loss of the nucleosome-binding protein hmgn1 affects the rate of n-nitrosodiethylamine-induced...
TRANSCRIPT

Chromatin, Gene, and RNA Regulation
Loss of the Nucleosome-Binding Protein HMGN1Affects the Rate of N-Nitrosodiethylamine-InducedHepatocarcinogenesis in Mice
Yuri V. Postnikov1, Takashi Furusawa1, Diana C. Haines3, Valentina M. Factor2, and Michael Bustin1
AbstractWe report that HMGN1, a nucleosome-binding protein that affects chromatin structure and function, affects
the growth of N-nitrosodiethylamine (DEN)-induced liver tumors. Following a single DEN injection at 2 weeks ofage, Hmgn1tm1/tm1 mice, lacking the nucleosome-binding domain of HMGN1, had earlier signs of livertumorigenesis than theirHmgn1þ/þ littermates. Detailed gene expression profiling revealed significant differencesbetween DEN-injected and control saline–injected mice, but only minor differences between the injectedHmgn1tm1/tm1 mice and their Hmgn1þ/þ littermates. Pathway analysis revealed that the most significant processaffected by loss of HMGN1 involves the lipid/sterol metabolic pathway. Our study indicates that in mice, loss ofHMGN1 leads to transcription changes that accelerate the progression of DEN-induced hepatocarcinogenesis,without affecting the type of tumors or the final total tumor burden of these mice.
Implications: Loss of HMGN1 leads to accelerated progression of DEN-induced hepatocarcinogenesis inmice. Mol Cancer Res; 12(1); 82–90. �2013 AACR.
IntroductionThe dynamic properties of the chromatin fiber play a key
role in transcriptional regulation and in the establishmentand maintenance of epigenetic marks. Disruption of theseprocesses can alter gene expression, potentially leading tovarious diseases including cancer. Indeed, increasing evi-dence links misexpression of tumor suppressors and onco-genes, and dysregulation of DNA repair processes, toalterations in chromatin structure and to changes in epi-genetic marks such as covalent modification in DNA andhistones (1–3). Factors that affect epigenetic marks in thegenome have been shown to contribute to the progressionof cancer (4) and epigenetic drugs targeting chromatinregulators show promise as anticancer agents (5). Becauseany factor that affects epigenetic processes could poten-tially contribute to tumorigenicity, it is important toexamine the role of various chromatin modifiers in theetiology of cancer. Here, we investigate the potential role ofthe chromatin-binding protein high mobility group N1(HMGN1) in hepatocarcinogenesis.
HMGN1 is a member of the high mobility group N(HMGN) family of chromosomal proteins. HMGN pro-teins are ubiquitously present in the nuclei of vertebrate cellsand bind specifically to nucleosome core particles, thebuilding blocks of the chromatin fiber (6). The chromatinresidence time of all HMGNs is short; these proteinscontinuously roam throughout the nucleus and reside onlytransiently on a specific site (7). Although HMGNs caninteract with all nucleosomes, their genome-wide organiza-tion is not random. Chromatin immunoprecipiation studiesrevealed that HMGN1 preferentially binds to gene promo-ters and enhancers where they tend to colocalize withDNaseI hypersensitive sites, a hallmark of chromatin regulatorysites (8).The binding of HMGN1 to nucleosomes induces struc-
tural changes in chromatin and alters the levels of posttrans-lational modifications of core histones raising the possibilitythat HMGN1 affects epigenetic regulatory processes. Sig-nificantly, the effects of HMGN1 on chromatin structureand function are contingent on the ability of the protein tobind to nucleosomes, HMGN1mutants that do not bind tonucleosomes do not affect chromatin structure or histonemodifications (9–13). We have generated the mice lackingfunctional HMGN1 (Hmgn1tm1/tm1 mice, formerly namedHmgn1�/�), that has been considered as Hmgn1 knockoutmice, although the mice express heavily truncated HMGN1that does not localize to nucleus. Studies with cells andtissues derived from genetically altered HMGN1 mice,which express HMGN1mutant that cannot bind to nucleo-somes, revealed that loss of HMGN1 function alters thecellular transcription profile (14, 15) and impairs the abilityto mount a proper response to cellular stress. Hmgn1tm1/tm1
Authors' Affiliations: 1Laboratory of Metabolism and 2Laboratory ofExperimental Carcinogenesis, National Cancer Institute, NIH, Bethesda;and 3Science Applications International Corporation, National CancerInstitute-Frederick, NIH, Frederick, Maryland
Note: Supplementary data for this article are available at Molecular CancerResearch Online (http://mcr.aacrjournals.org/).
Corresponding Author: Yuri V. Postnikov, NIH, 37 Convent Dr #3122,Bethesda, MD 20892-4255. Phone: 1-301-496-2885; Fax: 1-301-496-8419; E-mail: [email protected]
doi: 10.1158/1541-7786.MCR-13-0392
�2013 American Association for Cancer Research.
MolecularCancer
Research
Mol Cancer Res; 12(1) January 201482
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

mice and cells are hypersensitive to heat shock and theirability to repair DNA damaged by either UV or ionizingradiation is impaired (16–19). Faulty repair of damagedDNA could lead to genomic instability and increasedtumorigenicity. Taken together, the available informationsuggests that loss ofHMGN1may increase the susceptibilityto tumorigenesis, a possibility that has not yet been fullyexamined.Here, we examine the potential role of HMGN1 in
carcinogenesis, by comparing the progression of N-nitroso-diethylamine (DEN)-induced hepatocarcinogenesis (20) inHmgn1tm1/tm1 mice and Hmgn1þ/þ littermates. DEN is achemical carcinogen that has been extensively used foranalysis of factors that affect liver cancer development, oneof the most frequent human cancers (21). Young male miceare particularly susceptible to this carcinogen, as a singleinjection can result in hepatocellular carcinoma that issimilar to that seen in humans (22). We find that loss ofHMGN1 accelerates the development of liver cancers inDEN-injected mice. Transcription analysis of livers fromthese mice links loss of HMGN1 to alterations in severalpathways including the sterol/cholesterol/lipid metabolicprocess, raising the possibility that transcriptional changesresult in the accelerated carcinogenesis seen inHmgn1tm1/tm1
mice.
Materials and MethodsAnimal studiesHmgn1tm1/tm1 (previously namedHmgn1�/�) and control
Hmgn1þ/þmice were generated and genotyped as described(16). In these mice, exons II, III, and IV of theHmgn1 gene,which code for the nucleosome-binding domain of theprotein, have been excised. For genotyping, tail DNA wasextracted using REDExtract-N-Amp Tissue PCR Kit (Sig-ma-Aldrich) using three primers (see Supplementary TableS1). Since female mice are less sensitive than males to DEN-induced carcinogenesis, only male homozygous mice wereused for the experiments. Mice received a single intraperi-toneal injection of 10 mg/g body weight of N-nitrosodiethy-lamine (Sigma-Aldrich, Inc., Cat. 40334), or saline as acontrol, at 14 days of age.Mice were sacrificed at 23, 48, and73 weeks after the injection. Animals were housed at theNCI Animal Facility and NCI-Frederick SAIC facility andcared for in accordance with theNIHGuide for the Care andUse of Laboratory Animals.
Necropsy and histopathologyAll livers were harvested at necropsy, weighed, photo-
graphed, and thoroughly examined. The number of mac-roscopic nodules/masses � 1 mm was recorded for eachliver. Livers were then fixed in 10% neutral bufferedformalin, routinely processed to paraffin block, and sec-tioned at 5 mm. Hematoxylin and eosin (H&E)-stainedsections were evaluated microscopically for quantificationof foci, adenomas, and carcinomas. The areas occupied byfoci and neoplasms were measured using ImageJ software(NIH, Bethesda, MD).
Protein isolation and Western blot analysisLiver caudate lobes were homogenized by Dounce light
homogenizer in 1� PBS. The cell suspensions were washedin 1� PBS and centrifuged at 600 � g for 10 minutes. Thecellular pellet was dissolved in either 0.2 mol/L sulfuric acidor 5% perchloric acid, both containing a protease inhibitorcocktail (Roche), homogenized by Dounce tight pestlehomogenizer for 2 minutes, kept on ice for 5 minutes, andspun at 3,000� g for 10minutes. The supernatant wasmade25% in TCA, incubated on ice for 15 minutes, and cen-trifuged at 3,000� g for 20minutes. The pellet was stored at�20�C overnight in 100% ethanol, air-dried, and resus-pended with 50 to 100 mL of water. The preparations werereprecipitated by HCl/acetone, washed in 100% acetone,air-dried, and resuspended with 50 to 100 mL of water.Proteins were resolved on 15%Tris-glycine-SDS gels, trans-ferred to polyvinylidene difluoridemembrane, and subjectedto Western blotting.
Immunohistochemistry stainingImmunohistochemical staining was carried out on forma-
lin-fixed, paraffin-embedded tissue using the avidin-biotin-peroxidase complex method (Vector Laboratories) and 30-diaminobenzidine (DAB) for staining. Proliferating cellnuclear antigen antibody (PCNA; SantaCruzBiotechnology,sc-25280) and rabbit anti-Ki67 antibody (Abcam, ab15580)were used according to the manufacturer's recommendation.Sections were counter-stained with hematoxylin.
RNA isolationTotal RNA was isolated from frozen liver tissue with
TRIzol reagent (Invitrogen) followed by purification usingRNeasy Kit (Qiagen) according to the manufacturer's ins-tructions. Reverse transcription of total RNA (2.0 mg) intofirst strand cDNAusing oligo(dT) primers (SuperScript FirstStrand Synthesis System for RT-PCR; Invitrogen) has beenfollowed by PCR using Platinum PCR SuperMix (Invitro-gen) and the specific primers (see Supplementary Table S1).PCR products were resolved on 2% agarose gels and visu-alized by ethidium bromide staining.
Microarray analysisExpression profiling was conducted for six groups of mice.
GroupA consisted ofmice at 4weeks of age. Two other groupsconsisted of 12-week-old mice injected at the age of 4 weekseither with saline (group B) or DEN (group C). Each groupincludes both genotypes (Hmgn1þ/þ and Hmgn1tm1/tm1).For microarray analysis, total RNA was isolated as
described above. The cDNA synthesis, fluorescent labelingof the samples, andmouse 430.2 Affymetrix expression arrayhybridizations were done following the suppliers' recom-mendations. Arrays were scanned with Agilent scanneradjusted to achieve optimal signal intensity at both channelswith <1% saturated spots and normalized to the 50thpercentile of the median signal intensity. Normalized inten-sity values were used for further analysis.Mouse Genome 430 2.0 array has 45,101 probe sets
associated with approximately 20,000 Mouse Genome
HMGN1 Affects Hepatocarcinogenesis in Mice
www.aacrjournals.org Mol Cancer Res; 12(1) January 2014 83
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

Informatics (MGI) gene identifiers. Probe sets were mappedtoMGI identifiers using information provided by the JacksonLaboratory (http://www.informatics.jax.org/). GeneSpringGX software package (Agilent Technologies) was employedto evaluate the quality of the arrays and for analysis.Differentially expressed genes were selected using univariate
two-sample t test (P � 0.01) with a random variance model,using mean value for each of six groups (A-Hmgn1þ/þ, A-Hmgn1tm1/tm1, B-Hmgn1þ/þ, B-Hmgn1tm1/tm1, C-Hmgn1þ/þ,C-Hmgn1tm1/tm1). Unsupervised and supervised cluster anal-yses were done with GeneSpring GX analysis suite (version11.5.1, Agilent Technologies, Cat. G3784AA). FunctionalGO and other data mining tools for significant genes werebased on gene ontology annotations (Ingenuity PathwayAnalysis software, Ingenuity Systems). The raw data for all18 samples in can be found at GEO dataset # GSE44356.
Quantitative real-time PCRSelected gene expression microarray data were confirmed
by quantitative real-time PCR (qRT-PCR) using AppliedBiosystems 7900 HT. Sequences of primers used for qRT-PCR analyses are provided in Supplementary Table S1.GAPDH and actin primers were used for normalization ofexpression levels. qRT-PCR was performed using PowerSYBR Green RNA-to-CT 1-Step Kit (Applied Biosystems,Cat. No 4391178) and iScript One-Step RT_PCR Kit withSYBR Green (Bio-Rad, Cat. No 170-8893) on a 7900HTFast Real-Time PCR System instrument. A dissociationcurve program was employed after each reaction. The purityof the PCR products was validated by electrophoresis on 4%NuSieve 3:1 agarose (Lonza).
ResultsHMGN1 affects the latency of DEN-inducedhepatocarcinogenesisTo test whether loss of HMGN1 function affects the
course of DEN-induced hepatocarcinogenesis, 2-week-oldmaleHmgn1þ/þ andHmgn1tm1/tm1 littermate mice receiveda single intraperitoneal injection of N-nitrosodiethylamineat a dose of 10 mg/g body weight. Saline injected mice servedas controls. Mice were sacrificed at ages of 25, 50, and 75weeks and their livers examined for tumor incidence, size,multiplicity, and histologic appearance. Western blot anal-ysis of nuclear extracts from these livers indicated that duringthe course of the experiment the expression of HMGN1, orof its closely related protein HMGN2, did not change andthat in Hmgn1tm1/tm1 mice, which lack nuclear HMGN1,
there were no compensatory adjustment in the levels ofHMGN2 (Fig. 1).As expected, the mice injected with DEN developed
tumors, whereas the saline injected mice did not. Wefollowed the development of the tumors by sacrificing mice23, 48, and 73 weeks after DEN injection. At 23 weeks afterDEN administration, the total number of preneoplastic fociobserved in Hmgn1tm1/tm1 mice was not significantly differ-ent from that observed in their Hmgn1þ/þ littermates;however, the size of the foci was significantly larger inHmgn1tm1/tm1 mice (Fig. 2A). Thus, in Hmgn1þ/þ mice,43% of the foci were smaller than 50 mm2 and only 2%werelarger than 500 mm2, whereas in Hmgn1tm1/tm1 mice, lessthan 20% of the foci were smaller than 50 mm2 and almost30% of the foci were larger than 500 mm2 (Fig. 2A).Although the foci in Hmgn1tm1/tm1 mice were larger, therate of hepatocyte proliferation in the surrounding liver, asmeasured by mouse antigen Ki67 (Mki67) and PCNAimmunostaining did not differ between the mice (Fig.3A). Likewise, although qRT-PCR analyses reveal theexpected upregulation of cyclin D1 (Ccnd1), Mki67, andMyc, there were no significant differences between theDEN-injected Hmgn1þ/þ and Hmgn1tm1/tm1 littermatemice (Fig. 3B). Comparative evaluation of H&E-stainedsections from the liver samples at 23 weeks after injectionindicated that in both Hmgn1þ/þ and Hmgn1tm1/tm1 mice,the early preneoplastic lesions displayed a predominantbasophilic cell phenotype characteristic of theDEN-inducedcarcinogenesis (Supplementary Fig. S1).In contrast, at 48 weeks post-DEN, both the number and
size of preneoplastic and neoplastic lesions in Hmgn1tm1/tm1
livers were considerably bigger than inHmgn1þ/þmice (Fig.2B and C). The increased tumor frequency and size corre-lated with a higher liver-to-body weight ratio, reflecting theapparent differences in the growth rate of preneoplastic andneoplastic hepatic lesions between knockout and controlmice (Fig. 2D). However, at 75 weeks of age, no significantdifference between Hmgn1þ/þ and Hmgn1tm1/tm1 micecould be detected in any of the parameters of tumor growthexamined, including liver-to-body weight ratios, tumorburden, and malignancy (Fig. 2C and D). At this stage, thetumors were mostly hepatocellular adenomas and carcino-mas and the type of tumors observed was essentially the samein both wild-type and Hmgn1tm1/tm1 mice (Fig. 2E). Thus,loss of HMGN1 enhances the rate of DEN-induced hepa-tocarcinogenesis but does not alter the type of tumorsformed.
Figure 1. Expression of HMGN1and HMGN2 during the course ofthe experiment. Shown areWestern blotting analyses ofHMGN1, HMGN2, and histone H3in livers of 25-, 50-, and 75-week-old Hmgn1þ/þ and Hmgn1tm1/tm1
mice which were injected witheither DEN or saline at 2 weeks ofage.
Postnikov et al.
Mol Cancer Res; 12(1) January 2014 Molecular Cancer Research84
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

Effects of HMGN1 on the global gene expression patternin the livers of DEN-injected miceTo gain insights into the mechanisms whereby HMGN1
enhances the rate of tumorigenicity in the livers of DEN-injected mice, we examined the transcription profile inthe livers of control and DEN-injected Hmgn1þ/þ andHmgn1tm1/tm1 littermate mice. Since changes in gene expres-sion would precede the appearance of preneoplastic foci andtumors, we examined the hepatic expression profiles in thefollowing 3 groups, each consisting of 6mice. The first groupA which included 4-week-old untreated Hmgn1þ/þ andHmgn1tm1/tm1 littermates served to test whether loss ofHMGN1 affects the liver transcription profile before anytreatment. The group B contained 12-week-old Hmgn1þ/þ
and Hmgn1tm1/tm1 littermates that were injected with saline
at 4 weeks of age. This group served as controls for group C,which consisted of 12-week-oldHmgn1þ/þ andHmgn1tm1/tm1
littermates that were injected with DEN at 4 weeks of age.The raw data for all 18 samples can be found at GEO dataset# GSE44356.Principal component analysis (PCA) of the transcriptomes
of all livers revealed a clear separation between the 3 groups(Fig. 4A). The distinct clustering of group A from group Breflects the age-related changes in gene expression, whereasthe distinct clustering of B fromC reflects theDEN-inducedchanges in gene expression. Within each group, theHmgn1þ/þ mice formed a subcluster that was separate fromtheir Hmgn1tm1/tm1 littermates, the largest differencebetween these subclusters was observed in the youngestmice (group A). Supervised comparison based on asymptotic
Figure 2. Incidence and kineticsof growth of liver tumors inDEN-treated Hmgn1þ/þ andHmgn1tm1/tm1 mice. A, sizedistribution of hepatic lesions inDEN-injected Hmgn1þ/þ andHmgn1tm1/tm1 mice at 25 weeks.Areas of the foci have beendetermined using ImageJsoftware. B, representativeimages of livers from 50-week-oldDEN-injected Hmgn1þ/þ andHmgn1tm1/tm1 mice. Tumors areindicated by arrows. C, kineticsof tumor incidence in livers fromDEN-treated Hmgn1þ/þ andHmgn1tm1/tm1 mice. ��, P � 0.01,determined by Fisher test. D, liverto whole body weight ratios of 25-,50-, and 75-week-old Hmgn1þ/þ
andHmgn1tm1/tm1mice after salineor DEN treatment (�, P � 0.05). E,histograms of tumor types in liversof 75-week-oldDEN-injectedmice.
HMGN1 Affects Hepatocarcinogenesis in Mice
www.aacrjournals.org Mol Cancer Res; 12(1) January 2014 85
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

one-way ANOVA calculation using a P� 0.01 revealed thatin total the expression of 2,654 genes was altered. Of these,the expression of 520 genes was altered more than two-foldbetween at least two groups (Supplementary Table S2). Themicroarray expression data were validated by real-time PCRof 8 genes randomly selected in group A, using two sets ofprimers per each gene (Fig. 4B).Venn diagrams of the up- and downregulated genes
revealed a partial overlap between the 3 groups (Fig. 4C).We note that the total number of genes which expressionchanged due to lack of HMGN1 was similar among the 3groups. Thus, in the 4-week-old untreated mice (groupA), loss of HMGN1 led to the upregulation of 172 genesand downregulation of 152 genes; a total change of 324genes. Similarly, the total number of genes changed ingroup B and C was 322 and 334, respectively. Examina-tion of the 10 top genes in which expression was either up-or downregulated in each group (Table 1) did not revealcommon genes or pathways regulated by HMGN1. Like-wise, it did not contain any genes that could likelycontribute to shorten the latency or tumorigenicity. Thepartial overlap between the genes downregulated in groupsB and C suggests that the effect were due to loss ofHMGN1 rather than to DEN treatment. The data areconsistent with previous findings that HMGN1 altersslightly the expression of numerous genes but does notspecifically regulate the expression of a distinct subset ofgenes (14, 15).
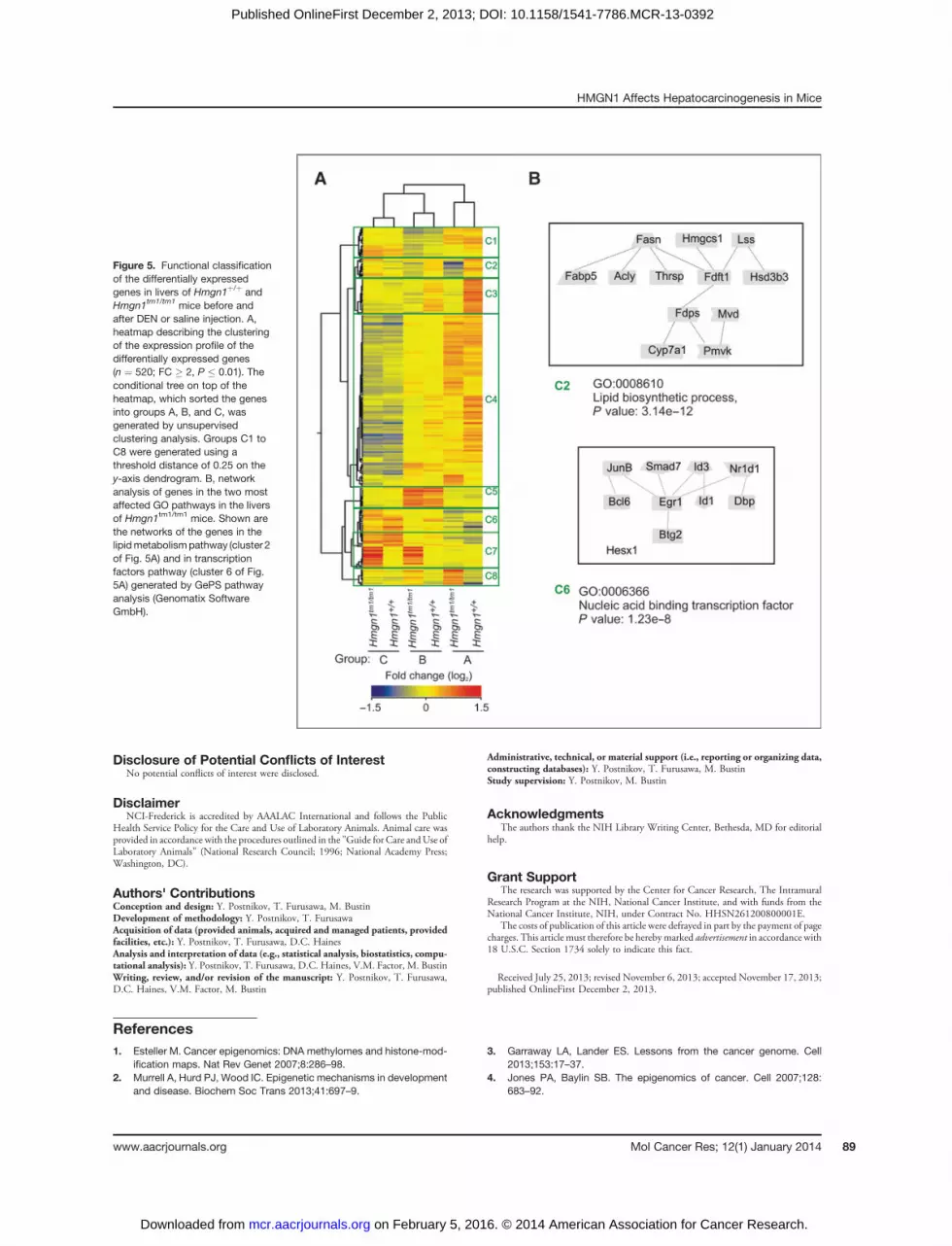
Hierarchical clustering defines groups of genes associatedwith DEN-induced altered expression patternsHierarchical condition tree of unsupervised clustering
analysis of the differentially expressed genes (n ¼ 520; FC� 2, P � 0.01) sorted groups A, B, and C (top of the heatmap) apart, without separating two genotypes in eachgroup (Fig. 5A). Using a threshold distance of 0.25 on
the y-axis dendrogram, the genes were classified into 8major clusters, each representing a specific gene expressionpattern (green boxes C1–C8, Fig. 3A). The overall patternof clustering revealed that the differences between groupsA, B, and C were larger than the differences between theHmgn1þ/þ and Hmgn1tm1/tm1 within each group, a furtherindication that loss of HMGN1 does not have a majorimpact on the expression levels of a specific subset of genes.Nevertheless, clusters C2, C3, C7, and C8 contained thesubsets of genes affected by the loss of HMGN1. Thus, inHmgn1tm1/tm1 mice, the genes in clusters C2, C3, and partof C4 were downregulated, the genes in clusters C7 and C8were upregulated. The differences between groups B and Cwere the most prominent in clusters C4, C5, and C6; thesegenes may be involved in the liver response to DENinjection.A search for the genes linked to hyperproliferation,
dysplasia, and other cancer-related genes in the variousgroups and clusters yielded JunB. This was the only can-cer-related gene which was overexpressed in Hmgn1tm1/tm1
mice at 4 weeks of age but not following DEN adminis-tration at 12 weeks, making it unlikely to contribute to thedifference in tumor development at the later stages. Like-wise, of the 65 genes associated with high risk of poorprognosis for hepatocellular carcinoma (23) only theTbx3transcription factor was differentially expressed inHmgn1tm1/tm1 livers, but its expression was downregulated.These results further support the notion thatHMGN1 doesnot specifically regulate the expression of genes involved inliver carcinogenesis.Functional analysis of all 520 differentially expressed
genes identified the sterol/cholesterol/lipid metabolic pro-cess pathway-associated genes as the most altered (P �4.09E-05; Supplementary Table S3). Similar analysis of the8 gene clusters identified by unsupervised clustering analysis(Fig. 5A) revealed that the sterol/cholesterol/lipid metabolic
Figure 3. Apoptotic and proliferative activity in livers of Hmgn1þ/þ and Hmgn1tm1/tm1 mice, 23 weeks after DEN injection. A, representative light microscopyimages of immunoperoxidase staining with Mki67 (top images) and PCNA (bottom images) of paraffin-embedded liver sections counterstained withhematoxylin. Positively stained cells are indicated by arrows. B, relativemRNA expression levels of genes involved in control of proliferation (cyclin D1 andKi-67) and apoptosis (caspase-9 andBcl2) asmeasuredby qRT-PCR (3mice per saline-injected andDEN-injected group ofmice of each genotype). The data areDDCt values (mean � SEM). The expression levels were normalized to that of saline-injected Hmgn1þ/þ mice, which was set to 1.
Postnikov et al.
Mol Cancer Res; 12(1) January 2014 Molecular Cancer Research86
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

process pathway-associated genes were most significantlyover-represented (P � 3.14E-12) in cluster C2 (Fig. 5B).This cluster contained several genes belonging to this path-way including Fasn, Cyp7a1, Mvd, and Thrsp (Fig. 5B). Inaddition, in cluster C6, the presence of transcription factoractivity pathway-oriented genes GO:0006366 is highlyenriched (P � 1.23E-8). This cluster contained genes thatwere upregulated by DEN treatment and differed betweenHmgn1þ/þ and Hmgn1tm1/tm1 littermates (Fig. 5B) andcould contribute to earlier signs of DEN-induced hepato-carcinogenesis which we observed in Hmgn1tm1/tm1 mice.
DiscussionIn the present study, we show that loss of HMGN1
protein in mice increases the rate of liver tumorigenesis afterDEN treatment. In Hmgn1tm1/tm1 mice, preneoplasticlesions seen at 23 weeks after DEN administration werelarger than in the Hmgn1þ/þ littermates. Likewise, thenumber of foci and the average size of tumors seen wassignificantly increased in the 50-week-old mice lacking
functional HMGN1 protein as compared with wild-typemice. However, at 73 weeks after DEN administration, thesize and number of tumors, and their histologic appearancewere similar in both Hmgn1þ/þ and Hmgn1tm1/tm1 mice.Thus, loss ofHMGN1 increased the growth rate, but not thetype of tumors or the tumor burden.The results are compatible with previous findings of
increased tumor incidence inHmgn1tm1/tm1mice suggestingthat loss of HMGN1 predisposes to increased tumor sus-ceptibility (17). Embryonic fibroblasts from Hmgn1tm1/tm1
mice were shown to proliferate faster than cells preparedfrom their Hmgn1þ/þ littermates and SV40-immortalizedHmgn1tm1/tm1 cells induced more tumors than SV40-trans-formedHmgn1þ/þ cells when injected into nude mice (17).In addition, HMGN1 increased the recruitment rate ofPCNA toUV-damagedDNA sites (24) and loss ofHMGN1impaired the ability of Hmgn1tm1/tm1 cells to repair DNAdamaged by either UV or ionizing irradiation (16, 17, 25),thereby predisposing Hmgn1tm1/tm1 mice to increased geno-mic instability and tumorigenicity (17, 26). In agreementwith these findings (17, 26), the cancer initiation was
Figure 4. Effects of HMGN1ongeneexpression in livers. A, PCA of thetranscription profiles in the liversof control and DEN-injectedHmgn1þ/þ andHmgn1tm1/tm1 livers.Each dot represents one mouse.The size of the dot reflects itsposition within the three-dimensional plot. B, validation ofmicroarray data by semi-quantitative PCR. Microarray dataare represented by logarithm base2 ratios of the average individualentity (gene) intensities forHmgn1þ/þ and Hmgn1tm1/tm1 mice(three mice of each genotype, non-treated by DEN at 4 weeks of age)and normalized for the totalintensity (GeneSpring GX 11.5.1 byAgilent Technologies). Ratiosbased on RT-PCR data werecalculated in a way to be comparedto microarray data (comparative Ct
method). DCt value was calculatedfor each primer set normalized tocontrol primers (GAPDH andb-actin) for every biologic sample.Next, the values were convertedinto relative amount of target byusing a formula "Normalizedamount of specificmRNA¼ 2DDCt".Normalized amounts of mRNAswere plotted as log2 ratio betweenwild-type and mutant samples. C,Venn diagrams of up - anddownregulated genes in the 3groups investigated. Group A, 4-week-old untreated Hmgn1þ/þ andHmgn1tm1/tm1 littermates. Group B,12-week-old littermates injectedwith saline at 4weeksof age.GroupC, 12-week-old littermates injectedwith DEN at 4 weeks of age.
HMGN1 Affects Hepatocarcinogenesis in Mice
www.aacrjournals.org Mol Cancer Res; 12(1) January 2014 87
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

notably faster in Hmgn1tm1/tm1 mice treated with DEN.However, at the later stages, the differences inDEN-inducedtumor development betweenHmgn1þ/þ and Hmgn1tm1/tm1
mice became less pronounced suggesting the differentialrequirements for HMGN1 function during multistep pro-cess of chemical hepatocarcinogenesis. As we have shown inthe past, several proto-oncogenes, such as JunB and c-Jun,have been downregulated in the Hmgn1tm1/tm1
fibroblasts(9). Thus, HMGN1 regulates the transcription of multiplegenes which may have opposing effects on the rate of tumorgrowth at different stages of tumor development.Our analysis of the liver transcription profiles did not
reveal significant effect of HMGN1 deletion on the expres-sion levels of DNA damage repair factors suggesting thatthe accelerated hepatocarcinogenesis in Hmgn1tm1/tm1 isnot due to faulty expression of these factors. In agreementwith previous analyses of a variety of tissues including liver,loss of HMGN disrupted the expression of multiple genes,but only mildly, suggesting that HMGN1 fine tunes the
fidelity of the cellular transcription profile (14, 15). DENinjection did not significantly increase the transcriptiondifferences between Hmgn1þ/þ and Hmgn1tm1/tm1 mice, afinding that is fully compatible with the similarity in thenumber and types of tumors seen in the mice. Thus, lossof HMGN1 does not fundamentally alter the cellularresponse to DEN.GO analysis identified the sterol/lipid metabolic pathways
as the most different category of genes between DEN-injected Hmgn1þ/þ and Hmgn1tm1/tm1 mice. Emergingevidence links altered sterol/lipid metabolic profile to chron-ic low-grade systemic inflammation (27), which is believedto contribute to metabolic disorders, and the stagewiseprogression to hepatic steatosis, fibrosis, cirrhosis, and finallyto carcinoma (28–30). Hepatocellular carcinoma has beenalso linked to nonalcoholic fatty liver disease (31). As anarchitectural element of chromatin, HMGN1 protein iscapable to change expression profile of the cell globally,affecting several pathways simultaneously (14, 15).
Table 1. Top 10 genes, differentially expressed in livers of Hmgn1þ/þ and Hmgn1tm1/tm1a mice
Genes, upregulated in Hmgn1�/�
Group A Group B Group C
Gene symbolEntrezgene
Expression log2
ratio, Hmgn1þ/þ
vs. Hmgn1�/�a)Genesymbol
Entrezgene
Expression log2
ratio, Hmgn1þ/þ
vs. Hmgn1�/�)Genesymbol
Entrezgene
Expression log2
ratio, Hmgn1þ/þ
vs. Hmgn1�/�)
Cyp7a1 13122 2.56 Xlr4a 434794 1.46 Myh1 17879 1.18Olig1 50914 2.40 Cyp7a1 13122 1.20 Creld2 22059 1.02Acta2 11475 1.61 Olig1 50914 1.16 Dbp 13170 0.92Pltp 18830 1.58 Mvd 192156 1.04 Tff3 21786 0.90Cdk5rap1 66971 1.54 Rgs16 19734 1.01 Mt2 17750 0.84Naip2 17948 1.54 Hspa8 15481 0.95 Scara5 71145 0.80Mvd 192156 1.52 Ccdc25 67179 0.94 Syvn1 74126 0.71Hspa8 15481 1.51 Fasn 14105 0.91 Mt1 17748 0.70Fabp5 16592 1.51 Thrsp 21835 0.90 Mtss1 211401 0.63Hhex 15242 1.48 BC005512 192885 0.88 Onecut1 15379 0.60
Genes, downregulated in Hmgn1�/�
Group A Group B Group C
Gene symbolEntrezgene
Expression log2
ratio, Hmgn1þ/þ
vs. Hmgn1�/�)Genesymbol
Entrezgene
Expression log2
ratio, Hmgn1þ/þ
vs. Hmgn1�/�)Genesymbol
Entrezgene
Expression log2
ratio, Hmgn1þ/þ
vs. Hmgn1�/�)
Scara5 71145 �3.52 Hsd3b1 15492 �2.20 Hsd3b1 15492 �2.15Mt2 17750 �3.12 Star 20845 �2.08 Star 20845 �2.03Id1 15901 �2.73 Akr1b7 11997 �2.02 Akr1b7 11997 �1.95Mt1 17748 �2.43 Chgb 12653 �1.70 Adipoq 9370 �1.88Slc15a2 57738 �2.24 Adipoq 9370 �1.69 Scg2 20254 �1.78Id2 15902 �2.15 Id1 15901 �1.55 Cyp11b1 110115 �1.73Dbp 13170 �1.94 Cyp11b1 110115 �1.45 Chga 12652 �1.71Skiv2l2 72198 �1.77 Id2 15902 �1.37 Chgb 12653 �1.66Egr1 13653 �1.74 Bcl6 12053 �1.36 Cyp11a1 13070 �1.62Hes1 15205 �1.64 Scg2 20254 �1.30 Hao2 56185 �1.52
aIn the table, the Hmgn1tm1/tm1 genotype is indicated as Hmgn1�/�.
Postnikov et al.
Mol Cancer Res; 12(1) January 2014 Molecular Cancer Research88
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
DisclaimerNCI-Frederick is accredited by AAALAC International and follows the Public
Health Service Policy for the Care and Use of Laboratory Animals. Animal care wasprovided in accordance with the procedures outlined in the "Guide for Care andUse ofLaboratory Animals" (National Research Council; 1996; National Academy Press;Washington, DC).
Authors' ContributionsConception and design: Y. Postnikov, T. Furusawa, M. BustinDevelopment of methodology: Y. Postnikov, T. FurusawaAcquisition of data (provided animals, acquired and managed patients, providedfacilities, etc.): Y. Postnikov, T. Furusawa, D.C. HainesAnalysis and interpretation of data (e.g., statistical analysis, biostatistics, compu-tational analysis): Y. Postnikov, T. Furusawa, D.C. Haines, V.M. Factor, M. BustinWriting, review, and/or revision of the manuscript: Y. Postnikov, T. Furusawa,D.C. Haines, V.M. Factor, M. Bustin
Administrative, technical, or material support (i.e., reporting or organizing data,constructing databases): Y. Postnikov, T. Furusawa, M. BustinStudy supervision: Y. Postnikov, M. Bustin
AcknowledgmentsThe authors thank the NIH Library Writing Center, Bethesda, MD for editorial
help.
Grant SupportThe research was supported by the Center for Cancer Research, The Intramural
Research Program at the NIH, National Cancer Institute, and with funds from theNational Cancer Institute, NIH, under Contract No. HHSN261200800001E.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be herebymarked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
Received July 25, 2013; revised November 6, 2013; accepted November 17, 2013;published OnlineFirst December 2, 2013.
References1. Esteller M. Cancer epigenomics: DNA methylomes and histone-mod-
ification maps. Nat Rev Genet 2007;8:286–98.2. Murrell A, Hurd PJ, Wood IC. Epigenetic mechanisms in development
and disease. Biochem Soc Trans 2013;41:697–9.
3. Garraway LA, Lander ES. Lessons from the cancer genome. Cell2013;153:17–37.
4. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007;128:683–92.
Figure 5. Functional classificationof the differentially expressedgenes in livers of Hmgn1þ/þ andHmgn1tm1/tm1 mice before andafter DEN or saline injection. A,heatmap describing the clusteringof the expression profile of thedifferentially expressed genes(n ¼ 520; FC � 2, P � 0.01). Theconditional tree on top of theheatmap, which sorted the genesinto groups A, B, and C, wasgenerated by unsupervisedclustering analysis. Groups C1 toC8 were generated using athreshold distance of 0.25 on they-axis dendrogram. B, networkanalysis of genes in the two mostaffected GO pathways in the liversof Hmgn1tm1/tm1 mice. Shown arethe networks of the genes in thelipidmetabolismpathway (cluster 2of Fig. 5A) and in transcriptionfactors pathway (cluster 6 of Fig.5A) generated by GePS pathwayanalysis (Genomatix SoftwareGmbH).
HMGN1 Affects Hepatocarcinogenesis in Mice
www.aacrjournals.org Mol Cancer Res; 12(1) January 2014 89
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

5. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism totherapy. Cell 2012;150:12–27.
6. Bustin M. Chromatin unfolding and activation by HMGN(�) chromo-somal proteins. Trends Biochem Sci 2001;26:431–7.
7. Catez F, Lim JH, Hock R, Postnikov YV, Bustin M. HMGNdynamics and chromatin function. Biochem Cell Biol 2003;81:113–22.
8. Cuddapah S, Schones DE, Cui K, Roh TY, Barski A, Wei G, et al.Genomic profiling of HMGN1 reveals an association with chromatin atregulatory regions. Mol Cell Biol 2011;31:700–9.
9. LimJH,Catez F,Birger Y,WestKL,Prymakowska-BosakM,PostnikovYV, et al. Chromosomal protein HMGN1 modulates histone H3 phos-phorylation. Mol Cell 2004;15:573–84.
10. Lim JH, West KL, Rubinstein Y, Bergel M, Postnikov YV, Bustin M.Chromosomal protein HMGN1 enhances the acetylation of lysine 14 inhistone H3. EMBO J 2005;24:3038–48.
11. Postnikov YV, Belova GI, Lim JH, Bustin M. Chromosomal proteinHMGN1 modulates the phosphorylation of serine 1 in histone H2A.Biochemistry 2006;45:15092–9.
12. Ueda T, Postnikov YV, Bustin M. Distinct domains in high mobilitygroup N variants modulate specific chromatin modifications. J BiolChem 2006;281:10182–7.
13. Kim YC, Gerlitz G, Furusawa T, Catez F, Nussenzweig A, Oh KS,et al. Activation of ATM depends on chromatin interactionsoccurring before induction of DNA damage. Nat Cell Biol 2009;11:92–6.
14. Kugler JE, Horsch M, Huang D, Furusawa T, Rochman M, Garrett L,et al. Highmobility groupN proteins modulate the fidelity of the cellulartranscriptional profile in a tissue- and variant-specific manner. J BiolChem 2013;288:16690–703.
15. Rochman M, Taher L, Kurahashi T, Cherukuri S, Uversky VN, Lands-man D, et al. Effects of HMGN variants on the cellular transcriptionprofile. Nucleic Acids Res 2011;39:4076–87.
16. Birger Y,WestKL, PostnikovYV, Lim JH, FurusawaT,Wagner JP, et al.Chromosomal protein HMGN1 enhances the rate of DNA repair inchromatin. EMBO J 2003;22:1665–75.
17. Birger Y, Catez F, Furusawa T, Lim JH, Prymakowska-Bosak M, WestKL, et al. Increased tumorigenicity and sensitivity to ionizing radiationupon loss of chromosomal protein HMGN1. Cancer Res 2005;65:6711–8.
18. Belova GI, Postnikov YV, Furusawa T, Birger Y, Bustin M. Chromo-somal protein HMGN1 enhances the heat shock-induced remodelingof Hsp70 chromatin. J Biol Chem 2008;283:8080–8.
19. Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockaynesyndrome A and B proteins differentially regulate recruitment of chro-matin remodeling and repair factors to stalled RNA polymerase II invivo. Mol Cell 2006;23:471–82.
20. Dragan YP, Xu YH, Pitot HC. The effect of the dose of diethylnitrosa-mine on the initiation of altered hepatic foci in neonatal female rats.Carcinogenesis 1993;14:385–91.
21. Soerjomataram I, Lortet-Tieulent J, Parkin DM, Ferlay J, Mathers C,FormanD, et al. Global burden of cancer in 2008: a systematic analysisof disability-adjusted life-years in 12 world regions. Lancet 2012;380:1840–50.
22. Verna L, Whysner J, Williams GM. N-nitrosodiethylamine mechanisticdata and risk assessment: bioactivation, DNA-adduct formation,mutagenicity, and tumor initiation. Pharmacol Ther 1996;71:57–81.
23. Kim SM, Leem SH, Chu IS, Park YY, Kim SC, Kim SB, et al. Sixty-fivegene-based risk score classifier predicts overall survival in hepato-cellular carcinoma. Hepatology 2012;55:1443–52.
24. Postnikov YV, Kurahashi T, Zhou M, Bustin M. The nucleosomebinding protein HMGN1 interacts with PCNA and facilitates its bindingto chromatin. Mol Cell Biol 2012;32:1844–54.
25. Subramanian M, Gonzalez RW, Patil H, Ueda T, Lim JH, Kraemer KH,et al. The nucleosome-binding protein HMGN2 modulates globalgenome repair. FEBS J 2009;276:6646–57.
26. Gerlitz G. HMGNs, DNA repair and cancer. Biochim Biophys Acta2010;1799:80–5.
27. Vongsuvanh R, George J, Qiao L, van der Poorten D. Visceral adiposityin gastrointestinal and hepatic carcinogenesis. Cancer Lett 2013;330:1–10.
28. Jou J, Choi SS, Diehl AM. Mechanisms of disease progression innonalcoholic fatty liver disease. Semin Liver Dis 2008;28:370–9.
29. Szabo G, Lippai D. Molecular hepatic carcinogenesis: impact ofinflammation. Dig Dis 2012;30:243–8.
30. Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol2012;56:704–13.
31. Starley BQ, CalcagnoCJ, Harrison SA. Nonalcoholic fatty liver diseaseand hepatocellular carcinoma: a weighty connection. Hepatology2010;51:1820–32.
Postnikov et al.
Mol Cancer Res; 12(1) January 2014 Molecular Cancer Research90
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392

2014;12:82-90. Published OnlineFirst December 2, 2013.Mol Cancer Res Yuri V. Postnikov, Takashi Furusawa, Diana C. Haines, et al. of N-Nitrosodiethylamine-Induced Hepatocarcinogenesis in MiceLoss of the Nucleosome-Binding Protein HMGN1 Affects the Rate
Updated version
10.1158/1541-7786.MCR-13-0392doi:
Access the most recent version of this article at:
Material
Supplementary
http://mcr.aacrjournals.org/content/suppl/2013/12/02/1541-7786.MCR-13-0392.DC1.html
Access the most recent supplemental material at:
Cited articles
http://mcr.aacrjournals.org/content/12/1/82.full.html#ref-list-1
This article cites 31 articles, 11 of which you can access for free at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications Department at
on February 5, 2016. © 2014 American Association for Cancer Research. mcr.aacrjournals.org Downloaded from
Published OnlineFirst December 2, 2013; DOI: 10.1158/1541-7786.MCR-13-0392