asymmetric hydrogenation of activated ketones
TRANSCRIPT

Asymmetric hydrogenation of activatedketones
Jozsef L. Margitfalvia and Emılia Talasa
DOI: 10.1039/9781847559630-00144
In this contribution key features of the asymmetric hydrogenation ofactivated ketones over cinchona–-Pt catalyst system are reviewed. Bothhistorical backgrounds and recent results are evaluated and discussed.The focus is laid on the peculiarities of these reactions, such as (i) methodsand approaches used, (ii) substrate specificity, (iii) rate acceleration,(iv) the form of conversion-ee dependencies; (v) inversion of ee,(vi) nonlinear phenomena, (vii) origin of enantio-differentiation and (viii)character of modifier – Pt, substrate-modifier and substrate-modifier-Ptinteractions.
1. Introduction
1.1 General information
In order to reduce the environmental and health hazards the demand foroptically active compounds in high enantiopurity is increasing in the field ofpharmaceutical, agrochemical and cosmetic products. Although the mostcommon applications are bio-related, there is also a great interest in the areaof materials science for chiral compounds, such as chiral polymers or chiralliquid crystals. For this reason the interest to produce chiral compounds inhighly pure form is expanded over the past decades. There are differentapproaches to obtain compounds in high enantiopurity. One of the mostenvironmentally friendly methods is the use of chiral catalysts.1–3 There aredifferent approaches in chiral catalysis, such as homogeneous,4 hetero-geneous,5 enzymatic,6 and ‘‘artificial catalytic antibodies’’.7 The commonfeature of all approaches is that relatively small amount of chiral auxilaritiesis required to produce chiral compounds in high enantiopurity. In this re-spect the term ‘‘multiplication of chirality’’8 is often quoted. When het-erogeneous catalysts are applied, the chiral auxilarities used are often calledas chiral modifiers.
In the past decades, significant progress has been achieved in homo-geneous enantioselective catalysis, what is reflected by the Nobel Prizeawarded in 2001 to Sharpless, Noyori, and Knowles. Variety of transitionmetal complexes containing unique chiral ligands have been developed toinduce enantioselectivity by homogeneous catalysts. These chiral metalcomplexes were used in various catalytic reactions, such as hydrogenation,9
dihydroxilation,10 epoxidation,11 Diels-Alder reaction,12 C–C bond for-mation12,13 Michaels reaction,14 etc. These reactions are considered havegreat importance for the production of fine chemicals and pharmaceuticalproducts.15
aInstitute of Surface Chemistry and Catalysis, Chemical Research Centre, Hungarian Academyof Sciences, POB 17, 1525, Budapest, Hungary
144 | Catalysis, 2010, 22, 144–278
�c The Royal Society of Chemistry 2010

Homogeneous catalytic reactions are highly selective and require rela-tively small amount of catalyst, however the chiral ligands and the metalcomplexes used are relatively expensive. Homogeneous catalysts are sensi-tive both to oxygen and moisture, for this reason the handling of thesecompounds is quite troublesome. Additional serious problem is the catalystrecovery. As the remaining trace impurities of metals have a definite en-vironmental and health hazard the development of complex methods for theremoval of traces of transition metals increases significantly the productioncosts in homogeneous catalytic reactions. The immobilization of metalcomplexes into inorganic or organic supports is one of the approaches toovercome the above problems as immobilized catalysts can be separated byfiltration or can be used in continuous-flow reactors.
Homogeneous catalysts can be immobilized both to inorganic16 andpolymeric supports.17 Most of the cases the solid contains a ligand, what isconsidered as an anchoring site.
In case of inorganic supports the reactivity of surface OH groups is usedto immobilize a given type of ligand upon using the reactive trimethoxy (ortrichloro) silane derivatives.18 However, in this type of immobilizedhomogeneous catalysts leaching of the metal-complex has often beenobserved.
Among the new strategies to heterogenize transition metal complexes theencapsulation of a chiral metal complex in micropores19 and the use oftethered type metal complexes have to be mentioned.20 The tethered metalcomplex catalysts were recently developed by Augustine and coworkers.21–23
These catalysts showed high activity and high chemo- and/or enantioselec-tivity in various hydrogenation reactions.
When metal-complexes (both homogeneous and immobilized one) areused the enantio-differentiation is controlled by the structure of the metalcomplex. In this respect the molecular character of the catalytic step has tobe emphasized, although the exact form or structure of the [metal complex –substrate] adduct is often unknown. In these enantioselective catalytic re-actions the chirality and the helicity of ligands has a great importance tocontrol the enantio-differentiation step.24 This control can be either ther-modynamic or kinetic in character. The distinction between these twomodes of control is often very difficult. Here we should like to refer to theclassical contribution made by Halpern and coworkers.25
The most important class of ligands used in asymmetric reactions has achiral organic backbone with tertiary phosphino, amino and alcoholicfunctional groups. Highly effective chiral ligands are often bidentate; i.e.they have two coordinating sites for the coordination of the substratemolecule. In chiral homogeneous catalysis, due to the high substrate spe-cificity high enantioselectivities can only be achieved for a defined class ofsubstrate molecules, i.e., the right combination of metal and ligands has tobe found for each individual catalytic reaction.26
Even though enantioselective homogeneous catalysis is still a relativelyyoung discipline, several enantioselective homogeneous catalytic processesare already used on an industrial scale.27,28
As concerns the performance (enantioselectivity), the mechanistic insightand general understanding heterogeneous enantioselective catalysis is far
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 145

behind to its homogeneous analogue. Only a relatively narrow range ofprochiral compounds with CQO and CQC bonds can be hydrogenatedwith high enantioselectivity. However, due to the above-mentioned draw-backs in homogeneous catalysis, such as separation, reuse, and stability, theinterest for heterogeneous asymmetric catalysis increases permanently.
The first publication related to asymmetric heterogeneous catalytichydrogenation was published by Schwab in the early thirties.29 In the firstattempts intrinsically chiral solids, such as quartz has been applied.30
In other approaches chiral biopolymers or natural fibers, such as silk fibroinwere used,31 but due to the low optical yield and severe reproducibilityproblems this approach has been almost completely discarded.
The discovery of the Ni/tartrate system for the enantioselective hydro-genation of beta-diketones or their analogs was the first real breakthroughin this field. This area has been recently reviewed.32–35 Optical yields over90% were obtained for various substrates using the Ni-NaBr/tartrate sys-tem.36 In general this catalytic system requires a pre-modification of theparent nickel catalyst with tartaric acid prior to the reaction in order toform chirally modified sites involved in enantio-differentiation. Recently,the use of in situ modification procedures for Ni/tartrate system has beendescribed.37
In this catalytic system reactive chemisorption of tartaric acid to thenickel surface resulting in some leaching of the nickel to the liquid phase hasbeen evidenced.38 Due to the interaction with tartaric acid the formation ofimprinted sites on the nickel surface has been suggested. The enantio-dif-ferentiation takes place over this type of chirally modified sites.
Since the discovery of the cinchona-platinum catalyst system by Orito’sgroup39–43 for the enantioselective hydrogenation of methyl pyruvate(MePy) or ethyl pyruvate (EtPy) the platinum-cinchona system has beensuccessfully applied in the enantioselective hydrogenation of a variety ofa-functionalized activated ketones, such as various a-ketoesters, keto-pantolactone (KPL), a-ketoamides, a-diketones, a-keto acetals, a-ketoe-thers, trifluoromethyl ketones, and pyrrolidine-2,3,5-triones. Recently themethod has been extended to other types of ketones.44 This type of asym-metric hydrogenation reaction is considered as the most intensively studiedheterogeneous enantioselective hydrogenation reaction.
Since the early eighties great amount of knowledge accumulated on thiscatalytic system. The topic has been reviewed by different authors.15,33,35,45–54
The most important characteristic features of the platinum-cinchona systemare as follows:� Variety of activated ketones can be used as substrate;� Under properly chosen conditions the cinchona alkaloids can induce
high enantioselectivity (ee W97%);55,56
� When a-keto esters are used the enantio-differentiation ability is lostwhen the basic nitrogen at the quinuclidine moiety of the cinchona alkaloidis blocked by alkylation;57
� Upon using various substrates the addition of the cinchona alkaloidresults in significant rate acceleration;58
� The amount of modifier required to induce high enantioselectivity is inthe range of 1� 10� 5M, or in other words the substrate/modifier ratio can
1
5
10
15
20
25
30
35
40
45
146 | Catalysis, 2010, 22, 144–278

exceed a value above 100.000 (in case of KPL the above ratio was276.000);59
� When a-keto esters are used the replacement of the quinoline ring ofthe modifier by phenyl or pyridyl resulted in complete loss of ee;60
� Inversion of ee is observed under various experimental conditions.In the last twenty years step-by-step progress has been made in the pro-
cess of understanding the peculiarities of the platinum-cinchona system.This progress covers the following main issues: (i) elucidation of both therate acceleration and the origin of enantio-differentiation, (ii) clarificationof the nature of species formed both in liquid phase and on Pt surface byusing various spectroscopic methods, (iii) establishment of general andspecific kinetic patterns, and (iv) theoretical calculations and relatedmodeling.
As far as several reviews have already been published on the enantiose-lective hydrogenation of activated ketones in the presence of cinchona-Ptcatalyst in this review an attempt was done focusing on (i) methods andapproaches used, (ii) recent achievements, and (iii) recalling historicalevents.
Contrary to earlier reviews in this contribution the mechanistic con-siderations will be treated without any preconception. It means that it willbe a priori not accepted that all phenomena involved in the rate accelerationand induction of enantio-differentiation can be related to events takingplace exclusively over Pt surface.61 Consequently, in this review we shallalso refer to the general aspects of chiral induction generated by cinchonaalkaloids. Possible interactions in the liquid phase will also be discussed.
Enantio-differentiation is a phenomenon characteristic mostly for organicreactions. There are various synthetic methods in organic chemistry to in-duce chirality. In this respect it has to be emphasized that cinchona alkaloidsare well-known chiral compounds used by organic chemists to bring aboutchiral induction.62 This issue will be briefly discussed in Chapter 2.1
We shall try to demonstrate that the enantioselective hydrogenation ofactivated ketones is a very complex reaction. Depending on the conditionsof catalyst pretreatment, the accomplishment of the hydrogenation reactionand the type of substrate and modifier used the key interactions responsiblefor the transformation of the chiral information can takes place both at thePt surface and in the liquid phase. Methods and approaches used in thisarea will also be discussed as these issues were not treated in previousreviews.
1.2 Orito’s followers
After Orito’s publications39,40–43 intensive research programs were startedby different research groups. First two groups in Switzerland, one at CibaGeigi in Basel under supervision by Dr. H.U. Blaser,63 the other in Zurich atETH with professor A. Baiker.64 Parallel to that professor P.B. Wells65 inHulls (Great Britain) started a program related to the use cinchona alkal-oids in heterogeneous catalytic asymmetric hydrogenation.
Later on other groups in the USA (Dr. D. Blackmond at Merck,66 Pro-fessor R. Augustine67 in Seaton Hall), in Hungary (Prof. Tungler68 at
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 147

Technical University in Budapest, Prof. J.L. Margitfalvi 69 at the HungarianAcademy of Sciences and Prof. M. Bartok70 at University of Szeged ), inFinnland ( professors T. Salmi and D. Murzin71at Abo Academy University)joined to this research area. Today there are around 15–20 independentresearch groups all around the word that are involved in the investigation ofone of the aspects of Orito’s reaction.
It is interesting to emphasize that the method developed by Orito wasquickly modified as the ‘‘pre-modification’’ approach was replaced by insitu modification technique. In this respect the pioneering works were doneby the two Swiss groups. Only one group has continued for a while to applythe ‘‘pre-modification’’ approach: (the group in Hulls), however today thisapproach is almost forgotten. Further details about modification pro-cedures will be given in Chapters 4 and 5.
In the first approaches mostly Pt/Al2O3 catalysts and EtPy were used inorder to elucidate the general kinetic patterns.58,73 Later on the focus waslaid on (i) the elucidation of the reaction mechanisms,61,65,67,74,75 (ii) the useof new substrates,76,77 (iii) the application of new modifiers,60,78 (iv) newtype of catalysts,79,80 (v) the formation of by-products,81 (vi) the role ofimpurities,82 (vii) modeling substrate modifier interaction.83–85 Today so-phisticated experimental and computational techniques, such as reactioncalorimetry,86 the AFM,87 NMR,88 FTIR,89 in situ reflection-adsorptioninfrared spectroscopy (RAIRS),90 attenuated total reflection infraredspectroscopy (ATRIR),91 surface-enhanced Raman spectroscopy (SERS),92
circular dichroism,93 electrochemical methods,94 Near-edge X-ray ab-sorption fine structures (NEXAFS)95,96 studies, DFT calculations97,98 areused to get as much as possible information about these unique asymmetrichydrogenation reactions.
Orito’s approach later on was extended to other type of activated ke-tones. The substrates were classified according to the observed rates andenantioselectivities. This classification is given in Fig. 1. Further discussionof substrate specificity will be given in Chapter 5.3.
From the above discussion it can be concluded that the enantioselectivehydrogenation of activated ketones is the most detailed studied asymmetrichydrogenation reaction. However, despite of the extensive studies there areplenty of unanswered questions related to the origin of enantio-differentiation.
1
5
10
15
20
25
30
35
40
45
High rate – high ee
(a) (b) (c)
Medium rate – medium ee Low rate – low ee
8 9 10
1 2 3 4
5 6 7
11 12 13
14 15
Fig. 1 Classification of substrates according to their ability to give high rate and high ee.(Reproduced from ref. 72 with permission.)
148 | Catalysis, 2010, 22, 144–278

1.3 Specificity of heterogeneous enantioselective catalysis
Heterogeneous catalysis is a complex field in physical chemistry. However,the state-of-the-art current knowledge in this area requires additionalknowledge in the field of materials science, surface chemistry, surface sci-ence, surface analysis, computational chemistry, chemical engineering, etc.Heterogeneous enantioselective hydrogenation requires an additionalknowledge, i.e. an education in the field of organic reactions. As far asenantioselective reactions are a specific area of organic catalysis theknowledge in this area should also be very specific.
It has to be emphasized that those who joined to this research area havedifferent scientific background and different research interest. In most of thecases we are witnessing the prevalence of approaches and views reflectingthe mode of thinking of a chemical engineer or a surface scientist.
Although excellent reviews were written in the last ten years,46–52 thosewho have a solid background in organic chemistry can realize that both thestructure and the conclusions of these reviews reflect the view of those whohave a ‘‘surface science’’ or ‘‘chemical engineering’’ background.
The investigation of heterogeneous catalytic reactions requires a complexknowledge and the use of various experimental techniques. Among thesemethods reaction kinetics and surface characterization has the greatestimportance. Detailed reaction kinetic studies can provide sufficient infor-mation to suggest or create a proposed reaction mechanism. However, evenif reaction kinetics can completely be described by the given system ofdifferential equations the elucidation of the exact reaction mechanismcannot be guaranteed. This would require the comparison of several pos-sible or potential kinetic equations derived from other possible reactionmechanisms. Unfortunately, such kind of comparison is only seldom isperformed in studies related to the elucidation of reaction mechanisms ofasymmetric hydrogenation of activated ketones.
In the enantioselective hydrogenation of activated ketones the accom-plishment of detailed reaction kinetic measurements is strongly hindered bythe peculiarities of the reaction, such as (i) the ability of modifiers to induceenantio-differentiation at very low modifier/substrate ratio, (ii) the highreactivity of the substrates resulting in the formation of various by-prod-ucts, (iii) the loss of alkaloid during the reaction, and (iv) the catalyst poi-soning. All these issues will be discussed latter in separate sub-paragraphs.
Due to the formation of byproducts, the loss of alkaloids and catalystpoisoning the analysis of the full experimental curve is almost impossible.Consequently, the use of initial (or maximum) reaction rates cannot providea proper background for exact kinetic analysis or for the establishment ofcorrect reaction mechanism. The use of in situ calorimetry is one of the mostprecise methods to obtain direct reaction rates,99 although this method isnot common. The preliminary analysis of results obtained by in situ cal-orimetry indicates that the rate follows the Michaelis-Menten mech-anism,100 what is characteristic to enzyme catalytic reactions.101
The fact, that only trace amount of cinchona alkaloid can result in eevalues above 90%, strongly indicate that this catalytic system is extremelyunique. In this respect the results obtained in the enantioselective
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 149

hydrogenation of KPL has to be emphasized, where the substrate/modifierratio is 276 000:159. In other studies related to the enantioselective hydro-genation of EtPy the above ratio is in the rage of 50 000 to 166 000.102 Sucha high value of ‘‘chiral amplification’’ is characteristic only to enzymes.Cinchona alkaloids, due to their high extent of ‘‘chiral amplification’’, theirdistinct substrate specificity and their flexible structure, can be considered asa ‘‘mini-enzyme’’.93 This aspect is often forgotten by those working in thisarea; although in a recent study the similarity to enzymes has already beenmentioned.103
The other key approach for the elucidation of reaction mechanism is theinvestigation of the formation of different surface intermediates by differentsurface analytical tools. These approaches will be described in details inChapter 6.2. In this respect the key issue is to answer the following question:‘‘can the given observed surface entity be involved in the reaction route ordoes it represent a dead-end in the given reaction scheme?’’ In the formercase we talk about ‘‘actors’’, while in the letter case about ‘‘spectators’’.Unfortunately, this kind of questions is only seldom raised. It has to beemphasized that the differentiation between ‘‘actors’’ and ‘‘spectators’’ is along dispute in the area of heterogeneous catalysis. Even in gas phase re-actions taking place at atmospheric pressure neither the ‘‘in situ’’ nor the‘‘operando’’ spectroscopy can always provide an unambiguous exact answerabout the involvement of a given surface species in the reaction network.When we are dealing with heterogeneous hydrogenation reactions takingplace in a three-phase system at high hydrogen pressure the accomplishmentof in situ or operando spectroscopy is very difficult.
Catalysis scientists like very much to refer to sophisticated spectroscopicdata and use these data in favor of their ideas with respect to the reactionmechanism. Even one of the authors of this contribution walked into thiscatch. In early eighties the direct route for the hydrogenation of acetylene toethane was confirmed by a sophisticated ‘‘double isotope labeling’’ techni-que.104 Based on surface science results we proposed that the formedethylidyne species (RC–CH3) are responsible for the direct route of ethaneformation. Of course, we did not detect these species; we just referred to oneof recently published LEED results.105 Later on it turned out that theethylidyne species are so stable that they cannot be removed from the Pdsurface by hydrogen at room temperature.106 Of course, several similarmisinterpretations can be found in the literature.
It will be discussed latter on that in the hydrogenation of activated pro-chiral ketones the presence of hydrogen at the Pt surface is very crucial.It strongly determines the performance of the catalyst. The hydrogenationof ketones requires relatively large abundance of hydrogen at the Pt surface.Probably, it is the reason that these enantioselective hydrogenation re-actions cannot be performed under conditions of transfer hydrogenation.Any disturbance in the amount of available hydrogen can result in signifi-cant alteration is the performance of Pt-cinchona catalytic system. It isespecially notable when the hydrogenation is performed in a continuous-flow reactor under trickle bed condition.107
However, too much hydrogen at the Pt sites will result in the hydro-genation of the quinoline ring of cinchona alkaloids. This leads to the loss of
1
5
10
15
20
25
30
35
40
45
150 | Catalysis, 2010, 22, 144–278

ee at low concentration of cinchona alkaloids.108,109 Contrary to thathydrogen ‘‘starving’’ conditions increases the chance for undesired side re-actions, such as oligomerization and polymerization. Consequently, if in situmeasurements cannot be performed under optimum hydrogen coverage thechance to detect ‘‘spectators’’ is very high. For this reason all spectroscopicdata presented so far should be treated with definite precaution.
2. Cinchona alkaloids
2.1 Characteristic features of cinchona alkaloids
Cinchona alkaloids are used in many fields of our everyday life. They arewidely used in the pharmaceutical and chemical industry. Quinine, derivedfrom the bark of Cinchona ledgeriana Moens ex Trimen is the oldest knownnatural antimalarial drug. Cinchona alkaloids as easy available chiralagents have great importance both in the academic research and in largescale industrial use. In this respect the classical separation of racemicnaproxene can be mentioned.110 An explanation why Cinchona alkaloidsare universal molecules for so many purposes was given by Wynberg.62 Thisis shown in Fig. 2.
Various parts of the molecule fill the following functions: (a) hydrogenbond formation (interaction with metals); (b) basic amine; (c) bulk aliphatichydrocarbon moiety; (d) ‘‘handle’’ to modify; (e,f) chiral pocket (epimersavailable; conformer formation); (g) bulk aromatic hydrocarbon, polariz-able, p-p interaction; (h) ‘‘handle’’ to modify; steric and polar influence. Inthis chapter the use of cinchona alkaloids in chiral separation and chiralcatalysis will be reviewed very briefly. More detailed reviews can be foundelsewhere.62,111,112
2.2 Use of cinchona alkaloids as chiral auxiliaries
2.2.1 Chiral separation. The first example of resolution through for-mation of diastereomeric salts was made by Pasteur113 who used quinicineand cinchonicine, derivatives of quinine and cinchonidine, respectively.Since that time Cinchona alkaloids have been largely employed for the
1
5
10
15
20
25
30
35
40
45
Fig. 2 Multifunctional nature of quinine as a catalyst. (Reproduced from ref. 62 withpermission, (Figure 19))
Catalysis, 2010, 22, 144–278 | 151

separation of various racemic mixtures.114 In the sixties quinine and someother Cinchona alkaloids were used to prepare chiral sorbents.115 Alkaloidsof this type were covalently bonded to a silica support via their olefinicgroup. In this way several functional groups in a bulky chiral system pro-vided multiple contact points with the racemate to be resolved.
Up to now big variety of separation techniques using cinchona alkaloidshas been reported. A two-dimensional liquid chromatography–mass spec-trometry (LC–MS) system was developed for the separation of both dia-stereomers and enantiomers of peptides.116 Generally the presence ofelectron-deficient aromatic N-acyl constituents and bulky, highly lipophilicside chains enhances enantioselective adsorption, reflecting the importanceof intermolecular p-donor-acceptor and hydrophobic interaction with thechiral selector.117 Mixed ternary ion associate formation between xanthenedye, cinchona-alkaloid and quaternary ammonium ion has been applied tothe determinate the trace amount of quaternary ammonium salts inpharmaceuticals by UV spectrophotometry.118
2.2.2 Chiral catalyst. In the field of chiral catalysis huge amount ofwork has been done and Cinchona alkaloids have been involved the in thefollowing areas:� chiral basic and nucleophilic catalysts in organo-catalytic reactions,� chiral ligands coordinating metals like osmium in homogeneous cata-
lytic reactions,� phase transfer catalysts in form of quaternary ammonium derivatives,� chiral modifiers (templates) in heterogeneous catalytic asymmetric
reactions.The cinchona alkaloid catalysed reaction of diethylzinc and aldehydes haslead to optically active alcohols having enantiomeric excess up to 92%.119
Cinchona alkaloids have been used both in solute form in liquid phaseand in bounded form immobilized into polymer or oxide type supports. Inorgano-catalysis based on cinchonas large variety of substrates and types ofthe reactions has been reported. In 1954 Prelog and Wilhelm described thebehaviour of different cinchona alkaloids and some of their derivatives inthe asymmetric cyanhydrin synthesis.120 A review of cinchona alkaloid-catalyzed reactions covering the period prior 1968 was given by Pracejus.121
Cyanohydrin reaction, the Michael reaction, the 1,4-thiol and thiolace-tate additions, selenophenol addition reactions, epoxidation of electron-poor olefins, formation of the phosphorus-carbon bond using chiral aminecatalysis, 1,2-additions in the presence of cinchonas has been detailed byWynberg in 1986.62
Highly enantioselective Reformatsky reaction of ketones has been ac-complished using cinchona alkaloids as chiral templates.122 Cinchona al-kaloid-derived chiral bifunctional thiourea organocatalysts weresynthesized and applied in the Michael addition between nitromethane andchalcones with high ee and chemical yield.123
The osmiumtetraoxide catalyzed asymmetric dihydroxilation (AD) is avery important field of cinchona utilization (see Fig. 3).10,124 Cinchona al-kaloid backbone is ideally suited for providing high ligand acceleration aswell as enantioselectivity in AD. It has been found that the
1
5
10
15
20
25
30
35
40
45
152 | Catalysis, 2010, 22, 144–278

enantioselectivity is influenced mainly by the nature of O9 substituent of thecinchona alkaloid backbone. Three different classes of ligands are veryeffective for the dihydroxylation of almost any olefin (PHAL-,125 PYR-126
and IND-class127). Large scale of substrates (monosubstituted, 1,1-di-substituted, 1,2-disubstituted, trisubstituted even tetrasubstituted olefins,enol ethers, cyclic olefins, amides, enones, sulfur-containing olefines, pro-tected divinyl ketones, conjugated dienes, trienes etc) can be successfullydihydroxylated.10
Phase transfer catalysis (PTC) based on cinchona alkaloids128–153 is acontinuously developing practical method for asymmetric synthesis becausethese catalysts are very selective. Enantioselective epoxidation of a,b-un-saturated ketones utilizing cinchona alkaloid-derived quaternary ammo-nium phase-transfer catalysts bearing an N-anthracenylmethyl functiongave also appropriate results.
Common to all successful applications of cinchona alkaloid derived phasetransfer catalysts is that the reaction conditions have to be optimized,consequently structures of substrate, reagent, and catalyst must ‘‘fit toge-ther’’: An attempt has been made to understand the role of differentstructure units of cinchona derivatives in PTC. The N-anthracenylmethylgroup introduced by Lygo139,143 and Corey141 has been good for an increasein selectivity, although 1-naphthylmethyl was almost as effective.144 Phasetransfer catalysts having diaryl substitution at the 3-and 4-positions of theN-benzyl group in cinchonidinium salts were prepared to check how sub-stituted aryl groups affect the asymmetric induction in the benzylation re-action as compared to those having flat linear aryl systems likenaphthylmethyl and anthracenylmethyl groups.149
Tremendous amount of work has aimed the preparation and investigationof supported cinchona alkaloids as catalyst.154–170 Polymer bound cinchonaalkaloids have been employed for a number of heterogeneous catalytic re-actions e.g. asymmetric Michael additions154–156,171 asymmetric synthesis ofa-amino acids,169 enantioselective a-chlorination of acid chlorides,170 asym-metric aminohydroxilation167 asymmetric dihydroxilation of alkenes.157,160
To recycle the alkaloid-OsO4 complex in asymmetric hydroxilation reaction
1
5
10
15
20
25
30
35
40
45
Fig. 3 Role of structural elements of Cinchona ligands in osmium-catalyzed asymmetricdihydroxilation (AD) reactions. (Reproduced from ref. 10 with permission (Figure 4))
Catalysis, 2010, 22, 144–278 | 153

Kim and Sharpless159 have synthesized four different polymers, Salvadoriet al. have examined copolymers of acrylonitrile and substituted quinidineand quinine and reported very low optical purity of diols (up to 45% ee).160
Lohray et al. prepared several copolymers of styrene and 4-phenylstyrenewith 10% DHQD-4-vinylbenzoate affording the most effective catalyst forheterogeneous AD.161 A nice example of immobilized quinine used as acatalyst for enantioselective a-chlorination of acid chlorides was given in ref.170. Large scale of different type of polymers has been reported for immo-bilization of cinchonas, copolymers of styrene, alkaloid–acrylonitrile co-polymers.155,157,158 Silica gel supported cinchona alkaloids have been usedalso as catalysts in asymmetric dihydroxylation163,165,166 and aminohydrox-ylation.167 Norcinchol supported on silica via ethoxy-silane compound hasbeen applied for enantioselective hydrogenation of a-keto esters with mod-erate success.47 For different purposes different cinchona alkaloids are suit-able. In methanolysis of different tricyclic anhydrides quinidine and quininehas given higher ee than cinchonidine or cinchonine.172
For asymmetric dihydroxylation also quinine (QN) and quinidine(QD) have been found as the most effective ligand.10 In a few cases etheraltype cinchona alkaloids are also successfully used as chiral template,e.g. b-isocupreidine has been used in the asymmetric Baylis-Hillmansreactions of aromatic imines with 1,1,1,3,3,3-hexafluoroisopropyl acrylategiving (S)-enriched, N-protected-a-methylene-b-amino acid esters. Incontrast to the corresponding aldehydes, imines have shown the oppositeenantioselectivity.173
Based on the above short review it can be concluded that cinchona al-kaloids and their derivatives due to their multifunctional structure and easyavailability have been widely used for chiral induction in asymmetric syn-theses as well as chiral separations for a century. When new problems inasymmetric techniques have to be solved the application of cinchonas oftenprovides the proper solution again.
All of these results clearly indicate that cinchona alkaloids have been usedby organic chemists in various areas in order to induce chiral induction orchiral recognition. Consequently, the use of these alkaloids in Orito’s re-actions is only one of the opportunities provided by the unique chemicalproperties of these natural compounds. Any attempt to describe the action ofcinchona alkaloids exclusive to surface phenomenon seems to us a mistake.
2.3 Structure of Cinchona alkaloids, conformational analysis, and
NMR studies
2.3.1 General information. Conformational investigations of cinchonaalkaloids based on computational or spectroscopic methods have beenmade with the aim of understanding of chiral discrimination process.88,174
The most frequently investigated cinchona alkaloids and Cinchona de-rivatives are summarized in Fig. 4. Cinchona alkaloids consist of two rigidcyclic systems, a heteroaromatic quinoline ring and a saturated cyclic qui-nuclidine ring connected by two carbon-carbon single bonds. They havefour asymmetric centers: C3, C4, C8, and C9. However, their configurationsdiffer from each other only at C9 and C8.
1
5
10
15
20
25
30
35
40
45
154 | Catalysis, 2010, 22, 144–278

N
H
N
R2
R4
R3
R1
4′
4′9
8
3N R
4
N
R2
R3
H
R1
8 94′
3
R1
R2
R3
R4
R2
R3
R4
quin
ine
C2H
3O
CH
3O
HH
cinc
honi
dine
C2H
3H
OH
Hdi
hydr
oqui
nine
C2H
5O
CH
3O
HH
dihy
droc
inch
onid
ine
C2H
5H
OH
Hep
iqui
nine
C2H
3O
CH
3H
OH
benz
oylq
uini
neC
2H5
OC
H3
OB
zH
(p-C
l)-be
nzoy
l-D
ihyd
roqu
inid
ine
C2H
5O
CH
3pC
lBzO
H
chlo
roqu
inin
eC
2H3
OC
H3
Cl
Hde
oxyc
inch
onid
ine
C2H
3H
HH
met
hoxy
dihi
dro-
cinc
honi
dine
C2H
3H
OC
H3
H
R1
Qui
nidi
neC
2H3
OC
H3
OH
HC
inch
onin
eC
2H3
HO
HH
dihy
droq
uini
dine
C2H
5O
CH
3O
HH
dihi
droc
inch
onin
eC
2H5
HO
HH
Epiq
uini
dine
C2H
3O
CH
3H
OH
epid
ihyd
roqu
inid
ine
C2H
5O
CH
3H
OH
(p-C
l)-be
nzoy
l-di
hydr
oqui
nidi
neC
2H5
OC
H3
PClB
zOH
acet
ildih
ydro
quin
idin
eC
2H5
OC
H3
OA
cH
(dim
ethy
lcar
bam
oyl)-
dihi
droq
uini
dine
C2H
5O
CH
3O
CO
NM
eH
met
hoxy
dihy
dro-
quin
idin
eC
2H5
OC
H3
OC
H3
H
Fig.4
Structure
andconfigurationoftheCinchonaalkaloidsmost
frequentlyinvestigated(R
eproducedfrom
ref.183withpermission)
Catalysis, 2010, 22, 144–278 | 155

2.3.2 Conformational analysis. Crystallographic structure of QN,175
QD,176 CD177 and cinchonine (CN) 178 is well described. In solution how-ever, existence of several other conformers can be supposed. Conformationof quinine and quinidine was a key issue in different studies62,179,180 and theC8-C9 and C40-C9 bonds were considered most important in determiningthe overall conformation of these compounds. Hiemstra and Wynberg181
have proposed that the most stable conformation of quinine have the largestsubstituent-the quinuclidine ring-on one side of the quinoline ring, andhydrogen at C8 and the hydroxyl at C9 on the other side. Prelog182 andMeurling180 found this conformation to be the most favorable too.
In their pioneer work, Dijkstra and coworkers have combined NMRstudy and molecular modeling approaches to elucidate the conformationalproperties of QN and QD.174,183,184 By use of using the molecular modelingprogram CHEMX, the conformational freedom with respect to the C8-C9and C9-C40 bond was investigated.184 Molecular mechanics studies showedthat cinchona alkaloids can in principle adopt four different conformations:two ‘‘open’’ one in which the quinuclidine nitrogen points away from thequinoline ring and two ‘‘closed ’’ one in which the quinuclidine nitrogenpoints toward the quinoline ring184(see Fig. 5). One of the calculated con-formations of QD (open conformation (3) in Fig. 5) has almost the samegeometry as the crystal structure.174 Different dihydroquinidine (DHQD)
1
5
10
15
20
25
30
35
40
45
Fig. 5 The four minimum energy conformations of quinidine (Reproduced from ref. 184 withpermission, Figure 2)
156 | Catalysis, 2010, 22, 144–278

derivatives as model substances behave in different way. Acetyldihy-droquinidine exhibits the closed conformation in CDCl3.
184
Hydroxy cinchona alkaloids exist in open conformation (3) (see Fig. 5) atleast in 90%, wherein some conformational freedom of the quinuclidinering exists. Methoxy derivatives predominantly adopt the open conform-ation (3) and to a lesser amount the closed conformation (2) in CDCl3.However, in CD2C12 the closed conformation (2) is found in excess.184
Hydroxy cinchona alkaloids exist in open conformation (3) (see Fig. 5)at least in 90%, wherein some conformational freedom of the quinuclidinering exists. Methoxy derivatives predominantly adopt the open conform-ation (3) and to a lesser amount the closed conformation (2) in CDCl3.However, in CD2C12 the closed conformation (2) is found in excess.184
The combination of variable temperature (þ 20 to �100 1C) NMR andcircular dichroism spectroscopy as well as molecular mechanics compu-tations have shown that in ether solution dihydroquinidine existed in twoconformations, the open184 conformation (3) (anti, open) (80–90%) andclosed conformation (1) (syn, closed) (10–20%) separated by a barrier of8.3 kcal/mol.
The results of computations favoured to the closed conformation (1) inthe gas phase and this discrepancy was explained by preferential solvatationof hydroxy group, which is sterically more available in anti conformation.Bulky substituents on the hydroxy group, such as in the p-chlorobenzoateester have the same effect.185
The conformation of cinchonidine in solution has been investigated byNMR techniques as well as with theoretical tools.88 Three conformers ofcinchonidine (CD) are shown to be substantially populated at room tem-perature, closed conformation (1), closed conformation (2), and openconformation (3). The latter is the most stable in apolar solvents. The sta-bility of the closed conformers relative to that of open conformer (3),however, increases with solvent polarity. In polar solvents the three con-formers have similar energies. The relationship between relative energiesand the dielectric constant of the solvent is not linear but resembles the formof an Onsager function.88
In o-dichlorobenzene or dimethyl sulfoxide solution the dihydroquinidine(DHQD) and (p-chlorobenzoyl)dihydroquinidine (p-ClBzDHQD) werefound to exist as an equilibrating mixture of two main conformers, seeTable 1.174 The relative amounts of these two conformers depend on con-centration as well as on solvent and temperature. Changes in the ratio of thetwo conformers of DHQD can explain the observed changes of the enan-tioselectivity in the indene rearrangement when the solvent was changedfrom o-dichlorobenzene to dimethyl sulfoxide.
Solute-alkaloid interactions are also able to influence the conformationalbehavior.184 In case of ester derivatives the energy difference between closedand open conformation is less and is probably of the same order of mag-nitude as the amount of stabilization caused by interactions with solutes,such as methanol or weak acids, or with strong electrophiles, such as os-mium tetraoxide.
In case of the methoxy derivatives the energy difference between closedconformation (2) and open conformation (3) has vanished. In non-
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 157

coordinating solvents like CD2C12, the methoxy derivatives are still pre-dominantly found in the closed conformation (2), but in the presence of anyelectrophile the equilibrium shifts towards the open conformation (3).Quinine and quinidine (and other hydroxy derivatives) by themselves al-ready possess a distinct preference for the open conformation (3) and thusdo not depend on extra stabilization caused by interactions with solute.184
Upon investigation of the above mentioned cinchona alkaloid catalyzedMichael addition of thiols to enones, it was found from the NOESY spectraof QD and QN in the presence of 4-methylbenzenethiol that the alkaloidconformations do not change on formation of an ion pair.174
When cinchona alkaloids are used as chiral bases, the main interactionwith the substrate comes from protonation of the tertiary nitrogen in thequinuclidine ring and a subsequent formation of an ion pair between theprotonated alkaloid and the deprotonated substrate molecule.184
When the alkaloids are used as chiral ligands, the main interaction is theformation of a dative bond between the tertiary nitrogen of the quinuclidinering and the metal atom of the substrate molecule (osmium tetraoxide).184
Investigation of the temperature effect has led the authors to come to animportant finding. Low temperature experiments at �20 1C and �60 1C inCDCl3, did not alter the 1H NMR spectra, no line broadening has beenobserved, and averaged spectra were still recorded at 40 1C. Thus, even atthese low temperatures, it was not possible to freeze out different con-formers. This was an indicative of a fast exchange between the differentconformations on the NMR time scale and thus of a low energy barrier.183
The syn-anti barrier was estimated ca 8 kcal/mol and the closed-openbarrier only half as this.185
The 1H NMR relaxation method was applied to QD. The proposedconformation had the following dihedral angles: o(C11–C10–C3–C4)=1501 and o(C40–C9–C8–C7)=701. The conformation of side-chaino(C11–C10–C3–C4) was found to be different from the one found forcrystalline form by X-ray analysis.187 Potential energy surface (PES) for QDhas been comprehensively investigated using the molecular mechanics
1
5
10
15
20
25
30
35
40
45
Table 1 Populations of open (A) and closed (B) conformers of dihydroquinidine (DHQD) and
(p-chlorobenzoyl) dihydroquinidine (p-ClBzDHQD) calculated from JH8�H9a (Reproduced
from Ref. 174 with permission, Table 7)
Base Solution (25 1C) J (A)a J (A)b J(obs) P(A)b P(B)b
DHQD CDCl3, 0.2M 2.6 8.29 4.2 0.72 0.28
DHQD CDCl3, 0.02M 2.6 8.29 5.0 0.58 0.42
DHQD THF-d8, 0.02M 2.6 8.29 4.2 0.72 0.28
DHQD o-DCB-d4, 0.005M 2.6 8.29 4.8 0.61 0.39
DHQD o-DCB-d4, 0.02M 2.6 8.29 5.0 0.58 0.42
DHQD dioxane-d8, 0.02M 2.6 8.29 5.0 0.58 0.42
DHQD acetone-d6, 0.02M 2.6 8.29 6.3 0.35 0.65
DHQD DMSO-d8, 0.02M 2.6 8.29 7.2 0.19 0.81
p-ClBzDHQD CDCl3, 0.2M 2.5 8.73 7.4 0.21 0.79
p-ClBzDHQD o-DCB-d4, 0.02M 2.5 8.73 7.8 0.15 0.85
a Based on AM1 structures, J values calculated with substituent corrections by Gandour et al.186;bPopulations of open (A) and closed (B) conformer.
158 | Catalysis, 2010, 22, 144–278

(MM) and quantum mechanical semi-empirical AM1 and PM3 methods.Theoretical results were in agreement with the experimental NMR data, i.e.,there are two conformations of the quinidine molecule in solution.188
Structures of etheral and D3,10 isomers of cinchona alkaloids were alsodetermined by NMR and supported with molecular mechanics.189 NOEinteractions in quinuclidine moiety of the cinchona ethereal isomers areshown in Fig. 6. Further structural information on cinchona derivatives willbe given in Chapter 6.1.
2.3.3 Solute-solute interaction. Intermolecular interaction of the alkal-oid molecules in solution can also be observed. Significant difference be-tween the NMR spectra of optically pure and racemic dihydroquinidineswas found under the same conditions (0.35M in CDCl3). The spectraldifferences were greatly reduced when CD3OD was used as solvent. Theacetates of optically active and racemic dihydroquinine showed significantlysmaller differences than those observed with dihydroquinine. The authorshave explained the observations by solute-solute interactions of theenantiomers.190
Osmometry was used to measure average molecular weight for quinine.Results of these experiments have indicated the presence of particles largerthan monomeric quinine at 37 1C for a 16mM solution in toluene. Forconcentrationso4mM the quinine was almost completely monomeric.181
The coexistence of monomer and dimers of quinidine was established inquinine-chloroform solutions by investigating the temperature and con-centration dependence of the NMR spectral parameters by combination of2D NOESY and proton-selective relaxation rate measurements. Similarconformation of the alkaloid was found both in the dimer and monomerforms. It was shown that the quinuclidine ring is on one side of the quin-oline ring and the CHOH moiety on the other, with the quinoline planealmost bisecting the angle between C-H8 and C-OH191 see Fig. 7.
1
5
10
15
20
25
30
35
40
45
Fig. 6 NOE interactions in quinuclidine moiety of the cinchona ethereal isomers. (Repro-duced from Ref. 189 with permission, Scheme 2)
Catalysis, 2010, 22, 144–278 | 159

Upon investigation the circular dichroism spectra of cinchona alkaloids,exciton type Cotton effect at 230 nm band of free bases (0.4mM) was foundin CH2Cl2 or dioxane, but not in MeOH. This effect was attributed to theweek association of alkaloid molecules in non-polar solvents via N?HOhydrogen bonds.192
3. Alkaloids used in Oritos’s reaction
Studies aimed at systematic variation of cinchona modifiers and theiranalogs have played a definite role in building up hypotheses for themechanism of Orito’s reaction. The structural units of cinchona alkaloidshave been discussed in previous chapter. There are different reviews46,193,194
related to the analysis of modifiers used in asymmetric hydrogenation ofactivated ketones. For this reason in this chapter only the key issues will bebriefly mentioned.
We shall apply the following classification for chiral modifiers applied (i)flexible cinchona alkaloids, (ii) flexible cinchona derivatives, (iii) rigid cin-chona derivatives, (iv) flexible cinchona analogues, and (v) other type ofchiral templates.
3.1 Flexible cinchona alkaloids and their derivatives
Chiral templates most often used in the heterogeneous catalytic asymmetrichydrogenation of activated ketones are natural cinchona alkaloids such as,
1
5
10
15
20
25
30
35
40
45
Fig. 7 Conformation of quinine dimer from NMR results (Reproduced from Ref. 191 withpermission, Figure 8)
160 | Catalysis, 2010, 22, 144–278

CD, CN, QN and QD (see Fig. 8).33,40,51 CD is the most frequently in-vestigated chiral template used in these reactions.
Upon hydrogenation of pyruvates QN and CD (C8(S), C9(R)) result in(R)-lactate while QD and CN (C8(R), C9(S)) give (S)-lactate.33,40,195,196 Ingeneral CD is a better modifier than CN. This difference is more pro-nounced in ethanol than in toluene, but in AcOH the difference is negligible.With the exception of epicinchona alkaloids197 and isocinchonines198 it is ageneral observation, that the configuration of C8 or C8 and C9 atoms of thecinchona alkaloid determines the product distribution.57,193,199 Already inone of the first studies it was evidenced that tertiary N in the quinuclidinemoiety of cinchonas plays crucial role195,199,200 although in recent studies itwas shown that in case of ketopantolactone201 even N-alkylated derivativesof CD can induce very slight enantioselection.
Surprisingly the N-oxide derivative of CD has also resulted in enantio-selection. A possible reason is that N-oxide can be reduced very fast underthe reaction conditions and than acts like 10,11-dihydrocinchonidine(DHCD)57 which is the most easily forming derivative of CD.63
Not only the vinyl group of cinchona alkaloids can be hydrogenated, butits quinoline ring. This is an undesired side reaction leading to the sub-stantial loss of enantioselectivity.57,196,200 It has been suggested that thedecrease in the ee values upon using CD derivatives with partially hydro-genated quinoline be attributed to a weaker adsorption of the alkaloid tothe Pt surface.193 The phenomenon can also be explained by the loss of theshielding effect of the aromatic p-system required for chemical shielding viap–p interaction83 (see Chapter 8.3).
In a detailed study (see ref18) different cinchona analogs and 8 differentsubstrates were investigated. The results indicated that no ‘‘best’’ chiraltemplate exists for all substrates.202 This finding indicates that interactions
1
5
10
15
20
25
30
35
40
45
Fig. 8 Structure of natural cinchonas used in Orito’s reactions.
Catalysis, 2010, 22, 144–278 | 161

between the substrate and the chiral modifier template depend on variousfactors.202
C9 substituted compounds represent an important group of flexible cin-chona alkaloids. 9–O-methyl-10,11-dihydrocinchonidine (MeODHCD) themost frequently used C9 derivative generally behaves slightly better thanCD in the hydrogenation of a-ketoesters.195,200,203,204 Similar positive re-sults were obtained upon using other substrates.44,200,205 However dike-tones, such as 1-phenyl-1,2-propanedione produces lower ee in the presenceof (MeODHCD) than in the presence of CD.206 Detailed studies on the useof these alkaloid derivatives can be found elsewhere.203,207 With respect tothe use of O-substituted derivatives the inversion of ee has to be mentioned.These results will be discussed in Chapter 5.6.4. The inversion of ee in thecase of bulky O-substituted derivatives of CD relative to DHCD has beenattributed to a tilted mode of adsorption of these chiral templates to the Ptsurface207 (see Chapters 6 and 8).
3.2 Rigid cinchona derivatives
C8(S) C9(R) type of cinchona alkaloids, such as CN, QD and cupreidinecan form inner ether derivatives. These derivatives are called ‘‘rigid’’ as inthese alkaloids the rotation around the bond C8–C9 as an axis is notpossible.
These alkaloids were used to demonstrate that the formation of closedconformation of alkaloids is not a prerequisite for the formation of sub-strate-modifier complex suggested by the ‘‘shielding effect’’ model.83
3.3 Flexible cinchona analogues
Synthetic analogues of alkaloids have all of the key elements of cinchonaalkaloids, such as aromatic ring, chiral moiety, and basic nitrogen. Im-portant feature of these new analogues is the presence of an aromatic groupin the close neighbourhood of the stereogenic center. Different types ofenantiomerically pure primary and secondary aminoalcohols78 and amineshave been tested as chiral templates in the hydrogenation of pyruvateesters,208,209 ketopantolactone,210 trifluoromethyl ketones,211 1,1,1-tri-fluoro-2,4-diketones, etc.44
3.3.1 Aminoalcohols. Series of enantiomerically pure 2-hydroxy-2-ary-lethylamines (see Fig. 9) has been prepared from the corresponding ole-fins.78 Upon using compound A in the hydrogenation of EtPy ee valueshigher than 75% was achieved.208,209 The replacement of the naphtyl ringby an anthracenyl one resulted in further increase of ee the up to 87%.60,212
However 1-9-triptycenyl)-2-1-pyrrolidinyl) ethanol resulted in significantdecrease in both ee (o5%)60 and reaction rate.
3.3.2 Amines. Upon using (R)-1-1-naphthyl) ethylamine as chiral tem-plate in the asymmetric hydrogenation of EtPy 82% ee has been achieved inAcOH. It has been shown that (R)-1-1-naphthyl) ethylamine is only aprecursor of the actual chiral template, which is a secondary amine (ami-noester) formed in situ from (R)-1-1-naphthyl) ethylamine and EtPy bycondensation to the corresponding imine and subsequent reduction of the
1
5
10
15
20
25
30
35
40
45
162 | Catalysis, 2010, 22, 144–278

CQN bond.46,200,213 The configuration at the stereogenic center a to theester group has no effect on the enantioselectivity.200 Substituent at theamino group of naphthylethylamine can influence the enantiodifferentiationability; in general, more bulky substituent at the N atom is detrimental toenantioselectivity in the hydrogenation reaction of EtPy.200 Furhter detailscan be found elsewhere.211,213
3.4 Other type of chiral templates
Other natural alkaloids and their derivatives were also applied as chiraltemplates in the asymmetric hydrogenation of activated ketones althoughthe ee values obtained were much lower than over CD and its derivatives.Blaser and coworkers tested about 100 different chiral auxiliaries, but theynever found any meaningful enantioselectivity.214 Ephedrine has given lowor moderate optical yields in the hydrogenation of a-ketoesters.215 Codeine,strychnine, and brucine have provided only 2–12% ee.216 Using tri-fluoromethylcyclohexyl ketone substrate brucine has not resulted in opticalyield.217 (� )-Dihydro-apovincaminic acid ethyl ester has also been appliedas chiral template for EtPy substrate (27–30% ee).68,218–220 Other vincaderivatives have also been tested but (� )-dihydro-apovincaminic acid ethylester has been found to be the most effective one.68,221,222 Upon using othercompounds as chiral templates in the hydrogenation of EtPy (S)-a,a-diphenyl-2-pyrrolidinemethanol223 resulted in moderate ee (max 25%) de-pending on the type of the solvent. (S)-proline chiral auxilary has also beentested.224 During the hydrogenation of EtPy in the presence of (S)-prolineresulted in the formation of N-alkylated proline while in case of isophoronesubstrate a diastereoselective oxazolidine type intermediate was formed in acondensation reaction. Hydrogenation reaction itself proved to be
1
5
10
15
20
25
30
35
40
45
Fig. 9 Preparation of enantiomerically pure 2-hydroxy-2-arylethylamines. (Reproduced fromRef. 78 with permission)
Catalysis, 2010, 22, 144–278 | 163

diastereoselective. Isophorone has produced ee up to 60% but (R)-ethyllactate has been formed in very low optical purity (1–5% ee).224 (S)-prolinederivatives, such as Z-(S)-proline 2-naphthyl ester, Z-(S)-proline 2-(2-naphthyl)-ethyl ester, Z-(S)-proline 3-ethyl-indole ester and (Z)-(S)-proline-3-ethyl-indolamide, (S)-proline-2-naphthylamide hydrochloride has alsobeen tested as chiral templates of new type in case of EtPy.225 a,a-Diphenyl-L-prolinol chiral template resulted in 14% (S) in the hydrogenation of tri-fluoromethylcyclohexyl ketone.217 Dextrocarbinol base has induced noenantioselectivity in the same reaction.217 ‘‘Troger’s base’’ ((5R,11S)-(þ )-2,8-dimethyl-6H,12H-5,11methanodibenzo[d,f][1,5] diazocin) as a chiraltemplates has given 65% ee using acetic acide solvent in the asymmetrichydrogenation of EtPy.226 (R)-(� )-2-phenylglycinol has induced poorenantioselectivity in the hydrogenation of 1,1,1-trifluoro-2,4-diketones.217
Other compounds tested in the hydrogenation of EtPy as ((R)-(þ )-N-(a-methylbenzyl) phtalic acid monoamide, (R)-(� )-1-1-naphthyl) ethyliso-cyanate has given moderate ee (23%, 59% respectively).226
It has been shown that 1-naphthyl-1,2-ethanediol227 is an effective chiralmodifier in the hydrogenation of KPL and ethyl-4,4,4-trifluoroacetoacetate.It is the first effective nonamine-type chiral template used in Orito’sreactions.
4. Methods and approaches used
4.1 General information
In this chapter methods and approaches applied in the enantioselectivehydrogenation of activated ketones will be described. One of the charac-teristic features of this catalytic system is the need for catalyst pretreatmentin hydrogen at 350–400 1C prior to the reaction. The omitting of pre-re-duction step resulted in low rates and low enantioselectivities. The need forcatalyst pretreatment has already been described by Orito’s group.40 In alater study it has been shown that the modification of the Pt surface by thealkaloid requires pure Pt sites.63 Recently a new type of Pt/Al2O3 catalysthas been developed by Degussa (catASium F214) which can be used withoutpre-reduction.228 This catalyst gives high rates and high ee values when it isused as received.
Some authors claimed that the aerobic treatment of the catalyst, i.e. theformation of chemisorbed oxygen on the Pt sites, is needed to improve boththe reaction rate and the ee values.65,229,230 This issue will be discussed inChapter 4.3. The use of ultrasound resulted in also an improved perform-ance of supported Pt catalyst.70,231 In a recent study microwave treatmentwas also described.232 The other important issue is the mode of introductionof the modifier. In Orito’s approach pre-modification has been used.40 Thediscovery of in situ modification was the next important finding.63 Uponusing in situ modification the ‘‘ligand acceleration’’ phenomenon has beendiscovered.58 However, it has to be mentioned that rate acceleration (RA)was not observed for all substrates and all modifiers investigated.
Based on this fact recently same groups questioned the validity of the rateacceleration phenomenon.233–235 This issue will be discussed in Chapter 5.1.
1
5
10
15
20
25
30
35
40
45
164 | Catalysis, 2010, 22, 144–278

Most of the authors are calculating either the initial rate or the maximumrate observed after a short induction period. Unfortunately, due to sidereactions and the transformation of the alkaloid during the hydrogenationreaction (see Chapter 5.1) the analysis of whole kinetic curve is very trou-blesome, although there were attempts to do that.100
We should like to emphasize that in order to get a full picture about thepeculiarities of these unique reactions reliable data with respect to both thereaction rates and the optical yields should be provided. One of the mostserious problems is that in large number of recent publications the rates arenot given at all.211,236–238 Only conversion or yields values measured at theend of the reaction are compared.
There were couples of papers devoted to the problems of reproducibilityand variation of the initial rates measured under identical conditions.47,61
It has been shown that initial rates depend not only on the purity, but theorigin of the substrate as well as on the batch number.69,93 Systematicstudy of this effect was done by the Ciba group. These results are shown inTable 2.95
Of course, the purity of the reactant and solvent has a great impact on thevalidity of kinetic results, consequently results obtained upon using un-purified substrates, especially ketoesters, has to be treated with greatconcern.
Researchers with sufficient background in organic chemistry realized veryearly that the purification of the substrates before the use is a must. The useof purified modifier is also very important.239 There were strong disputes inthe literature concerning to the use of unpurified or contaminated sub-strates.66,69,82,229 The lack of background in organic chemistry often re-sulted in strange and unreliable results. For instance, the reactionmechanism was investigated by different groups in alcoholic solvent, despitethe fact that the most investigated substrates, i.e., EtPy or MePy, react withlinear alcohols in a side reaction catalyzed by tertiary amines. The
1
5
10
15
20
25
30
35
40
45
Table 2 Effect of substrate origin and quality on initial rate (100�mol/g catalyst/min) and ee
values in the hydrogenation of EtPy in the presence of CD under different conditions (catalyst,
solvent, pressure in bar). Bold numbers show the highest and lowest rate or ee values. (Re-
produced from Ref. 195 with permission)
Origin
Undistilled Distilled
J, T, 20 J, T, 20 E, T, 20 J, Ac, 20 E, Et, 20 J,T,100 Average
rate ee rate ee rate ee rate ee rate ee rate ee Rate ee
Fluka91 3 63 12 69 10 69 12 83 25 74 56 82 20 73
Fluka92 4 78 44 80 50 80 64 88 90 74 96 83 58 81
Lancaster 7 71 14 78 14 77 26 87 36 77 48 85 24 79
ICN, Ohio 9 77 15 79 05 79 38 89 40 78 64 86 30 81
Sigma 9 76 24 80 20 80 46 87 40 78 114 87 42 81
Jansen 18 80 24 83 18 83 46 90 50 80 102 89 43 84
R.de Haen 5 73 30 81 21 79 46 87 50 78 148 87 50 81
Aldrich 50 84 70 85 36 85 76 91 70 84 132 90 72 87
TCI, 50 82 48 83 62 83 78 90 68 70 164 80 78 81
J=JMC, E=Engelhard, T=toluene, E=ethanol, Ac= AcOH, TCI=TCI, Tokyo
Catalysis, 2010, 22, 144–278 | 165

formation semi-ketals will be discussed in Chapter 5.1. Despite all disputesand argues even these days it is possible to find papers, where no words issaid how the substrate was purified or what even is worst, there are publi-cations where unpurified substrate has been used.234,240 These facts oftenresulted in misinterpretation of experimental results. These issues will bedescribed in Chapter 5.6.1 and 5.6.2.
In all pioneering studies, i.e., in early nineties, the determination of op-tical yield was not an easy task, especially at low conversion. For this rea-son, the changes in the ee values with conversion were not investigated,consequently the anomalous monotonic increase type ee-conversion de-pendencies were not discussed till 1995 (see details in Chapter 5.5.2.).
In the last 10 years sophisticated physical or physical-chemical methods,such as STM, ATR�IR, AFS, Raman spectroscopy, etc. has been used inorder to elucidate the reaction mechanism or the origin of RA and enantio-differentiation (ED) (see Chapter 6.2). The common problem related tothese studies is that conditions of these measurements are far away fromthose used in real hydrogenation reaction, although some measurementmethods, such as (ATR-IR) were performed under condition close to hy-drogenation.241 A typical misused situation is when the chemisorption ofCD was investigated by electrochemical methods in concentrated H2SO4.Based on these results it was concluded that the adsorption of CD on Pt(111) is irreversible.242 The problem with these results is that those who needsome additional proof with respect to ‘‘surface induced’’ RA and ED likethese results and refer to these false findings quite often.
In this respect there is one more problem what can be formulated in thefollowing way: How to distinguish between surface species what are in-volved in the catalytic step from those, what are formed on the surface ofplatinum, but are not involved in the catalytic act? The latter species areoften called as ‘‘spectators’’ in a given catalytic reaction. In many cases thesurface concentration of ‘‘spectators’’ can be much higher than that of the‘‘actors’’. In this respect let us remind the reader for the classical problem inhomogeneous catalysis discussed by Halpern.243 In his classical study it wasdemonstrated that in homogeneous catalytic enantioselective hydrogen-ation not the most stable [substrate-catalyst] complex is involved in the EDstep, but the less stable one, what reacts with hydrogen much faster than theformer.
In connection to the above discussion the use of sophisticated surfacetechniques for the elucidation of the origin of ED has to be mentioned.None of these methods can fully guarantee that the observed surface speciesis really involved in the given step of enantioselective hydrogenation.Consequently, it is almost impossible to distinguish, whether an identifiedsurface entity is an ‘‘actor’’ or just a ‘‘spectator’’.
4.2 Catalysts applied
4.2.1 Supported metal catalysts. In the enantioselective hydrogenationof activated ketones supported Pt is the most commonly used catalyst. Pt/Ccatalysts have been used by Orito in his original approach. Alumina sup-ported Pt catalysts containing around 5wt% metal are the most commonly
1
5
10
15
20
25
30
35
40
45
166 | Catalysis, 2010, 22, 144–278

used catalyst. Two industrial catalysts, E 4759 from Engelhard and JMC 94from Johnson Matthey, have been widely used by different research groups.The Pt dispersion of these catalysts is in the range of 0.2–0.3.57 E4759 hasrather small pores and a low pore volume, while JMC 94 is a wide-porecatalyst with a large pore volume. There are reports related to the use of Ptcolloids both as prepared79,100,244–247 or stabilized on a support.248
The use of other supported noble metals, such as Ir,249–251 Ru252,253 andRh254–258 is considered as a curiosity, although recent results using rhodiumis very promising.259 Supported iridium catalysts were used in the enan-tioselective hydrogenation of diketones in order to suppress the hydrogen-ation of the second carbonyl group.260 Palladium is not a suitable metal forthe hydrogenation of keto carbonyl group.
Pt supported on HNaAY261 and ZSM-5 zeolites,262 MCM-41263 meso-porous materials, clays264 and ion exchanges resins252 were also tested in theenantioselective hydrogenation of EtPy, however, the performance of thesecatalysts was lower than that of the alumina or silica supported Pt.
It is interesting to mention that most of the Pt/C catalysts resulted in lowee values (below 35%) and very moderate reaction rates.265 There are re-ports on the use of carbon nanotubes as support.266 We consider that allhigh surface area materials are inefficient supports for this reaction, due totheir high adsorption power resulting in high modifier concentration at thesupport and lowering the modifier concentration in the liquid phase.
In earlier studies it has been suggested that Pt dispersion has a decisiveinfluence on both the activity and ee and it was suggested that in order toobtain high optical yields the dispersion should be r0.2.63 It has beensuggested that an appropriate flat Pt surface be required to accommodatethe modifier or the modifier-substrate complex in order to get pronouncedED.267
Contrary to the above results and suggestions results upon using a Pt/SiO2 catalyst (EUROPT-1) relatively high ee values were also obtained,although the dispersion of Pt in this catalyst is around 0.6–0.7.229 Furtherresults on Pt nanocolloids prepared,79,245–248 indicated also that there is noreal need to have large flat Pt surface to get high ee values.
4.2.2 Pt colloids. The common feature of Pt colloids is that they arestabilized by nitrogen and oxygen containing ligands. Under properlychosen experimental condition these Pt colloids show high activity andrelatively high enantioselectivity.248 Pt colloids were also used in kineticinvestigations.100 It was demonstrated that the RA could also be observedwhen Pt colloids are used.
In this respect Pt colloids stabilized by cinchona alkaloids have thegreatest interest. The concept of using chiral stabilizing agent for thepreparation of Pt colloids has been applied by Bonnemann.79 These colloidswere used to hydrogenate EtPy. Upon using DHCD or CD as stabilizingagent the mean size of Pt colloids was in the range of 1.5–2.8 nm. It isinteresting to note that upon using these colloids in the hydrogenation ofEtPy ee values in the range of 75–80% were obtained. In a recent study Ptnanocolloids stabilized by cinchona alkaloids were used in enantioselective
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 167
hydrogenation of EtPy in as received form or immobilized on varioussupports.268
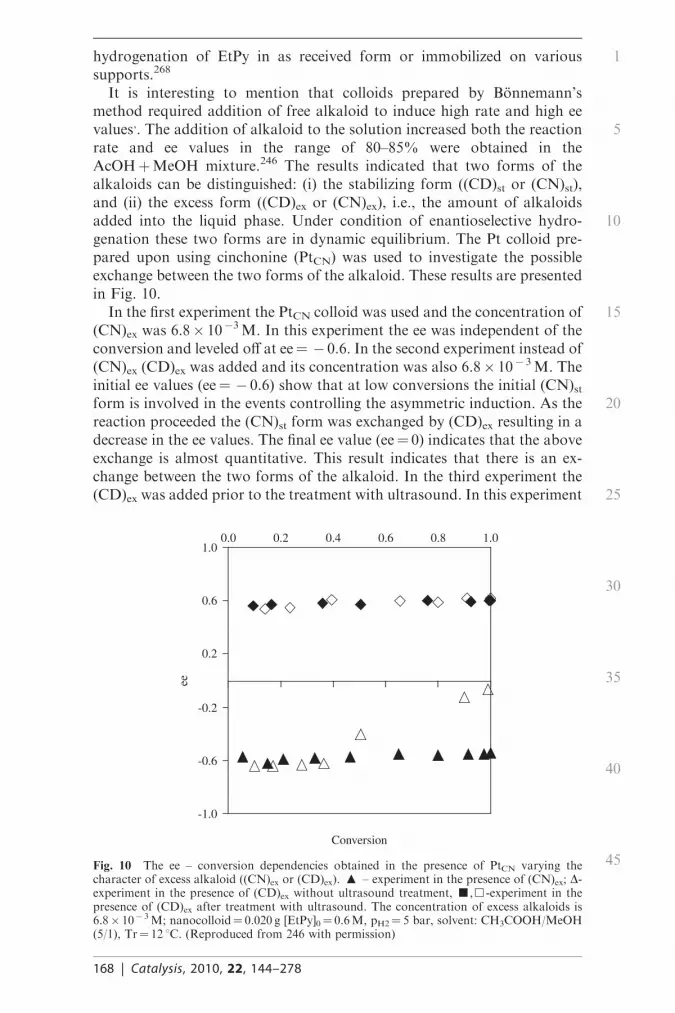
It is interesting to mention that colloids prepared by Bonnemann’smethod required addition of free alkaloid to induce high rate and high eevalues,. The addition of alkaloid to the solution increased both the reactionrate and ee values in the range of 80–85% were obtained in theAcOHþMeOH mixture.246 The results indicated that two forms of thealkaloids can be distinguished: (i) the stabilizing form ((CD)st or (CN)st),and (ii) the excess form ((CD)ex or (CN)ex), i.e., the amount of alkaloidsadded into the liquid phase. Under condition of enantioselective hydro-genation these two forms are in dynamic equilibrium. The Pt colloid pre-pared upon using cinchonine (PtCN) was used to investigate the possibleexchange between the two forms of the alkaloid. These results are presentedin Fig. 10.
In the first experiment the PtCN colloid was used and the concentration of(CN)ex was 6.8� 10�3M. In this experiment the ee was independent of theconversion and leveled off at ee=� 0.6. In the second experiment instead of(CN)ex (CD)ex was added and its concentration was also 6.8� 10� 3M. Theinitial ee values (ee=� 0.6) show that at low conversions the initial (CN)stform is involved in the events controlling the asymmetric induction. As thereaction proceeded the (CN)st form was exchanged by (CD)ex resulting in adecrease in the ee values. The final ee value (ee=0) indicates that the aboveexchange is almost quantitative. This result indicates that there is an ex-change between the two forms of the alkaloid. In the third experiment the(CD)ex was added prior to the treatment with ultrasound. In this experiment
1
5
10
15
20
25
30
35
40
45
-1.0
-0.6
-0.2
0.2
0.6
1.00.0 0.2 0.4 0.6 0.8 1.0
Conversion
ee
Fig. 10 The ee – conversion dependencies obtained in the presence of PtCN varying thecharacter of excess alkaloid ((CN)ex or (CD)ex).7 – experiment in the presence of (CN)ex; D-experiment in the presence of (CD)ex without ultrasound treatment, ’,&-experiment in thepresence of (CD)ex after treatment with ultrasound. The concentration of excess alkaloids is6.8� 10� 3M; nanocolloid=0.020 g [EtPy]0=0.6M, pH2=5 bar, solvent: CH3COOH/MeOH(5/1), Tr=12 1C. (Reproduced from 246 with permission)
168 | Catalysis, 2010, 22, 144–278

the ee value was constant but was opposite in sign, i.e., during the ultrasonictreatment full exchange between the two forms of the alkaloid ((CN)st and(CD)ex) took place (see Fig. 10). The phenomenon appeared to be fullyreproducible.
According to computer modeling the above Pt nanocolloids have particlesize in the range of 1.6 –2.8 nm, i.e., the size of accessible Pt (111) surface isvery small (3� 4 or 5� 5 Pt atoms). This small Pt colloid can accommodatethe ‘‘shielded’’ modifier-substrate complex, while the accommodation of theopen modifier-substrate complex would require much larger surface sites.
4.2.3 Characteristic features of supported Pt catalysts used. The resultsdiscussed so far indicated there is no need to have a preferred particle size ashigh ee values were obtained over catalyst having broad Pt dispersion range.However, in active and enantioselective catalysts the Pt sites should berelatively clean. Both the pre-treatment in hydrogen at 300–400 1C and thetreatment in ultrasound can provide clean Pt surface.
Another important issue is that the catalyst used has to be relatively inertrelated to in the hydrogenation of the quinoline ring of the alkaloid. Con-ditions for ring hydrogenation of modifiers were investigated in variousstudies.269–273
Although it has been shown that upon using Al2O3 support in AcOH Aloxonium ions and their adducts with the alkaloid has been detected274 theinvolvement of these species in the catalytic reaction is quite doubtful. Thesuggested ‘‘electrostatic acceleration’’275 needs further experimental proof.
As a rule the support should be relatively inert. Highly acidic supportscan induce acid catalyzed undesired side reactions. Both the high acidityand the high surface area of supports decrease the amount of alkaloidavailable for ED. It was shown that Cl containing alumina precursor andchlorine-containing platinum salts exhibit significantly higher optical yieldthan similar catalysts prepared from chlorine free starting-materials.267 Ithas also been demonstrated that the modification of alumina support byalkoxy-silanes decrease both the rate and ee values276 (see Chapter 5.7).
4.3 Catalyst pretreatment
In the first publication by Orito’s group Pt/C catalyst was applied and thebeneficial effect of preheating the catalyst in hydrogen at 300–400 1C priorto the modification was emphasized.40,41 The selection of a proper pre-treatment procedure for supported Pt catalysts is one of the basic issues.Several other pretreatment methods were applied and different explanationswere given for the favourable effect of reductive, aerobic and ultrasonictreatments. Fig. 11 shows the general scheme for catalysts pretreatment.76
The common feature is the reduction of the catalyst used at relatively hightemperature (300–400 1C). It is called reductive treatment. The catalyst canbe cooled either in hydrogen or in an inert atmosphere. In oxidativetreatment after the reductive treatment the catalyst is treated in air andcooled down in an inert atmosphere. Most of the authors agree thatupon using supported Pt catalyst a reductive treatment is a must and specialcare has to be done to prevent contamination of reduced catalyst with
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 169

oxygen. However, there are groups using pre-reduced catalysts kept orstored in air.277
4.3.1 Prereduction in hydrogen. In one of the first publications usingin situ modification of Pt/Al2O3 catalyst63,278 it was mentioned that thethermal treatment at 400 1C hydrogen has pronounced effect both on theactivity and the enantioselectivity. After thermal treatments in hydrogen at400 1C, 15–20% higher ee values were obtained than over untreatedcatalysts.
In a recent review196 the role of pretreatment was formulated as follows:(i) pretreatment cleans up the surface of the catalyst by removing oxygen aswell as impurities; (ii) residual Pt salts are converted to metallic Pt; (iii) theaverage particle size of Pt increases, (iv) the morphology of Pt particles, i.e.the distribution of exposed face, edge and corner atoms is also alteredfavourably; (v) it promotes adsorbate-induced surface restructuring.
Restructuring during pretreatment of Pt/alumina catalyst used in enan-tioselective hydrogenation of KPL was also studied.279 The influence ofreductive and oxidative heat treatment on the enantioselectivity of chirallymodified Pt/alumina has been reinvestigated. Enhancement in ee by39–49% has been observed after treatment in hydrogen at 250–600 1C, ascompared to untreated or pre-oxidized catalysts. The changes in ee afterreductive and oxidative treatments are reversible, and always the finaltreatment is decisive. A HRTEM study indicates that adsorbate-inducedrestructuring of Pt crystallites during hydrogen treatment at elevated tem-perature can play a role in the selectivity improvement, but the changes aresuperimposed by the strong structure-directing effect of the aluminasupport.
1
5
10
15
20
25
30
35
40
45
Fig. 11 A general scheme for catalyst pretreatment. (Reproduced from ref. 76 with permission)
170 | Catalysis, 2010, 22, 144–278

4.3.2 Influence of oxygen. The effect of oxygen on the performance ofcinchona – Pt catalyst system has been studied by different groups undervarious conditions.65,230 In these studies different solvents, different type ofsupported Pt catalysts and different experimental conditions were used. Forthis reason it is very difficult to make any right conclusion or interpretationrelated to the given observation; is a particular finding a general phenom-enon or an experimental artifact?
In a recent review33 a generalized comment was given, namely the effect ofmodification atmosphere of Pt-CD catalysts affects on the ee values. Forinstance under air higher ee values has been achieved in EtPy hydrogen-ation, whereas under anaerobic condition ee decreases drastically.230,280 Inref.230 it was demonstrated that the addition of oxygen during the enan-tioselective hydrogenation of EtPy has a positive effect both on the rate andthe ee values. The observed effect was attributed to restructuring of thesurface of Pt in the presence of oxygen. There is only one remark with re-spect to these findings, i.e., the final ee value (below 40%) is extremely lowfor the experimental conditions applied.
When anaerobic and aerobic treatments of Pt/SiO2 catalyst was com-pared after anaerobic treatment decreased enantioselectivity and greatlyreduced activity was observed using DHCD as modifier in the hydrogen-ation of MePy.65 It has to be mentioned that this pretreatment was per-formed in ethanol. In further studies it was shown that during this aerobictreatment ethanol was oxidized over platinum catalyst into acetic acid281
and probably the formed AcOH was responsible for the increased per-formance. Similar prove has been obtained in our laboratory.282
Table 3 shows the results obtained in the enantioselective hydrogenationof trifluoroacetophenone (TFAP).76 These results clearly show that eithertreatment in an oxygen atmosphere or stirring in air resulted in a decrease inthe enantioselectivity.
Bartok and coworkers have applied a reductive treatment prior to the useof catalyst, but the catalyst is stored in air before its final use. It was shownthat the increase of the storage time up to one week has no pronouncedeffect on the performance of the catalyst.277 With respect to the role ofoxygen it was also suggested that during this treatment PtO could beformed. During the hydrogenation reaction PtO is reduced to metallic Ptand water. It is not excluded that the presence of small amount of water canresult in some improvement in the performance.76 In the enantioselectivehydrogenation of MePy or butane-2,3-dione over Pt in the presence of CDthe coadsorption of oxygen with the alkaloid resulted in a positive effect
1
5
10
15
20
25
30
35
40
45
Table 3 Influence of catalyst pretreatment in the hydrogenation of TFAP (90mg 5w% Pt/
alumina, 1.28 g TFAP, 10 bar, 20 1C (Reproduced from ref. 76 with permission)
No Pretreatment Solvent (CD) mg Time (min) Conv. (%) ee (%)
1 – toluene 2 63 89 16
2 reductive A toluene 2 50 96 33
3 reductive B toluene 2 86 95 33
4 reductive A 1,2-dichlorobenzene 4 105 95 45
5 oxidative 1,2-dichlorobenzene 4 105 91 29
Catalysis, 2010, 22, 144–278 | 171

both on the rate and ee.280 It was suggested that the presence of a strong co-adsorbate, such as oxygen, the surface was not poisoned by CD. In a recentstudy the promoting effect of helium treatment was also mentioned.283
However, it was admitted that the effect is due to the small oxygen impurityin the helium used.
4.3.4 Use of ultrasound and microwave heating. The effect of ultrasoundradiation was investigated in details by Bartok’s group.55,70,284–286 Themethod appeared to be highly efficient as the ultrasound radiation resultedin and increase both in the reaction rate and the enantioselectivity. Table 4shows representative results using three different substrates and threemodifiers.55 The decrease in metal particle size was given as an explanationof improved performance.284 In another study TFAP was applied as asubstrate and the use of sonication resulted in positive effect.275 The resultsindicated also that both the frequency of ultrasound and the duration ofsonication have a strong influence of the enantioselectivity.55
However, it was also pointed out that the presence of the modifier isabsolutely crucial during sonication. It was proposed that the ultrasonicirradiation created a more effective surface modification, resulting in theformation of surface sites required for optimum enantio-differentiation.Besides it an additional positive effect of oxygen was also observed.275 Itwas suggested that ultrasonic irradiation helps the removal of the impuritiesfrom the Pt surface.196
1
5
10
15
20
25
30
35
40
45
Table 4 Sonochemical and silent enantioselective hydrogenation of a-ketoesters over 5% Pt/
Al2O3 in acetic acid using different cinchona modifiers under 10 bar hydrogen pressure. (Re-
produced from ref. 55. with permission)
Entry Substrate Modifier Catalyst Major product
Optical yield (ee %)
‘‘silent’’ ‘‘sonication’’
1 1 4 E40665 R 85 97
2 1 5 E40665 S 78 83
3 1 6 E40665 R 93 98
4 2 4 E4759 R 88 92
5 2 5 E4759 S 34 57
6 2 6 E4759 R 60 68
7 3 4 E4759 R 79 92
8 3 5 E4759 S 50 92
6 3 6 E4759 R 83 96
Substrates: 1=EtPy; 2-=PhGl, 3=Phenylethyl; Modifiers: 4-=CD, 5=CN; 6=O-Me-CD.
172 | Catalysis, 2010, 22, 144–278

In enantioselective hydrogenation of 1-phenyl-1,2-propanedione over5wt% Pt/SF (silica fiber) catalyst a notable enhancement of reaction rate, eeand rs was observed under ultrasound compared to silent conditions.232 Inmesitylene solvent four-fold increase in the reaction rate was observedunder ultrasound compared to identical silent conditions, while in methylacetate and in toluene the rate enhancement was only minor. Upon usingPt/SF catalyst it was suggested that surface smoothening and cleaning takeplace under ultrasound irradiation. However, no significant differences in Ptparticle size distribution between sonic and silent treated catalysts wereobserved by TEM.
Summing up results related to the use of ultrasound its positive effect hasbeen ascribed to the following facts:55 (i) ‘‘through the decrease in metalparticle size, the platinum dispersion becomes close to optimal using acatalyst of large metal particle size,’’ (ii) ‘‘the surface density of the modifierincreases as a result of insonation, providing more chiral sites for the hy-drogenation and, in parallel, suppresses the background reaction, i.e. ra-cemic hydrogenation’’.
Enantioselective and racemic hydrogenation of EtPy over Pt/Al2O3
catalyst was investigated under microwave dielectric and conventionalheating.287 A homemade laboratory microwave loop reactor was appliedallowing differentiating between dielectric and conventional heating. Theeffects of polar and non-polar solvents on enantioselective hydrogenation ofEtPy were studied in toluene and ethyl alcohol. In case of toluene, which ismicrowave transparent, no significant differences in the reaction rate andenantioselectivity were observed between dielectric and conventional heat-ing. In case of EtOH, the reaction rate remained unaffected. However, the eedramatically decreased from 60 to 40% under microwave heating. No sig-nificant improvement of the reaction rate with an increasing microwavepower input was observed. The authors suggested that this observation beprobably caused by the local superheating of the polar EtOH in the cavity,which is not possible in the non-polar toluene. Our explanation is different;namely the decreased ee is due to the formation of semi-ketal from thesubstrate and the alcohol used as solvent.
4.4 Premodification of the catalyst with the alkaloid
As it has already been discussed earlier that in Orito’s pioneering studies apremodification procedure was used to introduce the chiral modifier intothe Pt/C catalyst pretreated in hydrogen at 400 1C.40 It has to be emphasizedthat the premodification was performed at higher temperature than that ofthe hydrogenation reaction. During premodification the catalyst and thecinchona alkaloid has been stirred in a given solvent for a relatively longperiod (24 hours). This premodification procedure strongly resembled themodification process used for Ni/tartaric acid catalysts developed earlier.288
The premodification was followed by filtration and mild washing and thepremodified catalyst was introduced into the reactor containing the solventand the substrate.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 173

The Orito’s original approach was followed by P.B. Wells’ group.65,229,280
However, there were several drawbacks in the use of this method. Furtherresults indicated if the filtration and the washing is not carefully performedsolvated CD can be left in the pores of the catalyst support, for this reason,as it was pointed out in one of the comments,69 the exact amount of alkaloidintroduced was not really known. Probably it was the reason that in ref.289the authors admitted the ‘‘erratic variation of the initial rates’’. After suc-cessful introduction of the in situ methods57 and demonstration of the ad-vantage of premixing technique64 the premodification method was almostentirely forgotten. This approach is still used when the enantioselectivehydrogenation is investigated in the gas phase.290
4.5 In situ modification
4.5.1 General Information. In situ modification of the catalysts prior tothe hydrogenation of activated ketones can experimentally be performed indifferent way. These various approaches provides different surface coverageat t=0, i.e., prior to the start of the hydrogenation reaction.
Let us consider that the Pt catalyst is pre-reduced in hydrogen around400 1C and kept in an inert atmosphere prior to its use. The catalyst in thisform is introduced into the reactor containing the following components: (i)solvent, catalyst, substrate and modifier (premixing method); (ii) solvent,catalyst, substrate (injection of the modifier); (iii) solvent, catalyst, modifier(injection of the substrate). It is easy to propose that the above threemethods shall provide completely different surface coverage at t=0, whatcan have different influence both on the kinetics and ED.
4.5.2 Premixing technique. This method has been applied first by H.U.Blaser’s group.63 In this technique all components of the reaction are pre-mixed prior to the hydrogenation reaction. This method provides highcoverage of substrate and a relatively low coverage of modifier at the Pt site.This new approach resulted in the discovery of the ‘‘ligand acceleration’’phenomena.58 The rate increase was very pronounced and was proportionalto the amount of alkaloid used. This phenomenon will be discussed inChapter 5.6.1.
During premixing the substrate can decompose resulting in carbonmonoxide, which is considered as a strong catalyst poison. The substrateinteracts also with the modifier and induces the formation of high-molecularweight byproducts (see Chapter 5.1). These by-products have a negativeinfluence on the initial rate by their poisoning effects. If the amount ofhydrogen in the overall hydrogen pool is high (i.e., when the catalyst iscooled in the hydrogen atmosphere from the temperature of re-reduction) inthis case partial hydrogenation of both the cinchona alkaloid and thesubstrate can also take place prior to the introduction of hydrogen.
Consequently, upon using the premixing technique a new problem ap-peared, i.e. the reproducibility of the reaction rate. It was observed that therate of reaction depended on the duration of premixing, i.e. the time re-quired to close the high-pressure autoclave, purge the reactor with nitrogenand hydrogen, and pressurize the reactor and start stirring.64 Reliable andreproducible rate could only be obtained when the duration of premixing
1
5
10
15
20
25
30
35
40
45
174 | Catalysis, 2010, 22, 144–278

was kept constant.64 This problem is even more pronounced when alcoholshave been used as solvents.
4.5.3 Injection technique
Injection of the modifier. This method excludes all undesired interactionsbetween the substrate and modifier. It provides high coverage of the sub-strate at the Pt site. However, if the catalyst is not cooled in an inert at-mosphere racemic hydrogenation of the substrate can also take place beforestarting the reaction (i.e. before t=0). Neither the spontaneous oligomer-ization/condensation of the substrate or its decomposition into CO can beexcluded in this case.
Injection of the substrate. This method excludes also all undesired inter-actions between the substrate and modifier. It provides a definite surfaceconcentration of the modifier. No oligomers or condensation products existat t=0. However, partial hydrogenation of the modifier cannot be excludedif the catalyst has been cooled in a hydrogen atmosphere.
The injection method gives an opportunity to get reliable kinetic dataprovided a proper ratio between the batch and the injected volumes ischosen. In this way the influence of some of the undesired side reaction canbe eliminated, providing more chance to get intrinsic kinetic data. In thisapproach either the modifier or the substrate is injected by high-pressurehydrogen.69,93,196,291
4.6 Hydrogenation of a-keto esters in continuous-flow reactors
It is obvious that the separation, handling and reuse of the heterogeneouscatalysts become very efficient when the fixed-bed reactor is used; con-sequently it is very promising to introduce fixed bed reactors with the aim toindustrialize the asymmetric catalysis. However, up to the late nineties therewere only scarce data related to the use of continuous-flow reactors inasymmetric hydrogenation reactions.
As far as only trace amount of modifier is required to induce high ee anattempt was done to use a continuous fixed-bed reactor for the enantiose-lective hydrogenation a KPL.292 This approach resulted in significant pro-cess intensification; consequently upon using a small tubular reactor (size ofa pencil) more than 14 kg (R)-pantolactone per hour could be produced.Later on the approach was extended to use for other substrates.293–295
High reaction rates and high ee values were obtained by continuousfeeding of minute amounts of chiral modifier to the reactant stream. The eevalues for KPL and EtPy without optimization was 83.4 and 89.9%, re-spectively. Transient measurements by stopping of the flow of CD indicatethat continuous feeding of the modifier in ppm concentration is crucial.There was a short induction period prior reaching stable high ee values.
Knitted Pt/SiO2 was used in enantioselective hydrogenation of 1-phenyl-1,2-propanedione giving relatively high enantiomeric excesses.296 Theknitted silica fiber catalyst gave encouraging results in the continuous fixedbed operation with enantiomeric excesses comparable to those obtained inthe batch reactor.
EtPy was hydrogenated in a continuous-flow fixed-bed reactor and highee value up to 89% was obtained at modifier/substrate molar ratio of only
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 175

307 ppm.297 In another study298 the enantioselective hydrogenation of ethyl-2-oxo-4-phenylbutyrate (EOPB) on Pt/g-Al2O3 catalyst in the presence ofCD was investigated in a fixed-bed reactor with the aim to synthesizeenantiomerically pure (R)-(þ )-EHPB, a building block for the synthesisof several commercially important A.C.E. inhibitors. The highest ee valuearound 69% was obtained in toluene at 6MPa hydrogen pressure. Al-though stable conversion values were obtained in time on stream experi-ments, the ee values decreased in time.
The transformation of isopropyl-4,4,4-trifluoroacetoacetate to the cor-responding hydroxyester was studied in a fixed bed reactor over Pt/Al2O3 inthe presence of MeO-CD and trifluoroacetic acid.299 Around 0 1C and uponvarying the pressure, the total liquid flow rate and the feed composition eevalues up to around 90% were achieved. However, the time on streampattern showed quite notable activity decrease.
Enantioselective hydrogenation of EtPy was performed in a continuous-flow fixed-bed reactor using supercritical carbon dioxide and supercriticalethane (scC2H6).
300 In the latter solvent much higher catalytic activity wasobserved.
Ethyl benzoylformate (EBF) was hydrogenated in a continuous-flowfixed-bed reactor over Pt/Al2O3 catalyst in the presence of CD and CN.301
Variety of chemical and physico-chemical methods was applied to pretreator clean the chiral fixed bed between multiple hydrogenation reactions. Itwas observed that after an enantioselective hydrogenation with CD asmodifier at 0 1C, the continuous-flow reactor could be effectively cleaned at0 1C, and that a racemic unmodified hydrogenation could be performedthereafter. This implies the effective desorption of chiral species from thesurface during cleaning. Contrary to that cleaning of the reactor at 50 1Cresulted in a reproducible unmodified enantioselective hydrogenation, with amarked inversion of enantiomeric excess. The inversion of ee will be dis-cussed in Chapter 5.6.4.
In a recent study enantioselective hydrogenation of EtPy was also per-formed in continuous-flow reactor in the presence of CD over Pt/Al2O3.
302
All these results indicate that the use of continuous-flow reactors shouldbe applicable to different substrates based on the use of chiral modifiers andsupported metal hydrogenation catalysts. This approach provides moreefficient screening method for potential chiral modifiers establishing thebasis for future technical applications. Based on the use of continuous-flowreactors the so called ‘‘chiral switch’’ methodology has been developed forthe investigation of the relative adsorption strength or the competition ofchiral modifiers on a metal surface.239,303
4.7 Reuse and deactivation of catalysts
The reuse of the catalyst has a great practical significance. The reuse isstrongly connected to the catalyst deactivation phenomena. Deactivation willalso be discussed in Chapter 5.2. Due to catalyst deactivation for the reuse ofsupported Pt catalysts fresh modifier has to be added before each hydrogen-ation cycle,304,305 or the modifier is fed permanently in continuous man-ner108,292 to ensure good activity and high enantioselectivity.
1
5
10
15
20
25
30
35
40
45
176 | Catalysis, 2010, 22, 144–278

Typical setup for these experiments is as follows: after the first reaction‘‘the mixing was stopped, the reaction mixture was left to settle for30min, the liquid was removed, and solvent, EtPy and occasionally modifierwere added to the reactor. Hydrogenation was repeated as describedabove’’.270
An intelligent approach has been developed for the reuse of hydrogen-ation catalyst.306 Magnetic Pt/SiO2/Fe3O4 catalyst was prepared and suc-cessfully applied for the enantioselective hydrogenation of various activatedketones. This catalyst modified with CD showed catalytic performance(activity, enantioselectivity) in toluene comparable to the best-known Pt/alumina catalyst. The new catalyst can be easily separated by an externalmagnetic field and recycled several times with almost complete retention ofactivity and enantioselectivity.
Figs. 12A and B show two types of reuse experiments. In Fig. 12A freshmodifier has been added in each run, consequently the ee values are con-stant; however there is a significant catalyst deactivation. However, asshown in Fig. 12B both the activity and ee decrease on reuse of the catalystif no fresh modifier is added to the reaction mixture at the beginning of eachnew run. It is known from other studies248,250,263 that almost constant eevalues can be achieved in reuse experiments, where ‘‘fresh’’ modifier isadded in every reuse.
Interesting observation was described in ref.307. Stopping the enantio-selective hydrogenation of EtPy at a conversion of approximately 70%,long-term stability of the catalyst can be achieved. During 10 cycles ofhydrogenation, the activity and enantioselectivity of the repeatedly usedcatalyst remain constant at high values even without adding fresh modifierat the beginning of each new run. These observations indicate that the lossof modifier takes place only at high conversion, i.e., the presence of excess ofsubstrate prevents the hydrogenation of the quinoline ring of the modifier.These results are shown in Fig. 13.
1
5
10
15
20
25
30
35
40
45
Fig. 12 Repeated use of catalyst for the enantioselective hydrogenation of EtPy. A: 5ml ofroluene, [DHCD]=0.1mmol/l, fresh DHCD was added each time to the reaction solution. B:5ml of toluene, [DHCD]=0.01mmol/l, no DHCD was added for reuse of catalyst. (Repro-duced from ref. 270 with permission)
Catalysis, 2010, 22, 144–278 | 177

Different reuse methods and influence of the solvent were investigated byBartok et al.270 In the course of enantioselective hydrogenation of EtPy intoluene, ee increased with catalyst reuse. The increase was in the range of10–20%, however this increase was not observed in acetic acid. The authorsconsidered that the phenomenon of ‘‘increase in ee on reuse’’ is an intrinsicfeature of the catalyst system used, i.e. new chiral centers making higher eepossible are formed. It was suggested that during reuse due to the intensiveinteraction of a solid surface and the chiral modifier reconstruction ofthe Pt surface takes place. This is in a good agreement with recent find-ings308 that during reuse due to the intensive interaction of a solid surfaceand the chiral modifier reconstruction of the Pt surface takes place. Theauthors believe that ‘‘the surface atoms of the catalyst are continuouslyreorganized during the reaction as a result of adsorption/chemisorptionsteps. In this respect it was supposed that the presence of trace amounts ofoxygen might also play an important role in the reaction studied. The lackof increase in ee on reuse in acetic acid may indicate that in this solvent thereaction mechanism is different. The authors most important conclusionswith respect to the reuse of catalyst are as follows: ‘‘(i) Pt/Al2O3 catalystswith a Pt-dispersion of 0.2–0.3 and a mean Pt particle size of 3–5 nm are thebest; (ii) prior to use, the catalyst should be prereduced at 673K for 1–1.5 hin hydrogen flow; (iii) it is necessary to add fresh modifier for each reuse ofthe catalyst’’.270
5. Specificity of Orito’s reaction
As it has already been discussed that enantioselective hydrogenation ofactivated ketones has several specificities. The main specificities are as fol-lows: (i) side reactions, (ii) catalyst deactivation, (iii) solvent effect, (iv)substrate specificity, (v) rate acceleration (enhancement), (vi) enantioselec-tivity–conversion (time) dependencies, (vii) non-linear phenomenon, and(viii) inversion of enantioselectivity. In order to understand all peculiaritiesof these unique reactions the specificities have to be discussed separately.
1
5
10
15
20
25
30
35
40
45
Fig. 13 Activity and ee values of the modified catalyst used repeatedly for the stopping aftercomplete conversion; right: stopping at 70% conversion) without addition of fresh modifier.(Reproduced from ref. 307 with permission)
178 | Catalysis, 2010, 22, 144–278

5.1 Side reactions
The intrinsic reactivity of activated ketones is high. In this respect differenttype of side reactions, such as (i) semi-ketal formation, (ii) oligomerization(condensation, polymerization), (iii) hydrolysis, (iv) transesterification, (v)deuterium exchange, and (vi) decarbonylation, can be distinguished.
Side reactions have a great influence both on the rate and enantioselec-tivity. The occurrence of side reactions depends on the mode of introductionof the reaction components. The by-product formation can take place bothin racemic and enantioselective hydrogenation of activated ketones. In mostof the side reactions the substrates are involved. With respect to the modifierthe hydrogenation of the quinoline ring has to be mentioned.273
The first thorough information related to the side reactions and by-products formation was given in ref. 309 The formation of semi-ketals cantake place between the substrate and alcoholic solvent,281 the substrate andthe reaction product,234 the substrate and CD.109 It has been demonstratedin ref. 281 that CD catalyses the formation of semi-ketals. The extent ofsemi-ketal formation increases in the following order: t-BuOHoi-PrO-HoEtOHoMeOH. Semi-ketals can be hydrogenolized to the corres-ponding alcohols in a racemic reaction, i.e. this side reaction stronglydecreases to overall ee of the enantioselective hydrogenation.
Oligomerization and polymerization reactions have been discussed bydifferent authors.109,310,311 In these reactions both Pt and alumina sites canbe involved. A dimer of the substrate is formed in the condensation reactionof the enol and keto forms of EtPy.234 The keto-enol transformation ofMePy has been recently investigated by using thermal programmed de-sorption (TPD), STM and reflectance adsorbance infrared spectroscopy(RAIRS).312 It was shown that MePy undergoes CH bond scission at roomtemperature on clean Pt(111) leading to surface mediated enol formationand assembly into H-bonded superstructure. The latter was severely in-hibited by addition of hydrogen (10�6 Torr). STM data show no evidencefor an irreversible polymerization reaction.
In a recent study313 it was suggested that base-acid sites on the g-Al2O3
surface are responsible for the aldol reaction of EtPy to yield b-hydroxylketone, which is subsequently dehydrated to generate CQC containingspecies (see Scheme 5.1). The formed condensation product can be involvedin cyclization reactions as shown in Scheme 5.2. These cyclic products wereconsidered as one of the key compounds poisoning the catalyst during thehydrogenation of EtPy.81 In ref.313 side reactions with the involvement ofPt sites were also investigated. Scheme 5.3 shows these reactions and therole of hydrogen in their suppression.
The fact that the aldol condensation of EtPy on Al2O3 can be suppressedby adsorbed acetic acid may be interpreted as that the acetic acid adsorbsand blocks some basic sites on alumina.311 An additional set of side re-actions was discussed in ref.314. It was suggested that these reactions mighttake place over the Pt sites resulting in strong poisoning effect.314
Several adducts originating from the base-catalysed EtPy conden-sation were detected by ESI-MS method.315 Decarbonylation of both linear(EtPy, MePy)82,91 and cyclic a-ketoesters (KPL)316 was evidenced by using
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 179

ATR-FTIR method. It has been shown that both hydrogen and CD sup-presses the decomposition of EtPy taking place on Pt surface.91 The sta-bilization of MePy against decomposition has been suppressed bybenzene317 and 1-1-naphthyl)ethylamine.318
1
5
10
15
20
25
30
35
40
45
Scheme 1 Proposed mechanism for aldol condensation of EtPy catalyzed by the base-acidsites on the g-Al2O3 surface. (Reproduced from ref. 313 with permission)
O
O
OH
O
O
OO
O
OH
OH
O
O
OO
OO
O
O
O
OHO
O
-EtOH
2a 2b
1a 1b
Scheme 2 Further transformation of adduct formed in the condensation reaction of the ketoand the enol forms of EtPy. (Reproduced from ref 81 with permission)
180 | Catalysis, 2010, 22, 144–278

In a recent study it was shown that although adsorbed CD suppresses thedecomposition of EtPy it cannot suppress condensation and hydrolysis ofEtPy on g-alumina. Coexistence of CD and hydrogen is needed to suppressall side reaction of EtPy over Pt/g-Al2O3
313.Recently it has been suggested that the polymerization of EtPy takes
place preferentially at steps sites of Pt and CD or other tertiary aminesinhibits propagation of polymerization over Pt sites.234
It is known that most of the products of the above side reactions areconsidered as catalyst poison, consequently their presence significantly alterthe intrinsic kinetic patterns of these reactions. All these facts strongly in-dicate that experimental conditions have to be strictly standardized in orderto minimize the effect of by-products formed. The role of by-products to therate acceleration phenomena will be discussed in Chapter 5.6.1.
5.2 Catalyst deactivation
In the enantioselective hydrogenation of activated ketones catalyst de-activation takes place both in batch and continuous-flow reactors. Thedeactivation can be attributed to both of chemical and physical processes.
1
5
10
15
20
25
30
35
40
45
Scheme 3 Proposed mechanism for the side reactions of EtPy on Pt/Al2O3 inhibited by H2.(Reproduced from ref. 313 with permission).
Catalysis, 2010, 22, 144–278 | 181

These processes can be explained as follows: (i) poisoning, (ii) coking, (iii)sintering, (iv) restructuring and (v) phase transformation. Deactivation isinevitable, but it can be retarded or prevented and some of its consequencescan be avoided with clever process design.232 One of the main reasons forcatalyst deactivation can be ascribed to the formation of by-products asdescribed in the previous Chapter.
Catalyst deactivation cannot be directly investigated in a batch reactor;however the analysis of the time on stream behavior in continuous-flowreactors provides useful information with respect to catalyst deactivation.
It has been shown that that irreversible deactivation of Pt catalysts bycondensation and oligomerization polymerization can be related tohydrogen starvation.314 In a recent study313 it was found that only the co-existence of CD and H2 could thoroughly inhibit the side reactions of EtPyon Pt/g-Al2O3.
The decrease of the catalytic activity due to the use of unpurified sub-strates has been discussed in different studies.66,69,82,319 Consequently,purification of the reactant seems to be a necessary prerequisite to avoidcatalyst deactivation.320 With respect to catalyst deactivation the disputerelated to the origin of rate acceleration has to be mentioned.235,297 Furtherdiscussion of this dispute will be given in Chapter 5.6.1.
Catalyst deactivation has been observed in kinetic experiments performedin batch reactor.234,291 Deactivation was also evidenced in continuous-flowregime using various experimental designs.301 It was also observed onheating the platinum catalyst in either hydrogen or helium at 350 1C for twohours and then using it without exposure to oxygen.230
General principles of catalyst deactivation both in batch and continuousflow reactors were given in ref.321. In this work enantioselective hydro-genation of 1-phenyl-1,2-propanedione was studied. An elegant way topromote catalyst durability, activity and selectivity is to apply on-lineacoustic irradiation during the course of reaction.322 Ultrasound can retardcatalyst deactivation by (catalyst) surface cleaning and exposing fresh,highly active surface as well as by the reduction of diffusion length in thecatalyst pores by alteration of the surface of catalyst. Furthermore, stronglyabsorbed organic impurities that block active sites can also be removed bysonification. In ref. 232 the deactivation was studied both under con-ventional and microwave heating using EtPy as a substrate. No catalystdeactivation was observed in three consecutive experiments with re-usedcatalyst. Previously, it has been reported107,323 that during continuous hy-drogenation of EtPy and 1-phenyl-1,2-propanedione; a notable catalystdeactivation takes place.
Based on these results it can be concluded that in the enantioselectivehydrogenation of activated ketones deactivation is an inevitable phenom-enon. However, there are measures to decrease the extent of deactivation.These measures are as follows:
(i) application of acetic acid as solvent, which deactivates the aminemodifier and the alumina support for aldol reaction;297 (ii) decreasing themodifier/substrate ratio to reduce the rate of side reactions in solution; (iii)working at high surface hydrogen concentrations, that is, at high hydrogenpressure and in the absence of mass transport limitation; and (iv) carefully
1
5
10
15
20
25
30
35
40
45
182 | Catalysis, 2010, 22, 144–278

avoiding the contact of Pt with pyruvate in the absence of modifier at lowsurface hydrogen concentrations, and (v) minimizing the contact of thesubstrate and the modifier before entering them into the reactor.
5.3 Substrate specificity
It is known that various prochiral ketones can enantioselectively be hy-drogenated over Pt-cinchona catalyst system. This issue has been discussedin various earlier reviews.33,46,52,196,324 Characteristic feature of these sub-strates is the presence of an activating group with strong electron-with-drawing properties (ester, carbonyl, acetal, amido and a,a,a-trifluoro group)in the a-position to the prochiral keto group.76 As most of the substratesapplied so far were extensively discussed in earlier reviews in this chapter weshall give only a brief summary.
The first substrates successfully hydrogenated asymmetrically were linear and cyclic a-ketoesters,48,84,284,292,325 a-diketones,320,326–329 a-keto-acetals,330,331 aromatic a,a,a-trifluoro-ketones,286,332–335 and linear andcyclic a-ketoamides.336–338 The structure of these activated ketones is givenin Fig. 14.
Later on the studies were extended to non-aromatic a,a,a-trifluoro-ketones236 and substituted aromatic a,a,a-trifluoroketones,53,335,339 trifluorosubstituted a, b-ketones,44,340 b-ketoesters.341 Pt-cinchona system has alsobeen used in the hydrogenation of substituted deactivated aromatic ketones,such as 3,5-bis(trifluoromethyl) acetophenone.342
The types of substrates containing activated keto group and the highest eevalues in the presence of optimum modifier are shown in Table 5.5.150 Mostof the substrates, with the exception of the amido derivatives resulted in eevalues above 80%.
Additional results related to the influence of substituents in a-keto esterswere published in Refs. 340,343. In these studies both R1 and R2 substitu-ents (see Scheme 5.4) were systematically altered.
The Bartok’s group found that in AcOH when R1 was methyl, iso-propyl,terc.-butyl, phenyl and phenylethyl or the R2 group was methyl, ethyl andisopropyl the sense of the enantio-differentiation was not altered. The in-crease of the size of R1 and R2 resulted in slight decrease in the reaction rateand ee values.
Additional results presented by Baiker’s group are summarized inTable 6.340 In this series a of experiments the variation of the bulkiness bothat the keto and ester sides in nine different a-ketoesters was investigatedupon using CD and QN, as chiral modifiers. In toluene in the presence ofCD good to high ee values (eemax=94%) were achieved. Consequently, inthe presence of CD the enantio- differentiation is controlled by the estergroup, notwithstanding of the steric bulkiness or electronic structure of the
1
5
10
15
20
25
30
35
40
45
O
O
O
O
O
O
O
NH
O
O
R CF3
O
O
O
O
Fig. 14 Structure of first substrates successfully hydrogenated asymmetrically.
Catalysis, 2010, 22, 144–278 | 183

Table
5Hydrogenationofvariousactivatedketones
over
Pt/Al 2O
3-cinchonacatalysts.(R
eproducedfrom
ref.50withpermission)
No
Substrates
Modifier
Solvent
Substrate-M
odi-
fier
ratioa
Substrate-Ptratiob
ee(%
)Ref.
1O
O
O
QD
AcO
H1540
1640
98c
346
2Ethylbenzoylform
ate
CD
AcO
HþToluene
868
300
98
56
3
OH
O
O
MeO
CD
EtO
H/H
2O
350
440
82
347
4
O
O
OCD
Toluene
296000
1040
91d
325
5O
O
NHCD
AcO
H300
160
58
336
6butanedione
CD
Toluene
40
2100
63
328
7O
O
CD
EtO
Ac
143
66
94e
348
8
N
O
OCD
Toluene
320
210
91
349
184 | Catalysis, 2010, 22, 144–278

Table
5(C
ontinued
)
No
Substrates
Modifier
Solvent
Substrate-M
odi-
fier
ratioa
Substrate-Ptratiob
ee(%
)Ref.
9
OE
t
O
O
ODHCD
Toluene
720
710
86
350
10
O
OR
OR
MeO
CD
AcO
H1050
1320
97
332
CD
AcO
H130
180
97
330
11
CF 3
O
O
OMeO
CD
THF-TFA
290
180
96f
50
12
O
CF 3
CD
Toluene-TFA
290
180
92
334
asubstrate/m
odifier
molarratio.bsubstrate/Ptmolarratio.creaction
mixture
(withoutsubstrate)ultrasonic
treatm
ent.
d�81C.ekinetic
resolution.f�201C,CD:
cinchonidine,
DHCD:10,11-dihydrocinchonidine,
MeO
HCD:methoxy-H
CD,MeO
CD:methoxy-C
D,THF:tetrahydrofurane,
TFA:trifluoroaceticacid.
Catalysis, 2010, 22, 144–278 | 185

alkyl and functionalized aryl group on the other side of the keto group.Other studies reveal also that ester, carboxyl, amido, carbonyl, acetal, andtrifluoromethyl functions have similar directing effects. Results presented inTable 6 show that structurally more demanding substrates show significantdifferences between toluene and acetic acid. However, none of the mech-anistic models developed for the enantioselective hydrogenation of acti-vated ketones over Pt-cinchona catalyst system can explain the followinganomalies found in Table 6: (i) higher ee values in toluene in the presence ofCD for substrates (2), (4), (5) and (9); (ii) the strong increase of ee in aceticacid in the presence of QN for substrate (1); (iii) almost complete loss of eein AcOH in the presence of QN for substrates (3) and (9).
In the presence of CD the greatest drop both in the conversion and eevalues was measured for the hydrogenation of 9 (see Table 6) when chan-ging from toluene to AcOH. This observation was attributed by the authors‘‘to the strong, competing adsorption of AcOH’’. It seems to us that it is avery plausible explanation. We consider this observation as an artifact, as inour independent experiment no similar observation was found.344
There are additional controversial data with respect to the use of AcOH.In ref. 340 based on a relatively early study345 it has been mentioned that theenantioselectivities in toluene and acetic acid are similar.53 This statement isnot really correct as in various other studies73 it has been shown that inacetic acid both the rate and ee values are higher than in toluene. It hasalready been shown that the addition of a small amount of acetic acid eitherto toluene or ethanol has a very pronounced effect.84
Derivatives of trifluoroacetophenone with different substituents at thearomatic ring (CF3, N(Me)2 and Me) were hydrogenated over Pt/Al2O3 inthe presence of CD, CDXHCl and 9–O-methyl-CD.334 It was shown thatelectron-withdrawing substituents increased and electron-releasing one de-creased the rate and enantioselectivity in these reactions, although stericeffects (with m- or p-substituents) were also substantial. Cinchona alkaloidswere also used in the asymmetric hydrogenation of non-activated ketones.In this case the enantioselectivity is rather moderate as it was emphasized ina recent review.196
Upon using various nonactivated trifluoromethyl ketones, such as me-thyl-, adamantyl, and terc-butyl236 in the presence of CD low ee values wereobtained (eemax=44% for adamantyl derivatives). Positive effect of TFAwas also demonstrated, while the use of AcOH as a solvent resulted in lowyields and low ee values. In propanol inversion of the ee was observed.These results confirmed again that in the hydrogenation of non-activatedketones high ee values couldn’t be expected. Unfortunately, the initial rateswere not determined in this study. The authors claimed that their result‘‘indicates that enantioselectivity is guided by the trifluoromethyl
1
5
10
15
20
25
30
35
40
45
Scheme 4 R1 and R2 substituents systematically altered in the hydrogenation of a-keto esters.(Reproduced from refs. 340 and 343 with permission)
186 | Catalysis, 2010, 22, 144–278

substitution rather than by the relative bulkiness of the substituents at thetwo sites of the carbonyl group.
5.4 Solvent effect
In enantioselective hydrogenations of activated ketones both the rates andthe enantioselectivities are greatly influenced by the type of solvents used.
1
5
10
15
20
25
30
35
40
45
Table 6 Enantioselective hydrogenation of various a-ketoesters in toluene and acetic acid
(Reproduced from ref. 340 with permission)
Substrate Reaction in toluene Reaction in AcOH
R1O
R2
O
O
CD
ee(%)
[conv.]
QN
ee(%)
[conv.]
CD
ee(%)
[conv.]
QN
ee(%)
[conv.]
1 –CH3 –CH2CH3 80 (R) 21 (R) 88 (R) 92 (R)
[100] [100] [100] [100]
2 H3C
H3C CH3
–CH2CH3 56 (R) 12 (R) 27 (R) 2 (R)
[91] [61] [99] [99]
3 –CH2CH3 86 (R) 79 (R) 86 (R) 4 (R)
[100] [100] [100] [100]
4 H3C
H3C CH3
95 (R) 89 (R) 70 (R) 24 (R)
[94] [43] [73] [64]
5 –CH2CH3 92 (R) 75 (R) 80 (R) 31 (R)
[100] [197] [100] [100]
6
F
F –CH2CH3 87 (R) 76 (R) 86 (R) 76 (R)
[100] [99] [100] [100]
7 F3C
CF3
–CH2CH3 66 (R) 47 (R) 72 (R) 72 (R)
[94] [90] [98] [98]
8 O
O
–CH2CH3 94 (R) 84 (R) 94 (R) 84 (R)
[95] [100] [94] [91]
9 –CH2CH3 86 (R) 60 (R) 46 (R) 0 (R)
[100] [32] [31] [7]
Catalysis, 2010, 22, 144–278 | 187

Both protonic and aprotonic solvents have been applied. The solvent caninfluence the enantioselective hydrogenation in different ways; it changesthe solubility of the hydrogen99 and the substrate, the mass transportproperties of the reaction mixture, the adsorption behavior of substratesand modifier on the Pt active sites.
Solvents have also a great influence on the conformation of alkaloidused.88,183,184 Furthermore, with less rigid substrates, e.g. alkyl pyruvates,the solvent polarity can also affect the conformation of the substrate.49
Unfortunately, there are no general rules for the selection of an optimumsolvent as both the rate and ee values were affected not only by the solvent,but also by the substrate and the alkaloid applied.
In the first studies ethanol was the most common solvent used; howeveras it was shown latter the alcohols react with a-ketoester with the formationof semi-ketals (see Chapter 5.1). This effect was also discussed in a recentstudy related to the enantioselective hydrogenation of ethyl-4,4,4-tri-fluoroacetoacetate351 in ethanol and propanol.
All these results indicate that the use of alcohols in kinetic or mechanisticinvestigations65,249,352,353 should be avoided as it was emphasized in ref. 53.Upon using O-alkylated CD (R-O-CD) derivatives the use of AcOH orTFA is not recommended as in their presence hydrolysis of R-OCD cantake place. AcOH was not a proper solvent for KPL.77
In one of the first studies the reaction rates and enantioselectivities werecompared in ethanol, toluene and acetic acid.73,73 These results un-ambiguously show the advantage of using AcOH. In another studies it wasshown that upon hydrogenating EtPy AcOH is the best solvent as thehighest ee values (ee=98%)55 and highest reaction rates were obtained inthis solvent.
The influence of the two most commonly solvents, i.e., toluene and aceticacid, on the rates and ee in enantioselective hydrogenation of EtPy areshown in Table 7. These data clearly show the superior influence of AcOHon the ee values. Not only higher ee values were obtained in AcOH, but alsothe amount of modifier required to get high ee values is one order less inAcOH than in toluene. However, it is interesting to note that contrary toearlier observations at atmospheric hydrogen pressure the rate of hydro-genation in toluene is higher than in AcOH. In this respect it is worth for
1
5
10
15
20
25
30
35
40
45
Table 7 Hydrogenation of EtPy at atmospheric and at high pressure. Comparison of reaction
rates and ee values in toluene and acetic acid solvents. (Reproduced from refs. 269, 354 with
permission)
Modifier,
Mmol/l
Hydrogenation in
AcOH at 100
bar354Hydrogenation in
AcOH at 1 bar269
Hydrogenation in
toluene at 100
bar354Hydrogenation in
toluene at 1 bar269
rate, mmol/
sec ee, %
rate, mmol/
sec ee, %
rate, mmol/
sec ee, %
rate, mmol/
sec ee, %
0.001 35 83 16 69 8 35 35 16
0.01 80 92 55 91 20 75 57 64
0.1 105 94 85 92 48 82 96 78
1 135 94 63 92 25 83 71 77
188 | Catalysis, 2010, 22, 144–278

mentioning that in racemic hydrogenation of EtPy higher rates weremeasure in toluene than in AcOH.344
Other organic acids were also used to improve the enantioselectivity in Pt-cinchona system. Fig. 15 shows the influence of added trifluoroacetic acid(TFA) on the enantioselectivity in the hydrogenation of KPL in the pres-ence of synthetic modifier (R,R)-PNEA. The results showed that excessTFA was needed to get maximum ee values. It was suggested that part ofthe excess TFA be required to neutralize basic sites of the Al2O3 support.
Excellent correlation was found between the dielectric constant of thesolvents used and both the enantiomeric excess and the population ofconformer Open(3) as calculated by density functional theory in combin-ation with a reaction field model (POpen(3)).
88 This dependence is shown inFig. 16. Earlier results indicated that both reaction rates and ee values de-creased with the polarity of the solvent. Figs. 17A–C show the dependenceof ee on the empirical solvent parameter EN
T in three different systems. Inthe hydrogenation of EtPy (see Fig. 17A) good ee values are obtained inmoderately apolar solvents, in which the reactant and modifier dissolve.46
Interestingly, primary alcohols are also good solvent, although they reactrapidly with the substrate with the formation of corresponding semi-ketals.The highest ee that time was 95% obtained in acetic acid, while the lowestone in water. The former result was attributed to the protonation of thequinuclidine nitrogen of CD by AcOH and suggesting the alteration of thereaction mechanism in AcOH,283,283 while the low activity in water can beattributed to the side reaction between EtPy and water, i.e. to the formationof corresponding vicinal diol, and the racemic hydrogenolysis of the diolformed. The latter reaction is responsible for the loss of ee. Fig. 17A showsthat the solvent influence is notable, although the slope in this figure isrelatively moderate. Contrary to that in Rh/Al2O3-b-ICN system used in thehydrogenation of KPL the above slope is higher indicating that the solventhas more pronounced influence on the enantioselectivity (see Fig. 17B).342
1
5
10
15
20
25
30
35
40
45
Fig. 15 Enantioselective hydrogenation of KPL over Pt/Al2O3 in the presence of syntheticmodifier (R,R)-PNEA. The solvent was toluene with increasing amount of TFA. (Reproducedfrom ref. 210 with permission)
Catalysis, 2010, 22, 144–278 | 189

The third system behaves in a completely different way. The correlationbetween ee and EN
T is not really good, however it is more interesting that thecharacter of dependence is opposite compared to the systems given inFigs. 17A and B. Nevertheless, the striking effect of solvent properties onthe ee values is obvious. The highest ee to (S)-3,5-di(trifluoromethyl)phenylethanol was obtained in the weakly polar solvent toluene and ethylacetate. The ee decreased in polar solvents. In dimethylformamide, iso-propanol, and ethanol the ee inverted from the (S) to the (R) enantiomer.This behavior strongly indicates that in case of substituted acetophenonesthe reaction mechanism is strongly altered.
There is another unusual behaviour of this type of substrates. In theenantioselective hydrogenation of 3,5-bis(trifluoromethyl)acetophenone theaddition of trifluoroacetic acid (TFA) resulted in strong decrease in thereaction rate at TFA/CD=around 5 and full inversion of ee at TFA/CDW50.342
Among the solvents AcOH and its triflourinated derivative (TFA) hastheir own peculiarities. The difference in the rates in AcOH and othersolvents is well documented in one of the earlier results, where the additionof small amount of acetic acid strongly increased the overall performance ofthe reaction both in toluene and ethanol solvents84 as shown in Table 8.Results given in Table 8 reflect also the influence of semi-ketal formation inalcoholic solvents on the reaction rate and ee. The rate decreases in thefollowing order: n-butanolWethanolWmethanol, i.e. it follows the reactivitytrend of alcohols to form semi-ketals: n-butanoloethanolomethanol.
Very pronounced solvent effect was observed in the hydrogenation of1,1,1-trifluoro-5,5-dimethyl-2,4-hexanedione using Pt/Al2O3 in the presence
1
5
10
15
20
25
30
35
40
45
Fig. 16 Combined plot of the ee values obtained in the hydrogenation of KPL over Pt/Al2O3-CD system (left axis) and the population of conformer Open(3) as calculated by DFT incombination with a reaction field model (P Open(3), right axis) v.s. the dielectric constant of thesolvent (axis scale is arbitrarily chosen). Solvents: 1-cyclohexane, 2-hexane, 3-toluene, 4-diethylether, 5-tetrahydrofurane, 6-acetic acid, 7-ethanol, 8-water, 9-formamide. (Reproduced fromref. 88 with permission)
190 | Catalysis, 2010, 22, 144–278

of synthetic modifier, pantoyl-naphthylethylamine.211 The results show adramatic influence of solvents on the ee values. No correlation can beobtained between ee and relative permittivity (er,) or empirical solventparameter (EN
T). Surprisingly high ee values were obtained only in halogen
1
5
10
15
20
25
30
35
40
45
Fig. 17 Effect of solvent on enantiodifferentiation of in different enantioselective hydrogen-ation reactions. Dependence of the ee value on empirical parameter EN
T (reproduced fromrefs. 46, 356,342 with permission) A: Pt/Al2O3-CD and DHCD in EtPy hydrogenation.46
Solvents: 1=cyclohexane, 2= toluene, 3=chlorobenzene, 4=THF, 5=dichloromethane,6=propanol, 7=1–pentanol, 8=ethanol, 9=AcOH, 10=methanol, 11=water; B: Rh/Al2O3-b-ICN in KPL hydrogenation.355 Solvents: 1=toluene, 2=t-BuMe ether, 3=THF,4=dichloromethane, 5=DMF, 6=t-butanol, 7=acetonitrile, 8=2-propanol, 9=AcOH; C:Pt/Al2O3-CD in the hydrogenation of 3,5-bis(trifluoromethyl) acetophenone.342
Table 8 Solvent effect and influence of acetic acid. (Reproduced from ref. 84 with permission)
No Solvent k1, min� 1 k2, min� 1 Optical yieldc, %
1 Methanol 0.022 0.019 62.2
2 ethanol 0.057 0.034 72.0
3 n-butanol 0.099 k2Wk1 75.0
4 toluene (EtPy)a 0.057 k2Wk1 86.3
5 MCH (EtPy)a 0.106 0.106 78.1
6 toluene (AcOEt)a 0.063 k2Wk1 84.0
7 MCH (AcOEt)a 0.069 0.060 75.3
8 ethanolþAcOHb 0.074 0.048 91.4
9 tolueneþAcOHb 0.120 0.120 93.1
Reaction conditions: T=23 1C, P=50 bar, [EtPy]0=1.0M, [CD]=8.4� 10� 4M, CD in-jection. a Ethyl pyruvate or ethyl acetate (1.5 cm3) is added to dissolve cinchonidine in thesesolvents (8.5 cm3). b The solvent is mixed with acetic acid; [AcOH]0=5.0M;. c Measured at90–100% conversion.
Catalysis, 2010, 22, 144–278 | 191

containing solvents. Some of these solvents, for instance dichloromethane,are often called as ‘‘reactive solvent’’. A rapid loss of activity of Pt/aluminawas found in this solvent and steady-state conditions could not be reachedin the continuous-flow reactor at 10 bar.297
In one of the recent publications356 it was mentioned the use ofdichloromethane should be avoided in hydrogenation reactions. As it wasemphasized ‘‘dehalogenation of this solvent on Pt, particularly at highhydrogen pressure, affords HCl, which induces a new set of acid-catalyzedside reactions. It is a very correct remark what was addressed against the useof CH2Cl2 at high pressure by an English group.234
However, we consider also that the use of CH2Cl2 as a solvent by Baiker’sgroup in ATR-FTIR spectroscopic studies316 can also be criticized. TheseATR-FTIR studies are considered as one of the crucial proves for theformation of protonated CD in the absence of AcOH. However, as it wasmentioned in ref.356 HCl can be formed from CH2Cl2. Consequently, theuse of CH2Cl2 would result in the protonation of CD in the absence ofAcOH and hydrogen.
5.5 Kinetic aspects
5.5.1 Rate acceleration. The rate enhancement of enantioselective hy-drogenation of activated ketones has been observed by various researchgroups. The first results were published by the Ciba Group.57,58,63 Thephenomenon was also observed by others.65,67,291 In all of these studiesusing EtPy as a substrate the common observation was that the modifiedreaction is 20–100 times faster than the unmodified one.
The rate enhancement was also described as ‘‘ligand acceleration’’58
based on the analogous observation in homogeneous catalysis.357 It wasproposed that ‘‘a reaction is considered ligand accelerated if there is a slowerunmodified (unselective) cycle and a faster modified (selective) cycle’’.58
This term has been used for many years, although its chemical meaning isquite doubtful or even misleading.
Most of the authors use the term rate acceleration (RA) or rate en-hancement (RE). Not only cinchona alkaloids, but also other tertiaryamines, such as quinuclidine, triethylamine, etc. can induce RA63. Thisbehaviour was evidenced in various solvents.73,358 The addition of smallamount of acetic acid into ethanol or toluene resulted in even more pro-nounced RA84. This behaviour is very characteristic for a-keto esters(Etpy, MePy, KPL77) and was observed not only over Pt, but other metalssuch as Rh254,256 and Ir.359 In ref. 343 it was found that in a-ketoester theincrease of the size of R1 and R2 groups resulted in slight alteration in theextent of RA.
With respect to kinetics studies the reproducibility problems related todifferent impurities has to be mentioned.73,84 Different batches of ethylpyruvate can give completely different kinetic results.84,360 The impuritiesalter both the initial rates and the enantioselectivity. When unpurified EtPyis used in this case the rate of racemic hydrogenation is extremely low.234
For this reason, kinetic results using the given substrate without any purifi-cation65,66,229,234 should be treated with great precaution. However, even in
1
5
10
15
20
25
30
35
40
45
192 | Catalysis, 2010, 22, 144–278

this case the addition of cinchona alkaloids as modifiers or even differenttertiary amines increases the rate significantly.
The other problem in this respect is that initial rates or related kineticinformation are only seldom published. Most authors prefer to provideactivity data in the form of conversion values measured after a given re-action time. It is especially notable in recent publications.236,355,361 Con-sequently, less and less information is available with respect to the RAphenomena. It has to be mentioned that RA has been observed only in li-quid phase hydrogenations; in gas phase hydrogenation of MePy no RAwas measured.290
Direct measurement of the reaction rates by using reaction calorimetry ina transient experiment provided an unambiguous evidence for the RA100.The results show that upon injection of CD the rate increase is instant-aneous and the rate increase is in the range of 5–12 depending on the type ofcatalyst used. Similar pronounced RA was observed in other studies over5% Pt/Al2O3 catalyst using EtPy upon injecting cinchona type modifiers ortertiary amines at higher pressures.291,309
With respect to the RA one important question can be raised: is anydirect coupling between the reaction rate and the enantioselectivity. Thiscoupling was first described by Blaser et al. in their ‘‘ligand accelerationmodel’’.58 Although the above coupling was clearly presented recently moreand more evidences have been accumulated that this coupling is not aprerequisite to obtain high enantioselectivities.
The first evidences against the direct coupling were obtained in ourstudies.291,362 In ref. 362 it was shown that the modification of Pt/Al2O3 bySn(C2H5)x moieties strongly alter the reaction rates, but their effect on the eeis negligible. Based on the analysis of the form of conversion-selectivitydependencies in ref. 291 it was stated that, ‘‘at low concentrations of sub-strate and modifier, contrary to instantaneous rate acceleration, the maximumee values are obtained only after a certain time delay. The increase of the rateof enantioselective hydrogenation with respect to the racemic one was welldocumented in ref. 204 upon using four different substrates (EtPy, EOG,EBF, PADA) as shown in Fig. 18.
It should also be mentioned that there is a definite class of substrates thatdo not show any RA or even the rate of enantioselective reaction is slowerthan the rate of racemic one. In most of these substrates the prochiral ketogroup is not activated. These substrates are as follows: acetophenone,363
3,5-bis(trifluoromethyl)acetophone,342 alkylsubstituted trifluoromethylketones.363,364
Classification of substrates according to their extent of RA and ED wasgiven recently (see Fig. 1).72 In addition, the following experimental con-ditions are not favourable for RA: (i) MePy pyruvate in gas phase;290 (ii)EtPy in the presence of a-ICN;198,365 (iii) EBF in AcOH at 1 bar hydrogen;56
(iv) non-activated aromatic ketones363 and non-activated trifluoromethylderivatives;363 (v) hydrogenation of EtPy in the presence of CD at very lowsubstrate concentrations.235
The appearance and the disappearance of RA acceleration are welldocumented in a series of experiments using trifluoro acetophenone andcyclohexyl analog. In the case of the former substrate pronounced rate
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 193

acceleration was observed as shown in Fig. 19A. Contrary to that uponusing trifluoro cyclohexyl ketone in the presence of modifier the rate ofreaction decreased as presented in Fig. 19B.
Further insight on the origin of RA was obtained upon comparing thehydrogenation of acetophenone and trifluoromethyl derivatives of
1
5
10
15
20
25
30
35
40
45
Fig. 18 Conversion–reaction time dependencies in the enantioselective hydrogenations ofactivated ketones (standard conditions, DHCD concentration 0.01mmol l� 1, 0.16ml; EOG:diethyl 2-oxoglutarate EBF: ethyl benzoylformate, PADA: pyruvaldehyde dimethyl acetal, (�)racemic hydrogenations, (�) enantioselective hydrogenations). (Reproduced from ref. 204 withpermission).
Fig. 19 The effect of CD concentration on the enantioselective hydrogenation of trifluorocompounds. A: Substrate=trifluoroacetophenone; B: Substrate=trifluoromethylcyclohexylketone. (Reproduced from refs. 364,367 with permission)
194 | Catalysis, 2010, 22, 144–278

acetophenone over Pt/Al2O3-CD system.334,366 In these studies both the rateand the ee values strongly depended on the electronic and steric effect of thesubstituents and on the hydrogen-bonding interactions between the qui-nuclidine N atom of the alkaloid and the carbonyl group of the substrate.
In the hydrogenation of acetophenone and trifluoromethylacetophenonederivatives on CD-modified Pt/Al2O3, the conversion rates and enantio-selectivities varied strongly with the nature of the aromatic substitu-ents.334,366 Different reactivities were attributed to the electronic (and steric)effect of the substituents and to hydrogen-bonding interactions between thequinuclidine N atom of the alkaloid and the carbonyl group of the sub-strate. A linear correlation has been found between the logarithm of thereaction rate and the highest occupied molecular orbital and lowest un-occupied molecular orbital stabilization of the carbonyl compounds(DEorb), relative to the reference compound.363
The by-products are high-molecular weight compounds and are con-sidered as strong catalyst poisons reducing the number of available Pt sitesresulting in substantial decrease in the reaction rates. Consequently, inthis case the RA is masked by a catalyst poisoning effect. Recently theRA phenomenon has been questioned by two research groups.234,235,368
In ref. 234 it was concluded that ‘‘rate enhancement is now attributed toreaction occurring at a normal rate at an enhanced number of sites, not (aspreviously proposed) to a reaction occurring at an enhanced rate at aconstant number of sites’’.
The final conclusion was that the ‘‘rate enhancement in the presence of analkaloid modifier is attributed to the inhibition of the pyruvate ester poly-merization at the Pt surface’’. In another recent study369 it was emphasizedthat ‘‘the reaction rate was lower in all chirally modified reactions ascompared to the racemic reaction in the absence of modifier’’.
We believe that in references cited above experimental conditions werenot properly chosen as their findings strongly contradict to results observedearlier by several groups. In a recent paper356 the use of ‘‘reactive’’ solventin ref. 234, such as dichloromethane, were strongly criticized.
Recently, with respect to the RA phenomena an open dispute has beenemerged in Journal of Catalysis.297,356,370 In a recent study continuous–flowexperiments were performed providing clear evidence that the rate accel-eration exists and it was concluded that ‘‘it is not the suppression of catalystdeactivation by addition of chiral modifier, because under appropriateconditions catalyst deactivation is negligible in pyruvate hydrogenation’’.297
This statement was strongly opposed in ref. 370. Those who favor the roleof deactivation defended their view referring to their earlier results shown inFig. 20.235 This figure shows that the rate acceleration appears only at highconcentration of substrate, while at low concentration the rate of enantio-selective hydrogenation is lower than that of the racemic one. In this respectit has to be mentioned that the determination of reaction rate at low sub-strate concentration is very plausible. We consider that the minor differ-ences shown in Fig. 20 cannot be considered as a real prove for the lack ofRA.
We have to emphasize again that the decrease of the reaction rate inenantioselective hydrogenation of EtPy at low substrate concentration370
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 195

can be related to the decreased concentration of substrate-modifier com-plexes formed under condition of catalytic hydrogenation. In this respect itis irrespective where the above complex has been formed in the liquid phaseor at the Pt surface. The decrease of the concentration of intermediatecomplex can result in pronounced rate decrease; similar to the kinetic pat-terns observed in enzymatic kinetics.
In our recent study transient experiments with injection of CD duringracemic hydrogenation of EtPy were investigated using purified substratesand a ‘‘distillation residue’’. The ‘‘distillation residue’’ contained 20% ofcompound 1a (see Scheme 5.2 in Chapter 5). Fig. 21A shows that the in-crease in the time delay between the start of racemic hydrogenation and theinjection of CD has no influence on the rate of enantioselective
1
5
10
15
20
25
30
35
40
45
Fig. 20 Initial hydrogenation rates of enantioselective (D) and racemic hydrogenation (E) ofEtPy and the enantiomeric excess (K). (reproduced from refs. 235 with permission)
0.0
0.2
0.4
0.6
0.8
1.0A B
0 50 100 150
time, min
conv
ersi
on
0.0
0.2
0.4
0.6
0.8
1.0
0 50 100 150
time, min
conv
ersi
on
Fig. 21 Influence of the time delay in CD injection during raceme hydrogenation. Kineticcurves of EtPy hydrogenation upon using purified substrate; [CD]=5� 10� 5M, T=20 1C,PH2=50 bar, catalyst=5% Pt/Al2O3 (E 4759), 0.125 g; 7-CD injection at 0min; &-CD in-jection at 15min; � -CD injection at 30min;}-CD injection at 90min; (�) – no CD (racemichydrogenation); A: [EtPy]0=1.0M (purified by distillation prior to the use); B: [EtPy]0=1.0M(‘‘distillation residue’’ containing dimer 1a in the amount of 20%). (Reproduced from ref. 371with permission)
196 | Catalysis, 2010, 22, 144–278

hydrogenation. It means that racemic products formed up to 10% con-version have no measurable influence on the reaction rate, consequently thesize of free Pt surface available for enantioselective hydrogenation is notaltered during racemic hydrogenation. When similar series of experimentswere performed in ethanol in the above conversion range slight decrease inthe initial rates was observed.291
Results given in Fig. 21B clearly show that upon using the ‘‘distillationresidue’’ the rate of racemic hydrogenation decreases, the decrease in rate isaround eleven-fold compare to purified EtPy. The rate decrease in the ra-cemic hydrogenation is due to the strong poisoning effect induced bycompound 1a. The poisoning effect can also be observed in enantioselectivehydrogenation, its extent is around three-fold. The introduction of CDduring racemic hydrogenation of ‘‘distillation residue’’ resulted in also in-stantaneous rate acceleration in all cases (see Fig. 21B).
Contrary to results obtained in the previous series of experiments uponincreasing the time delay from zero to 90 minutes the rate of enantioselec-tive hydrogenation decreases. All these results unambiguously show that thestatement given in ref.234 ‘‘rate enhancement is now attributed to reactionoccurring at a normal rate at an enhanced number of sites, not (as previ-ously proposed) to a reaction occurring at an enhanced rate at a constantnumber of sites’’ cannot be valid. It is hard to suggest that the addition of5� 10� 5M modifier will compete with 0.2M high molecular weigh productand can remove their adsorbed forms instantaneously from the Pt surface.
In an analogous series of experiments shown in Fig. 22, upon usingmethyl-benzoyl formate (MBF) substrate371 similar trend in the concen-tration dependences was obtained as in the case of EtPy (see Fig. 20).However, the rate of the enantioselective hydrogenation was higher thanthat of the racemic one in the whole concentration range.
Results obtained in series of experiments using MBF shows that the RAeffect is maintained in a relatively broad concentration range. It is a goodexample for the appearance of RA for the class of substrate with decreasedability to form by-products. Finaly let us conclude that we completely
1
5
10
15
20
25
30
35
40
45
y = 0.007Ln(x) + 0.0306R2 = 0.9794
y = 0.0007Ln(x) + 0.0053R2 = 0.9206
0.000
0.005
0.010
0.015
0.020
0.025
0.030
0 0.2 0.4 0.6
Concentration MBF, M
d[R
+S]/
dt, M
/min
1
Fig. 22 Initial hydrogenation rates of enantioselective (&) and racemic hydrogenation (}) ofMBF (K). (Reproduced from ref. 371 with permission)
Catalysis, 2010, 22, 144–278 | 197

disagree with the new views advertised in refs. 234, 235. Both our earl-ier69,291,309 and recent results371 show that upon introduction of CD duringracemic hydrogenation of EtPy the RA are instantaneous. This conclusionhas been supported by recent results obtained in continuous flowreactor.297,372
5.5.2 Enantioselectivity–conversion dependencies. In enantioselectivehydrogenation of EtPy one of the most disputed kinetic pattern is the form ofthe enantioselectivity–conversion (time) dependencies (ECD). In the hydro-genation of EtPy monotonic increase (MI) type dependencies were obtainedat low conversion in various studies83,84,270,291,373 as shown in Fig. 23. TheMI type behaviour is often called as initial transient period (ITP).196
Further on incremental ee values (eeincr=eecalc or Dee) were also used.374
It was calculated using the following formula:
eeincr ¼ ½c2 � ee2=c� ee1=½c2=c1�
where c is the actual concentration of ethyl lactate and ee is the measuredoptical yield. The use of eeincr reflects the ee values in a given time interval.It is applied when two different types of modifiers are added to the reactionmixture or when the loss of the modifier during the enantioselective hy-drogenation is very pronounced. In addition kinetic ee values can also becalculated from the corresponding reactions rates:
eekin ¼ ð½rR� � ½rS�Þ=ð½rR� þ ½rS�Þ:
This kind of behaviour was also observed upon using other activatedketones, such as PADA,283 MBF283 and KPL.53 In earlier studies this be-haviour was not discovered as no attempt was done to determine the eevalues at low conversion.
1
5
10
15
20
25
30
35
40
45
Fig. 23 Changes in ee observed during the course of EtPy hydrogenation. A: hydrogenationover a DHCD-Pt catalyst;84 B: hydrogenation over CD-Pt catalyst373 (2, ’) in toluene,CDinj. [Etpy]0=1.0M, [CD]0 =8.4� 10� 4M, 3.4� 10� 2M, respectively; all other experi-
ments in ethanol, [Etpy]0=1.0M, (η)-[CD]0 =6.8� 10� 6M, (E)-[CD]0inj=3.4� 10� 5M,
(�)-[CD]0=0.4� 10� 4M, (CDþEtPy)inj, (&)-[CD]0=8.4� 10� 4M, (EtPy)inj, (CD)premixed.(Reproduced from refs. 84 and 373 with permission)
198 | Catalysis, 2010, 22, 144–278

In first publications integrated ee values were calculated from actualconcentration of (R) and (S) products according to the general formula
ee ¼ ð½R� � ½S�Þ=ð½R� þ ½S�Þ:
Other characteristic feature of ee-conversion (time) dependencies is thedecrease of the ee values at high conversion.108 This decrease was attributedto the loss of alkaloid during the enantioselective hydrogenation. Theaddition of further amount of alkaloid during the hydrogenation experi-ment resulted in almost constant ee values even at high conversion.108
Results shown in Figs 23A and 23B have one common feature, i.e., theuse of injection method for the introduction of reaction components.Fig. 23A shows the ee-time dependencies in EtPy hydrogenation in ethy-lacetate injecting the substrate into the mixture containing the catalyst andthe modifier. When CD was injected into the reactor using toluene orethanol as a solvent MI type enantioselectivity-conversion dependencieswere also observed provided the concentration of CD was less than 10� 4M(see Fig. 23B). In both solvents the appearance of MI character was in-dependent whether CD or the substrate was injected. In all cases the in-crease part was very pronounced in the first 15–25% of conversion.
Later on similar behaviour was also described by two other groups in thehydrogenation of EtPy in ethanol using the premixing technique.66,82 Therewas a very tough dispute between these two groups as they had completelydifferent view on this new kinetic phenomenon.82,240
Further results clearly indicated283,354,375 that the appearance of MI typeof enantioselectivity – conversion (time) dependencies strongly depends onthe following experimental conditions: (i) concentration of CD, (ii) themode of introduction of reaction components, (iii) the purity of substrates,(iv) the solvent used, and (v) conditions of catalyst pretreatment.
Figs. 5.24A and B show the eecalc-conversion dependencies obtained intoluene upon using premixing and injection techniques, respectively.93 The
1
5
10
15
20
25
30
35
40
45Fig. 24 Enantioselectivity (eecalc)-conversion dependencies during the hydrogenation of EtPyin toluene; &-[CD]0=1� 10� 4M; E-[CD]0=1.2� 10� 5M; T=23 1C, PH2= 50 bar,[Etpy]0=1.0M, catalyst: 5wt% Pt/Al2O3 (Engelhard, E4759); A: premixing, B: injection.(Reproduced from ref. 93 with permission)
Catalysis, 2010, 22, 144–278 | 199
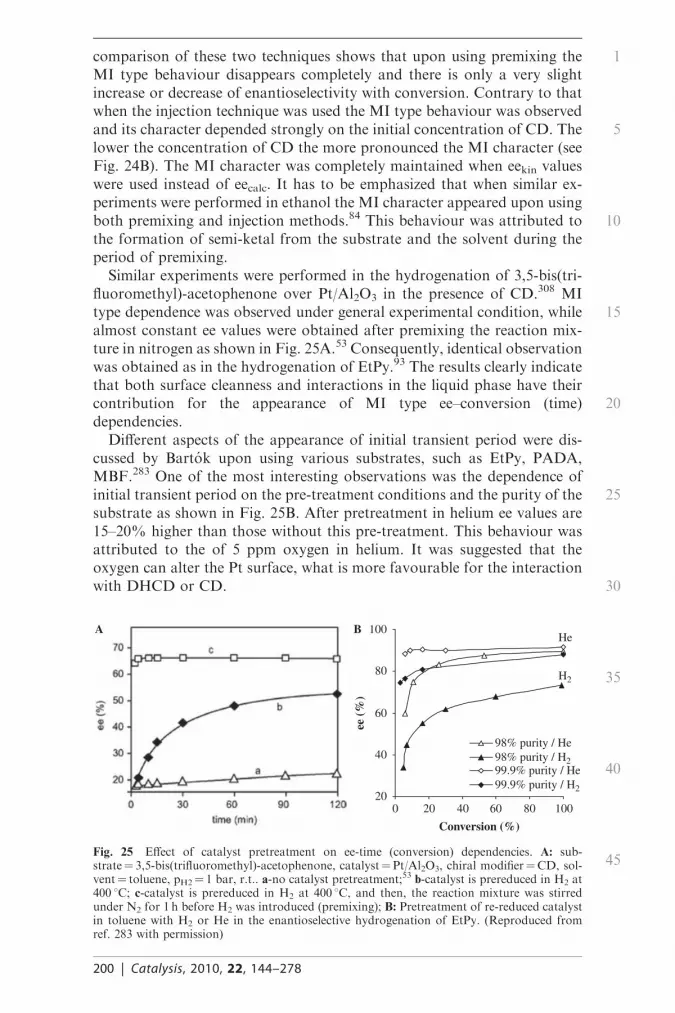
comparison of these two techniques shows that upon using premixing theMI type behaviour disappears completely and there is only a very slightincrease or decrease of enantioselectivity with conversion. Contrary to thatwhen the injection technique was used the MI type behaviour was observedand its character depended strongly on the initial concentration of CD. Thelower the concentration of CD the more pronounced the MI character (seeFig. 24B). The MI character was completely maintained when eekin valueswere used instead of eecalc. It has to be emphasized that when similar ex-periments were performed in ethanol the MI character appeared upon usingboth premixing and injection methods.84 This behaviour was attributed tothe formation of semi-ketal from the substrate and the solvent during theperiod of premixing.
Similar experiments were performed in the hydrogenation of 3,5-bis(tri-fluoromethyl)-acetophenone over Pt/Al2O3 in the presence of CD.308 MItype dependence was observed under general experimental condition, whilealmost constant ee values were obtained after premixing the reaction mix-ture in nitrogen as shown in Fig. 25A.53 Consequently, identical observationwas obtained as in the hydrogenation of EtPy.93 The results clearly indicatethat both surface cleanness and interactions in the liquid phase have theircontribution for the appearance of MI type ee–conversion (time)dependencies.
Different aspects of the appearance of initial transient period were dis-cussed by Bartok upon using various substrates, such as EtPy, PADA,MBF.283 One of the most interesting observations was the dependence ofinitial transient period on the pre-treatment conditions and the purity of thesubstrate as shown in Fig. 25B. After pretreatment in helium ee values are15–20% higher than those without this pre-treatment. This behaviour wasattributed to the of 5 ppm oxygen in helium. It was suggested that theoxygen can alter the Pt surface, what is more favourable for the interactionwith DHCD or CD.
1
5
10
15
20
25
30
35
40
45
20
40
60
80
100B
0 20 40 60 80 100
Conversion (%)
ee (
%)
98% purity / He98% purity / H299.9% purity / He99.9% purity / H2
H2
He A
Fig. 25 Effect of catalyst pretreatment on ee-time (conversion) dependencies. A: sub-strate=3,5-bis(trifluoromethyl)-acetophenone, catalyst=Pt/Al2O3, chiral modifier=CD, sol-vent=toluene, pH2=1 bar, r.t.. a-no catalyst pretreatment;53 b-catalyst is prereduced in H2 at400 1C; c-catalyst is prereduced in H2 at 400 1C, and then, the reaction mixture was stirredunder N2 for 1 h before H2 was introduced (premixing); B: Pretreatment of re-reduced catalystin toluene with H2 or He in the enantioselective hydrogenation of EtPy. (Reproduced fromref. 283 with permission)
200 | Catalysis, 2010, 22, 144–278

The influence of the reuse of the catalyst on the initial transient periodwas investigated by the Bartok’s group. Typical MI type dependencies areshown in Fig. 26.270 The results showed that the expression of MI characterdepended on the amount of modifier, but it was less pronounced after reuseof the catalyst. However, in repeated use, i.e. after removal of the reactionmixture and addition of fresh toluene, EtPy, and modifier resulted in10–20% increase in the ee values.
On the basis of these results it was concluded that the ‘‘the phenomenonof increase in ee on reuse is an intrinsic feature of the catalyst system used,i.e. new chiral centers making higher ee possible are formed’’.196 In thisrespect restructuring of the Pt surface was suggested based on analogousanalysis of results given in Refs. 55, 90, 279. It was also supposed thatoxygen plays a definite role in this process.
A sequential introduction of different substrates was performed in whichthe hydrogenation of MePy was carried out following the initial hydro-genation of EtPy using Pt/Al2O3-cinchona catalyst. In these experiments,the MI type character was obtained in both hydrogenation experiments (seeFig. 27A). The character of ee-conversion dependencies was maintainedafter reversed order of introduction of substrates (see Fig. 27B). The ob-servation that the initial transient effect is still observed with the sequentialhydrogenation of EtPy and MePy indicates that the phenomenon cannot beattributed to impurity effects. Consequently, it is more probable that thereaction-driven equilibrium of the chiral environment play a role in the MIcharacter of ee-conversion dependencies.
In one of the recent studies three different modifiers, such as CD, 9–O-phenyl-CD (PhOCD), 9–O-pyridil-CD (PyrOCD) were investigated in thehydrogenation of EtPy.376 Well-expressed MI type behaviour was obtainedfor all three modifiers. However, despite all the convincing results presented
1
5
10
15
20
25
30
35
40
45
20
40
60
80
100
0 20 40 60 80 100
Conversion (%)
ee (
%) 12
2r1r
3
3r
[DHCD](mmol/L)
Temp.(°C)
1
1r
2
2r33r
0.01 use
0.01 reuse
0.1 use
0.1 reuse0.01 use0.01 reuse
-10
-10
-10
-10-20-20
Fig. 26 Repeated use of catalyst in the enantioselective hydrogenation of EtPy: effect ofDHCD concentration and reaction temperature on the ee-conversion dependencies (freshDHCD was added for reuse, r=reuse). (Reproduced from ref. 270 with permission)
Catalysis, 2010, 22, 144–278 | 201

in this Chapter the following statement was done: ‘‘We assume that the slowremoval of surface impurities is the major reason for this behavior’’ (i.e. theMI type ee-conversion dependencies). Consequently, there are groups whodo not learn from results obtained by other groups and keeping their oldviews as a dogma.
Finally we can summarize the general views with respect to the origin ofthe initial transient behaviour of ECD: (i) it is related to impurities or otherexperimental artifacts,82 (ii) it is due to surface induced alterations,60,82,240
(iii) intrinsic kinetics.75,196
5.5.3 Non-linear phenomenon. Non-linear effects (NLE) in homo-geneous asymmetric catalysis have been investigated for many years sincethe pioneering work of H.B. Kagan.377 First the phenomenon was attrib-uted to the diastereomeric association inside or outside the catalyticcycle.378 Later on the approach was extended to the use of mixtures ofdiastereomeric ligands.379
Recently this approach was been extended to the Orito’s reaction. It wassuggested that the nonlinear behavior be due to the deviation from theexpected ideal behaviour assuming that the molar ratios of the modifiers insolution and on the metal surface are identical. Consequently, the nonlinearbehavior of mixtures of two diastereoisomers or two completely differentchiral modifiers has been attributed mainly to their different adsorptionstrength,289 however the contribution of the adsorption geometries on themetallic sites was also emphasized and new term ‘‘non-linear phenomenon’’(NLP) has been introduced.271 Besides it was also suggested271 that modi-fier–modifier interactions may also be involved in the NLP, but no ex-perimental evidence has been found yet. It was concluded that theinvestigation of NLP behavior of mixtures of two modifiers is a powerfultool in heterogeneous catalysis for characterizing the relative adsorptionstrength of modifiers under truly in situ conditions.
However, in this respect the controversy between catalytic and spectro-scopic investigations related to the evaluation of the relative adsorption
1
5
10
15
20
25
30
35
40
45
Fig. 27 Enantioselectivity-conversion dependencies in sequential hydrogenation experiments.a: EtPy hydrogenation to 100% conversion prior to addition of MePy; b: MePy hydrogenationto 100% conversion prior to addition of EtPy; ’-EtPy conversion, K-e.e. in (R)-EtLa,
7- MePy conversion,E-e.e. in (R)-MeLa. T=20 1C, reaction pressure 30 bar H2 for EtPyhydrogenation, 50 bar H2 for MePy introduction (Reproduced from ref. 375 with permission)
202 | Catalysis, 2010, 22, 144–278

strength of various chiral modifiers has to be mentioned.380–382 We have toadmit that this controversy can be attributed to the absence of substrate andhydrogen in spectroscopic investigations.
In a recent review based on the above results the following statement wasdone: ‘‘An essential conclusion from this study is that, although strongadsorption is a crucial requirement for an efficient modifier, there is nopositive correlation between the adsorption strength (AS) and the enan-tioselectivity achieved with the modifier alone’’.53
In a recent study it was emphasized that the NLP ‘‘can presumably beinterpreted on the basis of differences in the structure of the substrate-modifier complexes formed and in the adsorption-desorption processes ofthe complexes, thus the NLP is not solely dependent on the adsorption ofcinchona alkaloids, as suggested by earlier experimental data’’.383
The above view is very close to our one. In one of our study365 we turnedback to the original idea given by Kagan, namely to the formation of dia-stereomeric associations between the substrate and different competingchiral entities as modifiers in the liquid phase. Consequently, in our inter-pretation the nonlinear behaviour is due to different enantio-differentiationability of two modifiers acting simultaneously in the liquid phase resultingin different substrate-modifier complexes (associations). Of course theenantio-differentiation ability of two modifiers is further influenced bydifferent factors, such as the adsorption-desorption behaviour, the abun-dance and the reactivity of the formed associates.
It has to be added that kinetically the difference between the two inter-pretations (different adsorption strengths v.s. differences in the structureand stability of substrate-modifier complexes) for the NLP cannot be done.Only careful analysis of the chemical and surface properties can providesome hints inside the origin of these observations.
Different experimental techniques were used to investigate the NLP oftwo alkaloids: (i) variation of the initial ratio of two modifiers measuring theee values at the end of the reaction,289 (ii) applying a fixed initial ratio of twomodifiers and following the ee-conversion dependencies (our), (iii) usingtransient method in a batch reactor, where one of the modifiers is intro-duced at t=0, while the other one after a given time lap,271 (iv) usingcontinuous flow reactors and creating transient conditions by switchingfrom one modifier to another one.303
The deviation from the expected linear correlation was first observed inthe hydrogenation of EtPy in the presence of CD–CN and QN–QD mix-tures.289 At mole fraction of 0.5 of these alkaloids the ee value was higherthan zero indicating that the ED ability of CD and QN is higher than that ofthe CN and QD, respectively. Similar result was also obtained in otherpublications.60,383 In all of these studies the findings were attributed to thedifferences in the adsorption strength of the alkaloids.
The most striking NLP behaviour was observed in the hydrogenationof KPL over Pt/Al2O3 in the presence of CD-PhOCD mixtures (seeFig. 28A).239 It is known that PhOCD gives (S)-pantolactone, whereas CDaffords (R)-pantolactone as major enantiomer. The addition of smallamount of CD (XCDo0.05) to a reaction mixture containing PhOCD re-sulted in drastic change from (S)-pantolactone to (R)-pantolactone as the
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 203

major product. This non-linear behavior is attributed to the much strongeradsorption of CD compared to PhOCD. The weaker strengths of ad-sorption of PhOCD were related to its tilted form of adsorption as shown inFig. 28A.
Systematic investigation of NLP was done by two groups.198,271,382–385
Results obtained in a series of experiments using CD, CN, QN and QDis shown in Fig. 28B.384 Based on these results the following order wasestablished for the adsorption strength of cinchona alkaloids: CDW
CNWQNWQD. The above order was also supported by other studies.271,382
Contrary to that RAIRS measurements of adsorbed alkaloids resulted in adifferent order for the strength of adsorption:
QN,QDWCDWCN.380 In a more recent RAIRS experiments the order inthe adsorption equilibrium constants (Kads) the following sequence wasestablished: CNWQDWCDWQN.381 Probably based on these results in arecent review the following statement was done: ‘‘An essential conclusionfrom this study is that, although strong adsorption is a crucial requirementfor an efficient modifier, there is no positive correlation between the ad-sorption strength (AS) and the enantioselectivity achieved with the modifieralone’’.53
Upon investigating the behaviour of O-alkylated derivatives of CD it wasnicely demonstrated that the adsorption strength of this type of modifiers onPt decreases in the following order: CDWMeOCDWEtOCDWPhOCDETMSOCD.203
1
5
10
15
20
25
30
35
40
45
Fig. 28 Non-linear effect in enantioselective hydrogenation reactions over Pt/Al2O3 catalyst.A: substrate=KPL, chiral modifier=CD-PhOCD mixtures; schematic illustration of the ad-sorption of CD and PhOCD on an idealized flat Pt surface;239 B: substrate=EtPy, sol-vent=toluene, chiral modifier=mixtures of different modifiers, the 2nd modifier was added at10–20% conversion of EtPy384. (Reproduced from refs. 239 and 384 with permission)
204 | Catalysis, 2010, 22, 144–278

A continuous-flow fixed-bed reactor was applied in the enantioselectivehydrogenation of EtPy on Pt/Al2O3 using the principle of ‘‘chiralswitch’’.303 These time on stream experiment start with the introduction ofone of the modifiers and a given moment this modifier is switched to an-other one. The QD-CD and CD-QD switch is shown in the Fig. 29A andB.297 The results clearly show that the enantio-differentiation ability of CDis stronger than that of the QD. All of the above results strongly support thegeneral view that NLP and the adsorption strength of the modifiers arecoupled. However, there are experimental findings indicating that the originof NLP has more complex basis.
In this respect let us refer to a series of transient experiments performed ina batch reactor. Fig. 28B shows the influence of solvents when CD wasadded to the reaction mixture containing QN. In the opposite situationwhen QN was added to the reaction mixture containing CD only minorchanges in the ee values were observed. The most interesting finding is thatthe ‘‘rate of replacement’’ of QN by CD shows strong solvent dependency.No real explanation was given for this finding, although the possibility forthe involvement of solvent polarity and the formation of an alkaloid–acidion pair has been mentioned. Our view is that these experimental findingsindicate that not only the difference in the adsorption strength controlsNLP. Even more striking results related to NLP were observed when theamount of modifiers was varied in the above experiments. These results areshown in Figs. 30 A–C.271 When CD was added to the reaction mixturecontaining QD the direction of the enantioselectivity was immediatelyaltered. The time period required to reach the maximum Dee showedstrong concentration dependence, what was attributed to the fast hydro-genation of the quinoline ring of QN in the first 30 minutes at low con-centration of modifiers. In the opposite situation, i.e. when QD was addedto CD (see Fig. 30C) the decrease part was also explained by the fasthydrogenation of CD, however no acceptable explanation was given for theincrease part.
1
5
10
15
20
25
30
35
40
45
Fig. 29 Appearance of chiral switch. A: Chiral switch induced by replacing CN with CD (filledcircles) and vice versa (filled squares). The conversion is shown with open symbols (opensquares are barely seen due to overlapping). The second modifier reached the catalyst afterabout 45min (vertical dashed line); B: Influence of the modifier concentration on the dynamicsof the chiral switch. Conditions: 0.226mM CD, 0.226mM QD concentration for 1:1 ratio ofCD and QD (filled circles and squares) and 2.26mM for the 10 fold amount of QD relative toCD (open triangles) 100% conversion. (Reproduced from ref. 297 with permission)
Catalysis, 2010, 22, 144–278 | 205

The mixing of two different alkaloids was also applied to study theanomalous behaviour of both ICN. The results shown in Fig. 31A and Bindicate that the addition of a-ICN to CD, CN and b-ICN has no influenceon the enantio-differentiation ability of these alkaloids.365 Based on theseresults it was suggested that the origin of enantio-differentiation ability ofa-ICN is different than that of for CD, CN and b-ICN.
Results shown in Fig. 31B indicate also that in the presence of CN theaddition of a-ICN resulted in less loss of ee at high conversions. It is due tothe suppression of the hydrogenation of the quinoline ring of CN in thepresence of a-ICN. Consequently, a-ICN should be strongly adsorbed onthe Pt surface.
The first attempt to compare the behaviour of two substrates (EtPy andKPL) in NLP under identical conditions using CN and QN was done in arecent study. The investigations were performed in two solvents (tolueneand AcOH).383 Three different methods were applied. Here we show resultsobtained in a batch reactor using conventional and transient experiments.According to results given in Fig. 32A, in the hydrogenation of EtPy thedirection of enantio-selection changes almost linearly with the concen-trations of the two modifiers. On the contrary, in the hydrogenation of KPLthe direction of enantio-selection is affected to a much higher extent by CN
1
5
10
15
20
25
30
35
40
45
Fig. 30 Hydrogenation of EtPy over Pt/Al2O3; A: Solvent effect on the exchange of QD byCD. Addition of one equivalent CD after a 30-min reaction time; B: Influence of modifierconcentration on the transient behaviour. Addition of one molar equivalent CD after a 30-minreaction carried out in the presence of QD; C: Addition of one molar equivalent QD after a 30-min reaction carried out in the presence of CD. Standard conditions; acetic acid; amounts ofthe modifiers: 1.7, 0.17, and 0.017mm. (Reproduced from ref. 271 with permission)
206 | Catalysis, 2010, 22, 144–278

1
5
10
15
20
25
30
35
40
45
Fig. 31 Enantioselectivity-conversion dependencies in the presence of mixtures of a-ICN andflexible alkaloids. T=20 1C, pH2=50 bar, injection method, 500 rpm, reaction time=90min;A: ’-1.2� 10� 5MCD; &-1.2� 10� 5MCDþ 1.2� 10� 5Ma-ICN; K-1.2� 10� 5Mb-ICD;
�-1.2� 10� 5Ma-ICNþ 1.2� 10� 5Mb-ICN; B: x-1.2� 10� 5M a-ICN; 7-1.2� 10� 5MCN; D-1.2� 10� 5MCNþ 1.2� 10� 5Ma-ICN. (Reproduced from ref. 365 with permission)
Fig. 32 Comparing the behavior of EtPy (EP) and KPL in NLP. A: QN-CNmodifier mixture,total modifiers concentration: 0.1mM, solvent: toluene/AcOH=9/1; B: QN-CN modifiers;C: CN-QNmodifiers. In B and C concentration of each modifier=0.05mM, first abbreviation-modifier used first, second abbreviation-modifier added afterwards; solvent=toluene (T) andAcOH; modifiers. (Reproduced from ref. 383 with permission)
Catalysis, 2010, 22, 144–278 | 207

than by QN. This might indicate that in the presence of EtPy the adsorptionstrengths are identical, while in the presence of KPL the adsorption strengthof CN is higher than that of QN. Based on results shown in Fig. 32B, i.e.when QN was used as a first modifier, the following conclusions can bedrawn: (i) CN desorbs QN more readily in the hydrogenation of KPL thanin the hydrogenation EtPy, (ii) in the case of EtPy, CN cannot fully desorbQN from the surface; (iii) in the hydrogenation of KPL CN can nearly fullydesorb QN. Based on these findings the order of adsorption strength inthese two substrates is different, namely in EtPy CNBQN, while in KPLCNWQN. When CN was used as the first chiral modifier (see Fig. 32C), inthe hydrogenation of EtPy CN cannot be desorbed by QN, while in thehydrogenation of KPL under identical conditions CN acted as if QN wasnot present at all. Consequently, the order of the adsorption strength of thetwo cinchonas is different in these to substrates, in case of EtPy CNBQN,while in case of KPL CNWWQN. These findings are similar as those ob-served in ref.365, where CD, CN and b-ICN acted as if a-ICN was notpresent at all. Such observation was also made in another earlier study intransient experiments using CN and QN.198
Summing up all investigations related to the elucidation of the origin ofNLP we accept the conclusions given in a recent study that ‘‘the NLP de-pends not only on the chiral modifier but also on the substrate to be hy-drogenated. This observation can presumably be interpreted on the basis ofdifferences in the structure of the substrate-modifier complexes formed andin the adsorption-desorption processes of the complexes, thus the NLP isnot solely dependent on the adsorption of cinchona alkaloids, as suggestedby earlier experimental data.198 The statement is in full agreement with ourview related to the importance of the formation of substrate-modifiercomplexes.
5.5.4 Inversion of enantioselectivity. To find relationship betweenthe configuration of chiral centers of the modifier and the chirality of theproduct was one of the early tasks. It has been generally accepted that theconfiguration of C8 or C8 and C9 atoms of the cinchona alkaloid moleculedetermines the product distribution.57,193 Changes in the sense of enantio-selection were first observed by Augustine et al. in 1993. Upon varying theDHCD/ catalyst ratio in the hydrogenation of EtPy over Pt/Al2O3 catalyst(S)-ethyl lactate formed at low modifier concentrations and (R)-enantiomerat higher modifier levels.67 However, the extent of inversion is within theexperimental error. The other intriguing fact is that the ee values are ex-tremely low. It is unprecedented that in this reaction the ee values are lessthan 20%.
Analogous observation was found in gas phase hydrogenation of EtPyover Pt/SiO2 catalyst pre-modified with a series of C9 cinchona derivatives301
i.e., the sense of enantioselectivity has changed as a function of the modifierconcentration. The inversion of ee was found to be dependent on the natureof the substituent at C9.386 The appearance of inversion of enantioselectivitywas observed due to the changes in the modifier structure,193,203,207,239
variation of the solvent44,56,217,239,342 changes of the modifier concen-tration207 and even changes of the substrate.211 Inversion has been reported
1
5
10
15
20
25
30
35
40
45
208 | Catalysis, 2010, 22, 144–278

both on Pt56,203 and Rh256,258,355 catalysts. Summary of recent results wasgiven in ref. 387. Certain C9 substituted derivatives of cinchonas such 9–O-alky,203 -aryl193,203,340 and -silyl203,207,239 derivatives of cinchonidine andSharpless-ligands,193,207 furthermore b-isocinchonine56,198,388 (the rigid de-rivative of CN) have resulted in product with the opposite sense of ee thanthe underivatized alkaloid. Fig. 33 shows both the diminished enantios-electivity and its inversion with increasing bulkiness of the ether function ofthe modifiers.203
Upon using b-isocinchonine-Pt/Al2O3 catalyst system in the hydrogen-ation of EtPy ee decreases continuously and turn to opposite value withdecreasing of pH (see Fig. 34). Investigation of unexpected inversion hasgiven a new possibility for mechanistic studies.
In the hydrogenation of ethyl-4,4,4-trifluoroacetoacetate over O-methyl-cinchonidine-Pt/Al2O3 catalyst system a significant variation of ee value wasobserved with the conversion in the presence of even trace amounts of wateror catalytic amounts of a strong acid.351,389 This issue has been discussed inChapter 5.1. The explanation for the inversion of enantioselectivity is notcompletely clear at the molecular level. It is obvious that the inversion of eecan be related to increasing bulkiness of the substituent at C9 and the in-creased rigidity of the alkaloid molecule. However, these factors alone givenot sufficient answer why O-pyridoxy derivative376 of CD does not lead toinversion in spite of the fact that O-phenyl and O-pyridyl moieties havealmost identical van der Waals volumes. Further arguments are necessary toexplain why a-ICN198 and a-isoquinine387 owning similar rigid structure asb-ICN shows no inversion.
To understand the origin of inversion different physical chemical methodshave been applied. HPLC-MS and GC-MS measurements have shown that
1
5
10
15
20
25
30
35
40
45
Fig. 33 Hydrogenation of EtPy over Pt/Al2O3 in toluene, at pH2=1 bar. CD: cinchonidine,MeOCD: 9-O-methyl-cinchonidine, EtOCD: 9-O-ethyl-cinchonidine, TMSOCD: 9-O-tri-methylsylil-cinchonidine, PhOCD: 9-O-phenyl-cinchonidine, XylOCD: 9-O-(3,5-dimethylphe-nyl)-cinchonidine, HFXylOCD: 9-O-[3,5-bis(trifluoromethyl) phenyl]-cinchonidine,NaphOCD: 9-O-naphthyl-cinchonidine. (Reproduced from ref. 203 with permission)
Catalysis, 2010, 22, 144–278 | 209

b-isocinchonine modifier keeps its inner ether structure during the enan-tioselective hydrogenation.390 NMR measurements have proved that thehydrogenation of phenyl group in 9–O-phenyl-CD (PhOCD) is not re-sponsible for inversion.203 From ATR-FTIR spectroscopic measurementsand DFT calculations it has been concluded that the shape of the chiralspace formed by the adsorption of PhOCD onto the metal is altered com-pared to that formed by CD239,391 (see Fig. 28). The phenyl group has acomplex interaction with platinum; it can adsorb via its p system influencingthe strength of adsorption of the modifier. However, at the same time it cangenerate steric repulsion in the proximity of the chiral site.392 The presenceof the phenyl group in (PhOCD) can also hurt the efficiency of the shieldingeffect (see Chapter 8.3).
Chiral pocket393 has been defined as a physical space that is able to ac-commodate, via bonding and repulsive interactions, a pro-chiral adsorbate,and that is able to discriminate between its enantiomers. It was suggestedthat although CD and PhOCD display similar adsorption modes, the dif-ferent adsorption strengths and the change of the chiral pocket are sufficientto induce the inversion of enantioselectivity.391 The different role of thebulky ether groups i.e. repulsion by the phenoxy and attraction by the 2-pyridoxy group explains the different behaviour of these derivatives.376
Based on results obtained in the hydrogenation of EtPy198 and keto-pantolactone in the presence of b-ICN388 in toluene interactions responsiblefor the inversion were proposed.
The conformational rigidity of both the chiral modifier and the reactantmay inhibit the geometrical fit of the three components (modifier, reactant,and Pt), consequently the formation of the adsorbed intermediate respon-sible for enantio-selection is hindered. Beside the interaction between thenucleophilic N atom of the quinuclidine skeleton and the electrophilic Catom of the keto group of KPL or EtPy, H-bonded interaction verified byMcBreen are also suggested.72 It is proposed that the sense of enantio-se-lection is controlled by the conformation of the adsorbed reactant–chiral
1
5
10
15
20
25
30
35
40
45
Fig. 34 Hydrogenation of EtPy to (R)- and (S)-ethyl lactate on b-isocinchonine modifiedplatinum in toluene and AcOH mixtures. (Reproduced from ref. 56 with permission)
210 | Catalysis, 2010, 22, 144–278

modifier (1:1) complex which can be influenced by the solvent.388 It has beenconcluded that the adsorption mode and conformation of the modifierduring interaction with the substrate play the crucial importance in thechange of the sense of enantio-selection.53
In a recent study the inversion of enantioselectivity was investigated in thehydrogenation of of 1-phenyl-1,2-propanedione (PPD) using differentO-ether derivatives of CD.395 The data confirmed that the origin of inversionis related to depletion of the H-bonding interaction between the modifierOH-group and the carbonyl group of the reactant rather than to a decreasedpopulation of the Open(3) conformation in the solutions of O-ether de-rivatives when compared with the solution behavior of the parent alkaloid.
In another recent study different O-ethers of CD and CN were used inboth the enantioselective hydrogenation of PPD and the kinetic resolutionof the 1-hydroxyketones formed over Pt catalysts394 Characteristic resultsfor PPD are shown in Fig. 35. As emerges from these results all O-ethersshowed inversion of enantioselectivity. Similar trend was also observed inthe kinetic resolution of 1-hydroxyketones. These results are different fromthat obtained in the enantioselective hydrogenation of EtPy, where in-version was observed only in case of large substituents. Another importantfinding is that in the presence of AcOH the above modifiers showed onlyvery low enantio-differentiation ability (eeo5%).
Inversion of enantioselectivity has also been observed by Garland et al.using a continuous-flow three-phase reactor.301
5.6 Addition of other components
Several papers are devoted to the investigation of the influence of variousadditives on the behaviour of Pt/cinchona catalyst. These additives can beconsidered as modifiers either of the platinum or the support.
1
5
10
15
20
25
30
35
40
45
Fig. 35 Enantioselectivities of PPD hydrogenation in toluene over Pt/Al2O3 catalysts modifiedwith: (K) CD; (�) CN; (’) MeOCD; (&) MeOCN; (7) PhOCD; (D) PhOCN; (E)TMSOCD; (}) TMSOCN. (Reproduced from ref. 394 with permission)
Catalysis, 2010, 22, 144–278 | 211

5.6.1 Modification of Pt by tin tetraethyl. In one of our earlier studiesthe Pt sites in a Pt/Al2O3 catalyst were modified by Sn(C2H5)4.
362 Thismodification is a two step anchoring type surface reaction.396
Pt�Ha þ SnðC2H5Þ4��! Pt�SnðC2H5Þð4�xÞ þxC2H6 ðPSCÞ ð1Þ
Pt�SnðC2H5Þð4�xÞ þH2��! Pt�Snþðn� xÞC2H6 ðSBAÞ ð2Þ
In reaction (1) primary surface complexes (PSC) are formed. After tinanchoring the surface of Pt is covered by surface organometallic complexeswith general formula of Pt-SnR3 or Pt-SnR2. These PSCs are stable at roomtemperature. Upon heating in a hydrogen atmosphere (see reaction (2)) theydecompose with the formation of alkanes and stabilized bimetallic alloy(SBA) type surface species, i.e., supported Pt-Sn alloys are formed.397
Further details on this kind of modification of Pt can be found elsewhere.398
Results in the enantioselective hydrogenation using tin modified catalystsare summarized in Table 9. As emerges from this Table the modificationsresulted in both PSC and SBA forms of Pt-Sn/Al2O3 catalysts with differentSnanch/Pts ratios. The activity of modified catalyst was strongly altered bythe amount and the type of surface species. However, over catalysts con-taining PSC (see Exps. 5, 13, 14, 15, 16 in Table 9) the ee values were almostconstant, i.e. they were not affected by modification of Pt (ee=86–89%).
Striking observation was that upon using PSC the hydrogenation re-action was completely blocked at relatively small Snanch/Pts ratios. This wasattributed to the selective blocking of kink and corner sites responsible for
1
5
10
15
20
25
30
35
40
45
Table 9 Enantioselective hydrogenation using tin modified catalysts (Reproduced from ref.
362 with permission)
Exp.No
Catalyst
Code No
Temperature of
H2 treatment, 1C Sn/Pts, g
Rate
of hydrogenation,
mol(kgcatsec)� 1
Optical
yield, %
1. Pt no 0.000 0.83 64
2. Pt 150 0.000 1.70 82
3. Pt 200 0.000 1.66 87
4. Pt 400 0.000 2.00 88
5. PtSn-1 no 0.025 3.00 89
6. PtSn-1 100 0.025 2.60 84
7. PtSn-1 200 0.025 2.12 80
8. PtSn-1 200 0.025 2.17 86
9. PtSn-1 400 0.025 1.15 85
10. PtSn-2 200 0.036 1.06 72
11. PtSn-2 400 0.036 1.08 81
10. PtSn-3 200 0.056 0.02 72
11. PtSn-3 400 0.056 0.74 81
13. PtSn-4 no 0.030 1.77 86
14. PtSn-4 no 0.030 1.55 86
15. PtSn-5 no 0.008 2.00 89
16. PtSn-5 no 0.008 1.77 88
212 | Catalysis, 2010, 22, 144–278

hydrogen activation by Sn(R)(4� x) moieties. The highest rates in theenantioselective hydrogenation were obtained upon using modified catalystscontaining PSC. This finding was attributed to the suppression of the poi-soning effect induced by byproducts formed.
There is one interesting remark: in a related study it was confirmed that inthis kind of surface modification of Pt at low tin coverage tin prefers kinkand corner sites.399 Based in this old results it can be concluded that theinvolvement of kink and corner sites of Pt in the ED step, as it has beensuggested by different authors,234,373,400 is highly questionable. In additionthe above results can be considered as a first real hint that the reaction ratesand the ee values are not well correlated, i.e. relatively high ee values can beobtained even when the reaction rate is strongly suppressed. Unfortunately,this result was almost forgotten, as its conclusion did not fit into the conceptof ‘‘ligand acceleration model ’’. This model a priori suggests a definite re-lationship between reaction rate and enantioselectivity.
5.6.2 Addition of achiral amines. Based on the use of different experi-mental methods181,190–192,401,402 it has been suggested that cinchona alkal-oids can form dimmers. As far as any dimer would decrease the virtualconcentration of CD in the liquid phase attempts were done to use differentachiral tertiary amines (ATAs) with the aim to shift the equilibrium betweenthe dimer and the monomer form of CD as shown in the following scheme:
½CD�2 ! 2½CD� ð4Þ
½CD�2 þATA! ½CD�ATA� þ CD ð5Þ
This concept has been tested in the enantioselective hydrogenation ofEtPy and hexanedione.93,403–405 It is suggested that the modifier in the formof a dimer is a spectator in the asymmetric hydrogenation reaction.
Results in the presence of various ATAs at different experimental con-ditions are summarized in Tables 10 and 11. These results clearly show the
1
5
10
15
20
25
30
35
40
45
Table 10 Influence of different added achiral tertiary amines on the enantioselective hydro-
genation of EtPy in the presence of CD-Pt/Al2O3 catalyst system. (Reproduced from ref. 93
with permission)
Achiral tertiary
amines added
Concentration of
achiral amines (M)
Rate constant,
k1 (min� 1)
Rate constant,
k2 (min� 1)
Enantioselectivity
(eemax)
No – 0.0352 0.0465 0.750 (0.714)b
TEA 1.2� 10� 5 0.0407 0.0676 0.841 (0.793)b
DABCO 1.2� 10� 5 0.0886 0.1588 0.915b
QN 1.2� 10� 5 0.1289 0.1645 0.898b
QN 1.2� 10� 5 0.1297 0.1757 0.909b
QNc 1.2� 10� 5 0.0832 0.1346 0.926b
QNc 6.0� 10� 5 0.1267 n.m.d 0.936b
QNc 1.2� 10� 4 0.1219 n.m.d 0.946b
aReaction conditions, solvent: toluene; reaction temperature: 23 1C; hydrogen pressure: 50 bar;[Etpy]0=1.0M, [CD]0=1.2� 10� 5M, TEA - triethylamine; DABCO -1,4-diazabicyclo-[2.2.2]octane; QN - quinuclidine;. bee values measured at the end of reaction. creactions carriedout at 10 1C. d n.m.: not measurable.
Catalysis, 2010, 22, 144–278 | 213

strong effect of ATAs in toluene, however no effect has been observed inEtOH and AcOH. In the presence of quinuclidine unprecedented high eevalues were obtained at low CD concentration (1.2� 10� 5). The ee valueequal to 0.946 is close to those obtained in pure AcOH.
Further evidence with respect to the involvement of alkaloids dimers inthe ATA effect was obtained upon comparison of CD with 9-methoxy-CD(CH3OCD) in the hydrogenation of EtPy. As far as at C-9 position the OHgroup is replaced by a methoxy one, (CH3OCD) cannot interact with ATA.It is the reason that no increase in the ee is observed in case of(CH3OCD).404
Results of kinetic studies were supported by results of circular dichroismspectroscopy. In table 12 the intensities of the Cotton shift around 237 nmare shown in the presence of various ATAs. This Cotton shift has beenascribed to the dimer form.192 In case of CH3OCD no similar Cotton shift isobserved. The calculated De values well correlated with the ability of ATAsto increase the reaction rate and ee values. These results suggest that theATA added into the solution of CD be involved in new type of solute–soluteinteraction.
Summing up the ATA effect the following conclusions can be drawn: (i)ATA effect appears only at low concentration of CD; (ii) no ATA effect inEtOH and AcOH; (iii) the ATA effect depends on its concentration; (iv)ATAs containing bulky substituents show more pronounced effect; (v) theOH group of CD is involved in the interaction with ATAs.
5.6.3 Addition of nitrogen containing aromatic and condensed aromatic
compounds. In our recent study406 the influence of the addition of variousnitrogen containing aromatic and condensed aromatic compounds wasstudied. The aim of these studies was testing of the validity of the ‘‘surface
1
5
10
15
20
25
30
35
40
45
Table 11 Influence of different tertiary amines on the reaction rate and enantiomeric excess in
the enantioselective hydrogenation of EtPy. (Reproduced from ref. 93 with permission)
No ATAs k1, min� 1 k2, min� 1 eemax eeend
1 Noa 0.0045 0.0068 – –
2 No 0.0236 0.0747 0.838 0.819
3 quinuclidinea 0.0077 0.0109 – –
4 quinuclidine 0.0482 0.0997 0.901 0.882
5 Dabco 0.0486 0.1267 0.909 0.905
6 MPD 0.0322 0.0989 0.895 0.872
7 TEA 0.0300 0.0905 0.849 0.843
8 Edcha 0.0220 0.0735 0.850 0.785
9 Edipa 0.0243 0.1280 0.824 0.796
10 3-quinuclidinol 0.0468 0.0867 0.888 0.870
11 Nob 0.0409 0.1268 0.945 0.945
12 quinuclidineb 0.0699 0.1518 0.946 0.946
[EtPy]0=1.0 M, [CD]=1.2� 10� 5M, ATA=6� 10� 5M, T=20 1C, pH2=50 bar, solvent=toluene, coinjection of ATA, Dabco:1,4-diazabicyclo-[2.2.2]octane, MPD: 1-methylpiperidine,TEA: triethylamine, Edcha: N-ethyldicyclohexylamine, Edipa: N-ethyldiisopropylamine.k1, k2: first order rate constants calculated from experimental points measured in the first 10minutes and between 25–60 minutes, respectively a in the absence of cinchonidine.b solvent=1M acetic acid in toluene.
214 | Catalysis, 2010, 22, 144–278

model.’’ The ‘‘surface model’’ assumes that CD adsorbs by its aromaticquinoline ring almost parallel to the Pt surface.
The condensed ring system has been considered as the anchoring site (AS)of the modifier.46 Based on this view it was suggested if condition of com-petitive adsorption between CD and condensed aromatic compounds can beestablished, the number of ‘‘chirally modified sites’’ should decrease re-sulting in definite loss of enantioselectivity.
Table 13 shows the influence of added quinoline on the reaction kineticand ee values. These results unambiguously show that the addition ofquinoline increases both the rate and the ee values. The effect of quinoline isvery pronounced at low concentration of CD, while upon increasing the
1
5
10
15
20
25
30
35
40
45
Table 12 Effect of ATA on the circular dichroism data of cinchonidine (Reproduced from ref.
93 with permission)
No ATA added
Concentration
of ATA
(� 10� 3M)
ATA-CD
molar
ratio solvent De (M� 1cm� 1
1 no – – ethanol –
2 no – – CH2Cl2 0.94
3 quinuclidine 0.4 1 CH2Cl2 0.91
4 quinuclidine 0.8 2 CH2Cl2 0.74
5 quinuclidine 2.0 5 CH2Cl2 0.70
6 quinuclidine 4.0 10 CH2Cl2 0.61
7 quinuclidine 8.0 20 CH2Cl2 0.49
8 Dabco 2 5 CH2Cl2 0.92
9 Dabco 4 10 CH2Cl2 0.69
10 Dabco 8 20 CH2Cl2 0.39
11 TEA 2 5 CH2Cl2 0.94
12 TEA 4 10 CH2Cl2 0.90
13 TEA 8 20 CH2Cl2 0.79
[CD]=4� 10� 1M, T: 25 1C, cell length: 0.2 cm, time mode detection, wavelength: 237.6 nm,Dabco:1,4-diazabicyclo-[2.2.2]octane, TEA: triethylamine
Table 13 Effect of QN on the reaction rate and enantioselectivity in the enantioselective hy-
drogenation of EtPy (Reproduced from ref. 406 with permission)
No [CD], 10� 5M [QN], 10� 5M k1, min� 1 k2, min� 1 eemax eeend
1 0.6 no 0.027 0.015 0.573 0.235
2 0.6 6.0 0.040 0.069 0.832 0.699
3 0.9 no 0.032 0.058 0.719 0.575
4 0.9 6.0 0.045 0.090 0.867 0.813
5a 1.2 no 0.034 0.073 0.830 0.798
6 1.2 1.2 0.054 0.114 0.880 0.847
7 1.2 6.0 0.056 0.118 0.874 0.860
8 1.2 12.0 0.059 0.123 0.880 0.869
9 6.0 no 0.065 0.187 0.894 0.894
10 6.0 6.0 0.060 0.140 0.898 0.898
[EtPy]0=1M, treact=90min, catalyst: 0.125 g, 5wt% Pt/Al2O3, solvent: toluene, mode ofintroduction: Pr-I for QN followed by Inj-I of CD, conversionW99%. a average of five parallelexperiments.
Catalysis, 2010, 22, 144–278 | 215

concentration of CD the effect disappears. Due to the presence of quinolineunusually high ee values were obtained in toluene at [CD]0=1.2� 10� 5M.
Figure 36A–C shows the ee-conversion dependencies at different CDconcentration. Characteristic feature of these dependencies is that at lowCD concentration ee decreases at high conversion (see Fig. 36A). The loss ofee at high conversion is attributed to the loss of CD due to the hydrogen-ation of its quinoline ring. However, this decrease is strongly suppressed bythe addition of quinoline; consequently the results indicate that quinolinereplaces CD from the Pt surface. This replacement reduces the chance of CDto be hydrogenated by its quinoline ring.
Table 14 shows the influence of various nitrogen containing and con-densed aromatic compound on the rate and enantioselectivity. These resultswere obtained on highly dispersed Pt/SiO2 catalysts. Over this catalyst thering hydrogenation of the quinoline ring was relatively fast. It is the reasonthat over this catalyst the ee decreases with conversion. It is reflected by thelow value of eeend/eemax.
The results show that none of the additives used (see Table 14) resulted inmeasurable rate decrease. However, substantial rate increase was observed
1
5
10
15
20
25
30
35
40
45
A B C
0.0
0.2
0.4
0.6
0.8
1.0
0.0 0.2 0.4 0.6 0.8 1.0
Conversion
0.0 0.2 0.4 0.6 0.8 1.0
Conversion
0.0 0.2 0.4 0.6 0.8 1.0
Conversion
0.0
0.2
0.4
0.6
0.8
1.0
0.0
0.2
0.4
0.6
0.8
1.0
ee
Fig. 36 Influence of CD concentration on the hydrogenation of EtPy in the presence of QN.[EtPy]0=1.0M, PH2=50 bar, solvent: toluene, 500 rpm, catalyst: 0.125 g, 5wt.% Pt/Al2O3(Engelhard 4759), mode of introduction: Pr-I for QN followed by Inj-I of CD; A:[CD]0=0.6� 10� 5M; B: [CD]0=1.2� 10� 5M; C: [CD]0=6.0� 10� 5M, &-no QN; &-6� 10� 5M QN. (Reproduced from ref. 406 with permission)
Table 14 Hydrogenation of EtPy over cinchona-Pt/SiO2 catalyst system in the presence of
condensed aromatic compounds and aromatic nitrogen bases (Reproduced from ref. 406 with
permission)
No Additives k1, min� 1 k2, min� 1 convend, % eemax eeend eeend/eemax
1 – 0.031 0.053 98.8 0.666 0.343 0.515
2 Acridine 0.045 0.047 98.4 0.690 0.434 0.629
3 Quinoline 0.048 0.078 99.0 0.686 0.468 0.682
4 Pyridine 0.078 0.069 99.4 0.585 0.446 0.762
5 4-Picoline 0.042 0.059 99.5 0.575 0.447 0.780
6 Naphthalene 0.037 0.044 97.7 0.625 0.323 0.517
7 Antracene 0.033 0.049 98.3 0.620 0.323 0.521
8 Pyrene 0.039 0.048 98.4 0.619 0.337 0.544
[EtPy]0=1 M, [CD]=1.2� 10� 5M, [Additive]=1� 10� 4M, treact=90min, catalyst: 0.07 g2.7wt% Pt/SiO2, solvent: toluene, mode of introduction: Pr-I for additives followed by Inj-I ofCD.
216 | Catalysis, 2010, 22, 144–278

in the presence of acridine, quinoline and pyridine. Two classes of con-densed aromatic compounds can be differentiated: (i) nitrogen containingone resulting in increased eeend/eemax values and (ii) condensed aromaticcompounds having no influence on the eeend/eemax values.
Summing up these results it was concluded that in the enantioselectivehydrogenation of EtPy achiral condensed aromatic N-bases, as additives,are able to increase both the enantioselectivity and the reaction rate.Based on the concept of ‘‘chirally modified sites’’ the co-presence of CDand quinoline over Pt sites can be considered as a competition. Theconsequence of this competition is the decrease of the number of sites in-volved in ED. Therefore, one would expect a decrease in the ee values.However, it is not the case, the ee increases when the CD/quinolineratio is properly chosen and the CD concentration is low. The observedeffect appeared both on alumina and silica supported Pt catalysts and wasfound to be strongly concentration dependent. However, according to the‘‘Surface model’’ the co-adsorption of these compounds should result in adecrease in the enantioselectivity, what is not observed in our study. Con-sequently, our results might indicate that the ‘‘surface model’’ needs somecorrections.
5.6.4 Modification of the support. There are only scarce data on themodification of the support. In one of our studies the influence of themodification of alumina support by alkyl silanes was investigated. Afterdehydroxilation of the support at 400 1C it was modified by different alkylsilanes resulting in anchored –Si(CH3)3, or –Si(CH3)2C8H17 moieties276. Therate of this anchoring type surface reaction can be controlled by the con-centration of the modifier, the temperature of anchoring reactions and thelength of the R group in the alkyl silanes.
Catalysts modified in this way were used in the enantioselective hydro-genation of EtPy in the presence of CD. The above modification was notbeneficial for the above reaction as the modified catalysts showed pro-nounced decrease in reaction rates and slight loss in enantioselectivitiescompared to the unmodified Pt/Al2O3. These results are shown in Figs. 37 Aand B. The catalytic performance of these modified catalysts was
1
5
10
15
20
25
30
35
40
45
Fig. 37 The effect of the surface coverage of CH3(CH2)7Si(CH3)3 moieties on the reaction rate(A) and the enantioselectivity (B). (Reproduced from ref. 276 with permission)
Catalysis, 2010, 22, 144–278 | 217

significantly altered upon using various pre-activation procedures as shownin Table 15. The removal of the anchored –O–Si(CH3)2R moieties the ori-ginal activity and enantioselectivity was restored completely.
The catalytic performance of these modified catalysts was significantlyaltered upon using various pre-activation procedures as shown in Table 15.The removal of the anchored –O–Si(CH3)2R moieties the original activityand enantioselectivity was restored completely. The behaviour of thistype of modified catalysts was explained by the following phenomena:(i) partitioning or retention of CD or the [substrate-CD] complex by an-chored –Si(CH3)2R moieties, and (ii) decreasing the mobility of CD or the[substrate-CD] complex in the boundary layer.
We believe that the results obtained in the above study provided furtherindirect evidences that interactions in the liquid phase play a very importantrole. Results obtained in this study strongly indicate that in this enantio-selective hydrogenation reaction the enantio-differentiation cannot be at-tributed exclusively to the interaction between the half-hydrogenatedsubstrate and CD on the Pt surface.
5.6.5 Modification of Pt by other components. These studies were per-formed by English groups. These groups use the classical aerobic Pre-modification procedure (see Chapter 4.4), In the enantioselectivehydrogenation of MePy or butane-2,3-dione in the presence of cinchoni-dine-modified platinum catalysts it was shown that at the catalyst prepar-ation stage, the co-adsorption of the alkaloid with a strong co-adsorbate hasa strong positive effect.280 One of these coadsorbates was oxygen or airdissolved in reactant and solvent. In addition acetylene, methyl acetyleneand butadiene appeared to be effective co-adsorbates. It was suggested thatin the absence of a strong co-adsorbate the surface is poisoned bycinchonidine.
It has been shown that the modification under methylacetylene providesreaction rates and ee values excess under standard conditions (10 bar,293K) that are comparable to, or higher than those obtained with normalaerobic modification.
The importance of surface morphology of small supported Pt particleswas confirmed in Refs. 234, 407. In these studies Pt/C and Pt/SiO2 catalysts
1
5
10
15
20
25
30
35
40
45
Table 15 Influence of the catalytic performance of modified catalysts as a function of the
temperature of pre-activation (Reproduced from ref. 276 with permission)
Experiment No. Temperature of preactivation (1C) Rate constant, k1 (min� 1) ee (%)
1 150 o0.001 58.3
2 250 0.012 77.2
3 400 0.020 84.7
4 400, blanka 0.044 85.0
5 400b 0.054 84.3
6 400, parent 0.057 86.3
Catalyst tested: No.10 (see Table 1 in Ref. 276); a Catalyst No. 1 (see Table 1 in Ref.276). b Modified catalyst treated in air at 300 1C prior its preactivation in a hydrogen atmo-sphere. c Catalyst without modification.
218 | Catalysis, 2010, 22, 144–278

modified by bismuth and sulphur and were characterized by electrochemicalmethods.408 Their key findings are summarized in Table 16.
The authors’ main results and conclusions were as follows:� A cyclic voltametric analysis indicated that in poisoning by Bi king
sites, while in poisoning by sulfur terrace sites are involved� The reaction rates increased in bismuth modified catalysts; while de-
creased in sulfur modified one.� The formation of polymeric residues were strongly reduced over Bi
modified catalysts.� Both the enantioselective hydrogenation and the formation of poly-
meric residues are formed on king sites.� RE is now attributed to reaction occurring at a normal rate at an
enhanced number of sites, not (as previously proposed) to a reaction oc-curring at an enhanced rate at a constant number of sites.
It has to be stressed out that in this study EtPy was used without anypurification. It is the reason for extremely low rates in the racemic hydro-genation (see Table 16).
This fact strongly questions both the results and the conclusions. There isan additional serious drawback, i.e., the lack of information on racemichydrogenation over catalysts modified by Bi. In this respect it has tomention that the adsorption of Bi on the Pt can give different species, in-cluding metallic and ionic one.409 We suggest the rate increase and thedecrease of the ee over Bi modified catalysts is due to the acceleration effectin the racemic hydrogenation by bismuth cations created over the Ptsurface.
In this respect we should like to revert to our results on tin modifiedcatalysts (see Chapter 5.6.1). This method clearly indicated that the in-volvement of kink sites in the ee is excluded as tin is located on the kink siteand ee was independent on the amount of tin anchored to the platinum,while the rate showed a strong dependence.
1
5
10
15
20
25
30
35
40
45
Table 16 Variation of activity (rmax), enantiomeric excess (ee) and HMMP yield in reactions
over Pt/graphite, Pt–Bi/graphite and Pt–S/graphite modified by cinchonidine (CD) and qui-
nuclidine (QN) (Reproduced from ref. 234 with permission)
Surface Alkaloid modifier rmax (mmol h� 1gcat� 1) ee (%(R)) HMMP yield
Pt None –b 0 2c
Pt CD 850 41 100
Pt QN 440 0 40
Pt 1:1 CD:QN 1205 37 –
Pt-Bi ((YBi)ch=0.35) CD 1350 35 49
Pt-Bi ((YBi)ch=0.35) QN 1710 0 36
Pt-Bi ((YBi)ch=0.35) 1:1 CD:QN 4600 15 –
Pt-S ((YS)ch=0.19) CD 440 52 109
Pt-S ((YS)ch=0.19) QN 310 0 –
Pt-S ((YS)ch=0.19) 1:1 CD:QN 645 51 –
a Conditions: 65mmol ethyl pyruvate, 0.17mmol CD and/or 0.17mmol QN, 0.25 g catalyst,12.5ml dichloromethane, 30 bar hydrogen, 293K, 1000 rpm. b For this reactionrinitial=24mmolh� 1gcat
� 1. c ConversionW20%.
Catalysis, 2010, 22, 144–278 | 219

6. Spectroscopic investigations
6.1 NMR
NMR techniques related to substrates, reaction products and chiral modi-fiers have widely been used in the investigation of enantioselective hydro-genation of activated ketones. It has also been applied for the determinationof by-products, alternative reaction routes and intermediate complexes andattempts were also made to use NMR for elucidation of different hypo-thetical reaction-mechanisms.
To study the directing effect of ester group340 or trifluoromethyl group236
of the substrate a series of new compound were prepared and identified byNMR. It was also applied as a tool to check the purity of substrate.82 Inmany cases NMR gives an opportunity for identification of reactionproducts.204,211,236,333,338,340,410
It is known long before that cinchona alkaloids are extremely active chiralmodifiers in various organic reactions. Mapping the role of differentstructural elements of cinchonas in the enantio-selection several derivatives,especially C9 substituted cinchonas,203 inner ethers,411 N-alkyl deriva-tives316 have been prepared and checked by NMR,44,239,258,412 Upon iden-tification of the structure of ether derivatives of cinchonidine such as 9–O-phenyl-203,207 9–O-pyridyl-,376 9–O-sylil-cinchonidine203 etc. NMR was anindispensable tool. Beside of the aforementioned chiral modifiers, thestructure of several cinchona analogues, i.e. amines and amino alcohols44,382
and aryl alcohols227 prepared for chiral template in the Orito’s reaction hasalso been confirmed by NMR.
Chiral modifiers itself very often suffer changes during the enantioselec-tive hydrogenation. To follow the conversion of 9–O-pyrydil-cinchoni-dine376 and the saturation of naphthalene ring of 1-naphthyl ethylaminederivatives209,313 NMR was applied as well as to check the resistance ofphenyl group in 9–O-phenyl-cinchonidine203 and the stability of methoxy-cinchonidine376 and isocinchonines.355 NMR analysis of the reaction mix-ture showed that 1-naphthyl ethylamine derivatives is quantitatively con-sumed during the hydrogenation reaction and converted to the secondaryamine.213 It has been shown by NMR that quaternary ammonium deriva-tives of CD as new chiral modifiers remained stable during hydrogen-ation.201 Formation and structure of hexahydro-cinchonidines andhexahydro-cinchonines has been investigated by NMR.273,413 According toNMR analysis, at 36% saturation of the quinoline rings of CD in acetic acidthe ratio of homoaromatic and heteroaromatic hydrogenation products was2.5 to 1.271
Upon hydrogenation of b-trifluoro ketones the sense of enantioselectivitychanges when the polarity of the solvent changes. The phenomenon wasexplained by the shift of keto-enol equilibrium confirmed by NMR.44,389 Inalcoholic solvent ethyl-4,4,4-trifluoroacetoacetate has shown a reactionroute via semi-ketal.351 Semi-ketal was also detected by use of NMR inother cases.333,414 IR and NMR experiments have revealed that the enan-tioselective hydrogenation of EtPy in nonacidic solvents is complicated bythe simultaneously occurring self-condensation (aldol reaction) of thereactant.81 In the hydrogenation of ketopantolactone GC and NMR results
1
5
10
15
20
25
30
35
40
45
220 | Catalysis, 2010, 22, 144–278

has shown that no by-product formation appears.76 The reaction pathwayswere followed by NMR in the enantioselective reduction of isatin deriva-tives over cinchonidine modified Pt/alumina.415
In the study of hydrogenation of 1,1,1-trifluoro-2,4-diketones strongacid-base interaction has been revealed by NMR. The high chemo- andenantioselectivities in the above reaction were attributed to the formation ofan ion pair involving the protonated quinuclidine part of the chiral modifierand the enolate form of the substrate.410 In the hydrogenation of KPL theformation of dimer was confirmed by NMR.210,325
Recently a mechanistic model involving nucleophilic catalysis and zwitter-ionic adduct formation between a cinchona alkaloid and activated ketone hasbeen suggested.416 Upon investigation of hydrogenation of flouoroketones theformation of the ionic species in the solution was studied by 13C-NMRspectroscopy. Trifluoromethylcyclohexyl ketone was used as a strong elec-trophile agent and the complex was created by the addition of excess tertiaryamine quinuclidine. The adduct formation was studied in two different solventsystems such as deuterated chloroform and acetone but the formation of thezwitterionic species was observed only in acetone.217 13C-NMR confirmed theexistence of other adducts of the zwitterionic type.417 However an NMR studyhas indicated that the zwitterions model is probably based on erroneous in-terpretation of the experimental data; the NMR spectra that had been re-ported for zwitterion formation may arise from an aldol addition the a,a,a-trifluoromethyl ketone and the solvent acetone, and the reaction is catalyzedby the tertiary amine used as a model for the chiral amine modifier.418
Upon using NMR technique it was verified that the enol form of EtPy isnot the reacting species, but under condition of enantioselective hydro-genation deuterium exchange takes place not only at the quinoline ring, butat C9 carbon atom, too.419
6.1.1 Conformation analysis of the modifier. The study of the con-formation of cinchona alkaloids investigated by NMR has been brieflymentioned in Chapter 2.3. The question of which conformer of cinchonidineis involved in the enantio-differentiation step is regarded as a key issue.Baiker el al. have used ab initio calculations and NMR measurements toinvestigate the conformers of (dihydro)cinchonidine in different solvent(such as benzene, toluene, ethyl ether, acetone, etc.).88 The existence of agiven conformer has been rendered by nuclear Overhauser enhancementspectroscopy. NOESY experiments have suggested that Open (3) andClosed (1) and Closed (2) conformers appears. This observation is inqualitative agreement with their calculations. The dihedral angles for dif-ferent conformers have been calculated by ab initio methods. The measuredcoupling constants (13JH8H9(exp)) and the dihedral angles by applying theKarplus equation420 have given possibility for calculation of couplingconstants of different conformers (13JH8H9(i)). The above method is limitedto the determination of only two conformers from one coupling constantsmeasured, however the dihedral angle has been found very similar for thetwo closed conformers of CD, so the population of Open (3) and the sum ofpopulations of closed conformers has been possible to calculate. The resultsare represented in Table 17.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 221

According to the above approach the conformer Open (3) has been foundthe most stable one.88 Based on parallel solvent dependence of ee and popu-lation of Open (3) it has been suggested that Open (3) conformer plays crucialrole in the asymmetric induction. In case of 9-deoxy-CD derivatives similarmethod has been used.421 The coupling constants of 2-phenyl-9-deoxy-10,11-dihydrocinchonidine (13JH9H8a (5.0Hz); 13JH9H8b (9.3Hz)) is similar to thatof 9-deoxy-10,11-dihydrocinchonidine (5.5 and 8.5Hz) in CDCl3 indicatingthat the relative stability of conformers is also similar. NMR experiments andab initio calculations revealed that conformer Closed (1) is stabilized relativeto Open (3) when going from CD to 9-deoxy-10,11-dihydrocinchonidine.
In a recent study the effect of the protonation on the conformation of CDwas investigated.422 It was shown that protonation strongly hinders therotation around the C40–C9 and C9–C8 bond. Structures and conforma-tional behaviour of several cinchona alkaloid O-ethers in solution (NMRand DFT) were also investigated.395 It was demonstrated that the con-formation found to be abundant in the liquid phase has no direct correl-ation with the enantioselectivity of the PPD hydrogenation reaction.
The authors concluded that the driving force for production of one of theenantiomers in excess is due to the specific adsorption of the modifier on thecatalyst surface, a phenomenon that does not correlate with the populationof the conformers in the liquid phase.
6.1.2 Substrate-modifier interaction. In an early work NMR measure-ments have already shown an interaction between CD and EtPy in the liquidphase.84 It was shown that in CD3OD in the presence of 0.15M MePy thecharacteristic doublet of CD at 5.65 ppm was shifted to 5.85 ppm and a newsmall singlet was observed at 6.0 ppm. Upon increasing the concentrationof MePy to 0.6 or 1.0M the doublet vanished and only the new singletat 6.0 ppm was found. More noticeable shift of the C(9) proton, up to6.3–6.4 ppm with a formation of a singlet was observed in neat CD3COODor if small amount of CD3COOD was added into the solution of CD inC6D6. These NMR results suggested that the torsional angle between thehydrogen atoms at C(8) and C(9) carbon atom of CD has been changedresulting in a new conformer of CD.
1
5
10
15
20
25
30
35
40
45
Table 17 Vicinal 3JH8H9 coupling constants for cinchonidine and derived population of
conformer open (3) in different solvents. (Reproduced from ref. 88 with permission)
Solvent hT3JH8H9 Popen (3) PClosed
benzene 2,28 5.0 0.58 0.42
toluene 2,34 4,1 0,7 0,3
ethylether 4,3 4 0,71 0,29
tetrahydrofurane 7,6 4,7 0,62 0,38
acetone 20,7 6,4 0,4 0,6
dimethylfomamide 36,7 7 0,33 0,67
dimethylsulfoxide 40 7,5 0,27 0,73
water 78,5 7,2 0,3 0,7
ethanol 24,3 3,5 0,77 0,23
a For open(3) 3JH8H9 is calculated as 1.7Hz. For closed (1) and Closed (2), respectively,3JH8H9 is calculated as 9.6 and ) 4Hz. In this determination of P closed a value of 9.6 was used.
222 | Catalysis, 2010, 22, 144–278

In Pt-catalysed hydrogenation of 1,1,1-trifluoro-2,4-diketones the com-bined catalytic, NMR and FTIR spectroscopic, and theoretical study re-vealed that high chemo- and enantioselectivities are attributed to theformation of an ion pair involving the protonated amine function of thechiral modifier and the enolate form of the substrate.410
On the basis of NOE studies and theoretical calculations related to thehydrogenation of ketopantolactone in the presence of the (R,R) and (R,S)diastereomers of a new chiral modifier, pantoyl-naphthylethylamine, differ-ent properties of the above diastereomers were investigated, in particular theeffect of acid on the modifier structure.210 The results indicated that in case ofthe (R,R)-diastereomer in apolar solvent, a loose, extended structure changesto a compact one via an additional intra-molecular hydrogen bond, resultingin a more suitable ‘‘chiral pocket’’ available for the reactant on the Pt surface.
Standard 2D NMR spectroscopic methods and diffusion-ordered NMRspectroscopy combined with theoretical calculations has been used to verifythe formation of supramolecular complexes between the pairs O-methyl-cinchonine–ketopantolactone (KPL) and b-isocinchonine–KPL.423 WhenO-methyl-cinchonine or b-isocinchonine and KPL were mixed in dry deu-terobenzene solution, time-dependent chemical shift changes for the cin-chonas and new signals for KPL bound to the modifier have been detected.The spatial pattern of the chemical shift differences and the conformationsof the modifiers determined by NOESY demonstrated that the substratebinding occurs at the quinuclidine N atom, H9, and the quinoline H50 re-gion for O-methyl-cinchonine (H8 and H50 for b-isocinchonine. Based ondiffusion measurements hydrodynamic radii has been estimated which hasproved the co-diffusion of the cinchonas and KPL in a complex. The resultshave shown that not only 1:1, but also 2:1 cinchona-KPL complexes mustbe taken into account. NMR evidences has also been found for the cor-relation between the solution-state concentration of the nucleophilic 1:1modifier-substrate complex and the ee on enantioselective hydrogenation ofKPL using Pt–b-isocinchonine chiral catalyst.217,424
6.2 Circular dichroism
Vibrational circular dichroism (VCD) is a useful tool to determine the ab-solute configuration of the enantiomer produced in excess in an enantio-selective reaction when reference data on the enantiomer are not available.The absolute configurations of the enantiomers can be obtained by com-paring the theoretically calculated VCD spectrum of one enantiomer withthe experimental VCD spectrum of the product of the asymmetric reaction.It is important to know that VCD signal is about three orders of magnitudeless intensive than the corresponding signal in the ordinary transmissionspectrum.236 This method has been successfully applied for the determin-ation of product alcohols in the studies related to the directing effect oftrifluoromethyl group236 or ester group340 of the substrates. It was also usedupon investigating CD modified Rh/alumina catalyst425 in the hydrogen-ation of various aromatic ketones possessing an a-hydroxy or a-methoxygroup and, in case of the enantioselective reduction of isatin derivatives overCD modified Pt/alumina.415
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 223

Theoretical (DFT) and VCD spectroscopy study has been applied for theconformational analysis of the synthetic chiral modifier 9-O-phenyl-CD.426
According to these results 9-O-phenyl-cinchonidine behaves similarly toCD and shows four main conformers, denoted as Closed (1), Closed (2),Open (3), and Open (4). A combined theoretical-experimental VCD spec-troscopy approach has given possibility to increase the spectroscopic sen-sitivity toward changes in the distribution of conformers upon changing thesolvent polarity. The VCD spectra confirm that Open (3) is the most stableconformation in CCl4. Changing from CCl4 to CDCl3 the equilibrium be-tween the conformers does not change significantly. Upon increasing solv-ent polarity besides similar non-coordinating properties the fraction ofClosed (2) species increases considerably.
Relating the conformational results to the enantio-differentiation shownby this modifier (9-O-phenyl-CD) in the platinum-catalysed asymmetrichydrogenation of KPL the inversion of the sense of enantio-differentiationobserved cannot be traced to the conformational behaviour.426
HPLC and UV-vis/circular dichroism301 has been used to assess con-version and selectivity in chiral fixed bed reactor for stereoselective het-erogeneous catalysis.427 The UV-CD method and the HPLC-CD methodhave been used to simultaneously determine ee values and concentration ofeach enantiomer.428
Tungler and coworkers have described that the circular dichroism spec-trum of dihydrovinpocetine changes upon addition of both isophorone aswell as EtPy indicating interactions between these two substrates and thechiral modifier.68 The similar method has been applied in case of (S)-prolinebased chiral modifiers.225 The circular dichroism spectra of CD in toluenehas been found to change by addition of EtPy as shown in Fig. 38.93 Add-ition of EtPy to cinchonidine in chloroform has also resulted in changes inthe circular dichroism spectra of cinchonidine, although these changes wereless pronounced than those in toluene. The above results strongly indicatethat there is an interaction between CD and EtPy in the liquid phase.
The results of circular dichroism spectroscopy405 have provided furtherproof for dimer formation of CD in liquid phase. These results are related tothe addition of ATAs discussed in Chapter 5.7.2.
1
5
10
15
20
25
30
35
40
45
Fig. 38 The circular dichroism spectra of cinchonidine in toluene and its change by addition ofEtPy. (from ref. 93 with permission)
224 | Catalysis, 2010, 22, 144–278

The Cotton shift of CD was measured at different [CD]0/[QN]0 ratios.The corresponding circular dichroism spectra are shown in Fig. 39. TheCotton shift around 235 nm appeared to be very sensitive to the amount ofquinuclidine added. This Cotton shift is related to the dimer form ofCD.192,404 Analogous results using MeO-CD have shown that this alkaloidcannot form a dimer.
6.3 Characterization of the solid and the solid-liquid interface
6.3.1 Introduction. Various surface science techniques were used so farto investigate the interaction of substrates and modifiers or the substrate-modifier complex with the Pt surface. Most of these methods are usingconditions (often high-vacuum) far from those applied in catalytic hydro-genations. For this reason, although some important details of the ad-sorption behaviour of CD and substrates have been revealed, the resultshave to be treated with certain precaution.
Surface characterization methods applied so far are as follows: (i) near-edge absorption fine structure spectroscopy (NEXAFS),429 (ii) X-ray photo-electron spectroscopy430,431(XPS), (iii) low-energy electron diffraction(LEED,431 (iv) scanning tunnelling microscopy (STM),431–433 (v) reflection-absorption infrared spectroscopy (RAIRS),434,435 (vi) surface-enhancedRaman spectroscopy (SERS),436,92 (vii) attenuated total reflection infrared(ATR-IR) spectroscopy,303,437,438 and (viii) electrochemical polarization.242
It had to be emphasized that only ATR-IR spectroscopic method241,438
and its combination with modulation excitation spectroscopy (MES) in aflow-through cell439 can be considered as appropriate methods approachingalmost real in situ conditions. It has to be emphasized that above twotechniques have the advantage to obtain information about adsorptionprocesses at the solid-liquid interface. In this respect it is important tomention that exact vibrational assignments for adsorbed CD on Pt surfaceusing combination of experimental vibrational spectroscopic measurementsand it>/it> computational methods were also reported.92,440,441 Recently a
1
5
10
15
20
25
30
35
40
45
Fig. 39 Circular dichroism spectra of cinchonidine in the presence of different amount ofquinuclidine. [CD]0=1.2� 10� 5M (from Ref. 404 with permission)
Catalysis, 2010, 22, 144–278 | 225

new method for in situ spectroscopic investigation of heterogeneous cata-lysts and reaction media at high pressure have been developed.442
6.3.2 Investigation of the substrates. The adsorption of EtPy on Pt(111)at low temperature was investigated by XP and UP spectroscopy.430 Theresults indicated that EtPy adsorbed strongly to the Pt. The ketone carbonylis more strongly involved in the chemisorption bond than the carboxyl one.Further analysis showed that EtPy is predominantly adsorbed in a tiltedrather than a completely flat mode.
The behaviour of EtPy during adsorption on alumina-supported plat-inum films and on a commercial 5wt% Pt/ Al2O3 catalyst has been studiedin the absence and presence of coadsorbed CD.91 The in situ ATR-IR studyat room temperature using hydrogen-saturated CH2Cl2 as solvent demon-strated that upon adsorption on the Pt EtPy decomposes with the formationof strongly adsorbed CO and other organic residues. The presence of CD(10� 4M) strongly decrease the rate of decomposition of EtPy.
Upon using STM method self-condensation of MePy over Pt surface wasobserved.310 This reaction took place in the absence of cinchona modifier atlow hydrogen coverages. Based on this finding new set of side reactions withthe involvement of MePy was proposed and conditions to avoid the by-product formation was discussed (see Chapter 5.1).
Side reactions of EtPy during enantioselective hydrogenation on Pt/Al2O3 have been investigated using in situ ATR-IR and ex situ DRIFT.443
The studies revealed that EtPy can decomposed and polymerize (aldolcondensation) under conditions of hydrogenation. These side reactions takeplace both on the Pt site and the Al2O3 support.
Based on analysis of the RAIRS spectra of MePy it has been shown thaton Pt(111) at room temperature MePy undergoes ‘‘surface mediated enolformation’’ leading to an assembly of H-bonded superstructures.312 Thedecrease of the temperature and the use of low background hydrogenpressure suppress these surface reactions.
In a recent study adsorption and reaction of EtPy on Pt/g-Al2O3 wasstudied by IR spectroscopy.313 Several side reactions of EtPy were detected.These results were discussed in Chapter 5.1.
The adsorption mode of MePy and EtPy was studied under ultra-highvacuum conditions on Pt single-crystal surfaces using X-ray and UVphotoelectron spectroscopies (XPS and UPS),430 NEXAFS,96 and(RAIRS).444 The results indicated that alkyl pyruvates adsorbs via lonepair-metal interaction of both carbonyl groups, i.e., in cis conformationwith their plane oriented normal or tilted with respect to the surface. Athigh coverage, a minority species was assigned to an Z1-transconfiguration.444
The coadsorption of hydrogen resulted in significant influence on theadsorption of alkyl pyruvates by lowering the tilting angle of the adsorbedspecies445 and suppressing surface polymerization of the adsorbed enedio-late species observed earleir.310
6.2.3 Investigation of the modifiers. Adsorbed forms of cinchona al-kaloids display different IR spectra from each other and from the solutionform of the alkaloids. This fact makes vibrational spectroscopy a suitable
1
5
10
15
20
25
30
35
40
45
226 | Catalysis, 2010, 22, 144–278

method to investigate the adsorption of cinchona alkaloids on metal sur-faces. Although this method seems to be very powerful it could not answerthe key question, namely, which of these species interacts with the substratein the enantio-differentiating step.
The first information that the mode of adsorption of CD on Pt dependson its coverage was obtained by using in situ ATR-IR spectroscopy.241 Atlow coverage the flat mode, while at high one the tilted mode prevails.Further study revealed that abstraction of hydrogen form the quinoline ringcan also take place resulting in a so called a-H abstracted form.437 Study onPt/Al2O3 in the presence of an organic solvent and hydrogen revealed threedifferent adsorption modes of CD as shown in Fig. 42.
Infrared spectroscopy (IR), Raman spectroscopy, surface-enhancedRaman scattering (SERS) and reflection–adsorption infrared spectra(RAIRS) studies434,437,440 ratify the results discussed above (Fig. 40).
The adsorption of CD on Rh/Al2O3 has also been investigated usingATR-IR spectroscopy. The adsorption appears to be more complex thanthat observed on Pt and Pd. Strongly adsorbed flat form was observed onRh when adsorption was performed in the absence of dissolved hydrogen.This form is responsible for the fast hydrogenation of the quinoline ring anddoes not allow the detection of the flat form in the presence of dissolvedhydrogen.272 Contrary to Pt it has been discovered that on Rh hydrogen-ation of the heteroaromatic part of the quinoline ring takes place. AdsorbedCD in the flat geometry is the intermediate of the hydrogenation reaction,
1
5
10
15
20
25
30
35
40
45
Fig. 40 Suggested adsorption mechanism of cinchonidine on Pt/Al2O3 at 283K based on ATRexperiments; y represents the surface coverage. Species 1: p-bonded, 2: a-H abstracted and 3: Nlone pair bonded (tilted). (Reproduced from ref. 437 with permission)
Catalysis, 2010, 22, 144–278 | 227

Zaera and coworkers investigated the adsorption of CD at Pt using in situRAIRS technique.434 Good correlation has been found between the surfaceconcentration of flat lying CD and the enantioselectivity in EtPyhydrogenation.
In another study it was found that the oxygen present in most CD so-lutions from dissolved air blocks the surface toward any CD uptake, pre-sumably via its dissociation to atomic surface oxygen atoms (and maybe bypartial oxidation of the platinum surface), while Ar, N2, or CO2 has noinfluence on the adsorption of CD. Hydrogen plays a unique role, initiallyfacilitating the uptake of CD.90
Non-linear effects (see Chapter 5.6.3) has also been characterized byin situ ATR-IR spectroscopy comparing the behaviour of CD andPhOCD.441 It was shown that both alkaloids are adsorbed via the quinolinering and that the spatial arrangement of the quinuclidine ring is crucial forthe chiral recognition. The result helped to elucidate the role of the phenylgroup played in the creation of the chiral space responsible for the inversionof ED.
Surface-enhanced Raman spectroscopy has been applied to investigatethe adsorption of CD on polycrystalline Pt.92 The effects of liquid-phaseconcentration in ethanol and that of co-adsorbed hydrogen were studied.It was found that CD is strongly and irreversibly adsorbed through itsquinoline ring via p-bonding. Stronger adsorption of DHCD comparedwith CD was also suggested.
The room-temperature adsorption of four cinchona alkaloids and threereference quinoline-based compounds from CCl4 solutions onto a poly-crystalline Pt surface was characterized by in situ RAIRS.446 The results areshown in Fig. 41. Data show Langmuir type adsorption kinetics. The
1
5
10
15
20
25
30
35
40
45
Fig. 41 Adsorption uptakes for all the quinoline-derived compounds from CCl4 solutionsonto Pt as a function of concentration. (Reproduced from ref. 446 with permission)
228 | Catalysis, 2010, 22, 144–278

calculated adsorption equilibrium constants (Kads) are given in Table 18and were found to follow the sequence CNWQDWCDWQNW6–methoxy-quinolineWlepidineWquinoline. Some of this ordering can be explained bydifferences in solubility, but QD displays a much larger Kads than expectedon the basis of its large relative solubility.
Results indicated also that each alkaloid binds differently on Pt at sat-uration coverages. At low concentrations all alkaloids adsorb with theirquinoline ring flat on the surface and then tilt abruptly upon increasingcoverages, but the switch-over takes place at significantly different solutionconcentrations in each case. CD tilts mainly along its quinoline long axis,whereas CN does it along the short one. CN has also larger degree of ringdistortion. The most surprising result is the fact that CN shows a higherKads than CN, QN, or QD. In this In this respect results obtained in anearlier study has to be mentioned. As shown in Fig. 42 when in sequentialintroduction of CCl4 solutions of CN, CD, and back to CN was applied CNwas replaced by CD, while CN cannot replace CD.380 Similar conclusionscan be obtained from other studies using the ‘‘chiral switch’’ techni-que.239,383 The difference in the adsorption mode of CD and CN was in-vestigated in a recent study.447
The main message from these studies is that the solvent has to be takeninto consideration in the formation of the above discussed adsorbed formsof alkaloids.
The adsorption of 1-(1-naphthyl)ethylamine (NEA) on platinum surfaceshas also been characterized by RAIRS and temperature-programmed de-sorption (TPD) both under ultra-high vacuum and in situ from liquidsolutions.448
ATR-IR spectroscopy was also used to prove the flexibility of the qui-nuclidine moiety resulting in surface quinuclidine bound CD. It was done bycomparison of the ATR-IR spectra of CD and PhOCD adsorbed onPt.98,441 The difference in the intensity of the signal at 1458 cm� 1 (d(C–H)deformation modes of the quinuclidine skeleton) was attributed to thepossible interaction of the quinuclidine moiety of CD with the Pt surface.Based on the comparison of the ATR-IR spectra of CD and CD hydro-chloride adsorbed on Pt under similar condition the authors came to theconclusion that at the Pt the quinuclidine moiety of CD has identicalstructure as in the protonated quinuclidine of CD hydrochloride.98 Thisfinding was considered as an additional evidence that CD can be protonated
1
5
10
15
20
25
30
35
40
45
Table 18 Adsorption equilibrium constants (estimated from the data given in Fig. 41 and
expressed as Kads� 1) and solubilities in CCl4 for the quinoline-derived compounds. (Repro-
duced from ref. 446 with permission)
Compounds Kads� 1 mM Solubility in CCl4, mM Kads
� 1/solubility
Quinoline 30 infinite
lepidine 11 infinite
6-methoxyquinoline 6.5 infinite
QN 0.65 8.63 0.075
CD 0.5 1.56 0.32
QD 0.25 16.3 0.015
CN 0.1 0.30 0.33
Catalysis, 2010, 22, 144–278 | 229

by chemisorbed hydrogen in aprotic solvent. However, the authors admitthat ‘‘this conclusion is tempting, further studies are needed to confirm itsvalidity. Doubts may arise from the presence of HCl originating fromCH2Cl2 solvent decomposition on Pt’’.329 We also have serious doubt withrespect to these interpretations.
ATR-IR method was also used to give some new information about theformation of quinuclidine bonded form of alkaloids. However, these resultsare quite dubious. In this respect in a recent study the following informationwas given:272 ‘‘Recent results indicate that the quinuclidine moiety is alsoinvolved in adsorption on Pt.98,449 At low coverage, the energetically fa-voured geometry exhibits the aromatic ring parallel to the surface and thequinuclidine moiety oriented toward the metal surface in a geometry thathas been named surface quinuclidine bound (SQB).450
However, the careful analysis of references given above clearly indicatesthat there is no experimental evidence for the above statement. In ref. 449upon using molecular dynamics simulation ‘‘CD was found to adsorb withthe quinoline ring oriented largely parallel (ao61) to the surface. CD sur-face attachment was found to be through both p- bonding of the aromaticgroup and adsorption of the CQC double bond of the vinyl group’’. It wasalso mentioned that ‘‘we found that CD conformation at the surface wasnot only affected by the ethanol solvent, but also by the cinchonidine–cinchonidine steric interactions and their competition for surface sites’’.However, no words were given related to the involvement of the
1
5
10
15
20
25
30
35
40
45
Fig. 42 In-situ RAIRS from experiments: sequential introduction of CCl4 solutions of CN,CD, and back to CN (from top to bottom). (Reproduced from ref. 435 with permission)
230 | Catalysis, 2010, 22, 144–278

quinuclidine moiety in the adsorption. In ref. 98 only computational resultswere given, while ref. 450 contains scientific speculations and not real evi-dences. Consequently, there is no exact experimental evidence for the for-mation of surface quinuclidine bound CD.
The adsorption of CD and CN on Pt(111) and Pd(111) single crystals hasbeen investigated by means of time-lapse STM in an ultra-high vacuumsystem.451 CD and CN showed similar adsorption modes and diffusionbehaviour on Pt(111). The only exception is that the flatly adsorbed CNmolecules were significantly more mobile than CD.
NEXAFS and corresponding STEX calculations have been appliedto investigate the orientation of DHCD on Pt(111) at 298 and 323K.429
The results indicate that at 298K the quinoline ring is almost parallelto the Pt surface but is tilted up from the surface by 60� 101. However, theresults show that at higher temperatures the alkaloid dissociates toquinoline.
Various techniques, such as NEXAFS, XPS, STM, and temperatureprogrammed reaction was applied to investigate at 320K the molecularorientation, spatial distribution, and thermal behaviour of the powerfulchiral modifier precursor (S)-naphthylethylamine adsorbed on Pt(111)433
No formation of ordered arrays was observed in the presence or the absenceof coadsorbed hydrogen. Based on high resolution STM images somespeculation was done related to the formation of 1:1 docking complex be-tween MePy reactant and the chiral modifier.
NEXAFS revealed that the quinoline ring of 10,11-dihydrocinchonidineis orientated parallel to the surface at 298K, whereas at 323K the orien-tation is tilted about 601 to the surface.429 von Arx et al. used STM to revealthat the cinchonidine molecules are randomly distributed on the Pt (111)surface.452
Attard and co-workers studied the influene of surface structure andsurface chirality on the adsorption rate of several modifiers.453 However,probably due to the large adsorption energy of these systems, no differencein the adsorption rate was observed. It was also observed that in a hydro-gen-saturated solution, the alkaloid dihydrocinchonine is partially desorbedfrom a kinked, chiral Pt surface.
The adsorption of CD on polycrystalline Pt surfaces in H2SO4 was in-vestigated by cyclic voltammetry.242 The adsorption was found to be ir-reversible. The results indicated that at maximum coverage, 50% of the Ptatoms were still accessible for hydrogen adsorption. They calculated alsothe site requirement for CD equal to 13–14 Pt atoms. In another study it wascalculated that in the surface modification model each enantioselective siterequires 25 or so Pt atoms to achieve simultaneous adsorption of modifier,reactant, and hydrogen.454
The adsorption of quinoline and CN on Pt (111), Pt (332) and poly-crystalline Pt electrode has been studied by differential electrochemical massspectrometry (DEMS). It was shown that benzene is even able to displacesome of the alkaloid.455
Electrochemical method was applied to investigate the introduction ofcinchona alkaloids with R- and S-kink sites of the Pt(643) surface.455 Nointeraction was evidenced.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 231

6.2.4 Substrate modifier interactions. Interaction of KPL with CD wasinvestigated by ATRIR concentration modulation spectroscopy usingCH2Cl2 as a solvent.316 The results showed that in the presence of CD andKPL a new band appeared at 2580 cm� 1 as shown in Figs. 43 and 44.
This band was attributed to the formation of protonated quinuclidine bychemisorbed hydrogen. This experimental results is considered as a keyprove for the support of authors general view with respect to the reactionmechanism (see more details in Chapter 8), In this respect the use of CH2Cl2solvent has to mentioned. Due to its use it is not excluded that the observedprotonation is simple an artefact.
Recently the formation of a surface complex between adsorbedcis-EtPy and protonated CD has been suggested using ATRIR methodduring asymmetric platinum catalysed hydrogenation of EtPy in super-critical ethane solvent.456 These results are shown in Fig. 45. Basedon the shifts in the 1200–1300 cm� 1 region preferential adsorption ofEtPy as cis-conformer was suggested. The appearance of the band at1660 cm� 1 was tentatively ‘‘be attributed to carbonyl stretching vibrationsof EtPy’’.
1
5
10
15
20
25
30
35
40
45
Fig. 43 Demodulated ATR spectra of different concentration modulation experiments. TheKPL concentration was modulated (modulation period T (184 s) between 0 and 5� 10� 2mol/Lin CH2Cl2. Spectrum a: clean, uncoated Ge internal reflection element; spectrum b: a Pt/Al2O3
film in the absence of CD; spectrum c–e were recorded on a Pt/Al2O3 film in the presence of CD(5� 10� 4mol/L). Before the modulation experiments were started (c–e), the Pt/Al2O3 filmswere differently treated: (c) 30min N2 saturated CH2Cl2 only; (d) pretreatment with 5 min H2
saturated KPL solution; (e) directly contacted with modulation solutions. (Reproduced fromref. 316 with permission)
232 | Catalysis, 2010, 22, 144–278

The above shift to lower wave-numbers occurs ‘‘due to hydrogen bondingbetween the quinuclidine nitrogen and the keto oxygen atom of EtPy’’.However, in this respect it is necessary to mention the author’s followingstatement: ‘‘This interpretation is in line with our recently reported study on
1
5
10
15
20
25
30
35
40
45
Fig. 44 Comparison between two KPL concentration modulation ATR experiments. TheKPL concentration was modulated (modulation period T 184 s) between 0 and 5� 10� 2mol/Lin CH2Cl2. Use of modifiers: (a) N-Methyl CD (5� 10� 4mol/L), (b) CD (5� 10� 4mol/L).(Reproduced from ref. 316 with permission)
Fig. 45 ATR-IR spectra of adsorption/reaction of EP in ‘‘supercritical’’ ethane on (1) Al2O3
in absence of H2; (2) CD-premodified Pt-black; (3) unmodified Pt/Al2O3, but CD dissolved inEP; (4) CD-premodified Pt/Al2O3. Conditions in all experiments were 40 1C, and 95 bar. Molarratio EP:H2 : ethane =1:5:200. (Reproduced from ref. 457 with permission)
Catalysis, 2010, 22, 144–278 | 233

KPL adsorption on a CD-modified Pt/Al2O3 thin film’’.437 However, in theabove reference there is no words related to the assignment of any bandsaround 1660 cm� 1.
The conformational flexibility of the quinuclidine moiety was investigatedby ATRIR experiments under nearly in situ conditions, by comparing theadsorption behaviour of CD and O-phenyl-CD on platinum.98,441 It wasconcluded that the tertiary nitrogen of the quinuclidine moiety can par-ticipate in the anchoring of the alkaloid and can be protonated by surfacehydrogen. This study is related to the mechanism proposed by Baiker’sgroup whereby the tertiary nitrogen can promote charge polarization ofhydrogen and its transfer to the substrate (see more details in Chapter 8).
There is one more comment on these results. Even if we accept resultsshown in Figures 43–45 one question still remains: What is the proof thatsurface species assigned to the [CD-Hþ -substrate] complex is really in-volved in the ED?
6.2.5 HPLC-MS and HPLC-ESI-MS investigations. These studies wererelated to the investigation of the products formed from different alkaloidsduring enantioselective hydrogenation reactions. Upon investigation of theeffect of a-ICN it was demonstrated by HPLC–ESMS measurements thatthe cyclic ether structure of the alkaloid remained unchanged.457 In anotherstudy the product of isomerization of b-ICN, b-isocinchonicine (b-ICNN),was hydrogenated using supported Pt and Pd catalysts. The products wereanalyzed using HPLC-ESI-ion-trap MS measurements.458
Combined HPLC and ESI-MS method was used to investigation ofcinchona alkaloid derivatives formed in the hydrogenation of a-ICN and b-ICN.198 The products of reaction are shown in Fig. 46. The hydrogenatedcompounds were identified as 10,20,30,40- tetrahydro-a-ICN (A) and10,200,300,400-tetrahydro-b-ICN (B) and decahydro-a-ICN (C).
Upon investigating C9-O-substituted cinchona alkaloids in the enantio-selective hydrogenation of EtPy ESI-MSD-ion-trap method was applied tofollow and identify the hydrogenated derivatives of these cinchonaderivatives.459
HPLC-MS method was used to investigate products of H–D exchangemeasurements of different alkaloids.460 As revealed by these measurements,iso-alkaloids are not converted back to CN or QD; (v) in all alkaloidsstudied, H–D exchange takes place on the quinoline skeleton as well as oncarbon atom C9; (vi) H–D exchange on the quinuclidine skeleton appearssignificant only in the case of CN and a-ICN.231 Deuterium exchange in CDwas also studied in ref. 48.
1
5
10
15
20
25
30
35
40
45
Fig. 46 Cinchona alkaloid derivatives formed by hydrogenation of a-ICN and b-ICN.
234 | Catalysis, 2010, 22, 144–278

Potential products of hydrogenation of a-ICN were investigated in an-other study. The aim of this work was identifying hydrogenated cinchonasformed during enantioselective hydrogenation of EtPy. The target com-pound was DHCN which might possibly be formed from a-ICN. The resultsobtained by HPLC/ESI-MS measurements showed that of DHCN was notformed.387
7. Theoretical calculations
7.1 Introduction and first attempts
The proposed mechanisms for the asymmetric hydrogenation of activatedketones over cinchona-platinum catalyst system that leads to the observedED require supports using different theoretical-computational studies.These studies are related to the conformational analysis of both substratesand modifiers, adsorption of both substrates and modifiers into Pt andenergetic calculations for the whole complex system Pt-modifier-substrate.
It is generally accepted such that the CD or its natural or syntheticanalogue forms a 1:1 complex, what is hydrogenated on the metal surface.The question is where the chiral discrimination takes place on the Pt surfaceor in the liquid phase.
Both the structure and the conformational complexity of cinchona al-kaloids generate several possible interactions with the substrates. As a re-sults a ‘‘chiral pocket’’ is created for the ED either on the Pt surface or in theliquid phase.
The way to explore these properties one has to investigate or modeltheoretically the characteristic features of substrate molecules and the cin-chona alkaloids. Based on this knowledge, the modifier-Pt and the sub-strate-modifier and substrate-modifier-Pt interactions can be investigated.It is useful to extend all these calculations with solvent effect.
Various computational methods have been used so far, such as molecularmechanics461 and quantum chemical calculations.462 Molecular geometriescan be optimized on MMFF94 molecular mechanic level. Relativelyaccurate energies (mainly for energy differences) can be obtained e.g. onHF-SCF/6–31G* or B3LYP/6–31G* level single point energy calculations.The involvement of metal in these calculations requires the use DFTmethods.
In the next sub-chapters we shall review most of the relevant results re-lated to the computation on substrates, modifiers. Calculations related tothe substrate-modifier interactions and possible interactions of all thesecomponents with Pt surface will be discussed in Chapter 8 related to thereaction mechanism.
7.2 Characteristic features of substrate molecules
Among the substrates, for which the cinchona-Pt catalytic system yieldshigh ee values, the following groups of molecules are in the focus of ex-periments and computations: (i) pyruvate esters, (ii) ketopantolactone(KPL), (ii) diketones (PPD), (iii) trifluoro acetophenone (TFA), and (iv)trifluorodiketones. In addition, the fluor substituted derivatives of the first
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 235

and third groups of compounds were also included into computationalstudies.
It was also shown that s-trans conformer of MePy is more stable in thegas phase by 1–2 kcal/mol, but the relative stability could be strongly in-fluenced by the metal surface, especially because the s-cis conformer has aconsiderably larger dipole moment.463 With respect to the formation ofsubstrate CD complex the two conformers of MePy were compared,464 andit was found that the complex yielding (R)-methyl lactate upon hydrogen-ation was more stable than the corresponding pro-(S) complex by 1.8 kcal/mol (corresponds to an enantiomeric excess of 92%, in good agreement withexperiment), however, for the analogous complexes of s-cis-methyl pyruvatethe energy difference is only 0.2 kcal/mol in favour of pro-(R), corres-ponding to 17% ee value. The relation between the electronic structure ofa-substituted ketones and their reactivity465 in the racemic and enantiose-lective Pt-catalysed hydrogenation was also investigated. A correlationbetween the keto carbonyl orbital energy and the hydrogenation rate wasfound, which rationalizes the effect of the substituent on the rate of hy-drogenation (the often observed rate acceleration).
The first model calculations indicated that in the complex responsible forthe enantio-differentiation the a-keto ester existed in trans-conformation.466
Further studies revailed that in the enantio-differentiation complex thea-keto ester can also exist in its cis conformation464 The fact that in thehydrogenation of KPL to (R)-PL high enantioselectivites were obtainedindicated that the rigid cis conformation has no influence on the enantio-differentiation step.
Generally speaking, the number of substrate molecules with high ee is in arelatively narrow374,467 range: This strong substrate specificity has not beenanswered yet, neither by theory nor with computation. A-keto esters weremodelled in different studies.49,74,465,468 The trans conformer is more stablethan the cis one, the carbon atom in the keto group is partially positivelycharged, while the oxygen part is negatively.
In the hydrogenation of acetophenone and TFA derivatives on CD-modified Pt/Al2O3, the rates and ee values varied strongly with the natureof the aromatic substituents.334,367 The different reactivities were attributedto the electronic (and steric) effect of the substituents and to hydrogen-bonding interactions between the quinuclidine N atom of the alkaloid andthe carbonyl group of the substrate.363,465 Theoretical calculations revealeda linear correlation between the logarithm of the reaction rate and thehighest occupied molecular orbital and lowest unoccupied molecular orbitalstabilization DEorb of the carbonyl compounds, relative to the referencecompound (see Fig. 47.).53 The relative orbital stabilization is defined as thesum of two numbers: the difference between the energy of the anti-bondingorbital of the reference compound acetophenone and that of the substitutedacetophenone, and the corresponding energy difference for the bondingorbitals. The more stabilized the orbitals of the substituted acetophenoneare, the larger DEorb and the reactivity of the molecule are. According tothese calculations (where the metal surface was not involved), the origin of‘‘ligand acceleration’’ is the lowering of the p-orbitals in the diastereomericcomplex of the substrate and modifier. In the pro(R) and pro(S) complexes,
1
5
10
15
20
25
30
35
40
45
236 | Catalysis, 2010, 22, 144–278

the carbonyl -orbitals are differently stabilized, which results in differentintrinsic rates in the formation of the two enantiomers. It remains, however,to be proven that the concept can be extended to other substrates and re-action types.
The proton affinities of seven different ketones, vicinal diketones, and a-keto esters (acetophenone, TFA, 2,3-butanedione, PPD, MePy, EBF andKPL) have been evaluated theoretically using the conventional ab initio HFand several post-HF methods (MP2, MP4, CCSD), density functionalmethods with the B3LYP hybrid functional, as well as some ab initio modelchemistries [CBS-4M, G2(MP2), and G3(MP2)//B3LYP].469
In the most stable protonated species, the proton is bound to oneof the carbonyl oxygens in the molecule. The preferred site depends on themolecule. In two a-keto esters (MePy and KPL) the carbonyl oxygenof the ester group is protonated. In the case of EBF and the asym-metric vicinal diketone, PPD it is the carbonyl oxygen next to the phenylgroup, which forms a more stable bond with the proton. These pheno-mena can be understood in terms of resonance stabilization of theresulting cations. It was shown that the protonation of both the modifierand the reactant in acidic solvent hinders the formation of a reactant–modifier complex, which is believed to be crucial for enantio-discrimination,consequently in these cases the ee decreases. This decrease of eewas observed in case of butanedione (14% vs. 47%), KPL (35% vs. 79%)and PPD (6% vs. 65%) comparing results in AcOH and toluene,respectively.
It is know that trifluoro beta-diketones can also exist in enol form. Theadsorption of both the keto and enol forms of 1,1,1-trifluoro-2,4-diketoneinto Pt(111) was modelled and calculated410 DFT calculations including thesimulation of the interaction of a protonated amine with the enolate
1
5
10
15
20
25
30
35
40
45
Fig. 47 Linear correlation between the logarithm of the hydrogenation rate (mmol h� 1) ofacetophenone and TFA derivatives and the relative orbital stabilization DEorb. (Reproducedfrom ref. 53 with permission)
Catalysis, 2010, 22, 144–278 | 237

adsorbed on Pt revealed that only the C–O bond next to the CF3 group ofthe substrate is in direct contact with Pt and can be hydrogenated.
7.3 Characteristic features of cinchona alkaloids
Characteristic features of cinchona alkaloids have already been discussed inChapters 2 and 3. For this reason, we shall refer to information given inthese chapters. In the next sub-chapter we shall focus mainly on the con-formational analysis of cinchona alkaloids and their analogues.
7.3.1 Conformational behaviour of cinchonidine. Among the effectivechiral modifiers used for enantioselective hydrogenation of activated ke-tones over Pt/Al2O3 catalysts the most effective and widely used one is CD.The characteristic feature of this alkaloid is shown in Figs. 2 and 3.
It has already been mentioned earlier cinchona alkaloids were intensivelyinvestigated by NMR methods183,470 as described in Chapter 2. Con-formational behaviour of cinchonidine was calculated independently bydifferent groups.471,83,472,88 Most of these results are in accordance withearlier results discussed in Chapter 2.473,183 In our first study,84 the con-formational analysis was done by using rigid quinoline and quinuclidinemoieties. As a result, four stable conformations have been found. In oursubsequent study,472 all of these calculations were repeated in such a waythat only the phi and psi torsion angles were forced to be constant, while allother freedom of the molecule were left to relax. The conformational an-alysis indicated that CD might exist at least in nine different forms, howeveronly four of them are relatively stable (two open (A1 and A2) and twoclosed (C1 and C2) conformers). These results are shown in Fig. 54. Thesolid line in this figure gives the contour of the possible forms of CD within8 kcal/mol energy range. For CD the 2–D NOE spectra indicate454 that themajor conformation in solution is conformation A2, this is close to thatadopted by the molecules in the solid state.177 Thus, the conformationalanalysis strongly indicates that CD can exist both in open and closed formsand both forms of CD can be involved in the formation of substrate–modifier complex.
Detailed conformational analysis of CD in solutions using NMR tech-niques as well as theoretical calculations was done in ref. 88. Three con-formers of CD are shown to be stable at room temperature, cl(1), cl(2), andop(3), with the latter being the most stable in apolar solvents. The stabilityof the closed conformers relative to that of open(3), however, increased withsolvent polarity. In polar solvents the three conformers have similar energy(Fig. 48).
Structures and relative energies in kcal/mol of low energy CD conform-ations were calculated using hybrid density functional (B3LYP/6–31þG*/PCM B3LYP) and AMBER* optimization (AMBER*/GB/SA).416 Therelative stability of the conformers is as follows: op(3)Wcl(1)Wcl(2)Wop(4).
The effect of protonation on the conformation of CD was investigated ina recent study.474 It was shown that the protonation of cinchonidine ap-pears to hinder the rotation around the C40–C9 and C9–C8 bonds and tofavour only a narrow range of the conformational space of the molecule. Interms of the behaviour of CD and CN molecules in solution, 2D NMR
1
5
10
15
20
25
30
35
40
45
238 | Catalysis, 2010, 22, 144–278

experiments indicate a somewhat more restricted rotational conformationspace for cinchonine than for cinchonidine.475 Protonation of cinchonidinealso significantly restricts its rotational conformation space.474
7.3.2 Conformational behaviour of cinchona derivatives. Detailed NMRanalysis and ab initio quantum chemical calculations were performed on10,20,30,40,10,11-hexahydroderivatives of CD.413,414 The rotation around theC40–C9 and C9–C8 bonds led to conformers of close energies, providingevidence on the possible presence of other stable conformers in the solutionof these cinchonidine derivatives.
The conformational analysis of the synthetic chiral modifier O-phenyl-cinchonidine in vacuum has been performed at semi-empirical level and atDFT level with a medium-size basis set for energetics related to the parentalkaloid cinchonidine.426 The O-phenyl-cinchonidine behaves similarly tocinchonidine and owns the same main stable conformers as mentionedabove in vacuum and in CH2Cl2 and CCl4 solvents. Based on combinedtheoretical – experimental results, the open(3) appears to be the mostpopulated in these solvents, but indication was found that an excesscl(2) conformer has to be also expected in CD2Cl2 in comparison to CD.The authors suggest that the sterical constraints imposed by insertionof O-phenyl at the C9 position shows its effect when the substituted CDadsorbs on the surface via its quinoline part.
Isocinchonines belongs to the class of rigid alkaloids (see Chapter 3.2).In these molecules the rotation of the quinuclidine ring is restricted (seeFig. 55.). b-ICN was investigated in a recent study and its conformationalanalysis was performed. The results confirmed that the numerous con-formational changes possible for CD and CN are reduced to a single degreeof freedom, namely rotation around C(40)-C(9).476
1
5
10
15
20
25
30
35
40
45
Fig. 48 Conformational analysis of cinchonidine. The calculated energy map has been ob-tained by changing the torsion angles phi ((C30)–(C40)–(C9)–(C8)) and psi ((C40)–(C9)–(C8)–(C7)). The contours are given in steps 0.5 kcal/mol. (Reproduced from ref. 472 withpermission)
Catalysis, 2010, 22, 144–278 | 239

Recently detailed NMR, DFT, and X-ray investigation of some cinchonaalkaloid O-ethers related to the determination of their structure and con-formations was published.395
7.3.3 Other calculations. Conformational analysis of synthetic modi-fiers, such as (R)-2-1-pyrrolidinyl)-1-1-naphthyl)ethanol, (R)-2-1-pyrrolidi-nyl)-1-2-naphthyl)ethanol, and (R)-2-1-pyrrolidinyl)-1-1-8-methyl-naphthyl)]ethanol was also performed. Open–(3) conformers appeared to bethe most stable. Minimum energy structures of the pro-(R) and pro-(S)interaction complexes between (R)-2-1-pyrrolidinyl)-1-1-naphthyl)ethanoland trans ethyl pyruvate were also performed. Good quantitative agreementbetween calculated and experimental ee values has been found for theenantioselective hydrogenation of EtPy over Pt catalyst chirally modified bysynthetic pyrrolidinyl– naphthyl–ethanol modifiers, assuming that EtPyexists in the trans conformation in the adsorbed enantio-differentiatingcomplex. The destabilising repulsive interaction between EtPy and the an-choring aromatic moiety within the pro-(S) complex has been identified tobe important for ED.85
7.4 Substrate-Pt interaction
The adsorption of ketones on transition metals has been the topic of variousstudies.477–479 In general, ketones adsorb on transition metal surfaces via twodifferent bonding mode: as �1 (O) in an end-on adsorption configuration inwhich the oxygen atom is bonded by its lone pair orbital to the metal surface,
or as �2 (C, O), with both the carbon and the oxygen atoms of the keto-group
p-bonded to the metal and the CQO moiety lying parallel to the surface.The bonding interaction between an adsorbate and a surface is a very
complex process.480 To perform first principle calculation on the adsorptionof substrate molecule97 on a reasonably large (about 20–40 atom) Pt (or Pd)surface or cluster has become feasible only recently, however these resultsshould be handle cautiously. For example a drawback of using metalclusters of this size is that the Pt cluster is strongly paramagnetic (high spinstates) in the result of the computation,98 while experimentally it is notmagnetic et al.
The interaction of various ketones with Pt surface was investigated indetails.97 Fig. 49 shows the adsorption geometries of EtPy for both the cisand in the trans conformations. The cis Z2 adsorption appeared to be themost stable one (see Fig. 49a) In the Z1 adsorption mode only the transconformation showed an energy minimum (see Fig. 49c), whereas the cisconformer was not stable when Z1 adsorbed. When adsorbed Z2 the maininteraction the keto-carbonyl moiety interacts with the metal. Once thepreferred keto-carbonyl adsorption had taken place, the ester group inter-acts only weekly with the Pt surface.
The adsorption EBF and its derivatives onto Pt surface was also modelledand calculated.481 The results showed that the introduction of two o-sub-stituents into the aromatic ring completely eliminated the reactivity of theketone. The dramatic difference between EBF and ethyl mesithylglyoxylate(5) is their mode of adsorption. The o-substitution suppresses adsorption
1
5
10
15
20
25
30
35
40
45
240 | Catalysis, 2010, 22, 144–278

modes where the keto-carbonyl group is bound to the metal in Z2(C,O)mode involved in the hydrogenation reaction.
In a recent study the interaction of PPD with Pt surface was investigatedby DRIFT spectroscopic method and DFT calculations.482 Seven differentadsorption forms were suggested as shown in Fig. 50. The calculated ad-sorption energies are given in Table 19. DFT calculations demonstrated thatZ1-(O2) configuration is the most stable end-on adsorption mode of PPD.Tilted p-bonded adsorption mode of cinchonidine was revealed on theplatinum catalyst at higher concentration of CD.
7.5 Modifier-Pt interaction
The interaction of CD with Pt(111) both in ultrahigh vacuum (UHV) and inethanol solvent has been studied using molecular dynamics (MD) simu-lation. In UHV at low coverage (0.0125 molecules/Pt atom) and 298.15Kthe CD was found to adsorb with the quinoline ring oriented largely parallel(a=61) to the surface.449 Cinchonidine surface attachment was found to bethrough both p bonding of the aromatic group and adsorption of the CQCdouble bond of the vinyl group. The interactions between ethanol solutionsof CD (0.129 and 1.035M) and the platinum surface were also simulated.For the less concentrated solution (0.129M) two different equilibriumconformations were found, one in which only part of the quinoline is at-tached to the surface, and another slightly more stable conformation. In thelatter one the quinoline group is adsorbed parallel to the platinum surface.
1
5
10
15
20
25
30
35
40
45
Fig. 49 Adsorption modes of EtPy on Pt: (a) Z2-cis, (b) Z2-trans, (c) Z1-trans, and (d) semi-hydrogenated Z7.3 -cis. (Reproduced from ref. 97 with permission)
Catalysis, 2010, 22, 144–278 | 241

It was also observed that that CD conformation at the surface was affectedboth by the ethanol solvent and the CD-CD interactions and their com-petition for surface sites.
The conformations of CD adsorbed on a Pt(111) surface were also in-vestigated.450 Eight conformationally different adsorption states due todifferent degrees of rotation around the t1 and t2 degrees of freedom wereidentified. The possible role of these conformations in the formation ofchiral surface sites relevant to enantioselective hydrogenation was also in-vestigated. The comparison of the conformational behaviour of CD in so-lution and on Pt has revealed the effect of the metal surface on the internalmobility of the alkaloid. In the study the role of the adsorbed op(3) con-former, the observed conformational flexibility on the Pt surface revealedthe possibility that other conformers of CD also might be involved in ED.Closed conformations of CD are found to play an important role in the
1
5
10
15
20
25
30
35
40
45
Fig. 50 PPD adsorption modes. (Reproduced from ref. 482 with permission)
Table 19 Adsorption Energies for Different Adsorption Modes of 1-Phenyl-1,2-propanedione
(Reproduced from ref. 482 with permission)
adsorption configuration DE (kJmol� 1)
Z1(O1) � 19a
Z1(O2) � 36
pseudo- n1(O1) � 55
di- n1(O1, O2) � 3a
Z2(C1, O1) � 88b
Z2(C2, O2) � 38a
Z3(C1, O1, O2) � 59
a Taken from ref. 483. b Phenyl ring only partly adsorbed.
242 | Catalysis, 2010, 22, 144–278

conformational equilibria on the surface due to their stability and areidentified as precursors of the less stable, but probably more active, openconformers. Although the open and closed conformers are closely related tothe correspondent ones found in solution, surface species that are also ad-sorbed via quinuclidine moiety have been identified also as possible forms ofmetal–modifier interaction and can be involved in ED.
In a recent study the possible forms of adsorbed CD was given as shownin Fig. 51.482 Density functional theory (DFT) at the B3LYP/T(ON)DZPlevel was used to model one-to-one reactant-modifier interactions relevantto the enantioselective hydrogenation of PPD and MP over platinumcatalysts In an other study206 DFT calculations revealed that protonatedcinchonidine and 10,11-dihydrocinchonidine are more stable on Pt whenadopting the so-called QA-Open(4) conformation rather than the Open-(3)conformation. Thus, the QA-Open conformations may have some role inthe enantioselective hydrogenation over modified Pt catalysts. The results ofthese calculations are shown in Fig. 52.
8. Reaction mechanisms and related calculations
8.1 Introduction
In the first approaches related to the enantioselective hydrogenation ofactivated ketones over Pt-cinchona catalytic system mechanistic views de-veloped earlier for Ni-tartaric acid catalyst system.484 It also means that thefirst models were proposed without any solid knowledge about the reactionmechanism, i.e., the proposed reaction mechanism and schemes were basedon pure ‘‘presumption’’ related to the knowledge accumulated in studiesover the Ni-tartaric acid system.
However, careful analysis of these two enantioselective hydrogenationreactions shows definite differences as follows: (i) mode of introduction ofthe modifier, (ii) amount of modifiers, (iii) reaction rate, and (iv) reactiontemperature
In Ni/tartrate system the catalyst requires pre-modification under con-ditions different from those used in the hydrogenation reaction. Contraryto that the Pt/cinchona system the introduction of the chiral modifier
1
5
10
15
20
25
30
35
40
45
Fig. 51 The adsorption modes of cinchonidine: (a) parallel p-bonded; (b) tilted p -bonded; (c)a-H-abstracted quinolyl; (d) quinoline-N-lone pair. (Reproduced from ref. 482 withpermission)
Catalysis, 2010, 22, 144–278 | 243

into the reaction mixture during racemic hydrogenation instantaneouslyinduces ED.
In the Pt/cinchona system the substrate/modifier ratio is very high (in caseof KPL it is 276 000), but the optimum substrate/modifier ratio stronglydepends on the type of substrate. In Ni/tartrate system this value is severalorder lower.
In the Pt/cinchona system as it has already been discussed the cinchonaalkaloid induces not only enantio-differentiation but a well-pronounced rateacceleration. Contrary to that in the Ni/tartrate system the modifier de-creases the reactions rate. It has to be emphasized that the rate accelerationeffect has also been observed in homogeneous catalytic reactions in thepresence of cinchona alkaloids.357
In Ni/tartrate system the reaction takes place at moderate temperatureabove 60 1C, while the enantioselective hydrogenation of prochiral ketonesrequires low temperature around 0–10 1C. High temperatures above 40 1Care not favourable to get high ee values.
As it has been mentioned in a recent publication301 ‘‘two types ofmechanisms-modified catalyst,58,267,464,485 and shielding effect74 have beenproposed’’. Unfortunately, not all of the authors consider this way. Thosewho accepted the modified catalyst model (we shall call it ‘‘surface
1
5
10
15
20
25
30
35
40
45
Fig. 52 Side and top views of the RI-BP86/SV(P) optimized Open(3) and QA-Open(4) con-formations of cinchonidine, 10,11-dihydrocinchonidine, and their protonated counterparts onthe Pt38 cluster. The adsorption energies as defined in the text are given in parentheses (inkJmol� 1) (Reproduced from ref. 206 with permission)
244 | Catalysis, 2010, 22, 144–278

modification’’ model) did an enormous effort using various spectroscopicmethods and computational tools aimed to demonstrate that in fact onlyone reaction scheme is working, i.e., both in aprotic and protic reactionmedia the enantioselective hydrogenation reaction proceeds via the proto-nated form of cinchonidine and the surface entities responsible for theenantio-differentiation step has a N–Hþ–O bond.53 It also means that allkey events, i.e., the rate acceleration and enantio-differentiation are ex-clusively surface related phenomena.
Of course, the surface of Pt has a crucial role in this reaction. First or allPt provides landing sites for all participants in the given reaction, i.e., both‘‘actors’’ and ‘‘spectators’’ can land and react on the Pt sites. The question iswhat of these ‘‘surface induced interactions’’ shall have a direct contributioninto the rate acceleration and enantio-differentiation steps.
8.2 The ‘‘surface modification’’ model
The surface modification concept was first suggested in early nineties, whenthe number of publications in this area was very scarce. The distinctionbetween ‘‘modified’’ and ‘‘unmodified’’ sites over platinum was done byBlaser and coworkers.63 These terms have been widely accepted and used bythose who believe in the ‘‘surface modification’’ concept. The above dis-tinction was formulated into a kinetic equation describing the ‘‘ligand ac-celeration’’ phenomenon.58 It has to be mentioned that in the above studyno mechanistic views were given just a very simple reaction scheme. Ac-cording to this scheme enantioselective hydrogenation takes place over‘‘modified’’ sites, while racemic hydrogenation over ‘‘unmodified’’ sites.This model gives a relatively good correlation between rate and ee values,but it does not explain the variation of the ee values with the concentrationof the substrate.
The first mechanistic view or scheme was given by Wells and coworkers inearly nineties (‘‘template model’’).65,229 According to this the enantio-dif-ferentiating sites are created by an ‘‘ordered layer of the alkaloid’’ with theformation of a ‘‘chiral pocket’’, i.e., a free room between adsorbed chiralentities, where the enantio-differentiation can take place. However, theirconcept was not supported by surface spectroscopic methods454 and theoriginal idea was withdrawn45 very soon and a new idea based on the in-volvement of the half-hydrogenated form of a-keto ester in the enantio-dif-ferentiation step was proposed by the same research group.485 It has to beemphasized again that this new idea was suggested without any experi-mental prove or evidence.
The fact that in acetic acid the enantioselective hydrogenation of alkylpyruvates takes place with higher rates and higher enantioselectivity than inaprotic solvents the original idea given by Wells and coworkers was furtherextended to the involvement of the protonated form of cinchonidine.49 Inthis model the quinuclidine nitrogen atom is protonated and the substrate isstill in its original state, maintaining the double bond character of thecarbonyl group. Later on this model was accepted as a general one even inthe absence of acids.53 Baiker and coworkers have published severalexperimental85,97,394 and theoretical papers316,437,450 trying to convince the
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 245

readers that in the presence of cinchona alkaloids the atomic hydrogen isspontaneously ionized with the formation protonated form of alkaloid.There is a general view that the hydrogen-bonding interactions can triggerfor rate acceleration.363,465
8.3 ‘‘Shielding effect model’’83
8.3.1 The principle of chemical shielding.473 The basis for this approachis the shielding effect (SE) known in organic chemistry. If a prochiral moietyis preferentially shielded its further reaction can take place only from itsunshielded site resulting in an ED. A chiral template molecule can induceSE in a similar way, i.e. it preferentially interacts with one of the prochiralsites of the substrate leaving the unshielded site free for the reaction.
Intramolecular steric shielding of an a–keto ester moiety has been ob-served resulting in enantio-differentiation in the hydrogenation of thea-keto group.486 ED was observed only in the presence of large aromaticsubstituent, such as naphthyl, and it was completely lost if it was substitutedfor a phenyl one. Based on this finding the ED was attributed to the SEinduced by the large aromatic moiety. Similar phenomena was also de-scribed for the hydrogenation of an a,b- unsaturated ester moiety.487 Theabove two examples are shown in Fig. 53. Additional examples for chemicalshielding can be found elsewhere.488,489
8.3.2 Application of the principle of chemical shielding to the Orito’s
reaction.83,473 Both reacting groups shown in Fig. 69 have a common fea-ture, namely a conjugated double bond system. This feature is also char-acteristic for most of the substrates what can enantio-selectively behydrogenated in the presence of cinchona-Pt or cinchona- Pd catalyst490
systems.On the other hand it was also shown that in the hydrogenation of EtPy
over Pt/Al2O3 catalyst in the presence of new types of modifiers (derivativesof 2-1-pyrrolidinyl)-1-naphthyl)ethanol) the ED was completely lost if thenaphthyl ring was replaced by phenyl or pyridyl one.60 It should also bementioned that in the hydrogenation of a-keto esters over CD-Pt/Al2O3
1
5
10
15
20
25
30
35
40
45
R = Me, Et, Ph
OAc
O O
OR
O
CO2Me
O
CH2
OO
OAc
O O
R
CO2Me
Fig. 53 Intermolecular chemical shielding in the involvement of a-keto ester and a,b-un-saturated ester. (Reproduced from refs. 488 and 489 with permission)
246 | Catalysis, 2010, 22, 144–278

catalysts the ED was partially or fully lost if the quinoline ring of CD waspartially or fully hydrogenated.64
If the key p–p interactions given in Fig. 53 are compared with resultsobtained in the enantioselective heterogeneous hydrogenation experimentsusing cinchona-Pt systems the following very important elements of simi-larity can be found: (i) ED can only be observed in the presence of largearomatic shielding groups; (ii) the reactive prochiral group (both the ketocarbonyl and the CQC double bond) is activated by an electron with-drawing carbonyl group; (iii) the prochiral keto carbonyl group in most ofthe activated ketones is in a conjugation with the adjacent carbonyl group(or with the aromatic ring in TFA as it was shown earlier).472
As emerges from these comparisons the presence of a large aromaticsubstituents in the modifier and a conjugated double bond system in thesubstrate should play an important role to induce ED in these asymmetrichydrogenation reactions, i.e. these are the key elements responsible for thesubstrate specificity.
It has to be added that the shielding effect model suggest that substratemodifier interactions responsible for the ED take place in the liquid phaseand not on the Pt sites. The term substrate-modifier interaction in liquidphase was also mentioned by other authors. ‘‘The substrate-modifierinteraction exists, according to circular dichroism, in solution, probably inthe form of aggregates’’.68 Similar views were given in another study.491 Ithas also been suggested that for some substrates, the solvent is involved inthe substrate–modifier interaction.492 It has been suggested that the OHgroup of the alkaloid should be involved in the substrate–modifier inter-action which more likely occurs in the liquid phase.493
8.4 Character of substrate – modifier interaction
In a recent review it has been admitted that ‘‘in the absence of reliable ex-perimental evidence, most mechanistic ideas are based on assumptions and(at best) calculations. In most cases, the models assume two interactionsbetween the amine type modifier and the ketone: an N–H–3O52,77,468,494,495
or N-C type attractive interaction67,416,496 and a second attractive or re-pulsive interaction that directs the adsorption of the ketone on Pt.52,497
In the following section we shall follow the above consideration, i.e., weshall distinguish electrophilic and nucleophilic interactions between thesubstrate and the modifier. It has to be emphasized that all existing modelspostulate 1:1 type interactions between CD and the substrate. Bartok et al.498
and Augustine et al.373 proposed that not only the quinuclidine N, but alsothe OH function of CD would be involved in the interactions. However thisview can be questioned as neither the methylation nor the removal of the OHgroup in CD hinders the enantio-selection in the hydrogenation of EtPy.57
The first attempt aimed to elucidate the character of substrate-modifierinteractions was done in an early study468 related to the investigation ofinteraction between MePy with NH3 and NH4
þ . In this study the ammoniapart represented the quinuclidine nitrogen of CD. The results indicated thatMePy can interact with both NH3 and NH4
þ and the electrophilic inter-action is more favourable than the nucleophilic one. However, the
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 247

nucleophilic interaction provided interesting result, namely the reactionpocket of MePy is located between the two carbonyl groups. This findingindicated that under condition of enantioselective hydrogenation bothcarbonyl groups are activated. Consequently, if the enantioselective hy-drogenation of EtPy is performed in methanol trans-esterification reactioncan take place. This has been proved experimentally.309
In the next approaches two types of interactions were modelled, namely theBaiker’s group focused on the electrophilic interaction between the half-hy-drogenated substrate and CD.53,74,499,500 In our group, based on kinetic re-sults over cinchonidine-Pt/Al2O3, and the proposed ‘‘shielding effect’’ modelthe nucleophilic interaction between the modifier and substrate was favored.69
8.4.1 Electrophilic interaction. In the first theoretical study related to thesubstrate-modifier interaction the formation of a week complex betweenprotonated CD and MePy methyl pyruvate was investigated.74 In this studymolecular mechanics and AM1 semi-empirical methods were used. The cal-culated surface complex bifurcated electrophilic interaction between the pro-tonated quinuclidine and the keto carbonyl group was considered. The resultsrevealed that adsorption of the complex leading to (R)-methyl lactate is morefavorable than that of the corresponding system yielding (S)-methyl lactate.
In another study ab initio calculations were used to study the interactionbetween protonated amines (NH3, (CH3)3N and quinuclidine) and methylpyruvate (MP), as well as between protonated MP and these amines.501
Based on results it has been suggested that interactions mediated by aproton between the MP and the alkaloid are the main driving force leadingto enantiodifferentiation in the hydrogenation of a-ketoesters. MP interactswith protonated amines preferentially in the s-cis conformation, with aproton making two hydrogen bonds to the carbonyl oxygens. This protonmay be transferred to MP, forming a new complex in which the amines arebonded to the protonated MP. The last complex is approximately 10 kcalmol� 1 less stable than the first one. However, this energy difference de-creases to approximately 5 kcalmol� 1 when solvent effects are included.
Characteristic feature of these models is that both in the absence andpresence of acid in the key reaction intermediate the electrophilic interactionprevails with the involvement of N–H–O or N–Hþ–O bonds. This interactioncan be either monodentate or bidentate (bifurcated) as shown in Figs. 54.
1
5
10
15
20
25
30
35
40
45
Fig. 54 Schematic representation of mechanistic models suggested by Baiker and co-workers;A: monodentate interaction;49,98 B: monodentate interaction;340 C: bidentate interaction.340
(Reproduced from refs. 49, 98, 340 with permission)
248 | Catalysis, 2010, 22, 144–278

The key issue in this model the adsorption of the chrial modifier with itsquinoline ring parallel to the Pt surface as it has already been discussed inChapter 6. Several adsorbed conformations of CD were calculated.450
Sequence of events on the Pt surface related to the protonation of qui-nuclidine nitrogen is shown in Fig. 55, while the calculated substrate-modifier complex and its interaction with the Pt surface are given in Fig. 56.In this respect the waging motion of the quinuclidine part has been em-phasized. The overall route for proton transfer from protonated CD toadsorbed substrate supported by DFT calculations and in situ ATR-IRspectroscopy as shown in Fig. 57. It has be added that in the recent pub-lication of this group it was found that the most stable intermediate complexforms without adsorption of the substrate.103
We have serious objection against the exclusiveness of the electrophilicinteraction in the Orito’s reaction. Assuming the scheme given in Fig. 57.one would suggest that not only the quinuclidine nitrogen of the cinchonaalkaloid can be involved in the transformation of atomic hydrogen formedon the Pt site to a protonated nitrogen base. All tertiary nitrogen basesshould have similar ability. Consequently, the hydrogen transfer in thepresence of an achiral tertiary amines (ATA) should result in a racemic
1
5
10
15
20
25
30
35
40
45
Fig. 55 Simplified scheme for the interaction of the quinuclidine N atom with the Pt-H systemand the subsequent transfer of the H to the adsorbed ketone. (Reproduced from ref. 376 withpermission)
Fig. 56 Hydrogen uptake of CD from a platinum surface. (Reproduced from ref. 98 withpermission)
Catalysis, 2010, 22, 144–278 | 249

product. Thus, the simultaneous addition of cinchona alkaloids and ATAsshould result in a decrease in the ee values. However, our results discussed inChapter 5.7.2 showed a completely opposite effect.
According to Figs. 55–57 the key issues in the ‘‘surface modification’’model are as follows: (i) adsorption of the modifier with its condensedaromatic ring parallel to the Pt surface, (ii) stabilization of the modifier in itsopen form, (iii) adsorption of the substrate via its both carbonyl bonds in re-phase, (iv) formation of a hydrogen bond between protonated alkaloid andthe substrate and (v) transfer of the proton to the substrate. The condensedaromatic ring is often called as anchoring site.
As it has already be mentioned earlier the chiral C8 and C9 carbon atomsof the alkaloid play a vital role in the enantio-differentiation, i.e. theirconformation determines the position of the ‘‘chiral pocket’’ located in theneighbourhood of quinuclidine nitrogen. In addition the mode of ad-sorption of the chiral modifier (flat or tilted) has a decisive role in the EDstep as it has been shown in the series of O-substituted derivatives ofCD441.
In a recent study the role of the modifier structure in the reactant-modifierinteractions relevant to the heterogeneous enantioselective hydrogenationof PPD and MePy) was studied using DFT calculations.206 Two protonatedmodifiers, CD and MeO-CD, in different conformations were considered.So-called bifurcated and cyclic hydrogen-bonded reactant-modifier inter-action modes were investigated. The results showed that only the bifurcatedreactant-modifier(Open3) complexes were found to be relevant in the de-termination of enantioselectivity. Analysis of the orbital stabilization im-plies a notable decrease in the enantiomeric excess of the mainhydrogenation product of PPD when CD is replaced with MeO-CD. On theother hand, according to the theoretical calculations the hydrogenation ofMP over modified Pt is expected to yield an equal ee values in the presence
1
5
10
15
20
25
30
35
40
45
Fig. 57 Proposed relative surface structures of adsorbed CD and MePy on a Pt31 cluster(DFT calculations), which allow an H-bonding interaction (not shown). (Reproduced from ref.393 with permission)
250 | Catalysis, 2010, 22, 144–278

of both modifiers. DFT calculations revealed that protonated CD andDHCD are more stable on Pt when adopting the so-called QA-Open (4)conformation rather than the Open (3) conformation. These conformationsare shown in Fig. 58. Thus, the QA-Open conformations may have somerole in the enantioselective hydrogenation over modified Pt catalysts. TheQA-Open (4) conformation of a modifier is adsorbed on the surface via bothits quinoline and quinuclidine moieties, and a reactant may interact sim-ultaneously with the protonated quinuclidine nitrogen and the functionalgroup at the C(9) position of the modifier.
The interaction between KPL (Pro-(R) conformation) and adsorbed o-PyOCD over Pt surface was also modelled.376 In this case both the quinu-clidine and pyridine moieties in o-PyOCD were protonated. At the end ofthe simulation the hydrogen was transferred to the keto-carbonyl group ofketopantolactone, therefore forming a semi-hydrogenated surface species,while protonated o-pyridyl group coordinated to the ester carbonyl groupas shown in Fig. 59.
Modelling studies revealed also that there is no mode of docking of anylow energy conformation of epiquinidine with pyruvate ester that could leadto selective enantioface adsorption of the latter.502
1
5
10
15
20
25
30
35
40
45
Fig. 58 Calculated stabilized structure of CD over Pt in Open (3) and QA-Open (4). (Re-produced from ref. 206 with permission)
Fig. 59 Interaction of adsorbed o-PyOCD in the most stable position of the o-pyridoxymoiety, with KPL adsorbed in a Pro-(R) conformation. (Reproduced from ref. 376 withpermission)
Catalysis, 2010, 22, 144–278 | 251

Recently a new idea was suggested assuming a H-bond between thequinuclidine N of CD and the ester carbonyl or trifluoromethyl group of thesubstrate and a second, monodentate or bidentate H-bond involving two orone aromatic H atoms of the modifier at 50- and 60-positions and the O atomof the ketocarbonyl group.318,497,503
In addition, it was suggested that similar interactions might exist for allactivated ketones. General principles of this type of interactions are shownin Fig. 60.
This type of interactions has also been proposed in recent studies by theBartok’s goup388,504 (see Fig. 61). However, in this case the character ofsubstrate modifier interaction is nucleophilic.
However, these models strongly contradicts to experimental findings asring-substituted cinchona derivatives, such as QN and QD containing amethoxy group in the 60-position, are highly effective modifiers in the hy-drogenation of activated ketones (see Chapter 3.1). Consequently, theinteraction of the substrate molecule with the proton of the quinoline ring isnot a prerequisite for enantio-differentiation.
In addition, the proposed interaction of the keto group with the aromatichydrogen (see Figure cannot give any reasonable explanation for the acti-vation of the keto group resulting in rate acceleration.
Based on ESI-MS spectra of EtPy, DHCD and EtPy-DHCD mixturesinteresting equilibria (see Fig. 62) were suggested by Bartok et al.196 In thisrespect it interesting to note that the semi-ketal formed between the CD andEtPy was also evidenced in another study.109
1
5
10
15
20
25
30
35
40
45
Fig. 60 Two-point H-bonding model suggested by McBreen et al. A: interaction betweenprotonated CD and MePy, B: interaction between protonated CD and trifluoroacetophenone(TFA). (Reproduced from refs. 318 and 497 with permission)
Fig. 61 Hydrogen bounded substrate – modifier complex. (Reproduced from ref. 505 withpermission)
252 | Catalysis, 2010, 22, 144–278

8.4.2 Nucleophilic interaction. Nucleophilic interaction betweenthe substrate and the modifier has been suggested by differentauthors.83,84,373,416,468,506 In an early mechanistic view67 it was proposedthat ‘‘the hydrogenation of pyruvate over either modified or unmodifiedplatinum takes place on the more coordinatively unsaturated corneratoms or adatoms on the platinum surface’’. In their further study373
it was suggested that the substrate-modifier complex (association) ‘‘in-corporates some interaction between the quinuclidine nitrogen and theketone carbonyl group of the pyruvate. One such interaction is betweenthe electron pair on the nitrogen and the electron deficient carbon atomof the carbonyl group, which, as discussed above would account for theobserved increase in hydrogenation rate in these reactions’’. The involve-ment of coordinatively unsaturated platinum was also suggested by theBartok’s group.504,505
The ‘‘shielding effect’’ model is also based on the nucleophilic inter-actions. The key other key issue of this model is the involvement of closedconformer of CD in the substrate modifier interaction. The character ofthese interactions will be discussed in the next chapter. Upon investigatingenantioselective hydrogenation of KPL in the presence of b-ICN Bartok ancoworkers suggested two possible forms of surface complex representingeither electrophilic or nucleophilic interaction as shown in the next scheme(see Fig. 63.) The inversion of the ee was attributed to the change of thereaction mechanism from nucleophilic to electrophilic one.
It was also suggested that in the hydrogenation of activated ketones in thepresence of cinchona-Pt catalysts proceeds ‘‘through nucleophilic additionof a cinchona alkaloid to the ketone to form a zwitterionic adduct, which isthen hydrogenolyzed with inversion of configuration. The enantioselectivityof the reaction is determined by the relative stabilities of the diastereomericadducts adsorbed on platinum’’.416 However, this mechanism has beenruled out as it was pointed out that this approach does not take into accountsteric hindrance against the interaction of the amine modifier with cyclicketones and further critical point is the regioselectivity of the hydro-genolysis of the hypothetical zwitterionic intermediate.418
1
5
10
15
20
25
30
35
40
45
+
NH OH
NC
COOEt
OHMe
+
N
HOH
NH
C
COEt
Me
O
O
NO
HC COOEt
Me
OH
N
H
+
Fig. 62 Possible form of adducts between DHCH and EtPy (Reproduced from ref. 196 withpermission)
Catalysis, 2010, 22, 144–278 | 253

When the inversion of enantioselectivity in the presence of b-ICNs wasinvestigated the formation of different nucleophilic adducts were proposedas shown in Fig. 64.
8.4.3 Character of interactions in the ‘‘shielding effect’’ model. Cinchonaalkaloids (CA) have two rotational axises, which allow to rotate either thequinoline ring around the C(1) 0-C(9) axis or the quinuclidine ring aroundthe C(9)-C(8) axis (see Chapters 2.1 and 3.1). Molecular mechanics andab initio calculations performed by different groups that in liquid phase CAcan exist at least in three different stable forms. We suggest that the closedform of the modifier is required both for the RA and the ED. Only theclosed form of CA can provide the cooperative required for ED.
The possible arrangements of the substrate and the modifier in theshielded complex are shown in Fig. 65. In the hydrogenation of pyruvateesters the complex shown in Fig. 65A would result in the expected (R)-lactate ester, while the complex given in Fig. 65B would give the corres-ponding (S)-product. The major difference between the (R) and (S) com-plexes is the mode of interaction between the lone pair of electrons of the
1
5
10
15
20
25
30
35
40
45
Fig. 63 Enantioselective hydrogenation of KPL in the presence of b-ICN. (Reproduced fromref. 424 with permission)
Fig. 64 The proposed structures of adduct complexes of b-ICN (B) and CD (C) with esters ofphenylglyoxylic acids. (Reproduced from ref. 504 with permission)
254 | Catalysis, 2010, 22, 144–278

quinuclidine nitrogen and the keto carbonyl group. In complex (R), the‘directionality’507 of the nucleophilic attack by quinuclidine nitrogentowards the keto carbonyl group is very favourable to increase the reactivityof the keto carbonyl group because the electron- rich quinuclidine nitrogenand the keto-oxygen of the substrate are on the opposite sides of the ketocarbon atom. According to the orbital steering theory,508 a proper ‘reactionwindow’ or ‘reaction cone’ can result in perturbation of the reacting group.
In our case the proper ‘reaction window’ is determined by the relativeposition of the quinuclidine N1, pyruvate C2 and O3 atoms, i.e., by directN1–C2 interaction as shown in Fig. 68. In case of proper ‘reaction window’the overall reactivity of the keto group should increase. We suggest that theabove perturbation leads to a pronounced rate increase both in the hy-drogenation reaction and the formation of by-products, such as semi-ketal,transesterification and deuterium exchange products84,291,309 Thus, incomplex (R), the favourable directionality promotes the perturbation of theketo carbonyl group, resulting in the observed RA. Contrary to that incomplex (S), due to the misalignment of the interacting groups, i.e., due tothe lack of direct N1–C2 interaction, no RA can be expected, consequently,the hydrogenation of (S). complex is not accelerated.
Those who favour the modifier-surface or modifier-metal interactionssuggest that the quinoline ring is involved in the adsorption of the modifierto the metal.45,82,215 Contrary to that we suggest that the quinoline ring isinvolved in the p-p interaction with the substrate via ‘‘p-p stacking’’.83,84 Wesuggest that the RA is a cooperative effect with the involvement of both thequinuclidine nitrogen and the quinoline ring (Fig. 66).
Monte-Carlo simulation method was used to investigate the interactionof the [methyl pyruvate– CD]closed complex with Pt (111). surface. The resultshown in Fig. 67 indicates that the shielded complex retains its entity evenafter adsorption.
The above figure gives a good presentation of the SE provided by thelarge aromatic moiety. With respect to the explanation related to the use ofsmall Pt colloids we should like to refer to results of our calculations (MonteCarlo simulation). These results given in Fig. 68 shows that the closed[substrate – modifier] complex can be accommodated at the Pt(111) faceeven of a small Pt nanocluster.246
When the ‘‘shielding effect’’ model was proposed it was also mentionedthat in the enantioselective hydrogenation of activated ketones cinchonaalkaloids behave like an enzyme.93 This view has recently been emphasizedwithout reference to our original idea.103
1
5
10
15
20
25
30
35
40
45
Fig. 65 Shielded [methyl pyruvate-CDclosed] complexes. A-favourable alignment; B-un-favorable alignment. (Reproduced from ref. 472 with permission)
Catalysis, 2010, 22, 144–278 | 255

8.5 Pros and contras related to existing models
8.5.1 ‘‘Surface modification’’ model. The surface modification modelhas been strongly altered since the introduction of the first concept, i.e., thedivision of Pt surface into modified and unmodified sites. According to thegeneralized view the key issues in this model are as follows: (i) adsorption ofCD in its open (3) conformation via its quinoline ring parallel to the Ptsurface, (ii) conformational changes at the Pt surface to form quinuclidinebonded CD, (iii) formation of protonated quinuclidine moiety, (iv) trans-formation of the proton to the substrate to form half hydrogenated surfacespecies, and (v) direct addition of the second hydrogen form the Pt surfaceto get the chiral keto-alcohol. This scheme has been shown in Fig. 57.
However, based on the discussion in this contribution we can emphasizethat this model was not able to give appropriate answer to the following
1
5
10
15
20
25
30
35
40
45
Fig. 66 A-Simplified scheme for the [MePy–CDclosed ] complex; B-the ‘reaction window’ forthe substrate–modifier interaction in [MePy–CDclosed ] complex. (Reproduced from ref. 472with permission)
Fig. 67 Monte-Carlo simulation of the adsorption of the [MePy–CD]closed complex onto Pt(111) surface. (Reproduced from ref. 83 with permission)
256 | Catalysis, 2010, 22, 144–278

important observations: (i) rate acceleration (see Chapter 5.6.1), (ii) theappearance of the initial transient period in the conversion-selectivity de-pendencies (see Chapter 5.6.2); (iii) the influence of ATAs (see Chapter5.7.2), (iv) the influence of compounds with large aromatic ring (see Chapter5.7.3), (v) the influence of the modifiers of the Pt and support (see Chapter5.7.3), (vi) the stability of nucleophilic [substrate – modifier] complex (seeChapter 6.1.1) in the presence of AcOH, (vii) high enantioselectivities oververy small Pt nano-colloids (see Chapter 4.2.2).
Even if scheme shown in Fig. 57 is valid a simple question can be raised?Why the adsorption and subsequent strong distortion of the alkaloid at Ptsurface is needed to pick up a proton form the Pt surface if it could also bedone without any preadsorption via quinuclidine N-Pt interaction. Wouldnot be it more logical and energetically more favourable?
In this respect we have to address literature data related to the proto-nation of pyridine and its analogs observed under high vacuum and lowtemperatures.509,510 These references were cited in several papers as a directproof related to surface reactions given in Fig. 55. However, it has to bepointed out that none of the authors referred to experimental conditionsused in these earlier studies. It has to be emphasized that in refs. 510,511results obtained under ultra-high vacuum were presented. The key experi-ments were performed over Pt(110) surface under base pressure between5� 10� 11 and 1� 10� 10 Torr and the temperature was kept between 100–180K. The intensities of the 3450 cm� 1 EELS peak characteristic of cationformation showed strong temperature dependence and above 180K it washardly detected. Further peculiarities of ‘‘surface modification’’ model andsome recent mechanistic views will be discussed in Chapter 8.3.1.
Consequently, there are many speculations related to this model. Finally,it has to be added that this model does not take into account one of theimportant issues that these alkaloids are used by organic chemist for manyyears to induce ED or chiral separation.
1
5
10
15
20
25
30
35
40
45
Fig. 68 Monte-Carlo simulation of the adsorption of the [MePy–CD]closed complex onto thePt(111) surface of small Pt colloid. (Reproduced from ref. 246 with permission)
Catalysis, 2010, 22, 144–278 | 257

9.5.2 ‘‘Shielding effect’’ model (SEM). The shielding effect model canexplain the following experimental findings:74,83 (i) substrate specificity), (ii)inversion of enantioselectivity for enantiopairs CD-CN, QN-QD, (iii) rateacceleration, (iv) the MI character of the ee-conversion dependencies (v) theloss of ee in case of replacing the quinoline ring for pyridyl or phenyl, (vi)formation of transesterification and deuterium exchange products; (vii) ef-fectiveness of very small Pt colloids, (viii) the role of achiral tertiaryamines, (viii) the need for of large aromatic moieties in cinchona alkaloidsto induce ED.
The reaction network derived form the shielding effect model providedkinetic equations what can describe the following kinetic events:83 (i) rateacceleration, (ii) increase the reaction rate at the initial period of the re-action,66 (iii) the the MI character of the ee-conversion dependencies.
The strongest conflict with ‘‘shielding effect’’ model is the finding thatICNs can also induce enantio-differentiation. However, in this respect theanomalous behaviour of these rigid alkaloids365 has to be mentioned. Thebehaviour of these alkaloids needs further elucidation and probably the useof more pure alkaloids. In this respect the lack of rate acceleration in thepresence of a-ICN and the inversion of ee in the presence of b-ICN have tobe mentioned. Another unclear issue is that shielding effect model requiresnucleophilic interaction between the substrate and the modifier, while in thepresence of AcOH electrophilic interactions interaction prevails. Althoughin this relation recent NMR results has to be emphasized, what clearly in-dicated that in case of KPL even in the presence of AcOH the nucleophilicsubstrate-modifier adduct can be formed.424
Finally, we have to admit that the ‘‘shielding effect’’ model was notsupported by the scientific community in the field of heterogeneous cata-lysis. This fact can be attributed to the deficiency of the model. However, itcannot be excluded that due to the strong influence of those who favour the‘‘surface modification’’ model, the scientific community just simple followedthe main stream without any criticism.
9. Conclusions
In this review an attempt was done to give a retrospective overview aboutmethods, approaches and results obtained in the last three decades in thearea of enantioselective hydrogenation of activated ketones. Both practicaland theoretical aspects were discussed. Characteristic feature of this reviewis that the term ‘‘chirally modified surface’’ was not really used.
Although tremendous effort has been done so far to elucidate the pecu-liarities of this reaction there are still several open questions related to thesubstrate-modifier and substrate-modifier-platinum interactions involved inthe enantio-differentiation step.
It seems to us that starting from the beginning of early nineties there is apermanent desire to demonstrate and prove that in the presence of cin-chona-Pt catalyst system all interactions responsible for ED take place onthe Pt surface. In addition, last years the mainstream concentrated to provethat the protonated quinuclidine moiety is involved in the first step ofhydrogen transfer, with the involvement of adsorbed form of the alkaloid by
1
5
10
15
20
25
30
35
40
45
258 | Catalysis, 2010, 22, 144–278

its quinuclidine moiety and adsorbed hydrogen, i.e., the key mechanism inaprotic and protic solvents is the same. We consider that this mechanisticview is too general and in addition it is artificial and far-fetched. This viewneglects series of experimental evidences obtained by different researchgroups. Let us remind the reader only three of these neglecting facts:
Use of Pt colloid. As it was mentioned in Chapter 4.2.2 high rates andhigh enantioselectivities were obtained upon using very small Pt colloids. Innone of the reviews published so far by catalytic scientists this anomalousfindings were not really discussed. However, in a recent review written byorganic chemists it was emphasized that upon using Pt colloids in enan-tioselective hydrogenation of MePy in the presence of CD ‘‘the smallest Ptclusters gave the best results despite having no flat surface large enough forthe adsorption of cinchonidine’’.511
Addition of quinoline. In this respect one of the earlier results has also tobe mentioned.345 In this study it was shown that the addition of quinoline tothe reaction mixture at very low concentration (0.1 g/L) increased both therate and the ee values. The authors attributed this observation to some sortof base effect. Unfortunately, due to the dominance of the general view, i.e.the ‘‘formation of chirally modified surfaces’’ this result has completely beenforgotten and in the last eighteen years it was only very seldom cited. In thisrespect we should like to refer to our results discussed in Chapter 5.7.2).These results clearly indicated that the addition of quinoline has no negativeeffect either on the reaction rate and the ee values. Based on these findingsthe scheme shown in Fig. 69 has been suggested.
Fig. 69 shows four different situations described in ref. 102. A representsthe racemic hydrogenation in the absence of any additive, where due to thepoisoning effects of by-products the rate is controlled by free Pt sites left. Bcorresponds to the situation when quinoline is added prior to the additionof hydrogen. In this case the poisoning effect of by-products decreased re-sulting in a rate increase in racemic hydrogenation. C represents theenantioselective hydrogenation upon injecting CD, where the initial surfacecoverages are identical to those established in case A.
1
5
10
15
20
25
30
35
40
45
Fig. 69 Surface coverages in the presence of quinoline added to the reaction mixture.(Reproduced from ref. 102 with permission)
Catalysis, 2010, 22, 144–278 | 259

After injection of CD instantaneous RA and MI type the ee – conversiondependencies are evidenced. D corresponds to the situation when cincho-nidine is injected to surface B containing preadsorbed quinoline. In case D,due to the established competition between CD and quinoline the Pt sitesare covered by both CD and quinoline. However, the net results are un-expected, i.e., increased rate and increased ee. Consequently, these resultsmight indicate that the general view that ‘‘strongly bonded to the platinumCD via its quinoline ring’’ needs some corrections.
Spectroscopic results in liquid phase. Both NMR83,84,423,424 and CircularDichorism68,93 spectroscopic results clearly indicated that there is a complexformation between the substrates and cinchona alkaloids in the liquidphase. These facts were completely neglected by the scientific community. Inthis respect let us refer to a very recent results indicating that the nucleophiliccomplex between KPL and b-ICN can exist even in the presence of AcOH.The authors showed a nice dependence between the solution concentrationof the 1:1 substrate-modifier complex and the enantioselectivity as shown inFig. 70. In addition it was shown that there is nice correlation between theee values, the concentration of the 1:1 substrate/modifier complex and theamount of AcOH added as shown in Fig. 71.
It is known that in the enantioselective hydrogenation of KPL all polarsolvents have a negative effect.76 Results presented in Figs. 2 and 3 definitelyshow the importance of the complex formation in the liquid phase. How-ever, even in the light of these unambiguous evidences the authors of thisstudy were not brave enough as they made the following remark: ‘‘we didnot doubt the role of the protonated cinchona despite the fact that thespectroscopy data published previously, obtained under the conditions ofthe Orito reaction in toluene, are not totally convincing in terms of pro-tonation of the N atom of quinuclidine’’.
We believe that in the near future further high quality and unambiguousexperimental data will be obtained related to the character of substrate-modifier interactions as well to the formation of substrate-modifier complexat the Pt surface. We also hope that those who have different views on themechanism of Orito’s reaction in the future will get more wide open plat-forms to publish their results and ideas.
1
5
10
15
20
25
30
35
40
45
Fig. 70 Dependence of the ee of the concentration of substrate-modifier complex in the liquidphase. (Reproduced from ref. 424 with permission)
260 | Catalysis, 2010, 22, 144–278

Abbreviations used
AcOH acetic acidAS Anchoring sitesATR-IR attenuated total reflection infrared (spectroscopy)CD cinchonidineCI chiral inductionCN cinchonineDe diastereomeric excessDHCD 10,11-dihydrocinchonidineDHCN 10,11-dihydrocinchonineDRIFT diffuse reflectance FT infraredECD enantioselectivity–conversion dependenciesED enantio-differentiationED enantio-differentiationee enantiomeric excess (%)Et ethylEtLa ethyl lactateEtpy ethyl pyruvateHHCD hexahydrocinchonidineHRTEM high-resolution transmission electron microscopyICN isocinchonineIR Infrared spectroscopyKPL ketopantolactoneM metalM/S modifier–substrate molar ratioMe methylMBF methyl benzoylformateMeLa methyl lactateMeOCD O-methyl-cinchonidineMeODHCD O-methyl-10,11-dihydrocinchonidineMePy methyl pyruvateNED 1-naphthyl-1,2-ethanediol
1
5
10
15
20
25
30
35
40
45
Fig. 71 Comparison of the total concentration of the 1:1 b-ICN–KPL complexes measured byNMR (circles) with the enantioselectivities (diamonds) obtained at identical AcOH(CD3COOD for the NMR) concentrations (logarithmic scale). (Reproduced from ref. 424 withpermission)
Catalysis, 2010, 22, 144–278 | 261

NLP non-linear phenomenaPADA pyruvaldehyde dimethyl acetalPPD 1-phenyl-1,2-propanedionePh phenylPhOCD O-phenyl-cinchonidinePNEA pantoylnaphthylethylamineQ quinolineQD quinidineQN quinineRA rate accelerationRE rate enhancementRAIRS reflection absorption infrared spectroscopyRAIRS reflection adsorption infrared spectraSERS surface-enhanced Raman scatteringSTM scanning tunnelling microscopyTEA triethylamineTFA trifluoroacetic acidTMS trimethylsilaneTOF turnover frequency (h� 1)Y yield (%)
References
1 R. A. Sheldon, Chirotechnology, Marcel Dekker, New York, 1993.
2 Chiral Catalyst Immobilization and Recycling, D. E. De Vos, I. F. J. Vank-
elecom and P. A. Jacobs (Eds), Wiley-VCH, 2000.
3 Comprehensive Asymmetric Catalysis, E. N. Jacobsen, A. Pfaltz, and
H. Hisashi (Eds.) 2004, Springer, Berlin, Heidelberg, New York, LXXVIII,
1856.
4 Applied Homogeneous Catalysis by Organometallic Complexes, (second ed.),
B. Cornils, W. A. Herrmann (Eds.) Wiley-VCH, Weinheim, 2002
5 C. E. Song and S. Lee, Chem. Rev., 2002, 102, 3495.
6 M. T. Reetz, Adv. Catal., 2006, 49, 1.
7 M. T. Martin, Drug Discovery Today, 1996, 1, 239.
8 R. Noyori, Tetrahedron, 1994, 50, 4259.
9 W. S. Knowles, Angew. Chem. Int. Ed., 2002, 41, 1998.
10 H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94,
2483.
11 K. B. Sharpless, Angew. Chem. Int. Ed., 2002, 41, 2024.
12 T. T. L. Au-Yeung and A. S.C. Chan, Coord. Chem. Rev., 2004, 248, 2151.
13 J. M. Brown, Platinum Metals Rev., 1987, 31, 137.
14 H. Sasai, E. Emori, T. Arai and M. Shibasaki, Tetrahedron Letters, 1996, 37,
5561.
15 H. U. Blaser, B. Pugin and F. Spindler, J. Mol. Catal. A: Chem., 2005, 231, 1.
16 I. F. J. Vankelecom and P. A. Jacobs, in Chiral Catalyst Immobilization and
Recycling, D.E. de Vos, I.F.J. Vankelecom, P.A., Jacobs, (Eds.), Wiley-VCH,
Weinheim, 2000, p. 1.
17 H. U. Blaser, B. Pugin and M. Studer, in Chiral Catalyst Immobilization and
Recycling, D. E. de Vos, I. F. J. Vankelecom and P. A., Jacobs (Eds.), Wiley-
VCH, Weinheim, 2000, p.19.
18 S. J. Bae, S. W. Kim, T. Hyeon and B. M. Kim, Chem. Commun., 2000, 31.
1
5
10
15
20
25
30
35
40
45
262 | Catalysis, 2010, 22, 144–278

19 M. D. Jones, R. Raja, J. M. Thomas and B. F. G. Johnson, Top. Catal., 2003,
25, 71.
20 J. A. M. Brandts, C. V. Cavenaghi, A. Gerlach and M. J. Burk, Chimica Oggi,
2000, 18, 47.
21 S. K. Tanielyan, R. L. Augustine, US Patent 6,005,148 (1999) and 6,025,295
(2000) to Seton Hall University
22 R. L. Augustine, S. K. Tanielyan, N. Mahata, Y. Gao, A. Zsigmond and
H. Yang, Appl. Catal. A: Gen., 2003, 256, 69.
23 R. L. Augustine, P. Goel, N. Mahata, C. Reyes and S. K. Tanielyan, J. Mol.
Catal. A: Chem., 2004, 216, 189.
24 M. Reggelin, M. Schultz and M. Holbach, Angew. Chem. Int. Ed., 2002, 41,
1614.
25 C. R. Landis and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1746.
26 E. Burello, D. Farruseng and G. Rothenberg, Adv. Synth. Catal., 2004, 346,
1844.
27 H. U. Blaser, Chem. Rev., 1992, 92, 935.
28 A. M. Thayer, Chem. Eng. News, 2005, 83, 40.
29 G. M. Schwab and L. Rudolph, Naturwiss., 1932, 20, 362.
30 E. I. Klabunovskii, in Absolute asymmetric synthesis and asymmetric catalysis,
Aspects of the Origin of Life, M. Florkin (Ed.), Pergamon Press, N.Y.,
Oxford, 1960, p. 105.
31 S. Akabori, S. Sakurai, Y. Izumi and Y. Fuji, Nature, 1956, 178, 323.
32 A. Tai and T. Sigimura, in Chiral Catalyst Immobilization and Recycling, D. E.
de Vos, I.F.J. Vankelecom and P.A. Jacobs (Eds.), Wiley-VCH, Weinheim,
2000, p. 173.
33 D. Y. Murzin, P. Maki-Arvela, E. Toukoniitty and T. Salmi, Catal. Rev.-Sci,.
Eng., 2005, 7, 175.
34 T. Osawa, T. Harada and O. Takayashu, Top. Catal., 2000, 13, 155.
35 E. Klabunovskii, G. V. Smith and A. Zsigmond, in Heterogeneous Enantio-
selective Hydrogenation, Theory and Practice, Catalysis by Metal Complexes,
Vol 31, Springer, Dordrecht (The Netherlands), 2003.
36 S. Nakagawa, T. Sugimura and A. Tai, Chem. Lett., 1997, 859.
37 T. Osawa, A. Ozawa, T. Harada and O. Takayasu, J. Mol. Catal. A: Chem.,
2000, 154, 271.
38 A. Hoek and W. M. H. Sachtler, J. Catal., 1979, 58, 276.
39 Y. Orito, S. Imai and S. Niwa, Collected papers of the 43rd Catalysis Forum,
Japan, 1978, 30.
40 Y. Orito, S. Imai and S. Niwa, J. Chem. Soc. Jpn., 1979, 1118.
41 Y. Otito, S. Imai, S. Niwa and G.-H. Nguen, J. Synth. Org. Chem. Jpn., 1979,
37, 173.
42 Y. Orito, S. Imai and S. Niwa, J.Chem. Soc. Jpn., 1980, 670.
43 Y. Orito, S. Imai and S. Niwa, J.Chem. Soc. Jpn., 1982, 137.
44 R. Hess, S. Diezi, T. Mallat and A. Baiker, Tetrahedron: Asymmetry, 2004,
15, 251.
45 G. E. Webb and P. B. Wells, Catal. Today, 1992, 12, 319.
46 A. Baiker, J. Mol. Catal. A: Chem., 1997, 115, 473.
47 H. U. Blaser, H. P. Jalett, M. Muller and M. Studer, Catal. Today, 1997,
37, 441.
48 P. B. Wells and A. Wilkinson, Top. Catal., 1998, 5, 39.
49 A. Baiker, J. Mol. Catal. A: Chem., 2000, 163, 205.
50 M. Von Arx, T. Mallat and A. Baiker, Top. Catal., 2002, 19, 75.
51 M. Studer, H. U. Blaser and C. Exner, Adv. Synth. Catal., 2003, 345, 45.
52 T. Burgi and A. Baiker, Acc. Chem. Res., 2004, 37, 909.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 263

53 T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863.
54 H. U. Blaser, B. E. Pugin, F. Spindler and M. Thommen, Acc. Chem. Res.,
2007, 40, 1240.
55 B. Torok, B. Balazsik, M. Torok, Gy. Szoll +osi and M. Bartok, Ultrasonics
Sonochem., 2000, 7, 151.
56 M. Sutyinszki, K. Szori, K. Felfoldi and M. Bartok, Catal. Comm., 2002,
3, 125.
57 H. U. Blaser, H. P. Jalett, D. M. Monti, A. Baiker and J. T. Wehrli, Stud. Surf.
Sci. Catal., 1991, 67, 147.
58 M. Garland and H. U. Blaser, J. Am. Chem. Soc., 1990, 112, 7048.
59 A. Baiker, Catal. Today, 2005, 100, 59.
60 M. Schurch, T. Heinz, R. Aeschimann, T. Mallat, A. Pfaltz and A. Baiker,
J. Catal., 1998, 173, 187.
61 H. U. Blaser, H. P. Jalett, M. Garland, M. Studer, H. Thies and A. Wirth-
Tijani, J. Catal., 1998, 173, 282.
62 H. Wynberg, in: Topics in Stereochemistry, E. L. Eliel, S. H. Wilen and N. L.
Allinger (Eds.), Wiley, New York, 1986, Vol. 16, p. 87.
63 H. U. Blaser, H. P. Jalett, D. M. Monti, J. F. Reber and J. T. Wehrli, Stud.
Surf. Sci. Catal., 1988, 41, 153.
64 J. L. Margitfalvi, P. Marti, A. Baiker, L. Botz and O. Sticher, Catal. Lett.,
1990, 6, 281.
65 I. M. Sutherland, A. Ibbotson, R. B. Moyes and P. B. Wells, J. Catal., 1990,
125, 77.
66 J. Wang, Y. K. Sun, C. Leblond, R. N. Landau and D. G. Blackmond,
J. Catal., 1996, 161, 752.
67 R. L. Augustine, S. K. Tanielyan and L. K. Doyle, Tetrahedron: Asymmetry,
1993, 4, 1803.
68 A. Tungler, T. Mathe, T. Tarnai, K. Fodor, G. Toth, J. Kajtar, I. Kolossvary,
B. Herenyi and R. A. Sheldon, Tetrahedron: Asymmetry, 1995, 6, 2395.
69 J. L. Margitfalvi and M. Heged +us, J. Catal., 1995, 156, 175.
70 B. Torok, K. Felfoldi, G. Szakonyi and M. Bartok, Ultrasonics Sonochem.,
1997, 4, 301.
71 (a) P. Maki-Arvela, M. Kuzma, R. Sjoholm, R. Leino and T. Salmi, in:
Proceedings of the 9th International Symposium on Relations between Homo-
geneous and Heterogeneous Catalysis, July 20–24, 1998, Southampton, Book of
Abstracts, p. 23; and (b) E. Toukoniitty, P. Maki-Arvela, A. N. Villea, A. K.
Neyestanaki, T. Salmi, R. Leino, R. Sjoholm, E. Laine, J. Vayrynen,
T. Ollonquist and P. J. Kooyman, Catal. Today, 2000, 60, 175.
72 S. Lavoie, M.-A. Laliberte, I. Temprano and P. H. McBreen, J. Am. Chem.
Soc., 2006, 128, 7588.
73 H. U. Blaser, M. Garland and H. P. Jallet, J. Catal., 1993, 144, 569.
74 O. Schwalm, J. Weber, B. Minder and A. Baiker, J. Mol. Struct. (Theochem),
1995, 330, 353.
75 J. L. Margitfalvi, M. Heged +us and E. Tfirst, Tetrahedron: Asymmetry, 1996, 7,
571.
76 T. Mallat, M. Bodmer and A. Baiker, Catal. Lett., 1997, 44, 95.
77 M. Schurch, O. Schwalm, T. Mallat, J. Weber and A. Baiker, J. Catal., 1997,
169, 275.
78 K. E. Simon, G. Wang, T. Heinz, T. Giger, T. Mallat, A. Pfaltz and A. Baiker,
Tetrahedron: Asymmetry, 1995, 6, 505.
79 H. Bonnemann and G. A. Braun, Angew. Chem. Int. Ed. Engl., 1996, 35,
1992.
1
5
10
15
20
25
30
35
40
45
264 | Catalysis, 2010, 22, 144–278

80 S. Basu, H. Paul, C. S. Gopinath, S. Bhaduri and G. K. Lahiri, J. Catal., 2005,
229, 298.
81 D. Ferri, T. Burgi, K. Borszeki, T. Mallat and A. Baiker, J. Catal., 2000, 193,
139.
82 T. Mallat, Z. Bodnar, B. Minder, K. Borszeky and A. Baiker, J. Catal., 1997,
168, 183.
83 J. L. Margitfalvi, M. Heged +us and E. Tfirst, Stud. Surf. Sci. Catal., 1996,
101, 241.
84 J. L. Margitfalvi and M. Heged +us, J. Mol. Catal. A: Chem., 1996,
107, 281.
85 A. Vargas, T. Burgi and A. Baiker, J. Catal., 2001, 197, 378.
86 Y. Sun, J. Wang, C. Leblond, R. N. Landau, J. Laquidara, J. R. Sowa and
D. G. Blackmond, J. Mol. Catal. A: Chem., 1997, 115, 495.
87 D. Ferri, T. Burgi and A. Baiker, J. Phys. Chem. B, 2001, 105, 3187.
88 T. Burgi and A. Baiker, J. Am. Chem. Soc., 1998, 120, 12920.
89 D. Ferri, T. Burgi and A. Baiker, J. Chem. Soc., Perkin Trans., 2002,
2, 437.
90 Z. Ma, J. Kubota and F. Zaera, J. Catal., 2003, 219, 404.
91 D. Ferri, T. Burgi and A. Baiker, J. Phys. Chem. B, 2004, 108, 14384.
92 R. J. LeBlanc, W. Chu and C. T. Williams, J. Mol. Catal. A: Chem., 2004,
212, 277.
93 J. L. Margitfalvi, E. Talas, E. Tfirst, C. V. Kumar and A. Gergely, Appl. Catal.
A: Gen., 2000, 191, 177.
94 G. A. Attard, J. Phys. Chem. B, 2001, 105, 3158.
95 T. Evans, A. P. Woodhead, A. Gutierrez-Soza, G. Thornton, T. J. Hall, A. A.
Davis, N. A. Young, P. B. Wells, R. J. Oldman, O. Plashkevych, O. Vahtras,
H. Agren and V. Carravetta, Surf. Sci., 1999, 436, L691.
96 J. M. Bonello, E.C.H. J. Sykes, R. Lindsay, F. J. Williams, A. K. Santra and
R. M. Lambert, Surf. Sci., 2001, 482, 207.
97 A. Vargas, T. Burgi and A. Baiker, J. Catal., 2004, 222, 439.
98 A. Vargas, D. Ferri and A. Baiker, J. Catal., 2005, 236, 1.
99 Y. Sun, J. Wang, C. LeBlond, R. N. Landau and D. G. Blackmond, J. Catal.,
1996, 161, 759.
100 J. U. Kohler and J. S. Bradley, Langmuir, 1998, 14, 2730.
101 L. Michaelis and M. Menten, Biochem. Z., 1913, 49, 333.
102 E. Talas and J. L. Margitfalvi, Catal. Comm., 2008, 9, 984.
103 A. Vargas, G. Santarossa, M. Iannuzzi and A. Baiker, J. Phys. Chem. C, 2008,
112, 10200.
104 J. Margitfalvi, L. Guczi and A. H. Weiss, React. Kinet. Catal. Lett., 1980,
15, 475.
105 L. L. Kesmodel, L. H. Dubois and G. A. Somorjai, J. Chem. Phys., 1979,
70, 2180.
106 G. A. Somorjai, M. A. Van Hove and B. E. Bent, J. Phys. Chem., 1988,
92, 973.
107 E. Toukoniitty, P. Maki-Arvela, A. K. Neyestanaki, T. Salmi and D.Yu.
Murzin, Appl. Catal. A: Gen., 2002, 235, 125.
108 C. LeBlond, J. Wang, J. Liu, A. T. Andrews and Y.-K. Sun, J. Am. Chem.
Soc., 1999, 121, 4920.
109 E. Talas, L. Botz, J. Margitfalvi, O. Sticher and A. Baiker, J. Planar Chro-
matogr., 1992, 5, 28.
110 H. M. R. Hoffmann and J. Frackenpohl, Eur. J. Org. Chem., 2004, 4293.
111 K. Kacprzak and J. Gawronski, Synthesis, 2001, 961.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 265

112 S. K. Tian, Y. Chen, J. Hang, L. Tang, P. McDaid and L. Deng, Acc. Chem.
Res., 2004, 37, 621.
113 L. Pasteur, C.R. Acad. Sci., 1853, 37, 110.
114 Optical Resolution Procedures for Chemical Comounds, Optical Resolution
Information Center, Manhattan College, Riverdale, New York, 1981
115 S. Rogozhin and V. A. Davankov, Russ. Chem. Rev. (Uspekhi Khimii), 1968,
37, 1327.
116 C. Czerwenka, N. M. Maier and W. Lindner, J. Chromatography A, 2004,
1038, 85.
117 W. R. Oberleitner, N. M. Maier and W. Lindner, J. Chromatography A, 2002,
960, 97.
118 T. Sakai and A. Hirose, Talanta, 2003, 59, 167.
119 A. A. Smaardijk and H. Wynberg, J. Org. Chem., 1987, 52, 135.
120 V. Prelog and M. Wilhelm, Helv. Chim. Acta, 1964, 37, 1634.
121 H. Pracejus, Fortschr. Chem. Forsch., 1967, 8, 493.
122 A. Ojida, T. Yamano, N. Taya and A. Tasaka, Org. Lett., 2002, 4, 3051.
123 B. Vakulya, Sz. Varga, A. Csampai and T. Soos, Org. Letts., 2005,
7, 1967.
124 H. C. Kolb, P. G. Andersson and K. B. Sharpless, J. Am. Chem. Soc., 1994,
116, 1278.
125 K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K. S.
Jeong, H. L. Kwong, K. Morikawa, Z. M. Wang, D. Xu and X. L. Zhang,
J. Org. Chem., 1992, 57, 2768.
126 G. A. Crispino, K. S. Jeong, H. C. Kolb, Z. M. Wang, D. Xu and K. B.
Sharpless, J. Org. Chem., 1993, 58, 3785.
127 L. Wang and K. B. Sharpless, J. Am. Chem. Soc., 1992, 114, 7568.
128 U. H. Dolling, P. Davis and E. J. J. Grabowski, J. Am. Chem. Soc., 1984,
106, 446.
129 A. Bhattacharya, U. H. Dolling, E. Grabowski, S. Karady, K. Ryan and
L. Weinstock, Angew. Chem. Int. Ed. Engl., 1986, 25, 476.
130 R. S. E. Conn, A. V. Lovell, S. Karady and L. M. Weinstock, J. Org. Chem.,
1986, 51, 4710.
131 D. L. Hughes, U. H. Dolling, K. M. Ryan, E. F. Schoenewaldt and E. J. J.
Grabowski, J. Org. Chem., 1987, 52, 4745.
132 M. Masui, A. Ando and T. Shioiri, Tetrahedron Letters, 1988, 29, 2835.
133 M. J. O’Donnell, W. D. Benett and S. Wu, J. Am. Chem. Soc., 1989,
11, 2353.
134 W. Nerinckx and M. Vandewalle, Tetrahedron: Asymmetry, 1990, 1, 265.
135 K. B. Lipkowitz, M. W. Cavanaugh, B. Baker and M. J. O’Donnell, J. Org.
Chem., 1991, 56, 5181.
136 T. B. K. Lee and G. S. K. Wong, J. Org. Chem., 1991, 56, 872.
137 A. Ando, T. Miura, T. Tatematsu and T. Shioiri, Tetrahedron Letters, 1993,
34, 1507.
138 M. J. O’Donnell, S. Wu and J. C. Huffman, Tetrahedron, 1994, 50, 4507.
139 B. Lygo and P. G. Wainwright, Tetrahedron Letters, 1997, 38, 8595.
140 M. J. O’Donnell, Z. Fang, X. Ma and J. C. Huffman, Heterocycles, 1997, 46,
617.
141 E. J. Corey, F. Xu and M. C. Noe, J. Amer. Chem. Soc., 1997, 119,
12414.
142 B. Lygo and P. G. Wainwright, Tetrahedron Letters, 1998, 39, 1599.
143 B. Lygo, J. Crosby and A. J. Peterson, Tetrahedron Letters, 1999, 40, 8671.
144 E. V. Dehmlow, S. Wagner and A. Muller, Tetrahedron, 1999, 55, 6335.
145 B. Lygo and D. C. M. To, Tetrahedron Letters, 2001, 42, 1343.
1
5
10
15
20
25
30
35
40
45
266 | Catalysis, 2010, 22, 144–278

146 H. Park, B. S. Jeong, M. S. Yoo, J. H. Lee, M. K. Park, Y. J. Lee, M. J. Kim
and S. Jew, Angew. Chem., Int. Ed., 2002, 41, 3036.
147 B. Lygo and D. L. Humphreys, Tetrahedron Letters, 2002, 43, 6677.
148 R. Chinchilla, P. Mazon, C. Najera and F. J. Ortega, Tetrahedron: Asymmetry,
2004, 15, 2603.
149 S. Kumar and U. Ramachandran, Tetrahedron, 2005, 61, 7022.
150 S. Elango, M. Venugopal, P. S. Suresh and P. S. Eni, Tetrahedron, 2005, 61,
1443.
151 M. S. Yoo, B. S. Jeong, J. H. Lee, H. Park and S. Jew,Organic Letters, 2005, 7,
1129.
152 M. B. Andrus, Z. Ye and J. Zhang, Tetrahedron Letters, 2005, 46, 3839.
153 A. Siva and E. Murugan, J. Mol. Catal. A: Chem., 2005, 241, 111.
154 K. Hermann and H. Wynberg, Helv. Chim. Acta, 1977, 60, 2208.
155 N. Kobayashi and K. Iwai, J. Am. Chem. Soc., 1978, 100, 7071.
156 P. Hodge, E. Khoshdel and J. Waterhouse, J. Chem. Soc., Perkin Trans., 1983,
1, 2205.
157 M. Inagaki, J. Hiratake, Y. Yamamoto and J. Oda, Bull. Chem. Soc. Jpn.,
1987, 60, 4121.
158 A. Sera, K. Takagi, H. Katayama, H. Yamada and K. Matsumoto, J. Org.
Chem., 1988, 53, 1157.
159 B. M. Kim and K. B. Sharpless, Tetrahedron Letters, 1990, 31, 3003.
160 D. Pini, A. Petri, A. Nardi, C. Rosini and P. Salvadori, Tetrahedron Letters,
1991, 32, 5175.
161 B. B. Lohray, A. Thomas, P. Chittari, J. R. Ahuja and P. K. Dhal, Tetrahedron
Letters, 1992, 33, 5453.
162 B. B. Lohray, Tetrahedron: Asymmetry, 1992, 3, 1317.
163 H. Han and K. D. Janda, J. Am. Chem. Soc., 1996, 118, 7632.
164 H. Han and K. D. Janda, Tetrahedron Letters, 1997, 38, 1527.
165 E. Nandanan, A. Sudalai and T. Ravindranathan, Tetrahedron Letters, 1997,
38, 2577.
166 L. Canali, E. S. Song and D. C. Sherrington, Tetrahedron: Asymmetry, 1998, 9,
1029.
167 C. E. Song, C. R. Oh, S. W. Lee, S. Lee, L. Canali and D. C. Sherrington,
Chem. Commun., 1998, 2435.
168 R. Alvarez, M. A. Hourdin, C. Cave, J. d’Angelo and P. Chaminade, Tetra-
hedron Letters, 1999, 40, 7091.
169 R. Chinchilla, P. Mazon and C. Najera, Tetrahedron: Asymmetry, 2000, 11,
3277.
170 D. Bernstein, S. France, J. Wolfer and T. Lectka, Tetrahedron: Asymmetry,
2005, 16, 3481.
171 P. Hodge, E. Khoshdel, J. Waterhouse and J. M. J. Frechet, J. Chem. Soc.,
Perkin Trans., 1985, 1, 2327.
172 R. A. Aitken and J. Gopal, Tetrahedron: Asymmetry, 1990, 1, 517.
173 S. Kawahara, A. Nakano, T. Esumi, Y. Iwabuchi and S. Hatakeyama,
Organic Letters, 2003, 5, 3103.
174 M. Aune, A. Gogoll and O. Matsson, J. Org. Chem., 1995, 60, 1356.
175 L. Dupond, A. Konsur, K. Lewinski and B. Oleksyn, Acta Cryst., 1985, C41,
616.
176 S. Kashino and M. Haisa, Acta Cryst., 1983, C39, 310.
177 B. J. Oleksyn, Acta Cryst., 1982, B38, 1832.
178 B. J. Oleksyn, L. Lebioda and M. Ciechanowicz-Rutkowska, Acta Cryst.,
1979, B35, 440.
179 H. Pracejus, Fortschr. Chem. Forsch., 1967, 8, 493.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 267

180 L. Meurling, Chem. Scr., 1975, 7, 90.
181 H. Hiemstra and H. Wynberg, J. Am. Chem. Soc., 1981, 103, 417.
182 V. Prelog and M. Wilhelm, Helv. Chim. Acta, 1964, 37, 1634.
183 G. D. H. Dijkstra, R. M. Kellogg and H. Wynberg, J. Org. Chem., 1990, 55,
6121.
184 G. D. H. Dijkstra, R.M. Kellogg, H. Wynberg, J. S. Svendsen, I. Marko and
K. B. Sharpless, J. Am. Chem. Soc., 1989, 111, 8069.
185 U. Berg, M. Aune and O. Matsson, Tetrahedron Letters, 1995, 36, 2137.
186 W. J. Colucci, S. K. Jungk and R. D. Gandour,Magn. Reson. Chem., 1985, 23,
335.
187 T. Sai, N. Takao and M. Sugiura, Magn. Reson. Chem., 1992, 30, 1041.
188 T. H. A. Silva, A. B. Oliveira and W. B. De Almeida, Bioorg. Med. Chem.,
1997, 5, 353.
189 J. Thiel and P. Fiedorow, J. Mol. Struct., 1997, 405, 219.
190 T. Williams, R. G. Pitcher, P. Bommer, J. Gutzwiller and M. R. Uskokovic,
J. Am. Chem. Soc., 1969, 91, 1871.
191 G. Ucello-Barretta, L. Di Bari and P. Salvadori, Magn. Reson. Chem., 1992,
30, 1054.
192 J. Gawronsky, M. Brzostowska and J. Koput, Croatica Chemica Acta, 1989,
62, 97.
193 H. U. Blaser, H. P. Jalett, W. Lottenbach and M. Studer, J. Am. Chem. Soc.,
2000, 122, 12675.
194 E. Talas and J. L. Margitfalvi, Chirality, 2009, in press, DOI: 10.1002/
chir.20694
195 H. U. Blaser, H. P. Jalett and F. Spindler, J. Mol. Catal. A: Chem., 1996, 107,
85.
196 M. Bartok, Curr. Org. Chem., 2006, 10, 1533.
197 M. Bartok, K. Felfoldi, Gy. Szoll +osi and T. Bartok, React. Kinet. Catal. Lett.,
1999, 68, 371.
198 M. Bartok, M. Sutyinszki, I. Bucsi, K. Felfoldi, Gy. Szoll +osi, F. Bartha and
T. Bartok, J. Catal., 2005, 231, 33.
199 H. U. Blaser, in ‘‘Advances in Catalyst Design. Proceedings of the UNIDO
Workshop on Catalyst Design’’, Trieste, Italy (11-13 December 1990), M.
Grazini, C. N. R. Rao (Eds.), Singapore, World Scientific, 1991, p. 317
200 A. Pfaltz and T. Heinz, Top. Catal., 1997, 4, 229.
201 E. Orglmeister, T. Burgi, T. Mallat and Baiker, J. Catal., 2005, 233, 333.
202 C. Exner, A. Pfaltz, M. Studer and H. U. Blaser, Adv. Synth. Catal., 2003, 345,
1253.
203 S. Diezi, T. Mallat, A. Szabo and A. Baiker, J. Catal., 2004, 228, 162.
204 K. Felfoldi, K. Balazsik and M. Bartok, J. Mol. Catal. A: Chem., 2003, 202,
163.
205 K. Balazsik, K. Szori, K. Felfoldi, B. Torok and M. Bartok, Chem. Commun.,
2000, 555.
206 A. Taskinen, V. Nieminen, M. Hotokka and D.Yu. Murzin, J. Phys. Chem. C,
2007, 111, 5128.
207 Sz. Cserenyi, K. Felfoldi, K. Balazsik, Gy. Szoll +osi, I. Bucsi and M. Bartok,
J. Mol. Catal. A: Chem., 2006, 247, 108.
208 G. Wang, T. Heinz, A. Pfaltz, B. Minder, T. Mallat and A. Baiker, J. Chem.
Soc. Chem. Commun., 1994, 2047.
209 B. Minder, T. Mallat, A. Baiker, G. Wang, T. Heinz and A. Pfaltz, J. Catal.,
1995, 154, 371.
210 E. Orglmeister, T. Burgi, T. Mallat and A. Baiker, J. Catal., 2005, 232, 137.
1
5
10
15
20
25
30
35
40
45
268 | Catalysis, 2010, 22, 144–278

211 S. Diezi, M. Hess, E. Orglmeister, T. Mallat and A. Baiker, J. Mol. Catal. A:
Chem., 2005, 239, 49.
212 T. Mallat and A. Baiker, Appl. Catal. A: Gen., 2000, 200, 3.
213 B. Minder, M. Schurch, T. Mallat, A. Baiker, T. Heinz and A. Pfaltz, J. Catal.,
1996, 160, 261.
214 H. U. Blaser and M. Studer, Acc. Chem. Res., 2007, 40, 1348.
215 H. U. Blaser and M. Muller, Stud. Surf. Sci. Catal., 1991, 59, 73.
216 S. P. Griffiths, P. Johnston, W. A. H. Vermeer and P. B. Wells, J. Chem. Soc.
Chem. Commun., 1994, 2431.
217 K. Felfoldi, T. Varga, P. Forgo and M. Bartok, Catal. Lett., 2004, 97, 65.
218 K. Fodor, A. Tungler, T. Mathe, S. Szabo and R. A. Sheldon, in: Catalysis of
Organic Reactions, Chem. Ind., E.F. Herkes, (ed.), Vol. 75, New York, Marcel
Dekker, 1998, p. 115.
219 A. Tungler, T. Mathe, K. Fodor, R. A. Sheldon and P. Gallezot, J. Mol. Catal.
A: Chem., 1996, 108, 145.
220 G. Farkas, K. Fodor, A. Tungler, T. Mathe, G. Toth and R. A. Sheldon,
J. Mol., Catal. A: Chem., 1999, 138, 123.
221 G. Farkas,_ . Sıpos, A. Tungler, A. Sarkany and J. L. Figueiredo, J. Mol. Catal.
A: Chem., 2001, 170, 101.
222 A. Tungler and G. Fogassy, J. Mol. Catal. A: Chem., 2001, 173, 231.
223 E. Sıpos, A. Tungler and I. Bitter, in: Catalysis of Organic Reactions. Chemical
Industries, D.G. Morrell, (ed.), Vol 89, New York-Basel, Marcell Dekker,
2003, p. 653.
224 A. Tungler, M. Kajtar, T. Mathe, T. Toth, E. Fogassy and J. Petro, Catal.
Today, 1989, 5, 159.
225 E. Sıpos, A. Tungler and I. Bitter, J. Mol. Catal. A: Chem., 2003, 198, 167.
226 B. Minder, M. Schurch, T. Mallat and A. Baiker, Catal. Lett., 1995,
31, 143.
227 A. Marinas, T. Mallat and A. Baiker, J. Catal., 2004, 221, 666.
228 R. Hartung, J. G. E. Krauter, S. Seebald, P. Kukula, U. Nettekoven and
M. Studer, Asymmetric Hydrogenation of Activated Ketones with catASium
F214, http://www.degussacatalysts.com/asp/documents/79 Document.pdf.
229 P. A. Meheux, A. Ibbotson and P. B. Wells, J. Catal., 1991, 128, 387.
230 R. L. Augustine and S. K. Tanielyan, J. Mol. Catal. A: Chem., 1997, 118, 79.
231 M. Bartok, T. Bartok, G. Szoll +osi and K. Felfoldi, Catal. Lett., 1999,
61, 57.
232 B. Toukoniitty, E. Toukoniitty, J. Kuusisto, J.-P. Mikkola, T. Salmi and
D.Yu. Murzin, Chem. Eng. J., 2006, 120, 91.
233 D. Y. Murzin, E. Toukoniitty and J. Hajek, React. Kinet. Catal. Lett., 2004,
83, 205.
234 D. J. Jenkins, A. M. S. Alabdulrahman, G. A. Attard, K. G. Griffin, P.
Johnston and P. B. Wells, J. Catal., 2005, 234, 230.
235 E. Toukonitty and D.Yu. Murzin, J. Catal., 2006, 241, 96.
236 A. Vargas, F. Hoxha, N. Bonalumi, T. Mallat and A. Baiker, J. Catal., 2006,
240, 203.
237 K. Sz +ori, Gy. Szoll +osi and M. Bartok, J. Catal., 2006, 244, 255.
238 E. Toukoniitty, S. Franceschini, A. Vaccari and D.Yu. Murzin, Appl. Catal.
A: Gen., 2006, 300, 147.
239 S. Diezi, A. Szabo, T. Mallat and A. Baiker, Tetrahedron: Asymmetry, 2003,
14, 2573.
240 D. G. Blackmond, J. Catal., 1998, 176, 267.
241 D. Ferri, T. Burgi and A. Baiker, Chem. Commun., 2001, 1172.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 269

242 I. Bakos, S. Szabo, M. Bartok and E. Kalman, J. Electroanal. Chem., 2002,
532, 113.
243 C. R. Landis and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1746.
244 X. Zuo, H. Liu and M. Liu, Tetrahedron Lett., 1998, 39, 1941.
245 H. Bonnemann and G. A. Braun, Chem. Eur. J., 1997, 3, 1200.
246 J. L. Margitfalvi, E. Talas, L. Yakhyaeva, E. Tfirst and I. Bertoti, in: Catalysis
of Organic Reactions, D.G. Morrell, (Ed.), Chem. Ind., Vol. 89, Dekker, New
York, 2003, p. 393.
247 J. U. Kohler and J. S. Bradley, Catal. Lett., 1997, 45, 203.
248 A. Kraynov and R. Richards, Appl. Catal. A: Gen., 2006, 314, 1.
249 K. E. Simons, A. Ibbotson, P. Johnston, H. Plum and P. B. Wells, J. Catal.,
1994, 150, 321.
250 T. Marzialetti, M. Oportus, D. Ruiz, J. L. G. Fierro and P. Reyes, Catal.
Today, 2008, 133, 711.
251 C. F. Yang, H. Y. Jiang, J. Feng, H. Yan Fu, R. X. Li, H. Chen and X. J. Li,
J. Mol. Catal. A: Chem., 2009, 300, 98.
252 S. Bhaduri, V. S. Darshane, K. Sharma and D. Mukesh, J. Chem. Soc. Chem.
Commun., 1992, 1738.
253 M. Maris, D. Ferri, L. Konigsmann, T. Mallat and A. Baiker, J. Catal., 2006,
237, 230.
254 Y. Huang, J. Chen, H. Chen, R. Li, Y. Li, L.-E. Min and X. Li, J. Mol. Catal.
A: Chem., 2001, 170, 143.
255 H. X. Ma, H. Chen, Q. Zhang and J. X. Li, J. Mol. Catal. A: Chem., 2003, 196,
131.
256 M. Maris, T. Mallat and A. Baiker, J. Mol. Catal. A: Chem., 2005, 242, 151.
257 W. Xiong, H. X. Ma, Y. Y. Hong, H. Chen and X. J. Li, Tetrahedron:
Asymmetry, 2005, 16, 1449.
258 R. Hess, F. Krumeich, T. Mallat and A. Baiker, J. Mol. Catal. A: Chem., 2004,
212, 205.
259 F. Hoxha, N. van Vegten, A. Urakawa, F. Krumeich, T. Mallat and A. Baiker,
J. Catal., 2009, 261, 224.
260 T. Marzialetti, J. L. G. Fierro and P. Reyes, Catal. Today, 2005, 107–108, 235.
261 T. J. Hall, J. E. Halder, G. H. Hutchings, R. L. Jenkins, P. McMorn, P. B.
Wells and R. P. K. Wells, Top. Catal., 2000, 11–12, 351.
262 W. Reschetilowski, U. Bohmer and J. Wiehl, Stud. Surf. Sci. Catal., 1994, 84,
2021.
263 U. Bohmer, F. Franke, K. Morgenschweis, T. Bieber and W. Reschetilowski,
Catal. Today, 2000, 60, 167.
264 K. Balazsik, B. Torok, G. Szakonyi and M. Bartok, Appl.Catal. A: Gen., 1999,
182, 53.
265 M. A. Fraga, M. J. Mendes and E. Jordao, J. Mol. Catal. A: Chem., 2002, 179,
243.
266 L. Xing, F. Du, J. J. Liang, Y. S. Chen and Q. L. Zhou, J. Mol. Catal. A:
Chem., 2007, 276, 191.
267 J. T. Wehrli, A. Baiker, D. M. Monti and H. U. Blaser, J. Mol. Catal., 1990,
61, 207.
268 A. Kraynov, A. Suchopar and R. Richards, Catal. Lett., 2006, 110, 91.
269 M. Bartok, K. Balazsik, T. Bartok and Z. Kele, Catal. Lett., 2003, 87, 235.
270 K. Balazsik and M. Bartok, J. Mol. Catal. A: Chem., 2004, 219, 383.
271 W.-R. Huck, T. Burgi, T. Mallat and A. Baiker, J. Catal., 2003, 216, 276.
272 E. Schmidt, D. Ferri and A. Baiker, Langmuir, 2007, 23, 8087.
273 Gy. Szoll +osi, P. Forgo and M. Bartok, Chirality, 2003, 15, S82.
1
5
10
15
20
25
30
35
40
45
270 | Catalysis, 2010, 22, 144–278

274 M. Bartok, P. T. Szabo, T. Bartok, G. Szollosi and K. Balazsik, Rapid
Commun. Mass Spectrom., 2001, 15, 65.
275 M. Bartok, K. Balazsik, Gy. Szoll +osi and T. Bartok, Ultrasonics Sonochem.,
1999, 5, 149.
276 J. L. Margitfalvi and E. Talas, Appl. Catal. A: Gen., 1999, 182, 65.
277 M. Bartok, Gy. Szoll +osi, K. Balazsik and T. Bartok, J. Mol. Catal., A: Chem.,
2002, 177, 299.
278 H. U. Blaser, H. P. Jalett, D. M. Monti and J. T. Wehrli, Appl. Catal., 1989,
52, 19.
279 T. Mallat, S. Frauchiger, P. J. Kooyman, M. Schurch and A. Baiker, Catal.
Lett., 1999, 63, 121.
280 S. P. Griffiths, P. Johnson and P. B. Wells, Appl. Catal. A: Gen., 2000, 191,
193.
281 B. Minder, T. Mallat, P. Skrabal and A. Baiker, Catal. Lett., 1994, 29, 115.
282 J. L. Margitfalvi and M. Heged +us, unpublished results.
283 K. Balazsik and M. Bartok, J. Catal., 2004, 224, 463.
284 B. Torok, K. Felfoldi, G. Szakonyi, K. Balazsik and M. Bartok, Catal. Lett.,
1998, 52, 81.
285 B. Torok, K. Balazsik, Gy. Szollosi, K. Felfoldi, I. Kun and M. Bartok,
Ultrasonics Sonochem., 1999, 6, 97.
286 K. Balazsik, B. Torok, K. Felfoldi and M. Bartok, Ultrasonics Sonochem.,
1999, 5, 149.
287 B. Toukoniitty, O. Roche, J.-P. Mikkola, E. Toukoniitty, F. Klingstedt, K.
Eranen, T. Salmi and D. Yu. Murzin, Chem. Eng. J., 2007, 126, 103.
288 (a) A. Tai, T. Hiraki and S. Murakami, Bull. Chem. Soc. Japan, 1983, 56, 1414;
and (b) K. Ito, T. Harad and A. Tai, Bull. Chem. Soc. Japan, 1980, 53, 3367.
289 K. E. Simons, P. A. Meheux, A. Ibbotson and P. B. Wells, Stud. Surf. Sci.
Catal. (ICC Budapest), 1993, 75, 2317.
290 (a) M. Arx, N. Dummer, D. J. Willock, S. H. Taylor, R. P. K. Wells, P. B.
Wells and G. J. Hutchings, Chem. Commun., 2003, 1926; and (b) N. F.
Dummer, R. P. K. Wells, S. H. Taylor, P. B. Wells and G. J. Hutchings, in
Catalysis in Application (International Symposium on Applied Catalysis,
Glasgow, Jul. 16–18, 2003) 2003, p. 278.
291 J. L. Margitfalvi, B. Minder, E. Talas, L. Botz and A. Baiker, Stud. Surf. Sci.
Catal. (ICC Budapest), 1993, 75, 2471.
292 N. Kunzle, R. Hess, T. Mallat and A. Baiker, J. Catal., 1999, 186, 239.
293 N. Kunzle, T. Mallat and A. Baiker, Eur. Pat. Appl. 99112078.3.
294 N. Kunzle, J. W. Soler, T. Mallat and A. Baiker, J. Catal., 2002, 210, 66.
295 N. Kunzle, J. W. Soler and A. Baiker, Catal. Today, 2003, 79–80, 503.
296 E. Toukoniitty, P. Maki-Arvela, A. Kalantar Neyestanaki, T. Salmi, R. Sjo-
holm, R. Leino, E. Laine, P. J. Kooyman, T. Ollonqvist and J. Vayrynen,
Appl. Catal. A: Gen., 2001, 216, 73.
297 D. M. Meier, D. Ferri, T. Mallat and A. Baiker, J. Catal., 2007, 248, 68.
298 X. Li and C. Li, Catal. Lett., 2001, 77, 251.
299 N. Kunzle, T. Mallat and A. Baiker, Appl. Catal. A: Gen., 2003, 238, 251.
300 R. Wandeler, N. Kunzle, M. Schneider, T. Mallat and A. Baiker, J. Catal.,
2001, 200, 377.
301 F. Gao, L. Chen and M. Garland, J. Catal., 2006, 238, 402.
302 D. Ruizand and P. Reyes, J. Chilean Chem. Soc., 2008, 53, 1740.
303 D. M. Meier, T. Mallat, D. Ferri and A. Baiker, J. Catal., 2006, 244, 260.
304 J. T. Wehrli, A. Baiker, D. M. Monti, H. U. Blaser and H. P. Jalett, J. Mol.
Catal., 1989, 57, 245.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 271

305 W. Reschetilowski, U. Bohmer and K. Morgenschweis, Chem. Ing.Tech., 1995,
67, 205.
306 B. Panella, A. Vargas and A. Baiker, J. Catal., 2009, 261, 88.
307 V. Morawsky, U. Prusse, L. Witte and K. D. Vorlop, Catal. Comm., 2000, 1,
15.
308 R. Hess, F. Krumeich, T. Mallat and A. Baiker, Catal. Lett., 2004, 92, 141.
309 J. L. Margitfalvi, in: Catalysis of Organic Reactions, M.G. Scaros and M.L.
Prunier (Eds.), Chem. Ind., Vol 62, Dekker, New York, 1995, p. 189..
310 J. M. Bonello, R. M. Lambert, N. Kunzle and A. Baiker, J. Am. Chem. Soc.,
2000, 122, 9864.
311 D. Ferri, S. Diezi, M. Maciejewski and A. Baiker, Appl. Catal. A: Gen., 2006,
298, 165.
312 S. Lavoie, M. A. Laliberte, G. Mahieu, V. Derners-Carpentire and
P. McBreen, J. Am. Chem. Soc., 2007, 129, 11668.
313 Z. Liu, X. Li, P. Ying, Z. Feng and C. Li, J. Phys. Chem. C, 2007, 111, 823.
314 J. M. Bonello, F. J. Williams, A. K. Santra and R. M. Lambert, J. Phys. Chem.
B, 2000, 104, 9696.
315 M. Bartok, K. Balazsik, G. Szollosi and T. Bartok, J. Catal., 2002,
205, 168.
316 N. Bonalumi, T. Burgi and A. Baiker, J. Am. Chem. Soc., 2003, 125, 13342.
317 S. Lavoie and P. H. McBreen, J. Phys. Chem. B., 2005, 109, 11986.
318 S. Lavoie, M. A. Liberte and P. H. McBreen, Catal. Lett., 2004, 97, 111.
319 P. Johnston and P. B. Wells, J. Catal., 1995, 156, 180.
320 O. J. Sonderegger, T. Burgi and A. Baiker, J. Catal., 2003, 215, 116.
321 T. Salmi, D.Yu. Murzin, J.-P. Mikkola, J. Warna, P. Maki-Arvela, E. Tou-
koniitty and S. Toppinen, Ind. Eng. Chem. Res., 2004, 43, 4540.
322 B. Toukoniitty, J.-P. Mikkola, D.Yu. Murzin and T. Salmi, Appl. Catal. A:
Gen., 2005, 279, 1.
323 E. Toukoniitty and D.Yu. Murzin, Catal. Lett., 2004, 93, 171.
324 H. U. Blaser, B. Pugin and F. Spindler, J. Mol. Catal. A: Chem., 2005,
231, 1.
325 M. Schurch, N. Kunzle, T. Mallat and A. Baiker, J. Catal., 1998, 176, 569.
326 W. A. H. Vermeer, A. Fulford, P. Johnston and P. B. Wells, Chem. Commun.,
1993, 1053.
327 E. Toukoniitty, P. Maki-Arvela, J. Kuusisto, V. Nieminen, J. Paivarinta,
M. Hotokka, T. Salmi and D. Y. Murzin, J. Mol. Catal. A: Chem., 2003, 192,
116.
328 J. A. Slipszenko, S. P. Griffiths, P. Johnston, K. E. Simons, W. A. H. Vermeer
and P. B. Wells, J. Catal., 1998, 179, 267.
329 E. Toukoniitty, P. Maki-Arvela, M. Kuzma, A. Villela, A. K. Neyestanaki,
T. Salmi, R. Sjoholm, R. Leino, E. Laine and D. Y. Murzin, J. Catal., 2001,
204, 281.
330 M. Studer, S. Burkhardt and H. U. Blaser, Chem. Commun., 1999, 1727.
331 B. Torok, K. Felfoldi, K. Balazsik and M. Bartok, Chem. Commun., 1999,
1725.
332 M. Bodmer, T. Mallat and A. Baiker, in: Catalysis in Organic Reactions, F. E.
Herkes, (Ed.), Chemical Industries, Vol. 75, Marcel Dekker, New York, 1998,
p. 75.
333 M. von Arx, T. Mallat and A. Baiker, J. Catal., 2000, 193, 161.
334 M. von Arx, T. Mallat and A. Baiker, Tetrahedron: Asymmetry, 2001, 12,
3089.
335 M. von Arx, T. Mallat and A. Baiker, Catal. Lett., 2002, 78, 267.
336 G. Z. Wang, T. Mallat and A. Baiker, Tetrahedron: Asymmetry, 1997, 8, 2133.
1
5
10
15
20
25
30
35
40
45
272 | Catalysis, 2010, 22, 144–278

337 A. Szabo, N. Kunzle, M. Schurch, G. Z. Wang, T. Mallat and A. Baiker,
Chem. Commun., 1998, 1377.
338 A. Szabo, N. Kunzle, T. Mallat and A. Baiker, Tetrahedron: Asymmetry, 1999,
10, 61.
339 S. Diezi, M. Hess, E. Orglmeister, T. Mallat and A. Baiker, Catal. Lett., 2005,
102, 121.
340 S. Diezi, S. Reimann, N. Bonalumi, T. Mallat and A. Baiker, J. Catal., 2006,
239, 255.
341 K. Szori, Gy. Szollosi and M. Bartok, Adv. Synth. Catal., 2006, 348, 515.
342 R. Hess, A. Vargas, T. Mallat, T. Burgi and A. Baiker, J. Catal., 2004, 222,
128.
343 K. Szori K, B. Torok and K. Felfoldi, M., in: ‘‘Catalysis in Organic Reactions’’
M. E. Ford (Ed.) Chemical Industries Vol. 82, Marcel Dekker, New York,
2001. p. 489
344 E. Talas, and J. L. M. Margitfalvi, unpublished results.
345 H. U. Blaser, H. P. Jalett and J. Wiehl, J. Mol. Catal., 1991, 68, 215.
346 B. Torok, K. Balazsik, G. Szollosi, K. Felfoldi and M. Bartok, Chirality, 1999,
11, 470.
347 H. U. Blaser and H. P. Jalett, Stud. Surf. Sci. Catal., 1993, 78, 139.
348 E. Tukonitty, P. Maki-Arvela, J. Warna and T. Salmi, Catal. Today, 2001, 66,
411.
349 N. Kunzle, A. Szabo, M. Schurch, G. Wang, T. Mallat and A. Baiker, A.
Chem. Commun., 1998, 1377.
350 M. Studer, S. Burkhardt, A. F. Indolese and H. U. Blaser, Chem. Commun.,
2000, 1327.
351 M. von Arx, T. Mallat and A. Baiker, Journal of Catalysis, 2001, 202,
169–176.
352 S. D. Jackson, J. Willis, G. D. McLellan, G. Webb, M. B. T. Keegan, R. B. T.
Moyes, S. Simpson, P. B. Wells and R. Whyman, J. Catal., 1993, 139, 191.
353 M. von Arx, T. Mallat and A. Baiker, J. Catal., 2001, 202, 169.
354 H. U. Blaser, D. Imhof and M. Studer, Stud. Surf. Sci., 1997, 108, 175.
355 F. Hoxha, T. Mallat and A. Baiker, J. Catal., 2007, 248, 11.
356 T. Mallat and A. Baiker, J. Catal., 2007, 251, 246.
357 E. N. Jacobsen, I. Marko, W. S. Mungal, G. Schroder and K. B. Sharples,
J. Am. Chem. Soc., 1988, 110, 1968.
358 M. Sutyinszki, K. Szori, K. Felfoldi and M. Bartok, Catal. Lett., 2002,
81, 281.
359 X. B. Zuo, H. F. Liu and C. Yue, J. Mol Catal. A: Chem., 1999, 147, 63.
360 H. U. Blaser, H. P. Jalett and F. Spindler, J. Mol. Catal. A: Chem., 1996, 107,
85.
361 K. Balazsik, I. Bucsi, Sz. Cserenyi, Gy. Szoll +osi and M. Bartok, J. Mol. Catal.
A: Chem., 2008, 285, 84.
362 J. L. Margitfalvi, H. P. Jalett, E. Talas, A. Baiker and H. U. Blaser, Catal.
Lett., 1991, 10, 325.
363 A. Vargas, T. Burgi and A. Baiker, New J. Chem., 2002, 26, 807.
364 T. Varga, K. Felfoldi, P. Forgo and M. Bartok, J. Mol. Catal. A: Chem., 2004,
216, 181.
365 J. L. Margitfalvi and E. Talas, Appl. Catal. A: Gen., 2006, 301, 187.
366 R. Hess, T. Mallat and A. Baiker, J. Catal., 2003, 218, 453.
367 K. Felfoldi, T. Varga, P. Forgo and M. Bartok, Catal. Lett., 2004, 97, 65.
368 E. Toukoniitty and D. Y. Murzin, Catal. Lett., 2004, 93, 171.
369 I. Busygin, E. Toukoniity, R. Sillanpaa, D.Yu. Murzin and R. Leino, Eur.
Org. Chem., 2005, 2811.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 273

370 E. Toukoniity and D. Yu. Murzin, J.Catal., 2007, 251, 244.
371 E. Talas, J. L. Margitfalvi and O. Egyed, J.Catal., 2009, 266, 191.
372 Gy. Szoll +osi, Sz. Cserenyi, F. Fulop and M. Bartok, J. Catal., 2008,
260, 245.
373 R. L. Augustine and S. K. Tanielyan, J. Mol. Catal. A: Chem., 1996, 112, 93.
374 H. U. Blaser, H. P. Jalett, M. Muller and M. Studer, Catal. Today, 1997, 37,
441.
375 X. Li, R. P. K. Wells, P. B. Wells and G. J. Hutchings, Catal. Lett., 2003, 89,
163.
376 F. Hoxha, L. Konigsmann, A. D. Ferri, T. Mallat and A. Baiker, J. Am.
Chem. Soc., 2007, 129, 10582.
377 C. Puchot, O. Samuel, E. Dunach, S. H. Zhao, C. Agami and H. B. Kagan,
J. Am. Chem. Soc., 1986, 108, 2353.
378 D. Guillaneux, S. H. Zhao, O. Samuel, D. Rainford and H. B. Kagan, J. Am.
Chem. Soc., 1994, 116, 9430.
379 H. B. Kogan, C. Girard, D. Guillaneux, D. Rainford, O. Samuel, S. Y. Zhang
and S. H. Zhao, Acta Chemica Scandinavica, 1996, 50, 345.
380 Z. Ma and F. Zaera, J. Am. Chem. Soc., 2006, 128, 16414.
381 Z. Ma, I. Lee and F. Zaera, J. Am. Chem. Soc., 2007, 129, 16083.
382 L. Balazs, T. Mallat and A. Baiker, J. Catal., 2005, 233, 327.
383 K. Balazsik, Sz. Cserenyi, Gy. Szollosi, F. Fulop and M. Bartok, Catal. Lett.,
2008, 125, 401.
384 M. Bartok, M. Sutyinszki, K. Balazsik and G. Szollosi, Catal. Lett., 2005, 100,
161.
385 K. Balazsik, Gy. Szoll +osi and M. Bartok, Catal. Lett., 2008, 124, 46.
386 N. F. Dummer, R. Jenkins, X. Li, S. M. Bawaked, P. McMorn, A. Burrows,
C. J. Kiely, R. F. K. Wells, D. J. Willock and G. J. Hutchings, J. Catal., 2006,
243, 165.
387 K. Balazsik, T. A. Martinek, I. Bucsi, Gy. Szollosi, G. Fogassy, M. Bartok and
G. A. Olah, J. Mol. Catal. A: Chem., 2007, 272, 265.
388 M. Bartok, K. Balazsik, I. Bucsi and Gy. Szollosi, J. Catal., 2006, 239, 74.
389 M. von Arx, T. Mallat and A. Baiker, Angew. Chem. Int. Ed., 2001, 40, 2303.
390 M. Bartok, M. Sutyinszki and K. Felfoldi, J. Catal., 2003, 220, 207.
391 N. Bonalumi, A. Vargas, D. Ferri and A. Baiker, J. Phys. Chem. C, 2007, 111,
9349.
392 N. Bonalumi, A. Vargas, D. Ferri and A. Baiker, Chem. Eur. J., 2007, 13, 923.
393 A. Vargas, T. Burgi and A. Baiker, J. Catal., 2004, 226, 69.
394 I. Busygin, J. Warna, E. Toukoniitty, D. Yu. Murzin and R. Leino, J. Catal.,
2008, 254, 339.
395 I. Busygin, V. Nieminen, A. Taskinen, J. Sinkkonen, E. Toukoniitty, Rr.
Sillanpa, D. Yu. Murzin and R. Leino, J. Org. Chem., 2008, 73, 6559.
396 J. L. Margitfalvi, E. Talas and S. G +obolos, Catal. Today, 1989, 6, 73.
397 J. L. Margitfalvi, Gy. Vanko, I. Borbath, A. Tompos and A. Vertes, J. Catal.,
2000, 190, 474.
398 J. L. Margitfalvi, I. Borbath, M. Heged +us and A. Tompos, Appl. Catal. A:
Gen., 2002, 229, 35.
399 L.-Ch. de Menorval, A. Chaqroune, B. Coq and F. Figueras, J. Chem. Soc.,
Faraday Trans., 1997, 93, 3715.
400 S. J. Pratt, S. J. Jenkins and D. King, Surf. Sci., 2005, 585, L159.
401 M. Bartok, P. T. Szabo, T. Bartok and G. Szollosi, Rapid Commun. Mass
Spectrom., 2000, 14, 509.
402 D. Ferri, T. Burgi and A. Baiker, A. J. Chem. Soc. Perkin Trans., 1999, 2, 1305.
403 J. L. Margitfalvi, E. Talas and M. Hegedus, Chem. Comm, 1999, 656–646.
1
5
10
15
20
25
30
35
40
45
274 | Catalysis, 2010, 22, 144–278

404 J. L. Margitfalvi, E. Talas F. Zsila and S. Kristyan, Tetrahedron: Asymmetry,
2007, 18, 750.
405 J. L. Margitfalvi, E. Talas, N. Marın-Astorga N and T. Marzialetti, in:
Catalysis of Organic Reactions, Sowa, (ed.), Chem. Ind., Vol. 104, New York,
Taylor&Francis, 2005, p. 535.
406 E. Talas E and J. L. Margitfalvi, Catal. Commun., 2008, 9, 984.
407 G. A. Attard, D. J. Jenkins, O. A. Hazzazi, P. B. Wells, J. E. Gillies, K. G.
Griffin and P. Johnston, P., In: Catalysis in Application, S. D. Jackson, J. S. J.
Hardgreaves and D. Lennon, D., (Eds.), The Royal Society of Chemistry,
Cambridge, 2003, p. 71 (483).
408 G. A. Attard, J. E. Gillies, C. A. Harris, D. J. Jenkins, P. Johnston, M. A.
Price, D. J. Watson and P. B. Wells, Appl.Catal. A: General, 2001, 222, 393.
409 (a) S. Szabo and F. Nagy, J. Electroanal. Chem., 1978, 87, 261; and (b) S.
Szabo and F. Nagy, J. Electroanal. Chem., 1978, 88, 259.
410 S. Diezi, D. Ferri, A. Vargas, T. Mallat and A. Baiker, J. Am. Chem. Soc.,
2006, 128, 4048.
411 M. Bartok, M. Sutyinszki, K. Felfoldi and G. Szoll +osi, Chem Comm., 2002,
1130.
412 R. Hess, A. Vargas, T. Mallat, T. Burgi and A. Baiker, J. Catal., 2004, 222,
117.
413 G. Szoll +osi, A. Chatterjee, P. Forgo, M. Bartok and F. Mizukami, J. Phys.
Chem. A, 2005, 109, 860.
414 T. Mallat and A. Baiker, J. Catal., 1998, 176, 271.
415 O. J. Sonderegger, T. Burgi, L. K. Limbach and A. Baiker, J. Mol. Catal., A:
Chemical, 2004, 217, 93.
416 G. Vayner, K. N. Houk and Y. K. Sun, J. Am. Chem. Soc., 2004, 126, 199.
417 T. Varga, K. Felfoldi, P. Forgo and M. Bartok, J. Mol. Catal. A: Chemical,
2004, 216, 181.
418 E. Orglmeister, T. Mallat and A. Baiker, J. Catal., 2005, 234, 242.
419 A. Solladie0-Cavallo, F. Hoernel, M. Schmitt and F. Garin, Tetrahedron
Letters, 2002, 43, 2671.
420 M. J. Karplus, Chem. Phys., 1959, 30, 11.
421 T. Burgi, Zh. Zhou, N. Kunzle, T. Mallat and Alfons Baiker, J. Catal., 1999,
183, 405.
422 R. A. Olsen, D. Borchardt, L. Mink, A. Agarwal, L. J. Mueller and F. Zaera,
J. Am. Chem. Soc., 2006, 128, 15594.
423 T. A. Martinek, T. Varga, F. Fulop and M. Bartok, J. Catal., 2007, 246, 266.
424 T. A. Martinek, T. Varga, K. Balazsik, G. Szollosi, F. Fulop andM. Bartok, J.
Catal., 2008, 255, 296.
425 O. J. Sonderegger, G. M.-W. Ho, T. Burgi and A. Baiker, J. Catal., 2005, 230,
499.
426 A. Vargas, N. Bonalumi, D. Ferri and A. Baiker, J. Phys. Chem. A, 2006, 110,
1118.
427 Y. Zhao, F. Gao, L. Chen and M. Garland, J. Catal., 2004, 221, 274.
428 L. Chen, Y. Zhao, F. Gao and M. Garland, Appl. Spectrosc., 2003, 57, 797.
429 T. Evans, A. P. Woodhead, A. Gutierrez-Sosa, G. Thornton, T. J. Hall, A. A.
Davis, N. A. Young, P. B. Wells, R. J. Oldman, O. Plashkevych, O. Vahtras,
H. Agren and V. Carravetta, Surf. Sci., 1999, 436, L691.
430 T. Burgi, F. Atamny, R. Schlogl and A. Baiker, J. Phys. Chem. B, 2000, 104,
5953.
431 A. F. Carley, M. K. Rajumon, M. W. Roberts and P. B. Wells, J. Chem. Soc.,
Faraday Trans., 1995, 91, 2167.
432 T. E. Jones and C. J. Baddeley, Surf. Sci., 2002, 519, 237.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 275

433 J. M. Bonello, F. J. Williams and R. M. Lambert, J. Am. Chem. Soc., 2003,
125, 2723.
434 J. Kubota and F. Zaera, J. Am. Chem. Soc., 2001, 123, 11115.
435 F. Zaera, J. Phys. Chem., 2008, 112, 16196.
436 W. Chu, R. J. LeBlanc and C. T. Williams, Catal. Comm., 2002, 3, 547.
437 D. Ferri and T. Burgi, J. Am. Chem. Soc., 2001, 123, 12074.
438 D. Ferri, T. Burgi and A. Baiker, J. Catal., 2002, 210, 160.
439 T. Burgi and A. Baiker, J. Phys. Chem. B, 2002, 106, 10649.
440 W. Chu, R. J. LeBlanc, C. T. Williams, J. Kubota and F. Zaera, J. Phys.
Chem. B., 2003, 107, 14365.
441 N. Bonalumi, A. Vargas, D. Ferri, T. Burgi, T. Mallat and A. Baiker, J. Am.
Chem. Soc., 2005, 127, 8467.
442 J.-D. Grunwaldt and A. Baiker, Phys . Chem. Chem. Phys., 2005, 7, 3526.
443 D. Ferri, S. Diezi, M. Maciejewski, A. Baiker and Baiker, A., Appl. Catal. A:
Gen., 2006, 297, 165.
444 M. Castonguay, J.-R. Roy, A. Rochefort and P. H. McBreen, J. Am. Chem.
Soc., 2000, 122, 518.
445 T. Burgi, F. Atamny, A. Knop-Gericke, M. Havecker, T. Schedel- Niedrig, R.
Schlogl and A. Baiker, Catal. Lett., 2000, 66, 109.
446 Z. Ma, I. Lee and F. Zaera, J. Am. Chem. Soc., 2007, 129, 16083.
447 J. Lai, Z. Ma, L. J. Mueller and F. Zaera, J. Phys. Chem. B, 2009, 113, 11696.
448 I. Lee, Z. Ma, Sh. Kaneko and F. Zaera, J. Am. Chem. Soc., 2008, 130, 14597.
449 S. R. Calvo, R. J. LeBlanc, C. T. Williams and P. B. Balbuena, Surf. Sci., 2004,
563, 57.
450 A. Vargas and Baiker, J. Catal., 2006, 239, 220.
451 M. Wahl, M. von Arx, T. A. Jung and A. Baiker, J. Phys. Chem. B, 2006, 110,
21777.
452 M. von Arx, M. Wahl, T. A. Jung and A. Baiker, Phys. Chem. Chem. Phys.,
2005, 7, 273.
453 O. A. Hazzazi, G. A. Attard and P. B. Wells, J. Mol. Catal. A: Chem., 2004,
216, 247.
454 K. E. Simons, P. A. Meheux, S. P. Griffiths, I. M. Sutherland, P. Johnston,
P. B. Wells, A. F. Carley, M. K. Rajumon, M. W. Roberts and A. Ibbotson,
Recl. Trav. Chim. Pays-Bas., 1994, 113, 465.
455 N. Fietkau, R. Bussar and H. Baltruschat, Electrochimica Acta, 2006, 51, 5626.
456 M. S. Schneider, A. Urakawa, J. D. Grunwaldt, T. Burgi and A. Baiker, Chem.
Comm., 2004, 744.
457 M. Bartok, K. Felfoldi, B. Torok and T. Bartok, Chem. Comm., 1998, 2605.
458 I. Bucsi, M. Sutyinszki, K. Felfoldi and M. Bartok, Catal. Comm., 2006, 7,
104.
459 Sz. Cserenyi, K. Felfoldi, K. Balazsik, Gy. Szollosi, I. Bucsi and M. Bartok,
J. Mol. Catal A: Chem., 2005, 247, 108.
460 M. Bartok, K. Felfoldi, Gy. Szollosi and T. Bartok, Catal. Lett., 1999, 61, 1.
461 W. J. Hehre, W. W. Huang, P. E. Klunzinger, B. J. Deppmeier and A. J.
Driessen: Spartan Manual, Wavefunction, Inc., 18401 Von Karman Ave.,
Suite 370, Irvine, CA 92612
462 Gaussian 98, Revision A.6, J. A. Pople et al., Gaussian, Inc., Pittsburgh PA,
1998
463 D. Ferri, T. Burgi and A. Baiker, J. Chem. Soc., Perkin Trans., 2000, 2, 221.
464 T. Burgi and A. Baiker, J. Catal., 2000, 194, 445.
465 A. Vargas, T. Burgi, M. von Arx, R. Hess and A. Baiker, J. Catal., 2002, 209,
489.
466 O. Schwalm, B. Minder, J. Weber and A. Baiker, Catal. Lett., 1994, 23, 271.
1
5
10
15
20
25
30
35
40
45
276 | Catalysis, 2010, 22, 144–278

467 A. Baiker, Catal. Today, 2005, 100, 159.
468 O. Schwalm, J. Weber, J. L. Margitfalvi and A. Baiker, J.Mol.Struct., 1993,
297, 285.
469 A. Taskinen, V. Nieminen, E. Toukoniitty, D.Yu. Murzin and M. Hotokka,
Tetrahedron, 2005, 61, 8109.
470 T. Sai, N. Takao and M. Sugiura, Magnetic Resonance in Chemistry, 1992, 30,
1041.
471 O. Schwalm, Ph.D. Thesis, University of Geneva, No 2754, 1995
472 J. L. Margitfalvi and E. Tfirst, J. Mol. Catal. A: Chemical, 1989, 139, 81.
473 G. D. H. Dijkstra, R. M. Kellogg, H. Wynberg, J. S. Svendsen, I. Marko and
K. B. Sharpless, J. Am. Chem. Soc., 1989, 111, 8070.
474 R. A. Olsen, D. Borchardt, L. Mink, A. Agarwal, L. J. Mueller and F. Zaera,
J. Am. Chem. Soc., 2006, 128, 15595.
475 L. Mink, Z. Ma, R. A. Olsen, J. N. James, D. S. Sholl, J. L. Mueller and
F. Zaera, Topics in Catal., 2008, 48, 120.
476 E. Schmidt, D. Ferri, A. Vargas and A. Baiker, J. Phys. Chem. C, 2008, 112,
3866.
477 C. Houtman and M. A. Barteau, J. Phys. Chem., 1991, 95, 3755.
478 Z. M. Liu and M. A. Vannice, Surf. Sci., 1994, 316, 337.
479 M. Mavrikakis and M. A. Barteau, J. Mol. Catal. A: Chem., 1998, 131, 135.
480 F. Ruette, F. M. Poveda, A. Sierraalta and J. Rivero, Surf. Sci., 1996, 349, 241.
481 A. Vargas, S. Reimann, S. Diezi, T. Mallat and A. Baiker, J. Mol. Catal. A:
Chemical, 2008, 282, 1.
482 I. Busygin, O. P. Tkachenko, V. Nieminen, V. Yu. Borovkov, R. Sillanpa,
E. Toukoniitty, L. M. Kustov, D. Yu. Murzin and R. Leino, J. Phys. Chem. C,
2007, 111, 9374.
483 V. Nieminen, A. Taskinen, M. Hotokka and D. Yu. Murzin, J. Catal., 2007,
245, 228.
484 C. J. Baddeley, Top. Catal., 2003, 25, 17.
485 G. Bond, P. A. Meheux, A. Ibbotson and P. B. Wells, Catal. Today, 1991, 10,
371.
486 U. Maitra and P. Mathivanan, Tetrahedron: Asymmetry, 1994, 5, 1171.
487 B. Xiang, K. Snow and M. Belly, J.Org. Chem., 1993, 58, 993.
488 (a) J. F. Maddaluno, N. Gresh and C. Giessner-Prettre’d, J. Org. Chem., 1994,
59, 793; and (b) W. Oppolzer, M. Kurth, D. Reichlin, C. Chapuie, M.
Mohnhaupt and F. Moffatt, Helv. Chim. Acta, 1981, 64, 2802.
489 G. B. Jones and B. J. Chapman, Synthesis-Stuttgart, 1995, 475.
490 T. J Hall, P. Johnston, W. A. H. Vermeer, S. R. Watson and P. B. Wells, Stud.
Surf. Sci. Catal., 1996, 101, 221.
491 A. Tungler and G. Fogassy, J.Mol. Catal. A: Chemical, 2001, 173, 231.
492 V. Nieminen, A. Taskinen, Esa Toukoniitty, M. Hotokka and D.Yu. Murzin,
J. Catal., 2006, 237, 131.
493 I. Kun, B. Torok, K. Felfoldi and M. Bartok, Appl. Catal. A: General, 2000,
203, 71.
494 X. Zuo, H. Liu, D. Guo and X. Yang, Tetrahedron, 1999, 55, 7787.
495 J. W. D. Carneiro, C. D. de Oliveira, F. B. Passos, D.A. G. Aranda,
P. Rogerio, P. R. N. de Souza and O. A. Antunes, Catal. Today, 2005, 107, 31.
496 M. Bartok, K. Balazsik and F. Notheisz, React. Kinet. Catal. Lett., 2002, 77,
363.
497 S. Lavoie, G. Mahieu and P. H. McBreen, Angew. Chem., Int. Ed., 2006, 45,
7404.
498 M. B. Bartok, K. Felfoldi, G. Szollosi and T. Bartok, Catal. Lett., 1999, 61, 1.
499 O. Schwalm, B. Minder, J. Weber and A. Baiker, Catal. Lett., 1994, 23, 268.
1
5
10
15
20
25
30
35
40
45
Catalysis, 2010, 22, 144–278 | 277

500 O. Schwalm, J. Weber, B. Minder and A. Baiker, J. Quantum. Chem., 1994, 52,
1191.
501 J. Walkimar de M. Carneiro, C. da S. B. de Oliveira, F. B. Passos, D. A. G.
Aranda, P. R. N. de Souza and O. A.C. Antunes, Catal. Today, 2005, 31, 107.
502 K. E. Simons, Ph.D. thesis (University of Hull), 1994.
503 S. Lavoie, M. A. Laliberte, I. Temprano and P. H. McBreen, J. Am. Chem.
Soc., 2006, 128, 7588.
504 K. Szori, K. Balazsik, K. Felfoldi and M. Bartok, J. Catal., 2006, 241, 149.
505 K. Szori, K. Balazsik, K. Felfoldi, I. Bucsi, S. Cserenyi, G. Szollosi, E. Vass,
M. Hollosi and M. Bartok, J. Mol. Catal. A: Chemical, 2008, 294, 14.
506 J. W. D. Carneiro, C. D. B. de Oliveira, F. B. Passos, D. A. G. Aranda, P. R.
N. de Souza and O. A. C. Antunes, J. Mol. Catal. A Chem., 2005, 226, 221.
507 F. Manger, Tetrahedron, 1983, 39, 1013.
508 D. R. Storm and D. E. Koshland Jr, J. Am. Chem. Soc., 1972, 94, 5805.
509 I. C. Lee and R. I. Masel, J. Phys. Chem. B, 2002, 106, 368–373.
510 I. C. Lee and R. I. Masel, J. Phys. Chem. B, 2002, 106, 3902–3908.
511 F. Fache, E. Schulz, M. L. Tommasino and M. Lemaire, Chem. Rev., 2000,
100, 2159.
1
5
10
15
20
25
30
35
40
45
278 | Catalysis, 2010, 22, 144–278