morphology of mesophase and crystals of polyamide 6 prepared in a fast scanning chip calorimeter
TRANSCRIPT

at SciVerse ScienceDirect
Polymer 53 (2012) 3994e4001
Contents lists available
Polymer
journal homepage: www.elsevier .com/locate/polymer
Morphology of mesophase and crystals of polyamide 6 prepared in a fast scanningchip calorimeter
Daniela Mileva a,1, René Androsch a,*, Evgeny Zhuravlev b, Christoph Schick b
aMartin-Luther-University Halle-Wittenberg, Center of Engineering Sciences, D-06099 Halle/Saale, GermanybUniversity of Rostock, Institute of Physics, D-18051 Rostock, Germany
a r t i c l e i n f o
Article history:Received 27 April 2012Received in revised form18 June 2012Accepted 23 June 2012Available online 14 July 2012
Keywords:Polyamide 6Crystal morphologyFast scanning chip calorimetry
* Corresponding author. Tel.: þ49 3461 46 3762; faE-mail address: [email protected] (R.
1 Present address: Borealis Polyolefin GmbH, A-402
0032-3861/$ e see front matter � 2012 Elsevier Ltd.http://dx.doi.org/10.1016/j.polymer.2012.06.045
a b s t r a c t
Atomic force microscopy (AFM) has been employed to study the effect of the pathway of nucleation/crystallization on the morphology of the ordered phase of polyamide 6 (PA 6). Samples of PA 6 werecrystallized or ordered at different supercooling of the melt, to obtain either a-crystals or g-mesophase,respectively. For improved control of the nucleation pathway, samples for AFM analysis were for the firsttime prepared using a fast scanning chip calorimeter (FSC). It has been found that melt-crystallization atlow supercooling is connected with the formation of lamellae, while the mesophase formed at highsupercooling is of non-lamellar shape. In case of cold-crystallization, the final semicrystalline structuredepends on the heating rate; slow heating first leads to formation of mesophase nodules which thenreorganize to crystals at elevated temperature, while fast heating suppresses the formation of mesophaseand allows direct transformation of supercooled liquid to lamellae. The general approach of using FSC forAFM sample preparation has been confirmed by analysis of the effect of the thermal pathway of ordering/crystallization on the structure of an isotactic polypropylene.
� 2012 Elsevier Ltd. All rights reserved.
1. Introduction
The morphology and structure of crystals of polymers includingpolyamide 6 (PA 6) is controlled by the temperature of the phasetransformation/supercooling of the liquid phase. In general, thecritical size of nuclei decreases with increasing supercooling of themelt and crystals of lower size may be obtained in comparison tocrystallization at low supercooling [1,2]. Furthermore, it is expectedthat the number of nuclei and consequently of crystals increaseswith decreasing transformation temperature. At low supercooling,the crystallization process likely proceeds via heterogeneousnucleation initiated at impurities, additives or other heterogene-ities including incompletely randomized former crystals duringprior melting. With increasing supercooling of the melt, formationof heterogeneous nuclei may be replaced by formation of homo-geneous nuclei, that is, of nuclei formed by accidental alignment/orientation of segments of macromolecules at local scale. Homo-geneous nucleation is the main mechanism on crystallization nearand even below the glass transition temperature (Tg), as has beenconcluded from recent analyses of the dependence of the rates ofnucleation and crystallization on temperature using fast scanning
x: þ49 3461 46 3891.Androsch).1 Linz, Austria.
All rights reserved.
chip calorimetry (FSC) [3e7], or from investigation of crystalliza-tion in droplets of low size [8e10]. In detail, FSC analysis of thecrystallization rate as a function of temperature of poly(butyleneterephthalate) [3], poly(ε-caprolactone) (PCL) [4], or isotacticpolypropylene (iPP) [5e7] revealed a strong acceleration of theordering process at temperatures close to Tg which has beenattributed to an increase of the number of nuclei, despite reducedcooperative mobility of molecular segments. For PA 6 it has beenproposed, based on evaluation of the crystallization behavior ofsub-micrometer size droplets that homogeneous nucleation at lowtemperature is related to the formation of less ordered crystals/mesophase [10].
For the specific case of iPP, extensive research has been per-formed to identify the morphology of the ordered phase formed atdifferent supercooling of the melt, ultimately confirming a drasticincrease of the nuclei number with increasing supercooling. Thereis observed spherulitic growth of lamellae from a rather lownumber of heterogeneous nuclei at low supercooling. In contrast, athigh supercooling, close to Tg, lateral growth of a large number ofnuclei is suppressed, and lamellae and spherulites cannot form dueto unavailable space [11e15].
Similar systematic research of the effect of supercooling andtherefore nucleation on the morphology of the ordered phase hasnot been performed yet on PA 6. Early research on PA 6, in thecontext of the present work, focused on the evaluation of the

D. Mileva et al. / Polymer 53 (2012) 3994e4001 3995
crystal polymorphism, in particular the determination of theinternal structure of the ordered phases and the conditions of theirformation. Monoclinic a-crystals form at low supercooling of themelt at temperatures higher 420 K, while at lower temperaturesa pseudo-hexagonal mesophase of lower order develops [16e20].The arrangement of molecular segments in the a-phase and themesophase is described in detail in the literature [21e23]. In short,the a-phase is characterized by fully extended molecule segmentsand sheet-like hydrogen bonding between the neighbored, parallelaligned chains. The pseudo-hexagonal mesophase, in contrast, hasbeen described of consisting of small aggregates of parallel, con-formationally disordered but straight chain segments, with long-range order only holding for parallelism and pseudo-hexagonalarrangement of chain axes; hydrogen bonding within the meso-phase is almost complete though not restricted to a specific crys-tallographic direction as in the a-form. The mesophase transformsto stable a-crystals on heating or annealing at elevated temperature[24,25]. It is worthwhile noting that the pseudo-hexagonal meso-phase of PA 6, which develops from the non-oriented melt at highsupercooling, has in the past diversely been named in the literature,including b-phase [22,23], g-phase [26], or g*-phase [19,24]. Inorder to avoid confusion and to account for the lower degree oforder compared to a-crystals, we use in the following the term‘mesophase’.
While the lamellar morphology and spherulitic superstructureof the a-phase formed on slow cooling the melt is known [27e30],in-depth studies of the morphology of the mesophase formed athigh supercooling and of the a-phase formed on heating the glassor the mesophase are not available. This notwithstanding, there arefew reports showing that also in PA 6 qualitatively different semi-crystalline structures may be attained by variation of the conditionof solidification the relaxed melt. It has been found that injection-molded samples exhibit a non-spherulitic surface consisting ofpseudo-hexagonal mesophase while in the center layer theexpected spherulitically grown a-structure has been observed,demonstrating the effect of cooling rate/supercooling [29]. Ina different work about the structure of melt-cast PA 6 it was foundby transmission electron microscopy that quenching leads toincomplete spherulite formation only, and irregular arrangement ofcrystals in inter-spherulitic regions [30].
Following the idea of generation of different semicrystallinestructures of PA 6 by variation of the crystallization conditions, wefocused in an initial work on the determination of the morphologyof cold-crystallized PA 6, that is, on samples which initially werequenched to obtain a glass, and which subsequently were heated toa temperature slightly above Tg [31]. Heating of amorphous, glassyPA 6 to 343 K, indeed, led to the development of mesophase ofparticle-like shape without higher-order organization. Heating to453 Kwas connected with formation of a-crystals, however, did notqualitatively affect the morphology of the ordered phase and itssuperstructure, that is, heating the glass of PA 6 at 20 K min�1 to453 K did not allow the formation of lamellae and spherulites, as istypically observed on melt-crystallization at this temperature. Itwas concluded that the final semicrystalline structure formed at453 K is strongly affected by the specific pathway of reaching thetransformation temperature.
To achieve further progress in the field of obtaining specificsemicrystalline morphologies in crystallizable polymers, wesuggest in this work direct observation of the nanoscale structure ofspecimens which were precisely prepared regarding the thermalpathway of nucleation and crystallization using FSC. If emphasis isplaced on gaining structural information on samples solidified athigh supercooling in the temperature range of homogeneousnucleation then sample preparation requires rapid cooling of theequilibrium melt in order to avoid crystal nucleation and growth at
low supercooling. In the past, this only could be achieved byquenching using home-made devices, with the disadvantages oflimited control of the cooling rate, low reproducibility, and, perhapsmost important, decreasing cooling rate on approaching the targettemperature. Similar, in case of intended analysis of cold-crystallization by heating/devitrification the glass, the heatingrate used to reach the transition temperature is crucial to control,e.g., homogeneous nucleation, growth of nuclei, or reorganizationof existing ordered domains. FSC has successfully been applied foranalysis of crystallization of polymers in recent few years andpermits exact control of crystallization conditions in combinationwith quantitative calorimetric analysis of phase transitions at ratesof cooling and heating >104 K s�1, or, in isothermal experiments, ata time scale of milliseconds. Combining FSC analysis of polymercrystallization with supplementary investigation of the nanoscalestructure using atomic force microscopy (AFM), to allow estab-lishment of correlations between crystallization conditions andmorphology, has not been achieved so far, and is therefore object ofour research. The capability of AFM for investigation of the struc-ture of semicrystalline polymers at the nanometer length scale hasbeen documented in numerous work [32e34]; however, thefollowing presentation of experimental results provides evidencethat specimens of well-defined thermal history and with a mass offew nanograms and dimensions in the micrometer range, as aretypically required for FSC analyses, are also acceptable for subse-quent evaluation of the morphology using AFM. Primary focus is togain new information about the morphology of crystals and themesophase of PA 6 formed at different thermal conditions. In orderto demonstrate generality of the approach of probing the AFMnanoscale structure of polymers crystallized/ordered in an FSC atwell-defined conditions of cooling, heating and/or isothermalannealing, an isotactic propylene/butene-1 random copolymer hasadditionally been included in this study. Evaluation of the nano-scale structure of iPP including random copolymers with 1-alkenesas a function of the conditions of crystallization has been object ofintense research for long time, that is, there exists a comprehensivedata base for comparison with structures obtained in this work.
2. Experimental section
We employed a PA 6 grade of molar mass of 18 kDa from Pol-ysciences (Catalog Number 18180) for analysis of the morphologyof the ordered phase as a function of the thermal history. The as-received and dried material has been processed to a film witha thickness of 100 mm by compression molding, before furtherpreparation of specimens with specific geometry for FSC bymicrotoming and microscope-aided cutting. The particular poly-mer has been characterized regarding its crystallization behaviorby FSC in prior work with the literature containing further detailedand specific information about the sample preparation [35]. Forsupplementary analysis of the morphology of the mesophase andcrystals of iPP we used a random copolymer of propylene with10.9 mol% butene-1 from Sigma Aldrich. Preparation of specimensfor FSC analysis has been done in analogy to that in case of PA 6.
For preparation of PA 6 samples of different thermal history, weused a twin-type power-compensation Mettler-Toledo fast scan-ning chip calorimeter Flash DSC 1. The top left photograph in Fig. 1is a total view of the chip XI-400 from Xensor Integration(Netherlands), as it is supported by a ceramic frame of 600 mmthickness for user-friendly operation/handling. It contains twoseparated silicon nitride/oxide membranes with an area of1.7 � 1.7 mm2 and thickness of 2.1 mm each, and is surrounded bya silicon frame of 300 mm thickness. The circular sample-measurement area with a diameter of 500 mm in the center ofeach membrane is coated with 0.5 mm aluminum, to ensure

Fig. 2. Active area of the XI-39321 chip calorimeter from Xensor Integration witha sample of a random propylene/butene-1 copolymer [39].
Fig. 1. UFS 1 sensor of the twin-type power-compensation Mettler-Toledo Flash DSC 1 showing both the sample and reference calorimeters supported by a ceramic frame (top left).Sample of PA 6 located in the center of the heated area of the sample calorimeter (top right). Schematic of the cross-section of the UFS 1 sensor (bottom) [37].
D. Mileva et al. / Polymer 53 (2012) 3994e40013996
a homogeneous temperature field. The top right photograph showsa typical sample of PA 6, as it has been analyzed in this particularstudy. The diameter and thickness of the sample are around 100and 20 mm, respectively, and the mass is of the order of magnitudeof 200 ng. In the lower part of Fig. 1 is sketched the cross-section ofa membrane, which also shows the positions of resistive heatersand of an integrated thermopile; the hot-junctions are positionedclose to the sample area while the cold-junctions are placed on thesilicon frame. Further details of the instrument including infor-mation about its performance and calibration are provided else-where [36].
Preparation of samples of a random propylene/butene-1copolymer with different supermolecular structure has been doneusing a chip calorimeter XI-39321 from Xensor Integration. Theactive area of this particular sensor is 60 � 60 mm2 and is shown inFig. 2 together with the specific sample used in this work. In thecontext of this work it is important to note that the chip XI-39321 ismounted and pinned differently in comparison to the UFS 1 sensor.In case of the UFS 1 sensor, the membrane needed to be detachedfrom the silicon frame and the ceramic housing since otherwise theAFM tip cannot approach the surface of the specimen. Note thatremoval of the polymer from the membrane was not attempted, inorder to not destroy or affect its structure. In contrast, in case of theX I-39321 sensor, which is attached to a TO-5 housing, AFM analysisafter a specific thermal treatment is possible without destruction ofthe sensor. As such, repeated FSC thermal analysis and AFMnanoscale structure analysis is possible in the latter case. Further-more, it has been proven that the roughness of the membrane inchip calorimeters complies with the requirements for AFM analysesof attached thin polymer films [38].
FSC has exclusively been employed for preparation of partiallyordered/crystallized samples of well-defined thermal history, withemphasis put on structure formation at high supercooling. Albeitheat-flow-rate data have been recorded as a function of tempera-ture/time in the various experiments discussed below, these dataare not presented as it has been done in prior work focusing oncalorimetric analysis of the kinetics of ordering [7,35,40]. Followingthe thermal treatment of samples using FSC, the specimens wereanalyzed regarding the nanoscale structure using AFM. We used

Fig. 3. Temperatureetime profiles used for isothermal crystallization or ordering ofsamples of PA 6 at temperatures of 453 and 343 K, respectively. The transitiontemperatures have been approached by direct cooling of the melt (right), or by heatingthe glass (left).
D. Mileva et al. / Polymer 53 (2012) 3994e4001 3997
a Quesant USPM instrument with a 5 � 5 mm2 scanner and eitherMikroMasch DP14 or NSC14 tips for simultaneous observation ofphase- and height-mode images, collected in intermittent contactmode at ambient temperature. In this work, the gray scale of theimages represents the phase angle in arbitrary units; an exceptionis the right image in Fig. 4 in which the nanoscale structure isvisualized by height contrast. It is worthwhile noting that AFManalysis has been performed on the upper free surface of polymersamples which were solidified at rather extreme conditions by FSC.In other words, images must be considered as being obtained atnon-ideal conditions from point-of-view of surface flatness orcontaminations.
3. Results and discussion
3.1. Isothermal ordering/crystallization of PA 6 at high/lowsupercooling of the melt
In Fig. 3 are shown temperatureetime profiles used for melt-crystallization (right) and cold-crystallization (left) at
Fig. 4. AFM images of the surface of semimesomorphic samples of PA 6 prepared using FSC.at 343 K, respectively.
temperatures of 453 and 343 K. Melt-crystallization implied directcooling of the melt from a temperature higher than the equilibriummelting temperature (Tm,0) to the transformation temperature,while cold-crystallization involved temporary vitrification of theamorphous phase by cooling below Tg and approach of the transi-tion temperatures by subsequent heating. The cooling and heatingsegments were performed using a rate of temperature change of103 K s�1, that is, nucleation and growth of the ordered phaseoccurred strictly isothermal within the bold drawn segments. Theselection of the transition temperatures is based on the knowledgethat at 453 K monoclinic a-crystals form while at 343 K pseudo-hexagonal mesophase develops [16,17]. The duration of theisothermal segments was set to 40 s which has been proven suffi-cient to finish both the crystallization process at 453 K and theordering process at 343 K [35]. After completion of the crystalli-zation at 453 K or ordering at 343 K, the samples were cooled toambient temperature, that is, to below the glass transitiontemperature of 313 K [41], to perform analysis of the morphology ofcrystals/mesophase by AFM.
In Fig. 4 are shown surface structures of samples of PA 6 whichhave isothermally been ordered at 343 K. The left image representsthe structure obtained on a sample which in a first step was cooledat 103 K s�1 to below Tg, to obtain a glass, and immediately re-heated at identical rate to the transition temperature. The rightimage was obtained on a sample directly cooled to 343 K. In case ofordering at 343 K, former X-ray studies revealed formation ofmesophase [16,17], of to date unknown morphology. The obtainedstructures, regardless whether formed on heating the glass (left), oron cooling the melt (right), are heterogeneous and composed of anamorphous phase (dark areas) and a separate ordered phase (brightareas). Apparently, the morphology of the mesophase may best bedescribed as consisting of closely neighbored particle-like domainswith a size not larger than 5e10 nm, forming a loose network. Wesuggest that the ordering process at 343 K, that is, at a temperatureonly 20e30 K higher than Tg is based on homogenous nucleation,with the assumption justified with the observation of a largenumber of independently formed entities. There is no indicationthat the initially formed small domains grew to larger objects,presumably caused by missing cooperative mobility of molecularsegments due to formation of a rigid amorphous fraction [4].Though the left and right images of Fig. 4 are slightly different, suchthat isolated mesophase objects are difficult to identify in case ofthe cold-ordered sample (left), we do not suggest qualitativelydifferent mechanism of structure formation. Close inspection of the
The left and right images were collected on samples isothermally cold- or melt-ordered

Fig. 6. Temperatureetime profiles used for non-isothermal crystallization or orderingof samples of PA 6 at rates of cooling (CR) of 0.167, 5, and 50 K s�1. The inset tableprovides information about temperatures (Tc) and enthalpies of crystallization/ordering (Dhc), as obtained in a separate study [35].
D. Mileva et al. / Polymer 53 (2012) 3994e40013998
left image reveals structural heterogeneity at similar length scale asin case of melt-ordered PA 6 as well as absence of lamellae andhigher-order organizations of objects.
Fig. 5, correspondingly, shows the structure of PA 6 isothermallycrystallized at 453 K. The left and right images represent sampleswhich were cold- or melt-crystallized, respectively, according tothe temperatureetime profiles shown in Fig. 3. These samples aresemicrystalline and consist of amorphous phase and monoclinic a-crystals. In contrast to the mesophase formed at high supercoolingof the melt at 343 K, the a-crystals are of lamellar habit, suggestingdirectional lateral growth of initially formed nuclei. We assumethat such growth only is possible if there are no constraints byclosely neighbored different nuclei/crystals in the early stage of thecrystallization process, that is, the total number of nuclei likely islower than in case of ordering at high supercooling. The thicknessof lamellae is 10e15 nm in case of cold-crystallized PA 6 (left) and15e20 nm in case of melt-crystallized PA 6 (right); their lateraldimension is between 50 and 200 nm. While the lamellae appearrather curved in the cold-crystallized sample and not radiallygrown from an initial nucleus, melt-crystallized PA 6 even revealsformation of small spherulites of sub-micrometer size.
The inset in the left image of Fig. 5 shows the structure of semi-crystalline PA 6 which has slowly been heated from the glassy stateat 20 Kmin�1, instead of using a rate of 103 K s�1. Slow heating of theglass of PA 6first allows formation ofmesophase near Tg and then itsreorganization to crystals on continued heating [18e20,25]. Reor-ganizationof themesophase toa-crystals obviously is not connectedwith a changeof themorphologyas canbe concludedbycomparisonwith the mesophase morphology shown in Fig. 4. Fast heating at103 K s�1, in contrast, does not allow mesophase formation at lowtemperature rather than leads to direct formation of lamellar crys-tals from supercooled liquid. Furthermore, if nuclei have beenformed on fast cooling near or below Tg then these nuclei obviouslyget destroyed on fast heating to the transition temperature of 453 K.
3.2. Non-isothermal crystallization/ordering of PA 6 at low andmedium supercooling of the melt
In Fig. 6 are shown temperatureetime profiles applied for non-isothermal crystallization or ordering of PA 6 at rates of cooling of0.167, 5, and 50 K s�1. The cooling rates have been selected in order toallow generation of semicrystalline and semimesomorphic samples
Fig. 5. AFM images of the surface of semicrystalline samples of PA 6 prepared using FSC. Theat 453 K, respectively. The inset in the left image serves for demonstration of the effect ofcrystallization. In this case, the sample has been prepared by quenching in ice water and subbar in the left image also applies for the inset.
at different supercooling of the melt. In prior work, using X-rayscattering or FSC [17,35,42], it has been shown that cooling at0.167 K s�1 (10 K min�1) leads to crystallization at about 470 K andformationof rather perfecta-crystals. Similar, cooling at 5K s�1 is alsoconnected with crystallization; however, the phase transition occursat lower temperatureof 445Kand leads to formationof lessperfecta-crystals. Further increase of the cooling rate to 50K s�1 doesnot allowgeneration of a-crystals; instead, mesophase forms at about 420 K.Finally, if the cooling rate exceeds a critical value of 150K s�1 then anyordering is suppressed. The inset table in Fig. 6 additional providesinformation about the enthalpy of the crystallization/orderingof PA 6when cooling at 0.167, 5, or 50 K s�1, suggesting a distinct decrease ofthe ordered fraction with increasing cooling rate. It is worthremarking that the lowestobserved temperatureoforderingon linearcooling to below Tg is about 410K, coincidingwith the temperature ofmaximum crystallization/ordering rate.
Fig. 7 shows the nanoscale surface structure of samples PA 6linearly cooled from 523 K to below Tg at rates of 0.167 (top images),(bottom left image), and 50 K s�1 (bottom right image). Slowcoolingof the melt at a rate of 0.167 K s�1 led to formation of sheaf-like
left and right images were collected on samples isothermally cold- or melt-crystallizeda lower rate of heating on the approach of the transition temperature in case of cold-sequent heating to 453 K at a rate of 20 K min�1, using a microscope hot stage. The scale
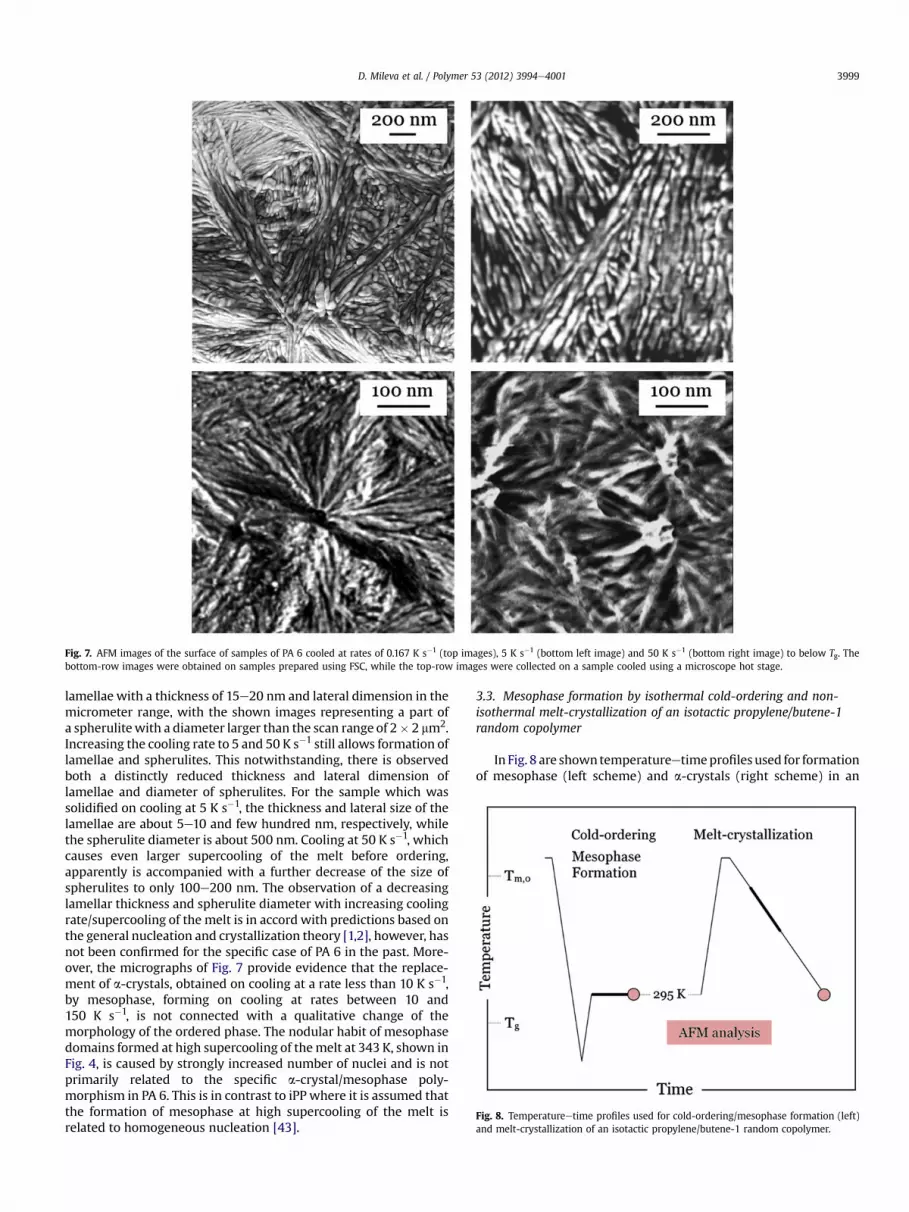
Fig. 7. AFM images of the surface of samples of PA 6 cooled at rates of 0.167 K s�1 (top images), 5 K s�1 (bottom left image) and 50 K s�1 (bottom right image) to below Tg. Thebottom-row images were obtained on samples prepared using FSC, while the top-row images were collected on a sample cooled using a microscope hot stage.
Fig. 8. Temperatureetime profiles used for cold-ordering/mesophase formation (left)and melt-crystallization of an isotactic propylene/butene-1 random copolymer.
D. Mileva et al. / Polymer 53 (2012) 3994e4001 3999
lamellae with a thickness of 15e20 nm and lateral dimension in themicrometer range, with the shown images representing a part ofa spherulitewith a diameter larger than the scan range of 2� 2 mm2.Increasing the cooling rate to 5 and 50 K s�1 still allows formation oflamellae and spherulites. This notwithstanding, there is observedboth a distinctly reduced thickness and lateral dimension oflamellae and diameter of spherulites. For the sample which wassolidified on cooling at 5 K s�1, the thickness and lateral size of thelamellae are about 5e10 and few hundred nm, respectively, whilethe spherulite diameter is about 500 nm. Cooling at 50 K s�1, whichcauses even larger supercooling of the melt before ordering,apparently is accompanied with a further decrease of the size ofspherulites to only 100e200 nm. The observation of a decreasinglamellar thickness and spherulite diameter with increasing coolingrate/supercooling of the melt is in accord with predictions based onthe general nucleation and crystallization theory [1,2], however, hasnot been confirmed for the specific case of PA 6 in the past. More-over, the micrographs of Fig. 7 provide evidence that the replace-ment of a-crystals, obtained on cooling at a rate less than 10 K s�1,by mesophase, forming on cooling at rates between 10 and150 K s�1, is not connected with a qualitative change of themorphology of the ordered phase. The nodular habit of mesophasedomains formed at high supercooling of themelt at 343 K, shown inFig. 4, is caused by strongly increased number of nuclei and is notprimarily related to the specific a-crystal/mesophase poly-morphism in PA 6. This is in contrast to iPP where it is assumed thatthe formation of mesophase at high supercooling of the melt isrelated to homogeneous nucleation [43].
3.3. Mesophase formation by isothermal cold-ordering and non-isothermal melt-crystallization of an isotactic propylene/butene-1random copolymer
In Fig. 8 are showntemperatureetimeprofiles used for formationof mesophase (left scheme) and a-crystals (right scheme) in an

Fig. 9. AFM images of the surface of samples of an isotactic propylene/butene-1 random copolymer, cold-ordered at high supercooling at 295 K (left) and non-isothermally melt-crystallized at low supercooling (right).
D. Mileva et al. / Polymer 53 (2012) 3994e40014000
isotactic propylene/butene-1 random copolymer. The formation ofmesophase included fast coolingof themelt at 104 K s�1 to 100K andimmediate re-heating at identical rate to ambient temperature. Thesamplewas then aged on the FSC sensor and analyzed regarding thenanoscale structure by AFM. Subsequently, both the FSC sensor andthe sample were heated to 473 K and slowly re-cooled to 295 K toallow melt-crystallization at rather low supercooling, usinga vacuum Heraeus oven, before repeated AFM analysis.
Fast cooling of the melt at 104 K s�1 to below Tg according to theleft scheme in Fig. 8 suppresses crystallization/ordering and leadsto complete vitrification of the melt. Subsequent devitrification ofthe glass and isothermal annealing at room temperature permitsformation of a mesophase, whose structure and morphology aredescribed in the literature [11e15,40,43]. The mesophase of iPP andrandom copolymers of propylene with low concentration on 1-alkene co-units is a conformationally disordered glass, and is ofparticle-like, nodular geometry. It has been suggested that theordering process at ambient temperature is connected withhomogeneous nucleation, justified with the observations of a rela-tively large number of independently grown ordered domains,a non-spherulitic superstructure, and high rate of ordering. Recentquantitative analysis of the temperature-dependence of the grossrate of crystallization/ordering of the specific copolymer used inthis work revealed a minimum half-time of ordering of less than 1 saround ambient temperature; crystallization, in contrast, even atthe temperature of the highest crystallization rate at around 330 Kis distinctly slower [7]. This notwithstanding, we do not assumethat homogeneous nucleation in general is a priori connected withdevelopment of less ordered phases, and that heterogeneousnucleation always leads to formation of crystals. Slow cooling of themelt of iPP and its random copolymers with low content on 1-alkenes according to the right scheme in Fig. 8 is expected to causeformation of spherulites and lamellae, with comprehensive back-ground information provided in the literature [44e47]. It is not theaim of the specific experiment to gain new structural informationabout iPP copolymers; rather than, it is the intention to providea further example of advantageous combination of FSC, to be usedfor precise preparation of structures of crystallizable polymers atextreme conditions, and subsequent AFM analysis, to characterizethe nanoscale morphology. While the experiments performed onPA 6 are novel since the morphology of the PA 6 mesophase has notbeen evaluated before, analysis of the morphology of the iPPcopolymer also serves for providing evidence that structureformation of thin polymer films on themembrane of the FSC sensordoes not affect the morphology.
In Fig. 9 are shown AFM images of the surface of the randompropylene/butene-1 copolymer prepared according to thetemperatureetime profiles shown in Fig. 8. The left image revealsthe typical semimesomorphic structure of iPP based materialsobtained after quenching and aging at room temperature, consist-ing of amorphous phase and occasionally string-like arranged, non-lamellar objects with a size of 5e10 nm. The right image wasobtained after heating of the initially semimesomorphic sample to473 K followed by slow cooling to room temperature. It shows theexpected structure of cross-hatched lamellae with a thickness of5e15 nm. The images of Fig. 9 prove that the nanoscale morphologyof samples with a mass of few nanograms and thickness of fewmicrometers, as are typically required in FSC analyses to allow fastheating and cooling, and formed as a result of a specific thermalhistory, is indifferent from the nanoscale morphology of rathermacroscopic samples, frequently analyzed in the past.
4. Conclusions
The recent introduction of FSC for analysis of polymer crystal-lization allows exact control of the thermal pathway of nucleationand crystallization/ordering in a wide temperature range. Priorwork about nucleation and crystallization of polymers at rather lowsupercooling is now completed by studying crystallization andordering at high supercooling, including temperatures close oreven below Tg. The calorimetric response of FSC samples subjectedto a specific thermal history for controlled crystallization/orderingcan precisely be detected to gain information about the kinetics ofphase transitions, phase compositions, or the stability of phases.However, in order to fully exploit the new capabilities achieved byFSC for imposing well-defined temperatureetime profiles to crys-tallizable polymers, we suggest to additionally collect valuableinformation about the morphology of phases; in particular in theshed of light that systematic research focusing on the effect of highsupercooling of the melt on the structure/morphology of crystal-lizable polymers at both the nanometer and micrometer lengthscales has not been performed yet. This includes PA 6 which formsa-crystals at low supercooling and a less stable mesophase at highsupercooling, with the morphology of the latter not known yet. Assuch the research presented here is twofold. First, we attempted toevaluate the general opportunity of AFM analysis of the nanoscalemorphology of specimens prepared in an FSC at conditions whichcannot be achieved by conventional instrumentation, and,secondly, we intended to gain new information about the structureof PA 6 formed at high supercooling.

D. Mileva et al. / Polymer 53 (2012) 3994e4001 4001
It was found that structure formation near Tg, regardlesswhether the transition temperature is approached on cooling theequilibrium melt or on heating the non-aged glass, leads toformation of non-lamellar mesophase. In case of crystallization atlow supercooling, the expected formation of lamellae is confirmed.However, if crystallization at low supercooling is performed onheating/devitrification of the glass, then the morphology of crystalsis controlled by the heating rate; slow heating first leads toformation of mesophase at high supercooling which then onfurther heating reorganizes to crystals with the initial mesophasemorphology preserved. It is concluded that the mesophaseecrystalphase transformation involves local changes of conformations onlyand not complete disordering and melt-recrystallization. Fastheating, in contrast, suppresses mesophase formation and allowsformation of lamellar crystals as on cooling themelt. The analysis ofthe structure of PA 6 cooled at different rate revealed the trivialinformation of decreasing spherulite size with increasing coolingrate/supercooling. Important is the observation that mesophaseformation on cooling at 50 K s�1 at about 410e420 K still involvesspherulitic growth of lamellae and is not primarily caused bydifferent type of nucleation at temperatures near Tg.
Acknowledgments
Financial support by the Deutsche Forschungsgemeinschaft(DFG) is greatly acknowledged.
References
[1] Hoffmann JD, Davis GT, Lauritzen JI. The rate of crystallization of linearpolymers with chain folding. In: Hannay HB, editor. Crystalline and non-crystalline solids. Treatise on solid state chemistry, vol. 3. New York: PlenumPress; 1976.
[2] Wunderlich B. Macromolecular physics. In: Crystal nucleation, growth,annealing, vol. 2. New York: Academic Press; 1976.
[3] Pyda M, Nowak-Pyda E, Heeg J, Huth H, Minakov AA, Di Lorenzo ML, et al.J Polym Sci Pol Phys 2006;44:1364e77.
[4] Zhuravlev E, Schmelzer JWP, Wunderlich B, Schick C. Polymer 2011;52:1983e97.
[5] De Santis F, Adamovsky S, Titomanlio G, Schick C. Macromolecules 2007;40:9026e31.
[6] Mileva D, Androsch R, Zhuravlev E, Schick C, Wunderlich B. Polymer 2012;53:277e82.
[7] Mileva D, Androsch R. Colloid Polym Sci 2012;290:465e71.[8] Burns JR, Turnbull D. J Appl Phys 1966;37:4021e6.[9] Koutsky JA, Walton AG, Baer E. J Appl Phys 1967;38:1832e9.
[10] Tol RT, Mathot VBF, Reynaers H, Goderis B, Groeninckx G. Polymer 2005;46:2966e77;Tol RT, Minakov AA, Adamovsky SA, Mathot VBF, Schick C. Polymer 2006;47:2172e8;Salmerón Sánchez M, Mathot V, Vanden Poel G, Groeninckx G, Bruls W.J Polym Sci Pol Phys 2006;44:815e25.
[11] Hsu CC, Geil PH, Miyaji H, Asai K. J Polym Sci Pol Phys 1986;24:2379e401.
[12] Wang ZG, Hsiao BS, Srinivas S, Brown GM, Tsou AH, Cheng SZD, et al. Polymer2001;42:7561e6.
[13] Ogawa T, Miyami H, Asai K. J Phys Soc Jpn 1985;54:3668e70.[14] Piccarolo S. J Macromol Sci Phys 1992;B31:501e11.[15] Zia Q, Androsch R, Radusch HJ, Piccarolo S. Polymer 2006;47:8163e72.[16] Kyotani M, Mitsuhashi S. J Polym Sci Pol Phys 1972;10:1497e508;
Kyotani M. J Macromol Sci Phys 1975;B11:509e25.[17] Brucato V, Piccarolo S, La Carruba V. Chem Eng Sci 2002;57:4129e43.[18] Hendus H, Illers KH, �Simak P. Kolloid Z Z Polym 1969;235:1244e6.[19] Illers KH, Haberkorn H. Makromol Chem 1971;142:31e67.[20] Gurato G, Fichera A, Grandi FZ, Zannetti R, Canal P. Makromol Chem 1974;
175:953e75.[21] Holmes DR, Bunn CW, Smith DJ. J Polym Sci 1955;17:159e77.[22] Ziabicki A. Kolloid Z 1959;167:132e41.[23] Auriemma F, Petraccone V, Parravicini L, Corradini P. Macromolecules 1997;
30:7554e9.[24] Fichera A, Malta V, Marega C, Zannetti R. Makromol Chem 1988;189:1561e7.[25] Androsch R, Stolp M, Radusch HJ. Acta Polym 1996;47:99e104.[26] Roldan LG, Kaufman HS. J Polym Sci Pol Lett 1963;1:603e8.[27] Geil PH. Polymer single crystals, polymer reviews, vol. 5. New York: Inter-
science; 1963.[28] Ferreiro V, Douglas JF, Coulon G, Karim A. Polym Prepr 2000;41:1427e8.[29] Russell DP, Beaumont PWR. J Mater Sci 1980;15:197e207.[30] Schaper A, Hirte R, Ruscher C, Hillebrand R, Walenta E. Colloid Polym Sci
1986;264:649e58.[31] Mileva D, Kolesov I, Androsch R. Colloid Polym Sci 2012;290:271e8.[32] Magonov SN, Reneker DH. Annu Rev Mater Sci 1997;27:175e222.[33] Hobbs JK, Farrance OE, Kailas L. Polymer 2009;50:4281e92.[34] Ivanov DA, Magonov SN. Atomic force microscopy studies of semicrystalline
polymers at variable temperature. In: Reiter G, Sommer JU, editors. Lecturenotes in Physics, vol. 606. Berlin: Springer; 2003.
[35] Kolesov I, Mileva D, Androsch R, Schick C. Polymer 2011;52:5156e65.[36] Mathot V, Pyda M, Pijpers T, Vanden Poel G, van de Kerkhof E, van
Herwaarden S, et al. Thermochim Acta 2011;522:36e45;van Herwaarden S, Iervolino E, van Herwaarden F, Wijffels T, Leenaers A,Mathot V. Thermochim Acta 2011;522:46e52;Iervolino E, van Herwaarden AW, van Herwaarden FG, van de Kerkhof E, vanGrinsven PPW, Leenaers ACHI, et al. Thermochim Acta 2011;522:53e9.
[37] Reprinted/adapted from Thermochim Acta, Vol. 522 van Herwaarden S,Iervolino E, van Herwaarden F, Wijffels T, Leenaers A, Mathot V. Design,performance and analysis of thermal lag of the UFS1 twin-calorimeter chip forfast scanning calorimetry using the Mettler-Toledo Flash DSC 1. p. 46e52[Copyright with permission from Elsevier]; 2008.
[38] La Spina L, Ovchinnikov D, Wien WHA, van Herwaarden AW, Goudena EJG,Loos J, et al. Sens Actuators, A 2008;144:403e9.
[39] Reprinted/adapted from Molecular crystals and liquid crystals, vol. 556Mileva D, Androsch R, Zhuravlev E, Schick C, Wunderlich B. Formation andreorganization of the mesophase of isotactic polypropylene. p. 74e83[Copyright with permission from Taylor & Francis Ltd]; 2012.
[40] Mileva D, Androsch R, Zhuravlev E, Schick C, Wunderlich B. Polymer 2011;51:1107e15.
[41] Advanced thermal analysis system ATHAS; Available from: http://athas.prz.rzeszow.pl.
[42] Cavallo D, Gardella L, Alfonso GC, Portale G, Balzano L, Androsch R. ColloidPolym Sci 2011;289:1073e9.
[43] Androsch R, Di Lorenzo ML, Schick C, Wunderlich B. Polymer 2010;51:4639e62.
[44] Binsbergen FL, De Lange BGM. Polymer 1968;9:23e40.[45] Norton DR, Keller A. Polymer 1985;26:704e16.[46] Olley RH, Bassett DC. Polymer 1989;30:399e409.[47] Lotz B, Wittmann JC. J Polym Sci Pol Phys 1986;24:1541e58;
Lotz B, Wittmann JC, Lovinger AJ. Polymer 1996;37:4979e92.