impaired megakaryocytopoiesis in type 2b von willebrand disease with severe thrombocytopenia
TRANSCRIPT

doi:10.1182/blood-2006-03-009449Prepublished online May 23, 2006;
Corvazier, Robert Combrie, Edith Fressinaud, Dominique Meyer, Alan T Nurden and Jocelyne EnoufPaquita Nurden, Najet Debili, William Vainchenker, Regis Bobe, Raymonde Bredoux, Elisabeth severe thrombocytopeniaImpaired megakaryocytopoiesis in type 2B von Willebrand disease with
(2497 articles)Hemostasis, Thrombosis, and Vascular Biology � (3368 articles)Clinical Trials and Observations �
(790 articles)Cell Adhesion and Motility �Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
digital object identifier (DOIs) and date of initial publication. theindexed by PubMed from initial publication. Citations to Advance online articles must include
final publication). Advance online articles are citable and establish publication priority; they areappeared in the paper journal (edited, typeset versions may be posted when available prior to Advance online articles have been peer reviewed and accepted for publication but have not yet
Copyright 2011 by The American Society of Hematology; all rights reserved.20036.the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

Impaired Megakaryocytopoiesis in Type 2B von Willebrand
Disease with Severe Thrombocytopenia
Paquita Nurden1, Najet Debili2, William Vainchenker2, Régis Bobe3, Raymonde
Bredoux3, Elisabeth Corvazier3, Robert Combrié1, Edith Fressinaud4, Dominique
Meyer4, Alan Nurden1, Jocelyne Enouf3.
From 1Centre de Référence des Pathologies Plaquettaires and Institut Fédératif
de Recherche N°4, Hôpital Cardiologique, Pessac, France; 2Inserm U.790,
Villejuif, France; 3Inserm U.689, Hôpital Lariboisière, Paris, France; 4Inserm
U.143, Le Kremlin-Bicêtre, France.
Short Title: Thrombocytopenia in type 2B VWD
Scientific heading: Hemostasis, Thrombosis and Vascular Biology
Abstract word count: 191
Text word count: 4996
Corresponding address: Paquita Nurden M.D., Ph.D., Centre de Référence des Pathologies
Plaquettaires, Laboratoire d’Hématologie, Hôpital Cardiologique, 33604 Pessac, France. Tel:
33 5 57 65 68 43; Fax: 33 5 57 65 65 31
E-mail. [email protected]
Blood First Edition Paper, prepublished online May 23, 2006; DOI 10.1182/blood-2006-03-009449
Copyright © 2006 American Society of Hematology
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

2
Abstract
In type 2B von Willebrand disease, there is spontaneous binding of mutated VWF multimers
to platelets. Here we report a family in which severe thrombocytopenia may also be linked to
abnormal megakaryocytopoiesis. A heterozygous mutation in the VWF A1 domain gave a
R1308P substitution in an interactive site for GPIbα. Electron microscopy showed clusters of
platelets in close contact. Binding of antibodies to the GPIbα N-terminal domain was
decreased, whereas GPIX and GPV were normally detected. In Western blotting (WB),
GPIbα, αIIb and β3 were normally present. Proteins involved in Ca2+ homeostasis were
analyzed by quantitating platelet mRNA or by WB. Plasma membrane Ca2+ ATPase (PMCA)-
4b and type III inositol trisphosphate receptor (InsP3-R3) were selectively increased. The
presence of degradation products of polyadenosine diphosphate (ADP)-ribose polymerase
protein (PARP) suggested ongoing caspase-3 activity. These were findings typical of
immature normal megakaryocytes cultured from peripheral blood CD34+ cells with TPO.
Significantly, megakaryocytes from the patients in culture produced self-associated and
interwoven proplatelets. Immunolocalization showed VWF not only associated with platelets,
but already on the megakaryocyte surface and within internal channels. In this family, type 2B
VWD is clearly associated with abnormal platelet production.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

3
Introduction
Von Willebrand disease (VWD) is the most common inherited disorder of the platelet-vessel
wall interaction and involves both quantitative and qualitative defects of von Willebrand
factor (VWF), a crucial mediator of platelet function and carrier of the FVIII protein. In type
1 and type 3 VWD, deficiencies or absence of VWF protein are responsible for the bleeding
syndrome; but in Type 2 disease a functionally abnormal protein or the specific lack of large
multimers account for the VWD phenotype (1,2). In hemostasis, GPIbα mediates platelet
attachment to exposed subendothelium by binding through its N-terminus to the A1 domain of
VWF exposed within the subendothelial matrix (3, 4). In normal subjects, soluble VWF
multimers in plasma fail to gain access to their binding site on GPIbα, accessibility being
controlled by a disulfide-linked double loop region just below the leucine-rich repeats of
GPIbα (5). In type 2B VWD, mutations giving rise to a selective number of amino acid
substitutions in the A1 domain provide gain of function to the VWF multimers which then
spontaneously bind to platelets in suspension through a direct interaction with GPIbα (6-8).
This often results in the loss of the largest multimers from plasma although these may be at
least partially preserved in some cases (9). Bleeding results from platelets having blocked
GPIb function despite a heightened ristocetin-induced platelet agglutination in platelet
function testing, and perhaps through the relative hemostatic inefficiency of the remaining
small multimers. The thrombocytopenia that accompanies this disorder in some although not
all patients may also be a factor in defining bleeding severity.
The gain-of-function mutations that give rise to type 2B VWD are clustered within
exon 28 (see ISTH SSC database at http://www.ragtimedesign.com/vwf/mutation) of the
VWF gene. Among the most affected codons is Arginine (R)1308 which is commonly
substituted by a cysteine (C), and in isolated cases by histidine (H), proline (P) or leucine (L)
residues (2, 10-15). Although the basic phenotype of type 2B VWD can be accounted for by
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

4
the ability of the mutated protein to spontaneously bind to platelets, some reports highlight
platelet abnormalities that are difficult to explain by this interaction alone. For example, the
degree of reported thrombocytopenia is highly variable, with sometimes giant and
morphologically abnormal platelets being observed (16, 17). We ourselves have previously
reported giant platelets in 3 unrelated patients with type 2B VWD (18). Here, we describe a
previously unreported family with an autosomal dominant trait and where severe
thrombocytopenia is associated with the presence of agglutinates of morphologically
abnormal platelets. Analysis of the VWF gene using DNA from both affected members of the
family identified a heterozygous R1308P substitution in VWF. Other studies on platelets of
both patients concentrated on two Ca2+-pump families, the plasma membrane Ca2+ATPases
(PMCAs) and the sarco/endoplasmic reticulum Ca2+ATPases (SERCAs) and in particular
SERCA3 of which several isoforms exist in platelets (19). We noted an increased presence of
PMCA4b and inositol trisphosphate receptor3 (InsP3-R3). As we also showed changes in
Ca2+-ATPase and InsP3-R3 distribution during proplatelet production by normal
megakaryocytes (MKs) in culture, we considered that abnormal megakaryocytopoiesis, might
be part of the phenotype in this family. Significantly, culture of peripheral blood CD34+ cells
of both patients resulted in MKs showing self-associated proplatelets, with VWF already
bound to the MK surface. In this family, type 2B VWD and familial thrombocytopenia due to
an altered megakaryocytopoiesis are clearly related.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

5
Methods:
Case reports
The patients are a brother (patient 1) and sister (patient 2), 58 and 57 years old respectively.
Relevant hemostatic parameters are summarized in Table I. Both have a lifelong bleeding
diathesis and a history of severe thrombocytopenia, with circulating platelet agglutinates a
constant feature (see Fig. 1). Single platelet counts for each patient were always < 25G/L
when performed with an Advia 120 counter (Bayer, Puteaux, France), values that were
confirmed by manual counting using a light microscope. For the brother, bleeding was
frequent during childhood with epistaxis several times a week. The sister additionally had
excessive bleeding at the time of menarche, and she experienced severe and prolonged
bleeding during surgery (appendectomy). The hemorrhagic tendency has decreased with age
for both patients and is now less frequently spontaneous. Arterial hypertension was first noted
for both patients at around 20 years old and they are treated with β-blockers. Although both
have received platelet transfusions, tests for anti-platelet antibodies are negative. Their now
deceased father had the same bleeding profile, with severe thrombocytopenia and agglutinated
platelets. There is no evidence of a bleeding syndrome in other family members although the
paternal grandfather died when very young. Patient 1 has one son in apparent good health but
who was unavailable for study. Controls were from hospital staff. All donors gave informed
consent according to the Declaration of Helsinki and studies were performed in line with local
ethical protocols.
Hemostatic tests and molecular diagnosis of type 2B VWD
Hemostatic tests were normal in routine testing unless stated otherwise. Prothrombin
consumption evaluated by measuring residual prothrombin with the Neoplastine CI plus kit
(Diagnostica Stago, Asnières, France) was abnormal, typical results being 22% for patient 1
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

6
and 38% for patient 2 (normal range 2-10%). Ristocetin cofactor activity (VWF:RCo) and
VWF:Ag were evaluated as described (18). Typical values are given in Table 1. FVIII:C was
measured using a one stage assay with Factor VIII:C-deficient plasma as the substrate
(Behring Diagnostica, Rueil Malmaison, France). It should be noted that results for the two
patients varied with blood group. Ristocetin-induced platelet agglutination (RIPA) was
hampered for the patient by the spontaneous agglutination; plasma from both patients caused
a weak agglutination of washed control platelets and this response was increased by low doses
of ristocetin (data not shown).
The Inserm network on molecular abnormalities in VWD (see 13) performed
specialized diagnosis. An absence of high molecular weight VWF multimers was shown by
SDS-polyacrylamide gel electrophoresis. ADAMTS 13 activity in the plasma was normal.
Genomic DNA was extracted from EDTA-anticoagulated whole blood using standard
methods and a heterozygous 3923 G->C transition was identified following PCR
amplification of the 5' portion of exon 28 of the VWF gene and direct sequencing. This gave
rise to a heterozygous R1308P substitution in the VWF protein for both patients. Sequencing
of the GPIbα gene according to ref 20 revealed no mutations (data not shown).
Table 1: Hemostatic parameters characterizing the patients Patient 1 Patient 2 Controls Platelet count < 20 < 20 150-400 G/L Platelet volume 8.5 8.5 6-8 µm3 Platelet agglutinates + + No VWF Ag 75-100 40-46 50-150% VWF RCo 100-115 46-55 50-150% VWF: HMW multimers Absent Absent Yes Blood group A+ 0+ RIPA 0.25 0.25 1-1.5 mg/ml Prothrombin consumption
32 32 <10%
ADAMTS 13 100 100 80-150% HMW = high molecular weight multimers
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

7
The VWF:RCo/VWF:Ag ratios were always > 0.7 for both patients, near normal values seen
for about 20% of type 2B patients in the French VWD network (13). The A blood group of
patient 1 with higher VWF:Ag and VWF:RCo was not associated with a more severe
thrombocytopenia or a clinically more severe form of the disease.
Platelet glycoprotein analysis
Unless stated otherwise, blood was withdrawn on 3.8 % sodium citrate (9:1 v/v). Platelet-rich
plasma (PRP) was prepared by centrifugation at 120 g for 10 min.
(a) Flow cytometry. PRP was incubated with MoAbs specific for αIIbβ3 (AP-2,
provided by Dr. T. Kunicki, Scripps Research Institute, La Jolla, USA), GPIbα (Bx-1,
prepared by the Bordeaux Laboratory; AP-1, from Dr. Kunicki; Alma-12, from Dr. F. Lanza,
EFS-Alsace, Strasbourg, France; SZ1, from Beckman Coulter, Marseille, France; WM23,
from Dr. M Berndt, Melbourne, Australia), GPIX (FMC25, Chemicon, Paris, France) and
α2β1 (Gi9, from Beckman Coulter) according to our standard procedures (21). In some
experiments, PRP prepared from ACD-A anticoagulated blood was supplemented with
apyrase (Sigma, St. Louis, MO, USA) and ACD-A as described (21), and the platelets
sedimented prior to resuspension in phosphate-buffered saline (PBS) buffer, pH7.2. These
platelets were incubated with the following antibodies: rabbit anti-VWF (Dako, Trappes,
France), or murine anti-LIBS MoAb, AP-6 (IgM, from T Kunicki); anti-RIBS, F26 (IgG,
originally given by Dr. H. Gralnick, NIH, Bethesda, USA), VH10 (IgG, anti-P-selectin,
prepared by the Bordeaux Laboratory). Platelets were sequentially incubated with primary
antibody followed by fluorescein isothiocyanate (FITC)-labeled F(ab')2 fragments of a goat
anti-mouse or by goat anti-rabbit immunoglobulins (Dako-France) (21). For the detection of
AP-6, dichlorotriazinylaminofluorescein (DTAF)–conjugated affinity-purified F(ab')2
fragments of donkey antimouse IgM (Jackson ImmunoResearch, West Grove, PA) were used.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

8
All antibodies were at pre-determined primary concentrations. Platelets were analyzed in a
Cytomics F 500 (Beckman Coulter,Villepinte, France) or a FACSCalibur (Becton Dickinson)
flow cytometer. Gating to select platelets was based on preliminary determinations of forward
and wide-angle light scatter, agglutinates were excluded. Mean fluorescence intensity (MFI)
was measured after passage through a 530 nm long-pass interference filter. Histograms were
generated from measurements of 10 000 cells, and data were analyzed using RXP software
(Beckman Coulter) or the Cell Quest software (Becton Dickinson).
(b) Western blotting. Washed platelets were prepared as described (21) and lysed in
buffer containing 10 mM Tris-HCl (pH 7.0), 3 mM EDTA (ethylenediaminetetraacetic acid),
5 mM N-ethylmaleimide, and 2% sodium dodecyl sulfate (SDS) (20). When performed,
disulfide reduction was with dithiothreitol. Then, 10 µg or 20 µg protein was separated by
SDS–polyacrylamide gel electrophoresis (SDS-PAGE) on minigels and transferred
electrophoretically onto nitrocellulose membrane using Trans-Blot Transfer Medium
(Amersham Life Science, Bucks, United Kingdom). Nonspecific binding of protein was
blocked by incubating the membrane for 1 h in a solution of 5% fat-free milk in 20 mM Tris-
HCl, pH 8.2, containing 0.05% (vol/vol) Tween 20 (TB-T). Individual membrane strips were
then incubated with MoAbs to GPIbα (Bx-1; 1 µg/mL), αIIb (SZ22, Beckman Coulter; 0.5
µg/mL), or β3 (Y2/51, Dakopatts, Glostrup, Denmark; 0.5 µg/mL) for 12 h at room
temperature. After washing, the membrane was further incubated with a 1:10,000 dilution of
horseradish peroxidase–conjugated antimouse or anti-rabbit IgG (Jackson ImmunoResearch)
and bound antibody was detected using a chemiluminescence procedure (Amersham Life
Science) (20).
Studies on calcium-binding proteins and caspase activities in platelets
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

9
Total RNA and protein lysates were simultaneously prepared from EDTA-anticoagulated
blood using TRIZOL solution as recommended by the manufacturer (Invitrogen, Cergy-
Pointoise, France).
(a) Western blotting. Electrophoresis of proteins and WB analysis was performed as
previously described (22,23). Individual nitrocellulose membrane strips were incubated with
MoAbs to total PMCA (5F10, Affinity BioReagents, NJ, USA), PMCA4b (JA3, Labvision,
Fremont, USA), total SERCA3 (PL/IM430, originally obtained from Dr. N Crawford,
London, UK), total SERCA2 (IID8, Affinity BioReagents; platelets only express SERCA2b),
caspase-9 (Upstate, Lake Placid, NY, USA), caspase-12 (Sigma), InsP3-R3 (Transduction
Laboratories, Lexington, KY, USA), µ-calpain (Calbiochem, San Diego, CA, USA) and
PARP (Calbiochem). Also used were rabbit antibodies to SERCA3a and SERCA3b (see ref
24), β3 (a gift from Dr. D. Pidard, Paris, France), calreticulin (Novus Biological Inc, Littleton,
CO, USA), InsP3-R1 (ABR) and InsP3-R2 (Santa Cruz Biotechnology). Anti-mouse and anti-
rabbit peroxidase-conjugated antibodies were from Jackson Immunoresearch.
(b) RT-PCR based analyses. These were essentially performed according to published
procedures (22, 23). The primers used to amplify mRNA for PMCA1b, PMCA4b, SERCA2b,
SERCA3a-c and InsP3-R1-R3 were detailed previously by us (22,25,26). PCR was performed
acording to Martin et al (22) where a touchdown-PCR was performed using 10 cycles with
annealing temperature decrements from 65°C to 55°C. PCR was conducted for different
cycles, each of them consisting of successive periods of denaturation at 95°C for 1 min,
annealing at 58°C for 1 min, and extension at 72°C for 1 min. GAPDH amplifications were
used as controls of the amounts of RNA. Other controls included amplifying SERCA2b in the
absence of RT. PCR products were visualized on ethidium bromide-stained gels. The gels
were scanned using ADOBE Photoshop and quantified by Molecular Analyst, version NIH
Image 1.62b7.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

10
In vitro liquid cultures of megakaryocytes from CD34+ cells
CD34+ cells were obtained from umbilical cord blood, from normal adult donors undergoing
leukapheresis, or from the peripheral blood of the patients. Precursor cells were separated
over a Ficoll-metrizoate gradient (Lymphoprep; Nycomed Pharma, Oslo, Norway) to obtain
an enriched fraction of mononuclear cells. CD34+ cells were purified using an
immunomagnetic cell sorting system according to the manufacturer's protocol (AutoMacs;
Miltenyi Biotec, Bergisch Gladbach, Germany) and cultivated in serum-free medium prepared
as previously reported and containing TPO (10 ng/ml; Kirin Brewery, Tokyo, Japan) (27).
Samples were taken between days 8 and 17 (see Results). Total RNA and protein lysate were
simultaneously prepared and treated for RT-PCR or WB analysis for Ca2+-ATPases or
calcium-binding proteins as described for platelets.
Analysis of proplatelet formation and immunofluorescence studies
Cultured megakaryocytes derived from CD34+ cells from the patients were plated on poly-L-
lysine-coated slides (Menzel-Glaser, Braunschweig, Germany) at day 12 of culture for 1 hour
in a cell incubator (37°C, 5% CO2 in air, 100 % humidity). Cells were then fixed with 2%
paraformaldehyde for 5 min, permeabilized with 0.1 % Triton X-100 for 5 min and incubated
with a mouse anti-P-selectin (VH10, see section on platelet glycoprotein analysis) and rabbit
anti-VWF (Dako) antibodies at predetermined saturating amounts for 1 hour at room
temperature. After 3 subsequent washes, cells were incubated with the appropriate secondary
antibodies conjugated with FITC or tetramethylrhodamine isothiocyanate (TRITC) (27).
When performed, counter staining for nuclei was with 4'-6-Diamidino-2-phenylindole
dihydrochloride (DAPI). The images were acquired using a Zeiss laser scanning microscope
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

11
(LSM 510 Zeiss, Jena, Germany) by using a 63X1/NA oil objective or an inverted Leica DM
IFBE microscope (Nikon Eclipse 600, Tokyo, Japan) again with a 63X1/NA oil objective.
Electron microscopy
PRP was prepared from citrated blood by centrifugation for 10 min at 80 g, incubated for 20
min at 37° C, and fixed in 1.25% glutaraldehyde (Fluka) for 1 h at room temperature. Samples
were processed for standard electron microscopy (EM) as previously described (18). Cultured
megakaryocytes from the patients or control donors were fixed under the same conditions for
1 h, washed, and transported to Bordeaux overnight in 0.5% glutaraldehyde. For immunogold
labeling, sucrose-loaded megakaryocytes were frozen in propane and then in liquid nitrogen
using a Reichert KF 80 freezing system (Leica, Vienna, Austria) (28),Ultrathin sections were
cut at –120° C with an Ultracut E ultramicrotome (Reichert, Villepinte, France) equipped with
an FC 4E cryokit attachment and placed on collodion-coated nickel grids (28). After washing,
sections were floated on buffer containing anti-VWF (10 µg/ml) (Dako). Bound antibody was
detected using goat anti-rabbit IgG conjugated to 10 nm gold particles (Amersham, Orsay,
France). The sections were stained with uranyl acetate and osmium according to our standard
procedures (28). Sections were embedded in a thin film of methylcellulose and observed with
a Jeol JEM-1010 transmission electron microscope (Jeol, Croissy-sur-Seine, France) at 80 kV.
Controls included the absence of primary antibody or its substitution with an irrelevant IgG of
the same species and at the same concentration.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

12
Statistics
Data are presented as mean ± S.D. Statistical significance was determined in multiple
comparisons among independent groups of data in which ANOVA indicated the presence of
significant differences. P < 0.01 was considered significant.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

13
Results
Platelet ultrastructure:
For the two patients, platelet agglutinates were observed on smears of whole blood. Results
were similar with blood anticoagulated with EDTA, ACD-A or citrate thus ruling out EDTA-
dependent pseudothrombocytopenia. Electron microscopy showed that platelets in PRP were
often round and somewhat enlarged, but not giant (Fig. 1). Interestingly, platelets were often
joined together by well-defined areas of contact. Sometimes, protein bridges could be seen
between the adjacent platelets (Fig 1B), but in others the contact was tight and fuzzy at high
power, as if fusion had occurred (Fig. 1C).. Although platelet agglutinates were present, there
were no signs of platelet activation; the α-granules were not centralized and pseudopodia
were rare. Platelet aggregation with physiologic agonists could not be tested, not only because
of the very low platelet count in PRP, but also because of continued spontaneous platelet
agglutination under stirring.
Studies on platelet glycoproteins by flow cytometry and western blotting:
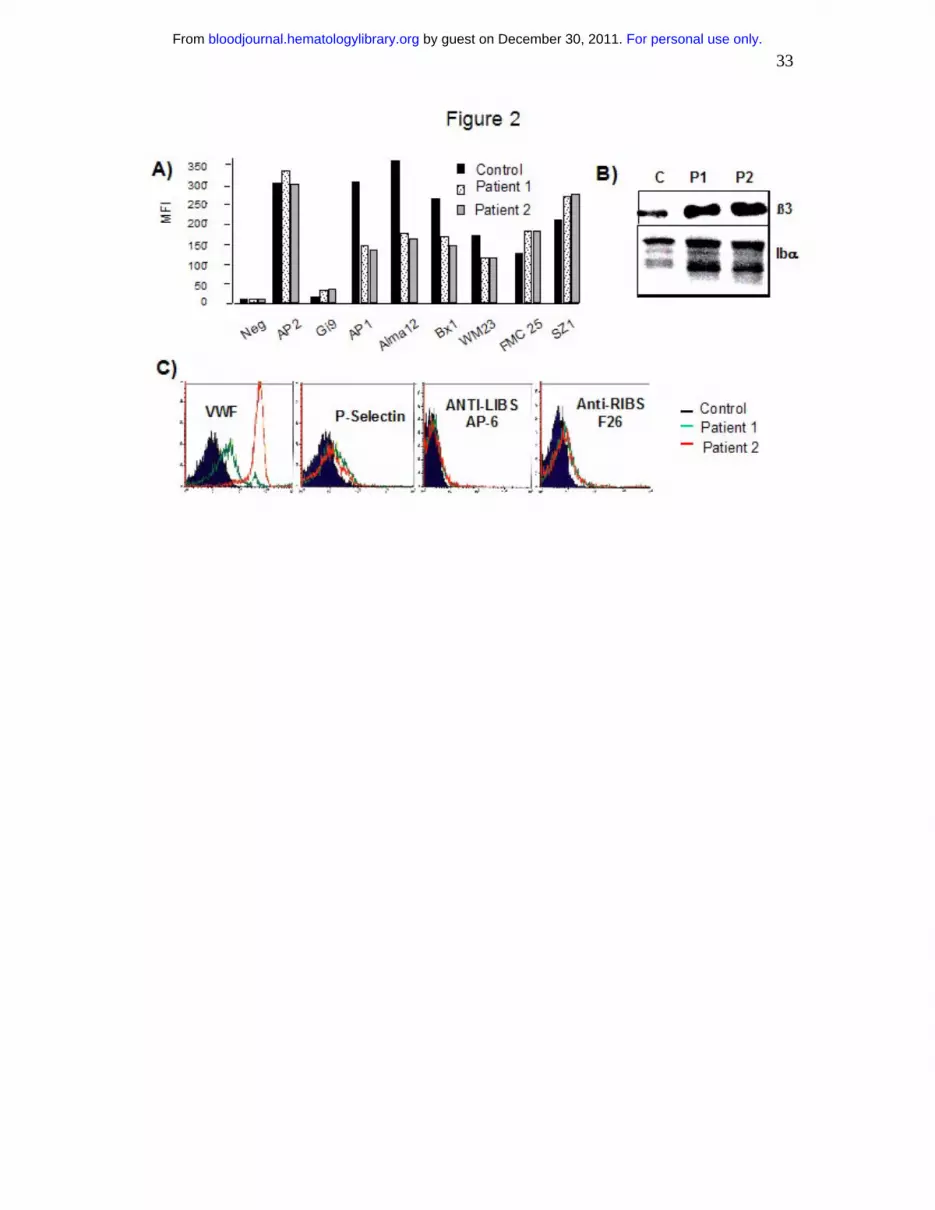
As shown in Fig 2A, platelets from both patients normally expressed α2β1 (Gi9) and αIIbβ3
integrins (AP2) by flow cytometry while both the αIIb (not shown) and β3 subunits were
normally seen by Western blotting (Fig 2B). A panel of MoAbs directed against different
epitopes of the GPIb-IX-V complex were tested in flow cytometry. Whereas MoAbs directed
against GPIbα (AP-1, Alma-12, Bx-1 and WM23) showed decreased binding, that of MoAbs
directed against GPIX (FMC25) or an epitope depending on the presence of the complex
(SZ1) were normal or increased. By testing platelets sedimented and resuspended in buffer,
we found an increased presence of VWF for both patients but more for patient 2. The use of
anti-RIBS and anti-LIBS MoAbs showed that αIIbβ3 showed little signs of activation (Fig.
2C). Expression of P-selectin at the platelet surface was also minimal thereby confirming the
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

14
absence of spontaneous secretion. Western blotting showed no evidence of platelet GPIbα
deficiency (Fig 2B) suggesting that the lower expression of GPIbα in flow cytometry was a
question of MoAb accessibility perhaps linked to the presence of VWF.
Studies on proplatelet production and VWF expression by megakaryocytes cultured in vitro
from CD34+ cells isolated from peripheral blood of the patients:
CD34+ cells were isolated from the blood of both patients and cultured in the presence of
TPO. As seen by phase contrast microscopy, control cultures contained megakaryocytes with
the typical long processes of mature cells (Fig. 3A). Megakaryocyte cultures were obtained
for the patients were clearly capable of producing proplatelets. Both short extensions and long
processes were seen. But to our surprise, and unlike those seen for control cultures, the latter
were interwoven with touching membranes (Fig. 3B). The distribution of VWF in the
megakaryocytes was next examined by fluorescence microscopy and compared to that of P-
selectin. Normally, VWF, which is synthesized in the megakaryocytes, is trafficked from the
trans-Golgi apparatus to the α-granules. This leads to a punctated and largely intracellular
distribution in confocal microscopy (illustrated in ref 29). For the megakaryocytes of both
patients, we unusually detected VWF on the cell surface including on the proplatelets (Figure
3C-K)). In most cells an accompanying intracellular distribution was also observed. In
contrast, P-selectin had a normal intracellular localization (Fig. 3G,J). After double labeling,
VWF was clearly not always co-localized with P-selectin as an α-granule marker, the VWF
surface pool being clearly distinguishable (Fig 3H,K). Immunogold labeling and electron
microscopy confirmed the presence of VWF on the surface of megakaryocytes (including on
protrusions), within the channels of the surface-connected system as well as in α-granules
(Fig. 4).
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

15
Studies of proteins involved in Ca2+ signaling in the patients' platelets:
A common feature of the binding of various ligands to their receptors is a modulation of Ca2+
homeostasis. Ca2+ signaling is controlled by many Ca2+ ATPases and InsP3-receptors, some
of them being involved in cell differentiation and apoptosis (30,31). This led us to compare
the expression of SERCA- and PMCA- type Ca2+-ATPases as well as InsP3-receptors in
platelet samples isolated from both patients and normal donors. Figure 5A compares the
expression of SERCA-type Ca2+-ATPases (2b, total 3 [lane 6], 3a [lane 7], 3b), total PMCA
(1b + 4b), PMCA4b and InsP3-R1-3 proteins by WB. Calreticulin and β3 were chosen as
control proteins. Quantitative comparisons of the expressions of the different proteins are
shown in Fig 5B. Expression of InsP3-R3 and PMCA4b was 149 ± 8 % and 308 ± 25 %
greater in platelets isolated from patient 1, and 192 ± 18 % and 368 ± 14 % greater in platelets
isolated from patient 2. RT-PCR was used to study mRNAs for these proteins in the platelets
from the controls and the patients. SERCA-type Ca2+-ATPases, InsP3-R1, InsP3-R2 and
PMCA1b showed only slight variations between the controls and the patients. In line with the
altered protein expression, platelets from both patients showed an increased content of InsP3-
R3 and PMCA4b mRNA (Fig 5C and D). Here increases amounted to 175 ± 36 % and 211 ±
23 % in platelets isolated from patient 1 and 221 ± 28 % and 228 ± 29 % in platelets
isolated from patient 2. Specific increases were also seen for mRNAs of some SERCAs with
SERCA 3a increased by 177 ± 22 % for patient 1 and 186 ± 26 % for patient 2. In the context
of our earlier work, these results suggested an abnormal megakaryocytopoiesis (26,31).
Studies of proteins involved in Ca2+-homeostasis in normal megakaryocytes:
Next, CD34+ cells isolated from cord blood or after leukapheresis were cultured in vivo.
Samples taken between days 8 and 17 were analyzed by WB or by RT-PCR for mRNA
content. Typical Western blots for protein expression in the megakaryocytes are shown in Fig.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

16
6A and compared to the expression of β3 and calreticulin. A quantitative evaluation of the
data is given in online Fig. 1. Whereas calreticulin levels remained stable, levels of β3
increased with megakaryocyte maturation. A specific increase in SERCA3a protein
expression was accompanied by a net decrease in PMCA4b expression during proplatelet
formation (days 11-17). The kinetics of this are shown in panel 6B and compared to β3
expression. The source of the CD34+CD41+ cells had little influence on the results. InsP3-
receptors were not studied here. Results for platelets are shown for comparison. In terms of
mRNA levels, while those of PMCA1b and InsP3-R1-2 showed modest regulations in the
megakaryocytes, significantly SERCA3-type Ca2+-ATPases are transitionally up-regulated
upon proplatelet formation and both PMCA4b and InsP3-R3 showed a net decrease from day
11 onwards (online Fig. 2).
Caspase activity in platelets of the patients and in normal megakaryocytes in culture:
Since platelet production is associated with caspase-3 activation and since PMCA4b is a
caspase-3 substrate (29, 30), we looked at possible variations in caspase-3 activation during
proplatelet formation and platelet release. We first looked for evidence of caspase-3 activity
in normal megakaryocytes by testing for cleavage of the caspase-3 substrate PARP. As
shown, in Fig. 6A, the native form of PARP (116 kDa) was cleaved from day 11 onwards and
in parallel with the loss of PMCA4b including its caspase-3 mediated proteolysis (typical 120-
kDa fragment) and the formation of proplatelets.
As sufficient cultured megakaryocytes were unavailable for WB, we re-examined
platelets from the patients and looked for an abnormal caspase-3 activity. Using the same
protein samples as those in Fig 5, we detected full-length PARP in platelets of normal donors
and in the two patients (Fig. 7). In addition, a cleaved 85-kDa PARP degradation product was
specifically observed in the platelets of the two patients (Fig. 7). In contrast, no trace of
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

17
cleaved PARP was detected in normal platelets. Because caspase-9 and caspase-12 are
activated in parallel to caspase-3 during megakaryocytopoiesis (32,33), we looked for their
presence in the platelets of patient 2. However, only procaspase-9 and pseudoprocaspase-12
were detected and without any degradation (Fig. 7). Finally, because µ-calpain may promote
apoptosis-like events during platelet activation and may degrade PMCA4b (although
proplatelet formation is not a calpain-dependent process) (31,32), we studied calpain
expression in the platelets of the same donors and patients. Again, no trace of autoproteolysis
could be detected under our experimental conditions (Fig 7).
Thus, the two patients studied here had an abnormal caspase-3 activity in their
platelets, and higher PMCA4b and InsP3-R3 expression. Comparison with the relative
expression and/or activity of the same proteins during megakaryocytopoiesis suggests that the
platelets may be produced from immature cells in an apoptotic phase of proplatelet formation.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

18
Discussion
Our family was characterized by the presence of a heterozygous R1308P substitution in the
VWF A1 domain, spontaneous binding of VWF to platelets, and autosomal dominant
inheritance that typifies type 2B VWD. Less typical was the constant presence of circulating
platelet agglutinates and a severe thrombocytopenia. Strikingly, the agglutinates were seen in
the presence of all anticoagulants and continued to be seen when blood was taken directly into
anticoagulant containing saturating amounts of blocking antibodies for αIIbβ3 (abciximab)
and GPIbα (alma-12) (data not shown). When characterized by electron microscopy, the
agglutinates were mostly composed of heterogeneously sized and often rounded platelets with
distinct zones of contact. It is probable that spontaneous binding of the mutated VWF is
causing the agglutination. Previous results from our laboratory have visualized VWF bridges
cross-linking platelets following RIPA or shear-induced platelet aggregation (34,35). The
finding that MoAbs to GPIbα bound less well to the patients' platelets than MoAbs to GPIX
or to a complex-dependent determinant on GPIb-IX, are also compatible with a masking
effect of surface-bound VWF.
Despite the above results, such a severe thrombocytopenia and the presence of platelet
agglutinates is rare in patients with type 2B VWD (1,2,13). One possibility is that the nature
of the mutation in this disease can influence phenotype, and studies with recombinant proteins
mutated at different residues within the A1 domain (aa1260-1479) have confirmed that they
bind with different affinities to GPIbα (36,37). While the presence of agglutinates and severe
thrombocytopenia is one variable in type 2B VWD, the presence of giant platelets is another
(16-18). Yet, 3 patients with thrombocytopenia and giant platelets possessed different
mutations in the VWF gene (1304insMet; V1316M, P1337L) showing that such morphologic
changes are not linked to a recurrent mutation (18). Although thrombocytopenia can be
greater during pregnancy or after DDAVP infusion in type 2B VWD, when plasma levels of
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

19
VWF are increased (1,38), in our family the severe thrombocytopenia was constant. Important
questions therefore arise as to why such platelet variability is seen in type 2B VWD, and what
factors control the 'platelet' phenotype.
It has previously been demonstrated that spontaneous binding of mutated type 2B
VWF induces a cytoplasmic Ca2+ rise in platelets (39). Thus we tested platelets of both
patients for their expression of a series of Ca2+-ATPases or Ca2+-binding proteins.
Significantly, PMCA4b and InsP3-R3 were increased for both protein and mRNA expression
while for SERCA-3a only mRNA was increased. PMCA4b is a substrate for caspase-3, an
enzyme involved in platelet production by megakaryocytes (31,32,40). So we studied its
expression compared to SERCAs during normal megakaryocytopoiesis. To do this,
CD34+CD41+ cells from umbilical cord blood or following leukapheresis were cultured with
SCF and TPO and examined between days 8 and 17. A specific increase in SERCA3 protein
expression in mature megakaryocytes paralleled a striking down-regulation of PMCA4b and
this in cells from each source. Thus a greater presence of PMCA4b in the patient's platelets
suggested that they were being produced from more immature megakaryocytes. This was
supported when the patients' platelets were unusually shown to contain a 85 kDa degradation
product of another caspase-3 substrate, PARP. Procaspase-3 but not caspase-3 has been
previously seen in control platelets (41). Comparison of the kinetics of PMCA4b and PARP
degradation in normal megakaryocytes suggested that both were linked with proplatelet
formation. As degradation of PMCA4b is associated with apoptosis in other cells in vitro (40),
and as InsP3-R3 has recently been shown to preferentially transmit apoptotic signals into
mitochondria (42), our results comfort the hypothesis that the defects in our family extend to
the apoptotic phase of megakaryocyte maturation. This were specific changes for no evidence
of caspase-9 or caspase-12 activities were seen in platelets of patient 2. Both caspase-9 (32)
and caspase-12 (33) are active during normal megakaryocytopoiesis.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

20
CD34+CD41+ cells from the peripheral blood of both patients normally gave rise to
megakaryocyte colonies. As cells matured they normally formed stunted processes possibly
resembling immature proplatelets (43), but a major surprise was that the long proplatelet-like
structures were intertwined and incompletely separated. Whereas we had previously shown
that cultured human and murine megakaryocytes gave a punctate pattern on
immunofluorescence analysis for VWF (29), for the patients there was an additional surface
staining for VWF. Confocal microscopy and dual staining with anti-P-selectin antibody
confirmed the normal formation of α-granules within the central megakaryocyte body, the
presence of an intracellular VWF pool and confirmed that P-selectin did not follow the VWF
to the surface. Immunogold staining for VWF confirmed that VWF was on the surface of the
megakaryocytes. One possibility is that it is being excluded from the megakaryocytes and
then rebinds. An intriguing alternative is that the mutated protein is spontaneously binding to
GPIbα during its passage through the Golgi apparatus and is carried to the surface by the
GPIb-IX-V complex.
The R1308P mutation (R545P in terms of an older nomenclature) has been located
previously in a family with type 2B VWD, the principal reported propositus had a platelet
count of 100 G/L (14) with plasma lacking high molecular weight multimers. In this same
study, recombinant mutated VWF with P1308 and C1308 substitutions bound spontaneously
to platelets and had greater reactivity with GPIb compared to substitutions at positions 1306
or 1341 (14). In fact, evidence obtained from the crystal structure of the A1 domain suggests
that R1308 is involved in salt bridge formation or intramolecular packing and the P1308
substitution changes A1 domain conformation allowing facilitated access of a closely
localized sequence directly involved in binding to GPIbα (44). As much evidence suggests
that GPIbα (or the GPIb-IX-V complex) is a signaling molecule through the association of
GPIbα or GPIbβ cytoplasmic domains with proteins such as 14-3-3_ζcalmodulin (a regulator
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

21
protein of PMCA4b) or FcRγ, one possibility is that GPIbα-regulated signaling is involved in
abnormal platelet production (45-47). Yet, in our study, platelets from both patients showed
few signs of activation in that secretion had not occurred and αIIbβ3 inetgrin was not in an
active conformation. Notwithstanding, αIIbβ3-independent shear induced platelet aggregation
has been observed with recombinant VWF mutated for another type 2B VWD mutation,
V1316M which led to pp125FAK phosphorylation (48).
The degree of thrombocytopenia in our family is severe, and while this is undoubtedly
in part due to the clearance of circulating agglutinates, we have presented evidence for
changes in the production of proplatelets during megakaryocytopoiesis. Perhaps the nearest
report in the literature to our family is the description of type 2B Tampa (49,50). Here,
chronic thrombocytopenia (9-46,000/µL for 3 patients) associated with in vivo platelet
aggregate formation and spontaneous platelet aggregation in vitro. Also of interest is type 2B
Hiroshima, where chronic thrombocytopenia was associated with normal plasma levels of
multimers (51). A platelet imbalance between PMCA and SERCA-type ATPases as shown
by us is a new marker of an abnormal megakaryocytopoiesis. This could in our family result
in the release of morphologically abnormal platelets, or even platelets that are incompletely
separated and which circulate as clusters. Abnormal processing of the mutated VWF in the
megakaryocytes could be a cause of this abnormality, although the fact that closely-related
mutations do not result in such a severe thrombocytopenia suggests that other contributory
factors are involved. Perhaps nonidentified modifier genes linked or not to the Ca2+ -binding
protein imbalance contribute to the phenotypic variability in type 2B VWD. Whatever the
cause, our results unambiguously show that this family with type 2B VWD may also be
considered as having familial thrombocytopenia.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

22
Acknowledgement: This work was performed in the context of the French network on
'Rare Diseases of Platelet Function and Production' financed by the GIS-Institut des Maladies
Rares and Inserm. We thank Jalil Abdelali, IFR54, Institut Gustave Roussy, for confocal
analysis.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

23
References
1. Sadler JE. New concepts in von Willebrand disease. Annu Rev Med. 2005;56:173-
191.
2. Mazurier C, Ribba AS, Gaucher C, Meyer D. Molecular genetics of von Willebrand
disease. Ann Genet. 1998;41:34-43.
3. Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-
receptor interactions in platelet thrombus formation under flow. Cell. 1998;94:657-666.
4. Dopheide SM, Maxwell MJ, Jackson SP. Shear-dependent tether formation during
platelet translocation on von Willebrand factor. Blood. 2002;99:159-167.
5. Uff S, Clemetson JM, Harrison T, Clemetson KJ, Emsley J. Crystal structure of the
platelet glycoprotein Ibα N-terminal domain reveals an unmasking mechanism for receptor
activation. J Biol Chem. 2002;277:35657-35663.
6. Ruggeri ZM, Pareti FI, Mannucci PM, Ciavarella N, Zimmerman TS. Heightened
interaction between platelets and factor VIII/von Willebrand factor in a new subtype of von
Willebrand's disease. N Engl J Med. 1980;302:1047-1051.
7. Huizinga EG, Tsuji S, Romijn RA, et al. Structures of glycoprotein Ibα and its
complex with von Willebrand factor A1 domain. Science. 2002;297:1176-1179.
8. Dumas JJ, Kumar R, McDonagh T, et al. Crystal structure of the wild-type von
Willebrand factor A1-glycoprotein Ibα complex reveals conformation differences with a
complex bearing von Willebrand disease mutations. J Biol Chem. 2004;279:23327-23334.
9. Federici AB, Mannucci PM, Stabile F, et al. A type 2b von Willebrand disease
mutation (Ile546-->Val) associated with an unusual phenotype. Thromb Haemost.
1997;78:1132-1137.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

24
10. Sadler JE. von Willebrand factor: two sides of a coin. J Thromb Haemost.
2005;3:1702-1709.
11. Cooney KA, Nichols WC, Bruck ME, et al. The molecular defect in type IIB von
Willebrand disease. Identification of four potential missense mutations within the putative
GpIb binding domain. J Clin Invest. 1991;87:1227-1233.
12. Randi AM, Rabinowitz I, Mancuso DJ, Mannucci PM, Sadler JE. Molecular basis of
von Willebrand disease type IIB. Candidate mutations cluster in one disulfide loop between
proposed platelet glycoprotein Ib binding sequences. J Clin Invest. 1991;87:1220-1226.
13. Meyer D, Fressinaud E, Gaucher C, et al. Gene defects in 150 unrelated French cases
with type 2 von Willebrand disease: from the patient to the gene. INSERM Network on
Molecular Abnormalities in von Willebrand Disease. Thromb Haemost. 1997;78:451-456.
14. Hilbert L, Gaucher C, Abgrall JF, Parquet A, Trzeciak C, Mazurier C. Identification of
new type 2B von Willebrand disease mutations: Arg543Gln, Arg545Pro and Arg578Leu. Br J
Haematol. 1998;103:877-884.
15. Baronciani L, Federici AB, Beretta M, Cozzi G, Canciani MT, Mannucci PM.
Expression studies on a novel type 2B variant of the von Willebrand factor gene (R1308L)
characterized by defective collagen binding. J Thromb Haemost. 2005;3:2689-2694.
16. Lopez-Fernandez MF, Lopez-Berges C, Martin-Bernal JA, et al. Type IIB von
Willebrand's disease associated with a complex thrombocytopenic thrombocytopathy. Am J
Hematol. 1988;27:291-298.
17. Moll S, Lazarowski AR, White GC, 2nd. Giant platelet disorder in a patient with type
2B von Willebrand's disease. Am J Hematol. 1998;57:62-67.
18. Nurden P, Chretien F, Poujol C, Winckler J, Borel-Derlon A, Nurden A. Platelet
ultrastructural abnormalities in three patients with type 2B von Willebrand disease. Br J
Haematol. 2000;110:704-714.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

25
19. Bobe R, Bredoux R, Corvazier E, et al. How many Ca2+ATPase isoforms are
expressed in a cell type? A growing family of membrane proteins illustrated by studies in
platelets. Platelets. 2005;16:133-150.
20. Hillmann A, Nurden A, Nurden P, et al. A novel hemizygous Bernard-Soulier
Syndrome (BSS) mutation in the amino terminal domain of glycoprotein (GP)Ibβ. Platelet
characterization and transfection studies. Thromb Haemost. 2002;88:1026-1032.
21. Nurden P, Savi P, Heilmann E, et al. An inherited bleeding disorder linked to a
defective interaction between ADP and its receptor on platelets. Its influence on glycoprotein
IIb-IIIa complex function. J Clin Invest. 1995;95:1612-1622.
22. Martin V, Bredoux R, Corvazier E, et al. Three novel sarco/endoplasmic reticulum
Ca2+ATPase (SERCA) 3 isoforms. Expression, regulation, and function of the members of the
SERCA3 family. J Biol Chem. 2002;277:24442-24452.
23. Bobe R, Bredoux R, Corvazier E, et al. Identification, expression, function, and
localization of a novel (sixth) isoform of the human sarco/endoplasmic reticulum Ca2+ATPase
3 gene. J Biol Chem. 2004;279:24297-24306.
24. Kovàcs T, Felfoldi F, Papp B, et al. All three splice variants of the human
sarco/endoplasmic reticulum Ca2+-ATPase 3 gene are translated to proteins: a study of their
co-expression in platelets and lymphoid cells. Biochem J. 2001;358:559-568.
25. Martin V, Bredoux R, Corvazier E, Papp B, Enouf J. Platelet Ca2+ATPases : a plural,
species-specific, and multiple hypertension-regulated expression system. Hypertension.
2000;35:91-102.
26. Lacabaratz-Porret C, Launay S, Corvazier E, et al. Biogenesis of endoplasmic
reticulum proteins involved in Ca2+ signaling during megakaryocytic differentiation: an in
vitro study. Biochem J. 2000;350:723-734.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

26
27. Debili N, Wendling F, Katz A, et al. The Mpl-ligand or thrombopoietin or
megakaryocyte growth and differentiation factor has both direct proliferative and
differentiative activities on human megakaryocyte progenitors. Blood. 1995;86:2516-2525.
28. Nurden P, Poujol C, Durrieu-Jais C, et al. Labeling of the internal pool of GP IIb-IIIa
in platelets by c7E3 Fab fragments (abciximab): flow and endocytic mechanisms contribute to
the transport. Blood. 1999;93:1622-1633.
29. Wilcox DA, Shi Q, Nurden P, et al. Induction of megakaryocytes to synthesize and
store a releasable pool of human factor VIII. J Thromb Haemost. 2003;1:2477-2489.
30. Khan AA, Soloski MJ, Sharp AH, et al. Lymphocyte apoptosis: mediation by
increased type 3 inositol 1,4,5-trisphosphate receptor. Science. 1996;273:503-507.
31. Pàszty K, Verma AK, Padanyi R, Filoteo AG, Penniston JT, Enyedi A. Plasma
membrane Ca2+ATPase isoform 4b is cleaved and activated by caspase-3 during the early
phase of apoptosis. J Biol Chem. 2002;277:6822-6829.
32. De Botton S, Sabri S, Daugas E, et al. Platelet formation is the consequence of caspase
activation within megakaryocytes. Blood. 2002;100:1310-1317.
33. Kerrigan SW, Gaur M, Murphy RP, Shattil SJ, Leavitt AD. Caspase-12: a
developmental link between G-protein-coupled receptors and integrin αIIbβ3 activation.
Blood. 2004;104:1327-1334.
34. Hourdillé P, Gralnick HR, Heilmann E, et al. von Willebrand factor bound to
glycoprotein Ib is cleared from the platelet surface after platelet activation by thrombin.
Blood. 1992;79:2011-2021.
35. Poujol C, Nurden A, Paponneau A, Heilmann E, Nurden P. Ultrastructural analysis of
the distribution of von Willebrand factor and fibrinogen in platelet aggregates formed in the
PFA-100TM. Platelets. 1998;9:381-389.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

27
36. de Romeuf C, Hilbert L, Mazurier C. Platelet activation and aggregation induced by
recombinant von Willebrand factors reproducing four type 2B von Willebrand disease
missense mutations. Thromb Haemost. 1998;79:211-216.
37. Hilbert L, Gaucher C, Mazurier C. Effects of different amino-acid substitutions in the
leucine 694-proline 708 segment of recombinant von Willebrand factor. Br J Haematol.
1995;91:983-990.
38. Pareti FI, Federici AB, Cattaneo M, Mannucci PM. Spontaneous platelet aggregation
during pregnancy in a patient with von Willebrand disease type IIB can be blocked by
monoclonal antibodies to both platelet glycoproteins Ib and IIb/IIIa. Br J Haematol.
1990;75:86-91.
39. Francesconi M, Casonato A, Pontara E, Dalla Via L, Girolami A, Deana R. Type IIB
von Willebrand factor induces phospholipase A2 activation and cytosolic Ca2+ increase in
platelets. Biochem Biophys Res Commun. 1995;214:102-109.
40. Pàszty K, Kovàcs T, Lacabaratz-Porret C, et al. Expression of hPMCA4b, the major
form of the plasma membrane calcium pump in megakaryoblastoid cells is greatly reduced in
mature human platelets. Cell Calcium. 1998;24:129-135.
41. Shcherbina A, Remold-O'Donnell E. Role of caspase in a subset of human platelet
activation responses. Blood. 1999;93:4222-4231.
42. Mendes CC, Gomes DA, Thompson M, et al. The type III inositol 1,4,5-trisphosphate
receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem.
2005;280:40892-40900.
43. Richardson JL, Shivdasani RA, Boers C, Hartwig JH, Italiano JE, Jr. Mechanisms of
organelle transport and capture along proplatelets during platelet production. Blood.
2005;106:4066-4075.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

28
44. Emsley J, Cruz M, Handin R, Liddington R. Crystal structure of the von Willebrand
Factor A1 domain and implications for the binding of platelet glycoprotein Ib. J Biol Chem.
1998;273:10396-10401.
45. Andrews RK, Munday AD, Mitchell CA, Berndt MC. Interaction of calmodulin with
the cytoplasmic domain of the platelet membrane glycoprotein Ib-IX-V complex. Blood.
2001;98:681-687.
46. Dai K, Bodnar R, Berndt MC, Du X. A critical role for 14-3-3zeta protein in
regulating the VWF binding function of platelet glycoprotein Ib-IX and its therapeutic
implications. Blood. 2005;106:1975-1981.
47. Ozaki Y, Asazuma N, Suzuki-Inoue K, Berndt MC. Platelet GPIb-IX-V-dependent
signaling. J Thromb Haemost. 2005;3:1745-1751.
48. Mekrache M, Bachelot-Loza C, Ajzenberg N, Saci A, Legendre P, Baruch D.
Activation of pp125FAK by type 2B recombinant von Willebrand factor binding to platelet
GPIb at a high shear rate occurs independently of αIIbβ3 engagement. Blood. 2003;101:4363-
4371.
49. Saba HI, Saba SR, Dent J, Ruggeri ZM, Zimmerman TS. Type IIB Tampa: a variant of
von Willebrand disease with chronic thrombocytopenia, circulating platelet aggregates, and
spontaneous platelet aggregation. Blood. 1985;66:282-286.
50. Takimoto Y, Imanaka F. Type 2B Hiroshima: a variant of von Willebrand disease
characterized by chronic thrombocytopenia and the presence of all von Willebrand factor
multimers in plasma. Int J Hematol. 1999;70:127-131.
51. Saba HI, Fujimura Y, Saba SR, et al. Spontaneous platelet aggregation in type IIB
Tampa von Willebrand disease is inhibited by the 52/48-kDa fragment of normal von
Willebrand factor, which contains the GPIb binding domain. Am J Hematol. 1989;30:150-
153.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

29
Figure Legends
Figure 1: Ultrastructure of platelets in PRP from patient 1 as seen by standard transmission
electron microscopy. To be noted are the presence of platelet agglutinates and platelets of
different size and shape that include round forms. Granules are randomly distributed and the
platelets show no signs of activation (A). Platelets are often attached through discrete areas of
surface contact, arrows highlight how at some interfaces protein bridges can be clearly seen
(B) while at other interfaces the membrane surfaces appear to be in direct contact (C). Bar in
A = 1 µm, in B and C = 200 nm.
Figure 2: Platelet glycoprotein analysis by flow cytometry (A,C) and Western blotting (B). In
(A), PRP from patients 1 and 2 or a control donor were incubated with a battery of MoAbs
recognizing αIIbβ3 (AP2), α2β1 (Gi9), GPIbα (AP1, Alma12, Bx1, WM23), GPIX (FMC25)
or the GPIb-IX complex (SZ1). In (C), platelets from patients 1 and 2 or a control donor were
sedimented and resuspended in washing buffer prior to incubation with a polyclonal antibody
to VWF, a MoAb to P-selectin (VH10), and anti-LIBS and anti-Ribs MoAbs respectively. In
(B) lysates of platelets from patients 1 (P1) and 2 (P2) and a control donor were analyzed by
SDS-PAGE and immunoblotted with MoAbs to β3 and GPIbα.
Figures 3: Phase contrast and immunofluorescence analysis of megakaryocytes cultured in
vitro from CD34+ peripheral blood samples from patient 1. Phase contrast microscopy shows
that megakaryocytes from a control donor at day 12 of culture have typical long and discrete
proplatelets (arrows) (A). In contrast, megakaryocytes from patient 1 give rise to proplatelets
that are intertwined (arrow heads). Fixed and permeabilized cells from the patient were
subjected to immunofluorescence labeling for VWF (green). Results show that at day 12 for
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

30
the patient, VWF has both intracellular and surface staining (white arrows) (C-E). Nuclei are
shown in blue by DAPI. Confocal microscopy in F-K shows the distribution of VWF and P-
selectin in cultured megakaryocytes from patient 1. Cells taken at day 12 were fixed and
permeabilized prior to being labeled for VWF (green fluorescence) (F,I) and P-selectin (red
fluorescence) (G, J). When the colors were merged (H,K), much of the VWF has a different
localization to the α-granule marker, P-selectin, with the labeling for VWF clearly extending
to the surface of the cell.
Figure 4: Immunogold labeling of VWF on cultured cells. Megakaryocytes at day 12 from
cultures of CD34+ cells isolated from the peripheral blood of patient 1 were fixed and frozen-
thin sections stained sequentially using a polyclonal anti-VWF antibody and a secondary
antibody to rabbit IgG adsorbed onto 10 nm gold beads. Transmission electron microscopy
revealed that VWF was present on the cell surface (arrow heads, A) as well as in intracellular
canals (arrows, A-C) and in well-developed α-granules (D, E). Multivesicular bodies (MVB)
are seen in (E). Bars = 500 nm
Figure 5: Abnormal expression of proteins involved in Ca2+ signaling in the patients.
Platelets isolated from 3 normal donors (C1-3) and the two patients P1 and P2 were treated
for simultaneous isolation of total lysate proteins and RNA. (A) Proteins were subjected to
SDS-PAGE and Western Blotting using the isoform-specific antibodies indicated to detect
PMCA, InsP3-R and SERCA-type proteins as well as β3 and calreticulin (internal controls).
In this Figure and the following, the size of proteins is given in kDa. (B) The intensity of the
bands was quantified by densitometry and the mean values obtained for the 3 normal donors
were arbitrarily taken as 100%. The mean ± SD values (n = 8) of relative intensity
(normalized to calreticulin) are shown in arbitrary units. *P < 0.01 compared with values
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

31
from normal donors. (C) Total RNA was subjected to specific RT-PCR to detect PMCA-,
InsP3-R- and SERCA- RNA species, as well as GAPDH ( internal control). Numbers indicate
the sizes of PCR products in base pairs (bp). (D) The intensity of the bands was quantified as
above except that the mean values (n = 6 to 10) of relative intensity were normalized to
GAPDH. *P < 0.01 compared with values from normal donors.
Figure 6: The proteins involved in Ca2+ signaling in the patients exhibit similarities with
immature megakaryocytes. (A) CD34+CD41+ cells isolated from both umbilical cord blood
(Left) and leukapheresis (Right) samples were FACS-purified, at day 8 of culture. Different
sorted cell samples (3) were cultured for either up to 14 or to 16-17 days and cells were
further treated as above. Proteins were treated for Western blotting to detect same SERCA
and PMCA-type Ca2+-ATPases and control proteins as in Fig. 6 and PARP (n = 5). The
intensity of the bands was quantified by densitometry and the mean values obtained at days 8
or 9 were arbitrarily taken as 100% (On-line supplemental Fig 1). (B) The summary of
densitometric measurements was used to draw the schematic representation of the relative
expressions of PMCA4b, SERCA3a and β3 along with megakaryocytopoiesis (Left) and to
position those of the platelets of the patients (Right).
Figure 7: Platelets of the patients exhibit abnormal caspase-3 processing.
Proteins from the same samples as those used in Fig. 5 were subjected to Western blotting
using the antibodies indicated, to detect the expression of proteins involved in the apoptotic
pathways. The Figure is typical of 3 independent experiments.
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

32
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom
33
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

34
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

35
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

36
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

37
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom

38
For personal use only. by guest on December 30, 2011. bloodjournal.hematologylibrary.orgFrom