mrna-lipoplex loaded microbubble contrast agents for ultrasound-assisted transfection of dendritic...
TRANSCRIPT

1
mRNA-Lipoplex Loaded Microbubble Contrast Agents for Ultrasound-Assisted
Transfection of Dendritic Cells
Marie-Luce De Temmermana,§, Heleen Dewittea,§, Roosmarijn E. Vandenbrouckeb,c, Bart
Lucasa, Claude Libertb,c, Jo Demeestera, Stefaan C. De Smedta,*, Ine Lentackera,† and
Joanna Rejmana,†,*
§Both authors contributed equally to this work
†Both senior authors contributed equally to this work
aLaboratory of General Biochemistry and Physical Pharmacy, Ghent Research Group on
Nanomedicine, Faculty of Pharmaceutical Sciences, Ghent University, Harelbekestraat
72, Ghent, B-9000 Belgium
bDepartment for Molecular Biomedical Research, VIB, Technologiepark 927, 9052
Ghent, B-9052 Belgium
cMolecular Mouse Genetics Unit, Department of Biomedical Molecular Biology, Ghent
University, Technologiepark 927, Ghent, B-9052 Belgium
*Corresponding authors: Tel +32 9 264 80 76; Fax +32 9 264 81 89; Email addresses:
[email protected] (S.C. De Smedt), [email protected] (J. Rejman)

2
ABSTRACT
In cancer immunotherapy the immune system should be triggered to specifically
recognize and eliminate tumor cells in the patient’s body. This could be achieved by
loading dendritic cells (DCs) with tumor associated antigens (TAAs). This can be
achieved by transfecting DCs with messenger RNA encoding a tumor-associated
antigen. Here we demonstrate transient transfection of dendritic cells by means of
mRNA lipoplexes bound to microbubbles. Microbubble-attached lipoplexes were
introduced into the cells by applying ultrasound. Our data demonstrate that ultrasound-
mediated delivery of mRNA complexes led to efficient transfection of DCs. When mRNA
encoding luciferase was used, maximal levels of the enzyme activity were detected 8h
after ultrasound application. Upon longer incubation protein expression gradually
declined. This treatment did not affect viability of the cells. Intracellular localisation of
mRNA-lipoplexes in DCs was determined by flow cytometry using fluorescently labelled
lipoplexes. Over 50 % of DCs contained fluorescently labelled mRNA-complexes. In the
absence of additional maturation signals, transfection of immature DCs with mRNA-
lipoplex loaded microbubbles and ultrasound application induced only a minor shift in the
expression level of maturation markers (CD40 and CD86). However, in the presence of
the activation stimulus (LPS), cells were able to further mature as shown by a significant
up-regulation of CD40 expression. Thus, our results demonstrate that mRNA-lipoplex
loaded microbubbles can serve as an applicable and safe tool for efficient mRNA-
transfection of cultured DCs.
KEYWORDS: mRNA, microsphere, gene therapy, controlled drug release

3
INTRODUCTION
Increasing knowledge of immunology and biology of dendritic cells (DCs) has
sparked interest in immunotherapy as a strategy for the treatment of cancer. DCs are
regarded as the most potent antigen presenting cells (APCs) that are able to initiate and
regulate T and B cell-mediated immunity [1]. Immature DCs function as sentinels
throughout the body where they recognize and capture antigens. These antigens are
subsequently processed and DCs migrate to the lymph nodes. On their way, they
mature from antigen-sampling cells into APCs. In the lymph nodes, they present the
antigen-derived peptides associated with MHC class I and II molecules to naive T cells,
thereby inducing an immune response [2]. For several reasons, vaccination strategies
based on modification of DCs to present tumor associated antigens (TAAs) offer an
attractive approach to stimulate the immune system of a patient to achieve therapeutic
immunity against tumors [3]. First of all, DCs are expected to give rise to tumor-specific
T cells that induce tumor regression or eradication. Moreover, DCs generate tumor-
specific memory cells that might help to prevent tumor relapse [4-6]. Although preclinical
and clinical studies demonstrated encouraging priming of T cells, clinical responses are
so far mainly limited to disease stabilization and prolonged survival. Complete remission
was only seen in low percentage of treated patients [7]. Therefore, the development of
novel vaccination strategies still remains a key issue.
In recent years, the focus of research has shifted from peptide and protein
loading of DCs towards genetic approaches, where plasmid DNA (pDNA) or messenger
RNA (mRNA) is delivered into isolated DCs [8-10]. Translation of the proteins by the DC
itself is attractive as it paves the way to antigen presentation not only by MHC class II

4
but also via MHC class I molecules involved in the generation of cellular immune
responses. Moreover, these strategies hold the potential to introduce multiple epitopes,
ultimately yielding an immune response against the broad antigenic repertoire of the
tumor. As compared to DNA-based approaches, mRNA-based vaccination presents
several advantages. Firstly, mRNA does not integrate into the genome, rendering its
application much safer. Secondly, while in non-dividing cells, the nuclear envelope
represents a serious obstacle for the delivery of pDNA into the nucleus, this obviously
does not apply to mRNA [11]. Finally, although mRNA generates only transient protein
expression, this should be adequate for antigen processing and presentation by DCs.
To deliver mRNA to DCs, numerous strategies have been explored. Despite the
appealing results obtained with viral vectors based on adenovirus, poxvirus and
lentivirus [12, 13], there are some considerable disadvantages associated with virus-
based approaches. Their application has been restricted due to toxicity and the
immunogenicity of the vector itself [14]. Moreover, the production of viral vectors is
costly and laborious and the size of the carried transgene is limited [15]. As a result,
non-viral approaches represent a promising alternative to deliver genetic material. At
present, electroporation is most frequently used to deliver mRNA to DCs [16-18]. The
transient permeabilization of the plasma membrane caused by the applied electric fields
allows direct entry of the mRNA into the cytoplasm, hence potentially leading to
immediate high protein expression. However, several aspects limit the practical
applicability of this strategy. It has only a limited range of applications in vivo as the
target tissue must be easily accessible (skin or muscle). As a consequence, in the
context of cancer immunotherapy, DCs have to be isolated from the patient’s blood and

5
subsequently transfected ex vivo. Apart from being expensive and laborious, this
approach has to deal with several additional issues such as the yield of DCs after
aphaeresis, the cell culture and freezing procedures and, ultimately, the re-
administration to the patient. In order to circumvent these drawbacks it would be
desirable to develop novel approaches which could potentially be used to deliver mRNA
to DCs in vivo.
In this study, we developed and characterized ultrasound-responsive
microbubbles loaded with mRNA-lipoplexes as an alternative physical method to deliver
mRNA to the cytosol of DCs. Apart from their clinical use as ultrasound contrast agents,
microbubbles have been investigated also as drug and gene delivery vehicles [19, 20].
They consist of a gas core surrounded by a stabilizing lipid or polymer layer. In response
to ultrasound waves, the microbubbles cavitate, and eventually implode [21]. Cavitation
and especially implosion is known to transiently open nearby cells by forming temporary
pores in their plasma membranes, which allows direct entry of larger molecules such as
nucleic acids into the cytosol, thus bypassing endocytic pathways [22]. Using an
ultrasound-based approach for DC transfection is particularly appealing as ultrasound is
a widely applied medical imaging and therapy tool, appreciated for its non-invasive and
safe character.
The specific aims of this study were (a) to evaluate the potential of ultrasound-
responsive microbubbles loaded with mRNA-complexes to transfect DCs, (b) to
measure the levels and kinetics of marker protein production following transfection, and
(c) to reveal the impact of ultrasound treatment on the viability and maturation status of
DCs.

6
MATERIALS AND METHODS
Cell culture
DC primary cultures were generated from bone marrow of C57Bl/6 mice. Female
C57BL/6 mice were purchased from Janvier (Le Genest Saint Isle, France) and housed
in a specified pathogen-free facility according to the regulations of the Belgian law and
the local Ethical Committee. Mice were sacrificed and bone marrow was flushed out of
the femur and tibia. After red blood cell lysis (Pharm Lyse Buffer, BD Biosciences), cells
were seeded at a cell density of 5x105 cells/ml in OptiCellsTM (Nunc, Thermo Scientific,
Aalst, Belgium) and incubated at 37°C in 5 % CO2. The cell culture medium was RPMI-
1640 (Gibco-Invitrogen, Merelbeke, Belgium) supplemented with 5 % FCS (Hyclone,
Pierce, Rockford, IL, USA), 1 % penicillin / streptomycin (Gibco-Invitrogen, Merelbeke,
Belgium), 1 % L-glutamine (Gibco-Invitrogen, Merelbeke, Belgium) and 50 µM β-
mercaptoethanol (Gibco-Invitrogen, Merelbeke, Belgium) and contained also 10 ng/ml
IL-4 (Peprotech, Rock Hill, NJ) and 10 ng/ml GM-CSF (Peprotech, Rock Hill, NJ). At day
2 and 6 of culture, the non-adherent cells were collected by centrifugation, resuspended
in fresh culture medium and seeded to the same OptiCellTM. The cells were treated 7
days after seeding.
Messenger RNA
To produce mRNA by in vitro transcription, plasmids encoding luciferase and
green fluorescent protein (EGFP) were purified using a QIAquick PCR purification kit
(Qiagen) and linearized using Dra I restriction enzyme (plasmid encoding firefly
luciferase) or Spe I (plasmid encoding EGFP). Linearized plasmids were used as

7
templates for the in vitro transcription reaction using the T7 mMessage mMachine kit
(Ambion). mRNAs were purified by DNase I digestion and precipitated with LiCl. This
was followed by washing with 70 % ethanol. The produced mRNAs were both capped
and polyadenylated. The mRNA concentration was determined by measuring the
absorbance at 260 nm. mRNAs were stored in small aliquots at -80°C at a concentration
of 1 µg/µl.
Bioluminescence assay
All cells were removed from OptiCellsTM and collected by centrifugation. After
removing the culture medium, the cells were washed once with PBS (Gibco/Invitrogen,
Merelbeke, Belgium). Subsequently, 100 µl of Cell Culture Lysis Reagent (Promega,
Leiden, The Netherlands) was added. After incubation for 30 min, the samples were
centrifuged (12,000 rpm at 4°C for 5 min) and 40 µl aliquots of the supernatants were
transferred to a 96-well plate. Luciferase activity of each sample was assayed in a
GloMaxTM 96 Luminometer (Promega, Leiden, The Netherlands). 100 µl of the substrate
solution was added to each well and the emitted light was measured over a 10 s period.
A standard Bradford assay was employed to determine the protein content of each
sample (Biorad, Nazareth Eke, Belgium). The results are expressed as relative light
units (RLU) per milligram of protein.
Lipoplexes
If not indicated differently, liposomes were composed of 42.5 % DOTAP (1,2-
dioleoyl-3-trimethylammonium-propane), 42.5 % DOPE (1,2-dioleoyl-sn-glycero-3-
phosphoethanolamine) and 15 % DSPE-PEG-2000-biotin [1,2-distearoyl-sn-glycero-3-

8
phosphoethanol-amine-N-[biotinyl(polyethylene glycol)-2000] (all from Avanti Polar
Lipids, Alabaster, AL). This was done by transferring the appropriate amounts of lipids,
dissolved in CHCl3 into a round-bottom flask. The CHCl3 was evaporated under nitrogen
and the resulting lipid film was rehydrated in RNase-free water (Ambion, Lennik,
Belgium). The resulting DOTAP/DOPE/PEG liposomes were sonicated. The total lipid
concentration in these liposomes was 1 mg/ml. Fluorescently labeled liposomes were
obtained by incorporation of 1 % NBD-PE (phosphatidylethanolamine-N-(7-nitro-1,3-
benzoxadiol-4-yl)) (Avanti Polar Lipids, Alabaster, AL).
Preparation and characterization of biotinylated microbubbles
Microbubbles were prepared starting from a solution of a mixture of DPPC (1,2-
dipalmitoyl-sn-glycero-3-phosphocholine) (Lipoid, Ludwigshafen, Germany) and DSPE-
PEG-biotin [1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[biotin(polyethylene
glycol)-2000] (Avanti Polar Lipids, Alabaster, AL) in a 1:2:7 glycerol-propyleneglycol-H2O
solvent (Sigma-Aldrich, Bornem, Belgium); the molar ratio of the lipids in the lipid
solutions was 85:15. The lipid solution was prepared as follows. Appropriate aliquots of
both lipids, dissolved in CHCl3, were transferred to a round bottom flask. After CHCl3
evaporation, the lipid film was dissolved in a 1:2:7 glycerol-propyleneglycol-H2O mixture
to obtain a clear solution with a final lipid concentration of 4.6x10-4 mmol/ml. Aliquots of
this lipid solution were transferred to 2.5 ml chromatography vials, the headspace of
which was filled with C4F10 gas (F2 chemicals, Preston, UK). Finally, microbubbles were
obtained by high-frequency shaking of the lipid solution in a Capmix™ device (3M-
ESPE, Diegem, Belgium) during 15 s. The size and the concentration of the

9
microbubbles in the dispersion (i.e. number of microbubbles per ml) were determined
with a Beckman-coulter Multisizer 4 (Beckman-coulter, Brea, CA).
Transfection
mRNA encoding luciferase or EGFP was complexed with cationic liposomes and
the resulting complexes were attached to the microbubble surface via avidin-biotin
interaction, as shown in Fig. 2A. The cells were transfected 7 days after seeding.
Different amounts of DOTAP/DOPE/PEG liposomes were dispersed in 50 µl of OptiMem
(Invitrogen, Merelbeke, Belgium). This solution was mixed with 50 µl of mRNA solution
in OptiMem. After 10 min of incubation at RT, 900 µl of OptiMem was added.
Subsequently, the mixture was added to the microbubble solution in OptiMem. After a
short incubation of the cells with microbubbles loaded with mRNA-complexes,
ultrasound was applied (Sonitron 2000, Artison, Inola, OK, USA) (1 MHz, 2 W/cm², 50 %
duty cycle, 30 s).
BM-DC staining for flow cytometric analysis and confocal microscopy
DCs were identified using anti-CD11c surface stain. Transfected cells were
stained for surface CD11c following the manufacturer’s protocols and instructions (BD
Pharmingen, Erembodegem, Belgium). Briefly, cells were surface stained for CD11c-
APC or CD11c-FITC (BD Pharmingen) in stain buffer (BD Pharmingen) for 1 h at 4°C.
Cells were washed twice and resuspended in flow buffer (PBS containing 1 % BSA and
0,1 % azide) for flow cytometric analysis or confocal microscopy.

10
Dendritic cell activation
The effect of the mRNA-lipoplex loaded microbubbles and ultrasound application
on the DC maturation status was assessed by evaluating up-regulation of the maturation
surface markers CD40 and CD86. Briefly, following transfection the cells were
transferred to 24 well plates and cultured with or without addition of E. coli derived
lipopolysaccharide (LPS, 1 µg/ml, Sigma Aldrich, Bornem, Belgium) to the culture
medium. 24 h after exposure of DCs to mRNA-lipoplexes, microbubbles and/or
ultrasound, the cells were washed with stain buffer (BD Pharmingen) and incubated on
ice for 30 min with 5 % goat serum in PBS. Then cells were stained for CD11c-FITC in
combination with staining for CD40-PE or CD86-PE (all BD Pharmingen) for 1 h on ice,
followed by washings with stain buffer. Untreated and LPS stimulated cells were used as
negative and positive controls respectively. All samples were analyzed on a BD flow
cytometer.
FACS analysis
FACS data were acquired using a FACSCaliburTM flow cytometer (BD
Pharmingen) and data were analyzed using CellQuestTM software. In all experiments,
DCs were identified based on their CD11c-APC or CD11c-FITC surface staining.
Intracellular localization of mRNA-lipoplexes was determined by using fluorescently
labeled liposomes. Transfection efficiency was evaluated by measuring EGFP
expression in gated DCs.

11
Confocal Microscopy
Confocal microscopy images of mRNA-lipoplex loaded microbubbles and
transfected DCs were recorded using a Nikon C1si confocal laser scanning module
attached to a motorized Nikon TE2000-E inverted microscope (Nikon Benelux, Brussels,
Belgium). A sample of microbubbles or cell suspension was placed on a cover glass and
analyzed with CLSM using a water immersion objective lens (Plan Apo 60X, NA 1.2,
collar rim correction, Nikon).
Toxicity assay
Cytotoxicity was evaluated 24 h after transfection of DCs in OptiCellsTM using a
SYTOX® green nucleic acid stain (Molecular Probes/Invitrogen, Merelbeke, Belgium).
24 h after treatment, the cells were washed in HEPES buffer and nuclei of non-viable
cells were stained with a 45 nM SYTOX® green solution for 30 min on ice. Prior to flow
cytometric analysis, samples were diluted in flow buffer.
RESULTS
Characterization of microbubbles loaded with mRNA-lipoplexes
To avoid degradation by nucleases and to achieve sustained circulation time in
vivo nucleic acids-based drugs are often complexed with cationic lipids and modified
with polyethylene glycol (PEG). The latter also serves to prevent complex clearance by
the reticulo-endothelial system [23] thus enhancing the chance of uptake by the target
cells.

12
As described in the material and methods section, complexes were prepared by
mixing mRNA with DOTAP/DOPE liposomes. Lipoplexes thus formed were attached to
microbubbles via biotin-avidin-biotin bridges as schematically depicted in Fig. 2A. To be
responsive to clinically applied ultrasound frequencies (1-5 MHz) and enable
microbubble implosion, microbubbles should ideally have a diameter between 1 and
7 µm [24]. We determined the size of microbubbles by coulter counter measurements.
The mean volume diameter of microbubbles employed in this study was 4.4 ± 1.6 µm
(consistent with a mean number diameter of 2.3 ± 1.2 µm), which meets clinical
requirements. The effective binding of mRNA-complexes to microbubbles was
demonstrated by confocal microscopy. As shown in Fig. 2B fluorescently labelled
mRNA-lipoplexes clearly mark the bubble surface.
Ultrasound-triggered delivery of mRNA-lipoplexes
Intracellular localisation of mRNA-lipoplexes in DCs was determined by flow
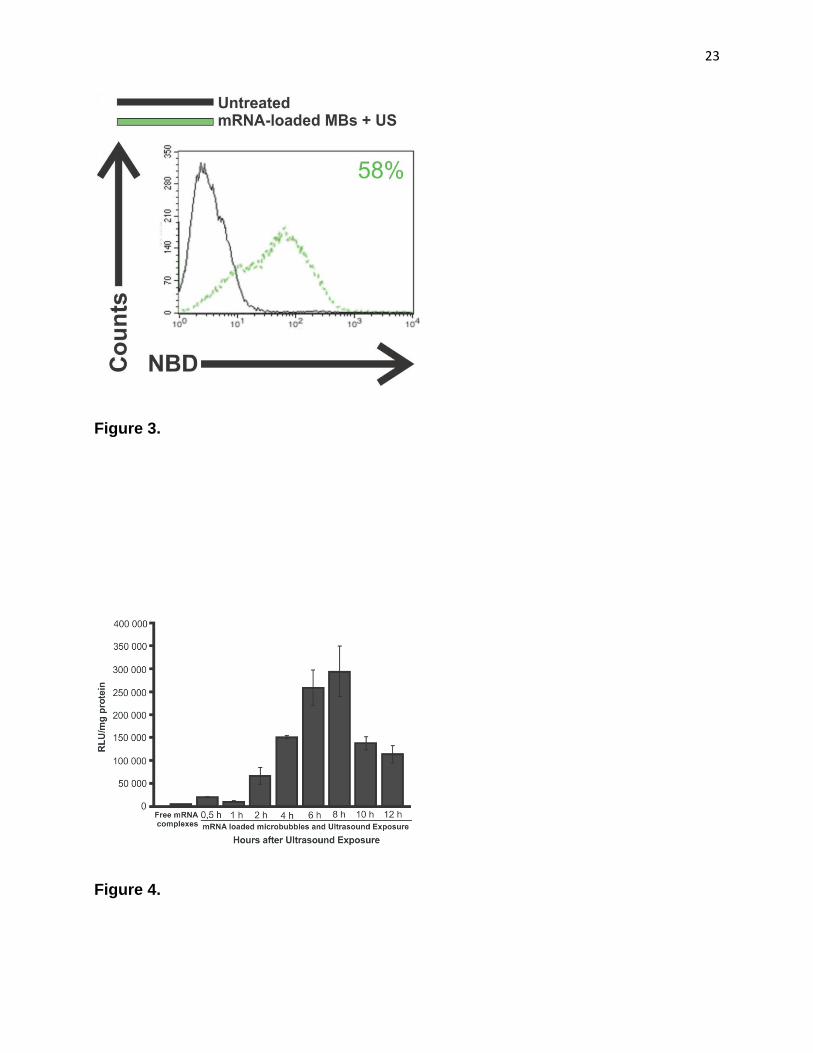
cytometry using fluorescently labelled lipoplexes. As shown in Fig. 3 over 50 % of DCs
contained fluorescently labelled mRNA-complexes. By contrast, negligible uptake of
complexes (< 1 %) was observed if cells were incubated with free mRNA-lipoplexes or
lipoplex-loaded microbubbles without application of ultrasound.
The insignificant uptake of free mRNA-complexes by DCs can be explained by
the high degree of PEGylation (15-20 %) of the complexes used in this study. It is known
that such a hydrophilic coating reduces uptake of the complexes because it shields their
positive charge thereby preventing interactions with the negatively charged cell surface
[25]. Moreover, it has been demonstrated that PEGylated particles, taken up by the cells

13
by endocytosis, get trapped in the endo-lysosomal compartment, which impedes their
release into the cytosol [26]. One possible way to overcome this problem is entry of
PEGylated particles directly into the cytosol as achieved by ultrasound-assisted delivery.
Our group has shown that microbubble implosion creates temporary cell membrane
perforations and at the same time pushes the complexes through such pores [27].
Transfection efficiency of mRNA-lipoplex loaded microbubbles
To assess the ability of mRNA-lipoplex loaded microbubbles to transfect DCs, an
mRNA encoding luciferase was employed. The expression kinetics was characterized by
determining the levels of luciferase at different time points. For this reason we used
firefly luciferase mRNA as this is known to have a very rapid intracellular turnover with a
half-life of 3 h [28]. As shown in Fig. 4, significant protein expression occurs already as
early as 30 min after mRNA-delivery. Maximal levels of luciferase were detected 8 h
after ultrasound application. Upon longer incubation, protein levels gradually declined.
Furthermore, no luciferase activity was detected when only mRNA-lipoplexes were
added to the cells, consistent with the insignificant uptake of mRNA-complexes by the
DCs if neither microbubbles nor ultrasound were applied. This proves that mRNA
transfection of dendritic cells can indeed be triggered by means of ultrasound and thus
opens possibilities towards an image-guided spatiotemporally controlled vaccination
system, as will be discussed later on.
In addition to measuring total levels of protein expression, we evaluated the
number of transfected cells. To that end DCs were transfected with complexes carrying
mEGFP. The proportion of DCs expressing EGFP was evaluated 24 h after transfection

14
by means of flow cytometry. The influence of the transfection protocol on the
background fluorescence signal of the DCs was verified by using mRNA encoding
luciferase. Application of ultrasound in combination with mRNA-lipoplex loaded
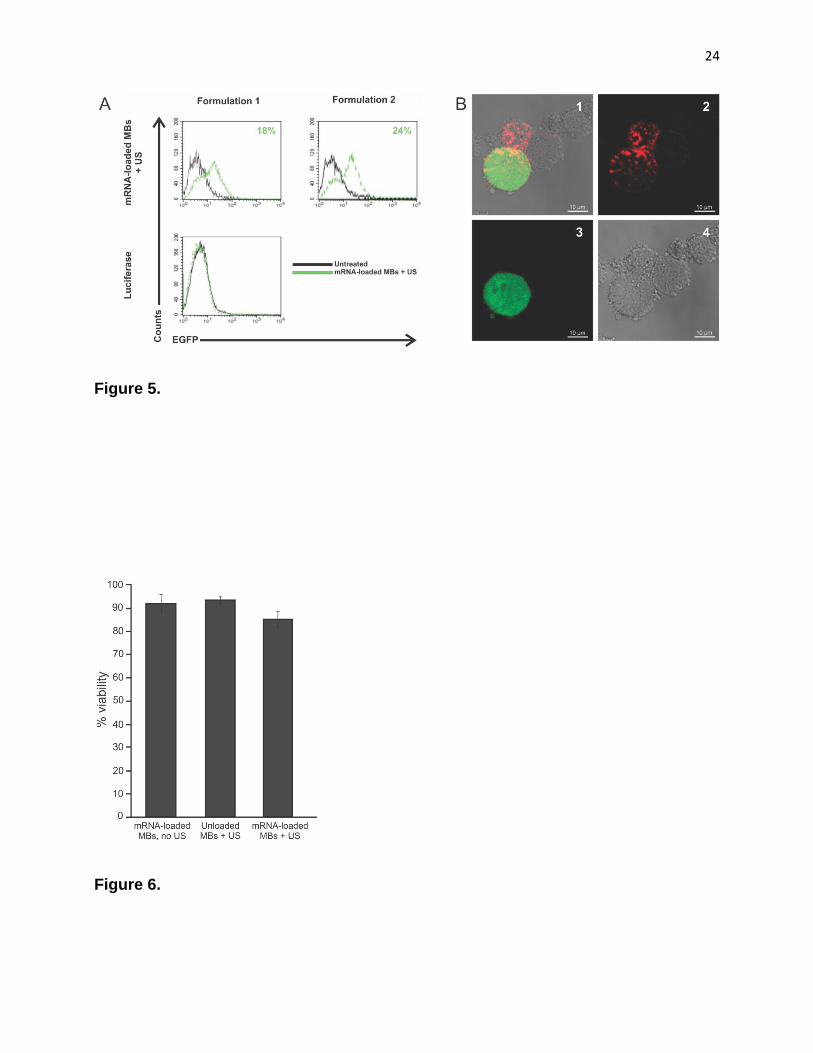
microbubbles resulted in transfection up to 24 % of DCs (Fig. 5A). Ultrasound-assisted
transfection of DCs with free mRNA-lipoplexes resulted in very low numbers of
transfected cells (< 1 %; data not shown). These results were confirmed by confocal
analysis of transfected DCs. Fig. 5B shows surface-stained DCs (anti-CD11c-APC)
expressing EGFP.
These transfection data provide proof of concept for the use of mRNA-lipoplex
loaded microbubbles to transfect DCs. Although 50 % of DCs were shown to contain
mRNA-complexes after this treatment (Fig. 3), only 24 % of the cells expressed EGFP
(Fig. 5A). This may be due to the incomplete release of the mRNA from the complexes.
mRNA electroporation has been shown to achieve transfection efficiencies of up
to 70 % for DCs [18]. However, Grünebach et al. proved that high transfection
efficiencies are not essential to elicit effective T cell responses. They even succeeded in
eliciting antigen-specific cytotoxic T cell (CTL) responses with low to hardly detectable
(< 1 %) transfection efficiencies [17]. Several suggestions explaining this phenomenon
were brought forward. Firstly, the CTL assy is possibly more sensitive than flow
cytometry in detecting low levels of antigen. Secondly, not all antigenic peptides that are
recognized by CTLs are necessarily derived from biologically functional proteins. Even
protein fragments translated from damaged mRNA could be processed into antigenic
peptides that can elicit CD8 positive T cells. Taking all this together, we may conclude

15
that the transfection results obtained with mRNA-lipoplex loaded microbubbles and
ultrasound are likely to be sufficient for effective stimulation of the immune system.
Cell viability of DCs following ultrasound exposure
The potential of mRNA delivery by means of ultrasound can be properly
evaluated only if potentially toxic effects on the cells are taken into consideration. To
assess the effect of the mRNA-complexes, microbubbles and ultrasound on DC viability,
we utilized a SYTOX® green nucleic acid stain. Flow cytometric analysis indicated a
survival of 93 % of the cells after exposure to mRNA-lipoplex loaded microbubbles, 91 %
after exposure to unloaded microbubbles combined with ultrasound and 84 % after
addition of mRNA-lipoplex loaded microbubbles with ultrasound application (Fig. 6). This
corresponds with data reported by Suzuki et al. and indicates that most of the dendritic
cells are indeed able to completely restore the ultrasound-induced cell membrane
damage within 24 h [29]. The percentage of dead cells after sonoporation is comparable
to that resulting from electroporation, where 80-90 % viability has been reported for
human monocyte-derived DCs, depending on the physical parameters used [16, 17].
Moreover, the 91 % toxicity observed when unloaded bubbles were used in combination
with ultrasound correlates with prior results of our group on other cell types [30]. This
indicates that DCs are not particularly sensitive to sonoporation-induced damage.
Maturation status of transfected DCs
In the context of DC-based vaccination strategies the DC maturation status
should be taken into consideration. This is a series of changes in the DC phenotype,
shifting DC function from antigen-sampling to antigen-presentation. As a result, this

16
maturation process is crucial for the ability of DCs to prime naive T cells [31, 32]. We
determined whether the mRNA-lipoplex loaded microbubbles and the ultrasound
treatment have an influence on DC maturation. To that end, DCs were evaluated with
respect to the expression of the co-stimulatory markers (CD40 and CD86) 24 h after
transfection. Moreover, we investigated the susceptibility of treated DCs to additional
maturation stimuli. Therefore, DCs were cultured in the absence or presence of a
maturation stimulus (lipopolysaccharide, LPS) added immediately after transfection.
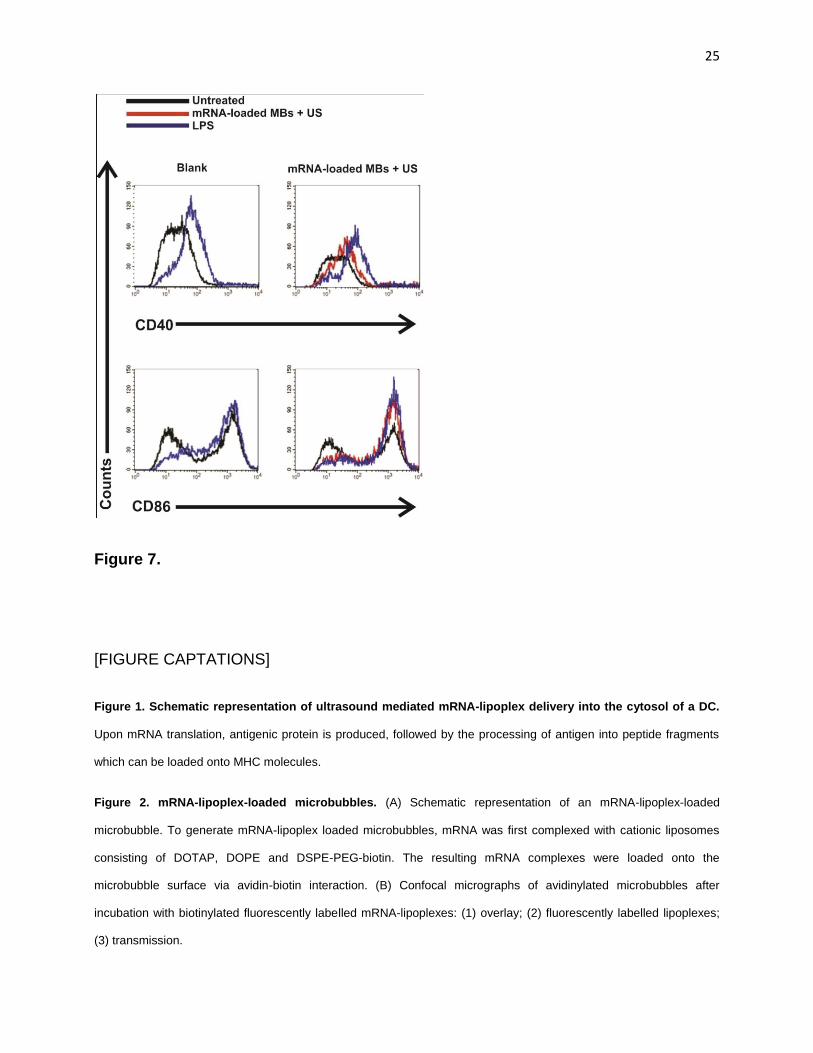
An overview of the phenotypic maturation of DCs in response to mRNA-lipoplex
loaded microbubbles and ultrasound is given in Fig. 7. In the absence of additional
maturation signals, transfection of immature DCs with mRNA-lipoplex loaded
microbubbles and ultrasound application induced only a minor shift in the expression
level of CD40 and a slightly higher increase of CD86, compared to that in the untreated
cells. However, in the presence of the activation stimulus LPS, cells were able to further
mature as shown by a significant up-regulation of CD40 expression. We take this to
indicate that the transfection procedure did not negatively affect the capacity of DCs to
respond to pathogen-associated molecular patterns (PAMPs) and to become activated.
Immature DCs exposed to either ultrasound or microbubbles did not express higher
levels of CD40 and CD86, while LPS stimulation of these cells induced expression of
these markers. Taken together, these observations indicate that the slight phenotypical
changes in DC maturation status were solely due to the mRNA-complexes which had
been delivered and that the transfection procedure itself did not induce phenotypical
changes in the treated DCs.

17
DISCUSSION
We were able to show that mRNA-lipoplexes can be attached to the surface of
ultrasound-responsive microbubbles via biotin-avidin-biotin bridges. Secondly, we
demonstrated an effective uptake of mRNA-lipoplexes by DCs resulting in a significant
expression of luciferase and EGFP by DCs after ultrasound-guided transfection with
mRNA-lipoplex loaded microbubbles, without compromising DC viability and maturation
capacities. In 2009 Suzuki and colleagues reported on the use of microbubbles and
ultrasound to pulse DCs with ovalbumine [29]. They showed that this resulted in MHC I
presentation and the development of a strong CD8+ T cell response. These results are
very encouraging as this indicates that the combination of microbubbles and ultrasound
can be a powerful vaccination tool. We believe that, compared to the system proposed
by Suzuki et al. [29] for vaccination purposes, the development of an mRNA-lipoplex
loaded microbubble offers several advantages. Firstly, when considering the use of
autologous tumor-derived antigens, even small tumor samples may yield sufficient
amounts of mRNA, while for protein extraction relatively large tumor samples are
required. Moreover, co-transfection with mRNA encoding maturation stimuli is possible,
leading to enhanced cytokine signaling, thus strengthening the desired immune
response [33].
The most important outcome of our findings is that ultrasound-assisted
transfection of DCs might eliminate the need of ex vivo procedures in the development
of a DC vaccine, as we will elaborate below. This is important because besides the
problems of costs and labor-intensiveness of current DC vaccination procedures, it has
been suggested that the capacity of the in vitro cultured and transfected DCs to migrate

18
to the lymph nodes is sub-optimal [34]. To address this drawback, the DC vaccine has
been injected intranodally [35, 36]. Alternatively, in vivo transfection of DCs could be
considered. The latter is particularly interesting, consindering that currently used ex vivo
generated monocyte-derived DCs are subject to debate. It has become more and more
clear that this type of DCs merely represents one of the many subsets that can be found
in vivo. As it remains unclear which particular DC subtypes are responsible for the most
efficient CTL priming, it would be advantageous to transfect several of these DC
subtypes in an in vivo setting [32, 37]. For this purpose, the ideal location would be the
lymph nodes, as these organs are populated by large numbers of both resident and
migratory DCs. In this connection, it has been reported that intranodal delivery of naked
pDNA resulted in an important increase of vaccine immunogenicity [38, 39]. Although
this is a promising observation, the unprotected DNA is rapidly cleared and still has to
make its way across the plasma membrane, avoid intralysosomal degradation and cross
the barrier of the nuclear membrane before it can be transcribed and translated into an
antigenic protein. Therefore, this approach is obviously amenable to substantial
improvement. Microbubbles and ultrasound could provide a possible solution, allowing
an increase in transfection efficiency of intranodally located DCs. We care to stress here
that besides the in vitro proof presented in the present work, satisfactory transfection
efficiencies with microbubbles and ultrasound have been shown also in vivo [40, 41].
Furthermore, our suggestion to use microbubbles for intranodal DC transfection is
supported by the consideration that microbubbles have already been used clinically as
ultrasound contrast agents in the lymphatics, and more specifically, as means to
visualize sentinel lymph node metastases [42, 43]. This would imply that a non-invasive
intradermal injection of the mRNA-lipoplex loaded microbubbles would be sufficient to

19
obtain intranodal localization of the nucleic acids. Intranodal location of the microbubbles
– and hence of the mRNA – can easily be visualized as the presence of the bubbles will
be revealed by enhanced ultrasound contrast. This way, the complete vaccination
procedure could be divided into a first phase of low intensity ultrasound, allowing
microbubble visualization and a second phase, where the ultrasound intensity is
increased to cause microbubble collapse and subsequent DC transfection.
CONCLUSIONS
Our results demonstrate that mRNA-lipoplex loaded microbubbles can be used as an
efficient ultrasound-triggered transfection tool for DCs, without compromising cell
viability or DC maturation capacities. These findings are especially important in the
context of a possible in vivo use of this technique. Needless to say that direct in vivo
transfection of DCs would not only eliminate the need for costly ex vivo DC handling and
transfection, but it could also allow transfection of several DC subsets which would
broaden the generated immune response.
ACKNOWLEDGEMENTS
Heleen Dewitte is a doctoral fellow of the Institute for the Promotion of Innovation
through Science and Technology in Flanders, Belgium (IWT-Vlaanderen). Ine Lentacker
is a postdoctoral fellow of the Research Foundation-Flanders, Belgium (FWO-
Vlaanderen). The support of both these institutions is gratefully acknowledged. We
would also like to thank this institution for granting the BRAINSTIM project. This
research was funded through the FWO research Grant G.0187.11.
REFERENCES

20
1. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245-52. 2. Sousa CRE. Activation of dendritic cells: translating innate into adaptive immunity. Curr Opin
Immunol. 2004;16:21-5. 3. Gilboa E. The makings of a tumor rejection antigen. Immunity. 1999;11:263-70. 4. Dallal RM, Lotze MT. The dendritic cell and human cancer vaccines. Curr Opin Immunol.
2000;12:583-8. 5. Banchereau J, Schuler-Thurner B, Palucka AK, Schuler G. Dendritic cells as vectors for therapy. Cell.
2001;106:271-4. 6. Steinman RM, Dhodapkar M. Active immunization against cancer with dendritic cells: the near
future. Int J Cancer. 2001;94:459-73. 7. Ridgway D. The first 1000 dendritic cell vaccinees. Cancer Invest. 2003;21:873-86. 8. Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-
presenting cells in vitro and in vivo. J Exp Med. 1996;184:465-72. 9. Kirk CJ, Mule JJ. Gene-modified dendritic cells for use in tumor vaccines. Hum Gene Ther.
2000;11:797-806. 10. Ponsaerts P, Van Tendeloo VFI, Berneman ZN. Cancer immunotherapy using RNA-loaded dendritic
cells. Clin Exp Immunol. [Review]. 2003;134:378-84. 11. Tavernier G, Andries O, Demeester J, Sanders NN, De Smedt SC, Rejman J. mRNA as gene
therapeutic: how to control protein expression. J Control Release. 2011;150:238-47. 12. Breckpot K, Corthals J, Heirman C, Bonehill A, Michiels A, Tuyaerts S, et al. Activation of monocytes
via the CD14 receptor leads to the enhanced lentiviral transduction of immature dendritic cells. Hum Gene Ther. 2004;15:562-73.
13. Aicher A, Westermann J, Cayeux S, Willimsky G, Daemen K, Blankenstein T, et al. Successful retroviral mediated transduction of a reporter gene in human dendritic cells: feasibility of therapy with gene-modified antigen presenting cells. Exp Hematol. 1997;25:39-44.
14. Hartman ZC, Appledorn DM, Amalfitano A. Adenovirus vector induced innate immune responses: impact upon efficacy and toxicity in gene therapy and vaccine applications. Virus Res. 2008;132:1-14.
15. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nature Reviews Genetics. 2003;4:346-58.
16. Van Tendeloo VFI, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, et al. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood. 2001;98:49-56.
17. Grunebach F, Muller MR, Nencioni A, Brossart P. Delivery of tumor-derived RNA for the induction of cytotoxic T-lymphocytes. Gene Ther. 2003;10:367-74.
18. Van Meirvenne S, Straetman L, Heirman C, Dullaers M, De Greef C, Van Tendeloo V, et al. Efficient genetic modification of murine dendritic cells by electroporation with mRNA. Cancer Gene Ther. 2002;9:787-97.
19. Tinkov S, Bekeredjian R, Winter G, Coester C. Microbubbles as ultrasound triggered drug carriers. J Pharm Sci. 2009;98:1935-61.
20. Lentacker I, De Smedt SC, Sanders NN. Drug loaded microbubble design for ultrasound triggered delivery. Soft Matter. 2009;5:2161-70.
21. Pitt WG, Husseini GA, Staples BJ. Ultrasonic drug delivery – a general review. Expert Opin Drug Deliv. 2004;1:37-56.
22. Schlicher RK, Radhakrishna H, Tolentino TP, Apkarian RP, Zarnitsyn V, Prausnitz MR. Mechanism of intracellular delivery by acoustic cavitation. Ultrasound Med Biol. 2006;32:915-24.

21
23. Eliyahu H, Servel N, Domb AJ, Barenholz Y. Lipoplex-induced hemagglutination: potential involvement in intravenous gene delivery. Gene Ther. 2002;9:850-8.
24. Schutt EG, Klein DH, Mattrey RM, Riess JG. Injectable microbubbles as contrast agents for diagnostic ultrasound imaging: the key role of perfluorochemicals. Angewandte Chemie-International Edition. [Review]. 2003;42:3218-35.
25. Song LY, Ahkong QF, Rong Q, Wang Z, Ansell S, Hope MJ, et al. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochimica Et Biophysica Acta-Biomembranes. 2002;1558:1-13.
26. Shi FX, Wasungu L, Nomden A, Stuart MCA, Polushkin E, Engberts J, et al. Interference of poly(ethylene glycol)-lipid analogues with cationic-lipid-mediated delivery of oligonucleotides; role of lipid exchangeability and non-lamellar transitions. Biochem J. 2002;366:333-41.
27. Lentacker I, Wang N, Vandenbroucke RE, Demeester J, De Smedt SC, Sander NN. Ultrasound exposure of lipoplex loaded microbubbles facilitates direct cytoplasmic entry of the lipoplexes. Mol Pharm. 2009;6:457-67.
28. Rejman J, Tavernier G, Bavarsad N, Demeester J, De Smedt SC. mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J Control Release. 2010;147:385-91.
29. Suzuki R, Oda Y, Utoguchi N, Namai E, Taira Y, Okada N, et al. A novel strategy utilizing ultrasound for antigen delivery in dendritic cell-based cancer immunotherapy. J Control Release. 2009;133:198-205.
30. Lentacker I, Geers B, Demeester J, De Smedt SC, Sanders NN. Design and evaluation of doxorubicin-containing microbubbles for ultrasound-triggered doxorubicin delivery: cytotoxicity and mechanisms involved. Mol Ther. 2010;18:101-8.
31. Sousa CR. Essay - dendritic cells in a mature age. Nature Reviews Immunology. 2006;6:476-83. 32. Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell
subsets in vivo. Nature Reviews Immunology. 2007;7:543-55. 33. Bonehill A, Van Nuffel AMT, Corthals J, Tuyaerts S, Heirman C, Francois V, et al. Single-step antigen
loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res. 2009;15:3366-75.
34. Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999;59:56-8.
35. Lambert LA, Gibson GR, Maloney M, Durell B, Noelle RJ, Barth RJ. Intranodal immunization with tumor lysate-pulsed dendritic cells enhances protective antitumor immunity. Cancer Res. 2001;61:641-6.
36. Bedrosian I, Mick R, Xu SW, Nisenbaum H, Faries M, Zhang P, et al. Intranodal administration of peptide-pulsed mature dendritic cell vaccines results in superior CD8+ T-cell function in melanoma patients. J Clin Oncol. 2003;21:3826-35.
37. Nestle FO, Farkas A, Conrad C. Dendritic-cell-based therapeutic vaccination against cancer. Curr Opin Immunol. 2005;17:163-9.
38. Maloy KJ, Erdmann I, Basch V, Sierro S, Kramps TA, Zinkernagel RM, et al. Intralymphatic immunization enhances DNA vaccination. Proc Natl Acad Sci U S A. 2001;98:3299-303.
39. Tagawa ST, Lee P, Snively J, Boswell W, Ounpraseuth S, Lee S, et al. Phase I study of intranodal delivery of a plasmid DNA vaccine for patients with Stage IV melanoma. Cancer. 2003;98:144-54.
40. Taniyama Y, Tachibana K, Hiraoka K, Aoki M, Yamamoto S, Matsumoto K, et al. Development of safe and efficient novel nonviral gene transfer using ultrasound: enhancement of transfection efficiency of naked plasmid DNA in skeletal muscle. Gene Ther. 2002;9:372-80.

22
41. Vannan M, McCreery T, Li P, Han ZG, Unger E, Kuersten B, et al. Ultrasound-mediated transfection of canine myocardium by intravenous administration of cationic microbubble-linked plasmid DNA. J Am Soc Echocardiogr. 2002;15:214-8.
42. Goldberg BB, Merton DA, Liu JB, Thakur M, Murphy GF, Needleman L, et al. Sentinel lymph nodes in a swine model with melanoma: contrast-enhanced lymphatic US. Radiology. 2004;230:727-34.
43. Sever AR, Mills P, Jones SE, Cox K, Weeks J, Fish D, et al. Preoperative sentinel node identification with ultrasound using microbubbles in patients with breast cancer. Am J Roentgenol. 2011;196:251-6.
[FIGURES]
Figure 1.
Figure 2.
23
Figure 3.
Figure 4.
24
Figure 5.
Figure 6.
25
Figure 7.
[FIGURE CAPTATIONS]
Figure 1. Schematic representation of ultrasound mediated mRNA-lipoplex delivery into the cytosol of a DC.
Upon mRNA translation, antigenic protein is produced, followed by the processing of antigen into peptide fragments
which can be loaded onto MHC molecules.
Figure 2. mRNA-lipoplex-loaded microbubbles. (A) Schematic representation of an mRNA-lipoplex-loaded
microbubble. To generate mRNA-lipoplex loaded microbubbles, mRNA was first complexed with cationic liposomes
consisting of DOTAP, DOPE and DSPE-PEG-biotin. The resulting mRNA complexes were loaded onto the
microbubble surface via avidin-biotin interaction. (B) Confocal micrographs of avidinylated microbubbles after
incubation with biotinylated fluorescently labelled mRNA-lipoplexes: (1) overlay; (2) fluorescently labelled lipoplexes;
(3) transmission.

26
Figure 3. Intracellular localisation of fluorescently labelled mRNA lipoplexes. Fluorescently labeled lipoplexes,
containing 1 % NBD-PE, were attached to the microbubbles and added to DCs. 15 min after ultrasound exposure, the
cells were thoroughly washed to remove non-internalized complexes, which was followed by anti-CD11c-APC surface
staining and flow cytometric analysis. Gated DCs were further analysed.
Figure 4. Transfection efficiency of mRNA lipoplex-loaded microbubbles. mRNA encoding luciferase was
complexed with DOTAP/DOPE/PEG lipoplexes and added to microbubbles and ultrasound was applied. Levels of
luciferase activity in DCs at different time points after ultrasound exposure were assayed by measuring
bioluminescence. Graphs represent means ± standard deviation (n=6). Transfection efficiency of free PEGylated
lipoplexes was assayed two hours after adding them to the cells.
Figure 5. Transfection efficiency of mRNA lipoplex-loaded microbubbles. (A) mRNA encoding GFP was
complexed with DOTAP/DOPE/PEG liposomes (15 % PEG – formulation 1; 20 % PEG – formulation 2) and added to
microbubbles and ultrasound was applied. Control cells were transfected with mRNA encoding luciferase. DCs were
analyzed for GFP expression 24 h after transfection by flow cytometry. DCs were gated based on CD11c-APC
staining. (B) Confocal images of DCs after transfection with mRNA lipoplex-loaded microbubbles and ultrasound. (1)
overlay; (2) CD11c stain; (3) EGFP expression; (4) transmission).
Figure 6. Cell viability. DCs were exposed to mRNA-lipoplex loaded microbubbles or microbubbles and ultrasound
or mRNA-lipoplex loaded microbubbles and ultrasound. Following the treatment, cells were stained with SYTOX®
green nucleic acid stain to identify dead cells. Viability of untreated cells was set as 100 %. Results are the mean ±
standard deviation (n=3).
Figure 7. Maturation of DCs. Representative histograms illustrating phenotypic maturation of DCs in response to
mRNA-lipoplex loaded microbubbles and ultrasound. 24 h after transfection DCs, cultured with or without LPS, were
stained for CD40 or CD86 maturation markers and assessed by flow cytometry. DCs were gated on CD11c. Untreated
DCs and LPS-stimulated DCs served as negative and positive controls, respectively.