In addition to the list of major thermodynamic studies given in Table 4, results of equilibrium calcula- tions for waste incineration processes have been pre- sented recently by Wu and Biswas s4 and Linak and Wendt. 5 s
5.2. Thermodynamic Stable Phases o f Trace Elements
In this section, results of the equilibrium characteri- zations performed in this work are presented and compared with results of the earlier studies. For some of the oxidizing systems, important reactions for the interpretation of the results are discussed.
5.2.1. Arsenic
At standard oxidizing conditions (in the As/O
system) arsenic is present as As2Os(cr) at tempera- tures up to 750 K (Fig. 3). Between 750 K and 900 K, As,Os(cr), As406(g) and AsO(g) coexist. As2Os(cr) is the dominant species between 750 K and 800 K, As406(g) is dominant between 800 K and 830 K and AsO(g) is dominant between 830 K and 900 K. Above 900 K only AsO(g) is present in significant amounts.
Moberg et al. 45 found As2Os(cr) to be the stable form of arsenic below 570 K. Between 570 K and 800 K arsenic was present as AsF3(g), and above 800 K as As406(g), As2(g ) and As(g), the latter being the main stable compound above 1800 K. At tempera- tures above 1800 K, small amounts of AsS(g) were formed in addition.
Malykh and Pertsikov 51 showed theoretically that during coal combustion above 1300 K all arsenic is reduced to AsO(cr) and sublimated. At temperatures between 800 K and 1200 K, the equilibrium form of arsenic was As204(cr), In addition, small amounts of AsF3(g) were formed with a maximum occurrence at 900 K.
In the present work no As204(cr) and AsF3(g) were formed in the As/O/F system, using two differ- ent sets of thermodynamic data for arsenic com- pounds, Barin 31 and Knacke et al. 35 If HF(g) was removed from the list of chemical species specified in the input to the program MINGTSYS, significant amounts of AsFs(g), but no AsF3(g), were formed. The lack of formation of AsF3(g) in our calculations may be explained by the fact that crystalline calcium fluoride, CaF2(cr), is not included in the As/O/F system. The formation of CaF2(cr) may change the equilibrium distribution of fluoride significantly. 47
Thus, in order to investigate the influence of CaF2(cr) formation on the equilibrium distribution of As in coal combustion flue gases, and the possible interaction between chemical species of As and Ca, a calculation was performed for the As/Ca/O/F system. It was shown that crystalline calcium arsenate, Ca3(AsO4)2(cr) was the major stable As species at temperatures up to 1270 K. Above 1270 K AsO(g) was the major stable form of As. Fluorine was present as CaF2(cr) below 730 K, and HF(g) above 730 K.
At standard reducing conditions (in the As/R system) arsenic is present as As2S2(cr,l) up to 550 K (Fig. 4). Between 550 K and 700 K As4(g) is the major stable form of arsenic. Above 700 K AsO(g) is the major stable form. In addition, between 550 K and 950 K Ase(g) and AsH3(g ) are formed, with a maximum occurrence of 13-14%(mol/mol) of the arsenic present as As2(g) and 2%(mol/mol) present as AsH3(g), at 750 K.
Malykh and Pertsikov 51 reported As4(g) to be common in reductive combustion flue gases at low temperatures. At temperatures above 1300 K arsenic appears as As2(g). The share of AsH3(g ), As(g) and AsO(g) in equilibrium products amounted to less than l%(mol/mol) of the total arsenic content. A similar pattern was observed during the course of

HBO2(cr)
H3BO3(g)
Q _
( / 3
I m~
t D
2 0 )
O._
HBOa(g)
BeSO4(a,cr)
o ~ 02(g) ~oo 8oo ~3oo 18oo
Temperature (K)
FIG. 5. Equilibrium distribution of B (~(moles B/moles B total)) at standard oxidizing conditions (in the B/O system) as a function of the temperature, in a flue gas from combus- tion ofa subbituminous coal: 2 = 1.2 and cB, o = 50 ppmw.
o o
(J 0 CL
CO
E) o OB~
EL
~ 5 0 ' , r , i . . . . i ,
850 1350 1850 Temperature (K)
BeO(cr,I) Be(OH)2lg) f
Trace elements from coal combustion 123
FIG. 6. Equilibrium distribution of Be (%(moles Be/moles Be total)) at standard oxidizing conditions (in the Be/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: :. = 1.2 and ea=,o =
1 ppmw.
coking; the temperature intervals of the existence of the various forms of arsenic coincide completely.
5.2.2. Boron
At standard oxidizing conditions (in the B/O system) boron exists as HBO2(cr) below 400 K, and as H3BO3(g ) up to 900 K (Fig. 5). Between 900 K and 1800 K H3BO3(g) progressively decomposes as the temperature is increased, forming HBO2(g) as the major boron carrying chemical species above 1300 K. Only small amounts ( < 1 ~(mol/mol) of the B present) of H3B306(g ) and BO2(g ) are formed; the first be- tween 350 K and 800 K, the latter above 1800 K.
Alvin 44 reported the formation of HBO2(cr) and HaBO3(cr ) between 300 K and 500 K, unaffected by changes in the sulfur content of the combustion gas
and variations in the air excess number above 1. Between 400 K and 1200 K HaBOa(g) was the major volatile phase. Above 1200 K HBO2(g ) was formed due to the release of water from H3BO3(g ).
Malykh et al. a7 found boron to be present mainly as H3B306(g) at low temperatures and as H3BO3(g) at high temperatures, where HBO2(g) and BO2(g) were also formed.
At standard reducing conditions (in the B/R system) the equilibrium distribution of boron is almost equal to that occurring at oxidizing condi- tions, with two exceptions. (1) The temperature range of stability of the minor
volatile compound H3BaOr(g ) is 700-1200K, compared with the range 350-800 K in oxidizing systems.
(2) No BO2(g ) is formed under reducing conditions. Kalfadelis and Magee .3 found H3BO3(g ) and
HBO2(g) to be the only boron carrying chemical species present at equilibrium at 1273 K and 1.5 atm in the COED-gasification process.
5.2.3. Beryll ium
At standard oxidizing conditions (in the Be/O system) the stable form of beryllium is ct-BeSO4(cr) up to 760 K, where BeO(cr,1) is formed (Fig. 6). Between 760K and 1240K the stable form is changed from BeO(cr, l) to Be(OH)2(g), being the only stable Be component above 1240 K.
If chlorine is present in the Be/O/CI system small amounts of BeCl2(g ) are formed. Alvin 44 found the same equilibrium distribution of beryllium. In addi- tion, it was found that if the SO=(g) concentration in a flue gas is very low (due to e.g., a high sulfur removal efficiency in a fluidized-bed), BeO(cr,l), rather than ~t-BeSO4(cr) is the stable form of beryl- lium in the low-temperature region.
Using a thermodynamic model with condensed compounds representing individual phases, Malykh et al. 48 found BeO(cr) to be the stable form of beryllium below I000 K. Above 1100 K, the equi- librium form of beryllium was BeO.Al203(cr); with a rise in temperature a significant amount of Be(OH)2(g) was formed.
In summary, at low temperatures, Be(cr) reacts with O2(g ) and SOx(g) (if present) to form the stable crystalline beryllium sulfate, BeSO4(cr). At tempera- tures between 700 and 800 K BeSO4(cr) dissociates to form BeO(cr,l):
BeSOa(cr) ~- BeO(cr,l) + SO2(g) + "~)2(g). (18)
Above 1100 K BeO(cr,I) reacts with gaseous water, H20(g), forming the stable Be(OH)2(g):
BeO(cr,1) + H20(g ) ~ Be(OH)2(g). (19)
Independent of the fuel composition and the air excess number, additional gaseous interactions be- tween Be(OH)2(g) and HCi(g) may occur, forming small amounts of BeCl2(g).

124 F. FrtANDSEN et al.
CdSO4(cr) o_
"6 (D_
( / 3
I q : ) o
~J
Cd(g)
C d O ( g ) 3 . . . . i . . . . , . . . . , ,
5 0 8 5 0 1 3 5 0 1 8 5 0
l Temperoture (K)
FIG. 7. Equilibrium distribution of Cd (~o(moles Cd/moles Cd total)) at standard oxidizing conditions (in the Cd/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and Ccd.o =
0.05 ppmw.
o CdSO,(cr) CdCI2(g}
I ,
i
{ 0 (D
I - 0 o ( _ ) t o -
® I! OJ
13_
o , ~ ¢ , 3 5 0
Cd(g)
~ _ ~ _ CdO(g) , . j . . . . j ,
8 5 0 1 . 5 5 O 1 8 5 0
Temperoture (K)
FIG. 8. Equilibrium distribution of Cd (~o(moles Cd/moles Cd total)) at standard oxidizing conditions (in the Cd/O/CI system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and Ccd,o =
0.05 ppmw and CcL o = 300 ppmw.
At standard reducing conditions (in the Be/R system) BeO(cr,l) is the stable form of beryllium below 950 K, while Be(OH)2(g) is the stable form above 1240 K. Between 950 K and 1250 K the stable form of beryllium changes from BeO(cr,l) to Be(OH)2(g) with increasing temperature, probably due to an equilibrium reaction between BeO(cr,l) and H20(g) as shown above.
If fluorine is present at reducing conditions (in the Be/R/F system) BeF2(cr,i) is stable at temperatures up to 380 K. Above 380 K, the equilibrium distribu- tion of beryllium is equal to that in a reducing system with no fluorine present.
Kalfadelis and Magee 43 found Be(OH)2(g ) to be the only beryllium compound present at equilibrium in the COED-gasification process (1273K and 1.5 atm).
5.2.4. C a d m i u m
At standard oxidizing conditions (in the Cd/O system) cadmium forms CdSO4(cr) at temperatures up to 950K (Fig. 7). Above 1000K Cd(g) and CdO(g) are the stable forms, Cd(g) accounting for more than 90%(mol/mol) of the Cd present at all temperatures. CdO(g) gradually decomposes when the temperature is raised above 950 K, forming Cd(g) and O2(g). If chlorine is present at oxidizing condi- tions (in the Cd/O/CI system), CdCl2(g ) is the major stable form of cadmium between 850 K and 1150 K (Fig. 8). CdSO4(cr) below 850 K and Cd(g) above 1150 K are the major stable forms of cadmium cover- ing the rest of the temperature range, In addition, CdO(g) is formed above 1050 K in small amounts ( < 5%(mol/mol) of the cadmium present).
Alvin 44 found almost the same equilibrium distribu- tion of cadmium, CdSO4(cr) being stable at tempera- tures a little higher than found in this study (probably due to a higher content of cadmium).
Studying the atmospheric fluidized-bed combus- tion of a low-sulfur coal, Mojtahedi et al. 46 found CdSO,(cr) to be stable up to 1000 K. Above 1100 K only Cd(g) is present. No formation of CdO(g) and CdCl2(g ) is reported (CdCl2(g) was not included in their calculations).
In summary, cadmium exists as CdSO4(cr) in com- bustion flue gases at temperatures below 950 K. At temperatures between 950 K and 1000 K, CdSO4(cr) dissociates:
CdSO4(cr) ~-- CdO(g) + SO2(g) + ½02(g) (20)
forming CdO(g). Dissociation of CdO(g) at tempera- tures above 1000 K follows the reaction:
CdO(g) ~- Cd(g) + ~02(g ). (21)
If chlorine is present, the distribution of Cd among Cd(g), CdO(g), and CdCl2(g) is governed by the reaction:
CdC12(g) + H20(g) ~ CdO(g) + 2HCI(g) (22)
simultaneous with the dissociation of CdO(g). At standard reducing conditions (in the Cd/R
system) CdS(cr) is stable at temperatures up to 650 K. Above 650 K only Cd(g) is formed. No hal- ides of cadmium are formed at reducing conditions.
Kalfadelis and Magee a3 found Cd(g) to be the only cadmium compound present at equilibrium in the COED-gasification process (1273 K and 1.5 atm).
When modeling atmospheric fluidized-bed gasifica- tion of a low-sulfur coal, Mojtahedi et al. 46 found CdS(cr) to be stable at temperatures up to 700 K: above 700 K only Cd(g) was formed.
5.2.5. Cobal t
At standard oxidizing conditions (in the Co/O system) cobalt forms a homologous series of hy-
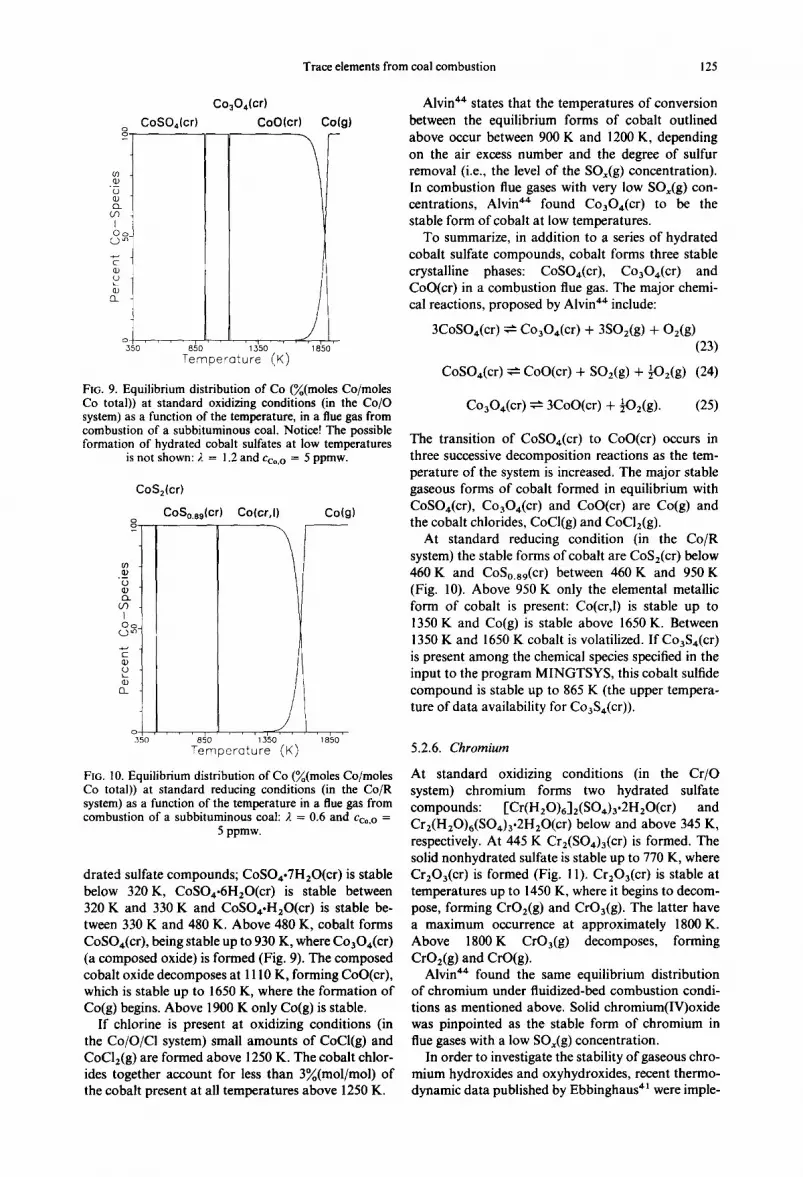
Trace elements from coal combustion 125
CoSO,(cr)
© Q _
O ' 3 I
O o
(D L )
L o , , , i 3 5 0 8 5 0
CozO4(cr) CoO(cr)
/ . . . . , , , , ~
1 . .350 1 8 5 0
Temperoture (K)
Co(g)
FIG. 9. Equilibrium distribution of Co (~(moles Co/moles Co total)) at standard oxidizing conditions (in the Co/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal. Notice! The possible formation of hydrated cobalt sulfates at low temperatures
is not shown: 2 = 1.2 and Cco.o = 5 ppmw.
CoS2(cr)
CoSo.a9lcrl
03
(1) Q . .
O 3 I
O o I C . D ~
~ 5 0 ' '
Co(cr,I) Co(g)
5 , - ~ , , i , '8~o ' ' ~5o ~sso
Temperoture (K)
Fro, I0. Equilibrium distribution of Co (%(moles Co/moles Co total)) at standard reducing conditions (in the Co/R system) as a function of the temperature in a flue gas from combustion of a subbituminous coal: 2 = 0.6 and Cco.o =
5 ppmw.
drated sulfate compounds; CoSO4.7H20(cr) is stable below 320 K, CoSO~.6H20(cr) is stable between 320 K and 330 K and CoSO4.H20(cr ) is stable be- tween 330 K and 480 K. Above 480 K, cobalt forms CoSO4(cr), being stable up to 930 K, where CoaO4(cr) (a composed oxide) is formed (Fig. 9). The composed cobalt oxide decomposes at 11 l0 K, forming CoO(cr), which is stable up to 1650 K, where the formation of Co(g) begins. Above 1900 K only Co(g) is stable.
If chlorine is present at oxidizing conditions (in the Co/O/CI system) small amounts of CoCi(g) and CoCl2(g ) are formed above 1250 K. The cobalt chlor- ides together account for less than 3%(mol/mol) of the cobalt present at all temperatures above 1250 K.
Alvin 4. states that the temperatures of conversion between the equilibrium forms of cobalt outlined above occur between 900 K and 1200 K, depending on the air excess number and the degree of sulfur removal (i.e., the level of the SOx(g) concentration). In combustion flue gases with very low SOx(g) con- centrations, Alvin 4. found Co304(cr) to be the stable form of cobalt at low temperatures.
To summarize, in addition to a series of hydrated cobalt sulfate compounds, cobalt forms three stable crystalline phases: CoSO,(cr), Co304(cr ) and CoO(c0 in a combustion flue gas. The major chemi- cal reactions, proposed by Alvin *4 include:
3CoSO4(cr) ~ Co304(cr ) + 3SO2(g) + O2(g) (23)
CoS04(cr) ~- CoO(cr) + S02(g) + -~Oz(g) (24)
Co304(cr) ~ 3CoO(cr) + ~O2(g). (25)
The transition of CoSO4(cr) to CoO(cr) occurs in three successive decomposition reactions as the tem- perature of the system is increased. The major stable gaseous forms of cobalt formed in equilibrium with CoSO4(cr), Co304(cr) and CoO(cr) are Co(g) and the cobalt chlorides, CoCl(g) and CoCl2(g).
At standard reducing condition (in the Co/R system) the stable forms of cobalt are CoS2(cr) below 460 K and CoSo.89(cr) between 460 K and 950 K (Fig. 10). Above 950 K only the elemental metallic form of cobalt is present: Co(cr,l) is stable up to 1350 K and Co(g) is stable above 1650 K. Between 1350 K and 1650 K cobalt is volatilized. If Co3S4(cr ) is present among the chemical species specified in the input to the program MINGTSYS, this cobalt sulfide compound is stable up to 865 K (the upper tempera- ture of data availability for Co3S4(cr)).
5.2.6. Chromium
At standard oxidizing conditions (in the Cr/O system) chromium forms two hydrated sulfate compounds: [Cr(H20)6]2(SO4)3.2HzO(cr) and Cr2(H20)6(SO~)3.2HzO(cr ) below and above 345 K, respectively. At 445 K Cr2(SO4)3(cr) is formed. The solid nonhydrated sulfate is stable up to 770 K, where Cr203(cr ) is formed (Fig. 11). CrzO3(cr) is stable at temperatures up to 1450 K, where it begins to decom- pose, forming CrO2(g) and CrO3(g). The latter have a maximum occurrence at approximately 1800 K. Above 1800 K CrO3(g) decomposes, forming CrO2(g) and CrO(g).
Alvin 44 found the same equilibrium distribution of chromium under fluidized-bed combustion condi- tions as mentioned above. Solid chromium(IV)oxide was pinpointed as the stable form of chromium in flue gases with a low SOx(g) concentration.
In order to investigate the stability of gaseous chro- mium hydroxides and oxyhydroxides, recent thermo- dynamic data published by Ebbinghaus 41 were imple-

126 F. FR^NDSEN etal.
0)
EL U3
I
O , n
q.)
[3_
~ 5 0 ' r
Cr2(SO,)a(cr) Cr2Oa(cr)
\ /
B50 1350 1850
Temperature (K)
CrO2(g)
CrOa(g)
CrO(g)
FIG. 11. Equilibrium distribution of Cr (%(moles Cr/moles Cr total)) at standard reducing conditions (in the Co/O system) as a function of the temperature, in a flue gas from combustion ofa subbituminous coal: The possible formation of hydrated chromium sulfates at low temperatures is not
shown: 2 = 1.2 and Cc,.o = 30 ppmw.
Cr2(SO4)a(cr) CrO2(OH)2(g) oo
I ~ 0 r 2 0 3 (
%0 . . . . 9~o' 1,oo ' ' '19bo' Temperature (K)
FIG. 12. Recalculated equilibrium distribution of Cr (%(moles Cr/moles Cr total)) at standard oxidizing condi- tions in the system Cr/O, with recent thermodynamic data from Ebbinghaus 41 implemented in the DGFDBASE
database.
mented in DGFDBASE and the minimization of Gibbs free energy in the system Cr/O was repeated.
It was shown that Cr2(SO4)3(cr), was stable up to 780 K (Fig. 12). At temperatures between 780 K and 1400 K CrOz(OH)2(g ) is the major stable form of chromium. Above 1400 K several gaseous chromium oxyhydroxides were formed, inlcuding CrO(OH)(g), CrO2(OH)(g) and CrO(OH)2(g) as the major chemi- cal species. Cr203(cr) is stable in a narrow tempera- ture range above 780 K, the dissociation temperature of Cr2(SO4)a(cr ).
In summary, chromium forms a series of hydrated sulfate compounds possessing a complex structure at low temperatures. At 445 K crystalline nonhydrated
q) EL
GO I
© o
©
(D [3_
o. 1200
Ga2Oz(cr)
\
17'00 Temperature (K)
Ga20(g) ~ GaO(g)
[ ~ Ga(g)
FIG. 13. Equilibrium distribution of Ga (%(moles Ga/moles Ga total)) at standard oxidizing conditions (in the Ga/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and c~=.o =
5 ppmw,
chromium sulfate is formed. This sulfate compound decomposes at 770 K:
Cr2(SO¢)a(cr) ~-~ CrzO~(cr) + 3SO2(g) + 3/202(g) (26)
forming crystalline chromium(III)oxide. Above 780 K several gaseous chromium species are stable.
At standard reducing conditions (in the Cr/R system) Cr2Oa(cr ) is the stable form of chromium up to 1800 K, where formation of CrOE(g), CrO(g) and Cr(g) begins. At 2000 K nearly 50%(mol/mol) of the chromium is present as Cr2Oa(cr), the rest is primarily bonded in the gaseous oxides and as elemen- tal gaseous chromium.
Kalfadelis and Magee 43 found Cr203(cr ) to be the only chromium-containing chemical species present at equilibrium at 1273 K and 1.5 atm in the COED- gasification process.
5.2.7. Gallium
At standard oxidizing conditions (in the Ga/O system) Ga203(cr ) is stable up to 1600 K, where form- ation of Ga20(g) and GaO(g) begins (Fig. 13). The relative concentration of Ga20(g) increases with in- creasing temperature up to 1800 K, where it begins to decompose forming GaO(g) and Ga(g). If chlorine is present at standard oxidizing conditions (in the Ga/O/CI system), formation of GaCl(g), at the ex- pense of Ga(g), GaO(g) and Ga20(g) begins at 1400 K (Fig. 14). The amounts of the gaseous oxides of gallium, GaO(g) and Ga20(g), formed in the Ga/O/CI system are decreased compared with the amounts formed in the Ga/O system (with no chlorine access). Above 1700 K GaCl(g) is the domi- nant species in the Ga/O/C1 system.
Alvin 44 found Ga203(cr ) to be the major stable compound of gallium in oxidative ltuidized-bed com- bustion processes. Less than I% of the initial feed-

Trace elements from coal combustion 127
O
E). LO I Oo
q) Q_
GazOa(cr)
"~aCl(g)
/ ~ GaO(g}
Ga(g) . . . . . . . . . . . . . Ga20(g)
850 1350 1850
Temperature (K)
0
U3
O o
C~
350 '
Ga=S3(cr) Ga2Oa(cr)
[ /
/ , , , r ,
850
Ga2S(g) , , f ,
1350
Z GaCl(g)
\
1850
Temperature (K)
FIo. 14. Equilibrium distribution of Ga (%(moles Ga/moles Ga total)) at standard oxidizing conditions (in the Ga/O/CI system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = !.2 and co,.o =
5 ppmw and Cc~.o = 300 ppmw.
FIG. 16. Equilibrium distribution of Ga (%(moles Ga/moles Ga total)) at standard reducing conditions (in the Ga/R/CI system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 -- 0.6 and e~a.o =
5 ppmw and Cc~,o = 300 ppmw.
g
£3- CO I
Oo
? Q~
o - 350
Ga2S3(cr) Ga203(cr)
850
~ Ga(g)
I ~ ~ GaOlg) 1350 1850
Temperature (K) FIG. 15. Equilibrium distribution of Ga (%(moles Ga/moles Ga total)) at standard reducing conditions (in the Ga/R system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 0.6 and ce,.o =
5 ppmw,
stock concentration of Ga was volatilized at 1300 K, the predominant gaseous species existing in equilib- rium with Ga203(cr) was GaCl(g). Variation in the operating parameters (the air excess ratio, sulfur removal efficiency, etc.) did not significantly affect the stability of the solid GazO3(cr).
In summary, Ga203(cr) is the stable form of gal- lium at temperatures up to 1400 K, where formation of GaCl(g) begins due to the equilibrium:
Ga2Oa(cr ) + 2HCI(g) ~- 2GaCl(g)
+ H20(g) + O2(g). (27)
Above 1750 K the equilibrium distribution of gallium may be controlled by the following reactions:
2GaCl(g) + H20 ~ Ga20(g ) 4- 2HCI(g) (28)
Ga20(g ) 4- ~rO2(g ) ~-~ 2GaO(g) (29)
GaO(g) ~ Ga(g) 4- {O2(g) (30)
where the first equilibrium accounts for the decompo- sition of GaCl(g) recognized in the calculations.
At standard reducing conditions (in the Ga/R system) gallium forms Ga2S3(cr), being stable up to 630K (Fig. 15). Between 630K and 1000K Ga203(cr) is the stable form of Ga. Above 1000 K, the equilibrium chemistry of gallium becomes very complex; Ga2S(g), Ga20(g), GaO(g) as well as Ga(g) occur in varying amounts. Formation of Ga2S(g) begins at 950 K. The amount of Ga2S(g) reaches a maximum occurrence at I150K. Formation of Ga20(g) begins at 1100 K. The amount of Ga20(g) reaches a maximum at 1600 K. Formation of Ga(g) begins at 1250 K. Above 1750 K Ga(g) is the major gallium-containing chemical species. Above 1700 K small amounts of GaO(g) are formed.
If chlorine is present at standard reducing condi- tions (in the Ga/R/CI system), formation of GaCl(g) begins at 800 K at the expense of Ga2S(g) and GazO(g) (Fig. 16). In addition, the amounts of Ga(g) produced at high temperatures in the Ga/R/C! system are significantly decreased compared with the amount produced in the Ga/R system. Between 1000 K and 1500 K GaCl(g) is the dominant chemi- cal species in the Ga/R/CI system. Above 1500 K GaCl(g) gradually decomposes (with increasing tem- peratures), forming Ga(g).
If fluorine is present at standard reducing condi- tions (in the Ga /R/F system) GaF3(cr) is stable below 360 K (Fig. 17). Above 1200 K GaF(g) is formed instead of Ga2S(g) and Ga20(g) (and to a smaller extent Ga(g)). GaF(g) is the dominant species be- tween 1200 K and 1800 K in the Ga /R/F system.

128 F. FRANDS~N et al.
5
I ~ o
~o_ i l ,-~J 350 850
GaF3(cr) Ga2Sa(cr)
Ga2Oa(cr)
Ga(g) GaF(g)
Ga20(g) GaO(g)
Temperoture (K)
FIG. 17. Equilibrium distribution of Ga (~(moles Ga/moles Ga total)) at standard reducing conditions (in the Ga/R/F system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: ). = 0.6 and c~,.o =
5 ppmw and CF.O = 100 ppmw.
o ° , NiSO4(cr) NiO(cr)
Q_ U3
I
(9 L
D_
5°
O(g)
~Ni (g )
. . . . . . . . . / / 1 / 850 1350 1850
Temperature (K)
F[G. 19. Equilibrium distribution of Ni (~o(moles Ni/moles Ni total)) at standard oxidizing conditions (in the Ni/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal. The possible formation of hydrated nickel sulfates at low temperatures is not
shown: 2 = 1.2 and cNi.o = 25 ppmw.
GeO2 (cr,I) GeO(g)
q
X,
©
. . . . J , , , . . . . . . . . . ~o 8~0 ~ 35o ~85o
Temperoture (K)
FIG. 18. Equilibrium distribution of Ge (~(moles Ge/moles Ge total)) at standard reducing conditions (in the Ge/R system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 0.6 and c~e.o =
5 ppmw.
5.2.8. Germanium At standard oxidizing conditions (in the Ge/O
system) GeO2(cr,l ) is stable up to 1100 K. Above 1300 K GeO(g) is stable. Alvin 44 states that the equilibrium distribution of germanium is nearly inde- pendent of variations in operating parameters such as the air excess number and the sulfur removal efficiency.
At standard reducing conditions (in the Ge/R system) GeO2(cr,1 ) is stable up to 700 K (Fig. 18). Between 800 K and 2000 K the equilibrium form of germanium gradually changes with increasing tem- perature; from GeS(g) as the major germanium- containing chemical species at 800 K to GeO(g) at 2000 K.
Kalfadelis and Magee 4a found GeS(g) and GeO(g) to be the only Ge containing chemical species present at equilibrium in the COED-gasification process at 1273 K and 1.5 atm.
5.2.9. Mercury
The equilibrium distribution of mercury at stand- ard oxidizing conditions (in the Hg/O and Hg]O/Ci systems) and at standard reducing conditions (in the Hg/R system) is outlined in Section 4.2.
5.2.10. Nickel
At standard oxidizing conditions (in the Ni/O system) nickel forms a series of hydrated sulfate compounds at low temperature. ~t-NiSO4.6H20(cr) is stable below 335 K; NiSO4.4H20(cr) is stable be- tween 335 K and 345 K and NiSO4.H20(cr) is stable between 345 K and 475 K. Above 475 K NiSOa(cr ) is stable up to 875 K, where NiO(cr) is formed by decomposition of NiSO4(cr) (Fig. 19). NiO(cr) is stable up to approximately 1600 K, where the forma- tion of NiO(g) and Ni(g) begins. NiO(g) is stable up to 1825 K, where the last trace amount of NiO(cr) disappears. Above 1825 K, an existing equilibrium between NiO(g) and Ni(g) is rapidly moved towards Ni(g) as the temperature is increased.
If chlorine is present at standard oxidizing condi- tions (in the Ni/O/CI system), the formation of NiCl(g) begins at 1550 K, and increases with increas- ing temperature towards a maximum of occurrence (8~(mol/mol) of the Ni present) at 1830 K. In addi- tion, small amounts of NiCl2(g ) are formed at high temperatures.
Alvin 44 reported NiSO4(cr ) to be the stable form of nickel up to 1100 K in fluidized-bed combustion

Trace elements from coal combustion 129
NiS,kr,l)
NiS,.,,lcr.l)
Ni,S,(cr, I)
I : Ni(cr,l) Ni(g)
I empefatufe (Kj
FIG. 20. Equilibrium distribution of Ni (O/,(moles Ni/moles Ni total)) at standard reducing conditions (in the Ni/R system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 1 = 0.6 and cNi,o =
25 ppmw.
H,PO&cr.l)
(P,O,),M PO,@\ 0”
\ /
FIG. 21. Equilibrium distribution of P (%(moles P/moles P total)) at standard reducing conditions (in the P/R system) as a function of the temperature, in a flue gas from combus- tion of a subbituminous coal: A = 1.2 and cp,, = 50 ppmw.
processes. At 1100 K NiO(cr) is formed. The conver- sion between NiS04(cr) and NiO(cr) depended on the degree of sulfur removal. Among the gaseous phases containing nickel, NiCl,(g) was the major one. In addition, secondary gaseous species such as Ni(OH),(g), NiCl(g), Ni(g), NiO(g) and NiH(g) were formed.
The major chemical equilibrium reactions of Ni include:
NiSO,(cr) + NiO(cr) + SO,(g) + $0,(g) (31)
and
NiO(cr) + NiO(g) (32)
NiO(cr) + 2HCI(g) G= NiCl,(g) + H,O(g) (33)
NiO(g) * W) + h(g). (34)
At standard reducing conditions (in the Ni/R system) nickel is present as NiS,(cr,l) up to 420 K (Fig. 20). Between 420 K and 550 K NiS,,&cr,l) is the stable form of nickel. Above 550 K Ni,S,(cr,l) is stable up to 1045 K, where Ni(cr,l) is formed. Above 1700 K only Ni(g) and small amounts of NiO(g) are present in the system.
Kalfadelis and Magee 43 found Ni(cr) to be the only Ni-containing chemical species present at equilib- rium at 1273 K and I .5 atm in the COED-gasification process.
5.2. I I. Phosphorus
At standard oxidizing conditions (in the P/O system) H,PO,(cr,l) is the stable form of phosphorus up to 670 K (Fig. 21). Between 670 K and 1250 K (P20J2(g) is stable. Above 1550 K only PO,(g) is present in significant amounts.
Alvin44 found the above outlined equilibrium dis- tribution to be nearly independent of the SO,(g) concentration in the flue gas from an oxidative fluidized-bed combustor. The stability of H,PO,(cr,l) was dependent of the air excess number. The equilib- rium distribution of phosphorus was found to be modified by the presence of magnesium.
The equilibrium distribution of phosphor in oxida- tive combustion flue gases is controlled by the princi- pal reactions:
4H,PO,(crJ) * (P20&(g) + 6&O(g) (35)
(P2O&(g) = 2PO,(g) + @z(g). (36)
Equation (35) is important between 316 K (the melt- ing point of H,PO,(cr)) and 800-900 K, while Eq. (36) is important between 1250 K and 1550 K.
At standard reducing conditions (in the P/R system) H,POl(cr,l) is stable up to 430 K (Fig. 22). Between 430 K and 1400 K (P,O,),(g) is stable. Around 1350 K, formation of PO,(g), PO(g) and PN(g) begins. The amounts of PO(g) and PO,(g) increase continuously with increasing temperature above 1350 K, while PN(g) has a maximum occur- rence (‘l%(mol/moI) of the phosphorus present) at 1750 K. At 2000 K the equilibrium distribution of phosphorus is 84%(mol/mol) PO,(g), 13%(mol/mol) PO (g) and 3%(mol/mol) PN(g).
Kalfadelis and Magee found (P,O,),(g) to account for all phosphorus present in the COED- gasification process at 1273 K and 1.5 atm.
5.2.12. Lead
At standard oxidizing conditions (in the Pb/O system) PbSO,(cr,l) is stable up to II00 K, where PbO(g) is formed (Fig. 23). Above II00 K an existing equilibrium between PbO(g) and Pb(g) slowly shifts towards Pb(g), with increasing temperatures.
JPECS 20:2-E

130 F. FRAND~N et aL
HzPO,(cr,I) (PzOa)2(g)
"6 O0
I o_I ©
(D Q_
350
/ 850 1550 Temperature (K)
PO2(g)
PO(g) ~ " ~ P N ( g ) 1850
FIG. 22. Equilibrium distribution of P (%(moles P/moles P total)) at standard oxidizing conditions (in the P/R system) as a function of the temperature, in a flue gas from combus- tion ofa subbituminous coal: 2 = 0.6 and Ca.o = 50 ppmw.
tO
U3 I t'~O.
t3_~
~6 q)
£1_
C l 350
PbSO4lcr,I) / ~ , ~ O ( g )
PbCl2(g)
850 1350 1850
Temperature (K)
Pb(g)
Fit. 24. Equilibrium distribution of Pb (%(moles Pb/moles Pb total)) at standard oxidizing conditions (in the Pb/O/CI system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and Cpb.O =
25 ppmw and Co.o = 300 ppmw.
'6 0) £L
U3 I
z3 o £L.o
~6
q) EL
PbSO4(cr,I) ,~bO(g)
~ P b ( g ) 50 850 1350 1850 Temperature (K)
Fro. 23. Equilibrium distribution of Pb (%(moles Pb/moles Pb total)) at standard oxidizing conditions (in the Pb/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and Cpb.o =
25 ppmw.
If chlorine is present at standard oxidizing condi- tions (in the Pb/O/CI system) PbCl2(g ) is formed in a narrow temperature range from 950 K to 1450 K, with a maximum occurrence of 52~(mol/mol) (of the lead present) at 1100 K (Fig. 24). PbCl(g) is present between 1050 K and 2000 K, with a maxi- mum occurrence of 5~(mol/mol) (of the lead present) at 1200 K. The amount of gaseous lead chlorides, including the PbCI4(g) formed was shown to be strongly dependent on the CI: Pb molar ratio.
Moberg e t al. 45 found PbSO4(cr ) to be the thermo- dynamically stable form of lead below 920 K. At tem- peratures between 920 K and 1170 K, lead was present as PbCl2(g), while PbO(g) was the dominant form of lead above I I70K. In addition, small amounts of Pb(g) and PbCl(g) were formed above 870 K.
Shpirt e t aL 49 showed that, assuming thermody- namic equilibrium, lead is present mainly as Pb(g) and PbO(g) in combustion processes. An increase in temperature results in a rise in the equilibrium concentration of Pb(g). The concentrations of other gaseous compounds were insignificant. The only solid lead compound found in significant amounts was PbO.SiO2(cr ) (a composed oxide compound). The nature of the distribution and formation of lead compounds did not change during the oxidation process for different concentrations of lead in coal. The distribution of lead compounds does not depend on the amount of the main ash-forming elements.
In summary, the thermodynamically stable form of lead at temperatures below 1000-1100K is PbSO4(cr). Above 1100 K PbO(g) formed by the reaction:
PbSO4(cr) ~ PbO(g) + SO2(g) + ~rO2(g) (37)
is the stable form. At high temperatures (T > 1400K) an equilibrium between Pb(g) and PbO(g) exists:
PbO(g) ~- Pb(g) + ~O2(g ). (38)
This equilibrium gradually moves towards the right when the temperature is increased from 1400 K to 2000 K.
Mojtahedi e t al. 46 found PbSO4(cr) to be stable up to 900 K in atmospheric fluidized-bed combustion. Above 900 K lead was present in the gaseous state, mainly as PbCI2(g), PbO(g) and Pb(g).
At standard reducing conditions (in the Pb/R system) PbS(cr, l) is stable up to 900 K (Fig. 25). Formation of PbS(g) begins at 750 K. The amount of PbS(g) reaches a maximum occurrence at 900 K and decreases with increasing temperature above 900 K, mainly forming Pb(g). In addition, small amounts of PbO(g) are present above 1700 K.

Trace elements from coal combustion 131
g
Q_
? 3:3o
(J
PbS(cr,I)
350 850
Temperature (K)
Pb(g)
PbSlg)
350 185(3
FIG. 25. Equilibrium distribution of Pb (%(moles Pb/moles Pb total)) at standard reducing conditions (in the Pb/R system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 0.6 and Cpb.O =
25 ppmw.
PbCI2(cr,I) PbS(cr,I) g. _ ~ eb(g)
© EL
03 I
£ ] o . EL""
E ©
(D 0 -
PbCl(g)
o350 . . . . 850 . . . . 13r50 . . . . 18'50'
Temae~ature (t4)
FIG, 26, Equilibrium distribution of Pb (~(moles Pb/moles Pb total)) at standard reducing conditions (in the Pb/R/CI system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 0.6 and cpb.o =
25 ppmw and Ccl.o = 100 ppmw.
If chlorine is present at standard reducing condi- tions (in the Pb/R/CI system), PbCl(g) is formed between 700 K and 1500 K (Fig. 26). The maximum amount of PbCl(g) present, 7-8%(tool/tool) (of the lead present), occurs around 900 K.
5.2.13. Antimony
At standard oxidizing conditions (in the Sb/O system) antimony forms Sb2(SO4),(cr) being stable up to 380 K, where Sb2Os(cr ) is formed (Fig. 27). Sb2Os(cr) decomposes at 720 K forming Sb2Ot(cr). Between 720 K and 810 K the stable form of anti- mony changes from Sb204(cr) to SbO(g), which is the only Sb-containing chemical species above 810K.
Sb2(SO4)3(cr) Sb2Os(cr) g
EL (/3
Uq~
G_
Tempercture (K)
SbO(g)
Sb204(cr)
8~0
FIG. 27. Equilibrium distribution of Sb (%(moles Sb/moles Sb total)) at standard oxidizing conditions (in the Sb/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and cBb.o =
1 ppmw.
Alvin .4 reports antimony to be present as SbO2(cr ) at temperatures up to 1200 K at fluidized-bed com- bustion conditions. Above 1200K antimony was present mainly as SbO(g). In addition, formation of small amounts of SbCl(g) and SbCla(g ) were reported.
In the present study SbO2(cr ) has not been re- garded, due to a lack of thermodynamic data. Only the dimeric form Sb20,(cr ) has been considered.
In summary, the equilibrium chemistry of anti- mony in oxidative combustion flue gases may be explained by the following three reactions: (1) a decomposition of Sb2(SO4)3(cr )
Sb2(SO4)3(cr) ~ Sb2Os(cr) + 3802(g) + ~)2(g) (39)
occurring around 380 K; (2) a decomposition of Sb2Os(cr)
Sb2Os(cr) ~ SbzO,(cr) + ~O2(g) (40)
occurring around 720 K; and (3) a decomposition of Sb204(cr)
Sb20,(cr ) ~ 2SbO(g) + O2(g). (41)
The change in equilibrium form from Sb20,(cr) to SbO(g) occurs with increasing temperatures between 720 K and 810 K.
At standard reducing conditions (in the Sb/R system) only SbS(g) is formed.
5.2.14. Selenium
At standard oxidizing conditions (in the Se/O system) an equilibrium between SeO2(cr) and SeO2(g) is established below 345 K. Above 345 K SeO2(g) is stable up to 1300 K (Fig. 28). Between

132 F. FR^m~S~N et al.
CD
O_ O0
~E G) t)
EL
550
~ 02(g)
// SeO(g)
/ ~ Selg)
850 ! 350 1850 7 e r - , , D L ~ c ~ u r e ~ / . { '
FIG. 28. Equilibrium distribution of Se (%(moles Se/moles Se total)) at standard oxidizing conditions (in the Se/O system) as a function of the temperature in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and Csc.o =
1 ppmw.
Sn(SO,~)2(cr} SnO2(cr) SnO(g) oo
q3 {3-
U3 I C o
COco
*d
I ©
%o . . . . . . . . . . . 4 . . . . . 850 1350 1850
Temperature (K)
FIG. 29. Equilibrium distribution of Sn (%(moles Sntmoles Sn total)) at standard oxidizing conditions (in the Sn/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and Cs..o =
5 ppmw.
1300 K and 2000 K the equilibrium form of selenium changes gradually from SeO2(g) to SeO(g) (and to a lesser extent to Se(g)) with increasing temperature. At 2000 K nearly equal amounts of SeO(g) and SeO2(g) (48.5%(mol/mol) of the Se present) are present together with a small amount of Se(g) (3%(tool/tool) of the Se present).
Moberg e t aL 4s found SeO2(g) to be the major stable form of selenium at temperatures up to 2000 K. Above 2000 K SeO(g) is the major stable form. Se(g) was found to be stable above 2000 K.
Malykh and Pertsikov 5. found that during the course of coal combustion, regardless of the air excess number (2e {1.0,1.2}), the most likely sele- nium compound to be formed is SeO2(g). In addition, small amounts of Se(g) and Sell(g) were formed above 1200 K.
The equilibrium chemistry of selenium at oxidizing conditions can be explained by the reactions:
SeO2(cr) ~ SeO2(g) (42)
SeO2(g) -~ SeO(g) + {O2(g ) (43)
SeO(g) ~,~ Se(g) + .~O2(g ). (44)
Equation (42) occurs below 345 K, while Eqs (43) and (44) become significant at temperatures above 1300 K.
At standard reducing conditions (in the Se/R system) only H2Se(g ) is formed.
Kalfadelis and Magee 43 found the following equi- librium distribution of selenium (unit:%(mol/mol) of the Se present): H2Se(g ) (97); COSe(g) (2.8); Se(g) (0.2), in a flue gas from the COED-gasification pro- tess (1273 K and 1.5 atm).
Gaseous carbonyl selenide, COSe(g), is not present in the database, DGFDBASE, used in this study, due to lack of thermochemical data.
Malykh and Pertsikov st found selenium to be present mainly in gaseous compounds with hydrogen, i.e., H2Se(g ) and Sell(g), in coal coking and gasifica- tion processes. Selenides e.g., SiSe(g), were formed during coal heating with no air access.
5.2.15. Tin
At standard oxidizing conditions (in the Sn/O system) Sn(SO,)2(er) is stable up to 530 K (Fig. 29). At 530 K the sulfate decomposes forming SnO2(cr), stable up to 1300 K, where the formation of SnO(g) begins. Above 1450 K only SnO(g) is formed.
Alvin 44 reported SnO2(cr) to be the major stable form of tin in oxidative flue gases from fluidized-bed combustion. In addition, small amounts of SnO(g) and SnCI2(g ) were formed. A maximum of 3%(mol/ mol) of the tin present was volatilized at atmospheric conditions. At pressurized conditions the degree of volatilization was lower (l%(mol/mol) of the Sn present). The air excess ratio and the initial amount of tin in the system caused only insignificant changes in the equilibrium distribution of tin.
In summary, the behavior of tin in oxidative com- bustion flue gases at equilibrium can be described by two reactions: (l) a decomposition of Sn(SO4)2(cr) occurring
around 500 K and forming SnO2(cr)
Sn(SO,)2(cr) ~-~ SnO2(cr) + 2SO2(cr) + O2(g); (45)
(2) a decomposition of SnO2(cr)
SnO2(cr) ~- SnO(g) + ~dD2(g ) (46)
occurring between 1300 K and 1450 K forming SnO(g), which is the stable form of tin at high temperatures (T > 1450 K).
At standard reducing conditions (in the Sn/R system) SnO2(cr ) is stable up to 650 K, where the
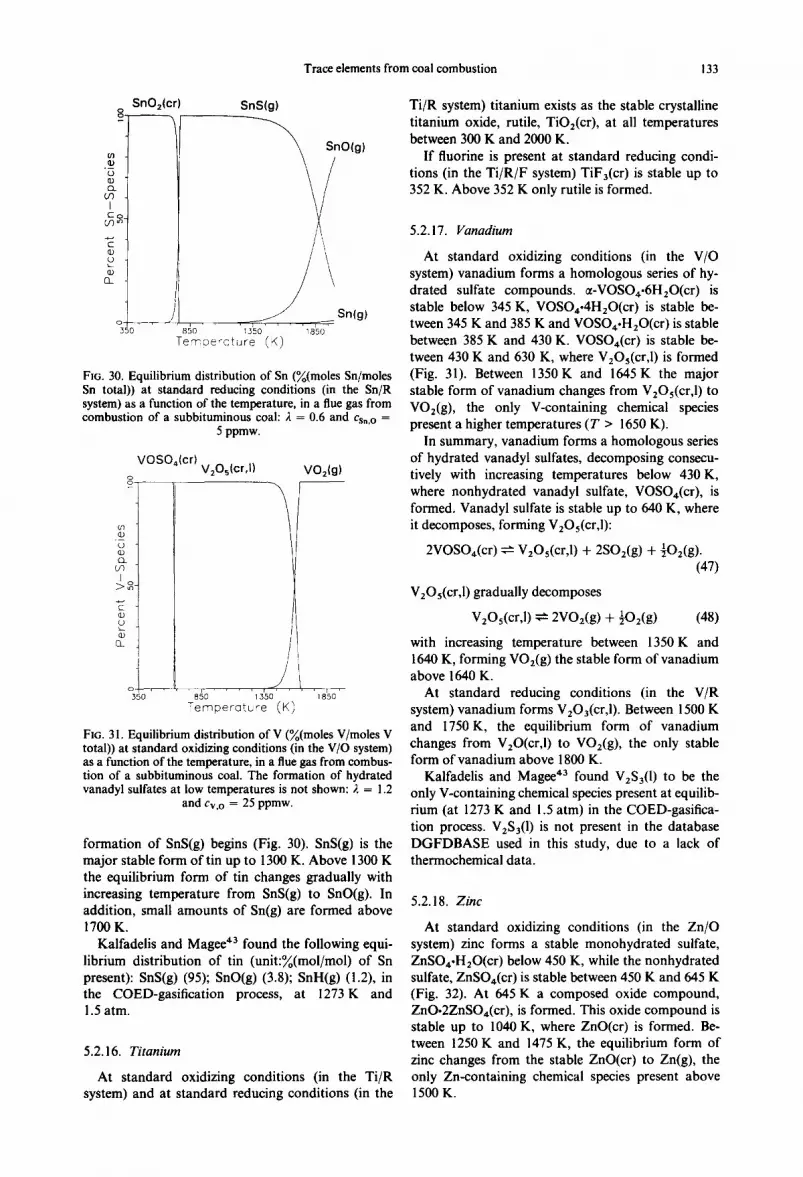
Trace elements from coal combustion 133
SnO2(cr) 8.
"~ .
I
C O _
n
35o
SnS(g)
SnOlg)
. / S n ( g ) 850 1350 1850 Temoe~cture (K)
Fro. 30. Equilibrium distribution of Sn (~o(moles Sn/moles Sn total)) at standard reducing conditions (in the Sn/R system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 0.6 and Cs,,o =
5 ppmw.
VOSO,(cr) V2Oslcr,i) VO2(g) o o _ r
Q_ UD
J
El_
J
o / , , , 350
' i . . . . 850 1350 Temperature (K) 1850
FIG. 3 I. Equilibrium distribution of V (%(moles V/moles V total)) at standard oxidizing conditions (in the V/O system) as a function of the temperature, in a flue gas from combus- tion of a subbituminous coal. The formation of hydrated vanadyl sulfates at low temperatures is not shown: 2 = 1.2
and Cv,o = 25 ppmw.
formation of SnS(g) begins (Fig. 30). SnS(g) is the major stable form of tin up to 1300 K. Above 1300 K the equilibrium form of tin changes gradually with increasing temperature from SnS(g) to SnO(g). In addition, small amounts of Sn(g) are formed above 1700 K.
Kalfadelis and Magee 43 found the following equi- librium distribution of tin (unit:~o(mol/mol) of Sn present): SnS(g) (95); SnO(g) (3.8); SnH(g) (1.2), in the COED-gasification process, at 1273 K and 1.5 atm.
5.2.16. Titanium
At standard oxidizing conditions (in the Ti/R system) and at standard reducing conditions (in the
Ti/R system) titanium exists as the stable crystalline titanium oxide, rutile, TiO2(cr), at all temperatures between 300 K and 2000 K.
If fluorine is present at standard reducing condi- tions (in the Ti/R/F system) TiF3(cr) is stable up to 352 K. Above 352 K only rutile is formed.
5.2. i 7. Vanadium
At standard oxidizing conditions (in the V/O system) vanadium forms a homologous series of hy- drated sulfate compounds. ~t-VOSO4.6H20(cr) is stable below 345 K, VOSO4.4H20(cr ) is stable be- tween 345 K and 385 K and VOSO4.H20(cr) is stable between 385 K and 430 K. VOSO4(cr) is stable be- tween 430 K and 630 K, where V2Os(cr,l) is formed (Fig. 31). Between 1350 K and 1645 K the major stable form of vanadium changes from V2Os(cr,l ) to VO2(g), the only V-containing chemical species present a higher temperatures (T > 1650 K).
In summary, vanadium forms a homologous series of hydrated vanadyl sulfates, decomposing consecu- tively with increasing temperatures below 430 K, where nonhydrated vanadyl sulfate, VOSO4(cr), is formed, Vanadyl sulfate is stable up to 640 K, where it decomposes, forming V20~(cr,l):
2VOSO4(cr) ~- V2Os(cr,1) + 2SO2(g ) + ½02(g ). (47)
VEOs(cr,1 ) gradually decomposes
V2Os(cr,l) ~- 2VO2(g) + ~)2(g) (48)
with increasing temperature between 1350K and 1640 K, forming VO2(g ) the stable form of vanadium above 1 640 K.
At standard reducing conditions (in the V/R system) vanadium forms V203(cr,i ). Between 1500 K and 1750 K, the equilibrium form of vanadium changes from VzO(cr,I ) to VO2(g), the only stable form of vanadium above 1800 K.
Kalfadelis and Magee 43 found V2S3(1) to be the only V-containing chemical species present at equilib- rium (at 1273 K and 1.5 atm) in the COED-gasifica- tion process. V2S3(I ) is not present in the database DGFDBASE used in this study, due to a lack of thermochemical data.
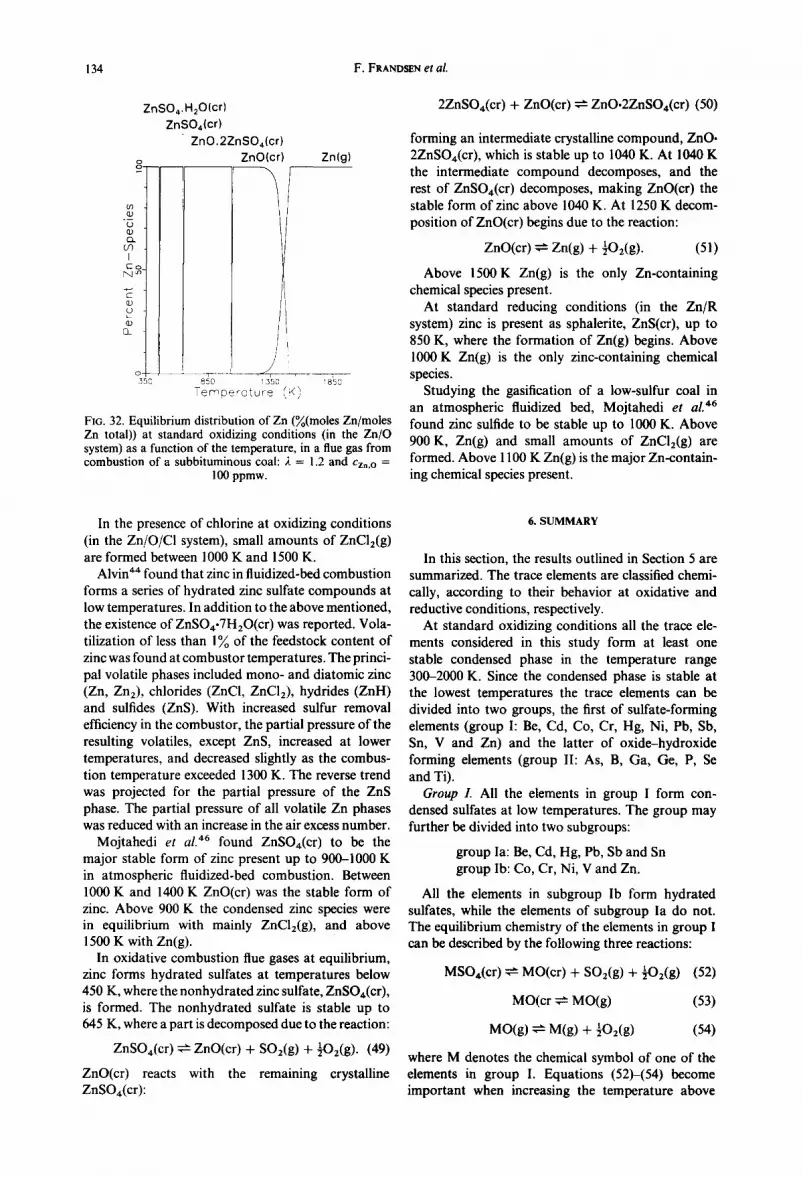
5.2.18. Zinc
At standard oxidizing conditions (in the Zn/O system) zinc forms a stable monohydrated sulfate, ZnSO4.H20(cr) below 450 K, while the nonhydrated sulfate, ZnSO4(cr) is stable between 450 K and 645 K (Fig. 32). At 645 K a composed oxide compound, ZnO.2ZnSO4(cr), is formed. This oxide compound is stable up to 1040 K, where ZnO(cr) is formed. Be- tween 1250 K and 1475 K, the equilibrium form of zinc changes from the stable ZnO(cr) to Zn(g), the only Zn-containing chemical species present above 1500 K.
134 F. FRANDSm4 et aL
© c-1
U3
I
r . . j ~-,
(1)
ZnSOa.H20(cr) ZnSO,,(cr)
ZnO.2ZnSO4(cr) ZnO(cr) Znlg)
r 0) 0_
H
i / ~50 850 1350 1850
Temmeroture (4)
FIG. 32. Equilibrium distribution of Zn (%(moles Zn/moles Zn total)) at standard oxidizing conditions (in the Zn/O system) as a function of the temperature, in a flue gas from combustion of a subbituminous coal: 2 = 1.2 and Cz..o =
100 ppmw.
2ZnSO4(cr) + ZnO(cr) ~ ZnO.2ZnSO4(cr) (50)
forming an intermediate crystalline compound, ZnO. 2ZnSO4(cr), which is stable up to 1040 K. At 1040 K the intermediate compound decomposes, and the rest of ZnSO4(cr) decomposes, making ZnO(cr) the stable form of zinc above 1040 K. At 1250 K decom- position of ZnO(cr) begins due to the reaction:
ZnO(cr) ~.~ Zn(g) + ~rO2(g ). (51)
Above 1500 K Zn(g) is the only Zn-containing chemical species present.
At standard reducing conditions (in the Zn/R system) zinc is present as sphalerite, ZnS(cr), up to 850 K, where the formation of Zn(g) begins. Above 1000 K Zn(g) is the only zinc-containing chemical species.
Studying the gasification of a low-sulfur coal in an atmospheric fluidized bed, Mojtahedi et al. 46
found zinc sulfide to be stable up to 1000 K. Above 900 K, Zn(g) and small amounts of ZnCl2(g) are formed. Above 1100 K Zn(g) is the major Zn-contain- ing chemical species present.
In the presence of chlorine at oxidizing conditions (in the Z n / O / C I system), small amounts of ZnCl2(g) are formed between 1000 K and 1500 K.
Alvin 44 found that zinc in fluidized-bed combustion forms a series of hydrated zinc sulfate compounds at low temperatures. In addition to the above mentioned, the existence of ZnSO4.7H20(cr) was reported. Vola- tilization of less than 1% of the feedstock content of zinc was found at combustor temperatures. The princi- pal volatile phases included mono- and diatomic zinc (Zn, Zn2), chlorides (ZnCI, ZnC12), hydrides (ZnH) and sulfides (ZnS). With increased sulfur removal efficiency in the combustor, the partial pressure of the resulting volatiles, except ZnS, increased at lower temperatures, and decreased slightly as the combus- tion temperature exceeded 1300 K. The reverse trend was projected for the partial pressure of the ZnS phase. The partial pressure of all volatile Zn phases was reduced with an increase in the air excess number.
Mojtahedi et al. 46 found ZnSO4(cr) to be the major stable form of zinc present up to 900-1000 K in atmospheric fluidized-bed combustion. Between 1000 K and 1400 K ZnO(cr) was the stable form of zinc. Above 900 K the condensed zinc species were in equilibrium with mainly ZnCl2(g), and above 1500 K with Zn(g).
In oxidative combustion flue gases at equilibrium, zinc forms hydrated sulfates at temperatures below 450 K, where the nonhydrated zinc sulfate, ZnSO4(cr), is formed. The nonhydrated sulfate is stable up to 645 K, where a part is decomposed due to the reaction:
ZnSO4(cr ) ~ ZnO(cr) + SO2(g) + ~dD:(g). (49)
ZnO(cr) reacts with the remaining crystalline ZnSO4(cr):
6. S U M M A R Y
In this section, the results outlined in Section 5 are summarized. The trace elements are classified chemi- cally, according to their behavior at oxidative and reductive conditions, respectively.
At standard oxidizing conditions all the trace ele- ments considered in this study form at least one stable condensed phase in the temperature range 300-2000 K. Since the condensed phase is stable at the lowest temperatures the trace elements can be divided into two groups, the first of sulfate-forming elements (group h Be, Cd, Co, Cr, Hg, Ni, Pb, Sb, Sn, V and Zn) and the latter of oxide-hydroxide forming elements (group II: As, B, Ga, Ge, P, Se and Ti).
Group I. All the elements in group I form con- densed sulfates at low temperatures. The group may further be divided into two subgroups:
group Ia: Be, Cd, Hg, Pb, Sb and Sn group Ib: Co, Cr, Ni, V and Zn.
All the elements in subgroup Ib form hydrated sulfates, while the elements of subgroup la do not. The equilibrium chemistry of the elements in group I can be described by the following three reactions:
MSO,(cr) ~,~ MO(cr) + SO2(g) + "~)2(g) (52)
MO(cr ~ MO(g) (53)
MO(g) ~ M(g) + .~O2(g ) (54)
where M denotes the chemical symbol of one of the elements in group I. Equations (52)-(54) become important when increasing the temperature above

Trace elements from coal combustion 135
the stability temperature of the pure (nonhydrated) sulfate. The following may be noted for the elements in group I:
Be: For beryllium, a reaction of the type given in Eq. (52) occurs between 700K and 800 K. Above 1100 K the behavior of beryllium deviates from the reactions given in Eqs (53) and (54): condensed beryllium oxide, BeO(cr,l), reacts with H20(g) to form the gaseous beryllium(II)hydrox- ide, Be(OH)z(g) (see Eq. (19), Section 5.2.3).
Cd." For cadmium, a reaction of the type given in Eq. (52) occurs between 950 K and 1000 K and a reaction of the type given in Eq. (54) occurs above 1000 K. In the case of cadmium, Eq. (53) is displaced completely toward the gaseous metal oxide, CdO(g). Cadmium forms significant amounts of the gaseous dichloride, CdClz(g), between 850 K and 1150 K, making a reaction such as Eq. (22) (see Section 5.2.4) necessary to interpret the equilibrium chemistry of cadmium in this temperature range.
Co: For cobalt, Eq. (23) (see Section 5.2.5) is a slightly modified form of the reaction type given in Eq. (52). A composed oxide, CosO4, is formed instead of the simple oxide, CoO(cr), being of the form MO(cr) suggested in Eq. (52). The composed oxide decomposes at 1110 K forming CoO(cr), in equilibrium with elemental gaseous cobalt, Co(g), at high temperatures. In fact this equilibrium is a combination of reactions of the type given in Eqs (53) and (54). Cobalt also forms the gaseous dichloride, CoCl2(g), adding further reactions to Eqs (52)-(54).
Cr: For chromium, Eq. (26) is a slightly modified form of the reaction type given in Eq. (52). Above the dissociation temperature of Cr2(SO4)a(cr), the equilibrium chemistry of Cr is very complex: chromium forms several oxyhy- droxides adding further reactions to the Eqs (52)-(54).
Hg: For mercury, a reaction of the type given in Eq. (52) occurs between 540 K and 600 K. Above 600 K the equilibrium chemistry of mercury is governed by reactions of the type given in Eqs (53) and (54). Between 380 K and 700 K mercury forms significant amounts of the gaseous dichlo- ride, HgC12(g), making a reaction such as that given in Eq. (16) (see Section 4.2.3) necessary to interpret the equilibrium distribution of mercury in this temperature range.
Ni: For nickel, reactions of the type given in Eqs (52)-(54) occur above 875 K (see Section 5.2.10). Reactions between crystalline nickel compounds, e.g., NiO(cr) and NiSO4(cr) and gaseous hydro- gen chloride result in the formation of small amounts of NiCI2(g).
Pb: For lead, reactions of the type given in Eq. (52) occur at 1100 K; above 1400 K reactions of the type given in Eq. (54) occur. Eq. (53) is displaced
completely toward the gaseous metal oxide, PbO(g). PbO(g) and PbSO4(cr) may react with hydrochloric acid, forming gaseous lead chlorides.
Sb: For antimony, a reaction of the type given in Eq. (52) is recognized in the set of reactions suggested to explain the equilibrium chemistry in oxidative combustion flue gases (see Section 5.2.13). The decomposition of Sb2(SO4)3(cr) occurs at 380 K. Above 380 K the behavior of antimony differs from Eqs (53) and (54).
Sn: For tin a modified version of Eq. (52) occurs around 500 K, while reactions of the type given in Eqs (53) and (54) in combination generate Eq. (46), the other reaction suggested to explain the equilibrium chemistry of tin (see Section 5.2.15).
V." For vanadium, a slightly modified reaction of the Eq. (52) forming V20~(cr,l ) occurs at 640 K. The decomposition of V2Os(cr,l ) can be ex- plained by combining reactions of the type given in Eqs (53) and (54) (see Section 5.2.17).
Zn: For zinc, the Eqs (49) and (51) suggested in Section 5.2.18 are similar to Eqs (52) and (53) combined with Eq. (54). A reaction like Eq. (50) combining ZnO(cr) and ZnSO,(cr) to form ZnO. 2ZnSO4(cr), and reactions accounting for the formation of ZnC12(g) are needed to explain the equilibrium chemistry in oxidative combustion flue gases.
In summary, the behavior of the group I elements may more or less be described by reactions of the types given in Eqs (52)-(54). Furthermore, reactions accounting for the decomposition of hydrated sul- fates (of Co, Cr, Ni, V and Zn) and for the formation of the gaseous dichlorides, MCI2(g), are needed. In the case of chromium the formation of gaseous oxy- hydroxides also occurs.
Group II. Except for arsenic in the presence of calcium, the trace elements in this group all form condensed oxides or hydroxides at low temperatures. Arsenic forms calcium arsenate, Cas(AsO4)2(cr) in the As/Ca/O/F system. No simple set of reactions can describe the behavior of these elements in oxida- tive combustion flue gases. A characteristic feature of the elements in group II is that gallium is the only element which forms a stable halide compound at oxidizing conditions. Gallium forms significant amounts of GaCl(g) (in the Ga/O/CI system) at temperatures above 1450 K.
At standard reducing conditions, the behavior of the trace elements considered in this work is complex. A simple classification of the elements as the one performed above for the oxidizing conditions is not found. Some features may be noted.
The pair of elements (Cd,Zn), (Co,N0 and (Ge,Sn) show similar chemical behavior mutually, as outlined below. (1) The pair (Cd,Zn). These two elements occur in two distinct forms in

136 F. FRANDSEN et aL
the temperature range considered: MS(cr) and M(g), M ~ {Cd,Zn}.
For cadmium the crystalline monosulfide, CdS(cr), is stable up to 650 K, where the equilibrium form changes to the elemental gaseous metal, Cd(g). For zinc the crystalline monosulfide, ZnS(cr) (in the sphalerite form) is stable up to 850 K, where the elemental gaseous metal, Zn(g) is formed. (2) The pair (Co,Ni). These two elements occur in at least four distinct forms, between 300 K and 2000 K: MS2(cr(,I)), MS,s- (cr(,I)), M(cr,l) and M(g), M e {Co,Ni}, where ns denotes nonstoichiometric, while (cr(,l)) denote crys- talline (cr) or condensed (cr,l) state.
Cobalt forms the chemical species CoS2(cr), CoSo.89(cr), Co(cr,l) and Co(g), while nickel forms the chemical species NiS2(cr,l), NiSo.8,(cr,l), Ni(cr,l) and Ni(g). In addition, nickel forms a sulfide com- pound, Ni3S2(cr,l), stable in a temperature range between the intervals where NiSo.a4(cr,I) and Ni(cr,l) are stable. The temperature ranges of stability for each of the chemical species of Co and Ni formed at reducing conditions are given in Section 5.2.5 and Section 5.2.10, respectively. (3) The pair (Ge,Sn). The elements germanium and tin occur in three differ- ent stable forms between 300K and 2000K: MO2(cr(,I)), MS(g) and MO(g), M ~ {Ge,Sn}.
Germanium forms GeO2(cr,I) below 700 K and GeS(g) and GeO(g) above 700 K. Tin forms SnO2(cr) below 650 K and SnS(g) and SnO(g) above 650 K.
The elements mercury, antimony and selenium each form only one chemical species between 300 K and 2000 K. Mercury forms the elemental gaseous metal, Hg(g), antimony forms the gaseous mono- sulfide, SbS(g), and selenium forms H2Se(g ).
Titanium forms TiO2(cr) (in the rutile form), as the single stable component. If fluorine is present at standard reducing conditions (in the Ti/R/F system) the stable form of titanium below 352 K changes from TiO2(cr) to TiFa(cr).
The elements boron, beryllium and chromium show similar chemistry at standard reducing and oxidizing conditions. For boron, the only differences in the equilibrium distribution at reducing and oxidiz- ing conditions are as follows. (1) The temperature range of stability of the minor
volatile compound H3BaO3(g ) is 700-1200 K in reducing systems, compared with the range 350- 800 K in oxidizing systems.
(2) No BO2(g) (a minor volatile compound) is formed at reducing conditions.
For beryllium, the only significant difference in the equilibrium chemistry, changing the redox condi- tions from oxidizing to reducing, in systems contain- ing no halogens, is the lack of BeSO,,(cr)--which forms in reducing systems. If fluorine is present at standard reducing conditions (in the Be/R/F system) BeF2(cr,l ) is stable up to 380 K.
For chromium, the equilibrium chemistry at low
temperatures (T < 1500K) is independent of the redox conditions considered in this work. At high temperatures (T > 1500 K) the amounts of gaseous chromium oxides formed are decreased at reducing conditions compared with oxidizing conditions.
The remaining elements arsenic, gallium, phos- phorus, lead and vanadium show individual behavior in reducing systems at equilibrium. These elements form both condensed and gaseous compounds and show significant changes in chemistry as a function of the redox conditions. They are not comparable with any of the other elements and do not fit into the classification of the behavior of trace elements in the reducing systems given above.
7. CONCLUSION
As shown in the previous text, thermodynamic calculations provide a useful guide to the chemical fate of trace elements in combustion and gasification systems.
The idea is to consider a combustion or gasi- fication process as a system with a known elemental composition, temperature and pressure. The thermo- dynamic stable phases present are then predicted by minimizing the total Gibbs free energy of the system. However, GEA has several limita- tions. (i) The temperature must be high ( > 1000 K) or the
time available for the system to reach equilibrium must be long enough to ensure the chemical processes occurring in the system reach equilibrium.
(2) In some combustion-gasification systems the mixing of reactants plays an important role for the chemical processes occurring. No limitations due to mixing are taken into account in GEA.
(3) The presence of particulate material, i.e., fly ash, in the system is not regarded. Some of the gaseous components may be adsorbed onto the surface of fly ash particles, e.g. mercury on unburnt material in fly ash. Another effect is the curvature of small particles, which may increase the partial pressure of the chemical species, thereby affecting the con- densational behavior (enrichment of submicro- meter fly ash particles).
In spite of these severe limitations, the method of pin-pointing stable phases, assuming global equilib- rium, is at present the only computational possibility for generating knowledge about the chemistry of trace elements in combustion and gasification systems.
In the present work, the behavior of 18 trace elements in flue gases, at reducing and oxidizing conditions, have been outlined one-by-one using simple tools (computer code for data handling) and assumptions to characterize various systems. Some similarity in the chemical behavior within groups of trace elements is recognized in oxidative systems. In

Trace elements from coal combustion 137
reductive systems the chemical behavior is more com- plex. The ultimate condensation o f all trace elements at low temperature in combustion flue gases should be verified experimentally.
The behavior of the 18 trace elements regarded in this study may in real systems, especially at low temperatures, differ significantly from the results ob- tained, due to the occurrence of kinetic limitations. However, to evaluate the validity of the equilibrium assumption at low temperatures, chemical kinetic data is needed to investigate the kinetic influence on the behavior of trace elements in flue gases.
Acknowledgements--This work was carried out as a part of the CHEC (Combustion and Harmful Emission Control) research programme. The CHEC programme is financially supported by ELSAM (the Jutland-Funen Electricity Con- sortium), ELKRAFT (the Zealand Electricity Consortium), STVF (the Danish Technical Research Council) and the Danish Ministry of Energy. Lone Aslaug Hansen is acknowl- edged for her valuable contribution in the work with the database.
REFERENCES
1. SWAINE, D. J., Trace Elements in Coal, Butterworth, London (1990).
2. KAUPPINEN, E. I. and PAKKANEN, T. A., Env. Sci. Technol. 24(12), 1811 (1990).
3. FISHER, G. L., PRENTICE, B. A., SILBERMAN, D., ONDOV, J. M., BIERMANN, A. H., RAGAINI, R. C. and MCFAR- LAND, A. R., Env. Sci. Technol. 12(4), 447 (1978).
4. DAVIDSON, R. L., NATUSCH, D. F. S., WALLACE, J. R. and EVANS, C. A., Jr., Eno. Sci. Technol. 8(13), ll07 (1974).
5. NORTON, G. A., DEKALB, E. L. and MALABY, K. L., Env. Sci. Technol. 20(6), 604 (1986).
6. GLADNEY, E. S., SMALL, J. A., GORDON, G. E. and ZOLLER, W. H., Arm. Env. 10, 1071 (1976).
7. COLES, D. G., RAGAINI, R. G., Ot,a~ov, J. M., FISHER, G. L., SILBERMAN, D. and PRENTICE, B. A., Enly. Sci. Technol. 13(4), 455 (1979).
8. HANSEN, L. D. and FISHER, G. L., Env. Sci. TechnoL 14(9), 1111 (1980).
9. ONDOV, J. M., RAGAINI, R. C. and BIERMANN, A. H., Env. Sci. TechnoL 13(8), 946 (1979).
10. SMITH, R. D., CAMPBELL, J. A., NIELSON, K. K., Env. Sci. Technol. 13(5), 553 (1979).
I I. MARKOWSKI, G. R. and F1LBY, R., Env. Sci. Technol. 19(9), 796 0985).
12. MEIJ, R., VAN DER KOOIJ, J., VAN DER SLUYS, J. L. G., SIEPMAN, F. G. C. and VAN DER SLOOT, H. A., KEMA Sci. Tech. Reports 2(I), 1 (1984).
13. HAYNES, B. S., NEVILLE, M., QUANN, R. J. and SAROF1M, A. F., J. Coll. Int. Sci. 87(1), 266 (1982).
14. LYONS, W. S., Trace Element Measurements at the Coal-Fired Steam Plant, CRC Press, Cleveland, OH (1977).
15. KLEIN, D. H., ANDREN, A. W., CARTER, J. A., EMERY, J. F., FELDMAN, C., FULKERSON, W., LYONS, W. S., OGLE, J. C., TALMI, Y., VAN HOOK, R. I. and BOLTON, N., Env. Sci. Technol. 9(10), 973 (1975).
16. KAAKINEN, J. W., JORDEN, R. M., LAWASAN1, M. H. and WEST, R. E., Env. Sci. Technol. 9(9), 862 (1975).
17. MEIJ, R. KEMA Rep. No. 32561-MOC 92-3641 (1992). 18. SANDER, B., Proc. 2rid Int. EP RI Conf. Managing Hazard-
ous Air Pollutants, Washington DC, July 13-15 (1993). 19. MAIER, H., VGB Kraftwerkstechnik (Eng. lss.) 70(10),
749 (1990).
20. MAIER, H., DAHL, P., GUTBERTLET, H. and DIECKMANN, A., VGB Kraftwerkstechnik (Eng. lss.) 72(5), 412 (1992).
21. CLARKE, L. B. and SLOGS, L. L., Trace Elements-Emis- sions from Coal Combustion and Gasification, IEA Coal Research, London, Report No. I EACR/49 (1992).
22. PAGE, A. L., ELSEEWl, A. A. and STRAUGHAN, I. R., Resid. Rev. 71, 83 (1979).
23. GLADNEY, E. S. and GORDON, G. E., J. Env. Sci. Health A13(7), 481 (1978).
24. BALE, C. W. and ERIKSSON, G., Can. Metall. Quart. 29(2), 105 (1990).
25. ERIKSSON, G. and HACK, K., Metall. Trans. B 21, 1013 (1990).
26. SMITH, W. R. and MlSSEN, R. W., Chemical Reaction Equilibrium Analysis: Theory and Algorithms, John Wiley & Sons, New York (1982).
27. BABUSHKIN, V. I., MATVEYEV, G. M. and MCHEDLOV- PETROSSYAN, O. P., Thermodynamics o f Silicates, Springer Verlag, Berlin, 239 (1985).
28. GREINER, H., CALculation o f PHAse Diagrams ( CAL- PHAD) 12(2), 143 (1988).
29. MICHELSEN, M. L., Fluid Phase Equilibria 53, 73 (1989).
30. STRAUSS, K., Kraftwerkstechnik--zur Nutzung fossiler, regenerativer und nuklearer Energiequellen, Springer Verlag, Berlin (1992).
31. BARIN, 1., Thermochemical Data of Pure Substances, Vols. 1 and 2, VCH Verlagsgesellschaft, Weinheim, Germany (1989).
32. CHANG, Y. A. and AHMAD, N., Thermochemical Data on Metal Carbonates and Related Oxides, The Metallur- gical Society of AIME (1982).
33. CHASE, M. W., Jr., DAVmS, C. A., DOWNEY, R., Jr., FRURIP, D. J., McDONALD, R. A. and SYVERUD, A. N., J. Phys. Chem. Ref. Data 14, Supplementary Vol. 1 (1985).
34. Joint Army and Navy Air Force, JANAF Thermochemi- cal Tables, National Institute of Standards and Technol- ogy, Standard Reference Database 13 (1985).
35. KNACKE, O., KUBACHEWSKI, O. and HESSELMANN, K., Thermochemicul Properties of Inorganic Substances, Vols. I and 2, Springer Verlag, Berlin (1991).
36. MILLS, K. C., Thermodynamic Data for Inorganic Sul- phides, Selenides and Tellurides, Butterworth, London (1974).
37. SCHICK, H. L., Thermodynamics of Certain Refractory Compounds, Academic Press, New York (1966).
38. SMITHELLS, C. J. and BRANDES, E. A., Metals Reference Handbook, pp. 231-241, Butterworth, London (1976).
39. PANKRATZ, L. B., STUVE, J. M. and GOKCEN, N. A., U.S. Bureau of Mines, Bulletin 677 (1984).
40. DEKOCK, C. W., U.S. Bureau of Mines, Information Circular 8910 (1984).
41. EBBINGHAUS, B. B., Combust. Flame 93, 199 (1993). 42. ALVIN, M. A., O'NEILL, E. P. and KEAIRNS, D. L.,
Proc. 5th Int. Conf. Fluidized-Bed Combustion, p. 526, Washington DC, December 12 14 (1977).
43. KALFADELIS, C. O. and MAGEE, E. M., Ind. Eng. Chem., Fund. 16(4), 489 (1977).
44. ALVIN, M. A., Trace and Minor Element Reactions in Fluidized-Bed Combustion Processes, EPA Contract No. 600/7-82-050, Report No. Pb 82-240219 (1982).
45. MOBERG, P.-O., WESTERMARK, M. and NOL~NG, B., Project Coal-Health-Environment, Technical Report No. 28 (1982).
46. MOJTAHEDI, W., BACKMAN, R. and LARJAVA, K., Techni- cal Research Centre of Finland, Espoo, Publication No. 42 (1987).
47. MALYKH, N. V., PERTSIKOV, I. Z. and BAEVA, T. V., Khimiya Tverdogo Topliva 22(4), 134 (1988).
48. MALYKH, N. V., PERTSIKOV, I. Z. and KATZOV, O. M., Khimiya Tverdogo Topliva 22(3), 116 (1988).
49. SHPIRT, N. YA., PERTSIKOV, I. Z., SAMUILOVA, L. N.

138 F. FRANDSEN et al.
and MALYKH, N. V., Khimiya Tverdogo Topliva 22(4), 126 (1988).
50. Morgantown Energy Technology Center Report, Va- porization of Trace Element Species from Coal under Gasification and Combustion Conditions, US Depart- ment of Energy, Morgantown, West Virginia (1989).
51. MALYKH, N. V. and PERTSIKOV, I. Z., Khimiya Tverdogo Tela 24(4), 50 (1990).
52. NORD1N, A., SCHAGER, P. and HALL, B., Proc. Finnish- Swedish Flame Days, Abo Academy University, Abo, Fin- land (1990).
53. YON FAHLKE, J., Chemie im Kraftwerk, V. G. B. (1992). 54. Wu, C. Y. and Biswas, P., Combast. Flame 93, 31
(1993). 55. LINAK, W. P. and WENDT, J. O. L., Prog. Energy
Combust. Sci. 19, 145 (1993).