miljanicthesis.pdf - index of
TRANSCRIPT

Synthetic and Structural Studies of Phenylenes and Dehydrobenzannulenes
by
Ognjen Scepan Miljanic
Diploma (University of Belgrade) 2000
A dissertation submitted in partial satisfaction of the
requirements for the degree of
Doctor of Philosophy
in
Chemistry
in the
GRADUATE DIVISION
of the
UNIVERSITY OF CALIFORNIA, BERKELEY
Committee in charge:
Professor K. Peter C. Vollhardt, Chair
Professor Robert G. Bergman
Professor Ronald Gronsky
Fall 2005

The dissertation of Ognjen Scepan Miljanic is approved:
Chair Date
Date
Date
University of California, Berkeley
Fall 2005

Synthetic and Structural Studies of Phenylenes and Dehydrobenzannulenes
Copyright 2005
by
Ognjen Scepan Miljanic

1
Abstract
Synthetic and Structural Studies of Phenylenes and Dehydrobenzannulenes
by
Ognjen Scepan Miljanic
Doctor of Philosophy in Chemistry
University of California, Berkeley
Professor K. Peter C. Vollhardt, Chair
This dissertation documents the findings on the syntheses of larger
dehydrobenzannulenes and [N]phenylenes and the exploration of their physical and
chemical properties.
Chapter One. This chapter summarizes the previous synthetic work on
[N]phenylenes. Their structural, magnetic, and energetic properties, as well as chemical
reactivity are reviewed, and comparisons are made between different phenylene
topologies.
Chapter Two. The synthesis of syn-doublebent [5]phenylene is presented.
Approaches to three novel phenylenes, U-shaped [7]- and [9]phenylenes and C-shaped
[7]phenylene are also discussed.
Chapter Three. The topic of this chapter is the development of a novel alkyne
metathesis-based route to ortho-dehydrobenzannulenes. Additionally, the application of
microwave irradiation to Sonogashira couplings with gaseous propyne is described.
Chapter Four. A versatile synthetic route based on a sequence of Sonogashira
couplings is described to access substituted dehydrobenzannulenes. CpCo-mediated

2
cycloisomerizations of these materials that produced partially cyclized phenylenes are
summarized.
Chapter Five. The finding that dehydrobenzannulenes substituted with
sufficiently bulky silyl-groups are conformationally locked at ambient temperatures is
presented. This result inspired the synthesis of the first chiral diphenylacetylene.
Variable-temperature NMR studies of both of these systems were undertaken to
determine the corresponding racemization barriers.
Chapter Six. The final chapter details the experimental procedures of the studies
presented in Chapters 2–5.

i
to Peace

ii
Table of Contents
Chapter One [N]Phenylenes: a Novel Class of Cyclohexatrienoid Hydrocarbons
1.1 Introduction......................................................................................................... 1
1.2 Preparation of Phenylenes................................................................................... 9
1.2.1 Early Synthetic Strategies ........................................................................... 9
1.2.2 Syntheses of New Phenylenes................................................................... 15
1.2.2.1 Angular and Helical Phenylenes ........................................................... 15
1.2.2.2 Zigzag Phenylenes ................................................................................ 19
1.2.2.3 Phenylenes with Mixed Topology: the “Bent” Isomers........................ 22
1.2.2.4 Branched Phenylenes ............................................................................ 27
1.2.2.5 Circular Phenylenes .............................................................................. 31
1.3 Comparative Reactivity of the Phenylenes ....................................................... 34
1.3.1 Hydrogenation........................................................................................... 34
1.3.2 Oxacyclopropanation and Cyclopropanation............................................ 37
1.3.3 [4+2]Cycloadditions.................................................................................. 41
1.3.4 Flash Vacuum Pyrolysis............................................................................ 46
1.3.5 Interaction with Organometallic Fragments.............................................. 48
1.4 Physical Properties of the Phenylenes .............................................................. 52
1.4.1 Structural Properties.................................................................................. 52
1.4.2 Magnetic Properties .................................................................................. 63
1.4.3 Energetic Properties .................................................................................. 67
1.5 Thesis Summary................................................................................................ 72

iii
Chapter Two Synthetic Approaches to Novel Phenylenes with Mixed Angular and
Linear Fusion
2.1 Introduction....................................................................................................... 73
2.2 Retrosynthetic Approach to 60, 118, 119, and 120........................................... 76
2.3 Synthesis of Doublebent [5]Phenylene 60 ........................................................ 78
2.4 Attempted Synthesis of C-Shaped [7]Phenylene (118) .................................... 82
2.5 Attempted Syntheses of U-Shaped [7]Phenylene (119).................................... 84
2.5.1 Intramolecular Approach .......................................................................... 84
2.5.2 Intermolecular Approach .......................................................................... 90
2.6 Attempted Synthesis of U-Shaped [9]Phenylene (120) .................................... 92
2.7 Calculated and Measured Properties of 60, 118, 119, and 120......................... 94
2.8 Summary ......................................................................................................... 102
Chapter Three A Novel Alkyne Metathesis-Based Route to Dehydrobenz-
annulenes
3.1 Introduction..................................................................................................... 103
3.2 Retrosynthetic Approach to Dehydrobenzannulenes ...................................... 105
3.3 Preparation of Iodinated Precursors ................................................................ 110
3.4 Classical and Microwave-Assisted Propynylations ........................................ 112
3.5 Dehydrobenzannulenes by Alkyne Metathesis ............................................... 115
3.6 Properties of Novel Dehydrobenzannulenes................................................... 119
3.7 Summary ......................................................................................................... 121

iv
Chapter Four Synthesis of Octaalkynylated Dehydrobenz[18]annulenes and
Attempted Cycloisomerization into Circular [8]Phenylene and Derivatives
4.2 Retrosynthetic Analysis of Circular [8]Phenylene.......................................... 126
4.3 Previous Attempts to Synthesize Circular [8]Phenylene ................................ 129
4.4 Synthesis and Properties of Octaalkynylated Dehydrobenz[18]annulenes 156
and 171b–c .................................................................................................................. 133
4.5 Attempted Cycloisomerization of 156 and 171b-c into Circular [8]Phenylenes
121 and 170b-c............................................................................................................ 137
4.6 Properties of Novel Phenylenes ...................................................................... 142
4.7 Summary and Future Directions ..................................................................... 143
Chapter Five Consequences of Steric Crowding Around Triple Bonds in Acyclic
and Cyclic Systems
5.1 Introduction..................................................................................................... 144
5.2 Previous Examples of Hindered Rotation in Phenylene Precursors ............... 149
5.3 Synthesis and Properties of the First Chiral 2,2’,6,6’-Tetrakisalkynyl
Diphenylacetylene....................................................................................................... 151
5.4 Stereochemical Properties of 171c.................................................................. 154
5.5 Proposed Mechanism of Interconversion between the Conformers of 171c .. 159
5.6 Summary and Future Directions ..................................................................... 174
Chapter Six Experimental and Computational Details
6.1 General Considerations ................................................................................... 175

v
6.2 Experiments and Calculations Related to Chapter 2....................................... 177
6.2.1 Calculated Structures of 60 and 118–120 ............................................... 203
6.3 Experiments Related to Chapter 3 .................................................................. 209
6.3.1 Crystallographic Information for 159 ..................................................... 226
6.4 Experiments Related to Chapter 4 .................................................................. 231
6.4.1 Calculated Structures of 121, 156, and 191–195 .................................... 254
6.5 Experiments Related to Chapter 5 .................................................................. 276
6.5.1 Calculated Structures of Transition States for the Inversion of 171c and
213–216 ................................................................................................................. 280

vi
Abbreviations and Acronyms
2D two dimensional
ASC algebraic structure count
BTMSA bis(trimethylsilyl)acetylene
Bu butyl
Cp cyclopentadienyl
Cp* pentamethylcyclopentadienyl
DFT density functional theory
DMF dimethylformamide
DMAD dimethyl butynedioate
DMDO dimethyldioxirane
DMSO dimethylsulfoxide
DMTS dimethylthexylsilyl
DMTSA (dimethylthexylsilyl)acetylene
Et ethyl
eth ethene
FVP flash vacuum pyrolysis
HOMO highest occupied molecular orbital
IC internal conversion
IR infrared
ISC intersystem crossing
LDA lithium diisopropylamide

vii
LUMO lowest unoccupied molecular orbital
Me methyl
MM molecular mechanics
NICS nucleus independent chemical shift
NMR nuclear magnetic resonance
PAH polycyclic aromatic hydrocarbon
Pr propyl
Ph phenyl
TBAF tetrabutylammonium fluoride
TBC tribenzocyclyne
TCNE tetracyanoethene
THF tetrahydrofuran
TIPS trisisopropylsilyl
TMS trimethylsilyl
TMSA trimethylsilylacetylene
UV ultraviolet
VT variable temperature

viii
Acknowledgements
Almost five years have passed since I set foot on American soil, intent to
commence this adventure that I am now finishing. Looking back at those years, I realize
how different my life has become, and I am enjoying the changes that have occurred.
Living and working in a place as diverse, as vibrant, and as intellectually stimulating as
Berkeley, brought about a part of these changes. The other, greater, part was precipitated
by the interaction with many amazing people, most of whom I met in Berkeley.
The person that undoubtedly deserves to be mentioned first is Professor Peter
Vollhardt, my doctoral advisor. Only now do I realize how risky was his decision to bring
me here, and I sincerely hope that some of that risk paid off. The influence Peter exerted
over me was, and still is, tremendous. Always approaching me as an advisor, rather than
a boss, he created a relaxed, yet stimulating atmosphere to work in. In such an
environment, absorbing some of his knowledge, as well as gaining my own, was a
seamless process. From him, I learned how to approach things systematically and with
scientific rigor. I learned to be open to new ideas and critical of them at the same time. It
is my firm intention to implement many of these principles in my independent career,
regardless of the direction in which it develops.
The faculty of the Department of Chemistry of the University of California at
Berkeley is an impressive collection of outstanding scientists and, simply put, pleasant
people. Clearly, I did not interact with all of them equally. First among those to be noted
is Professor Bob Bergman, whose fair judgment and sound advice helped to keep my
course straight during the times of doubt. Professors Dirk Trauner and Dean Toste were

ix
of great help in questions of synthetic methods and catalysis (causing me to nickname
them “walking SciFinders”). Professor Ken Raymond was the first to incite in me a
certain level of appreciation for inorganic chemistry (the scope of which I used to limit to
NaCl and CsF) and he is the one who placed the doctoral hood on my shoulders. Finally,
thanks are due to Professor Jeff Long for a primer of (American) football.
The staff scientists in the Department made everyday routine work exactly that –
routine. This is often taken for granted, although it certainly shouldn’t be. Trying to
correct this, I here express my deep gratitude to Kathy Durkin (Graphics Facility), Fred
Hollander and Allen Oliver (X–Ray), Rudi Nunlist and Herman van Halbeek (NMR) and
Ulla Andersen (Mass Spectral Facility).
A university is not a University without students, and Berkeley should certainly
be proud of its share. Several great guys shared my fate of a graduate student in the
Vollhardt group: Glenn Whitener, Michael Eichberg, Peter Dosa, David Barry, Phil
Leonard, Eli Rodriguez, Stephanie Chan, Mitch Garcia, Ken Windler, and Miles Carter. I
keep Glen and Dave in particularly fond memory, as great friends, drinking partners,
roommates (well, just Dave), and crystallographers (just Glenn).
Two dedicated undergraduate students, Sang-Yeul Lee and Nicole Plath, worked
with me for several months each. I sincerely hope they learned something and wish them
all the very best in their future careers. Renaud Paubelle, Hiu Fung Chu, and Nicolas
Agenet did not work with me, but were fun to have around nevertheless.
As I prepare myself for the carefree life of a postdoc, I cannot but remember some
of the postdocs that worked in the Vollhardt group. Yamato Miura and Sangdon Han
gave me the know-how on phenylenes, early in my work; Christian Cremer did the same

x
for organometallic chemistry and glove box work. Jürg Lehmann gave me his old TV on
one occasion, a present that I enjoyed until very recently. Patrick Betschmann was quite a
guide to the clubs of San Francisco. Yong Yu, Tobias Aechtner, Heiko Oertling, and
Elisa Paredes were good company in many outings, both in Berkeley and in San
Francisco. Ichiro Hisaki showed me a good time in Japan; I hope I reciprocated in
Berkeley. Kaspar Schärer, in whose capable hands I am leaving 640 Latimer, turned
writing this thesis into a fun, beer-filled, experience.
The above classification breaks down when it comes to the people that were
closest to me. Rebecca Abergel and Dorothea Fiedler, my long-time roommates, were
responsible for innumerable parties, lasagna dinners, and trips. They made our house on
1612 Edith St. my “home away from home”, and I will miss them greatly. The incredibly
educated Jens Röder opened my eyes in many ways, took me places (I probably shouldn’t
have been taken to), watched a million movies with me, and remains a good friend to this
day. Thomas Godet spiced up Jens’ and my life with the constant flow of on-the-edge-of-
good-taste jokes. Alex Shafir drank many a gallon of wine and tea with me, and spent
numerous evenings in heated discussions about science and other things. He also served
as a great liaison between my old Eastern European and new American identities. Emily
Dertz and Didier Pomeranc had a taste for alternative music and movies that made me
feel like I was in Belgrade again. Stefan Gradl was that nice guy on your floor you
always wish for. Mircea Dincă kept the Eastern European spirit alive, not the least by
supplying vişinată, Romanian sour-cherry brandy.
A number of my other friends, here, back in Belgrade, and around the world,
made my stay here even more pleasant through the time I spent with them, their letters,

xi
emails and phone calls. The space required to mention them all could easily be
transformed into another chapter; that’s why I will limit myself to an (incomplete) list:
Vladimir Šobajić, Nikola Mihajlović, Dejan Jovanović, Žarko Aćimović, Katarina
Vučićević, Mladen Marinković, Peđa Srejić, Ljubodrag Vujisić, Dejan Gođevac, Ivan
Vučković, Milica Počuča, Bojana Rakić, Jens Freese, María Proupín, Noemi Perez,
Alejandro Lago, Klara Štefflova, Jan Šmidrkal, Blake Farington, Ol'ga Medvedeva,
Adelina Smirnova, Pilar Vizcaíno, Marine Champsaur, Guillermo Rein, José María
Gonzalez, Tatjana Bolić, Vesna Rodić, Željka Čabrilo, Velimir Mimo Radmilović,
Vojislav and Tamara Stamenković, Radu Mihăescu, Ivana Ostojić, Vojislav and Gordana
Srdanov, Ivana Veljković, Petar Milošević, Carsten Dosche, Marina Rotanov, Alex
Krajete, Alessandro Pinto, Andrea Trave, Adam Shellhorse, Natalya Didenko, and others.
Somewhere in the midst of all this, I met a girl with the name of Olivia
Măciuceanu. This intelligent, attractive, cheerful, and kind person stood by me ever
since. Her company has been a true blessing, and I joyfully look forward to the years to
come by her side.
At last, but definitely not least, I need to thank my parents, Šćepan and Fatima
Miljanić, and my sister Bojana Miljanić. Their support, sometimes financial, but always
moral, was constant and fierce. Without them, I probably wouldn’t have written this
thesis; and even if I had done it, it would have had no meaning.
*
This work was sponsored by the National Science Foundation (CHE-0071887)
and the Director, Office of Energy Research, Office of Basic Energy Sciences, Chemical

xii
Sciences Division, of the U.S. Department of Energy, under Contract DE-AC03-
76SF00098. The Center for New Directions in Organic Synthesis is supported by Bristol-
Myers Squibb as a Sponsoring Member and Novartis as a Supporting Member.
I also acknowledge gratefully Arnold Schwarzenegger, the governor of the state
of California, for finding the time to carefully read this thesis and sign my doctoral
diploma.

xiii
VITA
July 8th, 1978 – Born – Belgrade, Yugoslavia
2000 Diploma in Chemistry, University of Belgrade
2000–2005 Research/Teaching Assistant, University of California, Berkeley
2005 Doctor of Philosophy in Chemistry, University of California, Berkeley
Publications
Fiedler, D.; Miljanić, O. Š.; Welch, E. J. “Dichlorooxo(N,N’,N”-trimethyl-1,4,7-
triazacyclononane–κ3N)vanadium(IV)” Acta Cryst., Sect. E 2002, E58, m347.
Miljanić, O. Š.; Vollhardt, K. P. C.; Whitener, G. D. “An Alkyne Metathesis-Based
Route to ortho-Dehydrobenzannulenes” Synlett 2003, 29.
Dosche, C.; Kumke, M. U.; Ariese, F.; Bader, A. N.; Gooijer, C.; Dosa, P. I.; Han, S.;
Miljanić, O. Š.; Vollhardt, K. P. C.; Puchta, R.; van Eikema Hommes, N. J. R.
“Shpol’skii Spectroscopy and Vibrational Analysis of [N]Phenylenes” Phys. Chem.
Chem. Phys. 2003, 5, 4563.

xiv
Bong, D. T.-Y.; Chan, E. W. L.; Diercks, R.; Dosa, P. I.; Haley, M. M.; Matzger, A. J.;
Miljanić, O. Š.; Vollhardt, K. P. C.; Bond, A. D.; Teat, S. J.; Stanger, A. “Syntheses of
Syn and Anti Doublebent [5]Phenylene” Org. Lett. 2004, 6, 2249.
Kumaraswamy, S.; Jalisatgi, S. S.; Matzger, A. J.; Miljanić, O. Š.; Vollhardt, K. P. C.
“Anatomy of a Cyclohexatriene: Chemical Dissection of the π and σ Frame of Angular
[3]Phenylene” Angew. Chem., Int. Ed. 2004, 43, 3711; Angew. Chem. 2004, 116, 3797.
Dosche, C.; Kumke, M. U.; Löhmannsröben, H.-G.; Ariese, F.; Bader, A. N.; Gooijer, C.;
Miljanić, O. Š.; Iwamoto, M.; Vollhardt, K. P. C.; Puchta, R.; van Eikema Hommes; N. J.
R. “Deuteration effects on the vibronic structure of the fluorescence spectra and the
internal conversion rates of D3h [4]phenylene: Α case for excited state π symmetrization
of a cyclohexatriene” Phys. Chem. Chem. Phys. 2004, 6, 5476.
Miljanić, O. Š.; Vollhardt, K. P. C. “[N]Phenylenes: a Novel Class of Cyclohexatrienoid
Hydrocarbons”, in Carbon-rich Compounds: From Molecules to Materials (Eds.: Haley,
M. M.; Tykwinski, R. R.), Wiley-VCH, Weinheim, 2005, in press.
Miljanić, O. Š.; Han, S.; Holmes, D.; Schaller, G. R.; Vollhardt, K. P. C. “Hindered
Rotation in an “Exploded” Biphenyl” Chem. Commun. 2005, 2606.

xv
Miljanić, O. Š.; Holmes, D.; Vollhardt, K. P. C. “1,3,6,9,12,14,17,20-
Octaethynyltetrabenz[a,b,f,j,k,o]-4,5,10,11,15,16,21,22-octadehydro[18]annulene: a
Carbon Rich Hydrocarbon” Org. Lett. 2005, 7, in press.
Zhu, B.; Miljanić, O. Š.; Vollhardt, K. P. C.; West, M. J. “Synthesis of 2,2’,3,3’-
Tetramethyl- and 2,2’,3,3’-Tetra-t-butylfulvalene: Attractive Platforms for Dinuclear
Transition Metal Fragments, as Exemplified by (η5:η5-2,2’,3,3’-tBu4C10H4)M2(CO)n (M =
Fe, Ru, Os, W). First X-ray Crystal Structures of Fulvalene Diiron and Diosmium
Complexes” Synthesis 2005, submitted.

1
Chapter One
[N]Phenylenes: a Novel Class of Cyclohexatrienoid Hydrocarbons1
1.1 Introduction
Aromaticity is one of the most frequently employed concepts in organic
chemistry.2 Despite the omnipresent use of the term, a unique definition is lacking to this
day. Aromaticity is most commonly viewed through the prisms of structural,2a,3
energetic,2a,4 and magnetic2a,5 properties of the systems under study. Structurally,
aromatic bond lengths lie between those of normal single and double bonds. Aromatic
rings are more stable than their open-chain counterparts, and their unusual magnetic
characteristics are reflected in the specific values of magnetic susceptibilities and 1H
NMR chemical shifts. Experimentalists often use qualitative chemical reactivity as
another measure of aromatic character. A unifying characteristic of aromatic compounds
is the preference for substitution versus addition reactions, which is a manifestation of
their tendency to retain the π-electronic skeleton. However, attempts to quantify this
effect have met with limited success.6 Krygowski and Cyrañski describe aromaticity as
an excess property, a deviation from an additive scheme.3 While there is a certain degree
of correlation between the various criteria given above,7 the issues are sufficiently
complex to have induced practitioners to treat aromaticity as a “multidimensional
phenomenon”.7,8
Two simple hydrocarbons, benzene and cyclobutadiene, stand at opposite ends of
the aromaticity continuum, regardless of the criterion chosen. All six C–C bonds in

2
benzene are equal in length (1.398 Å),9 contrasted by the distinctly single (1.526, 1.581
Å) and double (1.441, 1.359 Å) bonds in the crystallographically characterized
peralkylated and persilylated cyclobutadienes, respectively.10 The resonance energies of
these two compounds are also drastically different: relative to an isolated double bond,
benzene is stabilized by 32 kcal mol–1, cyclobutadiene destabilized by 48 kcal mol–1.11
The vastly different stabilities of the two molecules are reflected in the fact that benzene
has been known since Faraday’s times,12 whereas the first isolation of cyclobutadiene (in
an argon matrix) was reported only in 1973.13 This behavior, as well as the corresponding
alternating properties of the higher annulenes,14 is in accord with Hückel’s rule,15 which
states that fully conjugated systems with 4n+2 π-electrons should share the stabilization
of benzene, whereas those with 4n π-electrons should not be stabilized by cyclic
conjugation.
In light of this divergence, the juxtaposition of the benzene and cyclobutadiene
structural motifs fused in a single molecule is an intriguing topology. The simplest stable
system to have such a fusion is biphenylene (1, Figure 1.1), the five resonance forms of
which range from “[12]annulenoid” to increasingly “cyclobutadienoid”. Originally
prepared by Lothrop in 1941 by reacting 2,2’-dibromobiphenyl with Cu2O at 350 °C,16
biphenylene has since been synthesized in a multitude of ways17 and is now
1
Figure 1.1 The resonance forms of biphenylene (1).

3
commercially available.18 Most biphenylene syntheses can be classified into three
categories (Scheme 1.1, left): (i) dimerizations of arynes,17 (ii) oxidative dehalogenations
of 2,2’-dihalobiaryls,16,17,19 and (iii) small molecule extrusions from bridged biaryls.17,20
Despite the presence of cyclobutadienoid circuits, the chemical reactivity of 1
(Scheme 1.1, right) reflects considerable aromatic character: biphenylene undergoes
electrophilic substitution, rather than addition, almost exclusively at the β-positions and
at a rate that is comparable to that of naphthalene.17 The four-membered ring is
thermolyzed, most likely to the 2,2’-biphenyldiyl diradical, which dimerizes to
tetrabenzocyclooctatetraene.21 The aryl–aryl C–C bond in biphenylene is also readily
attacked by a number of metal complexes, and the organometallic intermediates thus
obtained can lead to a variety of ring-opened and insertion products.17,22 Biphenylene is
relatively inert in the Diels-Alder reaction: it does not react with tetracyanoethene,23
benzyne,24 or maleic anhydride.17a However, it functions as a dienophile with respect to
the more electron-deficient tetrachloro- and tetrafluorobenzynes, producing
monoadducts.23

4
R
A
B
XX
E
[M]
∆
R = -N=N-, -SO2-, -CO-
ox.
∆
products
E+
[M]
X4
X4
X = Br, I X = F, Cl
A = NH2, B = COOHA = Br, B = I
Scheme 1.1 General modes of biphenylene preparation (left) and reactivity (right).
The above reactivity notwithstanding, there are strong indications that the
cyclobutadienoid ring has a profound influence on the properties of the system. Thus, a
crystal structure25 highlights the reluctance of 1 to allow conjugation between the two
benzene nuclei, with relatively long aryl–aryl bonds (1.514 Å) and noticeably shorter
fused bonds (1.426 Å). Conversely, the six-membered rings are distorted in such a
fashion as to minimize cyclobutadienoid character in the center, exhibiting pronounced
bond alternation (long bonds 1.426 and 1.423 Å, short bonds 1.372 and 1.385 Å). In
short, the first resonance form in Figure 1.1 is a strong contributor to the description of
the molecule. Despite these distortions, the electronic spectrum of 126 is distinctively
different from that of biphenyl, with peaks that are strongly shifted bathochromically,
signaling a substantial narrowing of the HOMO–LUMO gap. Cross conjugation is also
evidenced by substituent effects on reactivity and IR absorptions.17a Perhaps most

5
informative, the 1H NMR spectrum of 1 exhibits relatively shielded resonances at δ =
6.60 (α-hydrogens) and 6.70 ppm (β-hydrogens),27 ascribed to the presence of a
paramagnetic ring current in the cyclobutadiene ring. 13C NMR spectroscopy is
diagnostic of σ-strain effects and reveals peaks at 117.8 (α-carbon), 128.4 (β-carbon) and
151.7 ppm (quaternary).28
The cumulative experimental data on 1 are to be viewed within the context of
recent advances in the understanding of how both σ- and π-effects impinge on the
aromaticity of benzene.29 To what extent are these effects operational in 1? Shaik,
Hiberty and coworkers have suggested that the D6h structure of benzene is the result of a
σ-π balance: while π-electrons tend to distort the molecule into the D3h symmetry of
cyclohexatriene, the rigidity of the σ-framework acts to enforce higher symmetry.30
Recently, Schaefer and Schleyer31 showed that, as a general rule, π-distortivity
overcomes σ-rigidity in higher annulenes - benzene is thus a fortuitous exception, rather
than a prototype! In this context, 1 is not readily classified as aromatic, non-, or
antiaromatic. Hückel’s rule seemingly does not apply to it2,32 as it would predict a
cyclically delocalized 12π-electron system to be unstable. In addition, the strain of the
four-membered ring complicates the picture, consequently making 1 an excellent subject
on which to study π- and σ-strain in polycyclic compounds.
Biphenylene is the simplest member of a novel class of polycyclic hydrocarbons
in which benzene rings are fused to cyclobutadiene moieties in an alternating manner.
The name [N]phenylenes was coined for these molecules, in which N equals the number
of benzene rings. Higher phenylenes exist as several isomers,33 due to the different modes

6
of fusion between the individual rings. A phenylene can be linear, angular, zigzag,
branched, or circular, based on the mode of fusion, and mixed topologies are possible.
Figure 1.2 exemplifies these designations.
(a) (b) (c) (d) (e) (f)
Figure 1.2 Simple phenylene topologies: (a) linear [4]–, (b) angular [4]–, (c) zigzag [4]–,
(d) branched [4]–, (e) (mixed) bent [4]–, and (f) circular [6]phenylene.
The various topologies of the [N]phenylenes offer the opportunity to test the
hypotheses advanced for the understanding of 1, significantly expand the range of
available strained ring aromatics in a systematic manner, and provide the opportunity to
explore new avenues in the area of electronic materials. For example, appropriate design,
as in angularly fused derivatives, should provide compounds in which benzene ring
distortion is enhanced compared to 1. Alternatively, linear fusion would enforce a
different, bisallylic type deformation, due to symmetry constraints. Moreover, Trinajstić
has suggested that the HOMO–LUMO gap along the linear series should drop rapidly,34
whereas the isomeric zigzag relatives should show much attenuated electronic activation.
Apart from the anticipated unusual physical properties, the reactivity of the phenylenes is

7
expected to be unique, due to the combination of electronic and ring-strain factors.
Synthetically, these structures pose a challenge, in large part due to the presence of
multiple cyclobutadiene rings, the cumulative ring strain of which (on the order of 50
kcal mol–1 per cyclobutadiene ring)35 seems prohibitive.
Phenylenes are closely related to the much larger family of the polycyclic
aromatic hydrocarbons (PAHs). The chemistry of PAHs has been studied
comprehensively with respect to synthesis,36 theory,32 and material science.37 Each
phenylene is correlated to a unique PAH (its “hexagonal squeeze”)38 by formal removal
of the cyclobutadiene cycles through fusion of the attached benzene rings.39 This
topological connection (Figure 1.3) is general, as it exists in one (linear phenylenes –
acenes), two (e.g., circular [6]phenylene sheets – graphite), and three dimensions (e.g.,
archimedene – fullerene). There are important differences, however, starting with the
incremental change in the number of π-electrons along the respective series. For example,
PAHs increase this count in increments of four, thus maintaining their 4n+2 π-character.
Phenylenes, on the other hand, are homologated by the addition of a C6 fragment and
accordingly alternate between (4n+2) and 4n π-electrons. Circular phenylenes preserve
the π-electron count of their open counterparts, whereas PAHs lose two electrons in this
formal transformation and switch from 4n+2 to 4n. Finally, both fullerenes40 and the
three-dimensional phenylenes alternate between 4n+2 and 4n electron count.
Gutman associated several theoretical parameters of the phenylenes with those of
the analogous PAHs.41 He showed that the algebraic structure count (ASC)42 of
phenylenes equals the number of Kekulé structures (K) of their hexagonal squeezes.38
ASC and K serve as measures of stability in nonbenzenoid and benzenoid hydrocarbons,

8
respectively.32,43 The stability of phenylenes therefore appears to parallel that of their
corresponding PAHs. The Wiener index, used to predict the boiling points of
hydrocarbons based on their structures,44 correlates linearly between the two classes.45 It
n n
n n
(a)
(b)
(c)
(d)
Figure 1.3 Phenylenes and topologically related PAHs: a) linear [N]phenylenes and
polyacenes; b) angular/zigzag [N]phenylenes and polyphenanthrenes/helicenes; c)
“circular [6]phenylene sheet” and graphite; d) archimedene (C120) and fullerene (C60).

9
has been proposed that six-membered rings in phenylenes follow the anti-Clar’s rule: if a
certain ring in phenylene is conjugated strongly, its analogue in the hexagonal squeeze is
conjugated weakly (i.e. is “empty” in Clar’s terminology) and vice versa.46 However, as
later Sections will show, this is not a general trend. The list of analogies is not exhausted
here,41,46 and future research may reveal new ties between the two classes.
This introductory Chapter will describe progress in the synthesis and the
exploration of the chemical and physical properties of the phenylenes, in that order. It is
written with the aim of placing all presently known members of this class of
hydrocarbons, including 1, on some comparative footing.47
1.2 Preparation of Phenylenes
1.2.1 Early Synthetic Strategies47
Although 1 had been constructed in a variety of ways17 attempts to extend these
methods to the synthesis of higher phenylenes either failed48 or were limited.
Nevertheless, Barton and coworkers managed to apply the extrusion of nitrogen from
benzodicinnolines by flash vacuum pyrolysis (FVP) (precedented for biphenylene)20 to
the relatively low-yielding preparation of angular and linear [3]phenylene.49 Application
of this technique to the isolation of branched [4]phenylene was unsuccessful,50 possibly
indicating the limits of this methodology.
The breakthrough that enabled the chemistry described in this account came
through the discovery of a new versatile biphenylene synthesis based on the

10
cyclotrimerization of alkynes catalyzed by [CpCo(CO)2].51 Thus, a variety of substituted
biphenylenes could be made by the cocyclization of 1,2-diethynylbenzene (2) with
alkynes, in the case of bis(trimethylsilyl)acetylene (BTMSA) yielding 3 in a remarkable
96% yield (Scheme 1.2).52 Exploiting the silyl substituents as masked ethynyl groups and
using tin instead of silicon, as appropriate,47 gave access to 4 and 6 and, hence, the linear
homologs 553 and 7
54 by iterative sequences involving up to three separate
cooligomerization steps (for 7). These linear [N]phenylenes were targeted first for
synthesis, because they are distinct from their angular isomers, as this topology (in which
cyclobutadienoid circuits cannot be completely avoided) imparts relative electronic
activation.55
(i)
TMS
TMSTMS
TMSN-2
2, N = 24, N = 36, N = 4
3, N = 25, N = 37, N = 4
N-1
+
Scheme 1.2 The last step in the preparation of linear [N]phenylenes 3, 5, and 7 by an
(iterative) single cocyclization strategy: N = 2, (i) [CpCo(CO)2], hν, ∆, 96%; N = 3, (i)
[CpCo(CO)2], hν, ∆, 36%; N = 4, (i) [CpCo(CO)2], THF, hν, ∆, 9h, 30%, then CO (1
atm), 90 °C, 16 h, 100%.
The increasingly long linear sequences necessitated by the single cocyclization
approach were significantly shortened by employing more convergent double
cocyclizations (Scheme 1.3). In this variant, a tetraethynylated arene precursor undergoes
biscycloadditions to generate four rings in a single operation, leading to 9,53,56 11,54 and

11
13.57 The power of the transition-metal-based approach is evident, when one recognizes
that eight of the nine rings in 13 are made by [CpCo(CO)2].
(i), (ii)
TMS
TMSTMS
TMSTMS
TMS
10 11
(i)
TMS
TMSR
RTMS
TMS
8 9a, R = TMS
9b, R = H
(i), (ii)
TMS
TMS
TMS
TMS
TMS
TMS
TMS
TMS
12
13
TMS
TMS
R
R
(ii)
(a)
(b)
(c)
Scheme 1.3 The last step in the preparation of linear [N]phenylenes 9b, 11, and 13 by a
double cocyclization strategy: (a) N = 3, (i) [CpCo(CO)2], PhCH3/DMF, hν, ∆, 6 h, 71%;
(ii) t-BuOK, t-BuOH, THF/DMSO, 85 °C, 6 h, 79%; (b) N = 4, (i) [CpCo(CO)2], THF,
hν, ∆, 13 h, 30%; (ii) CO (1 atm), 120 °C, 72 h, 99%; (c) N = 5, (i) [CpCo(CO)2], THF,
hν, ∆, 16 h, 20%; (ii) CuCl2•2H2O (4 equiv), 1,2-diethoxyethane, H2O/NEt3, 0 °C, 3 h,
40%.

12
The second topology addressed in this early work was the angular frame. In
contrast to their linear counterparts, angular [N]phenylenes possess one, presumably
dominant, resonance form that completely avoids double bonds in the four-membered
rings (Scheme 1.4). This simple representation should translate into increased bond
localization and alkene-like reactivity of the internal nuclei. Retrosynthetically, the
prototype angular [3]phenylene (15) can be unraveled by retrocyclization of the terminal
or the internal rings (Scheme 1.4a). The former strategy, while successful for derivatives
of 15,58 is not readily extendable to the higher homologs of 15, therefore only the latter is
described. This approach is distinct, in as much as it requires an intramolecular alkyne
cyclotrimerization (a cycloisomerization), initially deemed a dubious proposition
considering the large amount of ring strain that is generated during the process. In the
event, however, 15 could be made from 14 by [CpCo(CO)2]-mediated cyclization in 30%
yield.59 The generality of this transformation was evident with the biphenylenyl
substituted analogs of 14, namely 16 and 18, which isomerized successfully to angular
[4]– (17), and [5]phenylene (19), in 30 and 5% yield, respectively.60
Investigations since these early syntheses have brought about a marked
improvement in yields through a stepwise protocol. Thus, exposure of 1461 or 1662 to
[CpCo(eth)2]63 at low temperatures gave the corresponding
cobaltacyclopentadiene(alkyne) complexes, which, when heated in the presence of a
CpCo trap (e.g. 1,3-cyclohexadiene), furnished 15 and 17 in 70 and 51%, respectively.
The reasons for these improvements may be the use of stoichiometric cobalt at low
temperatures which serves to bind all the alkyne units, thus obviating adverse
polymerization or other processes, and the subsequent isomerization-demetallation under

13
conditions that bind CpCo irreversibly, thus avoiding strained ring opening by cobalt
fragments (see Section 1.3.5).
(i)
14 15
16 17
18 19
(i)
(i)
(a)
(b)
(c)
Scheme 1.4 Preparation of angular [N]phenylenes 15, 17, and 19: (a) (i) [CpCo(CO)2],
hν, ∆, 30% or [CpCo(eth)2], THF, –30 °C, followed by CO (8 atm), 100 °C, 70 %; (b)
[CpCo(CO)2], m-xylene, hν, ∆, 30% or [CpCo(eth)2], THF, –25 °C, 16 h, followed by
1,3-cyclohexadiene, THF, 100 °C, 2 h, 51 %; (c) [CpCo(CO)2], m-xylene, hν, ∆, 5%.

14
The third topology to be targeted early was the branched frame of 21b (Scheme
1.5). The central benzene ring of this system was expected to be maximally bond
localized, perhaps representing the first example of a 1,3,5-cyclohexatriene - a long-
sought experimental model for the estimation of the resonance energy in benzene. The
preparation of 21b was achieved via an ambitious triple cocyclization strategy, in which
2064 added three molecules of BMTSA to provide 21a in 39% yield, which could be
readily protodesilylated to the parent 21b.65 The construction of 21a is remarkable,
considering the explosive nature of 20, the regioselectivity of the individual
cotrimerizations, the fact that six rings are generated in one step, and, again, the strain in
the product.
(i)
20 21a, R=TMS
21b, R=H(ii)
R
R
R
R
R
R
Scheme 1.5 Preparation of branched [4]phenylene (21b) by triple cocyclization: (i)
BTMSA, [CpCo(CO)2], hν, ∆, 39%; (ii) CF3COOH, CHCl3, 77%.

15
1.2.2 Syntheses of New Phenylenes
The previous section summarized the essence of what was known at the time of
the last review of the subject.47a Since then, twelve new phenylenes of increasing size and
topological complexity have been prepared. With the exception of the linear series, forays
have been made into the assembly of all types of phenylenes depicted in Figure 1.2. The
following five subsections will describe, in order, the syntheses of angular [5] –
[9]phenylene, also dubbed ‘heliphenes”, because of their helical configuration;66 the
preparation of zigzag [4]– and [5]phenylene, through both intra- and intra/intermolecular
cyclizations; the construction of three phenylenes with new mixed topologies; new
branched phenylenes; and synthetic efforts towards the (still) elusive class of circular
phenylenes.
1.2.2.1 Angular and Helical Phenylenes
Molecular models indicate that, starting with angular [6]phenylene, the two ends
of the angular phenylenes suffer steric interactions that renders them helical, an
expectation that was quantified theoretically.67 The hexagonal squeezes of these helical
phenylenes (heliphenes) are helicenes, a class of PAHs that has received much scrutiny.68
As described in Section 1.2.1, the key step in the preparation of angular [3]– to
[5]phenylene employed a single cobalt-catalyzed cycloisomerization of the respective
precursor triynes 14, 16, and 18. Such a strategy was no longer feasible for the higher
analogs, as suitable building blocks based on functionalized angular [3]phenylene

16
derivatives are not (yet) readily available. Hence for the higher systems, multiple
cycloisomerizations had to be designed using the same building blocks. The already
known angular [5]phenylene (19) was chosen as a testing ground for a double cyclization
scheme (Scheme 1.6).66 Crucial for the success of the preparation of starting material 24
was the discovery that 1,2,3,4-tetrabromobenzene can be selectively alkynylated at the 1-
and 4-positions to give 22.69 Sonogashira coupling of 22 to the previously reported 2360
produced 24a (57%). The deprotected 24b was cyclized to 19 in 33% yield.66
(i)
22, R = DMTS
Br
Br
24a, R = DMTS
24b, R = H(ii)
R
R
R
2
23, R = DMTS
R
RR
R
(iii)19+
Scheme 1.6 Preparation of angular [5]phenylene (19) by double intramolecular
cyclization: (i) [PdCl2(PPh3)2], CuI, NEt3, ∆, 57%; (ii) TBAF, THF, (95%); (iii)
[CpCo(CO)2], m-xylene, hν, ∆, 33%.
Replacing the terminal benzene substituents in 24b once and twice by biphenylenyl
groups, in a manner analogous to that employed in the extension of the synthesis of 15 to
17 and 19 (Scheme 1.4), furnished hexaynes 25 and 27, respectively, both of which

17
underwent double cycloisomerization to [6]– (26, 12%) and [7]heliphene (28, 8%),
respectively (Scheme 1.7).66
25
(i)
26
2827
(i)
(a)
(b)
Scheme 1.7 Preparation of heliphenes 26 and 28 by double intramolecular cyclization:
(a) (i) [CpCo(CO)2], m-xylene, hν, ∆, 30 min, 12%; (b) (i) [CpCo(CO)2], m-xylene, hν,
∆, 30 min, 8%.
With the synthesis of 28, we have reached the limits of the double intramolecular
cyclization approach, and access to the next higher homologs required the execution of
even more ambitious triple cyclizations. The viability of such reactions was tested with
28 (Scheme 1.8).70

18
(i), (ii)
29, R = DMTS
I
R
RR
R
RR
3130, R = DMTS
(iii), (iv)
(v)
28
Scheme 1.8 Preparation of helical [7]phenylene (28) by triple intramolecular cyclization:
(i) TMSA, [PdCl2(PPh3)2], CuI, NEt3, 85 °C, 14 h 52%; (ii) K2CO3, THF/MeOH, 30 min,
92%; (iii) 29, [PdCl2(PPh3)2], CuI, NEt3, 65 °C, 14 h, 41%; (iv) TBAF, THF, 23 °C,
(95%); (v) [CpCo(CO)2], m-xylene, hν, ∆, 1 h, 2%.
Thus, starting with tetrayne 29,66 Sonogashira coupling with TMSA and selective
deprotection provided 30. This alkyne was reacted with another equivalent of 29 and the
resulting nonayne completely desilylated to give 31. Cobalt-catalyzed cyclization then
afforded 28 in 2% yield. While this yield is low, the reaction generates nine rings in one
step, including six four-membered rings with an estimated strain of over 300 kcal mol–1.
Having demonstrated the feasibility of triple cycloisomerizations, synthetic
schemes were once again developed that replaced the terminal benzene moieties with
biphenylene, giving rise to 32 and 34, respectively. The former then provided 33, the
latter 35 (both in 2% yield; Scheme 1.9).70 These two compounds represent the largest
phenylenes known.

19
32
(i)
33
3534
(i)
(a)
(b)
Scheme 1.9 Preparation of heliphenes 33 and 35 by triple intramolecular cyclization: (a)
(i) [CpCo(CO)2], m-xylene, hν, ∆, 30 min, 2%; (b) (i) [CpCo(CO)2], m-xylene, hν, ∆, 20
min, 2%.
1.2.2.2 Zigzag Phenylenes
The family of zigzag phenylenes is closely related to the angular isomers, in as
much as it has the same repeating angular fusion of benzocyclobutadiene units, although
“helical strain” is absent.67 The electronic properties of its members are thus expected to
be fairly similar. These phenylenes are also interesting as models for the one-dimensional
zigzag-phenylene polymer, with properties different from the infinite linear

20
[N]phenylene.71 Finally, both archimedene (Figure 1.2d)55a,72 and the octahedral C4873
contain zigzag phenylene subunits.
The topological analogy between the angular and the zigzag family of phenylenes
is reflected in the resemblance of the synthetic strategies to the two classes. The parent
zigzag [4]phenylene (38) was approached via 37, a regioisomer of 16 (Scheme 1.4), in
which the two alkynyl substituents on the biphenylene nucleus have traded places
(Scheme 1.10a). Compound 37 was in turn made via a three-step elaboration of 1,2-
diiodobiphenylene (36).60 Cobalt then converted 37 into 38 in 31% yield.74 An alternative
route (Scheme 1.10b) constituted the first example of a combination of intra- and
intermolecular cyclizations in a single reaction step. It started with tetrabromobenzene
39, which was elaborated with 23 (Scheme 1.6), followed by three-fold coupling with
TMSA and full deprotection, ultimately giving 41. This pentayne was cocyclized with
TMSA and subsequently protodesilylated to afford 38. This method was extended to the
synthesis of the bent [4]phenylenes (Section 1.2.2.3) and could, in principle, be used also
on a simplified route to angular [4]phenylene (17), a task yet to be tackled.

21
36
(i), (ii), (iii)
I
I
37
(iv)
Br
Br
Br
Br
Br
Br
Br
R
(i)
38
39 40, R = DMTS 41
(iv), (v)
(ii), (iii)(b)
(a)
Scheme 1.10 Two syntheses of zigzag [4]phenylene (38): (a) intramolecular approach, (i)
23, [PdCl2(PPh3)2], CuI, Et3N, 23 °C, 15 h; (ii) TMSA, [PdCl2(PPh3)2], CuI, Et3N, 50 °C,
2 d, 62% (over 2 steps); (iii) TBAF, THF, 23 °C, 40 min; (iv) [CpCo(CO)2], m-xylene,
hν, ∆, 18 h, 29% (over 2 steps); (b) mixed intra/intermolecular approach, (i) 23,
[Pd(PPh3)2Cl2], CuI, Et3N, 50 °C, 24 h, 66%; (ii) TMSA, [Pd(PPh3)2Cl2], CuI, piperidine,
100 °C, 7 d; (iii) TBAF, THF, 33% (over 2 steps); (iv) [CpCo(CO)2], BTMSA, hν, ∆, 10
h, 15%; (v) CF3CO2H/CHCl3, 23 °C, 12 h, 74%.

22
A variant of the double cycloisomerization route to angular [5]phenylene (Scheme
1.6) was used to prepare zigzag [5]phenylene (44, Scheme 1.11).74 Starting once more
with 39, double alkynylation with 23 assembled tetrayne 42, which was further
substituted with TMSA. Removal of all the silyl protecting groups provided 43, a
regioisomer of 24b (Scheme 1.6). Compound 43 was then cyclized to 44 in 2% yield.74
(i)39
Br
Br
R
R
(ii), (iii) (iv)
42, R = DMTS 43 44
Scheme 1.11 Synthesis of zigzag [5]phenylene (44): (i) 23, [Pd(PPh3)2Cl2], CuI, Et3N, 23
°C, 5 d, 66%; (ii) TMSA, [Pd(PPh3)2Cl2], CuI, piperidine, 80 °C, 3 d; (iii) TBAF, THF,
23 °C, 2 h, 80% (over 2 steps); (iv) [CpCo(CO)2], m-xylene, hν, ∆, 2 h, 2%.
1.2.2.3 Phenylenes with Mixed Topology: the “Bent” Isomers
All the phenylene topologies discussed so far contained only one mode of
repeating fusion: either linear or angular. The smallest molecule with mixed
linear/angular connectivity is bent [4]phenylene (48, Scheme 1.12), the last [4]phenylene
isomer to be made.54,60,65,74 This isomer is intriguing, in particular because of the unusual

23
nature of the two juxtaposed internal six-membered rings and their surroundings. Its
synthesis entailed application of a regioisomeric variation of the intramolecular approach
to 17, through 46 (Scheme 1.12a), formed by reaction of 2,3-diiodobiphenylene (45)53
with 23. Further ethynylation eventually resulted in triyne 47, which was cyclized to 48
in 33% yield.75 The 9,10-bis(trimethylsilyl) derivative of 48, 52, was made by the
combination of intramolecular cyclization and cocyclization with BTMSA, precedented
for 38 (Scheme 1.12b).74 The starting 1,2,4,5-tetrabromobenzene (49) was
desymmetrized into 50. A sequence of two Sonogashira couplings, first with 23 and then
with TMSA, was followed by the full deprotection to give 51. Cyclization proceeded in
19% yield, producing 52.75

24
(i)
46, R = DMTS
(ii), (iii)
48
I
I IR
(iv)
45
47
(a)
(i)
50
(ii), (iii), (iv)
52
Br
Br
I
Br
(v)
49 51
(b)
Br
Br
TMS
TMS
Br
Br
Scheme 1.12 The syntheses of bent [4]phenylenes 48 and 52: (a) (i) 23, [Pd(MeCN)2Cl2],
CuI, PPh3, piperidine, 90 °C, 40 h, 16%, (ii) TMSA, [Pd(PPh3)2Cl2], CuI, piperidine, 44
h, 93%, (iii) TBAF, THF, 20 min, (95%), (iv) [CpCo(CO)2], m-xylene, hν, ∆, 15 h, 33%;
(b) (i) BuLi, Et2O, –78 °C, followed by I2, Et2O, –78 °C, 93%, (ii) 23, [Pd(PPh3)2Cl2],

25
CuI, PPh3, Et3N, 23 °C, 15 h, (iii) TMSA, [Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 2.5 d, 29%
(over two steps), (iv) TBAF, THF, 2 h, (95%), (v) [CpCo(CO)2], BTMSA, hν, ∆, 16 h,
19%.
The success of Scheme 1.12 encouraged approaches to the higher homologues of
48, anti- (56, Scheme 1.13), and syn-doublebent [5]phenylene (60, Scheme 1.14). These
systems would allow an investigation of the effect of increasing bond localization of the
termini of the linear [3]phenylene fragment on the properties of the center piece.
Strategically, the approach to both systems was modeled after Schemes 1.6 (for 19) and
1.12, utilizing regioisomeric double intramolecular cyclizations.
(i)
53
BrI
Br I
Br
Br
R
56
R
(ii), (iii) (iv)
54 55
Scheme 1.13 The synthesis of anti-doublebent [5]phenylene (56): (i) 23, [Pd(PPh3)2Cl2],
CuI, Et3N, 72%; (ii) TMSA, [Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 70%; (iii) TBAF, THF, 2
h, (95%); (iv) [CpCo(eth)2], THF, –25 °C, 16 h, followed by 1,3-cyclohexadiene, THF,
110 °C, 2 h, 7%.

26
The synthesis of anti-doublebent [5]phenylene (56)76 commenced with the
tetrahalogenated C2h-symmetric 53.77 Another use of the versatile building block 23
provided 54. Subsequent coupling with TMSA and deprotection afforded 55. The
cyclization failed initially when attempted with [CpCo(CO)2] as the catalyst, but was
later rendered successful by the application of the milder [CpCo(eth)2] conditions.76 In an
analogous (but slightly altered) manner, the synthesis of 60 (Scheme 1.14) started with
1,3-dibromo-4,6-diiodobenzene (57)78 as a C2v-symmetric template. Reaction with TMSA
and deprotection gave 1,3-dibromo-4,6-diethynylbenzene. Another Sonogashira coupling,
this time with 1-bromo-2-iodobenzene, provided the tetrabrominated 58. This material
underwent a four-fold exchange of bromides with TMSA and, after fluoride-assisted
deprotection, yielded hexayne 59. The cyclization to 60 proceeded smoothly under the
conditions of [CpCo(eth)2] catalysis.
(i), (ii), (iii)
57
BrI
I Br
Br
Br
Br
60
Br
(iv), (v) (vi)
58 59
Scheme 1.14 The synthesis of syn-doublebent [5]phenylene (60): (i) TMSA,
[Pd(PPh3)2Cl2], CuI, Et3N, 23 °C, 2 h, 96%; (ii) KOH, Et2O/EtOH, (iii) 1-bromo-2-
iodobenzene, [Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 44% (over 2 steps); (iv) TMSA,

27
[Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 47%; (v) TBAF, THF, 2 h, (95%); (vi) [CpCo(eth)2],
THF, –25 °C, 16 h, followed by 1,3-cyclohexadiene, THF, 110 °C, 2 h, 14%.
1.2.2.4 Branched Phenylenes
Two other types of mixed topology are the branched/linear and branched/angular
motifs. To what extent can the bond localization of the central cyclohexatriene in the
branched [4]phenylene (21b) be manipulated by additional fusions? One might expect
linear fusion to increase it, whereas angular fusion should effect the opposite. To validate
this expectation, branched [5]phenylene 64b (Scheme 1.15), C3h-symmetric branched 66
(Scheme 1.16), and its D3h-symmetric isomer 71 (Scheme 1.17) were constructed.
The preparation of 64b relied on a modification of the iterative cocyclization
strategy to linear [N]phenylenes (Section 1.2.1).79 Thus, diyne 6165 was cocyclized with
bis(trisisopropylsilyl)-1,3,5-hexatriyne (62).57 The resulting 63 was deprotected and
subjected to a second cocyclization, this time with BTMSA, providing the Y-shaped 64a
in 33% yield (over 2 steps). Acid-catalyzed removal of the silyl groups produced the
parent branched [5]phenylene (64b, Scheme 1.15).79

28
61
(i)
TIPS
TIPS
TIPS
TIPS
R
R
62 63
(ii), (iii)
64a, R = TMS
64b, R = H(iv)
Scheme 1.15 The synthesis of branched [5]phenylene (64b): (i) [CpCo(CO)2], PhCH3,
hν, ∆, 16 h, 32%; (ii) TBAF, THF, 23 °C, 2 h, (95%), (iii) BTMSA, [CpCo(CO)2], THF,
hν, ∆, 16 h, 33%; (iv) CF3CO2H, CH2Cl2, 23 °C, 16 h, 65%.
The synthesis of C3h-symmetric branched 66 (Scheme 1.16) represents an
extension of Scheme 1.16. It starts with hexaethynylbenzene (20),64 which was
cocyclized with 62 in 38% yield. The resulting hexaalkynyl substituted 65a was treated
with TBAF to afford 65b. This material was cocyclized with BTMSA in 37% yield (over
2 steps), producing the C3-symmetric hexakis(trimethylsilyl)[7]phenylene (66).79

29
20(i)
R
R
TMS
TMS
66
R
R
R R
TMSTMS
TMS
TMS
65a, R = TIPS
65b, R = H(ii)
(iii)
Scheme 1.16 The synthesis of C3h-symmetric branched 66: (i) 62 (7 equiv),
[CpCo(CO)2], PhCH3, hν, ∆, 16 h, 38%; (ii) TBAF, THF, 23 °C, 30 min, (95%); (iii)
BTMSA, [CpCo(CO)2], THF, hν, ∆, 16 h, 37%.
For the preparation of 71 (Scheme 1.17), a strategy was necessary that
desymmetrized the sixfold symmetry of 20 to allow for the generation of angular fusion.
It started with trialdehyde 67,80 which was coupled with TMSA in 97% yield, to give 68.
A Corey-Fuchs dibromoolefination, followed by treatment with LDA provided the

30
hexayne 69. The remaining three benzene rings of 70 were introduced by reacting 69
with 1-iodo-2-(TMSethynyl)benzene.81 Base-catalyzed removal of all six TMS groups
was followed by threefold [CpCo(CO)2]-mediated cycloisomerization to 71 (2% yield).82
Br
BrBr
O
O
O
H
H
H O
O
O
H
H
H
TMS
TMS TMS TMS
TMS
TMS
TMSTMS
TMS
TMS
TMSTMS
(i) (ii), (iii), (iv)
(vi), (vii)
(v)
67 68 69
7071
Scheme 1.17 The synthesis of 71: (i) TMSA, [Pd(PPh3)2Cl2], CuI, Et3N, THF, 97%; (ii)
CBr4, Zn, PPh3, CH2Cl2, 99%; (iii) LDA, THF, –78 °C; (iv) aq. NH4Cl, 95% (over 2
steps); (v) 1-iodo-2-(TMSethynyl)benzene, [Pd(PPh3)2Cl2], CuI, i-Pr2NH, THF, 77%; (vi)
K2CO3, MeOH/THF, 61%; (vii) [CpCo(CO)2], m-xylene, hν, ∆, 1.2%.
Compound 71 has the distinction of representing the largest synthesized subunit of the
“Archimedean solid” archimedene (C120, Figure 1.2d).55,72 The successful conversion of

31
the nonayne precursor to 71 provides a valuable additional example of a triple
intramolecular cyclization, differing topologically from those employed on route to 28,
33, and 35 by the fact that six (of nine) reacting triple bonds reside on a single benzene
ring.
1.2.2.5 Circular Phenylenes
Circular phenylenes have the distinguishing characteristic of a resonance picture
that includes forms that encompass both the inner and outer peripheral loops, a
phenomenon described as superdelocalization.83 This class of phenylenes remains
elusive.69,84 The simplest member of this series that does not suffer from additional
“circular” strain is [6]phenylene 77d (Scheme 1.18), also christened antikekulene59 to
highlight its relationship to kekulene, its all-benzenoid relative with an equal number of
rings.85 In antikekulene, avoidance of (benzo)cyclobutadienoid local circuits is expected
to enhance the contribution of the potentially superdelocalized resonance form depicted
for the structure in Scheme 1.18, albeit with the added and destabilizing feature that both
inside and outside peripheries contain a 4n electron count.
An oligoalkyne polycyclization route to any circular phenylene is conceptually
different from those developed for the other topologies, as it requires the elaboration of a
suitably functionalized dehydrobenzannulene, a significant synthetic enterprise in its own
right. This is witnessed by the fact that even the preparation of the parent
dehydrobenz[12]annulene (also known as tribenzocyclyne, TBC) remains a challenging
task,84b,86 almost forty years after its original synthesis by Staab and Graf.87 In the case of

32
77d, the appropriate tribenzocyclyne is 74d (Scheme 1.18). Its synthesis commenced
with 39, which was manipulated into bromide 72a. Bromine–iodine exchange, followed
(i), (ii)39
(vii)
Br
R
TMS
R
I
R
R
(iii), (iv)
RR
R
R
RR
R
R
R
R
RR
R
R
RR
RR
72a, R = DMTS72b, R = CH2C6H1172c, R = Pr
73a, R = DMTS73b, R = CH2C6H1173c, R = Pr
74a, R = DMTS74b, R = CH2C6H1174c, R = Pr74d, R = H
(v), (vi)
(ix)
(viii)
75b, R = CH2C6H1175c, R = Pr75d, R = H
R
R
R
R
R
R
76b, R = CH2C6H1176c, R = Pr76d, R = H
77b, R = CH2C6H1177c, R = Pr77d, R = H
Scheme 1.18 Attempted syntheses of circular [6]phenylenes 77b–d: (i) RC≡CH,
[Pd(PPh3)2Cl2], CuI, Et3N, 23–60 °C, 3 d, 80% (72a), 58% (72b), 51% (72c); (ii) TMSA,
[Pd(PPh3)2Cl2], CuI, Et3N, 100 °C, 4 h–2.5 d, 49% (72a), 31% (72b), 27% (72c); (iii)
BuLi, Et2O, –78 °C, 30 min; (iv) I2, Et2O, from –78 °C to 23 °C; (v) K2CO3, CH3OH, 1 h,
91% (73a), 86% (73b), 73% (73c); (vi) CuCl, NH4OH, EtOH, 1 h, followed by pyridine,
∆, 6 h, 20% (74a), 36% (74b), 32 % (74c); (vii) TBAF, THF, CH3CN, 5 h, 95%; (viii)

33
[CpCo(CO)2], m-xylene, hν, ∆, 20 min, 45% (75b), 14% (75c), 0% (75d); (ix)
[CpCo(CO)2], 1,2,4-trichlorobenzene, hv, ∆, 20 min, 40% (76b), 14% (76c).
by TMS group removal delivered 73a in 91% overall yield. Attempted cyclocoupling
under Sonogashira conditions was complicated by irreproducibility. Switching to the
Stephens-Castro reaction gave better results, and cyclyne 74a emerged in 20% yield.
Deprotection with TBAF gave 74d in 95% yield.69 Compound 74d is the largest
synthesized substructure of the novel carbon allotrope88 graphyne89 and organizes into a
remarkable supramolecular framework in the crystal.84a
Unfortunately, attempted threefold cobalt-mediated cyclization of 74d gave only
insoluble dark brown materials. Suspecting that the insolubility of intermediates or 77d
itself might be the problem, the cyclohexylmethyl- and propyl-substituted materials were
prepared (74b and 74c, respectively; Scheme 1.18). Application of standard cyclization
conditions to these derivatives furnished the singly cyclized 75b and c, respectively.
Resubjecting these materials to the reaction conditions in the higher-boiling 1,2,4-
trichlorobenzene afforded the products of the double cyclization 76b and c, respectively.
Despite extensive efforts, the third cyclization did not take place even in sulfolane
(reaction temperature ~ 200 °C). This result is puzzling, especially in view of the ready
metallacycle formation from triyne 14 and [CpCo(eth)2].61 A possible explanation might
be the increasing distance between the reacting triple bonds along the series 74b–75b–
76b (all of which were crystallographically characterized). The notion that the problems
of the final cyclization are kinetic in nature is supported by the finding that the

34
conversion of 76d into 77d is calculated to be exothermic by –45.50 kcal mol–1
(B3LYP/6–31G*).69
To summarize this section, to date nineteen phenylenes have yielded to synthesis.
They can be divided broadly into 5 families (# of examples): linear (3), angular/helical
(7), zigzag (2), bent (3), and branched (4). Their topologies have been accessed through
26 different routes, 15 of which involved in the crucial step an all-intramolecular cobalt-
catalyzed cyclization, nine used intermolecular variants, and two a combination of the
two strategies.
1.3 Comparative Reactivity of the Phenylenes
The presence of strained cyclobutadiene moieties35 and cyclohexatrienoid rings
renders the phenylenes susceptible to various reactions. Thus, hydrogenation, metal
complexation, ring openings, and cycloadditions are all feasible. Early work focused on
the chemistry of linear [3]–53,90 and branched [4]phenylene65,91 and has been reviewed.47a
The following sections will concentrate on selected recent examples featuring the
comparative reactivity of angularly fused cyclohexatrienoid rings.
1.3.1 Hydrogenation
With the caveat of the mechanistic complexities of heterogenous catalytic
hydrogenations,92 the relative ease of hydrogenation of the cyclohexatrienoid rings in the

35
phenylenes (Scheme 1.19) can be used as a qualitative measure of reactivity. Thus, while
9b53 and 21b91 could be hydrogenated readily (Pd/C, 1 atm H2), 15 required more
stringent conditions (Pd/C, 10 atm H2),59 and 1 was inert or underwent hydrogenolytic
four-membered ring opening.17a
(i)
78
HH
H
HH
H
79
80
9b(a)
(i)15(b)
(i)21b(c)
H H
H
H
HH
HH
Scheme 1.19 Hydrogenation of phenylenes 9b, 15, and 21b: (a) (i) Pd/C, H2 (1 atm),
THF, 23 °C, 3 h, 74%; (b) (i) Pd/C, H2 (10 atm), THF, 23 °C, 99%; (c) (i) Pd/C, H2 (1
atm), THF, 23 °C, 18 h, 87%.
Preliminary observations thus suggested a reactivity order of 9b ≥ 21b > 15 > 1. The
ambiguities in the kinetics notwithstanding, thermodynamic measurements clearly point
to the fact that the central ring in 21b is more cyclohexatrienic than that in 15. Thus, the
measured heats of hydrogenation, corrected for the strain present in the respective all-cis-

36
hexahydroderivatives 80 and 79 (Scheme 1.19), are –(83.0 to 84.2) kcal mol–1 and –(68.1
to 73.6) kcal mol–1, respectively, revealing that the central ring in 15 enjoys more
resonance stabilization than that in 21b by at least ~ 10 kcal mol–1. Perhaps even more
interestingly, the corrected ∆Hhyd of 21b is remarkably close to that estimated for three
cyclohexene double bonds (–84.8 kcal mol–1), suggesting that the central ring is a true
cyclohexatriene, possibly devoid of any resonance interaction between the π bonds. Such
a picture has also been painted employing other methods.93 Finally, the estimated ∆Hhyd
of biphenylene (1), corrected for strain in the product, using a similar approach to that
described for 15 and 21b, has a value of –64.8 kcal mol–1, attesting to its expected
attenuated activation relative to the other two phenylenes, although still featuring benzene
rings that are less aromatic than benzene itself (∆Hhyd = –49.1 kcal mol–1).35
The relatively higher reactivity of 9b compared to 15 made the hydrogenation of
bent [4]phenylene (48) an interesting proposition: which one of the two internal rings is
the more reactive? On the basis of simple resonance arguments, the fusion of an
additional benzocyclobutadiene fragment should stabilize the linear and destabilize the
angular component of 48, and thus possibly invert the reactivity order observed for the
parents 9b and 15. Because 48 was not available in sufficient quantities, the problem was
addressed with its bis(trimethylsilyl) derivative 52 (Scheme 1.20).75 Upon subjecting 52
to the reaction conditions previously used on 9b and 21b (Pd/C, 1 atm H2), the B ring
was hydrogenated cleanly to give 81. This result was clearly in consonance with
expectation, even though the effect of the presence of the remote silyl groups in 52 may
have contributed to its outcome. More experimentation is in order to corroborate these
findings.

37
(i)TMS
TMS
52
TMS
TMS
81
H
D C B
A
H
HH
Scheme 1.20 The hydrogenation of 52: (i) H2 (1 atm), Pd/C, Et2O, 10 min, 44%.
In syn-doublebent [5]phenylene (60), the central linear moiety is stabilized even
further compared to 48, due to the presence of two angular fusions. The angular
components, in turn, are still destabilized compared to the parent 15, but to a lesser extent
than in 48 (since they “share” the destabilization caused by the linear fusion). An overall
decrease in reactivity of all rings, relative to 48 (or 52) is thus expected. Preliminary
results confirm this prediction, since, in contrast to 52, 60 remains inert to hydrogenation
(Pd/C, 1 atm H2, 2 h).94 Similarly, dipropyl substituted zigzag [5]phenylene resisted
hydrogenation even at increased pressures (Pd/C, 12.2 atm H2),74 in agreement with the
notion that extension of the angular/zigzag phenylene frame causes an (at least initial)
decrease in cyclohexatrienoid character of the internal rings.47a,60
1.3.2 Oxacyclopropanation and Cyclopropanation
In light of the difficulty to attach meaning to the relative kinetic reactivities of the
phenylenes in catalytic hydrogenations, it would be instructive to inspect their direct
reactions with electrophilic species capable of attacking the activated six-membered
rings. Indeed, and further corroborating the cyclohexatrienic character of the phenylenes,

38
it was possible to effect oxacyclopropanations of 1, 15, and 21b. Using
dimethyldioxirane (DMDO)95 as the oxidant, biphenylene (1) was converted sluggishly
into the corresponding trisoxacyclopropane 82 (Scheme 1.21a). Its stereochemistry was
(i)
82
84a, R = H84b, R = TMS
86
1(a)
(i)(b)
(i)21b(c)
O
O
O
O
O
O
85
(ii)
TMS
TMS
TMS
TMS
O
O
O
O
O
R
R
R
R
15, R = H83, R = TMS
R
R
R
R
Scheme 1.21 Oxacyclopropanation of 1, 9b, 83, and 21b: (a) (i) DMDO, acetone, 23 °C,
24 h, 30%; (b) (i) for 9b: DMDO, acetone, 23 °C, 30 min, (100%), for 83: DMDO,
acetone, 23 °C, 1 h, (100%); (ii) for 84b only: DMDO, acetone, 23 °C, 6 h, 26%; (c) (i)
DMDO, acetone, 23 °C, 84%.

39
assigned as trans on mechanistic grounds; however, a cis-geometry would also be
consistent with the spectral data.96 In contrast to the slow conversion of 1, angular
[3]phenylene (15) was oxidized comparatively quickly under these conditions, but only to
the moisture-sensitive (and hence difficult to completely characterize)
bisoxacyclopropane 84a (Scheme 1.21b). Switching to tetrakis(trimethylsilylated) 83
provided the more stable 84b, the connectivity of which could be proven by NMR
spectroscopy. Only on renewed oxidation of this compound was the trisoxacyclopropane
85 obtained in 26% yield.97 In the latter, the asymmetry of the trans,trans,cis-arrangement
manifests itself diagnostically in the 1H NMR spectrum. This stereochemical assignment
also corroborates the proposed trans-geometries of 84a and b, for which NMR data were
not definitive,97 and possibly provides further support for the proposed structure of 82.
Finally, and to complete the series, 21b underwent complete, but now all-cis,
oxacyclopropanation to 86 during the course of just one hour (84% yield; Scheme
1.21c).91 Its structure was ascertained by an X-ray crystallographic analysis (Figure 1.4).
The different stereochemical outcome of the oxidations of 1 and 15 compared to
21b may be a consequence of the unique all-benzofusion in 86, resulting in significant
steric hindrance to trans attack due to the outside rings, even after the first
oxacyclopropanation and pronouncedly so after the second.

40
Figure 1.4 X-ray crystal structure of 86 (thermal ellipsoids are shown at 50%
probability).
Compared to the results of the above oxidations, the picture is less clear for the
topologically seemingly analogous cyclopropanations. Thus, 1 transforms in the presence
of ethyl diazocarboxylate to 88 only at elevated temperature (Scheme 1.22a), presumably
through intermediate adduct 87.17a,98 On the other hand, while angular [3]phenylene (15)
was inert to modified Simmons-Smith conditions (Et2Zn, PhCH3, 60 °C),97,99 branched
21a,b responded to this reagent by providing the triscyclopropanated 89a and b in
excellent yields (Scheme 1.22b).91 In analogy to the trisoxacyclopropanation of 21b
(Scheme 1.21c), carbene addition occurs all-cis, as rigorously ascertained by an X-ray
crystal structure of 89a.

41
(i)
87
89a, R = TMS89b, R = H
1(a)
(i)21a,b(b)
COOEtCOOEt
88
R
R
R
R
R
R
Scheme 1.22 Cyclopropanation of 1, 21a, and 21b: (a) (i) ethyl diazoacetate
(N2CHCOOEt), 165 °C, 15%; (b) (i) Et2Zn, PhCH3, 60 °C, 78% (89a), 97% (89b).
1.3.3 [4+2]Cycloadditions
Another measure of the degree of diene character of phenylenes is their relative
susceptibility to undergo [4+2]cycloadditions. Such reactions would lead to highly
strained products, which might be expected to be labile. In addition, cycloadditions
should be regiocontrolled by the desire to avoid ensuing cyclobutadienoid circuits. In this
respect, singlet oxygen100 has proven to be an interesting dienophile. For example, the
oxidation of 1 with this species (Scheme 1.23a)96 was proposed to generate intermediate
endoperoxide 90, which underwent ring-opening to 91, followed by a series of skeletal
rearrangement and an ene-reaction with the reagent, ultimately giving hemiacetal
hydroperoxide 92 in 56% yield. Tetrasilylated linear [3]phenylene (9a) reacted with
atmospheric oxygen through an analogous endoperoxidation-ring opening sequence

42
giving the diketone 94. Interestingly, no irradiation, or added sensitizer were required for
this reaction to proceed; it has been proposed that phenylenes themselves act as
sensitizers for oxygen.101 Unlike the related 91, this compound could be isolated and
characterized (along with its E-isomer).101 In both cases, the regioalternative mode of
initial cycloaddition, which would have generated one (for 1) or two (for 9a)
benzocyclobutadiene subunits, was avoided.
90
1(a)
(i)9a(b)
91
OO
O
93
OO
TMS
TMS
TMS
TMS
94
TMS
TMS
TMS
TMSO
O
92
OOHHOO
O
(i)
Scheme 1.23 Reactions of 1 and 9a with singlet oxygen: (a) (i) O2,
tetraphenylporphyrine, hν, acetone, –40 °C, 5 d, 56%; (b) (i) O2, hν, C6H6, 23 °C, 1–2 h,
80% (Z:E = 3:1, by NMR).
On the basis of the above results, analogous endoperoxidation of the angular 15
was expected to be even more facile, as the subsequent skeletal rearrangement should
allow the opening of both four-membered rings. This expectation was confirmed by the
reaction of 15 with singlet oxygen (now requiring irradiation in the presence of a
sensitizer), which produced Z-dione 96 in 70% yield (Scheme 1.24a).97 The

43
corresponding conversion of dipropyl-substituted zigzag [5]phenylene 97 (again without
added sensitizer; Scheme 1.24b) provided 98, the structure of which was confirmed
(i)
95
15(a)
(b)
O O
O
O
96
97
Pr Pr Pr
(i)
OO
98
Pr
Scheme 1.24 Reactions of 15 and 97 with singlet oxygen: (a) (i) O2, methylene blue, hν,
CH2Cl2, 23 °C, 70%; (b) (i) O2, 23 °C, 12 h, 9%.
crystallographically (Figure 1.5).74 Unfortunately, no data are available that would allow
for an estimate of the relative reactivity of 1, 9a, 15, and 97. However, it is interesting to
note that the branched 21b, although containing the most highly cyclohexatrienic ring,
was recovered unchanged under these conditions. The reason must be that there is no
pathway available that does not generate a benzocyclobutadiene derivative.

44
Figure 1.5 X-ray crystal structure of 98 (thermal ellipsoids are shown at 50%
probability).
Considering the success of singlet oxygen cycloadditions, it seemed logical to
extend this chemistry to carbon-based dienophiles. Indeed, biphenylene, while generally
inert, even in the presence of o-benzyne, transforms to isolated Diels-Alder adducts with
more reactive benzyne derivatives (Scheme 1.1).17a,23,24 Angular phenylene 15 appears to
be more reactive, as expected, but undergoes further rearrangements driven by the release
of ring strain in the cycloadducts.97 Thus, on exposure to tetracyanoethene (TCNE), 15
formed a green charge-transfer complex, which, on heating, resulted in the
dibenzodehydro[10]annulene 99 (Scheme 1.25a). Mechanistically, this transformation
can be envisaged to proceed by a process similar to that leading to 96, except that double
bond isomerization has occurred (possibly during work-up).

45
(i) 15
(a)
99
NC
CN
NC
CN
(i)
(b)
101
CO2Me
CO2Me
CO2Me
CO2Me
100
Scheme 1.25 Cycloaddition reactions of 15: (a) (i) TCNE (1 equiv), CH3CN, ∆, 8 h,
78%; (b) (i) DMAD (1.6 equiv), AlCl3 (1 equiv), PhCH3, 23 °C, 1 h, 74%.
Remarkably, changing the dienophile to the alkyne dimethyl butynedioate (DMAD),
activated by added AlCl3, did not alter the course of the reaction, even though a highly
strained product is generated via 100 in the form of 101 (Scheme 1.25b). The
extraordinary structure of 101, the most distorted fully unsaturated [6]paracyclophane,
was confirmed by X-ray crystallography (Figure 1.6).97 In contrast, and again as
expected, branched 21b was unreactive to these reagents, with the exception of TCNE,
which produced a charge-transfer complex.
Figure 1.6 X-ray crystal structure of 101 (thermal ellipsoids are shown at 50%
probability).

46
1.3.4 Flash Vacuum Pyrolysis
The remarkable ring opening reactions in the preceding section herald the
phenylenes as “loaded springs”, not surprising in light of their ring strain and hence high
heat of formation (Section 1.3.1). One might therefore anticipate that, much like other
strained hydrocarbons,102 they would enter isomerization manifolds, ultimately ending in
PAHs as thermodynamic minima. Indeed, under flash-vacuum pyrolysis (FVP)
conditions, 1 had been shown to isomerize to acenaphtylene (104) as the major (Scheme
1.26a) and transient as-indacene (103) as a minor product, the existence of the latter
inferred through the isolation of a Diels-Alder adduct to 104 (Scheme 1.26a).103 Isomers
103 and 104 are derived from a common intermediate, benzopentalene 102, in this
cascade, which is generated by a sequential hydrogen shift/ring contraction from 1, as
indicated summarily in its structural drawing. A second such process leads to 103.
Acenaphthylene (104), in turn, is the result of a vinylidene carbene
deinsertion/reinsertion sequence from benzopentalene (wavy lines).104 These results
prompted an investigation of the behavior of the two isomeric [3]phenylenes 9b and 15
under these conditions. Aside from probing the kinds of PAHs that might be formed, it
was of interest to see whether the two compounds would interconvert prior to further
conversion, a possibility that, if realized, would shed experimental light on their relative
stability, a much debated issue.34,55 Recent calculations suggest that 15 is slightly more
stable than 9b.55

47
(i)
1021
(a)
(c)
103 104
105 (24 %) 106 (11 %) 107 (3 %) 108 (7 %)(i)
15
(b) 105 (10 %) 106 (4 %) 107 (1 %) 108 (1 %)(i)
9b 15 (1 %)
H
H HH
+
Scheme 1.26 FVP of 1, 9b, and 15: (a) (i) 900 °C, vacuum, 45% (104), 55% (adduct of
103 to 104); (b) (i) 1000 °C, 5x10–7 Torr; (c) (i) 1000 °C, 5x10–7 Torr.
In fact, 9b105 and 15105,106 gave not only the same mixture of PAHs on FVP, but
the linear isomer could be shown to isomerize to its angular relative at 1000 °C (Scheme
1.26b and c). A mechanism for this isomerization is patterned after a related
isomerization in the literature107 and proposes a four-membered ring opening, followed
by hydrogen shifts in the resulting biradical and ring closure. 13C-Labeling experiments
narrowed considerably the number of mechanistic pathways leading to the PAHs. Details,
too lengthy to be presented here, are reported in the original publication,105 and all
suggest initial hydrogen or carbon shift/ring contraction from 15.

48
1.3.5 Interaction with Organometallic Fragments
As σ and π activated hydrocarbons, phenylenes should be susceptible to
interaction with metal fragments. This notion is already borne out with 1, which readily
undergoes metal-promoted ring openings17a,22,108 and π complexation.17a,109 A systematic
comparison of the reactivity of higher phenylenes with transition metal complexes is yet
to be executed. Therefore, the following provides simply a summary of what has been
done so far.
(i)
109
9a
Fe(CO)3
TMS
TMS
TMS
TMS(OC)3Fe
Fe(CO)3
TMS
TMS
TMS
TMS(OC)3Fe
Fe(CO)3
TMS
TMS
TMS
TMS
Fe(CO)3
Fe(CO)3
110
111
Scheme 1.27 Reaction of 9a with [Fe2(CO)9]: (i) [Fe2(CO)9] (5.5 equiv), C6H6, ∆, 24 h,
67% (109), 18% (110), 14% (111).
In the linear phenylenes, the increased cyclobutadienoid character of the four-
membered rings manifests itself already in their synthesis, since both 11 and 13 were
formed initially as CpCo complexes. The use of an external ligand54 or oxidation of the
cobalt center57 was necessary to release the free phenylene (Scheme 1.3b and c). While

49
not recorded for CpCo, the C(aryl)–C(aryl) bond in 9a can be activated with [Fe2(CO)9]
to give rise to dibenzoferroles 109 and 110, as well as the bisallylic complex 111
(Scheme 1.27).53
The angular [3]phenylene (15) underwent double C–C activation by [CpCo(eth)2]
to afford 112 in 71% yield (Scheme 1.28a).97 The formation of an η4-complex between
the central benzene ring in 15 and CpCo was not observed in this reaction. This finding is
surprising, considering that the Cp*Co complex 113 can be prepared via a stepwise
sequence from the cyclization precursor 14 (Scheme 1.28b). In 113, the cobalt is attached
in such a way as to minimize cyclobutadienoid circuits (Scheme 1.28b, Figure 1.7, left).61
CpCo
(i)
112
15
CoCo
Cp Cp
CpCo
(a)
(i), (ii)
113
14(b)
CoCp*
Scheme 1.28 (a) C(aryl)–C(aryl) activation in 15: (i) [CpCo(eth)2] (10 equiv), C6H6, 70
°C, 6 h, 71%. (b) Preparation of the η4-complex of 15 to a Cp*Co-fragment: (i)
[Cp*Co(eth)2], THF, –20 °C, 16 h; (ii) ∆.
Finally, like biphenylene,109a,110 phenylenes appear to be readily complexed by
chromium tricarbonyl. For example, exposure of 15 to [Cr(CO)3(NH3)3] produced

50
complex 114 (Scheme 1.29a).111 The crystal structure of 114 (Figure 1.7, right) showed
that the three Cr–C–O axes are perpendicular to those of the formal single bonds of 15.112
Similarly, the branched skeleton of 21a is susceptible to metalation by Cr(CO)3, however,
Figure 1.7 X-ray crystal structures of 113 (left) and 114 (right). Thermal ellipsoids are
shown at 50% probability.
here giving rise to two regioisomeric complexes (Scheme 1.29b). Treatment with
[Cr(CO)3(NH3)3] resulted in the (so-called) exo-complex 115, while naphthalene–
Cr(CO)3 provided the endo isomer 116. The latter appears to be a kinetic product, as it
could be converted thermally to 115. Further complexation of 115 generated the
bischromium complex 117.113 It is clear from these cursory experiments that phenylenes
should be a rich source of new organometallic compounds.

51
Cr(CO)3
(i)
114
15 Cr(CO)3
(i) 115
21a
TMS
TMS
TMS
TMS
TMS
TMS
(ii)
117
Cr(CO)3
TMS
TMS
TMS
TMS
TMS
TMS
(a)
(b)
(ii)
116
TMS
TMS
TMS
TMS
TMS
TMS(iii)
Cr(CO)3
Cr(CO)3
Scheme 1.29 Complexation of six-membered rings in 15 and 21a: (a) (i)
[Cr(CO)3(NH3)3], dioxane, 100 °C, 4–5 h; (b) (i) [Cr(CO)3(NH3)3], dioxane, 100 °C, 14
h, 57%; (ii) naphthalene–Cr(CO)3, THF/Et2O, 60 °C, 14 h, 89% (116), 43% (117); (iii)
90 °C, (99%).

52
1.4 Physical Properties of the Phenylenes
As a novel class of hydrocarbons, an important aspect of the phenylenes lies in
their physical properties. The following sections will compare (to the extent that it is
possible) structural, spectroscopic, and calculated aspects of the known 19 phenylenes.
1.4.1 Structural Properties
As mentioned repeatedly in previous sections, the unique interplay of the π and σ
frame in the phenylenes gives rise to unusually distorted benzene rings, a feature that
manifests itself in experimental and calculated structural parameters. Generally, two
types of distortion are observed. The first is typical of linearly annulated systems, in
which, for reasons of symmetry, the inner six-membered rings cannot adopt a
cyclohexatrienoid configuration. Rather, the effect of fusion is to impart bisallylic
character, with long fused and shorter adjacent bonds. This is accompanied by a change
in the fusion angles in the six-membered rings to more obtuse.
The second and, at this point, more frequently encountered distortion of inner
rings is typical of angular and branched topologies and easier to understand, namely
cyclohexatrienoid bond alternation. Here, a simple descriptor of average bond length
alternation, i.e. (Σ 3 long bonds – Σ 3 short bonds)/3, can be employed for comparative
purposes. This number can also be expressed as the degree of bond alternation (in %), by
assigning a 0% value to benzene and choosing the exocyclic diene unit in 1,2-
dimethylenecyclobutene as the 100% standard. The difference between the 1.497 Å long

53
bond and 1.338 Å short bond in this reference equals 0.159 Å.114 This model was chosen
because of its appropriate geometry and the fact that the two exocyclic bonds show
almost no interaction, thus minimizing cyclobutadienoid resonance.
Keeping in tune with the experimental tenor of this Chapter, Figure 1.8
summarizes the available experimental 1H NMR and structural data (bond alternation
percentages) for all known parent phenylenes. Exceptions are 11, 13, and 66, for which
the parent systems have not been made. In these cases, calculated bond lengths of the
parent systems were employed in determining the extent of bond alternation. For 38, 44,
and 48, for which X-ray data could not be collected, the experimental geometries of
substituted derivatives were used, in conjunction with calculated data (parent) for the
substituted rings. Finally, the missing X-ray information for 60, 64b, and 71 has been
replaced by calculated values. Justification for blending experimental with calculated
information comes from the finding that the latter reproduces experimental trends
perfectly, although it tends to underestimate slightly bond alternation percentages. Figure
1.8 also lists NICS values (vide infra) for the parent systems. The following discussion
attempts to place these data on a comparative footing, focusing on selected illustrative
examples. As will be seen, a fairly consistent picture emerges.

54
28%-8.0
7.0
6.60
6.70
1
33%-7.5
7.3
6.42
9b
N/A-4.7
6.24
31%*-7.3
7.6
6.81
TMS
11, C6D6
N/A-5.2
5.89
TMS
TMS
TMS
7.5
32%*-7.3
7.6
6.81
TMS
13, C6D6
N/A-5.1
5.90
TMS
7.7
TMS
TMS
5.56
N/A-5.1
24%-9.5
3.1
6.96
6.98
15
6.996.90
6.18
64%-3.3
22%-9.1
4.2 6.89
6.94
17
6.93
6.82
6.31
53%-4.7
0.8
6.31
68%-4.3
1.5
6.51
6.29
19
7.02
48%-6.2
6.99
6.87
7.01
6.93 31%-9.2
4.0
55%-2.8
6.34
6.34
26
6.49
51%-4.5
6.49
6.84-6.82
7.00-6.95
6.84-6.82
6.88
15%-7.9
11.1
14.6
12.4
28
6.42
6.49
6.49
6.32
6.27
6.63 6.64
6.54
6.7753%-2.6
47%-4.7
23%-8.4
50%-3.9
11.1
12.0
14.3
6.63
33 35
6.69
6.92
6.69
6.545.98
6.13
6.49
6.47
6.44
6.44
6.95 6.61
6.61
6.59
5.996.04
6.06
6.23
6.39
6.39
6.43
54%-3.1
52%-4.4
29%-8.1
54%-4.1
53%*-3.0
48%*-5.0
23%*-8.2
48%*-3.8
49%*-4.2
12.0
11.0
11.7
14.410.9
12.0
11.7
29%*-7.5
7.5
6.85
48
N/A-6.46.46
2.6
67%c
-2.92.3
23%c
-9.8
6.68 6.68
6.50
6.39
5.94
6.07
6.90
6.90
26%a
-9.14.355%a
-4.7
0.8
57%a
-4.74.3
23%*-9.1
38
6.34
6.276.77-6.82
6.77-6.82
6.51
49%b
-6.2
1.5
54%*-4.3
3.9
28%b
-9.2
6.26
6.23
6.79-6.86
6.79-6.86
44, CD2Cl2 56, 1,2-dichlorobenzene-d4
N/A-7.5
2.9
66%-2.9
2.5
17%-9.7
6.52
5.96
6.04
6.78
6.86
60
N/A-7.5
6.73
2.9
60%*-2.9
2.522%*-9.7
6.58
6.06
6.12
6.92
6.97
7.016.97
6.89-6.93
14.4
6.86
6.86-6.90
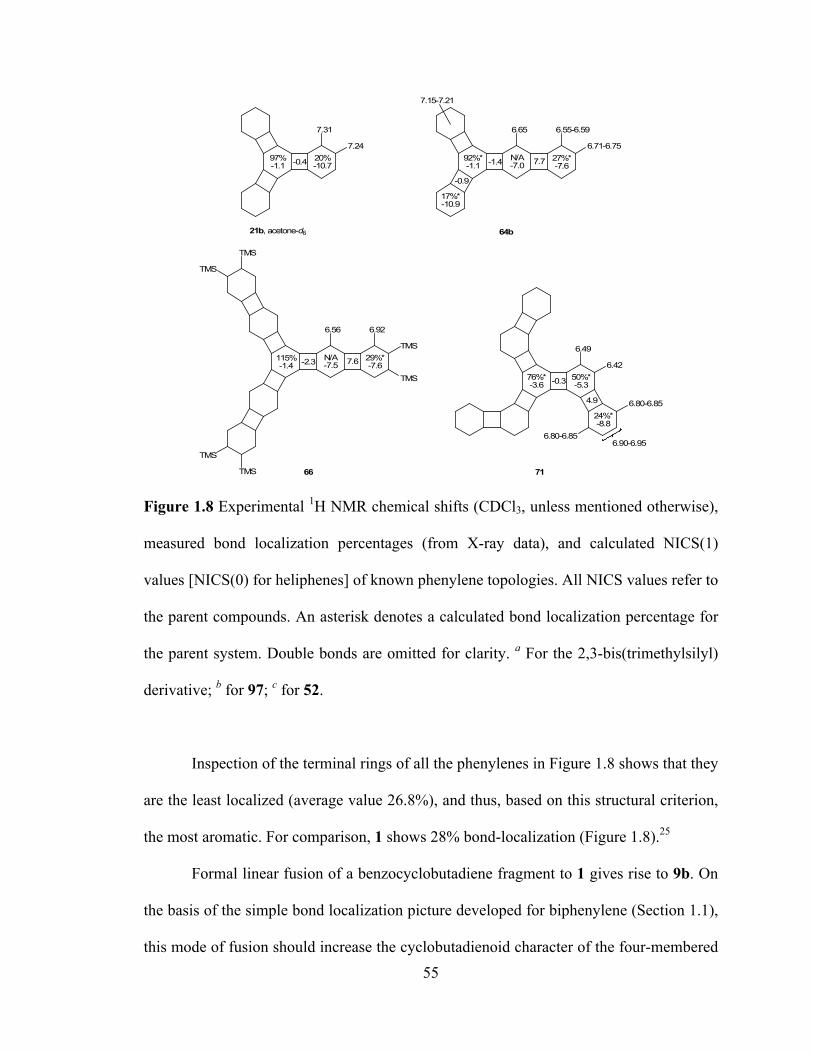
55
7.31
7.24
20%-10.7
-0.497%-1.1
21b, acetone-d6
6.65
N/A-7.0
-1.492%*-1.1
64b
-0.9
7.7 27%*-7.6
17%*-10.9
6.55-6.59
6.71-6.75
7.15-7.21
6.56
N/A-7.5-2.3
115%-1.4
66
7.6 29%*-7.6
6.92
TMS
TMS
TMS
TMS
TMS
TMS
6.49
50%*-5.3
-0.376%*-3.6
71
4.9
24%*-8.8
6.42
6.80-6.85
6.90-6.956.80-6.85
Figure 1.8 Experimental 1H NMR chemical shifts (CDCl3, unless mentioned otherwise),
measured bond localization percentages (from X-ray data), and calculated NICS(1)
values [NICS(0) for heliphenes] of known phenylene topologies. All NICS values refer to
the parent compounds. An asterisk denotes a calculated bond localization percentage for
the parent system. Double bonds are omitted for clarity. a For the 2,3-bis(trimethylsilyl)
derivative; b for 97; c for 52.
Inspection of the terminal rings of all the phenylenes in Figure 1.8 shows that they
are the least localized (average value 26.8%), and thus, based on this structural criterion,
the most aromatic. For comparison, 1 shows 28% bond-localization (Figure 1.8).25
Formal linear fusion of a benzocyclobutadiene fragment to 1 gives rise to 9b. On
the basis of the simple bond localization picture developed for biphenylene (Section 1.1),
this mode of fusion should increase the cyclobutadienoid character of the four-membered

56
ring and, in turn, force the terminal benzene ring of 9b to become more localized. This is
indeed the case, albeit subtly so: the termini of 9b are 33% localized.115 The core in 9b
has D2h symmetry, imposed upon it by the “conflicting” fusion of the adjacent
cyclobutadiene rings. In such cases, the bond localization analysis is not applicable, since
it assumes pseudo-three fold symmetry of the system. The central ring instead exhibits
the expected bisallylic character (Figure 1.9): two symmetric C3 fragments (bond lengths
1.392 Å) are connected by two long bonds (1.417 Å).115 Skeletal elongation in the linear
series apparently has little effect on bond alternation in the terminal rings, judged by the
calculated values of 31% (11) and 33% (13).
In contrast to 9b, 15 evolves from 1 by formal angular fusion of a
benzocyclobutadiene unit. This alteration acts to dramatically increase bond localization
in the center (64%),59 in turn decreasing the cyclobutadienoid character of the four-
membered rings and hence increasing delocalization of the terminal benzene nuclei
(24%). The effect of further angular fusions on the termini is quite small, as
corresponding values average 23.9% (Figure 1.8). However, such elongation imposes a
distinct pattern of oscillating bond alternation values on the internal rings in the angular
and, to the extent of available members, also the zigzag series.74 A simple rationale for
this phenomenon is that the most delocalized terminus enforces the highest degree of
bond localization in the first internal ring, which then allows for some “relaxation” of the
second internal ring, which in turn increases relative localization in the third and so on.
This pattern seems to be attenuating with size, as judged by the heliphene series,70
perhaps approaching a limiting value of ~ 50% in the corresponding polyheliphene.

57
The fusion of three benzocyclobutadienes to benzene, as in branched 21b,
maximizes its cyclohexatriene character (97% bond alternation), thus allowing the
termini to be maximally delocalized (20%).58
The bond-alternation approach can be used to evaluate the effects of mixed
fusions. For example, the skeleton of 48 can be built formally from 15 by linear
benzocyclobutadieno fusion. This change, by relay, appears to lead to increased
localization of the angularly fused six-membered ring (67% vs. 64% in 15).
Alternatively, regarding 48 as built by angular terminal benzocyclobutadieno fusion to
9b, the effect of increasing bond alternation in one of the termini of 9b “relaxes” the
other (29% of the linear end in 48 vs. 33% in 9b). Similar effects are observed in 56.76
While the numbers are small, the trends are consistent.
In the mixed branched examples, the elongation of 21b either linearly (as in 64b
or 66) or angularly (as in 71) has the expected localizing and delocalizing effects,
respectively, on the centers of branching. For example (experimentally), whereas the
central ring of 21b exhibits 97% localization, in 66, this value is increased to 115% -
larger than the reference 1,2-dimethylenecyclobutene! Taking recourse to calculated
structures for the other members in the branched family (and, for the sake of consistency,
using calculated numbers also for 21b and the parent of 66) confirms the experimental
trend: 89% (21b) – 92% (64b) – 92% (parent of 66) – 76% (71).76,82 One notes that the
juxtaposition of branched and angular fusion in 71 delocalizes not only the center (vs.
21b), but also the penultimate six-membered rings (50%, vs. 64% in 15).82

58
Figure 1.9 X-ray structures of selected [N]phenylenes - top and side views. Hydrogen
atoms omitted for clarity, thermal ellipsoids shown at 50% probability.

59
Figure 1.9 shows the X-ray structures of some of the phenylenes discussed. While
9b and 56 are essentially planar, 21b and the substituted 52, 66, 38(TMS)2, and 97 show
noticeable deplanarization. Such deplanarization is general and independent of topology,
size, and substitution pattern and is thought to be due to crystal-packing effects that are
comparable in energy to deformation energies (several kcal mol–1).58 VT NMR
experiments on a derivative of 21b bearing prochiral substituents revealed the absence of
decoalescence at the experimental low temperature limit of –93 °C, indicating either a
very low barrier to planarization, or a planar structure in solution, as indeed also
calculated for 21b. In addition, calculations showed that the phenylenes are more
deformable than their hexagonal squeezes. This flexibility was ascribed to ready
pyramidalization of the four-membered ring carbons as a result of two phenomena:
hyperconjugation of the low lying σ antibonding orbitals of the strained bonds with the
HOMO of the π system116 and minimization of antiaromatic overlap in the
cyclobutadiene nuclei.117 The observation of such facile deplanarization is encouraging in
view of projected syntheses of archimedene55,72 and circular [5]phenylene (the phenylene
analog of corannulene).55
In light of the preceding discussion, it is instructive to view the compilation of X-
ray structures of the heliphenes (Figure 1.10). Even in the absence of non-bonded
interactions, angular [5]phenylene (19) already shows a small, prehelical deviation from
planarity. The higher angular [N]phenylenes (N > 5) can no longer adopt planar structures
and are helical.66,70 The “helical strain” is not large, as determined by calculations, for
example, only 3.2, 5.4, and 7.0 kcal mol–1 for 26, 28, and 33, respectively.67 Table 1.1
summarizes some of the structural parameters of the heliphenes, highlighting the steady

60
increase in the helix climb and in-plane turn in the series. The angle between the planes
of the terminal benzene rings is the highest in [7]heliphene. The inner helix of the [6]–
and [7]helicene (hexagonal squeezes of 26 and 28) climbs more steeply than that in their
heliphene counterparts, due to the smaller diameters of the PAH systems.118,119
Molecule Terminal ring
centroid
distance (Å)
Terminal ring
interplanar
angle (°)
Inner
helix
climb (Å)
Inner
helix in-
plane turn
(°)
Racemiza-
tion barrier
(kcal mol–1)
Helical
strain
(kcal mol–1)
26 5.62 (5.87)a 22.8 (27.2)a 2.16
(2.30)a
337.3
(332.4)a
N/A (3.6)b (3.2)b
28 4.07 (4.54)a 30.1 (40.6)a 3.29
(3.64)a
361.3
(361.6)a
12.6±0.5a
(17.0)a
(5.4)b
33 4.41 (5.49)c 23.6 (41.1)c 3.35
(4.24)c
393.0
(389.1)c
13.4±0.4c (7.0)b
35 N/A (7.48)c N/A (33.2)c N/A
(5.07)c
N/A
(415.7)c
<12.0c
[6]heli-
cene
4.44 d 58.5d 3.20d 314.3d 36.2e
[7]heli-
cene
3.83f 32.3f 3.75f 380.8f 41.7e
Table 1.1 Comparison of the helix parameters of heliphenes and helicenes (calculated
values given in parentheses). a Ref. 66; b Ref. 67; c Ref. 70; d Ref. 118; e Ref. 68c–d,120; f
Ref. 119.

61
As the heliphenes are chiral, the possibility of enantiomer separation is intriguing.
Because of their extensive delocalization, the heliphenes should show remarkable
chiroptical properties.68c–d,120 In an attempt to probe the feasibility of their resolution,
configurational stability was probed by NMR experiments. For this purpose, a series of
heliphene derivatives bearing potentially diastereotopic substituents (isopropyl and
methoxymethyl, respectively) was prepared.66,70 In accord with the calculated low barrier
to enantiomerization of 26,67 decoalescence of the methyl group signals for an isopropyl
derivative was not evident at the limiting temperature of –75 °C. Turning to
methoxymethyl [7]heliphene, methylene decoalescence was recorded at –27 °C,
indicating a barrier of 12.6±0.4 kcal mol–1 for helix flipping66 - less than a third of the
value for the corresponding helicene.68c–d,120 Surprisingly, the analogous barrier for
methoxymethyl [8]heliphene was only slightly higher, 13.4±0.4 kcal mol–1 and
methoxymethyl substituted 35 showed no signal splitting for the methylene hydrogens on
cooling to its solubility limit at –45 °C.66,70 The flexibility of these systems is therefore
extraordinary, a consequence of both ready in- and out-of-plane deformations (vide supra
and infra).

62
Figure 1.10 X-ray structures of [N]heliphenes - top and side views. Hydrogen atoms
omitted for clarity, thermal ellipsoids shown at 50% probability.

63
1.4.2 Magnetic Properties
Proton NMR chemical shifts are highly diagnostic of whether a compound is
aromatic or not.5 Hydrogens located on the inside of aromatic rings exhibit relatively high
field chemical shifts, while those on the outside are relatively deshielded. Antiaromatic
circuits have the opposite effect. In the phenylenes, the generally observed shielding of
all protons (relative to alkylbenzenes) is the result of the simultaneous operation of two
effects: the decreased diatropism of the six-membered rings and the shielding influence
of the cyclobutadiene nuclei on the protons in their vicinity.
To better understand the relative contributions of the component rings to the
observed chemical shifts in the phenylenes, recourse was taken to nucleus-independent
chemical shift (NICS)121 calculations, which provide such data (in ppm) for a point
nucleus at any given position in a molecule. For cyclic polyenes this is typically 1.0 Å
above the center of the ring, chosen to minimize local perturbations.122 Negative NICS
values denote an aromatic ring (NICS(1)benzene = –12.5), whereas positive values indicate
an antiaromatic circuit (NICS(1)cyclobutadiene = 15.1).123 Used together, NICS and NMR are
useful tools in the following analysis of the corresponding entries in Figure 1.8. As will
be seen, in the phenylenes, the magnetic data correlate well with the structural criterion of
(anti)aromaticity.
As mentioned previously, the α- and β-protons in 1 resonate at δ 6.60 ppm and
6.70 ppm, respectively. The relative shielding of the α-proton is a consequence of the
residual paratropicity of the neighboring four-membered ring, applicable to all
phenylenes, with the exception of the branched isomers. The corresponding NICS values

64
are –8.0 and 7.0 for the six- and four-membered rings, respectively. In general, the
terminal rings exhibit the most negative NICS values and, correspondingly, the highest
NMR chemical shifts, in consonance with the occurrence of the smallest extent of bond
alternation.
In going from 1 to 9b, the paratropism (antiaromaticity) of the cyclobutadienes
(NICS = 7.3) is subtly increased, while the diatropism (aromaticity) of the terminal
benzene rings decreases, as evidenced by the lower NMR chemical shifts (δ = 6.42 and
6.63 ppm) and less negative NICS value (–7.5). The diatropic character is lowest in the
central ring (NICS = –4.7), which, in conjunction with the paratropism of the two
adjacent cyclobutadiene fragments, leads to strong shielding of its hydrogen (δ = 6.24
ppm).123 While the NICS values for the internal six-membered rings fluctuate somewhat
along the linear series 9b–11–13, the cyclobutadienes appear to become increasingly
paratropic, providing an explanation for the observation of incremental shielding of the
central hydrogens.123
The angular mode of fusion in 15 further reduces the diatropism of the center
(NICS = –3.3; δ = 6.18 ppm), in conjunction with decreased paratropism of the
cyclobutadienes (NICS = 3.1). As a consequence, the termini are more diatropic than in
biphenylene (NICS = –9.5 vs. –8.0).123 In the remainder of the angular series, the
arguments advanced previously for the rationalization of the trends in bond-localizations
are clearly augmented by the magnetic data. Thus, 19, as an example, shows the
alternation of diatropism of the six-membered rings: NICS = –9.2, –4.3, –6.2. The
paratropism of the cyclobutadiene moieties also oscillates: it is high in the outer ring
(NICS = 4.0) and less so in the inner one (NICS = 1.5), reflecting the interplay with the

65
neighboring six-membered rings and their respective aromaticity. For the heliphenes, the
use of NICS(1) values was abandoned, since the areas above and below the rings are now
inequivalent. Instead, NICS(0) data were computed, which, although numerically not
directly comparable with NICS(1) numbers, showed the same alternating trends, in
agreement with experimental NMR chemical shifts and bond localization numbers.66,70
The NMR and the NICS values of angular [4]– and [5]phenylene are essentially identical
to those of their zigzag counterparts, highlighting the similarity between the two
topologies.
In the branched 21b, the central six-membered ring becomes essentially atropic
(NICS = –1.1), as do the adjacent cyclobutadienes (NICS = –0.4), allowing for maximum
diatropism of the three terminal cycles (NICS = –10.7; δ = 7.24, 7.31 ppm). As such, the
system can be described essentially as an extended stilbene.123 In support of this view, the
signal for Hα (δ = 7.31 ppm) and Hβ (δ = 7.24 ppm) have traded their “normal” places,
appearing in the order observed for ordinary benzocycloalkanes, such as indane: Hα δ =
7.06 ppm, Hβ δ = 6.99 ppm.124
For the mixed topologies realized in 48 and 56/60, a component analysis is in
accord with the calculated magnetic behavior. Thus, if these systems are viewed as
perturbed linear [3]phenylenes, the added angular fusions serve to increase the
cyclohexatrienoid character of one or both termini of the linear substructure,
consequently reducing their diatropism, as observed. At the same time, the paratropism of
the four-membered rings (of the linear substructure) is also reduced, thus rendering the
center ring more diatropic (aromatic). The trends in the NICS values of the latter agree
with this analysis: –4.7 (9b), –6.4 (48), –7.5 (56/60), as do NMR chemical shifts δ 6.24

66
(9b), – 6.39 (48), – 6.58 (60) ppm. Along the same lines, completely removing the
diatropism of one terminus of 9b, as it occurs in 64b and 66, should have a similar
pronounced effect on the diatropism of the central benzene, as reflected by the
corresponding numbers for 64b (δ = 6.65 ppm, NICS = –7.0) and 66 (δ = 6.56 ppm,
NICS = –7.5). Turning to the mixed system 71 and viewing it as a perturbed 15, the
reduced diatropism of the core (the “perturbed” end of 15) goes with reduced paratropism
of the adjacent four-membered rings and increased diatropism of the next six-membered
cycle (δaverage = 6.58 ppm, NICS = –3.6). Alternatively, viewing 71 as a perturbed 21b,
the effect of reduced diatropism of the terminal rings of the substructure of branched
[4]phenylene is to increase the diatropism of the core (NICS = –3.6 vs. –1.1).
Carbon-13 NMR spectroscopy is not usefully diagnostic of ring currents, and a
typical 13C NMR spectrum of a phenylene exhibits four groups of signals. At the highest
chemical shifts (δ ~ 145–155) are the signals corresponding to the four-membered ring
carbon atoms without adjacent other four-membered rings. Those that are adjacent to a
second four-membered ring are relatively shielded125 and absorb at δ ~ 133–140 ppm.
The remaining carbons, namely the non-quaternary nuclei of the six-membered rings, are
also split into two categories - those adjacent to a cyclobutadiene (δ ~ 113–120 ppm) and
those distant from it (δ ~ 125–130 ppm). The averaged δ (over all six carbons) for the
internal six-membered rings in angular [3]–, [4]– and [5]phenylene changes very little
and lies between 132.5 and 134.5 ppm.

67
1.4.3 Energetic Properties
The determination of the ground-state energies of the [N]phenylenes is of crucial
importance in the evaluation of their aromaticity4 and strain. On the other hand, their
frontier orbital separation constitutes a measure of their kinetic stability126 and is central
to organic conductor applications.127 The excited states of the phenylenes are also of
interest for probing the changes in aromaticity that occur upon excitation and for
identifying the nature of radiative relaxation pathways (fluorescence and/or
phosphorescence).
Experimental enthalpies of formation for members of the series have been
obtained only for 1,128 15, and 21b, and the agreement between the calculated and
observed ∆H°f data is remarkable.35 For other phenylenes, only calculated data are
available, and the following will highlight some key findings.
Although the conjugated-circuit model34 suggested that the linear [N]phenylenes
are more stable than their angular isomers, the application of ab initio methods proved the
opposite.129 Schulman and Disch’s examination of the problem by modern DFT methods
placed the stabilization of 15 vs. 9b at 2.4 kcal mol–1.55 Branched [4]phenylene (21b) is
the most stable of the five [4]phenylenes, followed by 38 (relative energies: +4.1 kcal
mol–1), 17 (+4.3 kcal mol–1), 48 (+ 4.8 kcal mol–1), and linear [4]phenylene (+8.5 kcal
mol–1). The energies of zigzag [4]– (38) and [5]phenylene (44) are almost identical to
those of their angular isomers.55 A comparison of the relative energies of the twelve
[5]phenylenes reveals the same general trends: the linear isomer is the least (+10.7 kcal
mol–1), the branched 64b the most stable (0.0 kcal mol–1), surprisingly more stable than

68
the other branched isomer (+0.9 kcal mol–1), which is devoid of a linear [3]phenylene
substructure. Similarly, among the nonbranched [5]phenylenes, the two doublebent
isomers (56 and 60) have the lowest energies (+3.8 kcal mol–1), despite the presence of
the linear fragment.67 This disagreement with expectation, albeit associated with small
numbers, was attributed to the opposing energetic contributions of the σ- (stabilizing) and
π- (destabilizing) components of the linear frame.76
The “helical strain” in the smaller heliphenes is relatively small (Table 1.1), but
becomes substantial for larger systems. Thus, in order to model the limiting properties of
larger phenylenes, calculations have been executed on various illustrative topologies of
[19]phenylene.71 The results indicate that the helical topology is 26.9 kcal mol–1 (1.4 kcal
mol–1 per ring) less stable than its zigzag counterpart. Linear [19]phenylene is the least
stable, 40.4 kcal mol–1 (2.1 kcal mol–1 per ring) more energetic than the zigzag isomer.
The electronic spectra of the phenylenes feature two typical sets of bands: one at
lower wavelengths, with relatively large ε values, and a second at higher wavelengths,
with lower exctinction coefficients. The exact position of these absorptions, however,
depends on the phenylene topology (Table 1.2). In the linear series, the λmax value
increases dramatically in going from 1 to 9a (∆λmax = 75 nm), 11 (∆λmax = 54 nm), and
then 13 (∆λmax = 38 nm).47a,53,54,57 The extrapolation of this limited set of experimental
data to infinite N provides λmax = 662 nm, corresponding to a small band gap of 1.87 eV
for the linear polymer, boding well for potential electronic applications.

69
λmax (nm) HOMO–LUMO gap (eV)
N Angular Zigzag Linear Angular Zigzag Linear
2 363a (isooctane) 363a 363a 3.42 3.42 3.42
3 428b (THF) 428b 438b,d (THF) 2.90 2.90 2.83
4 448b (THF) 465c (THF) 492b,d (THF) 2.77 2.67 2.52
5 470b (THF) 484c (CH2Cl2) 530b,d (THF) 2.64 2.56 2.34
6 491e (CH2Cl2) – – 2.53 – –
7 503e (CH2Cl2) – – 2.47 – –
8 515f (CH2Cl2) – – 2.41 – –
9 524f (CH2Cl2) – – 2.37 – –
∞ 578f 587g 662g 2.14 2.11 1.87
Table 1.2 HOMO–LUMO gaps and λmax values in angular, zigzag and linear phenylenes.
a Ref. 47a; b Ref. 60; c Ref. 74; d for tetrasilylated compounds; e Ref. 66; f Ref. 70; g Ref.
130.
In contrast to the linear frame, the λmax values of angular phenylenes attenuate
more rapidly (N = ∞, λmax = 578 nm, band gap = 2.14 eV).47a,59,60,66,70 The same seems to
be true for the zigzag isomers, again with the caveat that only four experimental values
are available (N = ∞, λmax = 587 nm, band gap = 2.11 eV).74 The so-estimated band gaps
for the linear, angular, and zigzag family conform with theoretical predictions.34,71
In agreement with the virtual absence of antiaromatic circuits, the UV spectrum of
branched 21b, while still exhibiting the diagnostic phenylene pattern, is

70
hypsochromically shifted, with a highest wavelength absorption at 379 nm, reflecting a
HOMO–LUMO gap of 3.28 eV - the highest among the [4]phenylenes.65
The electronic spectra of the “mixed” phenylenes are strongly influenced by the
presence of the linear substructures, which cause strong bathochromic shifts. Thus, 48
exhibits a λmax of 486 nm, which is almost exactly equal to that of its linear relative and
higher than that of the remaining isomers.75 Doublebent [5]phenylenes 56 and 60 absorb
at 505 and 507 nm, respectively, at energies significantly lower than their isomers 19, 44,
and 64b, but higher than the all-linear 13 (Table 1.1).76 Finally, in the series of branched
phenylenes, the effect of the presence of linear annelation is highlighted by the changes
in λmax when going from 21b to 64b (∆λmax = 107 nm), 64b to 66 (∆λmax = 37 nm),79 and,
particularly, 71 to 66 (∆λmax = 67 nm).81
The exploration of the photophysics of the phenylenes, still in its infancy, has
focused on the smaller linear, angular, and branched systems, as well as 1. On the basis of
their rates of internal conversion of the first excited state (S1), the systems studied have
been labeled as either “fast IC compounds”, with kIC > 109 s–1, or “slow IC compounds”,
with kIC ≈ 107 s–1.131 Fast IC compounds, biphenylene and 9b, relax their S1 state
predominantly through internal conversion (ΦIC > 99%), since the rates of this reaction
are significantly greater than those of the competing intersystem crossing and
fluorescence processes. In angularly fused, slow IC compounds 15, 38, and 44, other
relaxation mechanisms gain in importance and become dominant in zigzag [5]phenylene
44, which releases 21% of its excited state energy through fluorescence and crosses over
efficiently (ΦISC = 45%) to the triplet state (T1). The corresponding quantum yields for
the branched 21b are ΦF = 15% and ΦISC = 3%.131

71
Shpol’skii spectroscopy132 of the phenylenes, together with DFT calculations,
have provided significant insights into the vibrational characteristics of their excited
states. The resemblance between the fluorescence emission and excitation spectra,
observed for 15 and 44, has indicated that, in phenylenes with angular substructures, the
S0 and S1 states have similar geometries.133 Studies performed on deuterated derivatives
of 21b allowed for the distinction between C–C and C–H vibrational modes in the S1
state and demonstrated that, while the terminal rings of 21b are essentially unperturbed in
the excited state, the central cyclohexatrienoid ring partly delocalizes, suggesting
rearomatization.134 This is an important result, in view of the fact that the opposite effect
- dearomatization of benzene in the first excited state - is well known.2a Photophysical
studies confirmed the presence of a theoretically invoked55,67 low-frequency (~ 35 cm–1)
out-of-plane vibration and also revealed a new in-plane vibration in angular phenylenes
(~ 100 cm–1), notably absent from the spectra of corresponding PAHs. The first vibration
deplanarizes the angular fragment by moving the terminal rings out of the molecular
plane in opposite directions. The second increases the angle between the centroids of the
three successive six-membered rings of an angular fragment, in turn pushing the terminal
rings away from each other.133 Both of these distortions have been proposed as
operational in the transition state for the racemization of [6]heliphene67 and their
magnitude provides a convincing rationale for the ease by which this process occurs for
the other heliphenes studied so far.

72
1.5 Thesis Summary
This dissertation will document the progress achieved in the syntheses of larger
dehydrobenzannulenes and novel [N]phenylenes with mixed modes of fusion. Along
these lines, the following Chapter 2 will present the synthesis of syn-doublebent
[5]phenylene and advances toward the (still elusive) U-shaped [7]– and [9]phenylenes, as
well as the C-shaped [7]phenylene. Building upon these results, an approach to circular
[8]phenylene will be described. As part of this effort, two routes to
dehydrobenzannulenes were developed. The first one, described in Chapter 3, utilized
alkyne metatheses of propynylated benzenes to produce dehydrobenzannulenes.
Although simple, this method was somewhat limited in scope, thus necessitating a
different strategy for the preparation of extensively substituted dehydrobenzannulenic
systems. An alternative procedure, to be presented in Chapter 4, relied on a sequence of
Sonogashira couplings to assemble a variety of substituted dehydrobenzannulenes. Some
of these systems, due to the appreciable bulk of their pendant substituents, were
conformationally constrained. This intriguing property will be more thoroughly discussed
in Chapter 5. This chapter will also feature the synthesis and stereochemical evaluation of
the first chiral diphenylacetylene. Finally, Chapter 6 will provide the experimental details
of the studies performed.

73
Chapter Two
Synthetic Approaches to Novel Phenylenes with Mixed Angular and Linear Fusion
2.1 Introduction
At the beginning of the work described in this dissertation only two phenylenes
with mixed angular and linear topologies were known: bent [4]phenylene (48, Section
1.2.2.3)75 and anti-doublebent [5]phenylene (56, Section 1.2.2.3).76 This chapter will
focus on the continued exploration of this mode of fusion through the attempted
syntheses of the four novel mixed phenylenes shown in Figure 2.1.
60 118 119 120
Figure 2.1 Novel phenylenes with mixed angular/linear fusion: syn-doublebent
[5]phenylene (60), C-shaped [7]phenylene (118), U-shaped [7]– (119), and [9]phenylene
(120).
Syn-doublebent [5]phenylene (60) and C-shaped [7]phenylene (118) feature a
central linear [3]phenylene fragment, extended by the double benzocyclobutadieno- and

74
biphenylenocyclobutadienofusion, respectively. Conversely, U-shaped phenylenes 119
and 120 share the central angular [5]phenylene unit, which is elongated by annelation of
two benzocyclobutadienes (119) or two biphenylenocyclobutadienes (120). These
kinships, between 60 and 118 on one, and 119 and 120 on the other side are reflected in
their proposed syntheses and expected properties.
The relevance of the investigations of 60 and 118–120 is manifold.
Methodologically, and as will be seen later, they represent a significant extension of the
CpCo-mediated approach to phenylenes. In terms of modes of fusion, syn-doublebent
[5]phenylene 60 is completely equivalent to its anti-relative 56, but with the added
feature of the “super-bay” region flanked by two benzocyclobutadieno groups. Fusing
benzocyclobutadienes to the termini of 60, as in 118, converts them from terminal
(relatively delocalized) into central rings of an angular fragment (relatively localized).
This modification is also expected to affect the terminal two rings of the linear fragment,
allowing them to delocalize relative to 60. Consequently, attenuation of “activity” of all
internal six-membered rings of 118 is expected, relative to the internal rings of linear and
angular [3]phenylene. In this context, the juxtaposition of 119 with its helical isomer 28
is particularly intriguing: switching the mode of fusion of the termini from angular (28) to
linear (119) is expected to invert the localization trends observed in 28, making the
central ring relatively delocalized and inverting the alternation of properties of the
subsequent rings. Finally, in addition to adopting the same features as those predicted for
119, 120 is hypothesized to adopt a helical conformation, thus opening up access to a
new class of chiral phenylenes, possibly with properties different from the heliphenes
made so far.

75
In the context of this dissertation, phenylenes 60, 118, and 119 have another,
common property: they constitute formally subunits of circular [8]phenylene (121, Figure
2.2). The structure of this elusive circular phenylene (Section 1.2.2.5) encompasses an
internal [18]annulene and an external [30]annulene circuit (shown in bold, Figure 2.2).
Both of these π-loops have 4n+2 electrons, and, according to Hückel’s rule, are therefore
aromatic. On the basis of the properties of the substructures in 121, one would anticipate
that the resonance structure shown in Figure 2.2 would contribute strongly to the
resonance picture of this molecule, thus enabling superdelocalization. The internal
protons of 121 would be an excellent probe for this phenomenon, as they should be
relatively shielded, as in [18]annulene itself.5 A more thorough treatment of the expected
properties of 121 is deferred until Chapter 4.
121
Figure 2.2 Circular [8]phenylene (121).
The topological resemblance between the above mentioned novel phenylenes and
121 makes the former ideal models for comparisons of physical properties discussed in
Section 1.4. Synthetically, they would enable the evaluation of the viability of preparative
route(s) to 121, including the stability and solubility of advanced precursors and

76
intermediates. Finally, the strategy to compound 120, which requires an unprecedented
four-fold CpCo-mediated cycloisomerization of an appropriate oligoalkyne precursor
(vide infra), would present a test for the viability of a similar approach to 121.
2.2 Retrosynthetic Approach to 60, 118, 119, and 120
Conceptually, 60 and 118 were targeted through a regioisomeric version of the
double intramolecular cyclization developed for angular [5]phenylene (19, Scheme 1.6)66
and helical [7]phenylene (28, Scheme 1.7b).66 Thus, while the 1,2,3,4-tetraalkynyl
substitution pattern of the heliphene precursors 24b and 27 translated into the angular
center of the products, the requirement for the central linear fragment in 60 and 118
dictated a related 1,2,4,5-relationship. This suggested 59 and 122 as plausible ultimate
cyclization precursors for these molecules (Scheme 2.1).
59
60
122
118(a) (b)
Scheme 2.1 Retrosynthetic analysis of 60 (a) and 118 (b).

77
Similarly, a topological variant of 27, namely attachment of 2,3-diethynylbiphenylene
units rather than their 1,2-alkynylated isomers, would suggest 123 as an ideal precursor to
119 (Scheme 2.2).
123
119
Scheme 2.2 Retrosynthetic analysis of 119.
Alternatively, 119 could be pursued through the more ambitious four-fold inter-
and intramolecular cyclization of 124, creating 10 rings in one step (Scheme 2.3a). In this
variant, the double-cyclization precursor to angular [5]phenylene, 24b, would have to be
functionalized by four additional ethynyl groups. The significance of this synthesis lies in
its potential extendibility, as exemplified by a possible access to 120 (Scheme 2.3b).
The remainder of Chapter 2 will discuss the attempted synthetic execution of the
above proposals, in the order in which they were introduced.

78
119(a)
120(b)
125
124
Scheme 2.3 Inter- and intramolecular retrosynthetic analysis of 119 (a) and the
retrosynthetic analysis of 120 (b).
2.3 Synthesis of Doublebent [5]Phenylene 60
Syn-doublebent [5]phenylene (60) has been targeted previously by our group,
through hexayne 59 as the ultimate cyclization precursor. The synthesis of 59 was
straightforward (Scheme 2.4): starting with the 1,5-dibromo-2,4-diiodobenzene (57)78 as
a C2v-symmetric template, treatment with 2360 and subsequently TMSA under
Sonogashira conditions provided tetrayne 126a. The complete, fluoride-assisted,
deprotection of this material yielded hexayne 59. Disappointingly, attempted cyclization

79
to 60 failed when [CpCo(CO)2] was used as the catalyst, prompting speculation that the
low solubility of 60 (imparted by its planar structure) plagued this synthetic route.135
57
BrI
I Br
126a, R = DMTS126b, R = Pr
R
TMS
TMS
R
(iii) (iv)
59, R = H127, R = Pr
R
R
R
R
60, R = H128, R = Pr
(i), (ii)
Scheme 2.4 The attempted synthesis of syn-doublebent [5]phenylene (60) and the
successful synthesis of its dipropyl derivative 128: (i) 23 (for 126a) or 1-ethynyl-2-(pent-
1-ynyl)benzene (for 126b), [Pd(PPh3)2Cl2], CuI, Et3N, 23–50 °C, 24–36 h, 65% (126a),
83% (126b); (ii) TMSA, [Pd(PPh3)2Cl2], CuI, Et3N, 120–135 °C, 18–72 h, 70% (126a),
86% (126b); (iii) for 59: TBAF, THF, 23 °C, 45 min, (95%); for 127: NaOH,
THF/MeOH, 23 °C, 30 min, 85%; (iv) [CpCo(CO)2], m-xylene, hν, ∆, 2 h, 0% (60), 1%
(128).
Such speculation gained some credibility through the successful completion of the
synthesis of 128, a derivative of 60 substituted with two solubilizing propyl groups
(Scheme 2.4). The preparation of 128 followed the same strategy as that projected for 60,
but employing 1-ethynyl-2-(pent-1-ynyl)benzene instead of 23 to install the propyl

80
substituents. Final cyclization of 127 proceeded successfully, albeit in a low 2% yield, to
give 128.
Subsequent to this preliminary work, investigations of the synthesis of other
phenylenes showed that [CpCo(eth)2],63 when used under certain conditions, could
provide a more successful catalytic system.61,62 Specifically, this complex allowed the
preparation of the parent anti-doublebent [5]phenylene 56 (Section 1.2.2.3, Scheme 1.13),
an effort that failed when using [CpCo(CO)2].76 Consequently, the synthesis of 60 was
revisited, including a new, more convergent, synthesis of 59 (Scheme 2.5). Starting again
(i), (ii), (iii)57
Br
Br
Br
60
Br
(iv), (v) (vi)
58 59
Scheme 2.5 The synthesis of syn-doublebent [5]phenylene (60): (i) TMSA,
[Pd(PPh3)2Cl2], CuI, Et3N, 23 °C, 2 h, 96%; (ii) KOH, Et2O/EtOH; (iii) 1-bromo-2-
iodobenzene, [Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 44% (over 2 steps); (iv) TMSA,
[Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 47%; (v) TBAF, THF, 23 °C, 2 h, (95%); (vi)
[CpCo(eth)2], THF, –25 °C, 16 h, followed by 1,3-cyclohexadiene, THF, 110 °C, 2 h,
14%.

81
with 57, Sonogashira coupling and base-assisted deprotection provided 1,5-dibromo-2,4-
diethynylbenzene. Another Sonogashira reaction between this material and 1-bromo-2-
iodobenzene gave the tetrabromide 58, the X-ray crystal structure of which is shown in
Figure 2.3.136 The molecule has a pseudo-C2 axis that passes through the two
unsubstituted carbons of the central benzene ring. The bromines are located anti, with
dihedral angles of 148.7 and 162.2 °, respectively. Compound 58 crystallizes in the P21/n
space group with four molecules in the unit cell.
Figure 2.3 ORTEP diagram of 58. Thermal ellipsoids shown at the 50% probability
level.
The use of 58 as an intermediate on route to 60 avoided the superfluous
differential silyl substitution of 126a. Reaction of 58 with an excess of TMSA, and
subsequent deprotection to 59 proceeded in a satisfactory 45% overall yield. Exposure of
59 to [CpCo(eth)2] was followed by treatment with 1,3-cyclohexadiene as an external
CpCo-trap and heating. Gratifyingly, the desired 60 was isolated in 14% yield (37% per
cyclization step).

82
2.4 Attempted Synthesis of C-Shaped [7]Phenylene (118)
Encouraged by the results described in Section 2.3, we embarked on the pursuit of
118, formally a derivative of 60 with two additional benzocyclobutadieno-fusions. As
discussed previously (Section 2.2), hexayne 122 was deemed a suitable precursor. Its
preparation could follow either of the two strategies precedented for 60, substituting a
suitable biphenylenyl building block for the corresponding phenyl fragment. However,
1,2-difunctionalized biphenylenes need several steps for their synthesis and are too
valuable to be introduced early in the synthesis. Therefore, the alternative Schemes 2.6
and 2.7 were executed. The central benzene ring of 122 was elaborated from 57 by two
(i), (ii)57
DMTS
(iii)
129
DMTS
TMS
TMS
DMTS
130
DMTS
Scheme 2.6 The synthesis of 130: (i) DMTSA, [Pd(PPh3)2Cl2], CuI, Et3N, 23 °C, 48 h,
92%; (ii) TMSA, [Pd(PPh3)2Cl2], CuI, Et3N, 110 °C, 48 h, 81%; (iii) K2CO3, Et2O/EtOH,
97%.
consecutive Sonogashira couplings. The first, with DMTSA, targeted the more reactive
iodinated positions; the second functionalized the brominated sites with TMSA. Thus
obtained tetrayne 129 was selectively deprotected into 130 (Scheme 2.6). Compound 130

83
underwent twofold Sonogashira coupling with biphenylene 131,60 giving the hexayne 132
in 54% yield (Scheme 2.7).
118
(ii)
(i)
DMTS
130
DMTS
IDMTS
R
132, R = DMTS
122, R = H
R
R
R
131
x
+
(iii)
Scheme 2.7 The attempted synthesis of 118: (i) [Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 16 h,
54%; (ii) TBAF, THF, 23 °C, 2 h, (95%); (iii) [CpCo(eth)2], THF, –25 °C, 16 h, followed
by 1,3-cyclohexadiene, THF, 100 °C, 90 min.
Bearing in mind the poor performance of [CpCo(CO)2] in the attempted
preparation of 60, we decided to use exclusively [CpCo(eth)2] in our efforts to produce
118. As precedented for 60, precursor 132 was subjected to an in-situ fluoride-assisted
deprotection into 122, which was followed by treatment with [CpCo(eth)2] at low
temperatures, and then with 1,3-cyclohexadiene at 100 °C. Although 1H NMR
spectroscopy indicated the formation of CpCo-cyclohexadiene, it could not detect any
phenylenic products. Additionally, neither mass spectral (EI, 70 eV), nor TLC analysis
showed indications of the presence of the desired material in the crude reaction mixture.

84
2.5 Attempted Syntheses of U-Shaped [7]Phenylene (119)
2.5.1 Intramolecular Approach
The intramolecular route to 119 bears close resemblance to the one developed for
the regioisomeric helical [7]phenylene (28).66 The hexayne 123 (Scheme 2.2), designed
as the ultimate precursor to 119, differs from its isomer 27 only in the substitution mode
of the biphenylenyl groups. It is understandable, therefore, that the most challenging part
of the preparation of 123 was the development of an efficient route to unsymmetrical 2,3-
substituted biphenylenes.
It was hoped that 2-alkynyl-3-iodobiphenylenes (134) could be prepared through
the exposure of the previously described 2,3-diiodobiphenylene (133)53 to one equivalent
of the alkyne (Scheme 2.8). In practice, almost statistical mixtures of the starting
(i)
133
I
I
134a, R = TMS134b, R = DMTS134c, R = Hex
I
135a, R = TMS135b, R = DMTS135c, R = Hex
R R
R
+
Scheme 2.8 Attempted monoalkynylations of 2,3-diiodobiphenylene (133): (i) R–C≡CH
(1 equiv), [Pd(PPh3)2Cl2], CuI, Et3N, 23 °C, 12 h. Yields: for R = TMS: 31% (133), 25%
(134a), 34% (135a); for R = DMTS: 31% (133), 25% (134b), 34% (135b); for R = Hex:
20% (133) 46% (134c), 9% (135c).

85
material, the desired product, and the dialkynylated derivative 135 were obtained. This
result was fairly independent of the nature of alkyne, indicating that the reactivity of the
monoalkynylated 134 is equal to, if not greater than, that of 133. This unsatisfactory
outcome suggested the use of (unknown) 2-bromo-3-iodobiphenylene (138) as an
alternative substrate for monoalkynylation. The preparation of 138 (Scheme 2.9)
followed closely that employed in the preparation of 2-bromo-3-iodobenzocyclobutene
from 2,3-bis(trimethylsilyl)benzocyclobutene.137 In such systems, steric strain renders
one silyl group much more reactive than the other in electrophilic aromatic substitutions.
Therefore, bis(trimethylsilyl)biphenylene (136)52 was treated briefly with bromine to give
monobrominated 137 as the major product (59%). The remaining TMS group was then
exchanged for iodide by treatment with ICl.137
(i)
136
TMS
TMS
137
TMS
Br
138
I
Br
(ii)
Scheme 2.9 Preparation of 2-bromo-3-iodobiphenylene (138): (i) Br2, pyridine, CH2Cl2,
0 °C, 3 min, 59%; (ii) ICl, CH2Cl2, 0 °C to 23 °C, 5 h, 99%.
Bromoiodide 138 behaved as expected in the Sonogashira reactions with TMSA,
DMTSA, or 1-octyne: no dialkynylation was observed and only a minor portion of the
starting material was left unreacted. The remaining bromine in the desired products

86
139a–c was converted successfully to iodide through halogen–lithium–halogen exchange
(Scheme 2.10).
(i)138
139a, R = TMS139b, R = DMTS139c, R = Hex
Br
134a, R = TMS134b, R = DMTS134c, R = Hex
(ii), (iii)
R
Scheme 2.10 Stepwise preparation of 2-iodo-3-alkynylbiphenylenes (134): (i) R–C≡CH,
[Pd(PPh3)2Cl2], CuI, Et3N, 23 °C, 12 h, 75% (for 139a, 78% converted yield), 60% (for
139b, 85% converted yield), 67% (for 139c, 91% converted yield); (ii) BuLi, Et2O, –45
°C, 30 min; (iii) I2, Et2O, from –45 °C to 23 °C, 2 h, 78 % (for 134a, over two steps),
82% (for 134b, over two steps), 85% (for 134c, over two steps).
With 134 in hand, the stage was set for the final couplings toward 123, namely
with 140, used previously in the synthesis of heliphenes (Scheme 2.11).66,69 While this
transformation worked reproducibly, the yields were disappointing, never exceeding
30%. Hexaynes 141 were deprotected (using methanolic K2CO3 for 141a and TBAF/THF
for 141b and c) into the ultimate cyclization precursors 142a and b, and the parent 123
(Scheme 2.12).

87
(i)134a, R = TMS134b, R = DMTS134c, R = Hex
DMTS
DMTS
140
DMTS
DMTSR
R
141a, R = TMS141b, R = DMTS141c, R = Hex
+
Scheme 2.11 Preparation of 141a–c: (i) [Pd(PPh3)2Cl2], CuI, Et3N, 120 °C, 36–48 h, 23%
(141a), 28% (141b), 30% (141c).
The parent 119 was targeted first: 123 was treated with [CpCo(eth)2] at low
temperature, followed by heating and exposure to 1,3-cyclohexadiene. As in the case of
118 (Scheme 2.7), analysis of the crude reaction mixture (by 1H NMR, MS, TLC) gave
no indication of the presence of the phenylene targets. At first, solubility problems were
invoked as a possible explanation for this failure. Since [5]phenylenes 56 and 60 are only
sparingly soluble in most of the common organic solvents, it was reasonable to assume
that the (presumably planar) [7]phenylene 119 would be even less soluble. However,
subjecting the two substituted precursors, 142a and b (bearing solubilizing DMTS and
hexyl groups, respectively) to [CpCo(CO)2] failed to produce even traces of phenylenes
143a and b, respectively (Scheme 2.12).

88
(i)
R'
R'R
R
143a, R' = DMTS, R = H119, R = R' = H143b, R' = H, R = Hex
141a, R = TMS141b, R = DMTS141c, R = Hex
(ii)
R
R'
R'
R
142a, R' = DMTS, R = H123, R = R' = H142b, R' = H, R = Hex
x
Scheme 2.12 Attempted preparation of 119 and 143a–b: (i) for 123 and 142b: TBAF,
THF, 23 °C, 1 h, (95%); for 142a: K2CO3, MeOH/Et2O, 23 °C, 1 h, (95%); (ii) for 119,
143a and b: [CpCo(CO)2], m-xylene, hν, reflux, 1 h, 0%; for 119 only: [CpCo(eth)2],
THF, –25 °C, 16 h, followed by 1,3-cyclohexadiene, THF, 90 min, 110 °C.
The failure of the synthetic routes to 119 and 118 (Section 2.4) is puzzling,
especially in view of the fact that the preparation of phenylenes by double CpCo-
cycloisomerization has precedence, most relevantly for the isomeric 28 (Section 1.2.2.1,
Scheme 1.7)66 and syn-doublebent [5]phenylene 60 (Section 2.3, see also Chapter 4).
During efforts to optimize the problematic Sonogashira coupling to 141 (Scheme
2.11), an unexpected result was obtained. Performing the reaction between 140 and 134c
in undried commercial piperidine provided the aldehyde 144 (Figure 2.4) as the sole
isolable product in 34% yield. Thus, while one of the reactive sites of 140 had been
functionalized in the expected Sonogashira fashion, the other terminal triple bond had
become hydrated in an anti-Markovnikov manner. The aldehyde was formed as an

89
exclusive product; not even a trace of Markovnikov-type methyl ketone could be detected
(by 1H NMR). This observation suggests stereochemical, rather than electronic control of
selectivity.
To place these results within the context of the literature, there are reports of anti-
Markovnikov palladium-catalyzed alkyne hydroesterification138 and
hydroalkoxylation,138a,139 but not of hydration. On the other hand anti-Markovnikov
alkyne hydrations have been catalyzed by other transition metals.138,140 Moreover,
Sonogashira reactions occur in water,141 suggesting that the formation of 144 was an
R
RR'
144, R = DMTS, R' = Hex
O
Figure 2.4 Aldehyde 144.
anomaly. Nevertheless, we tested our catalyst system in this respect by treating
phenylacetylene with iodobenzene in piperidine in the presence of added water (100
equivalents). The only product was diphenylacetylene. In addition, exposing simple
alkynes (1-octyne, phenylacetylene, TMSA, and DMTSA) to a 100-fold excess of water
and [Pd(PPh3)2Cl2]/CuI in piperidine at 120 °C. Again, hydration was not observed–the
main products (in the complex mixture) were the alkyne homocoupling dimers of the

90
type R–≡–≡–R. It is possible that steric hindrance in the singly coupled intermediate of
the reaction of 140 with 134c (Scheme 2.11) slows further reaction sufficiently to allow a
hydration pathway to take place. It remains to be tested whether other hindered alkynes
behave in the same way.
2.5.2 Intermolecular Approach
The intramolecular approach to 119 has biphenylene building blocks incorporated
into the final cyclization precursors 141. A more convergent synthetic alternative can be
envisioned, in which decayne 124 undergoes both intra- and intermolecular cyclizations
to generate all component four-membered rings in one step (Scheme 2.3a). This method
has been used previously in the syntheses of phenylenes 38 (Section 1.2.2.2, Scheme
1.10b)74 and 52 (Section 1.2.2.3, Scheme 1.12b).75
(i), (ii), (iii)57
Br
I
DMTS
145
DMTS
I
DMTS
146
DMTS TMS
(iv), (ii), (iii)
Scheme 2.13 The synthesis of 146: (i) DMTSA, [Pd(PPh3)2Cl2], CuI, Et3N, 23 °C, 48 h,
92%; (ii) BuLi, Et2O, –50 °C, 45 min; (iii) I2, Et2O, from –50 °C to 23 °C, 12 h, 99 %

91
(for 145, over two steps), 75% (for 146, over two steps); (iv) TMSA, [Pd(PPh3)2Cl2], CuI,
Et3N, 23 °C, 1 h, 79%.
To required dodecayne 124 was built through a Sonogashira coupling of 140 with
1,2,4,5-functionalized arene fragment 146 (Scheme 2.13). The latter was prepared as
shown in Scheme 2.13, starting with 57 via 145. The coupling between 140 and 146
(i)146140
R
RR
R
147, R = DMTS, R' = TMS124, R = R' = H
R
R'
R'
R
(iii)
148
TMS
TMS
TMS
TMS
(ii)
+
Scheme 2.14 Preparation of 148: (i) [Pd(PPh3)2Cl2], CuI, Et3N, reflux, 16 h, 87%; (ii)
TBAF, THF, 23 °C, 1 h, followed by EtOH, 23 °C, 1 h, (95%); (iii) BTMSA,
[CpCo(CO)2], m-xylene, hν, reflux, 1 h, traces.

92
proceeded uneventfully, providing 147 in 87% yield. The ensuing deprotection gave 124,
which was immediately subjected to the cobalt-mediated cocyclization with BTMSA
(Scheme 2.14). Analysis of the reaction mixture by thin-layer chromatography revealed
at least four products, the polarities of which were similarly low – possibly indicating that
all components of the mixture contained silyl groups. The 1H NMR spectrum was
difficult to interpret, as it showed at least six signals in the silyl region (δ 0–0.5 ppm) and
more than 15 weak signals between δ 6.4 and 7.6 ppm. The mass spectrum (EI, 70 eV)
was somewhat more insightful, as it revealed the presence of several weak ions in the
region of interest (m/z > 800), at m/z values of 810, 840, 923 and 934. The first one of
these signals is consistent with the presence of 148 (m/z 810), while the peak at m/z 934
can be interpreted as 148·CpCo. Repeated separation attempts by ordinary column
chromatography and HPLC failed to produce pure 148.
2.6 Attempted Synthesis of U-Shaped [9]Phenylene (120)
The last phenylene to be targeted as part of these investigations was the helical U-
shaped [9]phenylene 120, only the second [9]phenylene to be pursued to date.70 Its
proposed synthesis relied on the four-fold intramolecular cyclization of 125 (Scheme
2.3b).
As indicated in Section 2.2, 125 was built formally from 124 by tethering two
additional alkynes via an o-phenylene linker. For this purpose, the TMS groups of 147
(the synthetic equivalent of 124) were selectively deprotected. The resulting diterminal

93
alkyne was treated with 1-iodo-2-[(trimethylsilyl)ethynyl]benzene142 under Sonogashira
conditions, producing 149, the protected version of 125 (Scheme 2.15).
120(iii)
x
(i), (ii)
R
RR
R
147, R = DMTS, R' = TMS
R
R'
R'
RR
R
R
RR
RR'
R'
149, R = DMTS, R' = TMS
125, R = R' = H(iv)
Scheme 2.15 Attempted preparation of 120: (i) K2CO3, MeOH/Et2O, 23 °C, 90 min,
(95%); (ii) 1-iodo-2-(TMSethynyl)benzene, [Pd(PPh3)2Cl2], CuI, Et3N, reflux, 16 h, 88%;
(iii) TBAF, THF, 23 °C, 2 h, (95%); (iv) [CpCo(eth)2], THF, –25 °C, 16 h, followed by
1,3-cyclohexadiene, THF, 100 °C, 90 min.
In-situ fluoride-assisted deprotection of 149 was followed by the application of the
[CpCo(eth)2] cyclization conditions. Disappointingly, again no cyclization products were
observed. Given the ambitiousness of the four-fold CpCo-mediated cyclization and the
low yields (0.2–3.5%) observed in triple cyclizations (Section 1.2.2.1, Schemes 1.8 and
1.9),70,82 this negative outcome is perhaps not surprising. Additionally, the probable
instability of the octaterminal dodecayne 125 might have contributed to this negative
outcome.

94
2.7 Calculated and Measured Properties of 60, 118, 119, and 120
This section will focus on the experimentally observed properties of 60, as well as
on the calculated structural and energetic data for all the phenylenes discussed in this
chapter. Comparisons will be made, where applicable, between the novel phenylenes and
their previously synthesized homologues.
Syn-doublebent [5]phenylene (60) is an orange-red material, which decomposes
when exposed to air, both in the solid state and (significantly faster) in solution. The
system seems to be more inert to hydrogenation than either 9b or 15, as it remained intact
under comparable conditions (Pd/C, 1 atm H2, 5 h). Increasing the pressure of hydrogen
gas led to decomposition. Over the course of a few days, 60 reacted with atmospheric
oxygen (both as a solid and in CHCl3 solution). The mass spectrum of the product
mixture showed molecular ions consistent with the addition of one and two molecules of
oxygen, suggesting oxidation via initial single and double endoperoxidation. Analogous
results were observed in the oxidations of 15 and 97 (Section 1.3.3, Scheme 1.24, Figure
1.5).74,97 In an attempt to activate C(aryl)–C(aryl) bonds (as precedented for 15, Section
1.3.5, Scheme 1.28),97 60 was treated with an excess of [CpCo(eth)2], but this procedure
failed to produce any isolable products. No further experiments were carried out, because
of the small amounts of 60 prepared. A more efficient synthesis will be required to fully
assess its chemical reactivity.
Since it was impossible to obtain a sample of 60 suitable for X-ray analysis and
118–120 could not be made, a discussion of their structures has to rely on calculated

95
(B3LYP/6–31G*) data (Figure 2.5). Such calculations have been shown to reproduce
experimental values faithfully, most recently in the example of anti-doublebent
[5]phenylene 56 (with deviations ∆avg = 0.008 Å, ∆max = 0.016 Å, Section 1.4.1, Figure
1.8).76 In addition, since calculated bond lengths for 56 and 60 are essentially identical,
the extrapolation of the experimental bond lengths of the former to those of the latter is
justified.76
The central ring of 60 is calculated to adopt the characteristic bisallylic pattern
(average allyl bond length 1.393 Å), previously observed in 9b (1.392 Å, Section
1.4.1),115 bis(silyl)bent [4]phenylene 52 (1.389 Å, Section 1.4.1),75 and 56 (1.387 Å,
Section 1.4.1).76 The same is true for the central rings in the linear fragments of 118–120,
with the corresponding averaged allyl bond lengths being virtually identical (118: 1.395
Å, 119: 1.394 Å, 120: 1.394 Å).
Conversely, the angularly fused benzene ring in 60 is predicted to exhibit even
more bond fixation (experimentally in 56 - 66%, calculated in 60 - 60%) than the center
of 15 (64%, Section 1.4.1. Figure 1.8),35 but the same as the corresponding ring of
silylated 52 (67% Section 1.4.1. Figure 1.8). The localization in the first (50%) and
second (53%) internal cyclohexatriene ring of the C-shaped 118 is expected to be lower
compared to 60; this result is expected, since the replacement of a localized benzene ring
with a biphenylene moiety allows relative delocalization in the six-membered rings of the
latter. In 119, the central six-membered ring should be relatively delocalized (44%),
while the neighboring rings should show increased bond-alternation (56%). Phenylene

96
33%-7.5
7.3
6.42
9b
N/A-4.7
6.24
24%-9.5
3.1
6.96
6.98
15
6.996.90
6.18
64%-3.3
6.63
29%*-7.5
7.5
6.85
48a
N/A-6.46.46
2.6
67%-2.9
2.323%-9.8
6.68 6.68
6.50
6.39
5.94
6.07
6.90
6.90
56, 1,2-dichlorobenzene-d4
N/A-7.5
2.9
66%-2.9
2.5
17%-9.7
6.52
5.96
6.04
6.78
6.86
N/A-7.5
6.73
2.9
60%*-2.9
2.522%*-9.7
6.58
6.06
6.12
6.92
6.97
7.016.97
6.86
60
118 119 120
23%*
50%*
53%* 28%*
56%*
44%* 44%*
54%*
58%*
21%*
Figure 2.5 Measured bond localization percentages (from X-ray data), experimental 1H
NMR chemical shifts (CDCl3, unless mentioned otherwise), and calculated NICS(1)
values of phenylenes relevant to discussion in Section 2.6. All NICS values refer to the
parent compounds. An asterisk denotes a calculated bond localization percentage for the
parent system. Double bonds are omitted for clarity. a Localization values of 23% and
67% were crystallographically determined for the unsubstituted rings of 52 (the silylated
derivative of 48).

97
119 should thus feature the central angular [3]phenylene substructure with inverted
localization parameters compared to its counterpart in 28.66 The central [3]phenylene unit
in 120 behaves in the same way (44% and 54%, respectively). The terminal rings in all
systems are minimally distorted, with a somewhat higher localization predicted for 119
(28%), because of the linear fusion at the terminus. This expectation is in accord with the
higher localization of terminal rings in 9b vs. 15.
The calculated structures of phenylenes 60 and 118–120 are shown in Figure 2.7.
Compounds 60, 118, and 119 are predicted to be essentially planar. A structural feature
that is of interest in 119 and 120 is the “wing”-angle, defined as the angle between the
vertical projections of two linear[3] fragments onto the plane of the central ring
(equivalent to analyzing artificially flattened structures of the above mentioned
phenylenes). Because of the inherent distortion of the angular [3]phenylene frame, the
calculated value of this angle is not 0 °, but 11.2 ° (119) and 14.9 ° (120). The structure of
angular [3]phenylene (Figure 2.6) offers insight into the origins of this deformation.
Thus, focusing just on the bond angles of the central “bay” region, moving from the
center to the terminal ring, the first two neutralize each other’s effects (153 and 147 °).
However, the third is larger (123 °) than normal, a consequence of strain-induced
rehybridization, typical of phenylenes (Section 1.4.1).116,117 This widening effectively
moves the terminal rings of 15 away from each other, thus expanding the “bay” region.
As a consequence, the vectors (dashed lines in Figure 2.6) between the centroids of the

98
123 o
147 o
153 o
H
Figure 2.6 X-Ray structure of angular [3]phenylene 15 with bond angles responsible for
the “wing”-angle deformation in higher angular and U-shaped phenylenes.
terminal rings and the bonds shown in bold in Figure 2.6 are at an angle of 2.3 °. The
same distortion is evident in the crystal structure of the parent angular [5]phenylene (19,
9.1 °).66 Forcing 119 into a structure in which the “wing”-angle is 0 ° destabilizes the
system by 10.75 kcal mol–1. This result suggests that circular [8]phenylene (121) might
suffer from an additional form of strain caused by the constrained parallel arrangement of
the linear [3]phenylene fragments.
The calculated structure for 120 highlights its inherent helical nature (Figure 2.7).
A comparison between [7]heliphene (28) and 120 is particularly instructive, since the
latter can be viewed as 28 with “linear phenylene” spacers. The distance between the
centroids of the terminal rings is higher in 120 (5.23 Å, 28: 4.54 Å). On the other hand,
the corresponding interplanar angle is significantly larger in 28 (40.6 °, 120: 29.7 °),
hinting at the fact that the helix turns more sharply in 28. Indeed, analysis of the inner
helix shows that while the absolute climb is comparable (120: 3.51 Å, 28: 3.64 Å), the in-
plane turn has a much larger value in 28 (361.6 °, 120: 338.3 °).66 Clearly, the

99
displacement of the linear fragments in 120 away from each other (reflected in the
“wing” angle) also acts to separate the termini of the helix.
Figure 2.7 Calculated structures of phenylenes 60 and 118–120, top and side views.
The relatively low solubility of 60 precluded 13C NMR measurements. Its 1H
NMR spectrum, however, is revealing, especially in comparison with related phenylenes
(Figure 2.5). The assignments given in Figure 2.5 were made on the basis of signal

100
multiplicity, including the simplification of the spectra of the dipropyl derivative 128,
comparison to the NMR spectra of 9b, 15 and 48, and calculated chemical shifts.76 The
effect of additional annelation onto 48 (and 9b) is noticeably larger δ values (relative to
9b and 48) for the hydrogens attached to the central ring, in accord with the changes in
NICS values. Thus, the central protons in the linear fragment experience increasing
deshielding along the series 9b (δ = 6.24 ppm, NICS = –5.4) – 48 (δ = 6.46, 6.39 ppm,
NICS = –6.4) – 60 (δ = 6.73, 6.58 ppm, NICS = –7.5), the result of consecutive bond
fixation in the terminal rings, in turn subtly increasing aromaticity in the central ring and
attenuating paratropism of cyclobutadienoid nuclei. In contrast, the central protons of the
angular fragment, which are slightly shielded in 48 (δ = 5.94 and 6.07 ppm, NICS = –2.9)
relative to 15 (δ = 6.12 ppm, NICS = –3.3, also vide supra), seem to be unchanged in 60
(δ = 6.06 and 6.12 ppm, NICS = –2.9). The deshielding effect of the evolving “bay
region” manifests itself in the increased ∆δ of two protons on the central ring: 0.07 ppm
in 48, 0.15 ppm in 60. Finally, a long range effect of symmetrization in going from 48 to
60 seems absent, as indicated by the essentially identical NMR and NICS data for the
terminal and first internal rings.
Calculations of the heats of formation of the entire family of [5]phenylenes76
reveal that 56 and 60, despite the presence of the linear substructure, are not destabilized
relative to their all-angular isomers 19, 44 and 150 (Figure 2.8). This phenomenon has
been traced to the opposing effects of the σ- (relatively stabilizing) and π-frames
(relatively destabilizing) on the energetics of linear versus angular fusion.82 Since NICS
(1.0) values are reflective primarily of the properties of the π system, total NICS (i.e. the
sum of all NICS values)143 should rectify the relative ordering of these isomers. Indeed

101
(Figure 2.9), such is found, the entire series exhibiting a fairly good linear correlation
between ∆Hf and total NICS (R2 = 0.9816). Calculations on [7]phenylenes 118 and 119
favor the former by 1.56 kcal mol–1, understandably so in view of the fact that 118
possesses just one linear fusion, whereas 119 has two.
19, 0.65 (-22.12)
56, 0.01 (-21.96)
44, 0.36 (-22.30) 150, 0.48 (-22.29)
60, 0.00 (-21.91)
Figure 2.8 Calculated energies (relative to 60, in kcal mol–1) and total NICS values (in
parenthesis) of selected [5]phenylenes.
The electronic spectrum of 60 exhibits the typical two sets of absorptions at
higher (λmax ~ 340–390 nm) and lower energy (highest wavelength λmax = 507 nm). There
are no significant indications of the topological differences between the two doublebent
[5]phenylene isomers, unlike for the series of angular vs. zigzag phenylenes.60 In tune
with a trend emerging in the electronic spectra of the lower phenylenes, i.e. λmax

102
(branched/angular) < λmax (linear),1,47 these bands are at higher energy than that for the
linear [5]phenylene frame (530 nm),57 but bathochromically shifted from those in the
zigzag- (484 nm),74 Y-shaped (C2v) branched (486 nm),79 and angular isomers (470
nm).60
2.8 Summary
Four new phenylenes were targeted by CpCo-mediated cyclization; to this end,
five routes–four of which were all-intramolecular, and one mixed intra/intermolecular–
were developed to access the corresponding oligoyne precursors. Unfortunately, only one
of the envisioned cyclizations was successful.
Syn-doublebent [5]phenylene (60) showed properties that closely parallel those
previously reported for its anti-relative 56. The expected stabilization of linear [3]- and
destabilization of angular [3]phenylene fragments (relative to their parent molecules) was
indeed observed. The calculated structures and relative energies of 118–120 seem to
support predictions based on the arguments borne out in Section 1.4.
Future studies should focus on renewed attempts to synthesize 118–120. In this
respect, particularly appealing is the mixed intra/intermolecular route to 119, the
execution of which led to traces of compound that could be tetrasilylated 119.

103
Chapter Three
A Novel Alkyne Metathesis-Based Route to Dehydrobenzannulenes84b
3.1 Introduction
In an alkyne metathesis144 reaction, two triple-bonded carbon atoms swap their
substituent groups (Scheme 3.1). The first reports of a homogeneous catalytic version of
this transformation date back to the mid 1970’s, when Mortreux and coworkers145
showed that exposure of alkynes to mixtures of [Mo(CO)6]146 and resorcinol led to a
statistical scrambling of alkyl groups on the acetylene.
R1
R2
+
R1
R2
R1
R1
+
R2
R2
catalyst
Scheme 3.1 Schematic representation of alkyne metathesis.
It was not before the 1990’s, though, that the groups of Bunz and others
developed more practical variants of this reaction147 and started applying it
synthetically.148 In a typical example, a methyl alkyne (Scheme 3.1, R1 = Me) is treated
with an off-the-shelf mixture of [Mo(CO)6] and p-chlorophenol, in o-dichlorobenzene as
a solvent, at temperatures of 150–170 ºC. The use of methyl alkynes is crucial since it
leads to the formation of 2-butyne as the other product of the reaction. This material is
gaseous under the reaction conditions, and as such easily removed, either by a stream of
nitrogen, or by performing the reaction under a slight vacuum.

104
While the ease of manipulation certainly speaks in favor of the in situ
[Mo(CO)6]/phenol catalytic system, long reaction times and high temperatures required
are often incompatible with the sensitive functionalities of the starting materials. This
problem was particularly pronounced in natural product syntheses, necessitating the
development of a more gentle reaction system. A well-defined tungsten-alkylidyne
complex [(Me3CO)3W≡CCMe3] (151), first prepared by Schrock,149 was found to
catalyze alkyne metathesis under milder conditions (several hours, 80 ºC, toluene).
Fürstner’s group144a,150 was the first to apply 151 in synthesis. Because the resulting
alkynes can be reduced stereoselectively to cis- or trans-alkenes, an alkyne
metathesis/reduction sequence provides a solution to the problem of the poor E/Z
selectivity in the related alkene metathesis reaction.150a,c–e Other reports of use soon
followed.151 In 2005, 151 was made commercially available,152 paving the way for its
more extensive utilization in organic synthesis.
Recently, a third catalytic system, based on the mixture of [Mo{N(t-Bu)Ar}3] and
methylene chloride, was reported.153 This and related combinations constitute the most
active alkyne metathesis catalysts disclosed to date,154 although they have the drawback
of cumbersome preparation and low stability.
The general mechanism that is widely accepted as an explanation of the observed
trends in alkyne metathesis mediated by 151 (and related alkylidyne species) is given in
Scheme 3.2. It involves metallacyclobutadiene 152 as the key intermediate, which

105
R R
M R'M
R R
R'M
R R
R' R'
R
M
R
152
Scheme 3.2 Proposed general mechanism of the alkyne metathesis.
decomposes in solution to regenerate the reactive carbyne complex and yield the
metathesis product. The dissociation of an alkyne from the metallacyclobutadiene is often
the slowest step of the reaction.155
The mechanism of action of the [Mo(CO)6]/phenol catalytic system remains
largely obscure. Although it is tempting to speculate on the intermediacy of a
metallacarbyne as the active catalyst, recent investigations have suggested trinuclear
alkylidyne clusters as possible intermediates.144c,156
3.2 Retrosynthetic Approach to Dehydrobenzannulenes
This chapter will disclose the potential of alkyne metathesis in the construction of
dehydrobenzannulenes,157 as exemplified in Scheme 3.3. It was postulated that the
dehydrobenz[12]annulenes 153 might be constructed by the metathetic cyclotrimerization
of o-di(prop-1-ynyl)benzenes (154). Those would, in turn, be assembled from the readily
accessible diiodides 155.

106
153a-g
R2
R2
R1
R1
R2
R2
R1
R1
R2
R2
R1
R1
R2
R2
R1
R1 I
I
R2
R2
R1
R1
154a-g 155a-g
a: R1 = R2 = Hb: R1 = CH3, R2 = H
c: R1 = CH3O, R2 = Hd: R1 = R2 = CH3
e: R1 = Br, R2 = H
f: R1 = H, R2 = Brg: R1 = H, R2 = Cl
Scheme 3.3 Retrosynthetic analysis of dehydrobenz[12]annulenes 153a–g.
The class of dehydrobenzannulenes is interesting in several respects - it provides
attractive ligands to transition metal complexes,158 models for subunits of graphyne - a
novel allotrope of carbon,88,89 precursors to ordered carbon nanostructures,159 scaffolds
for molecules on which to study supramolecular phenomena,160 and materials with
interesting photophysical properties.161
Our need for an efficient synthetic entry into substituted derivatives stems from
our quest for the first members of the circular phenylenes (Section 1.2.2.5, Scheme 1.18),
such as antikekulene 77d69 and circular [8]phenylene (121, Figure 2.2).162 The synthesis
of these elusive phenylenes relies heavily on all-ortho alkynylated dehydrobenzannulenes
as precursors (exemplified for 121 in Scheme 3.4). Approaching circular phenylenes
through this method takes advantage of their higher symmetry (relative to other

107
phenylenes), and is therefore more elegant than the alternative based on Sonogashira
couplings (presented for 77d in Section 1.2.2.5, Scheme 1.18).
121156
Scheme 3.4 Retrosynthetic analysis of circular [8]phenylene 121.
Prior to the work described here, Bunz and coworkers reported the preparation of
a series of meta-fused dehydrobenz[30]annulenes (exemplified by 158, Scheme 3.5) by
[Mo(CO)6]/p-chlorophenol catalyzed alkyne metathesis. The desired products were
isolated in 0.5–6% yields.148e A recent improvement (Scheme 3.5) used molybdenum-
amido complexes in a precipitation-driven alkyne metathesis to produce 158 in a much
higher 61% yield (on a mg scale).154a
108
(i)
157 158
Scheme 3.5 An example of the preparation of meta-fused dehydrobenz[30]annulenes by
alkyne metathesis: (i) [Mo(CO)6], p-chlorophenol, 170 °C, 20 h, 6%,148e or
[EtC≡Mo{N(t-Bu)Ar}3], p-nitrophenol, 30 °C, 22 h, 61%.154a
As representative targets, we directed our initial efforts to examples of
tribenzocyclynes 153 (Scheme 3.3), and subsequently the more challenging
tetrabenzocyclyne 159, as well as the fused systems 160 and 161 (Figure 3.1). The parent
153a and its derivatives have received considerable attention,157 whereas a substituted
version of 159160f and parent 160163 have been prepared for the first time only recently,
and through relatively long sequences.

109
159 160
161
Figure 3.1 Dehydrobenzannulenes targeted via alkyne metathesis.
The following sections will detail the synthetic execution of the proposal given in
Scheme 3.3. The preparation of the requisite iodobenzenes will be described first,
followed by the Sonogashira coupling to the propynylated precursors. Finally, the results
of the desired metathesis reaction will be presented.

110
3.3 Preparation of Iodinated Precursors
As Scheme 3.3 outlines, a facile access to the iodides 155a–g was essential for the
evaluation of the proposed route. If successful, this initial pool of iodoarenes was to be
extended to several other systems (162–164, Figure 3.2) that would enable us to test the
viability of cross metathesis leading to targets 159–161.
I
XX
I
163a: X = H163b: X = I57: X = Br
I
I
162
I
I
I
I
I
I
164
Figure 3.2 Additional iodoarene starting materials.
Most of the starting materials were either available commercially (155a, 163a, 164) or
described previously in the literature.78,164 Simple iodination of 1,2-dibromobenzene with
I2/H5IO6,164d provided 1,2-dibromo-4,5-diiodobenzene (155e). The desymmetrized 1,2-
diiodo-3,5-dimethylbenzene (162) required utilization of 3,5-dimethylanthranilic acid in
an aprotic diazotization/iodination sequence, which proceeded through a substituted
benzyne intermediate (Scheme 3.6).165 An analogous sequence furnished 155g.

111
COOH
NH2
162
I
I(ii)(i)
Scheme 3.6 Preparation of 1,2-diiodo-3,5-dimethylbenzene (162): (i) isoamyl nitrite,
dioxane, 80 °C, 2 h; (ii) I2, 76% (over 2 steps).
In contrast, 1,4-dibromo-2,3-diiodobenzene (155f) had to be prepared by a significantly
more complicated synthetic route (Scheme 3.7).166 It started with 2,5-dibromoaniline,
which was converted into the corresponding isonitrosoacetanilide by treatment with
Br
Br
NH2
Br
Br
HN O
N
OH
Br
Br
HN
O
O
Br
Br
NH2
CO2H
Br
Br
I
I
(i) (ii) (iii) (iv)
155f
Scheme 3.7 Preparation of 1,4-dibromo-2,3-diiodobenzene (155f): (i) NH2OH·HCl,
Cl3CCHO·H2O, H2O, EtOH, reflux, 12 h, 84%; (ii) 86% H2SO4, 100 °C, 15 min, 47%;
(iii) NaOH, H2O2, 50 °C, 1 h, 47%; (iv) I2, isoamyl nitrite, ClCH2CH2Cl, 1 h, reflux,
58%.
chloral hydrate and hydroxylamine. Acid-catalyzed cyclization to 3,6-dibromoisatine
proceeded uneventfully167 and was followed by basic hydrolysis in aqueous hydrogen
peroxide to yield 3,6-dibromoanthranilic acid.168 Finally, the acid was converted to 155f
by employing an aprotic diazotization procedure.165 The overall yield of the reaction

112
sequence is a modest 11%, a fact that is compensated for by the simple workup, as no
purification of intermediates was needed.
3.4 Classical and Microwave-Assisted Propynylations
The requisite diynes 154a–g for the metathetical cyclization were accessed by
Sonogashira reaction of appropriately substituted iodoarenes (Scheme 3.8, Table 3.1).
The respective couplings of otherwise unsubstituted and of alkyl- or alkoxy-substituted
iodoarenes proceeded cleanly, in good to excellent yields (entries 1–4, 7–10, 12 in Table
3.1). Entry 12 in Table 3.1 deserves special mention as one of the rare examples of six-
fold Sonogashira coupling to hexaiodobenzene.169
I2-6 2-6
Scheme 3.8 Propynylation reactions: (i) propyne (1–2.5 atm), [PdCl2(PPh3)2], CuI, NEt3;
specific conditions for individual substrates are given in Table 3.1.
Entry Iodoarene Product Conditions Yield (%)
1 155a 154a 25 °C, 22 h 95
2 155b 154b 25 °C, 26 h 57
3 155c 154c 25 °C, 48 h 81
4 155d 154d 25 °C, 96 h 91
5a 155e 154e 110 °C, 3.75 min, microwave 71

113
Entry Iodoarene Product Conditions Yield (%)
6a 155f 154f 110 °C, 20 min, microwave 60
7 155g 154g 110 °C, 16 h 68
8 162
165
25 °C, 44 h 76
9 163a
166a
25 °C, 22 h 93
10 163b
166b
90 °C, 36 h 77
11a 57 BrBr
166c
100 °C, 2 min, microwave 64
12b 164
167
90 °C, 60 h 28
Table 3.1 Propynylation reactions. a DMF (10%) was used as a cosolvent. b Penta(prop-
1-ynyl)benzene was obtained as a side product in 32% yield.
To retain the potential of subsequent introduction of further alkyne substituents,69,162
tribenzocyclynes bearing bromine170 substituents were also of interest. Their preparation
required the selective alkynylation of bromoiodoarenes (entries 5,6,11 in Table 3.1).
While such is often achievable at room temperature,171 we noticed significant

114
overalkynylation in systems 155e, 155f, and 57. Monitoring the course of the reaction to
the point of optimal conversion proved difficult because of the use of closed systems
under the positive pressure of propyne (closed systems were chosen in order to ensure
economic use of the relatively expensive propyne gas).
We therefore turned our attention to microwave-assisted Sonogashira couplings172
executed in a Smith Synthesizer.173 Application of this technique allowed the
dipropynylation of three isomers of dibromodiiodobenzene at 100–110 °C (entries 5,6,11
in Table 3.1) with excellent selectivity in less than 20 minutes. Monitoring the change in
pressure during the course of the reaction (Figure 3.3) provided a convenient gauge of its
Figure 3.3 Pressure changes as a function of reaction time in Pd-catalyzed
propynylations of 155e in a Smith Synthesizer. There is an initial pressure increase, as
heating commences. The reaction starts at t ~ 5 min, causing the propyne pressure to
drop, and ends at t ~ 15 min. The heater was turned off at t ~ 44 min.
progress. The instrumental setup (heavy-walled sealed Smith Process vials, 5–10 mL
volume, pressurized with gaseous propyne) enabled this reaction to be performed only on

115
a relatively small scale; this procedure was thus restricted to the preparation of
propynylated benzenes for which classical conditions proved to be too hard to control.
The results of all propynylation reactions, both “classical” and microwave-
assisted, are summarized in Table 3.1.
3.5 Dehydrobenzannulenes by Alkyne Metathesis
With the well-developed route to o-dipropynylated benzenes in hand, the
feasibility of alkyne metathesis to tribenzocyclynes could be tested. Due to the simplicity
of the [Mo(CO)6]/p-chlorophenol system, this catalyst was used in our first experiments
with 154a as the substrate. Under a variety of conditions - performing the reaction at
decreased pressure, under a constant stream of nitrogen (both of which served to remove
2-butyne formed), or with variable catalyst loading - not even trace amounts of 153a
(GC/MS) were detectable. The only occasional product was the metathesis dimer, 1,1’-
(1,2-ethynediyl)bis[2-(prop-1-ynyl)benzene, generated in approximately 2% yield (by
GC/MS analysis).
Gratifyingly, turning to 151174 as the catalyst, the reaction proceeded cleanly to
give 153a in 54% yield as the only isolable product (Scheme 3.9, entry 1 in Table 3.2).
This preparation is superior in terms of yield and simplicity compared to other recently
published approaches that, for the most part, rely on sequences of Pd-catalyzed cross
couplings and proceed in yields ranging from 15% to 40%.86c,d,158a,b,175

116
R2
R2
R1
R1
R2
R2
R1
R1
R2
R2
R1
R1
(i)
R2
R2
R1
R1
Scheme 3.9 Dehydrobenz[12]annulenes by alkyne metatheses: (i) 151 (20 mol%),
PhCH3, 80 °C, specific conditions for individual substrates given in Table 3.2.
Entry Starting material Cyclyne Reaction time (h) Yield (%)
1a 154a 153a 8 54
2b 154b 153b 24 27
3c 154c 153c 140 28
4 154d 153d 96 0
5 154e 153e 120 12
6 154f 153f 96 0
7 154g 153g 36 0
Table 3.2 Dehydrobenz[12]annulenes by alkyne metatheses catalyzed by 151. a Ref. 176. b
Ref. 177. c Ref. 158a,d.
Encouraged by this result, scope and limitations were investigated, summarized in
Table 3.2. A rather simple trend was observed - sterically more crowded
bisorthosubstituted precursors did not undergo cyclization (entries 4,6,7 in Table 3.2),
whereas bismetasubstituted ones did (entries 2,3,5 in Table 3.2). This outcome was not

117
particularly dependent on electronic effects - both electron withdrawing (Br) and electron
donating (OMe and Me) substituents were tolerated as long as they were located in meta
positions; interestingly, substrates with resonance donors as substituents (Br and OMe)
reacted more slowly (~ 2–4 times) than their unsubstituted or alkylsubstituted
counterparts.
To provide an intramolecular test for the proposed steric trend, substrate 165 was
investigated, in which the two alkyne units are differentiated by bearing an ortho- and a
meta-methyl group, respectively. It should react only once, to give the product of
metathesis of the sterically less crowded triple bond. Indeed, the system produced solely
168 in 55% yield (Scheme 3.10).
(i)
165 168
Scheme 3.10 Regioselective alkyne metathesis reaction: (i) 151, PhCH3, 80 °C, 72 h,
55%.
While the yields of the products depicted in Table 3.2 are modest, the simplicity
and straightforward execution of the method would seem to make it that of choice for the
rapid synthesis of specific derivatives, in particular when such are endowed with
interesting novel topologies. As a consequence, and to explore the possibility of ring
closure cross metathesis, we targeted the parent hydrocarbons 159–161. To our delight,

118
equimolar proportions of 154a and 166a converted directly to the new tetrabenzocyclyne
159 in 19% yield (Scheme 3.11a)! Even more impressive was the finding that 154a and
166b (4:1) underwent six-fold metathesis to furnish 160 in 6% yield (Scheme 3.11b).
This compound, as previously reported,163 was extremely insoluble in common organic
solvents, probably to the detriment of the isolated yield. Finally, not unexpectedly in light
of the results described above, hexapropynylbenzene 165 was inert to metathesis with
154a (on route to 161) and even simple propynylbenzenes.
(i)154a
159
166a+(a)
(i)154a
160
166b+(b)
Scheme 3.11 Alkyne metathesis to 159 and 160: (i) 151, PhCH3, 80 °C, 60–84 h, 19%
(159), 6% (160).

119
3.6 Properties of Novel Dehydrobenzannulenes
Cyclyne 159 constitutes the parent of a di-tert-butyl derivative synthesized as part
of a series of phenylacetylene macrocycles adorned with solubilizing substituents.160f It
is, nonetheless, quite soluble in common organic solvents, exhibiting strong, purple
fluorescence. Its 1H NMR spectrum contains a characteristic peak due to the proton inside
the macrocycle at δ = 8.05 ppm (CDCl3). The less benzofused system 169 (Figure 3.4)
shows the analogous absorption at δ 7.82 ppm (CDCl3), possibly (but not necessarily) a
reflection of increased dehydro[18]annulenoid diatropism.178 The aromaticity of cyclynes
as measured by the ring current criterion is a topic of renewed current scrutiny.23
169
Figure 3.4 Cyclyne 169.
The X-ray crystal structure of 159,179 shown in Figure 3.5, is only slightly distorted from
ideal planarity–the dihedral angle between the planes of the respective meta- and ortho-
fused rings is 7.1 °. The intraannular hydrogen–hydrogen distance is 2.29 Å; in
comparison, in 169 this distance is 2.57 Å–a possible indication of the greater flexibility
of the system.178 The compound crystallizes in the C2/c space group, with four molecules
of 159 in the unit cell.

120
Figure 3.5 ORTEP diagram of 159. Thermal ellipsoids shown at the 50% probability
level.
Although 160 was described previously,163 its 1H NMR spectrum could not be
obtained due to seemingly poor solubility. We have found 160 sufficiently soluble in
CDCl3 to allow for such a measurement. The molecule gives rise to an AA’BB’ multiplet
for the peripheral aromatic hydrogens at δ = 7.19 and 7.44 ppm, instead of the expected
ABCD pattern, reflecting local symmetry, and a singlet at δ = 7.34 ppm for the protons
on the central benzene ring. These appear shielded relative to the corresponding ring
hydrogens in 1,2,4,5-tetraethynylbenzene, which resonate at δ 7.63 ppm (CDCl3)–an
indication of the effect of the two neighboring paratropic cyclyne moieties.180

121
3.7 Summary
We have shown that alkyne metathesis holds promise as a general tool for the
construction of benzocyclynes. Although some of the reactions proceed in relatively
modest yields, this is compensated for by a synthetic approach that is short and
straightforward. The feasibility of the reaction is dependent on the substitution pattern of
the starting materials; this effect is presumably steric in nature and could be explored in
terms of regioselective alkyne metathesis reactions. Furthermore, it is possible that some
of the systems we found unreactive in the presence of 151 will yield themselves to
metathesis with the new, more active, molybdenum-based catalysts.

122
Chapter Four
Synthesis of Octaalkynylated Dehydrobenz[18]annulenes and Attempted
Cycloisomerization into Circular [8]Phenylene and Derivatives181
4.1 Introduction: Circular [8]Phenylene
Notably missing among the simple phenylene topologies synthesized are the
circular isomers. As mentioned in Section 1.2.2.5, this class of phenylenes differs from
other topologies by the potential for delocalization not just within the six-membered
rings, but also of the internal and the external annulenoid loops. This phenomenon is
known as superdelocalization83 and has not been observed in hydrocarbons so far.85
Synthetically, our group has pursued actively circular [6]phenylene (77d,
antikekulene, Figure 4.1, Section 1.2.2.5). In addition to this system, circular [4]-, [5]-,
and [7]phenylenes have been theoretically scrutinized.55 The former two molecules are
bowl-shaped and significantly strained (e.g. circular [5]phenylene is destabilized by 46.8
kcal mol–1 due to its bowl shape).55 Circular [7]phenylene is planar, but still strained
relative to 77d.
The next higher strain-free homologue is circular [8]phenylene (121, Figure 4.1),
formally a derivative of 77d with two linear phenylene spacers. Both 77d and 121 are 4n
species (Section 1.1), with π-electron counts of 36 and 48, respectively. In 77d, the
internal and the external supercircuits also have 4n π-counts–12 and 24, respectively.
Each of the extra linear fragments present in 121 contributes one additional atom to the
internal and one less atom to the external loop, switching the respective π-counts to 18

123
and 30, both of which are 4n+2. Thus, while both superloops in 77d are formally
antiaromatic, in 121 they are aromatic, which is expected to impart some additional
stability to the system.
12177d
Figure 4.1 Circular [6]- (77d) and [8]phenylene (121).
If annulenoid aromaticity of the 18- and 30-electron supercircuits were indeed to
be present, it could manifest itself in the accentuation of the resonance form depicted
above for 121. For example, one might expect bonds shared between the six-membered
rings and each one of the supercircuits to become increasingly “double”, while radial
bonds connecting two supercircuits might become increasingly “single”. A comparison
between the calculated structures of 121 and non-circular models 118–120 (Section 2.7)
is given in Figure 4.2. The effects of the superloops appear to be too subtle to be visible,
at least structurally. For example, going from 119 to 121, one would expect a decrease in
the bond alternation in the peripheral six-membered rings of the linear fragment, since the
bond-localizing effect of the linear moiety is shared by two angular fragments in 121
(analogous to comparison between 48 and 56/60, Section 1.4.1, Figure 1.8).75,76

124
118 119 120
23%
50%
53% 28%
56%
44% 44%
54%
58%
21%
121
56%
46%
Figure 4.2 Bond localization percentages for 121 and some non-circular analogs
(calculated values, B3LYP/6-31G*).
However, this behavior is not observed, as both 119 and 121 show 56% localization. The
central ring in 119 is 2% more localized than its counterpart in 121. Seemingly in
agreement with the annulene-enforced localization is a comparison between 120 and 121.
In the former, the central ring is 44% localized, its neighbor 54%. The values for
analogous rings of 121 are 46 and 56%, respectively, although the first point of difference
between the two molecules is four (relative to the center of 121) six-membered rings
away!

125
The structure of 121 is shown in Figure 4.3. The molecule is almost planar, the
most significant deviations being in the positions of two internal hydrogen atoms. These
are oriented in a way that maximizes their interatomic distance (to 1.80 Å) – one of them
lying above the average molecular plane, the other positioned below it. These distortions
go hand in hand with a deformation of the carbon skeleton. Thus, the angle between the
planes of the central six-membered rings of the linear fragment of 121 and the central six-
membered rings of the angular [5]phenylene subunit (the “most right”/”most left” rings in
Figure 4.3) is 3.9 °. Previous molecular mechanics calculations placed this angle at 7.1 °
and the internal H–H distance at 2.30 Å.162 Finally, the bisallylic pattern calculated for
the central linear rings of 60 and 118–120 (Section 2.7) is predicted for 121 as well.
Curiously, the bonds of the linearly fused ring that are also part of the [18]annulene
circuit are much shorter (1.385 Å) than those shared with its [30]annulene counterpart
(1.401 Å). In 60, these values are virtually identical (1.393 and 1.394 Å, respectively).
Figure 4.3 Calculated structure of 121 (B3LYP/6-31G*, top and side views).

126
A much more sensitive probe of aromaticity are 1H NMR chemical shifts.2,5
Therefore, the potential superaromaticity of the aromatic superloops might manifest itself
in relative deshielding of the outside hydrogens and, most diagnostically, shielding of the
inside ones.5 In [18]annulene itself, the latter value is –2.88 ppm (THF-d8, –59.5 °C),182 a
number large enough to lead to the anticipation that even a strongly attenuated effect of
superdelocalization would be measurable.
This chapter will detail the synthesis of dehydrobenzannulenic precursors to 121
and substituted versions thereof. The discussion of the results of the attempted conversion
of these materials by CpCo-cycloisomerization will follow. The chapter will conclude
with the presentation of the properties of some new phenylenes prepared during these
attempts.
4.2 Retrosynthetic Analysis of Circular [8]Phenylene
Due to their unique structures devoid of terminal six-membered rings, circular
[N]phenylenes can be accessed only by intramolecular CpCo-mediated cyclizations of the
corresponding precursors (Section 1.2). Focusing on circular [8]phenylene (121) as the
target, the choice of potential precursors is even more limited, since the central rings of
linear [3]phenylene fragments in 121 have to be included in the structure of the
precursors (Section 1.2.1). Based on this notion, dehydrobenz[18]annulenes 156 and
171a–c (Scheme 4.1) were chosen as precursors to 121 (and its derivatives 170a–c).
Successful conversion into circular phenylenes would require an unprecedented four-fold
CpCo-cycloisomerization.183

127
170a, R = Pr170b, R = Hex170c, R = DMTS121, R = H
RR
RR
RRR
RR
R
R
R
R
RR
R
171a, R = Pr171b, R = Hex171c, R = DMTS156, R = H
Scheme 4.1 Retrosynthetic analysis of 121 and its derivatives.
Consideration of simpler cycloisomerization substrates suggested two alternatives
to 156/171, shown in Figure 4.4. In 172 (Figure 4.4, left), five of the eight six-membered
rings of 121 are already preformed, requiring only a triple cobalt-catalyzed
cyclization.70,82 In 173 (Figure 4.4, right), six of these rings are already present,
necessitating only a double cyclization.66,74,76 While these routes may seem tempting,
neither the 1,2,8,9-tetrahalogenated biphenylenes required for the assembly of 172, nor
appropriately substituted linear [3]phenylenes on route to 173 are known and are in fact
not easily accessible by current art. Thus, only the 121 → 156 disconnection was
considered.

128
172 173
Figure 4.4 Alternative (but impractical) precursors to 121.
The results presented in Chapter 3 indicated that the direct formation of all-ortho
brominated dehydrobenz[18]annulenes (followed by per-alkynylation) would not be
possible due to the steric limitations of alkyne metathesis. Therefore, an alternative
stepwise approach was envisioned, which produces 171 by the formal dimerization of
174 (Scheme 4.2). Compound 174, in turn, would be made through a Sonogashira
coupling of 1,2,4,5- and 1,2,3,4-substituted arene fragments 175 and 176. This
retrosynthetic proposal allowed for the easy modification of the nature of the substituents
R, which might become necessary as both solubilizing and protecting units.
129
I
R
RRR
174a, R = Pr174b, R = Hex174c, R = DMTS
Br
R
R
RR
I
TMS
+
RR
RR
RRR
R
171a, R = Pr171b, R = Hex171c, R = DMTS156, R = H
175a, R = Pr175b, R = Hex175c, R = DMTS
176a, R = Pr176b, R = Hex146, R = DMTS
Scheme 4.2 Retrosynthetic analysis of 171.
4.3 Previous Attempts to Synthesize Circular [8]Phenylene
A route to circular [8]phenylene had been executed previously along the
guidelines outlined in Section 4.2. In anticipation of solubility problems, the focus was on
the octapropylated derivative 170a via 171a. Thus, starting with 57,78 triyne 177 was
made in three steps (with the exception of the last step, the preparation paralleled that for
146, Section 2.5.2, Scheme 2.13).162,181 This material was deprotected and coupled with

130
the previously described 2-iodo-1,4-di(pent-1-ynyl)-3-[(trimethylsilyl)ethynyl] benzene69
to give the elaborated diphenylacetylene 178 in 72% yield (Scheme 4.3).
178177
PrPr
Br
PrPr
Br
TMS
Pr
Pr
TMS
(i), (ii)
Scheme 4.3 The synthesis of 178: (i) TBAF, THF, 23 °C, 30 min, (95%); (ii) 2-iodo-1,4-
di(pent-1-ynyl)-3-[(trimethylsilyl)ethynyl]benzene, [Pd(PPh3)2Cl2], CuI, Et3N, 70 °C, 16
h, 72%.
Half of the so-produced 178 was deprotected to 179, while the other was subjected to
bromine-iodine exchange that produced 180. These two fragments were coupled to 181
(Scheme 4.4).

131
180
PrPr
I
TMS
Pr
Pr
(i)
178
179
Pr Pr
Br
Pr
Pr
(ii), (iii)
(iv)
181
Pr
Pr
Br
Pr
Pr
Pr
Pr
Pr
Pr
TMS
Scheme 4.4 The synthesis of 181: (i) TBAF, THF, 23 °C, 30 min, 66%; (ii) BuLi, Et2O, –
78 °C, 30 min; (iii) I2, Et2O, –78 °C, 10 min, 94% (over two steps); (iv) [Pd(PPh3)2Cl2],
CuI, Et3N, 70 °C, 14 h, 80%.
So-obtained 181 was subjected to another bromine-iodine exchange, deprotection of the
TMS group and intramolecular Sonogashira reaction to provide the octa(pent-1-
ynyl)dehydrobenz[18]annulene 171a in 30% overall yield (Scheme 4.5). Cobalt-
catalyzed cycloisomerization of 171a stopped at doubly-cyclized 182 and 183,
derivatives of doublebent [5]- and angular [3]phenylene, respectively. These two
products were formed exclusively – not even traces of mono- or triscyclized phenylenes
were observed (by 1H NMR). Curiously, although a third double-cyclization product was

132
possible, it was not generated in these experiments. Resubjecting 182 and 183 to the
catalyst in higher-boiling solvents either left the starting materials unaffected (1,2-
dichlorobenzene) or completely destroyed them (1,2,4-trichlorobenzene). The synthesis
of octapropyl circular [8]phenylene 170a and 121 was abandoned at this point,162,181 until
the work to be described next commenced.
(i), (ii), (iii), (iv)180 171a
(v)
Pr
183
Pr
Pr
PrPr
Pr
Pr
Pr
+
Pr
182
Pr
Pr
Pr
Pr
PrPr Pr
Scheme 4.5 The synthesis of 171a and its cycloisomerization into 182 and 183: (i) BuLi,
Et2O, –78 °C, 30 min; (ii) I2, Et2O, –78 °C, 10 min; (iii) TBAF, THF, 23 °C, 30 min; (iv)
[Pd(PPh3)2Cl2], CuI, Et3N, 65 °C, 14 h, 30% (over 4 steps); (v) [CpCo(CO)2], m-xylene,
reflux, 45 min, 10% (182), 35% (183).

133
4.4 Synthesis and Properties of Octaalkynylated Dehydrobenz[18]annulenes 156
and 171b–c
The failure to reach 170a via 171a could be due to a variety of reasons. Among
them were still insufficient solubility of the target as well as the penultimate cyclization
intermediates derived from 182 and 183, and steric hindrance to cobalt-catalyzed
cyclization imparted by the terminal alkyl substituents. To probe the validity of these
considerations, the synthesis of 156 and 171b–c was planned, 171b as a system in which
solubility problems were thought to be truly irrelevant, and 171c as a protected precursor
to 156, the latter providing the opportunity to tackle the parent 121.
Within the context of a bigger picture, the dehydrobenzannulenes of the type 171
were of interest also as novel members of this class of hydrocarbons. As Chapter 3
mentioned, such constructs are subject to intense scrutiny because of their potential
applications as optoelectronic, liquid crystalline, conducting, and sensing materials, as
building blocks in the construction of allotropes of carbon, as scaffolds for
supramolecular assemblies, and as monomers in topochemical and other
polymerizations.157 In this connection, the novelty of compounds 171 lies in their new
topology, combining the 1,2,3,4- with the 1,2,4,5-tetraethynylbenzene motifs in an
elaborated octaalkynylated tetrabenz[a,b,f,j,k,o]-4,5,10,11,15,16,21,22-
octadehydro[18]annulene,84b endowed with internal hydrogens that can serve as probes
for the effect of peripheral alteration on the aromaticity of the central core. Parent 156 is
also the only second hydrocarbon of composition C48H16 to be targeted for synthesis.184

134
The triynes 184b–c, reported previously,69,185 were chosen as the precursors for
the 1,2,3,4-substituted moieties of 171b–c (Scheme 4.6). Selective TMS removal from
184b–c generated the corresponding terminal alkynes, which were coupled with iodides
176b/146 to give the highly functionalized diarylacetylenes 185b–c. Compounds 185b–c
were the source of both pieces needed to assemble 171b–c. Protodetrimethylsilylation
provided 186b–c; alternatively, another bromine-iodine exchange led to 187b–c (Scheme
4.7). Sonogashira coupling of 186b–c and 187b–c created 188b–c, possessing all the
carbon atoms of 171b–c. Finally, bromine-iodine exchange, followed by TMS
deprotection and an intramolecular Sonogashira coupling afforded 171b–c (Scheme 4.7).
In situ deprotection of 171c (TBAF/SiO2) provided the parent 156, which decomposed
within minutes, even in dilute solutions.
184b, R = Hex184c, R = DMTS
R
R
Br
+176b146
(ii) TMS
RR
185b, R = Hex185c, R = DMTS
R
R
Br
TMS
175b175c
(i)
Scheme 4.6 The synthesis of 185b–c and its cycloisomerization into 182 and 183: (i)
K2CO3 or KOH, MeOH/Et2O, 23 °C, 1 h; (ii) [Pd(PPh3)2Cl2], CuI, NEt3, reflux, 16 h,
90% (185b, over two steps), 95% (185c, over two steps).

135
The approach to 156 and 171b–c is a topological alternative to the scheme
executed on route to 171a, as the roles of the coupling partners in Schemes 4.3 and 4.6
were switched. The yields of the two routes are comparable.
Dodecaynes 171b and c are yellow-brown waxy solids, stable to air, both neat and
in solution. On the other hand, parent 156 decomposed quickly, even in solution, thus
precluding its isolation and full characterization. The electronic spectra are characterized
by a lowest-energy band (λmax = 369–377 nm) that is significantly shifted
bathochromically relative to that of the parent dehydrobenz[18]annulene (λmax = 341 nm),
a reflection of the extensive alkynyl substitution. The NMR spectra show the
characteristically deshielded intracyclic hydrogen signals at δ ~ 7.8 ppm84b and the
appropriate number of 1H and 13C peaks. An exception is 171c, which featured four
(instead of the expected two) sets of resonances of the methyl groups in the DMTS
residue. This was a consequence of conformational rigidity of 171c which rendered the
DMTS methyl groups diastereotopic, and thus inequivalent in the NMR. A more detailed
treatment of this phenomenon will be presented in Chapter 5.

136
187b, R = Hex187c, R = DMTS
RR
I
R
R
(i)
185b185c
186b, R = Hex186c, R = DMTS
R R
Br
R
R
(ii), (iii)
(iv)
188b, R = Hex188c, R = DMTS
R
R
R
R
R
R
Br
R
R
TMS
TMS
(i), (ii), (iii), (iv)
171b171c
(v)156
Scheme 4.7 The synthesis of 156 and 171b-c: (i) K2CO3 or KOH, MeOH/Et2O, 23 °C, 1
h, 98% (186b), 87% (186c); (ii) BuLi, Et2O, –45 °C, 30 min; (iii) I2, Et2O, –45 °C to 23
°C, 2 h, 94% (187b, over two steps), 92% (187c, over two steps); (iv) [Pd(PPh3)2Cl2],
CuI, Et3N, reflux, 16 h, 78% (188b), 78% (188c); (v) TBAF/SiO2, THF, 23 °C, 1 h. The
overall yield of steps (i)–(iv) for 171b and c was 62% and 45%, respectively.

137
4.5 Attempted Cycloisomerization of 156 and 171b-c into Circular [8]Phenylenes
121 and 170b-c
With systems 156 and 171b-c available, experiments were executed aimed at
accessing circular [8]phenylene (121), and its derivatives 170b and c. The parent 156 was
treated with [CpCo(eth)2] at low temperatures, followed by heating with 1,3-
cyclohexadiene–a procedure analogous to the one employed in the successful synthesis of
syn-doublebent [5]phenylene 60 (Section 2.3, Scheme 2.5).76 Disappointingly, even when
using in situ deprotection protocols,157 complete decomposition of 156 was observed.
Silyl-substituted 171c proved inert to [CpCo(CO)2], not surprisingly in light of the steric
bulk of the DMTS groups. Therefore, our efforts focused on the isomerizations of 171b
(Scheme 4.8). Unfortunately, only double cyclizations were achieved to the deep-red
double bent [5]- and yellow angular [3]phenylene derivatives 189 and 190, respectively.
The polarities of the two products were virtually identical, precluding their separation.
However, we were able to obtain 189 as the exclusive product by increasing the amount
of [CpCo(CO)2] to 6 equiv. These results parallel essentially those obtained with the
corresponding propyl system 171a, ruling out solubility as an issue. It is clear that some
factor is precluding complete cycloisomerization of the dodecaynes, in the case of 171a
stopping at 182 and 183, in the case of 171b at 189 and 190. It is possible that the parent
system 156 suffers the same fate, but that in this case the dangling terminal triple bonds
undergo intermolecular oligomerizations.

138
171b(i)
R
190, R = Hex
R
R
RR
R
R
R
+
R
189, R = Hex
R
R
R
R
RR R
Scheme 4.8 Cycloisomerization of 171b: (i) [CpCo(CO)2], PhCH3, reflux, hν, 19%
(inseparable mixture of 189 and 190). Isomer 189 was the only product (11%), when 6
equiv of [CpCo(CO)2] were used.
Stymied by the above failures, calculational efforts were made aimed at gaining a
deeper understanding of the reasons for them. Specifically, DFT calculations (B3LYP/6-
31G*) of all possible cyclization products derived from 156 were performed (Figure 4.5).
The most stable forms of all these compounds were predicted to be non-planar, although
planarization was energetically cheap. The respective energies and some of the important
(vide infra) structural results are summarized in Figure 4.5 and Tables 4.1 and 4.2 (for
planar and deplanarized structures, respectively). The data predict that a) the
cycloisomerization sequence is quite exothermic, ~ 50 kcal mol–1 per step; b) as the
cyclization progresses, the resulting phenylenes planarize more easily (evidenced by
smaller ∆E values), and c) the inner 18-membered ring becomes more compact (judged
by the internal hydrogen separation). Particularly diagnostic for our purposes is the
change in the geometry of the triple bonds in the series 156 → 191 → 192–4 → 195: as
the cyclization proceeds, the remaining triple bonds in each intermediate product are

139
Figure 4.5 Calculated structures of in the products of the cyclization of 156.

140
calculated to become increasingly separated, thus likely retarding the normally facile
cobaltacyclopentadiene(alkyne) formation.61 To some extent, the origins of this
distancing are in the “opening” of the phenylenic frame, analogous to that observed in
angular [3]- (15) and U-shaped phenylenes 119 and 120 (Section 2.7, Figures 2.6 and
2.7). This deformation moves the terminal triple bond on the phenylene into an “empty
space”, rather than towards the other uncyclized triple bonds. A similar phenomenon had
frustrated an approach to circular [6]phenylene.69
Compound
Relative
energy
[kcal/mol]
Distance between
internal hydrogens
[Å]
Distance between
terminal triple bonds
[Å]a
156 0.000 2.101 3.352–4.220
191 –50.557 2.004
3.393–4.269
3.471–4.369
3.374–4.240
192 –100.292 1.907 3.545–4.446
193 –101.557 1.871 3.508–4.406
194 –99.026 2.002 3.420–4.296
195 –148.908 1.853 3.578–4.481
121 –195.507 1.753 N/A
Table 4.1 Comparison of calculated structural parameters for planar forms of 121 and its
alkyne isomers. a Terminal carbon atoms–internal carbon atoms. For 191, which contains

141
three sets of different triyne units, the order is top right, bottom left, bottom right, when
viewing the preceding structural drawing.
Compound Energya
[kcal/mol]
Distance between
internal hydrogens
[Å]
Distance between
terminal triple bonds
[Å]b
156 –2.221 2.321 3.522–4.291
191 –1.829 2.114
3.531–4.358
3.822–4.537
3.688–4.410
192 –0.054 1.907 3.560–4.445
193 –1.036 2.026 3.741–4.508
194 –0.173 2.002 3.479–4.324
195 +0.019 1.882 3.612–4.483
121 –0.013 1.800 N/A
Table 4.2 Comparison of calculated structural parameters for nonplanar forms of 121 and
its alkyne isomers. a Relative to the corresponding planar conformer. b Terminal carbon
atoms–internal carbon atoms. For 191, which contains three sets of different triyne units,
the order is top right, bottom left, bottom right, when viewing the preceding structural
drawing.

142
4.6 Properties of Novel Phenylenes
Despite the failure of the final two cyclization steps, phenylene systems 189 and
190 (and their octapropyl relatives 182 and 183) represent the only third and fourth
examples of phenylenocyclynes. In these systems, the terminal rings of a phenylene are
linked by a conjugating bridge, and the changes in their properties as a result of this
feature are of interest. Indeed, the molecules appear more air sensitive than their
component phenylenes (Section 1.3.3), and solutions (CHCl3) of, e.g., 189 in air
decomposed within hours. In this case, the mass spectra of the complex product mixture
revealed molecular ions consistent with the addition of one and two molecules of oxygen,
and the IR spectrum exhibited a strong band at 1645 cm–1, suggesting oxidation via initial
single and double endoperoxidation, as observed for angular [3]- (15) and dipropyl
zigzag [5]phenylene (97, Section 1.3.3, Scheme 1.24).74,97 The electronic spectrum of 189
is, as expected, very similar to that of 182 and it provides a quantitative measure of
increased delocalization in longest wavelength maxima, which are shifted to lower
energies when compared to the parent phenylene substructures by ∆λmax = 37 nm
(Section 1.4.3).76,181 A similar comparison of the 1H NMR spectra of 189 and 190 with
those of their component phenylenes and of 171b illustrates the absence of any
significant “super ring current” effects. For example, the two relatively sharp singlets at δ
= 7.69 (inner H) and 7.49 ppm (outer H) of the tetraalkynylbenzene ring hydrogens in
189, compare well with the corresponding signals in 171b: δ = 7.81 and 7.43 ppm. The
more coupled186 central phenylene hydrogens at δ = 6.67 (inner H) and 6.47 ppm (outer
H) have counterparts in the parent [5]phenylene at δ = 6.73 and 6.58 ppm. The

143
assignments in 189 were corroborated by NOE experiments. Polarization was cleanly
transferred from the inner benzenoid proton at δ = 7.69 ppm to the phenylenic one at δ =
6.67 ppm. No such correlation was observed for the other pair.
4.7 Summary and Future Directions
A convergent and robust synthetic route to the octaalkynylated
dehydrobenz[18]annulenes 156 and 171b–c was developed. Compound 171b was partly
cyclized to the cyclically delocalized phenylenes 189 and 190 in which the remaining
alkyne units seem too distant to undergo CpCo-catalyzed cyclotrimerization to circular
[8]phenylene 170b. Future work will aim to modify chemically the phenylene skeletons
of 189 and 190, in order to circumvent this problem.

144
Chapter Five
Consequences of Steric Crowding Around Triple Bonds in Acyclic and Cyclic
Systems181,187
5.1 Introduction
The three-dimensional structures of molecules are not unambiguously defined by
composition and configuration alone. A complete description requires information about
the torsional angles around all of the single bonds in a molecule - a quality known as
conformation. Since torsional angles are defined by four points, any system with an A–
B–C–D arrangement can have multiple conformers. The simplest such stable molecule is
hydrogen peroxide (H–O–O–H) in general, and ethane (CH3–CH3) among organic
molecules. In a large majority of systems, the rotation around single bonds is unrestricted.
However, adequate crowding can increase the barrier to rotation to the point where the
rate of the interconversion of conformers becomes comparable to the time scale of
analytical techniques. To allow the observation of this hindered rotation, the conformers
cannot be superimposable images of each other; this translates into the necessity of
differential substitution around the single bond that is considered. For example, even if
rotation in ethane were to be completely shut down, conformers would have identical
structures, thus making hindered rotation unobservable.188
Atropisomerism (from Greek: α = not, and τροποσ = to turn) is a type of
rotational isomerism in which detection and even isolation of rotamers is possible due to
a sufficiently hindered rotation around a chirogenic axis of the molecule. Atropisomeric

145
systems have been conveniently classified on the basis of the identity of the chirogenic
axis. As Table 5.1 shows, the phenomenon has been observed with bonds to all types of
–C(sp3) –C(sp2) –C(sp)
C(sp3)– COOCH3
COOCH3
196
33.2 kcal mol–1a
H
Br
197
27.1 kcal mol–1b
198
15.6 kcal mol–1c
C(sp2)– OH
HO
199
13.0 kcal mol–1d
200
18.0 kcal mol–1e
C(sp)– –
Table 5.1 Examples of atropisomeric molecules for which rotational barriers have been
measured, classified by the identity of the chirogenic axis (shown in bold in the
structures). Numbers below the structures are the corresponding ∆G‡ values. a Ref. 189. b
Ref. 190. c Ref. 191. d Ref. 192. e Ref. 193.

146
hybridized carbons. In this area of investigation, tetrasubstituted biphenyls exemplified
by 199 (Table 5.1) have attracted the most attention.188,194
Rotation around bonds to an sp-carbon is special, in as much as this nucleus does
not bear any substituents but only another such carbon at a 180 ° angle, generating a large
spacer (~ 4.0 Å) that requires unusually bulky groups at the termini for this motion to be
sufficiently retarded to be measurable. Thus, rotation around the –C≡C– unit is normally
essentially “free” - for example, in the parent diphenylacetylene the barrier is less than 1
kcal mol–1.195 The idea that appropriate substitution may hinder this motion195b has been
verified only rarely: for example, in ditriptycenyl- (198, Table 5.1),191,196 tritylphenyl-
(202, Figure 5.1),197 dianthryl- (200, Table 5.1),193 and constrained macrocyclic
acetylenes (203, Figure 5.1).198 These efforts notwithstanding, observable hindered
rotation in a simple diphenylacetylene has remained elusive. Notably, a 2,2’,6,6’-tetra-p-
tolyl derivative retained conformational mobility on the NMR time scale at temperatures
as low as –100 ºC.199
Rotational isomerism in diaryl– and other alkynes is of interest
fundamentally,191,192,196,199 as well as in applications. Examples of novel molecular
devices197,198,200 that contain this structural fragment include Glass’s molecular “sensors”
(201, Figure 5.1),200a,b Garcia-Garibay’s gyroscopes (202),197,200d,e and Moore’s
“turnstiles” (203).198 Deplanarized diphenylacetylenes also constitute an essential model
for the novel phenyleneethynylene polymers.201

147
F
F O
O
R
R
R
R
R
R
201 202 203
Figure 5.1 Examples of molecular devices that feature a –C≡C– unit: “sensors” (left),
“gyroscopes” (center), and “turnstiles” (right). R = receptor moiety.
The following two sections will deal with the NMR-detection of hindered rotation
around the –C≡C– unit in alkynylated diphenylacetylenes, notably core 205b (Figure 5.2,
center), the first member of the class of chiral 2,2’,6,6’-tetrakisalkynyl
diphenylacetylenes (205a).202 These compounds relate to chiral biphenyls by the insertion
of a C≡C fragment into all five of the single bonds that bring about the chirality of 204
and are thus, in Houk-Scott terminology,203 “exploded” biphenyls. Constructs of the type
205a function as building blocks in the assembly of carbon rich materials, such as planar
metallacycles,202 substructures of graphyne and its relatives,163,204 and nanotubes.159
Previous chapters have amply demonstrated the importance of elaborated variants of
205a in the syntheses of the phenylenes.1,47 Additionally, they might also be viable as
new scaffolds for chiral atropisomeric ligand construction.205

148
R'
RR
R'
204 205a205b, R = TMS, R' = DMTS
R
R'
R
R'
DMTS = SiH
R
R
R
R R
R
R
Rσ
171c, R = DMTS
Figure 5.2 General structures of chiral biphenyls (204), diphenylacetylenes (205a), and
the structure of a conformationally locked dehydrobenzannulene 171c.
Section 5.4 will focus on the stereochemical properties of the macrocycle 171c
(Section 4.4, Figure 5.2, right). Overall, 171c is achiral, since it possesses a plane of
symmetry (σ in Figure 5.2, vide infra); however, its structure encompasses the chiral
2,2’-dialkynyldiphenylacetylene motif. As will be seen, steric hindrance around the triple
bonds in 171c shuts down the free conformational equilibration of the molecule. This is
an example of a conformationally locked dehydrobenzannulene in which the rigidity is
induced by substituents, rather than the macrocyclic skeleton itself.159c,206

149
5.2 Previous Examples of Hindered Rotation in Phenylene Precursors
The occurrence of atropisomerism in 205a first became apparent during the
course of the synthesis of the [N]heliphenes, N = 7–9, by triple cobalt-catalyzed
cycloisomerization of the corresponding nonaynes (Section 1.2.2.1, Schemes 1.8 and
1.9).70 In particular, the 500 MHz 1H NMR spectrum of the advanced intermediate 206
(Figure 5.3) on route to methoxymethyl [9]heliphene revealed two doublets (δ = 4.08,
4.15 ppm; AB, 2J = 15 Hz) for the methylene hydrogens at room temperature (500 MHz,
CDCl3), clearly signaling the presence of a configurationally stable chiral conformation.
Furthermore, gradual cooling of the sample to –53 °C in toluene-d8, caused further
decoalescence and the appearance of several broad signals for these hydrogens. At this
temperature, the corresponding methoxy singlet separated into two distinct peaks (∆ν =
36 Hz), the combined data indicating the rotational restriction of a second (and perhaps
third) stereogenic axis in the molecule giving rise to two, or perhaps three, diastereomers.
Because of the complexity of the NMR signals of unsymmetrical 206 and to
elucidate the nature of these dynamic processes, we turned to the symmetrical 20766 and
208,70 the former as a model for probing the hindered rotation of the “outside”
diphenylacetylene axis (●), the latter for doing so with respect to its inside counterpart
(■). In these molecules, the signals for the potentially diastereotopic pairs of methyls of
the DMTS group, especially the distinct silylmethyl absorptions, were sufficiently well
resolved to allow for variable temperature NMR studies.

150
R
R
OCH3
R
RR R
R
207, R = DMTS206, R = DMTS
R
R
R
R
R RR
208, R = DMTS
R
Figure 5.3 Precursors to helical [N]phenylenes that exhibit hindered rotation around
“internal” (■) and “external” (●) chirogenic axes (see text for details).
In 207, a precursor to [7]heliphene by double cyclization,66 restricted rotation may
give rise to only two diastereomers: the syn form, in which the biphenylenyl substituents
face each other (as shown in Figure 5.3, Cs symmetry), and the anti rotamer (C2), in
which they point in opposite directions. Molecular mechanics calculations favor the
former energetically by ~ 1 kcal mol–1. At room temperature, the 1H NMR spectrum of
207 displayed two sets of resonances for the two inequivalent DMTS groups, without any
indication of hindered rotation. Specifically, only two silylmethyl singlets were visible at
δ = 0.12 and 0.16 ppm (400 MHz, CD2Cl2). Upon cooling to –54 ºC, the latter
decoalesced into two singlets, while the former started to broaden. The aromatic region of
the spectrum remained unchanged. Similarly, at this temperature, the 13C signals for the
three types of methyl carbons appeared as four lines each, while the remaining carbons
gave rise to single resonances. These observations are consistent with the occurrence of
hindered rotation around the biphenylenyl-phenyl alkyne bond and the presence of only

151
one of the two possible diastereomers of 207, presumably the syn isomer. Simple peak
coalescence analysis provided a ∆G‡ of 11.5 kcal mol–1 for this process,207 its facility
suggesting that it is also responsible for the lower energy restricted movement(s) taking
place in 206 (● axes).
To support this hypothesis, a similar analysis was performed on 208. Indeed,
decoalescence of the three silylmethyl singlets (δ = 0.04, 0.10, 0.16 ppm, 40 ºC)
associated with the three distinct DMTS groups to six singlets occurred already at 28 ºC
(400 MHz, CDCl3). Analysis of the decoalescence of the low field signal furnished an
approximate activation barrier of 15.6 kcal mol–1.207 It therefore seems that the
substructure 205a is responsible for the higher energy conformational process observed
in 206 (■ axis).
5.3 Synthesis and Properties of the First Chiral 2,2’,6,6’-Tetrakisalkynyl
Diphenylacetylene
The observations summarized in Section 5.2 provided the impetus for the
synthesis of 205b, devoid of all the unessential elements present in 206–208. In this
system, there is only one stereogenic axis, and a variable temperature NMR analysis
would provide unambiguous data addressing the possibility of hindered rotation around a
diphenylacetylene triple bond. The preparation of 205b (Scheme 5.1) commenced with
the previously described 2,2’,6,6’-tetrabromodiphenylacetylene (209),202 which was
desymmetrized to 2,2’-dibromo-6,6’-diiodo-diphenylacetylene (210) by bromine–iodine
exchange. The selectivity of this exchange is rather remarkable, as the reaction proceeded

152
to give only the desired product, without even traces of singly or triply exchanged
material.208 The iodinated positions in 210 were alkynylated with DMTSA209 under
standard Sonogashira coupling conditions171 to furnish triyne 211 in 52% yield. A second
bromine–iodine exchange afforded the doubly iodinated 212, the
trimethylsilylethynylation of which proceeded with great difficulty to give only 15% of
205b, after a laborious purification sequence that involved column chromatography,
Kugelrohr distillation, and HPLC.
X
Br X
Br
X
X
DMTS
DMTS
205b
209, X = Br
210, X = I(i)
(ii)
(iii)
211, X = Br
212, X = I(i)
Scheme 5.1 Synthesis of 205b: (i) BuLi, Et2O, –45 °C, 1 h, then I2, Et2O, –45 °C to 23
°C, 2 h, 75% (for 210), 92% (for 212); (ii) DMTSA, [PdCl2(PPh3)2], CuI, NEt3, 23 °C, 20
h, 52%; (iii) TMSA, [PdCl2(PPh3)2], CuI, NEt3, 100 °C, 72 h, 15%.
Remarkably, restricted rotation in 205b was evident already at room temperature
in both the 1H and 13C NMR spectra. The former featured a doubling of all the signals
due to the diastereotopic methyls of the DMTS group, observable in dioxane-d8 (Figure
5.4a), CDCl3, and THF-d8. The latter (CDCl3) revealed such behavior only for the
carbon-bound methyl groups, the silylmethyl carbons apparently being accidentally

153
isochronous. In both cases, the remainder of the spectrum was as expected for a single
species.
Figure 5.4 1H NMR (500 MHz) spectra of 205b (methyl group region). Conditions: a)
dioxane-d8, 22 °C; b) THF-d8, –82 °C; c) dioxane-d8, 109 °C, sealed tube.
For the purpose of evaluating the barrier to rotation, the most diagnostic isopropyl
doublets (marked with “x” in Figure 5.4) were chosen. Unfortunately, a solvent covering
the entire temperature range within which spectral changes were occurring could not be
found. Thus, toluene did not provide clear peak separations and DMF did not dissolve
205b. Therefore, low temperature NMR measurements were undertaken in THF-d8,
whereas dioxane-d8 was employed at high temperatures. An example of a clearly
resolved low temperature spectrum is shown in Figure 5.4b. Coalescence of the isopropyl

154
signals occurred at 96 ºC, and increasing the temperature generated eventually a single
set of DMTS peaks above 100 ºC (Figure 5.4c). The ∆G‡ for the enantiomerization in
205b was calculated to be 18.7(±0.4) kcal mol–1.
The rotational barrier in 205b is remarkably high, in the high range of those
reported for more complex diarylacetylene systems. For example, the ∆G‡ values for
Toyota’s bis(1-phenyl-9-anthryl)acetylenes ranges between 10 and 18 kcal mol–1,193
while Moore’s “molecular turnstiles” have corresponding values of 13–20 kcal mol–1,198
depending on the size of substituents on the aryl rings. On the other hand, the
conformationally mobile 2,2’,6,6’-tetrakisaryldiphenylacetylene frame exhibits barriers
below 8 kcal mol–1,199 less than a half of that in 205b.
This work constitutes the first observation of hindered rotation in a simple
substituted diphenylacetylene. It also attests to the power of substituted alkyne units in
exerting remote steric influence, in spite of the ready deformability of the carbon–carbon
triple bond.
5.4 Stereochemical Properties of 171c
As the preceding work showed, sufficient encumbrance around –C≡C– units can
hinder rotation about these axles as observed in diphenylacetylenes (Section 5.3) and the
more complex acyclic oligo(phenyleneethylene)benzenes (Section 5.2). This section will
extend the above notion to the octaalkynyl dehydrobenz[18]annulenes 171a–c, which
were the focus of Chapter 4. The basic structural feature of these cyclic systems is a 2,2’-
dialkynyldiphenylacetylene, a close relative of the 2,2’,6,6’-motif in acyclic systems.

155
However, the four-fold incorporation of this structural moiety into the cyclic skeleton of
dehydrobenzannulenes complicates the conformer equilibration mechanism significantly.
Molecular mechanics studies aimed at probing this mechanism will be the subject of
Section 5.6.
Figure 5.5 Line-bond (left) and calculated (right) structures of 156.
DFT calculations presented in Section 4.5 revealed the nonplanar conformation of
the parent 156 as the most stable (Figure 5.5). In this conformation, the 1,2,4,5-
substituted benzene rings are distorted out of the plane defined by the 1,2,3,4-substituted
rings, in opposite directions (Figure 5.5, right). The shallow conformation energy curve
of 156 features a negligible (~ 2 kcal mol–1) barrier to planarization. Appropriate
substitution at the termini of the triple bonds in 156 was expected to exacerbate the
energy differences between the conformers, possibly rendering the equilibration process
observable by NMR. This expectation was supported by the preliminary molecular
mechanics calculations on 171c (the MM was chosen over the DFT method because of
the size of 171c, see Section 5.5), which singled out the two most stable conformers,
shown in Figure 5.6. In the C2h-symmetric structure (left), analogous to that calculated for

156
R
R
R
R R
R
R
Rσ
R
R
R
R R
R
R
Rσ1
σ2C2
Figure 5.6 Two possible diastereomers of 171c, shown with their key symmetry
elements: C2h-isomer (left) and C2v-isomer (right).
the parent, the two benzene rings intersected by the σ plane lie in parallel planes.
Alternatively, the same two rings could be oriented in a roof-like formation, forming the
second symmetry plane in the now C2v-symmetric molecule (σ2 in Figure 5.6, right). The
latter conformer was favored by a significant 12 kcal mol–1, somewhat surprisingly
considering that the C2h-structure was favored for the parent system.
Experimentally, compounds 171a–c (Sections 4.3 and 4.4) served as ideal NMR-
probes of conformational (in)flexibility, since all three compounds bore potentially
diastereotopic substituents (propyl, hexyl, and DMTS, successively). The 1H NMR
spectra of octapentynyl– and octaoctynyl-substituted dehydrobenz[18]annulenes 171a
and b (Section 4.3 and 4.4) featured three singlets in the aromatic region and two sets of
resonances corresponding to the propyl and hexyl groups, respectively. This observation
was consistent with structures that are either planar, or nonplanar but conformationally
mobile on the NMR time scale. On the other hand, the DMTS spectral region of the
analogously built 171c revealed not two, but four sets of resonances for each of the three
methyl groups, illustrated for the silylmethyls on top of Figure 5.7. In accord with the

157
calculations, the aromatic region of the spectrum confirmed the presence of a single
conformer (presumably C2v). Equilibration of this conformer with its superimposable
mirror image (and hence coalescence) appeared remarkably sluggish, as measured by VT
NMR at 62 and 90 ºC, respectively (Figure 5.7, bottom). Phenomenologically, the rapid
equilibration of the two degenerate conformers is equivalent to an averaged planar
structure; in the acyclic analogy, this would translate into the free rotation around the
core diarylalkyne bonds.187
Figure 5.7 Silylmethyl 1H NMR spectra (dioxane-d8) and the corresponding schematic
representations of 171c at 22 ºC (top) and 99 ºC (bottom), respectively. The black and
white spheres represent the (potentially) diastereotopic methyl substituents. A = DMTS–
C≡C–, B = (CH3)2CH(CH3)2C–.
Peak-shape analysis207 provided an activation barrier of 19.4(±0.4) kcal mol–1,
almost ten times greater than the one calculated for the parent! In contrast, both the

158
acyclic precursor analogs 147 (Figure 5.8, left, also Section 2.5.2, Scheme 2.14) and 149
(Figure 5.8, center, also Section 2.6, Scheme 2.15), and the related hexaalkynylated
dehydrobenz[12]annulene 74a (Figure 5.8, right, also Section 1.2.2.5, Scheme 1.18)69
remained mobile conformationally at temperatures as low as –80 ºC.210
R
RR
R
147, R = DMTS
R
TMS
TMS
RR
R
R
RR
RTMS
TMS
149, R= DMTS
R
RR
R
R
R
74a, R = DMTS
Figure 5.8 Acyclic (147 and 149) and cyclic (74a) models for the behavior of 171c.
Rotation around –C≡C– axes shown in bold in 147 and 149 remained free on cooling to
the limiting temperature of –80 °C. Similarly, in 74a, the macrocyclic skeleton stayed
flexible at this temperature.
The incorporation of a hindered diarylalkyne into a cyclic environment of
dehydrobenz[18]annulenes led to the unprecedented restriction of the conformational
freedom of the latter system. The following section will deal with the energetic and the
mechanistic intricacies of the conformer equilibration at elevated temperatures.

159
5.5 Proposed Mechanism of Interconversion between the Conformers of 171c211
The unique stereochemical behavior of 171c kindled our interest in the
mechanism through which the interconversion of its conformers occurs. Several
questions were addressed: a) what is the calculated inversion barrier; b) are the
movements of the four quadrants of 171c (Figure 5.6): independent, somewhat correlated,
or completely synchronized; c) which parts of the system “take the load” in the transition
state of inversion - the macrocyclic skeleton, the pendant substituted triple bonds, or
something else? Experimental models suitable for addressing these concerns were
lacking, prompting us to turn to calculations for guidance. Due to the appreciable cost of
DFT calculations for 171c, molecular mechanics (MM) was selected as the computational
method, a choice justified by the purely steric nature of the problem. This section will
describe the results of the calculations performed. To best depict the relevant issues,
graphics and colors will be used, deviating from the format of the preceding chapters and
sections.
In order to separate the movements within the individual quadrants, we performed
calculations on 171c and 213–216 (Figure 5.9), constructed formally from 171c by the
sequential removal of pairs of adjacent alkynyl substituents (within the context of this
section, “adjacent” signifies the closest alkynyl group on a different benzene ring). The
overall strategy of the calculations assumed that the change in the torsion angle a–b–c–d
(shown for 213 in Figure 5.9) featured as crucial in the inversion process. This angle was
constrained successively to a set of predetermined values and the remainder of the
molecule was optimized for each. The so-obtained energies were plotted against the

160
Si
SiSi
SiSi
SiSi
Si
Si
Si
a
b
d
c
Si Si
Si
A B
Si
Si
A
Si
B
Si
B
Si
A
Si
C
213 214
215 216
B
Si
C
SiSi
A
Si
D
171c
Si
Figure 5.9 Molecules analyzed by MM methods. The torsion angles a–b–c–d (defined in
213) were constrained in all systems. Capital letters A–D are used to denote molecular
quadrants.

161
torsion angle in the search for a meaningful transition state. Such was deemed
accomplished if an energy maximum was traversed as the geometrical constraints were
varied. Pathways that led to continually increasing energy were discarded as
nonproductive.
Ground-state calculations of 171c and 213–216 indicated that only the
orientations of the rings bearing DMTSethynyl groups are predicted to influence the
overall energy of the molecule. In other words, the unsubstituted rings could adopt a
number of different arrangements with miniscule energetic consequences (the conformers
of 213 and 214 in Figure 5.9 are therefore arbitrarily chosen). For 171c, 215, and 216, in
which both meta-fused rings are substituted, the roof-shaped conformation (pseudo-C2v,
Figure 5.6, right) was predicted to be more stable than the pseudo-C2h alternative (Figure
5.6, left), by ~ 6.5, 9, and 12.5 kcal mol–1, respectively.
The first studied molecule was 213 (Figure 5.9), with only two adjacent alkynyl
groups. Table 5.2 and Figure 5.10 show the dependence of the energy of the system on
the torsion angle. As expected, the calculations predict that the initial increase in energy
is followed by a sudden decrease, establishing this movement as a possible pathway for
inversion. The barrier was estimated at ~ 15 kcal mol–1.

162
Torsion angle a–b–c–d [°] Energy [kcal mol–1]
50.7 0.00
44.8 0.24
39.1 0.72
33.5 1.43
27.7 2.63
21.9 4.06
16.0 5.73
10.0 7.65
3.7 9.80
–3.0 12.19
–6.7 13.62
–10.9 14.81
–14.7 15.05
–55.7 –0.24
–56.2 –0.24
Table 5.2 Calculated dependence of the total energy on torsion angle a–b–c–d in 213.

163
-60 -40 -20 0 20 40 60-2
0
2
4
6
8
10
12
14
16
En
erg
y [
kc
al
mo
l-1]
Torsion angle a-b-c-d [o]
Figure 5.10 Graphical representation of the calculated dependence of the total energy on
torsion angle a–b–c–d in 213. The red curve shows a Gaussian fit of the calculated data
points.
Figure 5.11 depicts the conformation that corresponds to the energy maximum of the
curve given in Figure 5.10. This structure should closely resemble the predicted transition
state for the inversion. This model suggests that the benzene rings invert first, leaving the
DMTSethynyl groups behind. A consequence of this facet is a significant increase in the
strain energy of the transition state. Accordingly, the release of this strain causes the
DMTSethynyl groups to “flip over”, precipitating the inversion of the overall system.

164
Figure 5.11 Calculated structure of the transition state for the inversion of 213.
Out of the three possible isomers with two pairs of adjacent alkynyl substituents,
two were examined (Figure 5.9): 214, with two DMTSethynyl substituents on the ortho-
fused benzene ring, and 215, possessing two of the four DMTSethynyl group on its meta-
fused ring. In order to minimize the constraints imposed on the system, only one of the
two possible torsion angles in 214/215 was changed (denoted as A in Figure 5.8). Table
5.3 and Figure 5.12 show the dependence of the other angle (B) and the overall energy of
the system on the constraint imposed on angle A.

165
Torsion angle A [°] Torsion angle B [°] Energy [kcal mol–1]
57.9 –54.9 0.00a
48.1 –51.2 0.48
42.9 –48.9 1.19
37.8 –46.9 2.39
32.8 –44.9 3.82
27.8 –42.9 5.50
22.9 –41.1 7.65
18.0 –39.3 10.04
13.3 –37.7 12.66
8.6 –36.4 15.77
3.9 –35.3 18.88
–0.9 –34.1 22.22
–5.5 –33.4 26.05
–10.1 –32.4 29.63
–14.8 –31.7 33.69
–83.6 70.8 –0.48
Table 5.3 Calculated dependence of the total energy and the torsion angle B in 214 on
the torsion angle A. a Simulation started with the pseudo-C2h structure.

166
-100 -80 -60 -40 -20 0 20 40 60
-60
-40
-20
0
20
40
60
80
Torsion angle B [o]
Energy [kcal mol-1
]
Torsion angle A [o]
Figure 5.12 Graphical representation of the calculated dependence of the total energy
and the torsion angle B in 214 on the torsion angle A. The red curve shows a Gaussian fit
of the calculated data points.
Changes in the torsion angle A were reflected in the angle B, causing both quadrants to
invert simultaneously, rather than independently. The inversion barrier was estimated at ~
34 kcal mol–1. The presumed structure of the transition state (shown in Figure 5.13)
indicated that the motion of the central benzene ring preceded that of the attached
substituents. In other words, the benzene rings “overshot” by ~ 15 ° before the
DMTSethynyl groups inverted.

167
Figure 5.13 Calculated structure of the transition state for the inversion of 214. Hydrogen
atoms omitted for clarity.
In an alternative arrangement of 215 (Figure 5.9), the ortho-fused ring bears two
DMTSethynyl groups. Analysis analogous to the one elaborated for 214 shows that the
two units are now calculated to move independently. The constraints imposed on A are
influencing B (Table 5.4, Figure 5.12), but not dramatically; more significantly, the
inversion of A does not cause one in B. Relative to the more stable conformer, the
inversion barrier is calculated to be ~ 16 kcal/mol, fairly close to the value obtained for
213.

168
Torsion angle A [°] Torsion angle B [°] Energy [kcal mol–1]
40.1 43.1 6.45a
34.5 44.4 6.69
29.0 45.9 7.17
23.4 47.2 7.88
17.6 48.6 9.08
11.6 50.0 10.51
5.1 51.4 11.18
–1.8 52.8 14.10
–9.9 54.7 16.01
–52.6 59.6 0.00
Table 5.4 Calculated dependence of the total energy and the torsion angle B in 215 on
the torsion angle A. The inversion of one quadrant produced a different diastereomer of
the material, hence the different values for energies before and after the inversion. a
Simulation started with the pseudo-C2h structure.

169
-80 -60 -40 -20 0 20 40 60
0
40
60
80
Torsion angle B [
o]
Energy [kcal mol-1
]
Torsion angle A [o]
Figure 5.14 Graphical representation of the calculated dependence of the total energy
and the torsion angle B in 215 on the torsion angle A. As a consequence of their
definition, the initial signs of the two angles are different. The inversion of one quadrant
produced a different diastereomer of the material, hence the different values for energies
before and after the inversion. The red curve shows a Gaussian fit of the calculated data
points.
In 216 and 171c, the systems with three and four pairs of adjacent alkynyl groups,
a change in just one of the torsion angles failed to model the inversion. Conformations of
unreasonable geometries and very high energies were obtained, suggesting a more
ordered transition state. To address this requirement, we decided to change

170
simultaneously two of the torsion angles in the same direction and by the same
increments.212 In the triply substituted system 216 (Figure 5.9), three possible
combinations of the angles could, in principle, be restrained: A-B, A-C, and B-C. The
former two were examined. Constraining angles A and C simultaneously gave rise to
high-energy conformers. On the other hand, a simultaneous change in A and B proved
productive, as these calculations predicted that the two units should invert synchronously
with an energy barrier of ~ 40 kcal mol–1 (relative to the more stable conformer) and,
significantly, without causing the inversion of C (Table 5.5).

171
Torsion angle [°]
A B C Energy [kcal mol–1]
42.1 –43.4 44.4 8.84a
35.5 –36.1 45.6 9.32
29.9 –30.6 46.4 10.51
24.2 –25.2 47.1 12.42
18.3 –19.8 47.9 14.81
12.1 14.2 48.8 17.68
4.3 –7.9 50.7 21.03
–1.6 –25 51.5 25.09
–7.8 2.7 52.3 29.63
–14.1 8.0 53.2 34.65
–20.7 13.4 54.1 39.90
–64.3 65.3 56.2 0.00
Table 5.5 Calculated dependence of the total energy and the torsion angle C in 216 on
the torsion angles A and B. The inversion of quadrants A and B produced a different
diastereomer of the material, hence the different values for energies before and after the
inversion. a Simulation started with the pseudo-C2h structure.
Finally, the fully substituted 171c (Figure 5.9) had three possible combinations of
the angles to be constrained: A-B, A-C, and A-D. While constraint in the A-C and A-D
pairs gave rise only to high-energy maxima, the simultaneous change of A and B gave a
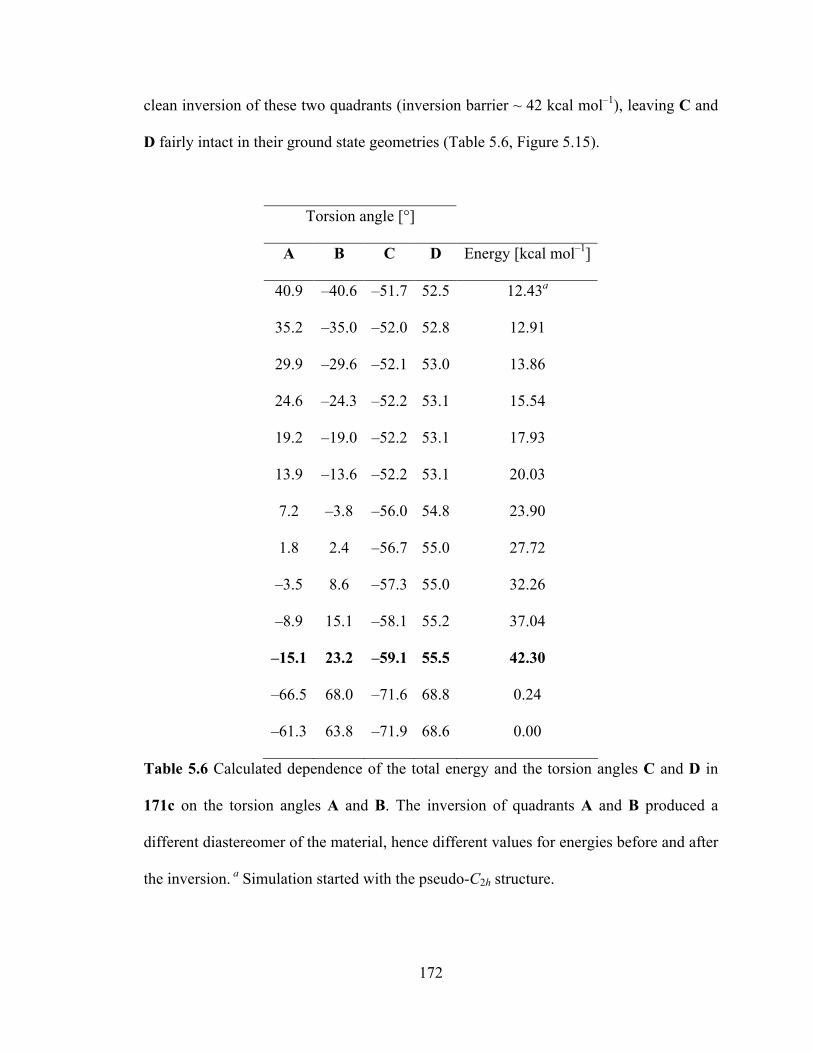
172
clean inversion of these two quadrants (inversion barrier ~ 42 kcal mol–1), leaving C and
D fairly intact in their ground state geometries (Table 5.6, Figure 5.15).
Torsion angle [°]
A B C D Energy [kcal mol–1]
40.9 –40.6 –51.7 52.5 12.43a
35.2 –35.0 –52.0 52.8 12.91
29.9 –29.6 –52.1 53.0 13.86
24.6 –24.3 –52.2 53.1 15.54
19.2 –19.0 –52.2 53.1 17.93
13.9 –13.6 –52.2 53.1 20.03
7.2 –3.8 –56.0 54.8 23.90
1.8 2.4 –56.7 55.0 27.72
–3.5 8.6 –57.3 55.0 32.26
–8.9 15.1 –58.1 55.2 37.04
–15.1 23.2 –59.1 55.5 42.30
–66.5 68.0 –71.6 68.8 0.24
–61.3 63.8 –71.9 68.6 0.00
Table 5.6 Calculated dependence of the total energy and the torsion angles C and D in
171c on the torsion angles A and B. The inversion of quadrants A and B produced a
different diastereomer of the material, hence different values for energies before and after
the inversion. a Simulation started with the pseudo-C2h structure.

173
Figure 5.15 Calculated structure of the transition state for the inversion of A and B
quadrants in 171c. Hydrogen atoms omitted for clarity.
With the caveat of the inaccuracy of absolute numbers produced by MM methods,
our calculations strongly suggest that the inversion of the
octa(DMTSethynyl)dehydrobenz[18]annulene 171c occurs via a stepwise “2+2”
mechanism. Of the four molecular quadrants, only pairs connected through the 1,2,4,5-
substituted rings can cooperate productively. Thus, using Figure 5.6 as reference, the C2v-
conformer (right) inverts its upper half first with a ~ 42 kcal mol–1 barrier, giving rise to
the energetically loaded (+12 kcal mol–1) C2h-isomer. This intermediate now inverts the
bottom two quadrants (+30 kcal mol–1), producing a superimposable mirror image of the
C2v-conformer.

174
In the transition state, both the triple bonds of the pendant DMTSethynyl groups
and the –C≡C– linkages in the macrocycle are significantly distorted. The transition state
geometries also reveal an interesting “catapult” behavior: The DMTSethynyl groups are
lagging behind the benzene ring to which they are attached as the system undergoes
inversion. Of course, it is likely that adding more flexibility to the structures employed in
these calculations will change the energetic values obtained and lead to more favorable
transition states. However, such optimizations were not pursued and probably would not
change the basic conclusions drawn in this section.
5.6 Summary and Future Directions
The first cases of hindered rotation around the triple bond in simple
diphenylacetylenes have been observed, including that in the simple chiral tetraethynyl
system 205b. The conformational barriers can be substantial, leading to the observation
of restricted rotation by NMR at room temperature. In cyclic systems, such as 171c, steric
crowding around triple bonds caused the loss of the conformational freedom. Molecular
mechanics calculations proposed a stepwise “2+2” mechanism for inversion of 171c at
elevated temperatures.
Future work will aim to gain insight into the effect of substituent size on the
flexibility of 205a with the ultimate goal of achieving resolution of suitable derivatives.
These investigations may lead to the development of 205a as a viable new tool in chiral
scaffold construction.

175
Chapter Six
Experimental and Computational Details
6.1 General Considerations
All reactions, except base-catalyzed silyl-group deprotections, the preparations of
starting iodoarenes, and microwave-assisted reactions, were performed under nitrogen
atmosphere in oven-dried glassware. Solvents were dried by distillation over the
corresponding drying agents: triethylamine (KOH pellets), ether (Na-benzophenone,
purple solution), THF (Na-benzophenone, purple solution), toluene (Na-benzophenone,
purple solution), and degassed by a 15 min nitrogen purge prior to use. All other
materials and solvents, unless noted otherwise, were purchased from commercial
suppliers and used without further purification. Bis(triphenylphosphine)palladium(II)
chloride213 was prepared according to previously published procedures. Flash
chromatography used silica gel, according to Still’s procedure.214 Microwave-assisted
reactions were run in a Smith Synthesizer single-mode microwave cavity, producing
continuous radiation at 2450 MHz. All manipulations of [(Me3CO)3W≡CCMe3] were
carried out in a glove box. Irradiation in [CpCo(CO)2]-mediated cyclizations was done
using a GE 300 W projector lamp, positioned ~ 5 cm away from the flask.
The identity of products was established by 1H NMR, 13C NMR, IR, and mass
spectrometry. Purity was confirmed by melting point and elemental analyses; in some
cases, due to the small amounts or the consistency (viscous oils) of prepared materials,
purity was assessed by NMR and high-resolution mass spectrometry, along with a

176
distillation/gas chromatography sequence (for some volatile compounds). Melting points
were taken in open capillary tubes, using a Thomas Hoover Unimelt apparatus, and are
uncorrected. Mass spectral measurements (FAB, EI of GC/MS incompatible compounds
and high resolution) and elemental analyses were supplied by the Micro Mass Facility of
the College of Chemistry, University of California at Berkeley, California. For
compounds containing polyisotopic elements, mass spectra give data only for the most
abundant isotopomer. In compounds that contain an odd number of bromines the two
most abundant isotopomers exist in roughly equal ratios, and data for both are given (in
M(81Br)+/M(79Br)+ format). NMR spectra were recorded on Bruker DRX-500, AVB-400,
AVQ-400, and AV-300 spectrometers, with working frequencies (for 1H nuclei) of 500,
400, 400, and 300 MHz, successively. All 13C NMR spectra were recorded with
simultaneous decoupling of 1H nuclei. 1H NMR chemical shifts are reported in ppm units,
relative to the residual signal of the solvent (CDCl3–7.26 ppm; C6D6–7.15 ppm; CD2Cl2–
5.32 ppm). IR measurements were performed on a Perkin Elmer System 2000 FT–IR
spectrometer. UV measurements were executed on an HP 8450A diode array
spectrometer and are reported in nm (logε). Gas chromatography utilized an HP 5890
Series II chromatograph. Column chromatography was carried out on silica gel 60, 32–63
mesh. Analytical TLC employed Merck aluminum-backed silica gel plates.
All calculations were carried out at the Molecular Graphics Facility in the College
of Chemistry, University of California at Berkeley. DFT calculations employed the ab
initio electronic structure software package Jaguar 5.5, while molecular mechanics
computations used MacroModel 8.1, both as modules of the Maestro 6.5 suite.215 All
DFT calculations were done at the B3LYP level, using the 6–31G* basis set.216 In most

177
cases, the geometries submitted to DFT scrutiny were preoptimized using molecular
mechanics, in order to minimize the time required for the higher level calculations.
6.2 Experiments and Calculations Related to Chapter 2
1,5-Dibromo-2,4-bis[(2-bromophenyl)ethynyl]benzene (58):
Br Br
TMS TMS
1,5-Dibromo-2,4-bis[(trimethylsilyl)ethynyl]benzene. A suspension of 1,5-
dibromo-2,4-diiodobenzene (57)78 (0.69 g, 1.40 mmol), TMSA (0.50 mL, 3.50 mmol),
[Pd(PPh3)2Cl2] (49.0 mg, 0.07 mmol), and CuI (13.0 mg, 0.07 mmol) in 100 mL of
triethylamine was stirred for 2 h at 23 °C. After removing the solids by filtration, the
solvent was evaporated in vacuo and the resulting crude product purified by column
chromatography (hexanes) to yield 1,5-dibromo-2,4-bis[(trimethylsilyl)ethynyl]benzene
as a yellow oil (581 mg, 96 %). UV-VIS (cyclohexane) λmax (logε) 254 (4.29), 275
(3.95), 284 (sh, 3.88), 306 (3.23), 335 (2.86) nm. IR (NaCl film): ~ν = 2960, 2898, 2159,
2068, 1451, 1340, 1250, 1174, 1058, 977, 843, 760, 696 cm–1. MS (EI, 70 eV) m/z (rel
intensity) 428 (M+, 33), 413 (100), 199 (21). 1H NMR (400 MHz, CDCl3) δ 7.81 (s, 1H),
7.61 (s, 1H), 0.28 (s, 18H). 13C NMR (100 MHz, CDCl3) δ 137.4, 135.6, 125.6, 124.5,
101.4, 101.3, –0.3. HR-MS Calcd for C16H20Br2Si2: 427.9450. Found: 427.9448. Anal.
Calcd for C16H20Br2Si2: C, 44.87; H, 4.71. Found: C, 44.51; H, 5.07.

178
Br Br
1,5-Dibromo-2,4-diethynylbenzene. A solution of 1,5-dibromo-2,4-
bis[(trimethylsilyl)ethynyl]benzene (520 mg, 1.21 mmol) in a mixture of ethanol (20 mL)
and ether (30 mL) was treated with solid KOH (800 mg). The mixture was stirred at 23
°C for 1 h and then filtered through a short plug of silica. After removal of the solvent,
white crystals of 1,5-dibromo-2,4-diethynylbenzene were obtained; since they appeared
to decompose quickly (darkened within 5 min), the material was used without any
purification in the subsequent step.
Br Br
Br Br
1,5-Dibromo-2,4-bis[(2-bromophenyl)ethynyl]benzene (58). A mixture of 1,5-
dibromo-2,4-diethynylbenzene (345 mg, 1.21 mmol), 1-bromo-2-iodobenzene (1.64 g,
5.80 mmol), [Pd(PPh3)2Cl2] (42.0 mg, 0.06 mmol), and CuI (12.0 mg, 0.06 mmol) in
triethylamine (100 mL) was stirred overnight at 23 °C. The solvent was removed in
vacuo and the desired material isolated by column chromatography (hexanes) as off-
white crystals, mp 156–158 °C (321 mg, 45 %). UV-VIS (cyclohexane) λmax (logε) = 299
(3.87), 308 (3.80), 320 (sh, 3.80), 354 (3.02), 371 (3.00), 406 (2.98) nm. MS (EI, 70 eV)
m/z (rel intensity) 594 (M+, 100), 516 (5), 514 (5), 434 (11), 274 (28), 137 (18). 1H NMR
(400 MHz, CDCl3) δ 7.92 (s, 1H), 7.81 (s, 1H), 7.63 (br t, J = 7.9 Hz, 4H), 7.33 (br t, J =
7.3 Hz, 2H), 7.23 (br t, J = 7.3 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 137.1, 136.0,

179
133.7, 132.6, 130.1, 127.1, 125.7, 125.6, 124.7, 124.6, 93.7, 90.7. HR-MS Calcd for
C22H10Br4: 593.7475. Found: 593.7467. Anal. Calcd for C22H10Br4: C, 44.49; H, 1.70.
Found: C, 44.78; H, 1.65.
1,5-Bis(ethynyl)-2,4-bis[(2-ethynylphenyl)ethynyl]benzene (59):
TMSTMSTMS
TMS
1,5-Bis[(trimethylsilyl)ethynyl]-2,4-bis[{(2-trimethylsilyl)ethynylphenyl}ethynyl]
benzene. A mixture of 58 (135 mg, 0.23 mmol), TMSA (0.32 mL, 2.27 mmol),
[Pd(PPh3)2Cl2] (8.0 mg, 0.011 mmol), and CuI (2.0 mg, 0.011 mmol) in triethylamine (40
mL) was sealed in a 200 mL Schlenk tube and heated at 100 °C for 8 d. The solvent was
removed in vacuo and the residue subjected to column chromatography
(hexanes/CH2Cl2), yielding 1,5-bis[(trimethylsilyl)ethynyl]-2,4-bis[{(2-
trimethylsilyl)ethynylphenyl}ethynyl]benzene contaminated by incompletely alkynylated
material, as a pale yellow oil (71 mg, 47 %), used as such in the subsequent steps. MS
(EI, 70 eV) m/z (rel intensity) 662 (M+, 28), 73 (100). 1H NMR (400 MHz, CDCl3) δ 7.74
(s, 1H), 7.66 (s, 1H), 7.52–7.49 (m, 4H), 7.30–7.26 (m, 4H), 0.24 (s, 18H), 0.23 (s, 18H).
13C NMR (100 MHz, CDCl3) δ 135.7, 135.3, 132.1, 131.7, 128.2, 128.0, 125.94, 125.86,
125.7, 125.0, 103.2, 102.3, 101.1, 99.1, 94.0, 90.8, 0.0, –0.2.

180
1,5-Bis(ethynyl)-2,4-bis[(2-ethynylphenyl)ethynyl]benzene (59). A solution of 1,5-
bis[(trimethylsilyl)ethynyl]-2,4-bis[{2-(trimethylsilyl)ethynylphenyl}ethynyl]benzene
(39 mg, 0.059 mmol) in THF (10 mL) was treated with 0.20 mL of 1M TBAF solution in
THF (0.20 mmol). The mixture turned brown immediately and was stirred at 23 °C for 1
h, after which water (0.20 mL) was added. The resulting solution was stirred for an
additional 20 min and subsequently filtered under N2 through a short plug of silica into a
50 mL Schlenk tube. The pad of silica was washed with two additional portions of dry
THF and the combined filtrates concentrated to 25 mL volume in vacuo.
syn-Doublebent [5]phenylene (60):
A solution of 59 was cooled to –25 °C, and [CpCo(eth)2]63 was added as an
ethereal solution (23.4 mg, 0.130 mmol; 10 mL of ether). The mixture was kept at –25 °C
for 18 h and then allowed to slowly warm. As soon as the temperature reached –10 °C,
1,3-cyclohexadiene (0.50 mL, 0.42 g, 5.25 mmol) was added in one portion. The Schlenk
tube was closed and heated at 100 °C for 90 min. The mixture was then preadsorbed on
silica and purified by column chromatography (hexanes/CH2Cl2) to elute a first, green
fluorescent fraction of 60 (3.0 mg, 14%), providing orange crystals, mp >220 °C (no

181
decomposition). UV-VIS (cyclohexane) λmax (logε) 304 (sh, 4.57), 312 (4.59), 329 (sh,
4.54), 343 (sh, 4.62), 355 (4.69), 373 (4.63), 393 (4.57), 414 (sh, 4.18), 442 (sh, 3.92),
475 (3.95), 507 (3.90) nm. IR (NaCl film): ~ν = 2921, 2850, 1739, 1635, 1464, 1262,
1106, 1091 cm–1. MS (EI, 70 eV) m/z (rel intensity) 374 (M+, 100), 187 (9), 186 (6). 1H
NMR (400 MHz, CDCl3) δ 7.03–6.99 (m, 4H), 6.97 (m, 2H), 6.92 (m, 2H), 6.75 (d, 5J =
1.5 Hz, 1H), 6.60 (d, 5J = 1.6 Hz, 1H), 6.14 (d, 3J = 6.8 Hz, 2H), 6.06 (d, 3J = 6.8 Hz,
2H). HR-MS Calcd for C30H14: 374.1096. Found: 374.1093.
1,5-Bis[(dimethylthexylsilyl)ethynyl]-2,4-bis[(trimethylsilyl)ethynyl]benzene (129):
BrBr
SiSi
1,5-Dibromo-2,4-bis[(dimethylthexylsilyl)ethynyl]benzene. A suspension of 5778
(2.84 g, 5.80 mmol), [Pd(PPh3)2Cl2] (0.20 g, 0.29 mmol), and CuI (55.0 mg, 0.29 mmol)
in triethylamine (100 mL) was degassed, and DMTSA209 (2.69 g, 16.0 mmol, 2.8 equiv)
was injected. The mixture was left to stir at 23 °C for 48 h. The suspension was filtered
and the filtrate concentrated in vacuo. The resulting crude oil was dissolved in hexanes
and passed through a short column of silica. Removal of the solvent in vacuo gave 3.04 g
(92%) of 1,5-dibromo-2,4-bis[(dimethylthexylsilyl)ethynyl]benzene as a yellow oil. IR
(NaCl film): ~ν = 2958, 2866, 2158, 1450, 1340, 1251, 1174, 1059, 838, 818, 776, 699
cm–1. MS (EI, 70 eV) m/z (rel intensity) 568 (M+, 2), 525 (3), 483 (100), 399 (31), 84
(44). 1H NMR (500 MHz, CDCl3) δ 7.79 (s, 1H), 7.56 (s, 1H), 1.72 (sept, 3J = 6.9 Hz,
2H), 0.97 (s, 12H), 0.94 (d, 3J = 6.9 Hz, 12H), 0.24 (s, 12H). 13C NMR (125 MHz,

182
CDCl3) δ 137.24, 135.50, 125.54, 124.62, 102.06, 101.23, 34.49, 23.48, 20.66, 18.66, –
2.65. HR-MS Calcd for C26H40Br2Si2: 568.1015. Found: 568.1021. Anal. Calcd for
C26H40Br2Si2: C, 54.92; H, 7.09. Found: C, 54.53; H, 7.19.
SiSi
TMSTMS
1,5-Bis[(dimethylthexylsilyl)ethynyl]-2,4-bis[(trimethylsilyl)ethynyl]benzene (129). A
mixture of 1,5-dibromo-2,4-bis[(dimethylthexylsilyl)ethynyl]benzene (1.97 g, 3.47
mmol), TMSA (1.13 mL, 8.00 mmol), [Pd(PPh3)2Cl2] (24 mg, 0.035 mmol), and CuI (7.0
mg, 0.035 mmol) in triethylamine (100 mL) was sealed in a 200 mL Schlenk tube and
heated at 110 °C for 48 h. The solvent was removed in vacuo and the residue subjected to
column chromatography (hexanes/ether), yielding 129 as a dark brown oil (1.69 g, 81 %).
IR (NaCl film): ~ν = 2959, 2164, 2070, 1480, 1378, 1250, 1186, 998, 876, 761, 674, 641
cm–1. MS (EI, 70 eV) m/z (rel intensity) 602 (M+, 29), 519 (80), 517 (88), 434 (38), 252
(62), 235 (50), 196 (66), 140 (88), 123 (100). 1H NMR (500 MHz, CDCl3) δ 7.56 (s, 1H),
7.48 (s, 1H), 1.71 (sept, 3J = 6.7 Hz, 2H), 0.98 (s, 12H), 0.94 (d, 3J = 6.7 Hz, 12H), 0.25
(br s, 30H). 13C NMR (125 MHz, CDCl3) δ 136.72, 136.11, 125.23, 125.02, 102.81,
102.12, 100.66, 100.39, 34.50, 23.44, 20.85, 18.68, –0.13, –2.41.

183
1,5-Bis[(dimethylthexylsilyl)ethynyl]-2,4-bis(ethynyl)benzene (130):
SiSi
A solution of 129 (105 mg, 0.174 mmol) in a mixture of methanol (30 mL) and
ether (30 mL) was treated with K2CO3 (74 mg, 0.533 mmol). The mixture was left to stir
at 23 °C for 2 h. The suspension was then filtered and the filtrate concentrated in vacuo.
The resulting crude oil was dissolved in hexanes and passed through a short column of
silica. Removal of the solvent in vacuo gave (80 mg, 97%) of 130 as a brown oil. The
crude material was used in the next step.
Compound 132:
DMTS
DMTS
DMTS
DMTS
A mixture of 130 (77 mg, 0.168 mmol), 1-iodo-2-
[(dimethylthexylsilyl)ethynyl]biphenylene60 (155 mg, 0.348 mmol), [Pd(PPh3)2Cl2] (6.3
mg, 0.009 mmol), and CuI (1.7 mg, 0.009 mmol) in triethylamine (15 mL) was sealed in
a 20 mL Schlenk tube and heated at 120 °C for 16 h. The solvent was removed in vacuo
and the residue subjected to column chromatography (hexanes/CH2Cl2), yielding 132 as a

184
dark brown oil (103 mg, 54 %). IR (NaCl film): ~ν = 2958, 2866, 2154, 1458, 1417,
1250, 1157, 873, 822, 775, 740 cm–1. MS (FAB) m/z (rel intensity) 1092 ([M+2H]+, 3),
1091 ([M+H]+, 3), 1090 (M+, 2), 960 (1), 765 (1), 252 (80), 235 (42), 140 (100), 124 (82).
1H NMR (400 MHz, CDCl3) δ 7.67 (s, 1H), 7.60 (s, 1H), 6.96 (d, 3J = 7.1 Hz, 2H), 6.83–
6.76 (m, 6H), 6.67–6.60 (m, 2H), 6.54 (d, 3J = 7.1 Hz, 2H), 1.72–1.51 (m, 4H), 0.90 (s,
12H), 0.88 (s, 12H), 0.86 (d, 3J = 6.8 Hz, 12H), 0.84 (d, 3J = 6.8 Hz, 12H), 0.20 (s, 12H),
0.17 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 153.35, 150.65, 149.97, 149.73, 136.02,
135.92, 133.26, 129.29, 128.99, 125.39, 124.70, 124.12, 118.60, 117.98, 116.60, 114.41,
104.29, 103.11, 101.06, 98.05, 92.49, 89.97, 34.57, 34.54, 23.51, 23.44, 20.73, 20.70,
18.66, 18.63, –2.25, –2.42. HR-MS Calcd for C74H90Si4: 1090.6120. Found: 1090.6100.
2-Iodo-3-[(trimethylsilyl)ethynyl]biphenylene (134a):
I
TMS
Method A. 2,3-Diiodobiphenylene (133)53 (1.40 g, 3.47 mmol) was dissolved in a
mixture of triethylamine (40 mL) and THF (10 mL). The solution was degassed in a 50
mL Schlenk flask, and [Pd(PPh3)2Cl2] (121 mg, 0.17 mmol) and CuI (34 mg, 0.17 mmol)
were added. The mixture was left to stir overnight at 80 °C (~ 16 h). After the reaction
was complete, solvent was removed in vacuo and the mixture chromatographed on silica
(hexanes) to give the starting material (436 mg, 31%), followed by the desired 134a (320
mg, 25%) as yellow crystals, mp 100 °C. The last fraction contained 413 mg (34%) of
2,3-bis(trimethylsilylethynyl)biphenylene (135a).53 IR (NaCl film): ~ν = 2959, 2152,
1422, 1250, 1154, 999, 876, 843, 739, 717 cm–1. MS (EI, 70 eV) m/z (rel intensity) 374

185
(M+, 100), 359 (65), 248 (16), 233 (24), 179 (18), 124 (6). 1H NMR (400 MHz,
CDCl3) δ 7.09 (s, 1H), 6.84–6.78 (m, 2H), 6.71 (s, 1H), 6.71–6.67 (m, 2H), 0.27 (s, 9H).
13C NMR (100 MHz, CDCl3) δ 151.66, 150.41, 149.92, 149.79, 129.37, 128.96, 128.66,
127.12, 120.50, 118.61, 118.57, 107.57, 101.18, 99.42, –0.15. HR-MS Calcd for
C17H15ISi: 373.9988. Found: 373.9988.
Method B. A solution of 139a (vide infra, 450 mg, 1.38 mmol) in THF (30 mL)
was cooled to –45 °C, and BuLi (0.70 mL of 2.5 M solution in hexane, 1.75 mmol) was
added via syringe. The dark brown solution was stirred at –45 ºC for 30 min. After that
time, a THF (20 mL) solution of iodine (508 mg, 2.00 mmol) was added dropwise via
syringe. The color of the solution lightened gradually with the addition of iodine. The
mixture was left to warm to 23 °C overnight, extracted with ether (2 x 100 mL), and
washed with aq. Na2S2O3 and then brine. Drying over MgSO4, followed by removal of
solvent in vacuo, gave 134a as a yellow oil (399 mg, 78%).
2-[(Dimethylthexylsilyl)ethynyl]-3-iodobiphenylene (134b):
I
Si
Method A. 2,3-Diiodobiphenylene (133)53 (100 mg, 0.25 mmol) was dissolved in
triethylamine (50 mL). The solution was degassed, and [Pd(PPh3)2Cl2] (9 mg, 0.013
mmol), CuI (3 mg, 0.016 mmol), and DMTSA209 (45 mg, 0.27 mmol) were added. The
mixture was stirred at 23 °C for 7 h, after which time an additional portion of DMTSA
(49 mg, 0.29 mmol) was injected through the septum. After an additional 2 h of stirring,

186
solvent was removed in vacuo and the mixture chromatographed on silica (hexanes) to
give the starting material (12 mg, 12%), followed by 134b (23 mg, 21%) as a yellow oil.
The last fraction contained 135b (74 mg, 63%) as yellow crystals, mp 106–108 °C. IR
(NaCl film): ~ν = 2958, 2154, 1735, 1464, 1378, 1248, 1113, 872, 830, 738 cm–1. MS
(EI, 70 eV) m/z (rel intensity) 444 (M+, 19), 359 (100), 283 (9), 233 (28), 161 (18), 123
(12). 1H NMR (400 MHz, CDCl3) δ 7.08 (s, 1H), 6.82–6.79 (m, 2H), 6.71 (s, 1H), 6.71–
6.66 (m, 2H), 1.75 (sept, 3J = 6.9 Hz, 1H), 0.97 (s, 6H), 0.95 (d, 3J = 6.9 Hz, 6H), 0.25 (s,
6H). 13C NMR (100 MHz, CDCl3) δ 151.50, 150.36, 149.95, 149.83, 129.34, 128.94,
127.12, 120.80, 118.57, 118.55, 108.19, 100.65, 100.01, 99.28, 34.63, 23.65, 20.84,
18.80, –2.42. HR-MS Calcd for C22H25ISi: 444.0770. Found: 444.0771.
Si
Si
Spectral data for 135b: IR (NaCl film): ~ν = 2959, 2865, 2147, 1465, 1424, 1249,
1108, 912, 874, 841, 773, 733 cm–1. MS (EI, 70 eV) m/z (rel intensity) 484 (M+, 33), 399
(8), 329 (78), 315 (100), 241 (17), 73 (55). 1H NMR (400 MHz, CDCl3) δ 6.83–6.81 (m,
2H), 6.69–6.67 (m, 2H), 6.67 (s, 2H), 1.72 (sept, 3J = 6.9 Hz, 2H), 0.97 (s, 12H), 0.96 (d,
3J = 6.9 Hz, 12H), 0.23 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 150.14, 150.04, 128.13,
126.01, 120.40, 118.35, 105.04, 99.17, 34.67, 23.57, 20.93, 18.79, –2.28. HR-MS Calcd
for C32H44Si2: 484.2982. Found: 484.2990.
Method B. A solution of 139b (vide infra, 350 mg, 0.88 mmol) in THF (30 mL)
was cooled to –45 °C, and BuLi (0.50 mL of 2.5 M solution in hexane, 1.25 mmol) was

187
added via syringe. The dark brown solution was stirred at –45 ºC for 30 min. After that
time, a THF (20 mL) solution of iodine (508 mg, 2.00 mmol) was added dropwise via
syringe. The color of the solution lightened gradually with the addition of iodine. The
mixture was left to warm to 23 °C overnight, extracted with ether (2 x 100 mL), and
washed with aq. Na2S2O3 and then brine. Drying over MgSO4, followed by removal of
solvent in vacuo, gave 134b as a yellow oil (320 mg, 82%).
2-Iodo-3-(oct-1-ynyl)biphenylene (134c):
I
Method A. 2,3-Diiodobiphenylene (133)53 (105 mg, 0.26 mmol) was dissolved in
triethylamine (5 mL). The solution was degassed, and [Pd(PPh3)2Cl2] (18 mg, 0.026
mmol), CuI (5 mg, 0.026 mmol), and 1-octyne (29 mg, 38 µL, 0.26 mmol) were added.
The mixture was stirred at 23 °C for 12 h, after which time the solvent was removed in
vacuo and the mixture was chromatographed on silica (hexanes) to give the starting 133
(21 mg, 20%), followed by 134c (46 mg, 46%) as a yellow oil. The last fraction
contained 135c (9 mg, 9%) as a yellow oil. IR (NaCl film): ~ν = 2927, 2855, 2225, 1567,
1423, 1377, 1346, 1256, 1155, 1112, 974, 872, 739 cm–1. MS (EI, 70 eV) m/z (rel
intensity) 386 (M+, 100), 315 (26), 260 (63), 216 (42), 202 (65), 189 (82), 165 (20). 1H
NMR (400 MHz, CDCl3) δ 7.07 (s, 1H), 6.80–6.79 (m, 2H), 6.70–6.65 (m, 3H), 2.44 (t,
3J = 7.0 Hz, 2H), 1.66–1.59 (m, 2H), 1.54–1.48 (m, 2H), 1.34–1.26 (m, 4H), 0.91 (t, 3J =
7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 150.67, 150.45, 149.99, 149.97, 129.61,
129.09, 128.84, 127.05, 120.64, 118.46, 118.30, 100.15, 95.60, 84.02, 31.40, 28.67,

188
28.51, 22.63, 19.71, 14.14. HR-MS Calcd for C20H19I: 386.0532. Found: 386.0535. Anal.
Calcd for C20H19I: C, 62.19; H, 4.96. Found: C, 62.87; H, 5.23.
Spectral data for 135c: IR (NaCl film): ~ν = 2929, 2857, 2221, 1466, 1427, 1378,
1327, 1279, 1154, 1112, 879, 739 cm–1. MS (FAB) m/z (rel intensity) 368 (M+, 33), 252
(100), 235 (56), 140 (89), 123 (73). 1H NMR (400 MHz, CDCl3) δ 6.79–6.77 (m, 2H),
6.67–6.65 (m, 2H), 6.62 (s, 2H), 2.44 (t, 3J = 7.1 Hz, 2H), 1.64–1.55 (m, 2H), 1.50–1.43
(m, 2H), 1.36–1.26 (m, 4H), 0.91 (t, 3J = 7.0 Hz, 3H). 13C NMR (100 MHz,
CDCl3) δ 150.24, 149.38, 128.78, 126.36, 120.05, 117.97, 94.94, 80.45, 31.49, 28.85,
28.66, 22.62, 19.82, 14.13. HR-MS Calcd for C28H32: 368.2504. Found: 368.2495.
Method B. A solution of 139c (vide infra, 400 mg, 1.18 mmol) in THF (30 mL)
was cooled to –45 °C, and BuLi (0.60 mL of 2.5 M solution in hexane, 1.50 mmol) was
added via syringe. The dark brown solution was stirred at –45 ºC for 30 min. After that
time, a THF (20 mL) solution of iodine (508 mg, 2.00 mmol) was added dropwise via
syringe. The color of the solution lightened gradually with the addition of iodine. The
mixture was left to warm to 23 °C overnight, extracted with ether (2 x 100 mL), and
washed with aq. Na2S2O3 and then brine. Drying over MgSO4, followed by removal of
solvent in vacuo, gave 134c as a yellow oil (388 mg, 85%).

189
2-Bromo-3-(trimethylsilyl)biphenylene (137):
Br
TMS
2,3-Bis(trimethylsilyl)biphenylene52 (3.62 g, 12.3 mmol) and pyridine (0.97 mL,
12.1 mmol) were dissolved in methylene chloride (35 mL), and the solution was cooled
to 0 °C. Neat bromine (0.65 mL, 12.7 mmol) was added via syringe. The originally
orange mixture immediately turned dark brown. The solution was stirred at 0 °C for 3
min and then treated with aq. NaHCO3, followed by dilution with ether and washing with
aq. Na2S2O3. The organic layer was dried over MgSO4, and the solvent was subsequently
removed in vacuo to give the crude material as an oily solid. Washing the solid with
hexanes extracted a yellow oil that contained 137 (2.20 g, 59%) that was used in the
following reaction without further purification. The remaining white crystals were
tentatively identified as impure 2,3-dibromobiphenylene (970 mg, 25%), which was not
fully characterized. IR (NaCl film): ~ν = 3066, 2955, 2898, 1932, 1653, 1587, 1418,
1322, 1248, 1196, 1075, 854, 739, 647 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.81 (s, 1H),
6.79–6.77 (m, 2H), 6.69 (s, 1H), 6.68–6.66 (m, 2H), 0.36 (s, 9H). 13C NMR (125 MHz,
CDCl3) δ 153.36, 151.23, 149.93, 148.89, 140.05, 130.18, 128.93, 128.32, 123.42,
122.30, 118.28, 117.75, –0.40.
2-Bromo-3-iodobiphenylene (138):
Br
I
Compound 137 (2.20 g, 7.26 mmol) was dissolved in CH2Cl2 (30 mL) and the
solution cooled to 0 °C. To this solution, ICl (1.19 g, 7.30 mmol) in CH2Cl2 (20 mL) was

190
added slowly via syringe. The mixture changed color from yellow to reddish-brown. The
solution was stirred for 2 h at 0 °C and an additional 3 h at 23 °C. Subsequently, it was
treated with aq. NaHCO3, followed by dilution with ether and washing with aq. Na2S2O3.
The organic layer was dried over MgSO4, and the solvent was removed in vacuo to give
the crude material, which was purified by column chromatography on silica (eluting with
hexanes). Final purification was achieved by Kugelrohr sublimation (250 °C, 0.3 Torr) to
give 138 as yellow crystals, mp 122–124 °C (2.57 g, 99%). MS (EI, 70 eV) m/z (rel
intensity) 358/356 (M+, 98/98), 312/310 (18/18), 229 (16), 150 (100), 75 (24). 1H NMR
(400 MHz, CDCl3) δ 7.04 (s, 1H), 6.85 (s, 1H), 6.82–6.80 (s, 2H), 6.69–6.65 (m, 2H). 13C
NMR (100 MHz, CDCl3) δ 152.29, 150.67, 149.63, 149.40, 129.28, 129.20, 128.60,
128.27, 121.79, 118.89, 118.64, 99.21. HR-MS Calcd for C12H679BrI: 355.8698. Found:
355.8694. Anal. Calcd for C12H6BrI: C, 40.37; H, 1.69. Found: C, 40.30; H, 1.82.
2-Bromo-3-[(trimethylsilyl)ethynyl]biphenylene (139a):
Br
TMS
A thoroughly degassed solution of 138 (700 mg, 1.96 mmol) in triethylamine (50
mL) was treated with [Pd(PPh3)2Cl2] (68 mg, 0.098 mmol), CuI (19 mg, 0.098 mmol),
and TMSA (0.35 mL, 2.50 mmol). After stirring for 12 h at 23 °C, an additional portion
of TMSA (0.30 mL, 2.14 mmol) was added. The mixture was left to stir for an additional
24 h, and the solvent was then removed in vacuo. Column chromatography of the residue
provided the starting material (29 mg, 4%), followed by the desired product (480 mg,
75%) as a yellow oil. IR (NaCl film): ~ν = 2958, 2153, 1868, 1489, 1250, 882, 844, 740

191
cm–1. MS (EI, 70 eV) m/z (rel intensity) 328/326 (M+, 100/92), 313/311 (85/83), 231 (15),
217 (18), 202 (31), 189 (76), 156 (15). 1H NMR (400 MHz, CDCl3) δ 6.84 (s, 1H), 6.84–
6.80 (m, 2H), 6.72–6.66 (m, 3H), 0.26 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 151.99,
149.81, 149.45, 149.33, 129.47, 128.99, 126.16, 123.82, 121.62, 121.13, 118.60, 118.38,
103.94, 100.23, –0.11. HR-MS Calcd for C17H1581BrSi: 328.0106. Found: 328.0103.
Anal. Calcd for C17H15BrSi: C, 62.39; H, 4.62. Found: C, 61.90; H, 4.96.
2-Bromo-3-[(dimethylthexylsilyl)ethynyl]biphenylene (139b):
Br
Si
A thoroughly degassed solution of 138 (700 mg, 1.96 mmol) in triethylamine (50
mL) was treated with [Pd(PPh3)2Cl2] (68 mg, 0.098 mmol), CuI (19 mg, 0.098 mmol),
and DMTSA (505 mg, 3.00 mmol). After stirring for 24 h at 23 °C, the solvent was
removed in vacuo. Column chromatography of the residue provided the starting material
(203 mg, 29%), followed by the desired product (467 mg, 60%) as a yellow oil. The
crude material was used in subsequent steps. IR (NaCl film): ~ν = 2959, 2866, 2152,
1421, 1250, 1010, 877, 818, 775, 739 cm–1. 1H NMR (500 MHz, CDCl3) δ 6.83 (s, 1H),
6.85–6.80 (m, 2H), 6.69 (s, 1H), 6.70–6.67 (m, 2H), 1.74 (sept, 3J = 6.9 Hz, 1H), 0.97 (s,
6H), 0.95 (d, 3J = 6.9 Hz, 6H), 0.24 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 151.75,
149.74, 149.39, 149.22, 129.37, 128.99, 125.96, 124.01, 121.56, 121.19, 118.50, 118.27,
104.63, 99.97, 34.63, 23.50, 20.70, 18.69, 18.57, –2.51.

192
2-Bromo-3-(oct-1-ynyl)biphenylene (139c):
Br
A thoroughly degassed solution of 138 (700 mg, 1.96 mmol) in triethylamine (50
mL) was treated with [Pd(PPh3)2Cl2] (68 mg, 0.098 mmol), CuI (19 mg, 0.098 mmol),
and 1-octyne (0.37 mL, 2.50 mmol). After stirring for 12 h at 23 °C, an additional portion
of 1-octyne (0.4 mL, 2.70 mmol) was added. The mixture was left to stir for an additional
24 h, and the solvent was then removed in vacuo. Column chromatography of the residue
provided the starting material (182 mg, 26%), followed by the desired 139c (446 mg,
67%) as a yellow oil. IR (NaCl film): ~ν = 3058, 2930, 2857, 2228, 1656, 1424, 1351,
1258, 1207, 1155, 1113, 1013, 872, 739 cm–1. MS (EI, 70 eV) m/z (rel intensity) 340/338
(M+, 100/100), 269/267 (34/34), 230 (21), 216 (48), 202 (38), 190 (66), 187 (62), 57 (10).
1H NMR (400 MHz, CDCl3) δ 6.81 (s, 1H), 6.81–6.79 (s, 2H), 6.68–6.64 (s, 2H), 6.64 (s,
1H), 2.45 (t, 3J = 7.0 Hz, 2H), 1.65–1.60 (m, 2H), 1.54–1.49 (m, 2H), 1.39–1.30 (m, 4H),
0.94 (t, 3J = 7.1 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 150.97, 149.89, 149.63, 149.41,
129.20, 128.88, 125.26, 124.79, 121.60, 121.26, 118.34, 118.24, 96.31, 80.45, 31.48,
28.68 (2C), 22.72, 19.81, 14.22. HR-MS Calcd for C20H1979Br: 338.0670. Found:
338.0664. Anal. Calcd for C20H19Br: C, 70.80; H, 5.64. Found: C, 71.10; H, 5.81.

193
Compound 141a:
TMSDMTS
TMSDMTS
In a 25 ml Schlenk flask, compounds 134a (331 mg, 0.885 mmol) and 14066 (196
mg, 0.428 mmol), along with [Pd(PPh3)2Cl2] (18 mg, 0.026 mmol) and CuI (5.1 mg,
0.026 mmol), were suspended in triethylamine (120 mL). The suspension was degassed
and the tube closed. The mixture was heated at 120 °C for 48 h. The solvent was removed
in vacuo and the mixture purified by column chromatography (hexanes/CH2Cl2) to give
141a as a yellow oil (93 mg, 23%). IR (NaCl film): ~ν = 2959, 2866, 2150, 1653, 1250,
1164, 942, 885, 841, 775, 761, 739 cm–1. MS (FAB) m/z (rel intensity) 950 (M+, 4), 877
(3), 307 (21), 154 (100), 136 (80). 1H NMR (500 MHz, C6D6) δ 7.11 (s, 2H), 6.79 (s,
2H), 6.54 (s, 2H), 6.42–6.41 (m, 4H), 6.30–6.29 (m, 2H), 6.19–6.17 (m, 2H), 1.73 (sept,
3J = 6.8 Hz, 2H), 1.01 (s, 12H), 0.94 (d, 3J = 6.9 Hz, 12H), 0.31 (s, 12H), 0.26 (s, 18H).
13C NMR (125 MHz, CDCl3) δ 150.16, 150.05, 150.00, 149.97, 131.10, 128.98, 128.95,
128.49, 126.75, 126.11, 125.96, 119.96, 119.83, 118.20, 118.19, 104.42, 103.71, 100.74,
99.69, 97.20, 91.21, 34.51, 23.44, 20.71, 18.65, –0.13, –2.37. HR-MS Calcd for
C64H70Si4: 950.4555. Found: 950.4560.

194
Compound 141b:
DMTSDMTS
DMTSDMTS
In a 25 ml Schlenk flask, compounds 134b (23 mg, 0.052 mmol) and 14066 (12
mg, 0.025 mmol), along with [Pd(PPh3)2Cl2] (3.5 mg, 0.005 mmol) and CuI (1.0 mg,
0.005 mmol), were suspended in triethylamine (20 mL). The suspension was degassed
and the tube closed. The mixture was heated at 120 °C for 48 h. The solvent was removed
in vacuo and the mixture purified by column chromatography (hexanes/CH2Cl2) to give
141b as a yellow oil (8 mg, 28%). IR (NaCl film): ~ν = 2958, 2866 , 2151, 1462, 1250,
1165, 943, 883, 838, 820, 775, 739 cm–1. MS (FAB) m/z (rel intensity) 1090 (M+, 4), 252
(82), 140 (100). 1H NMR (500 MHz, C6D6) δ 7.15 (s, 2H), 6.85 (s, 2H), 6.51 (s, 2H),
6.44–6.41 (m, 4H), 6.32–6.31 (m, 2H), 6.21–6.19 (m, 2H), 1.75 (sept, 3J = 6.8 Hz, 2H),
1.74 (sept, 3J = 6.8 Hz, 2H), 1.01 (s, 12H), 1.00 (s, 12H), 0.95 (d, 3J = 6.9 Hz, 12H), 0.94
(d, 3J = 6.9 Hz, 12H), 0.33 (s, 12H), 0.30 (s, 12H). 13C NMR (125 MHz,
CDCl3) δ 150.12, 150.05, 150.01, 149.77, 130.82, 128.90, 128.87, 128.86, 126.59,
126.22, 125.95, 119.96, 119.93, 118.11, 118.10, 105.07, 103.66, 100.62, 99.15, 97.10,
90.86, 34.50, 34.43, 23.41, 23.31, 20.64, 20.59, 18.63, 18.55, –2.35, –2.40. HR-MS Calcd
for C74H90Si4: 1090.6120. Found: 1090.6116.

195
Compound 141c:
DMTS
DMTS
In a 25 ml Schlenk flask, compounds 134c (388 mg, 1.01 mmol) and 14066 (359
mg, 0.57 mmol), along with [Pd(PPh3)2Cl2] (35 mg, 0.05 mmol) and CuI (5.0 mg, 0.025
mmol), were suspended in triethylamine (10 mL). The suspension was degassed and the
tube closed. The mixture was heated at 120 °C for 36 h. The solvent was removed in
vacuo and the mixture purified by column chromatography (hexanes/CH2Cl2) to give
141c as a brown oil (291 mg, 30%). IR (NaCl film): ~ν = 2958, 2866, 2225, 2155, 1717,
1458, 1428, 1378, 1250, 1157, 1120, 819, 776, 740 cm–1. MS (EI, 70 eV) m/z (rel
intensity) 976 (M+, 1), 252 (100), 235 (70), 140 (95), 123 (73). 1H NMR (400 MHz,
CDCl3) δ 7.33 (s, 2H), 6.81–6.79 (m, 6H), 6.70–6.65 (m, 6H), 2.24 (t, 3J = 7.3 Hz, 4H),
1.72 (sept, 3J = 6.8 Hz, 2H), 1.49–1.41 (m, 4H), 1.33–1.27 (m, 4H), 1.18–1.10 (m, 8H),
0.95 (s, 12H), 0.92 (d, 3J = 6.8 Hz, 12H), 0.79 (t, 3J = 7.1 Hz, 6H), 0.24 (s, 12H). 13C
NMR (100 MHz, CDCl3) δ 150.34, 150.27, 150.16, 148.77, 131.29, 128.99, 128.81,
128.56, 127.47, 126.19, 125.62, 120.46, 120.17, 118.18, 117.95, 103.97, 100.50, 97.92,

196
96.63, 90.79, 80.16, 34.63, 31.53, 28.88, 28.84, 23.56, 22.57, 20.82, 19.92, 18.71, 14.11,
–2.33. HR-MS Calcd for C70H78Si2: 974.5642. Found: 974.5650.
Compound 144:
DMTS
DMTS
O
In a 25 mL Schlenk flask, compounds 134c (129 mg, 0.334 mmol) and 14066 (77
mg, 0.168 mmol), along with [Pd(PPh3)2Cl2] (12 mg, 0.017 mmol) and CuI (3.2 mg,
0.017 mmol), were suspended in piperidine (15 mL). The suspension was degassed and
the tube closed. The mixture was heated at 120 °C for 24 h. The solvent was removed in
vacuo and the mixture purified by column chromatography (hexanes/CH2Cl2) to give 144
as a yellow oil (55 mg, 34%). IR (NaCl film): ~ν = 2958, 2927, 2860, 2153, 1728, 1465,
1428, 1379, 1250, 1157, 878, 822, 775, 739 cm–1. MS (FAB) m/z (rel intensity) 735
([M+H]+, 5), 734 (M+, 5), 252 (100), 235 (70), 140 (95), 123 (73). 1H NMR (400 MHz,
CDCl3) δ 9.70 (s, 1H), 7.38 (br s, 2H), 6.84–6.82 (m, 2H), 6.76 (s, 1H), 6.72–6.68 (m,
2H), 6.68 (s, 1H), 4.23 (s, 2H), 2.43 (t, 3J = 7.1 Hz, 2H) 1.75–1.67 (m, 2H), 1.61–1.54
(m, 2H), 1.45–1.37 (m, 2H), 1.26–1.22 (m, 4H), 0.96–0.92 (m, 24H), 0.87 (t, 3J = 6.9 Hz,
2H), 0.26 (s, 6H), 0.23 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 198.09, 150.77, 150.10,
150.03, 149.26, 137.22, 131.49, 131.42, 129.16, 129.02, 127.03, 126.90, 126.44, 124.70,
124.21, 120.41, 119.99, 118.42, 118.11, 104.16, 103.24, 101.62, 100.62, 98.37, 96.14,

197
89.98, 80.55, 47.85, 34.61 (2C), 31.42, 28.75, 28.73, 23.57, 23.50, 22.56, 20.83, 20.73,
19.88, 18.71, 18.66, 14.10, –2.29, –2.54. HR-MS Calcd for C50H62OSi2: 734.4367.
Found: 734.4358.
1-Bromo-2,4-bis[(dimethylthexylsilyl)ethynyl]-5-iodobenzene (145):
IBr
SiSi
A solution of 1,5-dibromo-2,4-bis[(dimethylthexylsilyl)ethynyl]benzene (2.90 g,
5.11 mmol) in ether (200 mL) was cooled to –50 °C, and BuLi (4.4 mL of 2.4 M solution
in hexane, 10.3 mmol) was added via syringe. The orange solution was stirred at –50 ºC
for 45 min. After that time, an ethereal solution (100 mL) of iodine (3.45 g, 13.5 mmol)
was added dropwise via syringe. The color of the solution lightened gradually with
addition of iodine. The mixture was left to warm to 23 °C overnight, extracted with ether
(2 x 100 mL), and washed with aq. Na2S2O3 and then brine. Drying over MgSO4,
followed by removal of solvent in vacuo, gave 145 as a yellow oil (3.11 g, 99%). IR
(NaCl film): ~ν = 2962, 2865, 2156, 1462, 1445, 1378, 1336, 1250, 1174, 1046, 970, 873,
818, 775, 690 cm–1. MS (EI, 70 eV) m/z (rel intensity) 616/614 (M+, 1/1), 601/599 (1/1),
573/571 (2/2), 531/529 (100/92), 451 (32), 405 (20), 319 (24), 85 (26). 1H NMR (500
MHz, CDCl3) δ 8.03 (s, 1H), 7.51 (s, 1H), 1.82–1.70 (m, 2H), 0.97 (s, 6H), 0.96 (s, 6H),
0.94 (d, 3J = 6.9 Hz, 6H), 0.94 (d, 3J = 6.9 Hz, 6H), 0.25 (s, 6H), 0.24 (s, 6H). 13C NMR
(125 MHz, CDCl3) δ 141.44, 136.37, 129.04, 125.40, 125.32, 105.42, 102.21, 101.48,
100.48, 100.28, 34.49 (2C), 23.53, 23.48, 20.73, 20.67, 18.71, 18.67, –2.61, –2.64. HR-

198
MS Calcd for C26H4081BrISi2: 616.0876. Found: 616.0867. Anal. Calcd for C26H40BrISi2:
C, 50.73; H, 6.55. Found: C, 50.99; H, 6.92.
1-Iodo-2,4-bis[(dimethylthexylsilyl)ethynyl]-5-[(trimethylsilyl)ethynyl]benzene
(146):
Br
SiSi
TMS
1-Bromo-2,4-bis[(dimethylthexylsilyl)ethynyl]-5-[(trimethylsilyl)ethynyl]benzene.
A suspension of 145 (1.45 g, 2.36 mmol), [Pd(PPh3)2Cl2] (83.0 mg, 0.12 mmol), and CuI
(23.0 mg, 0.12 mmol) in triethylamine (60 mL) was thoroughly degassed.
Trimethylsilylacetylene (4.00 mL, 28.3 mmol) was injected, the mixture stirred at 23 °C
for 1 h, the solids filtered off, the solvent removed in vacuo, and the resulting crude
mixture filtered through a short pad of silica (hexanes) to yield 1-bromo-2,4-
bis[(dimethylthexylsilyl)ethynyl]-5-[(trimethylsilyl)ethynyl]benzene as a yellow oil (1.03
mg, 75%). IR (NaCl film): ~ν = 2959, 2866, 2158, 1467, 1356, 1250, 1153, 862, 841, 775
cm–1. MS (EI, 70 eV) m/z (rel intensity) 586/584 (M+, 100/98), 518 (41), 485 (14), 405
(11). 1H NMR (500 MHz, CDCl3) δ 7.66 (s, 1H), 7.51 (s, 1H), 1.76–1.68 (m, 2H), 0.97
(br s, 12H), 0.94 (d, 3J = 6.8 Hz, 6H), 0.93 (d, 3J = 6.8 Hz, 6H), 0.25 (s, 9H), 0.24 (s, 6H),
0.24 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 136.71, 135.97, 126.25, 125.28, 124.73,
124.69, 102.66, 102.24, 101.92, 101.54, 101.36, 99.97, 34.51, 34.48, 23.50, 23.41, 20.83,
20.67, 18.68, 18.66, –0.17, –2.45, –2.61. HR-MS Calcd for C31H4981BrSi3: 586.2305.

199
Found: 586.2313. Anal. Calcd for C31H49BrSi3: C, 63.55; H, 8.43. Found: C, 63.23; H,
8.58.
I
SiSi
TMS
1-Iodo-2,4-bis[(dimethylthexylsilyl)ethynyl]-5-[(trimethylsilyl)ethynyl]benzene
(146). A solution of 1-bromo-2,4-bis[(dimethylthexylsilyl)ethynyl]-5-
[(trimethylsilyl)ethynyl]benzene (60.0 mg, 0.11 mmol) in ether (10 mL) was cooled to –
50 °C, and BuLi (0.2 mL of 2.34 M solution in hexane, 0.48 mmol) was added via
syringe. The orange solution was stirred at –50 °C for 45 min. After that time, an ethereal
solution (10 mL) of iodine (127 mg, 0.50 mmol) was added dropwise via syringe. The
color of the solution lightened gradually with addition of iodine. The mixture was left to
warm to 23 °C overnight, extracted with ether (2 x 10 mL), and washed with aq. Na2S2O3
and then brine. Drying over MgSO4, followed by removal of solvent in vacuo, gave 146
as a yellow oil (51.0 mg, 79%). IR (NaCl film): ~ν = 2958, 2867, 2157, 2068, 1463,
1378, 1350, 1250, 1191, 1149, 1036, 861, 775, 683 cm–1. MS (EI, 70 eV) m/z (rel
intensity) 632 (M+, 5), 589 (2), 547 (100), 421 (68). 1H NMR (500 MHz, CDCl3) δ 7.92
(s, 1H), 7.47 (s, 1H), 1.77–1.68 (m, 2H), 0.98 (s, 6H), 0.97 (s, 6H), 0.95 (d, 3J = 6.9 Hz,
6H), 0.94 (d, 3J = 6.9 Hz, 6H), 0.26 (s, 6H), 0.25 (s, 9H), 0.24 (s, 6H). 13C NMR (100
MHz, CDCl3) δ 142.23, 135.76, 129.64, 126.11, 125.43, 106.04, 102.39, 101.38, 101.30,
101.07, 100.25, 99.53, 34.53, 34.50, 23.59, 23.44, 20.86, 20.76, 18.73, 18.71, –0.13, –
2.42, –2.55. HR-MS Calcd for C31H49ISi3: 632.2187. Found: 632.2185. Anal. Calcd for
C31H49ISi3: C, 58.53; H, 7.80. Found: C, 58.84; H, 7.99.

200
Compound 147:
DMTS
TMS
DMTSDMTS
DMTS
TMS
DMTSDMTS
In a 250 ml Schlenk flask, compounds 14066 (343 mg, 0.75 mmol) and 146 (946
mg, 1.50 mmol), along with [Pd(PPh3)2Cl2] (26 mg, 0.04 mmol), and CuI (7.0 mg, 0.04
mmol) were suspended in triethylamine (100 mL). The mixture was set to reflux and left
to stir, with heating, overnight (~ 16 h). After the reaction was complete, solvent was
removed in vacuo and the mixture filtered through a short plug of silica (hexanes/EtOAc)
to give the desired product (950 mg, 87%) as a yellow oil. IR (NaCl film): ~ν = 2959,
2932, 2158, 1484, 1463, 1380, 1250, 1188, 930, 867, 840, 776, 673 cm–1. 1H NMR (400
MHz, C6D6) δ 7.84 (s, 2H), 7.57 (s, 2H), 7.09 (s, 2H), 1.71–1.59 (m, 6H), 0.97 (s, 12H),
0.92 (s, 12H), 0.91 (d, 3J = 6.9 Hz, 12H), 0.88 (s, 12H), 0.87 (d, 3J = 6.9 Hz, 12H), 0.84
(d, 3J = 6.9 Hz, 12H), 0.28 (s, 9H), 0.25 (s, 6H), 0.25 (s, 6H), 0.16 (s, 6H). 13C NMR (100
MHz, CDCl3) δ 136.06, 135.82, 131.11, 128.77, 125.89, 125.58, 125.54, 125.19, 124.99,
103.41, 103.18, 103.07, 102.40, 101.10, 100.87, 100.31, 100.07, 95.23, 91.78, 34.64,
34.56, 34.50, 23.53, 23.46, 23.38, 20.93, 20.66, 20.64, 18.78, 18.68, 18.60, 0.00, –2.32, –
2.37, –2.40.

201
Attempted preparation of compound 148:
TMS
TMS
TMS
TMS
A solution of 147 (90 mg, 0.061 mmol) in THF (50 mL) was treated with TBAF
(0.6 mL of 1.0 M THF solution, 0.60 mmol). The dark-colored mixture was stirred at 23
°C for 90 min, after which time ethanol (3 mL) was added via syringe. After an
additional 40 min of stirring, the solution was diluted with ether and washed with water.
The organic layer was dried over MgSO4. m-Xylene (20 mL) was added, and the volume
of the solution was reduced in vacuo to ~ 25 mL. The resulting solution was mixed with
[CpCo(CO)2] (0.2 mL) and injected over 5 min into a refluxing mixture of m-xylene and
BTMSA (with irradiation). Irradiation and heating were continued for 45 min. After
cooling, solvents were removed in vacuo, and the crude material was filtered through a
plug of silica (hexanes/CH2Cl2) to provide a complicated mixture of products. MS (EI, 70
eV) m/z (rel intensity) 933 (0.5), 922 (0.5), 840 (0.5), 810 (0.5, possibly 148), 711 (25),
623 (27), 294 (33), 125 (31), 111 (48), 97 (68), 83 (62), 71 (72), 57 (100).

202
Compound 149:
DMTS
DMTS
DMTS
DMTSDMTS
DMTSTMS
TMS
In a 100 mL round-bottom flask, compound 147 (475 mg, 0.324 mmol) was
dissolved in a mixture of ether (20 mL) and methanol (20 mL). Potassium carbonate (138
mg, 1.0 mmol) was added and the mixture left to stir at 23 °C for 90 min. Solids were
filtered off and the solvent removed in vacuo. The crude product was redissolved in
methylene chloride and the solution filtered through a short pad of silica. Solvent was
removed in vacuo. The resulting diterminal alkyne was transferred into a 250 mL Schlenk
flask and dissolved in triethylamine (100 mL). This solution was treated with
[Pd(PPh3)2Cl2] (11.2 mg, 0.016 mmol), CuI (3.0 mg, 0.016 mmol), and 1-iodo-2-
[(trimethylsilyl)ethynyl]benzene142 (203 mg, 0.700 mmol). The mixture was thoroughly
degassed, set to reflux, and left to stir, with heating, overnight (~ 16 h). After the reaction
was complete, solvent was removed in vacuo and the mixture filtered through a short
plug of silica (hexanes/EtOAc) to give the desired product (468 mg, 88%) as a yellow oil.
IR (NaCl film): ~ν = 2959, 2932, 2158, 1489, 1464, 1379, 1250, 930, 902, 874, 840, 776,
674 cm–1. MS (FAB) m/z (rel intensity) 1670 (M+, 58), 1585 (100), 1500 (71), 1416 (62).
1H NMR (500 MHz, C6D6) δ 7.97 (s, 2H), 7.72 (s, 2H), 7.49 (dd, 3J1 = 7.8 Hz, 4J2 = 0.6
Hz, 2H), 7.37 (dd, 3J1 = 7.8 Hz, 4J2 = 0.6 Hz, 2H), 7.12 (s, 2H), 6.84 (dt, 3J1 = 7.7 Hz, 4J2

203
= 1.2 Hz, 2H), 6.75 (dt, 3J1 = 7.7 Hz, 4J2 = 1.2 Hz, 2H), 1.70–1.63 (m, 6H), 0.94 (s, 12H),
0.92 (s, 24H), 0.89 (d, 3J = 6.9 Hz, 12H), 0.87 (d, 3J = 6.9 Hz, 12H), 0.86 (d, 3J = 6.9 Hz,
12H), 0.32 (s, 9H), 0.27 (s, 6H), 0.23 (s, 6H), 0.22 (s, 6H). 13C NMR (125 MHz,
CDCl3) δ 135.69, 134.98, 131.85, 131.75, 131.21, 128.22, 128.04, 127.85, 126.37,
126.07, 125.97, 125.85, 125.61, 128.28, 125.25, 103.41, 103.21 (2C), 103.06, 101.11,
100.98, 100.52, 99.26, 94.93, 93.49, 91.68, 91.05, 34.47 (2C), 34.39, 23.39, 23.34, 23.25,
20.69, 20.64, 20.59, 18.61 (2C), 18.54, –0.09, –2.38, –2.42 (2C). The high molecular
mass of 149 precluded HR-MS measurements.
6.2.1 Calculated Structures of 60 and 118–120
Calculated positional parameters for 60:
Atom x y z
C1 –0.0413513298 0.5413477993 5.8636121223 C2 –0.3092033702 3.2809890496 5.9994865076 C3 –0.1332418660 1.3270481111 4.6791300864 C4 –0.0809106068 1.1063130814 7.1202535966 C5 –0.2190847435 2.5130990264 7.1607367892 C6 –0.2682156736 2.6972635208 4.7120887729 C7 0.0308396940 –0.5551416246 1.1544397841 C8 0.1762396501 –1.9748646358 –1.1569207288 C9 0.1762396501 –1.9748646358 1.1569207288 C10 –0.0488828124 0.2199865013 0.0000000000 C11 0.0308396940 –0.5551416246 –1.1544397841 C12 0.2556451598 –2.7488282192 0.0000000000 C13 –0.1332418660 1.3270481111 –4.6791300864 C14 –0.2190847435 2.5130990264 –7.1607367892 C15 –0.0413513298 0.5413477993 –5.8636121223 C16 –0.2682156736 2.6972635208 –4.7120887729 C17 –0.3092033702 3.2809890496 –5.9994865076 C18 –0.0809106068 1.1063130814 –7.1202535966 C19 0.0752217707 –0.7109205642 5.0499839975 C20 0.1839570514 –1.9897099744 2.6526961077

204
C21 0.2140845876 –2.0681281615 5.0597906218 C22 –0.0185658986 0.0964210650 3.8385174058 C23 0.0354448344 –0.5411344328 2.6468986291 C24 0.2723310826 –2.7496877988 3.7827235826 C25 0.0752217707 –0.7109205642 –5.0499839975 C26 0.1839570514 –1.9897099744 –2.6526961077 C27 0.2140845876 –2.0681281615 –5.0597906218 C28 –0.0185658986 0.0964210650 –3.8385174058 C29 0.0354448344 –0.5411344328 –2.6468986291 C30 0.2723310826 –2.7496877988 –3.7827235826 H1 –0.0114672594 0.5259443880 8.0356174416 H2 –0.2560068873 3.0123736343 8.1252459797 H3 –0.1573752676 1.2998562463 0.0000000000 H4 0.3676986583 –3.8284323108 0.0000000000 H5 –0.0114672594 0.5259443880 –8.0356174416 H6 –0.2560068873 3.0123736343 –8.1252459797 H7 0.2819511805 –2.6420634789 5.9793310759 H8 0.3838560298 –3.8296237156 3.7532110711 H9 0.2819511805 –2.6420634789 –5.9793310759 H10 0.3838560298 –3.8296237156 –3.7532110711 H11 –0.3378598434 3.3099217750 –3.8180129970 H12 –0.4148014510 4.3588903003 –6.0870107319 H13 –0.3378598434 3.3099217750 3.8180129970 H14 –0.4148014510 4.3588903003 6.0870107319
Calculated positional parameters for 118:
Atom x y z
C1 1.8190162296 3.8598612631 0.4107420789 C2 2.9861222185 1.4100948500 0.3891866579 C3 3.1792765798 3.8018811710 0.5447765123 C4 0.9531544545 2.7063972912 0.2561901512 C5 1.5358910221 1.4780120245 0.2467054493 C6 3.7996027622 2.5013676200 0.5330518469 C7 –0.4534051883 –1.9831390690 –0.1325966739 C8 –1.7081555290 –4.4214111753 –0.3784292891 C9 0.2985986114 –3.1919972798 –0.1223174403 C10 –1.8230707838 –1.9710546873 –0.2636675256 C11 –2.4432586562 –3.2382107108 –0.3885740304 C12 –0.2985215803 –4.4272022193 –0.2429339644 C13 2.9275046915 –0.0748805783 0.3010819304 C14 1.5729570327 –2.4162087475 0.0429344977 C15 1.4823615908 –0.0128786502 0.1600466305 C16 3.6516394497 –1.2394630083 0.3110514742

205
C17 2.9350209135 –2.4789164099 0.1743188961 C18 0.8080584911 –1.1864685740 0.0316059838 C19 –1.2024564763 6.1682212563 0.2357174281 C20 –2.0687213758 5.0478683123 0.0759256651 C21 –1.6229975685 3.7247746269 0.0454662351 C22 –0.2370854226 3.6257348674 0.1858329940 C23 0.6227753556 4.7516741300 0.3405601958 C24 0.1804480264 6.0721950126 0.3735354789 C25 –4.1693958147 8.6905345016 0.0744322141 C26 –5.0136927439 7.6254009146 –0.0854891204 C27 –4.5826110276 6.2322483537 –0.1265599802 C28 –3.2541006355 5.9737516121 0.0041197380 C29 –2.3628973581 7.1060150237 0.1706153052 C30 –2.7621201680 8.4136572039 0.2096391312 C31 –8.7383363647 6.6022032447 –0.5789399064 C32 –8.3350045685 5.2937706700 –0.6196979169 C33 –6.9603801414 4.8575675041 –0.4882169238 C34 –6.0027242231 5.8068460205 –0.3144150483 C35 –6.4267421123 7.1964484932 –0.2717657407 C36 –7.7287305691 7.6096928494 –0.3944110110 C37 –9.4324737913 1.6042638852 –1.0083269543 C38 –8.1101128036 1.1847852530 –0.8795304801 C39 –7.0499156971 2.1045267383 –0.6887072676 C40 –7.4074592423 3.4324358809 –0.6384727493 C41 –8.7587508832 3.8613023749 –0.7688141481 C42 –9.7942823627 2.9725113988 –0.9552448245 H1 4.8772122391 2.4126435163 0.6366687650 H2 0.2569464199 –5.3606958918 –0.2366272956 H3 –2.2330051075 –5.3682840752 –0.4772296660 H4 4.7325920102 –1.2484197860 0.4188601290 H5 3.4748539907 –3.4216415461 0.1784117829 H6 3.7892774471 4.6935825324 0.6574908467 H7 –2.2802458238 2.8691443545 –0.0743855838 H8 0.8376110624 6.9278431352 0.4928846520 H9 –4.5261782597 9.7161687859 0.1001651794 H10 –2.0579341198 9.2309542653 0.3372590454 H11 –9.7805417468 6.8914112358 –0.6817950574 H12 –8.0103321362 8.6582370103 –0.3577960225 H13 –2.4119828374 –1.0582505996 –0.2729888745 H14 –3.5238427429 –3.2895906764 –0.4950063144 H15 –10.2099345277 0.8585511665 –1.1540160999 H16 –7.8864761415 0.1222626782 –0.9274970797 H17 –6.0240868339 1.7608364492 –0.5906470725 H18 –10.8313013993 3.2792815595 –1.0580793730

206
Calculated positional parameters for 119:
Atom x y z
C1 2.2552637652 3.7124088815 0.3938557760 C2 2.4476658018 1.0030523814 0.2877885071 C3 3.5008783042 3.1592088607 0.4837226979 C4 1.0266715358 2.9509936287 0.2445382669 C5 1.1189357743 1.5992069443 0.1884969107 C6 3.6030708977 1.7195780054 0.4279026405 C7 –2.0046956608 –0.8750344317 –0.2222010830 C8 –4.0662839769 –2.8416810904 –0.5049255426 C9 –1.7327445313 –2.3093106852 –0.2481139274 C10 –3.2952365837 –0.4744704022 –0.3416644409 C11 –4.3159187962 –1.4991180704 –0.4828974113 C12 –2.6934915976 –3.2723111436 –0.3805838689 C13 1.8420541908 –0.3526478711 0.1747772997 C14 –0.2780761471 –2.0317012945 –0.0941353372 C15 0.5227947524 0.2342873127 0.0727169445 C16 2.0916416121 –1.7053437115 0.1463586018 C17 0.9760908421 –2.5886752900 0.0055763745 C18 –0.5421677446 –0.6087302195 –0.0658431611 C19 0.2383624007 6.9433578405 0.3876919351 C20 –0.9681210043 6.1998258069 0.2436395641 C21 –1.0198742096 4.8132603671 0.1639889413 C22 0.2489829904 4.2321429428 0.2404669370 C23 1.4519263332 4.9747016365 0.3850649288 C24 1.5010163631 6.3701511976 0.4652680945 C25 –6.9907121059 1.2039622746 –0.6861287107 C26 –6.7221036406 –0.1587877241 –0.6977659034 C27 –5.3565235230 –0.4274630615 –0.5579457923 C28 –4.3610995898 0.5772697399 –0.4202618243 C29 –4.6361931373 1.9476719018 –0.4106581709 C30 –5.9936706527 2.2122057298 –0.5489630054 C31 –9.7157297145 3.9491830888 –0.9091656219 C32 –8.7449351330 4.9323516729 –0.7747350378 C33 –7.3667479683 4.6124431561 –0.6320591210 C34 –7.0594558508 3.2751845821 –0.6352827827 C35 –8.0551729256 2.2676666795 –0.7727159135 C36 –9.3877194664 2.5652628275 –0.9114355535 C37 –1.8069112522 10.2336850659 0.4034968563 C38 –2.9833468417 9.5105983152 0.2625226336 C39 –2.9868567398 8.0908600691 0.1808690571 C40 –1.7580817058 7.4841145956 0.2492697727 C41 –0.5524179941 8.2260538418 0.3932496948 C42 –0.5381001235 9.5955705595 0.4737364650

207
H1 4.5773485994 1.2444354319 0.4988816701 H2 –2.4560688904 –4.3321819008 –0.3959757186 H3 –4.8572584751 –3.5782731271 –0.6136483985 H4 3.0974625622 –2.1078100951 0.2273289374 H5 1.1331557460 –3.6632962377 –0.0207084630 H6 4.3979020619 3.7616285597 0.5963163818 H7 –1.9386624903 4.2449156005 0.0572982838 H8 –7.4819154654 –0.9266102425 –0.8041968758 H9 –3.8753816521 2.7154986642 –0.3089243193 H10 –10.7563162648 4.2444875733 –1.0158738285 H11 –9.0454198286 5.9769692153 –0.7787567893 H12 –6.6217872574 5.3966057947 –0.5289618862 H13 –10.1610336747 1.8099833417 –1.0179097479 H14 2.4195771503 6.9377678728 0.5771211467 H15 –1.8554267547 11.3179704282 0.4621216157 H16 –3.9294093325 10.0435140475 0.2133905572 H17 –3.9185626230 7.5427962562 0.0716937797 H18 0.3721685341 10.1780541437 0.5846218227
Calculated positional parameters for 120:
Atom x y z
C1 2.4432614782 3.6747979008 1.0437363054 C2 2.5539152919 0.9955562378 0.6740020788 C3 3.6313725584 3.0498572809 1.2977469025 C4 1.2324795062 3.0090995997 0.5927874592 C5 1.2782992480 1.6600268343 0.4300146268 C6 3.6918852194 1.6249065148 1.0959829787 C7 –1.7996593675 –0.6697127913 –0.7231820603 C8 –3.7840086704 –2.5553293523 –1.5726929853 C9 –1.5014047215 –2.0708363683 –1.0006484283 C10 –3.0916596325 –0.2654363044 –0.8459614290 C11 –4.0662328115 –1.2486892956 –1.2878224429 C12 –2.4205144125 –2.9946170100 –1.4152424341 C13 1.9607762955 –0.3003994928 0.2580004576 C14 –0.0857116669 –1.8472715647 –0.6111311663 C15 0.6791967718 0.3405855467 0.0424682539 C16 2.2151723080 –1.6341148977 0.0407879229 C17 1.1369617141 –2.4481125984 –0.4260502492 C18 –0.3614353619 –0.4467974513 –0.3624485146 C19 0.7438174657 7.0564429600 0.7053296124 C20 –0.4431972986 6.4244454392 0.2301337556 C21 –0.5937210594 5.0411957153 0.1084834471 C22 0.5529702928 4.3479264632 0.4971227672

208
C23 1.7438095351 4.9874021425 0.9532943133 C24 1.8930578478 6.3655677709 1.0831528160 C25 –6.8605495321 1.2928943491 –0.8533031761 C26 –6.5131859885 0.0031703685 –1.2506345157 C27 –5.1430324222 –0.2258799341 –1.1539953463 C28 –4.1977555640 0.7450694568 –0.7083201502 C29 –4.5481534749 2.0331763350 –0.3015542732 C30 –5.9228959792 2.2633580558 –0.3918179548 C31 –9.8682150094 3.6961513641 –0.2371850793 C32 –8.9787769280 4.6250525850 0.2179659296 C33 –7.5355863597 4.4212274501 0.2808106698 C34 –7.0426028310 3.2326060368 –0.1453174640 C35 –7.9916616684 2.2393568602 –0.6255832067 C36 –9.3425353539 2.4244783854 –0.6842215418 C37 –0.7509835364 10.6107022144 0.1234411590 C38 –1.8826967055 10.0112792972 –0.3473762824 C39 –2.0763964080 8.5676513477 –0.4251727674 C40 –1.0705642046 7.7673992540 0.0026862818 C41 0.1398034533 8.4057122915 0.4977641892 C42 0.3277457815 9.7557318652 0.5709271917 C43 –8.5119767863 8.1586380376 1.7135433064 C44 –7.1318035422 7.9633608791 1.7768446692 C45 –6.5268374696 6.7591154481 1.3465919841 C46 –7.3825656653 5.7951359544 0.8591185662 C47 –8.7911929265 5.9937236588 0.7942674913 C48 –9.3862135518 7.1649552770 1.2148320997 C49 –5.5676791891 9.1410867878 –1.9766725842 C50 –4.5789004437 8.2320341569 –1.5343040316 C51 –3.4305815274 8.7946421954 –1.0212205324 C52 –3.2434517017 10.2042999459 –0.9421238260 C53 –4.2035219025 11.0948080080 –1.3751228471 C54 –5.3852028730 10.5219651137 –1.8999433902 H1 4.6222887585 1.0902542155 1.2645157446 H2 –2.1566684833 –4.0302921944 –1.6091267812 H3 –4.5495522948 –3.2586091084 –1.8881572996 H4 3.1959827869 –2.0692247904 0.2099942637 H5 1.2936930420 –3.5058594171 –0.6179655238 H6 4.5151256016 3.5928995736 1.6204339424 H7 –1.4917362764 4.5621426035 –0.2696200698 H8 –7.2297431047 –0.7427408421 –1.5801460529 H9 –3.8357218346 2.7663746633 0.0641190953 H10 –10.9386529062 3.8789957362 –0.2661771372 H11 –10.0205939684 1.6559319775 –1.0443881067 H12 2.8065983999 6.8463727506 1.4187323889 H13 –0.6358283960 11.6902391567 0.1616425594 H14 1.2494330890 10.1969200229 0.9393650140

209
H15 –8.9262656171 9.1028990679 2.0574064346 H16 –6.5026057636 8.7600908447 2.1640605392 H17 –5.4502455926 6.6267927668 1.4038433734 H18 –6.4966804687 8.7526151904 –2.3849390633 H19 –4.7386180515 7.1598499893 –1.6028633123 H20 –4.0795041400 12.1730221799 –1.3265645144 H21 –6.1768335484 11.1770841180 –2.2547385692 H22 –10.4585760540 7.3343169940 1.1745790579
6.3 Experiments Related to Chapter 3
1,2-Dibromo-4,5-diiodobenzene (155e):
IBr
IBr
This compound was prepared by using a modified literature procedure.78 Periodic
acid (2.96 g, 13.0 mmol) was dissolved in concentrated H2SO4 (12 mL). The mixture was
cooled to 0 °C, and powdered KI (6.46 g, 39.0 mmol) was slowly added over 15 min.
After the addition of KI was complete, 1,2-dibromobenzene (6.00 g, 25.0 mmol) was
added dropwise. The reaction mixture was stirred at 23 °C for 1 h and then poured onto
crushed ice. The dark precipitate was recrystallized four times from benzene to yield 2.06
g (17%) of 155e as white needles, mp 173–175 °C. IR (CS2): ~ν = 2925, 1408, 1282,
1005, 877 cm–1. MS (EI, 70 eV) m/z (rel intensity) 488 (M+, 100), 361 (32), 234 (17), 153
(8), 74 (20). 1H NMR (400 MHz, CDCl3): 8.03 (s, 2H). 13C NMR (100 MHz, CDCl3):
142.5, 125.4, 106.9. HR-MS Calcd for C6H2Br2I2: 487.6592. Found 487.6596. Anal.
Calcd for C6H2Br2I2: C, 14.78; H, 0.41. Found C, 14.55; H, 0.43.

210
1,4-Dibromo-2,3-diiodobenzene (155f):
I
I
Br
Br
The preparation of 3,6-dibromoisatine used a slightly modified literature
procedure:167 chloral hydrate (9.93 g, 60.0 mmol), 2,5-dibromoaniline (12.6 g, 50.0
mmol), hydroxylamine hydrochloride (5.21 g, 75.0 mmol), and sodium sulfate (60.0 g)
were suspended in a mixture of water (300 mL) and ethanol (300 mL). The mixture was
stirred and kept at reflux for 12 h; after that time, it was concentrated by evaporation of
the ethanol and poured onto crushed ice, which caused precipitation of a white solid.
After 5 h at 0 °C, the suspension was filtered, and the crystals were air-dried to yield 13.5
g (84%) of crude 2,5-dibromoisonitrosoacetanilide. This isonitrosoacetanilide was
cyclised by heating at 100 °C in 86% sulfuric acid for 15 min. The resulting dark red
suspension was poured onto crushed ice to yield 5.98 g (47 %) of 3,6-dibromoisatine as
bright orange crystals, which were subsequently subjected to basic hydrolysis in aq.
H2O2168 to yield 2.72 g (47%) of off-white crystals of 3,6-dibromoanthranilic acid.
Finally, 3,6-dibromoanthranilic acid was converted to 1,4-dibromo-2,3-diiodobenzene by
employing the aprotic diazotization procedure of Nakayama.165 After column
chromatography (hexanes) the product was obtained in yield of 2.61 g (58%), as white
crystals, mp 97–99 °C. Crystals of 155f are stable for several months; however, its
solutions in CHCl3 and hydrocarbon solvents appear to decompose within days with the
loss of iodine. IR (CHCl3): ~ν = 2920, 1396, 1150, 1002, 810 cm–1. MS (EI, 70 eV) m/z
(rel intensity) 488 (M+, 100), 361 (29), 234 (21), 153 (18), 74 (24). 1H NMR (400 MHz,
CDCl3) δ 7.49 (s, 2H). 13C NMR (100 MHz, CDCl3) δ 132.8, 127.8, 117.4. HR-MS

211
Calcd for C6H2Br2I2: 487.6592. Found 487.6596. Anal. Calcd for C6H2Br2I2: C, 14.78; H,
0.41. Found C, 14.74; H, 0.04.
1,4-Dichloro-2,3-diiodobenzene (155g):165
I
I
Cl
Cl
A solution of iodine (620 mg, 2.43 mmol) and isoamyl nitrite (396 µL, 2.92
mmol) in 1,2-dichloroethane (50 mL) was brought to reflux. 3,6-Dichloroanthranylic acid
(500 mg, 2.43 mmol) was added as a dioxane solution (vide infra, 20 mL) over a period
of 20 min and mixture left at reflux for 3 h. After cooling, CH2Cl2 was added and the
resulting solution washed with two portions of aq. Na2S2O3, followed by brine. The
solution was subsequently dried over MgSO4; removal of the solvents in vacuo gave the
crude material, which was purified by column chromatography on silica (hexanes) to give
155g as a white powder, mp 96–97 °C (292 mg, 30%). MS (EI, 70 eV) m/z (rel intensity)
398 (M+, 100), 271 (21), 144 (18), 74 (11). 1H NMR (500 MHz, CDCl3) δ 7.42 (s, 2H).
13C NMR (125 MHz, CDCl3) δ 136.83, 129.10, 114.80. HR-MS Calcd for C6H2Cl2I2:
397.7623. Found: 397.7626. Anal. Calcd for C6H2Cl2I2: C, 18.07; H, 0.51. Found: C,
17.91; H, 0.45.

212
1,2-Diiodo-3,5-dimethylbenzene (160):
I
I
3,5-Dimethylanthranilic acid (165 mg, 1.00 mmol) was converted to a colorless
oil of 1,2-diiodo-3,5-dimethylbenzene according to the literature procedure.165 However,
we noticed that the low yield of this reaction–111 mg (31%)–was caused by significant
contamination of product by 1-chloro-2-iodo-3,5-dimethylbenzene and 2-chloro-1-iodo-
3,5-dimethylbenzene (by GC/MS). Separation of these side-products was achieved by
Kugelrohr distillation. We also found that the reaction could be performed successfully in
dioxane; this avoids the formation of chlorinated by-products, but does not increase the
yield significantly (32%). Data for 1,2-diiodo-3,5-dimethylbenzene: IR: reported
previously.217 MS (EI, 70 eV) m/z (rel intensity) 358 (M+, 100), 231 (16), 104 (17). 1H
NMR (400 MHz, CDCl3) δ 7.55 (s, 1H), 6.98 (s, 1H), 2.54 (s, 3H), 2.19 (s, 3H). 13C
NMR (100 MHz, CDCl3) δ 144.0, 139.6, 137.7, 129.8, 110.2, 109.7, 32.5, 20.4. HR-MS
Calcd for C8H8I2: 357.8716. Found 357.8721. Anal. Calcd for C8H8I2: C, 26.84; H, 2.25.
Found C, 27.10; H, 2.33.
General procedure for propynylations (method A):
To a 150 mL Schlenk flask, equipped with a Teflon-coated magnetic stirring bar,
aryl iodide (1.67 mmol, in the case of diiodides), [Pd(PPh3)2Cl2] (177 mg, 0.25 mmol),
CuI (32 mg, 0.17 mmol), and triethylamine (7.50 mL) were added. The flask was then
evacuated and filled with propyne gas to 1.5 atm (approx. 10 mmol, 3 equiv) of pressure.
Depending on the system, the reaction mixture was stirred for 22–96 h, at either room or

213
elevated temperature (Table 3.1). After that time, the reaction mixture was diluted with
ether, washed with two portions of aq. NH4Cl, and dried over MgSO4. Solvent was
removed in vacuo and the resulting crude product purified by Kugelrohr distillation or
sublimation (unless indicated otherwise). The following compounds were prepared using
this method:
1,2-Di(prop-1-ynyl)benzene (154a):
Starting from 155a, product was obtained as a pale yellow oil (distilled at 120
°C/2 Torr), 244 mg (95%). MS (EI, 70 eV) m/z (rel intensity) 154 (M+, 100), 152 (84), 76
(12). 1H NMR (400 MHz, CDCl3) δ 7.38 (dd, 2H, 4J1 = 3.2 Hz, 3J2 = 5.7 Hz), 7.18 (dd,
2H, 4J1 = 3.2 Hz, 3J2 = 5.7 Hz), 2.12 (s, 6H). Spectral data are in good agreement with
those reported previously.218
1,2-Dimethyl-4,5-di(prop-1-ynyl)benzene (154b):
Starting from 155b,164a product was obtained as yellow crystals (sublimed at 150
°C/2 Torr), mp 92–93 °C, yield 174 mg (57%). IR (CHCl3): ~ν = 2920, 2261, 1494, 887
cm–1. MS (EI, 70 eV) m/z (rel intensity) 182 (M+, 100), 165 (51), 152 (42). 1H NMR (400
MHz, CDCl3) δ 7.15 (s, 2H), 2.17 (s, 6H), 2.10 (s, 6H). 13C NMR (100 MHz, CDCl3) δ

214
136.1, 133.0, 123.4, 88.3, 78.7, 19.4, 4.7. HR-MS Calcd for C14H14: 182.1096. Found
182.1096. Anal. Calcd for C14H14: C, 92.26; H, 7.74. Found C, 92.02; H, 7.47.
1,2-Dimethoxy-4,5-di(prop-1-ynyl)benzene (154c):
O
O
Starting from 155c,164a product was obtained as yellow crystals (sublimed at 190
°C/2 Torr), mp 164–166 °C (dec), yield 121 mg (34%). The yield was probably lowered
due to some decomposition during sublimation. Purification by column chromatography
(hexanes/ethyl acetate) provided the material in 81% yield (289 mg). IR (CHCl3): ~ν =
2919, 2170, 1223, 1155, 862 cm–1. MS (EI, 70 eV) m/z (rel intensity) 214 (M+, 100), 128
(25). 1H NMR (400 MHz, CDCl3) δ 6.74 (s, 2H), 3.73 (s, 6H), 1.99 (s, 6H). 13C NMR
(100 MHz, CDCl3) δ 148.3, 118.9, 114.2, 87.8, 78.5, 55.8, 4.5. HR-MS Calcd for
C14H14O2: 214.0994. Found 214.0994. Anal. Calcd for C14H14O2: C, 78.48; H, 6.59.
Found C, 78.21; H, 6.62.
1,2,3,4-Tetramethyl-5,6-di(prop-1-ynyl)benzene (154d):
Starting from 155d,164a product was obtained as a very thick yellow oil (distilled
at 200 °C/2 Torr), yield 35.0 mg (10%). The yield was probably lowered due to some
decomposition during sublimation. Purification by column chromatography

215
(hexanes/ethyl acetate) provided the material in 91% yield (319 mg). IR (CHCl3): ~ν =
2917, 1704, 1214, 773 cm–1. MS (EI, 70 eV) m/z (rel intensity) 210 (M+, 100), 195 (27),
179 (30), 165 (42). 1H NMR (400 MHz, CDCl3) δ 2.39 (s, 6H), 2.19 (s, 6H), 2.16 (s, 6H).
13C NMR (100 MHz, CDCl3) δ 135.5, 134.5, 123.7, 91.7, 78.9, 18.6, 16.7, 4.8. HR-MS
Calcd for C16H18: 210.1409. Found 210.1415.
1,4-Dichloro-2,3-di(prop-1-ynyl)benzene (154g):
Cl
Cl
Starting from 155g, the product was obtained as a yellow oil (252 mg, 68%),
which solidified into off-yellow crystals, mp 58–59 °C, upon standing. IR (NaCl film): ~ν
= 2917, 2227, 1542, 1439, 1412, 1376, 1264, 1161, 993, 810, 762, 650 cm–1. MS (EI, 70
eV) m/z (rel intensity) 222 (M+, 100), 186 (33), 152 (94), 75 (18). 1H NMR (400 MHz,
CDCl3) δ 7.19 (s, 2H), 2.19 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 134.22, 128.39,
127.47, 96.97, 75.57, 4.89. HR-MS Calcd for C12H8Cl2: 223.9974. Found: 223.9968.
3,5-Dimethyl-1,2-di(prop-1-ynyl)benzene (165):
Starting from 162, the product was obtained as a yellow oil (distilled at 220 °C/4
Torr), which solidified upon standing (yellow crystals, mp 45–46 °C), 231 mg (76%). IR
(NaCl film): ~ν = 2920, 2246, 1455, 859 cm–1. MS (EI, 70 eV) m/z (rel intensity) 182

216
(M+, 100), 165 (57), 152 (47). 1H NMR (400 MHz, CDCl3) δ 7.06 (s, 1H), 6.91 (s, 1H),
2.36 (s, 3H), 2.24 (s, 3H), 2.16 (s, 3H), 2.11 (s, 3H). 13C NMR (100 MHz, CDCl3) δ
140.2, 136.7, 129.9, 129.6, 126.1, 123.0, 92.8, 88.5, 79.2, 77.4, 21.1, 21.0, 4.8, 4.7. HR-
MS Calcd for C14H14: 182.1096. Found 182.1100.
1,3-Di(prop-1-ynyl)benzene (166a):
Starting from 163a, the product was obtained as a pale yellow oil (distilled at 120
°C/2 Torr), 239 mg (93%). MS (EI, 70 eV) m/z (rel intensity) 154 (M+, 100), 152 (68),
115 (16), 76 (12). 1H NMR (400 MHz, CDCl3) δ 7.40 (s, 1H), 7.27 (d, 2H, 3J = 7.3 Hz),
7.17 (t, 1H, 3J = 7.4 Hz), 2.03 (s, 6H). Spectral data are in good agreement with those
reported previously.219
1,2,3,4-Tetra(prop-1-ynyl)benzene (166b):
Starting from 163b,164c the product was obtained as brownish crystals (sublimed
at 220 °C/1 Torr), mp 168–171 °C (dec), 148 mg (77%). IR (KBr): ~ν = 2914, 2238,
1485, 1396, 902 cm–1. MS (EI, 70 eV) m/z (rel intensity) 230 (M+, 100), 213 (20), 202
(20), 189 (11), 101 (6). 1H NMR (400 MHz, CDCl3) δ 7.36 (s, 2H), 2.08 (s, 12H). 13C

217
NMR (100 MHz, CDCl3) δ 135.4, 125.1, 91.0, 77.9, 4.7. HR-MS Calcd for C18H14:
230.1096. Found 230.1094.
General procedure for microwave-assisted propynylations (method B):172
A heavy-walled Smith process vial was charged with a magnetic stirring bar,
triethylamine (0.9 mL), DMF (0.1 mL), [Pd(PPh3)2Cl2] (19.6 mg, 0.028 mmol), CuI (5.4
mg, 0.028 mmol), and the respective dibromodiiodobenzene (55.0 mg, 0.113 mmol). The
vial was sealed, evacuated, and filled with propyne through a Teflon septum up to 2.5
atm pressure. It was then irradiated in the microwave cavity. It is of crucial importance to
stop these reactions immediately after irradiation by filtering through a short pad of silica
gel (hexanes/ethyl acetate) to remove the catalyst. So-obtained crude products were
further purified by column chromatography on silica gel (hexanes). The following
compounds were prepared using this method:
1,2-Dibromo-4,5-di(prop-1-ynyl)benzene (154e):
Br
Br
Starting from 155e, the product was obtained as a thick yellow oil, which slowly
crystallizes upon standing (yellow crystals, mp 67–69 °C), 25.1 mg (71%). IR (CHCl3):
~ν = 2957, 2363, 2321, 1461, 775 cm–1. MS (EI, 70 eV) m/z (rel intensity) 312 (M+, 100),
233 (11), 231 (11), 152 (80), 76 (18). 1H NMR (400 MHz, CDCl3) δ 7.60 (s, 2H), 2.09 (s,
6H). 13C NMR (100 MHz, CDCl3) δ 136.3, 126.5, 123.4, 91.9, 76.8, 4.7. HR-MS Calcd
for C12H8Br2: 311.8972. Found 311.8982.

218
1,4-Dibromo-2,3-di(prop-1-ynyl)benzene (154f):
Br
Br
Starting from 155f, the product was obtained as a viscous yellow oil, 21.2 mg
(60%). IR (CHCl3): ~ν = 2952, 1437, 1217, 757 cm–1. MS (EI, 70 eV) m/z (rel intensity)
312 (M+, 90), 233 (11), 231 (11), 152 (100), 76 (18). 1H NMR (400 MHz, CDCl3): 7.29
(s, 2H), 2.19 (s, 6H). 13C NMR (100 MHz, CDCl3): 131.8, 131.7, 124.3, 96.1, 77.7, 4.9.
HR-MS Calcd for C12H8Br2: 311.8972. Found 311.8975.
1,3-Dibromo-4,6-di(prop-1-ynyl)benzene (166c):
BrBr
Starting from 57,78 the product was obtained as pale yellow crystals, mp 78–80
°C, 22.5 mg (64%). IR (CHCl3): ~ν = 2922, 2242, 1454, 1059, 896 cm–1. MS (EI, 70 eV)
m/z (rel intensity) 312 (M+, 100), 233 (11), 231 (11), 152 (75), 76 (22). 1H NMR (400
MHz, CDCl3) δ 7.72 (s, 1H), 7.44 (s, 1H), 2.08 (s, 6H). 13C NMR (100 MHz, CDCl3) δ
137.0, 135.3, 125.2, 124.4, 92.3, 77.2, 4.9. HR-MS Calcd for C12H8Br2: 311.8972. Found
311.8976.
Utilizing a somewhat modified general propynylation procedure, the following
compounds were also prepared:

219
Hexa(prop-1-ynyl)benzene (167) and penta(prop-1-ynyl)benzene:
To a 200 mL Schlenk flask, equipped with a Teflon-coated magnetic stirring bar,
hexaiodobenzene (164, 528 mg, 0.63 mmol), [Pd(PPh3)2Cl2] (66 mg, 0.094 mmol), CuI
(36 mg, 0.189 mmol), and triethylamine (7.50 mL) were added. The flask was evacuated
and filled with propyne gas up to 1.5 atm (approx. 7.60 mmol, 12 equiv) of pressure. The
reaction mixture was stirred for 60 h at 90 °C. After that time, it was diluted with ether,
washed with two portions of aq. NH4Cl, and dried over MgSO4. Solvent was removed in
vacuo and the resulting crude product chromatographed on silica (hexane/ethyl acetate).
The first fraction contained penta(prop-1-ynyl)benzene (54 mg, 32%) and the second
hexa(prop-1-ynyl)benzene (167, 55 mg, 28%), both as dark red oily solids. The
separation of the two compounds was not complete; repeated efforts gave no
improvement. Therefore, materials were characterized only partly, and 167 was used as
such in further experiments.
Spectral data for penta(prop-1-ynyl)benzene: MS (EI, 70 eV) m/z (rel intensity)
268 (M+, 100), 250 (34), 239 (10), 226 (13). 1H NMR (400 MHz, CDCl3) δ 7.28 (s, 1H),
2.16 (br s, 9H), 2.09 (s, 6H). HR-MS Calcd for C21H16: 268.1252. Found 268.1258.
Spectral data for 167: MS (EI, 70 eV) m/z (rel intensity) 306 (M+, 100), 289 (40),
281 (34), 263 (29), 144 (13), 96 (8). 1H NMR (400 MHz, CDCl3) δ 2.15 (s, 18H). 13C

220
NMR (100 MHz, CDCl3) δ 127.80, 94.72, 77.71, 5.04. HR-MS Calcd for C24H18:
306.1409. Found 306.1411.
General procedure for [(Me3CO)3W≡CCMe3]-mediated alkyne metatheses:
A 25 mL Schlenk flask was charged, under an atmosphere of nitrogen, with
propynylated benzene (0.20–0.35 mmol), [(Me3CO)3W≡CCMe3] (20–40 mol %), and
toluene (20 mL). The solution was stirred at 80 °C for 8–96 h (Table 3.2). After the
reaction was complete, solvent was removed in vacuo and the residue subjected to flash
chromatography on silica (hexane/ethyl acetate). Utilizing this procedure, the following
compounds were prepared:
5,6,11,12,17,18-Hexadehydrotribenzo[a,e,i]cyclododecene (153a):
The reaction was carried out with 54.0 mg (0.35 mmol) of 154a; isolated yield of
153a was 18.7 mg (54%) as pale yellow crystals showing green fluorescence, mp 209 °C
(lit. 210–211 °C).163 MS (EI, 70 eV) m/z (rel intensity) 300 (M+, 100), 149 (18). 1H NMR
(400 MHz, CDCl3) δ 7.33 (dd, 6H, 4J1 = 3.3 Hz, 3J2 = 5.8 Hz), 7.18 (dd, 6H, 4J1 = 3.3 Hz,
3J2 = 5.8 Hz). Spectral data are in good agreement with those reported previously.87b,163

221
5,6,11,12,17,18-Hexadehydro-2,3,8,9,14,15-hexamethyltribenzo[a,e,i]-cyclododecene
(153b):
The reaction was performed with 49.0 mg (0.27 mmol) of 154b; isolated yield of
153b was 9.2 mg (27%) as pale yellow crystals showing green fluorescence, mp 334–336
°C (dec). The extremely low solubility of this compound prevented analysis by 13C NMR
spectroscopy. MS (EI, 70 eV) m/z (rel intensity) 386 ([M+2H]+, 7), 385 ([M+H]+, 33),
384 (M+, 100), 192 (M2+, 8). 1H NMR (400 MHz, CDCl3) δ 7.09 (s, 6H), 2.19 (s, 18H).
HR-MS Calcd for C30H24: 384.1878. Found 384.1884. Spectral data are in good
agreement with those reported previously.86d
5,6,11,12,17,18-Hexadehydro-2,3,8,9,14,15-hexamethoxytribenzo[a,e,i]-
cyclododecene (153c):
O
O
O
O
O
O

222
The reaction was run with 22.5 mg (0.105 mmol) of 154c. Column
chromatography (hexane/ethyl acetate) yielded 13.6 mg (60%) of starting material in the
first fraction, followed by 153c, 4.7 mg (28%), as yellow crystals showing green
fluorescence. We found this compound to be unstable, both as a solid and in solution–it
completely decomposed within 24 h (previous reports158a,d,220 do not indicate similar
problems). MS (EI, 70 eV) m/z (rel intensity) 482 ([M+2H]+, 8), 481 ([M+H]+, 33), 480
(M+, 100), 240 (M2+, 18). 1H NMR (400 MHz, CDCl3) δ 6.93 (s, 6H), 4.00 (s, 18H).
Performing this reaction for a shorter time (96 h), gave 3.1 mg (16%) of the metathesis
dimer as the major product:
1,1'-(1,2-Ethynediyl)bis[4,5-dimethoxy-2-(prop-1-ynyl)]benzene:
O
O O
O
Isolated as a brownish solid, mp 250–253 °C (dec), 3.1 mg (16%). IR (CHCl3): ~ν
= 2928, 2360, 1464, 1274, 1154 cm–1. MS (EI, 70 eV) m/z (rel intensity) 375 ([M+H]+,
18), 374 (M+, 100), 187 (M2+, 8). 1H NMR (400 MHz, CDCl3) δ 6.98 (s, 2H), 6.90 (s,
2H), 3.94 (s, 6H), 3.88 (s, 6H), 2.12 (s, 6H). HR-MS Calcd for C24H22O4: 374.1518.
Found 374.1519.

223
5,6,11,12,17,18-Hexadehydro-2,3,8,9,14,15-hexabromotribenzo[a,e,i]-cyclododecene
(153e):
Br
Br
Br
Br
Br
Br
The reaction was executed with 33.0 mg (0.106 mmol) of 154e to give 153e, 3.3
mg (12%), as brown crystals showing green fluorescence. MS (EI, 70 eV) m/z (rel
intensity) 774 (M+, 100), 695 (29), 614 (19). 1H NMR (400 MHz, CDCl3) δ 7.56 (s, 6H).
The material decomposed within several hours.
Utilizing a somewhat modified general metathesis procedure, the following
compounds were also prepared:
5,6,12,13,18,19,25,26-octadehydro-7,11:20,24-dimethenodibenzo[a,l]-cyclodocosene
(159):
A 25 mL Schlenk tube was charged with a magnetic stirring bar, 154a (52.6 mg,
0.34 mmol), 164a (46.5 mg, 0.30 mmol), [(Me3CO)3W≡CCMe3] (44.0 mg, 0.092 mmol),

224
and dry toluene (25 mL). The mixture was heated at 80 °C for 84 h. After that time,
solvent was removed in vacuo and the residue purified by column chromatography
(hexane/ethyl acetate) to yield 24.6 mg of a mixture of starting materials as the first
fraction, 5.4 mg (16%) of 153a and, as a third fraction, 5.8 mg (19%) of 159 as a
yellowish solid showing green fluorescence, mp 310–315 °C (dec). UV-VIS (CH2Cl2):
λmax (logε) = 265 (4.03), 272 (4.05), 278 (4.11), 280 (4.10), 317 (3.62), 341 (3.26) nm. IR
(CHCl3): ~ν = 2920, 2219, 1468, 1212, 892 cm–1. MS (EI, 70 eV) m/z (rel intensity) 401
([M+H]+, 31), 400 (M+, 100), 199 (11). 1H NMR (400 MHz, CDCl3) δ 8.05 (broad t, 4J =
1.5 Hz, 2H), 7.59 (dd, 3J1 = 5.7 Hz, 4J2 = 3.3 Hz, 4H), 7.54 (dd, 3J1 = 7.9 Hz, 4J2 = 1.6
Hz, 4H), 7.38 (t, 3J = 8.0 Hz, 2H), 7.32 (dd, 3J1 = 5.8 Hz, 4J2 = 3.4 Hz, 4H). HR-MS
Calcd for C32H16: 400.1252. Found 400.1248.
5,6,11,12,14,15,20,21,26,27,29,30-Dodecadehydrotetrabenzo[e,e',i,i']benzo-[1,2-a:4,5-
a']dicyclododecene (160):
A 25 mL Schlenk tube was charged with a magnetic stirring bar, 154a (116 mg,
0.76 mmol), 164b (35.0 mg, 0.15 mmol), [(Me3CO)3W≡CCMe3] (36.0 mg, 0.076 mmol),
and dry toluene (25 mL). The mixture was heated at 80 °C for 60 h. After that time, the
solvent was removed in vacuo and the residue purified by washing with hexanes, ether,
and acetone to yield 5.0 mg (6%) of 160 as an extremely insoluble yellow powder,

225
showing green fluorescence, mp 348–351 °C [dec, lit. mp 350 °C (dec)].163 Ether and
acetone washes contained tribenzocyclyne 153a as the main component, resulting from
the homometathesis of the excess 154a. The compound was too insoluble to provide
meaningful 13C NMR information. MS (EI, 70 eV) m/z (rel intensity) 524 ([M+2H]+, 10),
523 ([M+H]+, 42), 522 (M+, 100), 261 (M2+, 25). 1H NMR (400 MHz, CDCl3) δ 7.44 (dd,
3J1 = 6.0 Hz, 4J2 = 3.6 Hz, 8H), 7.34 (s, 2H), 7.19 (dd, 3J1 = 6.0 Hz, 4J2 = 3.6 Hz, 8H).
HR-MS Calcd for C42H18: 522.1408. Found 522.1428. Spectral data are in good
agreement with those reported previously.163
1,1'-(1,2-Ethynediyl)bis[3,5-dimethyl-2-(prop-1-ynyl)]benzene (168):
A 10 mL Schlenk tube was charged with a magnetic stirring bar, 165 (20.0 mg,
0.11 mmol), [(Me3CO)3W≡CCMe3] (10.0 mg, 0.021 mmol), and dry toluene (7.5 mL).
The mixture was heated at 80 °C for 72 h. After that time, solvent was removed in vacuo
and the residue filtered through short pad of silica gel (ethyl acetate). The unreacted
starting material (4.0 mg, 20%) was removed by distillation, which left 9.4 mg (55%) of
pure 168 as a brown solid. IR (CHCl3): ~ν = 2926, 2360, 1601, 1468, 1212 cm–1. MS (EI,
70 eV) m/z (rel intensity) 310 (M+, 100), 295 (32), 280 (65), 263 (21). 1H NMR (400
MHz, CDCl3) δ 7.18 (s, 2H), 6.96 (s, 2H), 2.38 (s, 6H), 2.28 (s, 6H), 2.17 (s, 6H). 13C

226
NMR (100 MHz, CDCl3) δ 140.1, 136.7, 130.3, 130.1, 129.2, 126.5, 109.7, 90.9, 77.2,
21.1, 21.0, 4.8. HR-MS Calcd for C24H22: 310.1722. Found: 310.1721.
6.3.1 Crystallographic Information for 159
Crystal data and collection parameters:
A. Crystal Data
Empirical Formula C32H16
Formula Weight 400.48
Crystal Color, Habit colorless, cubic
Crystal Dimensions 0.16 x 0.17 x 0.17 mm
Crystal System monoclinic
Lattice Type C-centered

227
No. of Reflections Used for Unit
Cell Determination (2θ range) 1520 (3.0–45.0 °)
Lattice Parameters a 28.905(4) Å
b 4.8000(6) Å
c 18.010(2) Å
β 124.107(2) °
V 2068.9(4) Å3
Space Group C2/c (#15)
Z value 4
Dcalc 1.286 g cm–3
F000 832.00
µ(MoKα) 0.73 cm–1
B. Intensity Measurements
Diffractometer SMART
Radiation MoKα (λ = 0.71069 Å)
graphite monochromated
Crystal to Detector Distance 60.0 mm
Temperature –132.0 °C
Scan Type ω (0.3 ° per frame)
Scan Rate 25.0 seconds per frame
2θmax 50.8 °

228
No. of Reflections Measured Total: 4635
Unique: 1923 (Rint = 0.036)
Corrections Lorentz-polarization
Absorption (Tmax = 1.00, Tmin = 0.64)
C. Structure Solution and Refinement
Structure Solution Direct Methods (SIR92)
Refinement Full-matrix least-squares
Function Minimized Σ w (|Fo| – |Fc|)2
Least Squares Weights 1/σ2(Fo) = 4Fo2/σ2(Fo
2)
p-factor 0.030
Anomalous Dispersion All non-hydrogen atoms
No. Observations (I>3.00σ(I)) 1049
No. Variables 145
Reflection/Parameter Ratio 7.23
Residuals: R; Rw; Rall 0.040; 0.047; 0.068
Goodness of Fit Indicator 1.29
Max Shift/Error in Final Cycle 0.00
Maximum peak in Final Diff. Map 0.17 e–/Å3
Minimum peak in Final Diff. Map –0.24 e–/Å3

229
Positional parameters and B(eq):
Atom x y z B(eq)
C(1) 0.5697(1) –0.5571(5) 0.4550(1) 1.84(8) C(2) 0.62573(9) –0.6578(5) 0.5111(1) 1.61(8) C(3) 0.64472(9) –0.8641(5) 0.4799(1) 1.84(8) C(4) 0.6994(1) –0.9572(5) 0.5318(1) 2.15(9) C(5) 0.73623(9) –0.8473(5) 0.6171(1) 2.00(9) C(6) 0.71804(9) –0.6446(5) 0.6502(1) 1.79(8) C(7) 0.66311(9) –0.5462(4) 0.5982(1) 1.55(7) C(8) 0.64453(8) –0.3358(5) 0.6324(1) 1.59(8) C(9) 0.62866(8) –0.1603(5) 0.6605(1) 1.60(8) C(10) 0.60748(8) 0.0508(4) 0.6903(1) 1.48(7) C(11) 0.63941(8) 0.1522(5) 0.7783(1) 1.65(8) C(12) 0.61777(8) 0.3559(5) 0.8049(1) 1.81(8) C(13) 0.56501(9) 0.4635(5) 0.7461(1) 1.80(8) C(14) 0.53225(8) 0.3642(5) 0.6577(1) 1.60(8) C(15) 0.4772(1) 0.4724(5) 0.5958(1) 1.79(8) C(16) 0.55404(9) 0.1590(5) 0.6307(1) 1.76(8) H(1) 0.6190 –0.9400 0.4208 2.2 H(2) 0.7119 –1.1043 0.5094 2.5 H(3) 0.7743 –0.9084 0.6534 2.3 H(4) 0.7431 –0.5719 0.7095 2.2 H(5) 0.6763 0.0797 0.8215 1.8 H(6) 0.6396 0.4219 0.8650 2.2 H(7) 0.5514 0.6117 0.7654 1.9 H(8) 0.5316 0.0913 0.5694 1.9
Intramolecular distances involving the nonhydrogen atoms:
Atom atom distance
C(1) C(2) 1.429(3) C(1) C(15) 1.203(3) C(2) C(3) 1.394(3) C(2) C(7) 1.419(3) C(3) C(4) 1.383(3) C(4) C(5) 1.392(3) C(5) C(6) 1.389(3) C(6) C(7) 1.398(3) C(7) C(8) 1.434(3) C(8) C(9) 1.199(3) C(9) C(10) 1.435(3) C(10) C(11) 1.400(3)

230
C(10) C(16) 1.396(3) C(11) C(12) 1.383(3) C(12) C(13) 1.380(3) C(13) C(14) 1.403(3) C(14) C(15) 1.435(3) C(14) C(16) 1.395(3)
Intramolecular bond angles involving the nonhydrogen atoms:
Atom atom atom angle
C(2) C(1) C(15) 176.8(2) C(8) C(9) C(10) 177.5(2) C(1) C(2) C(3) 120.1(2) C(9) C(10) C(11) 121.4(2) C(1) C(2) C(7) 120.8(2) C(9) C(10) C(16) 119.6(2) C(3) C(2) C(7) 119.1(2) C(11) C(10) C(16) 119.0(2) C(2) C(3) C(4) 121.0(2) C(10) C(11) C(12) 119.9(2) C(3) C(4) C(5) 120.0(2) C(11) C(12) C(13) 121.2(2) C(4) C(5) C(6) 119.9(2) C(12) C(13) C(14) 119.8(2) C(5) C(6) C(7) 120.7(2) C(13) C(14) C(15) 120.5(2) C(2) C(7) C(6) 119.2(2) C(13) C(14) C(16) 119.1(2) C(2) C(7) C(8) 120.2(2) C(15) C(14) C(16) 120.4(2) C(6) C(7) C(8) 120.6(2) C(1) C(15) C(14) 177.8(2) C(7) C(8) C(9) 179.6(2) C(10) C(16) C(14) 121.0(2)

231
6.4 Experiments Related to Chapter 4
2-Bromo-1,4-di(oct-1-ynyl)-3-[(trimethylsilyl)ethynyl]benzene (184b):
Br Br
2,3-Dibromo-1,4-di(oct-1-ynyl)benzene. 1,2,3,4-Tetrabromobenzene221 (5.00 g,
12.7 mmol), 1-octyne (4.50 mL, 30.5 mmol, 2.4 equiv), [Pd(PPh3)2Cl2] (0.89 g, 1.27
mmol), and CuI (0.24 g, 1.27 mmol) were suspended in triethylamine (250 mL). The
mixture was heated at 60 ºC for 96 h. After that time, the solids were filtered off, the
solvent removed in vacuo and the resulting crude mixture separated by column
chromatography (hexanes) to yield 2,3-dibromo-1,4-di(oct-1-ynyl)benzene as the second,
yellow, fraction (4.20 g, 73%). The first fraction consisted predominantly of
monooctynylated product (1.25 g, 22%), which could be resubjected to the reaction
conditions and converted to 2,3-dibromo-1,4-di(oct-1-ynyl)benzene (up to 80%). UV-
VIS (cyclohexane): λmax (logε) = 224 (3.94), 232 (3.95), 279 (4.00), 292 (4.12), 371
(2.83), 388 (2.75), 422 (2.15) nm. IR (NaCl film): ~ν = 3583, 2930, 2858, 2230, 1579,
1378, 1351, 1330, 1095, 827, 726, 666 cm–1. MS (EI, 70 eV) m/z (rel intensity) 452 (M+,
100), 409 (25), 302 (32), 294 (57), 165 (52). 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 2H),
2.47 (t, 3J = 7.0 Hz, 4H), 1.63 (m, 4H), 1.47 (m, 4H), 1.31 (m, 8H), 0.90 (t, 3J = 7.0 Hz,
6H). 13C NMR (100 MHz, CDCl3) δ 130.96, 128.31, 127.00, 97.47, 79.94, 31.28, 28.52,
28.36, 22.53, 19.62, 14.03. HR-MS Calcd for C22H28Br2: 452.0537. Found: 452.0543.

232
Br
TMS
2-Bromo-1,4-di(oct-1-ynyl)-3-[(trimethylsilyl)ethynyl]benzene (184b). A
suspension of 2,3-dibromo-1,4-di(oct-1-ynyl)benzene (4.70 g, 10.4 mmol),
[Pd(PPh3)2Cl2] (0.13 g, 0.21 mmol), and CuI (35.0 mg, 0.21 mmol) in triethylamine (150
mL) was degassed thoroughly in a 250 mL Schlenk tube. Trimethylsilylacetylene (1.60
mL, 12.5 mmol) was injected through the septum and the tube closed. The mixture was
heated at 90 ºC for 24 h. After that time, the solids were filtered off, the solvent removed
in vacuo, and the resulting crude mixture separated by column chromatography (hexanes)
to yield the starting material as the first, yellow, fraction (1.89 g, 40%), followed by 184b
(1.63 g, 33%) as an orange oil. UV-VIS (cyclohexane): λmax (logε) = 252 (3.82), 260
(4.02), 280sh (3.64), 288sh (3.67), 298sh (3.72), 321 (2.91), 336 (2.78) nm. IR (NaCl
film): ~ν = 2957, 2931, 2859, 2230, 2160, 1579, 1456, 1378, 1329, 1250, 1192, 1133,
956, 844, 760, 675 cm–1. MS (EI, 70 eV) m/z (rel intensity) 470/468 (M+, 58/56), 398
(20), 295 (100), 252 (38), 166 (40). 1H NMR (400 MHz, CDCl3) δ 7.23 (br s, 2H), 2.46
(t, 3J = 7.2 Hz, 4H), 1.61 (m, 4H), 1.48 (m, 4H), 1.32 (m, 8H), 0.90 (t, 3J = 6.8 Hz, 3H),
0.89 (t, 3J = 6.8 Hz, 3H), 0.29 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 131.72, 129.93,
128.37, 127.79, 127.32, 125.74, 103.49, 102.05, 96.98, 96.93, 79.46, 78.87, 31.25 (2C),
28.59, 28.56, 28.45, 28.33, 22.48, 22.43, 19.63, 19.58, 13.97 (2C), –0.22. HR-MS Calcd
for C27H3781BrSi: 470.1827. Found: 470.1834. Anal. Calcd for C27H37BrSi: C, 69.06; H,
7.94. Found: C, 68.84; H, 7.90.

233
2-Bromo-3-ethynyl-1,4-di(oct-1-ynyl)benzene (175b):
Br
A solution of 184b (1.63 g, 3.46 mmol) in ether (10 mL) was treated with a
saturated solution of KOH in a 1:1 mixture of ether and ethanol (100 mL). The resulting
brown solution was stirred for 1 h at 23 °C. The mixture was diluted with ether (200 mL)
and washed with saturated aq. NH4Cl. The aqueous layer was extracted with ether (3 x
100 mL) and the combined organic phases were washed with brine. After drying over
MgSO4 overnight, removal of solvent in vacuo gave 175b as a brown oil (1.37 g, 99%).
UV-VIS (cyclohexane): λmax (logε) = 246sh (4.56), 254sh (4.63), 283sh (4.40), 294sh
(4.56), 319 (3.31), 332 (3.29) nm. IR (NaCl film): ~ν = 3307, 2931, 2858, 2230, 1717,
1579, 1522, 1457, 1375, 1329, 1249, 1183, 1132, 1119, 830, 774, 748, 723, 694, 626 cm–
1. MS (EI, 70 eV) m/z (rel intensity) 398/396 (M+, 100), 318 (29), 277 (49), 189 (48). 1H
NMR (400 MHz, CDCl3) δ 7.20 (d, 3J = 8.4 Hz, 1H), 7.17 (d, 3J = 8.4 Hz, 1H), 3.49 (s,
1H), 2.38 (t, 3J = 6.8 Hz, 2H), 2.37 (t, 3J = 7.2 Hz, 2H), 1.53 (m, 4H), 1.40 (m, 4H), 1.24
(m, 8H), 0.82 (t, 3J = 6.8 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 132.16, 130.09,
128.50, 127.78, 126.80, 125.86, 97.31, 97.22, 85.23, 81.11, 79.34, 78.72, 31.28, 31.24,
28.47, 28.45, 28.39, 28.33, 22.49 (2C), 19.59 (2C), 13.99 (2C). HR-MS Calcd for
C24H2981Br: 398.1432. Found: 398.1433. Anal. Calcd for C24H29Br: C, 72.54; H, 7.36.
Found: C, 72.71; H, 7.49.

234
2-Bromo-1,4-bis[(dimethylthexylsilyl)ethynyl]-3-ethynylbenzene (175c):
Br
SiSi
A solution of 184c69 (1.50 g, 2.56 mmol) in a mixture of ether (150 mL) and
methanol (150 mL) was treated with solid K2CO3 (352 mg, 2.56 mmol). The resulting
orange solution was stirred for 3 h at 23 °C. After the reaction was complete, the mixture
was diluted with ether (200 mL) and washed with saturated aq. NH4Cl. The aqueous layer
was extracted with ether (2 x 100 mL), and the combined organic phases were washed
with brine. The solution was dried over MgSO4 overnight. Removal of solvent in vacuo
gave 175c as an orange oil (1.30 g, 99%). IR (NaCl film): ~ν = 3310, 2958, 2866, 2159,
1449, 1372, 1251, 1129, 1091, 962, 860, 836, 814, 776, 673 cm–1. MS (EI, 70 eV) m/z
(rel intensity) 514/512 (M+, 0.8), 499 (0.8), 471/469 (1.6), 429/427 (100/92), 346 (30). 1H
NMR (400 MHz, CDCl3) δ 7.35 (d, 3J = 8.1 Hz, 1H), 7.33 (d, 3J = 8.1 Hz, 1H), 1.73 (br
sept, 3J = 6.9 Hz, 2H), 0.97 (s, 6H), 0.96 (s, 6H), 0.94 (d, 3J = 6.9 Hz, 6H), 0.93 (d, 3J =
6.9 Hz, 6H), 0.25 (s, 6H), 0.24 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 132.53, 130.47,
128.96, 127.78, 127.44, 126.09, 103.44, 102.99, 101.90, 101.83, 86.14, 80.81, 34.59,
34.55, 23.62, 23.57, 20.76 (2C), 18.75, 18.73, –2.48, –2.55. HR-MS Calcd for
C28H4179BrSi2: 512.1930. Found: 512.1920.

235
1-Iodo-2,4-di(oct-1-ynyl) -5-[(trimethylsilyl)ethynyl]benzene (176b):
BrBr
1,5-Dibromo-2,4-di(oct-1-ynyl)benzene. A suspension of 5778 (6.72 g, 13.7
mmol), 1-octyne (4.50 mL, 30.5 mmol), [Pd(PPh3)2Cl2] (0.49 g, 0.69 mmol), and CuI
(0.13 g, 0.69 mmol) in triethylamine (150 mL) was degassed in a 250 mL Schlenk tube.
The tube was closed and the mixture heated at 120 ºC for 2.5 h. After that time, solids
were filtered off, solvent was removed in vacuo, and the resulting crude product purified
by column chromatography (hexanes) to yield 1,5-dibromo-2,4-di(oct-1-ynyl)benzene as
a pale yellow oil (5.98 g, 96%). UV-VIS (cyclohexane): λmax (logε) = 248sh (4.54),
268sh (4.23), 277sh (4.17), 296sh (3.87), 395 (2.80) nm. IR (NaCl film): ~ν = 3583,
2955, 2931, 2858, 2233, 1455, 1363, 1326, 1060, 893, 869, 725, 666 cm–1. MS (EI, 70
eV) m/z (rel intensity) 452 (M+, 100), 409 (27), 344 (14), 302 (21), 165 (24). 1H NMR
(400 MHz, CDCl3) δ 7.76 (s, 1H), 7.45 (s, 1H), 2.43 (t, 3J = 6.8 Hz, 4H), 1.63 (m, 4H),
1.47 (m, 4H), 1.30 (m, 8H), 0.90 (t, 3J = 6.8 Hz, 6H). 13C NMR (100 MHz,
CDCl3) δ 136.69, 135.15, 125.15, 124.28, 96.83, 78.06, 31.25, 28.45, 28.30, 22.49, 19.51,
13.98. HR-MS Calcd for C22H28Br2: 452.0537. Found: 452.0550.
IBr
1-Bromo-5-iodo-2,4-di(oct-1-ynyl)benzene. A solution of 1,5-dibromo-2,4-di(oct-
1-ynyl)benzene (2.78 g, 6.15 mmol) in ether (100 mL) was cooled to –45 ºC, and BuLi
(3.2 mL of 2.5 M solution in hexane, 8.00 mmol) was added via syringe. The dark brown

236
solution was stirred at –45 ºC for 30 min. After that time, an ethereal solution (100 mL)
of iodine (2.54 g, 10.0 mmol) was added dropwise via syringe. The color of the solution
lightened gradually with the addition of iodine. The mixture was left to warm to 23 °C
overnight, extracted with ether (2 x 100 mL), and washed with aq. Na2S2O3 and then
brine. Drying over MgSO4, followed by removal of solvent in vacuo, gave 1-bromo-5-
iodo-2,4-di(oct-1-ynyl)benzene as a yellow oil (3.00 g, 97%). UV-VIS (cyclohexane):
λmax (logε) = 250 (4.87), 266 (4.53), 274 (4.47), 282 (4.29), 307 (3.38), 363 (3.37) nm. IR
(NaCl film): ~ν = 2929, 2857, 2232, 1449, 1378, 1361, 1326, 1252, 1210, 1111, 1047,
892, 871, 819, 724, 605 cm–1. MS (EI, 70 eV) m/z (rel intensity) 500/498 (M+, 100/100),
456/454 (15/15), 421 (23), 374 (50), 295 (55), 165 (50), 91 (73). 1H NMR (400 MHz,
CDCl3) δ 8.00 (s, 1H), 7.40 (s, 1H), 2.43 (t, 3J = 6.8 Hz, 2H), 2.42 (t, 3J = 6.8 Hz, 2H),
1.61 (m, 4H), 1.47 (m, 4H), 1.32 (m, 8H), 0.90 (m, 6H). 13C NMR (100 MHz,
CDCl3) δ 141.04, 135.72, 129.58, 125.88, 124.24, 99.4, 97.06, 96.00, 81.60, 78.27, 31.31,
31.28, 28.58, 28.49, 28.33, 28.30, 22.55, 22.53, 19.58, 19.53, 14.06, 14.03. HR-MS Calcd
for C22H2881BrI: 500.0399. Found: 500.0407.
Br
TMS
1-Bromo-2,4-di(oct-1-ynyl)-5-[(trimethylsilyl)ethynyl]benzene. A solution of 1-
bromo-5-iodo-2,4-di(oct-1-ynyl)benzene (1.95 g, 3.90 mmol), trimethylsilylacetylene
(0.63 mL, 4.50 mmol), PdCl2(PPh3)2 (54.6 mg, 0.08 mmol), and CuI (14.9 mg, 0.08
mmol) in triethylamine (100 mL) was stirred for 2 h at 23 °C. After removing the solids
by filtration, the solvent was evaporated in vacuo and the resulting crude product purified

237
by column chromatography (hexanes) to yield 1-bromo-2,4-di(oct-1-ynyl)-5-
[(trimethylsilyl)ethynyl]benzene as a yellow oil (1.77 g, 96%). UV-VIS (cyclohexane):
λmax (logε) = 256 (4.48), 283 (4.06), 288 (4.09), 300 (4.19), 320 (3.09), 334 (3.00) nm. IR
(NaCl film): ~ν = 2930, 2558, 2232, 2157, 1475, 1428, 1376, 1327, 1250, 1178, 1111,
1055, 895, 844, 760, 725 cm–1. MS (EI, 70 eV) m/z (rel intensity) 470/468 (M+, 56/54),
398/396 (20/18), 374 (8), 295 (100), 252 (40), 73 (35). 1H NMR (400 MHz,
CDCl3) δ 7.63 (s, 1H), 7.41 (s, 1H), 2.43 (m, 4H), 1.61 (m, 4H), 1.47 (m, 4H), 1.31 (m,
8H), 0.90 (m, 6H), 0.26 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 135.78, 135.25, 125.98,
125.79, 125.41, 123.46, 102.06, 100.24, 97.60, 95.99, 78.70, 77.82, 31.29, 31.28, 28.60,
28.59, 29.47, 28.34, 22.51, 22.46, 19.55 (2C), 13.99 (2C), –0.23. HR-MS Calcd for
C27H3781BrSi: 470.1827. Found: 470.1824.
I
TMS
1-Iodo-2,4-di(oct-1-ynyl)-5-[(trimethylsilyl)ethynyl]benzene (176b). A solution of
1-bromo-2,4-di(oct-1-ynyl)-5-[(trimethylsilyl)ethynyl]benzene (4.07 g, 8.68 mmol) in
ether (100 mL) was cooled to –45 °C, and BuLi (8.0 mL of 2.4 M solution in hexane,
19.2 mmol) was added via syringe. The dark brown solution was stirred at –45 °C for 30
min. After that time, an ethereal solution (50 mL) of iodine (5.59 g, 22.0 mmol) was
added dropwise via syringe. The color of the solution lightened gradually with addition of
iodine. The mixture was left to warm to 23 °C overnight, extracted with ether (2 x 100
mL), and washed with aq. Na2S2O3 and then brine. Drying over MgSO4, followed by the
removal of solvent in vacuo, gave 176b as a yellow oil (3.98 g, 89%). UV-VIS

238
(cyclohexane): λmax (logε) = 260 (4.77), 285 (4.28), 291 (4.30), 297 (4.26), 303sh (4.33),
322 (3.47), 337 (3.36) nm. IR (NaCl film): ~ν = 2957, 2931, 2858, 2231, 2160, 1467,
1428, 1371, 1250, 1211, 1176, 1048, 878, 843, 760, 725, 700, 673, 633 cm–1. MS (EI, 70
eV) m/z (rel intensity) 516 (M+, 100), 73 (36). 1H NMR (400 MHz, CDCl3) δ 7.88 (s,
1H), 7.37 (s, 1H), 2.45 (t, 3J = 6.8 Hz, 2H), 2.42 (t, 3J = 7.2 Hz, 2H), 1.62 (m, 4H), 1.46
(m, 4H), 1.32 (m, 8H), 0.90 (m, 6H), 0.26 (s, 9H). 13C NMR (100 MHz,
CDCl3) δ 141.49, 134.77, 130.32, 126.52, 125.32, 101.84, 100.26, 98.46, 96.90, 96.31,
82.21, 77.94, 31.28 (2C), 28.59, 28.55 (2C), 28.29, 22.51, 22.45, 19.58, 19.55, 14.03,
14.00, –0.21. HR-MS Calcd for C27H37BrI: 516.1709. Found: 516.1703. Anal. Calcd for
C27H37BrI: C, 62.78; H, 7.22. Found: C, 63.01; H, 7.45.
Compound 185b:
Br
TMS
A solution of 175b (1.11 g, 2.79 mmol), 176b (1.57 g, 3.04 mmol),
[Pd(PPh3)2Cl2] (39.0 mg, 0.06 mmol), and CuI (15.0 mg, 0.09 mmol) in triethylamine
(100 mL) was heated at reflux for 16 h. After that time, the solids were filtered off,

239
solvent removed in vacuo, and the resulting crude mixture purified by column
chromatography (hexanes/ethyl acetate) to yield 10a as brown, highly fluorescent (at 354
nm) fraction (2.00 g, 91%). UV-VIS (cyclohexane): λmax (logε) = 239 (4.41), 268 (4.77),
276 (4.87), 290 (4.75), 333 (4.41), 358 (4.23), 371 (3.83) nm. IR (NaCl film): ~ν = 2956,
2858, 2360, 2230, 1577, 1457, 1249, 901, 843, 759, 669 cm–1. MS (EI, 70 eV) m/z (rel
intensity) 786 (M+, 100), 706 (8), 678 (8), 73 (13). 1H NMR (400 MHz, C6D6) δ 7.86 (s,
1H), 7.74 (s, 1H), 7.11 (d, 3J = 7.9 Hz, 1H), 7.08 (d, 3J = 7.9 Hz, 1H), 2.30–2.18 (m, 8H),
1.51–1.39 (m, 8H), 1.39–1.27 (m, 8H), 1.26–1.05 (m, 16H), 0.90–0.79 (m, 12H), 0.27 (s,
9H). 13C NMR (100 MHz, CDCl3) δ 135.88, 135.22, 131.90, 130.13, 128.04, 128.03,
127.55, 126.74, 126.27, 125.90, 124.24 (2C), 102.77, 99.44, 97.45, 97.33, 97.10, 97.05,
95.49, 91.97, 79.55, 79.10, 76.84, 76.82, 31.62, 31.45, 31.40, 31.37, 28.71 (5C), 28.63,
28.58, 28.46, 22.69, 22.62, 22.55, 22.54, 20.06, 19.85, 19.75, 19.71, 14.14, 14.10 (3C), –
0.07. HR-MS Calcd for C51H6579BrSi: 784.4039. Found: 784.4036. Anal. Calcd for
C51H65BrSi: C, 77.93; H, 8.33. Found: C, 78.95; H, 8.53.
Compound 185c:
DMTSDMTS
Br
DMTS
DMTS
TMS
In a 500 mL Schlenk flask, 146 (1.58 g, 2.51 mmol) and 175c (1.29 g, 2.51
mmol), along with [Pd(PPh3)2Cl2] (88 mg, 0.13 mmol) and CuI (24 mg, 0.13 mmol),

240
were suspended in triethylamine (300 mL) and degassed. The mixture was kept at reflux
for 16 h, the solvent removed in vacuo and the crude material filtered through a short
plug of silica (hexanes/ethyl acetate) to give 185c (2.44 g, 96%) as a brownish oil. IR
(NaCl film): ~ν = 2959, 2932, 2865, 2158, 1482, 1404, 1405, 1378, 1250, 1188, 1129,
1089, 873, 842, 819, 775, 674 cm–1. MS (FAB) m/z (rel intensity) 1019 (M+, 58), 849
(100). 1H NMR (500 MHz, C6D6) δ 7.78 (s, 1H), 7.76 (s, 1H), 7.00 (AB q, 2H), 1.76–
1.62 (m, 4H), 1.02 (s, 6H), 1.01 (s, 6H), 0.98 (d, 3J = 6.9 Hz, 6H), 0.93 (d, 3J = 6.9 Hz,
6H), 0.91 (br s, 12H), 0.86 (d, 3J = 6.9 Hz, 6H), 0.86 (d, 3J = 6.9 Hz, 6H), 0.29 (s, 9H),
0.28 (s, 6H), 0.26 (s, 6H), 0.25 (s, 6H), 0.22 (s, 6H). 13C NMR (100 MHz,
CDCl3) δ 136.48, 136.39, 132.16, 130.54, 128.99, 128.17, 127.31, 125.96, 125.53,
125.39, 125.08, 124.89, 103.59, 103.18, 102.98, 102.91, 102.12, 101.83, 101.70, 101.22,
100.86, 100.52, 95.39, 92.15, 34.60 (2C), 34.54, 34.49, 23.66, 23.54, 23.49 (2C), 20.95,
20.73 (3C), 18.77, 18.74, 18.68, 18.62, –0.04, –2.32, –2.36 (2C), –2.55. HR-MS Calcd
for C59H8979BrSi5: 1016.4994. Found: 1016.5006.

241
Compound 186b:
Br
A solution of 185b (1.00 g, 1.28 mmol) in ether (10 mL) was treated with a
saturated solution of KOH in a 1:1 mixture of ether and ethanol (20 mL). The resulting
brown solution was stirred for 1 h at 23 °C. After the reaction was complete, the mixture
was diluted with ether (50 mL) and washed with saturated aq. NH4Cl. The aqueous layer
was extracted with ether (3 x 100 mL), and the combined organic phases were washed
with brine. The solution was dried over MgSO4 overnight. Removal of solvent in vacuo
gave 186b as a thick brown oil (0.89 g, 98%). UV-VIS (cyclohexane): λmax (logε) = 238
(4.54), 265 (4.84), 273sh (4.94), 293 (4.71), 331 (4.50), 355 (4.36), 369 (3.98) nm. IR
(NaCl film): ~ν = 3305, 2930, 2858, 2362, 2229, 1579, 1488, 1457, 1328, 1106, 900, 828,
722, 642 cm–1. MS (FAB) m/z (rel intensity) 714 (M+, 100). 1H NMR (400 MHz,
C6D6) δ 7.81 (s, 1H), 7.69 (s, 1H), 7.11 (d, 3J = 8.2 Hz, 1H), 7.08 (d, 3J = 8.2 Hz, 1H),
3.00 (s, 1H), 2.30–2.18 (m, 8H), 1.49–1.37 (m, 8H), 1.37–1.24 (m, 8H), 1.23–1.05 (m,
16H), 0.91–0.79 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 136.18, 135.36, 131.97,
130.15, 128.06, 127.97, 127.54, 126.94, 126.73, 125.91, 124.33, 123.14, 97.52 (2C),

242
97.27, 97.14, 95.30, 92.11, 81.69, 81.51, 79.53, 79.03, 78.54, 78.50, 31.44, 31.40 (2C),
31.36, 28.75, 28.72 (2C), 28.70, 28.57, 28.55 (2C), 28.45, 22.61 (2C), 22.54 (2C), 20.06,
19.87, 19.71 (2C), 14.10 (4C). HR-MS Calcd for C48H5779Br: 712.3644. Found:
712.3659. Anal. Calcd for C48H57Br: C, 80.76; H, 8.05. Found: C, 80.83; H, 8.24.
Compound 186c:
DMTSDMTS
Br
DMTS
DMTS
A solution of 185c (1.13 g, 1.11 mmol) in a mixture of ether (70 mL) and ethanol
(70 mL) was treated with solid K2CO3 (306 mg, 2.22 mmol). After stirring for 2 h at 23
°C, the solvents were removed in vacuo, and the resulting oil was redissolved in 20 mL
of CH2Cl2 and filtered through a short plug of silica to give (0.91 g, 87%) of 186c as a
brown oil. IR (NaCl film): ~ν = 3309, 2959, 2867, 2156, 1482, 1464, 1379, 1250, 1129,
1090, 837, 776, 675 cm–1. MS (FAB) m/z (rel intensity) 946 (M+, 0.5), 777 (1.5), 252
(100), 235 (58), 140 (98), 123 (83). 1H NMR (500 MHz, C6D6) δ 7.65 (s, 1H), 7.64 (s,
1H), 6.99 (AB q, 2H), 2.97 (s, 1H) 1.80–1.56 (m, 4H), 1.00 (s, 6H), 0.98 (s, 6H), 0.94 (d,
3J = 6.9 Hz, 6H), 0.93 (d, 3J = 6.9 Hz, 6H), 0.89 (s, 6H), 0.88 (s, 6H), 0.83 (d, 3J = 6.9
Hz, 6H), 0.83 (d, 3J = 6.9 Hz, 6H), 0.24 (s, 6H), 0.24 (s, 6H), 0.22 (s, 6H), 0.19 (s, 6H).
13C NMR (100 MHz, CDCl3) δ 136.16, 136.01, 132.19, 130.51, 128.94, 128.22, 127.38,
126.20, 126.03, 125.98, 125.08, 124.39, 103.60, 103.19, 102.78 (2C), 101.89, 101.74,

243
101.44, 101.27, 95.27, 92.36, 82.77, 81.10, 34.72, 34.64, 34.60, 34.54, 23.67, 23.56,
23.50, 23.48, 20.78 (2C), 20.74, 20.69, 18.78, 18.75, 18.67, 18.62, –2.39 (2C), –2.42, –
2.54. HR-MS Calcd for C56H8179BrSi4: 944.4599. Found: 944.4589.
Compound 187b:
I
TMS
A solution of 185b (1.01 g, 1.28 mmol) in ether (40 mL) was cooled to –45 ºC,
and BuLi (0.8 mL of 2.4 M solution in hexane, 1.92 mmol) was added via syringe. The
dark brown solution was stirred at –45 ºC for 30 min. After that time, an ethereal solution
(20 mL) of iodine (762 mg, 3.00 mmol) was added dropwise via syringe. The color of the
solution lightened gradually with addition of iodine. The mixture was stirred at –45 ºC for
10 min, left to warm to 23 °C over 90 min, extracted with ether (2 x 100 mL), and
washed with aq. Na2S2O3 and then brine. Drying over MgSO4, followed by removal of
solvent in vacuo gave 187b as a brown oil (1.01 g, 94%). UV-VIS (cyclohexane): λmax
(logε) = 271 (4.26), 278 (4.24), 284 (4.20), 291 (4.14), 330 (3.76), 353 (3.64) nm. IR
(NaCl film): ~ν = 2930, 2858, 2229, 2157, 1454, 1329, 1249, 1111, 901, 844, 724, 700

244
cm–1. MS (FAB) m/z (rel intensity) 832 (M+, 100). 1H NMR (400 MHz, C6D6) δ 7.87 (s,
1H), 7.74 (s, 1H), 7.13 (d, 3J = 8.2 Hz, 1H), 7.04 (d, 3J = 8.2 Hz, 1H), 2.30–2.21 (m, 8H),
1.52–1.37 (m, 8H), 1.37–1.05 (m, 24H), 0.91–0.79 (m, 12H), 0.27 (s, 9H). 13C NMR (100
MHz, CDCl3) δ 135.87, 135.27, 132.59, 131.70, 131.00, 130.98, 130.68, 126.71, 126.68,
126.18, 124.24, 124.21, 106.82, 102.79, 99.43, 97.24, 97.01, 96.31, 95.69, 94.60, 83.44,
79.32, 78.83, 78.63, 31.46, 31.39 (3C), 28.70 (3C), 28.68 (3C), 28.63, 28.41, 22.62, 22.55
(2C), 22.52, 20.24, 19.85, 19.74, 19.71, 14.09 (4C), –0.07. HR-MS Calcd for C51H65SiI:
832.3900. Found: 832.3904.
Compound 187c:
DMTSDMTS
I
DMTS
DMTS
TMS
A solution of 185c (1.13 g, 1.11 mmol) in ether (100 mL) was cooled to –50 ºC,
and BuLi (0.95 mL of 2.34 M solution in hexane, 2.22 mmol) was added via syringe. The
dark brown solution was stirred at –50 ºC for 45 min. After that time, an ethereal solution
(100 mL) of iodine (1.02 g, 4.00 mmol) was added dropwise via syringe. The color of the
solution lightened gradually with addition of iodine. The mixture was left to warm to 23
°C overnight, extracted with ether (2 x 100 mL), and washed with aq. Na2S2O3 and then
brine. Drying over MgSO4, followed by removal of solvent in vacuo, gave 187c as a dark
brown oil (1.09 g, 92%). IR (NaCl film): ~ν = 2959, 2923, 2864, 2157, 1482, 1463, 1442,

245
1249, 869, 839, 775 cm–1. MS (FAB) m/z (rel intensity) 1066 (M+, 100). 1H NMR (300
MHz, C6D6) δ 7.62 (s, 1H), 7.61 (s, 1H), 7.05 (d, 3J = 8.1 Hz, 1H), 6.96 (d, 3J = 8.1 Hz,
1H), 1.81–1.54 (m, 4H), 0.99 (s, 6H), 0.98 (s, 6H), 0.93 (d, 3J = 6.9 Hz, 6H), 0.89 (d, 3J =
6.9 Hz, 6H), 0.87 (s, 6H), 0.84 (s, 6H), 0.80 (d, 3J = 6.9 Hz, 6H), 0.80 (d, 3J = 6.9 Hz,
6H), 0.24 (m, 21H), 0.18 (s, 6H), 0.13 (s, 6H). 13C NMR (125 MHz, CDCl3) δ 136.26,
136.18, 132.77, 131.37, 131.23, 130.71, 126.39, 125.45, 125.40, 125.03, 124.96, 107.22,
107.11, 103.34, 102.98, 102.94, 102.14, 101.60, 101.14, 100.72 (2C), 100.45, 95.80,
94.43, 34.59, 34.57, 34.48, 34.44, 23.66, 23.50, 23.41, 23.41, 20.91, 20.79, 20.67 (2C),
18.75 (2C), 18.63, 18.59, –0.06, –2.32, –2.36, –2.41, –2.55. HR-MS Calcd for C59H89ISi5:
1065.4964. Found: 1065.4934.
Compound 188b:
Hex
Hex
Hex
Hex
Hex
Hex
Br
Hex
Hex
TMS
A solution of 186b (0.89 g, 1.25 mmol), 187b (1.01 g, 1.21 mmol),
[Pd(PPh3)2Cl2] (17.5 mg, 0.025 mmol), and CuI (5.0 mg, 0.025 mmol) in triethylamine
(50 mL) was degassed thoroughly in a Schlenk tube. The tube was closed and the mixture
heated at 110 ºC for 11 h. After that time, the solids were filtered off, solvent was

246
removed in vacuo, and the resulting crude mixture purified by column chromatography
(hexanes/ethyl acetate) to yield 188b as a thick brown oil (1.34 g, 78%). UV-VIS
(cyclohexane): λmax (logε) = 224 (4.78), 276 (5.20), 292 (5.25), 324 (4.94), 362 (4.67),
384 (4.15) nm. IR (NaCl film): ~ν = 2930, 2857, 2230, 2157, 1578, 1458, 1249, 1113,
901, 843, 759, 725 cm–1. MS (FAB) m/z (rel intensity) 1420 (M+, 100). 1H NMR (400
MHz, C6D6) δ 8.16 (s, 1H), 8.00 (s, 1H), 7.77 (s, 1H), 7.70 (s, 1H), 7.16 (s, 2H), 7.11 (d,
3J = 7.9 Hz, 1H), 7.08 (d, 3
J = 7.9 Hz, 1H), 2.41–2.13 (m, 16H), 1.60–1.40 (m, 16H),
1.40–1.27 (m, 16H), 1.26–1.06 (m, 32H), 0.90–0.80 (m, 24H), 0.28 (s, 9H). 13C NMR
(125 MHz, CDCl3) δ 136.27, 135.99, 135.29, 135.12, 131.74, 131.01, 130.95, 130.07,
128.07, 127.95, 127.80, 127.74, 127.60, 126.51, 126.43, 126.36, 126.33, 126.24, 126.05,
125.71, 124.61, 124.45, 124.08, 124.02, 102.74, 99.07, 97.53, 97.48, 97.37, 97.05, 96.89,
96.80, 96.67, 96.64, 95.58, 95.27 (2C), 91.82, 91.75, 91.73, 79.55, 79.14, 79.08, 78.94,
78.73, 78.55 (2C), 78.47, 31.45 (2C), 31.37 (3C), 31.33 (3C), 28.77 (4C), 28.69 (4C),
28.64 (2C), 28.61 (3C), 28.58, 28.54 (2C), 22.66, 22.59 (2C), 22.53 (5C), 20.02, 19.88,
19.84, 19.79, 19.73 (2C), 19.68, 19.66, 14.08 (6C), 14.00 (2C), –0.10. HR-MS Calcd for
C99H12179BrSi: 1416.8421. Found: 1416.8405. Anal. Calcd for C99H121BrSi: C, 83.80; H,
8.59. Found: C, 84.16; H, 8.75.

247
Compound 188c:
DMTS
DMTS
DMTS
DMTS
DMTS
DMTS
Br
DMTS
DMTS
TMS
In a 200 mL Schlenk tube, 186c (641 mg, 0.68 mmol) and 187c (721 mg, 0.68
mmol) were dissolved in triethylamine (125 mL). [Pd(PPh3)2Cl2] (24 mg, 0.034 mmol)
and CuI (7 mg, 0.034 mmol) were added and the mixture was degassed. The tube was
closed and heated at 110 ºC for 17 h. Solvent was removed in vacuo and the remaining
crude material chromatographed on silica (hexanes/CH2Cl2) to give 188c as a viscous
brown oil (1.03 g, 78%). IR (NaCl film): ~ν = 2959, 2867, 2158, 1485, 1467, 1379, 1250,
1191, 1129, 868, 841, 775 cm–1. MS (FAB) m/z (rel intensity) 1884 (M+, 10), 1800 (13),
1716 (17), 1631 (10), 895 (25), 486 (100). 1H NMR (400 MHz, C6D6) δ 7.95 (s, 1H),
7.91 (s, 1H), 7.72 (s, 1H), 7.70 (s, 1H), 7.12 (s, 2H), 7.00 (AB q, 2H), 1.74–1.62 (m, 8H),
1.02–0.84 (m, 96H), 0.35–0.25 (m, 57H). 13C NMR (125 MHz, CDCl3) δ 136.06, 135.70,
135.68, 135.55, 134.76, 134.75, 132.02, 131.08, 130.20, 128.83, 128.56, 128.41, 127.32,
125.98, 125.97, 125.86, 125.73, 125.51, 125.50, 125.47, 125.40, 125.06, 124.91, 124.88,
103.60, 103.32, 103.29, 103.08, 102.95, 102.90, 102.22, 101.87, 101.58, 101.40, 101.05,
101.03, 100.95, 100.89, 100.76, 100.44, 100.16, 99.94, 95.39, 95.06, 94.90, 91.86, 91.69,
91.59, 34.52 (2C), 34.50, 34.44, 34.42, 34.40, 34.35, 34.35, 23.57, 23.41, 23.35, 23.34,

248
23.28, 23.26, 23.26, 23.23, 20.83, 20.65, 20.59, 20.57, 20.56, 20.52, 20.50, 20.47, 18.69,
18.65, 18.59, 18.58, 18.50 (2C), 18.48, 18.43, –0.06, –2.40, –2.46 (2C), –2.48, –2.48, –
2.49, –2.53, –2.62. The high molecular mass of 188c precluded HR-MS measurements.
Compound 171b:
Bromine–iodine exchange: A solution of 188b (1.10 g, 0.77 mmol) in ether (40
mL) was cooled to –45 ºC, and BuLi (0.6 mL of 2.5 M solution in hexane, 1.50 mmol)
was added via syringe. The dark brown solution was stirred at –45 ºC for 30 min.
Subsequently, an ethereal solution (20 mL) of iodine (762 mg, 3.00 mmol) was added
dropwise via syringe. The mixture was stirred at –45 ºC for 10 min, left to warm to 23 °C
over 90 min, extracted with ether (2 x 100 mL), and washed with aq. Na2S2O3 and then
brine. Drying over MgSO4, followed by removal of solvent in vacuo, gave the
intermediate iodide as a thick brown oil (1.11 g, 98%). The compound was partly
characterized and used without purification in the following step. MS (FAB) m/z (rel

249
intensity) 1466 (M+, 100). 1H NMR (400 MHz, C6D6) δ 8.16 (s, 1H), 7.98 (s, 1H), 7.76
(s, 1H), 7.69 (s, 1H), 7.16 (s, 2H), 7.14 (d, 3J = 8.0 Hz, 1H), 7.04 (d, 3J = 8.0 Hz, 1H),
2.41–2.12 (m, 16H), 1.83–0.98 (m, 64H), 0.91–0.79 (m, 24H), 0.28 (s, 9H). HR-MS
Calcd for C99H121SiI: 1464.8282. Found: 1464.8253.
Cleavage of the TMS group: A solution of the iodide (973 mg, 0.66 mmol) in
THF (100 mL) was treated with Bu4N+F– (2.00 mL of 1.0 M solution in THF, 2.00 mmol)
and stirred at 23 °C for 40 min. Ethanol (30 mL) was added via syringe and the resulting
brown solution stirred for an additional 2 h; subsequently, the mixture was filtered
through short plug of silica (ethyl acetate). Removal of solvent in vacuo gave the
deprotected material as a dark brown oil (876 mg, 95%), which was used immediately in
the next step.
Macrocyclization: A thoroughly degassed solution of the terminal alkyne (563
mg, 0.40 mmol) in triethylamine (50 mL) was injected slowly (syringe pump, 36 h) into a
refluxing solution of PdCl2(PPh3)2 (14.0 mg, 0.02 mmol) and CuI (4.0 mg, 0.02 mmol) in
triethylamine (100 mL). After the addition was complete, the mixture was left at reflux
for an additional 10 h. The solvent was removed in vacuo and the resulting crude product
purified by column chromatography (hexanes/ethyl acetate = 95/5) to yield 171b as dark
red waxy solid (344 mg, 67%).
Performing this reaction on a larger scale (0.63 mmol) and without the slow
addition of the alkyne gave 171b in somewhat lower yield (53%). UV-VIS
(cyclohexane): λmax (logε) = 229 (4.73), 271 (5.00), 286 (4.98), 307 (4.92), 351 (4.56),
370 (4.33) nm. IR (NaCl film): ~ν = 2929, 2857, 2228, 1646, 1541, 1466, 1378, 1117,
901, 829 cm–1. MS (FAB) m/z (rel intensity) 1266 (M+, 100). 1H NMR (400 MHz,

250
CD2Cl2) δ 7.81 (s, 2H), 7.43 (s, 2H), 7.32 (s, 4H), 2.42 (t, 3J = 7.0 Hz, 8H), 2.35 (t, 3J =
7.0 Hz, 8H), 1.60 (t, 3J = 7.6 Hz, 8H), 1.56 (t, 3J = 7.6 Hz, 8H), 1.46–1.36 (m, 8H), 1.36–
1.28 (m, 8H), 1.25–1.19 (m, 16H) 1.16–1.12 (m, 16H), 0.92–0.85 (m, 24H). 1H NMR
(500 MHz, C6D6) δ 7.88 (s, 2H), 7.66 (s, 2H), 7.18 (s, 4H), 2.40 (t, 3J = 7.0 Hz, 8H), 2.34
(t, 3J = 7.0 Hz, 8H), 1.61 (t, 3J = 7.6 Hz, 8H), 1.54 (t, 3J = 7.6 Hz, 8H), 1.46–1.38 (m,
8H), 1.38–1.29 (m, 8H), 1.23–1.17 (m, 16H) 1.17–1.11 (m, 16H), 0.90–0.82 (m, 24H).
13C NMR (125 MHz, CDCl3) δ 137.03, 135.02, 130.93, 128.60, 126.01, 125.47, 124.49,
96.86, 96.55, 95.29, 91.43, 79.00, 78.70, 31.39 (2C), 28.72 (2C), 28.60, 28.58, 22.49,
22.45, 19.90 (2C), 14.01, 14.00. HR-MS Calcd for C96H112: 1264.8764. Found:
1264.8762.
Compound 171c:
DMTS
DMTS
DMTSDMTS
DMTSDMTS DMTS
DMTS
Bromine–iodine exchange: A solution of 188c (1.00 g, 0.512 mmol) in ether (100
mL) was cooled to –50 ºC, and BuLi (0.44 mL of 2.34 M solution in hexane, 1.03 mmol)
was added via syringe. The dark brown solution was stirred at –50 ºC for 45 min.
Subsequently, an ethereal solution (100 mL) of iodine (523 g, 2.06 mmol) was added
dropwise via syringe. The mixture was left to warm to 23 °C overnight, extracted with
ether (2 x 100 mL), and washed with aq. Na2S2O3 and then brine. Drying over MgSO4,

251
followed by removal of solvent in vacuo gave the desired iodide as a dark brown oil (875
mg, 85%). IR (NaCl film): ~ν = 2983, 2956, 2914, 2866, 2157, 1485, 1463, 1378, 1253,
1190, 1090, 997, 930, 871, 844, 770, 675 cm–1. MS (FAB) m/z (rel intensity) 1932 (M+,
22), 1846 (26), 1762 (30), 1678 (29), 1595 (24), matrix peak (100). 1H NMR (500 MHz,
C6D6) δ 7.84 (s, 1H), 7.79 (s, 1H), 7.65 (s, 1H), 7.59 (s, 1H), 7.08 (br s, 2H), 7.03 (d, 3J =
8.1 Hz, 1H), 6.94 (d, 3J = 8.1 Hz, 1H), 1.74–1.52 (m, 8H), 1.02–0.76 (m, 96H), 0.27–0.15
(m, 57H). The crude material was used in the next step without further purification.
Cleavage of the TMS group: A solution of the above iodide (765 mg, 0.040
mmol) in a mixture of ether (50 mL) and ethanol (50 mL) was treated with solid K2CO3
(110 mg, 0.80 mmol). After stirring for 2 h at 23 °C, the solvents were removed in vacuo,
and the resulting oil was redissolved in a small amount of CH2Cl2 and filtered through a
short plug of silica to give (729 mg, 99%) of the deprotected material as a brown oil. This
material was used in the subsequent steps without further purification.
Macrocyclization: To a thoroughly degassed solution of the alkyne from the
previous step (631 mg, 0.34 mmol) in triethylamine (250 mL) were added [Pd(PPh3)2Cl2]
(12.0 mg, 0.017 mmol) and CuI (3.3 mg, 0.017 mmol). The mixture was brought to reflux
and stirred for 16 h, after which the solvent was removed in vacuo and the resulting crude
product purified by column chromatography (hexanes/CH2Cl2) to yield 171c as yellow
flaky crystals, m.p. 144–145 ºC (311 mg, 53%). UV-VIS (CH2Cl2): λmax (logε) = 249
(4.68), 274 (4.88), 305 (4.90), 330 (4.58), 377 (3.77) nm. IR (NaCl film): ~ν = 2959,
2928, 2867, 2156, 1486, 1463, 1378, 1250, 1192, 997, 929, 836, 775, 675 cm–1. MS
(FAB) m/z (rel intensity) 1732 (M+, 13), 1564 (10), 1396 (7), 503 (92), 488 (100). 1H
NMR (500 MHz, C6D6) δ 7.63 (s, 2H), 7.24 (s, 2H), 7.09 (s, 4H), 1.84 (sept, 3J = 6.9 Hz,

252
4H), 1.77 (sept, 3J = 6.9 Hz, 4H), 1.10 (s, 12H), 1.07 (s, 12H), 1.05 (s, 12H), 1.03 (s,
12H), 1.01 (d, 3J = 6.6 Hz, 12H), 0.99 (d, 3J = 6.6 Hz, 12H), 0.97 (d, 3J = 7.1 Hz, 12H),
0.95 (d, 3J = 7.1 Hz, 12H), 0.40 (s, 12H), 0.38 (s, 12H), 0.36 (s, 12H), 0.32 (s, 12H). 13C
NMR (100 MHz, CDCl3) δ 137.76, 135.88, 131.27, 129.39, 125.23, 125.05, 124.93,
103.37, 103.06, 101.24, 101.06, 95.76, 91.54, 34.68, 34.56 (2C), 34.48, 23.68 (2C), 23.53
(2C), 21.06, 20.85, 20.65, 20.60, 18.75, 18.68, 18.61, 18.55, –2.35, –2.38 (3C).
Compound 156:
A solution of 171c (17 mg, 0.01 mmol) in THF–d8 (0.5 mL) was treated with
TBAF (0.1 mL of 1.0 M solution in THF, 0.1 mmol). The mixture turned brown
immediately. After 2 h, water (0.5 mL) was added, along with diethyl ether (5 mL). The
layers were separated and the organic layer, containing 156, was analyzed by 1H NMR:
1H NMR (400 MHz, THF–d8/ether) δ 7.93 (s, 2H), 7.62 (s, 2H), 7.44 (s, 4H), 3.97 (s,
4H), 3.95 (s, 4H). UV-VIS (THF) λmax (rel. absorbance) = 235 (0.63), 271 (0.40), 288
(0.40), 306 (0.79), 340 (0.25), 373 (0.08), 401 (0.06) nm. Removal of solvent at high
vacuum gave an insoluble yellow-white powder, which darkened in less than 1 min.

253
Compounds 189 and 190 (CpCo-mediated cycloisomerization of 171b):
To 171b (46 mg, 0.036 mmol) in degassed toluene (10 mL) was added
[CpCo(CO)2] (26 mg, 0.144 mmol, 4 equiv). The resulting solution was added over 25
min to boiling, degassed toluene (100 mL), and irradiated with a slide projector lamp.
After 1 h, heating and irradiation were discontinued and the solvents removed under
reduced pressure. The residue was dissolved in CH2Cl2, preadsorbed on silica, and dried
overnight under high vacuum. Column chromatography (hexanes/ethyl acetate) gave a
deep red band that contained 189 and 190 in a ~ 1:1 ratio (by 1H NMR). The total yield
was 9.1 mg (19%).
In a separate attempt, treating 171b (140 mg, 0.11 mmol) with six equiv of
[CpCo(CO)2] (116 mg, 0.66 mmol) gave 15.5 mg (11%) of 189 only, as an orange oil.
Both pure 189 and the 189/190 mixture decomposed within several hours, when
left exposed to air.
189: UV-VIS (dichloromethane) λmax (logε) = 235 (4.50), 273 (4.81), 311 (4.90),
350sh (4.44), 369sh (4.40), 420sh (4.20), 444 (4.31), 508 (3.80), 544 (3.68) nm. IR
(KBr): ~ν = 2958, 2930, 2858, 2227, 2170, 1451, 1329, 1249, 1111, 1031, 844 cm–1. MS

254
(FAB) m/z (rel intensity) 1266 (M+, 5), 501 (100), 486 (91). 1H NMR (400 MHz, CD2Cl2)
δ 7.69 (s, 1H), 7.49 (s, 1H), 7.06 (d, 3J = 7.3 Hz, 2H), 6.72 (d, 3J = 7.3 Hz, 2H), 6.67 (d,
3J = 1.5 Hz), 6.47 (d, 3J = 1.5 Hz, 1H), 2.50–2.42 (m, 8H), 2.32–2.26 (m, 8H), 1.60–1.10
(m, 64H), 0.89–0.80 (m, 24H). HR-MS Calcd for C96H112: 1264.8764. Found: 1264.8774.
190 (based on the difference between 189/190 mixture and pure 189): 1H NMR
(400 MHz, CD2Cl2) δ 7.12 (s, 2H), 7.08 (d, 3J = 7.5 Hz, 2H), 6.87 (s, 2H), 6.72 (d, 3J =
6.6 Hz, 2H), 2.49–2.42 (m, 8H), 2.31–2.26 (m, 8H), 1.57–1.10 (m, 64H), 0.89–0.81 (m,
24H).
6.4.1 Calculated Structures of 121, 156, and 191–195
Calculated positional parameters for the planar structure of 121:
Atom x y z
C1 0.0000000000 5.7631791204 –1.3735933016 C2 0.0000000000 5.7631791204 1.3735933016 C3 0.0000000000 4.4946408315 –0.6793841882 C4 0.0000000000 6.9747603226 –0.7189211185 C5 0.0000000000 6.9747603226 0.7189211185 C6 0.0000000000 4.4946408315 0.6793841882 C7 0.0000000000 1.1473170575 2.7304484215 C8 0.0000000000 –1.1662414596 4.1554084185 C9 0.0000000000 0.0000000000 1.9560033812 C10 0.0000000000 1.1662414596 4.1554084185 C11 0.0000000000 0.0000000000 4.9318418836 C12 0.0000000000 –1.1473170575 2.7304484215 C13 0.0000000000 1.1473170575 –2.7304484215 C14 0.0000000000 –1.1662414596 –4.1554084185 C15 0.0000000000 1.1662414596 –4.1554084185 C16 0.0000000000 0.0000000000 –1.9560033812 C17 0.0000000000 –1.1473170575 –2.7304484215 C18 0.0000000000 0.0000000000 –4.9318418836 C19 0.0000000000 –4.4946408315 –0.6793841882 C20 0.0000000000 –6.9747603226 0.7189211185

255
C21 0.0000000000 –5.7631791204 –1.3735933016 C22 0.0000000000 –4.4946408315 0.6793841882 C23 0.0000000000 –5.7631791204 1.3735933016 C24 0.0000000000 –6.9747603226 –0.7189211185 C25 0.0000000000 5.0327142569 –2.6914196499 C26 0.0000000000 2.6830646757 –4.1249724903 C27 0.0000000000 5.1032287174 –4.0597570334 C28 0.0000000000 3.7710865561 –1.9681491057 C29 0.0000000000 2.6218474041 –2.6769955711 C30 0.0000000000 3.8649095917 –4.8161927194 C31 0.0000000000 –5.0327142569 2.6914196499 C32 0.0000000000 –2.6830646757 4.1249724903 C33 0.0000000000 –3.7710865561 1.9681491057 C34 0.0000000000 –5.1032287174 4.0597570334 C35 0.0000000000 –3.8649095917 4.8161927194 C36 0.0000000000 –2.6218474041 2.6769955711 C37 0.0000000000 –5.0327142569 –2.6914196499 C38 0.0000000000 –2.6830646757 –4.1249724903 C39 0.0000000000 –5.1032287174 –4.0597570334 C40 0.0000000000 –3.7710865561 –1.9681491057 C41 0.0000000000 –2.6218474041 –2.6769955711 C42 0.0000000000 –3.8649095917 –4.8161927194 C43 0.0000000000 2.6830646757 4.1249724903 C44 0.0000000000 5.0327142569 2.6914196499 C45 0.0000000000 2.6218474041 2.6769955711 C46 0.0000000000 3.8649095917 4.8161927194 C47 0.0000000000 5.1032287174 4.0597570334 C48 0.0000000000 3.7710865561 1.9681491057 H1 0.0000000000 7.9210776320 –1.2521911470 H2 0.0000000000 7.9210776320 1.2521911470 H3 0.0000000000 0.0000000000 0.8765635444 H4 0.0000000000 0.0000000000 6.0173474308 H5 0.0000000000 0.0000000000 –0.8765635444 H6 0.0000000000 0.0000000000 –6.0173474308 H7 0.0000000000 –7.9210776320 –1.2521911470 H8 0.0000000000 –7.9210776320 1.2521911470 H9 0.0000000000 6.0486558343 –4.5945629231 H10 0.0000000000 3.9075229634 –5.9015550425 H11 0.0000000000 –6.0486558343 4.5945629231 H12 0.0000000000 –3.9075229634 5.9015550425 H13 0.0000000000 –6.0486558343 –4.5945629231 H14 0.0000000000 –3.9075229634 –5.9015550425 H15 0.0000000000 3.9075229634 5.9015550425 H16 0.0000000000 6.0486558343 4.5945629231

256
Calculated positional parameters for the non planar structure of 121:
Atom x y z
C1 0.3239589662 –1.3350403605 5.7634717974 C2 –0.2883571257 1.3431261500 5.7643409283 C3 0.1244584234 –0.6679597477 4.4961852979 C4 0.2144374930 –0.6888421466 6.9746169901 C5 –0.1112219204 0.7116884265 6.9752291492 C6 –0.1597478968 0.6603684734 4.4965134250 C7 –1.0591502381 4.6956487788 3.8623328013 C8 –0.5681840298 2.6117412907 2.6218298696 C9 –0.5614226753 2.6658413340 1.1471936071 C10 –0.9022677227 4.0497654530 –1.1655458638 C11 –0.3595386931 1.9167098573 0.0000000000 C12 –0.9022677227 4.0497654530 1.1655458638 C13 –1.0848808022 4.8045332386 0.0000000000 C14 –0.5614226753 2.6658413340 –1.1471936071 C15 0.4729899003 –2.6881028313 1.1474625074 C16 0.8709495014 –4.0567686652 –1.1654654053 C17 0.8709495014 –4.0567686652 1.1654654053 C18 0.2378284529 –1.9488012127 0.0000000000 C19 0.4729899003 –2.6881028313 –1.1474625074 C20 1.0851294696 –4.8031131590 0.0000000000 C21 0.5064448825 –2.6263929469 –2.6221106834 C22 1.1167454627 –4.6809489502 –3.8600717640 C23 0.1244584234 –0.6679597477 –4.4961852979 C24 –0.1112219204 0.7116884265 –6.9752291492 C25 0.3239589662 –1.3350403605 –5.7634717974 C26 –0.1597478968 0.6603684734 –4.4965134250 C27 –0.2883571257 1.3431261500 –5.7643409283 C28 0.2144374930 –0.6888421466 –6.9746169901 C29 0.6137600983 –2.6196304918 5.0314520596 C30 0.8923997571 –4.0219409465 2.6809962303 C31 0.9633023631 –3.9426125657 5.0995819333 C32 0.3900835416 –1.9279787191 3.7719496907 C33 0.5064448825 –2.6263929469 2.6221106834 C34 1.1167454627 –4.6809489502 3.8600717640 C35 –0.5865894898 2.6263400001 –5.0330060226 C36 –0.8988197454 4.0211936160 –2.6816556380 C37 –0.4329438967 1.9182843357 –3.7722321561 C38 –0.8886994002 3.9608927649 –5.1018804137 C39 –1.0591502381 4.6956487788 –3.8623328013 C40 –0.5681840298 2.6117412907 –2.6218298696 C41 0.6137600983 –2.6196304918 –5.0314520596 C42 0.8923997571 –4.0219409465 –2.6809962303

257
C43 0.9633023631 –3.9426125657 –5.0995819333 C44 0.3900835416 –1.9279787191 –3.7719496907 C45 –0.8886994002 3.9608927649 5.1018804137 C46 –0.8988197454 4.0211936160 2.6816556380 C47 –0.4329438967 1.9182843357 3.7722321561 C48 –0.5865894898 2.6263400001 5.0330060226 H1 0.3674451868 –1.2009964371 7.9201722769 H2 –0.2042926656 1.2366614074 7.9215787610 H3 1.4071574788 –5.7268842984 –3.8988609688 H4 1.1351891004 –4.4523438701 –6.0432049281 H5 –1.0046945462 4.4845621520 6.0463955788 H6 –0.0631166648 0.8778911324 0.0000000000 H7 –1.3416664479 5.8592396283 0.0000000000 H8 –0.0973380193 –0.9217258854 0.0000000000 H9 1.3921141350 –5.8443389906 0.0000000000 H10 –1.3052117282 5.7528079016 3.9031149259 H11 0.3674451868 –1.2009964371 –7.9201722769 H12 –0.2042926656 1.2366614074 –7.9215787610 H13 1.1351891004 –4.4523438701 6.0432049281 H14 1.4071574788 –5.7268842984 3.8988609688 H15 –1.0046945462 4.4845621520 –6.0463955788 H16 –1.3052117282 5.7528079016 –3.9031149259
Calculated positional parameters for the planar structure of 156:
Atom x y z
C1 0.0000000000 5.9449587962 –1.4100330467 C2 0.0000000000 5.9449587962 1.4100330467 C3 0.0000000000 4.7064763409 –0.7135992462 C4 0.0000000000 7.1519697690 –0.6913791040 C5 0.0000000000 7.1519697690 0.6913791040 C6 0.0000000000 4.7064763409 0.7135992462 C7 0.0000000000 3.4842966984 –1.4361053498 C8 0.0000000000 3.4842966984 1.4361053498 C9 0.0000000000 2.4482656017 2.0753950775 C10 0.0000000000 2.4482656017 –2.0753950775 C11 0.0000000000 6.0243504772 –2.8316035951 C12 0.0000000000 6.2157424677 –4.0258340773 C13 0.0000000000 6.0243504772 2.8316035951 C14 0.0000000000 6.2157424677 4.0258340773 C15 0.0000000000 1.2308200723 2.8084212247 C16 0.0000000000 –1.2244438342 4.2327300655 C17 0.0000000000 0.0000000000 2.1334826427 C18 0.0000000000 1.2244438342 4.2327300655

258
C19 0.0000000000 0.0000000000 4.9071111447 C20 0.0000000000 –1.2308200723 2.8084212247 C21 0.0000000000 1.2308200723 –2.8084212247 C22 0.0000000000 –1.2244438342 –4.2327300655 C23 0.0000000000 1.2244438342 –4.2327300655 C24 0.0000000000 0.0000000000 –2.1334826427 C25 0.0000000000 –1.2308200723 –2.8084212247 C26 0.0000000000 0.0000000000 –4.9071111447 C27 0.0000000000 –2.4482656017 2.0753950775 C28 0.0000000000 –3.4842966984 1.4361053498 C29 0.0000000000 2.4134336114 5.0151903592 C30 0.0000000000 3.3555751919 5.7735963669 C31 0.0000000000 –2.4134336114 5.0151903592 C32 0.0000000000 –3.3555751919 5.7735963669 C33 0.0000000000 2.4134336114 –5.0151903592 C34 0.0000000000 3.3555751919 –5.7735963669 C35 0.0000000000 –4.7064763409 –0.7135992462 C36 0.0000000000 –7.1519697690 0.6913791040 C37 0.0000000000 –5.9449587962 –1.4100330467 C38 0.0000000000 –4.7064763409 0.7135992462 C39 0.0000000000 –5.9449587962 1.4100330467 C40 0.0000000000 –7.1519697690 –0.6913791040 C41 0.0000000000 –6.0243504772 2.8316035951 C42 0.0000000000 –6.2157424677 4.0258340773 C43 0.0000000000 –2.4482656017 –2.0753950775 C44 0.0000000000 –3.4842966984 –1.4361053498 C45 0.0000000000 –2.4134336114 –5.0151903592 C46 0.0000000000 –3.3555751919 –5.7735963669 C47 0.0000000000 –6.0243504772 –2.8316035951 C48 0.0000000000 –6.2157424677 –4.0258340773 H1 0.0000000000 8.0856984692 –1.2429588285 H2 0.0000000000 8.0856984692 1.2429588285 H3 0.0000000000 6.3701544067 –5.0789644667 H4 0.0000000000 6.3701544067 5.0789644667 H5 0.0000000000 0.0000000000 1.0506309370 H6 0.0000000000 0.0000000000 5.9908294781 H7 0.0000000000 0.0000000000 –1.0506309370 H8 0.0000000000 0.0000000000 –5.9908294781 H9 0.0000000000 4.1886234325 6.4364573440 H10 0.0000000000 –4.1886234325 6.4364573440 H11 0.0000000000 4.1886234325 –6.4364573440 H12 0.0000000000 –8.0856984692 –1.2429588285 H13 0.0000000000 –8.0856984692 1.2429588285 H14 0.0000000000 –6.3701544067 5.0789644667 H15 0.0000000000 –4.1886234325 –6.4364573440 H16 0.0000000000 –6.3701544067 –5.0789644667

259
Calculated positional parameters for the non planar structure of 156:
Atom x y z
C1 –0.3245961271 –1.3747351375 –5.9585905776 C2 0.3245961271 1.3747351375 –5.9585905776 C3 –0.1816701840 –0.6880128539 –4.7233364017 C4 –0.1518228387 –0.6760334977 –7.1644860953 C5 0.1518228387 0.6760334977 –7.1644860953 C6 0.1816701840 0.6880128539 –4.7233364017 C7 –0.4291223859 –1.3559235193 –3.4959061152 C8 0.4291223859 1.3559235193 –3.4959061152 C9 0.7199694284 1.8993690062 –2.4475610330 C10 –0.7199694284 –1.8993690062 –2.4475610330 C11 –0.6184234277 –2.7679550069 –6.0023004320 C12 –0.8379804395 –3.9542356902 –6.0928628756 C13 0.6184234277 2.7679550069 –6.0023004320 C14 0.8379804395 3.9542356902 –6.0928628756 C15 1.1307556647 2.4981333285 –1.2268261067 C16 2.0486715103 3.5879044924 1.2228804457 C17 0.6836641529 1.9854963044 0.0000000000 C18 2.0486715103 3.5879044924 –1.2228804457 C19 2.4800781466 4.1116337867 0.0000000000 C20 1.1307556647 2.4981333285 1.2268261067 C21 –1.1307556647 –2.4981333285 –1.2268261067 C22 –2.0486715103 –3.5879044924 1.2228804457 C23 –2.0486715103 –3.5879044924 –1.2228804457 C24 –0.6836641529 –1.9854963044 0.0000000000 C25 –1.1307556647 –2.4981333285 1.2268261067 C26 –2.4800781466 –4.1116337867 0.0000000000 C27 0.7199694284 1.8993690062 2.4475610330 C28 0.4291223859 1.3559235193 3.4959061152 C29 2.5687806488 4.1324647137 –2.4315966586 C30 3.0511777113 4.6097324853 –3.4330383622 C31 2.5687806488 4.1324647137 2.4315966586 C32 3.0511777113 4.6097324853 3.4330383622 C33 –2.5687806488 –4.1324647137 –2.4315966586 C34 –3.0511777113 –4.6097324853 –3.4330383622 C35 –0.1816701840 –0.6880128539 4.7233364017 C36 0.1518228387 0.6760334977 7.1644860953 C37 –0.3245961271 –1.3747351375 5.9585905776 C38 0.1816701840 0.6880128539 4.7233364017 C39 0.3245961271 1.3747351375 5.9585905776 C40 –0.1518228387 –0.6760334977 7.1644860953

260
C41 0.6184234277 2.7679550069 6.0023004320 C42 0.8379804395 3.9542356902 6.0928628756 C43 –0.7199694284 –1.8993690062 2.4475610330 C44 –0.4291223859 –1.3559235193 3.4959061152 C45 –2.5687806488 –4.1324647137 2.4315966586 C46 –3.0511777113 –4.6097324853 3.4330383622 C47 –0.6184234277 –2.7679550069 6.0023004320 C48 –0.8379804395 –3.9542356902 6.0928628756
H1 –0.2653900212 –1.2133065580 –8.0998068395 H2 0.2653900212 1.2133065580 –8.0998068395 H3 –1.0226098201 –5.0023276796 –6.1437670822 H4 1.0226098201 5.0023276796 –6.1437670822 H5 –0.0187688578 1.1601031974 0.0000000000 H6 3.1861670320 4.9339098514 0.0000000000 H7 0.0187688578 –1.1601031974 0.0000000000 H8 –3.1861670320 –4.9339098514 0.0000000000 H9 3.4735006073 5.0077995198 –4.3262399399 H10 3.4735006073 5.0077995198 4.3262399399 H11 –3.4735006073 –5.0077995198 –4.3262399399 H12 –0.2653900212 –1.2133065580 8.0998068395 H13 0.2653900212 1.2133065580 8.0998068395 H14 1.0226098201 5.0023276796 6.1437670822 H15 –3.4735006073 –5.0077995198 4.3262399399 H16 –1.0226098201 –5.0023276796 6.1437670822
Calculated positional parameters for the planar structure of 191:
Atom x y z
C1 2.8711963134 5.3815371523 0.0000000000 C2 0.1439896360 6.0974099118 0.0000000000 C3 1.8831513144 4.3593797614 0.0000000000 C4 2.4831389471 6.7311388050 0.0000000000 C5 1.1454132895 7.0824683101 0.0000000000 C6 0.5020892745 4.7220803233 0.0000000000 C7 2.2783022617 2.9972071455 0.0000000000 C8 –0.5119250690 3.7274249856 0.0000000000 C9 –1.4066823140 2.9011504132 0.0000000000 C10 2.6572918029 1.8392794183 0.0000000000 C11 4.2649359078 5.0904641976 0.0000000000 C12 5.4666432998 4.9524513937 0.0000000000 C13 –1.2113145603 6.5343918890 0.0000000000 C14 –2.3193846322 7.0194146117 0.0000000000 C15 –2.4249882171 1.9090721031 0.0000000000

261
C16 –4.4116404484 –0.1210896353 0.0000000000 C17 –2.0765539612 0.5494075624 0.0000000000 C18 –3.8070874408 2.2533045944 0.0000000000 C19 –4.7632031427 1.2319697624 0.0000000000 C20 –3.0325787140 –0.4768450424 0.0000000000 C21 3.0077346712 0.4639737052 0.0000000000 C22 3.6836116788 –2.2331963786 0.0000000000 C23 4.3681046180 0.0473573191 0.0000000000 C24 1.9542278745 –0.5015561928 0.0000000000 C25 2.3175118866 –1.8212987434 0.0000000000 C26 4.7170190315 –1.3332671209 0.0000000000 C27 –2.6205132837 –1.8371520554 0.0000000000 C28 –2.2607858967 –3.0001420461 0.0000000000 C29 –4.2710484523 3.5991841646 0.0000000000 C30 –4.7700513450 4.7010685247 0.0000000000 C31 –5.4527097569 –1.0918637292 0.0000000000 C32 –6.3932087652 –1.8523225246 0.0000000000 C33 5.4383920854 0.9858245765 0.0000000000 C34 6.4186685454 1.6954685247 0.0000000000 C35 –0.4790406512 –4.7060500739 0.0000000000 C36 –2.2709890818 –6.7900739497 0.0000000000 C37 –0.0312860877 –6.0557714646 0.0000000000 C38 –1.8194556794 –4.3491705840 0.0000000000 C39 –2.7375707951 –5.4667597644 0.0000000000 C40 –0.9021157709 –7.1219729783 0.0000000000 C41 –4.1470907629 –5.2610973953 0.0000000000 C42 –5.3531675713 –5.1634839961 0.0000000000 C43 3.2761158501 –3.6665123213 0.0000000000 C44 1.3919019855 –5.6063608531 0.0000000000 C45 1.8807590806 –3.2521347584 0.0000000000 C46 3.7058713861 –4.9640048219 0.0000000000 C47 2.7008969929 –5.9979020668 0.0000000000 C48 0.9361318104 –4.2246280967 0.0000000000 H1 3.2536054766 7.4944750580 0.0000000000 H2 0.8491833296 8.1257256127 0.0000000000 H3 6.5221940021 4.8149802720 0.0000000000 H4 –3.3008641232 7.4312776504 0.0000000000 H5 –1.0277010448 0.2831554941 0.0000000000 H6 –5.8134269533 1.4997265382 0.0000000000 H7 0.9268818217 –0.1605788481 0.0000000000 H8 5.7640567418 –1.6156886388 0.0000000000 H9 –5.2028887619 5.6737849818 0.0000000000 H10 –7.2160147298 –2.5280935644 0.0000000000 H11 7.2750350430 2.3279788140 0.0000000000 H12 –0.5807676796 –8.1585869141 0.0000000000 H13 –3.0134505171 –7.5813951224 0.0000000000

262
H14 –6.4137229715 –5.0702556913 0.0000000000 H15 4.7588420538 –5.2290576756 0.0000000000 H16 2.9962955503 –7.0427896743 0.0000000000
Calculated positional parameters for the non planar structure of 191:
Atom x y z
C1 3.0438399320 5.3172268616 0.5038322060 C2 0.3580148273 6.0060401215 –0.0359774031 C3 2.1214748683 4.2990076432 0.1388525005 C4 2.6122968090 6.6515649874 0.5811379659 C5 1.2952963590 6.9909603578 0.3144607597 C6 0.7665499679 4.6486876677 –0.1267212412 C7 2.5394413416 2.9461194125 0.0487169561 C8 –0.1757115457 3.6469614396 –0.4764251487 C9 –0.9980761403 2.8045443687 –0.7800483665 C10 2.8983540211 1.7845931381 –0.0204495599 C11 4.4004905270 5.0143218228 0.8151364588 C12 5.5570458941 4.8151610182 1.1091756565 C13 –0.9938186340 6.3859435208 –0.2789514027 C14 –2.1337012436 6.7460464718 –0.4657352039 C15 –1.9194198181 1.7852352220 –1.1383905484 C16 –3.6795506715 –0.3105865323 –1.8784010917 C17 –1.7252478800 0.4800393534 –0.6669700901 C18 –3.0270691318 2.0465600078 –1.9942834842 C19 –3.8855583888 0.9955093327 –2.3369818819 C20 –2.5681491842 –0.5786125140 –1.0289336858 C21 3.2418405294 0.4074083637 –0.0488396759 C22 3.9199632175 –2.2871221514 0.0428834942 C23 4.6030569334 –0.0077965414 –0.0480039463 C24 2.1882558202 –0.5567686263 –0.0462859849 C25 2.5544912703 –1.8757197814 –0.0044272652 C26 4.9537956838 –1.3865788935 0.0069852575 C27 –2.2723624665 –1.8985154910 –0.5951923469 C28 –1.9765037997 –3.0355229078 –0.2806528122 C29 –3.2641932347 3.3462319517 –2.5270649051 C30 –3.4803473783 4.4361968563 –3.0056283955 C31 –4.5657012341 –1.3480723028 –2.2875391904 C32 –5.3245079633 –2.2165112810 –2.6534917931 C33 5.6581321864 0.9464450749 –0.1154214637 C34 6.5971658377 1.7055115845 –0.2005432561 C35 –0.2505361073 –4.7352405447 0.1330418550 C36 –2.0824715944 –6.7749803809 0.3031962600

263
C37 0.1753868100 –6.0804206944 0.3076182653 C38 –1.5789570165 –4.3681794726 –0.0004003920 C39 –2.5221805185 –5.4574473169 0.1072858866 C40 –0.7189021721 –7.1241262048 0.3984279162 C41 –3.9213807132 –5.2010841624 0.0206835520 C42 –5.1129360680 –4.9963132778 –0.0388592665 C43 3.5075829840 –3.7187404462 0.1384247514 C44 1.6071447210 –5.6460189569 0.2896241173 C45 2.1152817664 –3.3028939079 0.0733841225 C46 3.9259915475 –5.0134672479 0.2724429752 C47 2.9144370689 –6.0386906491 0.3561116270 C48 1.1663129157 –4.2688814564 0.1300872110 H1 3.3299302278 7.4156274626 0.8599672233 H2 0.9694188101 8.0234040491 0.3805422086 H3 6.5685085359 4.6138219094 1.3752477745 H4 –3.1460342054 7.0364619992 –0.6270031553 H5 –0.8928537237 0.2833079978 –0.0026247514 H6 –4.7262243507 1.1959542631 –2.9914801289 H7 1.1598364599 –0.2188382182 –0.0683807212 H8 6.0011054583 –1.6680881957 0.0239061989 H9 –3.6493718884 5.4000565363 –3.4270283714 H10 –5.9782167558 –2.9947389725 –2.9733673921 H11 7.4084939593 2.3898799054 –0.2890768666 H12 –0.4203325756 –8.1590450665 0.5316837423 H13 –2.8380558432 –7.5505566606 0.3757255898 H14 –6.1588929400 –4.7985552367 –0.0848746411 H15 4.9764449905 –5.2834722647 0.3238187583 H16 3.2054533978 –7.0784161743 0.4713502702
Calculated positional parameters for the planar structure of 192:
Atom x y z
C1 –0.8136479177 5.9906811026 0.0000000000 C2 –3.4226643095 5.0649522227 0.0000000000 C3 –1.0842381701 4.5942989444 0.0000000000 C4 –1.8123679356 6.9384785426 0.0000000000 C5 –3.1285257033 6.4366406201 0.0000000000 C6 –2.3709316796 4.0711676539 0.0000000000 C7 –2.6481092760 2.6799063827 0.0000000000 C8 –2.8806713238 1.4840520964 0.0000000000 C9 –4.7929590803 4.6753311957 0.0000000000 C10 –5.9720241706 4.4031055281 0.0000000000 C11 –3.0549184727 0.0751533737 0.0000000000 C12 –3.3732699383 –2.6873666128 0.0000000000

264
C13 –1.8866055576 –0.7456062659 0.0000000000 C14 –4.3494825920 –0.5135874079 0.0000000000 C15 –4.5152159072 –1.9281128434 0.0000000000 C16 –2.0727741130 –2.1013922308 0.0000000000 C17 2.0727741130 2.1013922308 0.0000000000 C18 4.3494825920 0.5135874079 0.0000000000 C19 3.3732699383 2.6873666128 0.0000000000 C20 1.8866055576 0.7456062659 0.0000000000 C21 3.0549184727 –0.0751533737 0.0000000000 C22 4.5152159072 1.9281128434 0.0000000000 C23 2.8806713238 –1.4840520964 0.0000000000 C24 2.6481092760 –2.6799063827 0.0000000000 C25 –5.5245147005 0.2898252024 0.0000000000 C26 –6.5646268783 0.9082607044 0.0000000000 C27 5.5245147005 –0.2898252024 0.0000000000 C28 6.5646268783 –0.9082607044 0.0000000000 C29 2.3709316796 –4.0711676539 0.0000000000 C30 1.8123679356 –6.9384785426 0.0000000000 C31 3.4226643095 –5.0649522227 0.0000000000 C32 1.0842381701 –4.5942989444 0.0000000000 C33 0.8136479177 –5.9906811026 0.0000000000 C34 3.1285257033 –6.4366406201 0.0000000000 C35 4.7929590803 –4.6753311957 0.0000000000 C36 5.9720241706 –4.4031055281 0.0000000000 C37 0.6554712990 5.7299240157 0.0000000000 C38 2.7800211778 4.0555200394 0.0000000000 C39 1.9017392742 6.2899482176 0.0000000000 C40 0.3838520968 4.3002826465 0.0000000000 C41 1.4503232806 3.4616941766 0.0000000000 C42 3.0347521443 5.3981938333 0.0000000000 C43 –0.6554712990 –5.7299240157 0.0000000000 C44 –2.7800211778 –4.0555200394 0.0000000000 C45 –0.3838520968 –4.3002826465 0.0000000000 C46 –1.9017392742 –6.2899482176 0.0000000000 C47 –3.0347521443 –5.3981938333 0.0000000000 C48 –1.4503232806 –3.4616941766 0.0000000000 H1 –1.6239400570 8.0073058974 0.0000000000 H2 –3.9651843819 7.1277266413 0.0000000000 H3 –7.0080026891 4.1574556985 0.0000000000 H4 –0.9134758470 –0.2739515473 0.0000000000 H5 –5.5165967921 –2.3446938745 0.0000000000 H6 0.9134758470 0.2739515473 0.0000000000 H7 5.5165967921 2.3446938745 0.0000000000 H8 –7.4758573519 1.4593392966 0.0000000000 H9 7.4758573519 –1.4593392966 0.0000000000 H10 3.9651843819 –7.1277266413 0.0000000000

265
H11 1.6239400570 –8.0073058974 0.0000000000 H12 7.0080026891 –4.1574556985 0.0000000000 H13 2.0565252047 7.3647717636 0.0000000000 H14 4.0435012685 5.8001155537 0.0000000000 H15 –2.0565252047 –7.3647717636 0.0000000000 H16 –4.0435012685 –5.8001155537 0.0000000000
Calculated positional parameters for the non planar structure of 192:
Atom x y z
C1 0.0888459014 6.1284891366 0.6897145792 C2 –2.3274962291 5.4293222368 1.8485452784 C3 –0.2986336905 4.7635855986 0.7866286668 C4 –0.6989114519 7.1551985747 1.1599180766 C5 –1.9188902772 6.7678065621 1.7488610480 C6 –1.5037513466 4.3539465592 1.3405428477 C7 –1.9160114319 2.9980398054 1.3961431100 C8 –2.2784519102 1.8352255720 1.4150048368 C9 –3.5750453290 5.1521773424 2.4784774395 C10 –4.6335529568 4.9683551222 3.0359557840 C11 –2.6161369239 0.4562132299 1.4007473387 C12 –3.2537370596 –2.2514342311 1.3741775255 C13 –1.6219691948 –0.4736507985 0.9713781613 C14 –3.9055627304 0.0025317226 1.7943967609 C15 –4.2329653048 –1.3838751609 1.7867757769 C16 –1.9651700589 –1.7982659308 0.9624113592 C17 2.2728247892 2.0051361919 –0.5974835778 C18 4.1425534837 0.2146452741 –1.5968909523 C19 3.4850054475 2.4692695891 –1.1893107217 C20 1.9829742444 0.6729955500 –0.4808461843 C21 2.9426143970 –0.2518427182 –0.9917769378 C22 4.4251758915 1.6074742218 –1.6944998470 C23 2.6573536308 –1.6383476507 –0.8821926922 C24 2.3393753588 –2.8075029453 –0.7562929545 C25 –4.9149135341 0.9222621843 2.1971108812 C26 –5.8214635081 1.6493860363 2.5353319172 C27 5.0944020456 –0.6968386320 –2.1355968293 C28 5.9366570514 –1.4149171039 –2.6255021891 C29 1.9527797883 –4.1671554073 –0.6408814936 C30 1.1394394073 –6.9672420528 –0.4824066330 C31 2.8436591258 –5.2519311932 –0.9904490731 C32 0.6978965280 –4.5697648441 –0.2050754913 C33 0.2988165449 –5.9330736397 –0.1365885369

266
C34 2.4287236108 –6.5895173664 –0.9070186687 C35 4.1772496108 –4.9865196603 –1.4158817544 C36 5.3231218509 –4.8148677721 –1.7658702483 C37 1.3668789686 5.7413568030 0.0223433366 C38 3.0903176764 3.8857019494 –0.9335105477 C39 2.5334819831 6.1871298057 –0.5316818725 C40 0.9785034776 4.3435233495 0.1306157226 C41 1.8480191616 3.4145387696 –0.3379147818 C42 3.4511436389 5.1992701303 –1.0434652097 C43 –1.0530017546 –5.5356532049 0.3568237676 C44 –2.8573550277 –3.6682640629 1.1226716089 C45 –0.6438363096 –4.1407665606 0.2983905957 C46 –2.2821243147 –5.9724266852 0.7632224477 C47 –3.2449175266 –4.9781244841 1.1687920729 C48 –1.5446721577 –3.2071816995 0.6930731500 H1 –0.4201658238 8.2021394350 1.0956940721 H2 –2.5846305198 7.5257707739 2.1487815094 H3 –5.5589903091 4.7938863242 3.5328574867 H4 –0.6496146782 –0.1045024379 0.6751682573 H5 –5.2236162837 –1.6963311969 2.0987531189 H6 1.0776145549 0.2942075573 –0.0265995460 H7 5.3523850258 1.9291321246 –2.1561859347 H8 –6.6134530398 2.3010282505 2.8217753091 H9 6.6654704449 –2.0577188562 –3.0609594699 H10 3.1462396579 –7.3546488828 –1.1851830784 H11 0.8531122446 –8.0130925106 –0.4365479674 H12 6.3325375094 –4.6520710676 –2.0631782499 H13 2.7830998153 7.2421729639 –0.5925019794 H14 4.3899715041 5.5105293601 –1.4915612089 H15 –2.5458017265 –7.0254323554 0.7905941505 H16 –4.2336827092 –5.2823659526 1.4987729371
Calculated positional parameters for the planar structure of 193:
Atom x y z
C1 0.0000000000 5.8430029800 –1.5252391965 C2 0.0000000000 5.9759994888 1.2443807100 C3 0.0000000000 4.6557024572 –0.7398399064 C4 0.0000000000 7.0987879797 –0.9586608113 C5 0.0000000000 7.1332274077 0.4490942109 C6 0.0000000000 4.6587122259 0.6489946065 C7 0.0000000000 3.4705769611 1.4284677156 C8 0.0000000000 2.4452697948 2.0860869823

267
C9 0.0000000000 6.1435246687 2.6591621138 C10 0.0000000000 6.3574564345 3.8502459933 C11 0.0000000000 1.2324668305 2.8301734790 C12 0.0000000000 –1.2261842897 4.2545381712 C13 0.0000000000 0.0000000000 2.1600834089 C14 0.0000000000 1.2261842897 4.2545381712 C15 0.0000000000 0.0000000000 4.9287041858 C16 0.0000000000 –1.2324668305 2.8301734790 C17 0.0000000000 1.1516057756 –2.6502915473 C18 0.0000000000 –1.1570894335 –4.0770411097 C19 0.0000000000 1.1570894335 –4.0770411097 C20 0.0000000000 0.0000000000 –1.8731594791 C21 0.0000000000 –1.1516057756 –2.6502915473 C22 0.0000000000 0.0000000000 –4.8561614675 C23 0.0000000000 –2.4452697948 2.0860869823 C24 0.0000000000 –3.4705769611 1.4284677156 C25 0.0000000000 2.4249361447 5.0224306384 C26 0.0000000000 3.4000909275 5.7378993808 C27 0.0000000000 –2.4249361447 5.0224306384 C28 0.0000000000 –3.4000909275 5.7378993808 C29 0.0000000000 –4.6557024572 –0.7398399064 C30 0.0000000000 –7.1332274077 0.4490942109 C31 0.0000000000 –5.8430029800 –1.5252391965 C32 0.0000000000 –4.6587122259 0.6489946065 C33 0.0000000000 –5.9759994888 1.2443807100 C34 0.0000000000 –7.0987879797 –0.9586608113 C35 0.0000000000 –6.1435246687 2.6591621138 C36 0.0000000000 –6.3574564345 3.8502459933 C37 0.0000000000 5.0388773588 –2.7828503425 C38 0.0000000000 2.6543382062 –4.0839518892 C39 0.0000000000 5.0608594092 –4.1469534028 C40 0.0000000000 3.8234827848 –1.9764628450 C41 0.0000000000 2.6382204133 –2.6273448866 C42 0.0000000000 3.7898795510 –4.8412073638 C43 0.0000000000 –5.0388773588 –2.7828503425 C44 0.0000000000 –2.6543382062 –4.0839518892 C45 0.0000000000 –5.0608594092 –4.1469534028 C46 0.0000000000 –3.8234827848 –1.9764628450 C47 0.0000000000 –2.6382204133 –2.6273448866 C48 0.0000000000 –3.7898795510 –4.8412073638 H1 0.0000000000 8.0171925630 –1.5370160825 H2 0.0000000000 8.0902654477 0.9605934245 H3 0.0000000000 6.5389428787 4.8993268569 H4 0.0000000000 0.0000000000 1.0794435370 H5 0.0000000000 0.0000000000 6.0126249960 H6 0.0000000000 0.0000000000 –0.7919042756

268
H7 0.0000000000 0.0000000000 –5.9414399327 H8 0.0000000000 4.2624351160 6.3626187568 H9 0.0000000000 –4.2624351160 6.3626187568 H10 0.0000000000 –8.0171925630 –1.5370160825 H11 0.0000000000 –8.0902654477 0.9605934245 H12 0.0000000000 –6.5389428787 4.8993268569 H13 0.0000000000 5.9857583821 –4.7160490687 H14 0.0000000000 3.7686486876 –5.9269406817 H15 0.0000000000 –5.9857583821 –4.7160490687 H16 0.0000000000 –3.7686486876 –5.9269406817
Calculated positional parameters for the non planar structure of 193:
Atom x y z
C1 0.5356749502 –1.4133961270 5.8440951182 C2 0.2289541362 1.3441561500 5.9655574382 C3 0.3393615094 –0.6480696593 4.6609955776 C4 0.5802343260 –0.8293566712 7.0916776250 C5 0.4241579105 0.5715077268 7.1208074585 C6 0.1729727183 0.7278706347 4.6606551997 C7 –0.0581065196 1.4687114412 3.4724037429 C8 –0.2948140789 2.0684824998 2.4406698054 C9 0.1021851513 2.7575108478 6.0969910739 C10 0.0124175493 3.9574044243 6.2290518994 C11 –0.6317062317 2.7283469190 1.2280916937 C12 –1.4097474369 3.9215183484 –1.2246142586 C13 –0.2436173357 2.1752345060 0.0000000000 C14 –1.4097474369 3.9215183484 1.2246142586 C15 –1.7707556208 4.4950336619 0.0000000000 C16 –0.6317062317 2.7283469190 –1.2280916937 C17 0.3583384360 –2.5531847313 1.1528782401 C18 0.5762112903 –3.9636204741 –1.1564709547 C19 0.5762112903 –3.9636204741 1.1564709547 C20 0.2297806617 –1.7847960764 0.0000000000 C21 0.3583384360 –2.5531847313 –1.1528782401 C22 0.6929240090 –4.7336319822 0.0000000000 C23 –0.2948140789 2.0684824998 –2.4406698054 C24 –0.0581065196 1.4687114412 –3.4724037429 C25 –1.8490037617 4.5246651211 2.4378695440 C26 –2.2460731417 5.0491480116 3.4532545810 C27 –1.8490037617 4.5246651211 –2.4378695440 C28 –2.2460731417 5.0491480116 –3.4532545810 C29 0.3393615094 –0.6480696593 –4.6609955776 C30 0.4241579105 0.5715077268 –7.1208074585

269
C31 0.5356749502 –1.4133961270 –5.8440951182 C32 0.1729727183 0.7278706347 –4.6606551997 C33 0.2289541362 1.3441561500 –5.9655574382 C34 0.5802343260 –0.8293566712 –7.0916776250 C35 0.1021851513 2.7575108478 –6.0969910739 C36 0.0124175493 3.9574044243 –6.2290518994 C37 0.6199632293 –2.6701984715 5.0388871366 C38 0.6320660449 –3.9678706364 2.6515478029 C39 0.8100112704 –4.0211687636 5.0529644371 C40 0.4173497800 –1.8799910477 3.8291227174 C41 0.4180829695 –2.5278242997 2.6416026538 C42 0.8190079680 –4.7122951867 3.7806099368 C43 0.6199632293 –2.6701984715 –5.0388871366 C44 0.6320660449 –3.9678706364 –2.6515478029 C45 0.8100112704 –4.0211687636 –5.0529644371 C46 0.4173497800 –1.8799910477 –3.8291227174 C47 0.4180829695 –2.5278242997 –2.6416026538 C48 0.8190079680 –4.7122951867 –3.7806099368 H1 0.7279187536 –1.3890130054 8.0098201873 H2 0.4568273013 1.0923567394 8.0722726456 H3 –0.0580860336 5.0158742273 6.3271902489 H4 0.3713288356 1.2840014063 0.0000000000 H5 –2.3673051051 5.4002134479 0.0000000000 H6 0.0523966689 –0.7168329193 0.0000000000 H7 0.8695605413 –5.8044132381 0.0000000000 H8 –2.5952606019 5.4921416604 4.3570482049 H9 –2.5952606019 5.4921416604 –4.3570482049 H10 0.7279187536 –1.3890130054 –8.0098201873 H11 0.4568273013 1.0923567394 –8.0722726456 H12 –0.0580860336 5.0158742273 –6.3271902489 H13 0.9626373030 –4.5803247606 5.9712371339 H14 0.9823557410 –5.7854410927 3.7518467551 H15 0.9626373030 –4.5803247606 –5.9712371339 H16 0.9823557410 –5.7854410927 –3.7518467551
Calculated positional parameters for the planar structure of 194:
Atom x y z
C1 0.0000000000 –1.3696624865 5.8159966245 C2 0.0000000000 1.3696624865 5.8159966245 C3 0.0000000000 –0.6769273629 4.5463695612 C4 0.0000000000 –0.7197169631 7.0276400390 C5 0.0000000000 0.7197169631 7.0276400390 C6 0.0000000000 0.6769273629 4.5463695612

270
C7 0.0000000000 2.7603818264 1.1847902765 C8 0.0000000000 4.2806285674 –1.1257214087 C9 0.0000000000 2.0812298642 –0.0012063555 C10 0.0000000000 4.1857322714 1.2602277519 C11 0.0000000000 4.9614187870 0.1297172874 C12 0.0000000000 2.8599837212 –1.1987847791 C13 0.0000000000 –2.7603818264 1.1847902765 C14 0.0000000000 –4.2806285674 –1.1257214087 C15 0.0000000000 –4.1857322714 1.2602277519 C16 0.0000000000 –2.0812298642 –0.0012063555 C17 0.0000000000 –2.8599837212 –1.1987847791 C18 0.0000000000 –4.9614187870 0.1297172874 C19 0.0000000000 –2.1613276724 –2.4353313922 C20 0.0000000000 2.1613276724 –2.4353313922 C21 0.0000000000 1.4575651756 –3.4311698232 C22 0.0000000000 –1.4575651756 –3.4311698232 C23 0.0000000000 5.0878849051 –2.2983657957 C24 0.0000000000 5.8606943976 –3.2298682592 C25 0.0000000000 –5.0878849051 –2.2983657957 C26 0.0000000000 –5.8606943976 –3.2298682592 C27 0.0000000000 –0.7144955156 –4.6404635726 C28 0.0000000000 0.6918280290 –7.0893945830 C29 0.0000000000 –1.4089466154 –5.8824550538 C30 0.0000000000 0.7144955156 –4.6404635726 C31 0.0000000000 1.4089466154 –5.8824550538 C32 0.0000000000 –0.6918280290 –7.0893945830 C33 0.0000000000 2.8312058597 –5.9536040122 C34 0.0000000000 4.0295493097 –6.1183560570 C35 0.0000000000 –2.8312058597 –5.9536040122 C36 0.0000000000 –4.0295493097 –6.1183560570 C37 0.0000000000 –2.6836024062 5.0841446331 C38 0.0000000000 –4.1198727887 2.7600356499 C39 0.0000000000 –4.0527178788 5.1719158503 C40 0.0000000000 –1.9650624431 3.8228573458 C41 0.0000000000 –2.6752984414 2.6711046425 C42 0.0000000000 –4.8106187844 3.9447229481 C43 0.0000000000 2.6836024062 5.0841446331 C44 0.0000000000 4.1198727887 2.7600356499 C45 0.0000000000 4.0527178788 5.1719158503 C46 0.0000000000 1.9650624431 3.8228573458 C47 0.0000000000 2.6752984414 2.6711046425 C48 0.0000000000 4.8106187844 3.9447229481 H1 0.0000000000 –1.2545347832 7.9729898032 H2 0.0000000000 1.2545347832 7.9729898032 H3 0.0000000000 1.0012480122 –0.0750644879 H4 0.0000000000 6.0459559064 0.1397923398

271
H5 0.0000000000 –1.0012480122 –0.0750644879 H6 0.0000000000 –6.0459559064 0.1397923398 H7 0.0000000000 6.5339533895 –4.0547805157 H8 0.0000000000 –6.5339533895 –4.0547805157 H9 0.0000000000 –1.2434871437 –8.0232070412 H10 0.0000000000 1.2434871437 –8.0232070412 H11 0.0000000000 5.0864427774 –6.2446191172 H12 0.0000000000 –5.0864427774 –6.2446191172 H13 0.0000000000 –4.5753455106 6.1239414018 H14 0.0000000000 –5.8959382425 3.9847084762 H15 0.0000000000 4.5753455106 6.1239414018 H16 0.0000000000 5.8959382425 3.9847084762
Calculated positional parameters for the non planar structure of 194:
Atom x y z
C1 0.5932254918 5.7775545854 –1.3698624762 C2 0.5932254918 5.7775545854 1.3698624762 C3 0.4112288747 4.5207626356 –0.6772536870 C4 0.7552704555 6.9782056486 –0.7195154095 C5 0.7552704555 6.9782056486 0.7195154095 C6 0.4112288747 4.5207626356 0.6772536870 C7 0.1729429935 1.1656478665 2.7550096293 C8 0.1638252780 –1.1593498588 4.2518660498 C9 –0.0036338284 –0.0058773651 2.0739845042 C10 0.3206443229 1.2221160023 4.1733953118 C11 0.3287236484 0.0824627611 4.9368673535 C12 –0.0186788452 –1.2090265563 2.8419476243 C13 0.1729429935 1.1656478665 –2.7550096293 C14 0.1638252780 –1.1593498588 –4.2518660498 C15 0.3206443229 1.2221160023 –4.1733953118 C16 –0.0036338284 –0.0058773651 –2.0739845042 C17 –0.0186788452 –1.2090265563 –2.8419476243 C18 0.3287236484 0.0824627611 –4.9368673535 C19 –0.2336468289 –2.4283308346 –2.1465326241 C20 –0.2336468289 –2.4283308346 2.1465326241 C21 –0.4579835413 –3.4039625846 1.4511355470 C22 –0.4579835413 –3.4039625846 –1.4511355470 C23 0.2161879220 –2.3520134853 5.0277923203 C24 0.3040765135 –3.3215305338 5.7472723521 C25 0.2161879220 –2.3520134853 –5.0277923203 C26 0.3040765135 –3.3215305338 –5.7472723521 C27 –0.7514407799 –4.5806983881 –0.7138011322

272
C28 –1.3715975253 –6.9478101712 0.6923369219 C29 –1.0736138040 –5.7790541278 –1.4099823023 C30 –0.7514407799 –4.5806983881 0.7138011322 C31 –1.0736138040 –5.7790541278 1.4099823023 C32 –1.3715975253 –6.9478101712 –0.6923369219 C33 –1.1307834069 –5.8254256449 2.8324402008 C34 –1.2224989391 –5.9358525522 4.0336804208 C35 –1.1307834069 –5.8254256449 –2.8324402008 C36 –1.2224989391 –5.9358525522 –4.0336804208 C37 0.5644790473 5.0439832274 –2.6825071416 C38 0.4378340888 2.7185637547 –4.1134017017 C39 0.6811544141 5.1183015446 –4.0474886205 C40 0.3756271202 3.7961526093 –1.9654482719 C41 0.3018818192 2.6457639137 –2.6741988848 C42 0.6163093227 3.8906791980 –4.8022086014 C43 0.5644790473 5.0439832274 2.6825071416 C44 0.4378340888 2.7185637547 4.1134017017 C45 0.6811544141 5.1183015446 4.0474886205 C46 0.3756271202 3.7961526093 1.9654482719 C47 0.3018818192 2.6457639137 2.6741988848 C48 0.6163093227 3.8906791980 4.8022086014 H1 0.8981984369 7.9127345481 –1.2540241720 H2 0.8981984369 7.9127345481 1.2540241720 H3 –0.1332141653 –0.0631884533 1.0008267059 H4 0.4576441173 0.0767096113 6.0137075167 H5 –0.1332141653 –0.0631884533 –1.0008267059 H6 0.4576441173 0.0767096113 –6.0137075167 H7 0.3923739987 –4.1860391603 6.3630014646 H8 0.3923739987 –4.1860391603 –6.3630014646 H9 –1.6127361907 –7.8506348361 –1.2427926472 H10 –1.6127361907 –7.8506348361 1.2427926472 H11 –1.3072415065 –6.0030176207 5.0929371862 H12 –1.3072415065 –6.0030176207 –5.0929371862 H13 0.8302442505 6.0592692739 –4.5686682993 H14 0.7184184366 3.9200761632 –5.8829462072 H15 0.8302442505 6.0592692739 4.5686682993 H16 0.7184184366 3.9200761632 5.8829462072
Calculated positional parameters for the planar structure of 195:
Atom x y z
C1 0.8836671466 5.9290591622 0.0000000000 C2 3.4774461520 4.9589371166 0.0000000000 C3 1.1305946529 4.5256716396 0.0000000000

273
C4 1.8996756690 6.8593324486 0.0000000000 C5 3.2064450430 6.3356803144 0.0000000000 C6 2.4112565450 3.9809331424 0.0000000000 C7 2.6845395830 2.5869633996 0.0000000000 C8 2.9457490159 1.3957926327 0.0000000000 C9 4.8409564734 4.5460555593 0.0000000000 C10 6.0133123058 4.2462055748 0.0000000000 C11 3.1059162873 –0.0171614826 0.0000000000 C12 3.3505792884 –2.8026495017 0.0000000000 C13 1.9202907069 –0.8121516851 0.0000000000 C14 4.3820473958 –0.6451824038 0.0000000000 C15 4.5100700968 –2.0682631042 0.0000000000 C16 2.0716448866 –2.1703072321 0.0000000000 C17 –2.0350276033 2.0941973689 0.0000000000 C18 –4.2661667412 0.5324102284 0.0000000000 C19 –3.3422671061 2.6628333253 0.0000000000 C20 –1.7788960732 0.7306582931 0.0000000000 C21 –2.9481263978 –0.0138360315 0.0000000000 C22 –4.5188683362 1.9059852584 0.0000000000 C23 5.5790223311 0.1258060620 0.0000000000 C24 6.6320230468 0.7226114794 0.0000000000 C25 –2.4249295048 –3.8978746973 0.0000000000 C26 –2.1246915950 –6.7254817413 0.0000000000 C27 –3.5671602097 –4.7861544546 0.0000000000 C28 –1.1797408100 –4.4326663056 0.0000000000 C29 –1.0460570147 –5.8707607999 0.0000000000 C30 –3.4467996167 –6.1571919417 0.0000000000 C31 –0.5900729379 5.6977887221 0.0000000000 C32 –2.7470493142 4.0412584721 0.0000000000 C33 –1.8301972820 6.2670996708 0.0000000000 C34 –0.3402542349 4.2607484084 0.0000000000 C35 –1.4178553645 3.4427766086 0.0000000000 C36 –2.9805675349 5.3858195106 0.0000000000 C37 0.4515115098 –5.7189226539 0.0000000000 C38 2.6941788838 –4.1546103317 0.0000000000 C39 0.2915338394 –4.2756565974 0.0000000000 C40 1.6739521905 –6.3417343961 0.0000000000 C41 2.8574295423 –5.5160497194 0.0000000000 C42 1.4019500959 –3.5006442237 0.0000000000 C43 –4.4825139138 –3.5888612466 0.0000000000 C44 –4.8446153753 –0.8647020855 0.0000000000 C45 –5.7641187041 –3.1040012011 0.0000000000 C46 –3.3140106457 –2.7235556983 0.0000000000 C47 –3.4944364702 –1.3868969011 0.0000000000 C48 –5.9549424628 –1.6665810716 0.0000000000 H1 1.7292912200 7.9312432041 0.0000000000

274
H2 4.0548517825 7.0123137479 0.0000000000 H3 7.0430486157 3.9752320391 0.0000000000 H4 0.9591533221 –0.3180604104 0.0000000000 H5 5.5024077269 –2.5063264223 0.0000000000 H6 –0.7876046024 0.2994260875 0.0000000000 H7 –5.5140442905 2.3393800978 0.0000000000 H8 7.5563458217 1.2517289270 0.0000000000 H9 –4.3106537558 –6.8158075194 0.0000000000 H10 –2.0082894872 –7.8055068472 0.0000000000 H11 –1.9770615809 7.3431930637 0.0000000000 H12 –3.9817235893 5.8066350190 0.0000000000 H13 1.7764610098 –7.4230442560 0.0000000000 H14 3.8374826076 –5.9842505851 0.0000000000 H15 –6.6344941026 –3.7538881683 0.0000000000 H16 –6.9644962531 –1.2656683060 0.0000000000
Calculated positional parameters for the non planar structure of 195:
Atom x y z
C1 0.9170846128 5.9025314297 –0.3107939631 C2 3.4713977072 4.9373058949 0.1554948180 C3 1.1448308933 4.5059197285 –0.1480452280 C4 1.9334793752 6.8292633612 –0.2399313303 C5 3.2200255118 6.3083464545 –0.0040657750 C6 2.4027027212 3.9647339136 0.0937354551 C7 2.6489483986 2.5775562696 0.2712792217 C8 2.8828225611 1.3931328526 0.4429701096 C9 4.8180667784 4.5190991233 0.3592923282 C10 5.9774009118 4.2055830372 0.5099727436 C11 3.0214433331 –0.0109628907 0.6213051390 C12 3.2225031706 –2.7737599938 0.9987371970 C13 1.8903935759 –0.8288682515 0.3221685596 C14 4.2213692282 –0.6041928887 1.1038889522 C15 4.3302862133 –2.0166271075 1.2883168899 C16 2.0134229452 –2.1733779849 0.5362218327 C17 –1.9860749262 2.0614992830 –0.5106663851 C18 –4.1737729933 0.4826541338 –0.8796981561 C19 –3.2645603352 2.6178796069 –0.8101447035 C20 –1.7397760793 0.7023339525 –0.3748876800 C21 –2.8843594149 –0.0523264240 –0.5831581724 C22 –4.4184747746 1.8517320859 –1.0049509747 C23 5.3480792818 0.2003670857 1.4387430689 C24 6.3296786049 0.8336569578 1.7566880283 C25 –2.3902701393 –3.9306127735 –0.2735728445

275
C26 –2.1434894254 –6.7392409418 0.0951607242 C27 –3.5223617406 –4.8203517141 –0.4127060584 C28 –1.1769168606 –4.4570760655 0.0213823007 C29 –1.0726843689 –5.8839076374 0.2196170923 C30 –3.4268468996 –6.1825594700 –0.2415060499 C31 –0.5397971982 5.6658458071 –0.5303435666 C32 –2.6742677485 3.9984124057 –0.7875237790 C33 –1.7542607434 6.2238245560 –0.8042022066 C34 –0.3078129886 4.2359345516 –0.3625848082 C35 –1.3736695740 3.4124969946 –0.4876536449 C36 –2.8918862586 5.3368018848 –0.9424364364 C37 0.3849794477 –5.7174152305 0.5570270802 C38 2.5761608860 –4.1303695044 0.9596578527 C39 0.2589468009 –4.2882460990 0.3337333614 C40 1.5502303371 –6.3151926829 0.9652397924 C41 2.7045892060 –5.4768733790 1.1833053767 C42 1.3490798305 –3.5053014322 0.5115447765 C43 –4.4067070480 –3.6322072793 –0.6880413463 C44 –4.7454441369 –0.9157902612 –0.9190852416 C45 –5.6628047800 –3.1552313882 –0.9553299769 C46 –3.2516577090 –2.7631187797 –0.5291765588 C47 –3.4231130484 –1.4287318520 –0.6284061332 C48 –5.8404256977 –1.7219826830 –1.0822210828 H1 1.7794001321 7.8966683864 –0.3610863309 H2 4.0681727978 6.9827091963 0.0544115336 H3 6.9940758581 3.9136794603 0.6332017526 H4 0.9919962265 –0.3635748392 –0.0596701337 H5 5.2645482198 –2.4280556989 1.6549264816 H6 –0.7741419601 0.2827077354 –0.1279228411 H7 –5.3917676548 2.2753209102 –1.2314733681 H8 7.1818617574 1.4049969580 2.0417644237 H9 –4.2847166707 –6.8413416212 –0.3399255702 H10 –2.0511455246 –7.8100926596 0.2511695818 H11 –1.8896357313 7.2942205787 –0.9276922596 H12 –3.8707523308 5.7486902800 –1.1693285910 H13 1.6262318284 –7.3844057545 1.1395927393 H14 3.6341358485 –5.9239638697 1.5231756968 H15 –6.5234258543 –3.8077895990 –1.0697562150 H16 –6.8295511739 –1.3268599363 –1.2948424372

276
6.5 Experiments Related to Chapter 5
1,1'-(1,2-Ethynediyl)bis[2-bromo-6-iodo]benzene (210):
I
IBr
Br
A solution of 2,2’,6,6’-tetrabromotolane (209)202 (100 mg, 0.20 mmol) in ether
(60 mL) was cooled to –45 °C, and BuLi (0.28 mL of 1.6 M solution in hexane, 0.45
mmol) was added via syringe. The dark brown solution was stirred at –45 °C for 1 h.
After that time, an ethereal solution (10 mL) of iodine (178 mg, 0.70 mmol) was added
dropwise via syringe. The color of the solution lightened gradually with addition of
iodine. The mixture was left to warm to 23 °C overnight, and was then extracted with
ether (2 x 50 mL) and washed with aq. Na2S2O3 and brine. Drying over MgSO4, followed
by removal of solvent in vacuo, gave the crude 210 as yellow solid. After
recrystallization (CHCl3/MeOH), the product was obtained as off-white needles, mp 187–
190 °C (90 mg, 75%). MS (EI, 70 eV) m/z (rel intensity) 588 (M+, 72), 382 (18), 380
(18), 334 (15), 174 (100), 74 (14). 1H NMR (400 MHz, CDCl3) δ 7.85 (dd, 3J1 = 7.9 Hz,
4J2 = 0.9 Hz, 2H), 7.62 (dd, 3J1 = 8.0 Hz, 4J2 = 0.9 Hz, 2H), 6.87 (t, 3J = 8.0 Hz, 2H). 13C
NMR (125 MHz, CDCl3) δ 138.10, 132.43, 130.59, 130.56, 125.94, 101.22, 97.12. HR-
MS Calcd for C14H6Br2I2: 587.6905. Found: 587.6892. Anal. Calcd for C14H6Br2I2: C,
28.61; H, 1.03. Found: C, 28.90; H, 0.97.

277
1,1'-(1,2-Ethynediyl)bis[2-bromo-6-({dimethylthexylsilyl}ethynyl)]benzene (211):
Br
Br
DMTS
DMTS
A solution of 210 (114 mg, 0.19 mmol), [Pd(PPh3)2Cl2] (10.0 mg, 0.015 mmol),
and CuI (3.0 mg, 0.015 mmol) in triethylamine (25 mL) was degassed thoroughly.
DMTSA209 (372 mg, 2.21 mmol) was injected through a septum and the mixture stirred at
23 °C for 20 h. Solvent was removed in vacuo and the resulting crude product subjected
to sublimation (200 °C, 0.5 Torr), which removed DMTS–≡–≡–DMTS side product and
yielded pure 211 as a yellow oil (69 mg, 52%). IR (NaCl film): ~ν = 2959, 2865, 2164,
1544, 1459, 1442, 1249, 1130, 880, 838, 815, 731, 681 cm–1. MS (EI, 70 eV) m/z (rel
intensity) 668 (M+, 1.5), 584 (2), 531 (1), 499 (60), 73 (100). 1H NMR (300 MHz,
CDCl3) δ 7.56 (dd, 3J1 = 8.1 Hz, 4J2 = 1.1 Hz, 2H), 7.44 (dd, 3J1 = 7.8 Hz, 4J2 = 1.1 Hz,
2H), 7.11 (t, 3J = 7.9 Hz, 2H), 1.62 (sept, 3J = 6.9 Hz, 2H), 0.80 (s, 12H), 0.77 (d, 3J = 6.9
Hz, 12H), 0.14 (s, 12H). 13C NMR (100 MHz, CDCl3) δ 131.98, 130.85, 128.70, 128.20,
127.63, 125.60, 103.19, 99.97, 94.72, 34.41, 23.27, 20.51, 18.51, –2.52. HR-MS Calcd
for C34H44Br2Si2: 668.1328. Found: 668.1311.

278
1,1'-(1,2-Ethynediyl)bis[2-({dimethylthexylsilyl}ethynyl)-6-iodo]benzene (212):
I
I
DMTS
DMTS
A solution of 211 (50 mg, 0.07 mmol) in ether (20 mL) was cooled to –45 °C, and
BuLi (0.18 mL of 1.6 M solution in hexane, 0.29 mmol) was added via syringe. The
brownish solution was stirred at –45 °C for 1 h. After that time, an ethereal solution (10
mL) of iodine (101 mg, 0.40 mmol) was added dropwise via syringe. The color of the
solution lightened gradually with addition of iodine. The mixture was left to warm to 23
°C overnight, then extracted with ether (2 x 50 mL), and washed with aq. Na2S2O3 and
brine. Drying over MgSO4, followed by removal of solvent in vacuo gave a brown oil,
which was purified by filtration through a short plug of silica (eluting with CHCl3), to
yield 212 as a yellow oil (51 mg, 92%). IR (NaCl film): ~ν = 2958, 2925, 2865, 2161,
1540, 1452, 1391, 1378, 1250, 1037, 875, 837, 814, 779, 679 cm–1. MS (EI, 70 eV) m/z
(rel intensity) 762 (M+, 0.2), 592 (44), 467 (36), 341 (19), 73 (100). 1H NMR (500 MHz,
CDCl3) δ 7.83 (dd, 3J1 = 8.1 Hz, 4J2 = 1.1 Hz, 2H), 7.48 (dd, 3J1 = 7.9 Hz, 4J2 = 1.1 Hz,
2H), 6.95 (t, 3J = 7.9 Hz, 2H), 1.64 (sept, 3J = 6.9 Hz, 2H), 0.80 (s, 12H), 0.77 (d, 3J = 6.9
Hz, 12H), 0.14 (s, 12H). 13C NMR (125 MHz, CDCl3) δ 138.28, 131.85, 131.53, 128.74,
127.41, 103.46, 100.63, 99.73, 97.26, 34.36, 23.27, 20.51, 18.50, –2.49. HR-MS Calcd
for C34H44I2Si2: 762.1071. Found: 762.1057.

279
1,1'-(1,2-Ethynediyl)bis[2-({dimethylthexylsilyl}ethynyl)-6-({trimethylsilyl}ethynyl)]
benzene (205b):
DMTS
DMTSTMS
TMS
A solution of 212 (22 mg, 0.03 mmol), [Pd(PPh3)2Cl2] (2.1 mg, 0.003 mmol), and
CuI (0.6 mg, 0.003 mmol) in triethylamine (15 mL) was degassed in a 50 mL Schlenk
tube. TMSA (420 µL, 294 mg, 3.00 mmol) was added via syringe and the tube closed.
The mixture was heated at 100 °C for 72 h. The crude material was purified repeatedly by
column chromatography (petroleum ether/CH2Cl2) and then subjected to Kugelrohr
distillation (225 °C, 0.8 Torr), giving 205b as colorless oil (3.0 mg, 15%). IR (NaCl
film): ~ν = 2960, 2927, 2156, 1455, 1250, 979, 842, 797, 761, 740 cm–1. MS (EI, 70 eV)
m/z (rel intensity) 702 (M+, 0.5), 617 (2), 533 (8), 445 (28), 371 (18), 73 (100). 1H NMR
(500 MHz, CDCl3) δ 7.40 (t, 3J = 8.0 Hz, 4H), 7.17 (t, 3J = 7.9 Hz, 2H), 1.63 (sept, 3J =
6.8 Hz, 2H), 0.79 (s, 6H), 0.79 (s, 6H), 0.74 (d, 3J = 6.9 Hz, 6H), 0.73 (d, 3J = 6.9 Hz,
6H), 0.14 (s, 6H), 0.13 (s, 6H), 0.10 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 131.31,
130.71, 129.72, 127.15, 126.20, 126.12, 103.60, 103.21, 99.43, 98.73, 96.08, 34.35 (2C),
23.24 (2C), 20.43, 20.41, 18.50, 18.47, –0.33, –2.43 (2C). HR-MS Calcd for C44H62Si4:
702.3929. Found: 702.3926.

280
6.5.1 Calculated Structures of Transition States for the Inversion of 171c and 213-
216
Calculated positional parameters of the transition state for the inversion of 213:
Atom x y z
Si1 -0.02330 5.18210 -2.55140 Si2 1.62310 5.14290 2.28510 C1 -1.24410 0.32050 -1.04270 C2 -1.99320 1.50230 -1.19840 C3 -3.36990 1.44670 -1.45770 C4 -4.01690 0.21890 -1.56010 C5 -3.28540 -0.95820 -1.42150 C6 -1.90800 -0.91680 -1.15410 C7 1.19950 0.40980 -0.03710 C8 0.10260 0.38210 -0.53020 C9 -0.71670 -3.24220 -0.80430 C10 -1.23710 -2.17200 -0.96960 C11 1.11090 -6.97520 -0.04650 C12 -0.27800 -6.89230 -0.16800 C13 -0.88550 -5.66100 -0.42710 C14 -0.10360 -4.51340 -0.57580 C15 1.28440 -4.60180 -0.47370 C16 1.89140 -5.82800 -0.19550 C17 5.35230 0.19710 0.71730 C18 4.58850 -0.96530 0.61170 C19 3.20080 -0.87420 0.46150 C20 2.56320 0.37590 0.41550 C21 3.33940 1.53970 0.53070 C22 4.72870 1.44520 0.68940 C23 5.81900 -3.29330 0.61010 C24 5.23830 -2.24080 0.62000 C25 4.49930 -5.85340 0.13080 C26 3.30980 -5.88150 -0.03340 C27 -0.80350 3.78420 -1.71680 C28 -1.34300 2.77320 -1.35770 C29 2.25120 3.82090 1.22710 C30 2.72770 2.81800 0.76790 C31 8.04980 -6.89650 0.54230 C32 6.67090 -6.94140 0.32890 C33 5.91300 -5.76380 0.34910 C34 6.54520 -4.53230 0.58510

281
C35 7.92950 -4.49720 0.79820 C36 8.67850 -5.67550 0.77710 C37 -0.45070 4.90780 -4.65530 C38 -0.81330 6.81530 -2.06800 C39 1.71650 6.83290 1.49160 C40 2.59140 5.17080 3.90660 C41 -0.42380 4.72650 2.78860 C42 1.82340 5.23950 -2.31310 C43 -0.66170 5.23870 4.22820 C44 -1.28230 5.57710 1.81690 C45 -0.70580 3.18270 2.61430 C46 -0.01410 2.29100 3.65630 C47 -2.20450 2.83940 2.62580 C48 0.15910 6.11180 -5.40760 C49 -2.00040 4.97820 -4.75600 C50 0.11590 3.50870 -5.12310 C51 1.61990 3.50770 -5.43370 C52 -0.60590 2.95810 -6.36470 H1 -3.93350 2.36390 -1.61580 H2 -5.08320 0.17890 -1.76680 H3 -3.79780 -1.91470 -1.50630 H4 1.57620 -7.93350 0.17140 H5 -0.88770 -7.78440 -0.04970 H6 -1.96860 -5.60250 -0.50490 H7 1.89580 -3.71340 -0.60040 H8 6.43170 0.13970 0.83650 H9 2.61310 -1.77920 0.33940 H10 5.32640 2.34420 0.82290 H11 8.63200 -7.81450 0.52530 H12 6.19000 -7.90000 0.14710 H13 8.43200 -3.55000 0.98140 H14 9.75240 -5.63940 0.94340 H15 -1.89760 6.78720 -2.20650 H16 -0.61860 7.04190 -1.01660 H17 -0.40990 7.63530 -2.66870 H18 1.13700 6.87250 0.56730 H19 1.33110 7.60100 2.16880 H20 2.75290 7.08750 1.24970 H21 3.65370 5.34150 3.70750 H22 2.23430 5.97580 4.55540 H23 2.49220 4.22280 4.44180 H24 2.30160 5.88510 -3.05490 H25 2.07370 5.63650 -1.32630 H26 2.26590 4.24200 -2.38480 H27 -1.70380 5.08930 4.52950 H28 -0.02820 4.72900 4.96030

282
H29 -0.45010 6.31150 4.30790 H30 -2.34820 5.50100 2.05610 H31 -1.03120 6.64160 1.87600 H32 -1.14390 5.25570 0.78200 H33 -0.33520 2.87720 1.62870 H34 1.06590 2.44710 3.68640 H35 -0.41420 2.45850 4.66130 H36 -0.17340 1.23340 3.41560 H37 -2.68610 3.16360 3.55330 H38 -2.72870 3.29980 1.78310 H39 -2.35410 1.75760 2.53270 H40 -0.00520 6.01600 -6.48630 H41 -0.29910 7.05550 -5.09130 H42 1.23610 6.20480 -5.23730 H43 -2.32810 4.95490 -5.80100 H44 -2.47590 4.14180 -4.23240 H45 -2.39560 5.90640 -4.33030 H46 -0.05030 2.77690 -4.32300 H47 2.21730 3.88800 -4.60600 H48 1.96590 2.48700 -5.63410 H49 1.84840 4.10710 -6.32140 H50 -0.56300 3.66560 -7.19910 H51 -0.14810 2.01950 -6.69730 H52 -1.65570 2.73490 -6.15100
Calculated positional parameters of the transition state for the inversion of 214:
Atom x y z
Si1 1.09000 4.65790 -2.70140 Si2 0.48650 5.20590 2.48980 Si3 9.04620 1.15870 3.54370 Si4 10.91290 -0.93300 -0.55990 C1 -1.35760 0.53680 -0.50690 C2 -1.81300 1.69340 -1.16380 C3 -3.15840 1.80090 -1.53740 C4 -4.04730 0.75590 -1.28250 C5 -3.58930 -0.41710 -0.68280 C6 -2.24740 -0.53500 -0.30520 C7 1.12110 0.46110 0.40150 C8 -0.00140 0.45890 -0.02800 C9 -1.22390 -2.81000 0.51380 C10 -1.77320 -1.78970 0.19960 C11 1.28520 -6.09280 1.34790 C12 -0.03880 -6.10590 1.79690

283
C13 -0.88210 -5.02720 1.52290 C14 -0.40050 -3.94330 0.79030 C15 0.91840 -3.93950 0.32430 C16 1.76710 -5.00590 0.61370 C17 5.21390 0.74610 1.42530 C18 4.66140 -0.50890 1.13540 C19 3.29430 -0.57970 0.81790 C20 2.47640 0.55850 0.86030 C21 3.01540 1.78180 1.27700 C22 4.38540 1.87310 1.53880 C23 6.01890 -2.64690 0.41100 C24 5.46060 -1.65890 0.81050 C25 4.31090 -4.94680 -0.05380 C26 3.13670 -4.96320 0.20200 C27 -0.12790 3.55980 -1.94860 C28 -0.91190 2.74440 -1.54510 C29 1.50440 3.86630 1.83600 C30 2.19320 2.93360 1.52140 C31 7.65830 -6.49160 -0.40930 C32 6.28140 -6.29420 -0.43880 C33 5.73740 -5.01910 -0.21320 C34 6.58230 -3.90550 -0.01340 C35 7.96910 -4.13300 0.06910 C36 8.49880 -5.41480 -0.14720 C37 1.69660 3.69570 -4.53460 C38 0.31300 6.30120 -3.18500 C39 0.43810 6.68880 1.34530 C40 1.11180 5.73920 4.18410 C41 -1.52650 4.49690 2.74740 C42 2.59160 4.95730 -1.63990 C43 -2.12560 5.23860 3.96480 C44 -2.29550 4.93730 1.47360 C45 -1.51750 2.92680 2.90690 C46 -0.89540 2.42580 4.21870 C47 -2.91690 2.30130 2.78520 C48 2.76920 4.59170 -5.19300 C49 0.42130 3.69500 -5.42330 C50 2.19810 2.23050 -4.21600 C51 3.62960 2.15530 -3.66540 C52 2.12970 1.29870 -5.43780 C53 7.62330 0.99950 2.43950 C54 6.57110 0.87860 1.87340 C55 9.71570 -2.17400 -0.02530 C56 8.90570 -3.04610 0.13430 C57 8.27690 2.02990 5.36770 C58 7.64280 3.38880 4.99050

284
C59 7.15340 1.05900 5.82870 C60 10.35460 2.28240 2.83930 C61 11.95270 -1.71980 -2.23600 C62 13.19590 -0.81620 -2.45740 C63 12.46150 -3.12890 -1.81440 C64 11.09050 -1.83100 -3.55310 C65 10.83090 -0.50900 -4.28830 C66 9.76990 -2.59820 -3.41670 C67 12.21800 -0.63400 0.75990 C68 10.07830 0.66340 -1.04190 C69 9.73320 -0.51980 4.01520 C70 9.44320 2.14240 6.42620 C71 8.93200 2.16840 7.87680 C72 10.34750 3.37020 6.24260 H1 -3.51480 2.69530 -2.04410 H2 -5.08930 0.84410 -1.57990 H3 -4.27850 -1.24430 -0.52780 H4 1.93920 -6.92900 1.58440 H5 -0.40860 -6.95180 2.37110 H6 -1.90620 -5.03560 1.88720 H7 1.28390 -3.09450 -0.25390 H8 2.87620 -1.52480 0.48050 H9 4.80800 2.82070 1.86860 H10 8.07370 -7.48150 -0.57830 H11 5.62630 -7.14880 -0.59760 H12 9.57620 -5.56850 -0.13820 H13 -0.59390 6.15100 -3.77730 H14 0.04040 6.86900 -2.29020 H15 1.01310 6.90480 -3.76930 H16 0.05850 6.41350 0.35770 H17 -0.20570 7.47310 1.75450 H18 1.44070 7.10810 1.21790 H19 2.15000 6.07830 4.11470 H20 0.51220 6.56630 4.57520 H21 1.07090 4.91400 4.89990 H22 3.40560 5.40290 -2.21840 H23 2.35390 5.64690 -0.82510 H24 2.95750 4.03210 -1.18940 H25 -3.17010 4.95080 4.12350 H26 -1.57870 5.02840 4.88890 H27 -2.10610 6.32480 3.81710 H28 -3.35840 4.68340 1.54090 H29 -2.25110 6.02190 1.32690 H30 -1.88770 4.46160 0.57810 H31 -0.92600 2.50540 2.08600 H32 0.12540 2.78640 4.35900

285
H33 -1.48920 2.72420 5.08850 H34 -0.84510 1.33070 4.22040 H35 -3.60530 2.70230 3.53530 H36 -3.34820 2.46880 1.79430 H37 -2.86820 1.21530 2.92440 H38 3.12410 4.14620 -6.12860 H39 2.36910 5.58280 -5.43510 H40 3.63650 4.74530 -4.54370 H41 0.64050 3.31590 -6.42740 H42 -0.36960 3.07290 -4.99000 H43 0.01710 4.70390 -5.55780 H44 1.53710 1.79040 -3.45910 H45 3.77270 2.78510 -2.78870 H46 3.86630 1.12910 -3.36100 H47 4.36770 2.44790 -4.41970 H48 2.69830 1.70220 -6.28180 H49 2.54090 0.31140 -5.19830 H50 1.09650 1.13660 -5.75990 H51 8.35930 4.06010 4.50680 H52 6.80360 3.26140 4.29720 H53 7.25470 3.89390 5.88160 H54 6.40900 0.88940 5.04350 H55 7.55970 0.08400 6.11820 H56 6.60930 1.46740 6.68740 H57 10.01650 3.32210 2.81140 H58 11.27520 2.23230 3.42740 H59 10.61110 1.99230 1.81750 H60 13.89100 -0.86950 -1.61180 H61 13.74960 -1.13400 -3.34830 H62 12.92210 0.23650 -2.58350 H63 13.12350 -3.07550 -0.94260 H64 11.64370 -3.80860 -1.55750 H65 13.03360 -3.59030 -2.62750 H66 11.70090 -2.42250 -4.25240 H67 10.21640 0.17850 -3.70730 H68 11.76300 -0.00140 -4.55250 H69 10.30170 -0.69670 -5.23000 H70 9.01980 -2.02530 -2.86450 H71 9.34750 -2.79950 -4.40820 H72 9.90550 -3.56730 -2.93010 H73 12.76570 -1.55360 0.98440 H74 12.93550 0.12350 0.43170 H75 11.76030 -0.27970 1.68570 H76 10.78130 1.34890 -1.52340 H77 9.24340 0.48290 -1.72470 H78 9.66830 1.16690 -0.16280

286
H79 10.62100 -0.41650 4.64550 H80 8.99190 -1.10970 4.56160 H81 10.01550 -1.08690 3.12420 H82 10.07960 1.25310 6.34090 H83 8.21860 2.98380 8.03490 H84 8.44790 1.22510 8.14780 H85 9.76130 2.30770 8.57960 H86 9.81330 4.30030 6.46480 H87 11.20340 3.31720 6.92530 H88 10.75030 3.44580 5.23340
Calculated positional parameters of the transition state for the inversion of 215:
Atom x y z
Si1 -1.01990 4.30190 -2.52440 Si2 2.27490 5.86940 1.14630 Si3 -5.28850 -4.26640 3.58540 Si4 -4.47560 -4.66550 -1.49000 C1 -1.46570 0.50850 0.94790 C2 -2.25310 1.65170 0.72670 C3 -3.60150 1.66920 1.10060 C4 -4.18030 0.54560 1.68910 C5 -3.42820 -0.61960 1.86220 C6 -2.07290 -0.64790 1.47970 C7 1.17090 0.62860 0.90630 C8 -0.02920 0.58140 0.83990 C9 -0.74950 -2.92550 1.55260 C10 -1.32950 -1.87450 1.58070 C11 1.38570 -6.53790 1.01690 C12 0.10720 -6.40860 0.46700 C13 -0.61940 -5.22410 0.64010 C14 -0.07300 -4.17860 1.40170 C15 1.19820 -4.32020 1.96420 C16 1.93500 -5.48690 1.75120 C17 5.39830 0.33430 0.95740 C18 4.60770 -0.74040 1.36340 C19 3.21510 -0.61480 1.36410 C20 2.60440 0.59720 1.00120 C21 3.40500 1.68540 0.62360 C22 4.79990 1.54240 0.59340 C23 5.78550 -2.93710 2.21390 C24 5.23230 -1.95080 1.80550 C25 4.43300 -5.49270 2.55700 C26 3.28160 -5.55080 2.22140

287
C27 -1.43120 3.45530 -0.98790 C28 -1.76940 2.71810 -0.10270 C29 2.55390 4.16560 0.61570 C30 2.87760 3.01490 0.49460 C31 7.86510 -6.29200 3.76020 C32 6.51760 -6.42030 3.41890 C33 5.81290 -5.32560 2.90400 C34 6.46510 -4.09350 2.73030 C35 7.81730 -3.97470 3.07580 C36 8.51440 -5.07070 3.58860 C37 -2.11120 3.28470 -4.08850 C38 -1.60170 6.08580 -2.51860 C39 2.05320 7.05820 -0.27920 C40 3.72420 6.44330 2.21240 C41 0.52190 5.94060 2.38780 C42 0.79700 4.21320 -2.96110 C43 0.77700 6.99040 3.49430 C44 -0.62280 6.45640 1.47760 C45 0.21630 4.49650 2.94870 C46 1.22620 3.99910 3.99420 C47 -1.18900 4.37280 3.56010 C48 -1.40680 3.62560 -5.42130 C49 -3.52720 3.92370 -4.08390 C50 -2.18880 1.73610 -3.78240 C51 -0.83820 1.00650 -3.78110 C52 -3.12050 0.98600 -4.75050 C53 -4.58170 -2.75330 2.89350 C54 -4.07280 -1.77090 2.42720 C55 -2.93560 -4.94740 -0.58570 C56 -1.89160 -5.09430 -0.01060 C57 -4.00040 -4.69920 -3.58710 C58 -5.70450 -6.03950 -1.17170 C59 -5.17860 -2.98060 -1.07640 C60 -2.88750 -5.77310 -3.74480 C61 -3.41460 -3.31140 -3.93670 C62 -4.17080 -5.74120 3.29270 C63 -6.99800 -4.56640 2.89940 C64 -5.38920 -3.96750 5.71130 C65 -3.92080 -3.68820 6.14040 C66 -6.23430 -2.69600 5.95630 C67 -5.97680 -5.26040 6.40150 C68 -5.56150 -5.39130 7.87690 C69 -7.50860 -5.36310 6.35040 C70 -5.28050 -5.08440 -4.42820 C71 -6.40450 -4.03940 -4.40930 C72 -4.95470 -5.38680 -5.90150

288
H1 -4.21340 2.54710 0.90490 H2 -5.22940 0.57000 1.97540 H3 1.95590 -7.44730 0.84340 H4 -0.30620 -7.22270 -0.12410 H5 1.63200 -3.50370 2.53690 H6 6.48260 0.25090 0.95180 H7 2.59770 -1.46300 1.64830 H8 5.43010 2.38970 0.33100 H9 8.40760 -7.14530 4.15990 H10 6.02050 -7.37800 3.55610 H11 8.33460 -3.02630 2.94840 H12 9.56380 -4.97030 3.85490 H13 -2.65650 6.15520 -2.23700 H14 -1.02600 6.67820 -1.80240 H15 -1.47720 6.53660 -3.50750 H16 1.19490 6.78530 -0.89640 H17 1.90000 8.07810 0.08620 H18 2.94060 7.06060 -0.91960 H19 4.65580 6.39150 1.64090 H20 3.58060 7.47880 2.53410 H21 3.83240 5.81460 3.10030 H22 0.99110 4.70760 -3.91750 H23 1.39980 4.71750 -2.20260 H24 1.15230 3.18290 -3.03410 H25 -0.10060 7.09920 4.14020 H26 1.62650 6.72510 4.13070 H27 0.98940 7.97650 3.06470 H28 -1.54610 6.60940 2.04610 H29 -0.37890 7.42350 1.02490 H30 -0.83470 5.75170 0.67040 H31 0.24220 3.79050 2.10990 H32 2.25480 4.03310 3.62970 H33 1.17060 4.58240 4.91900 H34 1.01720 2.95520 4.25580 H35 -1.33900 5.08710 4.37550 H36 -1.96890 4.53360 2.81010 H37 -1.34730 3.36660 3.96470 H38 -1.94590 3.19670 -6.27210 H39 -1.36070 4.70920 -5.58070 H40 -0.37980 3.25010 -5.45340 H41 -4.14230 3.53460 -4.90170 H42 -4.04990 3.72740 -3.14120 H43 -3.48500 5.00850 -4.22680 H44 -2.62450 1.60630 -2.78360 H45 -0.17320 1.37710 -2.99940 H46 -0.98090 -0.06160 -3.57830
289
H47 -0.32770 1.08810 -4.74510 H48 -2.78960 1.08370 -5.78880 H49 -3.14580 -0.08320 -4.51120 H50 -4.15130 1.34380 -4.67840 H51 -5.31610 -6.99960 -1.52440 H52 -5.91130 -6.13080 -0.10110 H53 -6.65310 -5.84590 -1.68060 H54 -6.01750 -2.71380 -1.72200 H55 -5.54100 -2.96410 -0.04520 H56 -4.41460 -2.20380 -1.17790 H57 -2.01900 -5.55960 -3.11290 H58 -3.25710 -6.77090 -3.48380 H59 -2.51070 -5.80690 -4.77230 H60 -2.52800 -3.08950 -3.33100 H61 -3.10890 -3.26660 -4.98690 H62 -4.13270 -2.50440 -3.76260 H63 -3.16200 -5.55270 3.67130 H64 -4.56130 -6.63510 3.78690 H65 -4.09590 -5.95770 2.22340 H66 -6.98640 -4.50960 1.80640 H67 -7.36320 -5.56120 3.16940 H68 -7.71110 -3.82170 3.26410 H69 -3.47640 -2.86510 5.56990 H70 -3.86710 -3.39860 7.19560 H71 -3.28760 -4.57080 5.99800 H72 -5.76310 -1.80980 5.51580 H73 -7.23610 -2.77540 5.52210 H74 -6.34450 -2.50570 7.02930 H75 -5.57440 -6.14330 5.89010 H76 -4.48100 -5.53190 7.97780 H77 -5.85290 -4.50810 8.45460 H78 -6.03520 -6.26500 8.33870 H79 -7.98450 -4.59680 6.97160 H80 -7.90020 -5.26770 5.33830 H81 -7.83690 -6.33760 6.72980 H82 -5.69610 -6.01130 -4.01200 H83 -6.80030 -3.88370 -3.40500 H84 -6.07600 -3.07670 -4.81190 H85 -7.24950 -4.37540 -5.02160 H86 -4.48020 -4.53300 -6.39460 H87 -4.29670 -6.25500 -5.99890 H88 -5.86750 -5.62630 -6.45900

290
Calculated positional parameters of the transition state for the inversion of 216:
Atom x y z
Si1 -1.09520 4.66370 -2.29760 Si2 1.20130 5.91170 2.17110 Si3 -6.08470 -3.88240 3.10780 Si4 -4.83470 -4.97000 -1.71650 Si5 10.53410 -1.57390 0.32170 Si6 8.91460 2.21320 2.73850 C1 -1.87990 0.80070 0.87880 C2 -2.58560 2.01760 0.80650 C3 -3.94110 2.08240 1.14550 C4 -4.60790 0.93640 1.57370 C5 -3.93630 -0.28910 1.62250 C6 -2.57530 -0.36490 1.26510 C7 0.75160 0.86500 1.08960 C8 -0.43590 0.81420 0.90260 C9 -1.30730 -2.67560 1.29640 C10 -1.89350 -1.62830 1.31140 C11 1.10470 -6.13070 0.99370 C12 -0.16350 -6.13240 0.40620 C13 -0.98720 -5.00410 0.50070 C14 -0.54550 -3.88400 1.22040 C15 0.71640 -3.89500 1.82280 C16 1.54820 -5.00500 1.68910 C17 4.98950 0.99580 1.26590 C18 4.31360 -0.18300 1.61160 C19 2.90760 -0.19870 1.58630 C20 2.17570 0.95840 1.27130 C21 2.86420 2.12660 0.91220 C22 4.26240 2.13900 0.90890 C23 5.58650 -2.45160 2.08230 C24 5.01940 -1.41160 1.86720 C25 4.02920 -4.86540 2.53760 C26 2.88090 -4.94700 2.19710 C27 -1.60050 3.88190 -0.74830 C28 -2.01450 3.12650 0.08930 C29 1.72520 4.45700 1.24170 C30 2.19840 3.40110 0.91830 C31 7.36870 -5.93590 3.75110 C32 6.00620 -5.91380 3.46910 C33 5.42760 -4.79910 2.84400 C34 6.21580 -3.68370 2.49490 C35 7.59970 -3.74030 2.74020 C36 8.16510 -4.85950 3.37150

291
C37 -0.27070 3.07260 -3.50890 C38 -2.60050 5.30820 -3.23230 C39 0.00460 7.03210 1.27620 C40 2.69070 6.92570 2.72360 C41 0.20950 5.20950 3.95530 C42 0.16440 6.01910 -2.13440 C43 0.34250 6.32720 5.01470 C44 -1.28590 5.05820 3.56510 C45 0.81850 3.82440 4.40810 C46 2.28100 3.88400 4.86950 C47 0.00390 3.15300 5.52720 C48 0.06660 3.67390 -4.89140 C49 -1.43170 2.05180 -3.67040 C50 0.97490 2.45310 -2.75970 C51 2.28120 3.23760 -2.95170 C52 1.25350 0.99500 -3.15960 C53 -5.24960 -2.41060 2.47240 C54 -4.66350 -1.44230 2.07190 C55 -3.29150 -5.01360 -0.77630 C56 -2.25030 -5.01790 -0.17820 C57 -4.31230 -5.44670 -3.74920 C58 -6.02190 -6.29880 -1.13450 C59 -5.61670 -3.27880 -1.64560 C60 -3.61890 -6.83600 -3.66490 C61 -3.27650 -4.39630 -4.21320 C62 -4.96240 -5.38110 3.06390 C63 -7.67340 -4.18990 2.17790 C64 -6.51390 -3.48380 5.17760 C65 -5.12920 -3.18050 5.81720 C66 -7.38100 -2.20450 5.22770 C67 -7.20670 -4.74480 5.82790 C68 -7.03400 -4.79950 7.35550 C69 -8.71040 -4.86030 5.53660 C70 -5.61840 -5.48390 -4.63570 C71 -6.10950 -4.10300 -5.09520 C72 -5.46080 -6.35240 -5.89550 C73 7.47700 1.50760 1.90580 C74 6.39060 1.17940 1.51380 C75 9.32060 -2.19430 1.50500 C76 8.50970 -2.79400 2.15750 C77 8.17000 3.95200 3.79480 C78 9.67410 1.03610 3.96760 C79 9.75030 -0.37720 -0.88960 C80 10.19510 2.85720 1.52900 C81 11.21090 -3.26410 -0.84660 C82 7.03370 3.50600 4.79830

292
C83 7.63430 4.89230 2.67860 C84 9.38140 4.62200 4.48140 C85 6.05490 4.64390 5.13420 C86 7.55260 2.94560 6.13070 C87 12.06090 -0.85940 1.12710 C88 12.25850 -4.10380 -0.01620 C89 11.86200 -2.64720 -2.11500 C90 9.95180 -4.06070 -1.26000 C91 12.97920 -5.16100 -0.87060 C92 11.68300 -4.81710 1.21550 H1 -4.48870 3.01680 1.04460 H2 -5.66020 0.99700 1.84280 H3 1.74950 -6.99900 0.88090 H4 -0.49400 -7.00610 -0.15100 H5 1.06430 -3.01940 2.36540 H6 2.37960 -1.12570 1.79400 H7 4.78870 3.07540 0.72860 H8 7.81420 -6.80240 4.23290 H9 5.39270 -6.77150 3.73800 H10 9.23950 -4.90770 3.53740 H11 -3.35710 4.52750 -3.34890 H12 -3.05420 6.14090 -2.68630 H13 -2.31540 5.66780 -4.22490 H14 -0.84680 6.47430 0.87820 H15 -0.38020 7.80380 1.95000 H16 0.50350 7.53970 0.44620 H17 3.24850 7.27960 1.85110 H18 2.36980 7.80110 3.29580 H19 3.37140 6.33940 3.34490 H20 0.60480 6.27120 -3.10330 H21 -0.30310 6.92850 -1.74560 H22 0.97330 5.74210 -1.45500 H23 -0.19140 6.06350 5.93340 H24 1.38570 6.52290 5.28000 H25 -0.08230 7.27060 4.65250 H26 -1.89940 4.79750 4.43370 H27 -1.70220 5.98980 3.16800 H28 -1.42000 4.27880 2.80900 H29 0.77640 3.13750 3.55400 H30 2.94710 4.23510 4.08010 H31 2.40720 4.52780 5.74510 H32 2.63490 2.88430 5.14770 H33 -0.05700 3.78450 6.41870 H34 -1.01190 2.91500 5.19910 H35 0.46470 2.20390 5.82420 H36 0.51970 2.91670 -5.54050

293
H37 -0.83270 4.04100 -5.39860 H38 0.76100 4.51660 -4.81940 H39 -1.15480 1.24880 -4.36230 H40 -1.70300 1.59430 -2.71300 H41 -2.33170 2.51940 -4.08320 H42 0.76060 2.43800 -1.68530 H43 2.17900 4.28860 -2.68280 H44 3.07320 2.82080 -2.31930 H45 2.63430 3.18400 -3.98710 H46 1.40220 0.89860 -4.24000 H47 2.15490 0.62020 -2.66140 H48 0.43410 0.33440 -2.86010 H49 -5.57850 -7.29370 -1.23330 H50 -6.28050 -6.14900 -0.08240 H51 -6.94890 -6.27730 -1.71450 H52 -6.62780 -3.29240 -2.06180 H53 -5.69850 -2.93780 -0.60980 H54 -5.02400 -2.54010 -2.19260 H55 -2.78530 -6.83600 -2.95410 H56 -4.32180 -7.61600 -3.35300 H57 -3.19910 -7.12580 -4.63450 H58 -2.36170 -4.44160 -3.61120 H59 -2.98880 -4.57160 -5.25540 H60 -3.66060 -3.37420 -4.13570 H61 -4.04050 -5.19990 3.62400 H62 -5.45820 -6.25620 3.49320 H63 -4.68790 -5.62500 2.03400 H64 -7.49750 -4.15930 1.09830 H65 -8.08430 -5.17570 2.41290 H66 -8.42680 -3.43270 2.41270 H67 -4.59740 -2.38370 5.28560 H68 -5.23970 -2.84150 6.85300 H69 -4.48570 -4.06700 5.81720 H70 -6.84400 -1.33950 4.82170 H71 -8.30490 -2.30820 4.65000 H72 -7.65340 -1.96220 6.26060 H73 -6.72820 -5.64900 5.43250 H74 -5.98400 -4.92790 7.63550 H75 -7.41300 -3.89060 7.83410 H76 -7.57710 -5.65220 7.77850 H77 -9.27870 -4.06490 6.03050 H78 -8.93340 -4.82300 4.47100 H79 -9.09750 -5.81510 5.91070 H80 -6.42610 -5.93810 -4.04880 H81 -6.23830 -3.40800 -4.26630 H82 -5.41900 -3.64880 -5.81400

294
H83 -7.08230 -4.19180 -5.59240 H84 -4.61640 -6.01960 -6.50790 H85 -5.31090 -7.40570 -5.63960 H86 -6.36310 -6.30620 -6.51570 H87 8.90740 0.54020 4.56940 H88 10.23760 0.25160 3.45630 H89 10.36410 1.55280 4.64060 H90 9.25440 0.44150 -0.36330 H91 10.50370 0.05960 -1.55150 H92 9.00040 -0.88120 -1.50630 H93 10.99750 3.38250 2.05470 H94 10.64610 2.03670 0.96620 H95 9.74760 3.54660 0.80760 H96 6.43560 2.71760 4.32510 H97 6.77340 4.45410 2.16230 H98 7.32570 5.85840 3.09270 H99 8.39910 5.11030 1.92600 H100 9.06230 5.49590 5.05960 H101 9.90270 3.93960 5.15990 H102 10.11670 4.96930 3.74700 H103 5.49000 4.95850 4.25150 H104 5.31920 4.31810 5.87810 H105 6.57850 5.51520 5.54090 H106 8.03060 3.72350 6.73560 H107 6.72290 2.54000 6.72100 H108 8.27000 2.13740 5.99290 H109 12.82700 -0.63750 0.37830 H110 11.83210 0.06910 1.65510 H111 12.48700 -1.55820 1.85300 H112 13.03770 -3.41950 0.34340 H113 12.15920 -3.42470 -2.82650 H114 11.16830 -1.99240 -2.65260 H115 12.75120 -2.05970 -1.86160 H116 10.21790 -4.91120 -1.89590 H117 9.40320 -4.44870 -0.39690 H118 9.25490 -3.43600 -1.83050 H119 12.27460 -5.86810 -1.31860 H120 13.56600 -4.70080 -1.67070 H121 13.68430 -5.73710 -0.26050 H122 10.89710 -5.52920 0.94680 H123 12.46840 -5.38130 1.73190 H124 11.28360 -4.11290 1.94670

295
Calculated positional parameters of the transition state for the inversion of 171c:
Atom x y z
Si1 0.76540 5.44570 -2.26930 Si2 -0.07220 5.09600 2.94440 Si3 -5.32060 -4.39200 -2.23910 Si4 -5.02010 -3.46240 2.82910 Si5 9.99460 0.18180 0.43490 Si6 7.97580 -0.22410 5.22750 Si7 3.64680 -8.05770 5.00320 Si8 4.56820 -10.00580 -0.11420 C1 -1.19790 0.71410 -1.02960 C2 -1.75100 1.88450 -1.57950 C3 -2.98850 1.83290 -2.23140 C4 -3.67850 0.62460 -2.34180 C5 -3.13600 -0.54820 -1.80760 C6 -1.89890 -0.50850 -1.13990 C7 1.01040 0.77990 0.41890 C8 0.01780 0.77360 -0.25860 C9 -0.85090 -2.66140 -0.02790 C10 -1.34970 -1.70700 -0.56050 C11 1.25350 -5.57890 2.20790 C12 -0.10290 -5.35240 2.47340 C13 -0.82610 -4.41420 1.72590 C14 -0.19370 -3.71590 0.68680 C15 1.15040 -3.97350 0.39450 C16 1.88150 -4.88760 1.16140 C17 4.77380 0.64890 2.36080 C18 4.27430 -0.47460 1.68700 C19 3.01400 -0.41480 1.08250 C20 2.23430 0.74470 1.16610 C21 2.73110 1.86440 1.84880 C22 3.99640 1.81000 2.45030 C23 5.78000 -2.56790 1.15070 C24 5.09110 -1.62340 1.42680 C25 4.47740 -5.05110 0.76440 C26 3.28420 -5.02280 0.90050 C27 -0.33980 4.09460 -1.80520 C28 -1.01390 3.11620 -1.62910 C29 1.15610 3.90810 2.36300 C30 1.90970 3.01870 2.07530 C31 8.07110 -6.10490 0.68240 C32 6.67600 -6.16620 0.60720 C33 5.91550 -4.98960 0.72330 C34 6.56500 -3.74540 0.88670

296
C35 7.96770 -3.70020 0.97720 C36 8.71350 -4.87880 0.86780 C37 2.62660 4.52440 -2.85250 C38 0.11780 6.34720 -3.78830 C39 -1.56190 5.17970 1.80420 C40 0.64870 6.79780 3.19760 C41 -0.79430 4.32660 4.82600 C42 1.08010 6.65560 -0.89170 C43 -1.86940 5.30840 5.34350 C44 -1.47340 2.97500 4.46510 C45 0.42280 4.13680 5.81560 C46 0.87720 5.42810 6.51170 C47 0.14100 3.10000 6.91610 C48 3.51940 5.63210 -3.45550 C49 2.23930 3.52120 -3.97450 C50 3.25710 3.81160 -1.59100 C51 4.09460 4.74000 -0.69780 C52 4.14250 2.60980 -1.95590 C53 -4.47010 -2.80520 -2.07540 C54 -3.87360 -1.77010 -1.95590 C55 -3.32730 -3.90700 2.37900 C56 -2.19210 -4.15820 2.07960 C57 -4.96820 -2.93010 4.91230 C58 -5.60130 -1.94570 1.89530 C59 -6.17320 -4.90620 2.56890 C60 -3.94190 -1.76450 4.99710 C61 -4.40880 -4.14850 5.68280 C62 -7.07350 -4.29140 -1.58300 C63 -4.35610 -5.74880 -1.39700 C64 -5.45980 -4.78970 -4.34660 C65 -6.20640 -3.55890 -4.93480 C66 -4.01770 -4.81490 -4.90430 C67 -6.25300 -6.13490 -4.58070 C68 -6.89470 -6.21490 -5.97640 C69 -5.41060 -7.40570 -4.39480 C70 -6.40990 -2.49050 5.38280 C71 -7.35190 -3.65920 5.70860 C72 -6.38140 -1.57200 6.61640 C73 6.86160 0.31540 3.91720 C74 5.97330 0.54630 3.14260 C75 9.22990 -1.39230 0.87850 C76 8.67130 -2.44770 1.01160 C77 6.76240 -1.31940 6.64020 C78 9.35180 -1.35900 4.66590 C79 9.80280 1.48470 1.76540 C80 8.71200 1.24370 6.14260

297
C81 8.97730 0.91950 -1.31580 C82 5.68190 -2.18410 5.87530 C83 6.08670 -0.23520 7.52420 C84 7.72760 -2.15410 7.51190 C85 4.63590 -2.79990 6.82100 C86 6.25140 -3.32050 5.01480 C87 11.79840 -0.02250 -0.03070 C88 9.48910 0.15310 -2.59780 C89 9.32270 2.43210 -1.38720 C90 7.46130 0.76100 -1.05570 C91 9.01980 0.81030 -3.90750 C92 9.10070 -1.33130 -2.66000 C93 2.62910 -7.13130 3.83140 C94 1.98600 -6.45590 3.07510 C95 5.49470 -8.48800 0.21240 C96 6.05380 -7.44320 0.40610 C97 2.43350 -9.68420 5.71100 C98 4.14970 -6.96670 6.43390 C99 5.39860 -11.49440 0.66330 C100 5.14720 -8.81080 4.17520 C101 4.61560 -10.29750 -2.24300 C102 2.80290 -9.83130 0.46410 C103 6.12750 -10.34560 -2.60340 C104 3.87300 -11.64170 -2.61060 C105 4.32600 -12.22450 -3.96020 C106 2.34220 -11.52580 -2.65690 C107 3.98500 -9.04310 -2.89140 C108 1.18240 -9.06660 6.37750 C109 1.99610 -10.44310 4.42620 C110 3.28310 -10.59620 6.68030 C111 3.40230 -10.05580 8.11300 C112 2.74020 -12.03200 6.77920 H1 -3.40960 2.73090 -2.67890 H2 -4.63540 0.60030 -2.85890 H3 -0.58720 -5.87690 3.29500 H4 1.64440 -3.42220 -0.40350 H5 2.65260 -1.26530 0.50900 H6 4.35730 2.65850 3.02800 H7 8.66530 -7.01160 0.58940 H8 9.80040 -4.84440 0.90170 H9 -0.04510 5.65520 -4.61910 H10 -0.83530 6.83470 -3.56150 H11 0.82310 7.11760 -4.11260 H12 -1.95120 4.18090 1.58750 H13 -2.36350 5.77280 2.25310 H14 -1.29120 5.64890 0.85480

298
H15 0.79400 7.29900 2.23570 H16 -0.01360 7.42540 3.80030 H17 1.62420 6.74890 3.68930 H18 1.85410 7.37760 -1.16760 H19 0.17120 7.21780 -0.65580 H20 1.39800 6.14380 0.01880 H21 -2.25550 4.98160 6.31510 H22 -1.48070 6.32490 5.45930 H23 -2.72240 5.36640 4.65820 H24 -1.97390 2.54030 5.33720 H25 -2.24440 3.09780 3.69700 H26 -0.74520 2.24600 4.09280 H27 1.28050 3.75510 5.24780 H28 1.09570 6.22800 5.80520 H29 0.12210 5.79340 7.21590 H30 1.79440 5.24850 7.08450 H31 -0.75600 3.35750 7.48860 H32 0.01070 2.09740 6.49740 H33 0.98050 3.03920 7.61800 H34 4.50070 5.22910 -3.72900 H35 3.07620 6.05280 -4.36520 H36 3.67460 6.46350 -2.76050 H37 3.13150 3.07070 -4.42320 H38 1.60760 2.71200 -3.59220 H39 1.69450 4.01090 -4.78880 H40 2.44230 3.41410 -0.97490 H41 3.54890 5.63450 -0.39730 H42 4.39290 4.22140 0.21990 H43 5.01430 5.05800 -1.20070 H44 4.94760 2.90020 -2.63880 H45 4.60150 2.18000 -1.05870 H46 3.56030 1.80950 -2.42270 H47 -4.92760 -1.09970 2.05900 H48 -5.63460 -2.14660 0.82120 H49 -6.60580 -1.65060 2.21110 H50 -7.21870 -4.59760 2.65590 H51 -6.04030 -5.32690 1.56740 H52 -5.98360 -5.70430 3.29220 H53 -2.97590 -2.03580 4.55700 H54 -4.30560 -0.87220 4.47610 H55 -3.74180 -1.49110 6.03890 H56 -3.38500 -4.38510 5.37090 H57 -4.38200 -3.94640 6.75900 H58 -5.00890 -5.04990 5.52280 H59 -7.65070 -3.53100 -2.11660 H60 -7.58770 -5.25060 -1.69050

299
H61 -7.07140 -4.02880 -0.52110 H62 -4.08950 -5.45250 -0.37810 H63 -4.94560 -6.66680 -1.32350 H64 -3.42830 -5.97140 -1.93150 H65 -5.71710 -2.61600 -4.66700 H66 -6.22700 -3.59710 -6.02940 H67 -7.24080 -3.51000 -4.57770 H68 -3.52060 -3.84670 -4.77470 H69 -3.39450 -5.56420 -4.40600 H70 -4.02160 -5.03740 -5.97670 H71 -7.07860 -6.18330 -3.86020 H72 -7.66790 -5.45150 -6.10610 H73 -6.14820 -6.09210 -6.76790 H74 -7.38410 -7.18420 -6.12500 H75 -4.65220 -7.50440 -5.17890 H76 -4.90480 -7.43430 -3.43050 H77 -6.04860 -8.29550 -4.44750 H78 -6.87800 -1.90980 4.57860 H79 -7.42870 -4.37450 4.89050 H80 -7.02710 -4.20080 6.60350 H81 -8.36440 -3.28780 5.90440 H82 -5.85480 -2.04200 7.45330 H83 -5.89680 -0.61660 6.39310 H84 -7.39830 -1.33660 6.95020 H85 8.97480 -2.24370 4.14970 H86 10.02850 -0.83780 3.98430 H87 9.94760 -1.69570 5.51960 H88 10.23590 1.14410 2.70920 H89 10.31900 2.40730 1.48400 H90 8.74950 1.72290 1.93380 H91 9.25510 0.91210 7.03240 H92 9.41280 1.78090 5.49630 H93 7.93300 1.94620 6.45270 H94 5.12580 -1.51990 5.20210 H95 5.40800 0.39250 6.93620 H96 5.51220 -0.68780 8.33890 H97 6.82390 0.41970 8.00040 H98 7.18360 -2.69310 8.29430 H99 8.28270 -2.89090 6.92370 H100 8.46570 -1.51570 8.01120 H101 4.06340 -2.03020 7.34610 H102 3.90930 -3.39640 6.25730 H103 5.09840 -3.45740 7.56330 H104 6.81290 -4.04500 5.61210 H105 5.44110 -3.86790 4.51860 H106 6.89690 -2.94620 4.22010

300
H107 12.20630 0.91390 -0.42240 H108 12.38930 -0.31080 0.84390 H109 11.92370 -0.79730 -0.79260 H110 10.58570 0.19980 -2.60930 H111 8.77690 2.92630 -2.19770 H112 9.04230 2.95820 -0.46870 H113 10.39430 2.58930 -1.55200 H114 6.87970 1.14570 -1.89910 H115 7.17260 -0.28260 -0.90160 H116 7.14920 1.31730 -0.16430 H117 7.92830 0.86090 -3.96660 H118 9.42120 1.82190 -4.01700 H119 9.37280 0.24100 -4.77500 H120 8.01550 -1.47080 -2.66300 H121 9.48840 -1.78920 -3.57760 H122 9.52510 -1.90160 -1.83270 H123 3.29320 -6.71250 7.06440 H124 4.90890 -7.45130 7.05370 H125 4.58210 -6.03180 6.06380 H126 6.42130 -11.61240 0.29400 H127 5.44320 -11.38510 1.75110 H128 4.84610 -12.41180 0.44160 H129 5.76960 -9.34310 4.90010 H130 4.85120 -9.51750 3.39660 H131 5.76200 -8.03240 3.71320 H132 2.28540 -10.79450 0.45640 H133 2.77620 -9.45600 1.49100 H134 2.24390 -9.12870 -0.16030 H135 6.66420 -9.46280 -2.23890 H136 6.61280 -11.22980 -2.17590 H137 6.27300 -10.37020 -3.68890 H138 4.11970 -12.39530 -1.85280 H139 4.19240 -11.50320 -4.77290 H140 5.37720 -12.52720 -3.93320 H141 3.74840 -13.12170 -4.21040 H142 2.01060 -10.90050 -3.49270 H143 1.89030 -12.51510 -2.79330 H144 1.92680 -11.11100 -1.73930 H145 3.95930 -9.14290 -3.98180 H146 2.96190 -8.86790 -2.54390 H147 4.56120 -8.13960 -2.66090 H148 1.44200 -8.39100 7.19860 H149 0.59070 -8.48660 5.65990 H150 0.53080 -9.85060 6.77810 H151 1.50320 -9.77940 3.70750 H152 2.85220 -10.90430 3.92170

301
H153 1.27460 -11.23310 4.66120 H154 4.30020 -10.68040 6.27830 H155 3.77240 -9.03170 8.14350 H156 2.44020 -10.08480 8.63570 H157 4.10410 -10.66680 8.69240 H158 1.69230 -12.04290 7.09650 H159 2.81940 -12.55680 5.82220 H160 3.31600 -12.61700 7.50540

302
References
[1] This chapter constitutes a book chapter in press: Miljanić, O. Š.; Vollhardt, K. P. C.,
[N]Phenylenes: a Novel Class of Cyclohexatrienoid Hydrocarbons, in Carbon Rich
Compounds: From Molecules to Materials (Eds.: Haley, M. M.; Tykwinski, R. R.),
Wiley-VCH, Weinheim, 2005, in press. The large majority of the presented material is a
review of the field; my original work is limited to Section 1.2.2.3 (synthesis of syn-
doublebent [5]phenylene 60) and other minor results, properly acknowledged by the
given references.
[2] (a) Minkin, V. I.; Glukhovtsev, M. N.; Simkin, B. Ya. Aromaticity and
Antiaromaticity: Electronic and Structural Aspects, Wiley, New York, 1994; (b) Garratt,
P. J. Aromaticity, Wiley, New York, 1986; (c) Chem. Rev. 2001, 101, 1115, Special
Issue: Aromaticity (some of the reviews from this issue are also cited separately).
[3] Krygowski, T. M.; Cyrański, M. K. Chem. Rev. 2001, 101, 1385 and the references
cited therein.
[4] (a) Schaad, L. J.; Hess, B. A. Jr. Chem. Rev. 2001, 101, 1465 and the references cited
therein; (b) Slayden, S. W.; Liebman, J. F. Chem. Rev. 2001, 101, 1541 and the
references cited therein.
[5] (a) Mitchell, R. H. Chem. Rev. 2001, 101, 1301 and the references cited therein; (b)
Gomes, J. A. N. F.; Mallion, R. B. Chem. Rev. 2001, 101, 1349 and the references cited
therein.

303
[6] (a) Armit, J. W.; Robinson, R. J. Chem. Soc. 1925, 127, 1604; (b) Kruszewski, J.;
Krygowski, T. M. Tetrahedron Lett. 1970, 11, 319; (c) Krygowski, T. M. Tetrahedron
Lett. 1970, 11, 1311; (d) Dixon, W. T. J. Chem. Soc. B 1970, 612.
[7] Cyrañski, M. K.; Krygowski, T. M.; Katrizky, A. R.; Schleyer, P. v. R. J. Org. Chem.
2002, 67, 1333.
[8] (a) Jug, K.; Köster, A. M. J. Phys. Org. Chem. 1991, 4, 163; (b) Katrizky, A. R.;
Barczynski, P.; Masummarra, G.; Pisano, D.; Szafan, M. J. Am. Chem. Soc. 1989, 111, 7.
[9] Jeffrey, G. A.; Ruble, J. R.; McMullan, R. K.; Pople, J. A. Proc. R. Soc. London 1987,
A414, 47.
[10] (a) Irngartinger, H.; Nixford, M. Angew. Chem., Int. Ed. Engl. 1983, 22, 403; (b)
Sekiguchi, A.; Tanaka, M.; Matsuo, T.; Watanabe, H. Angew. Chem., Int. Ed. 2001, 40,
1675.
[11] Deniz, A. A.; Peters, K. S.; Snyder, G. J. Science 1999, 286, 1119.
[12] Faraday, M. Phil. Trans. R. Soc. London 1825, 115, 440.
[13] (a) Chapman, O. L.; McIntosh, C. L.; Pacansky, J. J. Am. Chem. Soc. 1973, 95, 614;
(b) Krantz, A.; Lin, C. Y.; Newton, M. D. J. Am. Chem. Soc. 1973, 95, 2744.
[14] Kennedy, R. D.; Lloyd, D.; McNab, H. J. Chem. Soc., Perkin Trans 1 2002, 1600
and the references cited therein.
[15] (a) Hückel, E. Grundzüge der Theorie ungesättigter und aromatischer
Verbindungen, Verlag Berlin, 1938; (b) Hückel, E. Z. Phys. 1931, 70, 204.
[16] Lothrop, W. C. J. Am. Chem. Soc. 1941, 63, 1187.

304
[17] (a) Shepherd, M. K. Cyclobutarenes: The Chemistry of Benzocyclobutene,
Biphenylene, and Related Compounds, Elsevier, New York, 1991; (b) Toda, F.; Garratt,
P. J. Chem. Rev. 1992, 92, 1685.
[18] Sigma-Aldrich Fine Chemicals Catalog, 2003–2004.
[19] Iyoda, M.; Kabir, S. M. H.; Vorasingha, A.; Kuwatani, Y.; Yoshida, M. Tetrahedron
Lett. 1998, 39, 5393.
[20] (a) Kanoktanapora, S.; MacBride, J. A. H. J. Chem. Res. (S) 1980, 203; (b)
MacBride, J. A. J. Chem. Soc., Chem. Commun. 1972, 1219.
[21] (a) Droske, J. P.; Stille, J. K. Macromolecules 1984, 17, 1; (b) Friedman, L.;
Lindow, D. F. J. Am. Chem. Soc. 1968, 90, 2324; (c) Lindow, D. F.; Friedman, L. J. Am.
Chem. Soc. 1967, 89, 1271.
[22] Perthuisot, C.; Edelbach, B. L.; Zubris, D. L.; Simhai, N.; Iverson, C. N.; Müller, C.;
Satoh, T.; Jones, W. D. J. Mol. Catal. A 2002, 189, 157 and the references cited therein.
[23] Jeany, H.; Mason, K. G.; Sketchley, J. M. Tetrahedron Lett. 1970, 11, 485.
[24] Heaney, H.; Lees, P. Tetrahedron Lett. 1964, 5, 3049.
[25] (a) Yokozeki, A.; Wilcox, C. F.; Bauer, S. H. J. Am. Chem. Soc. 1974, 96, 1026; (b)
Fawcett, J. K.; Trotter, J. Acta Crystallogr. 1966, 20, 87; (c) Mak, T. C. W.; Trotter, J. J.
Chem. Soc. 1962, 1.
[26] Yamaguchi, H.; Masafumi, A.; McOmie, J. F. W.; Barton, J. W.; Baumann, H. J.
Chem. Soc., Faraday Trans. 2 1983, 79, 599.
[27] Figeys, H. P.; Defay, N.; Martin, R. H.; McOmie, J. F. W.; Ayers, B. E.; Chadwick,
J. B. Tetrahedron 1976, 32, 2571.

305
[28] Jones, A. P.; Garratt, P. J.; Vollhardt, K. P. C. Angew. Chem., Int. Ed. Engl. 1973,
12, 241.
[29] (a) Maksić, Z. B.; Eckert-Maksić, M.; Mó, O.; Yáñez, M. in Pauling’s Legacy:
Modern Modelling of the Chemical Bond (Eds.: Maksić, Z. B., Orville-Thomas, W. J.),
Elsevier, Amsterdam, 1999, 47 and the references cited therein; (b) Stanger, A.; Ben-
Mergui, N.; Perl, S. Eur. J. Org. Chem. 2003, 2709; (c) Stanger, A.; Tkachenko, E. J.
Comp. Chem. 2001, 22 1377; (d) Stanger, A. J. Am. Chem. Soc. 1998, 120, 12034; (e)
Frank, N. L.; Siegel, J. S. in Advances in Theoretically Interesting Molecules, Vol. 3 (Ed.:
Thummel, R. P.), JAI, London, 1995, 209; (f) Siegel, J. S. Angew. Chem., Int. Ed. Engl.
1994, 33, 1721 and the references cited therein; (g) Jug, K.; Hiberty, P. C.; Shaik, S.
Chem. Rev. 2001, 101, 1477 and the references cited therein.
[30] (a) Shaik, S.; Shurki, A.; Danovich, D.; Hiberty, P. C. Chem. Rev. 2001, 101, 1501
and the references cited therein; (b) Hiberty, P. C.; Danovich, D.; Shurki, A.; Shaik, S. J.
Am. Chem. Soc. 1995, 117, 7760 and the references cited therein. For a popular account:
(c) Narahari Sastry, G. Curr. Sci. 2001, 81, 1288.
[31] Wannere, C. S.; Sattelmeyer, K. W.; Schaefer, H. F. III; Schleyer, P. v. R. Angew.
Chem., Int. Ed. 2004, 43, 4200.
[32] Randić, M. Chem. Rev. 2003, 103, 3449 and the numerous references cited therein.
[33] Gutman, I.; Cyvin, S. J.; Brunvoll, J. Monatsh. Chem. 1994, 125, 887. For
illustration, there are 12 [5]phenylenes, 122 [7]phenylenes and 6387 [10]phenylenes.
[34] Trinajstić, N.; Schmalz, T. G.; Živković, T. P.; Nikolić, S.; Hite, G. E.; Klein, D. J.;
Seitz, W. A. New J. Chem. 1991, 15, 27.

306
[35] Beckhaus, H.-D.; Faust, R.; Matzger, A. J.; Mohler, D. L.; Rogers, D. W.; Rüchardt,
C.; Sawhey, A. K.; Verevkin, S. P.; Vollhardt, K. P. C.; Wolff, S. J. Am. Chem. Soc.
2000, 122, 7819.
[36] (a) Watson, M. D.; Fechtenkötter, A.; Müllen, K. Chem. Rev. 2001, 101, 1267 and
the references cited therein; (b) Harvey, R. G. Polycyclic Aromatic Hydrocarbons,
Wiley-VCH, New York, 1995; c) Clar, E. The Aromatic Sextet, Wiley, London, 1972.
[37] Recent examples: (a) Debije, M. G.; Piris, J.; de Haas, M. P.; Warman, J. M.;
Tomović, Ž.; Simpson, C. D.; Watson, M. D.; Müllen, K.; J. Am. Chem. Soc. 2004, 126,
4641; (b) Wu, J. S.; Gherghel, L.; Watson, M. D.; Li, J. X.; Wang, Z. H.; Simpson, C. D.;
Kolb, U.; Müllen, K. Macromolecules 2003, 36, 7082; (c) Meng, H.; Bendikov, M.;
Mitchell, G.; Helgeson, R.; Wudl, F.; Bao, Z.; Siegrist, T.; Kloc, C.; Chem, C.-H. Adv.
Mater. 2003, 15, 1090; (d) Zhang, Y.; Petta, J. R.; Ambily, S.; Shen, Y.; Ralph, D. C.;
Malliaras, G. C. Adv. Mater. 2003, 15, 1632; (e) Dimitrakopoulos, C. D.; Kymissis, I.;
Purushothaman, S.; Neumayer, D. A.; Duncombe, P. R.; Laibowitz. R. B. Adv. Mater.
1999, 11, 1372.
[38] Gutman, I. J. Chem. Soc., Faraday Trans. 1993, 89, 2413.
[39] Alternatively, phenylenes can be treated as PAHs in which neighboring benzene
rings are separated by a two-bond spacer.
[40] Bühl, M.; Hirsch, A. Chem. Rev. 2001, 101, 1153 and the references cited therein.
[41] Tomović, Ž.; Gutman, I. Hemijski Pregled, 2001, 42, 29 and the references cited
therein.

307
[42] (a) Wilcox, C. F. Tetrahedron Lett. 1968, 9, 795; (b) Wilcox, C. F. J. Am. Chem.
Soc. 1969, 91, 2732.
[43] (a) Gutman, I.; Trinajstić, N.; Wilcox, C. F. Tetrahedron 1975, 31, 143; (b) Wilcox,
C. F.; Gutman, I.; Trinajstić, N. Tetrahedron 1975, 31, 147.
[44] (a) Nikolić, S.; Trinajstić, N.; Mihalić, Z. Croat. Chem. Acta. 1995, 68, 105; (b)
Gutman, I.; Yeh, Y. N.; Lee, S. L.; Luo, Y. L. Indian J. Chem. 1993, 32A, 651; (c)
Wiener, H. J. Am. Chem. Soc. 1947, 69, 17.
[45] Pavlović, Lj.; Gutman, I. J. Chem. Inf. Comput. Sci. 1997, 37, 355.
[46] Gutman, I.; Ivanov-Petrović, V. J. Mol. Struct. (THEOCHEM) 1997, 389, 227.
[47] For the two earlier summaries, see: (a) Vollhardt, K. P. C.; Mohler, D. L. in
Advances in Strain in Organic Chemistry, Vol. 5 (Ed.: Halton, B.), JAI, London, 1996,
121; (b) Vollhardt, K. P. C. Pure. Appl. Chem. 1993, 65, 153.
[48] Losey, E. N.; LeGoff, E. J. Org. Chem. 1973, 38, 3812.
[49] (a) Barton, J. W.; Walker, R. B. Tetrahedron Lett. 1978, 19, 1005; (b) Jamieson, N.
C.; Lewis, G. E. Aust. J. Chem. 1967, 20, 321.
[50] Barton, J. W.; Shepard, M. K. Tetrahedron Lett. 1984, 25, 4967.
[51] (a) Saito, S.; Yamamoto, Y. Chem. Rev. 2000, 100, 2901; (b) Aubert, C.; Buisine,
O.; Petit, M.; Slowinski, F.; Malacria, M. Pure. Appl. Chem. 1999, 71, 1463; (c)
Grotjahn, D. B. in Comprehensive Organometallic Chemistry II, Vol. 12 (Eds.: Abel, E.
W.; Stone, F. G. A.; Wilkinson, G.; Vol. Ed.: Hegedus, L. S.), Pergamon, Oxford, 1995,
741; (d) Vollhardt, K. P. C. Angew. Chem., Int. Ed. Engl. 1984, 23, 539; (e) Schore, N.
Chem. Rev. 1988, 88, 1081.

308
[52] Berris, B. C.; Lai, Y.-H.; Vollhardt, K. P. C. J. Chem. Soc., Chem. Commun. 1982,
953.
[53] Berris, B. C.; Hovakeemian, G. H.; Lai, Y.-H., Mestdagh, H.; Vollhardt, K. P. C. J.
Am. Chem. Soc. 1985, 107, 5670.
[54] Hirthammer, M.; Vollhardt, K. P. C. J. Am. Chem. Soc. 1986, 108, 2481.
[55] Schulman, J. M.; Disch, R. L. J. Am. Chem. Soc. 1996, 118, 8470 and the references
cited therein.
[56] Berris, B. C.; Hovakeemian, G. H.; Vollhardt, K. P. C. J. Chem. Soc., Chem.
Commun. 1983, 502.
[57] Blanco, L.; Helson, H. E.; Hirthammer, M.; Mestdagh, H.; Spyroudis, S.; Vollhardt,
K. P. C. Angew. Chem., Int. Ed. Engl. 1987, 26, 1246.
[58] Holmes, D.; Kumaraswamy, S.; Matzger, A. J.; Vollhardt, K. P. C. Chem. Eur. J.
1999, 5, 3399 and unpublished results.
[59] Diercks, R.; Vollhardt, K. P. C. Angew. Chem., Int. Ed. Engl. 1986, 25, 266.
[60] Schmidt-Radde, R. H.; Vollhardt, K. P. C. J. Am. Chem. Soc. 1992, 114, 9713.
[61] Diercks, R.; Eaton, B. E.; Gürtzgen, S.; Jalisatgi, S.; Matzger, A. J.; Radde, R. H.;
Vollhardt, K. P. C. J. Am. Chem. Soc. 1998, 120, 8247.
[62] Dosa, P. I.; Whitener, G. D.; Vollhardt, K. P. C.; Bond, A. D.; Teat, S. J. Org. Lett.
2002, 4, 2075.
[63] Jonas, K.; Deffense, E.; Habermann, D. Angew. Chem., Int. Ed. Engl. 1983, 22, 716.
[64] Diercks, R.; Armstrong, J. C.; Boese, R.; Vollhardt, K. P. C. Angew. Chem., Int. Ed.
Engl. 1986, 25, 268.

309
[65] Diercks, R.; Vollhardt, K. P. C. J. Am. Chem. Soc. 1986, 108, 3150.
[66] Han, S.; Bond, A. D.; Disch, R. L.; Holmes, D.; Schulman, J. M.; Teat, S. J.;
Vollhardt, K. P. C.; Whitener, G. D. Angew. Chem., Int. Ed. 2002, 41, 3223.
[67] Schulman, J. M.; Disch, R. L. J. Phys. Chem. A 1997, 101, 5596.
[68] (a) Hopf, H. Classics in Hydrocarbon Chemistry, Wiley-VCH, Weinheim, 2000,
321; (b) Vögtle, F. Fascinating Molecules in Organic Chemistry, Wiley, New York,
1992, 156; (c) Meurer, K. P.; Vögtle, F. Top. Curr. Chem. 1985, 127, 1; (d) Laarhoven,
W. H.; Prinsen, W. J. C. Top. Curr. Chem. 1984, 125, 63.
[69] Eickmeier, C.; Junga, H.; Matzger, A. J.; Scherhag, F.; Shim, M.; Vollhardt, K. P. C.
Angew. Chem., Int. Ed. Engl. 1997, 36, 2103.
[70] Han, S.; Anderson, D. R.; Bond, A. D.; Chu, H. V.; Disch, R. L.; Holmes, D.;
Schulman, J. M.; Teat, S. J.; Vollhardt, K. P. C.; Whitener, G. D. Angew. Chem., Int. Ed.
2002, 41, 3227.
[71] Schulman, J. M.; Disch, R. L. J. Phys. Chem. A 2003, 107, 5223.
[72] (a) Schulman, J. M.; Disch, R. L. Chem. Phys. Lett. 1996, 262, 813; (b) Haymet, A.
D. J. Chem. Phys. Lett. 1985, 122, 421.
[73] Dunlap, B. T.; Taylor, R. J. Phys. Chem. 1994, 98, 11018.
[74] Eickmeier, C.; Holmes, D.; Junga, H.; Matzger, A. J.; Scherhag, F.; Shim, M.;
Vollhardt, K. P. C. Angew. Chem., Int. Ed. Engl. 1999, 38, 800.
[75] Bong, D. T.-Y.; Gentric, L.; Holmes, D.; Matzger, A. J.; Scherhag, F.; Vollhardt, K.
P. C. Chem. Commun. 2002, 278.

310
[76] Bong, D. T.-Y.; Chan, E. W. L.; Diercks, R.; Dosa, P. I.; Haley, M. M.; Matzger, A.
J.; Miljanić, O. Š.; Vollhardt, K. P. C.; Bond, A. D.; Teat, S. J.; Stanger, A. Org. Lett.
2004, 6, 2249.
[77] Hart, H.; Katsumasa, K.; Du, C.-J. F. J. Org. Chem. 1985, 50, 3104.
[78] Goldfinger, M. B.; Crawford, K. B.; Swager, T. M. J. Am. Chem. Soc. 1997, 119,
4578.
[79] Boese, R.; Matzger, A. J.; Mohler, D. L.; Vollhardt, K. P. C. Angew. Chem., Int. Ed.
Engl. 1995, 34, 1478.
[80] Anthony, J. E.; Khan, S. I.; Rubin, Y. Tetrahedron Lett. 1997, 38, 3499.
[81] Baxter, P. N. J. Org. Chem. 2001, 66, 4170.
[82] Bruns, D.; Miura, H.; Vollhardt, K. P. C.; Stanger, A. Org. Lett. 2003, 5, 549.
[83] (a) Schleyer, P. v. R.; Jiao, H. Angew. Chem., Int. Ed. Engl. 1996, 35, 2383; (b)
Aihara, J. J. Chem. Soc., Faraday Trans. 1995, 91, 237; (c) Babić, D.; Trinajstić, N. J.
Mol. Struct. (THEOCHEM) 1994, 120, 321; (d) Gutman, I.; Cyvin, S. J. J. Mol. Struct.
(THEOCHEM) 1993, 107, 85; (e) Aihara, J. Bull. Chem. Soc. Jpn. 1993, 66, 57; (f)
Aihara, J. J. Am. Chem. Soc. 1992, 114, 865; (g) Cioslowski, J.; O’Connor, P. B.;
Fleischmann, E. D. J. Am. Chem. Soc. 1991, 113, 1086.
[84] (a) Matzger, A. J.; Shim, M.; Vollhardt, K. P. C. Chem. Commun. 1999, 1871; (b)
Miljanić, O. Š.; Vollhardt, K. P. C.; Whitener, G. D. Synlett 2003, 29.
[85] (a) Staab, H. A.; Diederich, F. Chem. Ber. 1983, 116, 3487; (b) Staab, H. A.;
Diederich, F.; Krieger, C.; Schweitzer, D. Chem. Ber. 1983, 116, 3504.

311
[86] (a) Campbell, I. D.; Eglinton, G.; Henderson, W.; Raphael, R. A. Chem. Commun.
1966, 87; (b) Solooki, D.; Ferrara, J. D.; Malaba, D.; Bradshaw, J. D.; Tessier, C. A.;
Youngs, W. J. Inorg. Synth. 1997, 31, 122; (c) Huynh, C.; Linstrumelle, G. Tetrahedron
1988, 44, 6337; (d) Iyoda, M.; Vorasingha, A.; Kuwatani, Y.; Yoshida, M. Tetrahedron
Lett. 1988, 39, 4701.
[87] (a) Staab, H. A.; Graf, F. Tetrahedron Lett. 1966, 7, 751; (b) Staab, H. A.; Graf, F.
Chem. Ber. 1970, 103, 1107.
[88] (a) Bunz, U. H. F.; Rubin, Y.; Tobe, Y. Chem. Soc. Rev. 1999, 28, 107; (b)
Diederich, F. Nature 1994, 369, 199.
[89] (a) Coluci, V. R.; Galvão, D. S.; Baughman, R. H. J. Chem. Phys. 2004, 121, 3228;
(b) Narita, N.; Nagai, S.; Suzuki, S. Phys. Rev. B 2001, 64, 245408/1; (c) Narita, N.;
Nagai, S.; Suzuki, S.; Nakao, K. Phys. Rev. B 2000, 62, 11146; (d) Narita, N.; Nagai, S.;
Suzuki, S.; Nakao, K. Phys. Rev. B 1998, 58, 11009; (e) Haley, M. M. Synlett 1998, 557.
[90] Hovakeemian, G. H.; Vollhardt, K. P. C. Angew. Chem., Int. Ed. Engl. 1983, 22,
994.
[91] Mohler, D. L.; Vollhardt, K. P. C.; Wolff, S. Angew. Chem., Int. Ed. Engl. 1990, 29,
1151.
[92] Augustine, R. L. Heterogeneous Catalysis for the Synthetic Chemist, Marcel Dekker,
New York/Basel/Hong Kong, 1996.
[93] (a) Soncini, A.; Havenith, R. W. A.; Fowler, P. W.; Jenneskens, L. W.; Steiner, E. J.
Org. Chem. 2002, 67, 4753; (b) Fowler, P. F.; Havenith, R. W. A.; Jenneskens, L. W.;
Soncini, A.; Steiner, E. Chem. Commun. 2001, 2386.

312
[94] Miljanić, O. Š.; Vollhardt, K. P. C. unpublished results.
[95] (a) Jeyaraman, R.; Murray, R. W. J. Am. Chem. Soc. 1984, 106, 2462; (b) Murray,
R. W.; Jeyaraman, R. J. Org. Chem. 1985, 50, 2847; (c) Adam, W.; Chan, Y.-Y.; Cremer,
D.; Gauss, J.; Scheutzow, D.; Schindler, M. J. Org. Chem. 1987, 52, 2800; (d) Adam, W.;
Curci, R.; Edwards, J. O. Acc. Chem. Res. 1989, 22, 205.
[96] Adam, W.; Balci, M.; Kiliç, H. J. Org. Chem. 1998, 63, 8544.
[97] Kumaraswamy, S.; Jalisatgi, S. S.; Matzger, A. J.; Miljanić, O. Š.; Vollhardt, K. P.
C. Angew. Chem., Int. Ed. 2004, 43, 3771.
[98] Kende, A. S.; MacGregor, P. T. J. Am. Chem. Soc. 1964, 86, 2088.
[99] Furukawa, J.; Kawabata, N.; Nishimura, J. Tetrahedron 1968, 24, 53.
[100] Sevin, F.; McKee, M. L. J. Am. Chem. Soc. 2001, 123, 4591 and the references
cited therein.
[101] Mestdagh, H.; Vollhardt, K. P. C. J. Chem. Soc., Chem. Commun. 1986, 281.
[102] (a) Gajewski, J. J. Hydrocarbon Thermal Isomerizations, Academic Press, New
York/London/Toronto/Syndey/San Francisco, 1981; (b) Harvey, R. G. Curr. Org. Chem.
2004, 8, 303; (c) Scott, L. T. Angew. Chem., Int. Ed. 2004, 43, 4994.
[103] Wiersum, U.; Jenneskens, L. W. Tetrahedron Lett. 1993, 34, 6615.
[104] For recent mechanistic reviews, see: (a) Necula, A.; Scott, L. T. J. Anal. Appl.
Pyrol. 2000, 54, 65; (b) Brown, R. F. C. Eur. J. Org. Chem. 1999, 3211.
[105] Dosa, P. I.; Schleifenbaum, A. S.; Vollhardt, K. P. C. Org. Lett. 2001, 3, 1017.
[106] Matzger, A. J.; Vollhardt, K. P. C. Chem. Commun. 1997, 1415.
[107] Preda, D. V.; Scott, L. T. Org. Lett. 2000, 2, 1489.

313
[108] (a) Oh, M.; Yu, K.; Li, H.; Watson, E. J.; Carpenter, G. B.; Sweigart, D. A. Adv.
Synth. Catal. 2003, 345, 1053 and the references cited therein; (b) Perthuisot, C.;
Edelbach, B. L.; Zubris, D. L.; Jones, W. D. Organometallics 1997, 16, 2016.
[109] (a) Oprunenko, Y.; Gloriozov, I.; Lyssenko, K.; Malyugina, S.; Mityk, D.;
Mstislavsky, V.; Günther, H.; von Firks, G.; Ebener, M. J. Organomet. Chem. 2002, 656,
27; (b) Dullaghan, C. A.; Carpenter, G. B.; Sweigart, D. A. Chem. Eur. J. 1997, 3, 75; (c)
Eischenbroich, C.; Schneider, J.; Massa, W.; Baum, G.; Mellinghoff, H. J. Organomet.
Chem. 1988, 355, 163.
[110] Ceccon, A.; Gambaro, A.; Romanin, A. M.; Venzo, A. J. Organomet. Chem. 1982,
239, 345.
[111] Nambu, M.; Siegel, J. S. J. Am. Chem. Soc. 1988, 110, 3675.
[112] Nambu, M.; Hardcastle, K.; Baldridge, K. K.; Siegel, J. S. J. Am. Chem. Soc. 1992,
114, 369.
[113] Nambu, M.; Mohler, D. L.; Hardcastle, K.; Baldridge, K. K.; Siegel, J. S. J. Am.
Chem. Soc. 1993, 115, 6138.
[114] Brown, R. D.; Godfrey, P. D.; Hart, B. T.; Ottrey, A. L.; Onda, M.; Woodruff, M.
Aust. J. Chem. 1983, 36, 639.
[115] Schleifenbaum, A.; Feeder, N.; Vollhardt, K. P. C. Tetrahedron Lett. 2001, 42,
7329.
[116] (a) Volland, W. V.; Davidson, E. R.; Borden, W. T. J. Am. Chem. Soc. 1979, 101,
533; (b) Allinger, N. L. J. Am. Chem. Soc. 1958, 80, 1953; (c) Allinger, N. L.; Sprague, J.
T. J. Am. Chem. Soc. 1972, 94, 5734; (d) Rogers, D. W.; Voitkenberg, H.; Allinger, N. L.

314
J. Org. Chem. 1978, 43, 360; (e) Mock, W. L. Tetrahedron Lett. 1972, 475; (f) Pople, J.
A.; Mock, W. L. Tetrahedron Lett. 1972, 479.
[117] (a) Petersson, E. J.; Fanuele, J. C.; Nimlos, M. R.; Lemal, D. M.; Ellison, G. B.;
Radziszewski, J. G. J. Am. Chem. Soc. 1997, 119, 11122; (b) Bremer, M.; Schleyer, P. v.
R.; Fleischer, U. J. Am. Chem. Soc. 1989, 111, 1147.
[118] De Rango, C.; Tsoucaris, G.; Declerq, J. P.; Germain, G.; Putzeys, J. P. Acta
Crystallogr. Sect. C 1973, 2, 189.
[119] (a) van den Hark, T. E. M.; Beurskens, P. T. Acta Crystallogr. Sect. C 1976, 5, 247;
(b) Beurskens, P. T.; Beurskens, G.; van den Hark, T. E. M. Acta Crystallogr. Sect. C
1976, 5, 241.
[120] Grimme, S.; Harren, J.; Sobanski, A.; Vögtle, F. Eur. J. Org. Chem. 1998, 1491.
[121] Schleyer, P. v. R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N. J.
R. J. Am. Chem. Soc. 1996, 118, 6317.
[122] Schleyer, P. v. R.; Manoharan, M.; Wang, Z.-X.; Kiran, B.; Jiao, H.; Puchta, R.;
van Eikema Hommes, N. J. R. Org. Lett. 2001, 3, 2465.
[123] Schulman, J. M.; Disch, R. L.; Jiao, H.; Schleyer, P. v. R. J. Phys. Chem. A 1998,
102, 8051.
[124] Adcock, W.; Gupta, B. D.; Khor, T. C.; Doddrell, D.; Kitching, W. J. Org. Chem.
1976, 41, 751.
[125] Thummel, R. P.; Nutakul, W. J. Org. Chem. 1978, 43, 3170.
[126] Fukui, K. Orientation and Stereoselection, Springer, Berlin, 1970.
[127] Heeger, A. J. Angew. Chem., Int. Ed. 2001, 40, 2591 (Nobel lecture).

315
[128] NIST Chemistry WebBook, NIST Standard Reference Database Number 69,
November 1998, National Institute of Standards and Technology, Gaithersburg, MD
20899 (http://webbook.nist.gov).
[129] Maksić, Z. B.; Kovaček, D.; Maksić-Eckert, M.; Böckmann, M.; Klessinger, M. J.
Phys. Chem. 1995, 99, 6410.
[130] (a) Wenz, G.; Müller, M. A.; Schmidt, M.; Wegner, G. Macromolecules 1984, 17,
837; (b) Martin, R. E.; Diederich, F. Angew. Chem., Int. Ed. 1999, 38, 1350; (c) Geerts,
Y.; Klärner, G.; Müllen, K. in Electronic Materials: The Oligomer Approach, Wiley-
VCH, Weinheim, 1998.
[131] Dosche, C.; Löhmannsröben, H.-G.; Bieser, A.; Dosa, P. I.; Han, S.; Iwamoto, M.;
Schleifenbaum, A.; Vollhardt, K. P. C. Phys. Chem. Chem. Phys. 2002, 4, 2156.
[132] Shpol’skii, E. V.; Il’ina, A. A.; Klimova, L. A. Dokl. Akad. Nauk SSSR 1952, 87,
935.
[133] Dosche, C.; Kumke, M. U.; Ariese, F.; Bader, A. N.; Gooijer, C.; Dosa, P. I.; Han,
S.; Miljanić, O. Š.; Vollhardt, K. P. C.; Puchta, R.; van Eikema Hommes, N. J. R. Phys.
Chem. Chem. Phys. 2003, 5, 4563.
[134] Dosche, C.; Kumke, M. U.; Löhmannsröben, H.-G.; Ariese, F.; Bader, A. N.;
Gooijer, C.; Miljanić, O. Š.; Iwamoto, M.; Vollhardt, K. P. C.; Puchta, R.; van Eikema
Hommes, N. J. R. Phys. Chem. Chem. Phys. 2004, 6, 5476.
[135] Haley, M. M. Postdoctoral Report, University of California, Berkeley, 1993.
[136] Westmoreland, I.; Que, E.; Miyagi, L. Acta Crystallogr. Sect. E 2005, manuscript
in preparation.

316
[137] Hillard III, R. L.; Vollhardt, K. P. C. J. Am. Chem. Soc. 1977, 99, 4058.
[138] Review: (a) Beller, M.; Seayad, J.; Tillack, A.; Jiao, H. Angew. Chem., Int. Ed.
2004, 43, 3368. Examples: (b) Alper, H.; Saldana–Maldonado, M.; Lin, L. J. B. J. Mol.
Catal. 1988, 49, L27; (c) El Ali, B.; Alper, H. J. Mol. Catal. A 1995, 96, 197.
[139] Camacho, D. H.; Saito, S.; Yamamoto, Y. Tetrahedron Lett. 2002, 43, 1085.
[140] Selected examples: (a) Ogo, S.; Uehara, K.; Abura, T.; Watanabe, Y.; Fukuzumi, S.
J. Am. Chem. Soc. 2004, 126, 16520; (b) Grotjahn, D. B.; Incarvito, C. D.; Rheingold, A.
L. Angew. Chem., Int. Ed. 2001, 40, 3884.
[141] Selected recent examples: (a) Liang, B.; Dai, M.; Chen, J.; Yang, Z. J. Org. Chem.
2005, 70, 391; (b) Wolf, C.; Lerebours, R. Org. Biomol. Chem. 2004, 2, 2161; (c)
Bhattacharya, S.; Sengupta, S. Tetrahedron Lett. 2004, 45, 8733.
[142] Nicolaou, K. C.; Dai, W. M.; Hong, Y. P.; Baldridge, K. K.; Siegel, J. S.; Tsay, S.
C. J. Am. Chem. Soc. 1993, 115, 7944.
[143] Moran, D.; Stahl, F.; Bettinger, H. F.; Schaefer, H. F. III, Schleyer, P. v. R. J. Am.
Chem. Soc. 2003, 125, 6746.
[144] Reviews: (a) Fürstner, A.; Davies, P. W. Chem. Commun. 2005, 2307; (b) Fürstner,
A. in Handbook of Metathesis, Vol. 2 (Ed.: Grubbs, R. H.), Wiley-VCH, Weinheim,
2005, 432; (c) Bunz, U. H. F. Science 2005, 308, 216.
[145] Selected references: (a) Mortreux, A.; Petit, F.; Blanchard, M. Tetrahedron Lett.
1978, 49, 4967; (b) Mortreux, A.; Dy, N.; Blanchard, M. J. Mol. Catal. 1976, 1, 101; (c)
Mortreux, A.; Blanchard, M. J. Chem. Soc, Chem. Commun. 1974, 786.
[146] Summary of the reactivity of [Mo(CO)6]: Marradi, M. Synlett 2005, 1196.

317
[147] Selected references: (a) Sashuk, V.; Ignatowska, J.; Grela, K. J. Org. Chem. 2004,
69, 7748; (b) Grela, K.; Ignatowska, J. Org. Lett. 2002, 4, 3747; (c) Brizius, G.; Bunz, U.
H. F. Org. Lett. 2002, 4, 2829; (d) Villemin, D.; Héroux, M.; Blot, V. Tetrahedron 2001,
42, 3701; (e) Pschirer, N. G.; Bunz, U. H. F. Tetrahedron Lett. 1999, 40, 2481; (f) du
Plessis, J. A. K.; Vosloo, H. C. M. J. Mol. Catal. A: Chem. 1998, 133, 305.
[148] Selected references: (a) Hellbach, B.; Gleiter, R.; Rominger, F. Synthesis 2003,
2535; (b) Brizius, G.; Billingsley, K.; Smith, M. D.; Bunz, U. H. F. Org. Lett. 2003, 5,
3951; (c) Brizius, G.; Kroth, S.; Bunz, U. H. F. Macromolecules 2002, 35, 5317; (d)
Pschirer, N. G.; Fu, W.; Adams, R. D.; Bunz, U. H. F. Chem. Commun. 2000, 87; (e) Ge,
P.-H.; Fu, W.; Herrmann, W. A.; Herdtweck, E.; Campana, C.; Adams, R. A.; Bunz, U.
H. F. Angew. Chem., Int. Ed. 2000, 39, 3607. Reviews: (f) Bunz, U. H. F. Acc. Chem.
Res. 2001, 34, 998; (g) Bunz, U. H. F.; Kloppenburg, L. Angew. Chem., Int. Ed. 1999, 38,
478.
[149] (a) Schrock, R. R. Acc. Chem. Res. 1986, 19, 342; (b) Schrock R. R.; Clark, D. N.;
Sancho, J.; Wengrovius, J. H.; Rocklage, S. M.; Pedersen, S. F.
Organometallics 1982, 1, 1645; (c) Wengrovius, J. H.; Sancho, J.; Schrock, R. R. J. Am.
Chem. Soc. 1981, 103, 3932.
[150] Selected references: (a) Fürstner, A.; Castanet, A.-S.; Radkowski, K.; Lehmann, C.
W. J. Org. Chem. 2003, 68, 1521; (b) Song, D.; Blond, G.; Fürstner, A. Tetrahedron
2003, 59, 6899; (c) Fürstner, A.; Dierkes, T. Org. Lett. 2000, 2, 2463; (d) Fürstner, A.;
Seidel, G. J. Organomet. Chem. 2000, 606, 75; (e) Fürstner, A.; Guth, O.; Rumbo, A.;

318
Seidel, G. J. Am. Chem. Soc. 1999, 121, 11108; (f) Fürstner, A.; Seidel, G. Angew.
Chem., Int. Ed. 1998, 37, 1734.
[151] (a) Bauer, E. B.; Hampel, F.; Gladysz, J. A. Adv. Synth. Cat. 2004, 346, 812; (b)
Bauer, E. B.; Szafert, S.; Hampel, F.; Gladysz, J. A. Organometallics 2003, 22, 2184; (c)
Ijsselstijn, M.; Aguilera, B.; van der Marel, G. A.; van Boom, J. H.; van Delft, F. L.;
Schoemaker, H. E.; Overkleeft, H. S.; Rutjes, F. P. J. T.; Overhand, M. Tetrahedron Lett.
2004, 45, 4379.
[152] Strem Chemicals, catalog number 74-1800, $295.00/500 mg (May 2005).
[153] (a) Fürstner, A.; Mathes, C.; Lehmann, C. W. Chem. Eur. J. 2001, 7, 5299; (b)
Fürstner, A.; Grela, K.; Mathes, C.; Lehmann, C. W. J. Am. Chem. Soc. 2000, 122,
11799; (c) Fürstner, A.; Mathes, C.; Lehmann, C. W. J. Am. Chem. Soc. 1999, 121, 9453.
[154] Selected references: (a) Zhang, W.; Moore, J. S. J. Am. Chem. Soc. 2004, 126,
12796 and the references cited therein; (b) Fürstner, A.; Mathes, C. Org. Lett. 2001, 3,
221.
[155] Collman, J. P.; Hegedus, L. S.; Norton, J. R.; Finke, R. G. in Principles and
Applications of Organotransition Metal Chemistry, University Science Books, Mill
Valley, 1987, Chapter 9.
[156] Bino, A.; Ardon, M.; Shirman, E. Science 2005, 308, 324.
[157] For a highlight, see: (a) Höger, S. Angew. Chem., Int. Ed. 2005, 44, 3806. Reviews:
(b) Tobe, Y.; Sonoda, M. in Modern Cyclophane Chemistry (Eds.: Gleiter, R.; Hopf, H.),
Wiley-VCH, Weinheim, 2004, 1; (c) Marsden, J. A.; Palmer, G. J.; Haley, M. M. Eur. J.
Org. Chem. 2003, 2355; (d) Haley, M. M. Synlett 1998, 557; (e) Haley, M. M.; Pak, J. J.;

319
Brand, S. C. Top. Curr. Chem. 1999, 202, 81; (f) Haley, M. M.; Wan, W. B. in Advances
in Strained and Interesting Organic Molecules, Vol. 8 (Ed.: Halton, B.), JAI Press, New
York, 2000, 1; (g) Kennedy, R. D.; Lloyd, D.; McNab, H. J. Chem. Soc., Perkin Trans. 1
2002, 1601; (h) Balaban, A.T.; Banciu, M.; Ciorba, V. Annulenes, Benzo-, Hetero-,
Homo-Derivatives and Their Valence Isomers, Vol. 2, CRC Press, Boca Raton, 1987,
146. For very recent work, see, inter alia: (i) Marsden, J. A.; Miller, J. J.; Shirtcliff, L. D.;
Haley, M. M. J. Am. Chem. Soc. 2005, 127, 2464; (j) Johnson, C. A.; Haley, M. M.;
Rather, E.; Han, F.; Weakley, T. J. R. Organometallics 2005, 24, 1161; (k) Baxter, P. N.
W.; Dali-Youcef, R. J. Org. Chem. 2005, 70, 4935; (l) Hisaki, I.; Eda, T.; Sonoda, M.;
Niino, H.; Sato, T.; Wakabayashi, T.; Tobe, Y. J. Org. Chem. 2005, 70, 1853.
[158] Review: (a) Youngs, W. J.; Tessier, C. A.; Bradshaw, J. D. Chem. Rev. 1999, 99,
3153. Selected references: (b) Iyoda, M.; Sirinintasak, S.; Nishiyama, Y.; Vorasingha, A.;
Sultana, F.; Nakao, K.; Kuwatani, Y.; Matsuyama, H.; Yoshida, M.; Miyake, Y. Synthesis
2004, 1527; (c) Nishinaga, T.; Kawamura, T.; Komatsu, K. Chem. Commun. 1998, 2263;
(d) Youngs, W. J.; Kinder, J. D.; Bradshaw, J. D.; Tessier, C. A. Organometallics 1993,
12, 2406; (e) Djebli, A.; Ferrara, J. D.; Tessier-Youngs, C.; Youngs, W. J. J. Chem. Soc.,
Chem. Commun. 1988, 548; (f) Ferrara, J. D.; Tessier-Youngs, C.; Youngs, W. J. J. Am.
Chem. Soc. 1988, 110, 3326.
[159] (a) Iyer, V. S.; Vollhardt, K. P. C.; Wilhelm, R. Angew. Chem., Int. Ed. 2003, 42,
4379; (b) Dosa, P. I.; Erben, C.; Iyer, V. S.; Vollhardt, K. P. C.; Wasser, I. M. J. Am.
Chem. Soc. 1999, 121, 10430; (c) Boese, R.; Matzger, A. J.; Vollhardt, K. P. C. J. Am.
Chem. Soc. 1997, 119, 2052.

320
[160] Selected references: (a) Pak, J. J.; Weakley, T. J. R.; Haley, M. M.; Lau, D. Y. K.;
Stoddart, J. F. Synthesis 2002, 1256; (b) Tobe, Y.; Utsumi, N.; Kawabata, K.; Nagano,
A.; Adachi, K.; Araki, S.; Sonoda, M.; Hirose, K.; Naemura, K. J. Am. Chem. Soc. 2002,
124, 5350; (c) Hosokawa, Y.; Kawase, T.; Oda, M. Chem. Commun. 2001, 1948; (d)
Nakamura, K.; Okubo, H.; Yamaguchi, M. Org. Lett. 2001, 3, 1097; (e) Höger, S.;
Enkelmann, V.; Bonrad, K.; Tschierske, C. Angew. Chem., Int. Ed. 2000, 39, 2268. See
also: (f) Moore, J. S. Acc. Chem. Res. 1997, 30, 402; (g) Zhang, J.; Pesak, D. J.; Ludwick,
J. L.; Moore, J. S. J. Am. Chem. Soc. 1994, 116, 4227; (h) Pickholz, M.; Stafström, S.
Chem. Phys. 2001, 270, 245.
[161] Selected references: (a) Sarkar, A.; Pak, J. J.; Rayfield, G. W.; Haley, M. M. J.
Mater. Chem. 2001, 11, 2943. (b) Wan, W. B.; Haley, M. M. J. Org. Chem. 2001, 66,
3893.
[162] Holmes, D. Ph. D. Dissertation, University of California, Berkeley, 1999.
[163] Kehoe, J. M.; Kiley, J. H.; English, J. J.; Johnson, C. A.; Petersen, R. C.; Haley, M.
M. Org. Lett. 2000, 2, 969.
[164] For 155b and 155d: (a) Kryska, A.; Skulski, L. J. Chem. Res.(S) 1999, 590. For
155c: (b) Lacour, J.; Monchaud, D.; Bernardinelli, G.; Favarger, F. Org. Lett. 2001, 3,
1407. For 161b: (c) Mattern, D. L. J. Org. Chem. 1983, 48, 4772. For 57, ref. 78.
[165] Nakayama, J.; Sakai, A.; Hoshino, M. J. Org. Chem. 1984, 49, 5084.
[166] Professor John Anthony, personal communication.
[167] Garden, S. J.; Torres, J. C.; Ferreira, A. A.; Silva, R. B.; Pinto, A. C. Tetrahedron
Lett. 1997, 38, 1501.

321
[168] Lisowski, V.; Robba, M.; Rault, S. J. Org. Chem. 2000, 65, 4193.
[169] Wan, W. B.; Haley, M. M. J. Org. Chem. 2001, 66, 3893.
[170] Recent reports opened the possibility of using chloroiodoarenes for the same
purpose: (a) Gelman, D.; Buchwald, S. L. Angew. Chem., Int. Ed. 2003, 42, 5993; (b)
Köllhofer, A.; Pullmann, T.; Plenio, H. Angew. Chem., Int. Ed. 2003, 42, 1056. Due to
the huge difference in Sonogashira reactivity between chlorides and iodides,
distinguishing these two groups was trivial in our propynylation experiments.
[171] (a) Sonogashira, K. in Handbook of Organopalladium Chemistry for Organic
Synthesis, Vol. 1 (Ed.: Negishi, E.-i.), Wiley, New York, 2002, 493 and the references
cited therein; (b) Sonogashira, K. in Metal-Catalyzed Cross-Coupling Reactions (Eds.:
Diederich, F., Stang, P. J.), Wiley-VCH, New York, 1998, Chapter 5; (c) Sonogashira, K.
in Comprehensive Organic Synthesis, Vol. 3 (Ed.: Trost, B. M.), Pergamon, New York,
1991, Chapter 2.4.
[172] Related applications of microwaves in Sonogashira reactions: (a) Petricci, E.; Radi,
M.; Corelli, F.; Botta, M. Tetrahedron Lett. 2003, 44, 9181; (b) Erdelyi, M.; Gogoll, A. J.
Org. Chem. 2001, 66, 4165. General reviews of microwave-assisted reactivity: (c) Kappe,
C. O. Angew. Chem., Int. Ed. 2004, 43, 6250; (d) Lidström, P.; Tierney, J.; Wathey, B.;
Westman, J. Tetrahedron 2001, 57, 9225.
[173] We are indebted to Personal Chemistry, Inc. (now Biotage, Inc.) for the loan of a
Smith SynthesizerTM.

322
[174] We would like to thank Prof. Alois Fürstner and Mr. Günter Seidel (Max-Planck-
Institut für Kohlenforschung, Mülheim/Ruhr, Germany) for catalyzing the initiation of
this effort with generous samples of [(Me3CO)3W≡CCMe3].
[175] Li, Y.; Zhang, J.; Wang, W.; Miao, Q.; She, X.; Pan, X. J. Org. Chem. 2005, 70,
3285.
[176] The spectral data are in good agreement with those in ref. 87b.
[177] The spectral data are in good agreement with those in ref. 86d.
[178] Srinivasan, M.; Sankararaman, S.; Dix, I.; Jones, P. G. Org. Lett. 2000, 2, 3849.
[179] See Experimental Section for complete details. Details of the crystal structure
determination (deposition number CCDC 192630) may also be obtained free of charge on
application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+44) 1223–336–
033; E-mail: [email protected]).
[180] (a) Alkorta, I.; Rozas, I.; Elguero, J. Tetrahedron 2001, 57, 6043; (b) Jusélius, J.;
Sundholm, D. Phys.Chem. Chem.Phys. 2001, 3, 2433; (c) Godard, C.; Lepetit, C.;
Chauvin, R. Chem. Commun. 2000, 1833; (d) Wan, W. B.; Kimball, D. B.; Haley, M. M.
Tetrahedron Lett. 1998, 39, 6795; (e) Matzger, A. J.; Vollhardt, K. P. C. Tetrahedron
Lett. 1998, 39, 6791.
[181] Miljanić, O. Š.; Holmes, D.; Vollhardt, K. P. C. Org. Lett. 2005, 7, in press.
[182] Gilles, J.-M.; Oth, J. F. M.; Sondheimer, F.; Woo, E. P. J. Chem. Soc. (B) 1971,
2177.
[183] For triple cyclizations of this type, see ref. 82 and references therein.

323
[184] For an isomeric dehydrotetrabenz[32]annulene, see: Haley, M. M.; Bell, M. L.;
Brand, S. C.; Kimball, D. B.; Pak, J. J.; Wan, W. B. Tetrahedron Lett. 1997, 43, 7483.
[185] While 184b and 176b were not known prior to this work, they were readily
prepared using the procedure described for 184c and 146 (see Section 6.4).
[186] Strained ring fusion increases Jpara in arenes. For a review, see: Frank, N. L.; Siegel,
J. S. in Advances in Theoretically Interesting Molecules, Vol. 3 (Ed.: Thummel R. P.),
JAI, London, 1995, 209.
[187] Miljanić, O. Š.; Han, S.; Holmes, D.; Schaller, G. R.; Vollhardt, K. P. C. Chem.
Commun. 2005, 2606.
[188] Eliel, E. L.; Wilen, S. H.; Mander, L. N. Stereochemistry of Organic Compounds,
Wiley, New York, 1994, 1142.
[189] Yamamoto, G.; Ōki, M. Chem. Lett. 1972, 45.
[190] Nakamura, M.; Ōki, M. Tetrahedron Lett. 1974, 15, 505.
[191] Mew, P. K. T.; Vögtle, F. Angew. Chem., Int. Ed. Engl. 1979, 18, 159.
[192] Meyer, W. L.; Meyer, R. B. J. Am. Chem. Soc. 1963, 85, 2170.
[193] Toyota, S.; Makino, T. Tetrahedron Lett. 2003, 44, 7775.
[194] First observation: Christie, G. H.; Kenner, J. H. J. Chem. Soc. 1922, 121, 614.
[195] (a) Seminario, J. M.; Zacarias, A. G.; Tour, J. M. J. Am. Chem. Soc. 1998, 120,
3970; (b) Stølevic, R.; Bakken, P. J. Mol. Struct. 1990, 239, 205; (c) Grumadas, A.;
Poskus, D. Zh. Fiz. Khim. 1987, 61, 2838; (d) Okuyama, K.; Hasegawa, T.; Ito, M.;
Mikami, N. J. Phys. Chem. 1984, 88, 1711.

324
[196] Toyota, S.; Yamamori, T.; Makino, T. Tetrahedron 2001, 57, 3521 and references
cited therein.
[197] Dominguez, Z.; Dang, H.; Strouse, M. J.; Garcia-Garibay, M. A. J. Am. Chem. Soc.
2002, 124, 2398.
[198] Bedard, T. C.; J. S. Moore J. Am. Chem. Soc. 1995, 117, 10662.
[199] Toyota, S.; Iida, T.; Kunizane, C.; Tanifuji, N.; Yoshida, Y. Org. Biomol. Chem.
2003, 1, 2298.
[200] (a) Raker, J.; Glass, T. E. J. Org. Chem. 2001, 66, 6505; (b) Glass, T. E. J. Am.
Chem. Soc. 2000, 122, 4522; (c) Yagi, S.; Kitayama, H.; Takagishi, T. J. Chem. Soc.,
Perkin Trans. 1 2000, 925; (d) Godinez, C. E.; Zepeda, G.; Mortko, C. J.; Dang, H.;
Garcia-Garibay, M. A. J. Org. Chem. 2004, 69, 1652; (e) Dominguez, Z.; Dang, H.;
Strouse, M. J.; Garcia-Garibay, M. A. J. Am. Chem. Soc. 2002, 124, 7719; (f) Joachim,
C.; Tang, H.; Moresco, F.; Rapenne, G.; Meyer, G. Nanotechnology 2002, 13, 330.
[201] Examples: (a) Toyota, S.; Goichi, M.; Kotani, M. Angew. Chem., Int. Ed. 2004, 43,
2248; (b) Brizius, G.; Billingsley, K.; Smith, M. D.; Bunz, U. H. F. Org. Lett. 2003, 5,
3951; (c) Levitus, M.; Schmieder, K.; Ricks, H.; Shimizu, K. D.; Bunz, U. H. F.; Garcia-
Garibay, M. A. J. Am. Chem. Soc. 2001, 123, 4259; (d) Miteva, T.; Palmer, L.;
Kloppenburg, L.; Neher, D.; Bunz, U. H. F. Macromolecules 2000, 33, 652.
[202] The parent 2,2’,6,6’-tetrakisethynyldiphenylacetylene (205a, R = R’ = H) is
known: Bradshaw, J. D.; Guo, L.; Tessier, C. A.; Youngs, W. J. Organometallics 1996,
15, 2582.

325
[203] Houk, K. N.; Scott, L. T.; Rondan, N. G.; Spellmeyer, D. C.; Reinhardt, G.; Hyun,
J. L.; DeCicco, G. J.; Weiss, R.; Chen, M. H. M.; Bass, L. S.; Clardy, J.; Jørgensen, F. S.;
Eaton, T. A.; Sarkozi, V.; Petit, C. M.; Ng, L.; Jordan, K. D. J. Am. Chem. Soc. 1985,
107, 6556.
[204] (a) Marsden, J. A.; O’Connor, M. J.; Haley, M. M. Org. Lett. 2004, 6, 2385; (b)
Sonoda, M.; Sakai, Y.; Yoshimura, T.; Tobe, Y.; Kamada, K. Chem. Lett. 2004, 33, 972.
[205] Reviews: (a) Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029; (b) Mikami, K.;
Aikawa, K.; Yusa, Y.; Jodry, J. J.; Yamanaka, M. Synlett 2002, 1561; (c) McCarthy, M.;
Guiry, P. J. Tetrahedron 2001, 57, 3809.
[206] For previous examples of conformationally locked dehydrobenzannulenes (and
analogs) in which this phenomenon is skeleton-induced, see: (a) Heuft, M. A.; Collins, S.
K.; Fallis, A. G. Org. Lett. 2003, 5, 1911; (b) Heuft, M. A.; Fallis, A. G. Angew. Chem.,
Int. Ed. 2002, 41, 4520; (c) Marsella, M.; Kim, I. T.; Tham, F. J. Am. Chem. Soc. 2000,
122, 974; (d) Boese, R.; Matzger, A. J.; Vollhardt, K. P. C. J. Am. Chem. Soc. 1997, 119,
2052.
[207] (a) Ōki, M. Applications of Dynamic NMR Spectroscopy to Organic Chemistry,
VCH, Weinheim, 1985; (b) Gasparro, F. P.; Kolodny, N. H. J. Chem. Ed. 1977, 54, 258.
[208] (a) Tobe, Y.; Ohki, I.; Sonoda, M.; Niino, H.; Sato, T.; Wakabayashi, T. J. Am.
Chem. Soc. 2003, 125, 5614; (b) Kowalik, J.; Tolbert, L. M. J. Org. Chem. 2001, 66,
3229.
[209] Müller, M.; Förster, W.-R.; Holst, A.; Kingma, A. J.; Schaumann, E.; Adiwidjaja,
G. Chem. Eur. J. 1996, 2, 949.

326
[210] Hisaki, I.; Vollhardt, K. P. C., unpublished results.
[211] We are grateful to Prof. Odile Eisenstein (University of Montpellier, France) for an
insightful discussion.
[212] The limitations of this approach are clear: two independent variables define a two-
dimensional conformation space, within which we meant to explore only a single
trajectory. While the software used had an option of a 2D conformational search, we
found the results much less accurate than those obtained by a sequence of single point
optimizations. As a compromise, we went ahead with our “single-trajectory” strategy,
and confirmed the reasonableness of the selected trajectory by the comparison with the
lowest-energy interconversion pathway suggested by the crude 2D conformational
search.
[213] Heck, R. F. Palladium Reagents in Organic Syntheses, Academic Press,
London/Orlando, 1985, 18.
[214] Still, W. C.; Kahn, M.; Mitra, S. J. Org. Chem. 1978, 43, 2923.
[215] Jaguar 5.5, Schrödinger, L.L.C., Portland, OR, 1991–2003; Macromodel 8.1,
Schrödinger, L.L.C., Portland, OR, 1991–2003; Maestro 6.5, Schrödinger, L.L.C.,
Portland, OR, 1991–2004.
[216] (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648; (b) Lee, C.; Yang, W.; Parr, R. G.
Phys. Rev. B 1988, 37, 785.
[217] Suzuki, H.; Maruyama, K.; Goto, R. Bull. Chem. Soc. Jpn. 1965, 38, 1590.

327
[218] (a) Bleckmann, W.; Hanack, M. Chem. Ber. 1984, 117, 3021; (b) Müller, E.;
Sauerbier, M.; Streichfuss, D.; Thomas, R.; Winter, W.; Zountsas, G.; Heiss, J. Justus
Liebigs Ann. Chem. 1971, 750, 63.
[219] Tanaka, Y.; Yamashita, H.; Tanaka, M. Organometallics 1995, 14, 530.
[220] Kinder, J. D.; Tessier, C. A.; Youngs, W. J. Synlett 1993, 149.
[221] Collins, I.; Suschitzky, H. J. Chem. Soc. C 1969, 17, 2337.