the evolution of reproductive isolation in the ectomycorrhizal hebeloma crustuliniforme aggregate...
TRANSCRIPT

1192
q 2000 The Society for the Study of Evolution. All rights reserved.
Evolution, 54(4), 2000, pp. 1192–1206
THE EVOLUTION OF REPRODUCTIVE ISOLATION IN THE ECTOMYCORRHIZALHEBELOMA CRUSTULINIFORME AGGREGATE (BASIDIOMYCETES) IN
NORTHWESTERN EUROPE: A PHYLOGENETIC APPROACH
DUUR K. AANEN,1,2,3 THOMAS W. KUYPER,4 TED H. M. MES,5,6 AND ROLF F. HOEKSTRA1,7
1Laboratory of Genetics, Wageningen Agricultural University, Dreyenlaan 2, 6703 HA Wageningen, The Netherlands2E-mail: [email protected]
4Department of Environmental Sciences, Section Soil Science and Plant Nutrition, Wageningen Agricultural University,P.O. Box 8005, 6700 EC Wageninge, The Netherlands
E-mail: [email protected] of Parasitology and Tropical Veterinary Medicine, Institute of Infectious Diseases and Immunology,
Faculty of Veterinary Medicine, University of Utrecht, P.O. Box 80.165, 3508 TD Utrecht, The Netherlands6E-mail: [email protected]
7E-mail: [email protected]
Abstract. To reconstruct the evolution of reproductive isolation in the ectomycorrhizal Hebeloma crustuliniformeaggregate (Basidiomycetes), phylogenetic relationships were determined between strains that belong to a clade con-sisting of nine intercompatibility groups (ICGs, biological species). Four of these nine ICGs are partially compatibleand belong to the H. crustuliniforme aggregate. Different levels of partial compatibility have been found between thesefour ICGs. Between ICGs 3 and 4, 15% of the combinations were compatible. One strain was compatible with allisolates of both ICGs 3 and 4 and also with one isolate of ICG 2. Both a nuclear phylogeny, based on ribosomal IGSsequence data, and a mitochondrial phylogeny, based on a group-I intron located in the large subunit ribosomal RNAgene (LrRNA), were reconstructed. The level of incompatibility was compared with the phylogenetic history ofindividuals belonging to this clade.
Different relationships were found between the level of compatibility and the relative age of the most recent commonancestor (MRCA) for different ICGs. On the one hand, the evolution of incompatibility between ICGs 2 and 3/4 ismost consistent with the class of ‘‘divergence- first’’ models because a positive correlation was found between therelative age of the MRCA and the level of incompatibility for ICG 2 versus 3/4. On the other hand, the lack of sucha correlation for ICGs 3 and 4 shows that (partial) incompatibility between these ICGs has arisen without strongdivergence. The ecological (and to a lesser extent geographical) differences found between ICGs 3 and 4 suggest thatselection for incompatibility, associated with host tree preference, has been important in the evolution of incompatibilitybetween these two ICGs. The incongruence between the nuclear and mitochondrial trees for ICG 1 could be explainedby a hybrid origin of this ICG, with different donors of the mitochondrial and nuclear sequences.
Key words. Basidiomycetes, ectomycorrhiza, Hebeloma crustuliniforme, reproductive isolation, sexual intercompa-tibility, speciation.
Received August 17, 1999. Accepted January 6, 2000.
Most Agaricales (Basidiomycetes) have a heterothallic lifecycle. In this life cycle, fusion between monokaryons, whichdevelop after spore germination, is an essential step. If fusionoccurs and a dikaryon develops, both monokaryons are con-sidered to be sexually compatible (hereafter referred to ascompatibility in this study). For most species the cells of thedikaryon have two separate nuclei. The dikaryon can formbasidiocarps (mushrooms), the fruit bodies. In the basidi-ocarp, nuclear fusion and meiosis take place followed byspore formation. Cell fusion and nuclear fusion are thus sep-arated in time. Sexual compatibility between monokaryonshas been used to delimit biological species (intercompati-bility groups, ICGs) within Basidiomycetes (e.g., Boidin1986; Petersen 1995). For ectomycorrhizal fungi, the use ofthe biological species concept has been restricted to a fewgenera (Laccaria, Fries and Mueller 1984; Mueller and Gar-des 1991; Suillus, Fries and Neumann 1990; Paxillus, Fries1985; Hebeloma, Aanen and Kuyper 1999). However, rela-tively few studies have explicitly addressed the question ofthe evolutionary origin of sexual incompatibility, which we
3 Present address: Zoological Institute, Universitetsparken 15,DK-2100 Copenhagen, Denmark.
define as speciation, in Basidiomycetes (Vilgalys 1991; Vil-galys and Sun 1994; Garbelotto et al. 1998).
Most models of speciation can be classified as belongingto one of two main classes. One class of models assumes thatthe origin of reproductive barriers between species is pre-ceded by a process of gradual genetic divergence. The clas-sical allopatric mode of speciation (Mayr 1963) belongs tothis class. Within Basidiomycetes, evidence has been foundin support of this class of models, incompatibility as a con-sequence of genetic divergence (Pleurotus, Vilgalys and Sun1995; Exidiopsis plumbescens, Wells and Wong 1989). In thisstudy we refer to this class of models as ‘‘divergence-first’’models.
The genetic basis of sexual incompatibility has been de-scribed by relatively simple models in some cases (Chaseand Ullrich 1990a,b). These simple models have led to hy-potheses about sexual incompatibility as a cause of geneticdivergence (Brasier 1987; Hallenberg 1991; Worrall 1997).Although agreement seems to exist that genetic reproductivebarriers can arise easily between populations of Basidio-mycetes (Bresinsky et al. 1987), the idea of ‘‘incompatibility-first’’ models is still controversial. Cases of so-called ABC-relationships, where two sympatric populations are incom-

1193REPRODUCTIVE ISOLATION IN HEBELOMA
patible but both of them are compatible with a third allopatricpopulation (Chamuris 1991; Hallenberg 1991; Petersen andRidley 1996; Garbelotto et al. 1998), have been interpretedas evidence for this incompatibility-first model, or at leastfor reinforcement, that is, the evolution of incompatibility insympatry after initial genetical divergence in allopatry. Phy-logenetic relationships between populations could help todiscriminate between those different classes of models be-cause these classes predict different combinations of evo-lutionary history and compatibility. However, detailedknowledge about phylogenetic relationships is usually lack-ing, especially between partially compatible populations (butsee Garbelotto et al. 1998).
The role of ecological specialization in speciation has beenstressed, both in theoretical models (Maynard Smith 1966;Diehl and Bush 1989) and in case studies, usually concerninghost switches in parasitic relationships (e.g., Heterobasidionannosum, Garbelotto et al. 1998). For ectomycorrhizal fungi,host tree switching can play this role in ecological special-isation (Kretzer et al. 1996; Aanen et al. 2000).
Sexual compatibility has been tested in the Hebeloma crus-tuliniforme (poison pie) complex to delineate biological spe-cies (ICGs; Aanen and Kuyper 1999). The mating system ofspecies in this complex was found to be bifactorial with mul-tiple mating types. A total of 20 ICGs have been found inthis group, some of which were only partially incompatible.These 20 ICGs formed two distinct monophyletic groups thatdid not form sister groups (Fig. 1; Aanen et al. 2000).
One of these clades (clade II) consisted of 17 ICGs, themajority of which form ectomycorrhizae with members ofthe Salicaceae. Within this clade, ICG 21 had a basal position.This basal position was independently supported by the lackof a unique three-base-pair deletion in ICG 21 that was foundin all other members of the clade (Fig. 1). Because ICG 21has not been found with Salicaceae, the switch to Salicaceaeappears to have preceded extensive speciation in this clade.Many of the phylogenetic relationships within this clade werenot resolved due to little sequence variation in the ribosomalITS 1 and 2. One monophyletic group within clade II (cladeIIa; circled in Fig. 1) consisted of nine ICGs, four of whichwere only partially incompatible. These four belonged to themorphospecies aggregate H. crustuliniforme together withICG 5, which was completely incompatible with these four.One isolate (605) could not be placed unambiguously intoone of the ICGs: It was fully compatible with all membersof ICGs 3 and 4. Of the other combinations between ICGs3 and 4, 7% were also compatible. Individual collections,however, could always be assigned to a single ICG. Isolate605 was also the single isolate of ICGs 3/4 that was com-patible with a member of ICG 2. Between ICGs 1 and 2 8%of the combinations were compatible. However, these com-binations usually showed signs of reduced performance in-dicated by lack of nuclear migration, aberrant morphology,and reduced growth rate (Aanen and Kuyper 1999). Com-patibility in this clade can be considered as a quantitativephenomenon with different levels of partial compatibility be-tween populations. Partial compatibility between some ICGsof the monophyletic group IIa suggests a relatively recentorigin of sexual incompatibility within this clade.
The interface between population genetics and systematics
is most informative for studies of speciation (Templeton1998; Coyne and Orr 1999; Schilthuizen 1999). The moredistant from the actual speciation event, the greater the ex-pected genetic differences among the species, but the moredifficult it becomes to infer which genetic differences werecausally involved in speciation versus which were conse-quences of speciation (Templeton 1998). More importantly,at this interface between population biology and systematicsexamples exist where incompatibility is not qualitative (all-or-nothing) but quantitative with different levels of partialincompatibility (Armillaria, Anderson et al. 1980; H. annos-um, Chase and Ullrich 1990a,b; Garbelotto et al. 1998; He-beloma, Aanen and Kuyper 1999). This provides the oppor-tunity to compare different levels of compatibility with phy-logenetic history of strains.
The aim of this study was to reconstruct the evolution of(partial) incompatibility in the H. crustuliniforme aggregate.We tried to find indications for either divergence-first or in-compatibility-first models for the origin of incompatibilityby reconstructing the phylogenetic history of strains belong-ing to ICGs of this aggregate that were partially or completelyreproductively isolated.
For the class of divergence-first models, genetic divergenceis followed by complete incompatibility via intermediatestages of partial incompatibility. The most important predic-tion is a correlation between the level of compatibility andthe relative age of the most recent common ancestor (MRCA).The incompatibility-first model does not predict this corre-lation between the level of compatibility and the relative ageof the MRCA.
Most studies on phylogenetic relationships between (bio-logical) species do not include taxa that show partial com-patibility (e.g., Anderson and Stasovski 1992; Vilgalys andSun 1994; Vilgalys et al. 1996; Piercey-Normore et al. 1998).Only Garbelotto et al. (1998) explicitly addressed phyloge-netic relationships between partially compatible strains of H.annosum. In this study different levels of partial compatibilitybetween ICGs within the H. crustuliniforme aggregate werecompared with phylogenetic history. We address four mainquestions: (1) Is there a positive correlation between the levelof (partial) compatibility and the relative age of the MRCA?(2) Does this correlation hold both for a nuclear and mito-chondrial phylogeny? (3) Do the data support divergence-first or incompatibility-first models? (4) Does ecological spe-cialization play a role in speciation events in this aggregate?
MATERIALS AND METHODS
Intercompatibility tests were performed between mono-karyons of collections belonging to the H. crustuliniformecomplex in a previous study (Aanen and Kuyper 1999). Thesecollections were made in a variety of habitats, with a varietyof host trees in northwestern Europe in 1994, 1995, and 1996.Only carpophores growing closely together were consideredto belong to the same mycelium and were given one collectionnumber. In cases of doubt, only one carpophore was col-lected. Data on ecology (including potential host trees), andmacro- and micromorphology for each collection were re-corded. For each collection, monokaryons were obtained us-ing spore germination. Sporocarps and monokaryons received

1194 DUUR K. AANEN ET AL.

1195REPRODUCTIVE ISOLATION IN HEBELOMA
←
FIG. 1. Phylogenetic relationships in the genus Hebeloma, based on ITS 1 and 2 sequences. Tree with the highest likelihood of 6527most parsimonious trees of length 357 (CI 5 0.63, RI 5 0.81; excluding uninformative characters, length 5 261, CI 5 0.49), foundwith PAUP*, using Agrocybe praecox as an outgroup. Two arrows mark the two single proposed deletion events for two indels of threebase pairs (not used as characters). Bootstrap values higher than 50% are indicated above branches. Decay indices (preceded by a d)are also indicated. On the right, ICGs to which isolates belong are indicated. The largest circled clade is clade II, the circled clade withinclade II is clade IIa, the subject of this study.
the same stock number. Within a stock, each monokaryonwas given an additional individual number. Voucher speci-mens are preserved in Wageningen (WAG) and cultures inBaarn (CBS). All isolates were collected and identified byD. K. Aanen. Nomenclature of species follows Kuyper andBoekhout (1995).
Nuclear and mitochondrial phylogenies were reconstructedbetween single spore isolates of all ICGs belonging to cladeIIa (Aanen et al. 2000), except for two ICGs that belong tothe morphospecies H. pusillum (ICGs 7 and 8). Because thisstudy focused on the partially incompatible ICGs 1, 2, 3, and4, three to five collections per ICG were used for these ICGs.Outgroups of clade IIa were based on the internal transcribedspacer (ITS) phylogeny (Fig. 1; Aanen et al. 2000). For thenuclear phylogeny, two ICGs (15 and 20) of the sister groupof clade IIa were used as outgroups. The sister-group statusof these two taxa was tested using another outgroup, ICG21, the basal position in clade II of which was strongly sup-ported (see introduction and Aanen et al., 2000). For themitochondrial phylogeny, one outgroup was used, a repre-sentative of the sister group of clade IIa, ICG 20. In all, 23isolates were used in the nuclear phylogeny and 21 in themitochondrial. In Table 1 data on all collections that havebeen used in this study are summarized. In Table 2, a sum-mary of ecological and geographical the data of all ICGs thatwere the subject of the present study are given as well (in-cluding data on collections that have not been used in thepresent study).
Choice of Sequences, DNA Isolation, Polymerase ChainReaction, and Sequencing
For methods of DNA isolation, polymerase chain reaction(PCR), and sequencing, we refer to Aanen et al. (2000). Asa nuclear sequence, the intergenic spacer (IGS; Anderson andStasovsky 1992), located between 25S rDNA and 5S rDNA,was used. This sequence has been found to be somewhat morevariable than the internal transcribed spacers 1 and 2 (ITS 1and 2; Erland et al. 1994). Primers 5SA and Cnl12 (Andersonand Stasovski 1992; Henrion et al. 1992) were used to amplifythis region. Both primers were also used as sequencing prim-ers.
To find a mitochondrial sequence with an appropriate levelof variation, several mitochondrial ribosomal primer pairsthat were available on the Bruns’s laboratory homepage (http://plantbio.berkeley.edu/;bruns/) were tested. The followingpairs were used: MS1 and 2 (part of the small ribosomalsubunit), ctb2 and 9, and CML5.5 and ML6 (both pairs partof the large subunit ribosomal RNA gene, LrRNA. To testthese different sequences, initially five sequences were de-termined from collections that belong to partially compatibleICGs. The level of variation found between these five se-
quences was used as a criterion to select one of those se-quences for a broader analysis. Ctb 2 and 9 gave a PCRproduct of about 650 base pairs, containing an intron. Thesesequences showed both single nucleotide position variationand length variation. This sequence was therefore chosen asa mitochondrial marker for a broader analysis. Ctb 9 wasused as a sequencing primer.
IGS 1 sequences have been deposited in GenBank underaccession numbers AF174437–AF174459 and CTB sequenc-es under accession numbers AF174460–AF174479. Thealigned sequences have been deposited to TreeBASE (http://herbaria.harvard.edu/treebase) as SN256–730 and SN256–731 or are available from D. K. Aanen upon request.
Phylogenetic Analysis
Sequences were aligned using CLUSTAL V (Higgins etal. 1989). The alignment was edited manually using a matrixcreated in PAUP* (Swofford 1998). Phylogenetic relation-ships were inferred from the aligned sequences using par-simony with PAUP*. Two analyses were performed, one withgaps as missing data and one with gaps coded according tothe coding scheme proposed by Hibbett et al. (1995). Underthis coding scheme, nucleotide positions with single-base in-dels are scored as characters, with gaps as fifth state; multiple-base indels are scored as binary characters; and single nu-cleotide positions aligned to multiple-base indel sites arescored as additional characters, with gaps as missing states.The goals of this coding scheme are that potentially infor-mative indels are included in the analyses, that overweightingof single indels is avoided and finally that phylogeneticallyinformative nucleotide variation in sequences aligned to gapsis preserved.
All transformations were unordered and equally weighyed.The branch-and-bound option in PAUP* was used to findmost parsimonious trees. The option ‘‘collapse branches ifminimum length is zero’’ was selected. Clade stability wasassessed by 1000 bootstrap replications (Hillis and Bull1993), using the branch- and bound-option. Decay indices(Bremer 1988; Donoghue et al. 1992) were calculated fromPAUP* tree files using the program Autodecay (Eriksson1998) and PAUP*. Other measures (tree length, sequencedivergence, consistency, and retention indices) were calcu-lated using PAUP*. To examine alternatives to the maximum-parsimony trees, a neighbor-joining tree (Saitou and Nei1987) was generated, based on the Kimura two-parametergenetic distance, using PAUP*. An additional test was per-formed with the constraint that ICGs 1, 2, 3, 4, and 5 (allbelonging to the morphospecies H. crustuliniforme) formeda monophyletic group in the nuclear phylogeny. Constrainedand unconstrained trees were compared by three criteria: treelength, Templeton’s (1983) nonparametric test, and the Kish-
1196 DUUR K. AANEN ET AL.
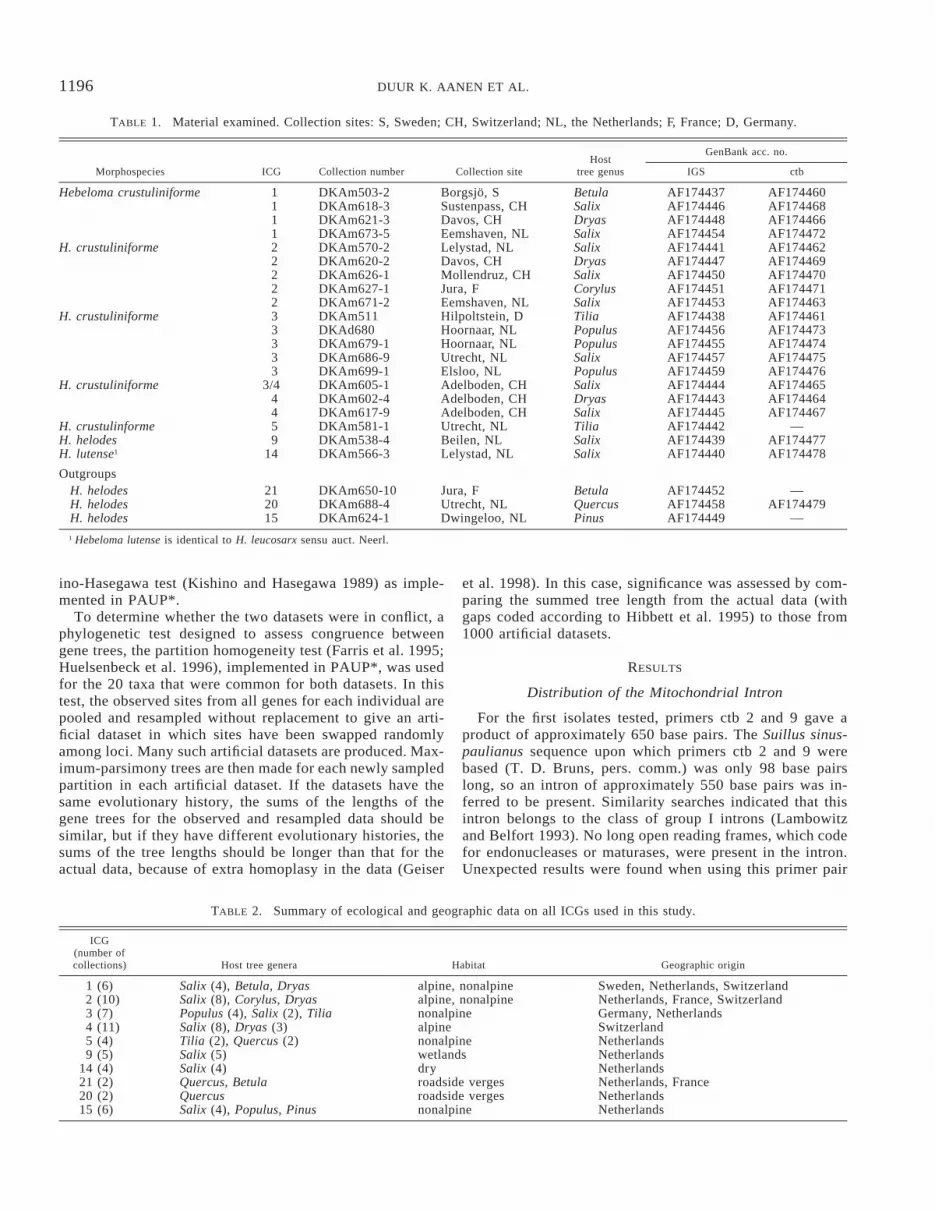
TABLE 1. Material examined. Collection sites: S, Sweden; CH, Switzerland; NL, the Netherlands; F, France; D, Germany.
Morphospecies ICG Collection number Collection siteHost
tree genus
GenBank acc. no.
IGS ctb
Hebeloma crustuliniforme 1111
DKAm503-2DKAm618-3DKAm621-3DKAm673-5
Borgsjo, SSustenpass, CHDavos, CHEemshaven, NL
BetulaSalixDryasSalix
AF174437AF174446AF174448AF174454
AF174460AF174468AF174466AF174472
H. crustuliniforme 22222
DKAm570-2DKAm620-2DKAm626-1DKAm627-1DKAm671-2
Lelystad, NLDavos, CHMollendruz, CHJura, FEemshaven, NL
SalixDryasSalixCorylusSalix
AF174441AF174447AF174450AF174451AF174453
AF174462AF174469AF174470AF174471AF174463
H. crustuliniforme 33333
DKAm511DKAd680DKAm679-1DKAm686-9DKAm699-1
Hilpoltstein, DHoornaar, NLHoornaar, NLUtrecht, NLElsloo, NL
TiliaPopulusPopulusSalixPopulus
AF174438AF174456AF174455AF174457AF174459
AF174461AF174473AF174474AF174475AF174476
H. crustuliniforme 3/444
DKAm605-1DKAm602-4DKAm617-9
Adelboden, CHAdelboden, CHAdelboden, CH
SalixDryasSalix
AF174444AF174443AF174445
AF174465AF174464AF174467
H. crustulinforme 5 DKAm581-1 Utrecht, NL Tilia AF174442 —H. helodes 9 DKAm538-4 Beilen, NL Salix AF174439 AF174477H. lutense1 14 DKAm566-3 Lelystad, NL Salix AF174440 AF174478
OutgroupsH. helodesH. helodesH. helodes
212015
DKAm650-10DKAm688-4DKAm624-1
Jura, FUtrecht, NLDwingeloo, NL
BetulaQuercusPinus
AF174452AF174458AF174449
—AF174479
—1 Hebeloma lutense is identical to H. leucosarx sensu auct. Neerl.
TABLE 2. Summary of ecological and geographic data on all ICGs used in this study.
ICG(number ofcollections) Host tree genera Habitat Geographic origin
1 (6)2 (10)3 (7)4 (11)5 (4)9 (5)
14 (4)21 (2)20 (2)15 (6)
Salix (4), Betula, DryasSalix (8), Corylus, DryasPopulus (4), Salix (2), TiliaSalix (8), Dryas (3)Tilia (2), Quercus (2)Salix (5)Salix (4)Quercus, BetulaQuercusSalix (4), Populus, Pinus
alpine, nonalpinealpine, nonalpinenonalpinealpinenonalpinewetlandsdryroadside vergesroadside vergesnonalpine
Sweden, Netherlands, SwitzerlandNetherlands, France, SwitzerlandGermany, NetherlandsSwitzerlandNetherlandsNetherlandsNetherlandsNetherlands, FranceNetherlandsNetherlands
ino-Hasegawa test (Kishino and Hasegawa 1989) as imple-mented in PAUP*.
To determine whether the two datasets were in conflict, aphylogenetic test designed to assess congruence betweengene trees, the partition homogeneity test (Farris et al. 1995;Huelsenbeck et al. 1996), implemented in PAUP*, was usedfor the 20 taxa that were common for both datasets. In thistest, the observed sites from all genes for each individual arepooled and resampled without replacement to give an arti-ficial dataset in which sites have been swapped randomlyamong loci. Many such artificial datasets are produced. Max-imum-parsimony trees are then made for each newly sampledpartition in each artificial dataset. If the datasets have thesame evolutionary history, the sums of the lengths of thegene trees for the observed and resampled data should besimilar, but if they have different evolutionary histories, thesums of the tree lengths should be longer than that for theactual data, because of extra homoplasy in the data (Geiser
et al. 1998). In this case, significance was assessed by com-paring the summed tree length from the actual data (withgaps coded according to Hibbett et al. 1995) to those from1000 artificial datasets.
RESULTS
Distribution of the Mitochondrial Intron
For the first isolates tested, primers ctb 2 and 9 gave aproduct of approximately 650 base pairs. The Suillus sinus-paulianus sequence upon which primers ctb 2 and 9 werebased (T. D. Bruns, pers. comm.) was only 98 base pairslong, so an intron of approximately 550 base pairs was in-ferred to be present. Similarity searches indicated that thisintron belongs to the class of group I introns (Lambowitzand Belfort 1993). No long open reading frames, which codefor endonucleases or maturases, were present in the intron.Unexpected results were found when using this primer pair

1197REPRODUCTIVE ISOLATION IN HEBELOMA
FIG. 2. Summary of gain and loss of intron(s) and/or mutation. (A) An explanation of the polymerase chain reaction (PCR) productsfound using different primers. Using primers ctb 2 and 9, no PCR products were obtained for ICGs 9, 14, and 20, but using mitD andctb 9, PCR products were obtained for those ICGs. The possible explanations for these observations are illustrated. I, the insertion presentin all ICGs except for ICG 5; II, the mutation/insertion present in ICGs 9, 14, and 20. (B) Most parsimonious explanation for the lossof the intron(s) and/or mutation, illustrated in Figure 2A, using ICG 20 as outgroup. I, the insertion present in all ICGs except for ICG5; II, the mutation/insertion present in ICGs 9, 14, and 20.
for other isolates. First, the intron was absent from ICG 5,resulting in a PCR product of 98 base pairs, the sequence ofwhich was identical with the partial LrRNA sequence of S.sinuspaulianus. Second, for ICGs 9, 14, and 20, no PCRproducts were obtained. Two possible explanations were: (1)another insertion is present somewhere in the mitochondrialDNA between ctb 2 and 9, making the sequence too long tobe amplified under the applied PCR conditions; (2) anotherinsertion or mutation is present in one of the two primerannealing sites ctb 2 or 9. To test whether the intron waspresent, several internal primers were tested in combinationwith either ctb 2 or 9. Primer mitD (GTTTATAAGAGAA-TATATCTA) located on the 59 side of the intron was usedin combination with ctb 9 and gave a product of 500 basepairs for ICGs 9, 14, and 20, which contains most of thephylogenetically informative variation. In Figure 2A, the pos-sible hypotheses are presented that explain these results. Us-ing ICG 20 as an outgroup, the most parsimonious expla-nation for the loss of the different introns and/or the mutationin the primer sites is given in Figure 2B.
Sequences
For the IGS, the total number of nucleotides used in thealignment ranged from 921 to 936. The length of the alignedsequences was 954. One region in the IGS sequences wasexcluded from the analysis because the alignment was am-biguous (nucleotide position 14–61). The length of thealigned sequences used in the phylogenetic analysis was 906.Under gap-as-missing coding, 819 of these 906 were constantand 87 variable, 33 of which were parsimony informative.Maximum sequence divergence (absolute number of base-pair differences) within the ingroup was 25 (2.7%) and be-tween in- and outgroup was 27 (2.9%). Under indel coding,the total number of characters used in the analysis was 914,with 812 of these were constant and 112 variable, 44 of which
were parsimony-informative. Base composition of the entireregion was approximately: A 5 0.32, C 5 0.23, G 5 0.22,and T 5 0.23. For the mitochondrial sequences, all variationwas within the intron. The following sequences were iden-tical: m503–2, m618–3, m621–3, and m673–5 (ICG 1);m679–1 and m686–9 (ICG 3); m511–1, m680–1, and m699–1 (ICG 3); and m602–4, m605–1, and m617–9 (ICG 4). Forthe sequences amplified with ctb 2 and 9 (ICGs 1, 2, 3, and4), the total number of nucleotides aligned ranged from 610to 636. For the sequences amplified with mitD and ctb 9(ICGs 9, 14, and 20), this number ranged from 455 to 504.The length of the aligned sequences was 696. Two regionsof the mitochondrial sequences were excluded from the anal-ysis because the alignment was ambiguous (nucleotide po-sitions 462–502 and 669–696). The length of the alignedsequences used in the phylogenetic analysis was 628. Undergap-as-missing coding, 608 of these 628 were constant and21 variable, 13 of which were parsimony informative. Max-imum sequence divergence (absolute number of base-pairdifferences) within the ingroup was 13 (2.1%), and betweenin- and outgroup was 20 (3.2%).
Under indel coding, the total number of characters was636, with 602 of these constant and 34 variable, 23 of whichwere parsimony informative. Base composition of the entireregion was approximately A 5 0.40, C 5 0.15, G 5 0.15,and T 5 0.30.
Phylogenetic Relationships
Nuclear Phylogeny
Using the IGS sequences, under gap-as-missing coding,one tree was found to have a length of 94 (CI 5 0.92, RI 50.93; using only informative variation, length 5 43, CI 50.81). Coding gaps according to Hibbett et al. (1995) gavetwo trees of length 124, which only differed in the relation-

1198 DUUR K. AANEN ET AL.
FIG. 3. One of two most parsimonious trees of length 124 (CI 5 0.92, RI 5 0.93, using all characters; using only parsimony-informativecharacters, length 5 57, CI 5 0.83) based on IGS 1 sequences, using ICG 21 as outgroup. Indels were coded according to Hibbett etal. (1995). Bootstrap values higher than 50% are indicated above branches. Decay indices (preceded by a d) are also indicated. On theright, ICGs to which isolates belong are indicated as well as host trees and geographic origin.
ship between ICGs 14 and 5 (CI 5 0.92, RI 5 0.93; usingonly informative variation, length 5 57, CI 5 0.83). One ofthe two most parsimonious trees found with gap-as-missingcoding is shown in Figure 3 (topology similar to tree foundwith gap5missing coding).
Using ICG 21 as an outgroup, ICGs 15 and 20 form a well-supported monophyletic group, which is the sister group ofgroup IIa. This relationship is in accordance with the ITStree (Aanen et al. 2000). The monophyly of clade IIa is sup-ported by a high bootstrap value (94%) and decay index (di5 3). The basal relationships of clade IIa are not resolved.One clade comprises ICG 9. The second clade consists of
ICGs 1, 5, and 14. Within this well-supported clade, ICG 1forms a monophyletic group. The sister group of ICG 1 isICG 5. The sister group of ICG 1 and 5 is ICG 14 (whichbelongs to the morphospecies H. lutense), whereas the twoICGs that constitute its sister group belong to the morphos-pecies H. crustuliniforme. The third clade within IIa consistsof members of ICGs 2, 3, and 4. Within this clade, threemonophyletic groups are found, the relationships betweenwhich are not resolved (a hard polytomy). One of these cladescomprises all isolates of ICG 2. Two other clades compriseisolates of ICG 3 and isolates of ICGs 3 and 4, includingisolate 605, respectively. Within the latter clade, one mono-

1199REPRODUCTIVE ISOLATION IN HEBELOMA
phyletic group consists of one member of ICG 4 and 605,which was compatible with both ICGs 3 and 4. The sistergroup of this clade comprises three members of ICG 3 andone of ICG 4.
The neighbor-joining tree is almost identical with one ofthe two most parsimonious trees, but the polytomy consistingof the three clades, that is, the clade consisting of ICG 2, theclade consisting of collections 511 and 680, and the cladeconsisting of the remaining collections of ICG 3 and 4, isresolved. In the neighbor- joining tree, the clade consistingof ICG 2 is basal to the remaining taxa, and the clade con-sisting of 511 and 680 is basal to the clade consisting of theother collections of ICG 3 and of ICG 4 (neighbor-joiningtree not shown).
Mitochondrial Phylogeny
When gaps were treated as missing data, one tree of 23steps was found (CI 5 0.96, RI 5 0.98; using only infor-mative sites, length 5 15, CI 5 0.93). The neighbor-joiningtree did not differ from the most parsimonious tree. UsingICG 20 as an outgroup, the basal taxon within the ingroupis ICG 14. ICG 9 is the sister group of a clade comprisingICGs 1, 2, 3, and 4. Within the clade consisting of ICG 1,2, 3 and 4, ICGs 1 and 2 together constitute a monophyleticgroup, as do ICGs 3 and 4. Within the clade consisting ofICGs 3 and 4, there is no resolution, because of a lack ofvariation. Within the clade with ICGs 1 and 2, 1 and 2 formmonophyletic groups.
Using the gap coding scheme as described by Hibbett etal. (1995), three trees of length 41 (CI 5 0.85, RI 5 0.93;using only informative sites, length 5 30, CI 5 0.80) werefound, which differ in the position of ICGs 14 and 9. Theneighbor-joining tree did not differ from one of the mostparsimonious trees. One of the most parsimonious trees undergap coding is depicted in Figure 4 (topology that is not inpositive conflict with tree found with gap-as-missing coding).The only other difference with the gap-as-missing tree is theresolution within the clade consisting of ICGs 3 and 4. ICGs3 and 4 in this tree form monophyletic groups.
Mitochondrial and Nuclear Phylogenies and CompatibilityCompared
The mitochondrial and nuclear phylogenies were incon-gruent in several respects. To determine the significance ofthis conflict, the partition homogeneity test was used for the20 taxa that were common to both datasets. The actual lengthof the summed trees was 135 steps, which was shorter than99.8% of the artificial datasets, indicating that the gene treeshave significantly different topologies (Fig. 5A).
In Figure 6, the two phylogenies are compared, togetherwith the pattern of compatibility, geographical origin, andhost tree genera. Strains of ICGs 1 and 2 form monophyleticgroups in both the nuclear and mitochondrial phylogeny. Inthe mitochondrial phylogeny, the strains of ICG 3 form amonophyletic group and two strains of ICG 4 form a cladewith strain 605, which was compatible with both ICGs 3 and4. In the nuclear phylogeny, however, strains of ICGs 3 and4 form paraphyletic groups. The partially compatible ICGs3 and 4 are sister groups in the mitochondrial phylogeny and
belong to a hard polytomy in the nuclear phylogeny. Treeswith ICGs 3 and 4 constrained as monophyletic groups arefour steps longer than unconstrained trees (both under gap-as-missing coding and under indel coding). The constrainedtrees found with gap coding cannot be rejected using Tem-pleton’s nonparametric test (P 5 0.22) or the Kishino-Has-egawa test (P 5 0.10). Under gap-as-missing coding, theconstrained trees cannot be rejected using Templeton’s non-parametric test (P 5 0.12), but they can be rejected usingthe Kishino-Hasegawa test (P 5 0.04). The position of ICG5 in the nuclear tree is not in contrast with the mitochondrialphylogeny, because we do not know its position there. In thenuclear phylogeny, ICGs 1, 2, 3, 4, and 5 form a paraphyleticgroup, but they form a monophyletic group with ICG 14.This is in contrast with the mitochondrial phylogeny, where14 has a basal position relative to a clade consisting of 1, 2,3, and 4 (and probably 5, according to the hypothesized gainand loss of the different introns/mutations, see Fig. 2B). How-ever, a tree that forces the monophyly of ICGs 1, 2, 3, 4,and 5 in the nuclear tree is only two steps longer (gaps coded)and cannot be rejected by the Kishino-Hasegawa test (P 50.41) or Templeton’s nonparametric test (P 5 0.69).
The main difference between the two phylogenies is theposition of ICG 1. In the mitochondrial phylogeny, ICG 1 isthe sister group of the partially compatible ICG 2, whereasin the nuclear phylogeny, ICG 1 belongs to a clade consistingof ICGs 5 and 14, which is basal to the clade consisting ofICGs 2, 3, and 4. Therefore, we also performed the partitionhomogeneity test on the dataset without the four strains ofICG 1. Significance was assessed by comparing the summedtree length from the actual data to those from 1000 artificialdatasets. The actual summed tree length of 123 was equal toor longer than 9.2% of the artificial data sets, indicating thatthe gene trees for the taxa other than ICG 1 do not havesignificantly different topologies (Fig. 5B). For these taxaother than ICG 1, a combined analysis was performed (gapscoded acoording to Hibbet et al. 1995). Two trees of length126 were found (CI 5 0.90, RI 5 0.89; using only informativevariation, length 5 63, CI 5 0.79). The trees only differedin the position of collection 617 of ICG 4. In one tree itbelonged to a clade consisting of the other collection of ICG4 and collection 605 (ICG 3/4) and in the other tree it wasbasal to a clade consisting of collections 679, 686, and 699of ICG 3. One of the two most parsimonious trees is shownin Figure 7.
DISCUSSION
Sexual Compatibility
ICGs in the H. crustuliniforme complex have a bifactorialmating system with multiple alleles (Aanen and Kuyper1999). The genetics of mating systems in heterothallic Ba-sidiomycetes have been well studied in other species, suchas Coprinus cinereus and Schizophyllum commune (Casseltonand Olesnicky 1998). Mating systems are usually uni- orbifactorial and extensively multi-allelic. In addition to thissystem of mating type incompatibility, Chase and Ullrich(1990a, b) described a model to explain incompatibility inH. annosum and posited the existence of a small number ofsterility genes, with two alleles each. In the present study an

1200 DUUR K. AANEN ET AL.
FIG. 4. One of three most parsimonious trees of length 41 (CI 5 0.85, RI 5 0.93, using all characters; using only parsimony-informativecharacters, length 5 30, CI 5 0.80) based on a group I intron located in the mitochondrial large subunit ribosomal RNA gene (LrRNA),using ICG 20 as an outgroup. Indels were coded according to Hibbett et al. (1995). Bootstrap values higher than 50% are indicatedabove branches. Decay indices (preceded by a d) are also indicated. On the right, ICGs to which isolates belong are indicated as wellas host trees and geographic origin.
attempt was made to measure the evolution of sexual incom-patibility in clade IIa of the H. crustuliniforme complex in-directly.
In a previous paper (Aanen and Kuyper 1999), we showedthat there are some cases of partial compatibility betweenICGs, that is, individual isolates usually could be placed intoICGs, but some combinations between ICGs were compatibleas well. Isolate 605–1, however, was compatible with allmembers of both ICGs 3 and 4. This multi-ICG-compatibilityof a single isolate has also been observed in the genus Pleu-rotus (Petersen and Ridley 1996). Seven additional siblingmonokaryons of this 605–1 were also compatible with mostisolates of both ICGs 3 and 4 (unpubl. results). Furthermore,sib monokaryons of 605–1 were also compatible with sib
monokaryons of 671–2, showing that the compatibility be-tween 605 and ICGs 2, 3, and 4 is not limited to the twomonokaryons that were originally used (unpubl. data).
Comparison of Mitochondrial and Nuclear Phylogenies andthe Level of Compatibility
In sexual Agaricales, nuclei migrate bidirectionally afterpairing, but mitochondria do not, resulting in dikaryotic my-celia that are mosaic for mitochondria from the two mono-karyons. Heteroplasmy only occurs in the region where thehyphae of the two monokaryons meet. Recombination be-tween different populations of mitochondrial genomes in anatural population of Armillaria gallica has been shown to

1201REPRODUCTIVE ISOLATION IN HEBELOMA
FIG. 5. Partition homogeneity test results. (A) Partition homogeneity test for all 20 strains for which both datasets were available. (B)PHT for the 16 strains other than ICG 1. See text for details.
occur (Saville et al. 1998). This implies that we cannot con-sider the mitochondrial sequences as strictly clonal; the fre-quency of recombination in Basidiomycete mitochondria innature remains to be determined.
In the present study, members of ICGs generally formedmonophyletic groups. ICGs 1 and 2 formed monophyleticgroups in the nuclear and the mitochondrial phylogenies.ICGs 3 and 4 formed monophyletic groups in the mitochon-drial phylogeny, but were paraphyletic in the nuclear. In otherphylogenies based on nuclear ribosomal DNA, paraphyleticICGs have been found as well (Pleurotus, Vilgalys and Sun1994; a different group of Hebeloma, Aanen et al. 2000). InPleurotus, these paraphyletic ICGs consisted of monophyleticpopulations from different continents. Future studies in theH. crustuliniforme aggregate should include strains from abroader geographic area to consider the possibility of findingother paraphyletic ICGs.
The main difference between the nuclear and mitochondrialtrees was the position of ICG 1. There are several possibleexplanations for this incongruence. One explanation is hor-izontal transmission of the mitochondrial intron sequences.At low taxonomic levels, good concordance has been foundbetween intron phylogenies and organismal phylogenies(Shinohara et al. 1996), but several unexpected cases of hor-izontal transfer have been documented (e.g., Cho et al. 1998;Nishida et al. 1998). However, because we did not find longopen reading frames coding for endonucleases or maturasesin this intron, it is probably not mobile (Shinohara et al. 1996;Cho et al. 1998). Therefore, horizontal transfer of the intronas an explanation for the found incongruence does not seemvery plausible. A different explanation is that ancestral poly-morphisms in the ribosomal DNA have been maintained withdifferent lineage sorting in the different clades. The nuclearsequence we have used is part of the tandemly repeated rDNAcluster. Although this repeated array of sequences is generallybelieved to be homogenized by the process of concerted evo-
lution (Elder and Turner 1995), ancestral polymorphisms canbe maintained and the majority type of alleles in an arraycan fluctuate (O’Donnell and Cigelnik 1997). As a conse-quence, incongruence between phylogenies based on ribo-somal sequences and phylogenies based on other sequenceshas been found (O’Donnell and Cigelnik 1997). One of thefactors that may contribute to the maintenance of ancestralpolymorphisms is that there are several repeats dispersed onnonhomologous chromosomes, because concerted evolutionis thought to be less efficient between chromosomal loci thanwithin a chromosomal locus (O’Donnell and Cigelnik 1997).However, basidiomycetes studied to date present a uniquerDNA locus (Selosse et al. 1996). Therefore, maintenance ofancestral polymorphisms is not a likely explanation for thedifferent position of ICG 1 in the nuclear and mitochondrialphylogenies.
Although intersterility barriers between most Agaricalesspecies are generally considered to be absolute and naturalhybridization is therefore considered to be rare or absent(Boidin 1986; Vilgalys and Sun 1994; Vilgalys et al. 1996),a different explanation for the incongruence between the nu-clear and mitochondrial trees is that ICG 1 has a hybrid origin,with different donors of the mitochondrial and nuclear se-quences. Under this scenario, the mitochondrial donor of ICG1 was the ancestral lineage leading to ICG 2, because ICG1 is the sister group of 2 in the mitochondrial phylogeny; thedonor of the nuclear sequence was the ancestral lineage lead-ing to ICG 5, because ICGs 1 and 5 are sister groups in thenuclear phylogeny. Recently, in several Ascomycete speciesin which a sexual state never had been observed, recombi-nation was found by comparing phylogenies based on singlegene sequences (Burt et al. 1996; Geiser et al. 1998). Com-paring phylogenies based on single gene sequences may bea strong tool to detect rare genetic exchange between ICGsin Basidiomycetes. The relative uniformity in both nuclearand mitochondrial sequences of ICG 1 (no differences in
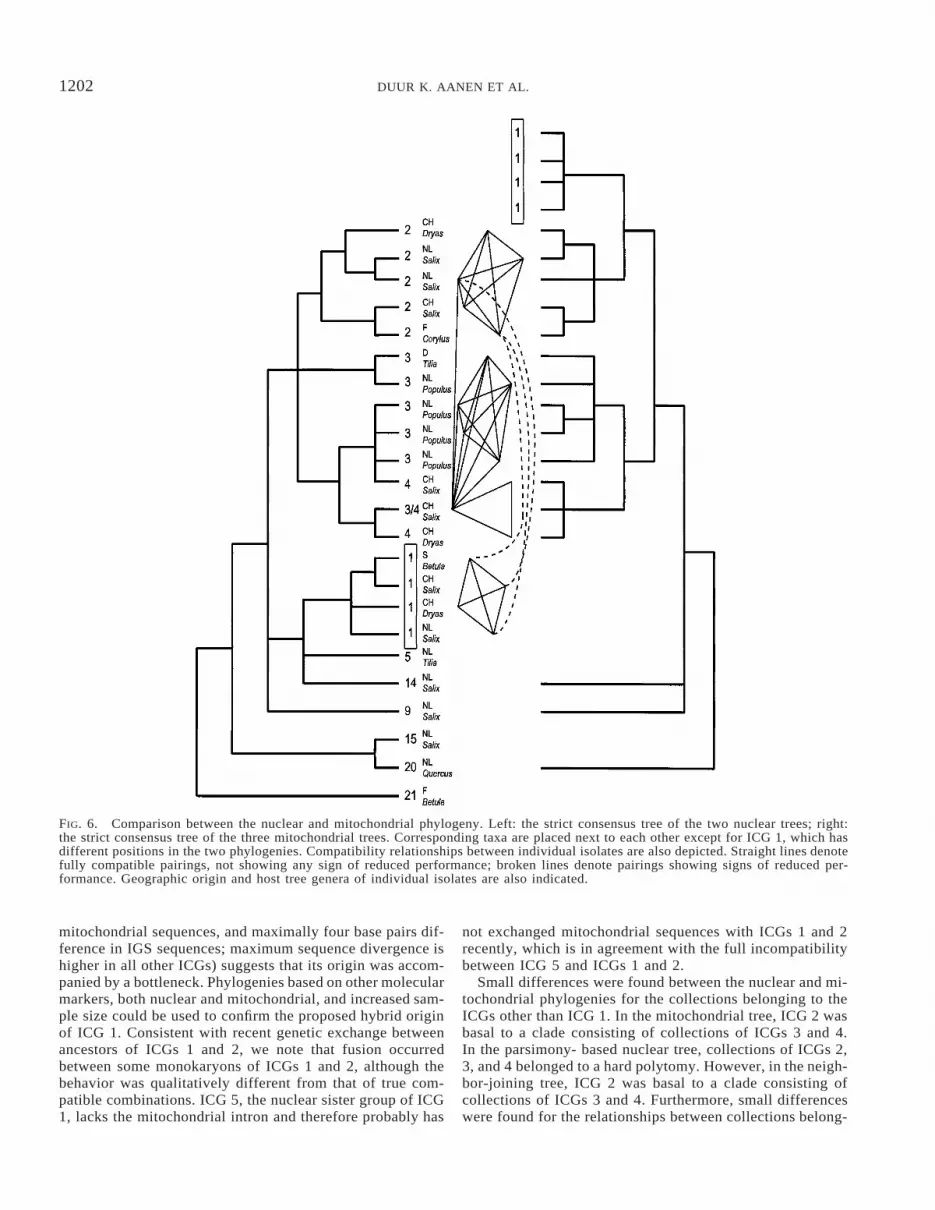
1202 DUUR K. AANEN ET AL.
FIG. 6. Comparison between the nuclear and mitochondrial phylogeny. Left: the strict consensus tree of the two nuclear trees; right:the strict consensus tree of the three mitochondrial trees. Corresponding taxa are placed next to each other except for ICG 1, which hasdifferent positions in the two phylogenies. Compatibility relationships between individual isolates are also depicted. Straight lines denotefully compatible pairings, not showing any sign of reduced performance; broken lines denote pairings showing signs of reduced per-formance. Geographic origin and host tree genera of individual isolates are also indicated.
mitochondrial sequences, and maximally four base pairs dif-ference in IGS sequences; maximum sequence divergence ishigher in all other ICGs) suggests that its origin was accom-panied by a bottleneck. Phylogenies based on other molecularmarkers, both nuclear and mitochondrial, and increased sam-ple size could be used to confirm the proposed hybrid originof ICG 1. Consistent with recent genetic exchange betweenancestors of ICGs 1 and 2, we note that fusion occurredbetween some monokaryons of ICGs 1 and 2, although thebehavior was qualitatively different from that of true com-patible combinations. ICG 5, the nuclear sister group of ICG1, lacks the mitochondrial intron and therefore probably has
not exchanged mitochondrial sequences with ICGs 1 and 2recently, which is in agreement with the full incompatibilitybetween ICG 5 and ICGs 1 and 2.
Small differences were found between the nuclear and mi-tochondrial phylogenies for the collections belonging to theICGs other than ICG 1. In the mitochondrial tree, ICG 2 wasbasal to a clade consisting of collections of ICGs 3 and 4.In the parsimony- based nuclear tree, collections of ICGs 2,3, and 4 belonged to a hard polytomy. However, in the neigh-bor-joining tree, ICG 2 was basal to a clade consisting ofcollections of ICGs 3 and 4. Furthermore, small differenceswere found for the relationships between collections belong-

1203REPRODUCTIVE ISOLATION IN HEBELOMA
FIG. 7. One of two most parsimonious trees of length 126 (CI 5 0.90, RI 5 0.89; using only informative variation, length 5 63, CI5 0.79) based on the nuclear and mitochondrial sequences used in this study, for the taxa other than ICG 1 of which both sequenceswere determined. ICG 20 was used as the outgroup. In the tree not shown, collection 617 was basal to the clade consisting of collections679, 686, and 699 of ICG 3. Indels were coded according to Hibbett et al. (1995). On the right, ICGs to which isolates belong areindicated.
ing to ICGs 3 and 4. The partition homogeneity test indicatedthat the gene trees for the taxa other than ICG 1 were notsignificantly different (Fig. 5B). Therefore, a combined anal-ysis was performed on the taxa other than ICG 1 (Fig. 7). Inthis tree, ICG 2 was basal to a clade consisting of collectionsof ICGs 3 and 4. This means that there is a positive correlationbetween the relative age of the MRCA and the level of in-compatibility between ICGs 2 and 3/4, because compatibilitybetween ICGs 2 and 3/4 was much lower than compatibilitybetween ICGs 3 and 4 (0.6% and 15%, respectively). There-fore, the origin of incompatibility between ICGs 2 and 3/4is consistent with the predictions of divergence-first models.
In the nuclear and combined analyses, ICGs 3 and 4 didnot form monophyletic groups. Nuclear trees, with the con-
straint that ICGs 3 and 4 formed a monophyletic group, werefour steps longer. The lack of a correlation between the rel-ative age of the MRCA and the level of incompatibility forICGs 3 and 4 shows that (partial) incompatibility betweenICGs 3 and 4 has arisen without strong divergence. The eco-logical (and to a lesser extent geographical) differences foundbetween ICGs 3 and 4 (see below) suggest that that selectionfor incompatibility, associated with host tree preference, mayhave been important in the evolution of incompatibility be-tween these two ICGs.
Ecological Differentiation
Ecological (and to a lesser extent geographical) differen-tiation between ICGs has been found to some degree (Table

1204 DUUR K. AANEN ET AL.
FIG. 8. A scenario for the evolution of partial compatibility in the Hebeloma crustuliniforme aggregate based on the mitochondrial andnuclear phylogenies. On the trees the different levels of compatibility (%) within and between the different ICGs are indicated. Thepercentage of partial compatibility between ICGs 1 and 2 has been placed in quotes because it is qualitatively different from the otherexamples of partial compatibility found (see text for details).
2), although the limited sampling does not allow firm con-clusions. From the partially compatible ICGs 3 and 4, ICG4 has been found exclusively in the alpine zone (as well asstrain 605 that could not be uniquely placed into ICG 3 or4), whereas ICG 3 has only been found in western Europe.Collections of ICG 4 have usually been found with the dwarfshrub Salix retusa, whereas collections of ICG 3 have mainlybeen found with Populus but also with tree-forming Salixspecies. The alpine collections all fructify early in the autumn(end of August–beginning of September), whereas the non-alpine collections all fructify later (end September–Novem-ber). These three modes of isolation, that is, temporal, eco-logical, and geographical, could have contributed to differ-entiation between ICGs 3 and 4. Alternatively, disruptiveselection for adaptation to one of these two niches could haveprovided the selection pressure for the evolution of incom-patibility between individuals adapted to the different niches.Collections of ICGs 1 and 2 were usually associated withSalix, both with dwarf shrubs (in alpine as well as in non-alpine habitats) as with larger trees. ICG 5, which is com-pletely incompatible with all other ICGs, has not been foundwith members of Salicaceae, whereas all other ICGs in cladeIIa show a preference for this family of hosts.
Conclusions
We have found different relationships between the levelof compatibility and the relative age of the MRCA for dif-ferent ICGs. On the one hand, the evolution of incompati-bility between ICGs 2 and 3/4 is most consistent with theclass of divergence- first models, because we found a positivecorrelation between the relative age of the MRCA and thelevel of incompatibility for ICG 2 versus 3/4. On the otherhand, the lack of such a correlation for ICGs 3 and 4 showsthat (partial) incompatibility between these ICGs has arisenwithout strong divergence. The ecological (and to a lesserextent geographical) differences found between ICGs 3 and
4 suggest that selection for incompatibility, associated withhost tree preference, may have been important in the evo-lution of incompatibility between these two ICGs. The in-congruence between the nuclear and mitochondrial trees forICG 1 could be explained by a hybrid origin of this ICG,with different donors of the mitochondrial and nuclear se-quences. Phylogenies based on other nuclear and mitochon-drial markers should confirm the proposed hybrid origin.
In Figure 8, a scenario for the evolution of (partial) in-compatibility in the H. crustuliniforme aggregate is illustrat-ed. In this scenario, ICG 1 has a hybrid origin and the in-compatibility between ICGs 2 and 3/4 is older than the in-compatibility between ICGs 3 and 4.
In summary, our data suggest that aspects of both classesof models, divergence first and incompatibility first, haveplayed a role in the evolution of incompatibility in this spe-cies complex.
ACKNOWLEDGMENTS
The authors wish to thank M. Schilthuizen, T. Boekhout,A. Debets, J. Bruggeman, and two anonymous reviewers forcritically reviewing this manuscript. These investigationswere supported by the Netherlands Organisation for ScientificResearch (NWO).
LITERATURE CITED
Aanen, D. K., and T. W. Kuyper. 1999. Intercompatibility tests inthe Hebeloma crustuliniforme complex in northwestern Europe.Mycologia 91:783–795.
Aanen, D. K., T. W. Kuyper, T. Boekhout, and R. F. Hoekstra.2000. Phylogenetic relationships in the genusHebeloma basedon ITS 1 and 2 sequences, with special emphasis on the He-beloma crustuliniforme complex. Mycologia 92:269–281.
Anderson, J. B., and E. Stasovsky. 1992. Molecular phylogeny ofnorthern hemisphere species of Armillaria. Mycologia 84:505–516.
Anderson, J. B., K. Korhonen, and R. C. Ullrich. 1980. Relation-

1205REPRODUCTIVE ISOLATION IN HEBELOMA
ships between European and North American biological speciesof Armillaria mellea. Exp. Mycol. 4:87–95.
Avise, J. C., and K. Wollenberg. 1997. Phylogenetics and the originof species. Proc. Natl. Acad. Sci. USA. 94:7748–7755.
Boidin, J. 1986. Intercompatibility and the species concept in thesaprobic Basidiomycotina. Mycotaxon 26:319–336.
Brasier, C. M. 1987. The dynamics of fungal speciation. Pp. 231–260 in A. D. M. Rayner, C. M. Brasier, and D. Moore, eds.Evolutionary biology of the fungi. Cambridge Univ. Press, Cam-bridge, U.K.
Bremer, K. 1988. The limits of amino-acid sequence data in an-giosperm phylogenetic reconstruction. Evolution 42:795–803.
Bresinsky, A., M. Fischer, B. Meixner, and W. Paulus. 1987. Spe-ciation in Pleurotus. Mycologia 79:234–245.
Burt, A., D. A. Carter, G. L. Koenig, T. J. White, and J. W. Taylor.1996. Molecular markers reveal cryptic sex in the human path-ogen Coccidioides immitis. Proc. Natl. Acad. Sci. USA 93:770–773.
Casselton, L. A., and N. S. Olesnicky. 1998. Molecular genetics ofmate recognition in basidiomycete fungi. Microbiol. Mol. Biol.Rev. 62:55–70.
Chamuris, G. P. 1991. Speciation in the Peniophors complex. My-cologia 83:736–742.
Chase, T. E., and R. C. Ullrich. 1990a. Genetic basis of biologicalspecies in Heterobasidion annosum: Mendelian determinants.Mycologia 82:67–72.
———. 1990b. Five genes determining intersterility in Heterobas-idion annosum. Mycologia 82:73–81.
Cho, Y., Y.-L. Qiu, P. Kuhlman, and J. D. Palmer. 1998. Explosiveinvasion of plant mitochondria by a group I intron. Proc. Natl.Acad. Sci. USA. 95:14244–14249.
Coyne, J. A., and H. A. Orr. 1999. The evolutionary genetics ofspeciation. Pp. 1–36 in A. E. Magurran and R. M. Ray, eds.Evolution of biological diversity. Oxford University Press, Ox-ford, U.K.
Diehl, S. R., and G. L. Bush. 1989. The role of habitat preferencein adaptation and speciation. Pp. 345–365 in D. Otte and J. A.Endler, eds. Speciation and its consequences. Sinauer, Sunder-land, MA.
Donoghue, M. J., R. G. Olmsted, J. F. Smith, and J. D. Palmer.1992. Phylogenetic relationships of Dipsacales based on rbcLsequences. Ann. MO Bot. Gard. 79:333–345.
Elder, J. F., Jr., and B. J. Turner. 1995. Concerted evolution ofrepetitive DNA sequences in eukaryotes. Q. Rev. Biol. 70:297–330.
Eriksson, T. 1998. Autodecay. Vers. 4.0. Program distributed bythe author, Department of Botany, University of Stockholm,Stockholm.
Erland, S., B. Henrion, F. Martin, L. A. Glover, and I. J. Alexander.1994. Identification of the ectomycorrhizal basidiomycete Ty-lospora fibrillosa Donk by RFLP analysis of the PCR-amplifiedITS and IGS regions of ribosomal DNA. New Phytol. 126:525–532.
Farris, J. S., M. Kallersjo, A. G. Kluge, and C. Bult. 1995. Testingsignificance of incongruence. Cladistics 10:315–319.
Fries, N., and G. M. Mueller. 1984. Incompatibility systems, cul-tural features and species circumscriptions in the ectomycor-rhizal genus Laccaria (Agaricales). Mycologia 76:633–642.
Fries, N. 1985. Intersterility groups in Paxillus involutus. Myco-taxon 24:403–409.
Fries, N., and W. Neumann. 1990. Sexual incompatibility in Suillusluteus and S. granulatus. Mycol. Res. 94:64–70.
Garbelotto, M., W. J. Otrosina, F. W. Cobb, and T. D. Bruns. 1998.The European S and F intersterility groups of Heterobasidionannosum may represent sympatric protospecies. Can. J. Bot. 76:397–409.
Geiser, D. M., J. I. Pitt, and J. W. Taylor. 1998. Cryptic speciationand recombination in the aflatoxin-producing fungus Aspergillusflavus. Proc. Natl. Acad. Sci. USA 95:388–393.
Hallenberg, N. 1991. Speciation and distribution in Corticiaceae(Basidiomycetes). Pl. Syst. Evol. 177:93–110.
Henrion, B., F. Le Tacon, and F. Martin. 1992. Rapid identification
of genetic variation of ectomycorrhizal fungi by amplificationof ribosomal RNA genes. New Phytol. 122:289–298.
Hibbett, D. S., Y. Fukumasa-Nakai, A. Tsuneda, and M. J. Dono-ghue. 1995. Phylogenetic diversity in shiitake inferred from nu-clear ribosomal DNA sequences. Mycologia 87:618–638.
Higgins, D. G., A. J. Bleasby, and R. Fuchs. 1992. CLUSTAL V:improved software for multiple sequence alignment. Comput.Appl. Biosci. 8:189–191.
Hillis, D. M., and J. J. Bull. 1993. An empirical test of bootstrappingas a method for assessing confidence in phylogenetic analysis.Syst. Biol. 42:182–192.
Huelsenbeck, J. P., J. J. Bull, and C. W. Cunningham. 1996. Com-bining data in phylogenetic analysis. Trends Ecol. Evol. 11:152–158.
Kishino, H., and M. Hasegawa. 1989. Evaluation of the maximumlikelihood estimate of the evolutionary tree topologies fromDNA sequence data, and the branching order in Hominoidea. J.Mol. Evol. 29:170–179.
Kretzer, A., Y. Li, T. Szaro, and T. D. Bruns. 1996. Internal tran-scribed spacer sequences from 38 recognized species of SuillusAWNAU Lro: phylogenetic and taxonomic consequences. My-cologia 88:776–785.
Kuyper, T. W., and T. Boekhout. 1995. Hebeloma (Fr.:Fr.) Kumm.Pp. 218–226 in E. J. M. Arnolds, T. W. Kuyper, and M. E.Noordeloos, eds. Overzicht van de paddestoelen in Nederland.Nederlandse Mycologische Vereniging, Wijster, The Nether-lands.
Lambowitz, A. M., and M. Belfort. 1993. Introns as mobile geneticelements. Annu. Rev. Biochem. 62:587–622.
Maynard-Smith, J. 1966. Sympatric speciation. Am. Nat. 100:637–650.
Mayr, E. 1963. Animal species and evolution. Harvard Univ. Press,Cambridge, MA.
Mueller, G. M., and M. Gardes. 1991. Intra- and interspecific re-lations within Laccaria bicolor sensu lato. Mycol. Res. 95:592–601.
Nishida, H., Y. Tajiri, and J. Sugiyama. 1998. Multiple origins offungal group I introns located in the same position of nuclearSSU rRNA gene. J. Mol. Evol. 46:442–448.
O’Donnell, K., and E. Cigelnik. 1997. Two divergent intragenomicrDNA ITS2 types within a monophyletic lineage of the fungusFusarium are nonorthologous. Mol. Phyl. Evol. 7:103–116
Peterson, R. H. 1995. There’s more to mushroom than meets theeye: mating studies in the Agaricales. Mycologia 87:1–17.
Petersen, R., and G. S. Ridley. 1996. A New Zealand Pleurotuswith multiple-species sexual compatibility. Mycologia 88:198–207.
Piercey-Normore, M. D., K. N. Egger, and J. A. Berube. 1998.Molecular phylogeny and evolutionary divergence of NorthAmerican biological species of Armillaria. Mol. Phyl. Evol. 10:49–66.
Saitou, N., and M. Nei. 1987. The neighbor-joining method: a newmethod for reconstructing phylogenetic trees. Mol. Biol. Evol.10:49–66.
Saville, B. J., Y. Kohli, and J. B. Anderson. 1998. mtDNA recom-bination in a natural population. Proc. Natl. Acad. Sci. USA 95:1331–1335.
Schilthuizen, M. 1999. Cloning Odysseus and the seed of speciation.Trends Ecol. Evol. 14:90–91.
Selosse, M.-A., G. Costa, C. Di Battista, F. Le Tacon, and F. Martin.1996. Meiotic segregation and recombination of the intergenicspacer of the ribosomal DNA in the ectomycorrhizal basidio-mycete Laccaria bicolor. Curr. Genet. 30:332–337.
Shinohara, M. L., K. F. LoBuglio, and S. O. Rogers. 1996. Group-I intron family in the nuclear ribosomal RNA small subunit genesof Cenococcum geophilum isolates. Curr. Genet. 29:377–387.
Swofford, D. L. 1998. PAUP*: Phylogenetic analysis using parsi-mony. Test vers.* 4.0. Smithsonian Institution, Washington, DC.
Templeton, A. R. 1983. Phylogenetic inference from restrictionendonuclease cleavage site maps with particular reference to theevolution of humans and the apes. Evolution 37:221–244.
———. 1998. The role of molecular genetics in speciation studies.Pp. 131–156 in R. Desalle and B. Schierwater, eds. Molecular

1206 DUUR K. AANEN ET AL.
approaches to ecology and evolution. Birkhauser, Basel, Swit-zerland.
Vilgalys, R. 1991. Speciation and species concepts in the Collybiadryophila complex. Mycologia 83:758–773.
Vilgalys, R., and B. L. Sun. 1994. Ancient and recent patterns ofgeographic speciation in the oyster mushroom Pleurotus re-vealed by phylogenetic analysis of ribosomal DNA sequences.Proc. Natl. Acad. Sci. USA 91:4599–4603.
Vilgalys, R., J.-M. Moncalvo, S.-R. Liou, and M. Volovsek. 1996.Recent advances in molecular systematics of the genus Pleu-
rotus. Pp. 91–101 in D. J. Royse, ed. Mushroom biology andmushroom products. Pennsylvania State Univ. Press, UniversityPark, PA.
Wells, K., and G. J. Wong. 1989. Partial intersterility and evidenceof allopatric speciation in Exidiopsis plumbescens (Exidiaceae).Mycologia 81:567–586.
Worrall, J. J. 1997. Somatic incompatibility in basidiomycetes. My-cologia 89:24–36.
Corresponding Editor: D. Waller