m · foki methylates adenine in both strands of its asymmetric recognition sequence
TRANSCRIPT

Gene, 77 (1989) l-10
Elsevier
GEN 02921
M. F&I methylates adenine in both strands of its asymmetric recognition sequence
(Flavobacterium okeanokoites; type-IIS restriction-modification system; DNA-methyltransferase; N6-methy-
ladenine)
David Landry, Mary C. Looney, George R. Feehery, Barton E. Slatko, William E. Jack, Ira Schildkraut and
Geoffrey G. Wilson
New England Biolabs, Inc., Beverly, MA 01915 (U.S.A.)
Received by G.N. Godson: 27 October 1988
Accepted: 21 November 1988
SUMMARY
M . FokI, a type-IIS modification enzyme from Flavobacterium okeanokoites, was purified, and its activity was
characterized in vitro. The enzyme was found to be a DNA-adenine methyltransferase and to methylate both
strands of the asymmetric FokI recognition sequence:
5’-GGATG
C CTAC-5’
M . FokI ____________+ 5’-GGm6 ATG
CCTm6 AC-5’.
M. FokI does not methylate single-stranded DNA, nor does it methylate double-stranded DNA at sequences
other than FokI sites.
INTRODUCTION
The FokI restriction-modification (R-M) system
from F. okeanokoites is a member of the type-IIS
class of R-M systems (see Roberts, 1988, for a
compilation of R-M systems). Like the enzymes of
type-II R-M systems, type-IIS enzymes have mini-
ma1 co-factor requirements; the ENases require only
magnesium ions, and the MTases require only
AdoMet. In contrast to the majority of type-II en-
Correspondence to: Dr. G.G. Wilson, New England Biolabs, Inc.,
32 Tozer Road, Beverly, MA 01915 (U.S.A.) Tel. (508)927-5054;
Fax (508)921-1350.
Abbreviations: A, adenine; A, absorbance (1 cm); AdoMet,
S-adenosylmethionine; buffers A, B, C, and M, see MATE-
RIALS AND METHODS, section b; bp, base pair(s); C, cy-
tosine; CD, catalytic domain; cpm, counts/min; dpm, disinte-
grations/min; ds, double-strand(ed); ENase (symbol R.), restric-
tion endonuclease; G, guanine; [3H]AdoMet, S-adenosyl-
[methyl-3H]methionine; HPLC, high-pressure liquid chromato-
graphy; kb, 1000 bp; m6A, N6-methyladenine; m6dA, N6-
methyldeoxyadenosine; m4C, N4-methylcytosine; m4dC, N“-
methyldeoxycytidine; m5C, 5-methylcytosine; m5dC, 5-methyl-
deoxycytidine; MTase (symbol M.), methyltransferase; nt, nu-
cleotide (s); oligo, oligodeoxyribonucleotide; PAGE, polyacry-
lamide gel electrophoresis; RF, replicative form; R-M, re-
striction-modification; solvents D and G, see Table I; ss, single-
strand(ed); T, thymine; TLC, thin-layer chromatography; TRD,
target recognition domain; u, unit(s).
0378.1119/89/$03.50 0 1989 Elsevier Science Publishers B.V. (Biomedical Division)

asymmetric nndeotide sequences and the ENases cleave outside of the recognition site. In these re- spects they resemble more closely the enzymes of type-III systems (reviewed by Yuan and Hamilton, 1984). The R + FokI ENase recognizes the sequence ~:~~~~~~~~ (Su~s~i and Kanazawa, 1981).
Type-II MTases methylate both strands of their recognition sequences (see Smith and Kelly, 1984; ~~~Ciell~d and Nelson, 1988, for reviews). Double- strand (ds) reactions are achieved by single enzyme moieties because the strands of sym,m~tri~ se- quences are equivalent to one another. The situation is not so simple for enzymes that methylate asymme- tric sequences; because the strands are different, a different reaction is probably needed to methylate each. An MTase requires a rni~irn~~ of three fu~~ti~~~ domains, (I) a TRD (Wilke et al., 19X8>, (2) au AdoMet-binding domain, and (3) a transme- thylation CD. For MTases that recognize symmetric sequences, one of each domain is suffricient, but for MTases that recognize as~~~~ sequences, at least two TRDs and one CD, or two CDs and one TRD, are needed. Conceivably, two of each domain may be needed, if domain-sharing cannot occur.
The type-1 MTases methylate both strands of their asyrn~~~~ic reco~ition sequences; accordingly, their sp~~i~city subunits possess two distinct TRDs (Gaugh and Murray, 1983; Nagaraja et al., 1985; Fuller-Pace and Murray, 1986). The multi-specific ~~~~~~~.~ phage MTases also possess one TRD for each sequence methylated; the TRDs share a com- mon CD (Iran-Bet&e et al., 1984; Behrens et al., 1987; Efalganesh et al., 1987; Wiie et al., 1988). The type-II1 MTases, in contrast, methyl&e only one strand of their asymmetric recognition sequences, and thus require only one TRD and CD (Schroeder et al,, 1986; H~n~belin et al., 1988). It is not clear whether the type-IIS MTases methylate one strand or both, nor, in the event that they methylate both, whether the methylated nucleotides are the same, and whether the MTase is a single protein or a com- plex of subunits.
The chara~te~ation of type-IIS R-M systems has not advanced as rapidiy as has the ~h~acterization of other types of R-M systems (Gann et al., 1987; Loenen et al., 1987; Chandrasegaran and Smith, 1988; ~~mbelin et al., 1988; Trautner et al., 1988). N~twiths~~d~g, type-IIS enzymes are useful rea-
gents for rn~jp~at~g DNA, and for certain appii- cations they are indispensable (Szybalski, 1985 ; Podhajska and Szybalski, 1985; Hasan et al., 1986; Mormeneo et al., 1987; Posfai and Szybalski, 1988b). Therefore, we have begun to clone and sequence the genes for several type-IIS systems, and to characterize the proteins that they encode (Nwankwo and Wilson, 1987). We report here that the MTase from the &‘&I system (M 1 F&f) methyl- ates both strands of its recognition sequence. Each strand ~o~t~~s a single adenine residue, and it is these adenines that become methyIated.
Earlier reports by Posfai and Szybalski (1988a,b) suggested that M * Fnkf methylates only the top strand of the recognition sequence. It appears that the enzyme used for these studies, desi~~~t~d M. FokIA, is not the complete MTase, but rather a truncated version which retains the domains neces- sary for the top-str,snd activity but not those required for bottom-strand activity (see RESULTS AND ix-
CUSSION, section I). The present enzyme, M 1 F&I,
exhibits both activities, although top-strand methy- lation predominates in low-ionic strength reaction conditions.
(a) Strains and reagents
F. ~k~~~~k~~~~~ was obtained from the Institute for Fermentation, Osaka, Japan, Cat. No. IF012S36. ~sc~e~c~~~ co& al,kaline phosphatase, T4 polynu- cleotide kinase, restriction enzymes, and DNA MTases were from New England Biolabs, Inc. Cro- t&s a~a~~~~~~~~ venom phosphodieste~~se I was purchased from Worthin~on Biochemic~s,
[ ‘H]Ad~M~t was purchased from New England Nuclear (80 Ci/mmol and 15 Ci/mmol, both at 0.55 mCi/ml), ~4-me~yldeoxy~~idine (m4dC) was synthesized by thiation of deoxyuridine followed by amination with me~yIarn~e (~Temp~n et al., 1961); m5dC, m”dA and the other standard d~xynu~ieos~d~s listed in Table I were obtained from Sigma Biochemical Co. Fluorescent cellulose TLC piates (DC-Fertigplatten Cellulose F) were purchased from Merck. Tryptone and yeast extract were purchased from Difco.

(b) Purification of M * F&I
3
NaCl/S mM 2-mercaptoethanol/lO mM EDTA (buffer M), containing 1 pg of phage A DNA (5 pmol of FokI sites) and 80 PM AdoMet. F. okeanokoites was grown to saturation at 37°C
in a loo-liter fermenter (New Brunswick) in medium containing, per liter, 10 g tryptone, 5 g yeast extract, 2 g glucose, 2 g NaCI, 4.4 g K,HPO,, and harvested by cent~ugation (Sharples). 400 g of cell paste was suspended in 800 ml of 10 mM K. phosphate buffer pH 7.6/l mM EDTA/lO mM 2-mercaptoethanol (buffer A), and the cells were lysed by four passages through a Gaulin press. KC1 was added to 150 mM, then the lysate was clarified by centrifugation (Sharples). 600 ml of the supernatant was diluted to 1600 ml with buffer A containing 150 mM KC1 (buffer B), then applied to a 750-ml radial-flow column containing phosphocellulose (~atm~) equilibrated with buffer B. The column was washed with 750 ml of buffer B, then eluted with a linear gradient of 150 mM to 1 M KC1 in buffer B. Fractions were assayed for DNA-MTase activity by incorporation of [3H]methyl groups from C3H]AdoMet into phage A DNA. Two peaks of activity were observed, the first (M + FokIA) at ap- prox. 600 mM KCl, the second (R/I. FokI) at 850mM KCl.
Fractions containing the second peak were diaiyz- ed against buffer A, then applied to a 15 ml DEAE Sepharose (Ph~acia) column equi~brat~ with buffer A. The column was eluted with a gradient of 0 to 1 M KC1 in buffer A. DNA-MTase activity eluted at approx. 500 mM KCl. Fractions containing activity were dialyzed against buffer A containing 5 mM KC1 (buffer C), then applied to a polycat A HPLC column (Custom LC) equilibrated with buffer C. The column was eluted with a gradient of 0 to 1 M KC1 in buffer A. DNA-MTase activity eluted at 300 mM KCl.
The fraction that contained rn~rn~ MTase activity was used for the M * FokI-methylation study reported here. The preparation was stored at -20 * C
in the presence of 50% glycerol. The yield was 7.5 x lo3 u, and the titer was 2.5 x lo3 u/ml. One unit is required to confer full resistance to R. FokI-
digestion on 1 pmol of FokI sites following incu- bation at 37°C for 1 h. The assays were performed in 25 ~1 of 50 mM Tris . HCl pH 7.5/50 mM
(c) Synthesis of oligod~xyribonucleotide substrates
Oligos S’-CTCGGGGATGCATCGAGTCAG- T (top strand, 22-mer) and 5’-CTGACTGAC- TCGATGCATCCCCGAG (bottom strand, 25- mer), were synthesized on a Biosearch 8600 auto- mated DNA synthesizer using P-cyanoethylphos- phorarnidite chemistry (Sinha et al., 1984). Synthesis was performed at the 1-pmol column-scale. The oligos were deprotected, then purified by 20% PAGE, followed by HPLC on an RCM C8 (Waters) cartridge with a binary elution system starting from 0.1 M aqueous ~ethyl~onium bicarbonate pH 7.0, and ending in 15% acetonitrile in the same buffer. Salts were removed by co-evaporation with water in a vacuum centrifuge (SpeedVac).
(d) In vitro methylation by M * F&I
Of each oligo, 2.5 pg was annealed in 30 ,ul of buffer M by heating to 72°C for 5 min, then incubat- ing at room temperature for 30 min. One ~1 of the annealed oligo (10 pmol of FokI sites) was diluted to 50 ,a1 with butler M, and incubated at 37°C for 1 h with 5 pl(l2.5 u) of M *FokI and 0.4 ~1 (2.8 pmol; 5 x IO5 dpm) of [ 3H]AdoMet (80 Ci/mmol). Ap- proximately 85% of the label was incorporated. Then 2.5 fig of each cold oligo was added and the strands were denatured and separated by 20% PAGE (40 : 1, acrylamide: bis-acrylamide) contain- ing 7 M urea, in running buffer containing 50 mM Tris-borate pH 8.3/l mM EDTA. The 22-mer and 25-mer bands were detected by UV-quen~h~g after placing the gel on a fluorescent cellulose TLC plate and illuminating the plate with a 254-nm UV lamp. Gel slices containing the oligos were extracted in 0.5 M ammonium acetate/l mM EDTA (Smith, 1980). The extracts were desalted by gel filtration through lo-ml Sephadex G-50 columns (Pharma- cia). The oligos were labeled to a specific activity of 2.5 x lo4 cpm/pg for the top strand, and 1.5 x 10” cpm/pg for the bottom strand.

RESULTS AND DISCUSSION TABLE I
Cellulose TLC of deoxynucleosides a
Compound Relative migration
distance b
Name (Symbol)
_--
Solvent DC Solvent
G*
Thymidine (dT) 1.00 1.00
Deoxycytidine (dC) 0.83 1.05
Deoxyadenosine (dA) 0.79 1.27
Deoxyguanosine (dG) 0.67 0.91
Deoxyinosine (dI) 0.61 0.72
5-Methyldeoxyc~idine (m5dC) 0.87 1.10
~4-~ethyldeoxycytidine (m4dC) 1.00 1.08
~6-Methyideoxyadenosine (m6dA) 1.00 1.30
S-Adenosylmethionine (AdoMet) 0 1.13
a Migration of 2’-deoxynucleosides and methylated 2’-deoxy-
nucleosides on cellulose TLC. Solvent D separates m5dC from
m4dC plus m6dA; soivent G separates m6dA from m4dC plus
m5dC. Deoxynucleotides do not migrate in either solvent.
b Values given are relative distances, measured with respect to
the migration of thymidine.
c Solvent D is 80 : 20 ethanol-water (v/v).
d Solvent G is 66 : 33 : 1 isobutyric acid-water-ammonium
hydroxide (v/v).
(a) Puri~~ation of M * FokI
A culture of F. okeanokoites was grown, the cells
were collected and lysed, and the clarified superna-
tant was chromatographed on phosphocellulose
(MATERIALS AND METHODS, section b). Two peaks
of MTase activity were observed. The second peak
was pooled and was purified further by DEAE-cellu-
lose chromatography and polycat-A HPLC. Methy-
lation by the purified activity completely protected
phage I DNA from digestion by R. FokI.
(b) TLC-characterization of methyiated deoxy-
nucleosides
Direct identification of methylated bases in DNA
requires that the DNA be degraded and that the
products be separated and identified. Degradation to
deoxynucleosides can be achieved enzymatically
(Ehrlich et al., 1985); degradation to purine and
pyrimidine bases can be achieved by hydrolysis with
perchloric acid (Butkus et al., 1985). The latter me-
thod can des~oy moor bases (Buryanov et al., 1974;
Iwanami and Brown, 1968), and so for the present
study we chose to use enzymatic degradation. Separ-
ation can be accomplished by HPLC or by TLC.
Since TLC is a simple and well-proven method
(R~derath, 1961; 1961/62; Mangold, 1969), it was
used here.
The TLC migration properties of a number of
methylated and unmethylated deoxynucleosides
were examined to ascertain the conditions needed to
d~erentiate them. Two solvents were chosen that,
when used in conjunction with one another, permitt-
ed differentiation among m6dh, m4dC and m5dC,
the degradative derivatives of the three products
commonly formed by DNA-MTases. In solvent D
(80 : 20 ethanol-water; Khorana et al., 1961) m6dA
and m4dC co-migrated, separate from m*dC. In
solvent G (66 : 33 : 1 isobutyric acid-water-ammon-
ium hydroxide) m4dC and m5dC migrated closely,
separate from m6dA (Table I).
(c) M. FokI methylates both strands of duplex DNA
Two complementary oligos, incorporating an FokI site (highlighted in bold type, below), were synthe-
sized and annealed to create a substrate for in vitro
methylation by M. FokI:
Top strand: 5’-CTCGGGGATGCATCGAGTCAGT- 3’
Bottom strand: 3’-GAGCCCCTACGTAGCTCAGTCAGTC-5’
The duplex has a calculated melting temperature
(T,) of 70°C (Gait, 1984), well above the methyla-
tion reaction temperature for M . FokI. The strands
were made of differing lengths to facilitate their sub-
sequent separation.
The duplex was incubated with M ’ FokI at 37 ‘C
in the presence of [ 3H]AdoMet to label the nucleo-
tides that become methylated during the reaction.
The strands were then separated and purified by
20% PAGE. Both strands were found to be labeled,
indicating that M - Fokl methylates both strands of
ds DNA, as do the type-II MTases. Methylation was
carried out under conditions of limiting AdoMet,
and in repeated experiments, incorporation was
found to be biased towards the top strand: on
average, 63 y0 of label was incorporated into the top
strand, and 37% was incorporated into the bottom
strand.

Incorporation into both strands of the oligo re-
quired a buffer with a combined Tris 3 HCl + NaCl
molarity of 100 mM, or more. In buffers with lower
combined molarities, such as 25 mM Tris * HCl, in-
corporation was found to be ten-fold lower and to be
confined to the upper strand, exclusively.
(d) M * FokI methyiates adenines not cytosines
A portion of each labeled strand was degraded by
incubation with phosphodiesterase and alkaline
phosphatase. The digests were mixed with cold me-
thyldeoxynucleosides to act as visual markers, then
the mixtures were separated by TLC. Spots corre-
sponding to the m’dA, m4dA and m5dC combi-
nations were analyzed for radioactivity. 80% of the
input radioactivity was recovered, and for both
strands it was found to be present only in m6dA
(Table II), indicating that ~6-me~yladenine is the
sole product of M - ~~~I-me~ylation.
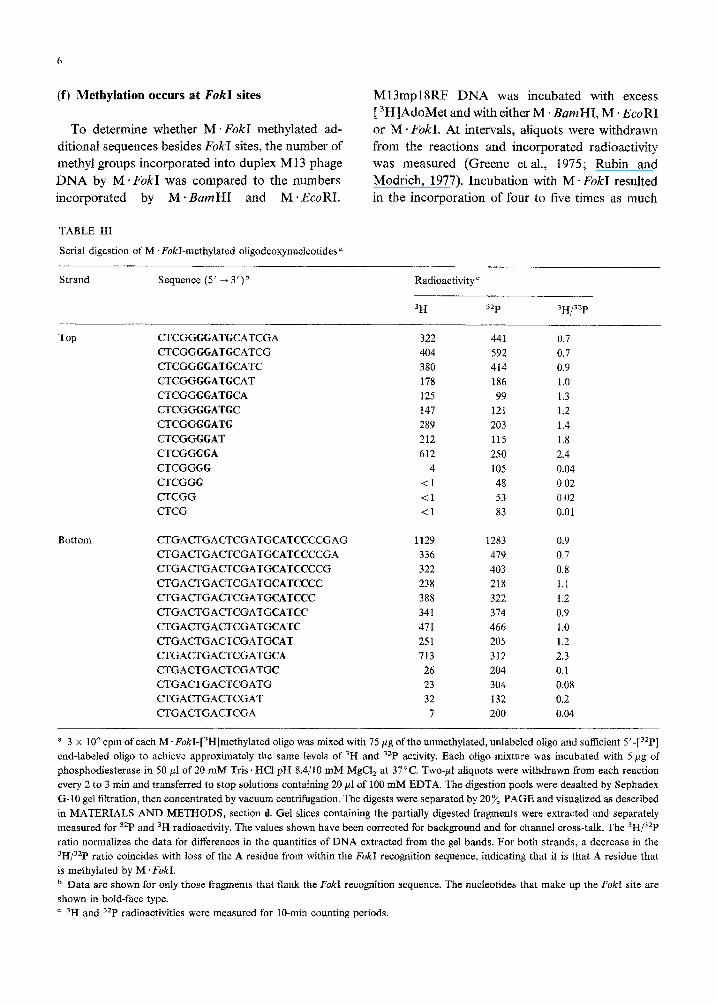
(e) Methylation occurs within the EbkI site
The two complementary oligos were resynthesized
with 5’PO, termini. The oligos were annealed, and
5
then methylated using M. F&I and [ 3H]AdoMet,
as described in RESULTS AND DISCUSSION, section
c. A portion of each labeled strand was exonucleoly-
tically digested from the 3’ terminus to form a set of
successively shorter fragments. The digests were
separated by 20 y0 PAGE, the fragments were excis-
ed and their radioactive incorporation was measur-
ed. To normalize for differences in the quantities of
DNA present in the bands and for differences in the
efficiencies with which the bands were extracted
from the gels, a small quantity of the appropriate
unmethylated oligo, which had been 5’ end-labeled
with 32P (by a T4 polynucleotide kinase reaction),
was added to each reaction before exonuclease di-
gestion. Following gel extraction, the bands were
counted for 3H, to monitor the presence of the
methylated nucleotide, and for 32P, to measure the
quantity of DNA present {Greene et al,, 1975). For
both strands, the 3H/32P ratios were observed to
drop precipitously when the A residue within the
F&I recognition site was removed by the exonu-
clease digestion (Table III). These results indicate
that the A residues methylated by M - FokI are those
that occur within the recognition sequence.
TABLE II
Cellulose TLC of M FokI-methylated deoxynucleosides ’
Solvent D Solvent G
Identity: m’dC m4dC + m6dA m6dA m4dC + msdC
Relative migration distance’ 0.9 1.0 1.3 1.1
Oligodeoxynucleotide 3H incorporation c (cpm)
Top strand, 22-mer
Bottom strand, 25-mer I_
26 400 435 16
22 420 390 26
a 1 x lo3 cpm each of the M .FokI-methylated, separated [3H]oligos, 5’-CTCGGGGATGCATCGAGTCAGT (top strand, 22-mer)
and 5’-CTGACTGACTCGATGCATCCCCGAG (bottom strand, 25-mer) were digested with excess bacterial alkaline phosphatase
and phosphodiesterase in 50 gl of 20 mM Tris 1 HCl pH 8.4/10 mM MgCl, at 37°C for 2 h. The digestions were desalted by Sephadex
G-10 gel filtration, then concentrated by vacuum cent~fugation. The samples were divided in half and each half was spotted onto a 20
cm x 20 cm fluorescent cellulose TLC plate. 0.1 A,,, u each of unlabeled msdC, m4dC and m%lA were spotted on top of the samples
to serve as internal standards. 0.1 A,,, u each of the deoxynucleosides listed in Table I were also spotted onto the plates, in separate
lanes, to act as external standards. One of each pair of plates was developed in solvent D and the other was developed in solvent G.
The plates were illuminated at 254 nM and the spots in the sample lane formed by the m5dC, m4dC and m6dA internal standard
combinations were scraped, extracted with water, and then measured for radioactivity. The results demonstrate that M. FokI methylates
adenine in both strands of the duplex.
b Migration distances are relative to thymidine (see Table I). Solvents D and G are defined in the footnotes to Table I. In this instance,
the data for solvent G, on their own, are sufficient to establish the nature of the methylated base, and the absence of strand-specificity.
The data for solvent D are included for completeness, however.
’ 3H incorporation was measured for IO-min counting periods; the data shown have not been reduced to compensate for the background
radiation (approx. 25 cpm).
6
(f) Methylation occurs at F&I sites
To determine whether M *Fold methylated ad-
ditional sequences besides FokI sites, the number of
methyl groups incorporated into duplex Ml3 phage
DNA by M * FokI was compared to the numbers
incorporated by M ~BamHI and M *EcoRI.
M13mp18RF DNA was incubated with excess
[ 3H]AdoMet and with either M ’ BarnHI, M * EcoRI
or M. Fold. At intervals, aliquots were with~awn
from the reactions and incorporated radioactivity
was measured (Greene et al., 1975; Rubin and
Modrich, 1977). Incubation with M * FokI resulted
in the incorporation of four to five times as much
TABLE III
Serial digestion of M . F&I-methylated oligodeoxynucieotides a
Strand Sequence (5’ -+ 3’)b Radioactivity ’
3H 32P 3H/32P
Top CTCGGGGATGCATCGA
CTCGGGGATGCATCG
CTCGGGGATGCATC
CTCGGGGATGCAT
CTCGGGGATGCA
CTCGGGGATGC
CTCGGGGATG
CTCGGGGAT
CTCGGGGG
CTCGGGG
CTCGGG
CTCGG
CTCG
Bottom CTGACTGACTCGATGCATCCCCGAG 1129 1283 0.9
CTGACTGACTCGATGCATCCCCGA 336 419 0.7
CTGACTGACTCGATGCATCCCCG 322 403 0.8
CTGACTGACTCGATGCATCCCC 238 218 1.1
~GA~GA~~GATGCATCCC 388 322 1.2
CTGACTGACTCGATGCATCC 341 314 0.9
CTGACTGACTCGATGCATC 471 466 1.0
CTGACTGACTCGATGCAT 251 205 1.2
CTGACTGACTCGATGCA 713 312 2.3
CTGACTGACTCGATGC 26 204 0.1
CTGACTGACTCGATG 23 304 0.08
CTGACTGACTCGAT 32 132 0.2
CTGACTGACTCGA 7 200 0.04
322 441 0.7
404 592 0.7
380 414 0.9
178 186 1.0
125 99 1.3
147 121 1.2
289 203 1.4
212 115 1.8
612 250 2.4
4 105 0.04
Cl 48 0.02
<I 53 0.02
<l 83 0.01
a 3 x 10“ cpm of each M. F&I-[3H]methylated oligo was mixed with 75 pg of the unmethylated, unlabeled oligo and sufficient 5’-[32P]
end-labeled oligo to achieve approximately the same levels of 3H and 32P activity. Each oligo mixture was incubated with 5 pg of
phosph~iesterase in 50 nl of 20 mM Tris ‘HCI pH 8.4/10 mM MgCl, at 37°C. TWO-PI ahquots were w~~dra~ from each reaction
every 2 to 3 min and transferred to stop soiutions containing 20 ~1 of 100 mM EDTA. The digestion pools were desalted by Sephadex
G-10 gel filtration, then concentrated by vacuum centrifugation. The digests were separated by 20% PAGE and visualized as described
in MATERIALS AND METHODS, section d. Gel slices containing the partially digested fragments were extracted and separately
measured for 32P and 3H radioactivity. The values shown have been corrected for background and for channel cross-talk, The 3H/32P
ratio normalizes the data for differences in the quantities of DNA extracted from the gel bands. For both strands, a decrease in the
3H/32P ratio coincides with loss of the A residue from within the F&I recognition sequence, indicating that it is that A residue that
is methylated by M . F&I.
b Data are shown for onfy those fragments that flank the F&I recognition sequence. The nucleotides that make up the FokokI site are
shown in bold-face type.
’ 3H and 3zP radioactivities were measured for lo-mm counting periods.
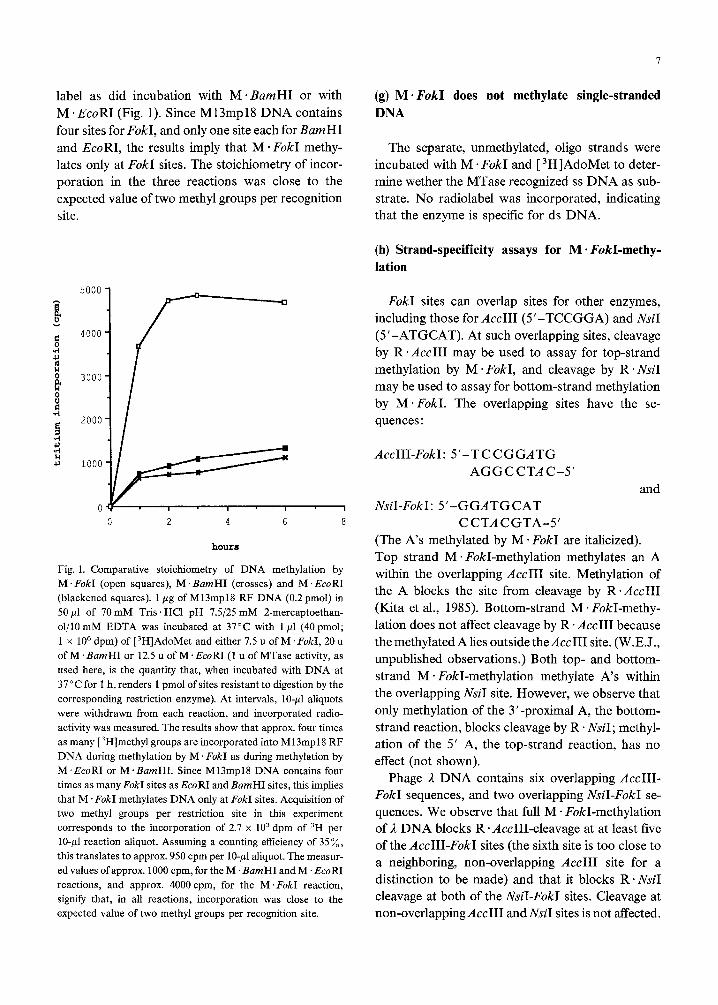
label as did incubation with M. BamHI or with
M .EcoRI (Fig. 1). Since M13mp18 DNA contains
four sites for FokI, and only one site each for BumHI
and EcoRI, the results imply that M - FokI methy-
lates only at FokI sites. The stoichiometry of incor-
poration in the three reactions was close to the
expected value of two methyl groups per recognition
site.
0 2 4 6 8 CCTACGTA-5’
hourrr
Fig. 1. Comparative stoichiometry of DNA methylation by
M. F&I (open squares), M .BamHI (crosses) and M. EcoRI
(blackened squares). 1 pg of M13mp18 RF DNA (0.2 pmol) in
50~1 of 70mM Tris. HCl pH 7.5/25 mM 2-mercaptoethan-
ol/lO mM EDTA was incubated at 37°C with 1 ~1 (40 pmol;
1 x lo6 dpm) of t3H]AdoMet and either 7.5 u of M. F&I, 20 u
of M ‘BarnHI or 12.5 u of M .EcoRI (1 u of MTase activity, as
used here, is the quantity that, when incubated with DNA at
37°C for 1 h, renders 1 pmol of sites resistant to digestion by the
corresponding restriction enzyme). At intervals, lo-p1 aliquots
were withdrawn from each reaction, and incorporated radio-
activity was measured. The results show that approx. four times
as many [3H]methyl groups are incorporated into M13mp18 RF
DNA during methylation by M . F&I as during methylation by
M.EcoRI or M.BamHI. Since M13mp18 DNA contains four
times as many F&I sites as EcoRI and BamHI sites, this implies
that M .FokI methylates DNA only at F&I sites. Acquisition of
two methyl groups per restriction site in this experiment
corresponds to the incorporation of 2.7 x 10’ dpm of 3H per
lo-p1 reaction aliquot. Assuming a counting efficiency of 35x,
this translates to approx. 950 cpm per lo-,nl aliquot. The measur-
ed values of approx. 1000 cpm, for the M ‘BarnHI and M .EcoRI
reactions, and approx. 4000 cpm, for the M .F&I reaction,
signify that, in all reactions, incorporation was close to the
expected value of two methyl groups per recognition site.
7
(g) M + I;okI does not methylate single-stranded
DNA
The separate, unmethylated, oligo strands were
incubated with M * FokI and [ 3H]AdoMet to deter-
mine wether the MTase recognized ss DNA as sub-
strate. No radiolabel was incorporated, indicating
that the enzyme is specific for ds DNA.
(h) Strand-specificity assays for M * F&I-methy-
lation
FokI sites can overlap sites for other enzymes,
including those for AccIII (5’-TCCGGA) and NsiI
(5’-ATGCAT). At such overlapping sites, cleavage
by R .AccIII may be used to assay for top-strand
methylation by M .FokI, and cleavage by R eNsi
may be used to assay for bottom-strand methylation
by M. FokI. The overlapping sites have the se-
quences:
AccIII-FokI: 5’-TCCGGATG
AGGCCTAC-5’
and
NsiI-FokI: 5’-GGATGCAT
(The A’s methylated by M . FokI are italicized).
Top strand M . FokI-methylation methylates an A
within the overlapping AccIII site. Methylation of
the A blocks the site from cleavage by R. AccIII
(Kita et al., 1985). Bottom-strand M - FokI-methy-
lation does not affect cleavage by R. AccIII because
the methylated A lies outside theAccII1 site. (W.E.J.,
unpublished observations.) Both top- and bottom-
strand M * FokI-methylation methylate A’s within
the overlapping NsiI site. However, we observe that
only methylation of the 3’-proximal A, the bottom-
strand reaction, blocks cleavage by R * NsiI; methyl-
ation of the 5’ A, the top-strand reaction, has no
effect (not shown).
Phage /z DNA contains six overlapping AccIII-
FokI sequences, and two overlapping NsiI-FokI se-
quences. We observe that full M. FokI-methylation
of 1 DNA blocks R - AccIII-cleavage at at least five
of the AccIII-FokI sites (the sixth site is too close to
a neighboring, non-overlapping AccIII site for a
distinction to be made) and that it blocks R. NsiI
cleavage at both of the NsiI-FokI sites. Cleavage at
non-overlappingAccII1 and NsiI sites is not affected.

M * FokI-methylation clearly causes nine fragments
to disappear from the R *Ace111 digestion pattern of
;1 DNA and four new fragments to appear. It clearly
causes two fragments to disappear from the R. NsiI
digestion pattern and two new fragments to appear
(Fig. 2). The other changes that take place are more
difficult to identify because of fragment superimpo-
sitions on the gels.
R *BspMII is an isoschizomer of R’ AccIII.
R . B~~MII-cleavage is blocked by methylation of the
C’s in the reco~ition sequence, but it is not blocked
by methylation of the A’s (Labbe et al., 1988). We
observe that, unlike R.AccIII, R * E3spMII does
cleave overlapping AccIII-FokI sequences in phage
,? DNA methylated by M. FokI.
(i) Two M + EbkI activities from FZavobacterium okennokoites
During an earlier preparation of DNA-MTase
activity from F. ~kea~oko~~es, the first peak of activity
from the phosphocellulos~ column was purified,
rather than the second (D.A. Wise, pers. commun.).
The first peak methylates only the top, 5’-GGATG,
strand of the FokI recognition sequence (M . FokIA;
Posfai and Szybalski, 1988a,b). The second peak,
the subject of this report, methylates both strands.
Methylation by either activity protects FokI sites
from cleavage by R * FokI. The relationship between
the two MTases is uncertain, but circumstantial evi-
dence suggests that the first enzyme lacks the
C-terminus of the second, possibly formed by
proteolytic de~adation or by premature te~ination
of transcription or translation.
(j) Structure of M - FokI
We have sequenced the genes for the Fokl MTase
and ENase (M.C.L., L.S. Moran, W.E.J., G.R.F.,
J.S. Benner, B.E.S. and G.G.W., submitted). The
gene for M * FokI is approx. 2 kb in length, longer
than is customary for the genes for type-II m6A-
MTases, which range from 0.7 kb (M . HhaII;
Schoner et al., 1983) to 1.6 kb (M sFaeR71; The-
riault et al., 1985). Perhaps its large size reflects the
presence of duplicate CDs in the MTase, one
domain for methylating each strand.
The tetrapeptide . . Asp-Pro-Pro-Tyr . ., or its
equivalents, occur once in all type-II m6A-MTases
1 2345678
R-Fokl R-RccIII R-NsiI none
- + - + - + - -t
Fig. 2. Resistance of M. FckI-methylated phage I, DNA to
restriction endonuclease cleavage. 1.6 ng of Dam-(Gm”ATC-
free) phage A DNA in 80 pl of 100 mM Tris HCl pH 7.6/25 mM
2-mercaptoethanol/2 mM EDTAjSO pM AdoMet was incubated
at 37°C for 2.5 h with 20 u of M. FokI (lanes marked ’ i ‘). A
second reaction, identical to the first except for the absence of
M .FokI, was incubated in parallel (lanes marked ‘- ‘). The
reactions were subsequently heated to 60°C for 10 mm, then 8 ~1
of 500mM NaCIjlOO mM MgCi, was added to each. Each
reaction was then divided into four equal parts; 4 u of R. FokI
was added to the first part of each, 2.5 u of R.AccIII to the
second, 5 u of R. NsiI to the third, and nothing (control) to the
fourth. The reactions were incubated for another hour at 37°C
(controls, R * Fokl-digestions and R. WI-digestions) or at 60°C
(R .AccIII-digestions). The eight reactions were then electro-
phoresed on a 0.7% agarose gel. Methylation by M .FokI does
not affect the integrity of the DNA (lanes 7 and 8), but it renders
the DNA completely resistant to cleavage by R. FokI (lane 2 vs.
lane 1). Several of the Ace111 and N&I sites in phage I DNA
overlap FokI sites. Methylation by M 1 FokI renders the overlap-
ping sites resistant to cleavage by R. AecIII (lane 4 vs. lane 3) and
R. NsiI (lane 6 vs. lane S), but it does not affect cleavage by these
enzymes at non-overlapping sites.
(Lauster et al., 1987; Chandrasegaran and Smith,
1988) and m4C-MTases (R.M. Blumenthal, pers.

9
commun*; J.E. Brooks, D.L. and B.E.S., unpub-
lished), and so it may be a necessary component of
CDs that methylate the extracyclic nitrogens of A
and C, The tetrapeptide occurs twice in M. FokI,
once in the N-terminal half of the protein, and again
in the C-terminal half. This might indicate that one
half of M * FokI forms the top-strand CT), and that
the other forms the bottom-strand CD. Perhaps the
present gene for M * FokI arose by fusion af adjacent
ancestral genes, which were originaily responsible for
me~yla~g one strand each of the reco~ition
sequence.
(k) Concfusions
(1) M 1 FokI, the type-IfS modi~cati~~ enzyme
from F. ok~a~oko~te~, has been purified.
(2) M - FokI is a DNA-adenine MTase; it methy-
lates the adenine residue (bold A’s) in both strands
of the recognition sequence:
5’-GGATG
CCTAC-5’
(3) M. FokI does not methylate ss DNA.
(4) In low-ionic-strength buffers, M * FukI methy-
lams only the top strand of the recognition sequence.
A presumed truncated form of the enzyme,
M * F&IA, performs only this reaction under all
ionic strength conditions,
(5) Only methylation of the top strand ofthe FokI
r~co~ition sequence protects overlapping FokI-
AccIII sites from cleavage by AccIII (but not from
cleavage by RI BspMII); only methyl~~ion of the
bottom strand of the FokI recognition sequence
protects overlapping FokI-MsiI sites from cleavage
by N&I. This allows independent assays of top-
strand and bottom-str~d methylation by M. FokI.
This paper is dedicated to the memory of our late
friend and colleague, David A. Wise. We thank
Donald Comb for support and encouragement, and
Elizabeth Van Cott for critical reading of the manus-
cript.
REFERENCES
Balganesh, T.S., Reiners, L., Lauster, R., Noyer-Weidner, M., Wilke, K. and Traumer, T.A.: Construction and use of chime& SPR/#3T DNA methyltransferases in the definition of sequence rec~)~~~~ng enzyme regions. EMBO J. 6 (1987) 3542-3549.
Behrens, B., Noyer-Weidner, M., Pawlek, B., Lauster, R., Balga- nesh, T.S. and Trautner, T.A.: Organization of multispecific DNA methyltransferases encoded by temperate Bacilhrs sub- tilkv phages. EMBO J. 6 (1987) 1137-l 142.
Buryanov, Y.I., Adreev, L.V. Eroshina, N.V. and Korsunskii, O.F.: The possible presence of rare deaminated nitrog~ous bases in DNA. ~iok~mia 39 (1974) 31-38.
Butkus, V.V., Klimasauskas, 5.3. and Jam&&is, A.A.: Analysis ofthe products ofDNA modification by methylascs: a prace- dure for the determination of 5 and @-methylcytosines in
DNA. Analyt. Biochem. 148 (1985) 194-198. ~~drase~~an, S. and Smith, H.O.: Ammo acid sequence
homologies among twenty-five restriction endonucleases and
methylases. In Sarma, M.H. and Sarma, R.H. (Eds.), From Proteins to Ribosomes, Vol. 1. Adenine Press, Guilderland, NY, 1988, pp. 149-156.
Ehrlich, M., Gama-Sosa, M.A., Carreira, L.H., Ljungdahl, LG., Kuo, K.C. and Gehrke, C.W.: DNA methylatian in thermo- philic bacteria: ~4-methylcytosine, 5-methylcytosine, and
~6-lnetllyl~deni~e. Nucleic Acids Res. 13 (1985) 1399-1411.
Fuller-Pace, F.V. and Murray, N.E.: Two DNA recognition
domains of the specificity polypeptides of a family of type I
restriction enzymes. Proc. Natl. Acad. Sci. USA 83 (1986)
9368-9372.
Gait, M.J.: I~tr~duct.ion to modern methods of DNA synthesis.
In Gait, M.J. (Ed.), Oligonucleotide Synthesis. IRL Press,
Oxford, 1984, pp, I-22.
Gann, A.A.F., Campbell, A.J.B., Collins, J.F., Co&on, A.F.W.
and Murray, NE.: Reassortment of DNA recognition domains and the evolution of new specificities. Mol. Mi-
crobioi. 1 (1987) 13-22.
Greene, P.J., Poonian, M.S., Nussbaum,A.L., Tobias, L., Garfin,
D.E., Boyer, H.W. and Goodman, H.M.: Restriction and
modification of a self-complementary octanucleotide
containing the EcoRI substrate. J. Mol. Biol. 99 (1975)
237-26 1. Gough, J.A. and Murray, N.E.: Sequence diversity among relat-
ed genes for re~~t~on of specific targets in DNA molecubs.
J. Mol. Biol. 166 (1983) I-19.
Hasan, N., Kim, SC., Podhajska, A.J. and Szybalski, W.: A
novel method for generating precise unidirectional deletions
using BspMI, a class-IIS restriction enzyme. Gene SO (1986)
55-62.
H~belin, M., Suri, B., Rao, D.N., Hornby, D.P., Eberle, II.,
Pripfl, T., Kenel, S. and Bickle, T.A.: Type III DNA re- striction and madification systems EcoPl and &~PlS. J.
Mol. Biol. 200 (1988) 23-29.
iwanami, Y. and Brown, G.M.: Methylated bases of ribosomal
ribonucleic acid from HeLa cells. Arch. Bioch. Biophys. 126
(1968) 8-15.
Khorana, HG., Turner, A.F. and Vizsolyi, J.P.: Experiments on

10
the pQIy~erj~atjon of monon~cleo~d~s: certain protected derivatives of deoxycytidined phosphate and the synthesis of d~oxyc~i~me polynu~leot~des~ f. Am. Chem. Sot. 83 (196X) 686-698.
Kita, R., Hiraoka, N., Oshima, A., Kadonishi, S. and Obayashi, A.: AccTII, a new restriction endonucleasc from Acinetobacter
calcoaceticw. Nucleic Acids Res. 13 (lQ85) 8685-8694.
Labbt, S., Xia, Y. and Roy, P.H.: BspMII and &cIII are an isoschizomr pair which differ in their sensitivity to cytosine methylation. Nucleic Acids Res. 16 (1988) 7184.
Lauster, R., Kriebardis. A. and Gusehibauer, W.: ?bc GA-
TATC-modi~cation enzyme EcuRV is closely related to the GATC-r~~~n~~ng methyltransf~r~es &nII and C&M ffom E. co& and &age T4. FEBS Lett. 2.20 (1987) 167-176.
Loenen, W.A.M., Daniel, AS., Braymert W.D. and Murray, NE.: Organization and sequence of the ?r& genes of&&eri- chin c& K-12. J. Mol. Eiof. 198 (1987) 159-170.
Mangold, ILK.: Thin-layer chromatography ofnucleic acids and their constituents. In Stahl, E. (Ed.), Thin-layer Chromato- graphy. A Laboratory Manual. Springer-Varlag, New-York, 1969, pp. 192-806.
McClelland, M. and Nefson, M.: The effect of site-specific methy- l&on on restriction endonncleases and DNA rn~j~icatio~l me~yi~~al~sfe~ases - a review. Gene 74 (lQ88) 291-304.
Mormeneo, S., Knott, R. and Perlman, D.: Precise nucleotide sequence m~~cations with bi~reetionally cleaving class- IXS excision linkers. Gene 61 (1987) Z-30.
Nagaraja, Y., Shepherd, J.C.W. and Bickle, T,A. : A hybrid recog- nition sequence in a recombinant rest&ion emyme and the evolution of DNA sequence specificity. Nature 316 (1985) 371-372.
Nwankwo, D.Q. and Wilson, G.G.: Cloning of two type-II methylase genes that recognise asymmetric nucleotide se- quences: F&I and Hgd. Mol. Gen. Genet. 209 (1987) 570-514.
Podhajska, A.J. and Szybalski, W.: Conversion oftbe F&I endo- nuclease to a universal restriction enzyme: cieavage ofphage M13mp7 DNA at Predete~ued sites. Gene 40 (1985) 175-182.
Pbsfai, G. and Szybalski, W.: A simple method for locating methylated bases in DNA as applied to detect asymmetric ~l~tbylation by MeFokIA. Gene 69 (1988a) 147-151.
Pbsfai, G. and Szybalski, W.: A simple methad for locating methylated bases in DNA using class-IIS restriction en- zymes. Gene 74 (1988b) 179-181.
Randerath, K.: Thin-layer chromatography on ion-exchange fums. Separation of nucleic acid derivatives. Angew. Chem. 73 (1961) 674-676.
Randerath, K.: A comparison between thin-layer chromatogra- phy and paper chromato~aPhy of nucleic acid derivatives. Biochem. Biophys. Res. Common. 6 (1961&2) 452-4s7.
Roberts, R.J.: Restriction enzymes and their isoschizomers. Nucleic Acids Res. 16 (19%) suppl. r271-r313.
Rubin, R.A. and Modrich, P.: EcoRI methylase: physical and catalytic properties of the homogeneous enzyme. 3. Biol. Chem. 252 (1977) 7265-7272.
Schroeder, C., lurks&at, H., MeiseI,A., Reich,J.-G. and Krliger, D.: Unusual occnrtence of EcoPl and EcoPl5 recognition sites and counterselection of type II methylation and rcstric- tion sequences in bacteriophage T7 DNA. Gene 45 (19%) 77-86.
Schoner, B., Kelley, S. and Smith, HO.: The nucleotide sequence of the HhalI restrictian and m~~fication genes from N~epno- @ills k~e~~5~lti~~~. Gene 24 (1983) 227-236.
Sinha, ND., Biernat, J., McManus, .I. and Roster, H.: Polymer support oligonucleotide synthesis, XVIII. Use of the & cy~oethyl-~-~~diai~o-~~-morpho~na phasphoramid- ite of deox~ucleos~des for the synthesis of DNA Fragments simplifying deprotection and isoiation of the &al product. Nncieic Acids Res. 12 (1984) 4539-4557.
Smith, H.O.: Recovery ofDNA from gels. Methods Enzymol. 65 (1980) 371-379.
Smith, H.O. and Kelly, S.V.: Methylases of the type II re- striction-modification systems. In Razin, A., Cedar, II. and Riggs, AD. (Bds.), DNA Methyiation, Biochemistry and Biological Significance. Springer-Verlag, New-York, 1981, pp. 39-71.
Szybalski, W,: Universal restriction endonucleases: designing novel cleavage spec%cities by combining adapter ohgodeoxy- nucleotide and enzyme moieties. Gene 40 (1985) 169-l 73.
Theriault, G., Roy, PH., Howard, K.A., Benner, J.S., Brooks, J.E., Waters, G.F. and Gingeras, T.R.: Nucleotide sequence of the PaeR7 restriction/modification system and partial characterization of its protein products. Nucleic Acids Res. 13 (1985) 8441-8461.
Tran-Bet&e. A., Behrens, B., Noyer-Weidner~ M. and Trautner, T.A,: DNA m?thyltr~sferas~ genes of &&l&s s~&iiis phages: comparison of their nncleotide sequences. Gene 42 (1986) 89-96.
Trautner, T.A., Balganesb,T.S. and Pawlek, B.: Chieric multis- pecific DNA me~~ltransferase with novei combinations of target re~~it~o~. Nucleic Acids Res. 16 (1988) ~4~-665~.
Wempen, I., Dnshinsky, R., Kaplan, L. and Fox, J.: Thiation of nucleosides, IV. The synthesis of S-fluoro-2’-deoxycytidine and related compounds. J. Am. Chem. Sot. 83 (,1961j 4755-4166.
Wilke, K., Rauhut, E., Noyer-Weidner, M., Lauster, R., Pawlek, B., Behrens, B,, and Trautner, T.A.: Sequential order of target-recognizing domains in multispecific DNA-methyl- transferases. EMBO J. 7 (i388) 2601-2609,
Yuan, R. and Hamilton, D.L.: Type I and type III restriction- mo~ca~on enzymes. In Razin, A., Cedar, H. and Riggs, AD. (Bds.), DNA Metb~la~on, Biochemistry and Biolagical Significance. Springer-Veriag, New-Yffrk, 1984, pp. 1 b-37.