s-allyl cysteine attenuates oxidative stress associated cognitive impairment and neurodegeneration...
TRANSCRIPT

B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te /b ra i n res
Research Report
S-allyl cysteine attenuates oxidative stress associatedcognitive impairment and neurodegeneration in mouse modelof streptozotocin-induced experimental dementia ofAlzheimer's type
Hayate Javeda, Mohd. Moshahid Khana,1, Andleeb Khana, Kumar Vaibhava,Ajmal Ahmada,2, Gulrana Khuwajaa,3, Md. Ejaz Ahmeda, Syed Shadab Razaa,Mohammad Ashafaqa, Rizwana Tabassuma, M. Saeed Siddiquia, O.M. El-Agnafb,Mohammed M. Safhic, Fakhrul Islama,⁎,4,§
aNeurotoxicology laboratory, Department of Medical Elementology and Toxicology (Fund for the Improvement of Science and Technologysponsored by DST and Special Assistance Programme sponsored by UGC), Jamia Hamdard (Hamdard University), Hamdard Nagar,New Delhi-110062, IndiabDepartment of Biochemistry, Faculty of Medicine and Health Sciences, United Arab Emirates University, P.O. Box No. 17666,Al-Ain, United Arab EmiratescDepartment of Pharmacology, Faculty of Pharmacy, Jazan University, Gizan, Saudi Arabia
A R T I C L E I N F O
⁎ Corresponding author at: Department of PhaE-mail address: [email protected] (
1 Present address: Department of Internal M2 Present address: Department of Neurolog3 Present address: Department of Pharmac4 Present address: Department of Pharmac§ Dedicated to my supervisor Prof. Dr. Mah
0006-8993/$ – see front matter © 2011 Elsevidoi:10.1016/j.brainres.2011.02.072
A B S T R A C T
Article history:Accepted 22 February 2011Available online 1 March 2011
S-allyl cysteine (SAC), a sulfur containing amino acid derived from garlic, has beenreported to have antioxidant, anti-cancer, antihepatotoxic and neurotrophic activity.This study was designed to examine the pre-treatment effects of SAC on cognitivedeficits and oxidative damage in the hippocampus of intracerebroventricularstreptozotocin (ICV-STZ)-infused mice. Mice pre-treated with SAC (30 mg/kg) andvehicle (intraperitoneal; once daily for 15 days) were bilaterally injected with ICV-STZ(2.57 mg/kg body weight), whereas sham rats received the same volume of vehicle. Thepre-treatment of this drug to Swiss albino mice has prevented the cognitive andneurobehavioral impairments. An increased latency and path length were observed inlesion, i.e. streptozotocin (STZ) group as compared to sham group and these wereprotected significantly in STZ group pre-treated with SAC. Levels of reduced glutathione(GSH) and its dependent enzymes (Glutathione peroxidase [GPx] and glutathionereductase [GR]) were decreased in STZ group as compared to sham group and pre-treatment of STZ group with SAC has protected their activities significantly. Conversely,the elevated level of thiobarbituric acid reactive substances (TBARS) in STZ group was
Keywords:Oxidative stressCognitive impairmentS-allyl cysteineStreptozotocinAntioxidant
rmacology, Faculty of Pharmacy, Jazan University, Gizan, Saudi Arabia. Fax: +96673217441.F. Islam).edicine, Carver College of Medicine, University of Iowa, IA 52242, USA.
y, Georgia Health Science University, Augusta, GA-30904.eutical Chemistry, Faculty of Pharmacy, Jazan University, Gizan, Kingdom of Saudi Arabia.ology, Faculty of Pharmacy, Jazan University, Gizan, Saudi Arabia.di Hasan on his PLATINUM JUBILEE BIRTHDAY.
er B.V. All rights reserved.
134 B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
attenuated significantly in SAC pre-treated group when compared with STZ lesionedgroup. Apoptotic parameters like DNA fragmentation, expression of Bcl2 and p53 wereprotected by the pre-treatment of SAC against STZ induced cognitive impairment. Thisstudy concludes that intervention of SAC could prevent free radicals associateddeterioration of cognitive functions and neurobehavioral activities.
© 2011 Elsevier B.V. All rights reserved.
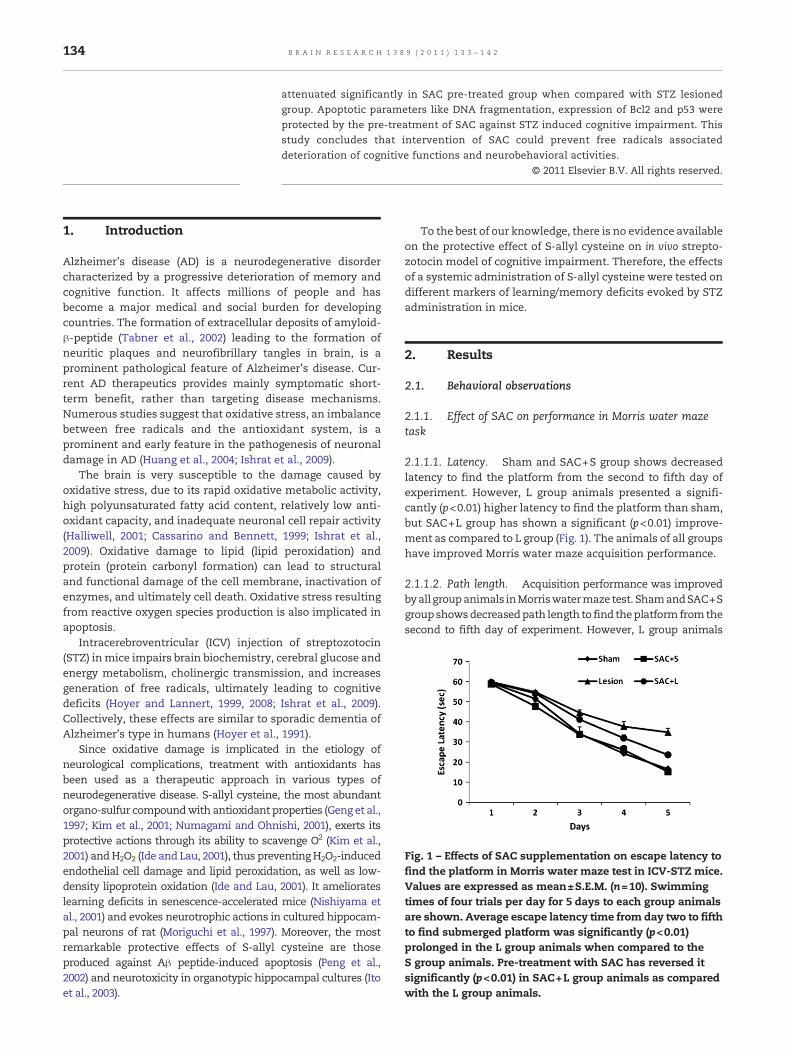
Fig. 1 – Effects of SAC supplementation on escape latency tofind the platform in Morris water maze test in ICV-STZ mice.Values are expressed as mean±S.E.M. (n=10). Swimmingtimes of four trials per day for 5 days to each group animalsare shown. Average escape latency time from day two to fifthto find submerged platform was significantly (p<0.01)prolonged in the L group animals when compared to theS group animals. Pre-treatment with SAC has reversed itsignificantly (p<0.01) in SAC+L group animals as comparedwith the L group animals.
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disordercharacterized by a progressive deterioration of memory andcognitive function. It affects millions of people and hasbecome a major medical and social burden for developingcountries. The formation of extracellular deposits of amyloid-β-peptide (Tabner et al., 2002) leading to the formation ofneuritic plaques and neurofibrillary tangles in brain, is aprominent pathological feature of Alzheimer's disease. Cur-rent AD therapeutics provides mainly symptomatic short-term benefit, rather than targeting disease mechanisms.Numerous studies suggest that oxidative stress, an imbalancebetween free radicals and the antioxidant system, is aprominent and early feature in the pathogenesis of neuronaldamage in AD (Huang et al., 2004; Ishrat et al., 2009).
The brain is very susceptible to the damage caused byoxidative stress, due to its rapid oxidative metabolic activity,high polyunsaturated fatty acid content, relatively low anti-oxidant capacity, and inadequate neuronal cell repair activity(Halliwell, 2001; Cassarino and Bennett, 1999; Ishrat et al.,2009). Oxidative damage to lipid (lipid peroxidation) andprotein (protein carbonyl formation) can lead to structuraland functional damage of the cell membrane, inactivation ofenzymes, and ultimately cell death. Oxidative stress resultingfrom reactive oxygen species production is also implicated inapoptosis.
Intracerebroventricular (ICV) injection of streptozotocin(STZ) inmice impairs brain biochemistry, cerebral glucose andenergy metabolism, cholinergic transmission, and increasesgeneration of free radicals, ultimately leading to cognitivedeficits (Hoyer and Lannert, 1999, 2008; Ishrat et al., 2009).Collectively, these effects are similar to sporadic dementia ofAlzheimer's type in humans (Hoyer et al., 1991).
Since oxidative damage is implicated in the etiology ofneurological complications, treatment with antioxidants hasbeen used as a therapeutic approach in various types ofneurodegenerative disease. S-allyl cysteine, the most abundantorgano-sulfur compoundwith antioxidant properties (Geng et al.,1997; Kim et al., 2001; Numagami and Ohnishi, 2001), exerts itsprotective actions through its ability to scavenge O2 (Kim et al.,2001) andH2O2 (Ide and Lau, 2001), thus preventingH2O2-inducedendothelial cell damage and lipid peroxidation, as well as low-density lipoprotein oxidation (Ide and Lau, 2001). It ameliorateslearning deficits in senescence-accelerated mice (Nishiyama etal., 2001) and evokes neurotrophic actions in cultured hippocam-pal neurons of rat (Moriguchi et al., 1997). Moreover, the mostremarkable protective effects of S-allyl cysteine are thoseproduced against Aβ peptide-induced apoptosis (Peng et al.,2002) and neurotoxicity in organotypic hippocampal cultures (Itoet al., 2003).
To the best of our knowledge, there is no evidence availableon the protective effect of S-allyl cysteine on in vivo strepto-zotocin model of cognitive impairment. Therefore, the effectsof a systemic administration of S-allyl cysteine were tested ondifferent markers of learning/memory deficits evoked by STZadministration in mice.
2. Results
2.1. Behavioral observations
2.1.1. Effect of SAC on performance in Morris water mazetask
2.1.1.1. Latency. Sham and SAC+S group shows decreasedlatency to find the platform from the second to fifth day ofexperiment. However, L group animals presented a signifi-cantly (p<0.01) higher latency to find the platform than sham,but SAC+L group has shown a significant (p<0.01) improve-ment as compared to L group (Fig. 1). The animals of all groupshave improved Morris water maze acquisition performance.
2.1.1.2. Path length. Acquisition performance was improvedbyall groupanimals inMorriswatermaze test. ShamandSAC+Sgroupshowsdecreasedpath length to find theplatformfromthesecond to fifth day of experiment. However, L group animals
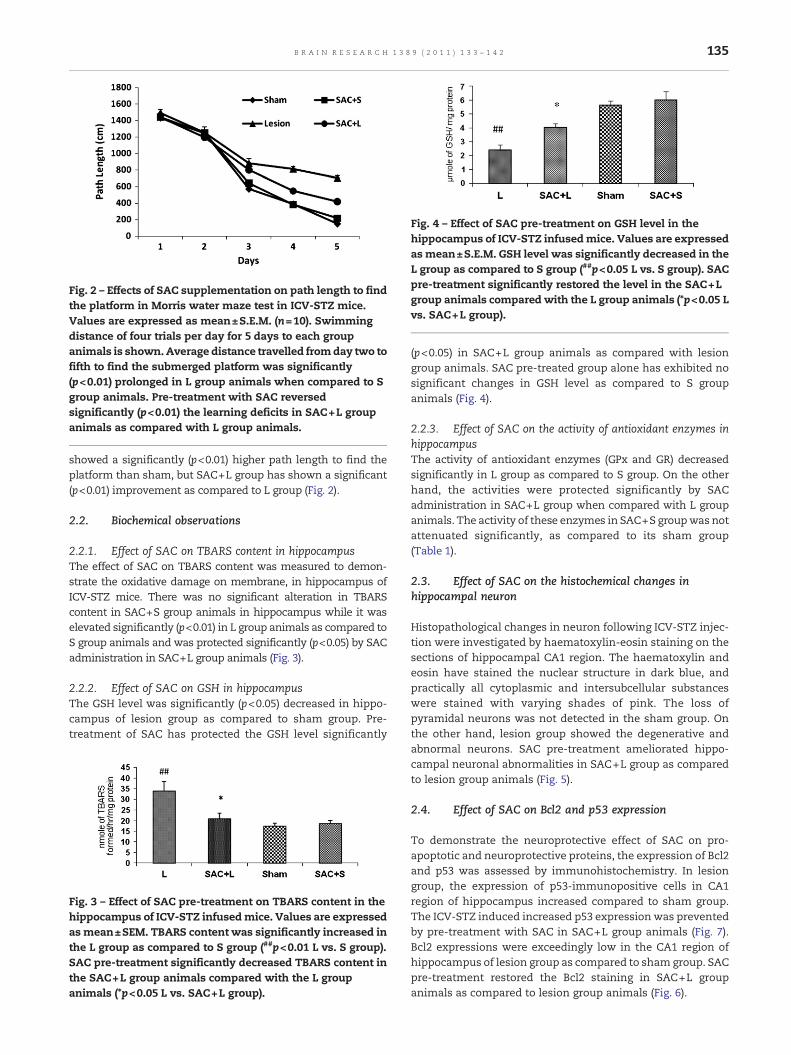
Fig. 2 – Effects of SAC supplementation on path length to findthe platform in Morris water maze test in ICV-STZ mice.Values are expressed as mean±S.E.M. (n=10). Swimmingdistance of four trials per day for 5 days to each groupanimals is shown.Average distance travelled fromday two tofifth to find the submerged platform was significantly(p<0.01) prolonged in L group animals when compared to Sgroup animals. Pre-treatment with SAC reversedsignificantly (p<0.01) the learning deficits in SAC+L groupanimals as compared with L group animals.
Fig. 4 – Effect of SAC pre-treatment on GSH level in thehippocampus of ICV-STZ infusedmice. Values are expressedas mean±S.E.M. GSH level was significantly decreased in theL group as compared to S group (##p<0.05 L vs. S group). SACpre-treatment significantly restored the level in the SAC+Lgroup animals compared with the L group animals (*p<0.05 Lvs. SAC+L group).
135B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
showed a significantly (p<0.01) higher path length to find theplatform than sham, but SAC+L group has shown a significant(p<0.01) improvement as compared to L group (Fig. 2).
2.2. Biochemical observations
2.2.1. Effect of SAC on TBARS content in hippocampusThe effect of SAC on TBARS content was measured to demon-strate the oxidative damage on membrane, in hippocampus ofICV-STZ mice. There was no significant alteration in TBARScontent in SAC+S group animals in hippocampus while it waselevated significantly (p<0.01) in L group animals as compared toS group animals and was protected significantly (p<0.05) by SACadministration in SAC+L group animals (Fig. 3).
2.2.2. Effect of SAC on GSH in hippocampusThe GSH level was significantly (p<0.05) decreased in hippo-campus of lesion group as compared to sham group. Pre-treatment of SAC has protected the GSH level significantly
Fig. 3 – Effect of SAC pre-treatment on TBARS content in thehippocampus of ICV-STZ infusedmice. Values are expressedas mean±SEM. TBARS content was significantly increased inthe L group as compared to S group (##p<0.01 L vs. S group).SAC pre-treatment significantly decreased TBARS content inthe SAC+L group animals compared with the L groupanimals (*p<0.05 L vs. SAC+L group).
(p<0.05) in SAC+L group animals as compared with lesiongroup animals. SAC pre-treated group alone has exhibited nosignificant changes in GSH level as compared to S groupanimals (Fig. 4).
2.2.3. Effect of SAC on the activity of antioxidant enzymes inhippocampusThe activity of antioxidant enzymes (GPx and GR) decreasedsignificantly in L group as compared to S group. On the otherhand, the activities were protected significantly by SACadministration in SAC+L group when compared with L groupanimals. The activity of these enzymes in SAC+S groupwas notattenuated significantly, as compared to its sham group(Table 1).
2.3. Effect of SAC on the histochemical changes inhippocampal neuron
Histopathological changes in neuron following ICV-STZ injec-tion were investigated by haematoxylin-eosin staining on thesections of hippocampal CA1 region. The haematoxylin andeosin have stained the nuclear structure in dark blue, andpractically all cytoplasmic and intersubcellular substanceswere stained with varying shades of pink. The loss ofpyramidal neurons was not detected in the sham group. Onthe other hand, lesion group showed the degenerative andabnormal neurons. SAC pre-treatment ameliorated hippo-campal neuronal abnormalities in SAC+L group as comparedto lesion group animals (Fig. 5).
2.4. Effect of SAC on Bcl2 and p53 expression
To demonstrate the neuroprotective effect of SAC on pro-apoptotic and neuroprotective proteins, the expression of Bcl2and p53 was assessed by immunohistochemistry. In lesiongroup, the expression of p53-immunopositive cells in CA1region of hippocampus increased compared to sham group.The ICV-STZ induced increased p53 expression was preventedby pre-treatment with SAC in SAC+L group animals (Fig. 7).Bcl2 expressions were exceedingly low in the CA1 region ofhippocampus of lesion group as compared to shamgroup. SACpre-treatment restored the Bcl2 staining in SAC+L groupanimals as compared to lesion group animals (Fig. 6).
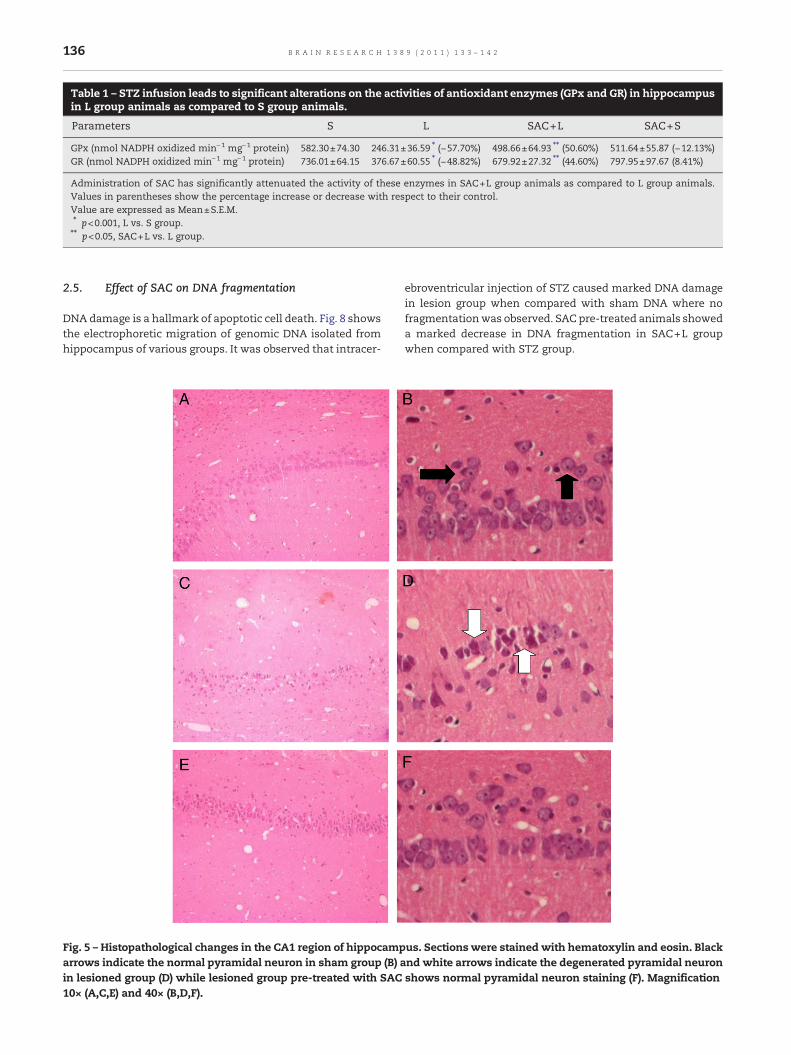
Table 1 – STZ infusion leads to significant alterations on the activities of antioxidant enzymes (GPx and GR) in hippocampusin L group animals as compared to S group animals.
Parameters S L SAC+L SAC+S
GPx (nmol NADPH oxidized min−1 mg−1 protein) 582.30±74.30 246.31±36.59 ⁎ (−57.70%) 498.66±64.93 ⁎⁎ (50.60%) 511.64±55.87 (−12.13%)GR (nmol NADPH oxidized min−1 mg−1 protein) 736.01±64.15 376.67±60.55 ⁎ (−48.82%) 679.92±27.32 ⁎⁎ (44.60%) 797.95±97.67 (8.41%)
Administration of SAC has significantly attenuated the activity of these enzymes in SAC+L group animals as compared to L group animals.Values in parentheses show the percentage increase or decrease with respect to their control.Value are expressed as Mean±S.E.M.⁎ p<0.001, L vs. S group.⁎⁎ p<0.05, SAC+L vs. L group.
136 B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
2.5. Effect of SAC on DNA fragmentation
DNA damage is a hallmark of apoptotic cell death. Fig. 8 showsthe electrophoretic migration of genomic DNA isolated fromhippocampus of various groups. It was observed that intracer-
Fig. 5 – Histopathological changes in the CA1 region of hippocamparrows indicate the normal pyramidal neuron in sham group (B) ain lesioned group (D) while lesioned group pre-treated with SAC10× (A,C,E) and 40× (B,D,F).
ebroventricular injection of STZ caused marked DNA damagein lesion group when compared with sham DNA where nofragmentationwas observed. SAC pre-treated animals showeda marked decrease in DNA fragmentation in SAC+L groupwhen compared with STZ group.
us. Sections were stained with hematoxylin and eosin. Blacknd white arrows indicate the degenerated pyramidal neuronshows normal pyramidal neuron staining (F). Magnification
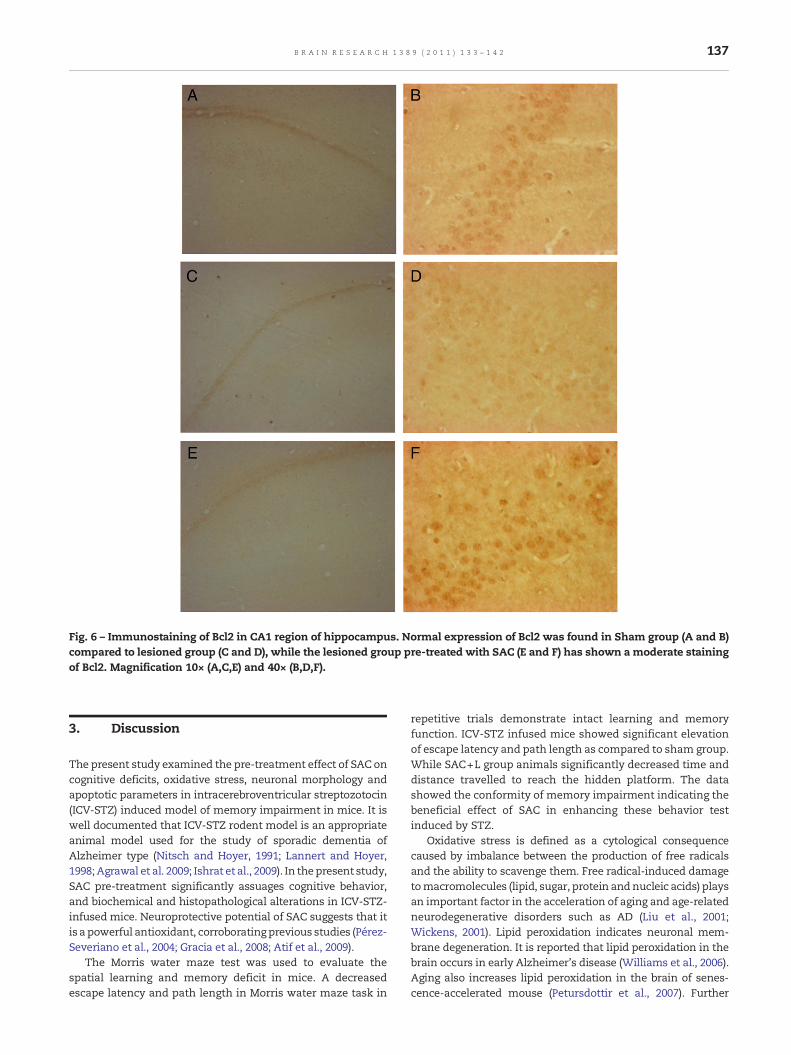
Fig. 6 – Immunostaining of Bcl2 in CA1 region of hippocampus. Normal expression of Bcl2 was found in Sham group (A and B)compared to lesioned group (C and D), while the lesioned group pre-treated with SAC (E and F) has shown a moderate stainingof Bcl2. Magnification 10× (A,C,E) and 40× (B,D,F).
137B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
3. Discussion
The present study examined the pre-treatment effect of SAC oncognitive deficits, oxidative stress, neuronal morphology andapoptotic parameters in intracerebroventricular streptozotocin(ICV-STZ) induced model of memory impairment in mice. It iswell documented that ICV-STZ rodent model is an appropriateanimal model used for the study of sporadic dementia ofAlzheimer type (Nitsch and Hoyer, 1991; Lannert and Hoyer,1998; Agrawal et al. 2009; Ishrat et al., 2009). In thepresent study,SAC pre-treatment significantly assuages cognitive behavior,and biochemical and histopathological alterations in ICV-STZ-infused mice. Neuroprotective potential of SAC suggests that itis a powerful antioxidant, corroboratingprevious studies (Pérez-Severiano et al., 2004; Gracia et al., 2008; Atif et al., 2009).
The Morris water maze test was used to evaluate thespatial learning and memory deficit in mice. A decreasedescape latency and path length in Morris water maze task in
repetitive trials demonstrate intact learning and memoryfunction. ICV-STZ infused mice showed significant elevationof escape latency and path length as compared to sham group.While SAC+L group animals significantly decreased time anddistance travelled to reach the hidden platform. The datashowed the conformity of memory impairment indicating thebeneficial effect of SAC in enhancing these behavior testinduced by STZ.
Oxidative stress is defined as a cytological consequencecaused by imbalance between the production of free radicalsand the ability to scavenge them. Free radical-induced damagetomacromolecules (lipid, sugar, protein andnucleic acids) playsan important factor in the acceleration of aging and age-relatedneurodegenerative disorders such as AD (Liu et al., 2001;Wickens, 2001). Lipid peroxidation indicates neuronal mem-brane degeneration. It is reported that lipid peroxidation in thebrain occurs in early Alzheimer's disease (Williams et al., 2006).Aging also increases lipid peroxidation in the brain of senes-cence-accelerated mouse (Petursdottir et al., 2007). Further

Fig. 7 – Immunostaining of p53 in CA1 region of hippocampus. Overexpression of p53 was found in lesioned group (A and B)compared to sham group (C and D), while the lesioned group pre-treated with SAC (E and F) has shown a moderate staining ofp53. However, the sham group has shown almost negligible staining. Magnification 10× (A,C,E) and 40× (B,D,F).
138 B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
results from the biochemical estimation indicate the significantincrease in TBARS content and noticeable decrease in the levelof reduced glutathione, and its dependent enzyme glutathioneperoxidase and glutathione reductase in the brains of ICV-STZinfused mice compared to sham group animals.
The antioxidant system requires reduced glutathione (GSH),a tripeptide and an essential antioxidant, which is responsibleto accept free radicals in the brain tissue (Dringen et al., 2000). Iteliminates H2O2 and organic peroxides by glutathione peroxi-dase (GPx) (Meister, 1988). During free radical clearance, oxy-radicals are reduced by glutathione peroxidase at the cost ofreduced glutathione to formglutathionedisulfide (GSSG). GSH isfurtherproducedby redoxrecycling, inwhichGSSG is reduced toGSH by glutathione reductase (GR) with an expenditure of oneNADPHmolecule. Reduced level of GSH impairs H2O2 clearanceand endorse formation of OH•, the most toxic molecule to thebrain, leading to more free radical level and oxidative stress(Sun, 1990; Dringen et al., 2000). There are several reports aboutmodulatory effect of SAC on lipid peroxidation and antioxidant
enzymes following injuries like hypoxia/ischemia and braininjuries (Pérez-Severianoetal., 2004;Graciaet al., 2008;Atif et al.,2009). In agreement with these findings, we also found thatSAC significantly reduced the TBARS level and increased inthe activity of antioxidant enzymes in hippocampus followingSTZ induction. This response of SAC could be attributed to itspotential antioxidant property (Kim et al., 2001). Changesobserved in oxidative stress parameters with cognitive dys-function in STZ infused mice parallel with the earlierreports (Saxena et al., 2008) suggesting that STZ inducedlearning and memory impairment is associated with oxidativestress in mice.
Several protein familieshighly conservedduringevolutionareconsidered to be specifically involved in regulating apoptotic celldeath such as Bax and Bcl-2, which form homo- and hetero-dimers in vivo. The relative expression of these two proteinsdetermines cell deathor survival.WhenBcl-2 is inexcess, theBcl-2 homodimer predominates and promotes cell survival. It hasbeen reported that overexpression of Bcl-2 protects a variety of

Fig. 8 – Genomic DNA agarose gel electrophoresis: No DNAladdering was observed in hippocampal tissue of the shamgroup or SAC+S group (figure not shown). DNA laddering wasdetected in the hippocampal tissue of lesion group (L) whichwasattenuatedby thepre-treatmentwithSAC inSAC+Lgroupanimals. Molecular weight (MW) lane showed 1 kb DNAstandard.
139B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
mammalian cells from apoptosis induced by diverse deathstimuli, including chemotherapeutic agents, radiation, tumornecrosis factor andglutamate (Zhonget al., 1993;Hartmannet al.,1999; Domen et al., 1998; Howard et al., 2002). Bcl-2 has beenproposed to prevent apoptosis by regulating an antioxidantpathway (Hockenbery et al., 1993). In the agreement of above fact,we observeddownexpression of bcl-2 protein in ICV-STZ infusedmice which is protected by the pre-treatment of SAC.
The tumorsuppressorandnuclear transcription factorp53 isatetramer phosphoprotein that can regulate severalmajor cellularfunctions including gene transcription, DNA synthesis, DNArepair, cell cycle regulation, senescence, and cell death (Sherr andMcCormick, 2002; Hofseth et al., 2004). The upregulation of p53 inresponse to a diverse array of cellular insults ranging fromischemia/hypoxia and excitotoxicity to oxidative stress inmultiple neuronal populations suggests that p53 is a key factorinvolved inneuronaldeath in response todifferent formsofacuteinsults and chronic neurodegenerative conditions. The regula-tion of p53 in a variety of neurodegenerative disorders raised thepossibility that inhibitors of p53 might prove effective insuppressing the associated mechanisms of neuron cell death.Our findings proposed that SAC reduced the expression of p53positive cells. This view is supported by the down-regulation ofp53 protein expression by SAC pre-treatment.
In neuropathological situations, apoptosis might contrib-ute to neuronal degeneration through a number of factorssuch as ischemia/hypoxia, radiation, oxidative stress, excit-atory, neurotoxicity, and so on (Thompson, 1995). It is wellrecognized that the DNA of apoptotic cells is cleaved intomultiple bands activated by endonucleases and pro-apoptoticmolecules like apoptotic protease activating factor-1 (Apaf-1)
and pro-caspase-9 which recruits caspase-3 leading to theformation of “DNA ladders.” However, these ladders are notthe only criterion for identifying apoptosis (Collins et al., 1992).The DNA ladders do not appear in some conditions becausethe apoptotic cells are easily and rapidly cleared by neighbour-ing cells; alternatively, as little as 2% of apoptotic cellsamongst necrotic cells can be detected as a ladder. In ourstudy, the DNA extracted from hippocampus of sham, lesionand SAC+L group was separated onto agarose gel electropho-resis showing fragmentation in lesion group whereas DNAfragmentation was not found in sham group and thisfragmentation was almost completely restore by pre-treat-ment of SAC in SAC+L group.
The beneficial effects of SAC in spatial memory processingmay be due to its ability to protect the cholinergic function,prevent neuronal damage possibly through its antioxidantpotential. Thus, these findings suggest that SAC is analternative for treating the cognitive impairment. Furtherinvestigation into the neuroprotective potential and mecha-nisms of SAC is required to determine, whether it can be aneffective cure for cognitive impairment.
4. Experimental procedures
4.1. Chemicals and reagents
Glutathione disulfide (GSSG), reduced glutathione (GSH), gluta-thione reductase (GR), nicotinamide adenine dinucleotidephosphate reduced form (NADPH), 5,5′-dithio bis-2-nitroben-zoic acid (DTNB), thiobarbituric acid (TBA), trichloroacetic acid(TCA), ethylene diamine tetraacetic acid (EDTA) and Streptozo-tocin (STZ) were purchased from Sigma-Aldrich, Chemicals Pvt.Ltd., India. Monoclonal Bcl-2 and p53 antibody were purchasedfromBio Vision, U.S.A. and anti-mouse IgGwas purchased fromJackson Immuno Research Laboratories Inc., West Groove, PA.S-allyl cysteine (SAC) was purchased from LGC Prochem,Bangalore, India. Other chemicals were of analytical reagentgrade.
4.2. Animals
The experiments were carried out in one-year-old Swissalbino mice, weighing 35–40 g, obtained from the CentralAnimal House facility of Hamdard University. They were keptin colony cages and maintained under standard housingconditions (room temperature of 25±2 °C and relative humid-ity 45–55%) with 12 h light/dark reverse cycles. The standardrodent pellet diet and water were available ad libitum.Experiments were conducted in accordance with the AnimalEthics Committee of the University.
4.3. Experimental protocol
4.3.1. Experiment IExperiment I was carried out to evaluate the pre-treatmenteffect of SAC (30 mg/kg body weight in normal saline [i.p.])supplementation for 15 days on the contents of TBARS,GSH, and activity of antioxidant enzymes. The mice weredivided into four groups of 10 animals each. Group I was sham

140 B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
operated vehicle treated control (S) group. Group II wassham operated and pre-treated with SAC (SAC+S) group.Group III was ICV-STZ infused and vehicle treated lesion (L)group. Group IV was ICV-STZ infused and pre-treated with SAC(SAC+L). The SACdoseused in this experimentwasdeterminedfrom previous study (Mostafa et al., 2000). Schematic figure fortime course in days of the experimental design timeline.
4.3.2. Experiment IIExperiment IIwas carriedout to evaluate thepre-treatment effectof SAC supplementation on DNA fragmentation, histopatholog-ical analysis, immunostaining for Bcl2 and p53. The mice weredivided into four groups and each group has 14 mice.
4.4. Intracerebroventricular (ICV) infusion of streptozotocin
After 15 days of SAC pre-treatment, Swiss albino mice wereanesthetized with 685.71 mg/kg body weight of chloralhydrates intraperitoneally (i.p.) and placed on a stereotaxicframe and skull was exposed. The stereotaxic coordinates forlateral ventricle were measured accurately as anterio-posteri-or −0.8 mm, lateral±1.0 mm and dorso-ventral −3.0 mmrelative to bregma and ventral from dura with the tooth barset at 0 mm. Through a skull hole, a 28-gauge Hamilton®syringe of 5 μl attached to a micro-injector unit and piston ofthe syringe was lowered manually into each lateral ventricle.The lesion groups received a bilateral ICV injection of STZ(2.57 mg/kg, body weight in saline, 2 μl/injection). The shamgroups underwent the same surgical procedures, but samevolume of saline was injected instead of STZ.
4.5. Behavioral testing
The behavioral tests were started 2 weeks after ICV-STZinfusion. The experiment was performed between 9.00 a.m. to4.00 p.m. at standard laboratory conditions. Behavioral testswere performed and analyzed by a researcher blind to theexperimental conditions.
4.5.1. Morris water maze testSpatial learning andmemory of animals were tested in Morriswater maze (Morris, 1984). It consisted of a circular water tank(132 cm diameter and 60 cm height) that was filled 30 cm withwater (25±2 °C). A non-toxic white paint was used to renderthe water opaque. The pool was divided virtually into fourequal quadrants, labeled north–south–east–west. An escapeplatform (10 cm in diameter) was hidden 2 cm below thesurface of water on a fixed location in one of the fourquadrants of the pool. The platform remained in the samequadrant throughout experiment. Before the training started,micewere allowed to swim freely into the pool for 60 swithoutplatform. They were given four trials (once from each startingposition) per session for 5 days, each trial having a ceiling timeof 60 s and a trial interval of approximately 30 s. After climbingon to the platform, the animal remained there for 30 s beforethe commencement of the next trial. If mice failed to reach theescape platform within the maximum allowed time of 60 s, itwas gently placed on the platform and allowed to remainthere for the same interval of time. An overhead video camerawas connected to a videomonitor and computer software was
used to track the animals's path and to calculate the escapelatency and travelled distance (path length).
4.6. Biochemical analysis
4.6.1. Tissue preparationAfter 3 weeks of ICV-STZ infusion, animals were sacrificedand their brains were taken out quickly on ice to dissecthippocampus. The dissected brain part were homogenized at4° in 10 mM phosphate buffer (PB, pH 7.0) having 10 μl/mlprotease inhibitor to get 5% w/v homogenate. The homoge-nate was centrifuged at 800g for 5 min at 4 °C to separate thenuclear debris. The supernatant S1 was used for estimation oflipid peroxidation in terms of TBARS content. The remainingS1 was further centrifuged at 10,500g for 30 min at 4 °C to getthe post-mitochondrial supernatant (PMS) which was used forestimation of reduced glutathione and antioxidant enzymes.
4.6.2. TBARS contentTBARS content was estimated by the method of Utley et al.(1967) asmodified by Islam et al. (2002). Homogenate 0.1 mlwaspipetted into a 15×100 mm test tube and incubated at 37 °C in ametabolic shaker for 1 h. An equal volume of homogenate waspipetted into a centrifuge tube and placed at 0 °C andmarked at0 h incubation.After 1 hof incubation, 0.45mlof 5% (w/v) chilledTCA and 0.45ml 0.67% TBAwere added after that centrifuged at4000g for 10min. Thereafter, supernatant was transferred toother test tubes and placed in a boiling water bath for 10 min.The absorbance of pink colour produced was measured at535 nm. The TBARS content was calculated by using a molarextinction coefficient of 1.56×105 M−1 cm−1 and expressed asnanomoles of TBARS formed per hour mg−1 of protein.
4.6.3. Reduced glutathione (GSH) contentGSH content wasmeasured by themethod of Jollow et al. (1974)with slightmodification. PMSwasmixedwith4.0%sulfosalisylicacid (w/v) in 1:1 ratio (v/v). The samples were incubated at 4 °Cfor 1 h, and centrifuged at 4000g for 10min at 4 °C. The assaymixture contained 0.1 ml of supernatant, 1.0 mM DTNB and0.1 M phosphate buffer pH 7.4 in a total volume of 3.0 ml. Theyellow colour developed was read immediately at 412 nm in aspectrophotometer (Shimadzu-1601, Japan). The GSH contentwas calculated as nanomoles GSH mg−1 protein, using a molarextinction coefficient of 13.6×103 M−1 cm−1.
4.6.4. Estimation of glutathione peroxidase (GPx)GPx activity was determined by the method of Mohandas et al.(1984). The reaction assay consisted of phosphate buffer (0.05 M,pH 7.0), EDTA (1 mM), sodium azide (1 mM), glutathione reduc-tase (1 EU/ml), glutathione (1 mM), NADPH (0.2 mM), hydrogenperoxide (0.25 mM) and 0.1 ml of PMS in the final volume of 2 ml.The disappearance of NADPH at 340 nm was recorded at roomtemperature. The enzyme activity was calculated as nmolNADPH oxidized/min/mg/protein by using molar extinctioncoefficient of 6.22×103 M−1 cm−1.
4.6.5. Glutathione reductase (GR)Glutathione reductase activity was measured by the methodof Carlberg and Mannervik (1975) as modified by Mohandaset al. (1984). The reactionmixture consisted of phosphate buffer

141B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
(0.1 M, pH 7.6), NADPH (0.1 mM), EDTA (0.5 mM) and oxidizedglutathione (1mM) and 0.05ml of PMS in total volume of 1 ml.The enzyme activity was quantified at room temperature bymeasuring the disappearance of NADPH at 340 nm andcalculated as nmol NADPH oxidized/min/mg protein usingmolar extinction coefficient of 6.22×103 M−1 cm−1.
4.7. DNA isolation and fragmentation
Genomic DNA was extracted from a modified method ofSimantov et al. (1996). Briefly, tissue samples (50 mg) werehomogenized in 0.5 ml of digestion buffer (10 mM Tris–HCl,10mM NaCl, 25mM EDTA, 1% SDS, 1 mg/ml proteinase K, pH7.4). Then, sampleswere incubated at 37 °C for overnight. At theend of incubation, 50 μl of 5 MNaCl was added to samples. DNAwas extracted twice with a mixture of phenol: chloroform:isoamyl alcohol (25:24:1 v/v) and centrifuged at 3000g for 4 min.After extensive washing with ethanol (70%), DNA was recen-trifuged at 3000g for 4 min and resuspended in buffer (10 mMTris–HCL, 1 mM EDTA, pH 7.4) and incubated with RNAse A(0.5 mg/ml) for 30min.Thereafter,DNA (10 μl) fromeachsamplewas loaded onto 1% agarose gel, containing 0.5 μg/ml ethidiumbromide and electrophoresed for the analysis of DNA fragmen-tation which was detected with UV transilluminator.
4.8. Hematoxylin and eosin (H&E) stain
The animals were anesthetized with chloral hydrate on 22 dayof lesioning and perfused transcardially through ascendingaortawith 100 ml ice cold phosphate buffered saline (PBS 0.1 MpH 7.4) followed by 4% paraformaldehyde in cold PBS (0.1 M pH7.4). Brains were removed quickly, post fixed in the parafor-maldehyde solution for 48 h, and embedded with wax.Coronal sections having hippocampus 5 μm thickness wasdewaxed and stained with hematoxylin and eosin.
4.9. Immunohistochemistry
Immunohistochemistry was performed to detect the expres-sion of Bcl2 and p53 proteins. Coronal sections (5 μm thick) atthe level of hippocampus were dewaxed and processed forimmunohistochemical staining. The sections were collectedserially on gelatin coated slides and placed in 3% H2O2 inmethanol for 20 min at room temperature to eliminate theendogenous peroxidase activity. Slides were washed with PBSfor several times and pre-incubated in 1% bovine serumalbumin for 45 min at room temperature; thereafter, the slideswere incubated with primary antibody, anti Bcl2 monoclonalmouse (dilution 1:200) or anti p53monoclonal mouse (dilution1:200) at 4 °C for overnight. Then, section was incubated withbiotinylated donkey anti-mouse IgG (Jackson ImmunoRe-search USA, dilution 1:500) and avidin biotin complex (ABCkit from Vector Laboratories Ltd. UK). The slides were treatedwith 3,4-diaminobenzidine (Vector Laboratories Ltd. UK) andobserved under light microscope.
4.10. Protein content
Protein content was determined by the method of Lowry et al.(1951) using bovine serum albumin (BSA) as a standard.
4.11. Statistical analysis
Results are expressed as mean±S.E.M. Statistical analysis ofthe data was done by applying the analysis of variance(ANOVA), followed by Tukey's test. The p-value<0.05 wasconsidered statistically significant.
Acknowledgments
The authors thank the Department of Ayurveda, Yoga andNaturopathy, Unani, Siddha and Homoeopathy (AYUSH),Ministry of Health and Family Welfare, Government of India,New Delhi, for the financial assistance. Technical assistanceof Late Anil Kumar, Dharamvir Singh and Abdul Fitr is greatlyacknowledged.
R E F E R E N C E S
Agrawal, R., Tyagi, E., Shukla, R., Nath, C., 2009. A study of braininsulin receptors, AChE activity and oxidative stress in ratmodelof ICV STZ induced dementia. Neuropharmacology 56, 779–787.
Atif, F., Yousuf, S., Agrawal, S.K., 2009. S-allyl L-cysteinediminishes cerebral ischemia-induced mitochondrialdysfunctions in hippocampus. Brain Res. 1265, 128–137.
Carlberg, I., Mannervik, B., 1975. Purification and characterizationof the flavoenzyme glutathione reductase from rat liver. J. Biol.Chem. 250, 5475–5480.
Cassarino, D.S., Bennett, J.P., 1999. An evaluation of the role ofmitochondria in neurodegenerative diseases: mitochondrialmutations and oxidative pathology, protective nuclearresponses, and cell death in neurodegeneration. Brain Res.Brain Res. Rev. 29, 1–25.
Collins, R.J.,Harmon,B.V.,Gobé,G.C.,Kerr, J.F., 1992. InternucleosomalDNA cleavage should not be the sole criterion for identifyingapoptosis. Int. J. Radiat. Biol. 61, 451–453.
Domen, J., Gandy, K.L., Weissman, I.L., 1998. Systemicoverexpression of bcl-2 in the hematopoietic system protectstransgenic mice from the consequences of lethal irradiation.Blood 91, 2272–2282.
Dringen, R., Gutterer, J.M., Hirrlinger, J., 2000. Glutathionemetabolism in brain metabolic interaction between astrocytesand neurons in the defense against reactive oxygen species.Eur. J. Biochem. 267, 4912–4916.
Garcia, E., Limon, D., Perez-De La Cruz, V., Giordano, M.,Diaz-Muñoz, M., Maldonado, P.D., Herrera-Mundo, M.N.,Pedraza-Chaverri, J., Santamaria, A., 2008. Lipid peroxidation,mitochondrial dysfunction and neurochemical andbehavioural deficits in different neurotoxic models: protectiverole of S-allylcysteine. Free Radic. Res. 42, 892–902.
Geng, Z., Rong, Y., Lau, B.H., 1997. S-allyl cysteine inhibitsactivation of nuclear factor kappa B in human T cells. FreeRadic. Biol. Med. 23, 345–350.
Halliwell, B., 2001. Role of free radicals in the neurodegenerativediseases: therapeutic implications for antioxidant treatment.Drugs Aging 18, 685–716.
Hartmann, B.L., Geley, S., Loffler, M., Hattmannstorfer, R.,Strasser-Wozak, E.M., Auer, B., Kofler, R., 1999. Bcl-2 interfereswith the execution phase, but not upstream events, inglucocorticoid-induced leukemia apoptosis. Oncogene 18,713–719.
Hockenbery, D.M., Oltvai, Z.N., Yin, X., Milliman, C., Korsmeyer, S.J.,1993. Bcl-2 functions in an antioxidant pathway to preventapoptosis. Cell 75, 241–251.

142 B R A I N R E S E A R C H 1 3 8 9 ( 2 0 1 1 ) 1 3 3 – 1 4 2
Howard, S., Bottino, C., Brooke, S., Cheng, E., Giffard, R.G.,Sapolsky, R., 2002. Neuroprotective effects of bcl-2overexpression in hippocampal cultures: interactionswith pathways of oxidative damage. J. Neurochem. 83,914–923.
Hoyer, S., Nitsch, R., Oesterreich, K., 1991. Predominantabnormality in cerebral glucose utilization in late-onsetdementia of the Alzheimer type: a cross-sectional comparisonagainst advanced late-onset and incipient early-onsetcases. J. Neural Transm. Park. Dis. Dement. Sect. 31,1–14.
Hoyer, S., Lannert, H., 1999. Inhibition of the neuronal insulinreceptor causes Alzheimer-like disturbances in oxidative/energybrain metabolism and in behavior in adult rats. Ann. N. Y. Acad.Sci. 893, 301–303.
Hoyer, S., Lannert, H., 2008. Long-term effects of corticosterone onbehavior, oxidative and energy metabolism of parietotemporalcerebral cortex and hippocampus of rats: comparison tointracerebroventricular streptozotocin. J. Neural Transm. 115,1241–1249.
Hofseth, L.J., Hussain, S.P., Harris, C.C., 2004. p53: 25 years after itsdiscovery. Trends Pharmacol. Sci. 25, 177–181.
Huang, R.D., Moir, R.E., Tanzi, A.I., Bush, Rogers, J.T., 2004.Redox-active metals, oxidative stress, and Alzheimer's diseasepathology. Ann. NY Acad. Sci. 1012, 153–163.
Ide, N., Lau, B.H., 2001. Garlic compounds minimize intracellularoxidative stress and inhibit nuclear factor-κ B activation.J. Nutr. 131, 1020S–1026S.
Ishrat, T., Hoda,M.N., Khan,M.B., Yousuf, S., Ahmad,M., Khan,M.M.,Ahmad, A., Islam, F., 2009. Amelioration of cognitive deficits andneurodegeneration by curcumin in rat model of sporadicdementia of Alzheimer's type (SDAT). EuropeanNeuropsychopharmacology 2009 (19), 636–647.
Islam, F., Zia, S., Sayeed, I., Zafar, K.S., Ahmad, A.S., 2002. Seleniuminduced alteration on lipids, lipid peroxidation, and thiol groupin circadian rhythm centers of rat. Biological Trace ElementResearch 90, 1–12.
Ito, Y., Ito, M., Takagi, N., Saito, H., Ishige, K., 2003.Neurotoxicity induced by amyloid beta-peptide andibotenic acid in organotypic hippocampal cultures:protection by S-allyl-L-cysteine, a garlic compound.Brain Res. 985, 98–107.
Jollow, D.J., Mitchell, J.R., Zampaglione, N., Gillette, J.R., 1974.Bromobenzene-induced liver necrosis. Protective role ofglutathione and evidence for 3,4-bromobenzene oxide as thehepatotoxic metabolite. Pharmacology 11, 151–169.
Kim, K.M., Chun, S.B., Koo, M.S., Choi, W.J., Kim, T.W., Kwon, Y.G.,Chung, H.T., Billiar, T.R., Kim, Y.M., 2001. Differentialregulation of NO availability from macrophages andendothelial cells by the garlic component S-allyl cysteine.Free Radic. Biol. Med. 30, 747–756.
Lannert, H., Hoyer, S., 1998. Intracerebroventricularadministration of streptozotocin causes long-termdiminutions in learning and memory abilities and in cerebralenergy metabolism in adult rats. Behav. Neurosci. 112,1199–1208.
Liu, R., Liu, I.Y., Bi, X., Thompson, R.F., Doctrow, S.R., Malfroy, B.,Baudry, M., 2001. Reversal of age-related learning deficits andbrain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc. Natl Acad. Sci. USA 100,8526–8531.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., Randall, R.J., 1951. Proteinmeasurement with the Folin phenol reagent. J. Biol. Chem. 193,265–275.
Morris, R., 1984. Developments of a water-maze procedure forstudying spatial learning in the rat. J. Neurosci. Methods 11,47–60.
Meister, A., 1988. Glutathione metabolism and its selectivemodification. J. Biol. Chem. 263, 1708–17205.
Mohandas, J., Marshall, J.J., Duggin, G.G., Horvath, J.S., Tiller, D.,1984. Differential distribution of glutathione andglutathione related enzymes in rabbit kidneys: possibleimplication in analgesic neuropathy. Cancer Res. 44,5086–5091.
Moriguchi, T., Matsuura, H., Kodera, Y., Itakura, Y., Katsuki, H.,Saito, H., Nishiyama, N., 1997. Neurotrophic activity oforganosulfur compounds having a thioallyl group on culturedrat hippocampal neurons. Neurochem. Res. 22, 1449–1452.
Mostafa, M.G., Mima, T., Ohnishi, S.T., Mori, K., 2000.S-allylcysteine ameliorates doxorubicin toxicity in the heartand liver in mice. Planta Med. 66, 148–151.
Nishiyama, N., Moriguchi, T., Morihara, N., Saito, H., 2001.Ameliorative effect of S-allylcysteine, a major thioallylconstituent in aged garlic extract, on learning deficitsin senescence-accelerated mice. J. Nutr. 131, 1093S–1095S.
Nitsch, R., Hoyer, S., 1991. Local action of the diabetogenic drug,streptozotocin, on glucose and energy metabolism in rat braincortex. Neurosci. Lett. 128, 199–202.
Numagami, Y., Ohnishi, S.T., 2001. S-allylcysteine inhibits freeradical production, lipid peroxidation and neuronal damage inrat brain ischemia. J. Nutr. 131, 1100S–1105S.
Peng, Q., Buz'Zard, A.R., Lau, B.H., 2002. Neuroprotective effect ofgarlic compounds in amyloid-beta peptide-induced apoptosisin vitro. Med. Sci. Monit. 8, BR328–BR337.
Pérez-Severiano, F., Salvatierra-Sánchez, R., Rodríguez-Pérez, M.,Cuevas-Martínez, E.Y., Guevara, J., Limón, D., Maldonado, P.D.,Medina-Campos, O.N., Pedraza-Chaverrí, J., Santamaría, A.,2004. S-allylcysteine prevents amyloid-beta peptide24 inducedoxidative stress in rat hippocampus and ameliorates learningdeficits. Eur. J. Pharmacol. 489, 197–202.
Petursdottir, A.L., Farr, S.A., Morley, J.E., Banks, W.A., Skuladottir,G.V., 2007. Lipid peroxidation in brain during aging in thesenescence-accelerated mouse (SAM). Neurobiol. Aging 28,1170–1178.
Saxena, G., Singh, S.P., Agrawal, R., Nath, C., 2002. Effect ofdonepezil and tacrine on oxidative stress in intracerebralstreptozotocin-induced model of dementia in mice. Eur. J.Pharmacol. 581, 283–289.
Simantov, R., Blinder, E., Ratovitski, T., Tauber, M., Gabbay, M.,Porat, S., 1996. Dopamine-induced apoptosisin human neuronal cells: inhibition by nucleic acidsantisense to the dopamine transporter. Neuroscience 74,39–50.
Sherr, C.J., McCormick, F., 2002. The RB and p53 pathways incancer. Cancer Cell 2, 103–111.
Sun, Y., 1990. Free radicals, antioxidant enzymes, and carcinogenesis. Free Radic. Biol. Med. 8, 583–599.
Tabner, B.J., Turnbull, S., El-Agnaf, O.M., Allsop, D., 2002.Formation of hydrogen peroxide and hydroxyl radicals fromAh and a-synuclein as a possible mechanism of cell death inAlzheimer's disease and Parkinson's disease. Free Radic. Biol.Med. 32, 1076–1083.
Thompson, C.B., 1995. Apoptosis in the pathogenesis andtreatment of disease. Science 267 (5203), 1456–1462.
Utley, H.G., Bernheim, F., Hockstein, P., 1967. Effect of sulfhydrylreagent on peroxidation in microsome. Arch. Biochem.Biophys. 260, 521–531.
Wickens, A.P., 2001. Ageing and the free radical theory. Respir.Physiol. 28, 379–391.
Williams, T.I., Lynn, B.C., Markesbery, W.R., Lovell, M.A., 2006.Increased levels of 4-hydroxynonenal and acrolein, neurotoxicmarkers of lipid peroxidation, in the brain in mild cognitiveimpairment and early Alzheimer's disease. Neurobiol. Aging27, 1094–1099.
Zhong, L.T., Sarafian, T., Kane, D.J., Charles, A.C., Mah, S.P.,Edwards, R.H., Bredesen, D.E., 1993. bcl-2 inhibits death ofcentral neural cells induced by multiple agents. Proc. NatlAcad. Sci. USA 90, 4533–4537.