minocycline in phenotypic models of huntington's disease
TRANSCRIPT

www.elsevier.com/locate/ynbdi
Neurobiology of Disease 18 (2005) 206–217
Minocycline in phenotypic models of Huntington’s disease
Kadiombo Bantubungi,a Carine Jacquard,b Anita Greco,c Annita Pintor,d Abdelwahed Chtarto,e,f
Khalid Tai,e,f Marie-Christine Galas,g Liliane Tenenbaum,e,f Nicole Deglon,h Patrizia Popoli,d
Luisa Minghetti,c Emmanuel Brouillet,b Jacques Brotchi,e,i Marc Levivier,e,i
Serge N. Schiffmann,a and David Blume,f,*
aLaboratory of Neurophysiology, ULB-Erasme, Brussels, BelgiumbURA CEA-CNRS 2210, Service Hospitalier Frederic Joliot, CEA, Orsay, FrancecDepartment of Cell Biology and Neuroscience, Istituto Superiore di Sanita, Roma, ItalydDepartment of Drug Research and Evaluation, Istituto Superiore di Sanita, Roma, ItalyeLaboratory of Experimental Neurosurgery, ULB-Erasme, CP602, Bat C-6/113, 808 Route de Lennik, B-1070
Brussels, BelgiumfIRIBHM, ULB-Erasme, Brussels, BelgiumgINSERM U422, Lille, FrancehDepartment of Medical Research and ImaGene Program, CEA, Orsay, FranceiDepartment of Neurosurgery, ULB-Erasme, Brussels, Belgium
Received 20 July 2004; revised 23 September 2004; accepted 30 September 2004
Available online 2 December 2004
Minocycline has been shown to be neuroprotective in various models of
neurodegenerative diseases. However, its potential in Huntington’s
disease (HD) models characterized by calpain-dependent degeneration
and inflammation has not been investigated. Here, we have tested
minocycline in phenotypic models of HD using 3-nitropropionic acid
(3NP) intoxication and quinolinic acid (QA) injections. In the 3NP rat
model, where the development of striatal lesions involves calpain, we
found that minocycline was not protective, although it attenuated the
development of inflammation induced after the onset of striatal
degeneration. The lack of minocycline activity on calpain-dependent
cell death was also confirmed in vitro using primary striatal cells.
Conversely, we found that minocycline reduced lesions and inflamma-
tion induced by QA. In cultured cells, minocycline protected against
mutated huntingtin and staurosporine, stimulations known to promote
caspase-dependent cell death. Altogether, these data suggested that, in
HD, minocycline may counteract the development of caspase-dependent
neurodegeneration, inflammation, but not calpain-dependent neuronal
death.
D 2004 Elsevier Inc. All rights reserved.
Keywords: Huntington’s disease; 3-Nitropropionic acid; Quinolinic acid;
Minocycline; Striatum; Cell death
0969-9961/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.nbd.2004.09.017
* Corresponding author. Laboratory of Experimental Neurosurgery,
Universite Libre de Bruxelles, Campus Erasme, CP602, Bat C-6/113, 808
Route de Lennik, B-1070 Brussels, Belgium. Fax: +32 2 555 46 55.
E-mail address: [email protected] (D. Blum).
Available online on ScienceDirect (www.sciencedirect.com).
Introduction
Huntington’s disease (HD) is a genetic neurodegenerative
disorder characterized by motor and cognitive impairments mainly
due to neuronal degeneration within the striatum and the cerebral
cortex (Blum et al., 2003a; Brouillet et al., 1999). The mutation
involved produces a polyglutamine expansion within the N-
terminal part of the huntingtin protein (The Huntington’s Disease
Collaborative Research Group, 1993), which leads to many
neuronal alterations such as impairments in transcription (Cha,
2000; Zuccato et al., 2003), calcium signaling (Tang et al., 2003),
or axonal transport (Gunawardena et al., 2003; Szebenyi et al.,
2003) and promotes mitochondrial complex II dysfunction (Beal,
2000), mitochondrial Ca2+ defects (Panov et al., 2002), NMDA
receptor sensitization (Song et al., 2003; Zeron et al., 2002), as well
as proapoptotic and pronecrotic protease activation (Wellington et
al., 2003). However, there is no efficient treatment that slows down
or halts the evolution of HD.
Minocycline is an antibiotic of the tetracycline family that
displays beneficial activity in various models of neurodegenera-
tion (Parkinson disease, amyotrophic lateral sclerosis, spinal cord
injury, ischemia) (Brundula et al., 2002; Kriz et al., 2002; Lee et
al., 2003; Stirling et al., 2004; Tomas-Camardiel et al., 2004; Wu
et al., 2002; Yrjanheikki et al., 1999; see also Blum et al., 2004,
for review), although recent studies pointed out that it may also
be detrimental (Diguet et al., 2004a; Tsuji et al., 2004; Yang et
al., 2003; see also for review Blum et al., 2004; Diguet et al.,
2004b). Beneficial activity of minocycline has been related to its
ability to inhibit both mitochondrial mechanisms leading to cell
death and inflammatory processes (Blum et al., 2004; Scarabelli

K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217 207
et al., 2004; Tikka et al., 2002; Wang et al., 2003, 2004; Zhu et
al., 2002).
Minocycline is being tested in HD patients (http://www.
huntington-study-group.org), and preliminary observations are en-
couraging (Bonelli et al., 2003; Thomas et al., 2004). However, the
potential of minocycline for treating HD is far from clear (for a
review, see Blum et al., 2004). While a former study (Chen et al.,
2000) demonstrated that i.p. injections of minocycline improve
motor alterations in the R6/2 transgenic mouse model of HD—that
is characterized by very limited neuronal loss even at very late
stages (Turmaine et al., 2000)—Smith et al. (2003) failed to
reproduce this effect in the same strain following continuous oral
administration. Additionally, although the inhibitory effect of
minocycline on caspase activation has been well described (Wang
et al., 2003), it is not known whether the antibiotic would be
effective against striatal degeneration involving brain calpains.
Indeed, increased calpain activity has been reported not only in the
striatum of HD patients, but also in transgenic and nontransgenic
rodent models of the disease (Bizat et al., 2003a; Gafni and Ellerby,
2002; Gafni et al., 2004). In the same way, the ability of
minocycline to modulate the development of striatal inflammation,
another important pathological feature of HD (Sapp et al., 2001), is
ill-defined. Hence, to further analyze the neuroprotective potential
of minocycline for HD, we tested whether it could attenuate striatal
neurodegeneration in phenotypic models of HD based on complex
II inhibition (Beal et al., 1993) or NMDA receptor overactivation
(excitotoxicity; Beal et al., 1986), both known to mimic numerous
behavioral, histological, neurochemical, and biochemical events
seen in HD patients (Brouillet et al., 1999).
Material and methods
Animals
We used adult male Lewis rats, 12 weeks of age, for the 3-
nitropropionic (3NP) model and adult Wistar rats (250 g) for the
quinolinic acid (QA) model and microdialysis experiments.
Animals were housed three per cage and maintained in a
temperature- and humidity-controlled room on a 12-h light/dark
cycle with food and water ad libitum. The number of animals was
kept to a minimum, and all efforts to avoid animal suffering were
made in accordance with the standards of the Institutional Ethical
Committees.
Treatments
3NP (Fluka, Belgium) was dissolved in 0.1 M PBS, pH 7.4,
adjusted to pH 7.3–7.4 with 5 N NaOH and quinolinic acid (Sigma,
Belgium) was dissolved in 2 N NaOH, the pH was adjusted to 7.4,
and the volume completed with PBS (pH 7.4) as previously
described (Bensadoun et al., 2001; Ouary et al., 2000). Minocy-
cline and doxycycline were dissolved in NaCl 0.9%. Fresh
solutions were prepared each day before injections. The volume
injected was adjusted according to the body weight of each rat (200
Al/100 g, i.p).
Chronic 3NP delivery (Fig. 1A)
Chronic treatments with 3NP (56 mg/kg/d) using 2mL1 (10 Al/h, 7 days) Alzet minipumps and neurological scoring were
performed as previously described (Blum et al., 2002a,b, 2003b;
Mittoux et al., 2002; Ouary et al., 2000). Rats were anesthetized
with a mixture containing xylazine hydrochloride (Rompun, Bayer;
4.5 mg/kg) and ketamine hydrochloride (Imalgene, Merial; 90 mg/
kg). An incision was made below the base of the neck and a 2mL1
Alzet osmotic minipump (delivering 10 Al/h for 7 days; IFFA
Credo, Belgium) containing 3NP (Fluka) was positioned under the
skin. The final concentration of 3NP in the pump was adjusted to
the weight of the rats on the day of implantation in order to exactly
deliver 56 mg/kg/d. Sham rats and animals treated with pharmaco-
logical compounds alone underwent all the surgical procedures
(without minipump implantation). Thirty-nine rats (sham/vehicle,
n = 5; 3NP/vehicle, n = 8; sham/minocycline 10 mg/kg, n = 5;
3NP/minocycline 10 mg/kg, n = 8; sham/minocycline 50 mg/kg,
n = 5; 3NP/minocycline 50 mg/kg, n = 8) were challenged for the
neuroprotective ability of minocycline. Animals were injected with
vehicle or minocycline just before minipump implantation and
each day until sacrifice. Controls and 3NP-treated animals were
evaluated every day for motor impairments. Briefly, behavioral
abnormalities were determined according to the presence and
severity of motor symptoms consisting of dystonia, gait abnormal-
ities, recumbency and also grasping and the ability to remain on a
small platform for N10 s. The final neurological score was assessed
as described (Mittoux et al., 2002; Ouary et al., 2000; minimum =
0, normal animal; score = 8, animal showing near-death
recumbency). All rats were killed after 5 days of 3NP subcutaneous
infusion (10 h after the last minocycline injection) according to the
known kinetics of striatal lesion occurrence in this model as we
previously reported (Bizat et al., 2003a; Blum et al., 2001, 2002a,b,
2003b; Ouary et al., 2000).
Chronic 3NP delivery and minipump removal (Fig. 1B)
We developed another 3NP protocol in which inflammation
was induced in addition to the striatal lesion (see also the Results
section) and determined the effect of minocycline on its develop-
ment. In this model, minipumps were positioned under the skin as
indicated above and removed under light anesthesia with halothane
5 days after implantation. Then, animals were injected daily with
vehicle or 10 mg/kg minocycline until the sacrifice for 5 days.
Thirty-two rats were used: sham/vehicle, n = 6; 3NP/vehicle, n =
10; sham/minocycline 10 mg/kg, n = 6; 3NP/minocycline 10 mg/
kg, n = 10. All rats were killed 5 days after minipump removal.
Quinolinic acid-induced striatal lesions (Fig. 1C)
Animals were anesthetized with xylazine/ketamine mixture and
received an intrastriatal stereotaxic injection of QA (1 Al; 180 nmol)
using the following coordinates: 1.0 rostral to bregma, 3.5 mm
lateral to midline and 5 mm ventral from the dural surface. The
toxin was injected over 4 min, and the needle was left in place for an
additional 2 min. Eighteen rats were used: QA/vehicle, n = 6; QA/
minocycline 10 mg/kg, n = 6; QA/doxycycline 10 mg/kg, n = 6. All
rats were killed 7 days after surgery.
Tissue postprocessing
All animals were killed by decapitation, and their brains were
quickly removed and frozen in 2-methylbutane cooled by dry ice
(�408C). The tissue was cut at 20-Am thickness on a cryostat
(Leitz), and serial coronal sections were mounted onto poly-l-
lysine and stored at �208C until use. In 3NP-treated rats, since
lesions are bilateral with similar extents (Mittoux et al., 2002;
Brouillet et al., unpublished results), hemispheres were separated at

K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217208
the time of sacrifice. One was used for histology and the other for
prostaglandin E2 (PGE2) dosage. Hematoxylin staining or succi-
nate dehydrogenase (SDH) histochemistry was used to reveal
striatal lesions. The latter was measured every 300 Am along the
anteroposterior axis of the striatum (bregma +1.6 to �0.9 mm), and
the lesional volume was calculated for each animal using
Cavalieri’s method as described (Coggeshall, 1992).
Semiquantitative measurement of succinate dehydrogenase activity
Measurement of SDH activity in control and 3NP-treated rats
was performed as described previously (Brouillet et al., 1998).
Frozen sections mounted on poly-l-lysine-coated slides were air
dried and then incubated for 15 min in 0.1 M PBS (pH 7.4, 0.9%
NaCl) at 378C followed by incubation in 0.3 mM nitroblue
tetrazolium (Sigma), 0.05 M sodium succinate (Sigma), and 0.05
M phosphate buffer, pH 7.6, for 30 min at 378C. Finally, sectionswere rinsed successively for 5 min in cold PBS and deionized
water and dried at room temperature. The image of each section
was acquired, and the quantification was performed as described
previously (Brouillet et al., 1998) using NIH image software.
In situ hybridization
The hybridization technique was adapted from previous reports
(Blum et al., 2002a,b). The sections mounted on RNAse-free poly-
l-lysine-coated slides were fixed in 4% freshly prepared parafor-
maldehyde for 30 min and rinsed in PBS 0.1 M. All sections were
dehydrated and dipped for 3 min in chloroform. After air drying, the
sections were incubated overnight at 428C with 0.35� 106 cpm per
section of 35S-labeled probes diluted in hybridization buffer, which
consisted of 50% formamide, 4� SSC (1� SSC: 0.15 m NaCl,
0.015 M sodium citrate, pH 7.4), 1� Denhardt’s solution (0.02%
each of polyvinylpyrrolidone, bovine serum albumin, Ficoll), 1%
sarcosyl, 0.02 M sodium phosphate at pH 7.4, 10% dextran sulfate,
yeast tRNA at 500 Ag/ml, salmon sperm DNA at 100 Ag/ml, and 60
mM dithiothreitol. Compounds were provided by Sigma. After
hybridization, the sections were rinsed for 4� 15 min in 1� SSC at
558C, dehydrated, and covered with Hyperfilm-gmax film (Amer-
sham, Belgium) for 2 or 3 weeks. The oligonucleotide probes were
synthesized by Eurogentech with the following sequences: GFAP
5V-CAGCTCCCGAAGTTCTGCCTGGTAAACGTCAGCCAG-TTTGGTGGG-3V and CD11b/MAC-1 5V-GACCTGGGGAG-GATCCCATATGGTCACCTTGTTGATCTGG-3V. They were
labeled with a-35S dATP (DuPont-NEN, Belgium) at it the 3V endby terminal DNA deoxynucleotidylexotransferase (Gibco, Bel-
gium) and purified with a G50 column (Pharmacia) according to the
manufacturer’s instructions. Digitalized images with 256 gray
levels were generated from the autoradiograms with the public
domain NIH image 1.61 program (National Institute of Health,
USA), a Power Macintosh G3, and a CCD video camera (Dage-
MTI, IN, USA) with fixed gain and black level as previously
described (Blum et al., 2002a,b). On each section, an averaged
optical density of the background level was subtracted from that of
the measured areas to obtain corrected values.
CD11b immunohistochemistry
Immunohistochemistry was performed on paraffin sections as
we previously described (Blum et al., 2002a). We used a
monoclonal mouse antirat CD11b antibody from Serotec (MCA
275R; France) at 1/100 dilution. For revelation, we used
biotinylated donkey antimouse secondary antibody (1/200 in 1%
normal horse serum; Jackson Immuno Research, USA) and the
ABC method (Vector Laboratories, Belgium) and diaminobenzi-
dine (Dako, Belgium).
Prostaglandin E2 (PGE2) dosage
PGE2 dosage was performed as previously described (Minghetti
et al., 2000). Striata were dissected out from frozen hemispheres of
3NP-treated animals, weighted, and homogenized in 50 mM Tris
buffer (pH 7.5) containing 10 Ag/ml indomethacin (Sigma) and 10
AM butylated hydroxytoluene (Sigma) to avoid ex vivo generation
of prostanoids. Homogenates were vigorously vortexed and
incubated for 5 min on ice, before centrifugation at 14,000 rpm
for 45 min at 48C. The recovery of the extraction procedure,
assessed by adding 5000 cpm of tritium-labeled PGE2 to the
homogenate and measuring the recovered radioactivity, was N70%.
Supernatants were collected and stored at �808C until use. PGE2
content was assessed using a specific high-sensitivity enzyme-
immunoassay (detection limit: 4 pg/ml; Assay Design, Ann Arbor,
MI, USA). Results were expressed as pg PGE2/mg of wet tissue.
Microdialysis experiments
Microdialysis and determination of glutamate concentrations by
high-performance liquid chromatography (HPLC) (electrochemical
detection) were performed as we previously reported (Blum et al.,
2003b; Popoli et al., 2002). Under Equithesin anesthesia (3 ml/kg
i.p.), Wistar rats were placed in a stereotaxic frame and implanted
with a concentric dialysis probe (mod CMA/12, 3-mm length,
Carnegie Medicine, Sweden) into the striatum. Stereotaxic
coordinates were as follows A = + 1; L = +3; V = �6.5 (anterior
striatum) according to the atlas of Paxinos and Watson. Twenty-
four hours later, the probe was perfused at a rate of 2 Al/min with a
Ringer solution (NaCl 147, CaCl2 2.3, and KCl 4.0 mM). After a
washout period of at least 90 min, samples were collected every 5
min into a refrigerated fraction collector (mod CMA/170) and then
frozen until assay. Glutamate outflow was induced by quinolinic
acid (QA) as previously shown (Popoli et al., 2002). Since the
intracerebral injection of QA induces tremors and convulsions in
rodents, these experiments were performed under general anes-
thesia (Equithesin). Results were expressed as percentage changes
of extracellular glutamate levels induced by probe perfusion with
QA (5 mM over 30 min) with respect to basal (predrug) values
(mean of three to four samples collected after the induction of
general anesthesia). Minocycline (10 mg/kg, i.p.) was given daily
for 3 or 7 days with the last injection 20 min before QA perfusion.
At the end of the experiments, each rat was sacrificed with an
overdose of Equithesin, and the brain was cut to verify the probe
location. The glutamate content of all samples was measured by
reverse-phase high-performance liquid chromatography coupled to
a fluorometric detector (Perkin Elmer LC240 at wavelength of 335
nm and emission cutoff filter of 425 nm), using a 15-min gradient
elution program (methanol from 20% to 80% with 50 mM
NaH2PO4 and CH3COONa) and automatic precolumn derivatiza-
tion with o-phthalaldehyde and h-mercaptoethanol. Cysteic acid
was used as internal standard. The concentration of the standard
was linear (r2 = 0.99) between 0.2 to 25 ng/10 Al. Basal glutamate
levels were calculated by comparison of sample peak height with
external standard peak height, both corrected for the internal

Fig. 1. Schematic drawing representing the experimental protocols used for
chronic intoxication with 3NP (protocols A and B), QA-induced lesions
(protocol C), and minocycline treatments.
K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217 209
standard peak height and expressed as ng/10 Al without probe
recovery correction.
Primary striatal cultures
Primary cultures of striatal neurons were obtained from 17- to
18-day-old Wistar rat embryos as described (Blum et al., 2002b,
2003b; Galas et al., 2004). Cells were exposed for 3 days to 100
AM 3NP with or without a daily treatment with 1–50 AMminocycline or doxycycline (Sigma; 10 mM stock in ethanol
50%) in 24-well plates. Cell viability was assessed by MTS assay
(Promega, Belgium) according to the manufacturer’s instructions.
The optical density was measured on a Titertek Multiskan MCC/
340 (ICN Biomedicals, Costa Mesa, CA).
Staurosporine treatment and transfection of HEK 293T cells
The HEK 293T immortalized human embryonic kidney cell line
was purchased from Q-One Biotech (Glasgow, UK). Cells were
cultured in Dulbecco’s modified Eagle medium (DMEM; Life
Technologies) supplemented with 10% FCS (Sigma), penicillin-
streptomycin, l-glutamine, nonessential amino acids, and sodium
pyruvate. Cells were seeded at 100,000 293T cells in 12-well tissue-
culture plates (Nunc). For staurosporine experiments, cells were
treated three times with minocycline (25–100 AM): 12 h after
seeding, the day after, and 2 h before staurosporine treatment (1 AMfrom a 1 mM stock solution in DMSO). MTS viability assay was
performed 24 h later. For huntingtin transfection experiments, we
used SIN-PGK-HD plasmids previously described (de Almeida et
al., 2002) encoding for 171-aa N-terminal fragments of huntingtin
carrying 19 or 82 glutamines. Twenty-four hours after seeding, the
medium was replaced by 500 Al DMEM 1% heated FCS containing
the transfection mixture which consisted DNA (1 Ag), 5 Alpolyethyleneimine (10 mM; 25 kDa, Aldrich, cat n8 4087270),
and 25 Al OptiMEM (Life Technologies) incubated 30 min at room
temperature. Twenty hours later, 500 Al DMEMmedium containing
heated 5% FCS (Sigma) was added to the wells. Cell viability was
analyzed 72 h later. Minocycline (100 AM) treatment was started 12
h after transfection and continued for the 2 following days.
Proteolytic activity assay using fluorogenic substrate for caspase-3
and calpain
Fluorescent assays for proteolytic activity of calpain and
caspase were performed according to previously described
methods (Bizat et al., 2003a). Calpain activity was determined
using N-succinyl-Leu-Tyr-(N-succinyl-LY)-AMC, a substrate pref-
erentially cleaved by A/m calpains. Caspase-3 activity was
determined using the substrate N-acetyl-Asp-Glu-Val-Asp-AFC
(DEVD-AFC) (Biomol). Enzyme activity was calculated using
standard curves of AFC or AMC and expressed as pmol AFC-
AMC released per min/mg of protein. It is noteworthy that
presence of minocycline in protein extracts led to apparent
decrease in calpain and caspase-3 activity. This occurred also with
recombinant caspase-3 and purified A-calpain. We found that this
apparent inhibition was due to the fact that minocycline produced
quenching of the fluorescence of AMC and AFC (concentrations
leading to 50% fluorescence quenching, 300 AM for AMC, and 1
mM for AFC). Thus, protease activity values determined in brain
homogenates from minocycline-treated animals could not reliably
be interpreted and were not included in the present results.
Analysis and statistics
Results were expressed as means F standard error. Depending
on the parameter studied, comparisons among groups were made
using Student t test or one-way analysis of variance (ANOVA)
followed by a LSD Fisher or Kruskal-Wallis post hoc tests using
GraphPad Instat and Statistica softwares.
Results
Minocycline is not neuroprotective against 3NP toxicity in vivo
We determined whether minocycline given daily at 10 and 50
mg/kg could protect striatal neurons from degeneration induced by
the chronic delivery of the complex II inhibitor 3NP in rats
(protocol A, Fig. 1). We first evaluated the evolution of motor
symptoms following 3NP treatment in animals receiving or not
minocycline. As shown on Fig. 2A, motor disabilities were not
significantly improved by minocycline. At the time of sacrifice,
that is, 5 days after the onset of intoxication, all animals displayed
gait abnormalities and dystonia. The neurological scores were
similar in all groups. Accordingly, using hematoxylin staining, we
found that the volume of the striatal lesions was similar between
groups despite a trend to a decrease in the 3NP/minocycline 10 mg/
kg group as compared to the 3NP/vehicle group (Fig. 2B). The lack
of significant effect was further assessed using quantitative
measurement of the activity of the complex II enzyme succinate
dehydrogenase (SDH), which active site is selectively inhibited by
3NP. The treatment with 3NP produces a loss of SDH activity in
the striatum that results from both 3NP-induced inactivation of the
enzyme at the catalytic site and degradation of the enzyme
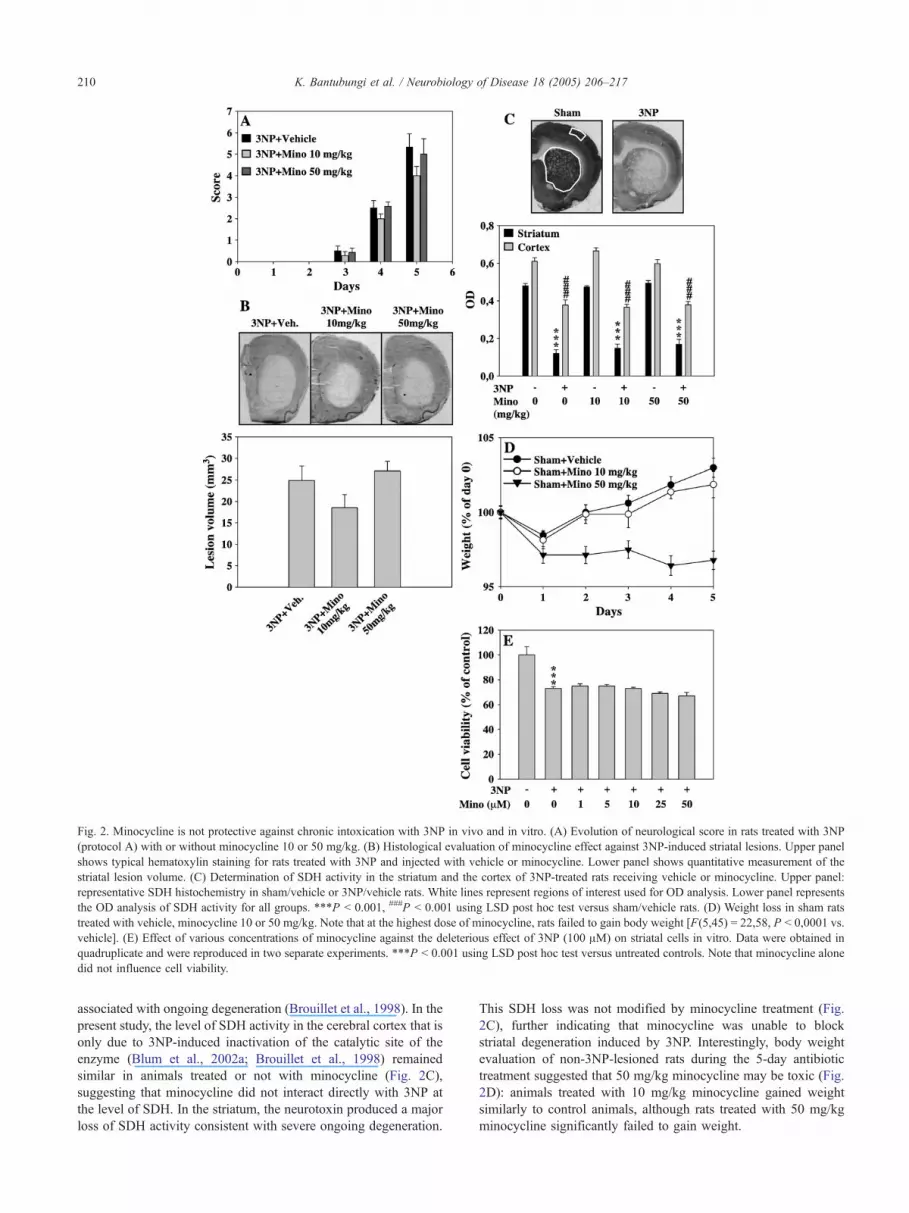
Fig. 2. Minocycline is not protective against chronic intoxication with 3NP in vivo and in vitro. (A) Evolution of neurological score in rats treated with 3NP
(protocol A) with or without minocycline 10 or 50 mg/kg. (B) Histological evaluation of minocycline effect against 3NP-induced striatal lesions. Upper panel
shows typical hematoxylin staining for rats treated with 3NP and injected with vehicle or minocycline. Lower panel shows quantitative measurement of the
striatal lesion volume. (C) Determination of SDH activity in the striatum and the cortex of 3NP-treated rats receiving vehicle or minocycline. Upper panel:
representative SDH histochemistry in sham/vehicle or 3NP/vehicle rats. White lines represent regions of interest used for OD analysis. Lower panel represents
the OD analysis of SDH activity for all groups. ***P b 0.001, ###P b 0.001 using LSD post hoc test versus sham/vehicle rats. (D) Weight loss in sham rats
treated with vehicle, minocycline 10 or 50 mg/kg. Note that at the highest dose of minocycline, rats failed to gain body weight [F(5,45) = 22,58, P b 0,0001 vs.
vehicle]. (E) Effect of various concentrations of minocycline against the deleterious effect of 3NP (100 AM) on striatal cells in vitro. Data were obtained in
quadruplicate and were reproduced in two separate experiments. ***P b 0.001 using LSD post hoc test versus untreated controls. Note that minocycline alone
did not influence cell viability.
K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217210
associated with ongoing degeneration (Brouillet et al., 1998). In the
present study, the level of SDH activity in the cerebral cortex that is
only due to 3NP-induced inactivation of the catalytic site of the
enzyme (Blum et al., 2002a; Brouillet et al., 1998) remained
similar in animals treated or not with minocycline (Fig. 2C),
suggesting that minocycline did not interact directly with 3NP at
the level of SDH. In the striatum, the neurotoxin produced a major
loss of SDH activity consistent with severe ongoing degeneration.
This SDH loss was not modified by minocycline treatment (Fig.
2C), further indicating that minocycline was unable to block
striatal degeneration induced by 3NP. Interestingly, body weight
evaluation of non-3NP-lesioned rats during the 5-day antibiotic
treatment suggested that 50 mg/kg minocycline may be toxic (Fig.
2D): animals treated with 10 mg/kg minocycline gained weight
similarly to control animals, although rats treated with 50 mg/kg
minocycline significantly failed to gain weight.

Fig. 3. Effect of minocycline on staurosporine (STS) and mutated
huntingtin-induced cell death. Viability was determined 24 h after treatment
of HEK293T cells with either STS (1 AM; A; ***P b 0.001 using LSD post
hoc test vs. untreated controls, ###P b 0.001 LSD post hoc test vs. STS-
treated cells) or 72 h following transfection with a plasmid encoding for N-
terminal part of huntingtin carrying 19 or 82 glutamines repetitions (B;
***P b 0.001 using LSD post hoc test vs. controls wells transfected with
the plasmid carrying 19 repetitions). Experiments were performed in
sixplicate for at least four times.
K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217 211
Minocycline is not protective against 3NP in vitro
As shown in Fig. 2E, 3NP significantly decreased neuronal
viability, an effect which was not prevented by 1 to 50 AMminocycline. In line with this, 3NP toxicity was not modified by
minocycline in primary cortical neurons (data not shown). We
previously demonstrated that chronic 3NP intoxication in Lewis
rats and in primary striatal neurons produced calpain-dependent
striatal cell death (Bizat et al., 2003a,b; Galas et al., 2004). In line
with our previous data, we found that on the fourth day of the
3NP treatment, activity of calpain was increased by 2.8-fold as
compared with sham (3NP animals: 91.78 F 4.56; sham animals:
32.25 F 4.67 pmol AMC released/min/mg of proteins; P b
0.0001), although caspase-3 was not activated (not shown). In
vitro, we observed that 48 h after 3NP treatment, calpain activity
was significantly increased by 1.5-fold (3NP-treated neurons:
40 F 1.82; control neurons, 60.22 F 4.12 pmol AMC released/
min/mg of proteins; P b 0.0001). Similarly to in vivo
observations, this effect was not accompanied by caspase-3
activation (not shown). The lack of protection we found with
minocycline against 3NP toxicity in vivo and in vitro thus
suggested that the antibiotic may not be protective against
calpain-dependent degeneration.
However, as previously suggested (Wang et al., 2003; Zhu et
al., 2002), we found that minocycline protected HEK 293T cells
from conditions (staurosporine and expression of an N-terminal
fragment of mutated huntingtin) known to promote caspase-
dependent cell death (Galas et al., 2004; Yu et al., 2003). In the
present studies, we also found that a 6-h staurosporine treatment
led to a significant 1.6-fold increase in caspase-3 activity (control:
30.47 F 0.86; staurosporine: 47.01 F 3.71 pmol AFC released/
min/mg of proteins; P b 0.0016). Calpain activity remained
unchanged during staurosporine treatment (data not shown). In
agreement with this, a 24-h treatment with 1 AM staurosporine
significantly decreased 293T cell viability, an effect dose-depend-
ently inhibited by minocycline (Fig. 3A). At a 100-AMconcentration, the antibiotic produced a complete protection
against staurosporine toxicity. We also found that this optimal
concentration of antibiotic significantly protected 293T cells from
death induced by ectopic expression of an N-terminal fragment of
mutated huntingtin carrying 82 glutamines (Fig. 3B). Similar
findings were observed using doxycycline, another tetracycline
analogue (data not shown).
Effect of minocycline on 3NP-induced striatal inflammation
Previous works have shown that minocycline can promote
neuroprotection through inhibition of inflammation and particu-
larly of microglial activation (Brundula et al., 2002; Tikka et al.,
2002; Tomas-Camardiel et al., 2004; Wu et al., 2002). It was also
recently reported that the antibiotic was able to reduce PGE2
produced by microglial cells (Kim et al., 2004).
We previously reported using MAC-1/CD11b immunohisto-
chemistry that on the fifth day of chronic 3NP treatment, no sign of
striatal inflammation was detected (Blum et al., 2003b). In the
present study, analysis of MAC-1 mRNA expression and PGE2
concentrations, two indices of inflammation, confirmed this
finding: none of these markers increased following 3NP treatment
(Figs. 4A and E), and 3NP treatment rather decreased both
markers, suggesting an inhibitory effect of the toxin on microglial
cells. A similar decrease was observed in rats cotreated with
minocycline (data not shown). In addition, on the fifth day of the
3NP treatment, the increase in GFAP mRNA expression seen
around the lesion and the cortex was not modified by the antibiotic
(Fig. 4B).
Given the lack of microgliosis in our chronic model and the
inhibitory effect exerted by 3NP on microglial cells (Ryu et al.,
2003), we performed an experiment where osmotic minipumps
were removed on the fifth day of 3NP treatment, and animals
were allowed to survive for 5 additional days, thus permitting
striatal inflammation to occur in absence of 3NP. In a pilot
experiment, we previously observed, using CD11b immunohis-
tochemistry, that such a protocol led to microglial activation
within the lesion core (Fig. 4C). We thus performed a trial in
which animals were treated with 10 mg/kg minocycline once
the minipumps were removed (protocol B, Fig. 1). Using such
a protocol, we did not observe a change in the volume of
striatal lesions (22.9 F 4.7 mm3 for the 3NP/vehicle group and
18.8 F 1.9 mm3 for the 3NP/minocycline 10 mg/kg group, ns
using Student t test). However, 5 days after removal of osmotic
pumps, striatal MAC-1 mRNA expression and PGE2 levels
were significantly increased, suggesting inflammation in the
striatum (Figs. 4D and E). In 3NP-treated animals receiving

Fig. 4. Effect of minocycline against inflammation induced by minipump removal following chronic 3NP intoxication. (A) Representative MAC-1 mRNA
expression in the cortex and the striatum of rats chronically treated for 5 days with 3NP (protocol A). Note that 3NP does not induce increase of MAC-1
expression and rather decrease it in the lesion area (star). (B) GFAP mRNA expression in the cortex and the striatum of rats chronically treated for 5 days with
3NP (protocol A). Upper panel: representative in situ hybridization signals. Lower panel: densitometric quantification of the signal within the area of interest,
indicated as a square in the upper panel, in sham or 3NP rats treated with vehicle or minocycline (**P b 0.01 using LSD post hoc test vs. sham/vehicle rats). (C)
CD11b/MAC-1 immunoreactivity in the lateral striatum of a sham rat (left) or a rat submitted to a 5-day 3NP intoxication in which the minipump has been
removed for additional 5 days (pilot experiment; protocol B). Note the increased number and size of positive microglial cells. (D) Quantification of MAC-1
mRNA expression in the striatum of rats chronically treated for 5 days with 3NP in which minipumps have been removed for additional 5 days and then injected
with vehicle or 10 mg/kg minocycline (protocol 1B). *P b 0.05 using LSD post hoc test versus sham/vehicle rats. Note that in the presence of minocycline,
MAC-1 mRNA expression decreased and remained nonsignificantly different from the controls. (E) Quantification of PGE2 levels in the striatum of rats
submitted to protocols A and B. ***P b 0.001 using LSD post hoc test versus sham/vehicle rats, #P b 0.05 using LSD post hoc test versus 3NP/vehicle rats.
K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217212
minocycline, striatal MAC-1 mRNA expression was similar to
control rats, suggesting an inhibitory effect of the antibiotic on
the microglial activation (Fig. 4D). In line with this, minocy-
cline significantly reduced striatal PGE2 levels in the 3NP/
minocycline group as compared to 3NP/vehicle animals (Fig.
4E). Conversely, minocycline did not reduce astrogliosis as

K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217 213
assessed by GFAP mRNA in situ hybridization (data not
shown).
Altogether, our results suggested that minocycline could not
counteract calpain-dependent degeneration but reduced the
development of inflammation in a lesioned striatum. To
Fig. 5. Minocycline is protective against QA-induced striatal lesions in vivo. (A) E
kg minocycline or doxycycline (protocol C). **P b 0.001 using LSD post hoc t
striatum in QA rats treated with 10 mg/kg minocycline or doxycycline. **P b 0.001
treated rats treated with vehicle, minocycline or doxycycline. (D) MAC-1 mR
minocycline or doxycycline. Upper panel: representative in situ hybridization sign
0.001 using LSD post hoc test versus QA/vehicle rats. (E) GFAP mRNA express
Lower panel: quantification of signal shown on the upper panel.
determine whether this property could be associated with
neuroprotection, we tested minocycline in the QA model of
HD in which inflammation is known to occur and contribute to
the development of the lesion (Dihne et al., 2001; Topper et al.,
1993).
valuation of the size of the striatal lesion core of QA rats treated with 10 mg/
est versus QA/vehicle rats. (B) Evaluation of the percentage of the spared
using Kruskal-Wallis test versus QA/vehicle rats (C) Weight curves in QA-
NA expression in the striatum of QA-injected rats treated with vehicle,
als. Lower panel: quantification of signal shown on the upper panel. **P b
ion in the striatum of QA-injected rats treated with vehicle or minocycline.

Fig. 6. Minocycline does not affect the basal and QA-evoked striatal
glutamate release. Evaluation of the influence of minocycline on the basal
and QA-evoked glutamate outflow in the striatum of rats chronically treated
for 3 (A) or 7 (B) days with vehicle or 10 mg/kg minocycline. Each
experimental group was made of three animals. The bar indicates the period
of QA perfusion through the probe.
K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217214
Minocycline is protective against QA-induced striatal lesions in
vivo
Animals unilaterally injected with QAwere treated with 10 mg/
kg minocycline for 7 days (protocol C, Fig. 1) and the size of the
striatal lesion core was then measured using SDH histochemistry.
We found that the volume of the lesion core was significantly
reduced in minocycline-treated animals (Fig. 5A). The percentage
of the spared striatum was then significantly increased in the
minocycline-treated animals (Fig. 5B). Noteworthy, at the dose
tested, minocycline did not alter body weight gain (Fig. 5C),
although higher dose of the antibiotic (50 mg/kg) prevented weight
gain of QA-treated animals and impaired the protective activity
found with the 10 mg/kg dose (not shown). Microglial activation
was assessed using MAC-1 mRNA in situ hybridization at the end
of the experiment. As compared to the unlesioned side, we found
that striatal MAC-1 mRNA expression was strongly increased in
animals treated by quinolinic acid (Fig. 5D). The MAC-1 mRNA
level was significantly decreased in the lesioned striatum of
minocycline-treated animals compared with the lesion striatum of
vehicle-treated rats. In contrast, minocycline did not exert an effect
on the increase of GFAP expression induced by QA (Fig. 5E). The
present results thus suggested that minocycline is beneficial in a
phenotypic model of HD in which striatal lesion involves
inflammatory processes. Noteworthy, the beneficial effects of
minocycline were not observed in animals injected with a similar
and nontoxic dose of doxycycline, another tetracycline analogue
(Figs. 5A–D).
Of interest, it was previously found that glutamate or glutamate
receptor agonists can induce microglial activation (Tikka and
Koistinaho, 2001). Given that QA neurotoxicity is mediated at least
in part by an increased glutamate release (Blum et al., 2003b; Popoli
et al., 2002), we determined whether the protective and anti-
inflammatory activity of minocycline could be related to a reduction
of evoked glutamate outflow. Using microdialysis, we found that
basal and QA-induced glutamate release remained unchanged in
animals chronically treated with minocycline at a protective dose
(Fig. 6). This thus suggested that the observed protective effect of
the antibiotic was not related to a presynaptic effect but rather to the
inhibition of events downstream glutamate release.
Discussion
In the present study, we assessed the effects of minocycline in
phenotypic models of HD. Our results showed that minocycline is
not beneficial against calpain-dependent striatal degeneration but is
able to counteract the development of striatal inflammation.
In a first attempt, we found that minocycline was not protective
against chronic 3NP intoxication both in vitro and in vivo. This
absence of protection was unlikely related to a lack of minocycline
activity which could arise from an insufficient dosage or to a
chemical inactivation of the molecule since minocycline was
prepared fresh before each injection, and the molecule displayed
anti-inflammatory and protective activities in the other models we
used. We previously showed that in both models, striatal cell death
was mediated through calpain but not caspase activation (Bizat et
al., 2003a,b; Galas et al., 2004), reproducing an important feature of
the human pathology (Gafni and Ellerby, 2002; Gafni et al., 2004).
Nonetheless, in line with recent studies (Scarabelli et al., 2004;
Wang et al., 2003; Zhu et al., 2002), we found that minocycline
protects HEK 293T cells against experimental conditions known to
promote caspase induction. These results indicate that minocycline
would not be protective against calpain-dependent degeneration. At
present, it is not clear which proteases are directly responsible for
neuronal degeneration in HD. Although caspases participate to the
pathological development in HD through, particularly, the cleavage
of mutated full-length huntingtin into shorter fragments (Goldberg
et al., 1996; Hermel et al., 2004; Kiechle et al., 2002; Ona et al.,
1999; Wellington et al., 2003), their role may not be directly related
to the death of striatal neurons. Indeed, it has been recently shown
that R6/2 mice displayed many features of mitochondrial-depend-
ent and independent apoptosis (Wang et al., 2003; Zhang et al.,
2003), despite lacking massive neuronal cell loss (Turmaine et al.,
2000) in contrast to what occurs in HD patients (Vonsattel et al.,
1985). Alternatively, calpains may play an important role as
suggested by recent studies performed on postmortem HD brains
and animal models of the disorder (Bizat et al., 2003a,b; Gafni and
Ellerby, 2002; Gafni et al., 2004). Then, benefits afforded by
minocycline in HD patients might depend on the cell death
processes involved. Furthermore, given that striatal and cortical
degeneration may involve differential mechanisms (Galas et al.,
2004), minocycline efficiency may be anatomically determined.
Using a paradigm of 3NP intoxication allowing the development
of a secondary microgliosis, we found that minocycline reduced
striatal inflammation. This finding was further confirmed using the

K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217 215
quinolinic acid model in which inflammation is concomitant to
degeneration (Dihne et al., 2001; Topper et al., 1993). In this model,
we found that minocycline reduced not only the MAC-1 mRNA
expression, but also the size of the lesion core produced by the
NMDA agonist. Conversely, minocycline did not act on the
increased GFAP mRNA expression associated with the lesion,
consistent with previous works demonstrating that minocycline does
not modify astrogliosis (Diguet et al., 2004a; Kriz et al., 2002;
Yrjanheikki et al., 1998, 1999). It remains also possible that the
neuroprotective activity ofminocycline against QA involves caspase
inhibition despite that its contribution has been poorly characterized
in this model (Nakai et al., 2000; Qin et al., 2000). However, our
results are in accordance with the known anti-inflammatory activity
of minocycline found in other models of degenerative diseases and
suggested in the R6/2 HD model (Amin et al., 1996; Chen et al.,
2000; Dommergues et al., 2003; He et al., 2001; Kriz et al., 2002;
Tikka and Koistinaho, 2001; Tomas-Camardiel et al., 2004). Given
that the presence of microglial activation in HD brains evenly
correlated with the neuropathological grade (McGeer et al., 1988;
Sapp et al., 2001), the ability of minocycline to inhibit striatal
inflammation could lead to clinical benefit in HD patients.
Previous observations reported that high doses of both
minocycline and doxycycline provided protection against cerebral
ischemia (Yrjanheikki et al., 1998). However, in vivo, we found that
at low doses (10 mg/kg/d), only minocycline displayed protective
potential, although in vitro, doxycycline displayed a similar
protective activity than minocycline. This difference may be due
to the lowest ability of doxycycline to cross the blood–brain barrier
as compared to minocycline (Smith et al., 2003). This suggests that,
clinically, minocycline may be more appropriated than doxycycline.
It is also important to note that, as reviewed by Diguet et al.
(2004b) and Blum et al. (2004), positive effects of minocycline
formerly found in models of Parkinson disease, HD, or ischemia
(see Blum et al., 2004; for review) have been recently questioned
regardless the mode of administration or the species involved
(Diguet et al., 2004a; Smith et al., 2003; Tsuji et al., 2004; Yang et
al., 2003). Additionally, repeated i.p. delivery of minocycline at
doses N45 mg/kg/d may even lead to unintended morbidity (Fagan
et al., 2004). In accordance, we found that high doses of
minocycline (or doxycycline, data not shown) impaired body
weight gain in rats as previously reported (Smith et al., 2003; Yang
et al., 2003). Together, this shows that minocycline may have
variable and contradictory effects and that neuroprotective mech-
anisms remain to be further clarified. Although it remains difficult to
extrapolate to humans observations made in models, these
controversies should encourage a careful evaluation of potential
minocycline toxicity at the peripheral (liver, kidney. . .) or immuno-
logical levels (Gottlieb, 1997; Knowles et al., 1996) in patients
enrolled in clinical trials (Blum et al., 2004; Diguet et al., 2004b).
In conclusion, our data suggest that minocycline, at low dose,
may be beneficial to HD patients but that its clinical efficiency will
depend on the relative contribution of caspase- and calpain-
dependent mechanisms and of inflammation in the development of
the pathology and neurodegeneration.
Acknowledgments
This work was supported by fundings from the Hereditary
Disease Foundation (DB), FNRS (DB/LT/JB, SNS), Region-
Bruxelles Capitale (JB/LT), Fondation Medicale Reine Elisabeth
(SNS), Action de Recherche Concertee (SNS), and Fondation
Universitaire David and Alice Van Buuren (DB/JB, SNS). KB and
AC hold grants from Televie. DB is bCharge de RecherchesQ of theFNRS.
References
Amin, A.R., Attur, M.G., Thakker, G.D., Patel, P.D., Vyas, P.R., Patel,
R.N., Patel, I.R., Abramson, S.B., 1996. A novel mechanism of action
of tetracyclines: effects on nitric oxide synthases. Proc. Natl. Acad. Sci.
U. S. A. 93, 14014–14019.
Beal, M.F., 2000. Energetics in the pathogenesis of neurodegenerative
diseases. Trends Neurosci. 23, 298–304.
Beal, M.F., Kowall, N.W., Ellison, D.W., Mazurek, M.F., Swartz, K.J.,
Martin, J.B., 1986. Replication of the neurochemical characteristics of
Huntington’s disease by quinolinic acid. Nature 321, 168–171.
Beal, M.F., Brouillet, E., Jenkins, B.G., Ferrante, R.J., Kowall, N.W.,
Miller, J.M., Storey, E., Srivastava, R., Rosen, B.R., Hyman, B.T.,
1993. Neurochemical and histologic characterization of striatal excito-
toxic lesions produced by the mitochondrial toxin 3-nitropropionic acid.
J. Neurosci. 13, 4181–4192.
Bensadoun, J.C., de Almeida, L.P., Dreano, M., Aebischer, P., Deglon, N.,
2001. Neuroprotective effect of interleukin-6 and IL6/IL6R chimera in
the quinolinic acid rat model of Huntington’s syndrome. Eur. J.
Neurosci. 14, 1753–1761.
Bizat, N., Hermel, J.M., Boyer, F., Jacquard, C., Creminon, C., Ouary, S.,
Escartin, C., Hantraye, P., Kajewski, S., Brouillet, E., 2003a. Calpain is
a major cell death effector in selective striatal degeneration induced in
vivo by 3-nitropropionate: implications for Huntington’s disease.
J. Neurosci. 23, 5020–5030.
Bizat, N., Hermel, J.M., Humbert, S., Jacquard, C., Creminon, C., Escartin,
C., Saudou, F., Krajewski, S., Hantraye, P., Brouillet, E., 2003b. In vivo
calpain/caspase cross-talk during 3-nitropropionic acid-induced striatal
degeneration: implication of a calpain-mediated cleavage of active
caspase-3. J. Biol. Chem. 278, 43245–43253.
Blum, D., Gall, D., Cuvelier, L., Schiffmann, S.N., 2001. Topological
analysis of striatal lesions induced by 3-nitropropionic acid in the Lewis
rat. NeuroReport 12, 1769–1772.
Blum, D., Galas, M.C., Gall, D., Cuvelier, L., Schiffmann, S.N., 2002a.
Striatal and cortical neurochemical changes induced by chronic
metabolic compromise in the 3-nitropropionic model of Huntington’s
disease. Neurobiol. Dis. 10, 410–426.
Blum, D., Gall, D., Galas, M.C., d’Alcantara, P., Bantubungi, K.,
Schiffmann, S.N., 2002b. The adenosine A1 receptor agonist adenosine
amine congener exerts a neuroprotective effect against the development
of striatal lesions and motor impairments in the 3-nitropropionic acid
model of neurotoxicity. J. Neurosci. 22, 9122–9133.
Blum, D., Hourez, R., Galas, M.C., Popoli, P., Schiffmann, S.N., 2003a.
Adenosine receptors and Huntington’s disease: implications for patho-
genesis and therapeutics. Lancet Neurol. 2, 366–374.
Blum, D., Galas, M.C., Pintor, A., Brouillet, E., Ledent, C., Muller, C.E.,
Bantubungi, K., Galluzzo, M., Gall, D., Cuvelier, L., Rolland, A.S.,
Popoli, P., Schiffmann, S.N., 2003b. A dual role of adenosine A2A
receptors in 3-nitropropionic acid-induced striatal lesions: implications
for the neuroprotective potential of A2A antagonists. J. Neurosci. 23,
5361–5369.
Blum, D., Chtarto, A., Tenenbaum, L., Brotchi, J., Levivier, M., 2004.
Clinical potential of minocycline for neurodegenerative disorders.
Neurobiol. Dis. 17 (3), 359–366.
Bonelli, R.M., Heuberger, C., Reisecker, F., 2003. Minocycline for
Huntington’s disease: an open label study. Neurology 60, 883–884.
Brouillet, E., Guyot, M.C., Mittoux, V., Altairac, S., Conde, F., Palfi, S.,
Hantraye, P., 1998. Partial inhibition of brain succinate dehydrogenase
by 3-nitropropionic acid is sufficient to initiate striatal degeneration in
rat. J. Neurochem. 70, 794–805.

K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217216
Brouillet, E., Conde, F., Beal, M.F., Hantraye, P., 1999. Replicating
Huntington’s disease phenotype in experimental animals. Prog. Neuro-
biol. 59, 427–468.
Brundula, V., Rewcastle, N.B., Metz, L.M., Bernard, C.C., Yong, V.W.,
2002. Targeting leukocyte MMPs and transmigration: minocycline as a
potential therapy for multiple sclerosis. Brain 125, 1297–1308.
Cha, J.H., 2000. Transcriptional dysregulation in Huntington’s disease.
Trends Neurosci. 23, 387–392.
Chen, M., Ona, V.O., Li, M., Ferrante, R.J., Fink, K.B., Zhu, S., Bian, J.,
Guo, L., Farrell, L.A., Hersch, S.M., Hobbs, W., Vonsattel, J.P., Cha,
J.H., Friedlander, R.M., 2000. Minocycline inhibits caspase-1 and
caspase-3 expression and delays mortality in a transgenic mouse model
of Huntington disease. Nat. Med. 6, 797–801.
Coggeshall, R.E., 1992. A consideration of neural counting methods.
Trends Neurosci. 15, 9–13.
de Almeida, L.P., Ross, C.A., Zala, D., Aebischer, P., Deglon, N., 2002.
Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats
induces a selective neuropathology modulated by polyglutamine repeat
size, huntingtin expression levels, and protein length. J. Neurosci. 22,
3473–3483.
Diguet, E., Fernagut, P.O., Wei, X., Du, Y., Rouland, R., Gross, C., Bezard,
E., Tison, F., 2004a. Deleterious effects of minocycline in animal
models of Parkinson’s disease and Huntington’s disease. Eur. J.
Neurosci. 19, 3266–3276.
Diguet, E., Gross, C.E., Tison, F., Bezard, E., 2004b. Rise and fall of
minocycline in neuroprotection: need to promote publication of
negative results. Exp. Neurol. 189, 1–4.
Dihne, M., Block, F., Korr, H., Topper, R., 2001. Time course of glial
proliferation and glial apoptosis following excitotoxic CNS injury.
Brain Res. 902, 178–189.
Dommergues, M.A., Plaisant, F., Verney, C., Gressens, P., 2003. Early
microglial activation following neonatal excitotoxic brain damage in
mice: a potential target for neuroprotection. Neuroscience 121,
619–628.
Fagan, S.C., Edwards, D.J., Borlongan, C.V., Xu, L., Arora, A., Feuerstein,
G., Hess, D.C., 2004. Optimal delivery of minocycline to the brain:
implication for human studies of acute neuroprotection. Exp. Neurol.
186, 248–251.
Gafni, J., Ellerby, L.M., 2002. Calpain activation in Huntington’s disease.
J. Neurosci. 22, 4842–4849.
Gafni, J., Hermel, E., Young, J.E., Wellington, C.L., Hayden, M.R., Ellerby,
L.M., 2004. Inhibition of calpain cleavage of Huntingtin reduces
toxicity: accumulation of calpain/caspase fragments in the nucleus.
J. Biol. Chem. 279, 20211–20220.
Galas, M.C., Bizat, N., Cuvelier, L., Bantubungi, K., Brouillet, E.,
Schiffmann, S.N., Blum, D., 2004. Death of cortical and striatal
neurons induced by mitochondrial defect involves differential molecular
mechanisms. Neurobiol. Dis. 15, 152–159.
Goldberg, Y.P., Nicholson, D.W., Rasper, D.M., Kalchman, M.A., Koide,
H.B., Graham, R.K., Bromm, M., Kazemi-Esfarjani, P., Thornberry,
N.A., Vaillancourt, J.P., Hayden, M.R., 1996. Cleavage of huntingtin by
apopain, a proapoptotic cysteine protease, is modulated by the
polyglutamine tract. Nat. Genet. 13, 442–449.
Gottlieb, A., 1997. Safety of minocycline for acne. Lancet 349, 374.
Gunawardena, S., Her, L.S., Brusch, R.G., Laymon, R.A., Niesman, I.R.,
Gordesky-Gold, B., Sintasath, L., Bonini, N.M., Goldstein, L.S., 2003.
Disruption of axonal transport by loss of huntingtin or expression of
pathogenic polyQ proteins in Drosophila. Neuron 40, 25–40.
He, Y., Appel, S., Le, W., 2001. Minocycline inhibits microglial activation
and protects nigral cells after 6-hydroxydopamine injection into mouse
striatum. Brain Res. 909, 187–193.
Hermel, E., Gafni, J., Propp, S.S., Leavitt, B.R., Wellington, C.L., Young,
J.E., Hackam, A.S., Logvinova, A.V., Peel, A.L., Chen, S.F., Hook, V.,
Singaraja, R., Krajewski, S., Goldsmith, P.C., Ellerby, H.M., Hayden,
M.R., Bredesen, D.E., Ellerby, L.M., 2004. Specific caspase inter-
actions and amplification are involved in selective neuronal vulner-
ability in Huntington’s disease. Cell Death Differ. 11, 424–438.
Kiechle, T., Dedeoglu, A., Kubilus, J., Kowall, N.W., Beal, M.F.,
Friedlander, R.M., Hersch, S.M., Ferrante, R.J., 2002. Cytochrome c
and caspase-9 expression in Huntington’s disease. Neuromol. Med. 1,
183–195.
Kim, S.S., Kong, P.J., Kim, B.S., Sheen, D.H., Nam, S.Y., Chun, W., 2004.
Inhibitory action of minocycline on lipopolysaccharide-induced release
of nitric oxide and prostaglandin E2 in BV2 microglial cells. Arch.
Pharm. Res. 27, 314–318.
Knowles, S.R., Shapiro, L., Shear, N.H., 1996. Serious adverse reactions
induced by minocycline. Report of 13 patients and review of the
literature. Arch. Dermatol. 132, 934–939.
Kriz, J., Nguyen, M.D., Julien, J.P., 2002. Minocycline slows disease
progression in a mouse model of amyotrophic lateral sclerosis.
Neurobiol. Dis. 10, 268–278.
Lee, S.M., Yune, T.Y., Kim, S.J., Park, d.W., Lee, Y.K., Kim, Y.C., Oh, Y.J.,
Markelonis, G.J., Oh, T.H., 2003. Minocycline reduces cell death and
improves functional recovery after traumatic spinal cord injury in the
rat. J. Neurotrauma 20, 1017–1027.
McGeer, P.L., Itagaki, S., McGeer, E.G., 1988. Expression of the
histocompatibility glycoprotein HLA-DR in neurological disease. Acta
Neuropathol. (Berl) 76, 550–557.
Minghetti, L., Greco, A., Cardone, F., Puopolo, M., Ladogana, A., Almonti,
S., Cunningham, C., Perry, V.H., Pocchiari, M., Levi, G., 2000.
Increased brain synthesis of prostaglandin E2 and F2-isoprostane in
human and experimental transmissible spongiform encephalopathies.
J. Neuropathol. Exp. Neurol. 59, 866–871.
Mittoux, V., Ouary, S., Monville, C., Lisovoski, F., Poyot, T., Conde, F.,
Escartin, C., Robichon, R., Brouillet, E., Peschanski, M., Hantraye, P.,
2002. Corticostriatopallidal neuroprotection by adenovirus-mediated
ciliary neurotrophic factor gene transfer in a rat model of progressive
striatal degeneration. J. Neurosci. 22, 4478–4486.
Nakai, M., Qin, Z., Wang, Y., Chase, T.N., 2000. NMDA and non-NMDA
receptor-stimulated IkappaB-alpha degradation: differential effects of
the caspase-3 inhibitor DEVD.CHO, ethanol and free radical scavenger
OPC-14117. Brain Res. 859, 207–216.
Ona, V.O., Li, M., Vonsattel, J.P., Andrews, L.J., Khan, S.Q., Chung, W.M.,
Frey, A.S., Menon, A.S., Li, X.J., Stieg, P.E., Yuan, J., Penney, J.B.,
Young, A.B., Cha, J.H., Friedlander, R.M., 1999. Inhibition of caspase-
1 slows disease progression in a mouse model of Huntington’s disease.
Nature 399, 263–267.
Ouary, S., Bizat, N., Altairac, S., Menetrat, H., Mittoux, V., Conde, F.,
Hantraye, P., Brouillet, E., 2000. Major strain differences in response
to chronic systemic administration of the mitochondrial toxin 3-
nitropropionic acid in rats: implications for neuroprotection studies.
Neuroscience 97, 521–530.
Panov, A.V., Gutekunst, C.A., Leavitt, B.R., Hayden, M.R., Burke, J.R.,
Strittmatter, W.J., Greenamyre, J.T., 2002. Early mitochondrial calcium
defects in Huntington’s disease are a direct effect of polyglutamines.
Nat. Neurosci. 5, 731–736.
Popoli, P., Pintor, A., Domenici, M.R., Frank, C., Tebano, M.T., Pezzola, A.,
Scarchilli, L., Quarta, D., Reggio, R., Malchiodi-Albedi, F., Falchi, M.,
Massotti, M., 2002. Blockade of striatal adenosine A2A receptor
reduces, through a presynaptic mechanism, quinolinic acid-induced
excitotoxicity: possible relevance to neuroprotective interventions in
neurodegenerative diseases of the striatum. J. Neurosci. 22, 1967–1975.
Qin, Z., Wang, Y., Chasea, T.N., 2000. A caspase-3-like protease is
involved in NF-kappaB activation induced by stimulation of N-methyl-
D-aspartate receptors in rat striatum. Brain Res. Mol. Brain Res. 80,
111–122.
Ryu, J.K., Nagai, A., Kim, J., Lee, M.C., McLarnon, J.G., Kim, S.U., 2003.
Microglial activation and cell death induced by the mitochondrial toxin
3-nitropropionic acid: in vitro and in vivo studies. Neurobiol. Dis. 12,
121–132.
Sapp, E., Kegel, K.B., Aronin, N., Hashikawa, T., Uchiyama, Y., Tohyama,
K., Bhide, P.G., Vonsattel, J.P., DiFiglia, M., 2001. Early and
progressive accumulation of reactive microglia in the Huntington
disease brain. J. Neuropathol. Exp. Neurol. 60, 161–172.

K. Bantubungi et al. / Neurobiology of Disease 18 (2005) 206–217 217
Scarabelli, T.M., Stephanou, A., Pasini, E., Gitti, G., Townsend, P.,
Lawrence, K., Chen-Scarabelli, C., Saravolatz, L., Latchman, D.,
Knight, R., Gardin, J., 2004. Minocycline inhibits caspase activation
and reactivation, increases the ratio of XIAP to smac/DIABLO, and
reduces the mitochondrial leakage of cytochrome c and smac/DIABLO.
J. Am. Coll. Cardiol. 43, 865–874.
Smith, D.L., Woodman, B., Mahal, A., Sathasivam, K., Ghazi-Noori, S.,
Lowden, P.A., Bates, G.P., Hockly, E., 2003. Minocycline and
doxycycline are not beneficial in a model of Huntington’s disease.
Ann. Neurol. 54, 186–196.
Song, C., Zhang, Y., Parsons, C.G., Liu, Y.F., 2003. Expression of
polyglutamine-expanded huntingtin induces tyrosine phosphorylation
of N-methyl-d-aspartate receptors. J. Biol. Chem. 278, 33364–33369.
Stirling, D.P., Khodarahmi, K., Liu, J., McPhail, L.T., McBride, C.B.,
Steeves, J.D., Ramer, M.S., Tetzlaff, W., 2004. Minocycline treatment
reduces delayed oligodendrocyte death, attenuates axonal dieback, and
improves functional outcome after spinal cord injury. J. Neurosci. 24,
2182–2190.
Szebenyi, G., Morfini, G.A., Babcock, A., Gould, M., Selkoe, K., Stenoien,
D.L., Young, M., Faber, P.W., MacDonald, M.E., McPhaul, M.J.,
Brady, S.T., 2003. Neuropathogenic forms of huntingtin and androgen
receptor inhibit fast axonal transport. Neuron 40, 41–52.
Tang, T.S., Tu, H., Chan, E.Y., Maximov, A., Wang, Z., Wellington, C.L.,
Hayden, M.R., Bezprozvanny, I., 2003. Huntingtin and huntingtin-
associated protein 1 influence neuronal calcium signaling mediated by
inositol-(1,4,5) triphosphate receptor type 1. Neuron 39, 227–239.
The Huntington’s Disease Collaborative Research Group, 1993. A novel
gene containing a trinucleotide repeat that is expanded and unstable on
Huntington’s disease chromosomes. Cell 72, 971–983.
Thomas, M., Ashizawa, T., Jankovic, J., 2004. Minocycline in Huntington’s
disease: a pilot study. Mov. Disord. 19, 692–695.
Tikka, T.M., Koistinaho, J.E., 2001. Minocycline provides neuroprotection
against N-methyl-d-aspartate neurotoxicity by inhibiting microglia.
J. Immunol. 166, 7527–7533.
Tikka, T.M., Vartiainen, N.E., Goldsteins, G., Oja, S.S., Andersen, P.M.,
Marklund, S.L., Koistinaho, J., 2002. Minocycline prevents neuro-
toxicity induced by cerebrospinal fluid from patients with motor
neurone disease. Brain 125, 722–731.
Tomas-Camardiel, M., Rite, I., Herrera, A.J., de Pablos, R.M., Cano, J.,
Machado, A., Venero, J.L., 2004. Minocycline reduces the lipopoly-
saccharide-induced inflammatory reaction, peroxynitrite-mediated nitra-
tion of proteins, disruption of the blood–brain barrier, and damage in the
nigral dopaminergic system. Neurobiol. Dis. 16, 190–201.
Topper, R., Gehrmann, J., Schwarz, M., Block, F., Noth, J., Kreutzberg,
G.W., 1993. Remote microglial activation in the quinolinic acid model
of Huntington’s disease. Exp. Neurol. 123, 271–283.
Tsuji, M., Wilson, M.A., Lange, M.S., Johnston, M.V., 2004. Minocycline
worsens hypoxic–ischemic brain injury in a neonatal mouse model.
Exp. Neurol. 189, 58–65.
Turmaine, M., Raza, A., Mahal, A., Mangiarini, L., Bates, G.P., Davies,
S.W., 2000. Nonapoptotic neurodegeneration in a transgenic mouse
model of Huntington’s disease. Proc. Natl. Acad. Sci. U. S. A. 97,
8093–8097.
Vonsattel, J.P., Myers, R.H., Stevens, T.J., Ferrante, R.J., Bird, E.D.,
Richardson Jr., E.P., 1985. Neuropathological classification of Hun-
tington’s disease. J. Neuropathol. Exp. Neurol. 44, 559–577.
Wang, X., Zhu, S., Drozda, M., Zhang, W., Stavrovskaya, I.G., Cattaneo,
E., Ferrante, R.J., Kristal, B.S., Friedlander, R.M., 2003. Minocycline
inhibits caspase-independent and -dependent mitochondrial cell death
pathways in models of Huntington’s disease. Proc. Natl. Acad. Sci.
U. S. A. 100, 10483–10487.
Wang, J., Wei, Q., Wang, C.Y., Hill, W.D., Hess, D.C., Dong, Z., 2004.
Minocycline up-regulates Bcl-2 and protects against cell death in the
mitochondria. J. Biol. Chem. 279, 19948–19954.
Wellington, C.L., Ellerby, L.M., Leavitt, B.R., Roy, S., Nicholson, D.W.,
Hayden, M.R., 2003. Huntingtin proteolysis in Huntington disease.
Clin. Neurosci. Res. 3, 129–139.
Wu, D.C., Jackson-Lewis, V., Vila, M., Tieu, K., Teismann, P., Vadseth,
C., Choi, D.K., Ischiropoulos, H., Przedborski, S., 2002. Blockade
of microglial activation is neuroprotective in the 1-methyl-4-
phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease.
J. Neurosci. 22, 1763–1771.
Yang, L., Sugama, S., Chirichigno, J.W., Gregorio, J., Lorenzl, S., Shin,
D.H., Browne, S.E., Shimizu, Y., Joh, T.H., Beal, M.F., Albers, D.S.,
2003. Minocycline enhances MPTP toxicity to dopaminergic neurons.
J. Neurosci. Res. 74, 278–285.
Yrjanheikki, J., Keinanen, R., Pellikka, M., Hokfelt, T., Koistinaho, J.,
1998. Tetracyclines inhibit microglial activation and are neuroprotec-
tive in global brain ischemia. Proc. Natl. Acad. Sci. U. S. A. 95,
15769–15774.
Yrjanheikki, J., Tikka, T., Keinanen, R., Goldsteins, G., Chan, P.H.,
Koistinaho, J., 1999. A tetracycline derivative, minocycline, reduces
inflammation and protects against focal cerebral ischemia with a wide
therapeutic window. Proc. Natl. Acad. Sci. U. S. A. 96, 13496–13500.
Yu, Z.X., Li, S.H., Evans, J., Pillarisetti, A., Li, H., Li, X.J., 2003. Mutant
huntingtin causes context-dependent neurodegeneration in mice with
Huntington’s disease. J. Neurosci. 23, 2193–2202.
Zeron, M.M., Hansson, O., Chen, N., Wellington, C.L., Leavitt, B.R.,
Brundin, P., Hayden, M.R., Raymond, L.A., 2002. Increased sensitivity
to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse
model of Huntington’s disease. Neuron 33, 849–860.
Zhang, Y., Ona, V.O., Li, M., Drozda, M., Dubois-Dauphin, M.,
Przedborski, S., Ferrante, R.J., Friedlander, R.M., 2003. Sequential
activation of individual caspases, and of alterations in Bcl-2 proapop-
totic signals in a mouse model of Huntington’s disease. J. Neurochem.
87, 1184–1192.
Zhu, S., Stavrovskaya, I.G., Drozda, M., Kim, B.Y., Ona, V., Li, M.,
Sarang, S., Liu, A.S., Hartley, D.M., Wu, d.C., Gullans, S., Ferrante,
R.J., Przedborski, S., Kristal, B.S., Friedlander, R.M., 2002. Minocy-
cline inhibits cytochrome c release and delays progression of
amyotrophic lateral sclerosis in mice. Nature 417, 74–78.
Zuccato, C., Tartari, M., Crotti, A., Goffredo, D., Valenza, M., Conti, L.,
Cataudella, T., Leavitt, B.R., Hayden, M.R., Timmusk, T., Rigamonti,
D., Cattaneo, E., 2003. Huntingtin interacts with REST/NRSF to
modulate the transcription of NRSE-controlled neuronal genes. Nat.
Genet. 35, 76–83.