preparation of the amphiphilic macro-rings of poly(ethylene oxide) with multi-polystyrene lateral...
TRANSCRIPT

Preparation of the Amphiphilic Macro-Rings ofPoly(ethylene oxide) with Multi-Polystyrene LateralChains and Their Extraction for Dyes
XINCHANG PANG, GUOWEI WANG, ZHONGFAN JIA, CHAO LIU, JUNLIAN HUANG
Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University,Shanghai 200433, China
Received 20 June 2007; accepted 30 July 2007DOI: 10.1002/pola.22333Published online in Wiley InterScience (www.interscience.wiley.com).
ABSTRACT: A novel method for synthesis of amphiphilic macrocyclic graft copolymerswith multi-polystyrene lateral chains is suggested, by combination of anionic ring-open polymerization (AROP) with atom transfer radical polymerization (ATRP). Theanionic ring-opening copolymerization of ethylene oxide (EO) and ethoxyethyl glycidylether (EEGE) was carried out first using triethylene glycol and diphenylmethylpotas-sium (DPMK) as coinitiators; the monomer reactivity ratio of them are r1(EO) ¼ 1.20 60.01 and r2(EEGE) ¼ 0.76 6 0.02 respectively. The obtained linear well-defined a,x-dihy-droxyl poly(ethylene oxide) with pendant protected hydroxylmethyls (l-poly(EO-co-EEGE)) was cyclized by reaction with tosyl chloride (TsCl) in the presence of solidKOH. The crude cyclized product containing the extended linear chain polymer washydrolyzed and then purified by treat with a-CD. The pure cyclic copolymer with mul-tipendant hydroxymethyls [c-poly(EO-co-Gly)] was esterified by reaction with 2-bro-moisobutyryl bromide, and then used as macroinitiators to initiate polymerization ofstyrene (St), and a series of amphiphilic macrocyclic grafted copolymers composed of ahydrophilic PEO as ring and hydrophobic polystyrene as side chains (c-PEO-g-PS)were obtained. The intermediates and final products were characterized by GPC,NMR and MALDI-TOF in detail. The experimental results confirmed that c-PEO-g-PSshows stronger conjugation ability with the dyes than the corresponding comb-PEO-g-PS. VVC 2007 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 45: 5824–5837, 2007
Keywords: anionic polymerization; ATRP; cyclization; graft copolymer; poly(ethyl-ene oxide); polystyrene
INTRODUCTION
Since the pioneering work done by Pedersenthat revealed the complexing potential of thecrown ethers for metal ions,1 some new cyclicmolecules with complicated structure derivedfrom the crown ethers have been synthesized
and the properties investigated. It is well knownthat cyclic molecules with some fascinatingproperties could find some actual applications inmany fields, such as the formation of organicnanotubes, ion complex, and ion transportacross membranes and so on.2 For example,cyclic polyamines bearing long paraffin chainsdisplayed a liquid crystal phase in which themacrocyclic units are stacked to form a tubularmesophase.3 A dibenzo-18-crown-6 with two poly(styrene) chains could be used to fabricate Agnanoparticles without using any templates.4
Correspondence to: J. Huang (E-mail: [email protected])
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 45, 5824–5837 (2007)VVC 2007 Wiley Periodicals, Inc.
5824

However, the macrocyclic polymers with longand endless chains are different from the afore-mentioned shape-persistent cycles, and theshapes of the former are always flexible. Previ-ous studies about cyclic polymers focused onlyon the preparation of single polymer ring includ-ing some hydrophobic homopolymers and copoly-mers such as cyclic PS, PE, PS-b-PI and PS-b-PI-b-PMMA,5 and the hydrophilic macrocyclicpoly(ethylene oxide)6; the investigation of the so-lution and crystallization properties of thesemacrocyclic polymers was also reported.7
However, in the family of these macrocycles,limited reports are published on the preparationof amphiphilic macrocyclic grafted copolymers.We have described the synthesis of a macrocylicgraft copolymer of poly(ethylene oxide) (PEO) asring and polystyrene as side chains by anioniccopolymerization of ethylene oxide (EO) and 4-glycidyloxy-2,2,6,6-tetramethylpieridinee-1-oxyl8
(G-TEMPO), but this method has two shortcom-ings:1 the monomers suited for TEMPO poly-merization are limited, so the method is not uni-versal;2 the separation and purification of themacrocyclic product is complicated and time-con-suming. In this article, a novel route to prepareamphiphilic macrocyclic graft copolymers withmulti-polystyrene lateral chains is provided, bya combination of anionic ring-open polymeriza-tion (AROP) with atom transfer radical polymer-ization (ATRP) using protected glycidol (Gly)[2,3-epoxypropyl-1-ethoxyethyl ether (EEGE)] ascomonomer, a series of macrocyclic graft copoly-mers composed of a hydrophilic PEO as ring,and hydrophobic polystyrene as side chains (c-PEO-g-PS) were obtained.
The interaction between polymer and dyeleading to polymer–dye complex formationexhibits many interesting and important practi-cal features. For example, poly(vinyl pyrroli-done) hydrogel can remove dyes from watereffectively.9 In addition, selective uptake of vari-ous oil-soluble dyes into an aggregate of amphi-hilic copolymer consisting of a hydrophilic linearpolyelectrolyte block and hydrophobic block car-rying pendant dendritic moiety has been investi-gated in water.10 In this work, the removal ofdyes from water through liquid–liquid extractionby the amphiphilic macrocyclic graft copolymerc-PEO-g-PS was investigated. The experimentsconfirmed that these closed macrocyclic copoly-mers showed stronger conjugation ability withthe dyes than opened comb-like amphiphilicgraft copolymers comb-PEO-g-PS.
EXPERIMENTAL
Materials
Glycidol (tech.) was purchased from Acros anddried over calcium hydride for 48 h, then dis-tilled under reduced pressure just before use. 2-Bromoisobutyryl bromide (98%), 2,20-bipyridyl(bpy; >99%), p-toluene sulfonic acid (TsOH;>98%), ethyl vinyl ether (98%), and pyridine(99.5%) were purchased from Aldrich and Sino-pharm Chemical Reagent, respectively, and usedas received. CuBr (95%) was stirred overnightin acetic acid and filtrated, washed with ethanoland diethyl ether successively, and driedin vacuo. a-Cyclodextrine (a-CD, Aldrich) was usedas received. p-toluenesulfonyl chloride (TsCl;98%, Aldrich) and potassium hydroxide (KOH;96%, Aldrich) were dried under vacuum prior touse. Styrene (>99.5%) was washed with 10%NaOH aqueous solution and water successively,and dried over anhydrous MgSO4, further driedover CaH2, and distilled under reduced pres-sure. Triethylene glycol was distilled from CaH2
under reduced pressure and the fraction at134 8C/90 Pa was collected. 1,1-Diphenylethy-lene (99%) was also distilled from CaH2 underreduced pressure and the fraction at 105 8C/80Pa was collected. THF (99%) was refluxed overpotassium wire and distilled from potassiumnaphthalenide solution. EO (Sinopharm Chemi-cal Reagent; 98%) was dried by calcium hydridefor 48 h and then distilled under N2 before use.All other reagents were purified by common pu-rification procedures.
Measurements
GC/MS (gas chromatographic-mass spectromet-ric) analysis was carried for EEGE using a Fin-nigan Voyager system with mass selective detec-tion operating in electronic ionization. The GC/MS parameters were as follows: ion source tem-perature was 200 8C, carrier gas helium, columnflow 1 mL/min; temperature program, from 100to 200 8C at 15 8C/min, splitless injection at250 8C; ionization at 70 eV. GPC was performedon an Agilent1100 with a G1310A pump, aG1362A refractive detector, and a G1314A vari-able wavelength detector. THF used as eluent at35 8C at 1.0 mL/min. One 5 lm LP gel column(500 E, molecular range 500 3 104 to 502 3 104
g/mol) and two 5 lm LP gel mixed bed column(molecular range 200 3 106 to 203 3 106 g/mol)
AMPHIPHILIC MACROCYCLIC GRAFT COPOLYMERS 5825
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

were calibrated with polystyrene standard sam-ples. For l-poly(EO43..5-co-EEGE) and c-poly(EO10.6-co-Gly), GPC analyses were performedin 0.1 M aqueous NaNO3 at 40 8C with an elu-tion rate of 0.5 mL/min on the same instrument,except that a G1315A diode-array detector wasused to substitute the G1314A variable wave-length detector. Three TSK-gel PW columns inseries (bead size: 6, 13, 13 lm; pore size: 200 A,greater than 1000 A, less than 100–1000 A; mo-lecular range: 0 3 104 to 5 3 104, 5 3 104 to8 3 106, 5 3 106 to 8 3 106 g/mol, respectively)were calibrated with PEO standard samples.The injection volume was 20 lL, and the concen-tration was 5 mg/mL. MALDI-TOF-MS analysiswas carried out on a Voyager DE-STR fromApplied Biosystems. The matrix a-cyano-4-hydroxycinnamic acid was dissolved in THF at aconcentration of 40 mg/mL. Potassium trifluor-acetate was used as cationization agent at typi-cal concentrations of 5 mg/mL. The sample wasdissolved in THF at �1 mg/mL. At last, matrix,salt, and polymer solutions were premixed in amolar ratio of 5:1:5. The premixed solutionswere hand-spotted on the target well and left todry. All mass spectra were recorded in thereflector mode and about 1000 laser shots werecollected per spectrum. 1H-NMR spectra wereobtained at a DMX 500 MHz spectrometer. Allthe samples were dissolved in CDCl3. IR spectrawere obtained on a Magna 550 Fourier trans-form infrared (FTIR) spectrometer. UV–Visdeterminations were taken on a 756 MC UV-Visspectrophotometer (Shanghai Third AnalyticalInstrument Factory, China), the concentration ofvarious dyes aqueous solution, before and after
extraction by the chloroform solution of theamphiphilic copolymers, was determined byUV–Vis spectrometer. UV/Vis (water): the kmax(e)¼ 435 (24,180) for cresol red (A), 596 (39,430)for thymol blue (B), 592 (57,680) for bromophe-nol blue (C), and 594 (15,170) for thymolphthal-ein (D).The ultrafiltration separator was pur-chased from the Shanghai Institute of NuclearResearch, Chinese Academy of Science, and thecutoff molecular weight of the poly(ether sul-fone) membrane was 20,000 g/mol (calibrated byglobular protein).
Synthesis of 2,3-Epoxypropyl-1-ethoxyethylether (EEGE)
The hydroxyl group of glycidol was protectedwith ethyl vinyl ether according to a previouslydescribed procedure,11 as shown in Scheme 1.Typically, the operation was carried out in a250-mL three-neck flask with a magnetic stirrer;1.25 g of TsOH was added in batch to 50 g(0.675 mol) of glycidol in 200 mL of ethyl vinylether solution, the temperature was kept below40 8C, and 100 mL of saturated NaHCO3 aque-ous solution was added after the mixture wasstirred for 3 h. The organic layer was separatedand dried with MgSO4. After filtration, the ethylvinyl ether was evaporated, the remainder wasdistilled under reduced pressure, and the frac-tion at 51 8C/80 Pa was collected. The productEEGE (bp 152–154 8C) was a colorless liquidand weighted 80.3 g (84%).
ELEM. ANAL. Calcd. for C7H14O3:C, 57.58%; H,9.66%. Found: C, 57.77%; H, 9.55%. GC (99.6%)/MS (70 eV)m/z (%): 131 (33) [M��CH3]
þ, 101 (27)
Scheme 1. Anionic copolymerization of EEGE with EO.
5826 PANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

[M��C2H5O]þ, 73 (88) [M��C4H9O]þ. 1H-NMR(CDCl3, d, ppm): 4.76 [m, ��O��CH(CH3) ��O�� ],3.35–3.90 (m, ��O��CH2CH3 and ��O�� CH2��C2H4O), 3.15 (m, methine CH of the epoxy ring),2.61–2.91 (m, methylene CH2 of the epoxy ring),1.33 [d, ��OCH(CH3) ��O�� ], 1.19 (t, ��O��CH2��CH3). FTIR (film, t): 1350, 1254 cm�1.
Anionic Copolymerization of EEGE with EO
The copolymerization of EEGE with EO isdescribed in Scheme 1. Diphenylmethyl potas-sium (DPMK) was prepared according to the lit-erature:12 to a 150-mL three-neck flask, 100 mLof dry THF and 7.7 g (0.06 mol) of naphthalenewere added. Then 2.34 g (0.06 mol) of potassiumwith fresh surface was added in nitrogen atmos-phere; after stirring for 4 h, 11.1 g (0.066 mol) ofdiphenylmethane was introduced by a syringeand the system was refluxed at 80 8C for 24 h.Titrated with 0.1 M HCl after filtration, the con-centration of DPMK was 0.57 M.
The typical procedure was as follows: A 150-mL kettle was vacuumed at 80 8C for 2 h, andcooled to room temperature, and then to 0 8C.Given volumes of initiator solution [triethyleneglycol (0.29 mL, 2.19 mmol) with DPMK (3.5mL, 2.0 mmol) in a mixture of THF and DMSO(10/40 v/v, 50 mL)], EEGE (11.1 g, 75.8 mmol)and EO (30.0 g, 681.8mmol) were introducedsuccessively into the kettle under magnetic stir-ring. Subsequently, it was heated to 60 8C understirring for 48 h. The reaction was terminatedby addition of a few drops of acidified methanol.After all the solvents were removed by reduceddistillation, the crude product was dissolved inCH2Cl2, dried over anhydrousMgSO4 and filtered.The yellowy viscous wax-like product, a linear a,x-dihydroxyl poly(ethylene oxide) with pendant pro-tected hydroxylmethyls [l-poly(EO10.6-co-EEGE)],was obtained at a yield of 94% after CH2Cl2 wasremoved. The model copolymer l-poly(EO43.5-co-EEGE) was synthesized by the samemethod.
1H-NMR (CDCl3, d, ppm): 4.70–4.73(m,��OCH(CH3)O��), 1.30 (d, ��OCH(CH3)O��), and1.19 (t, CH3CH2O��) for ring-opening EEGE unit,3.53–3.80 (m, ��CH2CH2O�� and ��CH2CHO��(connected to ethoxyethyl ether) for main chainof copolymer).
Determination of Reactivity Ratio of EO and EEGE
The copolymerization of EO and EEGE with dif-ferent feed ratio was carried out under the same
condition mentioned above, but the conversion isless than 10%. The copolymer composition wasmeasured by 1H-NMR data (see eq 2). The reac-tivity ratios were calculated by YBR13 methodby eq 1:
ðx= ffiffiffiy
p Þr1 � ð ffiffiffiy
p=xÞr2 þ ð1= ffiffiffi
yp � ffiffiffi
yp Þ ¼ 0 ð1Þ
where x is the feed ratio of EO to EEGE, y isthe molar ratio of EO to EEGE in the copolymer.r1 and r2 are reactivity ratio of EO and EEGE,respectively.
Cyclization of l-Poly(EO-co-EEGE) and Purification
In a flask, finely ground KOH (1.0 g) was dis-persed in a mixture of THF and heptane (70/30,v/v, 100 mL) and stirred under nitrogen at40 8C. The purpose of the poor solvent, heptane,was to improve ring closure by reducing theend-to-end distance.14,15 l-Poly(EO10.6-co-EEGE)(7.0 g) and TsCl (161 mg) were dissolved in 100mL of THF in a separate flask. This solutionwas then added dropwise to the KOH dispersionvia a syringe pump over 48 h. After a further 72 hunder reflux (40 8C) the mixture was filteredand the filtrate was evaporated under reducedpressure. The crude cyclized product wasobtained.
As the crude cyclized product with high con-tents of EEGE (l-poly(EO10.6-co-EEGE) as pre-cursor) was insoluble in the water, it was hydro-lyzed in acidic conditions, and then the hydroxylgroups were recovered by breaking of acetallinkage of EEGE units of copolymer chains. Thehydrolysis of EEGE segments of copolymer16
required two steps: (a) the crude cyclized prod-uct (6.0 g) was mixed with 80 mL of formic acid,and the solution was stirred at 20 8C for 30 minand then poured into methanol. The precipitatewas separated and dried in vacuo at 50 8C; (b)the dried product dissolved in a mixture of diox-ane (50 mL), methanol (30 mL), and KOH meth-anol solution (1 N, 20 mL) was refluxed for 24 h,then neutralized with 5% HCl. After removal ofsolvents under reduced pressure, the polymerwas dissolved in water and purified by ultrafil-tration. The typical procedure was as follows: Apolymer aqueous solution with concentration of4% (w/w) was added to the ultrafiltration sepa-rator fitted with the poly(ether sulfone) mem-brane under magnetic stirring, and then theformed salts were separated under nitrogenpressure. The filtrated aqueous solution was
AMPHIPHILIC MACROCYCLIC GRAFT COPOLYMERS 5827
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

concentrated to dryness, dissolved in CH2Cl2,dried over anhydrous MgSO4, and filtered. Thefiltrate was distilled in vacuum to removeCH2Cl2 and dried in vacuo at 50 8C. The hydro-lyzed product with pale yellow color wasobtained at a yield of 93%.
The crude hydrolyzed product (4.0 g) wasthen dissolved in 40 mL of distilled water (100g/L), to which an aqueous solution of a-CD (100mL, 100 g/L) was added at room temperature;the ratio of a-CD to linear byproducts (about25%) was estimated from the GPC was 10/1 (w/w).17 The resulting clear solution was ultrasoni-cally stirred for 30 min and then a turbid solu-tion appeared. It was allowed to stand overnightat room temperature and a white precipitateformed. The mixture was centrifuged and fil-tered to obtain a clear aqueous solution. The sol-vent was distilled and a solid mixture of thecyclic product, unthreaded a-CD or a smallquantity of linear byproducts remained. Thissolid mixture was dissolved in dichloromethaneand filtered to remove the unthreaded a-CD.The filtrate was distilled by rotating evaporationto obtain the product. The procedure wasrepeated twice to obtain the pure cyclized prod-uct [c-poly(EO10.6-co-Gly)]. It was a white waxysolid.
Preparation of the Cyclic Macroinitiator for ATRP
The esterification of hydroxymethyl groups onglycidol segments of c-poly(EO10.6-co-Gly) with2-bromoisobutyryl bromide18 was done to pre-pare the cyclic macroinitiator. A typical examplewas as follows: 1.7 g of c-poly(EO10.6-co-Gly) wasdissolved in 50 mL anhydrous pyridine underdry nitrogen, and 1.4 mL of 2-bromoisobutyrylbromide was added dropwise at 0 8C for 20 minunder vigorous stirring. The initial yellow colordisappeared immediately, and pyridinium bro-mide with red-brown color was precipitated. Itwas continuously stirred for another 15 min. Af-ter that, 5 g of K2CO3 was added to the systemat room temperature. The pyridine was removedby azeotropic distillation with dry toluene (33 40 mL). The residue was dissolved in waterand separated by ultrafiltration. The aqueoussolution was concentrated to dryness and dis-solved in CH2Cl2, dried over anhydrous MgSO4,and then filtered. The CH2Cl2 of filtrate wasremoved by distillation in vacuum. The remain-der was dried in vacuo at 50 8C and the yellowyproduct with a yield of 95% was obtained.
1H-NMR (CDCl3, d, ppm): 4.13–4.42(m,��CH2OOCC(CH3)2Br)), 1.86(s, ��OOCC (CH3)2Br), 3.35–3.90 (m, ��CH2CH2O�� and ��CH2
CHO�� for main chain of copolymer).
Synthesis of Amphiphilic Macrocyclic GraftCopolymer c-PEO-g-PS
The graft copolymerization of St was carried outusing c-poly(EO10.6-co-Gly) with pending bromoi-sobutyryl groups [c-poly(EO10.6-co-Gly) (ATRP)] asmacroinitiator: An ampoule charged with theCuBr (54.6 mg, 0.38 mmol), bpy (59.4 mg, 0.38mmol), c-poly(EO10.6-co-Gly) (ATRP) (Mn ¼ 11,800;0.2 g, 0.017 mmol), St (8 mL, 69.8 mmol) wasvacuumed by three freeze-thaw cycles at thetemperature of liquid nitrogen, then sealed andplaced in an oil bath at 90 8C. The ampouleswere taken out from the oil bath and dipped inliquid nitrogen at different time intervals tostop the polymerization. The polymerized prod-ucts were diluted by CH2Cl2 and passed througha neutral alumina column to remove the cata-lyst, and then precipitated in cold methanol. Af-ter filtration, the products were purified by dis-solution/precipitation with CH2Cl2/cold metha-nol twice and then dried at 35 8C undervacuum. The whole process is outlined inScheme 2.
Hydrolysis of c-PEO-g-PS
The grafted PS chains were hydrolyzed under abasic condition:19 0.2 g of c-PEO-g-PS (A1) wasdissolved in 50 mL of THF, 10 mL of a KOH so-lution (1 M in ethanol) was added, and the mix-ture was refluxed for 72 h. After evaporation todryness, the polymer was dissolved in CH2Cl2and then precipitated into acidified methanol;the products were purified twice by dissolution/precipitation with CH2Cl2/methanol and thendried at 50 8C. The molecular weight was deter-mined by GPC as Mn ¼ 1463; Mw/Mn ¼ 1.11.
Synthesis of Comb-PEO-g-PS
The comb-PEO-g-PS was synthesized to comparewith c-PEO-g-PS in the extraction for dyes. Alinear a,x-dihydroxyl poly(ethylene oxide) withpendant protected hydroxylmethyls l-poly(EO10.6-co-EEGE) was used, and the poly(EO-co-Gly) with pending bromoisobutyryl groups[poly(EO10.6-co-Gly) (ATRP)] was obtained afterthe hydrolysis and then esterification with 2-
5828 PANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

bromoisobutyryl bromide (the same as the syn-thesis route of c-poly(EO10.6-co-Gly)(ATRP)).
The graft copolymerization of St was carriedout with poly(EO10.6-co-Gly (ATRP) as the macro-initiator. Ampules charged with CuBr (27.6 mg,0.192 mmol), bpy (30.0 mg, 0.192 mmol), poly(EO-co-Gly)(ATRP) (Mn ¼ 13,800; 0.12 g, 0.0087mmol), and St (3.1 mL, 28.8 mmol) were vac-uumed by three freeze-thaw cycles at the tem-perature of liquid nitrogen and then sealed andplaced in an oil bath at 90 8C. The ampuleswere taken from the oil bath and dipped inliquid nitrogen at different times to stop thepolymerization. The polymerized productswere diluted with CH2Cl2 and precipitated incold methanol. After filtration, the productswere purified by dissolution/precipitation with
CH2Cl2/cold methanol twice and then dried at35 8C in vacuo.
Extraction Experiment
The c-PEO-g-PS and the comb-like PEO-g-PSwere dissolved in chloroform with the concentra-tion of 2 3 10�5 mol/L. All the dyes were dis-solved in deionized water with the concentrationof 1.0 3 10�4 mol/L. For thymol blue and thy-molphthalein, the aqueous solution was adjustedto pH ¼ 10. In a 25-mL conical flask, 1 mL ofchloroform solution and 2 mL of dye solutionwere added successively, stirred vigorously for10 min and the dye concentration in aqueousphase was determined by UV–Vis after phaseseparation.
Scheme 2. Schematic representation of the synthetic route of the amphiphilic mac-rocyclic graft copolymer consisting of a PEO ring and multi-polystyrene lateralchains.
AMPHIPHILIC MACROCYCLIC GRAFT COPOLYMERS 5829
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

RESULTS AND DISCUSSION
Characterization of Parent Copolymersl-Poly(EO-co-EEGE)
In the anionic copolymerization of EO and glyci-dol, the glycidol should be protected because theexchanging reaction between hydroxyl of glyci-dol and DMPK would take place, and so the sidereaction would be unavoidable. The glycidol(Gly) was protected with ethyl vinyl ether first,and then was copolymerized with EO using amixture of triethylene glycol and DPMK as ini-tiator. A linear a-methyl-x-hydroxyl poly(EO-co-EEGE) was formed. To control the polymeriza-tion reasonably, it was important that only 20–40% of the hydroxyl groups of the triethyleneglycol were activated; otherwise alkoxides wouldbe precipitated.20 A mixture of DMSO and THF(v/v: 4/1) was used as solvent for polymerizationinstead of THF because propagating alkoxideswould be aggregated in pure THF.21 Under suchconditions, all of the hydroxyl groups of triethy-lene glycol could efficiently initiate the copoly-merization of EO and EEGE because of therapid exchange of protons between dormanthydroxyls and propagating alkoxides, and allthe chains grew at the same rate.21 Two kindsof parent copolymers with different contents ofEEGE and different molecular weight could beprepared by variation of the monomer feed ratioand initiator volume (Table 1).
The copolymer composition can be readilyobtained by using following equation based onthe 1H-NMR data (see under Anionic Copolymer-ization of EEGE with EO in EXPERIMENTAL):
RT ¼ 4Af
Asum � 7Afð2Þ
where RT is the molar ratio of EEGE to EO inthe copolymer; Asum and Af represent the peakarea sum of the protons of the main chain andprotons of lateral chains methylene at 3.53–3.80ppm and the peak areas of the methine protonsof the EEGE moiety at 4.70–4.73 ppm, respec-tively. The RT values of copolymers A and B are1/10.6 and 1/43.5 respectively, which is nearlyequivalent to the monomer feed ratio of EEGEto EO (1/9 and 1/39). So, the linear a,x-dihy-droxyl poly(ethylene oxide)s with pendant pro-tected hydroxylmethyls (l-poly(EO-co-EEGE))can be described as l-poly(EO10.6-co-EEGE)(A)and l-poly(EO43.5-co-EEGE)(B). The number ofthe protected hydroxyls on the PEO chain couldbe evaluated by the combination of the molecu-lar weight determined by GPC and 1H-NMRdata using the following:
NEEGE ¼ Mn
ð146þ 44=RTÞ ð3Þ
in which Mn is the molecular weight of l-poly(EO-co-EEGE), 146 and 44 are the molar massesof EEGE and EO, and RT is the molar ratio ofEEGE units to EO units in l-poly(EO-co-EEGE);the calculated NEEGE values were about 20 and3, respectively.
According to the copolymer composition datafrom NMR (Table 2), the monomer reactivityratios of EO and EEGE could be derived byYBR method;13 the values are r1(EO) ¼ 1.206 0.01, r2(EEGE) ¼ 0.76 6 0.02, and r1 3 r2 � 1.Thus the anionic copolymerization of EO andEEGE is apt to the ideal nonazeotropic copoly-
Table 1. The Data for Parent Copolymersl-Poly(EO-co-EEGE)
Sample Rfa RT
b Mnc Mw/Mn
d NEEGEe
A 1/9 1/10.6 12,500 1.06 20B 1/39 1/43.5 6400 1.16 3
a The feed ratio of EEGE to EO.b The molar ratio of EEGE to EO in copolymer poly(EO-
co-EEGE) measured by 1H NMR.c Number-average molecular weight determined by GPC;
sample A: calibrated against PS standards using THF aseluent, sample B: calibrated against PEO standards using0.1 M aqueous NaNO3 as eluent.
d The polydispersity determined by GPC.e The number of EEGE units in poly(EO-co-EEGE) calcu-
lated by the integration of protons from 1H NMR.
Table 2. Determined and Analyzed Resultsof EO and EEGE Copolymerization
Sample Rfa RT
b Conversionc (%)
1 1/18 1/21.7 7.22 1/14 1/16.8 6.33 1/9 1/10.8 5.14 3/7 1/2.9 4.95 1/1 1/1.2 4.26 7/3 1/0.6 3.8
a The feed ratio of EEGE to EO.b The molar ratio of EEGE to EO in copolymer poly(EO-
co-EEGE) measured by 1H NMR.c Determined by a gravimetric method.
5830 PANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola
merization and the incorporation of the comono-mer is random.
Cyclization of Linear Copolymers and Purification
In the synthesis of macrocyclic molecules, theessential work is the closure of linear copoly-mers l-poly(EO-co-EEGE) and the purification ofclosed product. The ring closure of l-poly(EO-co-EEGE) was achieved via ether linkage by reac-tion with tosyl chloride (TsCl) in the presence ofsolid KOH. End-to-end intramolecular couplingwas promoted over intermolecular chain exten-sion by conducting the reaction at high dilution[C* < 10�5 mol/L]. This synthesis was based ona method reported by Booth and coworkers forpreparation of cyclic poly(ethylene oxide).14,15
They also reported a second method for ring clo-sure via an acetal linkage (reaction of the a,x-dialkoxide with CH2Cl2).
22
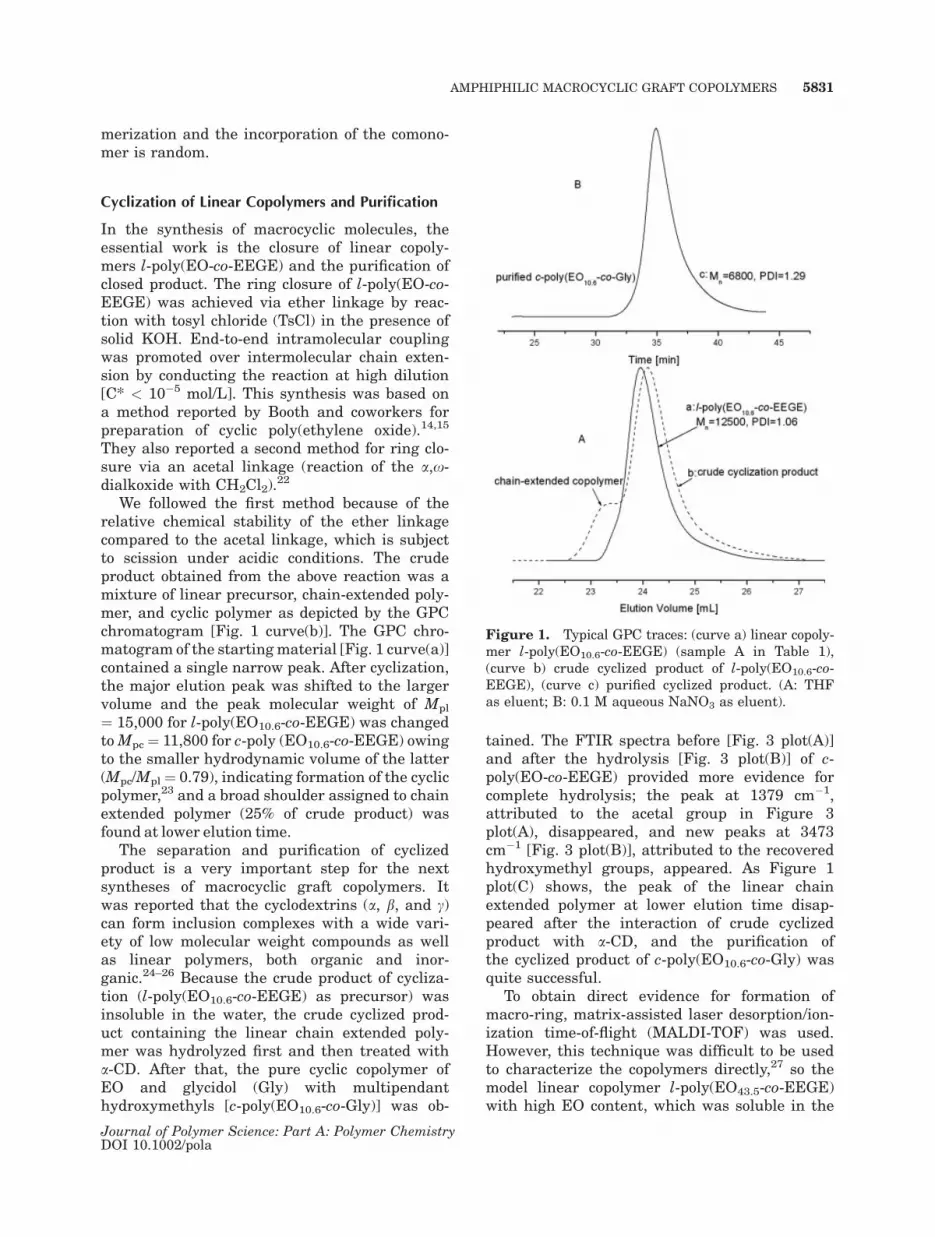
We followed the first method because of therelative chemical stability of the ether linkagecompared to the acetal linkage, which is subjectto scission under acidic conditions. The crudeproduct obtained from the above reaction was amixture of linear precursor, chain-extended poly-mer, and cyclic polymer as depicted by the GPCchromatogram [Fig. 1 curve(b)]. The GPC chro-matogram of the startingmaterial [Fig. 1 curve(a)]contained a single narrow peak. After cyclization,the major elution peak was shifted to the largervolume and the peak molecular weight of Mpl
¼ 15,000 for l-poly(EO10.6-co-EEGE) was changedtoMpc ¼ 11,800 for c-poly (EO10.6-co-EEGE) owingto the smaller hydrodynamic volume of the latter(Mpc/Mpl ¼ 0.79), indicating formation of the cyclicpolymer,23 and a broad shoulder assigned to chainextended polymer (25% of crude product) wasfound at lower elution time.
The separation and purification of cyclizedproduct is a very important step for the nextsyntheses of macrocyclic graft copolymers. Itwas reported that the cyclodextrins (a, b, and c)can form inclusion complexes with a wide vari-ety of low molecular weight compounds as wellas linear polymers, both organic and inor-ganic.24–26 Because the crude product of cycliza-tion (l-poly(EO10.6-co-EEGE) as precursor) wasinsoluble in the water, the crude cyclized prod-uct containing the linear chain extended poly-mer was hydrolyzed first and then treated witha-CD. After that, the pure cyclic copolymer ofEO and glycidol (Gly) with multipendanthydroxymethyls [c-poly(EO10.6-co-Gly)] was ob-
tained. The FTIR spectra before [Fig. 3 plot(A)]and after the hydrolysis [Fig. 3 plot(B)] of c-poly(EO-co-EEGE) provided more evidence forcomplete hydrolysis; the peak at 1379 cm�1,attributed to the acetal group in Figure 3plot(A), disappeared, and new peaks at 3473cm�1 [Fig. 3 plot(B)], attributed to the recoveredhydroxymethyl groups, appeared. As Figure 1plot(C) shows, the peak of the linear chainextended polymer at lower elution time disap-peared after the interaction of crude cyclizedproduct with a-CD, and the purification ofthe cyclized product of c-poly(EO10.6-co-Gly) wasquite successful.
To obtain direct evidence for formation ofmacro-ring, matrix-assisted laser desorption/ion-ization time-of-flight (MALDI-TOF) was used.However, this technique was difficult to be usedto characterize the copolymers directly,27 so themodel linear copolymer l-poly(EO43.5-co-EEGE)with high EO content, which was soluble in the
Figure 1. Typical GPC traces: (curve a) linear copoly-mer l-poly(EO10.6-co-EEGE) (sample A in Table 1),(curve b) crude cyclized product of l-poly(EO10.6-co-EEGE), (curve c) purified cyclized product. (A: THFas eluent; B: 0.1 M aqueous NaNO3 as eluent).
AMPHIPHILIC MACROCYCLIC GRAFT COPOLYMERS 5831
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

water, was synthesized and cyclized first, thenpurified directly by a-CD for the determinationof MALDI-TOF. Figure 2 shows the MALDI-TOF spectra of linear precursor l-poly(EO43.5-co-EEGE) (A) and cyclized product c-poly(EO43.5-co-EEGE) (B); the spacings 44.3 and 43.8 amubetween the peaks are ascribed to the molarmass of the EO unit and the spacings 146.1 and146.2 amu for the molar mass of EEGE unit.The spacings 14.7, 29.6, 15.2, and 29.9 betweenthe peaks are ascribed to the difference of themolar mass of different combination of EEGEand EO units, and the molecular-weight decreaseof 18 amu after cyclization reaction supports theformation of the ether linkage, consistent withloss of a watermolecule upon ring closure.
Synthesis of Cyclic ATRP Macroinitiator
The hydroxyl groups of the cyclized product c-poly(EO10.6-co-Gly) were esterified with 2-bro-moisobutyryl bromide to obtain cyclic macroini-tiators with narrow molecular weight distribu-tion (Table 3), and the complete esterification ofthe hydroxyl groups of c-poly(EO10.6-co-Gly) wasalso confirmed by 1H-NMR data (see underPreparation of the Cyclic Macroinitiator ForATRP in EXPERIMENTAL). The hydroxyl groupconversion can be calculated by
ET ¼�3þ 4
RT
�3 Ae
2Asum3 100% ð4Þ
where ET is the conversion efficiency of hydroxylgroups of c-poly(EO-co-Gly); Asum and Ae repre-sent the integral area of the protons of PEOmain ring (the peaks at d ¼ 3.35–3.90) and theintegral area of the protons linked to the ester(the peaks at d ¼ 4.13–4.42); and RT is themolar ratio of EEGE to EO in original linear co-polymer l-poly(EO10.6-co-EEGE) measured by1H-NMR. The ET value is nearly 100%, and thissuggests that hydroxyl groups are completelyconverted to bromoisobutyryl units. The FTIRspectra before and after esterification of c-poly(EO10.6-co-Gly) provided additional evidence forcomplete estification; the peak at 3473 cm�1
attributed to hydroxymethyl groups in Figure 3
Figure 2. MALDI-TOF mass spectra of model copoly-mer for (A) linear precursor l-poly(EO43.5-co-EEGE)(sample B in Table 1) and (B) cyclization product c-poly(EO43.5-co-EEGE).
Table 3. GPC Data for the Cyclic Macroinitiators
Samplea
c-poly(EO-co-Gly)c-poly(EO-co-Gly)(ATRP)
Mnb Mw/Mn
b Mnc Mw/Mn
c
A 6800 1.29 11,800 1.10B 5200 1.23 5400 1.12
a The samples A and B are coincident with the samplesin Table 1.
b Number average molecular weight (Mn) and molecularweight distribution (Mw/Mn) determined by GPC, calibratedagainst PEO standards using 0.1 M aqueous NaNO3 as elu-ent.
c Number average molecular weight (Mn) and molecularweight distribution (Mw/Mn) determined by GPC, calibratedagainst PS standards using THF as eluent.
Figure 3. FTIR spectra of copolymers (KBr): (plotA) sample A in Table 1, (plot B) c-poly(EO10.6-co-GLy)and (plot C) c-poly(EO10.6-co-Gly)ATRP (entry A inTable 3).
5832 PANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

plot(B) disappeared and new peaks at 1736cm�1 in Figure 3 plot(C) attributed to the ester-band appeared.
Synthesis of Amphiphilic Macrocyclic GraftCopolymer c-PEO-g-PS
In Scheme 2, the procedure for the synthesis ofamphiphilic macrocyclic graft copolymers isdescribed. The ATRP of styrene was carried outin bulk at 90 8C using the bpy/CuBr catalystsystem. The results of ATRP using two kinds ofc-poly(EO-co-Gly)ATRP with different bromoiso-butyryl groups density as cyclic macroinitiatorare presented in Table 4.
The obtained amphiphilic macrocyclic graftcopolymers c-PEO-g-PS composed of a hydro-philic PEO as ring and hydrophobic PS as lat-eral chains show the monomodal GPC eluo-grams (Fig. 4). The molecular weight of the mac-rocyclic graft copolymer increases with thepolymerization time and the molecular weightdistributions in all cases are low (Mw/Mn
< 1.15). That means in our system the interma-cromolecular coupling reactions and the ring-opening reaction during the polymerization didnot occur, and we also did not observe the homo-polymerization of St.
Figure 5 shows the typical 1H-NMR spectrumof the macrocyclic graft copolymer of c-poly(EO10.6-co-Gly)-g-PSt. The peaks at d ¼ 4.28–4.65 are assigned to the methylene protonslinked to ester, the chemical shift at d ¼ 6.33–7.31 are the protons of phenyl rings of polysty-rene chains, and the chemical shift at d ¼ 3.41–3.86 are the protons of the PEO main chain.Thus the molecular weight of PS side chains
and the initiation efficiency of cyclic ATRP mac-roinitiators can be obtained by eqs 5 and 6:
NPS ¼� 4RT
þ 3�3 Ai
5Asumð5Þ
ET ¼� 4RT
þ 3�3 Ai 3 MSt
5Asum 3 MnPS3 100% ð6Þ
where NPS is the average number of St on eachside PS (see Table 4), ET is the reaction effi-ciency of bromoisobutyryl for ATRP, Ai andAsum represent the integral area of phenyl pro-tons on the grafted PS chains and the integral
Table 4. The Data for the Amphiphilic Macrocyclic Graft Copolymers c-PEO-g-PS
Initiator Entry Time (h) Mna (3104) Mw/Mn
b MnNMRc (3104) NPS
d
Ae A1 2 2.19 1.10 4.05 13.8A2 4 2.50 1.11 4.92 18.0
Bf B1 2 1.11 1.10 1.24 15.8B2 4 1.20 1.08 1.47 23.1
a Number-average molecular weight determined by GPC, calibrated against PS standards.b The polydispersity determined by GPC.c MnNMR was calculated by the formula: MnNMR ¼ Mnc-poly(EO-co-Gly)(ATRP) þ NOH 3 NPS 3 MSt, where Mnc-poly(EO-co-Gly)(ATRP) is
the molecular weight of c-poly(EO-co-Gly)(ATRP) derived by GPC, NOH is the hydroxyl number on c-poly(EO-co-Gly), MSt is themolecular weight of St.
d The average number of the St on grafting chain of the final copolymers was calculated from the 1H NMR data.e c-poly(EO10.6-co-Gly)ATRP as macroinitiator.f c-poly(EO43.5-co-Gly)(ATRP) as macroinitiator.
Figure 4. GPC traces of the graft copolymer: c-PEO-g-PS (sample A as the macroinitiator c-poly(EO10.6-co-Gly)(ATRP), polymerization time for A1: 2 h,A2: 4 h).
AMPHIPHILIC MACROCYCLIC GRAFT COPOLYMERS 5833
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

area of all protons of the PEO main chainrespectively, and RT is the molar ratio of Gly toEO in copolymer c-poly(EO-co-Gly) measuredby 1H-NMR. Mn,PS is number-average molecu-lar weight of side PS chains derived from Nps
and mass of St, and the value is 1435 for thesample A1 in Table 4. The calculated ET valueis 98.1%. It suggests that nearly all the bromo-isobutyryl groups take part in the ATRP poly-merization of St.
Hydrolysis of c-PEO-g-PS
The molecular weight of a macrocyclic graft co-polymer obtained by GPC is apparent and unre-liable because of the sharp difference in thehydrodynamic volumes of the graft copolymerand linear PS standard. The detachment of thePS side chains from the c-poly(EO-co-Gly) back-bone by hydrolysis of the ester group under ba-sic conditions and the subsequent measurementof the molecular weight of the free PS sidechains can give real information about graftedPS chains. A monomodal peak with a narrowdistribution (Mw/Mn ¼ 1.11) can be observed forthe detached PS from GPC. Its molecular weightis Mn ¼ 1463, which is approximate to the 1435derived by 1H-NMR.
Extraction for Dyes
The interaction of the dye molecules withamphiphilic copolymer has been investigated byanother group.28 A variety of oil-soluble dyescan be selectively uptaken into aggregates of anamphiphilic copolymer in water phase. However,in the purification of water, it is very importantto remove the organic compounds as dyes fromthe water. Two samples, in Table 4, were usedfor extraction experiment. Figure 6 shows thephotos in which sample A1 was used, and theused dyes were Cresol red (A), Thymol blue (B),Bromophenol blue (C), and Thymolphthalein(D), as shown in Scheme 3. When the c-PEO-g-PS (A1) was dissolved in chloroform, it could effi-ciently extract the dye molecule from the aque-ous media (the right bottle in each group). As ablank experiment, the pure chloroform was usedto extract the dyes directly. However, as the leftbottle in each group shows, the chloroformphase was colorless and almost no dye wasextracted into the organic phase. The dye con-centration in aqueous media before and afterextraction could be estimated by measuring theabsorbance of dyes at its maximum wavelengthusing a UV–Vis spectrophotometer and then cal-culated the concentration from a calibration
Figure 5. 1H NMR spectrum of cyclic copolymers c-PEO-g-PS (A1 in Table 4, sol-vent: CDCl3).
5834 PANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

Figure 6. The extraction for dyes by c-PEO-g-PS (two bottles as a group, the leftwas blank and the right contained the c-PEO-g-PS. The dyes were Cresol red (A),Thymol blue (B), Bromophenol blue (C), and Thymolphthalein (D); the upper layerwas aqueous media and the lower was organic phase).
Scheme 3. Structure of Cresol red (A), Thymol blue (B), Bromophenol blue (C), andThymolphthalein (D).
AMPHIPHILIC MACROCYCLIC GRAFT COPOLYMERS 5835
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

curve. The amount of dye adsorbed onto theamphiphilic macrocyclic graft copolymers, qe(mol/mol), could be derived from
qe ¼ ðC0 � CeÞ 3 Vd
Cp 3 Vp
where C0 and Ce are the dye concentrations inaqueous media before and after extraction (mol/L), respectively, and Vd is the volume of the dyesolution (mL). Cp and Vp are the macrocyclicgraft copolymers chloroform solution concentra-tion (mol/L) and volume (mL). The extractiondata for c-PEO-g-PS are listed in Table 4.
As a comparison, the extraction data forcomb-PEO-g-PS are also listed in Table 5. Onlyless than 1 dye molecule was coordinated withthe opened comb-PEO-g-PS, which means theextraction ability of c-PEO-g-PS was muchhigher than that of the comb-PEO-g-PS, and 1–6 dye molecules could be trapped by one c-PEO-g-PS. As is well known, although the hydroxylgroups on dyes can be conjugated with the oxy-gen atom of either c-PEO-g-PS or comb-PEO-g-PS by the hydrogen bond and the phenyl ringson dyes can be interacted with PS of either c-PEO-g-PS or comb-PEO-g-PS, the cases are dif-ferent for them. For c-PEO-g-PS, as Figure 6shows, the whole macrocyclic molecule was con-sidered as a nanocage, so the dye moleculescould be trapped in it. For comb-PEO-g-PS, how-ever, one dye molecule was enveloped by severalcomb-like copolymers, and one dye moleculecould be conjugated with several comb-PEO-g-PS macromolecules to form the complex.
CONCLUSIONS
A novel method for synthesis of amphiphilicmacrocyclic graft copolymers with multi-polysty-
rene lateral chains was demonstrated. A seriesof amphiphilic macrocyclic graft copolymerscomposed of PEO ring and multi-polystyrene lat-eral chains (c-PEO-g-PS) were synthesized by acombination of AROP and ATRP. The intermedi-ates and object copolymers were well-character-ized by GPC, 1H-NMR, and MALDI-TOF. Theexperimental results of the extraction for dyesconfirmed that the c-PEO-g-PS has the strongerconjugation ability with the dyes than that ofthe comb-PEO-g-PS.
The authors acknowledge financial support fromNational Natural Science Foundation of China (No.20574010).
REFERENCES AND NOTES
1. (a) Pedersen, C. J. J Am Chem Soc 1967, 89,2495; (b) Pedersen, C. J. J Am Chem Soc 1967,89, 7017.
2. (a) Gokel, G. W.; Leevy, W. M.; Weber, M. E.Chem Rev 2004, 104, 2723; (b) Gibson, S. E.;Lecci, C. Angew Chem Int Ed 2006, 45, 1364.
3. Lehn, J. M.; Malthete, J.; Levelut, A. M. J ChemSoc Chem Commun 1985, 1794.
4. Gao, J. P.; Fu, J.; Lin, C. K.; Lin, J.; Han, Y. C.;Yu, X.; Pan, C. Y. Langmuir 2004, 20, 9775.
5. (a) Oike, H.; Hamada, M.; Eguchi, S.; Danda, Y.;Tezuka, Y. Macromolecules 2001, 34, 2776; (b)Bielawski, C. W.; Benitez, D.; Grubbs, R. H. Sci-ence 2041, 2002, 297; (c) Pantazis, D.; Schulz,D. N.; Hadjichristidis, N. J Polym Sci Part A:Polym Chem 2002, 40, 1476; (d) Takano, A.;Kadoi, O.; Hirahara, K.; Kawahara, S.; Isono, Y.;Suzuki, J.; Matsushita, Y. Macromolecules 2003,36, 3045.
6. (a) Sun, T.; Yu, G. E.; Price, C.; Booth, C. PolymCommun 1995, 36, 3775; (b) Sinnathamby, P.; Yu,G. E.; Price, C.; Booth, C. Chem Commun 1996,31; (c) Yang, Z.; Yu, G. E.; Attwood, D.; Price, C.;Booth, C. Macromolecules 1996, 29, 8479; (d)Tezuka, Y.; Mori, K.; Oike, H. Macromolecules
Table 5. The Extraction Ability of the Amphiphilic Macrocyclic Graft Copolymers and Comb-like Copolymer
Copolymer\Dye Cresol Red Thymol Blue Bromophenol Blue Thymolphthalein
c-PEO-g-PS1 (A1) 1.25a 1.96 4.18 6.23c-PEO-g-PS2 (A2) 2.73 2.12 2.91 4.42comb-PEO-g-PS1 (A1)
b 0.38 0.1 0.64 Ndc
comb-PEO-g-PS2 (A2)b 0.62 Ndc 0.73 Ndc
a The data in this table reflect the dye number extracted by the each copolymer molecule determined by the UV–Vis.b The parent copolymers l-poly(EO10.6-co-EEGE) as precursor (A1: NPS ¼ 10; A2: NPS ¼ 20).c Nd means that no obvious change was detected by UV–Vis.
5836 PANG ET AL.
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola

2002, 35, 5707; (e) Singla, S.; Zhao, T.; Beckham,H. W. Macromolecules 2003, 36, 6945.
7. (a) Yang, Z.; Cooke, J.; Viras, K.; Gorry, P. K.;Ryan, A. J.; Booth, C. J Chem Soc Faraday Trans1997, 93, 4033; (b) Cooke, J.; Viras, K.; Sun, T.;Yu, G. E.; Yonemitsu, T.; Ryan, A. J.; Price, C.;Booth, C. Macromolecules 1998, 31, 3030.
8. Jia, Z. F.; Fu, Q.; Huang, J. L. Macromolecules2006, 39, 5190.
9. Senkal, B. F.; Erkal, D.; Yavuz, E. Polym AdvTechnol 2006, 17, 924.
10. Tamano, K.; Imae, T.; Yusa, S.; Shimada, Y. JPhys Chem B 2005, 109, 1226.
11. Fitton, A.; Hill, J.; Jane, D.; Miller, R. Synthesis1987, 1140.
12. Normant, H.; Angelo, B. Bull Soc Chim Fr 1960,354.
13. Yezuielev, A. I.; Brohkina, E. L.; Roskin, Y. S.Vysokimil Soedin Ser 1969, A11, 1670.
14. Yan, Z. G.; Yang, Z.; Price, C.; Booth, C. Makro-mol Chem Rapid Commun 1993, 14, 725.
15. Yu, G. E.; Sinnathamby, P.; Price, C.; Booth, C.Chem Commun 1996, 1, 31.
16. Taton, D.; Borgne, A.; Sepulchre, M.; Soassky, N.Macromol Chem Phys 1994, 195, 139.
17. Harada, A.; Li, J.; Kamachi, M. Macromolecules1993, 26, 5693.
18. Maier, S.; Sunder, A.; Frey, H.; Mulhaupt, R.Macromol Rapid Commun 2000, 21, 226.
19. Cheng, G. L.; Bo1ker, A.; Zhang, M. F.; Krausch, G.;Muller, A.H. E.Macromolecules 2001, 34, 6883.
20. Angot, B.; Taton, D.; Gnanou, Y. Macromolecules2000, 33, 5418.
21. Feng, X. S.; Taton, D.; Chaikof, E. L.; Gnanou, Y.J Am Chem Soc 2005, 127, 10956.
22. Sun, T.; Yu, G. E.; Price, C.; Booth, C.; Cooke, J.;Ryan, A. J. Polymer 1995, 36, 3775.
23. Semlyen, J. A. Cyclic Polymers; Elsevier: NewYork, 1986.
24. Harada, A. Coord Chem Rev 1996, 148, 115.25. Harada, A.; Nishiyama, T.; Kawaguchi, Y.;
Okada, M.; Kamachi, M. Macromolecules 1997,30, 7115.
26. Harada, A.; Nishiyama, T.; Kawaguchi, Y.; Okada,M.; Kamachi, M. Macromolecules 2000, 33, 4472.
27. Willemse, R. X. E.; Staal, B. B. P.; Donkers, E. H.D.; van Herk, A. M. Macromolecules 2004, 37,5717.
28. Tamano, K.; Imae, T.; Yusa, S.; Shimada, Y. JPhys Chem B 2005, 109, 1226.
AMPHIPHILIC MACROCYCLIC GRAFT COPOLYMERS 5837
Journal of Polymer Science: Part A: Polymer ChemistryDOI 10.1002/pola