molecular investigation and long-term clinical progress in greek cypriot families with recessive...
TRANSCRIPT

Original Article
Molecular investigation and long-termclinical progress in Greek Cypriot familieswith recessive distal renal tubular acidosisand sensorineural deafness due to mutationsin the ATP6V1B1 gene
Feldman M, Prikis M, Athanasiou Y, Elia A, Pierides A, Deltas CC.Molecular investigation and long-term clinical progress in GreekCypriot families with recessive distal renal tubular acidosis and sensor-ineural deafness due to mutations in the ATP6V1B1 gene.Clin Genet 2006: 69: 135–144. # Blackwell Munksgaard, 2006
The spectrum of distal renal tubular acidosis (dRTA) includes agenetically heterogeneous group of inherited conditions of both auto-somal-dominant and recessive mode of inheritance. The basic defect islinked to the renal part of acid–base homeostasis, which is partlyachieved by the regulated luminal secretion of Hþ at the apical surfaceof the a-intercalated cells of renal collecting ducts. This is coupled tobicarbonate reabsorption with chloride counter transport across thebasolateral membranes Here, we describe the molecular findings of thefirst two Greek Cypriot families with recessive dRTA and the long-termclinical findings in four of five affected members. DNA linkage analysiswith four polymorphic markers flanking the ATP6V1B1 gene on chro-mosome 2 gave evidence for positive linkage; direct DNA analysis byautomated DNA sequencing revealed that patients in one family werehomozygous for mutation 229þ1G>T (IVS7þ1G>T) and that patientsin the second family were compound heterozygous for 229þ1G>T andR157C. The mutations were found on four different haplotypes. Boththe mutations were previously reported in patients of Turkish origin.Three known polymorphic variants were also identified. The fivepatients demonstrated the whole clinical spectrum of the diseaseincluding death in infancy, failure to thrive, rickets, nephrocalcinosis,nephrolithiasis, and episodes of hypokalemic paralysis. Some of thefamily members are now in their mid 30s and late 20s, and nephro-lithiasis with recurrent renal colics is their main problem. Renal func-tion has remained normal. In conclusion, early diagnosis in infancy andprompt treatment with alkali and potassium supplements is of greatbenefit to the patient with dRTA and ensures normal growth. Theidentification of responsible mutations facilitates antenatal or postnataldiagnosis in concerned families and improves the prognosis.
M Feldmana*, M Prikisb*,
Y Athanasioub, A Eliac,A Pieridesb and CC Deltasa,d
aThe Cyprus Institute of Neurology andGenetics, bDepartment of Nephrology,Nicosia General Hospital, cDepartment ofPediatric Nephrology, ArchbishopMakarios III Hospital, dDepartment ofBiological Sciences, University of Cyprus,Nicosia, Cyprus
Key words: ATP6V1B1 gene – Cyprus –distal renal tubular acidosis (dRTA) –founder effect – Greek Cypriot families –haplotypes
Corresponding author: C. ConstantinouDeltas, Professor, Department ofBiological Sciences, Faculty of Pure andApplied Sciences, University of Cyprus,Kallipoleos 75, 1678 Nicosia, Cyprus.Tel.: þ357 22 892880;fax: þ357 22 892881;e-mail: [email protected]
Received 31 August 2005, revised andaccepted for publication 3 November 2005
Normal cellular function as well as growth anddevelopment depend on strict acid–base home-ostasis which is achieved through precise lung
alveolar and renal tubular function. Bicarbonateis continuously ultrafiltered at the renal glomer-uli but reabsorbed at both the proximal (approxi-mately 90%) and the distal renal tubules(approximately 10%), while at the same time, a*These authors contributed equally to this work.
Clin Genet 2006: 69: 135–144 Copyright # Blackwell Munksgaard 2006
Printed in Singapore. All rights reservedCLINICALGENETICS
doi: 10.1111/j.1399-0004.2005.00559.x
135

daily net urinary Hþ secretion of approximately70 mmol, produced as non-volatile acid fromnormal daily catabolism, is also carried out.This net Hþ excretion is mediated largely viavacuolar-type Hþ-ATPases located in the apicalmembranes of the dynamic, type a-intercalatedcells of the collecting duct, responsible for acidsecretion (1, 2). This apical, luminal Hþ excretionis normally linked to bicarbonate reabsorptionwith chloride counter transport across the oppo-site basolateral membranes of these cells, whilethe HCO3
– is generated by cytosolic carbonicanhydrase II. Failure of any of these physiologi-cal mechanisms leads to distal renal tubularacidosis (dRTA) (1–5).Recent genetic advances have provided the
explanations for the various types of recessiveand dominant forms of dRTA (1–7). RecessivedRTA is a serious disease often but not alwaysassociated with early onset sensorineural deaf-ness, which unless recognized and treatedpromptly, will lead to lack of speech. Mostfamilies with recessive dRTA and sensorineuraldeafness are the result of mutations in theATP6V1B1 gene on chromosome 2p13, whichcodes for the B1 vacuolar subunit of the Hþ-ATPase pump which is located on the apicalmembrane of the type a-intercalated cells of thecollecting duct (1–5) and also in some specializedcells of the inner ear (8–10). This form of dRTAis a hypokalemic, hyperchloremic metabolicacidosis that unless treated promptly leads invari-ably to growth failure, possibly rickets, and alsonephrocalcinosis and extensive renal stone for-mation. Renal function appears to remain nor-mal for several decades. A small number of suchfamilies with recessive dRTA and a Hþ-ATPasedefect do not link to the ATP6V1B1 gene andmay exhibit no sensorineural deafness. Somesuch families link to a different gene, theATP6V0A4, on chromosome 7q33-34, whichencodes for the a4 subunit of the multimericHþ-ATPase pump (8–10).The dominant form of dRTA is a milder dis-
ease (5, 7, 11–13). It often develops in adulthoodand is not accompanied by deafness. It is causedby mutations in the anion exchanger-1 (AE1)
gene located on chromosome 17q21-q22 encod-ing the HCO3–/Cl– counter exchanger at thebasolateral membranes of the same acid secretinga-intercalated cells. In Thailand and adjacentareas, a recessive variety of the AE1 gene is alsodescribed (5, 7, 13). Both patterns of inheritance,recessive and dominant, include hypokalemiabecause of renal potassium wasting and nephro-calcinosis with renal stone formation because of alow urinary citrate excretion and hypercalciuria.A fourth type of dRTA associated with osteo-
petrosis, cerebral calcification and conductive-type deafness, has been linked to mutations ofthe cytosolic carbonic anhydrase II gene on chro-mosome 8q22. This is also inherited as a recessivetrait (14). Table 1 summarizes the genetics ofdRTA.A careful search in the literature shows a pau-
city of data regarding the long-term outcome ofchildren with recessive dRTA and sensorineuraldeafness in adult life (15, 16). In particular, thelong-term effect of the disease on glomerularfunction remains uncertain.In this work, we present molecular data as well
as long-term clinical findings in four patientswith dRTA and sensorineural deafness followedover 20–35 years. The patients belong to twoGreek Cypriot families that to our knowledgerepresent the first to be described on the islandCyprus.
Patients and long-term clinical progress
Two families with confirmed dRTA and sensor-ineural deafness are described.
Family 8051
In this family, there are three children, twoaffected and one normal, from two healthy, unre-lated parents (Fig. 1). Patient II-1, a girl, wasborn on 4 November 1971. At 3 months old,she was admitted to the pediatric departmentwith failure to thrive and was prescribedAlbright’s solution (alkali and potassium). Atage 2, she could not walk and was unable to
Table 1. Genetics of distal renal tubular acidosis (dRTA)
Gene localization Locus symbol Gene product
Primary dRTA (type 1)Autosomal dominant and recessive (S.E. Asia) 17q21-22 SLC4A1 KAE1Autosomal recessive with deafness 2p13 ATP6V1B1 B1 subunit of Hþ-ATPaseAutosomal recessive (variable hearing status) 7q33-34 ATP6V0A4 a4 subunit of Hþ-ATPase
Mixed, proximal, and distal (type 3)Autosomal recessive with osteopetrosis 8q22 CA2 Carbonic anhydrase
Feldman et al.
136
speak. No bone X-rays at that age are available.She grew on an erratic schedule of alkali supple-ments and was first seen in our adult nephrologyclinic in 1986, at age 15. She was deaf and mute.Her height was 146 cm and her weight 39 kg,both below the 5th percentiles. Blood pressure100/50 mmHg, serum creat: 0.72 mg/dl, urea:24 mg/dl, Na: 140 mmol/l, K: 3.5 mmol/l, Cl:108 mmol/l, bicarbonate: 17.0 mmol/l, urinepH 7.5. Thyroid function was normal. Kidney,ureter, and bladder X-rays (KUB) and a renalUS showed extensive nephrocalcinosis andnephrolithiasis. She was encouraged to take90 mmol of alkali with 90 mmol of potassiumas supplements daily, and she remained well for9 years until March 1995, when at age 24, shepresented with an episode of right renal colicbecause of a ureteric stone. She was treated with
extra corporeal lithotripsy. A few weeks later, shecomplained of painful muscle weakness of thearms and legs associated with hypokalemia(3.3 mmol/l), which was corrected with goodresults. In July 1995, she had another episode ofrenal colic because of a left ureteric stone thatpassed spontaneously. There were more episodesof renal colics in June 1997, April 1999, andJanuary 2000. In May 2001, she developed amajor left ureteric obstruction with several bigstones in the left ureter, giving rise to a stein-strasse appearance (Fig. 2). Two ureteric litho-tripsies within 4 weeks cleared all the stonematerial. This prolonged episode of left uretericobstruction associated with nausea and vomitingand failure to take her alkali and potassium sup-plements was complicated by further episodes ofpainful hypokalemic paralysis (K ¼ 2.5 mmol/l)
Family 8051(a)
(b)
2645
2245
5 464
61
3 5
1 2 3
D2S441D2S292D2S1394D2S286
ATP6V1B1
1 2
I
II
2 cM3 cM3 cM
5 26 24 43 5
4 26 61 45 5
4 26 61 45 5
Family 8052
1 2
1 2 3 4 5
2 54 63 45 1
622
65
45 5
D2S441D2S292D2S1394 D2S286
ATP6V1B12 cM3 cM3 cM
I
II
6 52 62 45 1
6 22 42 35 5
6 22 42 35 5
6 52 62 45 1
Fig. 1. Pedigrees of the two familiesstudied here along with the results ofDNA linkage analysis using the fourpolymorphic CA repeat markersshown on the left. Shown in (a) isalso the relative genetic position ofthe ATP6V1B1 gene and the geneticdistances between the markers. Thehaplotypes of individual I-2 in (a) hasbeen deduced (in brackets). Filledblack circles and squares representaffected females and males, respec-tively, whereas half-filled symbolsrepresent healthy carriers. Diagonallines above symbols indicate deceasedpersons.
ATP6V1B1 gene mutations in Cypriot dRTA families
137

requiring IV and oral potassium therapy. Sheremained well up to July 2005 when she presentedwith another episode of renal colic because ofseveral stones in the left lower ureter that clearedafter another session of lithotripsy. At age 34, shehas remained short below the 5th percentile.Kidney function remains essentially normal witha serum creatinine at 0.97 mg/dl and no hyper-calciuria at this stage, Ca/Cr ¼ 0.07 and 0.11(normal Ca/Cr < 0.24).Subject II-2 is a healthy lady in every respect.
She has developed normally without any hearingor speech problems and has had no renal calculior nephrocalcinosis.Patient II-3 was born on 19 March 1979. She
was first seen in our clinic in 1987 at age 8. Sheshowed nephrocalcinosis and nephrolithiasisdespite being prescribed alkali and potassiumsupplements. She was deaf and mute. Her weightwas 16 kg, below the 5th percentile, and herheight 119 cm, below the 10th percentile. Shewas treated systematically with 90 mmol ofpotassium and 90 mmol bicarbonate supple-ments daily, and gradually over the next 3years, she reached the 75th percentile for heightand weight. In October 1993, she had an unevent-ful tonsillectomy but a month later, presumablybecause she had stopped taking her alkali andpotassium supplements, she had an episode ofhypokalemic painful muscle paralysis with lowserum potassium at 2.8 and 3.0 mmol/l andHCO3
– 17.3 and 17 mmol/l. The acidosis andhypokalemia were corrected with promptimprovement of the paralysis. In September1995, she had an episode of right renal colic andpassed a kidney stone. At age 16.5, she migratedto the United Kingdom where she later married anon-syndromic deaf and dumb person of GreekCypriot origin, without dRTA or renal stones.
She has since given birth to two normal childrenwho are developing normally. In August 2004,she had another episode of left renal colic withspontaneous passage of stones. At age 25, shehad normal kidney function with a serum creati-nine of 0.8 mg/dl.
Family 8052
There are two healthy unrelated parents with fivechildren (Fig. 1). The first child, a boy, was bornon 27 February 1974 and died undiagnosed 6months later with failure to thrive and markedvomiting. Two boys, born in 1975 and 1980, havedeveloped normally and have no deafness, dRTAor renal stones. Two other sisters (patients II-3and II-5), born in 1976 and 1983, respectively,have sensorineural deafness, dRTA, renal stonesand nephrocalcinosis.Patient II-3 was born on 27 June 1976. She was
diagnosed with hypokalemic, distal, renal tubularacidosis at 6 months of age and received treat-ment with alkali and potassium supplements. In1983, at age 7, a radiological diagnosis of ricketswith genu valgus was made, and she was treatedeffectively with alkali and vitamin 1a-OH-chole-calciferol (Fig. 3a,b). She improved quickly. Shealso developed renal stones with nephrocalcino-sis, and in November 1983, she had a left renalstaghorn calculus removed surgically. She wasfirst seen in our adult nephrology clinic, a yearlater, in 1984 at age 8. Her height was 112 cm(below the 5th percentile) and weight 24 kg(below the 50th percentile). Her hearing wasmildly impaired, and she had problems with herspeech. She had a genu valgus deformity. Herrenal X-rays confirmed nephrocalcinosis andlithiasis. By June 1987, her left renal stag-horn had grown again, and this was treatedsuccessfully with extra corporeal lithotripsy. Herbilateral nephrocalcinosis and nephrolithiasisgradually increased over the next decade, andby the year 2004, a big right renal staghorn wasnoted. This was removed percutaneously. She isnow well on 90 mmol of alkali and potassiumsupplements daily, but she admits to takingthem erratically. At 29 years of age, she appearsto lead a normal life. Her latest laboratory resultsshowed a metabolic acidosis, HCO3
– 15.3 mmol/lwith an alkaline urine pH 7.27 and hypercal-ciuria, Ca/Cr ¼ 0.31 (normal <0.24). Renalfunction remains normal with a serum creatinineof 0.8 mg/dl.Patient II-5 was born on 16 November 1983.
She was treated systematically from birth withalkali and potassium supplements, and this has
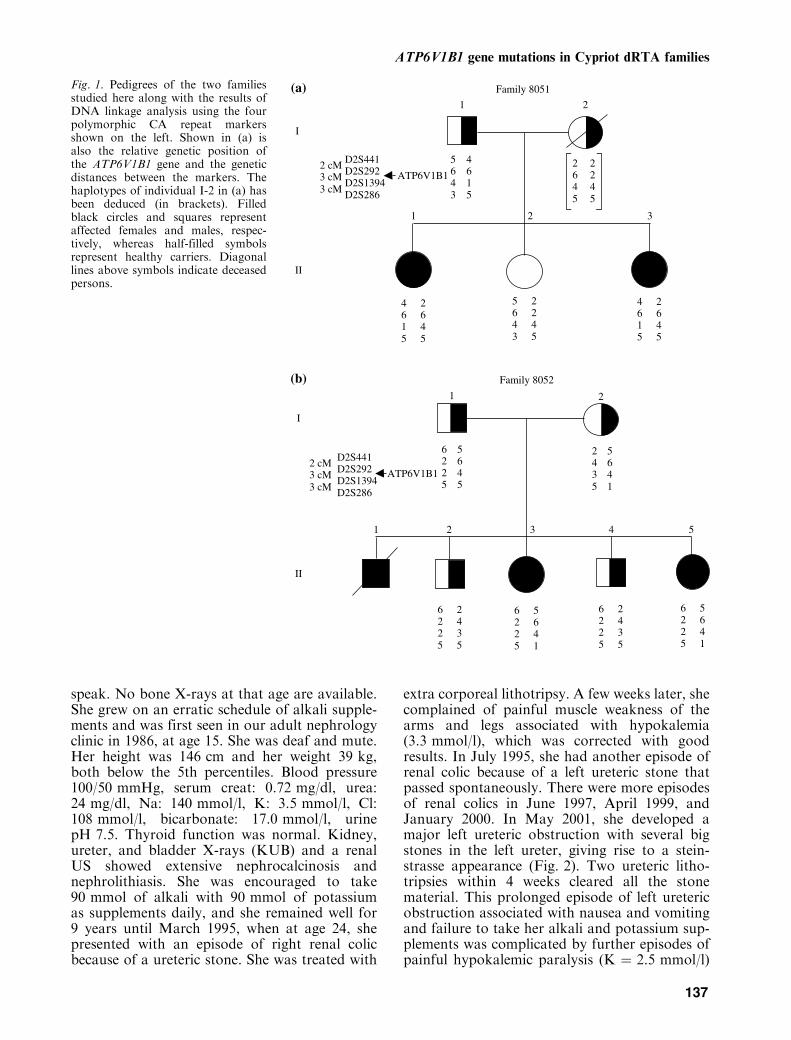
Fig. 2. Family 8051, patient II-1. May 2001, KUB showingextensive nephrocalcinosis and nephrolithiasis and a largenumber of stones in the left ureter, creating a steinstrasse.
Feldman et al.
138
resulted in normal growth along the 50th percen-tile for both height and weight. The treatment didnot prevent her sensorineural deafness, and shehas remained mute. At age 2 years and 2 months,her KUB was normal, but by age 7, she had devel-oped moderate nephrocalcinosis with nephro-lithiasis that have remained essentially unchangeduntil the present time. She is currently 22 years old,
and so far, she has had no episodes of renal colic.Her latest laboratory results showed a metabolicacidosis, HCO3
– 15.3 mmol/l with alkaline urinepH ¼ 7.4 and normal renal function, serum creat-inine, 0.8 mg/dl.
DNA extraction and genetic linkage analysis
Genomic DNA was prepared from peripheralblood leukocytes by the method of Miller et al.(17). A set of four polymorphic markers that flankthe ATP6V1B1 locus on chromosome 2 was usedfor genetic linkage analysis. Markers D2S441 andD2S1394 are tetranucleotide repeats, whereasD2S292 and D2S286 are dinucleotide repeats.The markers and their relative chromosomallocation are shown schematically in Fig. 1. Forlinkage analysis studies, one of the two polymer-ase chain reaction (PCR) amplification primerswas radioactively end-labeled with 33P-g-ATP(Amersham, Vienna, Austria) and T4 polynucleo-tide Kinase (New England Biolabs, Ipswich, MA,USA). The PCR products were fractionated on6% denaturing polyacrylamide gels and visua-lized through autoradiography, after overnightor longer exposure, if needed.
Mutation screening
The DNA sequences of the 14 coding exons ofthe ATP6V1B1 gene and the exon–intron bound-aries were PCR-amplified using as templategenomic DNA from affected and healthy familymembers. Oligonucleotide primer sequences aresummarized in Table 2. The PCR products weresubsequently used as substrates for direct cycleDNA sequencing using a kit from Beckman
(a)
(b)
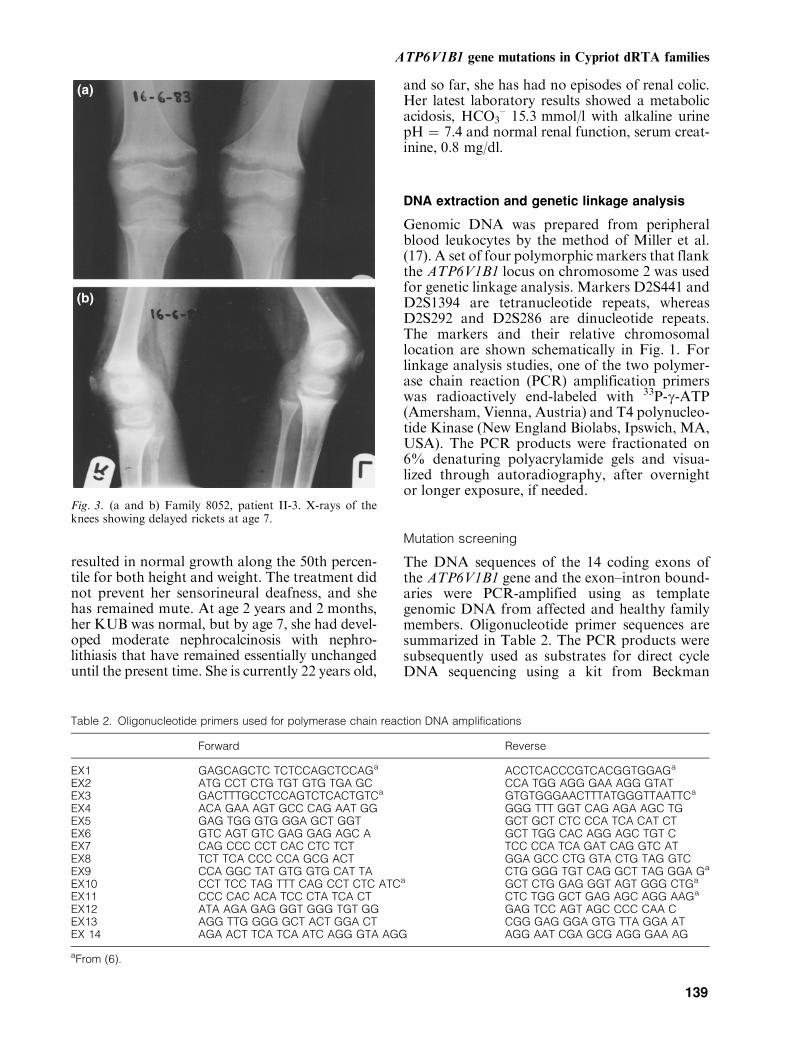
Fig. 3. (a and b) Family 8052, patient II-3. X-rays of theknees showing delayed rickets at age 7.
Table 2. Oligonucleotide primers used for polymerase chain reaction DNA amplifications
Forward Reverse
EX1 GAGCAGCTC TCTCCAGCTCCAGa ACCTCACCCGTCACGGTGGAGa
EX2 ATG CCT CTG TGT GTG TGA GC CCA TGG AGG GAA AGG GTATEX3 GACTTTGCCTCCAGTCTCACTGTCa GTGTGGGAACTTTATGGGTTAATTCa
EX4 ACA GAA AGT GCC CAG AAT GG GGG TTT GGT CAG AGA AGC TGEX5 GAG TGG GTG GGA GCT GGT GCT GCT CTC CCA TCA CAT CTEX6 GTC AGT GTC GAG GAG AGC A GCT TGG CAC AGG AGC TGT CEX7 CAG CCC CCT CAC CTC TCT TCC CCA TCA GAT CAG GTC ATEX8 TCT TCA CCC CCA GCG ACT GGA GCC CTG GTA CTG TAG GTCEX9 CCA GGC TAT GTG GTG CAT TA CTG GGG TGT CAG GCT TAG GGA Ga
EX10 CCT TCC TAG TTT CAG CCT CTC ATCa GCT CTG GAG GGT AGT GGG CTGa
EX11 CCC CAC ACA TCC CTA TCA CT CTC TGG GCT GAG AGC AGG AAGa
EX12 ATA AGA GAG GGT GGG TGT GG GAG TCC AGT AGC CCC CAA CEX13 AGG TTG GGG GCT ACT GGA CT CGG GAG GGA GTG TTA GGA ATEX 14 AGA ACT TCA TCA ATC AGG GTA AGG AGG AAT CGA GCG AGG GAA AG
aFrom (6).
ATP6V1B1 gene mutations in Cypriot dRTA families
139

Coulter and manufacturer’s instructions (CEQDTCS-Quick START Kit, Fullerton, CA).Automated DNA sequencing was performed onCEQ 2000 XL DNA analysis system (BeckmanCoulter). Sequencing was invariably carried outon DNA strands with both forward and reversePCR primers. To confirm the mutations detectedby sequencing, restriction analysis was performedaccording to manufacturer’s instructions, as onemutation abolished and the other created a novelrestriction recognition site for enzymes Hph1 andHpyCH4V, respectively.
Results
Molecular genetic studies
According to our records and the reportsobtained from family members, the two familiesare not related nor was there any known consan-guinity between spouses. Owing to the geneticheterogeneity observed with dRTA, we firstattempted genetic linkage analysis to identifythe gene at fault. The data with four polymorphicmarkers were consistent with linkage to theATP6V1B1 locus on chromosome 2, for boththe families (Fig. 1). On the basis of the putativeaffected haplotypes, we assumed that there wouldbe four mutations, associated with four differenthaplotypes. Genomic DNA sequencing revealedonly two mutations. In family 8051, the patientswere homozygous for a G>T transversion at theconserved splicing donor site of intron 7(229þ1G>T or IVS7þ1G>T) (Fig. 4). Thepatients in the second family were compoundheterozygotes, carrying the one mutation thatwas found in family 8051 and another mutationin exon 6 converting arginine to cysteine at posi-tion 157, R157C, as a result of a C to T transition(Fig. 5). Mutation IVS7þ1G>T abolished thesequence recognition site for restriction enzymeHph1 and mutation R157C created a new recog-nition site for restriction enzyme HpyCH4V(Figs 4 and 5). Both the mutations were subse-quently tested by restriction enzyme analysis on35 (IVS7þ1G>T) or 50 (R157C) unrelated DNAsamples from healthy subjects. None carriedeither of the mutations, thus providing geneticevidence that they are pathogenic. We shouldmention that both the mutations, IVS7þ1G>Tand R157C, were previously described by otherresearchers (6, 10).In addition to the disease causing mutations,
our genomic sequencing revealed the presence oftwo previously reported neutral polymorphismsin exons 2 (C>T, S46S) and 10 (C>T, R334R).
Both the polymorphisms were found in membersof the affected families and in some of the unre-lated samples tested. A missense change in exon 1(C>T, T30I) was detected in members of family8051. This apparently not pathogenic variant wasdetected also in 35% of the unrelated samplestested (10).
Discussion
Clinical features
The earliest clinical reports on this diseaseappeared in the mid-1930s (18, 19) in Londonand more clearly in the 1960s in Paris (20, 21).These and subsequent reports described the clin-ical aspects of familial, recessive dRTA with sen-sorineural deafness that include failure to thriveand vomiting, nephrocalcinosis with nephro-lithiasis, delayed rickets, and hypokalemic muscleparesis (22–26). The turning point for the under-standing of the pathophysiology of this group ofdisorders came in the late 1990s with the out-standing work on molecular genetics by Karetet al. (3, 6) that linked this disease to mutationsin the ATP6V1B1 gene on chromosome 2q13.This gene encodes the B1 subunit of the Hþ-ATPase pump on the luminal surfaces of thea-intercalated cells of the collecting duct and themutated Hþ-ATPase pump restricts Hþ loss inthe urine. This pump is also present in thecochlea and the endolymphatic sac and explainsthe presence of sensorineural deafness in thissyndrome. Subsequent reports confirmed thatATP6V1B1 is responsible for most cases ofdRTA (27, 28).Lack of normal acid–base homeostasis is
always significant, especially if it occurs in thesensitive, formative days of early infancy, anddRTA should be suspected in every infant failingto thrive or to grow normally. In family 8052, thefirst child of the family died undiagnosed at 6months of age, in 1974, while its fifth child,born in 1983, grew normally, after prompt diag-nosis soon after birth and the supplementationwith alkali and potassium. The administration ofalkali and potassium supplements does not, how-ever, appear to prevent deafness. What is import-ant, however, is the early recognition of deafnessin order that appropriate steps are takenpromptly to improve hearing and minimizefuture speech disabilities. In this respect, therecent availability of genetic testing offers thebest way of early diagnosis in suspected families.Rachitis and osteomalacia are also well knowncomplications of metabolic acidosis, and it is not
Feldman et al.
140
surprising that late rickets develop in such chil-dren, if left untreated (15, 23). The bone histolo-gical study by Domrongkitchaiporn et al. (29) in10 patients with untreated dRTA has confirmedthe therapeutic benefit of alkali supplementationin dRTA, while in the presence of rickets, theadditional administration of an active vitamin
D metabolite such as 1a-hydroxycholecalciferolhastens recovery. Nephrocalcinosis and nephro-lithiasis are the result of the untreated metabolicacidosis with low urinary citrate and hypercal-ciuria. Prompt alkali supplementation maycause the nephrocalcinosis to evolve more slowlyand into a milder form (30), but complete
Family 8051
3Homozygous 229 + 1G > T
1Heterozygous 229 + 1G > T
2
I
II
1Homozygous 229 + 1G > T
A T G GG G T T G AA T G GGGG T G A
229 + 1G > TWT
I.1 II.1 II.3 II.2 NR B/k φX
310 bp271–81 bp234 bp194 bp166 bp
148 bp
118 bp
79 bp69 bp 72 bp
79 bp69 bp
166 bp
1 314 bp *
HphI HphI
(a)
(b)
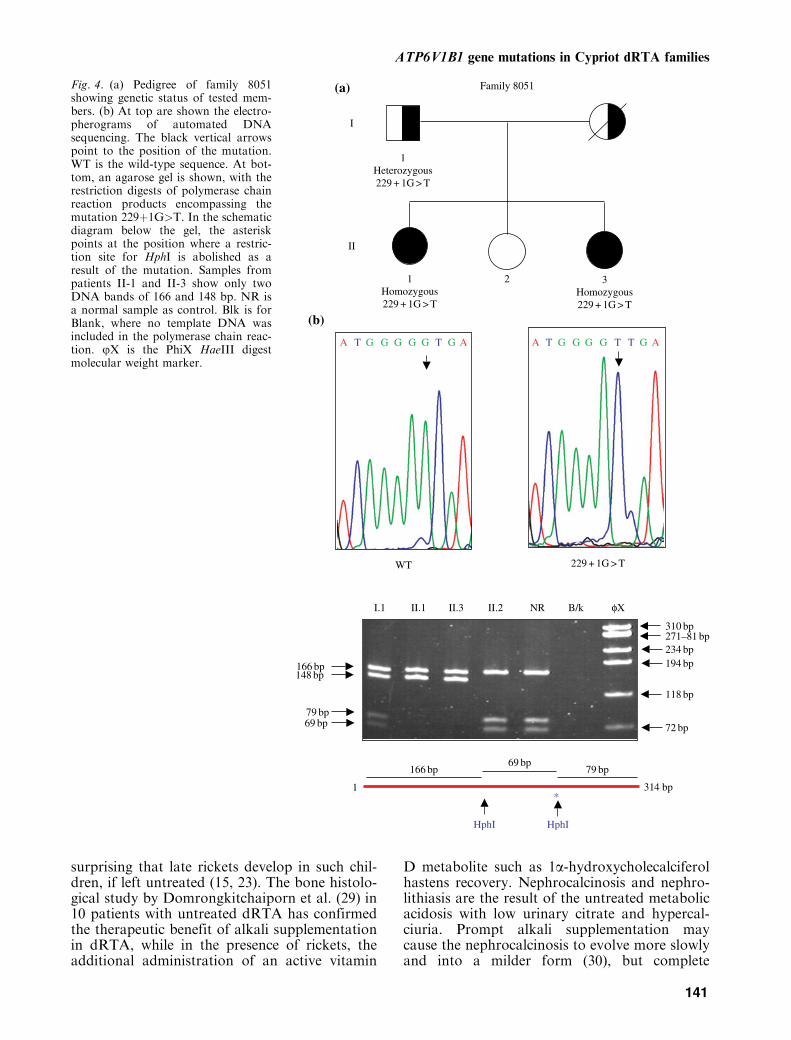
Fig. 4. (a) Pedigree of family 8051showing genetic status of tested mem-bers. (b) At top are shown the electro-pherograms of automated DNAsequencing. The black vertical arrowspoint to the position of the mutation.WT is the wild-type sequence. At bot-tom, an agarose gel is shown, with therestriction digests of polymerase chainreaction products encompassing themutation 229þ1G>T. In the schematicdiagram below the gel, the asteriskpoints at the position where a restric-tion site for HphI is abolished as aresult of the mutation. Samples frompatients II-1 and II-3 show only twoDNA bands of 166 and 148 bp. NR isa normal sample as control. Blk is forBlank, where no template DNA wasincluded in the polymerase chain reac-tion. jX is the PhiX HaeIII digestmolecular weight marker.
ATP6V1B1 gene mutations in Cypriot dRTA families
141
Family 8052
1 2
4321
I
II
HeterozygousR157C
R157C229 + 1G > T
Heterozygous R157C
R157C229 + 1G > T
Heterozygous 229 + 1G > T
Heterozygous R157C
A T G G G G G T G A A T G G G G K T G A
5
φx I.2 II.2 I.1II.3 II.4 II.5 B
φx I.2 II.2I.1 II.3 II.4 II.5 B
166 bp148 bp
79 bp69 bp72 bp
310 bp271–81 bp
234 bp194 bp
118 bp
310 bp271–81 bp
234 bp194 bp
118 bp
314 bp
79 bp69 bp
166 bp
*1
229 + 1G > TWT
C T C C C G C A T C C T C C Y G C A T C
218 bp
125 bp93 bp82 bp
*
300 bp1
82 bp125 bp93 bp
R157CWTHpyCH4V HpyCH4V
HphI HphI
(a)
(b)
(c)
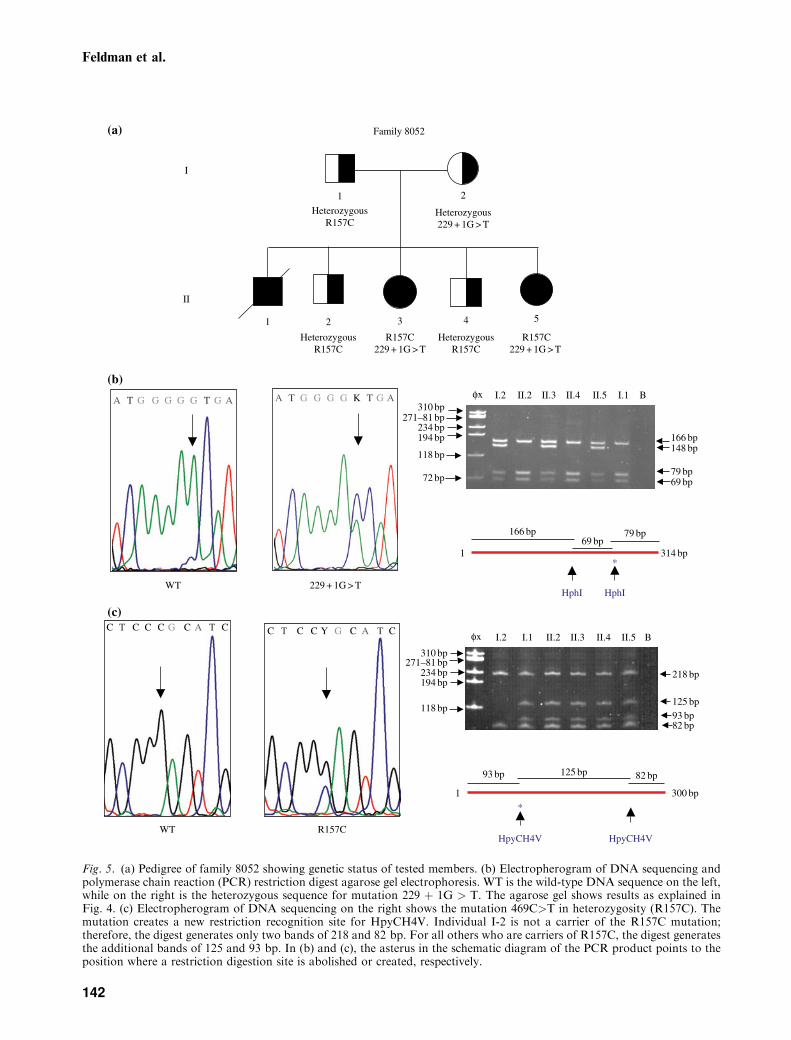
Fig. 5. (a) Pedigree of family 8052 showing genetic status of tested members. (b) Electropherogram of DNA sequencing andpolymerase chain reaction (PCR) restriction digest agarose gel electrophoresis. WT is the wild-type DNA sequence on the left,while on the right is the heterozygous sequence for mutation 229 þ 1G > T. The agarose gel shows results as explained inFig. 4. (c) Electropherogram of DNA sequencing on the right shows the mutation 469C>T in heterozygosity (R157C). Themutation creates a new restriction recognition site for HpyCH4V. Individual I-2 is not a carrier of the R157C mutation;therefore, the digest generates only two bands of 218 and 82 bp. For all others who are carriers of R157C, the digest generatesthe additional bands of 125 and 93 bp. In (b) and (c), the asterus in the schematic diagram of the PCR product points to theposition where a restriction digestion site is abolished or created, respectively.
Feldman et al.
142

prevention appears impossible, and nephrocalci-nosis with nephrolithiasis is invariably present,giving rise to the classical KUB appearance asillustrated by Fig. 2. Although these two compli-cations develop early in life, they seem to causeclinical problems after adolescence, when uretericstones and renal colics are a common clinicalproblem (15, 16, 31–33). Extracorporeal litho-tripsy is effective and has done away with thepreviously repeated ureterolithotomies. What isof clinical significance is that glomerular renalfunction in these patients appears to remain nor-mal, at least until the 4th decade and renal insuf-ficiency does not appear to become a problem.
Molecular studies
The two families reported here are, to our know-ledge, the first identified so far in the Greekpopulation in Cyprus. It is interesting to notethat the majority (32/62) of families reported byKaret et al. (6) were Turkish, one from Cyprus.Similarly all the six families initially used in thestudy by Ruf et al. (27) for linkage analysis werealso Turkish. We find it particularly interestingthat on an island where many times consan-guinity or even distant relationship accounts forcases of other recessive conditions in specificregions or isolates and small communities, ourfindings do not seem to follow this rule. To ourknowledge, the two families are not related norare the spouses in either family, and this is sup-ported by the fact that the mutations are locatedon four different genetic haplotypes. This couldbe explained by the fact that the markers testedare quite apart from each other. But still, withthis at hand and despite the rarity of this disease,the patients in family 8051 were homozygous forthe known mutation IVS7þ1G>T, whereaspatients in the other family carried the samemutation as well. The presence of the same muta-tion on three different haplotypes could beexplained by recurrence, as it might represent amutational hotspot. However, it could also beexplained by very ancient history, which gave ittime to recombine with various other chromo-somes. In support of this, we noticed that allthe three haplotypes that carry mutationIVS7þ1G>T share the same allele of markerD2S292 (allele 6), suggesting a possible foundereffect. But nevertheless this could also have hap-pened by chance. It is also interesting that boththe mutations identified here were previouslyfound in Turkish patients thereby allowing thehypothesis of a distant relationship of ourpatients with people of Turkish descent (6,10).
Also, this would not be unlikely considering theclose contacts of Turks and Greeks on the islandof Cyprus during the past centuries, and the factthat many Greek Cypriots had converted toIslam and joined the Turkish Cypriot communityduring the Ottoman occupation of Cyprus (34).Identification of the exact genetic defects and
the establishment of a quick and easy test byrestriction enzyme analysis permit prompt diag-nosis and genetic counseling of family members.Early diagnosis appears of paramount impor-tance because timely institution of therapyensures normal development and can preventsome complications.
Acknowledgements
This work was partly funded by the Cyprus Kidney Associationthrough a grant to CC Deltas. We wish to thank SabineHerterich for technical assistance with automated sequencing.
References
1. Batlle D, Hrishikesh G, Sheeja J, Mitra A. Hereditary distalrenal tubular acidosis: new understandings. Annu Rev Med2001: 52: 471–484.
2. Alper SL. Genetic diseases of acid-base transporters. AnnuRev Physiol 2002: 64: 899–932.
3. Karet FE. Inherited renal tubular acidosis. J Am SocNephrol 2002: 13: 2178–2184.
4. Stehberger PA, Schulz N, Finberg KE et al. Localizationand regulation of the ATP6V0A4 (a4) vacuolar Hþ-ATPase subunit defective in an inherited form of distalrenal tubular acidosis. J Am Soc Nephrol 2003: 14:3027–3038.
5. Nicoletta JA, Schwartz G. Distal renal tubular acidosis.Curr Opin Pediatr 2004: 16: 194–198.
6. Karet FE, Finberg KE, Nelson RD et al. Mutations in thegene encoding B1 subunit of Hþ-ATPase cause renal tubu-lar acidosis with sensorineural deafness. Nat Genet 1999:21: 84–90.
7. Toye AM. Defective kidney anion-exchanger I (AEI, band 3)trafficking in dominant distal renal tubular acidosis.Biochem Soc Symp 2005: 72: 47–63.
8. Karet FE, Finberg KE, Nayir A et al. Localization of agene for autosomal recessive distal renal tubular acidosiswith normal hearing (rd RTA2) to 7q33-34. Am J HumGenet 1999: 65: 1656–1665.
9. Smith AN, Skaug J, Choate KA et al. Mutations inATP6N1B, encoding a new kidney vacuolar proton pump116-kD subunit, cause recessive distal renal tubular acidosiswith preserved hearing. Nat Genet 2000: 26: 71–75.
10. Stover EH, Borthwick KJ, Bavalia C et al. NovelATP6V1B1 and ATP6V0A4 mutations in autosomal reces-sive distal renal tubular acidosis with new evidence forhearing loss. J Med Genet 2002: 39: 796–803.
11. Bruce LJ, Cope DL, Jones GK et al. Familial distal renaltubular acidosis is associated with mutations in the red cellanion exchanger (Band 3, AE1) gene. J Clin Invest 1997:100: 1693–1707.
ATP6V1B1 gene mutations in Cypriot dRTA families
143

12. Karet FE, Gainza FJ, Gyory AZ et al. Mutations in thechloride-bicarbonate exchanger gene AE1 cause autosomaldominant but not autosomal recessive distal tubular acid-osis. Proc Natl Acad Sci USA 1998: 95: 6337–6342.
13. Sritippayawan SS, Sumboonnanonda A, Vasuvattakul Set al. Novel compound heterozygous SLC4A1 mutationsin Thai patients with autosomal recessive distal renal tubu-lar acidosis. Am J Kidney Dis 2004: 44: 64–70.
14. Sly WS, Whyte MP, Sundaram V et al. CAII deficiency in12 families with the autosomal recessive syndrome of osteo-petrosis with RTA and cerebral calcification. N Engl J Med1985: 313: 139–145.
15. Peces R. Long-term follow-up in distal renal tubular acidosiswith sensorineural deafness. Pediatr Nephrol 2000: 15: 63–65.
16. Van Savage JG, Fried FA. Bilateral spontaneous stein-strasse and nephrocalcinosis associated with distal renaltubular acidosis. J Urol 1993: 150: 467–468.
17. Miller SA, Dykes DD, Polesky HF. A simple salting outprocedure for extracting DNA from human nucleated cells.Nucleic Acids Res 1988: 16: 1215.
18. Lightwood R. Communication no 1. Arch Dis Child 1935:10: 205.
19. Butler A, Wilson J, Farber S. Dehydration andacidosis with calcification at renal tubules. J Pediatr 1936:8: 489.
20. Royer P, Broyer M. L’acidose renale au cours des tubulo-pathies congenitales. In: Actualites Nephrologiques del’Hopital Necker (Hamburger J, Crosnier J, Funck-Bretano JL et al., eds.). Paris: Flammarion, 1967, 73.
21. Royer P, Broyer M, Nordman Y. Enzymatic anomalies andhuman nephropathies. Pathol Biol 1967: 15: 803–809.
22. Cohen T, Brand-Auraban A, Karshai C et al. Familialinfantile renal tubular acidosis and congenital nerve deaf-ness: an autosomal recessive syndrome. Clin Genet 1973: 4:275–278.
23. Donckerwolcke RA, van Biervliet JP, Koorevaar G et al.The syndrome of renal tubular acidosis with nerve deafness.Acta Paediatr Scand 1976: 65: 100–104.
24. Anai T, Yamamoto J, Matsuda I et al. Siblings with renaltubular acidosis and nerve deafness: the first family inJapan. Hum Genet 1984: 66: 282–285.
25. Bentur L, Alon U, Mandel H, Pery M, Berant M.Familial distal renal tubular acidosis with sensorineuraldeafness: early nephrocalcinosis. Am J Nephrol 1989: 9:470–474.
26. Stoll C, Gentine A, Geisert J. Siblings with congenital renaltubular acidosis and nerve deafness. Clin Genet 1996: 50:235–239.
27. Ruf R, Rensing C, Topaloglu R et al. Confirmation of theATP6B1 gene as responsible for distal renal tubular acido-sis. Pediatr Nephrol 2003: 18: 105–109.
28. Hyewon H, Kang HG, Ha IS, Cheong HI, Choi Y.ATP6B1 gene mutations associated with distal renal tubu-lar acidosis and deafness in a child. Am J Kidney Dis 2003:41: 238–243.
29. Domrongkitchaiporn S, Pongskul C, Sirikulchayanonta Vet al. Bone histology and bone mineral density after correc-tion of acidosis in distal renal tubular acidosis. Kidney Int2002: 62: 2160–2166.
30. Vandenberg CJ, Harrington TM, Bunch TW, Pierides AM.Treatment of renal lithiasis associated with renal tubularacidosis. Proc Eur Dial Transplant Assoc 1983: 20:473–476.
31. Peces R. Steinstrasse due to distal renal tubular acidosiswith sensorineural deafness. Nephrol Dial Transplant 2000:15: 1251–1252.
32. Homayoon K. Spontaneous steinstrasse due to renal tubu-lar acidosis. Br J Urol 1996: 77: 610–611.
33. Fedullo LM, Pollack HM, Banner MP, Amendola MA,Van Arsdalen KN. The development of steinstrasse afterESWL: frequency, natural history and radiologic manage-ment. AJR Am J Roentgenol 1988: 151: 1145–1147.
34. Deltas CC. Inherited diseases and Cyprus reality. A historico-genetic approach. In: Katsiaounis R, Koureas N, Louis K,eds. Annals of the Cyprus Research Center (Epetirida), Vol.XXX. 2004, 450–480 (Greek).
Feldman et al.
144