protective effect of creatine against 6-hydroxydopamine-induced cell death in human neuroblastoma...
TRANSCRIPT

Neuroscience 238 (2013) 185–194
PROTECTIVE EFFECT OF CREATINE AGAINST6-HYDROXYDOPAMINE-INDUCED CELL DEATH IN HUMANNEUROBLASTOMA SH-SY5Y CELLS: INVOLVEMENT OF INTRACELLULARSIGNALING PATHWAYS
M. P. CUNHA, a,b,d* M. D. MARTIN-DE-SAAVEDRA, a,b
A. ROMERO, a,b,e E. PARADA, a,b J. EGEA, a,b,c
L. DEL BARRIO, a,b A. L. S. RODRIGUES d ANDM. G. LOPEZ a,b,c
a Instituto Teofilo Hernando, Facultad de Medicina, Universidad
Autonoma de Madrid, 4-28029 Madrid, Spain
bDepartamento de Farmacologıa y Terapeutica, Facultad de
Medicina, Universidad Autonoma de Madrid, 4-28029 Madrid, Spain
c Instituto de Investigacion Sanitaria Hospital de la Princesa,
Madrid, Spain
dDepartamento de Bioquımica, Centro de Ciencias Biologicas,
Universidade Federal de Santa Catarina, Brazil
eDepartamento de Toxicologıa y Farmacologıa, Facultad de
Veterinaria, Universidad Complutense de Madrid, 28040 Madrid,
Spain
Abstract—The guanidine-like compound creatine exerts bio-
energetic, antiexcitotoxic, antioxidant and neuroprotective
properties; however, the intracellular mechanisms responsi-
ble for these effects are still not well established. The pur-
pose of this study was to investigate the protective effect
of creatine against 6-hydroxydopamine (6-OHDA)-induced
cell death in neuroblastoma SH-SY5Y cells and the possible
intracellular signaling pathways involved in such effect.
Exposure of SH-SY5Y cells to 100–300 lM of 6-OHDA for
24 h caused a significant concentration-dependent cell
death measured as a diminution of 3-[4,5-dimethylthiazol-
2-yl]-2,5-diphenyl-tetrazolium bromide (MTT) reduction and
as an increase in the extracellular release of lactate dehydro-
genase. SH-SY5Y cells incubated for 24 or 48 h with creatine
(10–5000 lM) was not cytotoxic. However, pre and co-treatment
with creatine (0.3–1000 lM) for 24 h reduced 6-OHDA-induced
0306-4522/12 $36.00 � 2013 IBRO. Published by Elsevier Ltd. All rights reservehttp://dx.doi.org/10.1016/j.neuroscience.2013.02.030
*Correspondence to: M. P. Cunha, Departamento de Bioquımica,Centro de Ciencias Biologicas, Universidade Federal de SantaCatarina, Brazil. Tel: +55-48-3721-5043; fax: +55-48-3721-9672.
E-mail address: [email protected] (M. P. Cunha).Abbreviations: 6-OHDA, 6-hydroxydopamine; ANOVA, analysis ofvariance; ATP, adenosine triphosphate; BDNF, brain-derivedneurotrophic factor; CaMKII, Ca2+/calmodulin-dependent proteinkinase II; CK, creatine kinase; CREB, cyclic adenosinemonophosphate response element-binding protein; DMSO, dimethylsulfoxide; EMEM, Eagle’s minimum essential medium; ERK1/2,extracellular signal-regulated kinases; FBS, fetal bovine serum; GSK-3b, glycogen synthase kinase-3b; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; LDH, lactate dehydrogenase; MEK1/2,mitogen-activated protein kinase kinase 1/2; MPP+, 1-methyl-4-phenylpyridinium ion; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine;MTT, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide;PCr, phosphocreatine; PD, Parkinson’s disease; PI3K,phosphatidylinositol-3 kinase; PKA, protein kinase A; PKC, proteinkinase C.
185
toxicity. The protective effect afforded by creatine against
6-OHDA-induced toxicity was reversed by inhibitors of
different protein kinases, i.e. phosphatidylinositol-3 kinase
(PI3K) (LY294002), Ca2+/calmodulin-dependent protein kinase
II (CaMKII) (KN-93), protein kinase A (H-89), mitogen-acti-
vated protein kinase kinase 1/2 (MEK1/2) (PD98059) and pro-
tein kinase C (PKC) (chelerythrine). Furthermore, creatine
prevented the 6-OHDA-induced dephosphorylation of glyco-
gen synthase kinase-3b (GSK-3b) at the serine 9 residue. In
conclusion, the results of this study show that creatine
can protect against 6-OHDA-induced toxicity and its protec-
tive mechanism is related to a signaling pathway that
involves PI3K, PKC, PKA, CaMKII, MEK1/2 and GSK-3b.� 2013 IBRO. Published by Elsevier Ltd. All rights reserved.
Key words: 6-OHDA, AKT, creatine, GSK-3b, neuroprotection,PKA.
INTRODUCTION
Parkinson’s disease (PD) is one of the most common
neurodegenerative disorders, affecting about 2% of the
population over the age of 60 years (Dorsey et al.,
2007; Davie, 2008; Gasser, 2009), with an annual direct
medical care cost attributable to PD of more than US
$10,000 per patient (Huse et al., 2005; Noyes et al.,
2006). This chronic disturbance causes severe motor
dysfunction, such as bradykinesia, resting tremor,
rigidity, postural instability, and also affects autonomic
function and cognition (Poewe et al., 2008; Lesage and
Brice, 2009). Pathologically, it is associated with the
profound loss of dopaminergic neurons in the substantia
nigra pars compacta with the subsequent decrease of
striatal dopamine (Schapira, 2008).
Accumulated evidence indicates that impairment of
cellular energy metabolism, particularly defective
mitochondrial function and oxidative stress, contributes
to neuronal death in PD (Gu et al., 1998; Mattson et al.,
1999). Furthermore, deregulations in signaling pathways
have been associated with PD and modulation of
several of the intracellular kinases could contribute
to the treatment of this neurodegenerative pathology.
In line with this, in mice treated with the dopami-
nergic toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP), Ca2+/calmodulin-dependent protein kinase II
(CaMKII) activity was reduced in the hippocampus
(Moriguchi et al., 2012). Furthermore, neuroprotective
d.

186 M. P. Cunha et al. / Neuroscience 238 (2013) 185–194
compounds were shown to protect cells against
dopaminergic injury in PD models by activating protein
kinase C (PKC) alpha and epsilon (Levites et al., 2002;
Tian et al., 2007; Tiong et al., 2010), extracellular
signal-regulated kinases (ERK1/2) (Levites et al., 2002;
Tian et al., 2007), AKT (Ge et al., 2010; Tiong et al.,
2010; Gong et al., 2012; Zhang et al., 2012) and protein
kinase A (PKA) (Carrasco et al., 2008). Moreover,
glycogen synthase kinase-3b (GSK-3b), a kinase
constitutively active that can be inactivated by
phosphorylation of its serine 9 residue (of its regulatory
amino-terminal domains) by several other kinases
(Frame and Cohen, 2001), was shown to be increased
in the neurons located in the substantia nigra and the
upper pons of PD patients (Nagao and Hayashi, 2009).
The neurotoxin 6-hydroxydopamine (6-OHDA) can
induce cell lineage apoptosis and is widely used to
mimic experimental models of PD (Gomez-Lazaro et al.,
2008; Ikeda et al., 2008; Mu et al., 2009). The
biochemical mechanism of the neurotoxic action of 6-
OHDA is not completely understood. However, because
this neurotoxin has similar structure to dopamine, it
shows high affinity for the dopamine transporter and for
this reason it selectively destroys dopaminergic/
catecholaminergic neurons (Lehmensiek et al., 2006).
Once inside the neuron, 6-OHDA accumulates and
undergoes non-enzymatic auto-oxidation, promoting
reactive oxygen species formation (Blandini et al.,
2008). Furthermore, 6-OHDA may provoke inhibition of
mitochondrial complexes I and IV, causing adenosine
triphosphate (ATP) depletion (Glinka et al., 1996;
Tirmenstein et al., 2005). These observations have led
to the hypothesis that a mitochondrial dysfunction is
responsible for the cell death induced by 6-OHDA.
A rapidly available alternative source for ATP
synthesis in brain is the creatine kinase/phosphocreatine
(CK/PCr) system, which can operate via substrate level
phosphorylation to equilibrate adenine nucleotides with
PCr and creatine. The CK/PCr system therefore plays
an important role in cells with high and fluctuating
energy demands like neurons (Wallimann et al., 1989,
1992; Hemmer and Wallimann, 1993). In the putamen
and midbrain of PD patients, a bilateral reduction of
high-energy phosphates such as ATP and PCr was
reported (Hattingen et al., 2009). Therefore, the
improvement of the metabolic state of neuronal cells
may have therapeutic potential in this disease (Beal,
2009).
Creatine is a guanidine compound that could
equilibrate ATP levels inside the cell by increasing the
PCr pool (Woznicki and Walker, 1979) and enhancing
the function of a cellular energy shuttle, coupling sites of
ATP production and ATP consumption (Bessman and
Geiger, 1981; Schlattner et al., 2006). This high-energy
phosphate precursor is synthesized from glycine,
arginine and S-adenosylmethionine in the kidneys, liver
and pancreas and to some extent in the brain (Braissant
et al., 2001). In vivo and in vitro studies indicate that
creatine exerts significant neuroprotection for
dopaminergic neurons against neurotoxic insults such
as 6-OHDA, MPTP/1-methyl-4-phenyl pyridinium ion
(MPP+) and rotenone; all compounds that are
specifically toxic to dopaminergic neurons and are
widely used to induce PD-like neurodegeneration
(Matthews et al., 1999; Andres et al., 2005a,b; Hosamani
et al., 2010; Yong-Kee et al., 2011).
The positive results obtained with creatine in
experimental studies prompted its use in clinical trials in
PD patients. In a pilot trial, creatine supplementation
improved mood and led to a smaller dose increase of
dopaminergic therapy in PD patients (Bender et al.,
2006). Furthermore, PD progression was slowed by
almost 50% over a one-year observation period in the
creatine-treated patients (NINDS NET-PD Investigators,
2006). Recently, the National Institutes of Health
announced a phase III clinical trial for PD, with a goal of
recruiting 1720 participants randomized to 10 g of
creatine or placebo (Bloom, 2007). However, more
compelling evidence is needed for elucidating its
neuroprotective role against PD disease. In this study,
we examined the protective action of creatine against
6-OHDA-induced cell death in SH-SY5Y neuroblastoma
cells and the signal transduction pathways regulating
its effect. Further elucidation of the protective effect of
creatine may lead to novel therapeutic strategies for PD.
EXPERIMENTAL PROCEDURES
Drugs and chemicals
Creatine monohydrate was obtained from Sigma (Madrid, Spain).
Chelerythrine, 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-
4-one (PD98059), 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-
4-one hydrochloride (LY294002), N-[2-[[[3-(4-chlorophenyl)-2-
propenyl]methylamino]methyl]phenyl]-N-(2-hydroxyethyl)-4-
methoxybenzenesulphonamide (KN-93), N-[2-[[3-(4-Bromophenyl)-
2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide hydrochloride
(H-89) were purchased from Tocris (Biogen Cientifica, Madrid,
Spain). Eagle’s minimum essential medium (EMEM), fetal
bovine serum (FBS), and penicillin/streptomycin were purchased
from GIBCO (Madrid, Spain).
The toxin 6-OHDA was dissolved in deionized water
containing 0.1% ascorbic acid, at a final concentration of 1 M.
The solution of creatine (100 mM) was always prepared fresh
on the day of the experiment and diluted in F-12/EMEM
supplemented with 1% FBS. The kinase inhibitors were
dissolved in 100% dimethyl sulfoxide (DMSO) and the final
concentration of DMSO in the well of the plates was 0.1%
diluted in the F-12/EMEM medium supplemented with 1% FBS.
The experimental control had 0.1% DMSO in the F-12/EMEM
medium supplemented with 1% FBS.
SH-SY5Y cell culture
The neuroblastoma cell line SH-SY5Y was a kind gift from the
Centro de Biologıa Molecular, Universidad Autonoma de
Madrid/Consejo Superior de Investigaciones Cientificas
(Madrid, Spain). SH-SY5Y cells were maintained in a 1:1
mixture of F-12 Nutrient Mixture (Ham12) (Sigma–Aldrich,
Madrid, Spain) and EMEM supplemented with 15 nonessential
amino acids, 1 mM sodium pyruvate, 10% heat-inactivated
FBS, 100 units/ml penicillin, and 100 lg/ml streptomycin
(reagents from Invitrogen, Madrid, Spain). Cultures were
seeded into flasks containing supplemented medium and
maintained in a monolayer at 37 �C in a humidified atmosphere

M. P. Cunha et al. / Neuroscience 238 (2013) 185–194 187
of 5% CO2 and 95% air. Stock cultures were passaged 1:3 twice
weekly; i.e., one plate was divided (subcultured or split) into three
plates. This procedure was performed twice a week. For assays,
SH-SY5Y cells were sub-cultured in 48-well plates (TPP,
Zellkutur and Labortechnologie, Trasadingen, Switzerland) at a
seeding density of 1 � 105 cells per well. Cells were treated
Fig. 1. Protective effect of creatine on cell death induced by 6-OHDA in SH-S
SH-SY5Y neuroblastoma cells. Cells were treated with F-12/EMEM alone or
reduction (Panel A) or LDH activity (Panel B) was analyzed 24 h after 6-OHD
24 h was not cytotoxic ‘‘per se’’ (Panel C). Co-incubation (24 h during 6-OHD
not alter the toxic effects induced by 6-OHDA (Panel D). Creatine at increa
(Panel E). Pre-incubation (24 h before 6-OHDA) and co-incubation (24 h durin
increased cell viability as compared to the group incubated with 6-OHDA alon
(Panel F). Data are shown as mean + SEM of 6–10 different and independen
control; ##P < 0.01 or ###P< 0.001 as compared to the 100 lM 6-OHDA g
with the drugs before confluence in F-12/EMEM medium
supplemented with 1% FBS. All treatments were performed
when cells were grown to about 65% confluence; at the end of
treatment, cells reached about 80–90% confluence. All cells in
this study were used at a low passage number (<13) and were
maintained in 10% FBS until the 6-OHDA treatment.
Y5Y cells. Concentration-dependent cell death induced by 6-OHDA in
F-12/EMEM containing four different concentrations of 6-OHDA. MTT
A addition. Creatine at increasing concentrations (100–5000 lM) for
A incubation) of the SH-SY5Y cells with creatine (100–5000 lM) did
sing concentrations (0.1–5000 lM) for 48 h did not alter cell viability
g 6-OHDA incubation) of SH-SY5Y cells with creatine (0.1–5000 lM)
e and maximum protection was achieved at a concentration of 10 lMt cell batches. ⁄P< 0.05 or ⁄⁄P< 0.01 or ⁄⁄⁄P< 0.001 with respect to
roup.
188 M. P. Cunha et al. / Neuroscience 238 (2013) 185–194
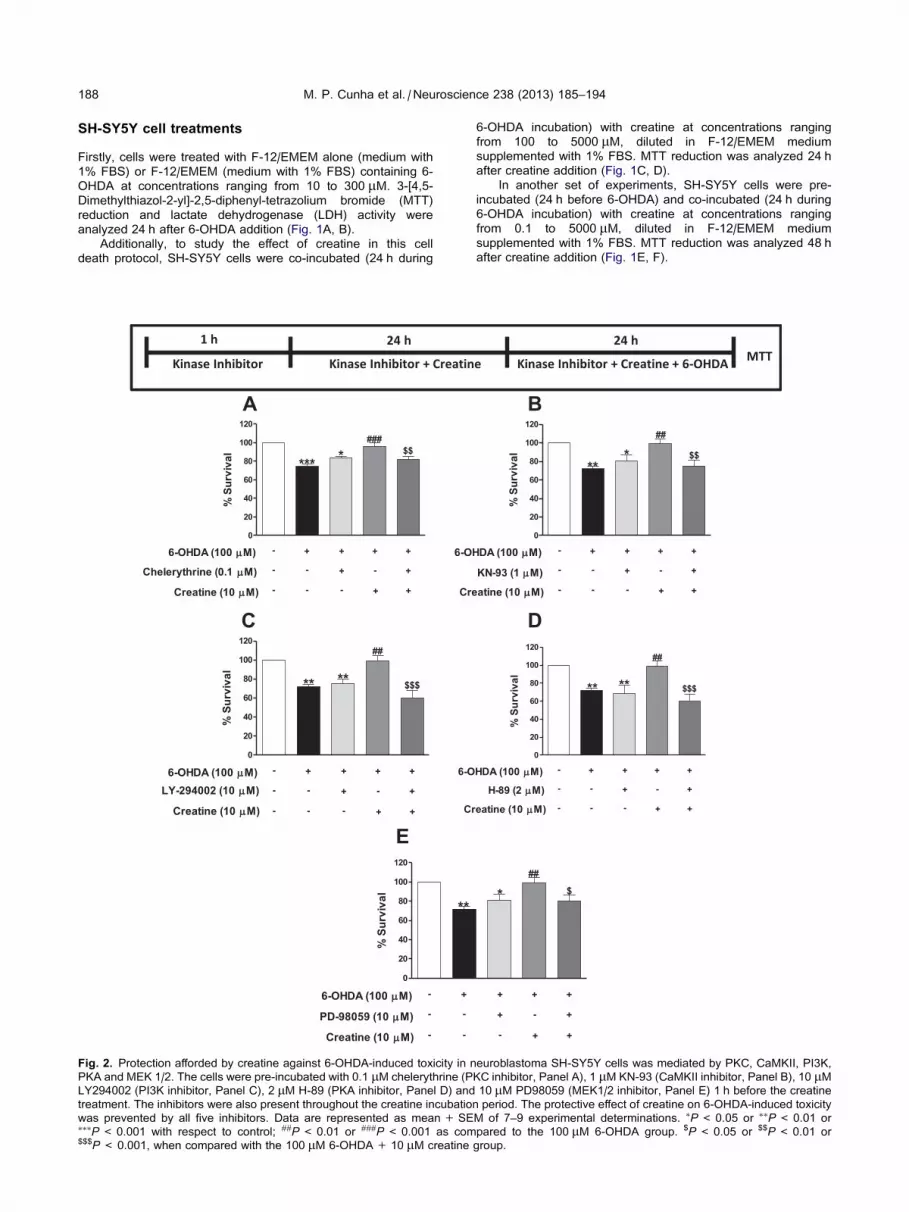
SH-SY5Y cell treatments
Firstly, cells were treated with F-12/EMEM alone (medium with
1% FBS) or F-12/EMEM (medium with 1% FBS) containing 6-
OHDA at concentrations ranging from 10 to 300 lM. 3-[4,5-
Dimethylthiazol-2-yl]-2,5-diphenyl-tetrazolium bromide (MTT)
reduction and lactate dehydrogenase (LDH) activity were
analyzed 24 h after 6-OHDA addition (Fig. 1A, B).
Additionally, to study the effect of creatine in this cell
death protocol, SH-SY5Y cells were co-incubated (24 h during
Fig. 2. Protection afforded by creatine against 6-OHDA-induced toxicity in n
PKA and MEK 1/2. The cells were pre-incubated with 0.1 lM chelerythrine (P
LY294002 (PI3K inhibitor, Panel C), 2 lM H-89 (PKA inhibitor, Panel D) and
treatment. The inhibitors were also present throughout the creatine incubatio
was prevented by all five inhibitors. Data are represented as mean + SE⁄⁄⁄P< 0.001 with respect to control; ##P< 0.01 or ###P< 0.001 as com$$$P< 0.001, when compared with the 100 lM 6-OHDA+ 10 lM creatine
6-OHDA incubation) with creatine at concentrations ranging
from 100 to 5000 lM, diluted in F-12/EMEM medium
supplemented with 1% FBS. MTT reduction was analyzed 24 h
after creatine addition (Fig. 1C, D).
In another set of experiments, SH-SY5Y cells were pre-
incubated (24 h before 6-OHDA) and co-incubated (24 h during
6-OHDA incubation) with creatine at concentrations ranging
from 0.1 to 5000 lM, diluted in F-12/EMEM medium
supplemented with 1% FBS. MTT reduction was analyzed 48 h
after creatine addition (Fig. 1E, F).
euroblastoma SH-SY5Y cells was mediated by PKC, CaMKII, PI3K,
KC inhibitor, Panel A), 1 lM KN-93 (CaMKII inhibitor, Panel B), 10 lM10 lM PD98059 (MEK1/2 inhibitor, Panel E) 1 h before the creatine
n period. The protective effect of creatine on 6-OHDA-induced toxicity
M of 7–9 experimental determinations. ⁄P< 0.05 or ⁄⁄P< 0.01 or
pared to the 100 lM 6-OHDA group. $P < 0.05 or $$P< 0.01 or
group.

M. P. Cunha et al. / Neuroscience 238 (2013) 185–194 189
In order to investigate the mechanisms underlying the
protective effect of creatine against 6-OHDA-induced cell death,
the cells were pre-incubated with 10 lM LY294002
(phosphatidylinositol-3 kinase, PI3K inhibitor), 1 lM KN-93
(Ca2+/calmodulin-dependent protein kinase II, CaMKII
inhibitor), 2 lM H-89 (protein kinase A, PKA inhibitor), 10 lMPD98059 (mitogen-activated protein kinase kinase, MEK1/2
inhibitor), 0.1 lM chelerythrine (protein kinase C, PKC inhibitor)
1 h before creatine addition and during creatine incubation
(Fig. 2).
MTT measurement
Cell viability was measured using the MTT reduction assay as
described previously (Mosmann, 1983). At the end of the
experimental protocol, MTT was added to each well at a final
concentration of 0.5 mg/ml in Krebs-HEPES solution (144 mM
NaCl, 5.9 mM KCl, 1.2 mM MgCl2, 2 mM CaCl2, 10 mM
HEPES, and 11 mM glucose; pH 7.3), and incubation at 37 �Cwas continued for an additional 2-h period. Then, the insoluble
formazan was dissolved with DMSO; colorimetric
determination of MTT reduction was measured at k 540 nm.
Control cells treated with vehicle (EMEM) were taken as
100% viability.
LDH activity measurement
Samples of incubation media were collected at the end of a 24-h
period with 6-OHDA exposure to estimate extracellular LDH, an
indication of cell death (Koh and Choi, 1987; Sobrado et al.,
2004). LDH activity was also measured in the cells after
treatment with 10% Triton X-100 (intracellular LDH). LDH
activity was measured spectrophotometrically at k 490–620 nm,
using a microplate reader (Labsystems iEMS reader MF;
Labsystems, Helsinki, Finland). Total LDH (intracellular plus
extracellular) was normalized as 100% and the amount of LDH
released to the extracellular medium was expressed as
percentage of this total value.
Western blot
The immunoblotting was performed as previously described by
our group (Parada et al., 2010; Dal-Cim et al., 2012). For
detection of proteins, SH-SY5Y cells were lysed in 100 ll ice-cold lysis buffer (1% Nonidet P-40, 10% glycerol, 137 mM
NaCl, 20 mM Tris–HCl, pH 7.5, 1 lg/ml leupeptin, 1 mM
phenylmethylsulfonyl fluoride, 20 mM NaF, 1 mM sodium
pyrophosphate, and 1 mM Na3VO4). A tablet of protease
inhibitor cocktail (complete Mini, Roche, Madrid, Spain) was
added for each 10 ml of buffer. Protein concentrations were
measured with the protein assay kit. Equivalent amounts of
protein (30 lg) were run in 10% sodium dodecyl sulfate
denaturing polyacrylamide gel electrophoresis (SDS–PAGE)
and transferred to an Immobilon-P Transfer Membrane
(Millipore, Bedford, MA, USA) at room temperature.
Membranes were blocked in Tris-buffered saline with 0.05%
Tween 20 (TTBS) containing 4% bovine serum albumin, and
incubated for 2 h at room temperature with primary antibodies
against p-GSK-3b-ser9, GSK-3b and b-actin (1:1000) (Santa
Cruz Biotechnology Inc., Santa Cruz, CA, USA) and then for
1 h with secondary antibodies conjugated with peroxidase
(1:10,000). The membrane was developed using the enhanced
chemiluminescence reagent (Amersham Biosciences, San
Francisco, CA, USA). Optical density was quantified using the
program Scion Image� Alpha 4.0.3.2. Control conditions were
taken as 1 and experimental variables were normalized with
respect to this value.
Statistical analysis
Data are represented as mean+ S.E.M. Comparisons between
experimental and control groups were performed by a one-way
analysis of variance (ANOVA) followed by the Newman–Keuls
post hoc test. Statistical difference was accepted when P 6 0.05.
RESULTS
Effect of 6-OHDA on the viability of SH-SY5Y cells
We first examined the effect of 6-OHDA on the cell
viability of cultured SH-SY5Y neuroblastoma cells. For
this purpose, cells were incubated without (control) or
with 6-OHDA at concentrations of 10–300 lM for 24 h.
The cellular viability was evaluated by the MTT
reduction assay and was expressed as percentage of
MTT metabolism in control cells, i.e. cells incubated for
24 h only with culture medium (100% cellular viability).
6-OHDA, in the range of 50–300 lM, significantly
decreased cell viability in a concentration-dependent
manner; cell viability was reduced by 20% at 50 lM and
by 60% at 300 lM (Fig. 1A). In agreement with this
reduction in cell viability, 6-OHDA increased cell death
measured as an augmentation in LDH release,
especially at the concentrations of 100 and 300 lM(Fig. 1B).
Effect of creatine on the cytotoxic effects of 6-OHDA
Creatine has been described to afford neuroprotection
against various cytotoxic stimuli (Brewer and Wallimann,
2000; Brustovetsky et al., 2001; Juravleva et al., 2005;
Andres et al., 2005a; Genius et al., 2012); hence, we
tried to evaluate if it could protect SH-SY5Y cells
against 6-OHDA-induced toxicity. For this purpose we
used two experimental conditions. The first protocol
consisted in co-incubating creatine with 6-OHDA
(100 lM) for 24 h. Exposure of SH-SY5Y cells for
24 h to increasing concentrations of creatine alone
(100–5000 lM) was not toxic (Fig. 1C). When creatine
(100–5000 lM) was co-incubated with 6-OHDA, no
protection was obtained (Fig. 1D).
The second protocol consisted in pre-incubating the
cells for 24 h with creatine (0.1–5000 lM), followed by
co-incubation with creatine plus 6-OHDA (100 lM) for
another 24 h. Initially, we evaluated whether 48 h
exposure to creatine (10–5000 lM) could be toxic per
se; the results indicated that it was not (Fig. 1E). When
creatine (0.3–1000 lM) was pre- and co-incubated with
the toxic stimuli, it presented a U-shaped protective
curve. Significant protection was achieved at 0.3 lM(24% increase in cell survival, as compared to the
6-OHDA group), and maximum protection at a
concentration of 10 lM (34% increase in cell survival)
(Fig. 1F).
Implication of kinases PI3K/AKT, PKA, PKC, MEK1/2and CaMKII in the protective effect of creatine
In order to investigate the mechanism underlying the
protective effect of creatine against 6-OHDA-induced

190 M. P. Cunha et al. / Neuroscience 238 (2013) 185–194
cell death, different kinase inhibitors were used employing
the protocol indicated in the top part of Fig. 2. Under these
experimental conditions, the PI3K/AKT inhibitor
(LY294002, 10 lM), the PKA inhibitor (H-89, 2 lM), the
PKC inhibitor (chelerythrine, 0.1 lM), the MEK1/2
inhibitor (PD98059, 10 lM) and CaMKII inhibitor (KN-93,
1 lM), all blocked the protective effect of creatine on
the neurotoxicity afforded by 6-OHDA in neuroblastoma
SH-SY5Y cells (Fig. 2A–E, respectively). The kinase
inhibitors per se did not exert any significant action on
the cell death caused by 6-OHDA.
Effect of creatine on GSK-3b phosphorylation
The results depicted in Fig. 3 show that 6-OHDA
incubation for 24 h decreases GSK-3b phosphorylation
at serine 9 residue (59% reduction). When creatine
Fig. 3. Protection elicited by creatine against 6-OHDA-induced toxicity in neu
(at serine 9). Representative immunoblot of P-GSK-3b-Ser9 and total GSK-3b3b phosphorylation (P-GSK-3b-Ser9/total GSK-3b) induced by 6-OHDA. ⁄⁄P100 lM 6-OHDA group.
(10 lM) was pre- and co-incubated with 6-OHDA, it
completely blocked the effect of this toxin on GSK-3bphosphorylation.
DISCUSSION
The main finding of this study is that the guanidine
compound creatine can afford neuroprotection in an
in vitro model of experimental PD induced by 6-OHDA
by a mechanism that implicates several intracellular
kinases and the prevention of the 6-OHDA-induced
dephosphorylation of GSK-3b at the serine 9 residue.
6-OHDA is a potent neurotoxin that causes
degeneration of dopaminergic neurons. It has been
used as a selective catecholaminergic neurotoxin to
produce cell and animal models of PD (Kostrzewa and
Jacobowitz, 1974; Cadet et al., 1989; Perumal et al.,
roblastoma SH-SY5Y cells is associated with GSK-3b phosphorylation
demonstrated that creatine (10 lM) prevented the decrease in GSK-
< 0.01, when compared with control. #P< 0.05 as compared to the

M. P. Cunha et al. / Neuroscience 238 (2013) 185–194 191
1989; Kumar et al., 1995). Several studies have indicated
that 6-OHDA induces cell death in a human
catecholaminergic cell line SH-SY5Y (Tirmenstein et al.,
2005; Bakos et al., 2012; Luo et al., 2012; Ossola et al.,
2012), in agreement with the present study in which
6-OHDA at the dose range of 100–300 lM caused
toxicity in SH-SY5Y neuroblastoma cells, measured as
a decrease in metabolic activity (assessed by MTT test)
and an increase in cell membrane permeability
(assessed by LDH assay; Fig. 1).
In the present study, creatine presented a protective
effect on cell death induced by 6-OHDA, suggesting a
neuroprotective effect of this compound in PD. To gain
information on the putative intracellular signaling
pathways implicated in the neuroprotective effect of
creatine, we considered different kinases. The first was
PI3K, a key kinase implicated in cell proliferation, growth,
survival, learning and memory (Dwivedi et al., 1998; Kelly
and Lynch, 2000; Huang et al., 2005; Yang et al., 2008).
Lipid products of PI3K act as second messengers by
recruiting proteins such as AKT and its activating kinases
(Hanada et al., 2004). Moreover, recent studies have
reported that the PI3K/AKT signaling pathway plays an
important role in the neuroprotective effect of several
compounds on cell death induced by 6-OHDA (Hwang
and Jeong, 2008, 2010; Kao et al., 2009; Mnich et al.,
2010; Deng et al., 2012). Our results also indicate that
exposure of SH-SY5Y cells to LY294002, a PI3K
inhibitor, suppressed the protective effect of creatine in 6-
OHDA-exposed cells. Furthermore, transient reporter
assays showed that LY294002 represses muscle CK
gene transcription, suggesting the important role of the
signaling pathway mediated by PI3K/AKT in creatine
metabolism (Jiang et al., 1998). In line with our results, a
previous study demonstrated that creatine incubation
increased the phosphorylation of AKT and of its
downstream target proteins, such as GSK-3b and
p70S6K, in C2C12 cells (Deldicque et al., 2007).
We also found that H-89 suppressed the protective
effect of creatine against 6-OHDA-induced toxicity. H-89
acts as a competitive inhibitor against ATP binding to
the catalytic subunit of PKA (Chijiwa et al., 1990), an
enzyme involved in neurotransmitter synthesis and
release, gene expression, synaptic plasticity, memory,
cell growth, differentiation, and cell survival. The major
mechanism of PKA-mediated function is through the
phosphorylation of specific substrates, which include
cyclic adenosine monophosphate response element-
binding protein (CREB) (D’sa and Duman, 2002; Gould
and Manji, 2002; Blendy, 2006). The phosphorylation of
CREB causes the expression of proteins such as brain-
derived neurotrophic factor (BDNF), which has been
implicated in the maintenance of neurons, cell survival,
and neuronal plasticity (D’sa and Duman, 2002). Our
results also indicate for the first time that exposure of
SH-SY5Y cells to H-89 abolished the protective effect of
creatine in 6-OHDA-exposed cells, suggesting that
modulation of PKA is implicated in the neuroprotective
effect of creatine.
Our results also indicate that the protective effect of
creatine against 6-OHDA cell death is dependent at
least in part on PKC activation, since chelerythrine was
able to abolish the protective effect of creatine.
Somewhat in agreement with our results, studies have
reported that the PKC signaling pathway plays an
important role in the protective effect of several
compounds on cell death induced by 6-OHDA (Tian
et al., 2007; Tiong et al., 2010; Quesada et al., 2011).
PKC activation can protect cultured neonatal neurons
from serum-deprivation-induced apoptosis (Behrens
et al., 1999). Furthermore, phorbol ester activation of
PKC protects hippocampal and cortical neuronal
cultures from H2O2-induced oxidative stress (Dore et al.,
1999; Maher, 2001).
Additionally, PD98059 (MEK1/2 inhibitor) was able to
block the protective action of creatine against 6-OHDA-
induced damage. Taking into account that MEK (MAP
kinase kinase) is the immediate upstream activator of
ERK (Lewis et al., 1998), these results suggest that
MEK/ERK pathway also participates in the protective
effect of creatine in this protocol of cell death. In line
with this evidence, a previous study demonstrated that
creatine incubation increased ERK1/2 phosphorylation
in C2C12 cells (Deldicque et al., 2007). Furthermore,
another study demonstrated that the rapid activation of
ERK1/2 in SH-SY5Y cells by oxidative stress induced
by 6-OHDA serves as a self-protective response,
reducing the content of reactive oxygen species and
caspase-3 activity and increasing downstream ERK1/2
substrates, suggesting that this pathway plays an
important role in protection against 6-OHDA toxicity (Lin
et al., 2008).
Besides, CaMKII is the most abundant protein kinase
in the brain involved in neuronal plasticity (Wang and
Maler, 1998; Hudmon and Schulman, 2002). Moreover,
CaMKII is altered after nigrostriatal denervation (Oh
et al., 1999). Our results show that KN-93 suppressed
the protective effect of creatine, suggesting that this
signaling pathway is involved in the protective
mechanism related to creatine in this cell death protocol.
Finally, GSK-3b is a serine/threonine kinase originally
identified as a regulator of glycogen metabolism, which is
now recognized as an important modulator of apoptosis.
GSK-3b activity is negatively regulated by the
phosphorylation at the serine 9 residue. In the present
study, we demonstrate that 6-OHDA decreased the
phosphorylation at the serine 9 residue of GSK-3b.Interestingly, 6-OHDA significantly inhibits
phosphorylation of GSK-3b at serine 9 in SH-SY5Y and
PC12 cells (Chen et al., 2004). Furthermore, knockdown
of GSK-3b attenuates 6-OHDA-induced apoptosis in
SH-SY5Y cells (Li et al., 2011). 6-OHDA also inhibits
phosphorylation of AKT at serine 473 (an important
upstream signaling component that regulates GSK-3binactivation) in SH-SY5Yand PC12 cells (Chen et al.,
2004).
The inactivation of GSK-3 can be induced by
phosphorylation at one of its N-terminal serine residues:
Serine 21 for GSK-3a and Serine 9 for GSK-3b (Plyte
et al., 1992) that can be pharmacological targets for
protective agents. The phosphorylation of GSK-3 can be
mediated by several kinases, including mitogen-activated

192 M. P. Cunha et al. / Neuroscience 238 (2013) 185–194
protein kinase (MAPK), AKT (protein kinase B), some
isoforms of PKC, cyclic AMP (cAMP)-dependent protein
kinase (PKA) and CaMKII (Rommel et al., 2001; Jope
and Roh, 2006; Song et al., 2010). Literature data have
reported that pretreatment with TDZD-8, lithium and
L803-mts (GSK-3 inhibitors) eliminates 6-OHDA-induced
cell death (Chen et al., 2004). In the present study,
creatine blocked the decrease in GSK-3b phosphorylation
at the serine 9 residue induced by 6-OHDA. Supporting
this finding, it has been described that creatine increases
GSK-3b phosphorylation at the serine 9 residue in
C2C12 cells (Deldicque et al., 2007).
Our results suggest that creatine could be activating
multiple signaling pathways that could probably be
cross-talking. At present, we cannot rule out whether
creatine is acting individually on each signaling pathway,
thus exerting a pleiotropic effect. Our interpretation is
that activation of the intracellular signaling pathways,
mediated by several kinases, could converge on GSK-3bphosphorylation at the serine 9 residue and that this
effect could actively participate in the protective effect of
creatine against 6-OHDA toxicity.
CONCLUSION
Our findings identify creatine as a rather potent natural
protective factor for catecholaminergic cell survival,
which may be of relevance for therapeutic approaches
in PD.
Acknowledgements—This work was supported by grants from
the Fundacao de Apoio a Pesquisa Cientıfica e Tecnologica do
Estado de Santa Catarina (FAPESC), CNPq and CAPES (Bra-
zil), Rede Instituto Brasileiro de Neurociencia IBN-Net/CNPq
and NENASC Project (PRONEX-FAPESC/CNPq). The Ministry
of Economy and compentence SAF2009-12150 and SAF2012-
32223 to M.G.L.
REFERENCES
Andres RH, Ducray AD, Perez-Bouza A, Schlattner U, Huber AW,
Krebs SH, Seiler RW, Wallimann T, Widmer HR (2005a) Creatine
supplementation improves dopaminergic cell survival and
protects against MPP+ toxicity in an organotypic tissue culture
system. Cell Transplant 14:537–550.
Andres RH, Huber AW, Schlattner U, Perez-Bouza A, Krebs SH,
Seiler RW, Wallimann T, Widmer HR (2005b) Effects of creatine
treatment on the survival of dopaminergic neurons in cultured fetal
ventral mesencephalic tissue. Neuroscience 133:701–713.
Bakos J, Strbak V, Ratulovska N, Bacova Z (2012) Effect of oxytocin
on neuroblastoma cell viability and growth. Cell Mol Neurobiol
32:891–896.
Beal MF (2009) Therapeutic approaches to mitochondrial dysfunction
in Parkinson’s disease. Parkinsonism Relat Disord 3:S189–S194.
Behrens MM, Strasser U, Koh JY, Gwag BJ, Choi DW (1999)
Prevention of neuronal apoptosis by phorbol ester-induced
activation of protein kinase C: blockade of p38 mitogen-
activated protein kinase. Neuroscience 94:917–927.
Bender A, Koch W, Elstner M, Schombacher Y, Bender J, Moeschl M,
Gekeler F, Muller-Myhsok B, Gasser T, Tatsch K, Klopstock T
(2006) Creatine supplementation in Parkinson disease: a
placebo-controlled randomized pilot trial. Neurology
67:1262–1264.
Bessman SP, Geiger PJ (1981) Transport of energy in muscle: the
phosphoryl creatine shuttle. Science 211:448–452.
Blandini F, Armentero MT, Martignoni E (2008) The 6-
hydroxydopamine model: news from the past. Parkinsonism
Relat Disord 14:S124–S129.
Blendy JA (2006) The role of CREB in depression and antidepressant
treatment. Biol Psychiatry 59:1144–1150.
Bloom MZ (2007) NIH announces phase III clinical trial of creatine for
Parkinson’s disease. Consult Pharm 22:378.
Braissant O, Henry H, Loup M, Eilers B, Bachmann C (2001)
Endogenous synthesis and transport of creatine in the rat brain:
an in situ hybridization study. Brain Res Mol Brain Res
86:193–201.
Brewer GJ, Wallimann TW (2000) Protective effect of the energy
precursor creatine against toxicity of glutamate and beta-amyloid
in rat hippocampal neurons. J Neurochem 74:1968–1978.
Brustovetsky N, Brustovetsky T, Dubinsky JM (2001) On the
mechanisms of neuroprotection by creatine and
phosphocreatine. J Neurochem 76:425–434.
Cadet JL, Katz M, Jackson-Lewis V, Fahn S (1989) Vitamin E
attenuates the toxic effects of intrastriatal injection of 6-
hydroxydopamine (6-OHDA) in rats: behavioral and biochemical
evidence. Brain Res 476:10–15.
Carrasco E, Werner P, Casper D (2008) Prostaglandin receptor EP2
protects dopaminergic neurons against 6-OHDA-mediated low
oxidative stress. Neurosci Lett 441:44–49.
Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J (2004) Glycogen
synthase kinase 3beta (GSK3beta) mediates 6-
hydroxydopamine-induced neuronal death. FASEB J
18:1162–1164.
Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T,
Naito K, Toshioka T, Hidaka H (1990) Inhibition of forskolin-
induced neurite outgrowth and protein phosphorylation by a newly
synthesized selective inhibitor of cyclic AMP-dependent protein
kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-
isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma
cells. J Biol Chem 265:5267–5272.
Dal-Cim T, Molz S, Egea J, Parada E, Romero A, Budni J, Martın de
Saavedra MD, del Barrio L, Tasca CI, Lopez MG (2012)
Guanosine protects human neuroblastoma SH-SY5Y cells
against mitochondrial oxidative stress by inducing heme
oxigenase-1 via PI3K/Akt/GSK-3b pathway. Neurochem Int
61:397–404.
Davie CA (2008) A review of Parkinson’s disease. Br Med Bull
86:109–127.
Deldicque L, Theisen D, Bertrand L, Hespel P, Hue L, Francaux M
(2007) Creatine enhances differentiation of myogenic C2C12 cells
by activating both p38 and Akt/PKB pathways. Am J Physiol Cell
Physiol 293:C1263–C1271.
Deng C, Tao R, Yu SZ, Jin H (2012) Sulforaphane protects against 6-
hydroxydopamine-induced cytotoxicity by increasing expression
of heme oxygenase-1 in a PI3K/Akt-dependent manner. Mol Med
Report 5:847–851.
Dore S, Takahashi M, Ferris CD, Hester LD, Guastella D, Snyder SH
(1999) Bilirubin, formed by activation of heme oxygenase-2,
protects neurons against oxidative stress injury. Proc Natl Acad
Sci U S A 96:2445–2450.
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway
RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A,
Tanner CM (2007) Projected number of people with Parkinson
disease in the most populous nations, 2005 through 2030.
Neurology 68:384–386.
D’Sa C, Duman RS (2002) Antidepressants and neuroplasticity.
Bipolar Disord 4:183–194.
Dwivedi Y, Janicak PG, Pandey GN (1998) Elevated [3H]inositol
1,4,5-trisphosphate binding sites and expressed inositol 1,4,5-
trisphosphate receptor protein level in platelets of depressed
patients. Psychopharmacology 138:47–54.
Frame S, Cohen P (2001) GSK3 takes centre stage more than
20 years after its discovery. Biochem J 359:1–16.

M. P. Cunha et al. / Neuroscience 238 (2013) 185–194 193
Gasser T (2009) Mendelian forms of Parkinson’s disease. Biochim
Biophys Acta 1792:587–596.
Ge KL, Chen WF, Xie JX, Wong MS (2010) Ginsenoside Rg1
protects against 6-OHDA-induced toxicity in MES23.5 cells via
Akt and ERK signaling pathways. J Ethnopharmacol
127:118–123.
Genius J, Geiger J, Bender A, Moller HJ, Klopstock T, Rujescu D
(2012) Creatine protects against excitoxicity in an in vitro model of
neurodegeneration. PLoS One 7:e30554.
Glinka Y, Tipton KF, Youdim MB (1996) Nature of inhibition of
mitochondrial respiratory complex I by 6-hydroxydopamine. J
Neurochem 66:2004–2010.
Gomez-Lazaro M, Galindo MF, Concannon CG, Segura MF,
Fernandez-Gomez FJ, Llecha N, Comella JX, Prehn JH, Jordan
J (2008) 6-Hydroxydopamine activates the mitochondrial
apoptosis pathway through p38 MAPK-mediated, p53-
independent activation of Bax and PUMA. J Neurochem
104:1599–1612.
Gong L, Zhang QL, Zhang N, Hua WY, Huang YX, Di PW, Huang T,
Xu XS, Liu CF, Hu LF, Luo WF (2012) Neuroprotection by urate
on 6-OHDA-lesioned rat model of Parkinson’s disease: linking to
Akt/GSK3b signaling pathway. J Neurochem 123:876–885.
Gould TD, Manji HK (2002) Signaling networks in the
pathophysiology and treatment of mood disorders. J Psychosom
Res 53:687–697.
Gu M, Cooper JM, Taanman JW, Schapira AH (1998) Mitochondrial
DNA transmission of the mitochondrial defect in Parkinson’s
disease. Ann Neurol 44:177–186.
Hanada M, Feng J, Hemmings BA (2004) Structure, regulation and
function of PKB/AKT – a major therapeutic target. Biochim
Biophys Acta 1697:3–16.
Hattingen E, Magerkurth J, Pilatus U, Mozer A, Seifried C, Steinmetz
H, Zanella F, Hilker R (2009) Phosphorus and proton magnetic
resonance spectroscopy demonstrates mitochondrial dysfunction
in early and advanced Parkinson’s disease. Brain
132:3285–3297.
Hemmer W, Wallimann T (1993) Functional aspects of creatine
kinase in brain. Dev Neurosci 15:249–260.
Hosamani R, Ramesh SR, Muralidhara (2010) Attenuation of
rotenone-induced mitochondrial oxidative damage and
neurotoxicty in Drosophila melanogaster supplemented with
creatine. Neurochem Res 35:1402–1412.
Huang TJ, Verkhratsky A, Fernyhough P (2005) Insulin enhances
mitochondrial inner membrane potential and increases ATP levels
through phosphoinositide 3-kinase in adult sensory neurons. Mol
Cell Neurosci 28:42–54.
Hudmon A, Schulman H (2002) Structure–function of the
multifunctional Ca2+/calmodulin-dependent protein kinase II.
Biochem J 364:593–611.
Huse DM, Schulman K, Orsini L, Castelli-Haley J, Kennedy S,
Lenhart G (2005) Burden of illness in Parkinson’s disease. Mov
Disord 20:1449–1454.
Hwang YP, Jeong HG (2008) The coffee diterpene kahweol induces
heme oxygenase-1 via the PI3K and p38/Nrf2 pathway to protect
human dopaminergic neurons from 6-hydroxydopamine-derived
oxidative stress. FEBS Lett 582:2655–2662.
Hwang YP, Jeong HG (2010) Ginsenoside Rb1 protects against 6-
hydroxydopamine-induced oxidative stress by increasing heme
oxygenase-1 expression through an estrogen receptor-related
PI3K/Akt/Nrf2-dependent pathway in human dopaminergic cells.
Toxicol Appl Pharmacol 242:18–28.
Ikeda Y, Tsuji S, Satoh A, Ishikura M, Shirasawa T, Shimizu T (2008)
Protective effects of astaxanthin on 6-hydroxydopamine-induced
apoptosis in human neuroblastoma SH-SY5Y cells. J Neurochem
107:1730–1740.
Jiang BH, Zheng JZ, Vogt PK (1998) An essential role of
phosphatidylinositol 3-kinase in myogenic differentiation. Proc
Natl Acad Sci U S A 95:14179–14183.
Jope RS, Roh MS (2006) Glycogen synthase kinase-3 (GSK3) in
psychiatric diseases and therapeutic interventions. Curr Drug
Targets 7:1421–1434.
Juravleva E, Barbakadze T, Mikeladze D, Kekelidze T (2005)
Creatine enhances survival of glutamate-treated neuronal/glial
cells, modulates Ras/NF-kappaB signaling, and increases the
generation of reactive oxygen species. J Neurosci Res
79:224–230.
Kao TC, Shyu MH, Yen GC (2009) Neuroprotective effects of
glycyrrhizic acid and 18beta-glycyrrhetinic acid in PC12 cells via
modulation of the PI3K/Akt pathway. J Agric Food Chem
57:754–761.
Kelly A, Lynch MA (2000) Long-term potentiation in dentate gyrus of
the rat is inhibited by the phosphoinositide 3-kinase inhibitor,
wortmannin. Neuropharmacology 39:643–651.
Koh JY, Choi DW (1987) Quantitative determination of glutamate
mediated cortical neuronal injury in cell culture by lactate
dehydrogenase efflux assay. J Neurosci Methods 20:83–90.
Kostrzewa RM, Jacobowitz DM (1974) Pharmacological actions of 6-
hydroxydopamine. Pharmacol Rev 26:199–288.
Kumar R, Agarwal AK, Seth PK (1995) Free radical-generated
neurotoxicity of 6-hydroxydopamine. J Neurochem 64:
1703–1707.
Lehmensiek V, Tan EM, Liebau S, Lenk T, Zettlmeisl H, Schwarz J,
Storch A (2006) Dopamine transporter-mediated cytotoxicity of 6-
hydroxydopamine in vitro depends on expression of mutant alpha-
synucleins related to Parkinson’s disease. Neurochem Int
48:329–340.
Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms
to genetic susceptibility factors. Hum Mol Genet 18:R48–R59.
Lewis TS, Shapiro PS, Ahn NG (1998) Signal transduction through
MAP kinase cascades. Adv Cancer Res 74:49–139.
Li Y, Luo F, Wei L, Liu Z, Xu P (2011) Knockdown of glycogen
synthase kinase 3 beta attenuates 6-hydroxydopamine-induced
apoptosis in SH-SY5Y cells. Neurosci Lett 487:41–46.
Levites Y, Amit T, Youdim MB, Mandel S (2002) Involvement of
protein kinase C activation and cell survival/cell cycle genes in
green tea polyphenol (�)-epigallocatechin 3-gallate
neuroprotective action. J Biol Chem 277:30574–30580.
Lin E, Cavanaugh JE, Leak RK, Perez RG, Zigmond MJ (2008) Rapid
activation of ERK by 6-hydroxydopamine promotes survival of
dopaminergic cells. J Neurosci Res 86:108–117.
Luo F, Wei L, Sun C, Chen X, Wang T, Li Y, Liu Z, Chen Z, Xu P
(2012) HtrA2/Omi is involved in 6-OHDA-induced endoplasmic
reticulum stress in SH-SY5Y cells. J Mol Neurosci 47:120–127.
Maher P (2001) How protein kinase C activation protects nerve cells
from oxidative stress-induced cell death. J Neurosci
21:2929–2938.
Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G,
Kaddurah-Daouk R, Beal MF (1999) Creatine and cyclocreatine
attenuate MPTP neurotoxicity. Exp Neurol 157:142–149.
Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S
(1999) Cellular and molecular mechanisms underlying perturbed
energy metabolism and neuronal degeneration in Alzheimer’s and
Parkinson’s diseases. Ann N Y Acad Sci 893:154–175.
Mnich K, Finn DP, Dowd E, Gorman AM (2010) Inhibition by
anandamide of 6-hydroxydopamine-induced cell death in PC12
cells. Int J Cell Biol 2010:818497.
Moriguchi S, Yabuki Y, Fukunaga K (2012) Reduced calcium/
calmodulin-dependent protein kinase II activity in the
hippocampus is associated with impaired cognitive function in
MPTP-treated mice. J Neurochem 120:541–551.
Mosmann T (1983) Rapid colorimetric assay for cellular growth and
survival: application to proliferation and cytotoxicity. J Immunol
Methods 65:55–63.
Mu X, He G, Cheng Y, Li X, Xu B, Du G (2009) Baicalein exerts
neuroprotective effects in 6-hydroxydopamine-induced
experimental parkinsonism in vivo and in vitro. Pharmacol
Biochem Behav 92:642–648.
Nagao M, Hayashi H (2009) Glycogen synthase kinase-3beta is
associated with Parkinson’s disease. Neurosci Lett 449:103–107.
NINDS NET-PD Investigators (2006) A randomized, double-blind,
futility clinical trial of creatine and minocycline in early Parkinson
disease. Neurology 66:664–671.

194 M. P. Cunha et al. / Neuroscience 238 (2013) 185–194
Noyes K, Liu H, Temkin-Greener H (2006) Cost of caring for
Medicare beneficiaries with Parkinson’s disease: impact of the
CMS-HCC risk-adjustment model. Dis Manag 9:339–348.
Oh JD, Vaughan CL, Chase TN (1999) Effect of dopamine
denervation and dopamine agonist administration on serine
phosphorylation of striatal NMDA receptor subunits. Brain Res
821:433–442.
Ossola B, Lantto TA, Puttonen KA, Tuominen RK, Raasmaja A,
Mannisto PT (2012) Minocycline protects SH-SY5Y cells from
6-hydroxydopamine by inhibiting both caspase-dependent and
-independent programmed cell death. J Neurosci Res 90:682–690.
Parada E, Egea J, Romero A, del Barrio L, Garcıa AG, Lopez MG
(2010) Poststress treatment with PNU282987 can rescue SH-
SY5Y cells undergoing apoptosis via a7 nicotinic receptors linked
to a Jak2/Akt/HO-1 signaling pathway. Free Radic Biol Med
49:1815–1821.
Perumal AS, Tordzro WK, Katz M, Jackson-Lewis V, Cooper TB,
Fahn S, Cadet JL (1989) Regional effects of 6-hydroxydopamine
(6-OHDA) on free radical scavengers in rat brain. Brain Res
504:139–141.
Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR (1992)
Glycogen synthase kinase-3: functions in oncogenesis and
development. Biochim Biophys Acta 1114:147–162.
Poewe W, Gauthier S, Aarsland D, Leverenz JB, Barone P,
Weintraub D, Tolosa E, Dubois B (2008) Diagnosis and
management of Parkinson’s disease dementia. Int J Clin Pract
62:1581–1587.
Quesada A, Ogi J, Schultz J, Handforth A (2011) C-terminal
mechano-growth factor induces heme oxygenase-1-mediated
neuroprotection of SH-SY5Y cells via the protein kinase Ce/Nrf2pathway. J Neurosci Res 89:394–405.
Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN,
Yancopoulos GD, Glass DJ (2001) Mediation of IGF-1-induced
skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/
Akt/GSK3 pathways. Nat Cell Biol 3:1009–1013.
Schapira AH (2008) Progress in Parkinson’s disease. Eur J Neurol
15:1.
Schlattner U, Tokarska-Schlattner M, Wallimann T (2006)
Mitochondrial creatine kinase in human health and disease.
Biochim Biophys Acta 1762:164–180.
Sobrado M, Roda JM, Lopez MG, Egea J, Garcia AG (2004)
Galantamine and memantine produce different degrees of
neuroprotection in rat hippocampal slices subjected to oxygen–
glucose deprivation. Neurosci Lett 365:32–136.
Song B, Lai B, Zheng Z, Zhang Y, Luo J, Wang C, Chen Y, Woodgett
JR, Li M (2010) Inhibitory phosphorylation of GSK-3 by CaMKII
couples depolarization to neuronal survival. J Biol Chem
285:41122–41134.
Tian LL, Zhou Z, Zhang Q, Sun YN, Li CR, Cheng CH, Zhong ZY,
Wang SQ (2007) Protective effect of (+/�) isoborneol against 6-OHDA-induced apoptosis in SH-SY5Y cells. Cell Physiol Biochem
20:1019–1032.
Tiong CX, Lu M, Bian JS (2010) Protective effect of hydrogen
sulphide against 6-OHDA-induced cell injury in SH-SY5Y cells
involves PKC/PI3K/Akt pathway. Br J Pharmacol 161:467–480.
Tirmenstein MA, Hu CX, Scicchitano MS, Narayanan PK, McFarland
DC, Thomas HC, Schwartz LW (2005) Effects of 6-
hydroxydopamine on mitochondrial function and glutathione
status in SH-SY5Y human neuroblastoma cells. Toxicol In Vitro
19:471–479.
Wallimann T, Schnyder T, Schlegel J, Wyss M, Wegmann G, Rossi
AM, Hemmer W, Eppenberger HM, Quest AF (1989) Subcellular
compartmentation of creatine kinase isoenzymes, regulation of
CK and octameric structure of mitochondrial CK: important
aspects of the phosphoryl-creatine circuit. Prog Clin Biol Res
315:159–176.
Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM
(1992) Intracellular compartmentation, structure and function of
creatine kinase isoenzymes in tissues with high and fluctuating
energy demands: the ‘phosphocreatine circuit’ for cellular energy
homeostasis. Biochem J 281:21–40.
Wang D, Maler L (1998) Differential roles of Ca2+/calmodulin-
dependent kinases in posttetanic potentiation at input selective
glutamatergic pathways. Proc Natl Acad Sci U S A 95:7133–7138.
Woznicki DT, Walker JB (1979) Formation of a supplemental long
time-constant reservoir of high energy phosphate by brain in vivo
and in vitro and its reversible depletion by potassium
depolarization. J Neurochem 33:75–80.
Yang PC, Yang CH, Huang CC, Hsu KS (2008) Phosphatidylinositol
3-kinase activation is required for stress protocol-induced
modification of hippocampal synaptic plasticity. J Biol Chem
283:2631–2643.
Yong-Kee CJ, Salomonczyk D, Nash JE (2011) Development and
validation of a screening assay for the evaluation of putative
neuroprotective agents in the treatment of Parkinson’s disease.
Neurotox Res 19:519–526.
Zhang Z, Cui W, Li G, Yuan S, Xu D, Hoi MP, Lin Z, Dou J, Han Y,
Lee SM (2012) Baicalein protects against 6-OHDA-induced
neurotoxicity through activation of Keap1/Nrf2/HO-1 and
involving PKCa and PI3K/AKT signaling pathways. J Agric Food
Chem 60:8171–8182.
(Accepted 15 February 2013)(Available online 26 February 2013)