recent advances in the synthesis of unnatural α-amino acids
TRANSCRIPT

Current Organic Chemistry, 2007, 11, 801-832 801
1385-2728/07 $50.00+.00 © 2007 Bentham Science Publishers Ltd.
Recent Advances in the Synthesis of Unnatural -Amino Acids
A. Perdih and M. Sollner Dolenc*
Faculty of Pharmacy, University of Ljubljana, Slovenia
Abstract: The synthesis of unnatural -amino acids is an area of research that has gained a lot of attention in recent years. The availability of synthetic methods for novel unnatural amino acid derivatives and related compounds will be a critical point in the de novo design of molecules that mimic the conformation of the natural, active peptides. These molecules (peptidomimetics) are designed to display high receptor affinity and selectivity, in addition to enhanced bioavailability and metabolic stability.
This review focuses on selected recent synthetic methodologies leading to unnatural -amino acids including chiral catalysts that enable enantioselective synthesis and microwave-assisted synthesis. Solid phase synthesis and construction of organometallic amino acids reveal the scope of influence that this field has in organic chemistry. Additionally, the article is aimed to provide a brief insight into the biosynthetic approaches to the synthesis of -amino acid derivatives, and to give the reader an appreciation of the field.
INTRODUCTION
Peptides are carriers of a variety of functions in living organisms. They act as neurotransmitters, neuromodulators, hormones, paracrine factors, cytokines and antigens, and influence essentially all vital physiological processes via inter- and intra-cellular communication and by signal trans-duction mediated through various classes of receptors [1, 2]. Peptides are generally poor drug candidates due to their limitations, characterized by fast hydrolytic cleavage, poor penetration of membranes, rapid photolytic degradation, conformational instability and unfavorable pharmacokinetics [3]. Numerous methods have been examined to overcome these problems and to improve the bioavailability of native and synthetic peptides, but these have met with only limited success to date [4]. For these reasons much effort has been expended to find ways to replace biologically active parts of peptides with non-peptide structures, termed peptido-mimetics, in the hope of obtaining orally active entities [5]. The scope of research in this field has expanded rapidly and at present constitutes an important approach to drug design and discovery [6, 7].
One of the possible strategies in the development of peptidomimetic agents, besides the popular bioisosteric amide bond replacements and mimetics of peptide secondary structure, is the incorporation of unnatural -amino acid derivatives. Conformationally restricted, non-proteinogenic
-amino acids have a great potential in elucidating the bio-active conformation of peptides. It must be emphasized that there are only a few amino acid analogs that facilitate suitable and predictable restrictions of conformational flexi-bility without drastically changing the stereo-electronic pro-perties of the initial peptide [8]. In order to retain biological activity, constraints must affect the conformation of the backbone and simultaneously enable crucial side-chain inter-actions with the receptor [1,6].
*Address correspondence to this author at the Faculty of Pharmacy, University of Ljubljana, Asker eva 7, 1000 Ljubljana, Slovenia, Phone: +386-1-4769-572; Fax: +386-1-4258-031; E-mail: [email protected]
Incorporation of unnatural -amino acids can specifically restrict the rotation of N - C , C -C(O), C(O)-NH bonds and side-chain conformations by covalent or noncovalent steric interactions [1,6]. There are many examples, ranging from simple -methylated amino acids to proline mimetics and various unsaturated , -amino acids [9].
Unnatural -amino acids incorporated into biologically active peptides and proteins modify their activity, stability, bioavailability and binding specificity [1,6]. Furthermore, incorporation of unnatural amino acids into various enzymes has been used to evaluate protein folding, protein function and signal transduction [10-12]. For this reason, incor-poration of constrained amino acids and related compounds into peptides is a critical component of efforts to understand the proteome and its relation to life, health and disease. The desire to develop enantiomerically pure unnatural amino acids, given the importance of the geometric distribution of side chain functional groups, pushes further not only the barriers of asymmetric synthesis but also the invention and development of enantioselective chromatographic separation methods that enable adequate separation of the amino acid enantiomers [13-15].
Interest in the design, synthesis and application of novel unnatural amino acids in the fields of peptide and com-binatorial chemistry is universal and captures the imagi-nation of many synthetic and peptide chemists. The aim of this review is to present selected recent synthetic approaches associated with unnatural -amino acid synthesis. As many procedures are published every year, our main aim is to provide the reader with an overall picture of this exciting field of peptidomimetic design. One must realize that, despite the myriad of syntheses that already exist, no single method is applicable to the design of every amino acid derivatives.
I. NOVEL SYNTHETIC METHODS FOR UN-NATURAL -AMINO ACIDS
1. Monosubstituted Unnatural Amino Acids
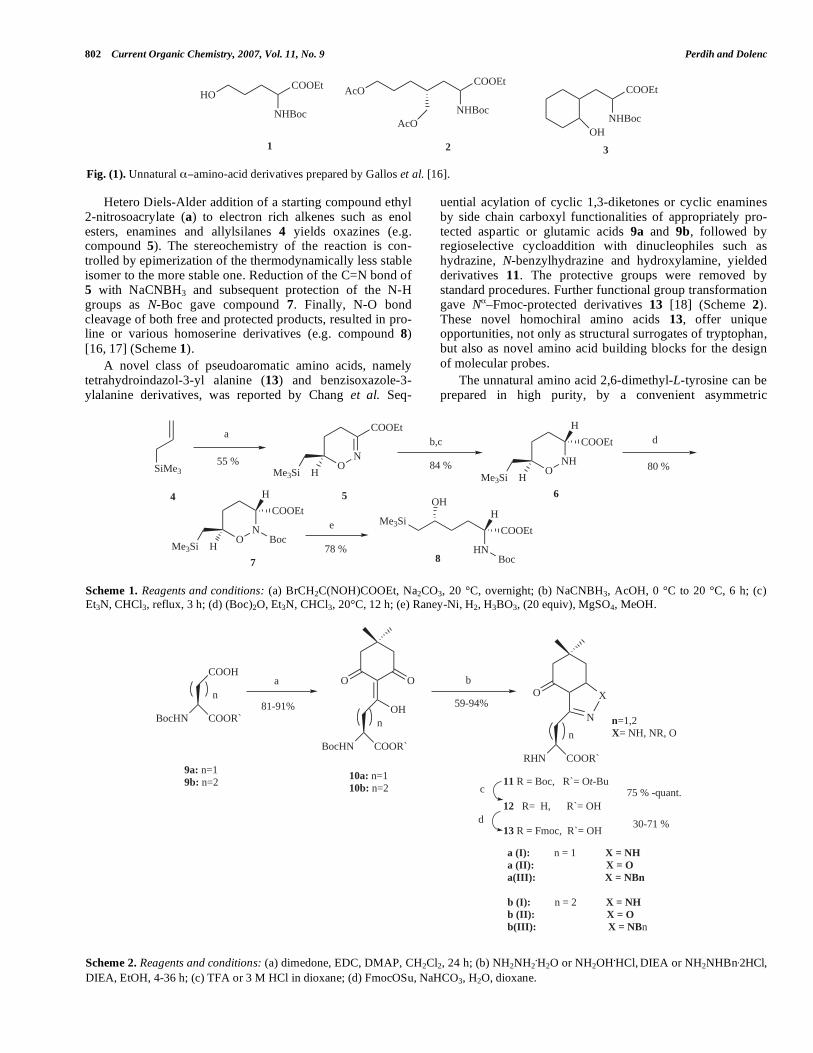
Gallos et al. reported the synthesis of racemic non-proteinogenic amino acids e.g. compounds 1-3 (Fig. 1).
802 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
Hetero Diels-Alder addition of a starting compound ethyl 2-nitrosoacrylate (a) to electron rich alkenes such as enol esters, enamines and allylsilanes 4 yields oxazines (e.g. compound 5). The stereochemistry of the reaction is con-trolled by epimerization of the thermodynamically less stable isomer to the more stable one. Reduction of the C=N bond of 5 with NaCNBH3 and subsequent protection of the N-H groups as N-Boc gave compound 7. Finally, N-O bond cleavage of both free and protected products, resulted in pro-line or various homoserine derivatives (e.g. compound 8) [16, 17] (Scheme 1).
A novel class of pseudoaromatic amino acids, namely tetrahydroindazol-3-yl alanine (13) and benzisoxazole-3-ylalanine derivatives, was reported by Chang et al. Seq-
uential acylation of cyclic 1,3-diketones or cyclic enamines by side chain carboxyl functionalities of appropriately pro-tected aspartic or glutamic acids 9a and 9b, followed by regioselective cycloaddition with dinucleophiles such as hydrazine, N-benzylhydrazine and hydroxylamine, yielded derivatives 11. The protective groups were removed by standard procedures. Further functional group transformation gave N –Fmoc-protected derivatives 13 [18] (Scheme 2). These novel homochiral amino acids 13, offer unique opportunities, not only as structural surrogates of tryptophan, but also as novel amino acid building blocks for the design of molecular probes.
The unnatural amino acid 2,6-dimethyl-L-tyrosine can be prepared in high purity, by a convenient asymmetric
Fig. (1). Unnatural amino-acid derivatives prepared by Gallos et al. [16].
Scheme 1. Reagents and conditions: (a) BrCH2C(NOH)COOEt, Na2CO3, 20 °C, overnight; (b) NaCNBH3, AcOH, 0 °C to 20 °C, 6 h; (c) Et3N, CHCl3, reflux, 3 h; (d) (Boc)2O, Et3N, CHCl3, 20°C, 12 h; (e) Raney-Ni, H2, H3BO3, (20 equiv), MgSO4, MeOH.
Scheme 2. Reagents and conditions: (a) dimedone, EDC, DMAP, CH2Cl2, 24 h; (b) NH2NH2.H2O or NH2OH.HCl, DIEA or NH2NHBn.2HCl,
DIEA, EtOH, 4-36 h; (c) TFA or 3 M HCl in dioxane; (d) FmocOSu, NaHCO3, H2O, dioxane.
HO
NHBoc
COOEt
NHBoc
COOEt
AcO
AcO
NHBoc
COOEt
OH1 2 3
ON
COOEt
Me3Si HSiMe3 ONH
COOEt
Me3Si H
H
Me3Si HBocO
N
COOEt
H
COOEtMe3Si
HN
H
Boc
OH
ab,c
78 %
4
55 % 84 % 80 %
d
e
5 6
7 8
BocHN COOR`
COOH
BocHN COOR`
OO
OH
RHN COOR`
XO
N
n
nn
11 R = Boc, R`= Ot-Bu
12 R= H, R`= OH
13 R = Fmoc, R`= OH
c
d
a (I): n = 1 X = NH
a (II): X = O
a(III): X = NBn
b (I): n = 2 X = NH
b (II): X = O
b(III): X = NBn
75 % -quant.
30-71 %
9a: n=19b: n=2
10a: n=110b: n=2
a b
81-91% 59-94%
n=1,2X= NH, NR, O

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 803
synthesis, from 3,5-dimethylphenol on the kilogram scale. The key steps are modified palladium-catalyzed coupling of an aryl iodide 14 with methyl 2-acetamidoacrylate (15) and stereoselective hydrogenation of the resulting dehydroamino acid 16 using [Rh (1,5-COD)-(R,R-DIPAMP)]BF4 as catalyst, to yield the desired compound 17 [19] (Scheme 3).
Enantioselective synthesis of malonylphenylalanyl 23 and malonylmethylphenylalanyl derivatives uses 4-bromo-benzaldehyde diethyl acetal (18) as a starting material and converts it to the corresponding products by a four step synthetic pathway as published by Garbay et al. The palladium catalyzed cross-coupling reaction with di-tert-butyl malonate, yielded the aldehyde 19. A Horner-Emmons type olefination of the resulting aldehyde 19 in anhydrous THF proceeded smoothly with methyl 2-(N-Boc-amino)-2-dimethylphosphonylacetate or methyl 2-(N-Cbz-amino)-2-dimethylphosphonylacetate using tetramethylguanidine as base. The (Z)-enamido esters 20a-b were formed and asymmetric hydrogenation using Burk's catalytic Rh(I)-(S,S)-
Me-Du-PHOS system - (-)-1,2-bis-(2S,5S)-2,5-dimethyl-phospholano benzene (cyclooctadiene) rhodium (21) - in deoxygenated MeOH, yielded the fully protected para-malonylphenylalanine derivatives 22a-b. The absolute configurations were assigned as S, based on the selectivity of the (S,S)-Me-Du-PHOS ligand. Saponification, performed under mild conditions in order to avoid concomitant partial deprotection of the malonyl tert-butyl ester, yielded the free acids 23a-b. Similar methodology was utilized in the preparation of the previously introduced malonylmethyl-phenylalanyl derivatives [20] (Scheme 4).
Arylglycine derivatives 24-28 were prepared in one step, starting from readily available serine derivatives. The method proposed by Boto et al. involves treating the starting protected serine with iodine and DIB (diacetoxyiodo benzene) at room temperature. The reaction mixture was then cooled and BF3
.Et2O, together with an excess of different nucleophiles, was added. These building blocks can be used to develop novel anti-neurodegenerative drugs, as
Scheme 3. Reagents and conditions: (a) Pd(OAc)2, Et3N, MeCN, (4-MeC6H4)P, reflux, 22 h; (b) H2, [Rh(1,5-COD)(R,R-DIPAMP)]BF4, 60 °C.
Scheme 4. Reagents and conditions: (a) tert-butyl malonate, Pd(dba)2, P(tBu)3, KO-tBu, citric acid; (b) methyl 2-(N-Boc-amino)-2-dimethylphosphonylacetate or methyl 2-(N-Cbz-amino)-2-dimethylphosphonylacetate, TMG; (c) H2, 10 atm Rh(COD)2OTf, (S,S)-Me-Du-PHOS; (d) Na2CO3, MeOH; (e) H2/Pd-C, FmocOSu, Na2CO3.
O Me
Me
IO
O Me
Me
O
AcHN COOMe
O Me
Me
O
AcHN COOMe
COOMe
NHAc
1416 17
15
a b
85% 87%
BrO
O
O
O
O
O
Ot-Bu
Ot-Bu
O
O
t-BuO
t-BuO
NH
O
OR
O
O
t- BuO
t- BuO
NH
O
OHR
O
t-BuO
t-BuO
NH
O
OR
O
P PRh+
-OTf
a
75 %
b
76-79 %
c d
94-96 % 86-89 %
1819 20a R=Boc
20b R=Cbz
22a R=Boc22b R=Cbz
23a R=Boc23b R=Cbz23c R=Fmoc
e
[(S,S)-Me-DuPHOS-Rh-COD]OTf
21
804 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
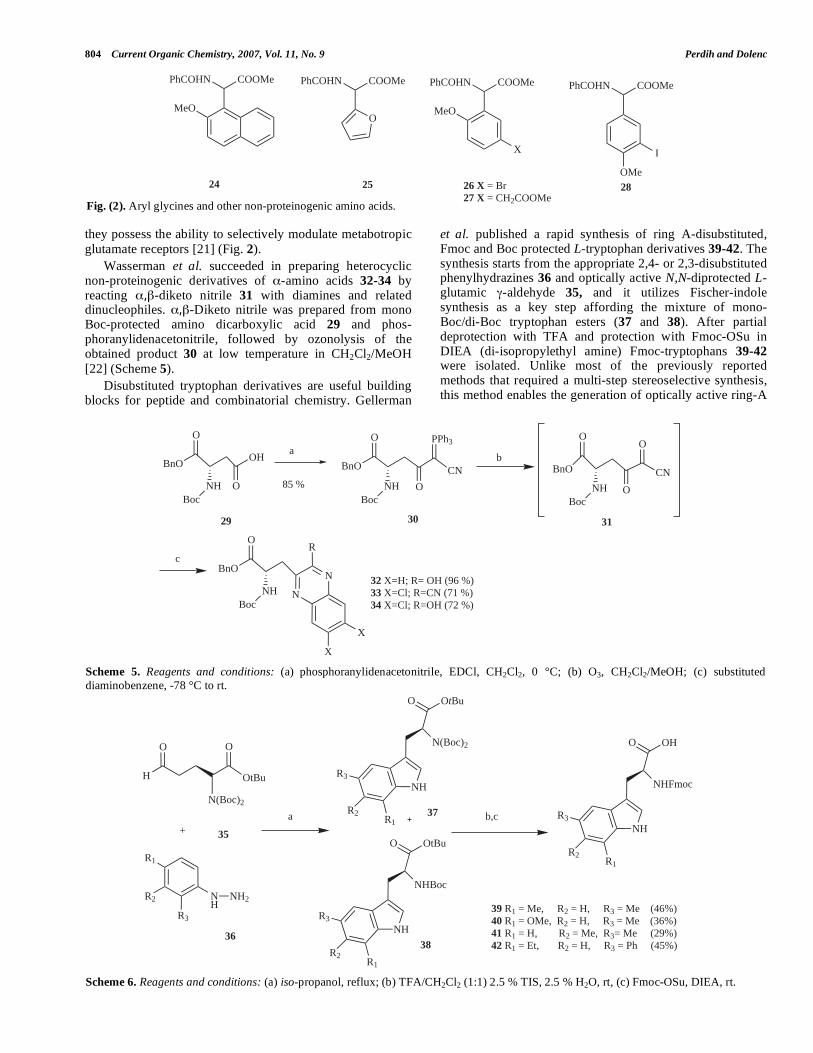
they possess the ability to selectively modulate metabotropic glutamate receptors [21] (Fig. 2).
Wasserman et al. succeeded in preparing heterocyclic non-proteinogenic derivatives of -amino acids 32-34 by reacting , -diketo nitrile 31 with diamines and related dinucleophiles. , -Diketo nitrile was prepared from mono Boc-protected amino dicarboxylic acid 29 and phos-phoranylidenacetonitrile, followed by ozonolysis of the obtained product 30 at low temperature in CH2Cl2/MeOH [22] (Scheme 5).
Disubstituted tryptophan derivatives are useful building blocks for peptide and combinatorial chemistry. Gellerman
et al. published a rapid synthesis of ring A-disubstituted, Fmoc and Boc protected L-tryptophan derivatives 39-42. The synthesis starts from the appropriate 2,4- or 2,3-disubstituted phenylhydrazines 36 and optically active N,N-diprotected L-glutamic -aldehyde 35, and it utilizes Fischer-indole synthesis as a key step affording the mixture of mono-Boc/di-Boc tryptophan esters (37 and 38). After partial deprotection with TFA and protection with Fmoc-OSu in DIEA (di-isopropylethyl amine) Fmoc-tryptophans 39-42 were isolated. Unlike most of the previously reported methods that required a multi-step stereoselective synthesis, this method enables the generation of optically active ring-A
Fig. (2). Aryl glycines and other non-proteinogenic amino acids.
Scheme 5. Reagents and conditions: (a) phosphoranylidenacetonitrile, EDCl, CH2Cl2, 0 °C; (b) O3, CH2Cl2/MeOH; (c) substituted diaminobenzene, -78 °C to rt.
Scheme 6. Reagents and conditions: (a) iso-propanol, reflux; (b) TFA/CH2Cl2 (1:1) 2.5 % TIS, 2.5 % H2O, rt, (c) Fmoc-OSu, DIEA, rt.
COOMePhCOHN
MeO
X
MeO
COOMePhCOHN PhCOHN COOMe
O
PhCOHN COOMe
I
OMe24 26 X = Br
27 X = CH2COOMe2825
O
BnO
ONHBoc
O
CN
O
BnOOH
ONHBoc
O
BnO
ONHBoc
PPh3
CN
N
N
O
BnO
NHBoc
R
X
X
ab
c
29 30 31
32 X=H; R= OH (96 %)33 X=Cl; R=CN (71 %)34 X=Cl; R=OH (72 %)
85 %
OtBu
NH
R1
R2
R3
N(Boc)2
O
OtBu
NH
R1
R2
R3
NHBoc
O
H
N(Boc)2
O
OtBu
O
R1
R2
R3
NH
NH2
OH
NH
R1
R2
R3
NHFmoc
O
+ 35
36
37
38
39 R1 = Me, R2 = H, R3 = Me (46%)40 R1 = OMe, R2 = H, R3 = Me (36%)41 R1 = H, R2 = Me, R3= Me (29%)42 R1 = Et, R2 = H, R3 = Ph (45%)
a b,c

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 805
disubstituted tryptophans from simple and common chiral precursor [23] (Scheme 6).
Tryptophan is superior to all other naturally occurring peptide residues in its ability to bind cations (the cation- interaction). In an effort to expand the toolbox of tryptophan-like amino acids, Carlier et al. reported a catalytic asym-metric synthesis of Trp regioisomers 44a-d, where the alanine unit is attached, not to C-3 of indole, but to C-2, C-4, C-5, C-6, or C-7. The most convenient catalyst was the pre-viously introduced Burk DuPhos system, with the EtDuPhos ligand affording the greatest enantiomeric selectivity (Scheme 7). The reactions were conducted in methanol, ethyl acetate, dichloromethane and acetone, all these solvents providing suitable reaction conditions [24] (Table 1).
Hanessian et al. developed an enantiomerically selective synthesis of allyl containing amino acids. The starting sultam derivatives of O-benzyl glyoxylic acid oximes 45 were reacted with allyl bromides 46 in the presence of zinc in aqueous ammonium chloride. After selective cleavage of the N-O bond in the presence of Mo(CO)6, the sultam auxiliary was removed by treatment with LiOH in THF/H2O solution to afford the corresponding free allylglycine derivatives (48-
51) without any loss of stereochemical purity (Scheme 8). Some of the derivatives obtained, with their corresponding yields, are listed in Table 2 [25].
A synthetic route to (R)-4-piperidinylglycine (59) was achieved by Shieh et al., which offers a promising alternative to the previously published 8-step synthesis. The Cbz-enamides 54a and 54b were prepared from commercially available N-Cbz-phosphonoglycine trimethyl ester 52 and N-
Boc-4-piperidone 53 using the Schmidt protocol. Rhodium catalyzed hydrogenation (modified Burk DuPhos system) of the Cbz-enamide 54a-b generated the (R)-enantiomer of protected (R)-4-piperidinylglycine 56 with 94 % enantio-meric excess. Removal of protective groups yielded the corresponding free unnatural amino acid 59 [26] (Scheme 9).
Scheme 7. Reagents and conditions: (a) 2-3 mol % [Rh(COD)/L]TfO (L=(S,S)-EtDuPhos), 25 °C, 3 days.
Table 1. Asymmetric Hydrogenation of Dehydroamino Acid Derivatives
Comp. Product e.e. Solvent
44a
NH NHAc
O
OMe
98.6 %
EtOAc
44b
NH
O
MeO
NHAc
99.1 %
MeOH
44c
NH
NHAc
O
MeO
98.0 %
MeOH
44d
NH
O
MeO
NHAc
96.7 %
MeOH
44e
NHO
MeO
NHAc
99.5 %
MeOH
Scheme 8. Reagents and conditions: (a) Zn powder, THF, NH4Cl (aq.), rt.
NHR2
MeOOC
N
R1
NHR2
MeOOC
N
R1
a2
45
6
7
2
45
6
7
43 44
3 3
R1 = H, BocR2 = Ac, Cbz
SO2
N
O
NOBn
R1
R2
R3
Br
SO2
N
O
HN
OBnR3
R2R1
a
45 46 47
90-95 %
806 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
Glycine enables the introduction of various substituents to the -carbon. The stereochemistry of the -substituted glycine derivatives is important, mainly if a biological response is expected. Fuji et al. performed a diastereo-selective alkylation of the (S)-glycine equivalent 60, which includes axially chiral binaphthol as an auxiliary, with several electrophiles yielding (R)- -amino acid derivatives
(66-68; Table 3) as depicted on Scheme 10 for the ally-glycine derivative 65. The alkylation of 60 proceeded smoothly with high diastereo-selectivity to give protected unnatural (R) - -amino acid 61 which was due to the instability of the diphenyl imine group converted to the N- benzoyl derivative 62 with an overall 66 % yield and 72 %
Table 2. Allyl glycine derivatives
Comp. Allyl bromide Temp. Product Yield
d.r.
Allylglycines
48 Br
rt
SO2
N
O
HNOBn
95 % 91:9 O
NH2
HO
49
Br
rt
SO2
N
O
HNOBn
90 % 99: 9 O
NH2
HO
50 Br
0 °C
SO2
N
O
HNOBn
93 % 81:19 O
NH2
HO
51 BrPh
rt
SO2
N
O
HNOBn
Ph
88 % 99:1 O
NH2
HO
Ph
Scheme 9. Reagents and conditions: (a) tetramethyl-guanidine; (b) H2, [(R,R)-Me-BPE-Rh-COD]OTf ; (c) LiOH, rt; (d) H2/PdC; (e) HCl.
N
O OR
O
OHNO
O
Cl-
O
O
NH
P
O
OO
O O
N
OR
O
O
N
O OR
O
OHNO
O
H2N
N
O OR
O
OH
O
OH+H3N
NH
P PRh+
N
O OR
O
OHHNO
O
+
ab
d e
52 53a R= t-Bu53b R= i-Pr
54a R= t-Bu54b R= i-Pr
56a R= t-Bu56b R= i-Pr
58a R= t-Bu58b R= i-Pr
59
[(R,R)-Me-BPE-RH-COD]OTf55
61 %99.8 %
82 %
57a R= t-Bu57b R= i-Pr
c
98 % 79 %
Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 807
Scheme 10. Reagents and conditions: (a) LDA, /THF-HMPA, allyl bromide, -78°C to rt, 0.5h; (b) (i) aq. HCl, THF; (ii) BzCl, i-Pr2Net, CH2Cl2, rt; (c) 0.5N KOH, MeOH, H2O, rt, 30 min; (d) CH2N2, MeOH.
Table 3. Diastereoselective Alkylation of Glycine Equivalent 59 Yielding (R)- Unnatural Amino Acids
Comp. Electrophile Product Yield
d.e. Configuration
66
Methyl iodide
O
OH NHBz
Me
62 %
88 %
R
67
Propargyl bromide
O
OH NH2
78 %
78 %
R
68
2-(bromomethyl)
naphthalene
O
OH NH2
71 %
69 %
R
Table 4. Arylalkyl and Heteroarylalkyl Aminoacid Methylesters
SO2Cl2
CH3COOH, Et2 O30 min
Ar
NH2
COOMen
Ar
NH2 HCl
COOMe
Cl
69 70
Comp. Substrate Comp. Product Yield e.e.
69a
SCOOMe
NH2
70a
SCOOMe
NH2 HClCl
78 %
90 %
69b
SCOOMe
NH2
70b
SCOOMe
NH2 HClCl
75 %
83 %
60c
COOMeH2N
MeO
70c
COOMe
NH2 HCl
MeO
Cl
89 %
>99 %
69d
COOMeH2N
MeO
70d
COOMe
NH2 HCl
MeO
Cl
86 %
> 99 %
OH
O
O
NPh
Ph
OH
O
O
NPh
Ph
H OBz
O
O
NHBz
H
OH
OH
O
HOH NHBz
O
OH NHBz
6061
62
ab
63 64
c d
65

808 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
diastereomeric excess. It was discovered that the free hydroxyl group of the auxiliary 60 played an important role in the induction of chiral selectivity. Hydrolysis of 62 and methylation with CH2N2, resulted in the final compound 65 (72% yield) [27].
Mono-chlorination of amino acids methylesters 69a-e was performed by sulfuryl chloride which resulted in heteroarylalkyl aminoacid methylesters 70a-d. Convenient one-pot synthesis afforded amino acid derivatives in high yields with a straightforward purification by direct filtration. Integrity of the chiral centres was affected minimally [28] (Table 4).
Kokotos et al. prepared enantiopure lipophilic -amino acids and also their -functionalized derivatives 73-75 and some bis -amino acids. The key intermediate was protected glutamic acid aldehyde 71 which was utilized in a Wittig reaction with -trityloxy alkylidene triphenylphosphoranes. After hydrogenation of 72 the obtained -hydroxy- -amino acid 73 was used as starting material in the synthesis of -functionalized -amino acids such as 74 and 75, depicted in Scheme 11 [29,30].
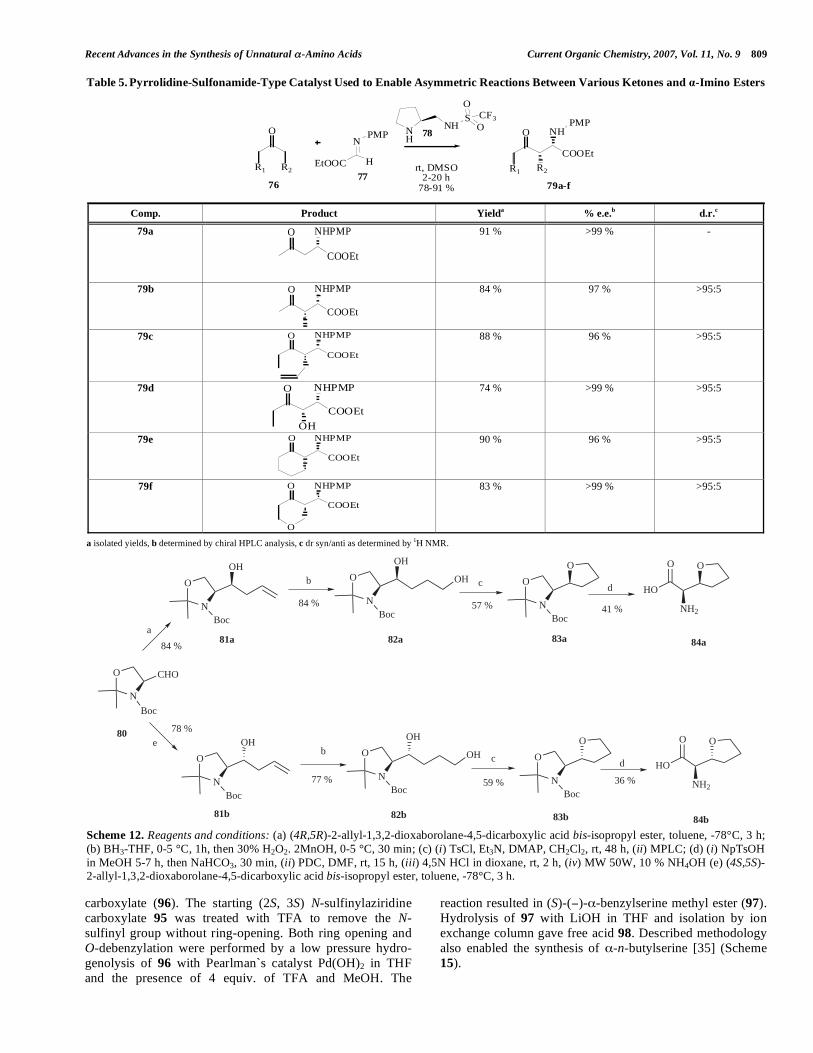
The novel pyrrolidine-sulfonamide 78 has been prepared and used successfully by Wang et al. to catalyze asymmetric Mannich-type reactions in DMSO between various ketones 76 and PMP (p-methoxyphenol) -imino ester 77. Other possible solvents were also explored and all could be employed in this reaction. The reaction is used to efficiently synthesize functionalized -amino acid derivatives (79a-f), most of them possessing lipophilic character with excellent levels of regio-, diastereo-, and enantio-selectivity as is evident from Table 5 [31].
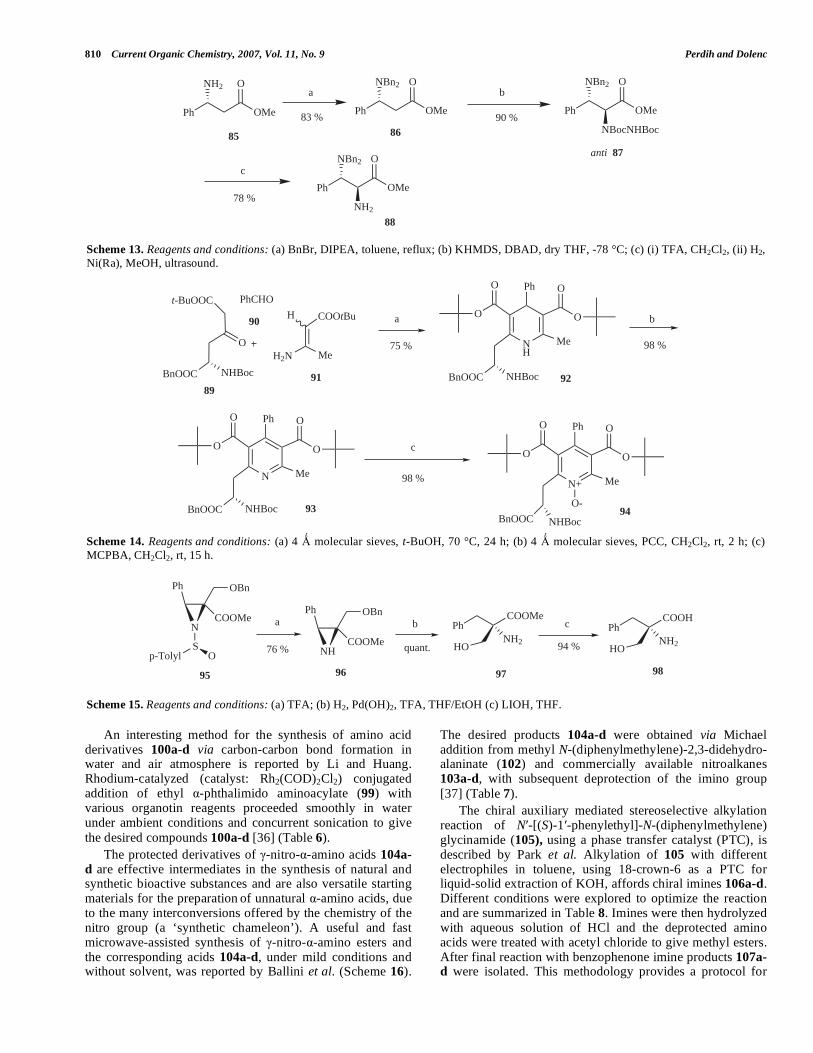
The 2-(tetrahydrofuran-2-yl)glycine (84a) is a con-formationally constrained amino acid. The first enantio-divergent synthesis of all four possible 2-(tetrahydrofuran-2-yl) glycine stereoisomers was described by Pellicciari et al. The key synthetic step is a highly stereocontrolled allyl-
boration of the (S)- or (R)-Garner's aldehydes 80 followed by oxidation with hydrogen peroxide under basic conditions, to give four chiral homoallylalcohols 82. Starting from these, the title compounds 84 are obtained in five classical steps. The synthetic route includes the application of microwaves in the final step. Since the synthetic routes for the selected compounds are analogous, only the synthesis of the (2R,2S) and (2R,2R) diastereomers is outlined in Scheme 12 [32].
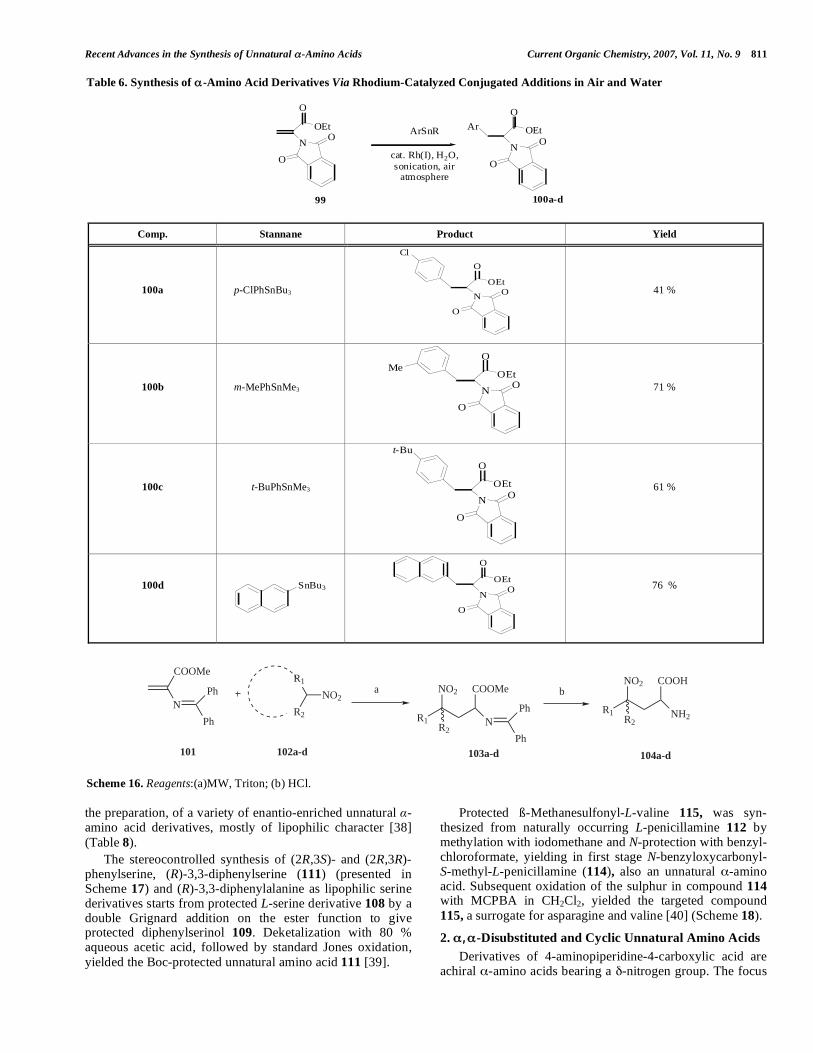
An orthogonally protected 2,3-amino acid 88 was recently reported by Pedatella et al. The starting enolates of N,N-dibenzylated ß3-amino esters 86 were treated with di-tert-butyl azodicarboxylate (DBAD) to give N`,N``-di-Boc-2-hydrazino derivatives 87 with excellent anti diastereomeric ratios. Removal of the Boc protective group and reductive cleavage of the hydrazine bond gave the expected compound 88. The main advantage of this reaction is that it does not require expensive chiral reagents and/or chiral auxiliaries and offers various options for incorporation into peptides [33] (Scheme 13).
A family of heterocyclic amino acids comprising highly functionalized ß-(2-pyridyl)- and ß-(4-pyridyl)-alanines 93,
and the corresponding N-oxide derivatives 94, have been developed by Dondoni et al. by a convenient one-pot thermal Hantzsch-type cyclocondensation of aldehyde–ketoester–enamine systems 89-91, in which one of the reagents (aldehyde or ketoester) carried the unmasked but protected chiral glycinyl moiety. Following this procedure 1,4-dihydro pyridine derivative 93 was obtained. The oxidation reactions to corresponding pyridine 93 and N-oxide derivative 94 were carried out almost quantitatively using PCC (pyridinium chlorochromate) and MCPBA (3-chlorobenzenecarbo-peroxoic acid) in CH2Cl2. The methodology was also successfully transferred to a solid support [34] (Scheme 14).
Another example of asymmetric synthesis was reported by Davis et al. for substituted serines 97 and 98 via the regioselective hydrogenolysis of 2-benzyloxyaziridine 2-
Scheme 11. Reagents and conditions: TrtO(CH2)10PPH3I, KHMDS, toluene; (b) H2, Pd/C, MeOH; (c) NaOCl, AcNH-TEMPO, Aliquat (tributylmethylammonium chloride solution in water), KBr, NaHCO3, CH2Cl2, H2O; (d) NaOCl, AcNH-TEMPO, NaBr, NaHCO3, EtOAc, toluene, H2O.
H
O
N(Boc)2
O
OMe
N(Boc)2
O
OMeOTr
O
OMe
N(Boc)2
HO
O
OMe
N(Boc)2
HO
O
O
OMe
N(Boc)2
H
O
a
b
9
c
d
71 72
73
74
75
84%
89%
78%
76%
Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 809
carboxylate (96). The starting (2S, 3S) N-sulfinylaziridine carboxylate 95 was treated with TFA to remove the N-sulfinyl group without ring-opening. Both ring opening and O-debenzylation were performed by a low pressure hydro-genolysis of 96 with Pearlman`s catalyst Pd(OH)2 in THF and the presence of 4 equiv. of TFA and MeOH. The
reaction resulted in (S)-( )- -benzylserine methyl ester (97). Hydrolysis of 97 with LiOH in THF and isolation by ion exchange column gave free acid 98. Described methodology also enabled the synthesis of -n-butylserine [35] (Scheme 15).
Table 5. Pyrrolidine-Sulfonamide-Type Catalyst Used to Enable Asymmetric Reactions Between Various Ketones and -Imino Esters
R1 R2
O
H
NPMP N
H
NHS
O
OCF3
rt, DMSO2-20 h
78-91 %
R1
COOEt
O
R2
NHPMP
EtOOC
7677
78
79a-f
Comp. Product Yield
a % e.e.
b d.r.
c
79a
COOEt
O NHPMP
91 % >99 % -
79b
COOEt
O NHPMP
84 % 97 % >95:5
79c
COOEt
O NHPMP
88 % 96 % >95:5
79d
COOEt
O NHPMP
OH
74 % >99 % >95:5
79e O NHPMP
COOEt
90 % 96 % >95:5
79f
COOEt
O NHPMP
O
83 % >99 % >95:5
a isolated yields, b determined by chiral HPLC analysis, c dr syn/anti as determined by 1H NMR.
Scheme 12. Reagents and conditions: (a) (4R,5R)-2-allyl-1,3,2-dioxaborolane-4,5-dicarboxylic acid bis-isopropyl ester, toluene, -78°C, 3 h; (b) BH3-THF, 0-5 °C, 1h, then 30% H2O2. 2MnOH, 0-5 °C, 30 min; (c) (i) TsCl, Et3N, DMAP, CH2Cl2, rt, 48 h, (ii) MPLC; (d) (i) NpTsOH in MeOH 5-7 h, then NaHCO3, 30 min, (ii) PDC, DMF, rt, 15 h, (iii) 4,5N HCl in dioxane, rt, 2 h, (iv) MW 50W, 10 % NH4OH (e) (4S,5S)-2-allyl-1,3,2-dioxaborolane-4,5-dicarboxylic acid bis-isopropyl ester, toluene, -78°C, 3 h.
O
N
CHO
Boc
O
NBoc
OHO
NBoc
OH
OH O
NBoc
O
HO
NH2
OO
O
NBoc
OHO
NBoc
OH
OH O
NBoc
O
HO
NH2
OO
a
b c d
80
81a 82a 83a 84a84 %
84 % 57 % 41 %
bc d
81b 82b 83b 84b
77 % 59 % 36 %
e
78 %
810 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
An interesting method for the synthesis of amino acid derivatives 100a-d via carbon-carbon bond formation in water and air atmosphere is reported by Li and Huang. Rhodium-catalyzed (catalyst: Rh2(COD)2Cl2) conjugated addition of ethyl -phthalimido aminoacylate (99) with various organotin reagents proceeded smoothly in water under ambient conditions and concurrent sonication to give the desired compounds 100a-d [36] (Table 6).
The protected derivatives of -nitro- -amino acids 104a-
d are effective intermediates in the synthesis of natural and synthetic bioactive substances and are also versatile starting materials for the preparation of unnatural -amino acids, due to the many interconversions offered by the chemistry of the nitro group (a ‘synthetic chameleon’). A useful and fast microwave-assisted synthesis of -nitro- -amino esters and the corresponding acids 104a-d, under mild conditions and without solvent, was reported by Ballini et al. (Scheme 16).
The desired products 104a-d were obtained via Michael addition from methyl N-(diphenylmethylene)-2,3-didehydro-alaninate (102) and commercially available nitroalkanes 103a-d, with subsequent deprotection of the imino group
[37] (Table 7). The chiral auxiliary mediated stereoselective alkylation
reaction of N -[(S)-1 -phenylethyl]-N-(diphenylmethylene) glycinamide (105), using a phase transfer catalyst (PTC), is described by Park et al. Alkylation of 105 with different electrophiles in toluene, using 18-crown-6 as a PTC for liquid-solid extraction of KOH, affords chiral imines 106a-d. Different conditions were explored to optimize the reaction and are summarized in Table 8. Imines were then hydrolyzed with aqueous solution of HCl and the deprotected amino acids were treated with acetyl chloride to give methyl esters. After final reaction with benzophenone imine products 107a-d were isolated. This methodology provides a protocol for
Scheme 13. Reagents and conditions: (a) BnBr, DIPEA, toluene, reflux; (b) KHMDS, DBAD, dry THF, -78 °C; (c) (i) TFA, CH2Cl2, (ii) H2, Ni(Ra), MeOH, ultrasound.
Scheme 14. Reagents and conditions: (a) 4 molecular sieves, t-BuOH, 70 °C, 24 h; (b) 4 molecular sieves, PCC, CH2Cl2, rt, 2 h; (c) MCPBA, CH2Cl2, rt, 15 h.
Scheme 15. Reagents and conditions: (a) TFA; (b) H2, Pd(OH)2, TFA, THF/EtOH (c) LIOH, THF.
O
OMe
NH2
Ph
O
OMe
NBn2
Ph
O
OMe
NBn2
Ph
NBocNHBoc
O
OMe
NBn2
Ph
NH2
85 86
88
a b
c
anti 87
83 % 90 %
78 %
PhCHO
H COOtBu
MeH2NNH
Ph
Me
NHBocBnOOC
O
O
O
O
N
Ph
Me
NHBocBnOOC
O
O
O
O
N+
Ph
Me
NHBocBnOOC
O
O
O
O
O-
t-BuOOC
NHBocBnOOC
O
a
75 %
b
98 %
c
98 %
89
90
91 92
93 94
N
OBn
COOMe
Ph
SOp-Tolyl
NH2HO
COOMePh
NH
OBn
COOMe
Ph
NH2HO
COOHPh
95 9796
a b
98
c
76 % 94 %quant.
Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 811
the preparation, of a variety of enantio-enriched unnatural -amino acid derivatives, mostly of lipophilic character [38] (Table 8).
The stereocontrolled synthesis of (2R,3S)- and (2R,3R)-phenylserine, (R)-3,3-diphenylserine (111) (presented in Scheme 17) and (R)-3,3-diphenylalanine as lipophilic serine derivatives starts from protected L-serine derivative 108 by a double Grignard addition on the ester function to give protected diphenylserinol 109. Deketalization with 80 % aqueous acetic acid, followed by standard Jones oxidation, yielded the Boc-protected unnatural amino acid 111 [39].
Protected ß-Methanesulfonyl-L-valine 115, was syn-thesized from naturally occurring L-penicillamine 112 by methylation with iodomethane and N-protection with benzyl-chloroformate, yielding in first stage N-benzyloxycarbonyl-S-methyl-L-penicillamine (114), also an unnatural -amino acid. Subsequent oxidation of the sulphur in compound 114 with MCPBA in CH2Cl2, yielded the targeted compound 115, a surrogate for asparagine and valine [40] (Scheme 18).
2. , -Disubstituted and Cyclic Unnatural Amino Acids
Derivatives of 4-aminopiperidine-4-carboxylic acid are achiral -amino acids bearing a -nitrogen group. The focus
Table 6. Synthesis of -Amino Acid Derivatives Via Rhodium-Catalyzed Conjugated Additions in Air and Water
NO
O
O
OEt
NO
O
O
OEtArArSnR
99 100a-d
cat. Rh(I), H2O,sonication, air
atmosphere
Comp. Stannane Product Yield
100a
p-ClPhSnBu3 N O
O
O
OEt
Cl
41 %
100b
m-MePhSnMe3 NO
O
O
OEtMe
71 %
100c
t-BuPhSnMe3
N O
O
O
OEt
t-Bu
61 %
100d
SnBu3
NO
O
O
OEt
76 %
Scheme 16. Reagents:(a)MW, Triton; (b) HCl.
COOMe
N
Ph
Ph NO2
R1
R2
COOMe
N
Ph
PhR1
R2
NO2COOH
NH2R1
R2
NO2a b
101 102a-d 103a-d 104a-d

812 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
on this amino acid has been due to the antimicrobial activity of its helical peptides. Tanaka et al. has prepared a new class of , -disubstituted -amino acids 121a-f bearing a pendent chiral centre. Bisalkylation of dimethyl malonate (116) by treatment with KOtBu and 2-bromomethyl-1,3-dioxane in DMSO afforded 117. Deprotection of the dioxolane group by HCl gave the crude dialdehyde which was then subjected to condensation with various amines to give cyclic diamines
118a-f. After hydrogenation and hydrolysis of the obtained diester 119a-f, the monocarboxylic acids underwent Curtius rearrangement, using DPPA (diphenylphosphoryl azide). The reaction was quenched with benzyl alcohol and further trans-formations resulted in novel amino acid derivatives 121a-f [41] (Scheme 19).
Another example of synthesizing sterically constrained - -symmetrically disubstituted -amino acids 124a-h was
Table 7. Microwave-Assisted Michael Addition of 102 to Starting Nitro Compounds
Compound Nitro compound Product Yield
104a
NO2
COOH
NH2H3C H
NO2
85 %
104b
NO2
COOH
NH2H
NO2
H3C
92 %
104c NO2
COOH
NH2
NO2
93 %
104d
O
O
NO2
COOH
NH2H
NO2
MeO
O
65 %
Table 8. Benzylation of N -[(S)-1 -phenylethyl]-N-(diphenylmethylene)glycinamide
Ph NH
N
OCH 3
C Ph 2
Ph NH
N
OC H3
CPh2
E +
KOH18 - C rown-6
toluen eMeO
N
O
CPh2
1) H +
2) AcCl, MeOH
3) HN=CPh2
EE
105106
107 a-d
S
S
Comp. Electrophile Structure Temp Time Yield
e.r. (product)
107a
ClBr
MeO
N
O
CPh2
Cl
- 40 °C
- 40 °C
4 h
3 h
52 %
43 %
75:25
78:22
107b
IBr
MeO
N
O
CPh2
I
-20 °C
-40 °C
0,5 h
5 h
40 %
55 %
76:24
76:24
107c
H3CBr
MeO
N
O
CPh2
CH3
-40 °C
7 h
49 %
76:24
107d
CH3CH2I
MeON
O
CPh2
H3C
-40 °C
3 h
65 %
83:17

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 813
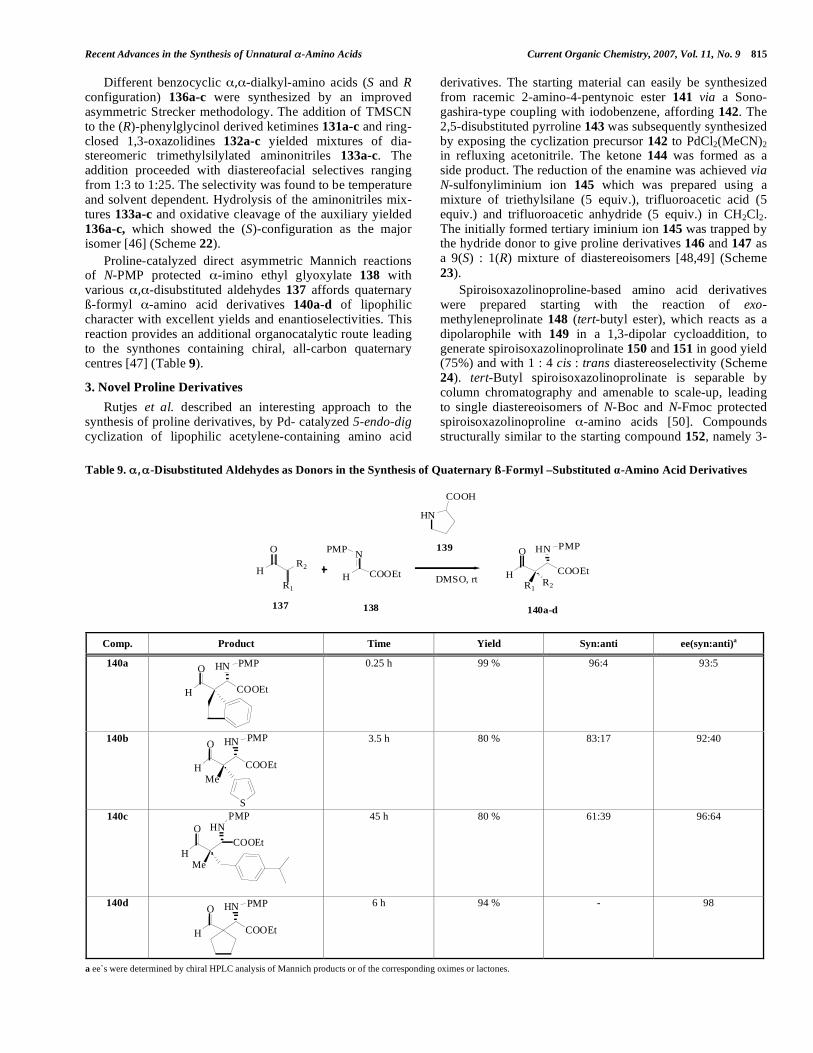
reported by Soloshonok et al. [42]. The interesting approach of dialkylating the Ni(II) complex of a glycine derivative 122 resulted in symmetrical- , -amino acids 124a-h (Scheme 20). he glycine Ni(II) complex 122 is inexpensive and can be synthesized in gram quantities [43]. The method was successful with alkyl halides; however it cannot be
extended to the use of the - or ß-branched alkyl halides or Michael acceptors as glycine equivalents. Nickel (II) com-plexes were also used for other phase transfer-catalyzed (PTC) alkylation reactions starting from glycine derived Schiff bases [44].
Scheme 17. Reagents and conditions: (a) PhMgBr, Et2O, 0 °C; (b) AcOH, 25 °C, 1 h; (c) CrO3 H2SO4 , acetone, 0 °C, 3 h; (d) HCl, EtOAc, 3 days.
Scheme 18. Reaction and conditions: (a) CH3I, NaOMe; (b) CbzCl; (c) MCPBA, CH2Cl2.
Scheme 19. Reagents and conditions: (a)KOtBu, 2-bromomethyl-1,3-dioxane, DMSO, 80 °C; (b) 10 % aq. HCl; (c) R-NH2,THF, rt, 12 h; (d) H2, Pd/C MeOH, rt, 12 h; (e) NaOH, MeOH, reflux, 3 h; (f) DPPA, MeOH, reflux, 3 h; (g) BzOH; (h) NaOH, reflux, 3 h .
Scheme 20. Reagents and conditions: (a) KOH or NaOtBu Alk-X (a-h); (b) MeOH/1 M HCl.
ON
Boc
COOMe
ON
Boc
OHPh
PhPh
HOOC
HN
OHPh
Boc
PhHOOC
-Cl+H3N
OHPh
da b,c
108 109 110 111
84 % 49 %quant.
HS
H2N COOH
S
H2N COOH
S
CbzHN COOH
OS
CbzHN COOH
O
a b c
112 113 114 115
MeOOC COOMe
OO O
O
MeOOC COOMe
H
N
MeOOC COOMe
R
N
MeOOC COOMe
R
N
ZHN COOBz
R R
N
P-HN COOH
a b c d e f
n-Hex
a b,c
d e,f,g h
R=
116 117118a-f
119a-f 120a-f 121a-f
47 % 36-60 %
67-86 % 30-61 %
H2N COOH
AlkAlkNH
N
O
NiO O
N NH
N
O
NiO O
N
Alk
Alk
122123a-h
124a-h
ab
Alk = a CH2-CH=CH2 e (CH2)2CH3 b CH2-C6H5 f (CH2)3CH3 c trans CH2-CH=CH-C6H5 g CH3 d CH2-CH3 h (CH2)4CH3

814 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
Besides nickel, other metals play an important role in , -amino acid synthesis. Takemoto et al. published a
tandem reaction yielding dehydroamino acid derivatives 127a-d and 129. The diethylzinc-promoted reaction of de-hydroamino acid derivatives 125 with -allyl palladium complex, formed by Pd(PPh3)4 and branched acetate 126,
afforded , -disubstituted amino acids 127a-d via radical and anionic carbon–carbon bond-forming processes. The authors also disclosed the reductive allylation reaction of N-phthaloyl dehydroalanine 128 with allyl acetate 126 which was accomplished by using Bu3SnH and Pd(PPh3)4 yielding 129 [45] (Scheme 21).
Scheme 21. Reagents and conditions: (a) (i) R-I, Et2Zn, Pd(PPh3)4, CH2Cl2, 0 °C; (ii) 1 M HCl, THF, 20 °C; (b) Bu3SnH, Pd(PPh3)4, CH2Cl2, 20 °C.
Scheme 22. Reagents and conditions: (a) (R)- -phenylamine, toluene, reflux, 6 h; (b) TMSCN, CH2Cl2, 0 °C, (c) conc. H2SO4/hexane (1:1), 3-5 days, rt; (d) (i) Pb(OAc)4, MeOH/ CH2Cl2 (1:2), 1 h; (ii) HCl, reflux, 3 h; (iii) EtOH, propylene oxide, reflux, 20 min.
Ph
Ph
N COO-tBu OAc
Ph
OAc
Ph
N
O
OCOOEt
Ph
COO-tBuH2N
R
Ph
N COOEt
O
O
+
+
a
b
125 126
127a R=i-Pr (56 %) 127b R=c-Hexyl (56 %) 127c R=c-Pentyl (58 %) 127d R=t-Bu (30 %)
128 126129
48 %
O
N
PhH
OHHN CN
PhH
OTMS
HNCN
PhH
OTMS
H2N COOH
H2N COOH
R
R
R
S
S
R
OHN
HPh
R
R
HN CONH2
PhH
OH
R
R
S
RO
HPh
O
O
HPh
O
R
S
R
R
RHN
CONH2
PhH
OH
n
n
a
n
130 a-c
(E/Z) 131a-c (1R, 1`R) 133a-c
(1S, 1`R) 133a-c
c
n
n
n
a: n=1, R=Hb: n=2, R=Hc: n=0, R=H
(R)-136a-c
(S)-136a-c
d
n
132a-c
n
(1R, 1`R) 134a-c
(1S, 1`R) 134a-c
n
n
(1R, 1`R) 135a-c
(1S, 1`R) 135a-c
n
b
Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 815
Different benzocyclic , -dialkyl-amino acids (S and R configuration) 136a-c were synthesized by an improved asymmetric Strecker methodology. The addition of TMSCN to the (R)-phenylglycinol derived ketimines 131a-c and ring-closed 1,3-oxazolidines 132a-c yielded mixtures of dia-stereomeric trimethylsilylated aminonitriles 133a-c. The addition proceeded with diastereofacial selectives ranging from 1:3 to 1:25. The selectivity was found to be temperature and solvent dependent. Hydrolysis of the aminonitriles mix-tures 133a-c and oxidative cleavage of the auxiliary yielded 136a-c, which showed the (S)-configuration as the major isomer [46] (Scheme 22).
Proline-catalyzed direct asymmetric Mannich reactions of N-PMP protected -imino ethyl glyoxylate 138 with various , -disubstituted aldehydes 137 affords quaternary ß-formyl -amino acid derivatives 140a-d of lipophilic character with excellent yields and enantioselectivities. This reaction provides an additional organocatalytic route leading to the synthones containing chiral, all-carbon quaternary centres [47] (Table 9).
3. Novel Proline Derivatives
Rutjes et al. described an interesting approach to the synthesis of proline derivatives, by Pd- catalyzed 5-endo-dig cyclization of lipophilic acetylene-containing amino acid
derivatives. The starting material can easily be synthesized from racemic 2-amino-4-pentynoic ester 141 via a Sono-gashira-type coupling with iodobenzene, affording 142. The 2,5-disubstituted pyrroline 143 was subsequently synthesized by exposing the cyclization precursor 142 to PdCl2(MeCN)2 in refluxing acetonitrile. The ketone 144 was formed as a side product. The reduction of the enamine was achieved via N-sulfonyliminium ion 145 which was prepared using a mixture of triethylsilane (5 equiv.), trifluoroacetic acid (5 equiv.) and trifluoroacetic anhydride (5 equiv.) in CH2Cl2. The initially formed tertiary iminium ion 145 was trapped by the hydride donor to give proline derivatives 146 and 147 as a 9(S) : 1(R) mixture of diastereoisomers [48,49] (Scheme 23).
Spiroisoxazolinoproline-based amino acid derivatives were prepared starting with the reaction of exo-methyleneprolinate 148 (tert-butyl ester), which reacts as a dipolarophile with 149 in a 1,3-dipolar cycloaddition, to generate spiroisoxazolinoprolinate 150 and 151 in good yield (75%) and with 1 : 4 cis : trans diastereoselectivity (Scheme 24). tert-Butyl spiroisoxazolinoprolinate is separable by column chromatography and amenable to scale-up, leading to single diastereoisomers of N-Boc and N-Fmoc protected spiroisoxazolinoproline -amino acids [50]. Compounds structurally similar to the starting compound 152, namely 3-
Table 9. , -Disubstituted Aldehydes as Donors in the Synthesis of Quaternary ß-Formyl –Substituted -Amino Acid Derivatives
H
O
R1
R2
H
N
COOEt
PMP
H COOEt
HN
R1R2
O PMP
137 138 140a-d
HN
COOH
139
DMSO, rt
Comp. Product Time Yield Syn:anti ee(syn:anti)a
140a
H COOEt
HNO PMP
0.25 h 99 % 96:4 93:5
140b
H COOEt
HN
Me
OPMP
S
3.5 h 80 % 83:17 92:40
140c
HCOOEt
HN
Me
OPMP
45 h 80 % 61:39 96:64
140d
H COOEt
HNO PMP
6 h 94 % - 98
a ee`s were determined by chiral HPLC analysis of Mannich products or of the corresponding oximes or lactones.

816 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
substituted-4-methylene prolines, can be prepared from chiral sulfoximine substituted acyclic ally-titanium (IV) complexes as reported by Gais et al. [51].
Vinyloxirane (153) can be used as a masked dienolate in a direct vinylogous Mannich-type reaction with -imino ester 152 which acts as an electrophile, using Sc(OTf)3 as a Lewis acid catalyst. The Mannich adduct 154 (amino alde-hyde) was then transformed to the corresponding proline-like derivative, 5-methylpipecolinic acid ester 155, under simple hydrogenation conditions, in high yield and diastereomeric purity [52] (Scheme 25).
The preparation of the unnatural, bicyclic proline deri-vatives 159 and 160, along with their utility as chiral ligands in the copper-catalyzed enantioselective allylic oxidation of cyclohexene with tert-butyl perbenzoate, is described by Andersson et al. Benzyl glyoxylate (156) was condensed with (+)-L-phenylethyl amine in the presence of molecular sieves. The resulting imine was treated with trifluoroacetic acid and BF3
.OEt2 to generate the free protonated imine, which was subjected to highly exo- and diastereo-selective cycloaddition, subsequently yielding the desired compound 159 and 160 [53] (Scheme 26).
Scheme 23. Reagents and conditions: (a) Iodobenzene, PdCl2(PPh3)2, CuI, Et2NH, Et2O, rt, 2 h; (b) PdCl2(MeCN)2, MeCN, reflux 2 h; (c) Et3SiH, TFA, TFAA, CH2Cl2, 0 °C to rt, 1,5 h.
Scheme 24. Reagent and conditions: (a) NaOCl (aq.), CH2Cl2.
Scheme 25. Reagents and conditions: (a) Sc(OTf)3, THF, 0 °C to 50 °C, 10 min; (b) H2, Pd/C, MeOH, rt, 4 h.
Scheme 26. Reagents and conditions: (a) (+)-L-phenylethyl amine, molecular sieves; (b) TFA, BF3.OEt2, -78 °C; (c) 1,3-cyclohexadiene and
1,3-cyclopentadiene, -78 °C; (d) H2, Pd/C.
HN COOMe
TsHN COOMe
Ts
N
Ts
COOMe
O
HN COOMe
Ts
N
Ts
COOMeN+
Ts
COOMe N
Ts
COOMe
+ab
c+
141142 143 (51 %) 144 (15 %)
145146
147
84 %
74 %
N
Boc
COO-tBu N
Boc
COO-tBu
ON
Cl
Cl
Cl
Cl
NOH N
Boc
COO-tBu
ON
Cl
Cl+a +
148 149 150 151
75 %
N
EtOOC H
CHPh2HN
EtOOC
CHPh2
CHONH
COOEtO
a b
152 153 154 155
74 % 84 %
BnO2C
O
H
COOH
NH
NPh
COOBn
159 n =1160 n =2
157 (n = 1) 83 %; 97% d.e.158 (n = 2) 71 %; 97% d.e.
nn
a,b,c d
156

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 817
4. N–Methyl -Amino Acids and Other Substitutions of the Amino Group
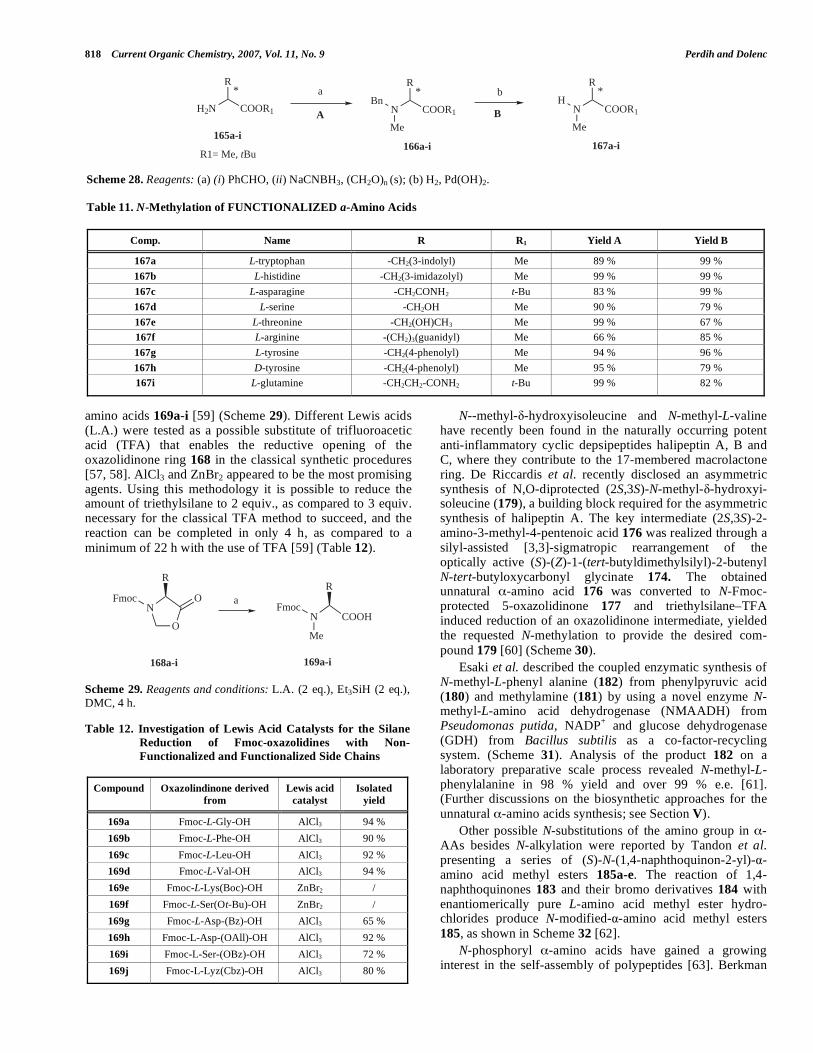
N-alkylation and other N-substitutions of the amino group in -amino acids are attractive synthetic modifications that lead to an important class of unnatural -amino acids. The most frequent N-alkylation modification involves N-methylation yielding N-methyl amino acids (NMA). NMA are found in a wide range of natural peptides and exhibit numerous biological effects, including antibiotic, anticancer, antiviral and immunosuppressive activity. Two of most widely utilized reactions for the preparation of NMA involve either direct N-methylation of N-protected- -amino acids or alternatively 5-oxazolidinone formation with the subsequent reduction. Hughes et al. recently published an extensive review discussing numerous synthetic preparations of N-methyl- -amino acids covering a broad time period for 1885 till 2003 [54].
In 2005, Kessler et al. reported a three-step synthesis of N -methyl-N -(o-nitrobenzenesulfonyl)- -amino acids 164a-
r without extensive purification (Scheme 27). The procedure is based on previously known N-alkylation of N -aryl-
sulfonylamino esters 162a-r, which was improved by utilizing dimethyl sulfate and DBU as base. Ester cleavage of 163a-r was efficiently achieved by using an SN2-type saponification with lithium iodide, avoiding racemization observed with lithium hydroxide hydrolysis (Table 10). Furthermore, compatibility of the synthesized N -methyl-amino acids with Fmoc solid-phase peptide synthesis was demonstrated by using normal coupling conditions to efficiently prepare several N-methyl dipeptides [55].
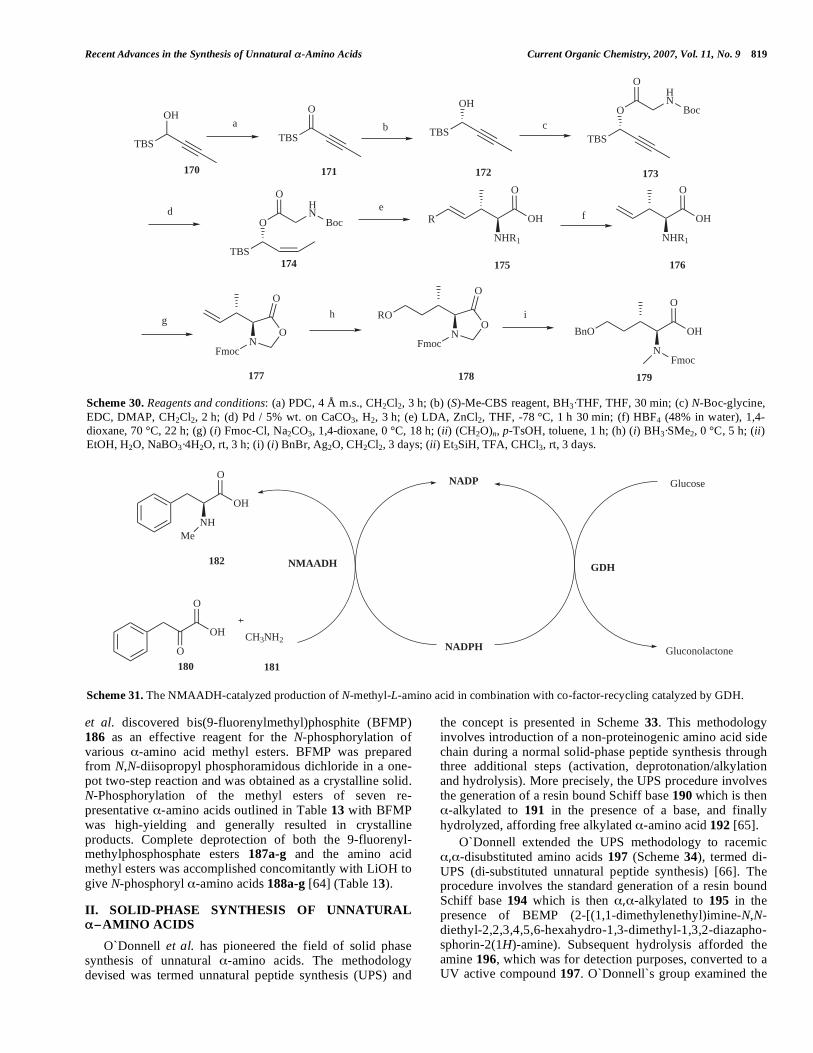
Konopelski et al. published a complementary method for the synthesis of optically pure NMA esters 167a-i that requires no protection of the functionalized amino acid side chain. The method comprises two consecutive reductive aminations, first with benzaldehyde, then with para-formaldehyde. An important feature of the reaction is that both sequences of imine formation and subsequent reduction were performed in the same flask and without isolation [56]. The results for the -amino acids 165a-i utilized in this study are presented in Table 11 (Scheme 28).
Starting from 5-oxazolidinenones 168a-i Arvidsson et al. disclosed an improved route leading to Fmoc-N-methyl- -
Scheme 27. Reagents and conditions: (a) o-NBSCl (1 equiv.), Et3N (2 equiv.), CH2Cl2, rt; (b) DBU (2 equiv.), (CH3)2SO4 (3 equiv.) DMF, 0 ºC, 15 min; (c) LiI (4 equiv.), AcOEt, reflux.
Table 10. Three-Step Synthesis of N -Methyl-N -o-NBS Amino Acids 164a-r with the Corresponding Yields
Comp. R -Amino acid o-NBS protection
(Yield)
N-methylation
(Yield)
Saponification
(Yield)
164a -CH2Ph Phe 97 % 98 % 96 %
164b -CH(CH3)CH2CH3 Ile 96 % 96 % 85 %
164c -CH3 Ala 90 % 99 % 90 %
164d -Ph PHg 95 % 98 % 94 %
164e -(CH2)4NHZ Lys(Z) 93 % 98 % 97 %
164f -(CH2)4NHBoc Lys(Boc) 96 % 99 % 93 %
164g -CH2OtBu Ser(tBu) 98 % 92 % 83 %
164h CH(CH3)OtBu Thr(tBu) 95 % 97 % 88 %
164i -CH2COOtBu Asp(tBu) 86 % 96 % 88 %
164j -CH2CH2COOtBu Glu(tBu) 95 % 93 % 91 %
164k -(CH2)3NHC(NH)NHPbf Arg(tBu) 98 % 98 % 95 %
164l -CH2CONHTrt Asn(Trt) 97 % 99 % 93 %
164m -CH2CH2CONHTrt Gln(tBu) 97 % 98 % 89 %
164n -CH2-indol-3-yl Trp 97 % 96 % 99 %
164o -CH2PhOtBu Tyr(tBu) 98 % 95 % 88 %
164p -CH2CH2S-CH3 Met 90 % 79 % 85 %
164r -CH2CH2SH Cys 95 % 65 % 35 %
+H3N
R
O
O NH
R
S
O O
O
O
NO2
N
R
S
O O
O
O
NO2
N
R
S
O O
O
OH
NO2
Cl-a b
c
161a-r 162a-r 163a-r
164a-r
818 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
amino acids 169a-i [59] (Scheme 29). Different Lewis acids (L.A.) were tested as a possible substitute of trifluoroacetic acid (TFA) that enables the reductive opening of the oxazolidinone ring 168 in the classical synthetic procedures [57, 58]. AlCl3 and ZnBr2 appeared to be the most promising agents. Using this methodology it is possible to reduce the amount of triethylsilane to 2 equiv., as compared to 3 equiv. necessary for the classical TFA method to succeed, and the reaction can be completed in only 4 h, as compared to a minimum of 22 h with the use of TFA [59] (Table 12).
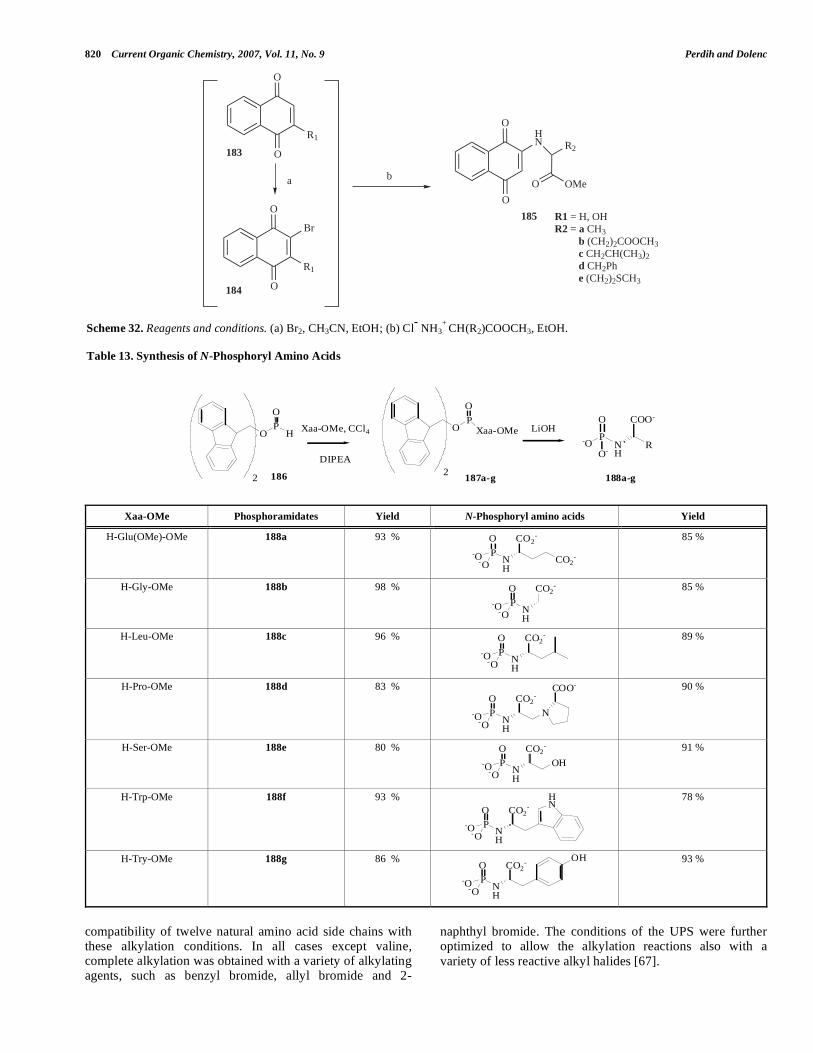
N--methyl- -hydroxyisoleucine and N-methyl-L-valine have recently been found in the naturally occurring potent anti-inflammatory cyclic depsipeptides halipeptin A, B and C, where they contribute to the 17-membered macrolactone ring. De Riccardis et al. recently disclosed an asymmetric synthesis of N,O-diprotected (2S,3S)-N-methyl- -hydroxyi-soleucine (179), a building block required for the asymmetric synthesis of halipeptin A. The key intermediate (2S,3S)-2-amino-3-methyl-4-pentenoic acid 176 was realized through a silyl-assisted [3,3]-sigmatropic rearrangement of the optically active (S)-(Z)-1-(tert-butyldimethylsilyl)-2-butenyl N-tert-butyloxycarbonyl glycinate 174. The obtained unnatural -amino acid 176 was converted to N-Fmoc-protected 5-oxazolidinone 177 and triethylsilane–TFA induced reduction of an oxazolidinone intermediate, yielded the requested N-methylation to provide the desired com-pound 179 [60] (Scheme 30).
Esaki et al. described the coupled enzymatic synthesis of N-methyl-L-phenyl alanine (182) from phenylpyruvic acid (180) and methylamine (181) by using a novel enzyme N-methyl-L-amino acid dehydrogenase (NMAADH) from Pseudomonas putida, NADP+ and glucose dehydrogenase (GDH) from Bacillus subtilis as a co-factor-recycling system. (Scheme 31). Analysis of the product 182 on a laboratory preparative scale process revealed N-methyl-L-phenylalanine in 98 % yield and over 99 % e.e. [61]. (Further discussions on the biosynthetic approaches for the unnatural -amino acids synthesis; see Section V).
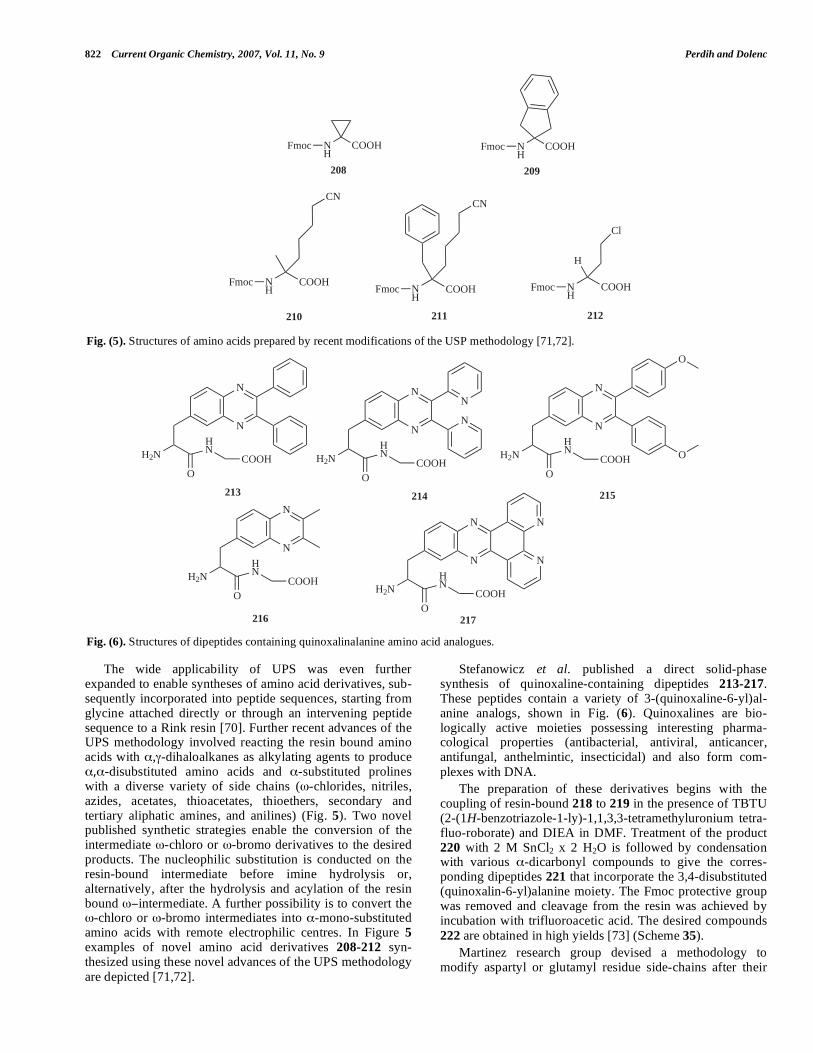
Other possible N-substitutions of the amino group in -AAs besides N-alkylation were reported by Tandon et al. presenting a series of (S)-N-(1,4-naphthoquinon-2-yl)- -amino acid methyl esters 185a-e. The reaction of 1,4-naphthoquinones 183 and their bromo derivatives 184 with enantiomerically pure L-amino acid methyl ester hydro-chlorides produce N-modified- -amino acid methyl esters 185, as shown in Scheme 32 [62].
N-phosphoryl -amino acids have gained a growing interest in the self-assembly of polypeptides [63]. Berkman
Scheme 28. Reagents: (a) (i) PhCHO, (ii) NaCNBH3, (CH2O)n (s); (b) H2, Pd(OH)2. Table 11. N-Methylation of FUNCTIONALIZED a-Amino Acids
Comp. Name R R1 Yield A Yield B
167a L-tryptophan -CH2(3-indolyl) Me 89 % 99 %
167b L-histidine -CH2(3-imidazolyl) Me 99 % 99 %
167c L-asparagine -CH2CONH2 t-Bu 83 % 99 %
167d L-serine -CH2OH Me 90 % 79 %
167e L-threonine -CH2(OH)CH3 Me 99 % 67 %
167f L-arginine -(CH2)3(guanidyl) Me 66 % 85 %
167g L-tyrosine -CH2(4-phenolyl) Me 94 % 96 %
167h D-tyrosine -CH2(4-phenolyl) Me 95 % 79 %
167i L-glutamine -CH2CH2-CONH2 t-Bu 99 % 82 %
Scheme 29. Reagents and conditions: L.A. (2 eq.), Et3SiH (2 eq.), DMC, 4 h. Table 12. Investigation of Lewis Acid Catalysts for the Silane
Reduction of Fmoc-oxazolidines with Non-
Functionalized and Functionalized Side Chains
Compound Oxazolindinone derived
from
Lewis acid
catalyst
Isolated
yield
169a Fmoc-L-Gly-OH AlCl3 94 %
169b Fmoc-L-Phe-OH AlCl3 90 %
169c Fmoc-L-Leu-OH AlCl3 92 %
169d Fmoc-L-Val-OH AlCl3 94 %
169e Fmoc-L-Lys(Boc)-OH ZnBr2 /
169f Fmoc-L-Ser(Ot-Bu)-OH ZnBr2 /
169g Fmoc-L-Asp-(Bz)-OH AlCl3 65 %
169h Fmoc-L-Asp-(OAll)-OH AlCl3 92 %
169i Fmoc-L-Ser-(OBz)-OH AlCl3 72 %
169j Fmoc-L-Lyz(Cbz)-OH AlCl3 80 %
H2N
*
COOR1
R
N
*
COOR1
R
Me
BnN
*
COOR1
R
Me
H
A B
a b
R1= Me, tBu
165a-i166a-i 167a-i
O
N
R
OFmoc
N
R
Fmoc
Me
COOH
a
168a-i 169a-i
Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 819
et al. discovered bis(9-fluorenylmethyl)phosphite (BFMP) 186 as an effective reagent for the N-phosphorylation of various -amino acid methyl esters. BFMP was prepared from N,N-diisopropyl phosphoramidous dichloride in a one-pot two-step reaction and was obtained as a crystalline solid. N-Phosphorylation of the methyl esters of seven re-presentative -amino acids outlined in Table 13 with BFMP was high-yielding and generally resulted in crystalline products. Complete deprotection of both the 9-fluorenyl-methylphosphosphate esters 187a-g and the amino acid methyl esters was accomplished concomitantly with LiOH to give N-phosphoryl -amino acids 188a-g [64] (Table 13).
II. SOLID-PHASE SYNTHESIS OF UNNATURAL AMINO ACIDS
O`Donnell et al. has pioneered the field of solid phase synthesis of unnatural -amino acids. The methodology devised was termed unnatural peptide synthesis (UPS) and
the concept is presented in Scheme 33. This methodology involves introduction of a non-proteinogenic amino acid side chain during a normal solid-phase peptide synthesis through three additional steps (activation, deprotonation/alkylation and hydrolysis). More precisely, the UPS procedure involves the generation of a resin bound Schiff base 190 which is then
-alkylated to 191 in the presence of a base, and finally hydrolyzed, affording free alkylated -amino acid 192 [65].
O`Donnell extended the UPS methodology to racemic , -disubstituted amino acids 197 (Scheme 34), termed di-
UPS (di-substituted unnatural peptide synthesis) [66]. The procedure involves the standard generation of a resin bound Schiff base 194 which is then , -alkylated to 195 in the presence of BEMP (2-[(1,1-dimethylenethyl)imine-N,N-diethyl-2,2,3,4,5,6-hexahydro-1,3-dimethyl-1,3,2-diazapho-sphorin-2(1H)-amine). Subsequent hydrolysis afforded the amine 196, which was for detection purposes, converted to a UV active compound 197. O`Donnell`s group examined the
Scheme 30. Reagents and conditions: (a) PDC, 4 Å m.s., CH2Cl2, 3 h; (b) (S)-Me-CBS reagent, BH3·THF, THF, 30 min; (c) N-Boc-glycine, EDC, DMAP, CH2Cl2, 2 h; (d) Pd / 5% wt. on CaCO3, H2, 3 h; (e) LDA, ZnCl2, THF, -78 °C, 1 h 30 min; (f) HBF4 (48% in water), 1,4-dioxane, 70 °C, 22 h; (g) (i) Fmoc-Cl, Na2CO3, 1,4-dioxane, 0 °C, 18 h; (ii) (CH2O)n, p-TsOH, toluene, 1 h; (h) (i) BH3·SMe2, 0 °C, 5 h; (ii) EtOH, H2O, NaBO3·4H2O, rt, 3 h; (i) (i) BnBr, Ag2O, CH2Cl2, 3 days; (ii) Et3SiH, TFA, CHCl3, rt, 3 days.
Scheme 31. The NMAADH-catalyzed production of N-methyl-L-amino acid in combination with co-factor-recycling catalyzed by GDH.
TBS
O
OHN
Boc R
NHR1
O
OH
NHR1
O
OH
TBS
OH
NO
O
Fmoc
RO
OH
O
NFmoc
BnO
TBS
O
TBS
OH
OHN
Boc
TBS
O
FmocN
O
O
a b c
d ef
gh i
170 171 172 173
174 175 176
177 178 179
O
O
OH
NHMe
O
OH
CH3NH2
NMAADH GDH
Gluconolactone
Glucose
180 181
182
NADP
NADPH
820 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
compatibility of twelve natural amino acid side chains with these alkylation conditions. In all cases except valine, complete alkylation was obtained with a variety of alkylating agents, such as benzyl bromide, allyl bromide and 2-
naphthyl bromide. The conditions of the UPS were further optimized to allow the alkylation reactions also with a variety of less reactive alkyl halides [67].
Scheme 32. Reagents and conditions. (a) Br2, CH3CN, EtOH; (b) Cl-
NH3+ CH(R2)COOCH3, EtOH.
Table 13. Synthesis of N-Phosphoryl Amino Acids
OP
O
HO
P
O
Xaa-OMe-O
P
O
NH
COO-
RO-
Xaa-OMe, CCl4
DIPEA
LiOH
22
186 187a-g 188a-g
Xaa-OMe Phosphoramidates Yield N-Phosphoryl amino acids Yield
H-Glu(OMe)-OMe 188a 93 %
CO2-
CO2-
NH
P-O
O
-O
85 %
H-Gly-OMe 188b 98 % CO2-
NH
P-O
O
-O
85 %
H-Leu-OMe 188c 96 % CO2-
NH
P-O
O
-O
89 %
H-Pro-OMe 188d 83 %
N
CO2-
NH
P-O
O
-O
COO-
90 %
H-Ser-OMe 188e 80 %
OH
CO2-
NH
P-O
O
-O
91 %
H-Trp-OMe 188f 93 % CO2
-
NH
P-O
O
-O
HN
78 %
H-Try-OMe 188g 86 % CO2
-
NH
P-O
O
-O
OH
93 %
HN
O
O
R2
O OMe
O
O
R1
O
O
R1
Br
b
185
183
184
R1 = H, OHR2 = a CH3 b (CH2)2COOCH3 c CH2CH(CH3)2 d CH2Ph e (CH2)2SCH3
a

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 821
UPS methodology is not only applicable to alkylation reactions. Conjugative addition of different Michael sub-strates can also be conducted using this methodology, opening up new possibilities for creating novel amino acid derivatives 198-203. Substituted derivatives of glutamic acid were prepared in this fashion and some are presented in Fig. (3) [68].
UPS methodology relies on generation of a resin bound glycine anion equivalent which acts as a nucleophile. An
attractive possibility is the opposite option, that of a bound glycine cation equivalent, which would enable many nucleo-philic reagents to be utilized. A resin bound glycine cation equivalent was prepared by reacting the usual Schiff base of a Wang resin bound glycine with tetraacetate in dichloro-methane. Subsequently, many organoboranes (e.g. 9-alkyl BBN) reacted with an electrophile produced in the presence of a phenoxide base to yield novel amino acid derivatives 204-207 [69] (Fig. 4).
Scheme 33. General Scheme of the UPS Methodology.
Scheme 34. Reagents and conditions: (a) ArCHO; (b) R2X, BEMP; (c) HONH3+Cl
-; (d) R3COOH, PyBrOP, TFA.
Fig. (3). Derivatives of glutamic acid prepared by USP methodology (R=quinadoyl).
Fig. (4). Amino acid derivatives prepared by a resin bound cation equivalent (R=quinadoyl).
H2N
O
Y
N
O
YPh
Ph
H2N
O
Y
R1
N
O
Y
Ph
Ph
R1Activate Alkylate Hydrolyse
189 190 191 192
H2N
O
O
R1
N
O
O
R1H
Ar N
O
O
R1H
Ar
R2
H2N
O
OR1 R2
HN
O
OHR1 R2O
R3
a b c
d
193194 195
196 197
R1= amino acid side chainR2= PhCH2 Allyl 2-NaphthylCH2R3 = Quinaldoyl
ROCHN COOH
O
ROCHN COOH
HOOC
ROCHN COOH
MeOOCNO2
ROCHN COOH
SO2Ph
ROCHN COOH
CN
ROCHN COOH
COOMe
COOMe
200
201 202 203
198 199
ROCHN COOHROCHN COOHROCHN COOH ROCHN COOH
204 205 206 207
822 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
The wide applicability of UPS was even further expanded to enable syntheses of amino acid derivatives, sub-sequently incorporated into peptide sequences, starting from glycine attached directly or through an intervening peptide sequence to a Rink resin [70]. Further recent advances of the UPS methodology involved reacting the resin bound amino acids with , -dihaloalkanes as alkylating agents to produce
, -disubstituted amino acids and -substituted prolines with a diverse variety of side chains ( -chlorides, nitriles, azides, acetates, thioacetates, thioethers, secondary and tertiary aliphatic amines, and anilines) (Fig. 5). Two novel published synthetic strategies enable the conversion of the intermediate -chloro or -bromo derivatives to the desired products. The nucleophilic substitution is conducted on the resin-bound intermediate before imine hydrolysis or, alternatively, after the hydrolysis and acylation of the resin bound intermediate. A further possibility is to convert the
-chloro or -bromo intermediates into -mono-substituted amino acids with remote electrophilic centres. In Figure 5
examples of novel amino acid derivatives 208-212 syn-thesized using these novel advances of the UPS methodology are depicted [71,72].
Stefanowicz et al. published a direct solid-phase synthesis of quinoxaline-containing dipeptides 213-217. These peptides contain a variety of 3-(quinoxaline-6-yl)al-anine analogs, shown in Fig. (6). Quinoxalines are bio-logically active moieties possessing interesting pharma-cological properties (antibacterial, antiviral, anticancer, antifungal, anthelmintic, insecticidal) and also form com-plexes with DNA.
The preparation of these derivatives begins with the coupling of resin-bound 218 to 219 in the presence of TBTU (2-(1H-benzotriazole-1-ly)-1,1,3,3-tetramethyluronium tetra-fluo-roborate) and DIEA in DMF. Treatment of the product 220 with 2 M SnCl2 x 2 H2O is followed by condensation with various -dicarbonyl compounds to give the corres-ponding dipeptides 221 that incorporate the 3,4-disubstituted (quinoxalin-6-yl)alanine moiety. The Fmoc protective group was removed and cleavage from the resin was achieved by incubation with trifluoroacetic acid. The desired compounds 222 are obtained in high yields [73] (Scheme 35).
Martinez research group devised a methodology to modify aspartyl or glutamyl residue side-chains after their
Fig. (5). Structures of amino acids prepared by recent modifications of the USP methodology [71,72].
Fig. (6). Structures of dipeptides containing quinoxalinalanine amino acid analogues.
COOH
CN
NH
Fmoc
Cl
NH
COOH
H
Fmoc
COOHNH
Fmoc
NH
COOH
CN
Fmoc
NH
COOHFmoc
210 211 212
209208
H2N
N
N
O
HN
COOH H2N
N
N
O
HN
N
COOH
N
H2N
N
N
O
HN
COOH
O
O
H2N
N
N
O
HN
COOHH2N
N
N
O
HN
COOH
N
N
213 214 215
216 217
Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 823
incorporation by solid phase synthesis into a peptide sequence. The concept of a Weinreb amide 223 was used which allowed the reduction of the amide side chain to an aldehyde 224 that could be further functionalized. In the case presented in Scheme 36 this methodology was validated by the synthesis of H-Phe[2-(4-ethoxycarbonyl-3-butene)-glycyl]-leucine amide (226) [74].
III. SYNTHESIS OF -AMINO ACIDS DERIVATIVES CONTAINING NON PROTEINOGENIC ATOMS
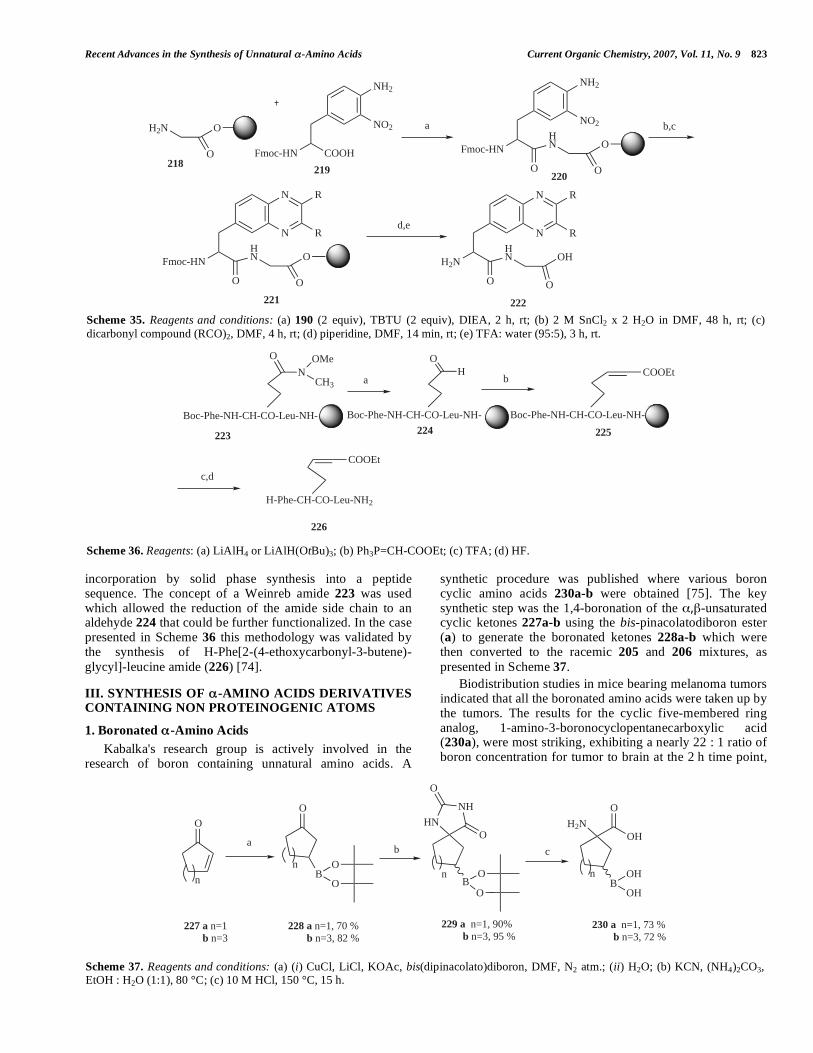
1. Boronated -Amino Acids Kabalka's research group is actively involved in the
research of boron containing unnatural amino acids. A
synthetic procedure was published where various boron cyclic amino acids 230a-b were obtained [75]. The key synthetic step was the 1,4-boronation of the , -unsaturated cyclic ketones 227a-b using the bis-pinacolatodiboron ester (a) to generate the boronated ketones 228a-b which were then converted to the racemic 205 and 206 mixtures, as presented in Scheme 37.
Biodistribution studies in mice bearing melanoma tumors indicated that all the boronated amino acids were taken up by the tumors. The results for the cyclic five-membered ring analog, 1-amino-3-boronocyclopentanecarboxylic acid (230a), were most striking, exhibiting a nearly 22 : 1 ratio of boron concentration for tumor to brain at the 2 h time point,
Scheme 35. Reagents and conditions: (a) 190 (2 equiv), TBTU (2 equiv), DIEA, 2 h, rt; (b) 2 M SnCl2 x 2 H2O in DMF, 48 h, rt; (c) dicarbonyl compound (RCO)2, DMF, 4 h, rt; (d) piperidine, DMF, 14 min, rt; (e) TFA: water (95:5), 3 h, rt.
Scheme 36. Reagents: (a) LiAlH4 or LiAlH(OtBu)3; (b) Ph3P=CH-COOEt; (c) TFA; (d) HF.
Scheme 37. Reagents and conditions: (a) (i) CuCl, LiCl, KOAc, bis(dipinacolato)diboron, DMF, N2 atm.; (ii) H2O; (b) KCN, (NH4)2CO3, EtOH : H2O (1:1), 80 °C; (c) 10 M HCl, 150 °C, 15 h.
Fmoc-HN COOH
NH2
NO2
Fmoc-HN
NH2
NO2
O
HN
O
O
Fmoc-HN
N
N
O
HN
O
O
R
R
O
OHH2N
N
N
O
HN
R
R
H2N
O
O a b,c
d,e
219220
221 222
218
O
NOMe
CH3
OH
COOEt
COOEta b
c,d
223
226
Boc-Phe-NH-CH-CO-Leu-NH- Boc-Phe-NH-CH-CO-Leu-NH- Boc-Phe-NH-CH-CO-Leu-NH-
224 225
H-Phe-CH-CO-Leu-NH2
O
BO
O BO
O
NH
HNO
O
BOH
OH
O
H2NOH
O
n
ab
n n
c
227 a n=1 b n=3
228 a n=1, 70 % b n=3, 82 %
229 a n=1, 90% b n=3, 95 %
230 a n=1, 73 % b n=3, 72 %
n

824 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
dropping to 7.3 after 6 h. Additionally, it was established that similar situation occurs between tumor vs. blood or skin ratios [76].
In the further search for novel 1-aminocyclobutane-1-carboxylic acid derivatives (ACBC), Kabalka and co-workers synthesized boronated ACBC derivatives [77,78]. In Scheme 38, the synthesis of 1-amino-3-[(dihydroxy-boryl)ethyl] cyclobutanecarboylic acid (241) is depicted. The 3-substituted cyclobutanone skeleton was constructed by a [2+2] cycloaddition reaction. The crude product 232 was used without purification, and reductive dechlorination with zinc in acetic acid gave compound 233 that was transformed into ketal 234. Further O-deprotection and subsequent bro-mination, using CBr4/PPh3, produced the bromo derivative 236. Dehydrobromination of 236 to generate the alkenyl group was realized using NaOH in PEG-600. After removal of the ketal protective group, the crude 238 was used directly for the Bücherer-Strecker reaction. Finally, hydroboration and hydrolysis steps produced 241 in good yield. All
presented boron-containing unnatural amino acids are currently being evaluated as potential agents for boron neutron capture therapy (BNCT) [79].
2. Phosphorus and Sulfur Containing -Amino Acids
Incorporation of phosphine containing amino acids into rigid secondary structures such as -helixes and ß-strands, allows the generation of large libraries of bis- and mono-phosphine ligands for screening in asymmetric catalytic reactions. Because these ligands are produced by a solid phase approach, a wide variety of ligands with different structures and chiral environments can be made. It is crucial for this procedure that the starting material is readily available. Diphenylphosphoserine 245 is one of key building blocks for this synthesis. Gilbertson et al. published a novel route leading to this unnatural amino acid that is superior to previously published methods. Starting from commercially available 3-iodo alanine methyl ester 242, following Knochel`s procedure, the iodo zinc amino acid 243 is
Scheme 38. Reagents and conditions: (a) Cl3CCOCl, POCl3, Zn-Cu, Et2O, reflux; (b) Zn, AcOH, 0 °C to reflux; (c) 1,2-hexanediol, PTSA, benzene, reflux; (d) H2, Pd/C (10 %), MeOH, rt; (e) CBr4, PPh3, CH2Cl2, rt; (f) NaOH (6 M), PEG-600, 90 °C; (g) 2 M HCl, EtOH, reflux; (h) (NH4)2CO3, KCN, EtOH : H2O (1:1), 60 °C; (i) (Ipc)2BH, THF, 0 °C to rt; (j) CH3CHO, THF, 0 °C to rt; (l) 12 M HCl, 160 °C.
Scheme 39. Reagents and conditions: (a) Zn, TMSCl, 1,2-dibromoethane, THF/DMF; (b) CuCN.2 LiCl, 0 °C; (c) Ph2PCl, 0 °C to rt; (d) S8; (e) LiOH, THF/H2O, 0 °C; (f) TFA, CH2Cl2; (g) Fmoc-OSu, NaHCO3, H2O/acetone.
BnOO
ClCl
BnOO
BnO
BnO
O
O
C4H9
HO
O
O
C4H9
Br
O
O
C4H9
O
O
C4H9
O
HN
O
NH
O
HN
O
NH
O(HO)2B NH2(HO)2B
COOH
a b c
d e
f g h
i,j,k l
231232 233
234 235 236
237 238239
240 241
BocHN COOMe
I
BocHN COOMe
Cu(CN)ZnI
BocHN COOMe
PPh2
O
FmocHN COOMe
PPh2
O
a,b c,d
e,f,g
242 243 244
245
75%
80%

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 825
obtained. Subsequent addition of stoichiometric amounts of CuCN. 2 LiCl and chlorodiphenylphosphine gave the desired product 244. Presumably, the reactive intermediate is the zinc/copper species in 243. Derivatives 244 and 245 can both be incorporated into peptides by standard solid phase peptide synthesis [80] (Scheme 39).
Notwithstanding the fact that the phosphonic and carboxylic acid groups differ substantially with respect to shape, size and acidity, -phosphonic acids are important structural analogs for -amino acids. Davis et al. reported the first example of asymmetric synthesis of -alkyl- -amino(arylmethyl)phosphonate derivatives 247 from enan-tiopure ketosulfinimines 246. The procedure involves the stereoselective addition of 2 equivalents of lithium diethyl phosphite, prepared in situ by treatment of diethyl phosphite
with LiHMDS (lithium hexa- methyldisilazide), to an enantiopure (S)-(+) ketosulfimine 246 at -78 °C [81] (Scheme 40).
3. Silane Containing -Amino Acids
Organosilanes were first incorporated into amino acids nearly 50 years ago [82]. These compounds provide opportunities for expanded evaluation of peptide con-formation. -Silyl amino acids can be prepared by several different procedures, and most of the recent publications originate from the Sieburth research group. For example, methylation between nitrogen and silicon resulted in compound 248 and further addition of ethyl chloroformate, gave 250. Another possibility is rhodium acetate-mediated insertion of diazoester 252 into the nitrogen- hydrogen bond which led to 253 in high yield [83] (Scheme 41).
Sieburth recently published a novel route starting from allylamine 254. Metalation of a racemic mixture of 255 with a mixture of sparteine and sec-butyllithium gave 256. The corresponding aldehyde 257 was formed with ozone in methylene chloride and subjected to Pinnick oxidation to yield the -silyl amino acid 258. Stereochemical integrity of the transformation was evaluated by coupling the acid with (R)- methyl benzyl amine to afford 259 (Scheme 42). The absence of change in the enantiomeric ratio indicated that oxidation had proceeded with little or no epimerization [84].
Scheme 40. Reagents and conditions: (a) LiP(O)(OEt)2, THF, -78 °C.
Scheme 41. Silane amino acid synthesis.
Scheme 42. Reagents: (a) Boc2O, TBSOTf; (b) sec-BuLi; (-)spartein; (c) O3, Ph3P; (d) NaClO2; (e) (R)-methylbenzylamine, CDI.
P
SNH
O
Ar
O
OR1
OR1
RArS
N
O R
97% de
246
(Ss, R)247
a
R = Me, EtR1= Et
p-Tolylp-Tolyl
HN
Boc NH
O
Ph
Si
H2N N
Si
Boc
Boc
HN
Si
HN HBoc
Si
O
Si
HN OHBoc
O
a b c
de
254 255 256
257 258259
N Li
Si
Bn
BocCl
O
OEt
N
Si
Boc
O
OEt
Bn
N2
Si
O
OEtHN
Si
Boc
O
OEtBoc-NH2
248 249250
251 252 253

826 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
4. Ferrocenyl Amino Acids
Unnatural amino acids can also be found in the field of organometallic chemistry. The organometallic compound ferrocene 262 is a promising candidate for incorporation in novel materials due to its stability, spectroscopic and electro-chemical properties. The incorporation of a ferrocene group into proteins has shown the mediation of electron transfer between electrodes and the protein redox site [85, 86]. Synthetic methodologies providing novel unnatural -amino acids containing ferrocene unit enable construction of a range of novel peptides possessing these properties [87].
Multi-step syntheses of the N- and C-protected ferrocene amino acids (e.g. 1 -(3-aminopropyl)ferrocene-1-carboxylic acid, 1 -amino-1-ferrocenebutyric acid) and their derivatives 260 were reported and described by Rapi et al. [88] and, recently, syntheses of N-ferrocenylglyoxyl amino acid derivatives 261 were published by Skoda-Földes et al. [89] (Fig. 7). Compounds 260 and 261 are formally not -amino acid derivatives, however they are included in this review to emphasize the importance of amino acid derivatives on a wider scale.
An equivalent case are N-meta-ferrocenyl benzoyl -amino acid esters 266a-d, which. have been prepared by
coupling meta-ferrocenyl benzoic acid 265 to the amino acid ethyl esters of glycine, L-alanine, L-leucine and L-phenyl-alanine using the conventional 1,3-dicyclohexylcarbodiimide (DCC) and 1-hydroxybenzotriazole (HOBt) protocol (Scheme 43). Using the same strategy novel N-ortho-ferrocenyl benzoyl amino acid esters were prepared and additionally the X-ray crystal structure of N-(ortho-ferrocenyl(benzoyl))-L-phenyl-alanine was published [90, 91].
5. Halogenated Amino Acids
The -amino acid (2S,4R)-5,5-dichloroleucine 269 is not a completely unnatural amino acid as it is found in sponges belonging to the genus Dysidea. Recently, Rodrigues et al. reported its first total synthesis from L-pyroglutamic acid. The key step outlined in Scheme 44 in an 11 step synthesis was the dichlorination on the hydrazone of aldehyde 267 with CuCl2 in triethylamine. Epimerization at C-4 was not observed and 268 was detected as the sole product. Final hydrolysis in the standard acid medium gave the desired amino acid 269 [92].
Fluorinated amino acids are attractive for synthesis due to their activity as suicide inhibitors of pyridoxal phosphate-dependent -amino acid decarboxylases and amino-transferases. The synthesis of (R)- -difluoroalanine (274) is depicted in Scheme 45, and additionally (S)- -difluoro serine (not shown) has been synthesized by Zanda et al. The hydrocyanation of enantiomerically pure N-Cbz -fluoro-alkyl ß-sulfinylenamine 270 occurs smoothly on treatment with KCN or on addition of trimethylsilylcyanide. The resulting diastereomeric -aminonitrile 271 can then be converted to the final compound 274 by standard synthetic procedures involving the application of microwaves in the final step [93].
The fluorinated derivatives of cysteine were of interest to a research group in China who reported the first four-step
Scheme 43. Reagents and conditions: (a) NaNO2, HCl, 5 °C; (b) NaOH/MeOH, H2O; (c) DCC, HOBt, Et3N, -amino acid ester.
Scheme 44. Reagents and conditions: (a) (i) NH2NH2.H2O, 2 molecular sieves, (ii) CuCl2, Et3N, MeOH, rt; (b) 4 N HCl, reflux.
Fig. (7). Structure of unnatural amino acids with an inserted 1,1-ferrocenylene unit.
FeH2N
O
OO
O
Fe
O
OH
Fe
O
HN
R
O
OH
Fe
ab
262 263 264
265
c
266a H b CH3 c CH3CH(CH3)3 d CH22Ph
COO-tBu
O
H
CH3 NBoc2
COO-tBu
Cl
Cl
CH3 NBoc2
Cl
Cl
CH3 NH2
COOH
a b
40% 75%
267 268 269
NH2
COOH
FeFe
O
NH
R
O
OMe
260 m=0-3; n=1-6
n
m
261 n=1,2
n

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 827
synthesis of both enantiomers of syn-(3-trifluoromethyl) cysteine derivatives 278, as outlined in Scheme 46 [94]. Starting from the previously synthesized trifluoromethylated alcohol 275 [95], treatment of a selected enantiomer (275) with trifluoromethanesulfonic anhydride and pyridine at -40°C, followed by cesium fluoride mediated SN2 dis-placement of trifluoromethanesulfonate with thiolacetic acid, led to the intermediate 276 in high yield. Reaction with boron trichloride and subsequent Jones oxidation afforded the desired product 278.
IV. BIOSYNTHETIC APPROACHES TO THE PRODUCTION AND RESOLUTION OF UNNATURAL
-AMINO ACIDS
In recent years there has been a veritable renaissance in the field of enzymology due to the realization that many enzymes will accept “unnatural” substrates. For synthetic chemists this “promiscuity” is very important since it offers a
novel tool for achieving stereoselective and high yield reactions. This is a field where synthetic organic chemistry and biotechnology go hand in hand. Furthermore, enzymes could be utilized in the enantiospecific separations. Enzymatic methods provide a convenient way to obtain products in large amounts so they are especially interesting from the industrial point of view. For this reason, a short section of this review is dedicated to presenting two basic biosynthetic systems for unnatural -amino acid production. More detailed information is available in the references cited.
L- -amino acid transaminases (LATs) are involved in the biosynthesis of most natural amino acids. D-amino acid transaminases (DATs) have also been identified in a number of bacterial species, most notably in the Bacilli. Both enzymes can be used for the preparation of unnatural amino acids. They are generally used in whole cell or immobilized systems in many industrial syntheses. The substrates that this system requires are appropriate -keto acids which are then
Scheme 45. Reagents and conditions: (a) KCN, THF/H2O; (b), MeOH : H2O (1 : 1), rt, 4 days; (c) Ni-Raney, EtOH, 60 °C, 5 days; (d) (i) Ba(OH)2, H2O, reflux, 24 h, (ii) HCl; (iii) DOWEX 50W.
Scheme 46. Reagents and conditions: (a) Tf2O, Py, CH2Cl2, -40 °C; (b) CH3COSH, CsF, DMF, 0 °C; (c) BCl3, CH2Cl2, -78 °C; (d) Jones Reagent, acetone, 0 °C.
Table 14. Aminotransferases Used in Amino Acid Processes. (Based on [96])
Transaminase Abbreviation Gene Source Products
Aspartate AAT aspC E. coli L-homophenylalanine
Branched chain BCAT ilvE E.coli L-tert-leucine
Tyrosine TAT tryB E.coli L-2-aminobutyrate
D-Amino acid DAT dat Bacillus sp YM 1
Bacillus sphaericus
D-glutamate D-leucine D-tyrosine D-phenylalanine
S
O.. NHCbz
CF2HS
O..NHCbz
CF2H
NC
S
O..
CF2H
HN
NH
O
O
HN
NH
O
O
CF2H CF2H
H2N COOH
a b
c d
274
270 271272
273
F3C OBnHR2N
OHH
F3C
R2N
OH
OBnH
AcS
F3C OHHR2N
OHAcS
HR2N
F3C
OHAcS O
OH
c
99%
275 276 277
278
R2N - phthalimide protection group
a,b
d
99%
87%
828 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
converted to the corresponding amino acid. Table 14 shows some of the unnatural amino acids and D-amino acids that have been produced using this family of enzymes [96].
Another promising route to enantiomerically pure un-natural amino acids is the hydantoinase/carbamoylase system. Here three enzymes, hydantoinase, N-carbamoylase and hydantoinacemarse, are used. Some of the micro-organisms from which these enzymes were isolated are
Arthrobacter aurens, Pseudomonas sp, E. coli, and Caenorhabditis elegans. These enzymes have different specificities so they enable the stereoselective synthesis of L- or D-amino acids 283 and 284. The enzymatic reactions are outlined in Scheme 47 [97,98]. The 5-substituted hydantoins act as starting substrates for this enzyme. The chemo-enzymatic synthesis of a rare amino acid D-citrulline has been described from L-arginine, utilizing this system [99].
7- aza-tryptophan is an unnatural amino acid with very strong fluorescence that is used for probing bioactive confor-mations. In a recent novel synthetic route for preparing the enantiopure compound, Aspergillus acylase was used for the effective separation of a racemic mixture of N-Ac-7-aza-tryptophan [101]. Another example of enzyme mediated chiral resolution was that of an unnatural bipyridyl amino acid 286, using alkaline protease to increase the proportion of enantiomer required for the subsequent peptide synthesis (Scheme 48) [101].
V. SYNTHESIS OF MISCELLANEOUS UN-
NATURAL -AMINO ACIDS
Without doubt unnatural amino acid synthesis has captured the imagination and creativity of many leading synthetic and peptide chemists, and all the examples listed in this review support this statement. In this section the authors wish to highlight some examples which can not be classified in the previously portrayed sections. Some of these amino acids cannot be classified as -amino acid derivatives, but they have been included due to their innovative structure.
The synthesis and evaluation of triazinyl-amino acids 287a-b is an interesting example. 1,3,5-triazine moiety served as the core and ethylenediamine and glycine moieties were connected to the nucleus by nucleophilic aromatic substitution. The third site (Fig. 8, R and R1) can be used to incorporate a wide variety of functional groups. The triazine core has the ability to participate in hydrogen bonding interactions analogous, if not identical, to conventional hydrogen bonding interactions involving the peptide backbone. These novel amino acids were then incorporated into microcyclic pseudopeptides that should be useful as clefts for molecular recognition studies or as models of ß-strand conformation. As in the case of ferrocene amino acids, these compounds 287a-b cannot be formally classified as -amino acids and are presented due to their innovative design [102].
Scheme 47. The hydantoinase/carbamoylase system.
Scheme 48. Reagents: (a) (i) alkaline phosphatase; (ii) di-tert-butyl pyrocarbonate.
H2N
O
N N
OMeHN
O
N N
OH
Boc
a
285286
HN NH
OR
O
HN NH
O
OR
HN NH2
O
R COOH+H2O
-H2O -H2O
+H2O
R COOH
NH2
H2O
HN NH2
O
R COOH
R COOH
NH2
H2O
pH > 8
L-Hydantoinase
L-amino acid
L-Carbamoylase
CO2, NH3
D-amino acid
CO2, NH3
D-Carbamoylase
D-Hydantoinase
Racemase
279 280 281 282
283 284

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 829
Oxazole-containing amino acids are suitable building blocks for the preparation of systems with well-defined secondary structure. Amino acids contain five-membered heterocyclic rings are usually found in naturally occurring cyclic peptides isolated from marine organisms and result from the intramolecular condensation of Cys, Thr or Ser side chains. The synthesis of a new family of densely functionalized oxazole-containing amino acids starts with the
treatment of the imine 288 with potassium tert-butoxide at low temperature, and acylation of the resulting anion with Boc-protected amino acid chloride. This is followed by in situ hydrolysis of the resulting Schiff base, giving the -amino-ß-keto esters 289 as stable hydrochloride salts in good yield (68-82 %). Subsequent coupling with other amino acids by mixed anhydride activation via isobutylchloroformate afforded intermediates 290a-b. Construction of the oxazole ring was performed with Ph3P, in the presence of I2 and Et3N to provide the desired cyclized products 291a-b, which were O-debenzylated to amino acids 292a-b [103] (Scheme 49).
These building blocks were employed for preparing macrocycles containing Lys and Glu residues by a combi-nation of solid- and solution-phase synthesis. The resulting structures 293 and 294 are presented in Fig. (9) as ortho-gonally protected scaffolds for supramolecular chemistry.
An interesting example is the construction of unnatural amino acids with the amino acid side chain connected to a sugar unit via an isometric triazole linkage. These triazole linked glycopeptides 297, 300 are superior in their chemical and metabolic stability and may possess biological activity such as inhibition of glycosidase. The preparation involves Cu(I)-catalyzed [3+2] cycloaddition of either azide-function-alized glycosides 296 and acetylenic amino acids 295 or
Fig. (8). Comparison of the dimensions of the triazinyl amino acids with those of the extended peptide chain.
Scheme 49. Reagents and conditions: (a) (ii) tert-BuOK, THF, -78 °C, 30 min, (ii) Fmoc-Phe-Cl; 3N HCl, -78 °C to rt; (b) BocNH(CH2)2COOH for 290a, or MeCOO(CH2)2COOH for 290b, iso-BuOCOCl, NMM, THF, -20 °C; (c) Ph3P, I2, Et3N, THF, 0 °C; (d) H2, Pd/C, MeOH, 1 h.
Fig. (9). Structures of macrocycles.
Ph
OO
R NH COOBn
NHFmoc
COOBn
NHFmoc
Ph
O
-Cl +H3NN COOBnPh2C
COOBn
NHFmoc
O
N
R
PhO
N
COOH
R
NHFmoc
Ph
c
288
289290
292
a b
a R= NHBocb R= COOMe
d
a R= NHBocb R= COOMe
a R= NHBocb R= COOMe
291
NH HN
N
O
HNNH
O
O
O
N
O
O
ZHN
NHZ
COOMe
MeOOC
NH HN
N
O
HNNH
O
O
O
N
O
O
BnOOC
COOBn
NHBoc
BocHN
293 294
N
NH
N
N
O
NH
H2N
NRR1
N
O
NH
O
HN
O
NH
O
287 a R=Hb R1= 3-methylPh

830 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
acetylenic glycosides 298 and azide-containing amino acids 299. The method was applied successfully to a variety of suitably functionalized saccharides and oligopeptides. Two examples of this synthetic route are presented in Scheme 50 [104].
Constrained glycosylated amino acids were prepared in order to probe the mechanism by which glycosyltransferases
recognize glycoproteins and assemble the core structures of O-linked oligosaccharides. Two constrained glycopeptides 301 and 302, based on the -N-acetylgalactosaminyl serine substructure, were chosen for synthesis. These compounds represent one of the two possible gauche conformations of the D-serine and L-serine configurations of the parent substructure [105] (Fig. 10).
Fullerene-containing amino acid derivatives were synthesized by Tomé et al. In the course of synthesis, 4-chloropyrimidine-fused 3-sulfones 303 were fused with lysine, yielding lysine derivative 304. Further, after thermo-lysis of sulfone in the presence of C60 fullerene, the mono adduct 306 was isolated in 40 % yield. The probable mecha-nism of reaction is a Diels-Alder reaction of the pyrimidine o-quinodimethane intermediate 305 to the corresponding C60 fullerene. If 2 equivalents of C60 are used, intramolecular bisadducts are formed [106] (Scheme 51).
Scheme 50. Cu(I)-catalyzed [3+2] cycloaddition leading to triazole linked glycopeptides.
Fig. (10). Constrained glycosylated amino acids.
Scheme 51. Reagents and conditions. (a) lysine, MeOH, K2CO3, (b) 214 °C; (c) C60.
OAcO
OAcN3
AcO
AcO
HN COOBn
Fmoc
OAcO
OAcN
AcO
AcO
NN
HN COOBn
Fmoc
HN COOH
N3
Boc
OBnO
OBn
BnO
BnO
OBnO
OBn
BnO
BnO
N
N N
HN COOH
Boc
Cu(OAc)2, Na-ascorbateH2O, t-BuOH
Cu(OAc)2, Na-ascorbateH2O, t-BuOH
295 297
298
296
299
300
N
N
NH
Me
NH2
COOH
bSO2
N
N
Cl
Me
N
N
Me
COOH
NH2NH
SO2
NH
Me
SO2
N
N
NH2
COOH
a
c
303 304 305
306
60 %
40 %
O
AcHN
OH
HO
O
OH
NH2
COOH
O
AcHN
OH
HO
O
OH
NH2
COOH
301 302

Recent Advances in the Synthesis of Unnatural -Amino Acids Current Organic Chemistry, 2007, Vol. 11, No. 9 831
CONCLUSION
The variety of synthetic methods and procedures presented in this review, as well as others that can be found in the literature, shows the remarkable creativity of those peptide and organic chemists involved in the design and pre-paration of novel amino acid derivatives. Besides classical wet chemistry procedures to synthesis, new technologies are being widely implemented, such as microwave-assisted synthesis and different chiral catalysts. Solid phase synthesis and implementation of organometallic -amino acids reveal the scope of influence that this field has in organic chemistry. The advances presented in this review enable the creation of superior peptidomimetics with improved biolo-gical properties and the ability to capture the bioactive con-formation of a native peptide sequence. Without doubt, novel synthetic methodologies to construct -unnatural amino acids will continue to provide new ways for peptidomimetic and consequently, drug design.
ACKNOWLEDGEMENTS
The authors wish to thank Prof. Roger Pain for critical reading of the manuscript.
ABBREVIATIONS
ACBC = 1-aminocyclobutane-1-carboxylic acid BBN = 9-Borabicyclo[3.3.1]nonane
BEMP = 2-tert-butylimino-2-diethylamino-1,3-dimethyl- perhydro- 1,3,2-diaza-phosphorine
BFMP = Bis(9-fluorenylmethyl)phosphite BNCT = Boron Neutron Capture Therapy Boc = tert-Butyloxycarbonyl CDI = N,N'-Carbonyldiimidazole COD = cycloocta-1,5-diene DBAD = di-tert-butyl azodicarboxylate DIB = diacetoxyiodo benzene DIEA = diisopropylethyl amine DMAP = dimethylamino pyridine DPPA = diphenylphosphoryl azide d.e. = diastereomeric excess e.e = enantiomeric excess EDCl = 1-(3-Dimethylaminopropyl)-3-ethyl-
carbodiimide hydrochloride ESI = Electrospray Ionization Mass Spectro-
metry Fmoc = 9-Fluorenylmethyloxycarbonyl GDH = glucose dehydrogenase (Ipc)2BH = diisopinocampheylborane
KHMDS = potassium hexamethyldisilazane LiHMDS = lithium hexamethyldisilazide MCPBA = 3-chlorobenzenecarboperoxoic acid MPLC = medium pressure liquid chromato-
graphy NMA = N-methyl amino acids NMAADH = N-methyl-L-amino acid dehydrogenase NMM = N-methylmorpholine
MW = microwave OSu = N-hydroxysuccinimide PCC = pyridinium chlorochromate PDC = pyridinium dichromate PEG = polyethylene glycol PMP = 4-methoxyphenol PTC = phase transfer catalyst PTSA = para-toluene sulfonamide TEMPO = tetramethyl-1-piperidinyloxy moiety TFA = trifluoroacetic acid TFAA = trifluoracetic anhydride TIS = tri-iso-propyl silane TMG = tetramethylguanidine TMSCN = cyanotrimethylsilane Ts = para-toluenesulphonic moiety UPS = unnatural peptide synthesis Z = benzyloxycarbonyl
REFERENCES
[1] Burger's Medicinal Chemistry and Drug Discovery, Volume 1, Drug Discovery, 6th Ed., John Wiley & Sons: New York, 2005.
[2] Bonner, G.G.; Davis, P.; Stropova, C.; Ferguson, R.; Yamamura, H.I.; Porreca, F.; Hruby, V.J. Peptides, 1997, 18, 93.
[3] Marshall, S.A.; Lazar, G.A.; Chirino, A.; Desjarlais, J.R. Drug Discov. Today, 2003, 8, 212.
[4] Szymkowski, D.E. Drug Discov. Today, 2004, 9, 381. [5] Adessi, C.; Soto, C. Curr. Med. Chem., 2002, 9, 963. [6] Gante, J. Angew. Chem. Int. Ed. Engl., 1994, 33, 1699. [7] Marshall, G.R. Tetrahedron, 1993, 49, 3547. [8] Wang, L.; Schultz, P.G. Chem. Commun., 2002, 43, 11623. [9] Maruoka, K.; Ooi, T. Chem. Rev., 2003, 103, 3013-3028. [10] Hendrickson,T.L.; Crécy-Lagard V.; Schimmel, P. Annu. Rev.
Biochem., 2004, 73,147. [11] Lu, V. Curr. Opin. Chem. Biol., 2005, 9, 118. [12] Beene, D.L.; Dougherty, D.A.; Lester, H.A. Curr. Opin.
Neurobiol., 2003, 13, 267. [13] Hyun, M.H.; Han, S.C.; Lipshutz, B.H.; Shin, Y.-J.; Welch, C.J. J.
Chromatogr. A, 2001, 910, 359. [14] Jin, J.S.; Stalcup, A.M.; Hyun, M.H. J. of Chromatogr. A, 2001,
933, 83. [15] Péter, A.; Árki, A.; Vékes, E.; Tourwé, D.; Lázár, L.; Fülöp, F.;
Armstrong , D.W. J. Chromatogr. A, 2004, 1031, 171. [16] Gallos, J.K.; Sarli, V.C.; Massen, Z.S.; Varvogli, A.C.;
Papadoyanni, C.Z.; Papaspyrou, S.D.; Argyropoulos, N.G. Tetrahedron, 2005, 46, 565.
[17] Gallos, J.K.; Sarli, V. C.; Varvogli, A.C.; Papadoyanni, C.Z.; Papaspyrou, S.D.; Argyropoulos, N.G. Tetrahedron, 2003, 44, 3905.
[18] Middleton, R.J.; Mellor, S.J.; Chhabra, S. R.; Bycroft, B. W.;
Chan, W.C. Tetrahedron Lett., 2004, 45, 123. [19] Dygos, J.H., Yonan, E.E.; Scros, M.G.; Goodmonson, O.J.;
Getman, D.P.; Periana, R.A.; Beck, G.R. Synthesis, 1992, 741. [20] Cheng, H.; Luzy, J.-P.; Garbay, C. Tetrahedron Lett., 2005, 46,
3319. [21] Boto, A.; Hernandez, R.; Montoya, A.; Suarez, E. Tetrahedron
Lett., 2005, 43, 8. [22] Wasserman, H.H.; Long, Y.O.; Parr, J. Tetrahedron Lett., 2003, 44,
361. [23] Gorohovsky, S.; Meir, S.; Shkoulev, V.; Byk, G.; Gellerman, G.
Synlett, 2003, 1411. [24] Carlier, P.; Lam, P.C.H.; Wong, D.M. J. Org. Chem., 2002, 67,
6256. [25] Hanessian, S.; Yang, R.-Y. Tetrahedron Lett., 1996, 37, 5273. [26] Shieh, W.-C.; Xue, S.; Reel, N.; Wu, R.; Fitt, R.; Repi , O.
Tetrahedron Asymmetry, 2001, 12, 2421. [27] Tanaka, K.; Ahn, M.; Watanabe, Y.; Fuji, K. Tetrahedron
Asymmetry, 1996, 7, 1771.

832 Current Organic Chemistry, 2007, Vol. 11, No. 9 Perdih and Dolenc
[28] Yu, G.; Mason, H.J.; Galdi, K.; Wu, X.; Cormelious, L.; Zhao, N.; Witkus, M.; Ewing, W.R.; Macor, J.E. Synthesis, 2003, 403.
[29] Padrón, J.M.; Kokotos, G.; Martín, T.; Markidis, T.; Gibbons, W.A.; Martín V.A. Tetrahedron Asymmetry, 1998, 9, 3381.
[30] Markidis, T.; Kokotos, G. J. Org. Chem., 2002, 67, 1685. [31] Wang, W.; Wang, J.; Li, H. Tetrahedron Lett., 2004, 45, 724. [32] Jingersons, A.; Marinozzi, M.; Pellicciari, R.; Tetrahedron, 2005,
61, 373. [33] Capone, S.; Guaragna, A.; Palumbo, G.; Pedatella, S. Tetrahedron,
2005, 61, 6575. [34] Dondoni, A.; Massi, A.; Minghini, E.; Bertolasi, V. Tetrahedron,
2004, 60, 2311. [35] Davis, F.A.; Zhang, Y.; Rao, A.; Zhang, Z. Tetrahedron, 2001, 57,
6345. [36] Huang, T.S.; Li, C. J. Org. Lett., 2001, 3 ,2037. [37] Ballini, R.; Balsamini, C.; Bartoccini, F.; Gianotti, M.; Martinelli,
C.; Savoretti, N. Synthesis, 2005, 2, 296. [38] Kim, H.J.; Lee, S.; Park, Y.S. Synlett, 2001, 5, 613. [39] Koskinen, A.M.P.; Hassilla, H.; Myllymäki, V.T.; Rissanen, K.
Tetrahedron Lett., 1996, 36, 5619. [40] Park, C.; Choi, H.; Son, Y.-C.; Lee, C.S.; Choy, N.; Koh, J.S.; Lee,
T.G.; Kwon, Y.D.; Kim, S.C.; Yoon, H. Biorg. Med. Chem., 1996, 6, 585.
[41] Oba, M.; Tanaka, M.; Takano, Y.; Suemune, H. Tetrahedron, 2005, 61, 593.
[42] Ellis, T.K.; Martin, C.H.; Tsai, G.H.; Ueki, H.; Soloshonok V.A. J. Org. Chem., 2003, 68, 6208.
[43] Ueki, H.; Ellis, T.K.; Collin, M.H.; Soloshonok, V.A. Eur. J. Org.Chem., 2003, 1954.
[44] Belokon, Y.N.; Bespalova, N.B.; Churkina, T.D.; Cisarova, I.; Ezernitskaya, M.G.; Harutyunyan, S.R.; Hrdina, R.; Kagan, H.B.; Kocovsky, P.; Kochetkov, K.A.; Larionov, O.V.; Lyssenko, K.A.; North, M.; Polasek, M.; Peregudov, A.S.; Prisyazhnyuk, V.V.; Vyskocil, S. J. Am. Chem. Soc., 2003, 125, 12860.
[45] Miyabe, H.; Asada, R.; Takemoto, Y. Tetrahedron, 2005, 61, 385. [46] Warmuth, R.; Munsch, T.E.; Stalker, R.A.; Li, B.; Beatty, A.
Tetrahedron, 2001, 57, 6383. [47] Chowdari, N.S.; Suri, J.T.; Barbas III, C.F. Org. Lett., 2004, 6,
2507. [48] van Esseveldt, B.C.J.; van Delft, F.L.; Smits, J.M.M.; de Gelder,
R.; Rutjes, F.P.J.T. Synlett, 2003, 15, 2354-2358. [49] van Esseveldt, B.C.J.; van Delft, F.L.; de Gelder, R; Rutjes,
F.P.J.T. Org. Lett., 2003, 5, 1717. [50] Cheng, W.-C.; Liu, Y.; Wong, M.; Olmstead, M.M.; Lam, K.S:;
Kurth M.J. J. Org. Chem., 2002, 67, 5673. [51] Tiwari, S.K.; Schneider, A.; Koep, S.; Gais, H.-J. Tetrahedron
Lett., 2004, 45, 8343. [52] Lautens, M.; Tayama, E.; Nguyen, D. Org. Lett., 2004, 6, 345. [53] Södergren, M.J.; Andersson P.G.; Tetrahedron Lett., 1996, 37,
7577. [54] Aurelio, L.; Brownlee, R.T.C.; Hughes, A.B. Chem. Rev., 2004,
104, 5823. [55] Biron, E.; Kessler, H. J. Org. Chem., 2005, 70, 5183. [56] White, K.N., Konopelski, J.P. Org. Lett., 2005, 7, 4111. [57] Freidinger, R.M.; Hinkle, J. S.; Perlow, D.S.; Arison, B.H. J. Org.
Chem., 1983, 48, 77. [58] Aurelio, L.; Brownlee, R.T.C.; Hughes, A.B.; Sleebs, B.E. Aust. J.
Chem., 2000, 53, 425. [59] Zhang, S.; Govender, T.; Norström, T.; Arvidsson, P.I. J. Org.
Chem., 2005, 70, 6918. [60] Izzo, I.; Avallone, E.; Della Corte, L.; Maulucci, N.; De Riccardis.
Tetrahedron Asymmetry, 2004, 14, 1181. [61] Muramatsu, H.; Mihara, H.; Kakutani, R.; Yasuda, M.; Ueda, M.;
Kurihara, T.; Esaki, N. Tetrahedron Asymmetry, 2004, 15, 2841. [62] Tandom, V.K.; Yadav, D.B.; Singh, R.V.; Chaturvedi, A.K.;
Shukla, P.S. Biorg. Med. Chem., 2005, 15, 5324. [63] Jiang, Y.; Tan, B.; Chen, Z.Z.; Liu, T.; Zhong, R.G.; Li, Y.M.;
Stewart, D.J.; Zhao Y.F.; Jiang, H.L. Int. J. Quantum Chem., 2003, 94, 232.
[64] Wu, L.Y.; Berkman, C.E. Tetrahedron Lett., 2005, 46, 5301. [65] O'Donnell, M.J.; Zhou, C.; Scott, W.L.; J. Am. Chem. Soc., 1996,
118, 6070.
[66] Scott, W.L.; Zhou, C.; Fang, Z.; O'Donnell, M.J. Tetrahedron Lett., 1997, 38, 3695.
[67] O'Donnell, M.J.; Lugar, C.W.; Pottorf, R.S.; Zhou, C. Tetrahedron
Lett., 1997, 38, 7163. [68] Domínguez, E.; O'Donnell, M.J.; Scott, W.L. Tetrahedron Lett.,
1998, 39, 2167. [69] O'Donnell, M.J.; Delgado, F.; Drew, M.D.; Pottorf, M.D.; Zhou,
C.; Scott, W.L. Tetrahedron Lett., 1999, 40, 5831. [70] Scott, W.L.; Delgado, F.; Lobb, K.; Pottorf, M.D.; O'Donnell, M.J.
Tetrahedron Lett., 2001, 42, 2073. [71] Scott W.L.; O'Donnell, M.J.; Delgado, F.; Alsina, J. J.Org.Chem.
2002, 67, 2960-2969. [72] O'Donnell, M.J.; Alsina, J.; Scott, W.L. Tetrahedron Lett., 2003,
44, 8403 [73] Staszewska, A.; Stefanowicz, P.; Szewczuk, Z. Tetrahedron Lett.,
2005, 46, 5525. [74] Paris, M.; Douat, C.; Heitz A.; Gibbons, W.; Martinez, J.; Fehrentz
J.-A. Tetrahedron Lett., 1999, 40, 5179. [75] Kabalka, G.W.; Das, B.C.; Das, S. Tetrahedron Lett., 2001, 42,
7146. [76] Kabalka, G.W.; Wu, Z.Z.; Yao, M.-L.; Natarajan, N. Appl. Radiat.
Isot., 2003, 61, 1111. [77] Kabalka, G.W.; Yao, M.-L. Synthesis, 2003, 2890. [78] Kabalka, G.W.; Yao, M.-L. J. Org. Chem., 2004, 69, 8280. [79] Kabalka, G.W.; Yao, M.-L.; Navarane, A. Tetrahedron Lett., 2005,
46, 4915. [80] Greenfield, S. J.; Gilbertson, S. R. Synthesis, 2001, 10, 2337-2340. [81] Davis, F.A.; Lee, S.; Yan, H.; Titus, D.D. Org. Lett., 2001, 3, 1757. [82] Birkofer, L.; Ritter, A. Angew. Chem., 1956, 68, 461. [83] Sieburth, S. McN.; Somers, J.J.; O`Hare, H.K. Tetrahedron, 1996,
52, 5669. [84] Liu, G.; Sieburth, S.M. Org. Lett., 2003, 5, 2677. [85] Degani V.; Heller, A. J. Am. Chem. Soc., 1988, 110, 2615. [86] Kira, M.; Matsubara, T.; Shinohara, H.; Sisido, M. Chem. Lett.,
1997, 89. [87] Brosch, O.; Weyhermuller, T.; Metzler-Nolte, N. Eur. J. Inorg.
Chem., 2000, 2, 323 [88] Bari i , L.; Rapi , V.; Pritzkow, H.; Pavlovi , G.; Nemet, I. J.
Organomet. Chem., 2003, 682, 131. [89] Kuik, A.; Skoda-Földes, R.; Balogh, J.; Kollár, L. J. Organomet.
Chem., 2005, 690, 3237. [90] Savage, D.; Malone, G.; Alley, S.R.; Gallagher, J.F.; Goel, A.;
Kelly, P.N.; Bunz, H.M.; Kenny, P.T.M. J. Organomet. Chem.,
2005, 691, 463. [91] Savage, D.; Neary, N.; Malone, G.; Alley, S.R.; Kenny, P.T.M.
Inorg. Chem. Commun. 2005, 8, 429. [92] Ardá, A.; Jiménez, C.; Rodríguez, J. Tetrahedron Lett., 2004, 45,
3241. [93] Bravo, P.; Capelli, S.; Meille, S.V.; Seresini, P.; Volonterio, A.;
Zanda, M. Tetrahedron Asymmetry, 1996, 7, 2321. [94] Jiang, Z.-X.; Liu, X.-P.; Qui, X.-L.; Qing, F.-L. J. Fluor. Chem.,
2005, 126, 497. [95] Jiang, Z.-X.; Qing, F.-L. J. Org. Chem., 2004, 69, 5486. [96] Taylor, P.P.; Pantaleone, D.P.; Senkpeil, R.F. Fotheringham I.G.
TIBTECH, 1998, 16, 412. [97] Bommarius, A.S.; Schwarm, M.; Drauz, K. J. Mol. Catal. B Enzym,
2001, 5, 1. [98] Altenbuchner, J.; Siemann-Herzberg, M. Curr. Opin. Biotechol.,
2001, 12, 559-563. [99] Bommarius, A.S.; Drauz, K. Biorg. Med. Chem., 1994, 2, 617. [100] Lecointe, L.; Rolland-Fulcrand, V.; Roumestant, M.L.; Viallefont,
P.; Martinez, J. Tetrahedron Asymmetry, 1998, 9, 1753. [101] Kise Jr., K.J.; Bowler, B.E. Tetrahedron Asymmetry, 1998, 9, 3319. [102] Zerkowski, J.A.; Hensley, L.M.; Abramowitz, D.M. Synlett, 2002,
29, 557. [103] Mann, E.; Kessler, H. Org. Lett., 2003, 5, 4567. [104] Kuijpers, B.H.M.; Groothuys, S.; Keereweer, A.B.R.; Quaedflieg,
P.J.L.M.; Blaauw, R.H.; van Delft, F.L.; Rutjes, F.P.J.T. Org. Lett., 2004, 6, 3126.
[105] Lane, J.W.; Halcomb, R.L. Tetrahedron, 2001, 57, 6531. [106] Enes, R.F.; Tomé, A.C.; Cavaleiro, J.A. Tetrahedron, 2005, 61,
1423.