evidence why paroxetine dose escalation is not effective in major depressive disorder: a randomized...
TRANSCRIPT

Downloaded from UvA-DARE, the Institutional Repository of the University of Amsterdam (UvA)http://dare.uva.nl/document/124010
Description ThesisVersion Final published version (publisher's pdf)File ID 124010Filename thesis.pdf
SOURCE, OR PART OF THE FOLLOWING SOURCE:Type DissertationTitle Dose-escalation in the picture : pharmacological and imaging studies in depressionAuthor H.G. RuhéFaculty Faculty of MedicineYear 2008Pages 300
FULL BIBLIOGRAPHIC DETAILS: http://dare.uva.nl/record/283611
Copyrights It is not permitted to download or to forward/distribute the text or part of it without the consent of the copyright holder(usually the author), other then for strictly personal, individual use. UvA-DARE is a service provided by the Library of the University of Amsterdam (http://dare.uva.nl)

Do
se-escalation
in th
e pictu
reH
.G. R
uh
éPharmacological and Imaging studies in depression
H.G. Ruhé
Dose-escalation in the picture
Dose-escalation in the picture
deprssion
Omslag Ruhe.indd 1 17-9-2008 15:56:58

DOSE-ESCALATION IN THE PICTURE
PHARMACOLOGICAL AND IMAGING STUDIES IN DEPRESSION
Eric Ruhé

The work described in this thesis was mostly performed at the Academic Medical Center, Programfor Mood Disorders in the Netherlands. The studies were conducted in close collaboration withthe departments of Nuclear Medicine, General Practice, Pharmacology and Pharmacotherapy,Clinical Epidemiology, Biostatistics and Bioinformatics and Radiology.
Most of the studies in this thesis were financially supported by grants from the Academic MedicalCenter (SFA.07.012), the Netherlands Organisation for Health Research and Development(ZonMw), program Mental Health, education of investigators in mental health (OOG; #100-002-002), and from the Dutch Brain Foundation (14F06.45).
Additional support for the completion of this thesis was provided by Amicale Facel Holland.
Dose-Escalation in the Picture. Pharmacological and Imaging studies in depressionThesis, University of Amsterdam, The Netherlands. With a summary in Dutch.
Copyright © 2008, Henricus G. Ruhé, Amstelveen, The Netherlands [email protected]
All rights reserved. No part of this thesis may be reproduced, stored or transmitted in any form orby any means, without prior permission of the author.
Layout: ProBook with FLE technology by GDS Cross Media Group BV, Nieuw Vennep, TheNetherlands
Cover: Inge E. Kos, Medical Photography and Illustration Academic Medical Center, TheNetherlands
Printed by : PrintPartners Ipskamp BV, Enschede, The Netherlands
ISBN: 978-90-9023559-2

Dose-Escalation in the PicturePharmacological and Imaging studies
in depression
ACADEMISCH PROEFSCHRIFT
ter verkrijging van de graad van doctoraan de Universiteit van Amsterdamop gezag van de Rector Magnificus
prof. dr. D.C. van den Boomten overstaan van een door het college voor promoties
ingesteldecommissie,in het openbaar te verdedigen in de Aula der Universiteit
op woensdag 5 november 2008, te 10:00 uur
door
Henricus Gerardus Ruhé
geboren te Amsterdam

Promotiecommissie
Promotor: Prof. dr. A.H. Schene
Co-promotor: Dr. J. Booij
Overige leden: Prof. dr. D.A.J.P. DenysProf. dr. R. DierckxProf. dr. B.L.F. van Eck-SmitProf. dr. C.G. KruseProf. dr. W.A. Nolen
Faculteit der Geneeskunde

‘The brain is not a chemical factory but an extremely complicated survival machine’
Arvid Carlsson, Nobel lecture 2000

Contents
Part I. General introduction 9
1. General introduction and outline of the thesis 11
2. Design and methods of the DELPHI-study 31
Part II. Studies to guide clinical treatment of Major Depressive Disorder 41
3. Clinical use of the Hamilton Depression Rating Scale: Is increased efficiency possible? A post hoc comparison of HDRS, Maier and Bech subscales, CGI and SCL-90 scores. 43Comprehensive Psychiatry 2005; 46: 417-427
4. Dose-escalation for insufficient response to a standard dose of a selective serotonin reuptake inhibitor in major depressive disorder: a systematic review. 59British Journal of Psychiatry 2006; 189: 309-316
5. Switching antidepressants after a first selective serotonin reuptake inhibitor in major depressive disorder: a systematic review 73Journal of Clinical Psychiatry 2006; 67: 1836-1855
Part III. Serotonin in the etiology of Major Depressive Disorder 99
6. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies 101Molecular Psychiatry 2007; 12: 331-359
7. Serotonin transporter binding with [123I]β-CIT SPECT in major depressive disorder versus controls: effect of season and gender. 139Submitted

Part IV. Neurobiological effects of paroxetine in the treatment of Major Depressive Disorder 155
8. Serotonin transporter gene promoter polymorphisms modify the association between paroxetine serotonin transporter occupancy and clinical response in major depressive disorder 157Pharmacogenetics and Genomics 2008; in press
9. Evidence why paroxetine dose-escalation is not effective in major depressive disorder: a randomized-controlled trial with assessment of serotonin transporter occupancy 173Neuropsychopharmacology 2008; in press
10. Successful pharmacological treatment of major depressive disorder attenuates amygdala activation to negative facial expressions. An fMRI study 195Submitted
11. Effect of the selective serotonin reuptake inhibitor paroxetine on platelet function is modified by a SLC6A4 serotonin transporter polymorphism 217Journal of Thrombosis and Haemostasis 2008; in press
Part V. Summary and general discussion 231
12. Summary, conclusions and general discussion 233
13. Samenvatting en conclusies 253
Part VI. Abbreviations, color figures, publications, acknowledgement and curriculum vitae 269
Abbreviations 271
Color figures 275
Publications 285
Dankwoord 291
Curriculum Vitae 297


GENERAL INTRODUCTION
PART
General introduction

10

CHA
PTER
GENERAL INTRODUCTION AND OUTLINE OF THE THESIS

12

ChapterGeneral introduction and outline
1
13
General introduction
The present thesis concerns the pharmacological treatment of Major Depressive Disorder (MDD).It especially focuses on what to do when patients do not respond to a standard dose of anantidepressant.
In this thesis, the results of studies that were performed since 2001, while working at theProgram for Mood Disorders of the department of Psychiatry of the Academic Medical Center willbe reported. These studies first aimed at systematically reviewing the existing literature abouttreatment strategies for non-response to the first antidepressant (included in Part II). Thesereviews identified an important gap between equivocal evidence for dose-escalation and the firmrecommendation of this strategy in clinical guidelines. This was the starting point for theexperimental studies to further investigate the clinical efficacy and the mechanism behind dose-escalation, described in Part IV. Meanwhile, the findings, and further reading of conflicting resultsof others raised questions about the pathophysiological origin of MDD. Therefore, the generallyknown serotonin hypothesis for MDD, will be addressed in two additional studies (Part III). Part Vof the thesis comprises the general discussion and will address directions for future research.
In this introduction chapter, the reader will be introduced to some backgrounds of MDD, itspharmacological treatment, the monoamine hypothesis about the etiology of MDD, and briefly onthe mechanisms of action of antidepressant drugs. After a summary, the separate studies of thethesiswill be introduced. In chapter 2, the methods of the Dose-Escalation Legitimate?Pharmacology and Imaging studies in depression (DELPHI-study) will be described. However, let’sstart with some history.
MELANCHOLY, the subject of our present discourse, is either in disposition, or habit. In dispositionis that transitory melancholy which comes and goes upon every small occasion of sorrow, need,sickness, trouble, fear, grief, passion or perturbation of the mind, any manner of care, discontent orthought, which causeth anguish, dullness, heaviness and vexation of spirit, any wayes opposite topleasure, mirth, joy, delight, causing forwardness in us, or a dislike. In which equivocal and impropersense, we call him melancholy, that is dull, sad, sowr, lumpish, ill disposed, solitary, any way moved ordispleased. And from these melancholy dispositions, no man living is free. […] Melancholy, in thissense, is the character of mortality. […]
It falleth out oftentimes that these dispositions become habits, and many affects contemnedmake a disease. […]for that which is but a flea-biting to one, causeth unsufferable torment toanother; and which one by his singular moderation and well composed carriage can happilyovercome, a second is no whit able to sustain; but upon every small occasion of mis-conceived abuse,injury, grief, disgrace, loss, cross, rumour, etc. (if solitary, or idle) yields so far to passion, that hiscomplexion is altered, his digestion is hindered, his sleep gone, his spirits obscured, and his heartheavy, his hypocondries mis-affected; wind, crudity, on a sudden overtake him, and he himselfovercome with melancholy. […] If any discontent seise upon a patient, in an instant, all otherperturbations will set upon him; and then, like a lame dog or broken-winged goose, he droops, andpines away, and is brought at last to that ill habit or malady of melancholy it self. […]
But all these melancholy fits, […] displeasing, violent and tyrannizing over those whom theyseise on for the time–yet these fits, I say, or men affected, are but improperly so called, because theycontinue not, but come and go, as by some objects they are moved. This melancholy, of which we areto treat, is an habit, morbus sonticus, or chronicus, a chronick or continuate disease, a settledhumour, not errant, but fixed; and as it was long increasing, so, now being (pleasant or painful)grown to an habit, it will hardly be removed. […]
INVETERATE melancholy, howsoever it may seem to be a continuate, inexorable disease, hard tobe cured, accompanying them to their graves most part, yet many times it may be helped, even thatwhich is most violent, or at least it may be mitigated and much eased. Nil desperandum. It may behard to cure, but not impossible for him that is, most grieviously affected, if he be but willing to behelped.”1

Chap
ter
Chapter 1
1
14
Burton’s work The anatomy of melancholy* (first published in 1621) was a best-seller.1 Itdescribes many aspects of the disease that we currently recognize as depression:2 the sometimesdifficult distinction of the illness melancholy (depression) from ‘normal’ sadness, pain or sorrowafter misery in life, the relation between stressful life-events and the development of the illness,the interaction of life-events and coping styles to develop the illness or remain well, the clinicalsymptoms, the enormous impact of the illness on a depressed person’s life, social dysfunction andthe tendency of recurrence and chronicity.1
Nowadays, MDD is one of the most prevalent and disabling illnesses in psychiatry, afterischemic heart disease the second most common cause of disability worldwide and expected tobe the world’s second cause of disability by 2030, behind HIV/AIDS.3 MDD is diagnosed by theoccurrence of a cluster of different symptoms affecting mood, pleasure, attention, activities, vitalsomatic functions (eating, sleep), and (ruminative) thoughts over oneself, guilt or suicide over aprolonged period of time. For clinical and scientific uniformity, MDD is currently defined bycriteria specified in the Diagnostic and Statistical Manual (4th edition; DSM-IV),4 which is a non-theoretical classification-system.
Epidemiology of MDDThe 12-month prevalence of MDD is estimated to be 5.8% in the Netherlands,5 and 5.3% in theUnited States.6 Prevalence rates are two times higher in women,5;7-10 and higher in widowed,divorced, unemployed or disabled people, people with somatic disease and first degree relativesof patients with MDD.6;9 In the general Dutch population, the median duration of MDD is 3months, with 63% of new MDD episodes recovering within 6 months (regardless whethertreatment is offered or the natural course of the disease is awaited), however, this prognosis isless favourable for people who seek help.11 Estimations of the lifetime prevalence are 15.4% in theNetherlands,5 >16% in Europe,8 and 13.2%-16.2%6 in the USA, with the same 2:1 female-maledistribution. Most epidemiological studies indicate that nowadays 51-69% of depressed patientsseek or receive treatment,8;9 which has increased over the last decade relative to the nineties ofthe previous century.10 However, only 50% of patients who are treated for MDD meet diagnosticcriteria of MDD or meet indication criteria for treatment,10;12 and it is estimated that only 21.7% ofMDD-patients in the USA receive adequate treatment.9 Therefore, depending on one’s view, MDDis frequently over-treated, under-treated or mis-treated.
MDD is a recurrent and potentially chronic disease. After the first episode, 50-80% of patientshave a recurrence within 10 year.13;14 Of incident episodes of MDD 15-20% will become a chronicdepression (defined as a duration of ≥2 years), although higher percentages (46%) have beenreported.11;15;16
MDD is associated with high direct treatment costs as well as indirect costs of loss ofproductivity and quality of life.8;17 In the Netherlands, treatment costs of MDD (€250 million) in1994 were estimated to comprise 1% of the total healthcare budget, and 13.6% of the total mentalhealth budget.18 In 2003, MDD treatment costs in the Netherlands had increased to €660 million,still consuming 1.1% of the total healthcare budget.19 In 1999 10% of newly assigned disability-payments (WAO) in the Netherlands was due to MDD.20
Treatment of MDDIn order to treat major depressive disorder effectively, many national clinical guidelines weredeveloped.13;21-30 Besides various forms of psychotherapy, pharmacotherapy is often applied asmonotherapy or in combination with psychotherapy. Antidepressants are effective drugs forMDD when compared to placebo, although effect sizes are moderate,24;26;31-33 and some criticsclaim that effects are exaggerated by pharmaceutical companies.34-37 Antidepressants areprescribed for MDD, but also for other psychiatric disorders, and are of considerable financialinterest. In 2006, approximately 780.000 inhabitants (4.7%) of the Netherlands used
* Melancholy is derived from the Greek Μελαγχολια (melancholia), which refers to the black choler (bile) that was seen asthe material cause of the illness those days.

ChapterGeneral introduction and outline
1
15
antidepressants, mostly Selective Serotonin Reuptake Inhibitors (SSRIs). Annual prescriptions of antidepressants increased over the last decade by ~6% every year. Thecosts of prescribed antidepressants were €156 million in 2006,38 again, most costs represent theprescription of SSRIs.
A range of antidepressants have been developed, all aiming at the stimulation of serotonergicand/or noradrenergic neurotransmission.39;40 Four classes of antidepressants exist: SSRIs, Tricyclicantidepressants (TCAs), Monoamine-oxidase inhibitors (MAOIs) and a miscellaneous group. TCAsand SSRIs, but also ‘dual action’ antidepressants block the transporters at the pre-synaptic nerveending, which causes inhibition of the reuptake of either serotonin (5-HT), noradrenaline (NA; =norepinephrine), or both. The classic MAOIs irreversibly inhibit the enzymes mono-amine oxidaseA and B, which degradeserotonin, noradrenaline and dopamine. The miscellaneous groupharbours ‘dual action’ antidepressants (e.g. venlafaxine or duloxetine), and antidepressants withpre- and post-synaptic targets different from serotonin or noradrenaline transporters.39;40
By nature of their largely similar, but also slightly distinct pharmacological affinities fortransporters and receptors, antidepressants have (almost) comparable efficacy,32;41-43 but aredifferent in their adverse effects. Because SSRIs are tolerated better than TCAs,32;44;45 SSRIs havebecome the antidepressants of first choicein most countries.38 Within their class, different SSRIsalso have comparable efficacy, but different side effect profiles.46;47
Achieving remission is the aim of treatment,because persistence of (residual) symptomsincreases the risk of a future relapse or recurrence and is associated with ongoing psychosocialdysfunction.48;49 If remission is achieved, patients are advised to continue their antidepressants(at least 6 months) in order to prevent relapse,21;50 although guidelines’ recommendations areinconsistent.13;21;23-25 Nevertheless, only 50% of the patients respond (defined as a 50% decrease indepression severity)40 to the initial antidepressant, and remission rates (defined as a depressionseverity score below a certain cut-off) are lower (28-35%).28;29;51 Therefore, clinicians who treatMDD are often confronted with non-response, for which evidence-based approaches arenecessary.52
Insufficient response to antidepressants: A challenge for the clinician.The clinician faces several dilemmas when a patient shows insufficient response to anantidepressant. First, how does one ascertain clinical improvement?
A variety of measurement scales is available to measure depression severity and improvementof therapy.53 Psychopathology rating scales with clear cut-off scores provide a more directrepresentation of the effects of treatment than quality of life scales or a functional outcome.Thus, the use of a symptom-rating scale is highly recommended to assess treatment effects,either clinician-rated (e.g. the Hamilton Depression Rating Scale (HDRS),54 Montgomery ÅsbergDepression Rating Scale (MADRS)55 or Inventory for Depressive Symptoms (IDS-C))56 or self-rated(Inventory for Depressive Symptoms (IDS-SR)56, Quick-IDS (QIDS-SR)57 or Beck DepressionInventory (BDI)).58 Despite criticism on internal validity, selection and rating of items, andsensitivity to change,59-64 most scales correlate with each other.57;65 Frank et al.66 proposedtentative definitions for response (≥50% decrease of pretreatment rating scale scores) andremission (e.g. HDRS ≤7, MADRS ≤7, IDS-SR ≤13, BDI ≤9). Therefore, measurements should beroutinely performed before the start of treatment and at consecutive critical decision points.67;68
However, scales are not routinely implemented in daily practice. Apparently, the currentchallenge of implementation is to persuade clinicians to measure MDD severity repeatedly withappropriate scales.
The next dilemma is when to measure the effects of the antidepressants. Although a recentmeta-analysis indicated that the effects of antidepressants on depressive symptoms can beobserved within the first weeks of treatment,69 patients may need 6-10 weeks to achievesubstantial improvement.51;70 Therefore, the initiated treatment should not be changed too early,neither too late. Changing a regimen early may forestall a potentially late effect of the currenttreatment, but may also offer an increased chance of response. Contrary, unchanged treatmentmay increase despair due to the failure of response,but may prevent the (possible premature)

Chap
ter
Chapter 1
1
16
start of a more aggressive treatment with increased side effects. Over the last decades, the timeto declare that an antidepressant trial failed, increased from 3 weeks71 to 6-8 weeks.70;72
Interestingly, recommendations on this lag-time were never derived from studies primarilydesigned to quantify the timing of the determination of response. Current guidelines fordepression differentially recommend a minimal duration of antidepressant therapy between 4and 6 weeks.13;21;23;25;26;28;29;67;73 At these time points (‘critical decision points’)67 a next treatment-strategy must be considered.
A final dilemma is to adequately re-evaluate the diagnosis, without the (counter-) transferenceof helplessness. Critical decision points are important for the reassessment of somatic orpsychiatric co-morbidity, re-evaluation of persistent psychosocial problems and treatmentadherence. However, when none of these issues apply, the patient may be accused of ‘a failure tocooperate’, or blamed for the non-response by ‘having a personality disorder’ or ‘not beingmotivated for treatment’. Although these reproaches occasionally may be justified, they couldalso indicate the patient’s and/or clinician’s helplessness, which is often transferred from adepressed person to the persons surrounding him or her. Most depressed patients want toimprove, but are indeed disabled in their coping-styles, their motivation and their problem solvingcapacities. As such, routinely administered behavioural interventions (registration, planning andgradual reactivation)74 are valuable in the pharmacological treatment of MDD, also deferringincreased helplessness.
Insufficient response to antidepressants: What to do when the miracle doesn’thappen?Five pharmacological strategies for insufficient response to an antidepressant can be distinguished: 1. prolongation, 2. dose-escalation, 3. switching, 4. augmentation and 5. combina tion, allregarding the antidepressant initially given. Of course, a sixth strategy could be the addition ofpsychotherapy to the monotherapy of pharmacotherapy,75;76 but as this is not a topic of thisthesis, only the first 5 strategies are described.
1 Prolongation of the trial for another 2-4 weeks. This strategy is applicable only in case of doubtof the nature of the non-response (e.g. amendable psychosocial stressors, non-adherance),but is not favourable given the relative importance assigned to critical decision points.
2 Dose-escalation. With this strategy the dose of the prescribed antidepressant is increased tothe maximal tolerable dose. Dose-escalation assumes a dose-response relationship, which isequivocal for SSRIs. The advantage of dose-escalation is that it is easy and quick to apply.
3 Switching of the antidepressant. With this strategy the prescribed antidepressant isdiscontinued (eventually after tapering and a wash-out period) and the antidepressant isswitched to another antidepressant either belonging to the same class, or to a different classcompared to the initial antidepressant. The obvious advantage of switching is that it facilitatesa new pharmacological approach, possibly targeting different neurotransmitter systems. Itsdisadvantage is that it requires some time, and that one may unwontedly discard achievedimprovement when the chosen antidepressant appears to be a less effective one for thispatient.
4 Augmentation of the prescribed antidepressant. With this strategy a drug is added that has nostrong antidepressant effects by itself, but is known to increase the effects of antidepressants(e.g. lithium). The advantage of augmentation is that the effect of the antidepressant initiallyprescribed is maintained. Furthermore augmentation might result in faster responses thanswitching (as tapering is not required, nor a wash-out). Disadvantage is the risk of poly-pharmacy and the related interactions.
5 Combination of antidepressants. With this strategy another antidepressant drug, mostly withdifferent pharmacological properties compared to the antidepressant already prescribed, isadded to the initial antidepressant. This strategy has the same advantages and disadvantagesas augmentation strategies, but the risks of severe interactions are higher.

ChapterGeneral introduction and outline
1
17
Neurotransmission, the serotonergic system and neuronal networksThe brain passes information through electrical signals in neurons. Synapses form the functionalcontacts between neurons and mainly communicate through neurotransmitter secretion.Neurotransmitters are stored in synaptic vesicles. The vesicles are released into the synaptic cleftin response to presynaptic depolarisation. After the release of neurotransmitters in the synapse,they are either metabolized or transported back into the terminal to be used again.77
Neurotransmitters bind to specific receptor proteins on the membrane of the pre- andpostsynaptic neurons. Postsynaptically, receptors either induce a change in postsynaptic ionchannels (ionotropic receptors), causing a depolarisation, or they activate G-protein mediatedcomplexes (metabotropic receptors), which activate one or more metabolic steps (‘secondmessengers’). Activation of metabotropic receptors is generally responsible for forming enzymesthat regulate gene expression, neurotransmitter synthesis, receptors and neuroplasticity. Whichof the mechanisms is used depends on the particular postsynaptic receptor type.78 Three major‘monoamine’ neurotransmitters are associated with psychiatric disorders: serotonin,noradrenaline and dopamine, of which the latter is traditionally related to psychotic disorders.SSRIs target the serotonergic system and increase serotonergic neurotransmission. From here,the serotonergic system and the neuronal networks that are believed to be involved in MDD willbe described.79
SerotoninThe serotonin neurons of the brain start in the raphe nuclei in the midbrain and project to theneocortex, basal ganglia, temporolimbic zones, hypothalamus, cerebellum and the brain stem(Figure 1.1).80;81 Serotonin is involved in several functions: sleep and wakefulness, appetite, nausea,migraine, headaches and regulation of mood.77 The serotonin receptors comprise of 5-HT1A, 1B,
1C, 1D, 1E, 1F, 5-HT2A, 2B, 2C, 5-HT4, 5-HT5, 5-HT6 and 5-HT7 (metabotropic) and the 5HT3 (ionotropic)receptors.78 Serotonin plays an important role in brain development via regulation of neuriteoutgrowth, synaptogenesis and cell survival.82;83 Serotonin that is released into the synaptic cleftis either taken up back into the presynaptic nerve ending by the serotonin transporter (SERT), ordegraded by MAO-A.77;78
The serotonin (and noradrenaline) deficiency hypothesisIn the late fifties of the previous century, TCAs appeared to be effective in treating MDD byincreasing serotonergic and noradrenergic neurotransmission. This discovery led to themonoamine hypothesis: MDD might etiologically be explained by a deficiency in monoamineneurotransmitters: serotonin ornoradrenaline. Since then, the working mechanism of AD isbelieved to be by (1) increased neurotransmission by increased synaptic levels of serotonin,noradrenaline and/or (2) specific agonistic effects on serotonin or noradrenaline (sub-)receptors.
Figure 1.1. Serotonergic pathways through the human brain.
Serotonergic system. From the raphe nuclei in the midbrain, neurons project to the neocortex, basal ganglia, temporolimbiczones, hypothalamus, cerebellum and the brain stem.
����������� � � �������� �
���������������������������
��������������� ������ ����� ������������������
��� �����������������������!���������
��!������� �������������� ��
������������

Chap
ter
Chapter 1
1
18
Depletion of the available serotonin andnoradrenaline is used as a model to test theinvolvement of monoaminergic systems in MDD. Serotonin depletion can be achieved by rapidlylowering the essential amino-acid tryptophan which cannot be synthesized by the body and mustbe ingested to enable formation of serotonin. To achieve depletion, a tryptophan free amino-acidmixture is administered (acute tryptophan depletion).84 Depletion ofnoradrenaline anddopamineoccurs simultaneously, and uses the same concept (acute depletion of the essentialamino-acids phenylalanine and tyrosine).85 As an alternative to induce a state of depletion,enzyme-blocking agents decrease the production of the monoamines. Para-chlorophenylalanineblocks serotonin synthesis,86 and Alpha-methyl-para-tyrosine blocks noradrenaline and dopaminesynthesis.87
Since 1975 an increasing number of depletion studies have been conducted, with differenteffects in different study-populations. In general in healthy controls no clear mood-effects werefound, unless they had relatives with MDD. In remitted MDD patients who used antidepressants(or shortly after tapering) approximately 50% of the patients experienced a relapse afterdepletion. However this occurred only after depletion of the monoamine that theirantidepressant targeted. In depressed patients no consistent deterioration of the mood effectswere found.88-93
Thus, the monoamine-deficiency theory, in its purest form, states that depression can becured by the increase of serotonergic and/or noradrenergic neurotransmission. However, thereverse train of thought, that depression is bio-etiologically caused by a deficiency of monoamines(e.g. serotonin and/or noradrenaline) has attractive face-validity, but probably is an untenable,superficial simplification.75;94 Therefore, the current, less pertinent view is that the monoaminehypothesis only partially explains MDD and the response to AD.95-98
The limbic-cortical dysregulation hypothesisIn a more multidimensional, systems-level model, MDD can be viewed as a disorder affectingdiscrete but functionally integrated pathways; neural networks, which can be identified byneuroimaging techniques.† In such a network, dysfunction in one or more of the elements (e.g.after cognitive or somatic stress), will initially be tried to be influenced (or compensated) byother, remaining parts of the network, that try to maintain homeostatic emotional control.Therefore, results from neuroimaging studies investigating differences between healthy controlsand MDD patients, must be considered as the identification of regions to be either etiologicallyabnormal or regions involved in (mal-)adaptive compensatory processes.99
Because MDD is an affective disorder, the neurobiology of emotion processes is likelyinvolved. For the processing of emotions two systems are important: a ventral system (consistingof amygdala, insula, ventral striatum, ventral anterior cingulate gyrus, and ventral prefrontalcortex) and a dorsal system (consisting of hippocampus, dorsal anterior cingulate gyrus, anddorsal prefrontal cortex). The ventral system serves to identify the emotional significance of astimulus, the production of mood states, and automatic regulation of emotional responses, whilethe dorsal system serves to effortfully regulate mood states and subsequent behaviour.100
Initial lesion-deficit studies, early Positron Emission Tomography (PET) studies (measuringregional resting state glucose metabolism or blood flow) and later functional MagneticResonance Imaging (fMRI)-studies identified several brain regions to be affected by MDD: thelimbic structures (amygdala, hippocampus, hypothalamus and brainstem), the subcortical (basalganglia and thalamus) and the cortical (dorsolateralprefrontal, ventrolateral prefrontal andorbitofrontalcortex (DLPFC, VLPFC, OFC respectively) structures. In these, the most consistentfinding is a hypoactivity of the (dorsal) frontal lobe, while often a hyperactivity in the (ventral)VLPFCand OFC is found.99;101 Additionally, an increased activity of the (rostral, subgenual) anteriorcingulated gyrus99;101 and the amygdala, anterior insula, and ventral striatum was found, althoughless consistently.101 Furthermore, fMRI studies point to a increased sensitivity of the amygdala for
† Single Photon Emission Computed Tomography (SPECT) and Positron Emission Tomography (PET) use a variety ofradioligands with various half-lives to quantify different targets (transporters, receptors, or blood-flow or metabolism).Functional Magnetic Resonance Imaging (fMRI) is a technique to image brain activity (hemodynamic response) related toa specific task or stimulus.

ChapterGeneral introduction and outline
1
19
emotional stimuli, and a bias to interpret these stimuli in a negative context.101 Interestingly,induction of sad mood in healthy volunteers also produces increased blood flow in the insula,subgenual anterior cingulate gyrus and decreased blood flow in the dorsomedial prefrontalcortex (DMPFC).102
The above abnormalities were combined in the limbic-cortical dysregulation model, whichincludes a dorsal neocortical hypofunction, which results in ventral (para)limbic hyperactivity,with a reciprocity in this inverse relationship.103;104
Which effects in the brain cause antidepressants to be antidepressants? The various treatment forms available to relieve MDD and their moderate efficacy poses thequestion: How do antidepressants bring about their therapeutic effects? For this question, threelevels of action must be distinguished: 1. direct neurotransmission effects, 2. second messengereffects, and 3. change in neuronal networks. For levels 1. and 2. see Figure 1.2. At all three levels,there appears to be a time dependent differentiation of the effects that occur, e.g. not all effectsare the same in the consecutive weeks after the initiation of treatment. This may explain why itsometimes takes several weeks before the therapeutic effects become apparent. Because mostof the recent research was done with SSRIs and SNRIs, the effects of these drugs are describedhereafter, unless indicated differently.
Direct effects of antidepressants on neurotransmission When an SSRI is ingested, the blockade of the target transporter (i.e. the serotonin transporter(SERT)) occurs within several minutes.105;106 Therefore, the antidepressant effects cannot only bebased on increased neurotransmission by decreased serotonin reuptake, as these effects takemuch longer than this direct pharmacological effect. Further research revealed that after twodays of treatment, the firing-rate of SERT-containing neurons in rats decreased, but that thisfiring-rate was restored within 2 weeks of continued treatment. This was attributed tosomatodendritic 5-HT1A autoreceptors, which normally have a negative feedback on the neuron’sfiring rate. These 5-HT1A autoreceptors appeared to desensitize.105 Microdialysis-experiments inrats showed that the restored firing-rate after 14 days was responsible for a 6-fold increase inintrasynaptic serotonin level, while after the acute blockade of the SERT this increase of theserotonin level was only small and transient.107 Furthermore, in humans, serotonin autoreceptors(5-HT1B/1D) (and also noradrenergic α2 autoreceptors) in the synapse normally inhibit serotoninrelease by feedback-mechanisms as well. Prolonged treatment with SSRIs again desensitize thesereceptors, resulting in increased serotonin release in the synaptic cleft.105;108 As such,desensitisation of HT1A autoreceptors in the raphe nuclei in the midbrain may have effects onserotonergic neurotransmission in critical brain areas where these serotonergic neurons projectto.109 Finally, in rats, after prolonged administration of SSRIs the SERT itself is downregulated by80-90%.110-112 This is probably caused by trafficking and internalization of the SERTs instead ofaltered SERT-gene-regulation, because mRNA expression in the cells studiedwas unaltered by thetreatment.106
Other antidepressants have rather different effects. TCAs (except clomipramine) do notchange the pre-synaptic serotonin-containing neurons, but appear to sensitize the postsynaptic 5-HT receptors for serotonin.105 Along with serotonin, the responsiveness for noradrenaline wasalso found to be enhanced, likely due to enhanced α-adrenoreceptor-mediated transmission.MAOIs also increase serotonergic transmission by desensitisation of 5-HT1A autoreceptors, but donot desensitize other 5-HT autoreceptors, and desensitize noradrenergic α2 autoreceptors, whichindirectly enhances serotonergic transmission.

Chap
ter
Chapter 1
1
20
Figure 1.2. Transporters, receptors and second messenger systems involved in the effects of antidepressants.
Figure adapted from Belmaker and Agam.79 The left half of the presynaptic neuron represents a serotonergic neuron, theright half a norepinephrinergic neuron. For color figure see page 277.In the presynaptic neuron, serotonin is synthesized from tryptophan by tryptophan hydroxylase and stored in vesicles.Likewise, norepinephrine is synthesized from tyrosine by tyrosine hydroxylase. These vesicles merge with the cell membranewhen the neuron is depolarized, thereby releasing their contents into the synaptic cleft. After release, serotonin and norepinephrine are transported back into the presynaptic neuron by serotonine andnorerepinephrine transporters. Furthermore, serotonin and norepinephrine are catabolized by the monoamine-oxidase A(MAO-A) enzyme. In the synaptic cleft, serotonin and norepinephrine affect both the pre-and post-synaptic neuron. The pre-synaptic 5-HT1A and 5-HT1B auto-receptors decrease serotonin release by inhibitory feedback; the α2-adrenergic receptordoes the same for the release of norepinephrine. Post-synaptically, serotonin and norepinephrine bind to G-protein-coupled monoamine receptors (MARs): the cyclicAMP(cAMP)-coupled receptor, which activates protein kinase A (PKA), and the Phosphatidylinositol (PI)-coupled receptor, whichactivates phospholipase C (PLC) which thereafter form inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 and DAGactivate protein kinase C (PKC). Both PKA and PKC finally activate cAMP responsive element binding (CREB) protein, whichstimulates DNA transcription. For example, this might result in the production of brain derived neurotrophic factor (BDNF).

ChapterGeneral introduction and outline
1
21
Second messenge reffects of antidepressants The secondary effects of antidepressants can be divided in post-synaptic effects on thesensitivityand availability of receptors, and the effects caused by the activation of post-synaptic receptors.79
Effects on post-synaptic receptors include a decrease of 5-HT1A and 5-HT2Areceptor density,113 and5-HT1D
114 and 5-HT2C/2B responsivity.113;115 Because these effects are either investigated in animals orby using indirect measures (e.g. growth-hormone release), it is important that Meyer et al. onlydemonstrated 10% in-vivo 5-HT2A receptor downregulation in MDD patients between 20-30 years,but not in older patients.116 The desensitisation of post-synaptic receptors might explain thetolerance to adverse effects that occurs after some days of exposure to antidepressants.109
Interestingly, in rodents, the activation of post-synaptic 5-HT1A receptors in the hippocampusstimulates neurogenesis, which appears to be required for therapeutic effects ofantidepressants.117
After the activation of the G-protein metabotropic receptors (either by serotonin ornoradrenaline), second messenger systems mediate signals to the neuron’s nucleus, where cAMPresponsive element binding protein (CREB) regulates CREB-directed gene transcription.79;118 Mostevidence indicates that CREB is upregulated by chronic antidepressant use, but not for allantidepressants, and results of studies appear to be biased by the cell type investigated, the brainregion where these cells originate from and the timing after the first antidepressant exposure.118
One of the genes that is (positively) influenced by CREB is the brain derived neurotrophic factor(BDNF) gene.119 BDNF is a plethoric growth factor, which regulates neuronal survival, migration,differentiation, axonal and dendritic growth and synapse formation. The genomic structure ofBDNF is complex, which facilitates differential activation by diverse and variable stimuli, which canbe different in different brain regions and even in different parts of the cell.83
Changes in neuronal networksAt the neuro-anatomical level, the novel neuroimaging techniques have ‘opened’ the brain tostudy the in-vivo effects of psychiatric treatment.104 In MDD, early PET studies found timedependent changes during the treatment with fluoxetine.120 After 1 week, glucose metabolismincreased in the hippocampus and brainstem, and decreased in posterior cingulate gyrus, striatumand thalamus compared to the pre-treatment scan. After 6 weeks, patients who responded to thetreatment had a decrease in metabolism in the subgenual cingulate gyrus, the hippocampus, thepallidum and insula and an increase in the anterior and posterior cingulate gyrus, prefrontal andparietal cortex. However, the non-responders showed a persistent, unchanged pattern of changeas seen in week 1. The changes in prefrontal cortex and subgenual cingulate gyrus correlated bestwith symptomatic improvement.99;101;120 These findings were replicated in patients treated withparoxetine.121 Moreover, patients who responded while on placebo-treatment showed somehowsimilar but also distinct‡ changes in brain metabolism compared to fluoxetine responders.99;122 Thisindicates that in neuroimaging studies, response andtreatment effects may coincide, but bothmay also have their specific, distinguishable effects.
In recent fMRI studies, the increased activation of the amygdala to negative (sad, fearful,angry) and happy faces have been investigated after treatment of SSRIs (fluoxetine,123;124
sertraline125), venlafaxine126;127 and bupropion.128 These studies rather consistently founddecreased activation of the amygdala, insula and increased cortical activity after treatment.However,most patients in these studies were treatment responders at the end of the study. Incontrast with previous PET-studies, none of the fMRI studies used a placebo-comparisonmeasured twice, so no distinction between specific drug and response effects in fMRI can bemade yet.
‡ Similar changes included increased metabolism in frontal, parietal and posterior cingulate, and decreased metabolism insubgenual cingulate gyrus. Distinct changes included no changes in subcortical brainstem, hippocampal, and caudatemetabolism in placebo-responders.

Chap
ter
Chapter 1
1
22
(Pharmaco-)genetic effects relevant for antidepressantsSeveral genetic polymorphisms have been investigated in relation to treatment response toSSRIs. The polymorphism studied most is the SERT promoter gene (5-HTTLPR), for which a long(L) and a short (S) variant were identified, with a recently discovered functional tri-allelic variant(rs25531).129 The 5-HTTLPR is associated with the transcriptional activity of the SERT gene.130 Cellshomozygous for the L-allele produce higher concentrations of SERT mRNA, and the rate ofserotonin uptake by the transporter is >2-fold higher than in cells containing one or two copies ofthe S-allele. A meta-analysis of 15 studies showed a pooled association between the 5-HTTLPR-polymorphism and SSRI efficacy,131 with MDD patients with at least one L-allele having higherresponse rates to SSRIs. However, in a large sample of patients treated with citalopram (an SSRI),treatment response was not associated with the tri-allelic 5-HTTLPR-polymorphism.132;133
Furthermore, individuals carrying the S-allele experience increased adverse events after SSRItreatment,132;134 have elevated risk of depression in relation to life events,135 but alsoshowincreased amygdala reactivity to fearful stimuli.136;137 A large MRI-study in healthy controls showedassociations of the 5-HTTLPR S/S polymorphism with unfavourable alterations in anatomy andfunction of the amygdala-cingulate feedback circuit.138 Thesefindings strongly argue for animportant role of the 5-HTTLPR-polymorphism in the development and functioning of emotionalnetworks involved in MDD. Other pharmacogenetic associations with clinical response have beeninvestigated, but will not be further addressed in this thesis.
Summary and questions addressed in this thesisMajor Depressive Disorder is a prevalent and disabling illness, which potentially recurs and maybecome a chronic disease. It is the second most common cause of disability worldwide, and has a12-month prevalence of ±5.5%, with lifetime prevalences of 12-14% in males and 22-24% in females.3;5
MDD is associated with high direct treatment costs as well as indirect costs of loss of productivityand quality of life. Besides various forms of psychotherapy, pharmacotherapy with antidepressantdrugs is often applied.13;21-25
Antidepressants are grouped in four different classes: Selective serotonin reuptake inhibitors(SSRIs), tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs) and amiscellaneous group including so-called dual action antidepressants. SSRIs, dual-actionantidepressants and TCAs are used most often.39 Most antidepressants increase serotonergic and/or noradrenergic neurotransmission. When considered more precisely, different classes ofantidepressants appear to have distinct additional pre- and postsynaptic effects.105;106 Secondaryto increased serotonergic and/or noradrenergic neurotransmission, complex second messengerpathways are activated, which are only partly understood.79 Macroscopically, presumably relevantchanges in neuronal networks following antidepressant treatment have been identified.99;101;104
Nevertheless, which changes are required and specific for the improvement of symptomsremainsan enigma. Furthermore, the effects of increased neurotransmission might be affected by thegenetic make-up of patients (i.e. the 5-HTTLPR polymorphism129). Genetic polymorphisms appearto be associated with treatment effects,131 but might also influence the development andconnectivity of the neuronal networks.138
Despite comparable efficacy of antidepressants, only 50% of the MDD-patients respond to thefirst antidepressant trial given, while fewer achieve full remission of symptoms.48;51 Therefore,insufficient response to the first antidepressant is a relevant clinical problem that challengesclinicians.
The clinician’s first dilemma is how to adequately and efficiently measure the changes insymptom severity, for which the further development and implementation of short, easy to useand valid clinician rated questionnaires would improve clinical decision making. The seconddilemma is to time the change of the treatment initiallystarted: this should not be changed tooearly, neither too late.70 The third dilemma is to balance helplessness, impaired functioning by thedisease and (counter-) transference by ‘blaming the victim’, i.e. reproaching the patient of themoderate efficacy of antidepressants as ‘being unmotivated’ or ‘having a personality disorder’.

ChapterGeneral introduction and outline
1
23
When ‘the miracle doesn’t happen’, 5 strategies for non-response are possible options:prolongation of the initial trial, dose-escalation, switching, augmentation and combination ofantidepressants. For all these options the current evidence is either equivocal or fragmentary. Inorder to develop evidence based treatment algorithms for the 50% of MDD-patients who turn outto be non-responsive to the first antidepressant trial, systematic overviews of the literature arerequired and gaps in the evidence-base need to be addressed by new research-projects.
More fundamentally, the perceived delayed onset of symptom improvement withantidepressants, their moderated efficacy, and their extensive effects in the brain beyond anincrease in serotonergic or noradrenergic neurotransmission, summons the question what is theetiopathogenetic model for MDD. Or, more specifically, what is the evidence that corroboratesthe corner-stone of most antidepressant’s action: the monoamine hypothesis and theinvolvement of the serotonergic system as a causal explanation for MDD.
Research projects underlying this thesisThis thesis incorporates three research projects that were initiated and conducted by the Programfor Mood Disorders of the Academic Medical Center (AMC). One additional project was initiatedby the department of vascular medicine of the AMC.
First, a guideline-project was initiated with a grant from the Academic Medical Center (NoSFA.07.012). Aim of this guideline project was to develop an evidence based guideline for non-response after a first SSRI.
Second, as a result of this guideline project, two grants were obtained from the NetherlandsOrganization for Health Research and Development (ZonMw), program Mental Health, educationof investigators in mental health (Geestkracht-OOG; projects #100-002-001 and #100-002-002).These grants were applied to initiate the DELPHI-study (Dose-Escalation Legitimate?Pharmacology and Imaging studies in depression). The DELPHI-study was set up as amethodologically sound trial to study clinical effectiveness of dose-escalation as a strategy fornon-response. As a novel extension to existent dose-escalation trials, we aimed to investigate themolecular neurobiological target of dose-escalation: the occupancy of the serotonin transporter(SERT) by paroxetine (a SSRI). This was done by the acquisition of two or three single photonemission computed tomography (SPECT) scans in a subgroup of drug-free patients of the totalDELPHI-cohort.
A third project was started as an extension to the DELPHI-SPECT study. This third project couldbe planned after a grant from the Dutch Brain Foundation (Hersenstichting) was obtained(project #14F06.45). The DELPHI-fMRI aimed to investigate the neurobiological changes in brainactivation by treatment with paroxetine in a functional Magnetic Resonance Imaging (fMRI)study. This study was superimposed on the SPECT-imaging of the last 22 drug-free patientsparticipating in DELPHI.
The final fourth research project in this thesis was initiated after an almost fatal bleedingcomplication, which occurred in a patient who underwent surgery, and lost 12 litres of blood. Thiscomplication was afterwards attributed to fluoxetine use. This project was funded internally bymutual contributions of both participating AMC-departments.
The questions addressed in this thesis
1 Is a short, easy to use clinician rated questionnaires as effective and precise as the routineHamilton depression rating scale (HDRS)?The HDRS54 is most frequently used as the golden-standardin clinical trials, but is probably tooextensive to use in clinical practice.61 To facilitate clinical measurements of depression severityand objectivity for critical decision points, we reanalyzed the treatment-outcomes of twoantidepressant-psychotherapy trials, which were performed by the Mentrum researchgroup.139;140 We therefore investigated whether the effect-sizes of two 6-item subscales of theHDRS (17-items) – the Maier141 and Bech59 subscales – were comparable to the original HDRS inthe measurement of depression severity and the sensitivity to measure changes.Furthermore, we investigated whether this comparability was stable across the full range of

Chap
ter
Chapter 1
1
24
response to treatment, and across different treatments and for different baseline severity ofdepression. We also determined cut-off points for remission for these subscales compared toconventional HDRS definitions.66;142;143 See chapter 3.
2 What is the evidence for dose-escalation as a strategy for non-response to a first SSRI?For this question, we performed a systematic review of the evidence for the dose responserelationship for SSRIs in MDD. See chapter 4.
3 What is the evidence for switching antidepressants as a strategy for non-response to a firstSSRI?For this question, we performed a systematic review of the evidence for switching afterfailure of a first SSRI in MDD. Part of this study was a meta-analysis of three switch-studies.See chapter 5.
4 Does the depletion of monoamine (5-HT and NA/DA) systems lower mood in humans, and isthis lowering of mood different across different populations?For this question, we performed a systematic review of monoamine depletion studiesreporting mood effects of depletion. As an extension of previous systematic reviews ofmonoamine depletion studies,88-93 we aimed to pool the results ofthe small-sized depletionstudies, because they might not have detected small differences by a lack of power, andpooling would quantify the balance of positive versus negative studies. Therefore, we applieda pooling technique (modified from conventional meta-analyses of randomized controlledtrial data and including an adjustment for small sample bias) to handle the statistically pairedcross-over designs of these studies in formal, stratified meta-analyses.144 See chapter 6.
5 Do MDD-patients and healthy controls differ in the number of central serotonin transporters,and is the amount of available SERTs correlated with depression severity?Despite the fact that the working-mechanism of antidepressants supports the monoaminedeficiency theory, the pathogenesis of MDD remains unclear.95 Therefore, differences in SERTavailability in patients and healthy controls have been studied previously, with conflictingresults. Additionally, significant effects on SERT availability have been reported for gender,145
smoking behavior,145 aging146;147 and season of scanning.148 Therefore, we analyzed thebaseline SPECT-scans of the DELPHI-SPECT participants versus age and sex-matched healthycontrols. Because our sample size was large, we were able to properly account for potentialconfounders and possible interactions, of which the multivariate effects are reported. Seechapter 7.
6 Does a common genetic polymorphism of the promoter region of the serotonin transportergene (SLC6A4) modify the association between the SERT occupancy by paroxetine and theclinical response?We performed this study because SSRI-response is likely associated with 5-HTTLPRpolymorphisms, 5-HTTLPR polymorphisms might influence SERT availability (the target forSSRIs), and it is unclear how occupancy of the available SERTs is related to clinical response.Thus, we aimed to investigate the paroxetine treatment by genotype interaction regardingclinical response on the molecular level of SERT occupancy. We quantified the relationbetween SERT occupancy and clinical response, and studied how the 5-HTTLPR -polymorphism affected this SERT occupancy-response relationship. We performed this studyin the open phase of the DELPHI-SPECT study, when patients were treated with paroxetine 20mg/day for 6 weeks. See chapter 8.
7 Is dose-escalation of paroxetine an effective clinical strategy for non-response in MDD?The systematic review of dose-escalation (chapter 4), identified methodological flaws inprevious dose-escalation trials. In this study we reevaluated the clinical efficacy of dose-escalation of paroxetine without these flaws, and, considering the molecular target of SSRIs,we also tested whether paroxetine dose-escalation increasedSERT occupancy more thanplacebo dose-escalation. We therefore performed a 6 week, multicenter, randomized study indepressed patients not responding to 6 weeks of paroxetine at 20 mg/day. As a novelextension to previous clinical trials, and in order to elucidate the neurobiological basis for an

ChapterGeneral introduction and outline
1
25
expected lack of benefit of dose-escalation, we included a SPECT imaging approach.Herewith, we quantified whether paroxetine dose-escalation increased SERT occupancy morethan placebo dose-escalation. This enabled us to relate clinical findings to the neurobiologicalcorrelate of SERT occupancy. See chapter 9.
8 Does treatment with paroxetine normalize amygdala hyperactivation in MDD?We initiated this study after the first reports of attenuated amygdala activation aftertreatment with sertraline.125 Thereafter several groups replicated a baseline hyperactivation ofthe amygdala but less consistently reported the attenuation of amygdala-hyperactivationafter treatment.123;126;128 We therefore investigated whether: activation of the amygdala by(negative) facial expressions differed from healthy controls, this activation of the amygdalachanged after 6 and 12 weeks of treatment with paroxetine, the activationof the amygdalaand other brain areas merely changedby paroxetine treatment or in relation with clinicalresponse, and whether dose-escalation of paroxetine in week 6 non-responders affectedactivations, compared to placebo-dose-escalation. For this study, we performed an fMRI studyin 22MDD patients who participated in the DELPHI-fMRI study. Patients were treated withparoxetine (20 mg/day followed by a randomized dose-escalation for non-responders) andwere scanned at baseline, 6 weeks and 12 weeks of treatment. We obtained a baseline scanfor 21 matched controls, to contrast baseline amygdala activation in MDD-patients. Seechapter 10.
9 What are the changes in hemostasis and blood platelet parameters when patients are treatedwith paroxetine, and are these changes modified by dose-escalation or a geneticpolymorphism of the promoter region of the serotonin transporter gene?In this study, we evaluated the effects of standard and increasing dosages of paroxetine onthe bleeding tendency and hemostatic functions of platelets in patients who were drug-freebefore the start of paroxetine. In addition, we assessed whether these effects are modified bythe 5-HTTLPR polymorphism. See chapter 11.
References
1. Burton R. The anatomy of melancholy, what it is, with all the kinds, causes, symptoms, prognostics, and several cures of it. Inthree partitions. With their several sections, members, and subsections, philosophically, medecinally, historically opened andcut up. 6th ed. London: J. Cuthell; J. Nunn; Longman and Co.; J. Mawman; Baldwin and Co.; Lackington and Co.; Black andCo.; S. Bagster; Edwards and Knibb; Ogle, Duncan, and Co. R. Saunders; J. Sheldon; Wilsons, York; 1651:1-461.
2. Schene AH, Huyser J, Ruhe HG. Validiteit van de diagnose depressie. In: Koerselman F, Vergouwen AC, eds. Degecompliceerde kliniek 2005: over de geldigheid van diagnoses. Amsterdam: Benecke; 2005.
3. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006; 3: e442.4. American Psychiatric Association. Diagnostic and statistical manual of mental disorders, fourth edition. 4 ed. Washington
DC: American Psychiatric Association; 1994.5. Bijl RV, Zessen Gv, Ravelli A. Psychiatrische morbiditeit onder volwassenen in Nederland: het NEMESIS-onderzoek. II.
Prevalentie van psychiatrische stoornissen. NedTijdschrGeneesk. 1997; 141: 2453-2460.6. Hasin DS, Goodwin RD, Stinson FS, Grant BF. Epidemiology of major depressive disorder: results from the National
Epidemiologic Survey on Alcoholism and Related Conditions. Arch Gen Psychiatry. 2005; 62: 1097-1106.7. Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S et al. Lifetime and 12-month prevalence of DSM-III-
R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry. 1994; 51:8-19.
8. Lepine JP, Gastpar M, Mendlewicz J, Tylee A. Depression in the community: the first pan-European study DEPRES(Depression Research in European Society). Int Clin Psychopharmacol. 1997; 12: 19-29.
9. Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR et al. The epidemiology of major depressive disorder:results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003; 289: 3095-3105.
10. Kessler RC, Demler O, Frank RG, Olfson M, Pincus HA, Walters EE et al. Prevalence and treatment of mental disorders,1990 to 2003. N Engl J Med. 2005; 352: 2515-2523.
11. Spijker J, de Graaf R, Bijl RV, Beekman AT, Ormel J, Nolen WA.Duration of major depressive episodes in the generalpopulation: results from The Netherlands Mental Health Survey and Incidence Study (NEMESIS). Br J Psychiatry. 2002; 181:208-213.
12. Kendrick T, King F, Albertella L, Smith PW. GP treatment decisions for patients with depression: an observational study.Br J Gen Pract. 2005; 55: 280-286.

Chap
ter
Chapter 1
1
26
13. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
14. Beekman AT, Ormel J. Depressie. In: Jong Ad, Brink Wvd, Ormel J, Wiersma D, eds. Handboek psychiatrischeepidemiologie. 1 ed. Maarssen: Elsevier/ De Tijdstroom; 1999:300-328.
15. Keller MB, Lavori PW, Mueller TI, Endicott J, Coryell W, Hirschfeld RM et al. Time to recovery, chronicity, and levels ofpsychopathology in major depression. A 5-year prospective follow-up of 431 subjects. Arch Gen Psychiatry. 1992; 49: 809-816.
16. Wells KB, Burnam MA, Rogers W, Hays R, Camp P. The course of depression in adult outpatients. Results from theMedical Outcomes Study. Arch Gen Psychiatry. 1992; 49: 788-794.
17. Greenberg PE, Stiglin LE, Finkelstein SN, Berndt ER. Depression: a neglected major illness. J Clin Psychiatry. 1993; 54: 419-424.
18. Ruysbroek JM. Aan welke ziekten en aandoeningen wordt het geld besteedt? 1.2 (25 juni 2001). 2001. Bilthoven, RIVM.Volksgezondheid Toekomst Verkenning, Nationaal Kompas Volksgezondheid.
19. Slobbe LC, Kommer GJ, Smit JM, Groen J, Meerding WJ, Polder JJ. Kosten van ziekten in Nederland 2003. Zorg vooreuro's. [RIVM rapport# 270751010]. 2008. Bilthoven, RIVM. Volksgezondheid Toekomst Verkenning, Nationaal KompasVolksgezondheid.
20. Ziektendiagnosen bij uitkeringen voor arbeidsongeschiktheid. Statistische informatie over medische classificaties inWAO, WAZ en Wajong 1999.2001. Amsterdam, Landelijk instituut sociale verzekeringen.
21. Richtlijn Depressie. Multidisciplinaire Richtlijn Depressie. Richtlijn voor de diagnostiek en behandeling van volwassencliënten met een depressie. Brandt-Dominicus JC.2005. Utrecht, Trimbos-instituut, in opdracht van de LandelijkeStuurgroep Multidisciplinaire Richtlijnontwikkeling in de GGZ.
22. Marwijk HWv, Grundmeijer HG, Bijl D, Gelderen MGv, Haan Md, Weel-Baumgarten EMv et al. NHG-Standaard Depressievestoornis (depressie) (eerste herziening). Huisarts Wet. 2003; 46: 614-623.
23. NICE. Clinical Guideline 23. Depression: management of depression in primary and secondary care. December 2004. 23, 1-63. 2004. London, National Institute for Clinical Excellence.
24. Kennedy SH, Lam RW, Cohen NL, Ravindran AV, CANMAT Depression Work Group. Clinical guidelines for the treatment ofdepressive disorders. IV. Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.
25. Anderson IM, Nutt DJ, Deakin JFW. Evidence-based guidelines for treating depressive disorders with antidepressants: Arevision of the 1993 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2000; 14: 3-20.
26. Mulrow CD, Williams JW, Jr., Trivedi M, Chiquette E, Aguilar C, Cornell JE et al. Evidence report on: Treatment ofdepression - Newer pharmacotherapies. Psychopharmacol Bull. 1999; 34: 409-795.
27. Centraal Begeleidingsorgaan voor de Intercollegiale Toetsing. Consensusbijeenkomst depressie bij volwassenen: vrijdag 16september 1994 Utrecht. Utrecht: Centraal Begeleidingsorgaan voor de Intercollegiale Toetsing; 1994.
28. Depression Guideline Panel. Depression in Primary Care: Volume 1. Detection and Diagnosis. Clinical Practice Guideline,Number 5. AHCPR Publication No. 93-0550. 1993. Rockville, MD, U.S. Department of Health and Human Services, PublicHealth Service, Agency for Health Care Policy and Research.
29. Depression Guideline Panel. Depression in Primary Care: Volume 2. Treatment of Major Depression. Clinical PracticeGuideline, Number 5. AHCPR Publication No. 93-0551. 1993. Rockville, MD, U.S. Department of Health and HumanServices, Public Health Service, Agency for Health Care Policy and Research.
30. Jonghe Fde, Swinkels JA, Vergouwen AC, Brink Gvan de. Antidepressiva 2008. Een leidraad voor het rationeel omgaan metantidepressiva bij de behandeling van patiënten met een depressie. Amsterdam: Benecke NL; 2008.
31. Anderson IM, Tomenson BM. The efficacy of selective serotonin re-uptake inhibitors in depression: A meta-analysis ofstudies against tricyclic antidepressants. J Psychopharmacol. 1994; 8: 238-249.
32. Anderson IM. Meta-analytical studies on new antidepressants. Br Med Bull. 2001; 57: 161-178.33. Anderson IM. Drug treatment of depression: Reflections on the evidence. AdvPsychiatrTreat. 2003; 9: 11-20.34. Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a
meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 2008; 5: e45.35. Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its
influence on apparent efficacy. N Engl J Med. 2008; 358: 252-260.36. Barbui C, Furukawa TA, Cipriani A. Effectiveness of paroxetine in the treatment of acute major depression in adults: a
systematic re-examination of published and unpublished data from randomized trials. CMAJ. 2008; 178: 296-305.37. Moncrieff J, Cohen D. Do antidepressants cure or create abnormal brain states? PLoS Med. 2006; 3: e240.38. Stichting Farmaceutische Kengetallen. Data en feiten 2007.2007. Den Haag, Stichting Farmaceutische Kengetallen. 39. Ruhe HG, Rijswijk Evan, Verkes RJ. Biologische interventies bij depressie. In: Huyser J, Schene AH, Sabbe B, Spinhoven Ph,
eds. Handboek depressieve stoornissen. Utrecht: De Tijdstroom; 2008: 201-218.40. Thase ME, Rush AJ. Treatment-Resistant Depression. In: Bloom FE, Kupfer DJ, eds. Psychopharmacology: The Fourth
Generation of Progress. New York: Raven Press Ltd.; 1995:1081-1097.41. Freemantle N, Anderson IM, Young P. Predictive value of pharmacological activity for the relative efficacy of
antidepressant drugs. Meta-regression analysis. Br J Psychiatry. 2000; 177: 292-302.42. Smith D, Dempster C, Glanville J, Freemantle N, Anderson I. Efficacy and tolerability of venlafaxine compared with
selective serotonin reuptake inhibitors and other antidepressants: a meta-analysis. Br J Psychiatry. 2002; 180: 396-404.43. Papakostas GI, Thase ME, Fava M, Nelson JC, Shelton RC. Are antidepressant drugs that combine serotonergic and
noradrenergic mechanisms of action more effective than the selective serotonin reuptake inhibitors in treating majordepressive disorder? A meta-analysis of studies of newer agents. Biol Psychiatry. 2007; 62: 1217-1227.

ChapterGeneral introduction and outline
1
27
44. Arroll B, MacGillivray S, Ogston S, Reid I, Sullivan F, Williams B et al. Efficacy and tolerability of tricyclic antidepressantsand SSRIs compared with placebo for treatment of depression in primary care: a meta-analysis. Ann Fam Med. 2005; 3:449-456.
45. Anderson IM. Selective serotonin reuptake inhibitors versus tricyclic antidepressants: a meta-analysis of efficacy andtolerability. J Affect Disord. 2000; 58: 19-36.
46. Kroenke K, West SL, Swindle R, Gilsenan A, Eckert GJ, Dolor R et al. Similar effectiveness of paroxetine, fluoxetine, andsertraline in primary care: a randomized trial. JAMA. 2001; 286: 2947-2955.
47. Simon G. Choosing a first-line antidepressant: equal on average does not mean equal for everyone. JAMA. 2001; 286:3003-3004.
48. Thase ME. Evaluating antidepressant therapies: remission as the optimal outcome. J Clin Psychiatry. 2003; 64 Suppl 13: 18-25.
49. McIntyre RS, O'Donovan C. The human cost of not achieving full remission in depression. Can J Psychiatry. 2004; 49: 10S-16S.
50. Geddes JR, Carney SM, Davies C, Furukawa TA, Kupfer DJ, Frank E et al. Relapse prevention with antidepressant drugtreatment in depressive disorders: a systematic review. Lancet. 2003; 361: 653-661.
51. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram fordepression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006; 163: 28-40.
52. Rush AJ, Fava M, Wisniewski SR, Lavori PW, Trivedi MH, Sackeim HA et al. Sequenced treatment alternatives to relievedepression (STAR*D): rationale and design. Control Clin Trials. 2004; 25: 119-142.
53. Yonkers KA, Samson J. Mood disorders measures. In: Rush AJ, Pincus HA, First MB, et al., eds. Handbook of psychiatricmeasures. 1st ed. Washington DC: American Psychiatric Association; 2000:515-548.
54. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.55. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979; 134: 382-
389.56. Rush AJ, Giles DE, Schlesser MA, Fulton CL, Weissenburger J, Burns CT. The Inventory for Depression Symptomatology
(IDS): preliminary findings. Psychiatry Res. 1986; 18: 65-87.57. Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN et al. The 16-Item Quick Inventory of Depressive
Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients withchronic major depression. Biol Psychiatry. 2003; 54: 573-583.
58. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch GenPsychiatry. 1961; 4:561 571: 561.
59. Bech P, Gram LF, Dein E, Jacobsen O, Vitger J, Bolwig TG. Quantitative rating of depressive states. Acta Psychiatr Scand.1975; 51: 161-170.
60. Jonghe F de. Leidraad voor het scoren van de Hamilton Depression Rating Scale: HDRS leidraad. Amsterdam: BeneckeConsultants; 1994.
61. Faries D, Herrera J, Rayamajhi J, DeBrota D, Demitrack M, Potter WZ. The responsiveness of the Hamilton DepressionRating Scale. J Psychiatr Res. 2000; 34: 3-10.
62. Gibbons RD, Clark DC, Kupfer DJ. Exactly what does the Hamilton Depression Rating Scale measure? J Psychiatr Res. 1993;27: 259-273.
63. Moeller H-J, Mueller H, Volz HP. How to assess the onset of antidepressant effect: Comparison of global ratings andfindings based on depression scales. Pharmacopsychiatry. 1996; 29: 57-62.
64. Santor DA, Coyne JC. Examining symptom expression as a function of symptom severity: Item performance on theHamilton Rating Scale for Depression. Psychol Assess. 2001; 13: 127-139.
65. Vittengl JR, Clark LA, Kraft D, Jarrett RB. Multiple measures, methods, and moments: a factor-analytic investigation ofchange in depressive symptoms during acute-phase cognitive therapy for depression. Psychol Med. 2005; 35: 693-704.
66. Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW et al. Conceptualization and rationale for consensusdefinitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48: 851-855.
67. Crismon ML, Trivedi M, Pigott TA, Rush AJ, Hirschfeld RMA, Kahn DA et al. The Texas medication algorithm project:Report of the Texas consensus conference panel on medication treatment of major depressive disorder. J Clin Psychiatry.1999; 60: 142-156.
68. Trivedi MH. Using treatment algorithms to bring patients to remission. J Clin Psychiatry. 2003; 64 Suppl 2: 8-13.69. Taylor MJ, Freemantle N, Geddes JR, Bhagwagar Z. Early onset of selective serotonin reuptake inhibitor antidepressant
action: systematic review and meta-analysis. Arch Gen Psychiatry. 2006; 63: 1217-1223.70. Quitkin FM, Petkova E, McGrath PJ, Taylor B, Beasley C, Stewart J et al. When should a trial of fluoxetine for major
depression be declared failed? Am J Psychiatry. 2003; 160: 734-740.71. Katz MM, Koslow SH, Maas JW, Frazer A, Bowden CL, Casper R et al. The timing, specificity and clinical prediction of
tricyclic drug effects in depression. Psychol Med. 1987; 17: 297-309.72. Quitkin FM, McGrath PJ, Stewart JW, Ocepek-Welikson K, Taylor BP, Nunes E et al. Chronological milestones to guide
drug change. When should clinicians switch antidepressants? Arch Gen Psychiatry. 1996; 53: 785-792.73. Kennedy N, McDonough M. Pharmacological management of treatment resistant depression: A clinical review.
IrJPsycholMed. 2003; 20: 18-23.74. Boer NAde. Verpleegkundige interventies bij depressie. 1 ed. Amsterdam: Programma Stemmingstoornissen AMC/De
Meren; 2003.75. Friedman MA, Detweiler-Bedell JB, Leventhal HE, Horne R, Keitner GI, Miller IW. Combined psychotherapy and
pharmacotherapy for the treatment of major depressive disorder. Clin Psychol Sci Prac. 2004; 11: 47-68.

Chap
ter
Chapter 1
1
28
76. Pampallona S, Bollini P, Tibaldi G, Kupelnick B, Munizza C. Combined pharmacotherapy and psychological treatment fordepression: a systematic review. Arch Gen Psychiatry. 2004; 61: 714-719.
77. Siegel GJ, Agranoff BW, Albers RW, Fisher SK, Uhler MD. Basic neurochemistry, molecular, cellular, and medical aspects. 6ed. Philadelphia: Lippincott, Williams & Wilkins; 1999.
78. Purves D, Augustine G, Fitzpatrick D, Katz L, LaMantia A. Neuroscience. 2 ed. Sunderland: Sinauer Associates, Inc.; 2001.79. Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008; 358: 55-68.80. Andreasen NC. Brave new brain: conquering mental illness in the era of the genome [Schitterend nieuw brein: psychiatrie in
het tijdperk van het genoom]. 1 ed. Amsterdam: Nieuwezijds B.V.; 2002.81. Marieb E. Human anatomy & Physiology. 5 ed. Redwood City: The Benjamin/ Cummings Publishing Company, Inc.; 1999.82. Gaspar P, Cases O, Maroteaux L. The developmental role of serotonin: news from mouse molecular genetics. Nat Rev
Neurosci. 2003; 4: 1002-1012.83. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;
33: 73-83.84. Young SN, Smith SE, Pihl RO, Ervin FR. Tryptophan depletion causes a rapid lowering of mood in normal males.
Psychopharmacology (Berl). 1985; 87: 173-177.85. Moja EA, Lucini V, Benedetti F, Lucca A. Decrease in plasma phenylalanine and tyrosine after phenylalanine-tyrosine free
amino acid solutions in man. Life Sci. 1996; 58: 2389-2395.86. Shopsin B, Gershon S, Goldstein M, Friedman E, Wilk S. Use of synthesis inhibitors in defining a role for biogenic amines
during imipramine treatment in depressed patients. Psychopharmacol Commun. 1975; 1: 239-249.87. Delgado PL, Miller HL, Salomon RM, Licinio J, Heninger GR, Gelenberg AJ et al. Monoamines and the mechanism of
antidepressant action: effects of catecholamine depletion on mood of patients treated with antidepressants.Psychopharmacol Bull. 1993; 29: 389-396.
88. Bell C, Abrams J, Nutt D. Tryptophan depletion and its implications for psychiatry. Br J Psychiatry. 2001; 178: 399-405.89. Booij L, Van der Does AJ, Riedel WJ. Monoamine depletion in psychiatric and healthy populations: review. Mol Psychiatry.
2003; 8: 951-973.90. Van der Does AJ. The effects of tryptophan depletion on mood and psychiatric symptoms. J Affect Disord. 2001; 64: 107-
119.91. Moore P, Landolt HP, Seifritz E, Clark C, Bhatti T, Kelsoe J et al. Clinical and physiological consequences of rapid
tryptophan depletion. Neuropsychopharmacology. 2000; 23: 601-622.92. Booij L, Van der DW, Benkelfat C, Bremner JD, Cowen PJ, Fava M et al. Predictors of mood response to acute tryptophan
depletion. A reanalysis. Neuropsychopharmacology. 2002; 27: 852-861.93. Van der Does AJ. The mood-lowering effect of tryptophan depletion: possible explanation for discrepant findings. Arch
Gen Psychiatry. 2001; 58: 200-202.94. Damasio AR. Descartes' error - emotion, reason and the human brain [De vergissing van Descartes - gevoel, verstand en het
menselijk brein]. 2nd ed. Amsterdam: Wereldbibliotheek; 1994.95. Owens MJ. Selectivity of antidepressants: from the monoamine hypothesis of depression to the SSRI revolution and
beyond. J Clin Psychiatry. 2004; 65 Suppl 4: 5-10.96. Burke WJ. Selective versus multi-transmitter antidepressants: are two mechanisms better than one? J Clin Psychiatry.
2004; 65 Suppl 4: 37-45.97. Fava M, Kendler KS. Major depressive disorder. Neuron. 2000; 28: 335-341.98. Delgado PL. How antidepressants help depression: mechanisms of action and clinical response. J Clin Psychiatry. 2004; 65
Suppl 4: 25-30.99. Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based
algorithms for diagnosis and optimised treatment. Br Med Bull. 2003; 65: 193-207.100. Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception I: The neural basis of normal emotion
perception. Biol Psychiatry. 2003; 54: 504-514.101. Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatric
disorders. Biol Psychiatry. 2003; 54: 515-528.102. Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA et al. Reciprocal limbic-cortical function and
negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999; 156: 675-682.103. Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 1997; 9: 471-
481.104. Malhi GS, Lagopoulos J. Making sense of neuroimaging in psychiatry. Acta Psychiatr Scand. 2008; 117: 100-117.105. Blier P, De Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci. 1994; 15: 220-
226.106. Nemeroff CB, Owens MJ. Neuropharmacology of paroxetine. Psychopharmacol Bull. 2003; 37 Suppl 1: 8-18.107. Bel N, Artigas F. Chronic treatment with fluvoxamine increases extracellular serotonin in frontal cortex but not in raphe
nuclei. Synapse. 1993; 15: 243-245.108. Davidson C, Stamford JA. Effect of chronic paroxetine treatment on 5-HT1B and 5-HT1D autoreceptors in rat dorsal raphe
nucleus. Neurochem Int. 2000; 36: 91-96.109. Vaswani M, Linda FK, Ramesh S. Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive
review. Prog Neuropsychopharmacol Biol Psychiatry. 2003; 27: 85-102.110. Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG et al. Effects of chronic antidepressant
treatments on serotonin transporter function, density, and mRNA level. J Neurosci. 1999; 19: 10494-10501.

ChapterGeneral introduction and outline
1
29
111. Benmansour S, Owens WA, Cecchi M, Morilak DA, Frazer A. Serotonin clearance in vivo is altered to a greater extent byantidepressant-induced downregulation of the serotonin transporter than by acute blockade of this transporter.JNeurosci. 2002; 22: 6766-6772.
112. Pineyro G, Blier P, Dennis T, De Montigny C. Desensitization of the neuronal 5-HT carrier following its long-term blockade.J Neurosci. 1994; 14: 3036-3047.
113. Maj J, Bijak M, Dziedzicka-Wasylewska M, Rogoz R, Rogoz Z, Skuza G et al. The effects of paroxetine given repeatedly onthe 5-HT receptor subpopulations in the rat brain. Psychopharmacology (Berl). 1996; 127: 73-82.
114. Whale R, Clifford EM, Bhagwagar Z, Cowen PJ. Decreased sensitivity of 5-HT1D receptors in melancholic depression. Br JPsychiatry. 2001; 178: 454-457.
115. Kennett GA, Lightowler S, de B, V, Stevens NC, Wood MD, Tulloch IF et al. Effect of chronic administration of selective 5-hydroxytryptamine and noradrenaline uptake inhibitors on a putative index of 5-HT2C/2B receptor function.Neuropharmacology. 1994; 33: 1581-1588.
116. Meyer JH, Kapur S, Eisfeld B, Brown GM, Houle S, DaSilva J et al. The effect of paroxetine on 5-HT(2A) receptors indepression: an [18F]setoperone PET imaging study. Am J Psychiatry. 2001; 158: 78-85.
117. Djavadian RL. Serotonin and neurogenesis in the hippocampal dentate gyrus of adult mammals. Acta Neurobiol Exp(Wars). 2004; 64: 189-200.
118. Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006; 59: 1144-1150.119. Angelucci F, Brene S, Mathe AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry.
2005; 10: 345-352.120. Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S et al. Regional metabolic effects of fluoxetine in
major depression: serial changes and relationship to clinical response. Biol Psychiatry. 2000; 48: 830-843.121. Kennedy SH, Evans KR, Kruger S, Mayberg HS, Meyer JH, McCann S et al. Changes in regional brain glucose metabolism
measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158: 899-905.
122. Mayberg HS, Silva JA, Brannan SK, Tekell JL, Mahurin RK, McGinnis S et al. The functional neuroanatomy of the placeboeffect. Am J Psychiatry. 2002; 159: 728-737.
123. Fu CH, Williams SC, Cleare AJ, Brammer MJ, Walsh ND, Kim J et al. Attenuation of the neural response to sad faces inmajor depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imagingstudy. Arch Gen Psychiatry. 2004; 61: 877-889.
124. Fu CH, Williams SC, Brammer MJ, Suckling J, Kim J, Cleare AJ et al. Neural responses to happy facial expressions in majordepression following antidepressant treatment. Am J Psychiatry. 2007; 164: 599-607.
125. Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA. Increased amygdala response to maskedemotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 2001; 50:651-658.
126. Davidson RJ, Irwin W, Anderle MJ, Kalin NH. The neural substrates of affective processing in depressed patients treatedwith venlafaxine. Am J Psychiatry. 2003; 160: 64-75.
127. Schaefer HS, Putnam KM, Benca RM, Davidson RJ. Event-related functional magnetic resonance imaging measures ofneural activity to positive social stimuli in pre- and post-treatment depression. Biol Psychiatry. 2006; 60: 974-986.
128. Robertson B, Wang L, Diaz MT, Aiello M, Gersing K, Beyer J et al. Effect of bupropion extended release on negativeemotion processing in major depressive disorder: a pilot functional magnetic resonance imaging study. J Clin Psychiatry.2007; 68: 261-267.
129. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD et al. Serotonin transporter promoter gain-of-functiongenotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. 2006; 78: 815-826.
130. Lesch KP, Gutknecht L. Pharmacogenetics of the serotonin transporter. Prog Neuropsychopharmacol Biol Psychiatry.2005; 29: 1062-1073.
131. Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry. 2007; 12:247-257.
132. Hu XZ, Rush AJ, Charney D, Wilson AF, Sorant AJ, Papanicolaou GJ et al. Association between a functional serotonintransporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Arch GenPsychiatry. 2007; 64: 783-792.
133. Kraft JB, Peters EJ, Slager SL, Jenkins GD, Reinalda MS, McGrath PJ et al. Analysis of association between the serotonintransporter and antidepressant response in a large clinical sample. Biol Psychiatry. 2007; 61: 734-742.
134. Murphy DL, Lerner A, Rudnick G, Lesch KP. Serotonin transporter: gene, genetic disorders, and pharmacogenetics. MolInterv. 2004; 4: 109-123.
135. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H et al. Influence of life stress on depression: moderation bya polymorphism in the 5-HTT gene. Science. 2003; 301: 386-389.
136. Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D et al. Serotonin transporter genetic variation and theresponse of the human amygdala. Science. 2002; 297: 400-403.
137. Hariri AR, Drabant EM, Munoz KE, Kolachana BS, Mattay VS, Egan MF et al. A susceptibility gene for affective disordersand the response of the human amygdala. Arch Gen Psychiatry. 2005; 62: 146-152.
138. Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS et al. 5-HTTLPR polymorphismimpacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8: 828-834.
139. Jonghe F de, Kool S, Aalst G van, Dekker J, Peen J. Combining psychotherapy and antidepressants in the treatment ofdepression. J Affect Disord. 2001; 64: 217-229.

Chap
ter
Chapter 1
1
30
140. Dekker J, Molenaar PJ, Kool S, Aalst G van, Peen J, Jonghe F de. Dose-effect relations in time-limited combined psycho-pharmacological treatment for depression. Psychol Med. 2005; 35: 47-58.
141. Maier W, Philipp M. Improving the assessment of severity of depressive states: a reduction of the Hamilton DepressionScale. Pharmacopsychiatry. 1985; 18: 114-115.
142. Prien RF, Carpenter LL, Kupfer DJ. The definition and operational criteria for treatment outcome of major depressivedisorder. A review of the current research literature. Arch Gen Psychiatry. 1991; 48: 796-800.
143. Ballenger JC. Clinical guidelines for establishing remission in patients with depression and anxiety. J Clin Psychiatry. 1999;60 Suppl 22: 29-34.
144. The Cochrane Collaboration. RevMan 4.2 User Guide, version 4.2 for Windows. 4.2 ed. Oxford, UK: The CochraneCollaboration; 2002.
145. Staley JK, Krishnan-Sarin S, Zoghbi S, Tamagnan G, Fujita M, Seibyl JP et al. Sex differences in [123I]beta-CIT SPECTmeasures of dopamine and serotonin transporter availability in healthy smokers and nonsmokers. Synapse. 2001; 41: 275-284.
146. Pirker W, Asenbaum S, Hauk M, Kandlhofer S, Tauscher J, Willeit M et al. Imaging serotonin and dopamine transporterswith 123I-beta-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000; 41: 36-44.
147. van Dyck CH, Malison RT, Seibyl JP, Laruelle M, Klumpp H, Zoghbi SS et al. Age-related decline in central serotonintransporter availability with [123I]beta-CIT SPECT. Neurobiol Aging. 2000; 21: 497-501.
148. Buchert R, Schulze O, Wilke F, Berding G, Thomasius R, Petersen K et al. Is correction for age necessary in SPECT or PET ofthe central serotonin transporter in young, healthy adults? J Nucl Med. 2006; 47: 38-42.

CHA
PTER
DESIGN AND METHODS OF THE DELPHI-STUDY

32

ChapterDesign of the DELPHI study
2
33
The DELPHI-study
DELPHI is an acronym for Dose-Escalation Legitimate? Pharmacology and Imaging studies indepression. The DELPHI-study is the backbone of the studies described in Part IV of this thesis.Because the DELPHI study in fact comprised three research projects, with slightly differentpatient populations and different numbers of participants, this chapter will describe the design ofthe DELPHI-study and indicate how the nested sub-studies relate to the major DELPHI-study.Furthermore the patient disposition over these projects will be given.
Objectives, rationale and research questionsThe major aims of the DELPHI-study were toinvestigate the clinical efficacy of and theneurobiological mechanism behind dose-escalation. In order to determine the neurobiologicaleffects of dose-escalation, we intended to study different biological measures during treatment:paroxetine serum concentrations (PSC), serotonin transporter (SERT) occupancy, functionalactivations in the cortico-limbic-network, awakening cortisol changes and changes in ω-3/ω-6 polyunsaturated fatty acids (PUFAs). Furthermore, we planned to study the effects of geneticpolymorphisms on outcomes (i.e. the serotonin transporter gene promoter region (5-HTTLPR)).Results related to some of these neurobiological measures are described in this thesis.
As a study drug, we choseparoxetine for three reasons. First, paroxetine is the selectiveserotonin reuptake inhibitor (SSRI) which is prescribed most frequently in the Netherlands.1
Second, paroxetine is a potent inhibitor of the cytochrome P450 2D6 sub-enzyme, which also isthe enzyme that is responsible for its metabolism. Therefore paroxetine inhibits its ownmetabolism, which causes an exponential rise in blood serum concentrations after dose-escalation.2 Third, when we initiated this study, Gilmor et al. reported noradrenergic reuptakeinhibition by paroxetine, suggesting that paroxetine, like venlafaxine and duloxetine, in fact was a‘dual action’ antidepressant, especially at higher doses.3
Our main research-questions were:1 Is a 6-week true dose-escalation of paroxetine (up to 30-50 mg/day) in patients non-responsive
to a 6-week trial with standard dose (20 mg/day) more effective than placebo dose-escalation?2 Does a 6 week true dose-escalation of paroxetine increase SERT occupancy more than a
placebo dose-escalation?3 What is the relation between SERT occupancy and clinical response to paroxetine (either at a
standard dose or after dose-escalation)? Is this relation modified by other neurobiologicalparameters (i.e. genetic polymorphisms)?
4 Which changes in the functional activation of the cortico-limbic brain regions correlate withclinical response to paroxetine? Is there a common and/or differential effect of paroxetineexposure and/or clinical response?
5 Does a 6 week true dose-escalation of paroxetine generate additional changes (e.g. innoradrenergic reuptake inhibition, cortisol awakening responses, or ω-3/ω-6 PUFAs) comparedto a placebo dose-escalation, and are these changes related to clinical response?
Only questions 1-4 will be discussed in this thesis, and the methods used for these questions willbriefly be discussed.However, for specific technical we refer to the method-sections in therelevant chapters.
Design and interventionsThe DELPHI-study was set up as a randomized clinical trial (ISRCTN register nr. ISRCTN44111488;http://www.trialregister.nl/trialreg/admin/rctview.asp?TC=193). The study was approved by theAcademic Medical Center (AMC) medical ethical committee (03/120, 03/287 and addendum to 03/120), and all participants provided written informed consent.

Chap
ter
Chapter 2
2
34
Between October 2003 and February 2007 patients were recruited from primary care, ourAMC Program for Mood Disorders, and public psychiatric settings. Patients were treated by theirreferring physician or were referred to our outpatient department. In DELPHI two nested sub-studies were embedded, the DELPHI-SPECT and the DELPHI-fMRI (Figure 2.1). These sub-studieshad slightly different in- and exclusion criteria (see below).
All eligible patients were treated open-label with paroxetine 20 mg/day for 6 weeks. Whensevere adverse effects occurred, dosages were reduced to 10 mg/day and again increased to 20mg/day after one week. After 6 weeks of treatment, patients who responded (defined as ≥50%reduction of the pretreatment Hamilton depression rating scale (HDRS)) continued paroxetine 20mg/day. Treatment non-responders were randomized to a true dose-escalation or a placebo dose-escalation, added to paroxetine 20 mg/day in a double blind design. Dose-escalation was providedin blue capsules containing 10 mg paroxetine or placebo. Randomization was stratified fortreatment setting (SPECT/fMRI-group, outpatient department AMC, primary care, and publicpsychiatry), gender and age. Within strata, we applied a minimization method to achieve abalanced distribution. We concealed allocation by using an independently operated computerprogram.
Dose-escalation consisted of incremental steps of one capsule every 5 days towards amaximum of 50 mg/day (20 mg + 3 capsules). Patients were allowed to increase at a slower pace(e.g. by 7 days) or stop further escalation (e.g. 20 mg + 2 capsules) according to adverse effects.4
No dosage adjustments were allowed during the last 3 weeks of the study. We checkedadherence by pill-counts and anamnesis.5
Figure 2.1. Design of the DELPHI-study.
DELPHI-SPECT and DELPHI-fMRI represent sub-studies nested within the total study. For these 2 sub-studies drug-freepatients were recruited and treated in the Academic Medical Center. For color figure see page 280.
������������� ����
������ �
������������������
�� ����� �� ����� !" �����
����������������
� �#�$����% #& #� ' &��� �'��(")
�������������
����� ����� ��
����� *���(�+�,���(� ���"������"����(�
�� -.�� �� -.�� !" -.��

ChapterDesign of the DELPHI study
2
35
Patients Figure 2.2 summarizes patient disposition in the DELPHI-study and the DELPHI-SPECT/fMRIneuroimaging sub-study.
General in- and exclusion criteriaEligible patients met the following inclusion criteria: Age between 18 and 70 years,majordepressive disorder (MDD) determined by the structured clinical interview for DSM-IV (SCID),6
and a HDRS (17 items)7 score above 18. All participants were either drug-free and/or hadundergone no more than one antidepressant treatment (other than paroxetine) at an effectivedose for ≥ 6 weeks for the present MDD-episode. By the latter criterion, we avoided treatmentresistance as potential bias for inefficacy of dose-escalation. Exclusion criteria, apart frompregnancy (or wish to become pregnant), were bipolar disorder, psychotic features, neurologicalcognitive impairments (i.e. dementia), primary anxiety and/or substance abuse disorders andacute, severe suicidal ideation. Contrary, we allowed secondary co-morbid anxiety and/orsubstance abuse to increase applicability of our findings. In total, 278 patients were referred, ofwhom 107 patients started treatment with paroxetine 20 mg/day. Twenty patients withdrew fromthe study before week 6, and 27 patients were responder by then. Sixty non-responders wererandomized.
Figure 2.2. Patient enrolment and disposition over sub-studies.
The patients in DELPHI-SPECT and DELPHI-fMRI sub-studies are combined (left). For disposition of patients randomized seechapter 9.
�������� ��������� �������
��������� �������� �� �������� �!������ ���"�#� �$���$%�&��'��� �� �� (� %�#�( �������
'��������� �) �#� �#�� �������
*��#���� #�"�&�� � $�# ���$� � �������
�!������ � %�# #��&��& ����+��������� *$���,%�&� �������
�������� �#�"�&�� -,�./ ������
*$���$%�&� -,�./�"01'����� �� *$���$%�&� 2���-,�./����� ��
3#�$�� �$���$%�&�
����#&� �""�� & ���� �
3#�$�� �$���$%�&�
���""����) ������
����#&� �""�� & ������
� %�# #��&��& ���� �
(� %�#�(� �" �# �� �#�4� ������
3#�$�� #����4�5� ��� $%�&�
���""����) ������
����#&� �""�� & �����
3#�$�� #����4�5� ��� $%�&�
���""����) ������
����#&� �""�� & ������
#�"�&�� �" �# #����4�5 � ������
67 ���.�4$�� �#�� #����4�5�� � �������
� ����)5���� �������� �� �� �������
� �!������ "�# ����)&�& �������
����������
���� ��
����� ��������
��������
�� �����
67 ���.�4$�� �#�� #����4�5�� � �������
� ����)5���� �������� �� ���������
� ����)5���� -,�./�"01' �������
�!������ "�# -,�./�"01'� ����)&�& ���� ��
67 ��.�4$�� �#�-,�./�"01'��������
� 67 ��1�&$����# -,�./�"01'��������
� 67 ��21 -,�./�"01'� #����4�5�� �������
67 ��.�4$�� �#�2���-,�./�"01'��������
� 67 ��1�&$����# �������
� 67 ��21 #����4�5�� �������
�������� ����� �������
�������������

Chap
ter
Chapter 2
2
36
DELPHI-SPECT and fMRIPatients who were drug-free (for >4 weeks and ≥5 half-lives of a previous antidepressant) wereasked to additionally participate in the neuroimaging sub-studies. Patients were initially asked toparticipate in DELPHI-SPECT only, but from august 2005 onwards, when DELPHI-fMRI wasapproved, we asked them for both DELPHI-SPECT and DELPHI fMRI. Participation in only one ofthe neuroimaging studies was also possible. We limited age to 25-55 years to reduce variability inSERT-measurements by age.8 For participation in the fMRIs, patients had to be free of metalobjects in their body. None of the included patients reported past or present use of 3,4-methylenedioxymethamphetamine. We treated DELPHI-SPECT and DELPHI-fMRI patients attheoutpatient department of the Program for Mood Disorders. We supplied medication inpillboxes.
We included 51 patients in the DELPHI-SPECT study, of whom 33 non-responders wererandomized. For DELPHI-fMRI we included 22 patients, of whom 16 were randomized. Twentypatients participated both in the DELPHI-SPECT and DELPHI-fMRI. Unfortunately not all(repeated) scans were analyzable adequately, the reason why in chapters 8-10 different numbersof patients are described.
Healthy controlsWe recruited 53 healthy controls as reference for the study-entry scans. We individually matchedeach patient in DELPHI-SPECT and DELPHI-fMRI by gender and age (±2.5 years). Healthy controlswere in good physical health, and had never used psychotropic medication. Exclusion criteriawere current or lifetime psychiatric disorder(s) according to the SCID (including abuse oraddiction disorders), a Beck Depression Inventory (BDI) score >9, alcohol use >4 units per day (lastmonth) or a 1st-degree relative with psychiatric disorder(s). We allowed healthy controls to haveincidentally used illicit drugs, unless criteria for a DSM-IV disorder was met, but we prohibited illicitdrug use the month prior to scanning. Twenty healthy controls also participated both in theDELPHI-SPECT and DELPHI-fMRI.
Measurements Time points and questionnairesWe administered the HDRS17,7 Inventory for depressive symptoms (IDS-SR30),9 the occurrence ofadverse effects and health-related quality of life (MOS-SF36)10 at study-entry, randomization (T0),and 6 weeks after randomization (T1). Adverse effects and depressive symptoms were alsomonitored in the weeks 1, 2 and 4 after the initial start of treatment, and after randomization,using the Maier and Bech subscales11 and IDS-SR30
9 (Figure 2.1). Three trained investigatorsadministered clinician-rated questionnaires. Agreement between raters was good (intraclasscorrelation coefficient = 0.98). Raters and patients were blinded for treatment.
Definition of primary and secondary outcomesPrimary clinical outcomes were HDRS17-scores and the proportion of patients achieving response(≥50% decrease in HDRS17) or remission (HDRS17 ≤7). Secondary outcomes were total and specific(adverse effects / inefficacy) dropout rates, the Maier and Bech subscales and IDS-SR30-scores,the occurrence of adverse effects and health-related quality of life.
Neurobiological measurementsParoxetine serum concentrations: See chapters 8 and 9 for details.SERT-gene promoter polymorphism: See chapters 8 and 11 for details.Saliva and additional blood specimens
At study-entry, at T0, and T1, we collected two saliva specimens at awakening and 30 minutesthereafter, to determine salivary cortisol, dehydroepiandrosterone-sulphate, and α-amylase.12-15
Furthermore, we collected blood-specimens to quantify [3H]Noradrenaline and [3H]Serotonineuptake in ex-vivo models.3;16;17 Finally we collected blood-specimens to determine levels of plateletand plasma ω-3/ω-6 PUFAs.18 These neurobiological measurements remain to be analysed indifferent papers not in this thesis.

ChapterDesign of the DELPHI study
2
37
Neuroimaging Measurement of SERT occupancy We performed SPECT imaging at study-entry, T0 and T1 (Figure 2.1), between 2 to 10 pm accordingto previously described procedures.19 We made all scans 230 ±18 (SD) minutes after intravenousinjection of approximately 100 MBq [123I]β-CIT, when the radioligand is at equilibrium for SERTbinding in brain areas expressing high densities of SERTs.20 We performed SPECT imaging using a12-detector single slice brain-dedicated scanner (Neurofocus 810, Strichmann Medical Equipment;Cleveland, OH). After attenuation correction and reconstruction in 3D mode (http://www.neurophysics.com), we defined regions of interest (RoIs) for midbrain, diencephalon(regions rich of SERT) and cerebellum (as a reference) by using validated templates (Figure 2.3).19
For further details of SPECT-procedures see chapters 7-10.
Magnetic resonance imaging of functional activation of thecortico-limbic-network We acquired fMRI scans at study-entry, T0 and T1 (Figure 2.1), between 2 to 10 pm. FMRI-sessionslasted 50-60 minutes, each including a cognitive task (Tower of London),21 a structural scan and afacial expression task, reported in this thesis.22;23 We used a 3Tesla Intera MRI scanner (Philips,Eindhoven, NL), with a 6 channel head-coil for radiofrequency reception. Two magnet compatibleresponse boxes were used to record subject’s performance and reaction times. For further detailsof fMRI-settings and the parameters of the faces paradigm: see chapter 10.
Power and interim analysisFor the randomization-phase of the total DELPHI-study, we performed a-priori power-calculationsfor two co-primary endpoints. We planned an interim analysis after SPECT data had been collectedon at least 30 patients. Stopping criteriawere predetermined using the O'Brien and Fleming24
approach, and were p<0.0026 in case of superiority and p>0.50 for futility. See chapter 9 forfurther details.
Figure 2.3. Regions of Interest (RoI) for Midbrain, Cerebellum and Diencephalon.
Example of SPECT images after 3D reconstruction. Templates with fixed RoIs are shown. For color figure see page 281.A. Midbrain (circle) and cerebellum. B. Striatum (for demarcation midbrain-diencephalon) and diencephalon (circle). RoIswere positioned by hand and situated based on anatomy and maximum concentration of activity/ml in the RoI.

Chap
ter
Chapter 2
2
38
References
1. Stichting Farmaceutische Kengetallen. Data en feiten 2007.2007. Den Haag, Stichting Farmaceutische Kengetallen. 2. Anonymous. Paxil® Prescribing information and medication guide PXL:47PI (available at http://us.gsk.com/products/
assets/us_paxil.pdf). 1-44. 2008. Research Triangle Park, North Carolina, GlaxoSmithKline. 3. Gilmor ML, Owens MJ, Nemeroff CB. Inhibition of norepinephrine uptake in patients with major depression treated with
paroxetine. Am J Psychiatry. 2002; 159: 1702-1710.4. Baker CB, Woods SW. Is there a SSRI dose response in treating major depression? The case for re-analysis of current data
and for enhancing future study design. Depress Anxiety. 2003; 17: 10-18.5. Saunders K, Simon G, Bush T, Grothaus L. Assessing the feasibility of using computerized pharmacy refill data to monitor
antidepressant treatment on a population basis: a comparison of automated and self-report data. J Clin Epidemiol. 1998;51: 883-890.
6. First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders Patient Edition(SCID-I/P version 2.0). Translated in Dutch by. Groenestijn MAC, Akkerhuis GW, Kupka RW, Schneider N, and NolenWA.1999. Lisse, the Netherlands, Swets & Zeitlinger B.V.
7. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.8. van Dyck CH, Malison RT, Seibyl JP, Laruelle M, Klumpp H, Zoghbi SS et al. Age-related decline in central serotonin
transporter availability with [123I]β-CIT SPECT. Neurobiol Aging. 2000; 21: 497-501.9. Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The Inventory of Depressive Symptomatology (IDS):
psychometric properties. Psychol Med. 1996; 26: 477-486.10. Aaronson NK, Muller M, Cohen PD, Essink-Bot ML, Fekkes M, Sanderman R et al. Translation, validation, and norming of
the Dutch language version of the SF-36 Health Survey in community and chronic disease populations. J Clin Epidemiol.1998; 51: 1055-1068.
11. Ruhe HG, Dekker JJ, Peen J, Holman R, Jonghe F de. Clinical use of the Hamilton Depression Rating Scale: is increasedefficiency possible? A post hoc comparison of Hamilton Depression Rating Scale, Maier and Bech subscales, ClinicalGlobal Impression, and Symptom Checklist-90 scores.Compr Psychiatry. 2005; 46: 417-427.
12. Nater UM, Rohleder N, Schlotz W, Ehlert U, Kirschbaum C. Determinants of the diurnal course of salivary alpha-amylase.Psychoneuroendocrinology. 2007; 32: 392-401.
13. Pruessner JC, Wolf OT, Hellhammer DH, Buske-Kirschbaum A, von Auer K, Jobst S et al. Free cortisol levels afterawakening: a reliable biological marker for the assessment of adrenocortical activity. Life Sci. 1997; 61: 2539-2549.
14. Rohleder N, Nater UM, Wolf JM, Ehlert U, Kirschbaum C. Psychosocial stress-induced activation of salivary alpha-amylase:an indicator of sympathetic activity? Ann N Y Acad Sci. 2004; 1032: 258-263.
15. Schmidt-Reinwald A, Pruessner JC, Hellhammer DH, Federenko I, Rohleder N, Schurmeyer TH et al. The cortisol responseto awakening in relation to different challenge tests and a 12-hour cortisol rhythm. Life Sci. 1999; 64: 1653-1660.
16. Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile ofantidepressants and their metabolites. J Pharmacol Exp Ther. 1997; 283: 1305-1322.
17. Owens MJ, Krulewicz S, Simon JS, Sheehan DV, Thase ME, Carpenter DJ et al. Estimates of Serotonin and NorepinephrineTransporter Inhibition in Depressed Patients Treated with Paroxetine or Venlafaxine. Neuropsychopharmacology. 2008.
18. Dacremont G, Cocquyt G, Vincent G. Measurement of very long-chain fatty acids, phytanic and pristanic acid in plasmaand cultured fibroblasts by gas chromatography. J Inherit Metab Dis. 1995; 18 Suppl 1: 76-83.
19. Win MM de, Habraken JB, Reneman L, Brink W van den, Heeten GJ den, Booij J. Validation of [123I]β-CIT SPECT to assessserotonin transporters in vivo in humans: a double-blind, placebo-controlled, crossover study with the selective serotoninreuptake inhibitor citalopram. Neuropsychopharmacology. 2005; 30: 996-1005.
20. Pirker W, Asenbaum S, Hauk M, Kandlhofer S, Tauscher J, Willeit M et al. Imaging serotonin and dopamine transporterswith [123I]β-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000; 41: 36-44.
21. Heuvel OAvan den, Veltman DJ, Groenewegen HJ, Cath DC, Balkom AJ van, Hartskamp Jvan et al. Frontal-striataldysfunction during planning in obsessive-compulsive disorder. Arch Gen Psychiatry. 2005; 62: 301-309.
22. Hariri AR, Bookheimer SY, Mazziotta JC. Modulating emotional responses: effects of a neocortical network on the limbicsystem. Neuroreport. 2000; 11: 43-48.
23. Wolfensberger SPA, Veltman DJ, Hoogendijk WJG, Boomsma DI, Geus EJC de. Amygdala responses to emotional faces intwins discordant or concordant for the risk for anxiety and depression. Neuroimage. 2008; E-pub: doi: 10.1016/j.neuroimage.2008.01.053.
24. O'Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979; 35: 549-556.

ChapterDesign of the DELPHI study
2
39


STUDIES TO GUIDE CLINICAL TREATMENT OF MAJOR DEPRESSIVE DISORDER
PART
Studies to guide clinical treatment of Major Depressive Disorder

42

CHA
PTER
CLINICAL USE OF THE HAMILTON DEPRESSION
RATING SCALE: IS INCREASED EFFICIENCY
POSSIBLE? A POST HOC COMPARISON OF HDRS, MAIER AND
BECH SUBSCALES, CGI AND SCL-90 SCORES.
Comprehensive Psychiatry 2005; 46: 417-427
Henricus G. Ruhé1,2
Jack J. Dekker1,3
Jaap Peen1
Rebecca Holman4
Frans de Jonghe1,2
1 Mentrum, Amsterdam, The Netherlands2 Department of Psychiatry, Academic Medical Center, University of Amsterdam, Amsterdam, TheNetherlands3 Department of Clinical Psychology, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands4 Department of Clinical Epidemiology and Biostatistics, Academic Medical Center, University ofAmsterdam, Amsterdam, The Netherlands

Chap
ter
Chapter 3
3
44
Abstract
BackgroundThe 17-item Hamilton Depression Scale (HDRS) is used as a semi gold standard in research. Intreatment guidelines, the HDRS-measurements serve to determine response, remission and guideclinical decision-making for non-responders. However, its use in clinical practice is limited, possiblybecause the HDRS is time-consuming. Additionally, the multidimensional HDRS is criticized for notmeasuring a unidimensional aspect as depression severity. The Maier and the Bech, two 6-itemseverity subscales extracted from the HDRS, are relatively unknown.
AimTo investigate whether the measurements obtained with Maier and Bech subscales arecomparable with the original HDRS-measurements.
MethodsData from two randomized controlled trials in 482 male and female patients, diagnosed with amajor depression (with or without dysthymia) according to DSM-III-R, of whom 219 participated inthe trials, were reanalyzed. A standardized stepwise psychopharmacological treatment wascompared with a combination of pharmacotherapy with Short Psychodynamic SupportivePsychotherapy in a psychiatric outpatient department. Outcome measures were internalconsistency and concurrent validity of HDRS, Maier, Bech, Clinical Global Impression (CGI) scalesand Symptom CheckList depression subscale. Effect sizes of HDRS, Maier and Bech were used tocompare measured treatment effects for the randomized subjects participating in the trials. ItemResponse Theory was used to obtain conversion tables for the HDRS, Maier, Bech and SCL-90depression subscale.
ResultsWe found moderate internal consistence (Cronbach α ≈ 0.6-0.7) and high correlations of the Maierand Bech subscales with overall HDRS-scores. Overall there were no clinically relevant differencesin effect sizes between Maier, Bech and HDRS, although some differences were statisticallysignificant. Receiver operating characteristic curves showed no difference between Maier andBech to define remission, but showed the CGI ratings to be unreliable. A cut-off ≤4 correspondedwith a HDRS ≤ 7 criterion in both subscales.
ConclusionsIn clinical practice, both Maier and Bech scales can be used as equivalents of the HDRS, but will bemore efficient.

ChapterClinical use of HDRS subscales
3
45
Introduction
Major depressive disorder is a severe, disabling illness, expected to be the world’s second health-problem in 2020.1 Depression is associated with high costs, regarding direct treatment andindirect costs of loss of productivity and quality of life.2 Several clinical guidelines were developedto guide the treatment of this disorder, both psychotherapy and pharmacotherapy (or incombination) appear effective.3-10
The use of self-report or clinician-rated symptom-scales is recommended to assess severityand response to treatment.8;11;12 Some experts claim clinician-rated symptom scales to have alarger validity and reliability than self-reporting scales, especially in patients with cognitiveimpairment, and more severe or psychotic depressions.11;13;14 Specific symptom-scales are morereliable than global rating scales.11;13;15 Especially, rating scales can be used to objectivelydetermine specific cut-off points for response and remission.12;16;17
In most clinical trials the Hamilton Depression Rating Scale (HDRS)18;19 – a clinician ratedsymptom-scale – is used as a standard to determine severity and response.5;8;11;15;20-23 Many versionsof the HDRS exist, with the number of items usually varying between 17 and 24,11;18;19;22 however upto 36 items have been described.23 Longer versions were especially developed to cover reverseneurovegetative (atypical) symptoms.23 The Clinical Global Impression (CGI)24 – a clinician-ratedglobal scale – is also frequently used.5;8;15;25 In clinical practice, although recommended, rating-scales are not used routinely. Explanations for this discrepancy could be ignorance of existingscales, a strong belief in one’s clinical judgment, an unsystematic approach of depression, but alsothe amount of time needed for rating-scales (e.g. 15-20 minutes for the HDRS11) and the necessityof training.20;26
The HDRS is criticized as being sensitive to somatic symptoms (e.g. somatic illness or side-effects of drugs),11;15;27;28 for not rating all 9 DSM-IV domains, its unequal weightings of differentsymptoms and for the multidimensionality of the HDRS total score.13;21;29-31 Multidimensionality isimportant to cover the maximum range of clinical features of major depressive disorder, but doesnot necessarily measures depression severity. Multidimensional scales can be misleading whenmeasurement of severity and treatment response is concerned,13;21;28 especially when themeasured depressive symptoms do not change proportionally with depression severity.Finally,some reports emphasize that the HDRS systematically favors (sedative) Tricyclic Antidepressants(TCAs) above Selective Serotonin Reuptake Inhibitors (SSRIs).27;32-35 Sleep and somatic items mayappear to be 'improved' by side-effects of TCAs, but worsened by side-effects (e.g. insomnia,gastrointestinal complaints, agitation) of SSRIs.
In order to overcome the problems of the multidimensional HDRS mentioned above, a moreunidimensional subscale from the HDRS covering core-symptoms of severity is desired. Also, froma clinical point of view, fewer items will be less time consuming for application by busy clinicians.However, for the purpose of reference, subscale scores must remain anchored to the originalHDRS. To identify shorter unidimensional subscales, Maier et al.28 used Rasch- and Mokken-analyses and Gibbons et al.29 used Factor-analysis. Bech and collegues developed another 6-itemsubscale. This scale initially emerged from an analysis with experienced psychiatrists as a validitycriterium,36 and was validated psychometrically thereafter using Rasch-analyses.37;38 This Bechsubscale was combined with four items of the Cronholm-Ottosson Depression Scale to form theBech-Rafaelsen Melancholia Scale.39 Santor and Coyne examined the score-performances ofindividual HDRS-items as a function of depression severity with a nonparametric Item ResponseTheory (IRT) approach, retaining 14 items.21 These 14 items included all 6 items of the Maiersubscale and all 8 items of the Gibbons subscale. However one item from the Bech subscale (13,somatic symptoms) was not included.
In a meta-analysis of individual patient-data, Faries et al. evaluated the responsiveness of totalHDRS and subscale scores in TCA and SSRI pharmacotherapy trials, finding a maximal sensitivityfor the Maier subscale.40 In a similar reanalysis, Entsuah et al. found larger effect sizes for theBech, Maier and Gibbons subscales compared to the HDRS in trials comparing SSRIs orvenlafaxine.41 O'Sullivan et al. found comparable sensitivity to detect changes for the six-item

Chap
ter
Chapter 3
3
46
Bech subscale compared to the 17-item HDRS.20 Hooper et al. found equal sensitivity to changeduring treatment for the 6-item Bech subscale compared to the HDRS 17 item version.26 Möllerand Bech et al. used the Bech subscale to reexamine treatment efficacy of SSRIs and mirtazapine(versus TCAs or placebo).32;42-44 The latter publications did not provide data for the Maier subscale.
In this paper we describe a secondary analysis of our trial data, in order to answer thefollowing questions:
1) Are the Maier, Bech and HDRS comparable in the measurement of depression severity andthe sensitivity to measure changes in severity? 2) Is this comparability stable across the full rangeof response to treatment (e.g. non-response, partial and full response), across differenttreatments and different baseline severity of depression? and 3) What are clinical cut-off pointsfor the subscales to determine remission compared to conventional definitions.12;16;17
We hypothesized that the differences between Maier, Bech and HDRS-scales would be smalland that there would be no apparent effect modification across neither treatments nor baselineseverity. In contrast, we hypothesized that for non-responders and partial responders the effectsizes would be smaller than for responders. This would additionally prove the hypothesis ofsensitivity to change.
Methods
Patient selectionIn the present analyses we use data from two published randomized controlled trials conductedbetween 1993 and 1998 which were published or accepted for publication.45;46 The first trial aimedat efficacy and effectiveness of pharmacotherapy versus the combination of pharmacotherapywith Short Psychodynamic Supportive Psychotherapy (SPSP)47-50 (16 sessions).45 The second trialinvestigated efficacy and effectiveness of a combination of pharmacotherapy with 8 versus 16sessions of SPSP.46 Pharmacotherapy in both trials consisted of three successive steps in case ofintolerance or inefficacy. Both trials started with fluoxetine (20 mg/ day), when this wasunsuccessful (CGI-I >2, only 'minimally improved' or worse) after 6 weeks amitriptyline (≥150 mg/day, dependent of plasma-levels) was initiated in trial 1 and nortriptyline (≥150 mg/day, dependentof plasma-levels) in trial 2. If again unsuccessful after 6 weeks, moclobemide (300-600 mg/day)was started in trial 1 and mirtazapine (30-45 mg/day) in trial 2.
Inclusion criteria for participation in the trials were age between 18 and 60 years, DSM-III-Rdefined Major Depression (with or without dysthymia) assessed in a structured clinical interview,a 17-item HDRS baseline score of at least 14 points and written informed consent. Patients wereexcluded in case of psycho-organic or psychotic or dissociative disorders, drug abuse, or when thepatient was considered to be too unreliable to participate in a clinical trial. Other axis 1 co-morbidity was not excluded. Further exclusion criteria were if there was a serious communicativeor practical problem (e.g. language barrier or the patient will soon leave the country), if there wasa contraindication for one of the antidepressants used, if the patient was adequately treated withantidepressants during the present depressive episode, if the patient used other psychotropicmedication, or if the patient was or planned to become pregnant. Additional exclusion criteriawere of the usual kind in drug research: “too ill” (e.g. antidepressants must be startedimmediately) and/or “too suicidal” (e.g. hospitalization is unavoidable) to participate in a clinicaltrial. The study was approved by the medical ethics committee. After complete description of thestudy to the subjects, written informed consent was obtained.
Of 3226 newly registered outpatients, 988 patients had a depressive disorder. By initialscreening 503 of these 988 patients were excluded by the above exclusion-criteria leaving 485subjects (including patients that later refused to participate or had a HDRS below 14; furtherreferred to as the diagnostic sample). To enter the trials, a second exclusion check was performedby a psychiatrist (excluding 73 patients), and 142 subjects with a HDRS-17 <14 were excluded,

ChapterClinical use of HDRS subscales
3
47
leaving 270 patients for randomization. After randomization 51 patients refused participation,leaving 219 patients who started the proposed therapy (further referred to as the per protocolsample).45;46
In this manuscript we used the diagnostic sample for most cross-sectional analyses, and therandomized patients in the per protocol sample for analyses of sensitivity of response-data. Fornon-completers, the last observation was carried forward (LOCF).
Outcome measuresPrimary outcome-measures were the 17-item HDRS,18;19 the Maier subscale of the HDRS(containing items 1,2,7-10),28 the Bech subscale of the HDRS (items 1,2,7,8,10 and 13),37 the ClinicalGlobal Impression Severity (CGI-S) and Improvement (CGI-I) scale,24 and the Symptoms check list,90 items (SCL-90) depression subscale (SCL-90dep).51;52 Thus, three levels of information wereobtained: data from 1) an independent, trained, supervised and blinded research-assistant (HDRS-17 & Maier, Bech), 2) the treating clinician (CGI-S/I) and 3) the patient (SCL-90dep). The HDRS wasadministered using a semi-structured interview.53 Before participating in the study, the reliabilityof the HDRS-assessments was established. During the study, in order to avoid slippage,audiotaped assessments were discussed monthly.
In the analyses of treatment-efficacy, response was defined as a ≥50% HDRS-score reduction,partial response as ≥ 20-50% reduction in HDRS-score and remission as a HDRS score of 7 points orless.16;54
StatisticsCronbach-α coefficients and mean inter-item correlations were used to express internalconsistency. To check whether the increased number of items in the HDRS accounted for a higherCronbach-α coefficient than in the subscales (with only 6 items), we applied the Spearman-Brownformula.55 Next we calculated concurrent validity as Pearson correlation coefficients betweentotal HDRS, Maier and Bech subscale scores and SCL-90dep scores. Linear regression modelscalculated variance of HDRS-scores explained by the subscales.56 These analyses were performedin our diagnostic sample. Concurrent validity between CGI-S/I and HDRS subscale-ratings wasdetermined also, however, to avoid low correlations due to limited dispersion, this was done forthe last observation in the per protocol sample. The CGI improvement scale was compared withchanges expressed as percentages of the baseline score.
In order to compare differences in sensitivity to measure treatment effects (also referred toas responsiveness), in data from the per protocol sample effect sizes (E-S) for HDRS, Maier andBech subscales were calculated per subject as the within-subject changes in scale-scores dividedby the pooled standard deviation of the mean change in scale score .20 In this way,differences in effect sizes could be tested and 95% confidence intervals (95% CI) could becalculated. Differences in E-S between the scales were tested by paired T-tests. In order todetermine significant effect-modification, the above analyses were repeated while data werestratified. For stratification we used initial HDRS scores of at least 19 for severe depression,11
criteria for response as described above and treatment-condition. Differences in E-S betweenstrata were tested by Analysis of variance-models (ANOVAs).
The Partial Credit Item Response Theory (IRT) model57 was used to estimate the relationshipsbetween total scores on the HDRS and total scores on the Maier and Bech subscales of the HDRS.The scores were those obtained at exit (per protocol sample). The computer program OPLM58
was used to obtain a set of weights for each item in the HDRS using conditional maximumlikelihood methods. The same software and the item weights were used to obtain estimates ofthe latent trait associated with each score on the HDRS, the Maier subscale and the Bechsubscale. The total scores for the pairs of scales were equated by matching the total scores forwhich the latent trait scores were most similar.59 These methods are very similar to those used in aprevious publication about the Quick Inventory for Depressive Symptomatology IDS.60 The rangeof SCL scores associated with each HDRS score was obtained directly from the original data.
⎟⎠⎞
⎜⎝⎛ −
differencepooledSDTT LOCFend
_
)(0

Chap
ter
Chapter 3
3
48
Finally, Receiver operator characteristic (ROC) curves were constructed to summarize validityof cut-off points. Differences in areas under the curve (AUC) were tested with attention forinterrelation (because we studied these tests within the same subjects) as described by Hanley.61
For all data analysis except the IRT analysis, SPSS for Windows version 10.1 was used.62 For all teststwo-tailed significance levels were applied.
Table 3.1. Studied populations.*
Diagnostic sample (n= 485†)
Per protocol sample (n= 219)‡
Trial I Trial II CombinedI + II AD
(n= 57) AD+SPSP(16) (n= 72)
AD+SPSP(8)(n= 45)
AD+SPSP(16)(n= 45)
Sex (% female) 60.3 63.2 61.1 60.0 68.9 63.0
Age 35.3 ±9.9 34.9 ±8.2 34 ±9.4 38.1 ±10.5 36.2 ±10.5 35.5 ±9.7
Marital status (%): married divorced widowed never married other
97 (20.2)60 (12.5)3 (0.6)318 (66.1)3 (0.6)
12 (21.1)7 (12.3)-38 (66.7)-
10 (13.9)8 (11.1)-54 (75.0)-
19 (42.2) 5 (11.1) 1 (2.2) 20 (44.4) -
9 (20.5)9 (20.5)-26 (59.1)-
50 (22.9)29 (13.3)1 (0.5)138 (63.3)-
Educational level (%): low intermediate high
72 (15) 179 (37.3) 229 (47.7)
11 (19.3) 21 (36.8) 25 (43.9)
13 (18.3)22 (31.0)36 (50.7)
8 (17.8)20 (44.4) 17 (37.8)
8 (18.2)16 (36.4)20 (45.5)
40 (18.4)79 (36.4)98 (45.2)
Occupational (%): job on sickness social security disabled student other
166 (34.7)134 (28.0)84 (17.5)27 (5.6)41 (8.6)27 (5.6)
18 (31.6)14 (24.6)12 (21.1)3 (5.3)5 (8.8)5 (8.8)
22 (30.6)23 (31.9)10 (13.9)5 (6.9)10 (13.9)2 (2.8)
19 (42.2)13 (28.9)8 (17.8)1 (2.2)2 (4.4)2 (4.4)
14 (31.8)16 (36.4)3 (6.8)3 (6.8)3 (6.8)5 (11.4)
73 (33.5)66 (30.3)33 (15.1)12 (5.5)20 (9.2) 14 (6.4)
Duration of episode < 1 year 1-2 years > 2 years
314 (67.0) 70 (14.9) 85 (18.1)
39 (70.9)6 (10.9)10 (18.2)
49 (70.0) 9 (12.9) 12 (17.1)
27 (61.4) 7 (15.9)10 (22.7)
25 (56.8) 14 (31.8) 5 (11.4)
140 (65.7)36 (16.9)37 (17.4)
Psychiatric treatment during this episode (%)
95 (20.2) 13 (23.6) 9 (12.9) 5 (11.4) 10 (22.7) 37 (17.4)
Antidepressants ≤3 mths before (%) adequate (%)
77 (16.4)1 (0.2)
6 (10.9)-
10 (14.3)-
9 (20.5) -
9 (20.5) -
34 (16.0) -
Depressive episodes (prev. 5 years) none 1-2 3 or more
296 (63.1)138 (29.4) 35 (7.5)
26 (47.3)21 (38.2)8 (14.5)
4 (58.6) 22 (31.4)7 (10.0)
33 (75.0)10 (22.7) 1 (2.3)
28 (65.1)13 (30.2)2 (4.7)
128 (60.4)66 (31.1)18 (8.5)
Ethnicity (%) North-west Europe Mediterranean Caribbean Other
414 (86.3)18 (3.8)22 (4.6)26 (5.4)
51 (89.5)3 (5.3)1 (1.8)2 (3.5)
63 (87.5)2 (2.8)2 (2.8) 5 (6.9)
38 (84.4) 1 (2.2) 5 (11.1) 1 (2.2)
40 (90.9) 1 (2.3) 2 (4.5) 1 (2.3)
192 (88.1)7 (3.2) 10 (4.6) 9 (4.1)
Baseline scores of rating scales
HDRS-17 17.1 ±6.5 21.0 ±4.8 20 ±4.9 19.4 ±3.8 20.3 ±4.4 20.2 ±4.6§
Maier 9.2 ±3.6 11.0 ±2.9 10.9 ±2.8 10.7 ±2.3 10.9 ±2.9 10.9 ±2.8§
Bech 9.4 ±3.7 11.5 ±2.8 11.2 ±2.7 10.7 ±2.3 11.0 ±3.0 11.1 ±2.3§
CGI-S ¶ 4.7 ±0.7 4.8 ±0.6 4.7 ±0.7 4.5 ±0.7 4.6 ±0.6 4.7 ±0.7
SCL-90 depression subscale 45.9 ±11.8 48.7 ±11.7 47.8 ±9.8 49.3 ±8.7 52.0 ±10.1 49.2 ±10.2§
* Data represent means (±SD) unless indicated. Denominators of percentages vary due to missing values.† Total diagnostic sample.‡ No significant differences between treatment-groups (per protocol sample) (ANOVA or χ2).§ Significant differences (p< 0.05) between in- and excluded patients (indep. T-Test).¶ n= 241 References to studies: Trial I45 and Trial II46

ChapterClinical use of HDRS subscales
3
49
Results
Patient characteristicsTable 3.1 shows demographics for the diagnostic and per protocol samples. There were nosignificant differences observed between the diagnostic and per protocol sample (tested asexcluded versus included), except from a lower mean HDRS-score (and Maier, Bech and SCL90depression scores) in the diagnostic sample. This difference was due to application of the entrancecriterion (HDRS ≥14) for randomization. No significant differences existed between the differenttreatment-groups. The studied population existed of mainly unmarried, mid-thirty, moderately tohighly educated, female, Caucasian adults, with moderate to severe depressive episodes of lessthan 1 year of duration. More than 75% of the subjects were not treated for the current depressiveepisode before, 16% percent received an inadequate trial of an antidepressant.
Internal and concurrent validityData for internal and concurrent validity are presented in Table 3.2. Cronbach-alphas were slightlylower for the Maier and Bech subscales. If a 17-item scale is reduced to 6 items, the expectedalpha is 0.49 (Spearman-Brown formula). Thus the observed values of 0.62 and 0.60 showincreased internal validity for the subscales. The mean inter-item correlation was markedly higherfor the Maier and Bech subscales. The correlation between Maier and Bech subscales was high.Both Maier and Bech subscales explained about 75% of the variance of the total HDRS-score. Theself-rated SCL-90dep was reasonably well correlated with the HDRS (r= 0.67) and the Maier andBech subscales (r= 0.64). Concurrent validity of the scales was overall slightly less in the moredepressed sub-group (HDRS ≥19; n= 194) compared to moderately depressed subjects, except forthe correlation between HDRS and Maier subscale.The CGI-S at study-endpoint was moderatelycorrelated with the HDRS (r= 0.57), as with the Maier and Bech subscales. The CGI-S showedhigher correlation with the Bech subscale, especially in those severely depressed. The CGI-I atstudy-endpoint was less well correlated with the percentage change in HDRS (r= 0.42) and thesubscales.
Table 3.2. Internal validity and concurrent validity of HDRS-17, Maier, Bech, SCL-90 depression subscale and CGI-S.
Internal consistency Concurrent validity: Pearson's r (% explained variance)
Cron-bach-α Mean Inter-item corr.
Overall Moderate depression* Severe depression*
Maier Bech Maier Bech Maier Bech
Diagnostic sampleMaier†
0.62 0.21 - - - - - -
Bech† 0.67 0.25 0.95(91%)
- 0.91(83%)
- 0.90(81%)
-
HDRS-17† 0.73 0.13 0.86(75%)
0.86(73%)
0.57(57%)
0.76(58%)
0.65(42%)
0.60(36%)
SCL-90 depression subscale†
0.88 0.33 0.64(41%)
0.64(40%)
0.45(21%)
0.45(20%)
0.40(16%)
0.42(18%)
Per protocol sampleCGI-S endpoint‡ NA 0.54
(29%) 0.58(34%)
0.55(31%)
0.55(30%)
0.59(34%)
0.65(42%)
CGI-I endpoint§ NA 0.42 (18%)
0.43(18%)
0.38(14%)
0.37(13%)
0.48(23%)
0.49(24%)
* Severe depression defined as initial HDRS-17 ≥19 (n= 221).† Maier, Bech, HDRS: n= 482; SCL-90dep: n= 473.‡ CGI-S: n= 229. § Compared with change expressed as percentage of baseline rating

Chap
ter
Chapter 3
3
50
Sensitivity to changeIn Table 3.3 and 3.4, overall and stratified E-S in the per protocol sample are presented. In thesetables the 95% CI of the E-S indicates whether the E-S significantly deviates from 0 (no effectmeasured). Comparisons between E-S may produce significant differences between E-S, evenwhen the 95% CIs between the two E-S overlap.
Of the 9 comparisons between the Maier and Bech subscales made in these tables, 5 were notsignificant. The Maier was significantly different from the HDRS in 4 out of 9 comparisons, whilethe Bech was significantly different from the HDRS in only 1 of the 9 comparisons. Differencesbetween E-S were small.
Table 3.3. Pre- and post-treatment Maier, Bech and HDRS-scores with corresponding effect sizes in per protocol sample. Stratification by depression-severity and final treatment response.
Mean ±SD baseline
Mean ±SD endpoint (LOCF)
Mean decrease (95% CI)
SD of decrease
Effect size [E-S](95% CI)
All subjects (n= 219)
Maier 10.9 ±2.75 6.2 ±4.46 4.7 (4.1 - 5.3) 4.54 1.03 (0.89 - 1.16)*†
Bech 11.1 ±2.69 6.2 ±4.50 4.9 (4.3 - 5.5) 4.54 1.08 (0.95 - 1.22)
HDRS-17 20.2 ±4.56 12.0 ±7.62 8.2 (7.2 - 9.2) 7.45 1.10 (0.96 - 1.23)
Moderate depression (initial HDRS-17 <19; n= 93)‡
Maier 8.9 ±2.13 5.3 ±3.86 3.7 (2.8 - 4.5) 4.19 0.81 (0.62 - 1.00)†
Bech 9.2 ±2.12 5.1 ±3.89 4.1 (3.2 - 5.0) 4.28 0.91 (0.71 - 1.10)
HDRS-17 16.2 ±1.42 9.8 ±6.15 6.4 (5.1 - 7.8) 6.47 0.86 (0.68 - 1.04)
Severe depression (initial HDRS-17 ≥19; n= 126)‡
Maier 12.3 ±2.25 6.9 ±4.75 5.4 (4.6 - 6.2) 4.66 1.19 (1.01 - 1.37)*
Bech 12.5 ±2.16 7.0 ±4.75 5.5 (4.7 - 6.3) 4.65 1.21 (1.03 - 1.39)
HDRS-17 23.1 ±3.79 13.7 ±8.18 9.5 (8.1 - 10.8) 7.88 1.27 (1.08 - 1.46)
Final Non-responders (n= 65)§
Maier 10.6 ±2.82 10.6 ±2.86 0.0 (-0.7 - 0.6) 2.65 -0.01 (-0.15 - 0.14)¶
Bech 11.1 ±2.76 10.7 ±2.89 0.4 (-0.2 - 1.1) 2.66 0.09 (-0.05 - 0.24)¶
HDRS-17 19.5 ±4.25 19.9 ±4.55 -0.4 (-1.2 - 0.3) 3.08 -0.06 (-0.16 - 0.05)¶
Final Partial-responders (n= 64)§
Maier 11.5 ±2.86 7.4 ±3.07 4.1 (3.4 - 2.7) 2.58 0.90 (0.76 - 1.04)
Bech 11.4 ±2.59 7.4 ±3.19 4.1 (3.4 - 4.7) 2.71 0.90 (0.75 - 1.04)
HDRS-17 21.4 ±5.06 14.2 ±4.41 7.2 (6.7 - 7.7) 1.88 0.96 (0.90 - 1.03)
Final Responders (n= 90)§
Maier 10.6 ±2.59 2.1 ±2.00 8.5 (7.8 - 9.1) 3.12 1.87 (1.72 - 2.01)**
Bech 10.9 ±2.71 2.1 ±1.89 8.8 (8.1 - 9.4) 3.14 1.93 (1.79 - 2.08)
HDRS-17 19.9 ±4.28 4.8 ±3.46 15.1 (14.1 - 16.1) 4.89 2.02 (1.89 - 2.16)**
Note that the overlap of two 95%CI of E-S does not rule out a statistical significant difference between these E-S (see text).* Significantly different from E-SHDRS (paired T-test; p <0.05) † Significantly different from E-SBech (paired T-test; p <0.05) ‡ Significant differences of E-SMaier, E-SBech and E-SHDRS between moderate and severe depression (ANOVA; p< 0.05)§ Response criteria: decrease in HDRS-scores: <20%= Non-response, 20-50%= Partial response and ≥50%= Response. Significant differences of E-SMaier, E-SBech and E-SHDRS between categories of response (ANOVA; p< 0.001)¶ Significant differences between E-SMaier-E-SBech, E-SHDRS-E-SMaier and E-SHDRS-E-SBech (paired T-test; p <0.05)** Significant difference between E-SHDRS-E-SMaier (paired T-test; p <0.05)

ChapterClinical use of HDRS subscales
3
51
In the total per protocol sample the Maier subscale was significantly less powerful to observetreatment effects: the E-S assessed by the Maier was significantly lower than the E-S of the Bechand HDRS. When stratified for depression severity, the E-S of Maier, Bech subscales and HDRSwere larger in severe compared to moderate depression. A significant difference between thesestrata was observed for all E-S (ANOVA). Within the group of severely depressed subjects, theMaier was significantly less sensitive compared to the HDRS (paired T-test). Within themoderately depressed group, the Bech outperformed the Maier (paired T-test). Across differentstrata of final response significant differences in E-S were found (ANOVA). Within strata, the Bechsubscale performed less in final non-responders, while the Maier performed significantly less thanthe total HDRS in final responders (paired T-tests).
In Table 3.4 it is shown that no overall differences in effect sizes were found betweentreatment modalities (ANOVA). Within the group of patients treated with anti-depressants only,the Maier subscale was significantly less sensitive to detect treatment differences than the HDRS,however the Maier did not differ significantly from the Bech subscale (paired T-test).
Conversion of HDRS scores, criteria for remission and depression severityTable 3.5 shows the conversion between HDRS scores and Maier, Bech and SCL90dep scores.Maier and Bech cut-off scores to define remission (bold), mild, moderate and severe depression)11
can be identified (italic). Figure 3.1 shows the ROC-curves for Maier, Bech CGI-S, CGI-I and SCL-90dep cut-off scores, with HDRS ≤7 as the reference criterion. The difference in AUC for the Maierand Bech subscales was not significant (z= 1.25; p= 0.21). The difference in AUC between Maierand Bech subscales compared to SCL90dep and both CGIs was highly significant (z> 3.8; p< 0.001).In the table below Figure 3.1 sensitivity and specificity for cut-off scores ≤3 and ≤4 for the Maierand Bech subscales are given.
Table 3.4. Pre- and post-treatment Maier, Bech and HDRS-scores with corresponding effect sizes in per protocol sample. Stratification by treatment modality.
Mean ±SD baseline Mean ±SD endpoint (LOCF)
Mean decrease(95% CI)
SD of decrease
Effect size [E-S](95% CI)
All subjects (n= 219)
Maier 10.9 ±2.75 6.2 ±4.46 4.7 (4.1 - 5.3) 4.54 1.03 (0.89 - 1.16)*†
Bech 11.1 ±2.69 6.2 ±4.50 4.9 (4.3 - 5.5) 4.54 1.08 (0.95 - 1.22)
HDRS-17 20.2 ±4.56 12.0 ±7.62 8.2 (7.2 - 9.2) 7.45 1.10 (0.96 - 1.23)
AD (n= 57)‡
Maier 11.0 ±2.95 7.2 ±4.97 3.8 (2.4 - 5.1) 5.12 0.83 (0.53 - 1.13)§
Bech 11.5 ±2.78 7.1 ±5.01 4.4 (3.1 - 5.7) 5.01 0.97 (0.68 - 1.26)§
HDRS-17 21.0 ±4.77 13.9 ±8.36 7.1 (4.9 - 9.3) 8.38 0.95 (0.65 - 1.25)
AD + 8 SPSP (n= 45)‡
Maier 10.7 ±2.34 5.9 ±4.31 4.8 (3.6 - 5.9) 3.81 1.05 (0.80 - 1.31)
Bech 10.7 ±2.28 6.0 ±4.21 4.6 (3.5 - 5.8) 3.87 1.02 (0.76 - 1.28)
HDRS-17 19.4 ±3.80 11.1 ±6.80 8.3 (6.4 - 10.2) 6.37 1.12 (0.86 - 1.38)
AD + 16 SPSP (n= 117)‡
Maier 10.9 ±2.82 5.8 ±4.21 5.1 (4.2 - 5.9) 4.47 1.12 (0.93 - 1.30)
Bech 11.1 ±2.78 5.8 ±4.32 5.3 (4.4 - 6.1) 4.54 1.16 (0.98 - 1.35)
HDRS-17 20.1 ±4.69 11.5 ±7.44 8.6 (7.3 - 10.0) 7.37 1.16 (0.98 - 1.34)
AD= Antidepressants, SPSP = Short Psychodynamic Supportive Psychotherapy Note that the overlap of two 95%CI of E-S does not rule out a statistical significant difference between these E-S (see text).* Significantly different from E-SHDRS (paired T-test; p <0.05) † Significantly different from E-SBech (paired T-test; p <0.05) ‡ No significant differences of E-SMaier, E-SBech and E-SHDRS between treatment modalities (ANOVA)§ Significant differences between E-SMaier-E-SBech (paired T-test; p <0.05)

Chap
ter
Chapter 3
3
52
Discussion
Major findingsThis study examined the relative effectiveness of the HDRS subscales as developed by Maier etal.28 and Bech et al.37 in monitoring severity and treatment effects in depression. We found thatthe Maier and Bech subscales gave results comparable to the original 17-item HDRS, with highconcurrent validity and increased mean inter-item correlations and internal consistency. Maierand Bech subscales were highly comparable to each other in the measurement of treatmentchanges. Differences between E-S were rather small, and clinically irrelevant. For interpretation aconversion table linking HDRS-scores and Maier and Bech scores are provided. The Maier had aslightly (non-significant) higher sensitivity and specificity to predict the reference criterion forremission (HDRS ≤7). Both Maier and Bech subscales differentiated non-responders from partialand final responders.
A significant difference in sensitivity to change existed between the Bech and Maier withinthe group treated with antidepressants only. We were unable to find the reason for thisdifference compared to other treatment modalities, where the difference between Maier andBech was not found or was not significant. The question arises whether there is a difference insensitivity between the Maier and Bech subscales across different treatment-modalities or thatother (post-randomization) differences between the groups or mere chance explain thisobservation. Because this difference was not found in the other groups (treated with bothantidepressants and psychotherapy) we think it cannot be ascribed to a difference in detectingpharmacological (side-) effects. If a Bonferroni correction would be applied for the number ofcomparisons tested (p < 0.01), the observed difference would not remain its significance.
Figure 3.1. Receiver operating characteristic curves for Maier and Bech subscales, SCL90 depression and CGI-S/I at endpoint compared to HDRS-17 'Remission' in per protocol sample.
Reference: HDRS-17 score ≤7 (Remission). Sens.= sensitivity, Spec.= specificityAUC (SE): Maier 0.972 (.009), Bech 0.963 (.011), SCL-90dep 0.862 (.028), CGI-S 0.743 (.036), CGI-I 0.738 (.035)
����� ��������� ��� ��� ��� ��� ������������ �� � �� � �� � �� ������������ �� � �� � �� � �� �
�����
����
����
�� � �� �
!�!�

ChapterClinical use of HDRS subscales
3
53
The relevance of the difference between the Maier and Bech subscalesThe only difference between the Maier and Bech subscales is the inclusion of agitation (e.g.running thoughts or restlessness, 0-4 points) in the Maier versus the inclusion of general somaticsymptoms (e.g. tiredness, 0-2 points) in the Bech. It could be argued that one scale is comparablewith the other scale without the different item, e.g. the Maier subscale would then becomparable to the Bech subscale minus the 'general somatic' item. In our (diagnostic) sample theitem agitation contributed 1.3 (SD= 0.9) points to the total Maier-score (9.2, SD= 3.6). The generalsomatic item contributed 1.5 (SD= 0.8) points to the total Bech-score (9.4, SD= 3.7). Thus, overall
Table 3.5. The conversion between the HDRS total scores and the Maier subscale, Bech subscale and the range of SCL scores using IRT analysis (per protocol sample).*
HDRS Maier Bech Range SCL90dep
0 0 0 -
1 – 2 1 1 -
3 – 4 2 2 -
5 3 3 -
6 3 4 -
7 (remission†) 4 4 -
8 – 9 5 5 -
10 – 11 6 6 -
12 7 7 -
13 (mild†) 7 8 -
14 8 8 31 – 61
15 8 9 22- 59
16 9 9 26 - 61
17 9 10 25 - 59
18 (moderate†) 10 10 30 - 62
19 10 10 28 - 60
20 11 11 38 - 67
21 11 11 30 - 72
22 12 12 39 - 61
23 12 12 36 - 61
24 (severe†) 13 13 38 - 67
25 (very severe†) 13 13 45 - 64
26 13 13 47 - 71
27 14 14 46 - 72
28 14 14 42 - 79
29 15 14 55 - 71
30 15 15 43 - 43
31 15 15 63 - 70
32 16 15 62 - 65
33 16 16 57 - 71
34 – 35 17 16 -
36 17 17 -
37 – 39 18 17 -
40 – 42 19 18 -
43 – 44 20 19 -
45 21 19 -
46 21 20 -
47 – 48 22 20 - * The only valid conversions that can be made from this table are between 1) HDRS and Maier, 2) HDRS and Bech and 3) HDRS and SCL90dep.† Cut-offs as provided by Yonkers.11

Chap
ter
Chapter 3
3
54
tiredness was more present than agitation in this sample, and agitation was not rated near itsmaximum like tiredness. Both items occurred intraindividually at the same time, but were notinterchangeable. This means that the Maier and Bech subscales show different perspectives ondepressive symptomatology. In this respect, it is noteworthy to mention that the agitation itemwas dropped beforehand when the Bech subscale was developed and validated, because thisitem showed limited variance (i.e. was not found to be scored) in the two studied samples.36;37
Furthermore, in the Maier subscale the items psychomotor agitation and psychomotorretardation are included, which -at first sight- seem to represent two opposed polarities.However, these items also co-occurred within the same individuals. This can be explained by thebroad definitions of agitation (both restlessness or running thoughts) and retardation (bothretardation in activities or in thinking) in our semi-structured interview.
The original HDRS is often criticized to measure these somatic symptoms.11;15;27;28 Although theBech subscale was designed as an unidimensional scale, the 'general somatic' item is still amongthe 6 items. However, in the Rasch-analysis this item was the least contributive and showed aceiling effect for moderate and severe depression.37 Though both a DSM-IV and ICD-10 criterionfor diagnosis,the aspecific 'tiredness' symptom may also be caused by physical illnesses. TheMaier subscale does not include this item. Thus, the Maier subscale might especially be useful inpatients suffering from somatic complaints or illnesses. Additional, methodological supportcomes from the exclusion of this somatic item by Santor et al.21 This hypothesis of betterperformance in patients with somatic complaints or illness needs further investigation, e.g. incomparison with the Hospital Anxiety and Depression Scale.63
Previous comparative studiesOur findings are in line with findings of previous studies,11;20;26;40-44 and extends the evidence tosupport the Maier and Bech subscale as a valid alternative for the HDRS. This is relevant for theplanning and conduction of clinical trials,40-42 but also for clinical practice.20;26 Hooper et al. 26 foundequal performance of the Bech subscale compared to the Montgomery Asberg Depression RatingScale (MADRS).30 Because the MADRS was not used in our trials, we were unable to examine theperformance of the Maier subscale compared to the MADRS. Hooper et al. questioned whether apossible ceiling effect in the Bech subscale would limit its usefulness in severely depressedpatients.26 In our study more than 57% of the per protocol patients had an initial HDRS greater than18 (indicative for severe depression).11 We did not find a ceiling effect in our diagnostic sample(data not shown), and found consequently higher effect sizes for the Maier and Bech subscales ininitially severely depressed patients, indicative for an adequate sensitivity to measure (larger)changes due to treatment. In addition to the observed ability to predict remission,41 we proposedcut-off scores for remission and various ranges for classification of depression severity.
In two publications Bech proposed the Bech subscale as an alternative measure to overcomethe confounding influence of drug related side-effects in the comparison with placebo or activedrugs.43;44 However this problem is not fully solved, as tiredness may be induced by histaminergiceffects from antidepressants (e.g. tricyclics and mirtazapine).43 On the other hand, agitation(included in the Maier subscale) is known as a (mostly transient) SSRI-induced side-effect .
An extra dimension of our study is that it extends the data for use of the Maier and Bechsubscales in populations treated with psychotherapy. Hooper et al. and O'Sullivan et al. alreadydemonstrated the usefulness of the Bech subscale in pharmacological treatment of melancholia,dysthymia and typical and atypical depression.20;26
An alarming point of our study is the moderate correlation of the Maier, Bech and HDRS withthe CGI-S and the CGI-I. Previous reports mentioned correlations between HDRS and CGI varyingbetween 0.65 and 0.90.11;28;40 Our results underscore the need of a HDRS or subscale rating insteadof the CGI. We consider the validity of the CGI to be questionable, as most CGI-raters (subjectively)evaluate their own treatment. Apparently, the clinician’s judgment does not coincide with scalescores.In this respect, the performance of the (self-rated) SCL-90dep is better. This was alsoillustrated in the ROC-curves regarding the criterion of remission. Further research is needed toinvestigate whether correlations with the HDRS of other self-rated scales (e.g. the Beck

ChapterClinical use of HDRS subscales
3
55
Depression Inventory64) are higher than the SCL-90dep. In addition to this, a major limitation in ourstudy and in any study investigating depression-'severity', is that there is no definite goldstandard. We used HDRS data as the gold standard, which means that scales under investigationcan never be judged to be better than the HDRS, however this would be reversed if the CGI wasused as a gold standard.65
ConclusionWe conclude that both Maier and Bech subscales of the HDRS are equivalent to the HDRS, andcan easily be used to increase efficiency to measure treatment response in clinical practice. Ontheoretical grounds we have a slight preference for the Maier subscale. The use of subscaleswould improve the efficiency and objectivity of measuring response in clinical practice, whereoften no scale (instead of a Clinical Global Impression) is used at all. This would further bridge thegap between clinical practice and research based treatment-recommendations for non-responsein depression. Maier and Bech subscales should be compared in patients suffering from co-morbidsomatic illnesses, or patients treated with psychotherapy only. The impact of the difference of theone somatic item versus the agitation-item between the Maier and Bech subscales and theconsequences for their applicability in clinical sub-groups needs further research.
AcknowledgementThe original randomized controlled trials were supported by an unrestricted educational grantfrom Eli Lilly Netherlands. All studies were carried out by the Mentrum Depression ResearchGroup. We thank all psychotherapists, psychiatrists and residents for their excellent work.
Conflicts of interestExternal funding did not support these post-hoc analyses.
References
1. Murray CJ, Lopez AD. Evidence-based health policy--lessons from the Global Burden of Disease Study. Science. 1996; 274:740-743.
2. Greenberg PE, Stiglin LE, Finkelstein SN, Berndt ER. Depression: a neglected major illness. J Clin Psychiatry. 1993; 54: 419-424.
3. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
4. Anderson IM, Nutt DJ, Deakin JFW. Evidence-based guidelines for treating depressive disorders with antidepressants: Arevision of the 1993 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2000; 14: 3-20.
5. Mulrow CD, Williams JW, Jr., Trivedi M, Chiquette E, Aguilar C, Cornell JE et al. Evidence report on: Treatment ofdepression - Newer pharmacotherapies. Psychopharmacol Bull. 1999; 34: 409-795.
6. Centraal Begeleidingsorgaan voor de Intercollegiale Toetsing. Consensusbijeenkomst depressie bij volwassenen: vrijdag 16september 1994 Utrecht. Utrecht: Centraal Begeleidingsorgaan voor de Intercollegiale Toetsing; 1994.
7. Depression Guideline Panel. Depression in Primary Care: Volume 1. Detection and Diagnosis. Clinical Practice Guideline,Number 5. AHCPR Publication No. 93-0550. 1993. Rockville, MD, U.S. Department of Health and Human Services, PublicHealth Service, Agency for Health Care Policy and Research.
8. Depression Guideline Panel. Depression in Primary Care: Volume 2. Treatment of Major Depression. Clinical PracticeGuideline, Number 5. AHCPR Publication No. 93-0551. 1993. Rockville, MD, U.S. Department of Health and HumanServices, Public Health Service, Agency for Health Care Policy and Research.
9. Rush AJ, Crismon ML, Toprac MG, Trivedi MH, Rago WV, Shon S et al. Consensus guidelines in the treatment of majordepressive disorder. J Clin Psychiatry. 1998; 59: 73-84.
10. Trivedi MH, Rush AJ, Crismon ML, Kashner TM, Toprac MG, Carmody TJ et al. Clinical results for patients with majordepressive disorder in the Texas Medication Algorithm Project. Arch Gen Psychiatry. 2004; 61: 669-680.
11. Yonkers KA, Samson J. Mood disorders measures. In: Rush AJ, Pincus HA, First MB, et al., eds. Handbook of psychiatricmeasures. 1st ed. Washington DC: American Psychiatric Association; 2000:515-548.
12. Ballenger JC. Clinical guidelines for establishing remission in patients with depression and anxiety. J Clin Psychiatry. 1999;60 Suppl 22: 29-34.

Chap
ter
Chapter 3
3
56
13. Biggs MM, Shores-Wilson K, Rush AJ, Carmody TJ, Trivedi MH, Crismon ML et al. A comparison of alternative assessmentsof depressive symptom severity: a pilot study. [republished from Psychiatry Res 2000 Jul 24;95(1):55-65]. Psychiatry Res.2000; 96: 269-279.
14. Carroll BJ, Fielding JM, Blashki TG. Depression rating scales. A critical review. Arch Gen Psychiatry. 1973; 28: 361-366.15. Anonymous. Scales for assessment of diagnosis and severity of mental disorders. Acta Psychiatr Scand Suppl. 1993; 372: 1-
87.16. Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW et al. Conceptualization and rationale for consensus
definitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48: 851-855.
17. Prien RF, Carpenter LL, Kupfer DJ. The definition and operational criteria for treatment outcome of major depressivedisorder. A review of the current research literature. Arch Gen Psychiatry. 1991; 48: 796-800.
18. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.19. Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol. 1967; 6: 278-296.20. O'Sullivan RL, Fava M, Agustin C, Baer L, Rosenbaum JF. Sensitivity of the six-item Hamilton Depression Rating Scale. Acta
Psychiatr Scand. 1997; 95: 379-384.21. Santor DA, Coyne JC. Examining symptom expression as a function of symptom severity: Item performance on the
Hamilton Rating Scale for Depression. Psychol Assess. 2001; 13: 127-139.22. Bagby RM, Ryder AG, Schuller DR, Marshall MB. The Hamilton Depression Rating Scale: has the gold standard become a
lead weight? Am J Psychiatry. 2004; 161: 2163-2177.23. Williams JB. Standardizing the Hamilton Depression Rating Scale: past, present, and future. Eur Arch Psychiatry Clin
Neurosci. 2001; 251 Suppl 2: II6-12.24. Guy W, Clearly PA, Close JH, Conners CK, Covi L, et al. ECDEU Assessment manual for psychopharmacology. DHEW
publication (ADM) 76-338. 1976. Washington, DC, US Department of Health, Education, and Welfare. 25. American Psychiatric Association. Handbook of psychiatric measures. 1st ed. Washington DC: American Psychiatric
Association; 2000.26. Hooper CL, Bakish D. An examination of the sensitivity of the six-item Hamilton Rating Scale for Depression in a sample of
patients suffering from major depressive disorder. J Psychiatry Neurosci. 2000; 25: 178-184.27. Linden M, Borchelt M, Barnow S, Geiselmann B. The impact of somatic morbidity on the Hamilton Depression Rating
Scale in the very old. Acta Psychiatr Scand. 1995; 92: 150-154.28. Maier W, Philipp M. Improving the assessment of severity of depressive states: a reduction of the Hamilton Depression
Scale. Pharmacopsychiatry. 1985; 18: 114-115.29. Gibbons RD, Clark DC, Kupfer DJ. Exactly what does the Hamilton Depression Rating Scale measure? J Psychiatr Res. 1993;
27: 259-273.30. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979; 134: 382-
389.31. Maier W, Philipp M. Comparative analysis of observer depression scales. Acta Psychiatr Scand. 1985; 72: 239-245.32. Moller HJ. Methodological aspects in the assessment of severity of depression by the Hamilton Depression Scale. Eur
Arch Psychiatry Clin Neurosci. 2001; 251: 13-20.33. Hughes JR, O'Hara MW, Rehm LP. Measurement of depression in clinical trials: an overview. J Clin Psychiatry. 1982; 43: 85-
88.34. Walczak DD, Apter JT, Halikas JA, Borison RL, Carman JS, Post GL et al. The oral dose-effect relationship for fluvoxamine:
a fixed-dose comparison against placebo in depressed outpatients. Ann Clin Psychiatry. 1996; 8: 139-151.35. Moller HJ, Glaser K, Leverkus F, Gobel C. Double-blind, multicenter comparative study of sertraline versus amitriptyline in
outpatients with major depression. Pharmacopsychiatry. 2000; 33: 206-212.36. Bech P, Gram LF, Dein E, Jacobsen O, Vitger J, Bolwig TG. Quantitative rating of depressive states. Acta Psychiatr Scand.
1975; 51: 161-170.37. Bech P, Allerup P, Gram LF, Reisby N, Rosenberg R, Jacobsen O et al. The Hamilton depression scale. Evaluation of
objectivity using logistic models. Acta Psychiatr Scand. 1981; 63: 290-299.38. Bech P, Allerup P, Reisby N, Gram LF. Assessment of symptom change from improvement curves on the Hamilton
depression scale in trials with antidepressants. Psychopharmacology (Berl). 1984; 84: 276-281.39. Bech P. The Bech-Rafaelsen Melancholia Scale (MES) in clinical trials of therapies in depressive disorders: a 20-year review
of its use as outcome measure. Acta Psychiatr Scand. 2002; 106: 252-264.40. Faries D, Herrera J, Rayamajhi J, DeBrota D, Demitrack M, Potter WZ. The responsiveness of the Hamilton Depression
Rating Scale. J Psychiatr Res. 2000; 34: 3-10.41. Entsuah R, Shaffer M, Zhang J. A critical examination of the sensitivity of unidimensional subscales derived from the
Hamilton Depression Rating Scale to antidepressant drug effects. J Psychiatric Res. 2002; 36: 437-448.42. Bech P, Tanghoj P, Andersen HF, Overo K. Citalopram dose-response revisited using an alternative psychometric
approach to evaluate clinical effects of four fixed citalopram doses compared to placebo in patients with majordepression. Psychopharmacol. 2002; 163: 20-25.
43. Bech P. Meta-analysis of placebo-controlled trials with mirtazapine using the core items of the Hamilton Depression Scaleas evidence of a pure antidepressive effect in the short-term treatment of major depression. Int J Neuropsychopharmacol.2001; 4: 337-345.
44. Bech P, Ciadella P, Haugh MC, Birkett MA, Hours A, Boissel JP et al. Meta-analysis of randomised controlled trials offluoxetine v. placebo and tricyclic antidepressants in the short-term treatment of major depression. Br J Psychiatry. 2000;176: 421-428.

ChapterClinical use of HDRS subscales
3
57
45. De Jonghe F, Kool S, Van Aalst G, Dekker J, Peen J. Combining psychotherapy and antidepressants in the treatment ofdepression. J Affect Disord. 2001; 64: 217-229.
46. Dekker J, Molenaar PJ, Kool S, Van Aalst G, Peen J, De Jonghe F. Dose-effect relations in time-limited combined psycho-pharmacological treatment for depression. Psychological Medicine. 2005; 35: 47-58.
47. Werman DS. The practice of supportive psychotherapy. New York: Brunner/Mazel; 1984.48. Strupp HH, Binder JL. Psychotherapy in a new key. A guide to Time-limited Dynamic Psychotherapy. New York: Basic Books;
1984.49. Rockland LH. Supportive Therapy. A psychodynamic approach. New York: Basic Books; 1989.50. De Jonghe F, Rijnierse P, Janssen R. Psychoanalytic supportive psychotherapy. J Am Psychoanal Assoc. 1994; 42: 421-446.51. Derogatis LR, Lipman RS, Covi L. SCL-90: an outpatient psychiatric rating scale--preliminary report. Psychopharmacol Bull.
1973; 9: 13-28.52. Arindell WA, Ettema JM. Handleiding bij een multidimensionele psychopathologie-indicator.1986. Lisse, Swets &
Zeitlinger. 53. de Jonghe F. Leidraad voor het scoren van de Hamilton Depression Rating Scale: HDRS leidraad. Amsterdam: Benecke
Consultants; 1994.54. Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. Psychiatr Clin North Am. 1996; 19:
179-200.55. Drenth PJ, Sijtsma K. Testtheorie. Inleiding in de theorie van de psychologische test en zijn toepassingen. Houten: Bohn
Stafleu Van Loghum; 1990.56. van Straten A, de Haan RJ, Limburg M, Schuling J, Bossuyt PM, van den Bos GA. A stroke-adapted 30-item version of the
Sickness Impact Profile to assess quality of life (SA-SIP30). Stroke. 1997; 28: 2155-2161.57. Masters GN. A Rasch model for partial scoring. Psychometrika. 1982; 47: 149-174.58. Verhelst ND, Glas CAW, Verstralen HHFM. OPLM: computer program and manual.1995. Arnhem, The Netherlands, CITO. 59. Orlando M, Sherbourne CD, Thissen D. Summed-score linking using item response theory: application to depression
measurement. Psychol Assess. 2000; 12: 354-359.60. Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN et al. The 16-Item Quick Inventory of Depressive
Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients withchronic major depression. Biol Psychiatry. 2003; 54: 573-583.
61. Hanley JA, McNeil BJ. A method of comparing the areas under receiver operating characteristic curves derived from thesame cases. Radiology. 1983; 148: 839-843.
62. SPSS. SPSS for Windows. [Release 10.1]. 2000. Chicago, Ill., SPSS Inc. 63. Zigmond AS, Snaith RP. The Hospital Anxiety and Depression Scale. Acta Psychiatr Scand. 1983; 67: 361-370.64. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961; 4:
561 571: 561.65. Mulder RT, Joyce PR, Frampton C. Relationships among measures of treatment outcome in depressed patients. J Affect
Disord. 2003; 76: 127-135.

Chap
ter
Chapter 3
3
58

CHA
PTER
DOSE-ESCALATION FOR INSUFFICIENT RESPONSE
TO A STANDARD DOSE OF A SELECTIVE SEROTONIN
REUPTAKE INHIBITOR IN MAJOR DEPRESSIVE
DISORDER: A SYSTEMATIC REVIEW.
British Journal of Psychiatry 2006; 189: 309-316
Henricus G. Ruhé1
Jochanan Huyser1
Jan A. Swinkels1
Aart H. Schene1
1 Program for Mood Disorders, Department of Psychiatry, Academic Medical Center, University ofAmsterdam, Amsterdam, The Netherlands

Chap
ter
Chapter 4
4
60
Abstract
BackgroundAlthough SSRIs are frequently used for major depressive disorder, only 50-60% of patientsrespond to a standard dose. For non-responders dose-escalation is often applied.
AimTo systematically review the evidence for dose-escalation of SSRIs.
MethodsA systematic literature search in MEDLINE, EMBASE, CINAHL and PsycInfo was performed.Randomised controlled trials and meta-analyses investigating dose-escalation of SSRIs wereidentified. Relevant articles were retrieved and critically appraised. Results were summarized inan evidence table. Pooling was not justified due to heterogeneity of the identified studies.
ResultsEight true dose-escalation studies and 3 meta-analyses were identified. The available dataprovided no unequivocal base for dose-escalation. Dose-escalation before 4 weeks of treatmentat a standard dose appeared to be ineffective.
ConclusionsDose-escalation of SSRIs is equivocally supported by evidence of RCTs, but methodologicaldifficulties in the studies may account for this lack of evidence. Clinical implications andmethodological considerations for future studies are discussed.

ChapterReview of SSRI dose-escalation in MDD
4
61
Introduction
Many countries developed national clinical guidelines for the treatment ofMajor DepressiveDisorder (MDD).1-8 In these guidelines pharmacotherapy is among the most important treatments,while in many of the countries, Selective Serotonin Reuptake Inhibitors (SSRIs) have become thefirst line antidepressants. It is less clear what should be done in those 40-50% of the patients whodo not respond to the first antidepressant given.9;10 Strategies in case of non-response have beenpublished in several narrative reviews11-24 and in one systematic review.25 Three major strategiesfor non-response are recommended: 1) increase of the dose of the antidepressant (dose-escalation), 2) augmenting the antidepressant by adding a second drug, 3) switching to anotherantidepressant of the same or a different class.
Available dose-finding studies do not provide evidence to initiate pharmacotherapy for MDDwith SSRIs in higher than standard doses.26-30 For non-responders, all guidelines recommend doseescalation as the appropriate strategy, instead of continuing an apparently insufficient regimen.1-7
Only the recent NICE guideline is less pronounced in this recommendation,8 and advises that if‘there are no significant side effects, a gradual increase in dose should be considered’. Moreover,surprisingly little systematic evidence is provided to underscore these recommendations.
Due to the above recommendations and its simplicity, dose-escalation is widely practiced andoften the first strategy applied.31-34 The aim of our study was to systematically review the evidencefor dose-escalation of SSRIs in MDD.
Methods
Design of studies to be includedIdeally the design of dose-escalation studies is randomisation of non-responders to higher dosesof an antidepressant or placebo after some weeks of a standard dose. In this review we considerthree other methodological requirements for those studies. First, dose-escalation should bedeferred, e.g. 3-6 weeks after the initiation of the treatment because antidepressants needseveral weeks to have a clinical effect.35 The practice of dose-escalation and the possibility todemonstrate a dose-response relationship is based upon a selection of ‘true’ non-responders.36 Asthis might take 6-10 weeks,37 dose-escalation studies with early randomisation unintentionallydiminish the possibility to prove the usefulness of dose-escalation. The inclusion of unidentifiedlate responders in both arms of the study reduces the contrast between the intervention andcontrol. Second, an outstanding study will have sufficient power to be able to demonstrate aclinically relevant difference (e.g. 20%) between treatment arms, and third will describe themethod of dose-escalation and describe the early drop-out rates due to dose-escalation.
Identification and selection of articlesFirst, systematic literature searches (updated until February 10th 2005) were performed in fourdatabases (MEDLINE, EMBASE, CINAHL, PsychInfo; all indexed years). As there are no specifickeywords for dose-escalation studies, ‘sensitive’ searches were performed with the followingterms: (((dose[textword(tw)] or dosage[tw]) and increase[tw]) or ((dose[tw] or dosage[tw]) andmaxim*[tw]) or (upward[tw] and titrat*[tw])) OR dose-response relationship, drug[MeSH] incombination with the Cochrane Collaboration search-filter for RCTs and systematic reviews, theCochrane Collaboration Depression Anxiety and Neurosis group (CCDAN) search-filter for MDDand MeSH-terms and text words for SSRIs. Primary selection (independently by the first andsecond author) was based on design and focus on dose-response relationships for SSRIs, byscreening title and abstract of the article. Agreement on predominantly exclusion of irrelevantarticles was 99.1%, with Cohen’s Kappa for interrater agreement of 0.62 (a 'substantial'agreement).38 Discrepancies between initial selection was resolved by discussion and concensus.

Chap
ter
Chapter 4
4
62
Second, all potentially relevant articles were judged according to specific inclusion andexclusion criteria (criteria available on request). In case of doubt an article was read fully andassigned afterwards. Additionally, relevant cross-references were retrieved. Double-publicationswere considered together to reveal the maximum of available information.
Critical Appraisal and summary Selected articles were critically appraised and abstracted by the first author, using standardizedforms derived from the Dutch Institute of Healthcare Improvement39 and the AHCPR.4 The itemsused for critical appraisal were the same as proposed by SIGN40 and Sackett et al.41 Each study wasassigned a ‘level of evidence’ (LoE; Table 4.1).39 Levels of evidence are based upon themethodological robustness of studies. For the results, the highest LoE of the supporting scientificevidence (A1-D) is indicated.
To assess judgement-bias by one person who performed the critical appraisal, interratervariation was determined in a slightly different set of 12 publications. Every other author criticallyappraised 4 publications, agreement for the appraisal-items was expressed by Cohen's Kappa.Kappa values were 0.49 (for 'validity of the study'), 0.86 (for 'concealment of allocation'), whilecomplete agreement existed for the appraisal-items 'randomization of the study', 'level ofevidence' and 'data extraction' (Kappa = 1.0). This is in line with other reports of interrateragreement in appraisal of psychiatric research.42
A qualitative summary with discussion of the results, restrictions, methodological flaws andexternal validity of the studies was described in an evidence table and a separate document, ofwhich a summary is provided in this paper. Because of the apparent heterogeneity in timing of thedose-escalation between the studies, results were not pooled in a meta-analysis.
Results
Search results and selection of studies are presented in Figure 4.1. The 11 studies selected for thisreview are summarized in Table 4.2. A table of excluded studies is available on request.
Table 4.1. Levels of Evidence: Therapeutic studies.
A1 Systematic review including at least some studies of A2-level. Consistent results (homogeneity) across the included trials.
A2 Randomized controlled (double-blind) trial of good methodological quality, adequate size and consistence of results.
B Randomized clinical trial of lower methodological quality or inadequate size. Other comparative research (e.g. non-randomized trial, comparative cohort-study, case-control study)
C Uncontrolled, open study
D Expert opinion, e.g. guideline-panel members
Source: Dutch Institute of Healthcare Improvement.39
Figure 4.1. Selection of reported studies.
Articles identified by systematicsearches (n= 1876)
Articles retrieved for moredetailed evaluation (n= 42)
Appropriate studies to beincluded in the review (n= 11)
Included dose-escalationstudies (n= 8)
Excluded, not meeting in- & ex-clusion criteria, based on title
and abstract (n= 1834)
Excluded articles:narrative review (n= 8);different objective (n= 5);no dose-response data (n= 3);retrospective data (n= 4);fixed-dose study (n= 11)
Included systematic reviews(n= 3)

ChapterReview of SSRI dose-escalation in MDD
4
63
Characteristics of the studies Our searches identified 8 dose-escalation studies that increased dosages after at least 3 weeks ofstandard dosages.43-50 We further found 3 systematic reviews about dose response relationships,which included respectively three,51 three,52 and four53 of the eight identified dose-escalationstudies.
Across the studies different outcome definitions for end-points were used. In 7 articlesresponse was defined as a reduction of ≥50% in the Hamilton depression rating scale(HDRS).43;44;47;50;53 A Clinical global impression (CGI) improvement or severity score ≤2 was used forresponse in one study.48 Partial response was used in 3 studies and defined as 25-50% decrease inHDRS.45;46;49 In 7 studies remission-rates were reported. These were defined as a HDRS ≤7,46;49;50 orHDRS ≤8.48
Different criteria were used to decide whether a patient should be randomized: non-response(by CGI,47 <50% decrease in HDRS43;44;46;49), or no remission (HDRS ≤848). In the present studies nogenetic information of the CYP P450 system, nor drug blood levels were reported.
The three previous reviews all had some methodological problems: Bollini et al. pooled studieswith completely different designs and drug-classes, and applied a dose-equivalence strategy thatlumped differential doses of SSRIs together.51 Baker et al. also pooled heterogeneous studies withdifferent moments of dose-escalation, and used an unusual low reference dose of fluoxetine(5mg).53 Corruble et al. did not use an adequate search strategy and only described the dose-response relationships found in their identified studies as ‘flat’ ‘curvilinear’ or ‘linear’.52
Outline of dose-escalation studiesWe will briefly outline the dose-escalation studies. Dorseif et al. first investigated week 3 non-responders (n= 371 outpatients) to fluoxetine who were randomized to continuation with 20 mgor increase to 60 mg/day for 5 weeks. Response rates were 40.5% and 44.7% respectively andremission rates 33.3% and 36.2%. Drop-out rates due to side-effects were significantly differentwith 5.3% and 11.6% respectively.43 Schweizer et al. investigated 77 non-responsive outpatientsafter 3 weeks of fluoxetine 20 mg/day, with a randomization to placebo-increase or dose-escalation up to 60 mg/day for 5 weeks. Response rates were 51.2% and 50% respectively, withnonsignificant drop-out rates of 4.9% versus 16.7%.44 In a similar study Schweizer et al. studieddose-escalation of sertraline in outpatient nonremitters after 3 weeks of sertraline 50 mg/day (n=75). Doses were randomly either kept at 50 mg/day or increased to 150 mg/day. Remission ratesafter 5 weeks were 32% and 47% respectively (non-significant). Specified drop out rates due to sideeffects were not reported.48
Fava et al. first openly treated 15 outpatients (who were week 8 non-responders to fluoxetineat 20 mg/day) with increased doses of fluoxetine titrated up to 80 mg/day for 4 weeks. Noresponse rates were given, but the mean HDRS17-score decreased 6.2 points in week 8 non-responders and 10.1 points in partial responders.45 In a second study Fava et al. randomized week8 non-responders to fluoxetine 20 mg/day (n= 41) to either fluoxetine 40-60 mg, desipramineaddition or lithium-addition for 4 weeks. No placebo-increase was practised. Remission rates were53%, 25% and 29% respectively, but these differences were nonsignificant. Initial partial respondersappeared to benefit most from fluoxetine dose-increases (data nonsignificant). Drop-out ratesfor side effects were 0%, 17%, and 7% respectively.46 In a third study Fava et al. repeated the 3-armrandomized design from their 1994 study with a stratification for partial or non-response at week8 (n= 101). After 4 weeks, the high-dose fluoxetine group showed increased but nonsignificantremission rates (42.4%) compared to desipramine addition (29.4%) and lithium-addition (23.5%).Again initial partial responders appeared to benefit more from fluoxetine dose-increasescompared to initial non-responders (differences nonsignificant). No specific data on dropout dueto side effects were given.49

64
Chap
ter
Chapter 4
4
Tabl
e 4.
2. E
ffec
tiven
ess
of in
crea
sing
the
dose
: Sel
ecte
d st
udie
s.
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Com
paris
on*
Out
com
e†Re
mar
ks
4.2a
. Dos
e-es
cala
tion
stu
dies
Benk
ert e
t al.
(199
7)47
B 54
4
MD
D, M
inD
Out
P
RCT
of w
eek
3 no
nres
pond
ers‡
(n=
86)
(3 w
eeks
)
PAR
40m
gPA
R 2
0mg
Res
pons
e (≥
50%
in H
DR
S 17)
all:
NN
T PA
R40m
g=
100
(5.1-∞
) For
MD
D o
nly:
NN
T PA
R40
mg
= 8
(2.4
- ∞
) For
bas
elin
e H
DR
S ≥2
4: N
NT P
AR4
0mg
= 6
(1.7
- ∞
)
Stud
y in
clud
es M
inD
(42%
of r
ando
miz
ed
patie
nts)
. Dos
e-tit
ratio
n re
sulte
d in
18-2
0% n
ew
SE. N
o sp
ecif
ied
rate
s of
Dro
p-ou
t. R
espo
nse
rate
in ‘p
lace
bo-g
roup
s’ 7
5%. S
tudy
als
o in
vest
igat
es m
apro
tilin
e do
se-e
scal
atio
n (d
ata
N/A
)
Dor
nsei
f et a
l. (1
989)
43B
572
MD
D
Out
P
RCT
of w
eek
3 no
nres
pond
ers§
(n=
371)
(5
wee
ks)
FLX
60m
g FL
X 20
mg
Res
pons
e (≥
50%
in H
DR
S 21)
NN
T =
25 (6
.5-∞
) R
emis
sion
(HD
RS21≤7
) NN
T =
36 (7
.3-∞
) R
espo
nse
(CG
I-I ≤
2) N
NT
= 20
(6.5
-∞) D
rop-
out S
E N
NH
16 (8
.3-14
4)
Blin
ding
is u
ncle
ar. M
ore
SE in
FLX
60m
g (n
s).
Resp
onse
rate
in ‘p
lace
bo-g
roup
’ 40.
5%.
Fava
et a
l. (1
992)
45C
15
MD
D
Out
P
Ope
n tr
ial o
f no
nres
pond
ers§ to
8-12
w
eeks
of F
LX 2
0mg
(4
wee
ks)
FLX
40-8
0mg
(if
tole
rate
d)
- D
ecre
ase
in H
DR
S 17-
scor
es in
NR
(-6.
2) a
nd P
R
(-10
.1) (
p<0.
05) D
ecre
ase
in C
GI-S
NR
(-0.
9; n
.s.)
an
d PR
(-2.
0 p<
0.05
)
Hig
hly
sele
cted
pop
ulat
ion
(ter
tiary
car
e). N
o pl
aceb
o co
ntro
l. Li
mite
d po
wer
.
Fava
et a
l. (1
994)
46B
41
MD
D
Sett
ing?
RCT
of n
onre
spon
ders
§ to
8 w
eeks
of F
LX
20m
g (4
wee
ks)
FLX4
0-60
mg
(if
tole
rate
d)
FLX
20m
g +
DES
25-
50m
g FL
X 20
mg
+ Li
30
0-60
0mg
Rem
issi
on (H
DRS
17≤7
) NN
T all =
4 (1
.6-∞
) N
NH
NR
= 6
(1.5
-∞) (
vs. L
i) N
NT P
R =
2 (0
.9-2
.8)
(vs.
Li)
Dro
p-ou
t SEN
NH
= 6
(2.6
-∞) (
vs. L
i)
Lim
ited
pres
enta
tion
of s
tudy
-pop
ulat
ion.
No
plac
ebo
cont
rol.
Lim
ited
pow
er, e
spec
ially
in
subg
roup
ana
lyse
s.
Fava
et a
l. (2
002)
49B
101
MD
D
Out
P
RCT
of n
onre
spon
ders
§ to
8 w
eeks
of F
LX
20m
g (4
wee
ks)
FLX4
0-60
mg
(if
tole
rate
d)
FLX
20m
g +
DES
25-
50m
g FL
X 20
mg
+ Li
30
0-60
0mg
Rem
issi
on (H
DRS
17≤7
) NN
T all =
6 (2
.4-∞
) (vs
. Li)
NN
T NR
= 5
(2.0
-∞) (
vs. L
i) N
NT P
R =
6 (2
.0-∞
) (vs
. Li
) Dro
p-ou
t SE
NA
No
plac
ebo
cont
rol.
Lim
ited
pow
er, e
spec
ially
in
sub
grou
p an
alys
es.
Lich
t et a
l. (2
002)
50A
2 16
29
MD
D
Out
P
RCT
of w
eek
6 no
nres
pond
ers§
(n=
295)
(5
wee
ks)
SER
200
mg
SER
100
mg
+ PL
AC
Res
pons
e (≥
50%
in H
DRS
17) N
NH
=7
(3.6
-74.
4)
Res
pons
e (C
GI-I
≤2)
NN
H =
6 (3
.4-16
.4)
Rem
issi
on (H
DRS
17≤7
) NN
H =
12 (4
.5-∞
) Dro
p-ou
t SE
not s
ign.
diff
eren
t
Dos
age
SER
was
incr
ease
d fr
om 5
0mg
to
100m
g 2
wee
ks p
rior t
o ra
ndom
izat
ion.
R
espo
nse
rate
in p
lace
bo-g
roup
70.
4%.
Schw
eize
r et a
l. (1
990)
44B
108
MD
D
Out
P
RCT
of w
eek
3 no
nres
pond
ers§
(n=
77)
(5 w
eeks
)
FLX
60m
g FL
X 20
mg
Res
pons
e (≥
50%
in H
DR
S 17)
NN
T =
82 (4
.2-
∞)D
rop-
out S
E N
NH
= 9
(3.9
-∞)S
E in
crea
sed
in
FLX
60m
g (t
rend
)
Gen
eral
izat
ion
to o
ther
SSR
Is m
ight
be
diff
icul
t be
caus
e of
long
hal
f-life
of F
LX a
nd it
s m
etab
olite
. Res
pons
e ra
te in
pla
cebo
-gro
up
51.2
%.
Schw
eize
r et a
l. (2
001)
48B
91
MD
D
Out
P
RCT
of w
eek
3 no
n-re
mitt
ers¶
(n
= 75
) (5
wee
ks)
SER
150m
g SE
R 50
mg
Rem
issi
on (H
DRS
17≤8
) NN
T 7
(2.7
-∞) R
espo
nse
(CG
I-I ≤
2) N
NT
5 (2
.3-6
1.5)
Dro
pout
NN
H =
34
(8.5
-∞) S
E bo
th in
crea
sed
or d
ecre
ased
(t
rend
s) in
SER
150m
g
Stud
y is
mar
gina
lly d
escr
ibed
. Blin
ding
unc
lear
. Re
mis
sion
-rat
e in
pla
cebo
-gro
up 3
2%.

ChapterReview of SSRI dose-escalation in MDD
4
65
4.2b
. Sys
tem
atic
revi
ews
and
met
a-an
alys
es
Bake
r et a
l. (2
002)
53A
2 11
02 +
57
3 M
DD
Se
ttin
g?
Met
a-an
alys
1s o
f 1) 4
fix
ed d
ose
RCT
s (3
-7
wee
ks) a
nd 2
) 4 d
ose-
esca
latio
n RC
Ts (3
-5
wee
ks) o
f NR
in w
eek
3-8.
(SSR
Is o
nly)
.
1) M
ediu
m /
Hig
h do
se 2
) D
ose-
esca
latio
n
1) L
ow d
ose
2)
addi
ng P
LAC
(or L
i/DES
)
Incr
ease
in R
espo
nse
rate
(≥50
% in
HD
RS)
acro
ss d
ose-
rang
e: 1)
ITT:
-9.5
% (n
.s.)
DT:
7.8
% (p
<0.0
1) 2
) ITT
: 6%
(n.s
.). D
T: 9
.3%
(n.s
.) C
hang
e of
HD
RS-d
ecre
ase
acro
ss d
ose-
rang
e: 1)
ITT:
-2.
0 (p
<0.0
001)
. DT:
una
vaila
ble.
2) I
TT: 1
.93
(p<0
.01)
. DT:
una
vaila
ble.
Dat
e of
sys
tem
atic
sea
rch
not g
iven
. No
appr
aisa
l of s
tudi
es. H
eter
ogen
eity
of d
ose-
esca
latio
n st
udie
s ig
nore
d in
met
a-an
alys
is.
Dos
e-re
spon
se re
latio
nshi
p ca
lcul
ated
as
regr
essi
on s
lope
usi
ng S
SRI m
g eq
uiva
lent
s (S
ME)
. Low
sta
ndar
d-do
se (5
mg)
for F
LX u
sed
as re
fere
nce
for S
ME.
ITT
popu
latio
n us
ed to
es
timat
e ex
pres
sed
dose
-resp
onse
re
latio
nshi
p. P
oten
tial d
ose-
resp
onse
re
latio
nshi
p es
timat
ed in
a d
ose-
tole
rant
(DT)
sa
mpl
e, o
mitt
ing
drop
outs
due
to s
ide
effe
cts
36.
Bolli
ni e
t al.
(199
9)51
A2
5844
M
DD
In
P &
Out
P
Met
a-an
alys
is o
f 33
RCTs
(197
5-19
97) w
ith
vario
us
antid
epre
ssan
ts. (
3-15
6 w
eeks
)
Hig
her d
oses
IM
I-equ
ival
ent
201-
250
(a) a
nd
>250
mg
(b)
Ave
rage
dai
ly
dose
(IM
I-eq
uiva
lent
100-
200m
g)
Effi
cacy
in IT
T an
alys
is: 5
3.3%
(ref
), 4
6.3%
(a),
48.3
% (b
) Com
plet
ers
anal
ysis
: 69%
(ref
), 6
7.3%
(a
), 7
6% (b
) Dro
pout
rate
s: 2
2% (r
ef),
28%
(a),
35
% (b
). S
E: 3
0% (r
ef),
36%
(a),
48%
(b)
Met
a-an
alys
is u
sing
regr
essi
on m
odel
s. H
ighl
y he
tero
geno
us s
tudi
es (i
.e. d
esig
ns) p
oole
d, n
o se
para
tion
of v
ario
us a
ntid
epre
ssan
t cla
sses
, no
nsys
tem
atic
bia
s es
peci
ally
for S
SRIs
by
conv
ersi
on to
IMI-e
quiv
alen
ts.
Corr
uble
et a
l. (2
000)
52C
? MD
D
Sett
ing
?
Revi
ew o
f lite
ratu
re o
f RC
Ts in
vest
igat
ing
dose
-res
pons
e re
latio
n fo
r SSR
Is
Hig
h do
se S
SRI
Stan
dard
dos
e Q
ualit
ativ
e de
scrip
tion
of d
ose-
resp
onse
re
latio
nshi
p pe
r dru
g. O
nly
for C
IT a
nd F
LX
poss
ibly
cur
vilin
ear,
oth
er S
SRIs
flat
dos
e-re
spon
se re
latio
nshi
p.
Sear
ch o
f MED
LIN
E on
ly. N
o ap
prai
sal o
f st
udie
s. N
o po
olin
g or
exp
lora
tion
of
diff
eren
ces
betw
een
iden
tifie
d st
udie
s an
d do
se-re
spon
se re
latio
nshi
p-da
ta p
er d
rug.
*All
dosa
ges
in m
g/da
y. †
Inte
ntio
n to
trea
t (IT
T)-r
esul
ts u
nles
s sp
ecif
ied.
‡ CG
I-eff
icac
y in
dex:
min
imal
or n
o ch
ange
in d
epre
ssio
n w
ith n
o or
non
-inte
rfer
ing
side
eff
ects
. § <
50%
redu
ctio
n in
HD
RS-
scor
e.
¶ H
DR
S 17
>8
Abb
revi
atio
ns: C
GI-I
/S =
Clin
ical
glo
bal i
mpr
essi
on im
prov
emen
t/se
verit
y, D
T =
Dos
e to
lera
nt s
ampl
e, H
DR
S xx
= H
amilt
on d
epre
ssio
n ra
ting
scal
e (x
x de
note
num
ber o
f ite
ms
used
), In
P =
Inpa
tient
s, IT
T =
Inte
ntio
n to
trea
t, M
DD
= M
ajor
dep
ress
ive
diso
rder
, Min
D=
Min
or d
epre
ssio
n, N
/A =
not
app
licab
le, N
NH
/T =
Num
ber n
eede
d to
har
m/t
reat
, NR
= N
onre
spon
ders
, Out
P =
Out
patie
nts,
PR
= Pa
rtia
l re
spon
ders
, RCT
= R
ando
miz
ed c
ontr
olle
d tr
ial,
SE =
sid
e ef
fect
s.D
rugs
: CIT
= c
italo
pram
, FLX
= fl
uoxe
tine,
IMI =
imip
ram
ine,
PA
R =
par
oxet
ine,
PLA
C =
plac
ebo,
SER
= s
ertr
alin
e, T
CA =
Tric
yclic
ant
idep
ress
ants
.
Tabl
e 4.
2. E
ffec
tiven
ess
of in
crea
sing
the
dose
: Sel
ecte
d st
udie
s. (C
ontin
ued)
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Com
paris
on*
Out
com
e†Re
mar
ks

Chap
ter
Chapter 4
4
66
Benkert et al. investigated dose-escalation of paroxetine 20 mg/day in outpatients that weredepressed or had minor depression. Those who did not respond after 3 weeks of treatment (n=86) were randomized to receive 40 mg paroxetine for 3 additional weeks or placebo-increase.Response rates were 75% in the placebo-increased group and 74% in the 40 mg group.47 Licht et al.investigated randomized dose-escalation of sertraline (up to 200 mg/day) versus sertraline 100mg/day (placebo-increase) or mianserin addition in 295 outpatiens non-responsive to sertraline 50mg for 4 weeks and additionally increased to 100 mg for 2 more weeks. Response rates 5 weeksafter randomization were significantly lower in the dose-increase group (56%) than in thesertraline 100 mg group (70%) and the mianserin-addition group (67%). Drop-outs due to sideeffects were not specified.50
Strengths, flaws and other details of all selected studies are provided in table 4.2. In summarywe mention several methodological problems we encountered: absence of placebocontrols,45;46;49 inclusion of minor depression,47 insufficient data-presentation,44;46 insufficientpower,44-49 uncertainty about blinding,43;48 earlier dose-escalation before the randomisation,50
inadequate pooling of heterogeneous data51;53 and problems with conversion to dose-equivalents.51;53 None of the studies provided information about the method of dose-escalationnor described the early drop-out rates due to dose-escalation.
Evidence for dose-escalation?From 4 of the 8 dose-escalation studies it appeared that dose-increments before 4 weeks werenot effective (LoE: A2).43;44;47;48;51-53 However, in the meta-analysis of some of these studies byBaker et al., a potential dose-response relationship was found for dose-escalation if dropouts dueto side effects were excluded from the analysis (a so called dose-tolerant sample).53 Baker et al.proposed that differential dropout due to side effects in the dose-escalation group (compared toplacebo-increase) gave a substantial (negative) bias of the potential dose-response relationship.They argued that by applying a last-observation carried forward approach (often used in theoriginal studies), more early drop-outs (due to side-effects) in the high-dose groups wouldunequally increase average severity scores and decrease response rates compared to the lowerdose (or placebo) groups. This methodological problem could be overcome by analysing onlydose-tolerant subjects (those not dropping out due to side effects).36
In the well performed study with sertraline by Licht et al. (not included in the three reviews),dose-escalation after 6 weeks was found to be less effective than continuation of the standarddose, or augmentation with mianserin (LoE: A2).50 After 8 weeks of treatment, increased dosagesof fluoxetine were more effective than augmentation with lithium or desipramine,46;49 although inthe latter study this was not significant (LoE: B). In these studies no placebo dose-escalation wasperformed. Both studies showed a non-significant trend of increased efficacy of dose-escalationcompared to augmentation (lithium or desipramine) especially for partial responders(LoE: B).46;49
Across all studies, higher doses were related to increased dropout rates, which wereassociated with more side effects in some studies (LoE: A2).51 It appeared that the occurrence ofside effects did not increase equally when dosages were gradually escalated in initial non-responders compared to fixed-dose trials. However, this could not be comparedstraightforwardly between the studies, and was not investigated specifically.
Additional concerns for cliniciansWe identified no evidence to recommend on how dose-increase should be practiced. Also themaximum dosage to be achieved was not investigated well.

ChapterReview of SSRI dose-escalation in MDD
4
67
Discussion
Our systematic review provided 8 studies about dose-escalation in SSRIs. Only one of theseapproached our rather stringent criteria.50 We found no evidence for increased efficacy of dose-escalation within the first 4 weeks. Dose-escalation after 6 weeks appeared less effective thancontinuing the same dose. We found some, but limited evidence for efficacy of dose-escalationafter 8 weeks, particularly in partial responders. This effect was seen within 4 weeks after dose-escalation. Irrespective of efficacy, dose-escalation unequivocally increased side-effects, buteffects on drop-out rates due to side effects were less straightforward. Thus, in the absence ofmethodologically well designed studies we can neither unequivocally state that dose-escalation isuseful, nor discard it as useless.
These findings may challenge the current beliefs and recommendations about dose-escalation, underscored by the fact that dose-escalation is generally practiced.31-34 Contrarily tothis challenge, many patients that only partially responded are too often treated with long-termobviously insufficient treatments (e.g. standard doses of SSRIs). For these patients, one couldargue that it is better to try dose-escalation than to continue inadequate treatment. Presumably,in the absence of clear guidance by trial-data, clinicians do not have many alternatives for non-responders or partial responders, and clinicians all have their case-histories of improvement afterdose-escalation. A more sophisticated question must therefore also be asked: which subgroup ofpatients will benefit from dose-escalation?
So far, only the NICE guideline displayed some reserve in the general recommendation aboutdose-escalation.8 The British Medicines and Healthcare products Regulatory Agency’s Committeeon Safety of Medicines examined the available evidence for dose-escalation as provided bypharmaceutical companies, and recommended the lowest efficacious dose (MHRA, 2004Internet: www.mhra.gov.uk). From this report it was unclear which studies were taken asevidence. Three previous reviews concerning higher doses of antidepressants were published,51-53
of which the methodological shortcomings were already mentioned. The findings in thesereviews previously challenged the belief of a dose-response relationship, but Baker et al.proposed a potential dose-response relationship, based on their dose-tolerant analysis. All reportssummarized studies performed until 1997, thereafter the study by Licht et al. further challengedthe efficacy of dose-escalation.50
Limitations of the identified studiesFour major issues of concern in the 8 identified studies should be mentioned. First, themethodological quality of these studies varied between poor and good according to ourclassification. We summarized these methodological problems in the results section and table 4.2.
Second, and more in general, all dose-escalation studies (except the studies of Fava andcollegues, that lacked a placebo-control)45;46;49 suffered a methodological problem of the timingof dose-escalation.36 Even the most robust study by Licht et al. hampered its own design by anonrandomized dose-increase 2 weeks prior to randomization.50 This problem might explain thehigh placebo response rates in some of the dose-escalation studies (up to 75%).
Third, in most studies no data were provided on the selective drop-out, nor the schedule ofdose-increments.36 Because patients randomised to true dose-escalation might drop out morefrequently and earlier after randomisation (with associated high severity scores) compared tothose receiving placebo, this might have biased the intention to treat analyses in which last-observations are usually carried forward to study endpoints. This especially happens when dose-escalation is performed rapidly. The analysis of a dose-tolerant sample in such studies wouldindeed provide additional information, but these data were not provided.
Fourth, in the selected trials, mostly response was used as primary outcome, while currentlyremission of depression is the clinical aim of treatment.54 If dose-escalation would be effective,the question remains whether dose-escalation will also further improve initial responders thatwere nonremitters. So far only Schweizer et al. addressed this topic with equivocal effects ofdose-escalation.48

Chap
ter
Chapter 4
4
68
Possible explanations for a dose-response relationshipA possible explanation of the clinical observation that response might occur after dose-escalation,can be initial lower blood levels. This may be related to increased metabolism due to geneticpolymorphisms of the cytochrome P450 (CYP P450) enzyme system.55-58 The incidence ofincreased metabolism by (multi-)duplicated genes of the CYP P450 2D6 genes varies between 1-2%in Swedish Caucasians, 3.6% in Germany and 7-10% in Spain and Sicily, and varies between ethnicgroups (e.g. 29% in Black Ethiopians).59 A few studies showed equivocal evidence for theinvolvement of CYP P450 polymorphisms (responsible for rapid metabolism) as an explanation ofnon-response to a standard dose of SSRIs.56;58-61 However, a clear relationship between bloodlevels of SSRIs and response was never found.26;62-66 Perhaps, genetic variability of the centraltarget of these drugs, the serotonin reuptake transporter, may be responsible more directly forthe effects of SSRIs.67;68
From in vitro and ex-vivo studies it appears that at higher doses selective antidepressants likeSSRIs may become dual action agents that will also affect other monoamine systems likenorepinephrine.69-71 From the current data of dose-escalation in SSRIs this theoretical hypothesiscannot be falsified nor proven. In addition, we are unaware of an acceptable method to testwhether specific sites of action are responsible for the observed treatment-effects.
Limitations of the review No meta-analysis was performed because the differences in timing of dose-escalation betweenthe identified studies introduced substantial heterogeneity. An extension of the meta-regressionapproach as performed by Baker et al.53 was considered impossible to acknowledge this problemas the number of studies to explore this heterogeneity will lack power, moreover as age, sex,outcome definition and type of SSRI ideally should also be included in such a model.
The grading system for studies is not representing the appraised methodological dimensionsof evidence. This improved the applicability of the results for busy clinicians, but reduced theirstrength.
Finally, patients studied in trials are generally selected populations, reducing external validityfor the 'real world' clinical practice. All identified studies excluded psychotic depression, bipolardepression, depression in children or adolescents, and depressive disorder with severe psychiatricand somatic comorbidity.
Future dose-escalation studies For future dose-escalation trials methodological issues should be considered. First, for optimalcontrast in the study, an appropriate group of non-responders should be selected by postponingrandomization and refraining from (additional) interventions before dose-escalation is applied.Six weeks is the minimum which can be reconciled with recommendations in current guidelinesand is acceptable for clinical practice. Second, studies should have enough power to detectsignificant differences. This implies a large sample to start with, as approximately 50% of patientswill show a response in the first 6 weeks. Third, the method of dose-escalation should bedescribed and applied in a way that few patients drop-out. Fourth, adequate results should bepresented: response and remission rates in intention to treat analyses and for the group thatcould be described as dose tolerant. Fifth, efficacy should be tested in predefined subgroups (e.g.partial responders at week 6). Sixth, genetic sampling (e.g. CYP P450 and SERT polymorphisms)and plasma SSRI level sampling would be interesting to further examine potential explanationsfor the clinically observed efficacy of dose-escalation and to identify potential prognosticvariables.
Clinical implications and conclusionFor clinicians this review may challenge the belief in dose-escalation of SSRIs for patients whoshow insufficient response to a standard dose. This review points out that dose-escalation ofSSRIs in a standard dose before the fourth week of treatment does not seem to be more

ChapterReview of SSRI dose-escalation in MDD
4
69
beneficial for patients. Thereafter dose-escalation is one of the easiest thing to do, but – to ouropinion – should not be performed obligatory, as the evidence for this strategy is not compelling.Contrary dose-escalation appears more adequate to try than to continue an inadequatetreatment, maybe partial responders will benefit most. Side-effects will most likely increase andmight induce drop-out of treatment. For more clinical guidance well-performed new dose-escalation studies are needed. In order to fill this gap, for the introduction and registration of(new) antidepressants, clinical studies with an adequate dose-escalation design should berequired by the U.S. Food and Drug Administration (FDA) and the European Agency for theEvaluation of Medicinal Products (EMEA).
AcknowledgementsThis systematic review was realised by a grant for the development of a local evidence-basedclinicalpractice guideline (No SFA.07.012) from the Academic Medical Center, Amsterdam, theNetherlands. This guideline considering strategies for non-response to a standard dose of a firstSSRI is available on request.
Conflicts of interestNone
References
1. Kennedy SH, Lam RW, Cohen NL, Ravindran AV, CANMAT Depression Work Group. Clinical guidelines for the treatment ofdepressive disorders. IV. Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
3. Anderson IM, Nutt DJ, Deakin JFW. Evidence-based guidelines for treating depressive disorders with antidepressants: Arevision of the 1993 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2000; 14: 3-20.
4. Mulrow CD, Williams JW, Jr., Trivedi M, Chiquette E, Aguilar C, Cornell JE et al. Evidence report on: Treatment ofdepression - Newer pharmacotherapies. Psychopharmacol Bull. 1999; 34: 409-795.
5. Depression Guideline Panel. Depression in Primary Care: Volume 1. Detection and Diagnosis. Clinical Practice Guideline,Number 5. AHCPR Publication No. 93-0550. 1993. Rockville, MD, U.S. Department of Health and Human Services, PublicHealth Service, Agency for Health Care Policy and Research.
6. Depression Guideline Panel. Depression in Primary Care: Volume 2. Treatment of Major Depression. Clinical PracticeGuideline, Number 5. AHCPR Publication No. 93-0551. 1993. Rockville, MD, U.S. Department of Health and HumanServices, Public Health Service, Agency for Health Care Policy and Research.
7. Rush AJ, Crismon ML, Toprac MG, Trivedi MH, Rago WV, Shon S et al. Consensus guidelines in the treatment of majordepressive disorder. J Clin Psychiatry. 1998; 59: 73-84.
8. NICE. Clinical Guideline 23. Depression: management of depression in primary and secondary care. December 2004. 23, 1-63. 2004. London, National Institute for Clinical Excellence.
9. Thase ME, Rush AJ. Treatment-Resistant Depression. In: Bloom FE, Kupfer DJ, eds. Psychopharmacology: The FourthGeneration of Progress. New York: Raven Press Ltd.; 1995:1081-1097.
10. Kroenke K, West SL, Swindle R, Gilsenan A, Eckert GJ, Dolor R et al. Similar effectiveness of paroxetine, fluoxetine, andsertraline in primary care: a randomized trial. JAMA. 2001; 286: 2947-2955.
11. Anderson IM. Drug treatment of depression: Reflections on the evidence. Adv Psychiatr Treat. 2003; 9: 11-20.12. Kennedy N, McDonough M. Pharmacological management of treatment resistant depression: A clinical review. Irish J
Psychol Med. 2003; 20: 18-23.13. Nelson JC. Managing treatment-resistant major depression. J Clin Psychiatry. 2003; 64 Suppl 1: 5-12.14. McIntyre RS, Muller A, Mancini DA, Silver ES. What to do if an initial antidepressant fails? Can Fam Physician. 2003; 49: 449-
457.15. Hirschfeld RM, Montgomery SA, Aguglia E, Amore M, Delgado PL, Gastpar M et al. Partial response and nonresponse to
antidepressant therapy: current approaches and treatment options. J Clin Psychiatry. 2002; 63: 826-837.16. Kennedy S, McIntyre R, Fallu A, Lam R. Pharmacotherapy to sustain the fully remitted state. J Psychiatry Neurosci. 2002;
27: 269-280.17. Marangell LB. Switching antidepressants for treatment-resistant major depression. J Clin Psychiatry. 2001; 62 Suppl 18: 12-
17.18. Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry. 2001; 62 Suppl 6: 22-29.19. Fava M. New approaches to the treatment of refractory depression. J Clin Psychiatry. 2000; 61: 26-32.20. Fava M. Management of nonresponse and intolerance: Switching strategies. J Clin Psychiatry. 2000; 61: 10-12.

Chap
ter
Chapter 4
4
70
21. O'Reardon JP, Brunswick DJ, Amsterdam JD. Treatment-resistant depression in the age of serotonin: Evolving strategies.Curr Opin Psychiatry. 2000; 13: 93-98.
22. Crismon ML, Trivedi M, Pigott TA, Rush AJ, Hirschfeld RMA, Kahn DA et al. The Texas medication algorithm project:Report of the Texas consensus conference panel on medication treatment of major depressive disorder. J Clin Psychiatry.1999; 60: 142-156.
23. Nelson JC. Treatment of antidepressant nonresponders: augmentation or switch? J Clin Psychiatry. 1998; 59 Suppl 15: 35-41.
24. Thase ME, Rush AJ. When at first you don't succeed: sequential strategies for antidepressant nonresponders. J ClinPsychiatry. 1997; 58: 23-29.
25. Stimpson N, Agrawal N, Lewis G. Randomised controlled trials investigating pharmacological and psychologicalinterventions for treatment-refractory depression. Systematic review. Br J Psychiatry. 2002; 181: 284-294.
26. Beasley CM, Jr., Bosomworth JC, Wernicke JF. Fluoxetine: relationships among dose, response, adverse events, andplasma concentrations in the treatment of depression. Psychopharmacol Bull. 1990; 26: 18-24.
27. Altamura AC, Montgomery SA, Wernicke JF. The evidence for 20mg a day of fluoxetine as the optimal dose in thetreatment of depression. Br J Psychiatry Suppl. 1988; 109-112.
28. Tignol J, Stoker MJ, Dunbar GC. Paroxetine in the treatment of melancholia and severe depression. Int ClinPsychopharmacol. 1992; 7: 91-94.
29. Dunner DL, Dunbar GC. Optimal dose regimen for paroxetine. J Clin Psychiatry. 1992; 53 Suppl: 21-26.30. Montgomery SA, Pedersen V, Tanghoj P, Rasmussen C, Rioux P. The optimal dosing regimen for citalopram--a meta-
analysis of nine placebo-controlled studies. Int Clin Psychopharmacol. 1994; 9 Suppl 1: 35-40.31. Byrne S, Rothschild AJ. Psychiatrists' responses to failure of maintenance therapy with antidepressants. Psychiatr Serv.
1997; 48: 835-837.32. Fredman SJ, Fava M, Kienke AS, White CN, Nierenberg AA, Rosenbaum JF. Partial response, nonresponse, and relapse
with selective serotonin reuptake inhibitors in major depression: a survey of current "next-step" practices. J ClinPsychiatry. 2000; 61: 403-408.
33. Mischoulon D, Nierenberg AA, Kizilbash L, Rosenbaum JF, Fava M. Strategies for managing depression refractory toselective serotonin reuptake inhibitor treatment: a survey of clinicians. Can J Psychiatry. 2000; 45: 476-481.
34. Shergill SS, Katona CL. Pharmacological choices after one antidepressant fails: a survey of UK psychiatrists. J AffectDisord. 1997; 43: 19-25.
35. Mischoulon D. Why do antidepressants take so long to work? Am Soc Clin Psychopharmacology Progress Notes. 1997; 8: 9-11.
36. Baker CB, Woods SW. Is there a SSRI dose response in treating major depression? The case for re-analysis of current dataand for enhancing future study design. Depress Anxiety. 2003; 17: 10-18.
37. Quitkin FM, Petkova E, McGrath PJ, Taylor B, Beasley C, Stewart J et al. When should a trial of fluoxetine for majordepression be declared failed? Am J Psychiatry. 2003; 160: 734-740.
38. Munoz SR, Bangdiwala SI. Interpretation of Kappa and B statistics measures of agreement. J Applied Stat. 1997; 24: 105-111.
39. Richtlijnontwikkeling binnen het Kwaliteitsinstituut voor de Gezondheidszorg CBO. Handleiding voor werkgroepleden.Kwaliteitsinstituut voor de Gezondheidszorg CBO.2000. Utrecht, Kwaliteitsinstituut voor de Gezondheidszorg CBO.
40. SIGN 50: A guideline developers' handbook. 50. 2001. Edinburgh, Scottish Intercollegiate Guideline Network. 41. Sackett DL, Straus SE, Richardson WS, Rosenberg W, Haynes RB. Evidence-Based Medicine. How to practice and teach EBM.
2 ed. Edinburgh: Churchill Livingstone; 2000.42. Moncrieff J, Churchill R, Drummond DC, McGuire H. Development of a quality assessment instrument for trials of
treatments for depression and neurosis. Int J Methods Psychiatr Res. 2001; 10: 126-133.43. Dornseif BE, Dunlop SR, Potvin JH, Wernicke JF. Effect of dose escalation after low-dose fluoxetine therapy.
Psychopharmacol Bull. 1989; 25: 71-79.44. Schweizer E, Rickels K, Amsterdam JD, Fox I, Puzzuoli G, Weise C. What constitutes an adequate antidepressant trial for
fluoxetine? J Clin Psychiatry. 1990; 51: 8-11.45. Fava M, Cohen L, Rosenbaum JF, Reiter S, et a. High-dose fluoxetine in the treatment of depressed patients not
responsive to a standard dose of fluoxetine. J Affect Disord. 1992; 25: 229-234.46. Fava M, Rosenbaum JF, McGrath PJ, Stewart JW, Amsterdam JD, Quitkin et al. Lithium and tricyclic augmentation of
fluoxetine treatment for resistant major depression: a double-blind, controlled study. Am J Psychiatry. 1994; 151: 1372-1374.
47. Benkert O, Szegedi A, Wetzel H, Staab HJ, Meister W, Philipp M. Dose escalation vs. continued doses of paroxetine andmaprotiline: a prospective study in depressed out-patients with inadequate treatment response. Acta Psychiatr Scand.1997; 95: 288-296.
48. Schweizer E, Rynn M, Mandos LA, DeMartinis N, Garcia-Espana F, Rickels K. The antidepressant effect of sertraline is notenhanced by dose titration: results from an outpatient clinical trial. Int Clin Psychopharmacol. 2001; 16: 137-143.
49. Fava M, Alpert J, Nierenberg A, Lagomasino I, Sonawalla S, Tedlow J et al. Double-blind study of high-dose fluoxetineversus lithium or desipramine augmentation of fluoxetine in partial responders and nonresponders to fluoxetine. J ClinPsychopharmacol. 2002; 22: 379-387.
50. Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first-line sertralinetreatment: A randomised study of extended duration of treatment, dose increase or mianserin augmentation.Psychopharmacology (Berl). 2002; 161: 143-151.
51. Bollini P, Pampallona S, Tibaldi G, Kupelnick B, Munizza C. Effectiveness of antidepressants. Meta-analysis of dose-effectrelationships in randomised clinical trials. Br J Psychiatry. 1999; 174: 297-303.

ChapterReview of SSRI dose-escalation in MDD
4
71
52. Corruble E, Guelfi JD. Does increasing dose improve efficacy in patients with poor antidepressant response: a review.Acta Psychiatr Scand. 2000; 101: 343-348.
53. Baker CB, Tweedie R, Duval S, Woods SW. Evidence that the SSRI dose response in treating major depression should bereassessed: A meta-analysis. Depress Anxiety. 2003; 17: 1-9.
54. Nierenberg AA, DeCecco LM. Definitions of antidepressant treatment response, remission, nonresponse, partialresponse, and other relevant outcomes: a focus on treatment-resistant depression. J Clin Psychiatry. 2001; 62 Suppl 16: 5-9.
55. Bertilsson L, Aberg-Wistedt A, Gustafsson LL, Nordin C. Extremely rapid hydroxylation of debrisoquine: a case report withimplication for treatment with nortriptyline and other tricyclic antidepressants. Ther Drug Monit. 1985; 7: 478-480.
56. Brosen K. Some aspects of genetic polymorphism in the biotransformation of antidepressants. Therapie. 2004; 59: 5-12.57. Charlier C, Broly F, Lhermitte M, Pinto E, Ansseau M, Plomteux G. Polymorphisms in the CYP 2D6 gene: association with
plasma concentrations of fluoxetine and paroxetine. Ther Drug Monit. 2003; 25: 738-742.58. Steimer W, Muller B, Leucht S, Kissling W. Pharmacogenetics: a new diagnostic tool in the management of antidepressive
drug therapy. Clin Chim Acta. 2001; 308: 33-41.59. Bertilsson L, Dahl ML, Dalen P, Al Shurbaji A. Molecular genetics of CYP2D6: clinical relevance with focus on psychotropic
drugs. Br J Clin Pharmacol. 2002; 53: 111-122.60. Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with
persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J ClinPharmacol. 2004; 59: 803-807.
61. Bertilsson L, Dahl ML, Tybring G. Pharmacogenetics of antidepressants: clinical aspects. Acta Psychiatr Scand Suppl. 1997;391: 14-21.
62. Amsterdam JD, Fawcett J, Quitkin FM, Reimherr FW, Rosenbaum JF, Michelson D et al. Fluoxetine and norfluoxetineplasma concentrations in major depression: a multicenter study. Am J Psychiatry. 1997; 154: 963-969.
63. Baumann P. Pharmacokinetic-pharmacodynamic relationship of the selective serotonin reuptake inhibitors. ClinPharmacokinet. 1996; 31: 444-469.
64. Bourdeaux R, Pannetier P, Younos C, Desor D, Lehr PR, Capolaghi B. Fluoxetine : Relationships among plasmaconcentrations and therapeutic effects in the treatment of 32 patients with major depressive disorder at 20 mg/day.Encephale. 1998; 24: 57-61.
65. DeVane CL. Translational pharmacokinetics: Current issues with newer antidepressants. Depress Anxiety. 1998; 8: 64-70.66. Norman TR, Gupta RK, Burrows GD, Parker G, Judd FK. Relationship between antidepressant response and plasma
concentrations of fluoxetine and norfluoxetine. Int Clin Psychopharmacol. 1993; 8: 25-29.67. Hahn MK, Blakely RD. Monoamine transporter gene structure and polymorphisms in relation to psychiatric and other
complex disorders. Pharmacogenomics J. 2002; 2: 217-235.68. Smits KM, Smits LJ, Schouten JS, Stelma FF, Nelemans P, Prins MH. Influence of SERTPR and STin2 in the serotonin
transporter gene on the effect of selective serotonin reuptake inhibitors in depression: a systematic review. MolPsychiatry. 2004; 9: 433-441.
69. Gorman JM, Sullivan G. Noradrenergic approaches to antidepressant therapy. J Clin Psychiatry. 2000; 61 Suppl 1: 13-16.70. Gilmor ML, Owens MJ, Nemeroff CB. Inhibition of norepinephrine uptake in patients with major depression treated with
paroxetine. Am J Psychiatry. 2002; 159: 1702-1710.71. Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of
antidepressants and their metabolites. J Pharmacol Exp Ther. 1997; 283: 1305-1322.

Chap
ter
Chapter 4
4
72

CHA
PTER
SWITCHING ANTIDEPRESSANTS AFTER A FIRST
SELECTIVE SEROTONIN REUPTAKE INHIBITOR IN
MAJOR DEPRESSIVE DISORDER: A SYSTEMATIC
REVIEW
Journal of Clinical Psychiatry 2006; 67: 1836-1855
Henricus G. Ruhé1
Jochanan Huyser1
Jan A. Swinkels1
Aart H. Schene1
1 Program for Mood Disorders, Department of Psychiatry, Academic Medical Center, University ofAmsterdam, Amsterdam, The Netherlands

Chap
ter
Chapter 5
5
74
Abstract
BackgroundSelective Serotonin Reuptake Inhibitors (SSRIs) are frequently used as a first antidepressant formajor depressive disorder, but have response-rates of 50-60% in daily practice. For patients withinsufficient response to SSRIs, switching is often applied.
AimTo systematically review the evidence for switching pharmacotherapy after a first SSRI.
MethodsA systematic literature search (updated until Feb. 10, 2005) in MEDLINE, EMBASE, CINAHL andPsycInfo (all indexed years) identified randomised controlled trials (RCTs) and open studiesinvestigating switching strategies. In the absence of specific keywords for switching, weperformed “sensitive” searches using free text words with w0ildcards ($): “switch$” or(“alternat$” adj5 “treat$”) or (“alternat$” adj5 “therap$”) in combination with the CochraneCollaboration search filter for RCTs, the Cochrane Collaboration Depression Anxiety and NeurosisGroup search filter for major depressive disorder, and MeSH terms for antidepressants (incombination with additional text words for all antidepressive agents). Additionally, we included 4recent STAR*D publications. We limited searches to adults and humans but did not applylanguage restrictions. Relevant articles were retrieved and critically appraised. The methodologyof the studies, the results on efficacy and dropout s due to side effects, and remarks weresummarized in an evidence table. Three studies comparing a switch tovenlafaxine or SSRIs werepooled.
ResultsEight RCTs and 23 open studies were identified, studying populations with different levels oftreatment resistance. Definitions of response and remission rates varied between studies.Observed response rates after switching to any of the classes of antidepressants varied between12% and 86%. Remission rates varied between 7% and 82%. The number of previous treatments withantidepressants was negatively correlated with treatment outcome. Rates of dropout due to side-effects varied considerably across agents (5-39%). Switching to venlafaxine showed a modest andclinically equivocal benefit over SSRIs (Number Needed to Treat = 13 (9.1-25.0))
ConclusionsAfter a first SSRI any switch within or between classes of antidepressants appear legitimate(second SSRI, novel dual acting antidepressants, selective noradrenergic or noradrenergic/dopaminergic agents, or TCA or mianserin). No unequivocal evidence is available to prove anadvantage of a between class switch. More guidance by randomized empirical studies is needed.Clinical implications and methodological considerations for future studies are discussed.

ChapterReview of switching in MDD
5
75
Introduction
Major depressive disorder (MDD) is one of the most prevalent and disabling illnesses inpsychiatry.1 For the treatment ofMDD, several national clinical guidelines were developed.2-9 Inthese guidelines, pharmacotherapy is among the most important treatments; mostly SelectiveSerotonin Reuptake Inhibitors (SSRIs) are the antidepressants of first choice. However, only 50 to60% of patients respond to the first antidepressant given.10;11 In a case of non-response, alltreatment guidelines recommend three major strategies: 1) increasing the dose of theantidepressant (dose-escalation), 2) switching to another antidepressant of the same or differentclass, and 3) augmenting the antidepressant by adding a second drug that by itself is not anantidepressant. By various authors, a fourth strategy of combination of antidepressants isproposed.12-15
Surprisingly very little systematic evidence exists to date to underscore the recommendationsfor non-responders. One Cochrane review summarizes randomized, controlled trials (RCTs) ofstrategies in patients non-responsive to at least 4 weeks of an antidepressant at therecommended dose.16 With a thorough methodology, 16 RCTs were selected. Unintentionally, thestudies included in this review represented more heterogeneous, difficult-to-treat populations,referred to as treatment-resistant depression (TRD). Although little information on previoustreatments was found, the included studies especially considered tricyclic antidepressant (TCA)non-responders. The switch-options that were investigated in the included studies did not reflectclinical practice of switching to another antidepressant (one of the above recommendations), butused a variety of other drugs (oestrogen, benzodiazepines, ketoconazole, olanzapine). For theaugmentation studies, meta-analyses were performed with 2 trials of lithium-augmentation and 3pindolol-trials. A clinically significant benefit was found only for lithium augmentation. Thus, thisreview does not provide helpful information for clinicians in the case of non-response to a (first)SSRI.
Strategies for non-response have been summarized in several narrative reviews, focusing onall strategies together,17-28 switching,12;13;29-32 augmentation,12;13;31;32 or combination.12-15;33 Doseescalation was summarized in 2 meta-analyses,34;35 1 narrative 36 and 1 recent systematic review.37
The evidence for lithium augmentation was also summarized in meta-analyses by Bauer et al.38;39
After dose-escalation, switching antidepressants is widely practiced.40-42 Switching to adifferent pharmacological class seems to be preferred by clinicians.43 The above narrative reviewsof switching strategies altogether provided a substantial overview. However, each reviewindividually was limited in its presentation, predominantly by a lack of a well defined searchstrategy, and none of the reviews presented data on critical appraisal of the identified studies asproposed by the Cochrane collaboration.44 The general conclusion today is that there is limitedevidence available for switching antidepressants, and that there is no clear proven advantage ofone switch option over the others. Additionally, recently the results of a large study designed toelucidate sequential treatment strategies after non-response became available (SequencedTreatment Alternatives to Relieve Depression, STAR*D).45 This study provided prospective dataon response and remission rates after randomized treatment allocations in patients who were notin remission after one to four sequential steps of treatment (further referred to as STAR*D level I-IV).
Therefore, our primary objective was to systematically review and appraise the availableresearch focusing on switching strategies for SSRI non-responders in MDD, including the recentSTAR*D results. A secondary aim was to acknowledge and investigate the expected differentlevels of TRD as a source of variation between studies. Our principal question was whether theavailable evidence justifies distinct recommendations for next-step strategies after non-responseto a first SSRI. We performed a systematic review following the Cochrane methodology andperformed a meta-analysis of two switch options after a first SSRI: a second SSRI versus aserotonin-norepinephrin reuptake inhibitor (SNRI).

Chap
ter
Chapter 5
5
76
Methods
Studies included in the reviewWe expected very few randomised, controlled, switch-studies a-priori, despite the widespreadavailability of SSRIs during the last decade. As best-available evidence, we included open andrandomized studies in which at least 50% of participants used an SSRI previously in the currentdepressive episode. Thus, we excluded studies describing switching from TCAs to SSRIs. Studiesperformed in populations with TRD were also included if previous use of an SSRI (in ≥50% ofsubjects) was unambiguously documented.
Identification and selection of articlesWe performed systematic literature searches (updated until February 10th 2005) in four databases(MEDLINE, EMBASE, CINAHL, and PsychInfo; all indexed years). In the absence of specifickeywords for switching, we performed ‘sensitive’ searches using free text words with wildcards($): “switch$” or (“alternat$” adj5 “treat$”) or (“alternat$” adj5 “therap$”) in combination withthe Cochrane Collaboration search-filter for RCTs, the Cochrane Collaboration Depression Anxietyand Neurosis group search-filter for MDD and MeSH-terms for antidepressants (in combinationwith additional text words for all antidepressive agents). We limited searches to adults andhumans, but did not apply language restrictions. Full queries are available on request. In addition,we included four identified studies released after these searches, including three studies from theSTAR*D trial.46-49
The first and second authors (H.G.R., J.H.) independently screened titles and abstracts andselected articles on the basis of design and focus on switching antidepressants after SSRI-treatment. Agreement on exclusion of irrelevant articles was 99.1%, with a Cohen’s κ for interrateragreement of 0.62 (κ values between 0.45-0.75 indicate 'substantial' agreement; values above0.75 indicate 'almost perfect' agreement).50 We resolved discrepancies between initial selectionby discussion and consensus.
The first author (H.G.R.) judged all potentially relevant articles according to specific inclusionand exclusion criteria (full criteria available on request). In case of doubt the article was fully readand assigned thereafter. We retrieved additional cross-references, and checked reference lists ofidentified narrative reviews. We considered double-publications together to reveal the maximumof available information.
Critical Appraisal and summary The first author (H.G.R.), a certified epidemiologist, critically appraised and abstracted thearticles, using standardized forms derived from the Dutch Institute of Healthcare Improvement51
and the Agency of Healthcare Policy and Research (AHCPR).5 We used the same items for criticalappraisal as proposed by the Scottish Intercollegiate Guidelines Network52 and Sackett et al.53 Weassigned a ‘level of evidence’ (LoE; see table 4.1) to each study.51 Levels of evidence are basedupon the methodological robustness of studies. In the results section, the LoE of the supportingscientific evidence (A1-D) is indicated. We extracted data on efficacy and tolerability from eachstudy. As primary efficacy outcome we took the percentage response or remission on anintention to treat (ITT) basis. If several scales were used, we a priori preferred data for theHamilton depression rating scale (HDRS;54 17-item version or other versions); otherwise we useddata from the Montgomery Åsberg depression rating scale (MADRS),55 clinical global impressionsscale (CGI),56 or other applied scales (e.g. the 16-item quick inventory for depressive symptoms-self-rated (QIDS-SR16).57 For tolerability, we took the dropout rate due to side effects as primarymeasure, followed by the overall dropout rate.
To assess judgement-bias by one person who performed the critical appraisal, we measuredinterrater variation in a slightly different set of 12 publications. Every other author (J.H., J.A.S.,A.H.S.) critically appraised 4 publications. Cohen's κ values for the appraisal-items were 0.49 (for'validity of the study') and 0.86 (for 'concealment of allocation'), while complete agreement

ChapterReview of switching in MDD
5
77
existed for the appraisal-items 'randomization of the study', 'level of evidence' and 'dataextraction' (κ= 1.0). These results are in line with other reports of interrater agreement inappraisal of psychiatric research.58
We first described a qualitative summary with discussion of the results, restrictions,methodological flaws and external validity of the studies in an evidence table and a separatedocument, of which a summary is provided in this article. For each study we indicated the level oftreatment resistance as proposed by Thase et al.10;24 If possible, we calculated risk-differences andcorresponding Numbers Needed to Treat (NNT) and Harm (NNH), with 95% confidence intervals(95% CIs). Because of the lack of homogenous randomised studies, we refrained from pooling in ameta-analysis, except for the three studies comparing the venlafaxine (SNRI) vs. second SSRIswitch. We grouped antidepressants into six classes following the classification of the AHCPR.5
Results
We selected thirty-one studies for this review. Figure 5.1 shows the search results and selection ofstudies. Table 5.1 summarizes the included studies. A table of 8 excluded studies59-66 is available onrequest.
Figure 5.1. Selection of reported studies.
Abbreviations: BUP = bupropion, MAO-I= irreversible inhibitor of monoamine-oxidase, MIAN = mianserin, MIR = mirtazapine,NEF = nefazodone, REB = reboxetine, RIMA = reversible inhibitor of MAO, SSRI = selective serotonin reuptake inhibitors, TCA= tricyclic antidepressant, VLX = venlafaxine
�������� "#$%&"'"$#�(L�)L)&$*+&",�)$+-,.$)���������
�������� ���M�� �������������M���������������
���������� ����� ������������������M�N������� ��
���������� �� �����������
�O����� �������!��� " #�O���$���������� ���$��������������$�����������������%��
�O��������������� ����������&� ����������� &���� �����������'�()��������'�*�+�������'�,-.�����%�'�/�0 "������%�������������������� &���M�����������'-������1�N��� � %����������2�'��������������$�!��������
��������/�� �345 �*�+�����%��
��������()� �/��*������ P�
� (N������������������$ P ���$������$���O�%�$��LQ �������2�O�������������$�N���������������$$������$�� � ��$L$�����$����$�
����������, �,-.������ P�
��������/�0 "���������
����������/��������
�O���345 �/���������Q
�O���,-.�����%� Q �O���/�0"������%�Q
�O���()������%� Q�O�������������� Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q
�O���/�0"������%�Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q
�O���,-.�����%� Q �O���/�0"������%�Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q
�O���345 �/���������Q
�O���,-.�����%� Q �O���/�0"������%�Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q
�O���345 �/���������Q
�O�������������� Q �O���()������%� Q
�O���345 �/���������Q
�O�������������� Q
�O���3456/������%�Q
�O���()������%� Q
�O���345 �/���������Q
�O�������������� Q
�O���/�0"������%�Q
�O���3456/������%�Q
�O���()������%� Q
�O���345 �/���������Q
�O�������������� Q
�O���,-.�����%� Q �O���/�0"������%�Q
�O���()������%� Q
�O���345 �/���������Q
�O�������������� Q�O�������������� Q �O���()������%� Q�O�������������� Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q
�O���/�0"������%�Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q
�O���,-.�����%� Q �O���/�0"������%�Q
�O���3456/������%�Q
�O���()������%� Q�O�������������� Q

78
Chapter 5
Chap
ter
5
Tabl
e 5.
1. Ef
fect
iven
ess
of s
witc
hing
aft
er ≥
1 SSR
I: Se
lect
ed s
tudi
es.
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Co
mpa
rison
* O
utco
me†
R
emar
ks
5.1.A
. to
2nd
SSRI
Bald
omer
o et
al.
(200
5)46
See
unde
r 5.2
.C.
Brow
n et
al.
(199
5)71
C 11
2 M
DD
Out
P TR
D-I
Ope
n m
ultic
ente
r tria
l of
FLX
into
lera
nt
subj
ects
(8
wee
ks)
SER
50-2
00m
g -
Res
pons
e (C
GI-I
≤2)
71.
8%
Dro
pout
ove
rall =
21.4
% D
ropo
ut S
E =
9.8%
No
plac
ebo
cont
rol.
FLX-
into
lera
nce
befo
re
switc
h on
ly.
Cala
bres
e et
al.
(200
3)73
C 55
M
DD
Out
PTR
D-I
Ope
n m
ultic
ente
r tria
l of
FLX
into
lera
nt
subj
ects
(6
wee
ks)
CIT
20-4
0mg
- R
espo
nse
(CG
I-I ≤
2) 6
5.5%
Dro
pout
ove
rall =
5%
Dro
pout
SE
= 0%
No
plac
ebo
cont
rol.
FLX-
into
lera
nce
befo
re
switc
h on
ly. N
o m
inim
um le
vel o
f HD
RS
requ
ired
for s
tudy
-ent
ranc
e. S
tart
of C
IT
afte
r pla
cebo
was
h-ou
t of 2
-4 w
eeks
.
Joff
e et
al.
(199
6)67
C 55
M
DD
O
utP
TRD
-I
Ret
rosp
ectiv
e st
udy
of
patie
nts
who
wer
e tr
eate
d w
ith a
sec
ond
SSRI
in 2
yea
rs. (≥5
w
eeks
)
FLX
(n=
12)
FLV
(n=
9)
PAR
(n=
11)
SER
(n=
23)
- R
espo
nse
(CG
I-I ≤
2) 5
1% N
o si
gnif
ican
t di
ffer
ence
s be
twee
n va
rious
com
bina
tions
of
sw
itchi
ng. N
o SE
repo
rted
.
No
plac
ebo
cont
rol.
Met
hodo
logi
cally
poo
r:
retr
ospe
ctiv
e st
udy,
unc
lear
def
initi
on o
f in
itial
non
-rep
onse
, sm
all n
umbe
rs, n
o ch
arac
teris
tics
of p
opul
atio
n. U
ncle
ar a
fter
ho
w m
any
wee
ks re
spon
se w
as d
eter
min
ed.
Poiri
er e
t al.
(199
9)74
;87
See
unde
r 5.1.
C.
Rus
h et
al.
(200
6)47
A2
727
MD
D
Out
P TR
D-I
3 ar
m M
ulti
cent
er
unbl
inde
d R
CT
(14
wee
ks)
1. VL
X-XR
75-
375m
g 2.
BU
P 15
0-40
0mg
SER
50-
200m
g R
emis
sion
(HD
RS17
≤7)
VLX
= 2
4.8%
, BU
P =
21.3
%, S
ER =
17.6
% N
NT V
LX-S
ER =
14 (7
.0-∞
),
NN
T BU
P-SE
R =
28 (9
.3-∞
) NN
T VLX
-BU
P =
29 (9
.2-
∞) R
espo
nse
(≥50
% in
QID
S 16)
VLX
= 2
8.2%
, BU
P =
26.1%
, SER
= 2
6.7%
NN
T VLX
-SER
= 6
6 (1
0.6-∞
), N
NT B
UP-
SER
= 18
9 (1
3.4-∞
) NN
T VLX
-BU
P =
49 (1
0.1-∞
) Dro
pout
SE V
LX =
21.2
%, B
UP
= 27
.2%,
SER
= 2
1.0%
(Non
sign
ific
ant
diff
eren
ces)
Unb
linde
d st
udy,
blin
ded
asse
ssor
s, n
o pl
aceb
o-gr
oup.
Met
hodo
logi
cally
wel
l pe
rfor
med
eff
ectiv
enes
s tr
ial.
Part
icip
ants
all
rece
ived
CIT
20-
60m
g as
prio
r tre
atm
ent
(leve
l II S
TAR
*D).
Due
to h
igh
dose
s of
CIT
in
‘leve
l 1’ 4
07/7
27 (5
6%) o
f sub
ject
s in
this
stu
dy
wer
e cl
assi
fied
as C
IT-in
tole
rant
. No
was
hout
ap
plie
d.
Thas
e et
al.
(199
7)68
C 10
6 M
DD
O
utP
TRD
-I
Ope
n m
ultic
ente
r tria
l of
SER
non
resp
onde
rs
or in
tole
rant
sub
ject
s (6
wee
ks)
FLX
20-6
0mg
- R
espo
nse
(≥50
% in
HD
RS 1
7) O
vera
ll 62
%;
initi
alin
tole
rant
sub
ject
s 71
%, in
itial
no
nres
pond
ers
58%.
No
plac
ebo
cont
rol.
Non
resp
onse
to S
ER w
as
dete
rmin
ed re
tros
pect
ivel
y (in
mos
t pa
tient
s).
Thas
e et
al.
(200
1)69
C 57
M
DD
O
utP
TRD
-I
Ope
n st
udy
of
pros
pect
ivel
y de
term
ined
FLX
no
nres
pond
ers
(12
wee
ks)
CIT
20m
g (d
osag
es
coul
d be
incr
ease
d to
60m
g)
- R
espo
nse
(CG
I-I ≤
2) 6
3% (≥
50%
in H
DRS
24)
46%
Dro
pout
ove
rall =
18%;
Dro
pout
SE
= 5%
W
ell p
erfo
rmed
ope
n st
udy.
Unk
now
n pl
aceb
o re
spon
se ra
te. T
oler
ance
for C
IT
afte
r FLX
is g
ood,
des
pite
dire
ct s
witc
h. N
o in
crea
sed
rate
of S
E in
firs
t wee
ks o
f stu
dy.

Review of switching in MDD
79
Chapter5
Thas
e et
al.
(200
2)72
C 61
M
DD
O
utP
TRD
-I
Ope
n st
udy
in P
AR
into
lera
nt p
atie
nts
(6 w
eeks
)
CIT
20m
g (d
osag
es
coul
d be
incr
ease
d to
40m
g)
- R
espo
nse
(CG
I-I ≤
2) 5
6% D
ropo
ut o
vera
ll =
13%;
D
ropo
ut S
E =
10%
Ope
n st
udy,
no
min
imal
HD
RS-
scor
e on
en
tran
ce. U
nkno
wn
plac
ebo
resp
onse
rate
. To
lera
nce
for C
IT a
fter
PA
R is
goo
d, 1
wee
k w
asho
ut. R
ecur
renc
e of
the
sam
e SE
as
durin
g PA
R tr
eatm
ent i
s 5-
30%.
Zara
te e
t al.
(199
6)70
C 39
M
DD
,Bi
pD,
SchA
, O
CD
InP
TRD
->I
Ret
rosp
ectiv
e st
udy
of
patie
nt c
hart
s in
pa
tient
s pr
evio
usly
us
ing
FLX
(var
ious
du
ratio
ns o
f fol
low
-up)
SER
mea
n do
se 9
3 ±6
2 m
g -
Res
pons
e (C
GI-I
≤2)
41.
9% o
n di
scha
rge,
26%
at
follo
w-u
p.M
etho
dolo
gica
lly v
ery
poor
stu
dy:
retr
ospe
ctiv
e, n
onsy
stem
atic
follo
w-u
p, S
ER
was
not
pre
scrib
ed fo
llow
ing
FLX,
he
tero
gene
ous
stud
ypop
ulat
ion
with
he
tero
geno
us h
isto
ry o
f pre
viou
s tre
atm
ents
(in
clud
ing
MA
O-I,
ECT
). C
onfo
undi
ng b
y no
n-co
mpl
ianc
e an
d ad
ditio
nal p
harm
acot
hera
py
(in 6
9% o
f pat
ient
s). P
ossi
ble
reca
ll-bi
as.
5.1.B
. to
a TC
A o
r mia
nser
in
Fava
et a
l (20
06)48
A2
234
MD
D
Out
P TR
D-II
Mul
ti ce
nter
unb
linde
d RC
T (1
4 w
eeks
)
NO
R 50
-200
mg
MIR
15-6
0mg
Rem
issi
on (H
DRS
17 ≤
7) N
OR
= 19
.8%,
MIR
=
12.4
% N
NT N
OR-
MIR
= 14
(6.0
-∞) N
ot d
iffer
ent
in le
vel I
I int
oler
ant g
roup
. Res
pons
e (≥
50%
in
QID
S 16)
NO
R =
16.5
%, M
IR =
13.5
% N
NT N
OR-
MIR
= 3
2 (8
.1-∞
) Dro
pout
SE N
OR
= 34
.7%,
MIR
=
33.3
%
Unb
linde
d st
udy,
blin
ded
asse
ssor
s, n
o pl
aceb
o-gr
oup.
Met
hodo
logi
cally
wel
l pe
rfor
med
eff
ectiv
enes
s tr
ial.
Part
icip
ants
re
ceiv
ed C
IT 2
0-60
mg
and
VLX
or B
UP
or S
ER
or a
ugm
enta
tion
of C
IT w
ith B
UP
or B
US
or
CBT
(leve
l III
STA
R*D
). 5
2.1%
was
con
side
red
leve
l II i
ntol
eran
t. B
lood
-leve
ls o
f NO
R al
low
ed b
ut n
ot o
blig
ator
y fo
r dos
ing
(33.
9%
mea
sure
d). N
o w
asho
ut a
pplie
d.
Ferr
eri e
t al.
(200
1)75
B 10
3 M
DD
In
P +
Out
P TR
D-I
3 ar
m m
ultic
ente
r RCT
of
FLX
20m
g-N
R (6
wee
ks)
1. M
IAN
60m
g 2.
FLX
20m
g +
MIA
N
60m
g
3. F
LX 2
0mg
Res
pons
e (≥
50%
in H
DR
S 17)
Mia
nser
ine
= 48
.5%,
Flu
oxet
ine
= 37
% N
NT M
IAN
-FLX
=9 (2
.9-
∞);
NN
T MIA
N+F
LX-F
LX=4
(2.1-
34.1)
Rem
issi
on
(HD
RS 1
7≤8)
Mia
nser
ine
= 36
%, F
luox
etin
e =
18%
NN
T MIA
N-F
LX=6
(2.6
-∞);
NN
T MIA
N+F
LX-
FLX=
4 (2
.2-2
3.9)
Dro
pout
SE
NN
HM
IAN
-FLX
=5
(2.6
-10.4
); N
NH
MIA
N+F
LX-F
LX=1
6 (6
.8-∞
) D
ropo
ut o
vera
ll NN
HM
IAN
-FLX
=6 (2
.6-∞
);
NN
HM
IAN
+FLX
-FLX
=64
(4.9
-∞)
Met
hodo
logi
cally
sou
nd, s
mal
l sam
ple
size
fo
r thr
ee a
rms,
lim
ited
pow
er; i
n M
IAN
-gro
up
due
to lo
ng h
alf-l
ife o
f FLX
, firs
t wee
ks a
lso
sort
of ‘
com
bina
tion’
-the
rapy
. Low
dos
e of
co
ntin
ued
FLX
in re
fere
nce
grou
p. N
o w
asho
ut a
pplie
d, d
irect
sw
itch
asso
ciat
ed
with
incr
ease
d in
tole
ranc
e an
d dr
opou
t .
Nie
renb
erg
et a
l. (2
003)
76C
92
MD
D
Out
P TR
D->
II
Ope
n ph
ase
of N
OR
tr
eatm
ent p
rece
ding
a
2nd R
CT14
0
(6 w
eeks
)
NO
R 10
0mg
(adj
uste
d to
ac
hiev
e 10
0 ng
/ml)
- R
espo
nse
(≥50
% in
HD
RS 1
7) =
42.
4%
Rem
issi
on (H
DRS
17≤7
) = 11
.9%
Dro
pout
ove
rall
= 34
.7%
TRD
def
ined
as
1-5 fa
iled
adeq
uate
tria
ls
durin
g cu
rren
t epi
sode
(mea
n ±S
D 2
.3
±1.5
). 9
5.7%
of p
atie
nts
was
trea
ted
with
≥1
SSR
I. U
nkno
wn
plac
ebo
resp
onse
rate
.
Nol
en e
t al.
(198
8)77
C 31
M
DD
InP
TRD
-III
Blin
ded
cons
ecut
ive
ther
apy
afte
r 4 w
eeks
of
FLV
(4
wee
ks)
OXA
100-
300m
g -
Res
pons
e (≥
50%
in H
DRS
17) =
38.
7% R
elap
se
= 19
.4%
with
in 6
mon
ths
Mod
ifie
d IT
T an
alys
is, e
xclu
ding
pat
ient
s w
ho d
ropp
ed-o
ut in
firs
t 2 w
eeks
. TR
D:
patie
nts
used
≥1 T
CA b
efor
e tr
eatm
ent w
ith
FLV.
No
data
on
drop
outs
.
Tabl
e 5.
1. Ef
fect
iven
ess
of s
witc
hing
aft
er ≥
1 SSR
I: Se
lect
ed s
tudi
es. (
Cont
inue
d)
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Co
mpa
rison
* O
utco
me†
R
emar
ks

80
Chapter 5
Chap
ter
5
Pese
low
et a
l. (1
989)
78C
15 +
10
MD
D
Out
P TR
D-I
Blin
ded
cros
s-ov
er
desi
gn w
ith o
rigin
al
rand
omiz
atio
n (6
wee
ks ?)
IMI 6
5-27
5mg
- R
espo
nse
(≥50
% in
HD
RS u
nkno
wn
or C
GI-I
≤2
) PA
R
IMI =
73%
M
etho
dolo
gica
lly p
oor:
unc
lear
des
crip
tion
of s
tudi
ed p
opul
atio
n, li
mite
d pr
esen
tatio
n of
dat
a. D
ata
of in
itial
non
resp
onde
rs to
IMI
switc
hed
to P
AR
als
o pr
ovid
ed.
Thas
e et
al.
(200
2)79
;80
C 11
7 Ch
ron
MD
D,
MD
D +
D
ys
Out
P TR
D-I
Blin
ded,
mul
ticen
ter
cros
s-ov
er d
esig
n w
ith
orig
inal
rand
omiz
atio
n(1
2 w
eeks
)
IMI 5
0-30
0mg
- R
espo
nse
(CG
I-I ≤
2, ≥
50%
in H
DRS
24,
HD
RS 2
4≤15
and
CG
I-S ≤
3) S
ER
IMI =
44.
4%
Rem
issi
on (H
DRS
24≤7
and
CG
I-I ≤
2) S
ER
IM
I = 2
3% D
ropo
ut o
vera
ll = 2
5%; D
ropo
ut S
E =
9%
Wel
l-per
form
ed s
tudy
.Unk
now
n pl
aceb
o re
spon
se ra
te. D
ata
of in
itial
non
resp
onde
rs
to 12
wee
ks IM
I sw
itche
d to
SER
als
o pr
ovid
ed. D
ue to
abs
ence
of s
econ
d ra
ndom
izat
ion
only
tent
ativ
e co
mpa
rison
s w
ith s
witc
h IM
I S
ER a
vaila
ble.
5.1.C
. to
mir
taza
pine
, nef
azod
one,
ven
lafa
xine
(dua
l act
ion
agen
ts)
Bald
omer
o et
al.
(200
5)46
B 30
97
MD
D +
M
inD
+
Dys
O
utP
TRD
-I
Mul
ticen
ter,
ope
n de
sign
; RCT
of V
LX v
s CA
in S
SRI-N
R or
in
tole
rant
pat
ient
s (2
4 w
eeks
)
VLX
75-2
25m
g CA
: FLX
, PA
R,
CIT
20-6
0mg
SER
50-
200m
g M
IR 15
-45m
g
Res
pons
e (≥
50%
in H
DR
S 17)
: VLX
= 7
7.3%
, SS
RIs
= 7
1.1%
NN
T VLX
-SSR
I = 17
(10.
5-35
.0)
Rem
issi
on (H
DRS
17 ≤
7): V
LX =
59.
3%, S
SRIs
=5
2.1%
NN
T VLX
-SSR
I = 14
(9.1-
29.3
) HD
RS17
-sc
ores
diff
er s
igni
fica
ntly
but
clin
ical
ly
irrel
evan
t at w
eek
12 a
nd 2
4Dro
pout
ove
rall:
VL
X =
19.6
%, C
A =
23.
3% N
NH
VLX-
CA =
27
(15.
1-12
0) D
ropo
ut S
E: V
LX =
2.3
%, C
A =
1.7%
N
NH
VLX-
CA 16
1 (62
.1-∞
)
Larg
e ra
ndom
ized
but
unb
linde
d st
udy.
So
me
met
hodo
logi
cal p
robl
ems.
63.
3% o
f in
clud
ed p
atie
nts
prev
ious
ly u
sed
a SS
RI.
Incl
usio
n of
8.7
% M
inD
. No
diff
eren
tia
tion
of
firs
t-SS
RI i
ntol
eran
t and
unr
espo
nsiv
e pa
tient
s. M
odif
ied
ITT
anal
ysis
of ≥
wee
k 4
com
plet
ers;
in C
A-t
reat
ed s
witc
h gr
oup
22.7
% re
ceiv
ed a
non
-SSR
I. Ba
selin
e H
DR
S-sc
ores
si
gn. h
ighe
r in
VLX-
grou
p; d
iffer
entia
l los
s to
fo
llow
up
26.2
% VL
X vs
36.
2% C
A O
nly
3 tim
e-po
ints
ove
r 24
wee
ks. N
o se
para
te
dich
otom
ous
data
for w
eek
12 re
spon
se/
rem
issi
on
Fava
et a
l. (2
001)
81C
94
MD
D
Out
P TR
D-I
Mul
ticen
ter,
ope
n de
sign
(RCT
of d
irect
sw
itch
vs. w
asho
ut)
(8 w
eeks
)
MIR
15-4
5mg
- R
espo
nse
(≥50
% in
HD
RS 1
7) S
SRI N
R =
48%,
SS
RI I
ntol
.= 5
3% D
ropo
ut o
vera
ll = 4
3%; D
ropo
ut
SE =
26%
Met
hodo
logi
cally
sou
nd. U
nkno
wn
plac
ebo
resp
onse
rate
. Was
hout
-pha
se o
ffer
s no
ad
vant
ages
.
Fava
et a
l (20
06)48
See
unde
r 5.1.
B
Kapl
an (2
002)
82C
73
MD
D
Out
P TR
D-I
Ret
rosp
ectiv
enat
ural
istic
stu
dy o
f SSR
I-NR
or
non-
sust
aini
ng S
SRI
resp
onde
rs
(6-8
wee
ks +
follo
w-
up)
VLX
50-4
00m
g -
Res
pons
e (H
DR
S 25≤
10 a
nd P
GI-2
1 ≥5)
= 8
6%
Rem
issi
on (H
DRS
25≤8
) = 8
2% D
ropo
ut S
E =
5.5%
Met
hodo
logi
cally
ver
y po
or: o
pen,
unb
linde
d de
sign
, 1 re
sear
cher
, ret
rosp
ectiv
e da
ta
obta
ined
. Mild
dep
ress
ion
incl
uded
als
o.
Unc
lear
, but
pro
babl
e se
lect
ion-
bias
. R
ecru
itmen
t of S
SRI r
espo
nder
s w
ho d
id n
ot
sust
ain
thei
r res
pons
e (5
2%) m
ight
incr
ease
re
spon
se ra
te. I
TT-r
esul
ts n
ot m
entio
ned
in
stud
y.
Tabl
e 5.
1. Ef
fect
iven
ess
of s
witc
hing
aft
er ≥
1 SSR
I: Se
lect
ed s
tudi
es. (
Cont
inue
d)
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Co
mpa
rison
* O
utco
me†
R
emar
ks

Review of switching in MDD
81
Chapter5
Mis
chou
lon
et a
l. (2
004)
83C
13
MD
D
Out
P TR
D->
I
Ope
n de
sign
of S
SRI-
NR
or i
n to
l era
nt
patie
nts
(12
wee
ks)
NEF
300
-600
mg
- R
espo
nse
(>50
% H
DRS
6 an
d/or
CG
I-S ≤
2) 3
1%
Dro
pout
SE
= 39
%Sm
all s
ampl
e (p
ilot s
tudy
). U
nkno
wn
plac
ebo
resp
onse
rate
; 61.5
% at
triti
on;
espe
cial
ly in
pre
viou
s FL
X-us
ers.
Was
hout
of
4-7
days
app
lied.
Het
ero
ge n
eous
gro
up o
f TR
D a
pp. 4
6% ≥
stag
e II.
No
sign
. diff
eren
ces
in re
spon
se ra
tes
and
side
eff
ects
com
pare
d to
13 p
atie
nts
trea
ted
with
NEF
as
first
ap
plie
d an
tidep
ress
ant (
but l
ow p
ower
).
Mitc
hell
et a
l. (2
000)
84C
312
MD
D
Sett
ing?
TR
D-≥
I
Mul
ticen
ter,
ope
n un
blin
ded
desi
gn
(8 w
eeks
)
VLX
75-3
00m
g-
Res
pons
e (>
50%
in M
AD
RS) =
52.
6%
Rem
issi
on (M
AD
RS
<12)
= 4
0.7%
Dro
pout
SE
= 11
%
Met
hodo
logi
cally
wel
l. U
ncle
ar s
ettin
g.
Unk
now
n pl
aceb
o re
spon
se ra
te. U
ncle
ar
whi
ch p
ropo
rtio
n us
ed ≥
1 SSR
I (41
-68%
). Pr
obab
ly c
hron
ical
ly d
epre
ssed
sub
ject
s.
De
Mon
tigny
et a
l. (1
999)
85C
152
MD
D In
P &
O
utP
TRD
-I
Mul
ticen
ter,
ope
n de
sign
(8
wee
ks)
VLX
75-3
75m
g -
Res
pons
e (≥
50%
in H
DR
S 21)
= 5
8%; (≥5
0%
in
MA
DR
S) =
62%
; (CG
I-S ≤
3) =
66%
Rem
issi
on
(≥75
% in
HD
RS21
) = 2
1% D
ropo
ut S
E =
7.9%
TRD
: at l
east
1 pr
evio
us T
CA o
r SSR
I or M
OC
or T
RA
Z or
NEF
, maj
ority
of p
atie
nts
used
a
SSR
I. M
ean
dura
tion
of e
piso
de 2
yea
rs
(ran
ge 2
mon
ths
- 12.
5 ye
ars)
. Unk
now
n pl
aceb
o re
spon
se ra
te.
Nie
renb
erg
et a
l. (1
994)
86C
70
MD
DIn
P &
O
utP
TRD
-III
2 ce
nter
, ope
n de
sign
(1
2 w
eeks
) VL
X 50
-450
mg
- R
espo
nse
(≥50
% in
HD
RS21
) = 3
2.9%
; (≥5
0%
in
MA
DR
S) =
30%
; (CG
I-I ≤
2) =
40%
Rem
issi
on
(HD
RS 2
1≤8)
= 15
.7%;
(MA
DRS
≤12
) = 18
.6%;
(C
GI-I
= 1)
= 2
2.9%
Dro
pout
SE
= 9.
6%
TRD
: at l
east
≥3
drug
s of
≥2
diff
eren
t cla
sses
, ≥1
TCA
and
≥1 a
ugm
enta
tion.
Unc
lear
wha
t pr
opor
tion
of p
atie
nts
used
≥1 S
SRI.
Chro
nica
lly d
epre
ssed
gro
up (m
edia
n du
ratio
n 2.
5 ye
ars)
. Unk
now
n pl
aceb
o re
spon
se ra
te.
Poiri
er e
t al.
(199
9)74
;87
A2
123
MD
DIn
P &
O
utP
TRD
-II
Mul
ticen
ter R
CT
(4 w
eeks
) VL
X 75
-300
mg
PAR
20-4
0mg
Res
pons
e (≥
50%
in H
DR
S 17)
: VLX
= 4
5.0%
, PA
R =
29.
0% N
NT V
LX-P
AR
= 7
(3.0
-∞)
Rem
issi
on (H
DRS
17≤1
0) :
VLX
= 36
.7%,
PA
R =
17
.7%
NN
T VLX
-PA
R =
6 (2
.9-2
8.9)
Dro
pout
SE:
VL
X =
8.2%
, PA
R =
4.8
%NN
HVL
X-PA
R =
30 (8
.3-
∞)
Met
hodo
logi
cally
wel
l. Sh
ort f
ollo
w-u
p du
ring
stud
y. D
osin
g-sc
hedu
les
are
diff
eren
t be
twee
n VL
X an
d PA
R.
Reyn
aert
-Dup
uis
et a
l. (2
002)
88C
688
MD
DIn
P &
O
utP
TRD
-I
Mul
ticen
ter,
na
tura
listic
, ope
n de
sign
(6
wee
ks)
VLX
75-3
75m
g -
Res
pons
e (≥
50%
in H
DR
S 21)
= ±
61%
(inpr
evio
us S
SRI-t
reat
ed p
atie
nts)
86
.3%
of p
atie
nts
wer
e sw
itche
d to
VLX
due
to
inef
fica
cy, o
f 41.7
% w
ho w
ere
switc
hed
from
a S
SRI s
epar
ate
resp
onse
rate
s ar
e gi
ven.
Imm
edia
te s
witc
hing
app
lied
(exc
ept
from
MA
O-Is
). U
ncle
ar p
rese
ntat
ion
of d
ata.
D
ropo
ut ra
te n
ot m
entio
ned.
Typ
e of
pr
evio
us S
SRI n
ot s
igni
fica
ntly
aff
ecte
d VL
X-ef
ficac
y.
Rus
h et
al.
(200
6)47
See
unde
r 5.1.
A
Tabl
e 5.
1. Ef
fect
iven
ess
of s
witc
hing
aft
er ≥
1 SSR
I: Se
lect
ed s
tudi
es. (
Cont
inue
d)
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Co
mpa
rison
* O
utco
me†
R
emar
ks
82
Chapter 5
Chap
ter
5
Saiz
-Rui
z et
al.
(199
8)89
C 69
M
DD
O
utP
TRD
-I
Mul
ticen
ter,
na
tura
listic
, ope
n de
sign
(2
4 w
eeks
)
VLX
75-3
75m
g -
Res
pons
e (≥
50%
in H
DR
S 17)
= 6
9.6%
; (CG
I-I
≤2) =
63.
8% D
ropo
ut o
vera
ll = 3
0.4%
Dro
pout
SE
= 8.
7% S
E oc
cur i
n 54
%
Sele
ctio
n of
NR
to a
pre
viou
s SS
RI in
at l
east
a
stan
dard
dos
e fo
r 4 w
eeks
. Lim
ited
pres
enta
tion
of d
ata.
In th
e pa
per a
mod
ifie
d IT
T an
alys
is is
app
lied
for w
eek
4 co
mpl
eter
s.
Endp
oint
of s
tudy
is o
nly
repo
rted
for w
eek
24.
Wan
et a
l. (2
003)
90C
24
MD
D
Sett
ing?
TR
D-≥
II
Retr
ospe
ctiv
e ch
art
revi
ew o
f con
secu
tive
sub
ject
s w
ho fa
iled
resp
onse
to ≥
1 TCA
and
≥1
SSR
I (2
wee
ks -
3 ye
ars)
MIR
15-4
5mg
- R
espo
nse
(CG
I-I ≤
2) =
16.7
% Pa
rtia
l res
pons
e (C
GI-I
=3)
= 2
0.8%
Dro
pout
SE =
20.
8%
Met
hodo
logi
cally
ver
y po
or. I
n 17
.2%
of
elig
ible
pat
ient
s da
ta w
ere
insu
ffic
ient
for
incl
usio
n. H
ighl
y tr
eatm
ent r
esis
tant
po
pula
tion
(ave
rage
7 p
revi
ous
drug
-tria
ls
[ran
ge 2
-13])
. Unc
lear
wha
t res
pons
e in
dica
ted
switc
h to
MIR
. CG
I-dat
a de
term
ined
by
char
t rev
iew
. Chr
onic
de
pres
sion
in 4
5.8%
. Hig
h le
vel o
f co
mor
bidi
ty w
ith a
nxie
ty d
isor
ders
. Co
med
icat
ion
with
ant
idep
ress
ants
and
an
tipsy
chot
ics
in 4
1.7%.
5.1.D
. to
rebo
xeti
ne o
r bup
ropr
ion
(sel
ecti
ve N
RI o
r no
radr
ener
gic
& d
opam
iner
gic
agen
ts)
Fava
et a
l. (2
003)
94C
128
MD
D
Out
P TR
D-I
Mul
ticen
ter,
ope
n de
sign
of F
LX-N
R (8
wee
ks)
REB
8-10
mg
- R
espo
nse
(≥50
% in
HD
RS 2
5 and
CG
I-I ≤
2) =
44
.5%
Resp
onse
(≥50
% in
HD
RS 1
7) =
45.
3%
Dro
pout
SE =
13.3
%
Met
hodo
logi
cally
wel
l per
form
ed o
pen
stud
y; u
nkno
wn
plac
ebo
resp
onse
rate
. FLX
-N
R de
term
ined
at e
nd o
f FLX
-tre
atm
ent;
di
rect
sw
itch
to R
EB w
ell t
oler
ated
. Due
to
long
hal
f-life
of F
LX, f
irst 4
wee
ks
‘com
bina
tion’
-the
rapy
.
Fava
et a
l. (2
003)
92C
29
MD
D
Out
P TR
D-I
Two-
cent
er o
pen
desi
gn o
f pr
ospe
ctiv
ely
dete
rmin
ed F
LX-N
R
(8 w
eeks
)
BUP-
SR 15
0-40
0mg
- M
odif
ied
ITT
(n=
26) R
espo
nse
(≥50
% in
H
DR
S 17)
= 3
4.6%
Par
tial r
espo
nse
(25-
49%
in
HD
RS 1
7) =
30.
8% R
emis
sion
(HD
RS 1
7 ≤7)
=
23.1%
Met
hodo
logi
cally
wel
l per
form
ed, b
ut s
mal
l op
en st
udy;
unk
now
n pl
aceb
o re
spon
se ra
te.
No
spec
ifie
d dr
opou
trat
es. F
LX-N
R
pros
pect
ivel
y de
term
ined
; dire
ct s
witc
h to
BU
P; n
o do
cum
enta
tion
of e
ffec
ts o
f thi
s sw
itch.
Due
to lo
ng h
alf-l
ife o
f FLX
, firs
t 4
wee
ks ‘c
ombi
natio
n’-t
hera
py.
Rus
h et
al.
(200
6)47
See
unde
r 5.1.
A
Wal
ker e
t al.
(199
3) 9
3 C
39
MD
D
Out
P TR
D-I
Ope
n de
sign
of
patie
nts
with
sex
ual
side
-eff
ects
on
FLX
(par
tially
FLX
-NR
; n=
16)
(8 w
eeks
)
BUP
150-
450m
g -
All
(n=
36):
H
DR
S 28:
16.6
(±7.
8)
8.4
(±
8.3)
Dro
pout
SE =
10.3
% D
ropo
utin
effi
cacy
=
10.3
% Bs
l HD
RS28≥1
8 (n
= 16
):
HD
RS28
: 25.
4 (±
5.8)
10
.9 (±
10.8
)
Initi
al d
esig
n of
sw
itchi
ng d
ue to
sex
ual s
ide-
effe
ct; l
imite
d da
ta p
rovi
ded
for d
epre
ssed
su
bjec
ts; e
.g. n
o re
spon
se-r
ates
; unk
now
n pl
aceb
o re
spon
se ra
te. I
mpr
ovem
ent o
f or
gasm
func
tion
(84%
), sa
tisfa
ctio
n (7
8%) a
nd
libid
o (7
8%) a
fter
sw
itch.
2 w
eeks
was
hout
ap
plie
d; d
isap
pear
ance
of s
exua
l dys
func
tion
linea
r with
FLX
-was
hout
Tabl
e 5.
1. Ef
fect
iven
ess
of s
witc
hing
aft
er ≥
1 SSR
I: Se
lect
ed s
tudi
es. (
Cont
inue
d)
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Co
mpa
rison
* O
utco
me†
R
emar
ks

Review of switching in MDD
83
Chapter5
5.1.E
. to
a M
AO
-I
McG
rath
et a
l. (2
006)
49B
109
MD
D
Out
P TR
D-≥
II
Mul
ticen
ter u
nblin
ded
RCT
(12
wee
ks)
TCP
10-6
0mg
VLX-
XR 7
5-30
0mg
+ M
IR 15
-45
mg
Rem
issi
on (H
DRS
17≤7
) TCP
: 6.9
%, V
LX-M
IR:
13.7
% N
NH
= 15
(5.5
-∞)
Res
pons
e (≥
50%
in
QID
S-SR
) TCP
: 12.
1%, V
LX-M
IR: 2
3.5%
NN
H =
9
(3.9
-∞) D
ropo
utSE
TCP
: 41.4
%, V
LX-M
IR: 2
1.6%
NN
H =
6 (2
.7-3
5.2)
Unb
linde
d st
udy,
blin
ded
asse
ssor
s.
Part
icip
ants
rece
ived
at l
east
1 SS
RI,
a 2nd
SS
RI o
r VLX
or B
UP
or C
IT-a
ugm
enta
tion
(BU
P or
BU
S) a
nd s
ome
CBT
and
a 3rd
tr
eatm
ent w
ith N
OR
or M
IR (l
evel
IV
STA
R*D
).
Nol
en e
t al.
(198
5)96
B 26
M
DD
InP
TRD
-≥II
Rand
omiz
ed
unbl
inde
d cr
osso
ver
desi
gn
(4 w
eeks
)
TCP
20-10
0mg
L5H
TP 2
0-20
0mg
Res
pons
e (≥
50%
in H
DR
S 17)
TCP
: 42.
9%,
L5H
TP: 0
% N
NT
= 3
(1.5
-5.9
) tra
nylc
ypra
min
e si
de e
ffec
ts =
62%
car
diov
ascu
lar;
15%
inso
mni
a
Smal
l stu
dy, a
lloca
tion
of in
itial
trea
tmen
t no
t cle
arly
des
crib
ed. S
tudy
-gro
ups
diff
er
sign
ifica
ntly
in b
asel
ine
HD
RS-s
core
. Sta
ge II
-III
TR
D-p
atie
nts.
In th
e pa
per d
ata
of s
econ
d 4
wee
ks (c
ross
-ove
r pha
se) a
re a
lso
give
n.
Lim
ited
pres
enta
tion
of d
ata.
Nol
en e
t al.
(198
8)95
B 21
MD
D
InP
TRD
-≥I
I
RCT
with
sec
onda
ry
cros
s-ov
er
(4 w
eeks
)
TCP
40-1
00m
g N
OM
150-
250m
g R
espo
nse
(≥50
% in
HD
RS 1
7) T
CP: 4
5.5%
N
OM
: 10.
0% N
NT
= 3
(1.4
-154
.8) T
CPSE
= 58
% ca
rdio
vasc
ular
, 33%
inso
mni
a
Stag
e II-
III T
RD
-pat
ient
s. U
ncle
ar m
etho
d of
ra
ndom
izat
ion.
In th
e pa
per d
ata
of s
econ
d 4
wee
ks (c
ross
-ove
r pha
se) a
re a
lso
give
n.
Lim
ited
pres
enta
tion
of d
ata.
* A
ll do
sage
s in
mg/
day.
† In
tent
ion
to tr
eat (
ITT)
-res
ults
unl
ess
spec
ified
. A
bbre
viat
ions
: AD
= a
ntid
epre
ssan
t, C
A =
‘Con
vent
iona
l ant
idep
ress
ants
’ (PA
R, F
LX, S
ER, C
IT, M
IR a
nd o
ther
ant
idep
ress
ants
not
spe
cifi
ed),
CBT
= Co
gniti
ve B
ehav
ior t
hera
py, C
GI-I
= C
linic
al g
loba
l im
pres
sion
–im
prov
emen
t sca
le, D
ys =
dys
thym
ia, H
DRS
? = H
amilt
on d
epre
ssio
n ra
ting
scal
e (?
indi
cate
s nu
mbe
r of i
tem
s), I
nP =
In-p
atie
nts,
MA
DR
S= M
ontg
omer
y Å
sber
g D
epre
ssio
n R
atin
g Sc
ale,
MD
D
= m
ajor
dep
ress
ive
diso
rder
,Min
D =
Min
or D
epre
ssio
n, N
NH
/T =
num
ber n
eede
d to
har
m/t
reat
, NR
= n
onre
spon
ders
, OCD
= O
bses
sive
com
puls
ive
diso
rder
, Out
P =
Out
-pat
ient
s, P
GI =
Pat
ient
glo
bal
impr
ovem
ent s
cale
, QID
S-SR
= Q
uick
inve
ntor
y of
dep
ress
ive
sym
ptom
atol
ogy
self-
rate
d, S
chA
= S
chiz
oaff
ectiv
e di
sord
er, S
E =
Side
-eff
ects
, TRD
= tr
eatm
ent r
esis
tant
dep
ress
ion,
sta
ges
I: fa
ilure
of 1
ad
equa
te tr
ial o
f 1 m
ajor
AD
cla
ss; I
I: St
age
I + fa
ilure
of a
n ad
equa
te tr
ial o
f 1 d
istin
ctly
diff
eren
t AD
cla
ss ;
III: s
tage
II +
failu
re o
f an
adeq
uate
tria
l of T
CA; I
V: S
tage
III +
failu
re o
f an
adeq
uate
tria
l of a
n M
AO
I; V:
Sta
ge IV
+fa
ilure
of a
cou
rse
of b
ilate
ral E
CT.10
;24 D
rugs
: BU
P =
bupr
opio
n, B
US
= bu
spiro
ne, C
IT =
cita
lopr
am, F
LV=
fluvo
xam
ine,
FLX
= fl
uoxe
tine,
IMI =
imip
ram
ine,
L5H
TP =
L-5
-hyd
roxy
-tr
ypto
phan
, MA
O-I
= irr
ever
sibl
e in
hibi
tor o
f mon
oam
ine-
oxid
ase,
MIR
= m
irtaz
apin
e, M
OC
= m
oclo
bem
ide,
NEF
= n
efaz
odon
e, N
OM
= n
omife
nsin
e, N
OR
= n
ortr
ipty
line,
NR
I = n
orad
rene
nalin
e re
upta
ke
inhi
bito
r, O
XA =
oxa
prot
iline
, PA
R =
par
oxet
ine,
PLA
C =
plac
ebo,
REB
= re
boxe
tine,
SER
= s
ertr
alin
e, S
SRI =
sel
ectiv
e se
roto
nine
reup
take
inhi
bito
r, T
CA =
tric
yclic
ant
idep
ress
ants
, TCP
= tr
anyl
cypr
omin
e,
TRA
Z =
traz
odon
e, V
LX =
ven
lafa
xine
.
Tabl
e 5.
1. Ef
fect
iven
ess
of s
witc
hing
aft
er ≥
1 SSR
I: Se
lect
ed s
tudi
es. (
Cont
inue
d)
Stud
y (y
ear)
refe
renc
eLo
E N
D
esig
n (f
ollo
w-u
p)
Inte
rven
tion*
Co
mpa
rison
* O
utco
me†
R
emar
ks

Chap
ter
Chapter 5
5
84
Second SSRIWe identified 7 open studies investigating a switch to a second SSRI.67;68;68-73 In one of thesestudies, the non-response to the initial SSRI was determined prospectively, and switching wasapplied immediately.69 In 4 studies, intolerance was determined retrospectively, with an (unclear)interval between the end of the previous SSRI and the next.67;68;70;72 In the remaining 2 (SSRI-intolerance) studies, patients either started a second SSRI soon after the first SSRI or had an SSRI-free interval.71;73
Response rates of switching in SSRI non-responders varied between 46% and 58% in threeuncontrolled studies of variable methodological quality.67-69 The response rate was lower (42%) ina fourth study with a heterogeneous group of inpatients.70 However, response rates to a secondSSRI varied between 56% and 72% when patients were intolerant to the first SSRI (4 studies).68;71-73
Dropout rates due to side effects were between 5% and 21%, in studies with initial non-responders69and between 0% and 10% in SSRI-intolerant samples71-73 (LoE: C).
In the SSRI-arms of three RCTs, response rates varied between 26.7% and 71.1%, while remissionrates were between 17.6-52.1%.46;47;74 Dropout rates due to side effects varied between 4.8-21.0%.For results on the comparisons with other arms see below (LoE: A2-B).
In summary, the data from the open studies and one of the RCTs46 suggest that, after 1 SSRI,non-responders and, notably, also SSRI-intolerant patients can benefit from a switch to a secondSSRI with response rates of approximately 50% and 70%, respectively. However, the results in twoRCTs47;74 indicated much less advantageous response and remission rates for a second SSRI (26.7-29.0% and ~17.6% respectively).
Tricyclic Antidepressants and mianserinWe identified 2 RCTs with a switch to a TCA,48;75 with one having limited power due to arandomization into 3 arms.75 Four open studies investigated a switch from an SSRI to a TCA.76-80
The methodology of the open studies varied: one large cross-over study was methodologicallysound,79;80 one small study was unequivocally poor,78 and two studies were of reasonable quality(investigating populations with TRD).76;77
In the RCT of Ferreri et al. switching to mianserin (a noradrenergic tetracyclic) versuscontinuation of fluoxetine was investigated, with a third arm for their combination.75 Nosignificant difference was found between switching to mianserin and continuation of fluoxetine(response 48.5% and 36.8% respectively; NNT= 9 (95% CI 2.9-∞)) in an ITT analysis. The combinationof fluoxetine and mianserin performed better than continuation of fluoxetine (response 62.5% inthe combination group; NNT= 4 (95% CI 2.1-34.1)). Dropouts due to side effects were highest in theswitch-group (24%; NNH vs continuation= 5 (95% CI 2.6-10.4)) (LoE: B).
The STAR*D level III study compared a switch to nortriptyline versus mirtazapine in arandomized unblinded design.48 All participants received citalopram plus either a switch tosertraline, venlafaxine or bupropion or citalopram augmentation with buspirone or bupropion.Response rates (≥50% decrease in QIDS-SR16 score) were 16.5% for nortriptyline and 13.5% formirtazapine (NNT= 32 (95% CI 8.1-∞)). Remission rates (HDRS17 ≤7) were 19.8% vs12.4% fornortriptyline and mirtazapine, respectively (NNT= 14 (95% CI 6.0-∞)). There were no differences inremission rates for those intolerant to the level II treatments versus those who tolerated theirsecond trial of antidepressants. Dropoutrates due to side effects were high both for nortriptyline(34.7%) and mirtazapine (33.3%) (LoE: A2).
Thase et al. investigated a switch to imipramine in non-responders to sertraline in chronicdepressive out-patients. They found a 44% ITT response rate, with a dropout rate due tointolerable side effects of 9%.79;80 The methodologically poor study by Peselow et al. (includingalso SSRI-intolerant patients) found a 73% response rate after a switch to imipramine inoutpatients.78 In the studies that recruited TRD populations, response rates after switching tonortriptyline76 and oxaprotiline77 decreased to 39% in in-patients77 and 42% in outpatients,76 with a35% overall dropout rate in the latter study (LoE: C).

ChapterReview of switching in MDD
5
85
In summary, for the switch to a TCA response rates of approximately 16.5 to 48.5% werefound. Lower response rates were observed in studies that included more treatment resistantpatients.
Mirtazapine, nefazodon or venlafaxine (novel dual acting agents)We identified 13 switch studies to novel dual acting agents.46-48;74;81-90 The methodological qualityvaried. Four studies were randomized controlled trials: Poirier and Boyer compared a switch toparoxetine versus venlafaxine,74 Baldomero et al. compared a switch to venlafaxine extendedrelease versus switching to any other antidepressant (77% of these switches used paroxetine,citalopram, sertraline, or fluoxetine) in an unblinded design,46 and the level II and III STAR*Dswitch studies, which were also unblinded studies, compared a switch after citalopram tovenlafaxine extended release, bupropion, or sertraline47 and the switch thereafter to nortriptylineor mirtazapine.48 Other studies described open studies with mirtazapine,81;90 nefazodone,83 andvenlafaxine.82;84-86;88;89 In seven of the studies, all patients received an SSRI before switching.47;48;81-
83;89;90 Five studies included patients with variable but higher levels of treatmentresistance;48;74;83;86;90 in 2 studies, this was unclear.81;84 In contrast, one study included patients(52%) who initially responded to an SSRI but did not sustain their response.82
In the RCT performed by Poirier and Boyer, switching to venlafaxine was more efficaciousthan paroxetine when remission (HDRS17≤10) was considered (remission rates: 36.7% and 17.7%respectively), with a NNT of 6 (95% CI 2.9-28.9).74 For a response criterion (≥50% reduction inHDRS17), the difference was insignificant (response rates: venlafaxine= 45% and paroxetine= 29%;NNT= 7 (95% CI 3.0-∞)). Dropout rates due to side effects were comparable (8.2% for venlafaxineand 4.8% for paroxetine; NNH= 30 (95% CI 8.3-∞)) (LoE: A2).
In the randomized, unblinded study by Baldomero et al., venlafaxine showed a significantlyincreased remission (HDRS17≤7) rate (59.3%) compared with conventional antidepressants (51.5%)after 24 weeks of treatment, with a NNT of 13 (95% CI 8.9-23.7).46 In the conventionalantidepressants group, 77.3% of the patients used a second SSRI; for SSRIs, the remission rate was52.1% (NNT= 14 (95% CI 9.1-29.3)). Response (≥50% reduction in HDRS17) rates also showed amodest but significant advantage: 77.3% for venlafaxine versus 71.1% for SSRIs (NNT= 17 (95% CI10.5-35.0)). Overall dropout was slightly lower in the venlafaxine group when compared with allconventional antidepressants (28.3% vs 32.8%; NNH= 27 (95% CI 15.1-120). Dropout rates due to sideeffects were not significantly different between venlafaxine and conventional antidepressants(12% vs 7.3% respectively; NNH= 161 (95% CI 62.1-∞)) (LoE: B).
The level II STAR*D trial did not find significant differences between the switches tovenlafaxine, bupropion, and sertraline.47 Before the switch, all participants received citalopram(20-60 mg for a maximum of 14 weeks). Patients were randomized over different randomizationpossibilities for which they were at equipoise.91 The assessors of the primary outcome (HDRS17)were blind to the treatment. After 14 weeks of treatment, response rates (≥50% decrease in QIDS-SR16) were 28.2% for venlafaxine, 26.1% for bupropion and 26.7% for sertraline (not significant).Remission rates (HDRS17 ≤7) were not significantly different for venlafaxine, bupropion, andsertraline (24.8%, 21.3% and 17.6% respectively). For corresponding NNTs see table 5.1. The dropoutrate due to side effects was not statistically different for venlafaxine (21.2%), bupropion (27.2%),and sertraline (21.0%) (LoE: A2).
The level III stwitch study was described earlier.48 Mirtazapine response, remission and side-effects related dropout rates were 13.5%, 12.4% and 33.3%, respectively (LoE: A2).
In open studies, mirtazapine, nefazodone, and venlafaxine showed response rates between17% and 86%, with decreased response rates at increased levels of treatment resistance (LoE:C).81-86;88-90 Dropout rates due to adverse effects varied between 5.5% and 11% for venlafaxine,between 20.8% and 26% for mirtazapine and was 39% in one study with nefazodone (LoE: A2, C).

Chap
ter
Chapter 5
5
86
We performed a meta-analysis of the three RCTs that compared switching to venlafaxineversus SSRIs,46;47;74 although the differences in duration of follow-up introduced someheterogeneity (ranging from 4 weeks by Poirier and Boyer74 to 24 weeks by Baldomero et al.46). Asshown in figure 5.2, the weighted difference in remission-rates (fixed effects model) was 8% (4 –11%) in favour of venlafaxine (NNT= 13 (95% CI 9.1 – 25.0), and for response 6% (1 – 10%), (NNT= 17(95% CI 10.0 – 100.0)). Omission of the methodologically poorer study of Baldomero et al.increased the difference in remission rates (10% (95% CI 3-16%) fixed effects model; NNT= 10 (95% CI6.3-33.3)), but decreased the difference in response rates (4% (-3-12%) fixed effects model; NNT=25 (95% CI 8.3-∞)). The dropoutrate due to side effects was only reported in two studies47;74 ; theweighted difference was 1% (95% CI -5-7%) (fixed effects model) with more dropouts forvenlafaxine.
In summary, heterogeneous studies considering switching to mirtazapine, nefazodone andvenlafaxine showed response rates of approximately 28-50% in subjects without obvious TRD,while in subjects with increased levels of TRD response percentages dropped (investigated forvenlafaxine and mirtazapine). Pooling of results showed a modest and clinically equivocallyadvantageous increased remission rate for venlafaxine over SSRIs (NNT= 13 (95% CI 9.1-25.0).
Bupropion and Reboxetine (agents specifically affecting dopaminergic and/ornoradrenergic neurotransmission)We identified one RCT and two small open studies of switching to bupropion.47;92;93 The STAR*Dlevel II switch study including bupropion was described earlier.47 There were no significantdifferences in remision or response rates for bupropion compared to venlafaxine or sertraline. Inthis study, bupropion had the (statistically unsignificant) highest dropout rate (27.2%) due to sideeffects (LoE : A2).
Figure 5.2. Meta-analysis of switch-studies comparing a switch to venlafaxine versus a second SSRI.
A: Remission. B: Response. VLX = venlafaxine. References to studies: Poirier et al.,74 Baldomero et al.,46 Rush et al.47
�
�

ChapterReview of switching in MDD
5
87
In two open studies with bupropion, Fava et al prospectively determined fluoxetinenoneresponse in a small but well performed study,92 and Walker et al. recruited patients that wereprimarily suffering sexual side-effects of fluoxetine, and only reported a decrease in 28-itemHDRS-scores.93 One larger, well-performed, open study investigated the switch to reboxetine influoxetine non-responders.94
Thus, switching from fluoxetine was investigated, with reported response rates of 34.6% forbupropion92 and 45.3% for reboxetine.94 For bupropion, specified dropout rates were not reportedin one study.92 The side effect-related dropoutrate was 10.3% in subjects with sexual dysfunctionwhile taking fluoxetine.93 For reboxetine, the dropout rate due to side effects was 13.3% (LoE: C).
In summary, switching to bupropion or reboxetine was scarcely studied but was a possibleoption with response-rates of 26.1%-34.6% and 45.3% respectively. The remission rate of switchingto bupropion was not different compared to venlafaxine or sertraline.
Reversible Inhibitor of Monoamine-oxidase A We identified no studies that investigated switching from a SSRI to a reversible inhibitor ofmonoamine-oxidase A.
Monoamine-oxidase A inhibitorWe identified one RCT from STAR*D49 (level IV) and two small, interrelated randomized studiesafter 4 weeks of treatment with at least one SSRI (fluvoxamine) and oxaprotiline.95;96 Weidentified no studies of SSRI non-responders in atypical depression. Two studies were RCTs49;95
and one an unblended, randomized, cross-over study.96 The STAR*D study investigatedoutpatients; the studies by Nolen et al.95;96 were performed in treatment resistant in-patients.
Nolen et al. found tranylcypromine to be more efficacious than nomifensine,95 in both studiesthe response rate for tranylcypromine was 42.9 and 45.5%.95;96 All patients previously received atleast fluvoxamine and oxaprotiline. Fifty-eight to 62% had side effects affecting their bloodpressure levels (LoE: B).
The STAR*D level IV study included patients that had not been in remission after citalopram(level I), either venlafaxine, bupropion, sertraline or citalopram augmentation with buspirone orbupropion (level II), and additionally received nortriptyline or mirtazapine (level III).49 Thesepatients were randomized between tranylcypromine and a combination of venlafaxine withmirtazapine. Of the included patients 32.1% were intolerant for the level III medication. Remissionrates (HDRS17 ≤7) were low for tranylcypromine (6.9%) and the combination treatment (13.7%;NNH= 15 (95% CI 5.5-∞)). Response rates (≥50% decrease in QIDS-SR16) were also not significantlydifferent: 12.1% vs 23.5% for tranylcypromine and venlafaxine with mirtazapine, respectively (NNH=9 (95% CI 3.9-∞)). Dropout rates due to side effects were higher for tranylcypromine: 41.4% versus21.6% for venlafaxine with mirtazapine (NNH= 6 (95% CI 2.7-35.2)).
Additional concerns for clinicians regarding switchingLittle evidence is available about the optimal way to switch.20;29;69;88;97 Abrupt reduction ordiscontinuation of SSRIs may produce somatic and psychological withdrawal symptoms, of whichoccurrence is inversely related to the plasma half-life of the initial SSRI.98;99 Overlap ofantidepressants during switching is generally avoided.20;29
Direct switching (without a washout phase) from an initial SSRI (fluoxetine at the standarddose or citalopram at high dosages) to another SSRI (paroxetine, citalopram, sertralin),47;69;97
nortriptyline,48 mirtazapine,48;81 bupropion,47 reboxetine,94 or venlafaxine47;88) was well tolerated.Also, direct switching reduced the emergence of side effects compared with placebo in a 1-weekwashout phase (which might have been discontinuation symptoms).97
In case of higher than standard doses of SSRIs, some data for tolerance of direct switchingwere generated by STAR*D.47-49 However, the results published so far do not specify dropoutrates in the first 2 weeks after switching. Also, because tapering of high doses of previousantidepressants was not applied in STAR*D, this trial was not designed to examine the optimal

Chap
ter
Chapter 5
5
88
switch strategy if higher than standard doses were used before the switch. Thus, if necessary,direct switching of high-dose antidepressant therapy appears possible after citalopram as a firstSSRI.47
In a case of switching from an SSRI to a TCA, other reviewers did not recommend a washoutperiod.20;29 In one included study a direct switch to mianserin was less well tolerated.75 Forswitching to nefazodone, in one study a 4-day to 7-day washout was applied but notinvestigated.83 A 1-week washout period is suggested for switching to a reversible inhibitor ofmonoamine-oxidase A, and a 1-week to 2-week washout period is recommended for switching to aMAO-I.20;29 For fluoxetine these washout-periods should be prolonged to 5 weeks because of thelong half-life of fluoxetine and norfluoxetine.20 The inhibition of cytochrome P450 subenzymes bySSRIs may increase the levels of some TCAs during the first to fifth (fluoxetine) week.20
Discussion
This report systematically reviewed and appraised the available research focusing on switchingstrategies for SSRI non-responders in MDD, including the recent STAR*D results. We found thatthe available evidence does not justify distinct recommendations for next-step strategies afternon-response to a first SSRI. Thepooled difference in remission rates of switching to venlafaxine(an SNRI) versus a second SSRI showed a modest and clinically equivocal advantage ofvenlafaxine (NNT= 13 (95% CI 9.1-25.0)), this difference increased when the largest andmethodologically poorest study was omitted (NNT= 10 (95% CI 7-34)).
In summary, after a first SSRI, switching to any of the current classes of antidepressants hasapproximately a 50% chance of response. Still, a direct comparison of the rates across thepredominantly open studies is methodologically not justified. In STAR*D response and remissionrates were lower (respectively 26.8% and 21.3% at level II,47 15% and 16.2% at level III,48 and 17.4% and10.1 at level IV49). Rush et al. attributed these lower remission rates to the inclusion of patientswho were more chronically depressed, had lower socio-economic status and suffered from moreco-morbid somatic and psychiatric diseases.47 In general, the level of TRD10 of included studies wasinversely correlated with treatment outcome. Although this finding carries the risk of anecological fallacy, it is worrisome, which further appears from the STAR*D results. After thesecond antidepressant the chances of response or remission by switching again are becomingrather low, challenging us to find new approaches.100-102
Dropout rates due to side-effects varied between 5-21% for a second SSRI and venlafaxine; 10-35% for TCAs, bupropion, and reboxetine; 20-33% for mirtazapine; 39% for nefazodone and 41.4%for tranylcypromine. It should be noted that these percentages cannotsimply be compared witheach other because of heterogeneous populations and open-study designs. In randomizedcomparisons, no significant differences in side-effect related dropoutwere found, except fortranylcypromine versus a combination of venlafaxine with mirtazapine.49
With eight RCTs,46-49;74;75;95;96 switching-options after a first SSRI were generally investigatedwith open studies. In these open studies, switching to a second SSRI (7 studies) and venlafaxine (7studies) were studied most frequently. Furthermore, the studies were of variable methodologicalquality. In our opinion, the available evidence for switching strategies allows generalrecommendations only. Switching is open to all studied antidepressant classes (second SSRI,novel dual-acting antidepressants, selective noradrenergic and noradrenergic/dopaminergicagents, or TCA or mianserin) without clear recommendations other than those that apply for theselection of initial treatment. In the choice of an initial antidepressant, some reports promotedTCAs for treatment of inpatients;103-106 however, it is unclear what special feature is associatedwith inpatients (e.g. severity), and studies investigating switching strategies after an SSRI ininpatients were not identified. From the available studies it must be emphasized that side-effectsto a first SSRI did not reduce the chance of response or increase the chance of intolerance for a

ChapterReview of switching in MDD
5
89
second SSRI. Because of side effects, we think that MAO-I should not be prescribed as a secondantidepressant after a first SSRI. A possible exception – but not investigated after a first SSRI – isfor atypical depression.
Switching from a failed TCA treatment was reviewed earlier.7;10;32;80;107 The response rates forwithin-class switching with SSRIs appear more favourable than a TCA-TCA switch: in two smalltrials,108;109 response-rates of a within-class TCA switch were 9108 and 30%.109 The SSRI resultschallenge the belief that any within class switch should be considered illogical. The betweenclasses switching strategies from a TCA to an SSRI (investigated in 10 trials;77-79;109-115 response ratesvarying between 4% (inpatients) and 75% (out-patients)), to a heterocyclic antidepressant (e.g.bupropion, trazodone, nomifensine, oxaprotiline; 6 studies;77;95;116-119 response rates between 10%-56%) and to a MAO-I (6 trials;95;96;120-123 response rates between 29-83%) showed similar broadranges of response rates. These ranges reflect differences in heterogeneous study-populations aswell. Again, it is inappropriate to simply compare these rates determined in different studies.
On theoretical grounds, it is logical (and often recommended) to switch to an antidepressantwith different or combined sites of action (e.g. norepinephrine uptake inhibition afterunsuccessful serotonergic uptake inhibition).124-126 Others pointed out the complex interaction ofmonoamine systems alone, proposed other possible etiologic mechanisms, and considered themonoamine hypothesis only partially explanative for depression and the response toantidepressants.127-131 Six RCTs so far compared different pharmacologic approaches in non-responders (venlafaxine versus paroxetine,74 venlafaxine versus an SSRI46, venlafaxine versussertraline or bupropion,47 nortriptyline versus mirtazapine,48 fluoxetine versus mianserin or amianserin-fluoxetine combination75, and tranylcypromine versus a venlafaxin-mirtazapinecombination49). These RCTs found equivocal superiority of dual-action pharmacotherapy.However, in STAR*D the empirical proof of this theoretical strategy was not found.
Apart from switching, augmentation or combination, and addition of (or switching to)psychotherapy are possible options. Only Ferreri et al.75 and McGrath et al.49 compared switchingversus combination (the latter at a higher level of TRD). In STAR*D, a switch to or augmentationwith cognitive behaviour therapy was possible after citalopram (unpublished yet), andaugmentation of citalopram with buspirone or bupropion was also studied.132 A direct comparisonbetween switching and augmentation after citalopram was not feasible.47 Therefore, clearrecommendations about choosing one of these strategies relative to each other are not possible.In most countries, SSRIs are generally prescribed as first line treatment, often provided in primarycare. We think that switching-strategies after a first SSRI will be preferred, especially in primarycare, in which augmentation and combination strategies may be unfamiliar to physicians. Thishypothesis is supported by audits, even among psychiatrists.40-42
Limitations of the identified studiesWell-designed switch-studies are difficult to carry out, and therefore, it does not surprise that theevidence to date is limited in several ways. We found predominantly open, uncontrolled studies,with a risk of more positive results than in blinded studies and without a possibility to activelycompare strategies. There were few studies that clearly described the inclusion ofprospectivelydetermined SSRI non-responders.47-49;69;75;79;92 This finding is of importance, as in retrospectivelydetermined non-responders, current depression may cause recall-bias. Furthermore, in somestudies non-responders were not treated directly after cessation of the unsuccessful drug, whichmight have biased results; for example depression worsened after cessation, or –the other wayaround– depression may have improved because of the natural course of depression.133;134
Several other problems were encountered: unclear criteria for initial non-response,67;72;73;82;90
inclusion of mild or minor depression,46;82 possible selection-bias,82 limited presentation ofresults,46;78;88;89;93;95;96 absence of ITT-data,82 small sample sizes (n< 40),70;77;78;83;90;92;93;95;96 and lowstatistical power.75;83 In general, less robust studies found more positive results for the drug ofinterest. Table 5.2 presents a summary of these problems.

Chap
ter
Chapter 5
5
90
The STAR*D trials47-49 were randomized but unblinded effectiveness trials. The primaryoutcome (remission by HDRS17) was determined by blind assessors; the secondary outcomes bythe QIDS-SR16 were self-rated by the unblinded patients. The a priori definition of nonremission formissing data will have decreased remission rates because of attrition, but this a priori definitionwas considered noninfluential after sensitivity analyses. The aggressive dose increases in STAR*Dtrials prevented undertreatment, but might have increased attrition, and definitely increased thepercentages of treatment-intolerant patients at all levels. Especially in the level IV trials thetreating physicians might have been unfamiliar with the prescribed medication (tranylcypromine,venlafaxine-mirtazapine combination), reducing the vigour of the applied pharmacologicintervention.49
Future switching studies After the STAR*D trials, the question arises as to whether many randomized direct comparisonsbetween switches among drug classes are fruitful to develop fully evidence-basedrecommendations for switching. Results of some studies might still be published.135-137 Also, thepredictors of poor response and nonremission need to be further clarified. In order to structuredirections of research, the recommended approaches in guidelines should be evaluated for eachtreatment step. The Texas Medication Algorithm project proved that algorithms are beneficial forpatient care;138 however, our next challenge is to investigate which steps within these algorithmsare better compared with each other.
Ideally, three or more armed studies should be designed. Switching within the same class or todifferent classes of drugs should be compared with an augmentation or new approach, while alsoan arm for continuation of the initial therapy should be included. The latter arm would thenrepresent a form of placebo control.Naturally, these studies are hard to carry out, may have toovercome resistance and doubts concerning the ethics of the continuation arm, or may sufferfrom selective patient withdrawal from this continuation arm. The STAR*D-project has been amajor step in this direction, especially by proving the feasibility of such large multicenter trials andthe methodology of (equipoise) randomization. At the same time the effectiveness approachwith many centers, high levels of co-morbidity, chronicity and many arms of treatment might havereduced the ability to find differences.
We found that the response rates in switch-studies decreased with increased levels of TRD.Therefore, future studies must consider the level of TRD as an important effect-modifyingvariable. Ideally, in future research, clear populations of prospectively determined treatmentresistance should be selected, or analyzed in a priori defined subgroups to increase ourknowledge about confounding or effect-modifying variables. Finally, to improve the acceptanceof switching in daily clinical practice, more studies of patients’ perspectives of switching ofantidepressants are needed.
Limitations of the reviewSeveral limitations of this review should be mentioned. First, a review like this cannot overcomethe paucity of high-quality evidence to date. The Cochrane Collaboration primarily rejects openstudies as high-quality evidence. If this criterion had been applied, only 8 studies would havequalified for the review, obviously limiting its applicability. The majority of included studies hadmethodological flaws, two studies were excluded for clear invalidity.61;62 We decided a priori toinclude open studies, and–even more–to include studies in which 50-100% of patients initially usedan SSRI, introducing different levels of TRD. Of course, the latter decision is debateable from amethodological point ofview.
Second, in the selected trials, mostly response was used as the primary outcome, whilecurrently remission of depression is the clinical aim of treatment.139 Only 13 of 31 studies (42%)included remission as an outcome criterion.46-49;74-76;79;82;84-86;92 Only STAR*D primarily investigatedthe practice of switching in order to achieve remission.47-49

ChapterReview of switching in MDD
5
91
Third, patients studied in the included trials represented selected populations, reducing thegeneralizability of the findings to the 'real world' clinical practice; as an effectiveness trial, theSTAR*D results overcame this problem. Fourth, critical appraisal was performed by one reviewer(H.G.R.), while ideally this should have been performed by two raters. However, we found ourinterobserver agreement to be moderate to good and no worse than in previous interraterattempts in psychiatry.58 Fifth, the grading system for studies does not represent the appraisedmethodological dimensions of evidence. This improved the applicability of the results for busyclinicians, but reduced their strength.
Strengths of the reviewThis is the first review that applied the thorough methodology to search for, identify, andappraise articles as used in Cochrane reviews. The applied methodology and transparentpresentation of data allow clinicians to make their own judgements and, if necessary, to retrievethe source of data. Apart from the relevant up-to-date information for clinicians, this review couldwell serve national guideline committees as a building stone for the development oftreatmentguidelines for MDD.
ConclusionThis systematic review about switching identified 8 RCTs and mostly open switch studies ofvariable methodological quality in heterogeneous populations. The STAR*D results largelyincreased the amount and quality of the available evidence, but did not show differential classeffects to guide switching. After a first SSRI switching is open to all studied antidepressant classes(except irreversible MAO-inhibitors), without clear recommendations other than those that applyfor the selection of initial treatment. For recommendations about when to choose betweenswitching, augmentation, combination, or psychotherapeutic strategies as a next step, hardly anyevidence of comparisons of these strategies relative to each other exists. Future algorithm-basedswitch studies and studies of patient perspectives regarding switching will have to improve ourknowledge to guide treatment for SSRI non-responders.
Acknowledgements
This systematic review was realised by a grant for the development of a local evidence-basedclinicalpractice guideline (No SFA.07.012) from the Academic Medical Center, Amsterdam, theNetherlands. This guideline considering strategies for non-response to a standard dose of a firstSSRI is available on request from the first author.
Conflicts of interest
None

Chap
ter
Chapter 5
5
92
References
1. Murray CJ, Lopez AD. Evidence-based health policy--lessons from the Global Burden of Disease Study. Science. 1996; 274:740-743.
2. Kennedy SH, Lam RW, Cohen NL, Ravindran AV, CANMAT Depression Work Group. Clinical guidelines for the treatment ofdepressive disorders. IV. Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.
3. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
4. Anderson IM, Nutt DJ, Deakin JFW. Evidence-based guidelines for treating depressive disorders with antidepressants: Arevision of the 1993 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2000; 14: 3-20.
5. Mulrow CD, Williams JW, Jr., Trivedi M, Chiquette E, Aguilar C, Cornell JE et al. Evidence report on: Treatment ofdepression - Newer pharmacotherapies. Psychopharmacol Bull. 1999; 34: 409-795.
6. Depression Guideline Panel. Depression in Primary Care: Volume 1. Detection and Diagnosis. Clinical Practice Guideline,Number 5. AHCPR Publication No. 93-0550. 1993. Rockville, MD, U.S. Department of Health and Human Services, PublicHealth Service, Agency for Health Care Policy and Research.
7. Depression Guideline Panel. Depression in Primary Care: Volume 2. Treatment of Major Depression. Clinical PracticeGuideline, Number 5. AHCPR Publication No. 93-0551. 1993. Rockville, MD, U.S. Department of Health and HumanServices, Public Health Service, Agency for Health Care Policy and Research.
8. Rush AJ, Crismon ML, Toprac MG, Trivedi MH, Rago WV, Shon S et al. Consensus guidelines in the treatment of majordepressive disorder. J Clin Psychiatry. 1998; 59: 73-84.
9. NICE. Clinical Guideline 23. Depression: management of depression in primary and secondary care. December 2004. 23, 1-63. 2004. London, National Institute for Clinical Excellence.
10. Thase ME, Rush AJ. Treatment-Resistant Depression. In: Bloom FE, Kupfer DJ, eds. Psychopharmacology: The FourthGeneration of Progress. New York: Raven Press Ltd.; 1995:1081-1097.
11. Kroenke K, West SL, Swindle R, Gilsenan A, Eckert GJ, Dolor R et al. Similar effectiveness of paroxetine, fluoxetine, andsertraline in primary care: a randomized trial. JAMA. 2001; 286: 2947-2955.
12. Nelson JC. Managing treatment-resistant major depression. J Clin Psychiatry. 2003; 64 Suppl 1: 5-12.13. Hirschfeld RM, Montgomery SA, Aguglia E, Amore M, Delgado PL, Gastpar M et al. Partial response and nonresponse to
antidepressant therapy: current approaches and treatment options. J Clin Psychiatry. 2002; 63: 826-837.14. Lam RW, Wan DD, Cohen NL, Kennedy SH. Combining antidepressants for treatment-resistant depression: a review. J Clin
Psychiatry. 2002; 63: 685-693.15. Shelton RC. The use of antidepressants in novel combination therapies. J Clin Psychiatry. 2003; 64 Suppl 2: 14-18.16. Stimpson N, Agrawal N, Lewis G. Randomised controlled trials investigating pharmacological and psychological
interventions for treatment-refractory depression. Systematic review. Br J Psychiatry. 2002; 181: 284-294.17. Anderson IM. Drug treatment of depression: Reflections on the evidence. Adv Psychiatric Treat. 2003; 9: 11-20.18. Kennedy N, McDonough M. Pharmacological management of treatment resistant depression: A clinical review. Irish J
Psychol Med. 2003; 20: 18-23.19. McIntyre RS, Muller A, Mancini DA, Silver ES. What to do if an initial antidepressant fails? Can Fam Physician. 2003; 49: 449-
457.20. Kennedy S, McIntyre R, Fallu A, Lam R. Pharmacotherapy to sustain the fully remitted state. J Psychiatry Neurosci. 2002;
27: 269-280.21. Trivedi MH, Kleiber BA. Algorithm for the treatment of chronic depression. J Clin Psychiatry. 2001; 62 Suppl 6: 22-29.22. Fava M. New approaches to the treatment of refractory depression. J Clin Psychiatry. 2000; 61: 26-32.23. Crismon ML, Trivedi M, Pigott TA, Rush AJ, Hirschfeld RMA, Kahn DA et al. The Texas medication algorithm project:
Report of the Texas consensus conference panel on medication treatment of major depressive disorder. J Clin Psychiatry.1999; 60: 142-156.
24. Thase ME, Rush AJ. When at first you don't succeed: sequential strategies for antidepressant nonresponders. J ClinPsychiatry. 1997; 58: 23-29.
25. Thase ME. Therapeutic alternatives for difficult-to-treat depression: A narrative review of the state of the evidence. CNSSpectr. 2004; 9: 808-821.
26. Parikh RM, Lebowitz BD. Current perspectives in the management of treatment-resistant depression. Dial Clin Neurosci.2004; 6: 53-60.
27. Pridmore S, Turnier-Shea Y. Medication options in the treatment of treatment-resistant depression. Aust N Z J Psychiatry.2004; 38: 219-225.
28. Mann JJ. The medical management of depression. N Engl J Med. 2005; 353: 1819-1834.29. Marangell LB. Switching antidepressants for treatment-resistant major depression. J Clin Psychiatry. 2001; 62 Suppl 18: 12-
17.30. Fava M. Management of nonresponse and intolerance: Switching strategies. J Clin Psychiatry. 2000; 61: 10-12.31. O'Reardon JP, Brunswick DJ, Amsterdam JD. Treatment-resistant depression in the age of serotonin: Evolving strategies.
Curr Opin Psychiatry. 2000; 13: 93-98.32. Nelson JC. Treatment of antidepressant nonresponders: augmentation or switch? J Clin Psychiatry. 1998; 59 Suppl 15: 35-
41.33. Rojo JE, Ros S, Aguera L, de la GJ, de Pedro JM. Combined antidepressants: clinical experience. Acta Psychiatr Scand
Suppl. 2005; 25-31, 36.

ChapterReview of switching in MDD
5
93
34. Bollini P, Pampallona S, Tibaldi G, Kupelnick B, Munizza C. Effectiveness of antidepressants. Meta-analysis of dose-effectrelationships in randomised clinical trials. Br J Psychiatry. 1999; 174: 297-303.
35. Baker CB, Tweedie R, Duval S, Woods SW. Evidence that the SSRI dose response in treating major depression should bereassessed: A meta-analysis. Depress Anxiety. 2003; 17: 1-9.
36. Corruble E, Guelfi JD. Does increasing dose improve efficacy in patients with poor antidepressant response: a review.Acta Psychiatr Scand. 2000; 101: 343-348.
37. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard-dose selectiveserotonin reuptake inhibitors in major depressive disorder: Systematic review. Br J Psychiatry. 2006; 189: 309-316.
38. Bauer M, Dopfmer S. Lithium augmentation in treatment-resistant depression: meta-analysis of placebo-controlledstudies. J Clin Psychopharmacol. 1999; 19: 427-434.
39. Bauer M, Forsthoff A, Baethge C, Adli M, Berghofer A, Dopfmer S et al. Lithium augmentation therapy in refractorydepression-update 2002. Eur Arch Psychiatry Clin Neurosci. 2003; 253: 132-139.
40. Byrne S, Rothschild AJ. Psychiatrists' responses to failure of maintenance therapy with antidepressants. Psychiatr Serv.1997; 48: 835-837.
41. Mischoulon D, Nierenberg AA, Kizilbash L, Rosenbaum JF, Fava M. Strategies for managing depression refractory toselective serotonin reuptake inhibitor treatment: a survey of clinicians. Can J Psychiatry. 2000; 45: 476-481.
42. Shergill SS, Katona CL. Pharmacological choices after one antidepressant fails: a survey of UK psychiatrists. J AffectDisord. 1997; 43: 19-25.
43. Fredman SJ, Fava M, Kienke AS, White CN, Nierenberg AA, Rosenbaum JF. Partial response, nonresponse, and relapsewith selective serotonin reuptake inhibitors in major depression: a survey of current "next-step" practices. J ClinPsychiatry. 2000; 61: 403-408.
44. Bero L, Rennie D. The Cochrane Collaboration. Preparing, maintaining, and disseminating systematic reviews of theeffects of health care. JAMA. 1995; 274: 1935-1938.
45. Rush AJ, Fava M, Wisniewski SR, Lavori PW, Trivedi MH, Sackeim HA et al. Sequenced treatment alternatives to relievedepression (STAR*D): rationale and design. Control Clin Trials. 2004; 25: 119-142.
46. Baldomero EB, Ubago JG, Cercos CL, Ruiloba JV, Calvo CG, Lopez RP. Venlafaxine extended release versus conventionalantidepressants in the remission of depressive disorders after previous antidepressant failure: ARGOS study. DepressAnxiety. 2005; 22: 68-76.
47. Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006; 354: 1231-1242.
48. Fava M, Rush AJ, Wisniewski SR, Nierenberg AA, Alpert JE, McGrath PJ et al. A comparison of mirtazapine andnortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am JPsychiatry. 2006; 163: 1161-1172.
49. McGrath PJ, Stewart JW, Fava M, Trivedi MH, Wisniewski SR, Nierenberg AA et al. Tranylcypromine versus venlafaxineplus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry.2006; 163: 1531-1541.
50. Munoz SR, Bangdiwala SI. Interpretation of Kappa and B statistics measures of agreement. J Applied Stat. 1997; 24: 105-111.
51. Richtlijnontwikkeling binnen het Kwaliteitsinstituut voor de Gezondheidszorg CBO. Handleiding voor werkgroepleden.Kwaliteitsinstituut voor de Gezondheidszorg CBO.2000. Utrecht, Kwaliteitsinstituut voor de Gezondheidszorg CBO.
52. SIGN 50: A guideline developers' handbook. 50. 2001. Edinburgh, Scottish Intercollegiate Guideline Network. 53. Sackett DL, Straus SE, Richardson WS, Rosenberg W, Haynes RB. Evidence-Based Medicine. How to practice and teach EBM.
2 ed. Edinburgh: Churchill Livingstone; 2000.54. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.55. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979; 134: 382-
389.56. Guy W, Clearly PA, Close JH, Conners CK, Covi L, et al. ECDEU Assessment manual for psychopharmacology. DHEW
publication (ADM) 76-338. 1976. Washington, DC, US Department of Health, Education, and Welfare. 57. Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN et al. The 16-Item Quick Inventory of Depressive
Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients withchronic major depression. Biol Psychiatry. 2003; 54: 573-583.
58. Moncrieff J, Churchill R, Drummond DC, McGuire H. Development of a quality assessment instrument for trials oftreatments for depression and neurosis. Int J Methods Psychiatr Res. 2001; 10: 126-133.
59. Posternak MA, Zimmerman M. Switching versus augmentation: a prospective, naturalistic comparison in depressed,treatment-resistant patients. J Clin Psychiatry. 2001; 62: 135-142.
60. Smeraldi E. Amisulpride versus fluoxetine in patients with dysthymia or major depression in partial remission: a double-blind, comparative study. J Affect Disord. 1998; 48: 47-56.
61. Emrich HM, Berger M, Riemann D, von Zerssen D. Serotonin reuptake inhibition vs. norepinephrine reuptake inhibition: adouble-blind differential-therapeutic study with fluvoxamine and oxaprotiline in endogenous and neurotic depressives.Pharmacopsychiatry. 1987; 20: 60-63.
62. Weintraub D. Nortriptyline in geriatric depression resistant to serotonin reuptake inhibitors: case series. J GeriatrPsychiatry Neurol. 2001; 14: 28-32.
63. Keller MB, McCullough JP, Klein DN, Arnow B, Dunner DL, Gelenberg AJ et al. A comparison of nefazodone, the cognitivebehavioral-analysis system of psychotherapy, and their combination for the treatment of chronic depression. N Engl JMed. 2000; 342: 1462-1470.
64. Sajatovic M, DiGiovanni S, Fuller M, Belton J, DeVega E, Marqua S et al. Nefazodone therapy in patients with treatment-resistant or treatment-intolerant depression and high psychiatric comorbidity. Clin Ther. 1999; 21: 733-740.

Chap
ter
Chapter 5
5
94
65. Lam RW, Hossie H, Solomons K, Yatham LN, Lam RW, Hossie H et al. Citalopram and bupropion-SR: combining versusswitching in patients with treatment-resistant depression. J Clin Psychiatry. 2004; 65: 337-340.
66. Birkenhager TK, van den Broek WW, Mulder PG, Bruijn JA, Moleman P. Efficacy and tolerability of tranylcypromine versusphenelzine: a double-blind study in antidepressant-refractory depressed inpatients. J Clin Psychiatry. 2004; 65: 1505-1510.
67. Joffe RT, Levitt AJ, Sokolov STH, Young LT. Response to an open trial of a second SSRI in major depression. J ClinPsychiatry. 1996; 57: 114-115.
68. Thase ME, Blomgren SL, Birkett MA, Apter JT, Tepner RG. Fluoxetine treatment of patients with major depressivedisorder who failed initial treatment with sertraline. J Clin Psychiatry. 1997; 58: 16-21.
69. Thase ME, Feighner JP, Lydiard RB. Citalopram treatment of fluoxetine nonresponders. J Clin Psychiatry. 2001; 62: 683-687.
70. Zarate CA, Kando JC, Tohen M, Weiss MK, Cole JO. Does intolerance or lack of response with fluoxetine predict the samewill happen with sertraline? J Clin Psychiatry. 1996; 57: 67-71.
71. Brown WA, Harrison W. Are patients who are intolerant to one serotonin selective reuptake inhibitor intolerant toanother? J Clin Psychiatry. 1995; 56: 30-34.
72. Thase ME, Ferguson JM, Lydiard RB, Wilcox CS. Citalopram treatment of paroxetine-intolerant depressed patients.Depress Anxiety. 2002; 16: 128-133.
73. Calabrese JR, Londborg PD, Shelton MD, Thase ME. Citalopram treatment of fluoxetine-intolerant depressed patients. JClin Psychiatry. 2003; 64: 562-567.
74. Poirier MF, Boyer P. Venlafaxine and paroxetine in treatment-resistant depression. Double-blind, randomisedcomparison. Br J Psychiatry. 1999; 175: 12-16.
75. Ferreri M, Lavergne F, Berlin I, Payan C, Puech AJ. Benefits from mianserin augmentation of fluoxetine in patients withmajor depression non-responders to fluoxetine alone. Acta Psychiatr Scand. 2001; 103: 66-72.
76. Nierenberg AA, Papakostas GI, Petersen T, Kelly KE, Iacoviello BM, Worthington JJ et al. Nortriptyline for treatment-resistant depression. J Clin Psychiatry. 2003; 64: 35-39.
77. Nolen WA, van de Putte JJ, Dijken WA, Kamp JS, Blansjaar BA, Kramer HJ et al. Treatment strategy in depression. I. Non-tricyclic and selective reuptake inhibitors in resistant depression: a double-blind partial crossover study on the effects ofoxaprotiline and fluvoxamine. Acta Psychiatr Scand. 1988; 78: 668-675.
78. Peselow ED, Filippi AM, Goodnick P, Barouche F, Fieve RR. The short- and long-term efficacy of paroxetine HCL: B. Datafrom a double-blind crossover studyand from a year-long term trial vs. imipramine and placebo. Psychopharmacol Bull.1989; 25: 272-276.
79. Thase ME, Rush AJ, Howland RH, Kornstein SG, Kocsis JH, Gelenberg AJ et al. Double-blind switch study of imipramine orsertraline treatment of antidepressant-resistant chronic depression. Arch Gen Psychiatry. 2002; 59: 233-239.
80. Thase ME, Keller MB, Gelenberg AJ, Hirschfeld RM, Schatzberg AF. Double-blind crossover antidepressant study:sertraline versus imipramine [Abstract]. Psychopharmacol Bull. 1995; 31: 535.
81. Fava M, Dunner DL, Greist JH, Preskorn SH, Trivedi MH, Zajecka J et al. Efficacy and safety of mirtazapine in majordepressive disorder patients after SSRI treatment failure: an open-label trial. J Clin Psychiatry. 2001; 62: 413-420.
82. Kaplan EM. Efficacy of venlafaxine in patients with major depressive disorder who have unsustained or no response toselective serotonin reuptake inhibitors: An open-label, uncontrolled study. Clin Ther. 2002; 24: 1194-1200.
83. Mischoulon D, Opitz G, Kelly K, Fava M, Rosenbaum JF, Mischoulon D et al. A preliminary open study of the tolerabilityand effectiveness of nefazodone in major depressive disorder: comparing patients who recently discontinued an SSRIwith those on no recent antidepressant treatment. Depress Anxiety. 2004; 19: 43-50.
84. Mitchell PB, Schweitzer I, Burrows G, Johnson G, Polonowita A. Efficacy of venlafaxine and predictors of response in aprospective open-label study of patients with treatment-resistant major depression. J Clin Psychopharmacol. 2000; 20:483-487.
85. de Montigny C, Silverstone PH, Debonnel G, Blier P, Bakish D. Venlafaxine in treatment-resistant major depression: aCanadian multicenter, open-label trial. J Clin Psychopharmacol. 1999; 19: 401-406.
86. Nierenberg AA, Feighner JP, Rudolph R, Cole JO, Sullivan J. Venlafaxine for treatment-resistant unipolar depression. J ClinPsychopharmacol. 1994; 14: 419-423.
87. Poirier MF. The concept of resistant depression and therapeutic strategies, particularly with venlafaxine. Encephale. 1999;25: 55-57.
88. Reynaert-Dupuis C, Zdanowicz N, Leyman S, Mignon A, Seghers S. Efficacy and tolerance of venlafaxine in depressedpatients switched from prior antidepressant treatment. Prim Care Psychiatry. 2002; 8: 63-68.
89. Saiz-Ruiz J, Ibanez A, Diaz-Marsa M, Arias F, Padin J, Martin-Carrasco M et al. Efficacy of venlafaxine in major depressionresistant to selective serotonin reuptake inhibitors. Prog Neuropsychopharmacol Biol Psychiatry. 2002; 26: 1129-1134.
90. Wan DD, Kundhur D, Solomons K, Yatham LN, Lam RW. Mirtazapine for treatment-resistant depression: a preliminaryreport. J Psychiatry Neurosci. 2003; 28: 55-59.
91. Lavori PW, Rush AJ, Wisniewski SR, Alpert J, Fava M, Kupfer DJ et al. Strengthening clinical effectiveness trials: Equipoise-stratified randomization. Biol Psychiatry. 2001; 15 NOV0150: 792-801.
92. Fava M, Papakostas GI, Petersen T, Mahal Y, Quitkin F, Stewart J et al. Switching to bupropion in fluoxetine-resistantmajor depressive disorder. Ann Clin Psychiatry. 2003; 15: 17-22.
93. Walker PW, Cole JO, Gardner EA, Hughes AR, Johnston JA, Batey SR et al. Improvement in fluoxetine-associated sexualdysfunction in patients switched to bupropion. J Clin Psychiatry. 1993; 54: 459-465.
94. Fava M, McGrath PJ, Sheu WP. Switching to reboxetine: an efficacy and safety study in patients with major depressivedisorder unresponsive to fluoxetine. J Clin Psychopharmacol. 2003; 23: 365-369.
95. Nolen WA, van de Putte JJ, Dijken WA, Kamp JS, et a. Treatment strategy in depression: II. MAO inhibitors in depressionresistant to cyclic antidepressants: Two controlled crossover studies with tranylcypromine versus L-5-hydroxytryptophanand nomifensine. Acta Psychiatr Scand. 1988; 78: 676-683.

ChapterReview of switching in MDD
5
95
96. Nolen WA, van de Putte JJ, Dijken WA, Kamp JS. L-5HTP in depression resistant to re-uptake inhibitors. An opencomparative study with tranylcypromine. Br J Psychiatry. 1985; 147: 16-22.
97. Kreider MS, Bushnell WD, Oakes R, Wheadon DE. A double-blind, randomized study to provide safety information onswitching fluoxetine-treated patients to paroxetine without an intervening washout period. J Clin Psychiatry. 1995; 56:142-145.
98. Rosenbaum JF, Fava M, Hoog SL, Ascroft RC, Krebs WB. Selective serotonin reuptake inhibitor discontinuation syndrome:a randomized clinical trial. Biol Psychiatry. 1998; 44: 77-87.
99. Judge R, Parry MG, Quail D, Jacobson JG. Discontinuation symptoms: Comparison of brief interruption in fluoxetine andparoxetine treatment. Int Clin Psychopharmacol. 2002; 17: 217-225.
100. Fava M, Rush AJ. Current status of augmentation and combination treatments for major depressive disorder:a literaturereview and a proposal for a novel approach to improve practice. Psychother Psychosom. 2006; 75: 139-153.
101. George MS, Rush AJ, Marangell LB, Sackeim HA, Brannan SK, Davis SM et al. A one-year comparison of vagus nervestimulation with treatment as usual for treatment-resistant depression. Biol Psychiatry. 2005; 58: 364-373.
102. Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamines. Nat Rev Neurosci. 2006; 7:137-151.
103. Anderson IM. SSRIS versus tricyclic antidepressants in depressed inpatients: A meta- analysis of efficacy and tolerability.Depress Anxiety. 1998; 7: 11-17.
104. Anderson IM. Meta-analytical studies on new antidepressants. Br Med Bull. 2001; 57: 161-178.105. Bruijn JA, Moleman P, Mulder PG, van den Broek WW, van Hulst AM, van der Mast RC et al. A double-blind, fixed blood-
level study comparing mirtazapine with imipramine in depressed in-patients. Psychopharmacology (Berl). 1996; 127: 231-237.
106. van den Broek WW, Birkenhager TK, Mulder PG, Bruijn JA, Moleman P. A double-blind randomized study comparingimipramine with fluvoxamine in depressed inpatients. Psychopharmacology (Berl). 2004; 175: 481-486.
107. Howland RH, Thase ME. Switching strategies for the treatment of unipolar major depression. In: Rush AJ, ed. Mooddisorders: systematic medication management. Basel, Switzerland: Karger; 1997:56-65.
108. Charney DS, Price LH, Heninger GR. Desipramine-yohimbine combination treatment of refractory depression.Implications for the beta-adrenergic receptor hypothesis of antidepressant action. Arch Gen Psychiatry. 1986; 43: 1155-1161.
109. Reimherr FW, Wood DR, Byerley B, Brainard J, Grosser BI. Characteristics of responders to fluoxetine. PsychopharmacolBull. 1984; 20: 70-72.
110. Amsterdam JD, Maislin G, Potter L. Fluoxetine efficacy in treatment resistant depression. Prog NeuropsychopharmacolBiol Psychiatry. 1994; 18: 243-261.
111. Beasley CM, Jr., Sayler ME, Cunningham GE, Weiss AM, Masica DN. Fluoxetine in tricyclic refractory major depressivedisorder. J Affect Disord. 1990; 20: 193-200.
112. Delgado PL, Price LH, Charney DS, Heninger GR. Efficacy of fluvoxamine in treatment-refractory depression. J AffectDisord. 1988; 15: 55-60.
113. White K, Wykoff W, Tynes LL, Schneider L, Zemansky M. Fluvoxamine in the treatment of tricyclic-resistant depression.Psychiatr J Univ Ott. 1990; 15: 156-158.
114. Gagiano CA, Mueller PG, Fourie J, LeRoux JF. The therapeutic efficacy of paroxetine: (a) An open study in patients withmajor depression not responding to antidepressants; (b) a double-blind comparison with amitriptyline in depressedoutpatients. Acta Neurol Scand. 1989; 80: 130-131.
115. Ghaziuddin N, Naylor MW, King CA. Fluoxetine in tricyclic refractory depression in adolescents. Depression. 1995; 2: 287-291.
116. Ferguson J, Cunningham L, Merideth C, Apter J, Feighner J, Ionescu-Pioggia M et al. Bupropion in tricyclic antidepressantnonresponders with unipolar major depressive disorder. Ann Clin Psychiatry. 1994; 6: 153-160.
117. Stern WC, Harto-Truax N, Bauer N. Efficacy of bupropion in tricyclic-resistant or intolerant patients. J Clin Psychiatry. 1983;44: 148-152.
118. Cole JO, Schatzberg AF, Sniffin C, Zolner J, Cole JP. Trazodone in treatment-resistant depression: an open study. J ClinPsychopharmacol. 1981; 1 (Suppl.): 49-54.
119. Schmauss M, Laakmann G, Dieterle D. Nomifensine: a double-blind comparison of intravenous versus oral administrationin therapy-resistant depressed patients. Pharmacopsychiatry. 1985; 18: 88-90.
120. Nolen WA, Haffmans PM, Bouvy PF, Duivenvoorden HJ. Monoamine oxidase inhibitors in resistant major depression. Adouble-blind comparison of brofaromine and tranylcypromine in patients resistant to tricyclic antidepressants. J AffectDisord. 1993; 28: 189-197.
121. McGrath PJ, Stewart JW, Harrison W, Quitkin FM. Treatment of tricyclic refractory depression with a monoamine oxidaseinhibitor antidepressant. Psychopharmacol Bull. 1987; 23: 169-172.
122. McGrath PJ, Stewart JW, Nunes EV, Ocepek-Welikson K, Rabkin JG, Quitkin FM et al. A double-blind crossover trial ofimipramine and phenelzine for outpatients with treatment-refractory depression. Am J Psychiatry. 1993; 150: 118-123.
123. Thase ME, Frank E, Mallinger AG, Hamer T, Kupfer DJ. Treatment of imipramine-resistant recurrent depression, III:Efficacy of monoamine oxidase inhibitors. J Clin Psychiatry. 1992; 53: 5-11.
124. Leonard BE. Neuropharmacology of antidepressants that modify central noradrenergic and serotonergic function: Ashort review. Hum Psychopharmacol. 1999; 14: 75-81.
125. Shelton RC. The dual-action hypothesis: does pharmacology matter? J Clin Psychiatry. 2004; 65 Suppl 17: 5-10.126. Stahl SM. Symptoms and circuits, part 1: major depressive disorder. J Clin Psychiatry. 2003; 64: 1282-1283.127. Hindmarch I. Beyond the monoamine hypothesis: mechanisms, molecules and methods. Eur Psychiatry. 2002; 17 Suppl 3:
294-299.

Chap
ter
Chapter 5
5
96
128. Owens MJ. Selectivity of antidepressants: from the monoamine hypothesis of depression to the SSRI revolution andbeyond. J Clin Psychiatry. 2004; 65 Suppl 4: 5-10.
129. Burke WJ. Selective versus multi-transmitter antidepressants: are two mechanisms better than one? J Clin Psychiatry.2004; 65 Suppl 4: 37-45.
130. Fava M, Kendler KS. Major depressive disorder. Neuron. 2000; 28: 335-341.131. Delgado PL. How antidepressants help depression: mechanisms of action and clinical response. J Clin Psychiatry. 2004; 65
Suppl 4: 25-30.132. Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D et al. Medication augmentation after the failure of
SSRIs for depression. N Engl J Med. 2006; 354: 1243-1252.133. Keller MB, Lavori PW, Mueller TI, Endicott J, Coryell W, Hirschfeld RM et al. Time to recovery, chronicity, and levels of
psychopathology in major depression. A 5-year prospective follow-up of 431 subjects. Arch Gen Psychiatry. 1992; 49: 809-816.
134. Mueller TI, Leon AC. Recovery, chronicity, and levels of psychopathology in major depression. Psychiatr Clin North Am.1996; 19: 85-102.
135. Lenox-Smith AJ, Schaeffer P, Reynolds A, Williard L. A double blind trial of venlafaxine XR versus citalopram in patientswith severe depression. [Abstract]. J Psychopharmacol. 2001; 15 Suppl 3: A11.
136. Thase ME, Kremer C, Rodrigues HE, et al. Mirtazapine versus sertraline after SSRI non-response. [Abstract] 39th annualmeeting of the American College of Neuropsychopharmacology. Dec 10-14, 2000.San Juan, Puerto Rico. 2000.
137. Thase ME, Kremer C, Rodrigues H. Mirtazapine versus sertraline after SSRI nonresponse [Abstract]. American PsychiatricAssociation.2001 annual meeting: New Orleans LA . 2001.
138. Trivedi MH, Rush AJ, Crismon ML, Kashner TM, Toprac MG, Carmody TJ et al. Clinical results for patients with majordepressive disorder in the Texas Medication Algorithm Project. Arch Gen Psychiatry. 2004; 61: 669-680.
139. Nierenberg AA, DeCecco LM. Definitions of antidepressant treatment response, remission, nonresponse, partialresponse, and other relevant outcomes: a focus on treatment-resistant depression. J Clin Psychiatry. 2001; 62 Suppl 16: 5-9.
140. Nierenberg AA, Papakostas GI, Petersen T, Montoya HD, Worthington JJ, Tedlow J et al. Lithium augmentation ofnortriptyline for subjects resistant to multiple antidepressants. J Clin Psychopharmacol. 2003; 23: 92-95.

ChapterReview of switching in MDD
5
97


SEROTONIN IN THE ETIOLOGY OF MAJOR DEPRESSIVE DISORDER
PART
Serotonin in the etiology of Major Depressive Disorder

100

CHA
PTER
MOOD IS INDIRECTLY RELATED TO SEROTONIN, NOREPINEPHRINE AND DOPAMINE LEVELS IN HUMANS: A META-ANALYSIS OF MONOAMINE DEPLETION STUDIES
Molecular Psychiatry 2007; 12: 331-359
Henricus G. Ruhé1
Nada S. Mason1
Aart H. Schene1
1Program for Mood Disorders, Department of Psychiatry, Academic Medical Center, University ofAmsterdam, Amsterdam, The Netherlands

Chap
ter
Chapter 6
6
102
Abstract
BackgroundDysfunction in the monoamine systems of serotonin (5-HT), norepinephrine (NE) and dopamine(DA) may causally be related to Major Depressive Disorder (MDD). Monoamine depletion studiesinvestigate the direct effects of monoamines on mood. Acute tryptophan depletion (ATD) orpara-chlorophenylalanine (PCPA) deplete 5-HT, acute phenylalanine/tyrosine depletion (APTD) oralpha-methyl-para-tyrosine (AMPT) deplete NE/DA. Available depletion studies found conflictingresults in heterogeneous populations: healthy controls, patients with previous MDD in remissionand patients suffering from MDD.
AimTo review and quantify the decrease in mood after 5-HT and NE/DA depletion in humans.
MethodsSystematic search of MEDLINE and EMBASE (1966-October 2006) and cross-references.Randomized studies applying ATD, PCPA, APTD or AMPT versus control depletion were included.Pooling of results by meta-analyses was stratified for studied population and design of the study(within or between subjects).
ResultsSeventy-three ATD, 2 PCPA, 10 APTD and 8 AMPT studies were identified of which 45 ATD and 8APTD studies could be meta-analyzed. 5-HT or NE/DA depletion did not decrease mood in healthycontrols. 5-HT or NE/DA depletion slightly lowered mood in healthy controls with a family historyof MDD. In drug-free patients with MDD in remission, a moderate mood decrease was found forATD, without an effect of APTD. ATD induced relapse in patients with MDD in remission who usedserotonergic antidepressants.
ConclusionsMonoamine depletion studies demonstrate decreased mood in subjects with a family history ofMDD and in drug-free patients with MDD in remission, but do not decrease mood in healthyhumans. Although depletion studies usefully investigate the etiological link of 5-HT and NE withMDD, they fail to demonstrate a causal relation. They presumably clarify a vulnerability trait tobecome depressed. Directions for further investigation of this vulnerability trait are proposed.

ChapterMonoamine depletion in humans
6
103
Introduction
Major depressive disorder (MDD) is characterized by a lowered mood. MDD is a disabling diseasewhich affects 20% of the world’s population.1 MDD is often treated with antidepressants (AD),mostly Selective Serotonin Reuptake Inhibitors (SSRIs), Serotonin Norepinephrin Reuptakeinhibitors (SNRIs), Norepineprine Reuptake Inhbitors (NERIs) or Tricyclic Antidepressants (TCAs).2-5
The working mechanism of AD is believed to be either by (1) increased neurotransmission byincreased synaptic levels of serotonin, norepinephrine and dopamine (monoamines) or (2)specific agonistic effects on serotonin or norepinephrine (sub-)receptors. The increased levels ofmonoamines were discovered in the late fifties, when the TCAs and Monoamine-oxidase Ainhibitors (MAO-I) appeared to effectively treat MDD. This discovery led to the monoaminehypothesis: MDD might etiologically be explained by a deficiency in monoamineneurotransmitters: serotonin (5-HT), norepinephrine (NE) or (to a lesser degree) dopamine (DA).The monoamine systems in the brain have complex interactions. Therefore, the current, lesspertinent view is that the monoamine hypothesis only partially explains MDD and the response toAD.6-10
Depletion of the available 5-HT, NE and/or DA is used as a model to test the involvement ofmonoaminergic systems in MDD. Two reviews recently described and reviewed the techniques ofmonoamine precursor depletion and enzyme-blocking methods.11;12 In brief, 5-HT depletion can beachieved by rapidly lowering the levels of the essential amino-acid tryptophan which cannot besynthesized by the body and must be ingested to enable formation of 5-HT. To achieve depletion,a tryptophan free amino-acid mixture is administered (acute tryptophan depletion (ATD)).13
Depletion of NE and DA uses the same concept (acute depletion of the essential amino-acidsphenylalanine/tyrosine (APTD)).14 As an alternative to induce a state of depletion, enzyme-blocking agents decrease the production of the monoamines. Para-chlorophenylalanine (PCPA)blocks 5-HT synthesis,15;16 and alpha-methyl-para-tyrosine (AMPT) NE and DA synthesis.17 Since1975 an increasing number of depletion studies have been conducted. Monoamine depletionshowed differential effects in different study populations: healthy controls, healthy controls witha family-history of MDD, patients with MDD in remission using AD or after cessation of AD, andpatients who had a current episode of MDD.
Previous reviews summarized the methodology of depletion tests.18 Others reviewed thefindings of specific depletion tests across different psychiatric illnesses,11;18-20 or the prediction ofresponse to depletion.21;22 Booij et al. presented a study that pooled results across studiesindividual subject data of six ATD studies ('mega-analysis') in order to investigate the mediatingrole of clinical, demographic and biochemical characteristics in the mood response to ATD inremitted subjects who previously had MDD.21 However, a clear summary of the mood-effects ofmonoamine depletion across different populations is lacking. Except from the study of Booij et al.,we are unaware of any attempt of pooling studies. Pooling is important because the small-sizeddepletion studies might not have detected small differences by a lack of power. Finally–as in drugresearch–pooling will quantify the weight of positive versus negative studies.
Therefore, we aimed to review the evidence for mood lowering properties of monoaminedepletion studies as a model for MDD. Our main question was: does the depletion of monoamine(5-HT and NE/DA) systems lower mood in humans? A secondary question was: is the lowering ofmood different across different populations? This paper reports our systematic review with astratified meta-analysis of the mood effects of monoamine depletion studies.

Chap
ter
Chapter 6
6
104
Methods
Design of the study We included all available randomized prospective monoamine depletion studies (tryptophan andphenylalanine/tyrosine) and enzyme blocking studies (PCPA or AMPT) in humans. Included studiesmeasured a change in mood after depletion. Two study designs were included. First, randomizedwithin-subjects studies, where each subject was exposed to a true depletion versus a shamintervention at a second occasion. This way, each subject served as his/her own control. Second,randomized between-subjects studies, where two groups of subjects were compared, with theintervention applied to one group and a control intervention to the other group. We excludedanimal studies and studies that selected patients with apparent co-morbidity (e.g. substanceabuse or dependence, studies with only smokers, psychotic disorders, anxiety disorders).Additionally, we excluded studies in depression subtypes with a supposed different etiology (e.g.seasonal affective disorder, bipolar disorder). We also excluded studies that combined depletionwith other interventions (e.g. sleep deprivation) or studies that did not report mood effects.Finally, we excluded studies recruiting a patient group with both unipolar and bipolar depressionwhen >25% of subjects had bipolar depression and when no separate data for the unipolar groupwere provided.
Literature searches and selectionWe searched PubMed from 1966 to October 1st 2006 and EMBASE from 1980 to October 1st 2006using a comprehensive search strategy (Table 6.1). We retrieved additional references identified inprevious reviews11;12;18-20;23-25 and cross-references from identified studies. Two reviewers (H.G.R.and N.S.M.) independently selected articles based on the a priori inclusion and exclusion criteriaas stated above. In case of doubt an article was retrieved and the full content was considered. In85.4% the reviewers agreed on selection instantly. The initial kappa for agreement was 0.61 (95%confidence interval (95% CI): 0.52 - 0.71). Agreement was mainly influenced by a differentialinclusion of research letters, studies in depression subtypes with a supposed different etiologyand studies without apparent mood-scores in the abstract. Discrepancies in included studies werediscussed between both reviewers until full consensus was reached.
Data extraction and Assessment of the studies Two authors (H.G.R. and N.S.M.) abstracted the retrieved studies as follows: population studied,study design (within-subjects or between subjects), applied mood scale(s), number of subjects(and–if applicable–number of subjects for each sequence of intervention and control depletion),male/female ratio, the intervention and its control condition (including dosage and duration).Furthermore, we extracted plasma levels of tryptophan, tyrosine or relevant levels ofmonoamine-metabolites before and after intervention and control condition and other relevant
Table 6.1. Search terms.
# Search terms used
1 Depressive disorder[MeSH] OR depression[MeSH] OR (depressive[TW] AND symptoms[TW]) OR mood[TW]
2 Indoleamine$ OR serotonin[MeSH] OR 5-hydroxytryptamine[TW] OR 5-HT[TW]
3 ((Tryptophan[MeSH] OR Tryptophan[TW]) AND depletion[All Fields]) OR (fenclonine[MeSH] OR PCPA[TW] OR parachlorophenylalanine[TW])
4 #1 AND #2 AND #3
5 Catecholamines[MeSH] OR norepinephrine[MeSH] OR epinephrine[MeSH] OR Dopamine[MeSH]
6 ((Tyrosine[MeSH] OR Tyrosine[TW]) AND depletion[All Fields]) OR (alpha-methyltyrosine[MeSH] OR AMPT[TW] OR alpha-methyl-para-tyrosine[TW])
7 #1 AND #5 AND #6
8 (#4 OR #7) NOT (Animal[MeSH] NOT Human[MeSH])
MeSH = Medical Subject Heading, TW = textword Note: PubMed search terms are provided; these terms were slightly modified for EMBASE-searches.

ChapterMonoamine depletion in humans
6
105
data that could influence the outcome of the study (e.g. different consistency of control drink,unclearly reported data, combined interventions). Primary outcome variables were changes inmood-scale scores before and after intervention and control, and the number of relapses in theintervention and control group. If more than one mood-scale was used, we primarily used theProfile of Mood States (POMS).26 Otherwise, we used the Multiple Affect Adjective Checklist(MAACL),27 Visual Analogue [Mood] Scales (VA[M]S),28 Hamilton-depression rating-scale (HDRS),29
and Montgomery-Åsberg Depression Rating Scale (MADRS).30 However, in studies in patientswith previous MDD in remission we primarily chose the HDRS or MADRS followed by the POMS,because most of these studies used depression rating scales to measure effects of depletion. Ifsubscales were used, we took the subscale representing depressed mood.
We validated the abstracted studies (criteria available on request). We based criteria onprevious reviews and the Cochrane handbook.11;12;18;31 We assessed studies as poor, moderate, orgood in the context of this meta-analysis. We particularly assessed quality as good when studiesapplied a randomized double-blind design, when the achieved depletion was judged to besufficient, and when mood-scale ratings were provided.11;18 If more than one of these items wasrated inadequate, we assessed the study as moderate. If studies had one of these items missingand at least one other aspect of the study was not well reported, we also assessed the study asmoderate. If crucial data (e.g. scores and SDs) were missing the study was assessed as poor.
Data synthesisWe qualitatively summarized all included studies in Tables 6.2-6.5 (ATD, PCPA, APTD and AMPTrespectively), irrespective whether the studies were pooled in the meta-analysis. Weacknowledged three clinically heterogeneous study populationsapriori: A. healthy controls (A1negative/ A2 positive family history of MDD), B. patients with a previous MDD currently inremission (B1 currently not using AD / B2 currently using AD) and C. patients with a currentepisode of MDD (with or without AD). These populations were considered and analyzedseparately.
Statistical pooling Mostly, continuous scores for changes in mood rating scales were reported. Some studies inpatients with MDD in remission presented relapse rates. In order to pool continuous effectestimates from different scales, we applied a standardization (providing Hedges’ g). The effect-estimates (1 per study) with the corresponding standard error (SE) were entered in the inverse-variance statistics for pooling in Review Manager 4.2.32;33 The supplementary appendix gives theformulas used to determine Hedge’s g, the difference in relapse rates and corresponding SEs fordifferent study designs.
Different study designs were not combined in pooling. Especially the within subjects designneeds attention in meta-analysis. In this design, differences between the experimental andcontrol condition are statistically paired. As paired data improve power, calculations with datafrom a within subjects (or cross-over) design in a weighted mean differences model would not bejustified.33
Heterogeneity, effect modification, sensitivity-analysis and assessment ofpublication biasWe first performed the meta-analyses with fixed effects models. We assumed morehomogeneous results after our a priori attempt to reduce clinical heterogeneity by stratification.However, if effect-estimates and 95% CI for the individual studies showed graphical poor overlapor consistency, we interpreted this an indication of statistical heterogeneity. We used the chi-squared test and I2 in addition. I2 represents a chi-squared statistic relative to its degree offreedom. An I2 value >50% is indicative of heterogeneity.34 We applied a conservative randomeffects model35 when we suspected statistical heterogeneity.

Chap
ter
Chapter 6
6
106
We investigated effect modification by gender, and the influence of a positive family historyof MDD in healthy controls. Furthermore, we investigated whether different mood scales causeddifferences in outcomes. Therefore, we stratified analyses for these variables, and presentedstratified Hedges’ g, with 95% CI. Differences between strata were tested by subtracting the chi-squared heterogeneity statistic per stratum from the total chi-squared heterogeneity statistic.This residual (Qres) has a chi-squared distribution, with the total number of strata-1 degrees offreedom.
We imputed R= 0.5 to calculate missing pooled SDs within each intervention/control and forthe changes in mood-scores between interventions (supplementary appendix). We checked theimpact of this imputation on the calculated effect estimates. Therefore, we increased R to 0.8(less conservative) and decreased R to 0.2 (more conservative) to compare the new effectestimates with the original findings for R = 0.5.
We assessed publication bias graphically with a funnel-plot, plotting Hedges’ g versus theprecision of the study (the inverse of the standard error of Hedges’ g). Additionally, we testedpublication bias with Galbraith’s radial plot, which regresses the standard normal deviate(Hedges’ g divided by its standard error) against the precision. For a set of studies not distortedby selection bias the intercept of the regression model will be close to zero.36
Results
Identified studiesOur systematic searches identified 392articles. In total we selected 90 studies. Three studiesapplied both ATD and APTD and a control.37-40 Therefore, 73 studies with ATD, 2 with PCPA, 10 withAPTD and 8 with AMPT as monoamine depletion method were identified (summarized in Tables6.2-6.5 respectively). Several studies investigated contrasts in different populations.41-51 A list ofexcluded studies is available on request. Most studies had a within-subjects design (n= 74). Themajority of studies investigated healthy controls (n=64).
Qualitative summary The majority of the 90 studies also investigated other effects of monoamine depletion: 24 studiesmeasured effects on cognitive functions,13;38;43;45;52-67;68-72 11 measured effects on other behavioralmeasures (pain, impulsivity, panic attacks, appetite, noise-stress, aggression),73-83 8 measuredeffects on neuroendocrine parameters,37;60;84-89 11 measured electric encephalogram (EEG)alterations and/or sleep effects,72;90-99 14 measured changes in brain function with PositronEmission Tomography (PET),47;49;51;100-104 Single Photon Emission Computed Tomography(SPECT),105 or Magnetic Resonance Imaging (MRI).66;68-70;106-108 Two studies stratified moodresponse to ATD by genetic polymorphism of the 5-HT transporter promoter region.46;51
We judged the overall methodological quality of the identified studies as good, withappropriate application of the ATD, APTD or AMPT depletion. Two studies with PCPA reportedcase series only and were rated ‘poor’.15;109 In 8 studies no data on the adequacy of the achieveddepletion was reported.15;73;81;88;97;103;109;110 In 2 studies insufficient depletion was attained.63;111 In 10studies no clear SDs or SEs were given for the observed mood-effects.13;76;85;88;92;103;110;112-115 Sixstudies included patients (<25%of total) with Bipolar I or II Disorder.15;65;86;94;114;115 In total, this were14 patients. The reason that several apparently highquality studies were rated as moderate in thisreview was due to the lack of presentation of the mood effects, which in those studies was notthe primary outcome.
In 34 of 90 studies the POMS was used, in 2studies the MAACL, in 34 studies a visual analoguescale, in 29 studies a version of the HDRS, and in 5 of the 90 studies the MADRS. In 5 studies onlyother or undefined mood-scales were used.79;80;97;98;113 Especially for the POMS and the VAS thedirection of a decrease of mood in subjects was not always clearly reported.53;57;77;113 In our meta-analysis no differential effect of the applied mood-scale was observed (Qres= 3.89, df= 2, p>0.05;data not shown).

Chapter6
107
Monoamine depletion in humans
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§
A1.
Hea
lthy
con
trol
s
Abb
ott (
1992
)73R
and.
, DB,
Be
tw. S
S2*
30
MN
ot c
lear
ly
repo
rted
POM
S-de
pres
sion
: ATD
: BL:
2.5
0 (±
0.58
SE)
, pos
t-te
st: 1
.73
(±0.
71).
CO
NT:
BL:
2.5
0 (±
0.58
), p
ost-
test
: 1.9
8 (±
0.58
).St
udy
prim
arily
inve
stig
ates
eff
ect o
f ATD
on
anal
gesi
a.M
oder
ate
Alle
n (2
006)
66R
and.
, DB,
W
ithin
SS,
FH
-10
M
+FG
ood
VAS:
No
exac
t sco
res
repo
rted
, no
sign
. diff
eren
ce in
m
ood
foun
dSt
udy
inve
stig
ates
eff
ects
of A
TD o
n co
gniti
on (v
erba
l flu
ency
and
wor
king
m
emor
y) a
nd p
refr
onta
l act
ivity
by
fMRI
Mod
erat
e
Am
in (2
006)
67R
and.
, DB,
W
ithin
SS
19
FG
ood**
POM
S an
d H
DR
S: N
o ex
act s
core
s re
port
ed, n
o si
gn.
diff
eren
ce in
moo
d fo
und
on to
tal P
OM
S or
HD
RS
Stud
y in
vest
igat
es e
ffec
t of A
TD o
n co
gniti
on (m
emor
y, v
isuo
spat
ial l
earn
ing)
an
d th
e pr
otec
tive
effe
cts
of e
stro
gen
ther
apy
(ET)
in p
eri-/
post
- men
opau
sal
wom
en. O
nly
pre
ET-d
ata
used
.
Mod
erat
e
Barr
(199
7)14
5R
and.
, DB,
W
ithin
SS,
FH
-6 M
+FG
ood
VAS-
happ
y: A
TD: B
L: 5
1.2 (±
9.1)
, pos
t-te
st: 4
2.7
(±13
.8).
CO
NT:
51.7
(±25
.9),
pos
t-te
st: 6
2.7
(±12
.2);
PO
MS
not
repo
rted
Stud
y in
vest
igat
es e
ffec
t of f
luox
etin
e tr
eatm
ent o
n AT
D in
hea
lthy
cont
rols
, onl
y pr
e-flu
oxet
ine
data
use
d.
Goo
d
Benk
elfa
t (19
94)41
Ran
d., D
B,
With
in S
S, F
H-
19 MG
ood
POM
S-de
pres
sed:
ATD
: BL:
57.
2 (±
2.0
SE),
pos
t-te
st: 5
8.9
(±2.
0), c
hang
e: 1.
7 (±
1.4).
CON
T: B
L: 5
6.3
(±2.
6), p
ost-
test
: 60
.0 (±
2.1)
, cha
nge:
4.3
(±2.
3). n
o on
e re
ache
d 10
poi
nt
decl
ine
in P
OM
S-D
.
POM
S ch
ange
d in
FH
+ sa
mpl
e, n
ot in
FH
- sa
mpl
e in
sam
e st
udy
(dat
a pr
ovid
ed
sepa
rate
ly).
Goo
d
Bhat
ti (1
998)
90R
and.
, DB,
W
ithin
SS,
FH
-10
M
Goo
dPO
MS-
depr
esse
d: A
TD: B
L:0.
9 (±
1.3),
post
-tes
t: 0
.7 (±
3.3)
, CO
NT:
BL:
0.9
(±1.3
), p
ost-
test
: 0.4
(±1.0
).
Cont
rol-d
rink
is 2
5% in
terv
entio
n dr
ink,
ATD
an
d CO
NT
diff
er >
48hr
s. S
tudy
inve
stig
ates
ef
fect
s of
ATD
on
slee
p EE
G.
Mod
erat
e
Crae
n (2
002)
74R
and.
?, D
B?,
With
in S
S,85
% FH
-
20
M
Goo
d PO
MS-
depr
esse
d: N
o ex
act s
core
s re
port
ed, n
o si
gn.
diff
eren
ce in
moo
d fo
und
Stud
y in
vest
igat
es d
iffer
ence
in im
puls
ivity
af
ter A
TD b
etw
een
cont
rols
with
FH
+ an
d FH
- for
alc
ohol
ism
Poor
Dan
jou
(199
0)84
Ran
d., D
B,
Betw
.SS,
2*
10
MG
ood**
VAS
and
adap
tatio
n of
MA
ACL
, exa
ct s
core
s no
t rep
orte
d,
no s
ign.
diff
eren
ce in
moo
d fo
und
Stud
y fo
cuse
s on
psy
chom
otor
and
ne
uroe
ndoc
riene
eff
ects
of A
TDM
oder
ate
Deb
ener
(200
2)97
Ran
d., D
B,
With
in S
S, F
H-
18
F N
ot te
sted
M
ood
‘que
stio
nnai
res’
did
not
reve
al d
iffer
ence
s in
aff
ect
for A
TD
Stud
y in
vest
igat
es th
e ef
fect
of A
TD o
n au
dito
ry e
voke
d po
tent
ials
with
EEG
Po
or
Elle
nbog
en (1
996)
134
Ran
d., D
B,
With
in S
S, F
H-
21
FG
ood
POM
S-de
pres
sed**
: ATD
: cha
nge
-4 (±
1.6 S
E), C
ON
T:
chan
ge: 0
.8 (±
1.55)
; VA
MS:
Stud
y al
so e
xam
ines
sta
bilit
y of
ATD
re
spon
se w
hich
app
ears
to b
e po
orG
ood
Ever
s (2
005)
107;
108
Ran
d., D
B,
With
in S
S, F
H-
12
M
Goo
d VA
S sa
dnes
s: A
TD: B
L: 1.
8 (±
1.6),
pos
t-te
st: 2
.0 (±
1.6).
CO
NT:
BL:
1.0
(±0.
7), p
ost-
test
: 1.8
(±1.6
).
Stud
y in
vest
igat
es e
ffec
t of A
TD o
n am
ygda
le re
activ
ity to
fear
ful f
aces
with
fM
RI
Mod
erat
e

108
Chap
ter
Chapter 6
6
Ever
s (2
006a
)68;6
9R
and.
, DB,
W
ithin
SS
13
M
Goo
d PO
MS:
No
exac
t sco
res
repo
rted
, no
sign
. diff
eren
ce in
m
ood
foun
d St
udy
inve
stig
ates
eff
ects
of A
TD o
n co
gniti
on (p
erfo
rman
ce m
onito
ring
and
resp
onse
inhi
bitio
n an
d m
emor
y) a
nd
pref
ront
al /
hipp
ocam
pal a
ctiv
ity b
y fM
RI
Mod
erat
e
Ever
s (2
006b
)70R
and.
, DB,
W
ithin
SS,
FH
- 15
F
Goo
d PO
MS:
No
exac
t sco
res
repo
rted
, no
sign
. diff
eren
ce in
m
ood
foun
d St
udy
inve
stig
ates
eff
ects
of A
TD o
n a
com
bine
d co
gniti
ve a
nd e
mot
iona
ltask
by
fMRI
Mod
erat
e
Gal
lagh
er (2
003)
52R
and.
, DB,
W
ithin
SS
15 MG
ood
VAM
S-co
nten
tedn
ess-
scal
e: s
core
s no
t com
plet
ely
repo
rted
. ATD
: pos
t-te
st: 8
3.4
(±7.
5),C
ON
T: p
ost-
test
: 86.
5 (±
7.8)
No
sign
. cha
nges
in m
ood.
Stud
y fo
cuse
s on
exe
cutiv
e ne
uroc
ogni
tive
func
tions
Mod
erat
e
Har
riso
n (2
002)
37R
and.
, DB,
W
ithin
SS,
FH
-13 F
Goo
d PO
MS-
depr
esse
d:AT
D: B
L: 2
.07
(±0.
63 S
E), p
ost-
test
: 0.7
6 (±
0.63
) CO
NT:
BL:
1.61
(±0.
52),
post
-tes
t: 1.
15 (±
0.56
).
Stud
y al
so re
port
s ab
senc
e of
eff
ects
of A
TD
and
APT
D o
n in
terle
ukin
-6 a
ctiv
atio
nG
ood
Har
rison
(200
4)38
Ran
d., D
B,
With
in S
S, F
H-
13 FG
ood
VAM
S: e
xact
sco
res
not r
epor
ted,
no
sign
. diff
eren
ces
in
moo
d fo
und
Stud
y al
so in
vest
igat
es m
emor
y an
d co
gniti
ve e
ffec
ts o
f ATD
and
APT
DM
oder
ate
Hay
war
d (2
005)
43R
and.
, DB,
Be
tw. S
S, 2
0 FH
-2*
12
M+F
G
ood
HD
RS: A
TD: B
L: 0
.17
(±0.
11 S
E), p
ost-
test
: 0.6
7 (±
0.19
),
CON
T: B
L: 0
.33
(±0.
19),
pos
t-te
st: 0
.41 (
±0.19
). B
DI:
ATD
: BL
: 1.5
(±0.
56 S
E), p
ost-
test
: 0.5
8 (±
0.33
), C
ON
T: B
L: 2
.13
(±0.
57),
pos
t-te
st: 2
.25
(±0.
80).
PO
MS
and
VAS:
exa
ct
scor
es n
ot re
port
ed.
No
sign
ific
ant e
ffec
t on
any
scal
e fo
und.
Stud
y al
so in
vest
igat
es c
ogni
tive
proc
essi
ng
in fo
rmer
ly d
epre
ssed
ver
sus
heal
thy
cont
rols
dur
ing
ATD
Mod
erat
e
Hug
hes
(200
0)98
Ran
d., D
B,
With
in S
S, F
H-
20
M
Goo
d U
ndef
ined
sca
le: n
o si
gnif
ican
t cha
nges
in m
ood
foun
d St
udy
inve
stig
ates
eff
ects
of A
TD o
n au
dito
ry e
voke
d po
tent
ials
on
EEG
Po
or
Hug
hes
(200
3)53
Ran
d., D
B,
With
in S
S, F
H-
20 MG
ood
VAS-
sadn
ess:
ATD
: BL:
90.
7 (±
3.5
SE),
pos
t-te
st: +
6.7
(±4.
7),
CON
T: B
L: 9
4.8
(±2.
6), p
ost-
test
: -1.6
(±1.9
). N
o si
gnifi
cant
m
ain
effe
cts
on a
ny V
AM
S sc
ale.
Stud
y in
vest
igat
es e
ffec
ts o
f ATD
on
verb
al
and
visu
ospa
tial l
earn
ing
and
mem
ory,
at
tent
ion
and
exec
utiv
e fu
nctio
n
Goo
d
Kähk
önen
(200
5)91
Ran
d., D
B,
With
in S
S14
M
+FG
ood
VAS-
depr
essi
on: A
TD: c
hang
e 41
.5 (±
20.7
), C
ON
T: c
hang
e 21
.5 (±
17.6
)St
udy
inve
stig
ates
eff
ects
of A
TD o
n au
dito
ry re
spon
se, M
EG a
nd E
EG s
igna
lsM
oder
ate
Klaa
ssen
(199
9a)14
6R
and.
, DB,
W
ithin
SS
13 M+F
Goo
d PO
MS-
depr
esse
d:AT
D: B
L: 0
.6 (±
1.3),
pos
t-te
st: 1
.5 (±
3.3)
, CO
NT:
BL:
0.5
(±1.0
), p
ost-
test
: 0.2
(±0.
6). V
AS-
depr
esse
d:AT
D: B
L: 2
.7 (±
6.6)
, pos
t-te
st: 8
.2 (±
18.2
), CO
NT:
BL
: 3.2
(±6.
3), p
ost-
test
: 4.5
(±13
.0).
Stud
y in
vest
igat
es s
peci
fici
ty o
f ATD
vs
LYS-
depl
etio
n on
moo
d-ef
fect
s an
d m
emor
y.
Goo
d
Klaa
ssen
(199
9b),
44
Klaa
ssen
(200
2)45
Ran
d., D
B,
With
in S
S, F
H+
& F
H-
11 F
H-
M +
FG
ood
POM
S-de
pres
sed:
sco
res
not c
ompl
etel
y re
port
ed. A
TD-
CON
T di
ffer
ence
s: F
H- 0
.0 (±
0.45
)N
o ba
selin
e PO
MS-
scor
es p
rovi
ded
(‘no
n-si
gnifi
cant
diff
eren
t ces
’). S
tudy
com
pare
d FH
+ gr
oup
with
FH
-. Th
e FH
+ ve
s re
spon
ded
to tr
ypto
phan
dep
letio
n w
ith a
sig
nifi
cant
lo
wer
ing
of P
OM
S sc
ores
, but
FH
- ves
did
no
t. A
lso
mem
ory
effe
cts
mea
sure
d.
Mod
erat
e
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

Chapter6
109
Monoamine depletion in humans
Kosz
ycki
(199
6)75
Ran
d., D
B,
Betw
. SS,
FH
-2*
20M
Goo
d VA
MS-
depr
esse
d:AT
D: B
L: 1.
5 (±
1.0 S
E), p
ost-
test
: 0.8
(±
0.6)
. CO
NT:
BL:
3.4
(±1.8
), p
ost-
test
: 4.5
(±1.4
).
Stud
y in
vest
igat
es e
ffec
t of a
dmin
istr
atio
n of
cho
lecy
stin
e-te
trap
eptid
e af
ter A
TD o
n oc
curr
ence
of p
anic
-att
acks
Goo
d
Leyt
on (1
999)
,39
Leyt
on (2
000)
40R
and.
, DB,
Be
tw. S
S, F
H-
15 A
TD
14 C
ON
T F
Goo
d PO
MS**
: ATD
: cha
nge:
-3.2
8 (±
1.4 S
E). C
ON
T: c
hang
e -1.
04
(±1.2
SE)
. VA
MS-
depr
esse
d: A
TD: B
L: 1.
0 (±
0.9)
, pos
t-te
st:
0.8
(±0.
8), c
hang
e: -0
.3 (±
0.8)
. CO
NT:
BL:
0.4
(±0.
5), p
ost-
test
: 0.4
(±0.
4), c
hang
e: 0
.1 (±
0.4)
.
POM
S; n
o ex
act s
core
s re
port
ed. T
hird
arm
w
ith A
PTD
incl
uded
Mod
erat
e
McA
llist
er-W
illia
ms
(200
2)72
Ran
d., D
B,
With
in S
S, F
H-
14
M
Goo
d PO
MS:
No
exac
t sco
res
repo
rted
, no
sign
. diff
eren
ce in
m
ood
foun
d St
udy
inve
stig
ates
eff
ects
of A
TD o
n ev
ent
rela
ted
brai
n po
tent
ials
on
EEG
dur
ing
an
epis
odic
mem
ory
task
Mod
erat
e
Mill
er (2
000)
76R
and.
, DB,
W
ithin
SS,
FH
-19
M
+FG
ood
POM
S**: A
TD: B
L: 1.
8, p
ost-
test
: 0.3
. CO
NT:
BL:
2.9
, pos
t-te
st: 0
.7.V
AS-
sad**
: ATD
: BL:
8.0
, pos
t-te
st: 1
0.1.
CON
T: B
L:
12.2
, pos
t-te
st: 1
0.9.
No
SDs
repo
rted
, no
sign
. diff
eren
ces
in m
ood
foun
d
No
clea
r pre
sent
atio
n of
moo
d-sc
ores
; stu
dy
com
pare
s AT
D+
CO2-
chal
leng
e in
hea
lthy
cont
rols
vs.
pat
ient
s w
ith p
anic
dis
orde
r
Poor
Mor
eno
(199
9),11
6 M
oren
o (2
000)
130
Ran
d., D
B,
With
in S
S, F
H-
12 M+F
Goo
d H
DRS
-25,
PO
MS
and
IDS
scor
es n
ot c
lear
ly re
port
ed. A
TD
did
not c
ause
sig
nifi
cant
incr
ease
s in
dep
ress
ive
sym
ptom
s in
hea
lthy
cont
rol s
ubje
cts.
Stud
y co
mpa
red
ATD
eff
ects
in h
ealth
y co
ntro
ls v
s. p
atie
nts
with
MD
D in
rem
issi
on.
Inst
ead
of p
lace
bo a
25%
str
engt
h de
plet
ion
drin
k w
as u
sed
as a
con
trol
.
Poor
Mur
phy
(200
2)54
Ran
d., D
B,
With
in S
S11
F
Goo
d Sc
ores
repo
rted
sep
arat
ely
for g
roup
s w
ith A
TD f
irst a
nd
CON
T fi
rst.
VA
MS-
sadn
ess:
ATD
firs
t:AT
D: c
hang
e: -1
.4
(±5.
5), C
ON
T: c
hang
e: -0
.2 (±
0.5)
. CO
NT
firs
t: A
TD: c
hang
e:
0.8
(±26
.6),
CO
NT:
cha
nge:
-12.
5 (±
26.1)
Wei
ghte
d av
erag
ed s
core
(with
hig
hest
SD
s) u
sed
for m
eta-
anal
ysis
Stud
y in
vest
igat
es c
ogni
tive
and
emot
iona
l pr
oces
sing
aft
er A
TDG
ood
Neu
mei
ster
(200
2)46
Ran
d., D
B,
With
in S
S, F
H-
24
FG
ood
HD
RS-2
1: s/
s sub
type
(n=
4): A
TD: c
hang
e 8.
5 (±
2.9)
. CO
NT:
0.
3 (±
0.5)
s/l
subt
ype
(n=
10):
ATD
: cha
nge
4.6
(±2.
9).
CON
T: 0
.5 (±
0.9)
l/l s
ubty
pe**
(n=
10):
ATD
: cha
nge
-0.1
(±1.
3). C
ON
T: 0
.0 (±
1.1)
Stud
y in
vest
igat
es e
ffec
t of b
i-alle
lic
sero
toni
n tr
ansp
orte
r pro
mot
er (5
-HTT
PR)
geno
type
and
eff
ect o
f ATD
Goo
d
Neu
mei
ster
(200
4)47
Ran
d., D
B,
With
in S
S, F
H-
19
M+F
Goo
dH
DRS
-24**
: ATD
: BL:
0.5
(±0.
1), p
ost-
test
: 3.4
(±0.
6). C
ON
T:
BL: 0
.5 (±
0.1)
, pos
t-te
st: 2
.6 (±
0.5)
.St
udy
mai
nly
inve
stig
ates
regi
onal
cer
ebra
l bl
ood-
flow
cha
nges
dur
ing
ATD
with
PET
-sc
ans
Goo
d
Neu
mei
ster
(200
6)51
Ran
d., D
B,W
ithin
SS,
FH
- 26
M
+F
Goo
d H
DRS
**:
s/s
subt
ype
(n=
7): A
TD: B
L: 0
.4 (±
0.4
SE),
post
-te
st: 3
.6 (±
1.4).
CON
T: B
L: 0
.8 (±
0.4)
, pos
t-te
st: 2
.4 (±
0.8)
. s/
l sub
type
(n=
12):
ATD
: BL:
0.4
(±0.
4), p
ost-
test
: 5.2
(±1.1
).
CON
T: B
L: 0
.4 (±
0.4)
, pos
t-te
st: 2
.5 (±
0.6)
. l/l
subt
ype
(n=
7): A
TD: B
L: 0
.4 (±
0.4)
, pos
t-te
st: 2
.4 (±
1.3).
CO
NT:
BL:
0.2
(±
0.4)
, pos
t-te
st: 1
.2 (±
0.8)
.
Stud
y in
vest
igat
es e
ffec
t of t
ri-al
lelic
se
roto
nin
tran
spor
ter p
rom
oter
(5-H
TTPR
) ge
noty
pe a
nd e
ffec
t of A
TD o
n m
ood
and
regi
onal
cer
ebra
l met
abol
ism
with
PET
Goo
d
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

110
Chap
ter
Chapter 6
6
Old
man
(199
4)77
Ran
d., D
B,
With
in S
S12 F
Goo
d PO
MS-
depr
esse
d: A
TD: B
L: 1.
1 (±0
.52
SE),
post
-tes
t: 0
.9
(±0.
58).
CO
NT:
BL:
1.9
(±0.
92),
pos
t-te
st: 2
.3 (±
0.89
).VA
S-sa
d:AT
D: B
L: 15
.0 (±
4.99
SE)
, pos
t-te
st: 9
.2 (±
4.5)
. CO
NT:
BL
: 11.7
(±4.
73),
pos
t-te
st: 8
.3 (±
3.87
).
In th
is s
tudy
a s
econ
d pl
aceb
o-ar
m (p
lain
w
ater
) is
used
; com
paris
ons
here
are
for
bala
nced
drin
k as
CO
NT.
Eff
ects
of A
TD o
n ap
petit
e in
vest
igat
ed a
lso.
Goo
d
Park
(199
4)55
Ran
d., D
B,
With
in S
S12 M
Goo
d VA
MS-
sad:
ATD
: BL:
6.7
(±5.
7 SE
), po
st-t
est:
6.6
(±8.
1).
CON
T: B
L: 3
.2 (±
3.0)
, pos
t-te
st: 5
.6 (±
6.8)
. St
udy
inve
stig
ates
cog
nitiv
e pe
rfor
man
ce
afte
r ATD
Goo
d
Pras
chak
-Rie
der
(200
5)10
0R
and.
, DB,
W
ithin
SS,
FH
-14
M
+FG
ood
No
exac
t VA
S-ra
tings
pro
vide
d in
stu
dy; “
none
of t
he 14
su
bjec
ts e
xper
ienc
ed a
tran
sien
t det
erio
ratio
n in
moo
d [..
] le
vels
.
Stud
y pr
imar
ily in
vest
igat
es e
ffec
ts o
f ATD
on
ser
oton
in tr
ansp
orte
r den
sity
/aff
inity
du
ring
ATD
Mod
erat
e
Ravi
ndra
n (1
999)
,85
Knot
t (19
99)92
Ran
d., D
B,
Betw
. SS,
FH
-2*
13
MG
ood
POM
S-ch
ange
:ATD
-1.2
9 (±
0.94
) vs
CON
T +1
.41(
±0.5
5).
Sign
ific
ant c
hang
es in
moo
d, b
ut th
is o
ccur
red
in 5
of t
he
13 s
ubje
cts
only
, whi
le in
oth
ers
in th
e AT
D-g
roup
no
decr
ease
of m
ood
was
obs
erve
d.
No
clea
r pre
sent
atio
n of
dat
a on
moo
d-sc
ores
; pro
babl
y SD
s re
port
ed. S
tudy
als
o in
vest
igat
es e
ffec
t of f
enflu
ram
ine
adm
inis
trat
ion
afte
r ATD
and
eff
ect o
f ATD
on
imm
une
mea
sure
s an
d EE
G
Mod
erat
e
Ric
hell
(200
5)78
Ran
d., D
B,
Betw
. SS
15 +
13
M+F
Goo
dN
o ba
selin
e da
ta o
n PO
MS
or V
AS
repo
rted
, onl
y m
easu
res
durin
g th
e no
ise-
stre
ss p
arad
igm
repo
rted
aft
er
ATD
, no
sign
. diff
eren
ces
in m
ood
foun
d.
Stud
y es
peci
ally
inve
stig
ates
diff
eren
ce in
re
spon
se to
noi
ce-s
tres
s by
ATD
vs
CON
T.Po
or
Rubi
a (2
005)
106
Ran
d., D
B,
With
in S
S,FH
-9 M
+FG
ood
No
VAS-
scor
es re
port
ed, n
o si
gn. c
hang
es o
f moo
d ob
serv
ed b
y AT
DSt
udy
inve
stig
ates
func
tiona
l bra
in a
ctiv
ity
by fM
RI w
ith a
resp
onse
inhi
bitio
n-ta
skPo
or
Rub
insz
tein
(200
1)56
Ran
d., D
B,
Betw
. SS,
80%
FH
-2*
15
M+F
Goo
d PO
MS-
depr
esse
d: n
o ex
act s
core
s re
port
ed. S
core
s w
ere
sim
ilar b
etw
een
test
and
pla
cebo
gro
ups,
als
o on
VA
MS,
no
sig
n. d
iffer
ence
s in
moo
d fo
und.
Stud
y in
vest
igat
es e
ffec
ts o
f ATD
on
cogn
itive
test
con
cern
ing
atte
ntio
nPo
or
Schm
eck
(200
2)79
Ran
d., D
B,
Betw
. SS,
FH
-12 M
+FG
ood
Eige
nsch
afts
wör
terli
ste:
no
exac
t sco
res
repo
rted
. Hig
h ag
gres
sive
wom
en w
ere
sign
. mor
e de
pres
sed
afte
r ATD
th
an o
ther
gro
ups.
Hig
h ag
gres
sive
men
did
not
sho
w a
n ef
fect
of A
TD o
n m
ood.
The
moo
d of
non
-agg
ress
ive
men
an
d w
omen
did
not
cha
nge
afte
r ATD
.
Stud
y lo
oked
at d
iffer
ent r
eact
ion
to A
TD in
hi
gh a
nd lo
w-a
ggre
ssiv
e m
ales
and
fem
ales
.Po
or
Schm
itt (
2000
)57R
and.
,DB,
W
ithin
SS,
FH
-17
M
+FG
ood
POM
S-de
pres
sion
: ATD
: BL:
0.9
2 (±
0.11
SE)
,pos
t-te
st: 0
.93
(±0.
12).
CO
NT:
BL:
0.9
5 (±
0.11
), p
ost-
test
: 0.9
6 (±
0.13
) St
udy
inve
stig
ates
cog
nitiv
e pe
rfor
man
ce
afte
r ATD
Goo
d
Shan
sis
(200
0)58
Ran
d.,D
B,
With
in S
S, 7
/12
FH-
12 MG
ood
POM
S-de
pres
sed
& V
AS:
no
exac
t sco
res
repo
rted
. Moo
d ch
ange
s on
con
trol
day
not
sig
n. d
iffer
ent t
han
on A
TD
day
for a
ny o
f the
six
dim
ensi
ons
in th
e PO
MS
and
VAS.
Prev
ious
moo
d-di
sord
er in
one
sub
ject
(not
ex
clud
ed).
Stud
y in
ves
tigat
es e
ffec
ts o
f ATD
on
att
entio
n an
d m
emor
y
Poor
Smith
(198
7)11
2R
and.
,DB,
W
ithin
SS
80
MG
ood
MA
ACL
**:A
TD: B
L: 10
.9, p
ost-
test
: 18.
2. C
ON
T: B
L: 9
.5,
post
-tes
t: 8
.2. N
o SD
s re
port
ed. S
igni
fica
nt lo
wer
ing
of
moo
d sc
ores
by
ATD
.
Stud
y in
vest
igat
es A
TD in
neg
ativ
e an
d po
sitiv
e en
viro
nmen
t. B
oth
cond
ition
s sh
ow
a si
gnif
ican
t cha
nge
on m
ood
scor
es.
Mod
erat
e
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

Chapter6
111
Monoamine depletion in humans
Smit
h (1
997b
)147
Ran
d.,D
B,
With
in S
S, F
H-
11
M+F
Goo
d VA
S-sa
dnes
s:no
exa
ct s
core
s re
port
ed. N
o si
gnif
ican
t m
ain
effe
cts
wer
e se
en. V
AS-
happ
y fo
r fem
ales
(n=6
) re
port
ed s
epar
atel
y**: A
TD: B
L: 6
9.7
(±5.
6 SE
), po
st-t
est:
61
.6 (±
6.4)
. CO
NT:
BL:
71.4
(±5.
3), p
ost-
test
: 66.
1 (±6
.1).
For m
ales
(n=5
)**: A
TD: B
L: 7
2.2,
pos
t-te
st: 6
7.8.
CO
NT:
BL:
77
.2, p
ost-
test
: 70.
6. n
o SE
s re
port
ed
Stud
y al
so in
vest
igat
es e
ffec
t of A
TD w
hen
sad-
moo
d is
indu
ced.
Dat
a fo
r fem
ales
VA
S-ha
ppy
used
in m
eta-
anal
ysis
Mod
erat
e
Stew
art (
2002
)50R
and.
,DB,
W
ithin
SS,
FH
-27
M
+FG
ood
POM
S-de
pres
sion
:ATD
: BL:
1.7
(±2.
6), p
ost-
test
: 1.1
(±2.
3),
chan
ge: -
0.6
(±2.
1). C
ON
T: B
L: 3
.4 (±
6.0)
, pos
t-te
st: 2
.4
(±4.
8), c
hang
e: -1
.0 (±
4.6)
.
Stud
y al
so in
vest
igat
es e
ffec
t mod
ifica
tion
by h
igh
vs lo
w n
euro
ticis
m s
core
s du
ring
ATD
(on
moo
d an
d ps
ycho
met
ric
perf
orm
ance
).
Goo
d
Talb
ot (2
006a
)71R
and.
, DB,
Be
tw. S
S, F
H-
17 +
15
M+F
G
ood
VAM
S: N
o ex
act s
core
s re
port
ed, n
o si
gn. d
iffer
ence
in
moo
d fo
und
Stud
y in
vest
igat
es e
ffec
ts o
f ATD
on
deci
sion
m
akin
g an
d le
arni
ng
Mod
erat
e
Talb
ot (2
006b
)105
Ran
d., D
B,
With
in S
S, F
H-
16
M
Goo
d VA
MS:
ATD
: cha
nge
-0.5
(±6.
1). C
ON
T: c
hang
e -5
.7 (±
8.1)
BD
I: AT
D: c
hang
e -0
.0 (±
1.4)
. CO
NT:
cha
nge
0.4
(±1.
5)
Stud
y in
vest
igat
es e
ffec
t of A
TD o
n R
egio
nal
Cere
bral
Blo
od F
low
by
SPEC
T G
ood
Vode
rhol
zer
(199
8)99
Ran
d., D
B,
With
in S
S, F
H-
12
M+F
G
ood
HD
RS-6
: ATD
: BL:
0.8
3 (±
1.8),
pos
t-te
st: 0
.42
(±1.2
). CO
NT:
BL
: 0.17
(±0.
4), p
ost-
test
: 0.4
2 (±
0.7)
. St
udy
inve
stig
ates
eff
ects
of A
TD o
n sl
eep
EEG
G
ood
Wel
tzin
(199
4)11
3††
Ran
d.,D
B,
With
in S
S9 F
Goo
dO
nly
peak
-cha
nges
for A
TD a
nd C
ON
T ar
e pr
ovid
ed fo
r an
uncl
early
def
ined
moo
d-sc
ale:
ATD
: 0.7
8. C
ON
T 0.
0. S
Ds
not r
epor
ted.
SD
of d
iffer
ence
est
imat
ed c
onse
rvat
ivel
y fr
om p
< 0.
05: ±
1.01.
Stud
y in
vest
igat
es e
ffec
t of A
TD o
n m
ood
in
bulim
ic p
atie
nts
(n=1
3) a
nd h
ealth
y co
ntro
ls
(n=
9).
Mod
erat
e
Wel
tzin
(199
5)80
††R
and.
,DB,
W
ithin
SS
10
FG
ood
Onl
y pe
ak-c
hang
es b
etw
een
ATD
and
CO
NT
are
prov
ided
fo
r an
uncl
early
def
ined
moo
d-sc
ale:
0.0
(±1.3
), th
is v
alue
us
ed in
met
a-an
alys
is
Stud
y in
vest
igat
es e
ffec
t of A
TD o
n m
ood
and
shor
t-te
rm e
atin
g be
ha v
iour
in b
ulim
ic
patie
nts
(n=1
0) a
nd h
ealth
y co
ntro
ls (n
= 10
).
Mod
erat
e
Yath
am (2
001)
104
Ran
d., D
B,
With
in S
S, F
H-
10
F G
ood
HD
RS-2
0, P
OM
S: N
o ex
act s
core
s re
port
ed, n
o si
gn.
diff
eren
ce in
moo
d fo
und
Stud
y in
vest
igat
es e
ffec
ts o
f ATD
on
5-H
T 2
rece
ptor
s w
ith P
ETM
oder
ate
Youn
g (1
985)
13R
and.
, DB,
Be
tw. S
S3*
12
MG
ood
No
clea
r MA
ACL
-sco
res
or S
Ds
repo
rted
per
gro
up fo
r ATD
vs
CO
NT.
Sig
n. d
ecre
ase
of m
ood
afte
r ATD
. St
udy
inve
stig
ates
eff
ects
of A
TD o
n pr
oofr
eadi
ng ta
sk w
ith o
r with
out
dist
ract
ion
Poor
A2.
Hea
lthy
con
trol
s w
ith
+ve
fam
ily h
isto
ry fo
r MD
D
Benk
elfa
t (19
94)41
Ran
d., D
B,
With
in S
S, F
H+
20 MG
ood
POM
S-de
pres
sed:
ATD
: BL:
55.
2 (±
1.7 S
E), p
ost-
test
: 51.2
(±
1.6),
cha
nge:
-4.0
(±1.
6). C
ON
T: B
L: 5
4.0
(±1.6
), p
ost-
test
: 57
.3 (±
1.1),
cha
nge:
3.3
(±1.6
). S
ix (3
0%) o
f peo
ple
show
ed a
de
clin
e of
10 p
oint
s or
gre
ater
on
POM
S-D
.
POM
S ch
ange
d in
FH
+ sa
mpl
e, n
ot in
FH
- sa
mpl
e in
sam
e st
udy.
G
ood
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

112
Chap
ter
Chapter 6
6
Elle
nbog
en (1
999)
135
Ran
d., D
B,
With
in S
S, F
H+
13
FG
ood
POM
S-de
pres
sed**
: 1st
ATD
: cha
nge:
-0.3
(±1.
7 SE
). CO
NT:
ch
ange
: -1.1
(±1.5
). V
AM
S:no
exa
ct s
core
s re
port
ed. N
o si
gnif
ican
t cha
nge
in m
ood
on a
ny o
f the
PO
MS
or V
AM
S ite
ms.
Poss
ible
sel
ectio
n-bi
as b
y ex
clus
ion
of m
any
form
er p
atie
nts
men
tione
d: e
xtre
mel
y FH
+ sa
mpl
e, b
ut n
ever
dep
ress
ed. S
tudy
foun
d po
or te
mpo
ral s
tabi
lity
of A
TD in
repe
ated
te
stin
g.
Goo
d
Klaa
ssen
(199
9b),
44
Klaa
ssen
(200
2)45
Ran
d., D
B,
With
in S
S, F
H+
& F
H-
16 F
H+
M+F
Goo
d PO
MS-
depr
esse
d: s
core
s no
t com
plet
ely
repo
rted
. ATD
-CO
NT
diff
eren
ces:
FH
+ 1.9
4 (±
3.17
)Se
e ab
ove
in s
ectio
n “h
ealth
y co
ntro
ls”
Mod
erat
e
Neu
mei
ster
(200
2)46
Ran
d., D
B,
With
in S
S, F
H+
21
FG
ood
HD
RS-2
1: s
/s s
ubty
pe (n
= 5)
: ATD
: cha
nge
9.2
(±2.
1). C
ON
T:
0.8
(±0.
5) s
/l su
btyp
e (n
= 7)
: ATD
: cha
nge
10.0
(±4.
5).
CON
T: 0
.1 (±
0.4)
l/l s
ubty
pe**
(n=
9): A
TD: c
hang
e 0.
4 (±
1.2).
CON
T: -0
.2 (±
1.3)
Stud
y in
vest
igat
es e
ffec
t of b
i-alle
lic
sero
toni
n tr
ansp
orte
r pro
mot
er (5
-HTT
PR)
geno
type
and
eff
ect o
f ATD
Goo
d
Stew
art (
2002
)50R
and.
,DB,
W
ithin
SS,
FH
-5 M
+FG
ood
POM
S-de
pres
sion
:ATD
: BL:
5.2
(±10
.0),
pos
t-te
st: 5
.6
(±10
.4),
cha
nge:
0.4
(±0.
5). C
ON
T: B
L: 6
.4 (±
12.7
), p
ost-
test
: 4.
2 (±
7.4)
, cha
nge:
-2.2
(±5.
5).
See
abov
e in
sec
tion
“hea
lthy
cont
rols
”G
ood
B1. P
atie
nts
wit
h M
DD
pre
viou
sly,
but
cur
rent
ly in
rem
issi
on (w
itho
ut m
edic
atio
n)
Cass
idy
(199
7)11
7R
and.
, DB,
W
ithin
SS
1-4 d
ays
afte
r EC
T
5
M+F
R
x-fr
ee
<3 m
ths
Goo
dM
AD
RS:
ATD
: BL:
4.2
(±3.
5), p
ost-
test
: 3.6
(±3.
2). C
ON
T: B
L:
4.8
(±2.
9), p
ost-
test
: 4.0
(±3.
5).
Stud
y in
vest
igat
es re
curr
ence
of M
DD
aft
er
ATD
in p
atie
nts
succ
essf
ully
trea
ted
with
ECT
Goo
d
Del
gado
(199
1)42
Ran
d., D
B,
With
in S
S ≥2
wee
ks a
fter
‘s
tabi
lity’
69
M+F
R
x-fr
ee
?? m
ths
Goo
d H
DRS
-25:
exa
ct s
core
s no
t rep
orte
d; n
o si
gn. d
iffer
ence
by
ATD
. 30%
exp
erie
nced
an
impr
ovem
ent o
f moo
d th
e da
y af
ter A
TD. D
epre
ssiv
e re
laps
e: N
o ex
act d
ata
prov
ided
.
In o
rigin
al s
tudi
es B
ipol
ar p
atie
nts
incl
uded
; un
clea
r whe
ther
thes
e w
ere
excl
uded
in th
is
pool
ed a
naly
sis
Poor
Hay
nes
(200
4)93
Ran
d., D
B,
With
in S
S 1-2
wee
ks
resp
onse
13
M+F
Po
st- C
BT
Goo
d PO
MS-
depr
essi
on: A
TD: B
L: 6
.4 (±
7.4)
, pos
t-te
st (8
hr):
4.9
(±
6.4)
. CO
NT:
BL:
5.5
(±7.
7), p
ost-
test
: 5.0
(±7.
8).
HD
RS-
19:
ATD
: BL:
4.7
(±3.
7), p
ost-
test
: 4.9
(±3.
9). C
ON
T: B
L: 3
.3
(±2.
5), p
ost-
test
: 3.5
(±4.
3).
Stud
y in
vest
igat
es re
curr
ence
of M
DD
aft
er
ATD
in p
atie
nts
succ
essf
ully
trea
ted
with
CB
T, a
nd e
ffec
ts o
n RE
M-s
leep
Mod
erat
e
Hay
war
d (2
005)
43R
and.
, DB,
Be
tw. S
S, 11
FH
- ≥6
mon
ths
rem
issi
on
2*12
M
+F
Rx-
free
≥3
mth
s
Goo
dH
DRS
: ATD
: BL:
1.0
(±0.
41 S
E), p
ost-
test
: 1.3
8 (±
0.36
), CO
NT:
BL:
0.4
2 (±
0.23
), p
ost-
test
: 0.5
0 (±
0.19
). B
DI:
ATD
: BL
: 2.2
5 (±
0.76
SE)
, pos
t-te
st: 1
.75
(±0.
68),
CO
NT:
BL:
2.17
(±
0.63
), p
ost-
test
: 1.6
3 (±
0.53
). P
OM
S an
d VA
S: e
xact
sc
ores
not
repo
rted
. N
o si
gnif
ican
t eff
ect o
n an
y sc
ale
foun
d.
Stud
y al
so in
vest
igat
es c
ogni
tive
proc
essi
ng
in fo
rmer
ly d
epre
ssed
ver
sus
heal
thy
cont
rols
dur
ing
ATD
Mod
erat
e
Mor
eno
(199
9),11
6 M
oren
o (2
000)
130
Ran
d., D
B,
With
in S
S ≥3
mon
ths r
e m
is
sion
(ave
rage
29
mon
ths)
12
M+F
R
x-fr
ee
≥3 m
ths
Goo
dH
DRS
-25,
PO
MS
and
IDS
scor
es n
ot c
lear
ly re
port
ed. A
TD
caus
ed n
on-s
igni
fican
t inc
reas
es in
dep
ress
ive
sym
ptom
s in
his
tory
-pos
itive
sub
ject
sPos
t-te
st: m
ean
peak
HD
RS
scor
es A
TD: 1
4 (±
6), ‘
CON
T’: 1
0 (±
7).
Stud
y co
mpa
red
heal
thy
cont
rols
with
pa
tient
s w
ith M
DD
in re
mis
sion
. Ins
tead
of
plac
ebo
a 25
% st
reng
th d
eple
tion
drin
k w
as
used
as
a co
ntro
l.
Poor
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

Chapter6
113
Monoamine depletion in humans
Neu
mei
ster
(200
4)47
Ran
d., D
B,
With
in S
S ≥3
mon
ths r
e m
is
sion
27
M+F
R
x-fr
ee
≥3 m
ths
Goo
dH
DRS
-24**
: ATD
: BL:
1.2
(±0.
2), p
ost-
test
: 11.5
(±10
.3).
CON
T: B
L: 0
.7 (±
0.2)
, pos
t-te
st: 2
.1 (±
0.3)
. 16/
27 (5
9%)
patie
nts
expe
rienc
ed tr
ansi
ent r
etur
n of
MD
D d
urin
g AT
D
vs 0
/27
durin
g CO
NT
See
abov
e in
sec
tion
“hea
lthy
cont
rols
”G
ood
Neu
mei
ster
(200
6)51
Ran
d., D
B,
With
in S
S ≥3
mon
ths r
e m
is
sion
27
M+F
R
x-fr
ee
≥3 m
ths
Goo
d H
DRS
**:
s/s
subt
ype
(n=
7): A
TD: B
L: 0
.4 (±
0.4
SE),
post
-te
st: 7
.0 (±
1.4)
. CO
NT:
BL:
0.6
(±0.
4), p
ost-
test
: 2.6
(±0.
8).
s/l s
ubty
pe (n
= 12
): AT
D: B
L: 0
.8 (±
0.4)
, pos
t-te
st: 1
1.4
(±1.0
). C
ON
T: B
L: 0
.8 (±
0.4)
, pos
t-te
st: 2
.4 (±
0.6)
. l/l
subt
ype
(n=
8): A
TD: B
L: 2
.0 (±
0.3)
, pos
t-te
st: 1
4.6
(±1.2
).
CON
T: B
L: 0
.9 (±
0.4)
, pos
t-te
st: 1
.7 (±
0.6)
.
Stud
y in
vest
igat
es e
ffec
t of t
ri-al
lelic
se
roto
nin
tran
spor
ter p
rom
oter
(5-H
TTPR
) ge
noty
pe a
nd e
ffec
t of A
TD o
n m
ood
and
regi
onal
cer
ebra
l met
abol
ism
with
PET
Goo
d
O’R
eard
on (2
004)
48R
and.
, DB,
W
ithin
SS
5 m
onth
s re
mis
si
on
10 M+F
Po
st-C
BT
Goo
d H
DRS
-24**
: ATD
: cha
nge:
4.9
7 (±
1.0
SE).
CO
NT:
cha
nge:
3.
22 (±
1.0)
0/1
0 pa
tient
s re
laps
ed in
eith
er c
ondi
tion
Curr
ently
not
usi
ng a
ntid
epre
ssan
t m
edic
atio
n, re
spon
ders
to C
BT tr
eatm
ent.
G
ood
Smit
h (1
997a
)118
Ran
d., D
B,
With
in S
S ≥6
mon
ths
rem
issi
on
15 F Rx-
free
≥6
mth
s
Goo
d
HD
RS-12
: ATD
: cha
nge
(7hr
): 7
.3. C
ON
T: c
hang
e: 0
.1. S
D o
f di
ffer
ence
ATD
-CO
NT
calc
ulat
ed fr
om 9
5% C
I: 7.
2 (±
4.9)
5/
15 p
atie
nts
expe
rienc
ed re
laps
e du
ring
ATD
, 0/1
5 du
ring
CON
T
Curr
ently
not
usi
ng a
ntid
epre
ssan
t m
edic
atio
n (r
ecov
ered
MD
D)
Mod
erat
e
B2. P
atie
nts
wit
h M
DD
pre
viou
sly,
but
cur
rent
ly in
rem
issi
on (c
urre
ntly
usi
ng m
edic
atio
n)
Abe
rg-W
iste
dt
(199
8)11
1R
and.
, DB,
Be
tw. S
S A
fter
resp
onse
12+8
M
+F
CIT
Insu
ffic
ient
M
AD
RS:
exa
ct s
core
s no
t rep
orte
d. F
ive
of th
e 12
pat
ient
s w
ith A
TD s
how
ed a
Wor
seni
ng o
f dep
ress
ive
sym
ptom
s in
AT
D: 5
/12
(MA
DR
S 12
-71%
). C
ON
T: 0
/8.
Stud
y al
so in
vest
igat
es re
latio
n w
ith re
laps
e du
e to
ATD
and
oth
er v
aria
bles
(pla
sma
cort
isol
, pla
tele
t-se
roto
nin)
Mod
erat
e
Booi
j (20
05a)
59R
and.
, DB,
W
ithin
SS
Aft
er ≥
par
tial
rem
issi
on
20
M+F
SS
RI
SNR
I
Goo
dM
AD
RS:
ATD
: BL:
4.6
(±0.
9 SE
), po
st-t
est:
7.9
(±1.8
). C
ON
T:
BL: 3
.7 (±
0.9)
, pos
t-te
st: 3
.7 (±
0.9)
. Rel
apse
of M
DD
: ATD
: 7/
20; C
ON
T: 0
/20
assu
med
.
Incl
usio
n of
sub
ject
s w
ith H
DRS
<15
(par
tial
rem
issi
on).
A 2
5% s
tren
gth
tryp
toph
an
defi
cien
t mix
ture
was
use
d as
CO
NT.
Goo
d
Booi
j (20
06)83
Ran
d., D
B,
With
in S
S A
fter
≥ p
artia
l re
mis
sion
19
M+F
SS
RI S
NRI
Goo
d H
DRS
-17 s
core
s si
gn. l
ower
aft
er A
TD:
SI+
(n=
8): A
TD: B
L:
2.5
(±0.
8 SE
), p
ost-
test
: 7.6
(±2.
0). C
ON
T: N
ot g
iven
SI-
(n=
11):
ATD
: BL:
2.9
(±0.
7), p
ost-
test
: 3.4
(±0.
6). C
ON
T: N
ot
give
n M
AD
RS s
core
s si
gn. l
ower
aft
er A
TD:
SI+:
ATD
: BL:
5.
0 (±
1.2 S
E), p
ost-
test
: 12.
00 (±
2.4)
. CO
NT:
Not
giv
en S
I-:
ATD
: BL:
5.4
(±1.3
SE)
, pos
t-te
st: 9
.3 (±
1.7).
CO
NT:
Not
gi
ven
Incl
usio
n of
sub
ject
s w
ith H
DRS
<15
(par
tial
rem
issi
on).
A 2
5% s
tren
gth
tryp
toph
an
defi
cien
t mix
ture
was
use
d as
CO
NT.
No
data
fo
r ATD
moo
d-ra
tings
aft
er C
ON
T pr
ovid
ed.
Stud
y al
so in
vest
igat
es e
ffec
t of A
TD o
n H
eart
Rat
e Va
riabi
lity
(HRV
) and
impu
lsiv
ity
and
the
inte
ract
ion
by +
ve/-v
e hi
stor
y of
su
icid
al id
eatio
n (S
I+/S
I-)
Mod
erat
e
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§
114
Chap
ter
Chapter 6
6
Brem
ner
(199
7)10
1R
and.
, DB,
W
ithin
SS
Ave
rage
rem
is
sion
45
wee
ks
21 M+F
FL
X PA
R
Goo
dR
elap
se o
f MD
D: A
TD: 7
/21;
CO
NT:
1/21
. HD
RS-
scor
es a
re
split
bet
wee
n re
laps
e an
d no
n-re
laps
e gr
oup.
With
rela
pse
(n =
7):
ATD
: BL:
6.8
6 (±
4.98
), po
st-t
est:
15.5
7 (±
6.92
). CO
NT:
BL:
7.0
0 (±
5.45
), p
ost-
test
: 6.7
1 (±6
.07)
. With
out-
rela
pse
(n =
14):
ATD
: BL:
4.7
9 (±
3.45
), p
ost-
test
: 6.3
6 (±
4.25
), C
ON
T: B
L: 6
.21 (
±4.6
9), p
ost-
test
: 9.2
1 (±5
.99)
.
19/2
1 of p
atie
nts u
sed
FLX.
Stu
dy in
vest
igat
es
brai
n m
etab
olis
m w
ith P
ET d
urin
g AT
D a
nd
asso
ciat
ions
with
MD
D re
laps
e
Goo
d
Del
gado
(199
1)42
Ran
d., D
B,
With
in S
S ≥2
wee
ks a
fter
‘s
tabi
lity’
46 M+F
va
rious
Goo
d H
DRS
-25:
exa
ct s
core
s no
t rep
orte
d. D
epre
ssiv
e re
laps
e:
ATD
: 24/
46, C
ON
T: 0
/46.
H
eter
ogen
eous
pop
ulat
ion
curr
ently
usi
ng
vario
us A
D. B
UP
or D
ES-t
reat
men
t low
er
chan
ce o
f rel
apse
In
orig
inal
stu
dies
Bip
olar
pa
tient
s in
clud
ed; u
ncle
ar w
heth
er th
ese
wer
e ex
clud
ed in
this
poo
led
anal
ysis
Mod
erat
e
Del
gado
(199
9)11
4R
and.
, DB,
W
ithin
SS ≥2
wee
ks
resp
onse
30
M+F
FL
X D
ES
Goo
dH
DRS
-25:
FLX
-tre
ated
pat
ient
s: A
TD: B
L: 7
.7, p
ost-
test
17.1.
CO
NT:
not
repo
rted
Rel
apse
: ATD
8/1
5, C
ON
T 0/
15. D
ES-
trea
ted
patie
nts:
ATD
: BL:
8.6
, pos
t-te
st 10
.7. C
ON
T: n
ot
repo
rted
. Rel
apse
: ATD
1/15
, CO
NT
0/15
.
Two
bipo
lar p
atie
nts
incl
uded
. Stu
dy
repl
icat
es p
revi
ous
findi
ng th
at D
ES-t
reat
ed
rem
itter
s do
not
suf
fer a
rela
pse
if im
prov
ed
on D
ES, b
ut d
id a
fter
FLX
-indu
ced
rem
issi
on.
Mod
erat
e
Evan
s (2
002)
94R
and.
, DB,
W
ithin
SS
Aft
er ≥
par
tial
rem
issi
on
8 M+F
BU
P
Goo
d**PO
MS-
depr
esse
d: A
TD: B
L: 5
.25
(±1.6
2 SE
), p
ost-
test
: 3.12
(±
1.37)
. CO
NT:
BL:
5.2
5 (±
1.62)
, pos
t-te
st: 3
.50
(±1.
80)
HD
RS-19
: ATD
: BL:
4.3
8 (±
1.21
SE)
, pos
t-te
st: 5
.00
(±1.0
2).
CON
T: B
L: 4
.38
(±1.2
1), p
ost-
test
: 4.12
(±0.
81)
Stud
y al
so in
vest
igat
es e
ffec
ts o
f ATD
on
slee
p-EE
G. O
ne B
ipol
ar II
pat
ient
incl
uded
. Fi
ve p
atie
nts
in p
artia
l rem
issi
on (H
DR
S 7-
9)
Goo
d
Land
olt (
2003
)95R
and.
, DB,
W
ithin
SS,
PHZ
Aft
er
rem
issi
on
5 M+F
PH
Z
Goo
dPO
MS-
depr
essi
on: n
o BL
mea
sure
s w
ere
give
n se
para
tely
fo
r ATD
and
CO
NT:
BL:
0.4
(± 0
.4 S
E). A
TD: p
ost-
test
: 2.0
(±
1.5).
CO
NT:
pos
t-te
st: 1
.0 (±
0.4)
. Pos
t-te
st m
easu
res
take
n ne
xt m
orni
ng a
fter
ATD
or C
ON
T at
16:0
0h.
Stud
y pr
imar
ily in
vest
igat
es e
ffec
t of A
TD o
n re
curr
ence
of R
EM-s
leep
whi
le u
sing
PH
ZM
oder
ate
Moo
re (1
998)
96R
and.
, DB,
W
ithin
SS ≥2
mon
ths
rem
issi
on
10 M
FLX
SER
PAR
Goo
d PO
MS-
depr
esse
d:AT
D: B
L: 3
.0 (±
1.3 S
E), p
ost-
test
: 2.7
(±
1.4),
CO
NT:
BL:
3.0
(±1.3
), p
ost-
test
: 3.3
(±1.6
). H
DR
S-25
ra
tings
not
exa
ctly
repo
rted
but
did
not
cha
nge
sign
ific
antly
Stud
y al
so in
vest
igat
es e
ffec
ts o
f ATD
on
REM
-sle
ep. A
25%
str
engt
h tr
ypto
phan
de
fici
ent m
ixtu
re w
as u
sed
as C
ON
T.
Subj
ects
in re
mis
sion
>2
mon
ths
Goo
d
Mor
ris
(199
9)14
8R
and.
, DB,
W
ithin
SS
Full
rem
issi
on
8 M
SSR
I A
MI
TRA
Z PH
Z TC
P Li
Goo
dM
odifi
ed H
DRS
-12, B
DI.
No
exac
t sco
res
give
n. H
DRS
in
crea
se >
5 po
ints
(rel
apse
): A
TD: 7
/8, C
ON
T: 0
/8
Stud
y in
vest
igat
es b
rain
met
abol
ism
with
PE
T du
ring
ATD
and
ass
ocia
tions
with
MD
D
rela
pse.
Six
of 8
par
ticip
ants
rece
ived
≥6
mon
ths
antid
epre
ssan
ts. N
o in
dica
tion
whi
ch d
rugs
wer
e cu
rren
tly u
sed.
Mod
erat
e
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

Chapter6
115
Monoamine depletion in humans
O’R
eard
on (2
004)
48R
and.
, DB,
W
ithin
SS
5 m
onth
s re
mis
sion
9 M+F
FL
X PA
R
Goo
d H
DRS
-24**
: ATD
: cha
nge:
8.2
2 (±
1.2),
CON
T: c
hang
e: 2
.56
(±1.2
). 3/
9 pa
tient
s re
laps
ed d
urin
g AT
D v
s 0/
9 du
ring
cont
rol
Curr
ently
usi
ng a
ntid
epre
ssan
t med
icat
ion,
re
spon
ders
to S
SRI t
reat
men
t.
Goo
d
Pras
chak
-Rie
der
(200
4)49
Ran
d., D
B,
With
in S
S 2
mon
ths
rem
issi
on
8 M
CIT
Goo
dH
DRS
-21:
ATD
: BL:
3.8
8 (±
2.6)
, pos
t-te
st: 1
2.3
(±5.
4).
CON
T:no
sco
res
repo
rted
. Rel
apse
: ATD
: 6/8
pat
ient
s,
CON
T: n
o nu
mbe
rs re
port
ed 0
/8 a
ssum
ed fo
r ana
lysi
s
Stud
y pr
imar
ily in
vest
igat
es e
ffec
ts o
f ATD
on
regi
onal
5-H
T 1A
bin
ding
pot
entia
l with
PE
T.
Poor
Smit
h (1
999)
102
Ran
d., D
B,
With
in S
S ≥6
mon
ths
rem
issi
on
8 M
vario
us
Goo
dH
DRS
: no
exac
t sco
res
repo
rted
Rel
apse
: ATD
: 6/8
pa
tient
s, C
ON
T: 1/
8St
udy
prim
arily
inve
stig
ates
eff
ects
of A
TD
on re
gion
al b
rain
act
ivity
with
PET
. Sub
ject
s in
rem
issi
on >
6 m
onth
s. 2
pat
ient
s di
d no
t us
e an
tidep
res-
sant
s, 6
use
d va
rious
type
s (S
SRI,
TCA
, MA
O-I)
Mod
erat
e
Spill
man
n (2
001)
119
Ran
d., D
B,
With
in S
S ≥3
mon
ths
rem
issi
on
10
M+F
FL
X SE
R VL
X PA
R
Goo
d H
DRS
-6:A
TD: B
L: 1.
1 (±1
.2) p
ost-
test
(7h)
:4.7
(±3.
4), c
hang
e:
3.6
(±3.
1); C
ON
T: B
L: 1.
4 (±
1.7)
pos
t-te
st: 1
.0 (±
1.4),
chan
ge:
-0.4
(±1.2
). In
crea
se in
sco
res
on th
e H
DRS
-6 w
as
sign
ific
antly
hig
her f
or A
TD th
an fo
r CO
NT.
In re
mis
sion
aft
er S
SRI,
curr
ently
usi
ng
antid
epre
ssan
t med
icat
ion.
5 in
itial
pa
rtic
ipan
ts d
ropp
ed o
ut.
Goo
d
C. P
atie
nts
curr
entl
y w
ith
MD
D (w
ith
or w
itho
ut a
ntid
epre
ssan
ts)
Booi
j (20
05b)
120
Ran
d., D
B,
With
in S
S14
M
+F
VLX
MIR
TC
A
Li
Goo
dH
DRS
: ATD
: BL:
12.9
(±1.5
SE)
, pos
t-te
st: 1
3.1 (
±1.9
). CO
NT:
BL
: 11.4
(±1.4
), p
ost-
test
: 13.
6 (±
1.3).
MA
DRS
: ATD
: BL:
22.
7 (±
3.7)
, pos
t-te
st: 2
3.1 (
±4.7
). CO
NT:
BL:
19.6
(±3.
3), p
ost-
test
: 22.
9 (±
2.0)
.
A 2
5% s
tren
gth
tryp
toph
an d
efic
ient
mix
ture
w
as u
sed
as C
ON
T. Im
prov
emen
t of m
ood-
ratin
gs 2
4hr a
fter
full
ATD
in V
LX/M
IR tr
eate
d pa
tient
s, b
ut n
ot in
pat
ient
s tr
eate
d ot
herw
ise.
Goo
d
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

116
Chap
ter
Chapter 6
6
Del
gado
(199
4)12
1R
and.
, DB,
W
ithin
SS
43
M+F
≥2
wks
R
x-fr
ee
Goo
dH
DRS
-25:
No
exac
t moo
d-sc
ores
pro
vide
d O
ne d
ay a
fter
AT
D 16
/43
patie
nts
expe
rienc
ed im
prov
emen
t of m
ood,
10/
46 d
ecre
ase
in m
ood
rela
tive
to C
ON
T
Impr
ovem
ent o
f moo
d-ra
tings
24h
r aft
er fu
ll AT
D a
ssoc
iate
d w
ith tr
eatm
ent r
espo
nse
afte
r ant
idep
ress
ant t
reat
men
t
Poor
Pric
e (1
997)
86R
and.
, DB,
W
ithin
SS
22
M+F
R
x-fr
ee
Goo
dVA
S, H
DR
S-25
: No
exac
t sco
res
prov
ided
; sco
res
unaf
fect
ed b
y AT
D.
Stud
y in
vest
igat
es th
e (b
ioch
emic
al) e
ffec
ts
of m
-Chl
oro
phen
yl p
iper
azin
e (m
CCP;
se
roto
nin-
agon
ist)
on
HD
RS,
cor
tisol
, pr
olac
tin a
nd g
row
th-h
orm
one
durin
g AT
D
Poor
Pric
e (1
998)
87R
and.
, DB,
W
ithin
SS
36
M+F
≥3
wks
Rx-
free
Goo
dH
DRS
-25:
ATD
: BL:
28
(±7)
, pos
t-te
st 2
8 (±
7). C
ON
T: 2
7 (±
9),
post
-tes
t 26
(±9)
Stud
y in
clud
ed 4
pat
ient
s w
ith B
D. S
tudy
in
vest
igat
es th
e (b
ioch
emic
al) e
ffec
ts o
f ra
pid
tryp
toph
an in
fusi
on o
n H
DRS
, cor
tisol
, pr
olac
tin a
nd g
row
th-h
orm
one
afte
r ATD
Goo
d
AD
= a
ntid
epre
ssan
ts; A
MI =
Am
itrip
tylin
e; A
MPT
= A
lpha
-met
hyl-p
ara-
tyro
sine
;ATD
= A
cute
Try
ptop
han
Dep
letio
n; B
D =
Bip
olar
Dis
orde
r; B
DI =
Bec
k D
epre
ssio
n In
vent
ory;
Bet
w.S
S =
Betw
een-
subj
ects
pa
ralle
l gro
ups
desi
gn; B
L =
Base
line;
BU
P =
bupr
oprio
n; C
BT =
Cog
nitiv
e Be
havi
our t
hera
py; C
IT =
cita
lopr
am; C
ON
T =
Cont
rol;
DB
= do
uble
-blin
d; D
ES =
des
ipra
min
e; E
CT =
Ele
ctro
-con
vuls
ive
ther
apy;
EEG
=
elec
tro-
ence
phal
ogra
phy;
FLX
= fl
uoxe
tine;
HA
MA
= H
amilt
on A
nxie
ty S
cale
; HD
RS
= H
amilt
on D
epre
ssio
n Sc
ale;
IMI =
imip
ram
ine;
LYS
= ly
sine
; Li =
lith
ium
; MA
ACL
= M
ultip
le A
ffec
t Adj
ectiv
e Ch
eckL
ist;
M
AD
RS =
Mon
tgom
ery
Åsb
erg
Dep
ress
ion
Rat
ing
Scal
e; M
AO
-I =
Mon
oam
ine
oxid
ase
inhi
bito
r; M
DD
= M
ajor
Dep
ress
ive
Dis
orde
r; M
EG =
mag
neto
-enc
epha
logr
aphy
; MIR
= m
irtaz
apin
e; N
OR
=
nort
ripty
line;
PA
R =
paro
xetin
e; P
HZ
= ph
enel
zine
; PO
MS
= Pr
ofile
of M
ood
Stat
es; R
and.
= ra
ndom
ized
; SD
= S
tand
ard
devi
atio
n; S
E =
Stan
dard
Err
or; T
CA =
tric
yclic
ant
idep
ress
ants
; TCP
=
Tran
ylcy
prom
ine;
TRA
Z =
Traz
odon
e; V
A(M
)S =
Vis
ual A
nalo
gue
(Moo
d) S
cale
; VLX
= v
enla
faxi
ne;
With
in S
S =
With
in-s
ubje
cts
cros
s ov
er d
esig
n*
stud
ies
incl
uded
in th
e m
eta-
anal
ysis
in b
old
† Pla
sma
tryp
toph
an d
eclin
ed b
y at
leas
t 50%
‡ S
D g
iven
unl
ess
indi
cate
d ot
herw
ise
(e.g
. SE)
§
Judg
emen
t in
the
cont
ext o
f app
licab
ility
for t
his
met
a-an
alys
is
** E
stim
ated
from
figu
re
†† U
ncle
ar w
heth
er p
artia
lly th
e sa
me
popu
latio
n is
repo
rted
(Wel
tzin
(199
4 an
d 19
95))
Tabl
e 6.
2. In
clud
ed s
tudi
es A
cute
Try
ptop
han
Dep
letio
n (A
TD).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
q. o
f de
plet
ion†
Res
ults
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
emen
t§

Chapter6
117
Monoamine depletion in humans
Tabl
e 6.
3. In
clud
ed s
tudi
es w
ith P
ara-
chlo
roph
enyl
alan
ine
(PCP
A).
Aut
hor
(yea
r)
Des
ign
N
Ade
quac
y of
de
plet
ion
Resu
lts
moo
d sc
ores
(sca
le, s
core
s (±
SD))
Re
mar
ks
Judg
emen
t§
Shop
sin
(197
5)15
With
in S
S, M
DD
in
rem
issi
on b
y IM
I1
Not
mea
sura
ble;
poo
r PC
PA g
iven
in s
mal
l dos
es fo
r rel
ativ
ely
shor
t per
iods
cau
sed
a re
vers
al o
f the
ant
idep
ress
ant e
ffec
t of I
MI.
HD
RS, n
o ex
act
moo
d sc
ores
repo
rted
.
Not
e: n
= 1,
1 pa
tient
with
BD
and
3
patie
nts
trea
ted
with
AM
PT n
ot
incl
uded
her
e.
Poor
Shop
sin
(197
6)10
9W
ithin
SS,
MD
D in
re
mis
sion
by
TCP
3 N
ot re
port
ed; p
oor
All
patie
nts
expe
rienc
ed a
rela
pse.
HD
RS,
no
exac
t moo
d sc
ores
re
port
ed.
2 pa
tient
s w
ith B
D n
ot in
clud
ed
here
. Po
or
PCPA
= P
ara-
chlo
roph
enyl
alan
ine.
For
oth
er s
ymbo
ls a
nd a
bbre
viat
ions
see
foot
note
in T
able
6.2
.

118
Chap
ter
Chapter 6
6
Tabl
e 6.
4. In
clud
ed s
tudi
es A
cute
Phe
nyla
lani
ne/T
yros
ine
Dep
letio
n (A
PTD
).
Aut
hor
(yea
r)*
Des
ign
N
Ade
quac
y of
de
plet
ion†
Resu
lts
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
e m
ent§
A1.
Hea
lthy
con
trol
s
Coup
land
(200
1)81
Rand
., D
B,
With
in S
S5
MN
ot re
port
edPO
MS-
depr
esse
d:A
PTD
: cha
nge:
-0.2
(±2.
7) C
ON
T:
chan
ge: 0
.2 (±
2.6)
.St
udy
inve
stig
ates
eff
ect o
f pen
taga
strin
e to
indu
ce
anxi
ety
afte
r APT
DM
oder
ate
Har
mer
(200
1)60
Rand
., D
B,
With
in S
S12
M+F
Goo
dVA
S-de
pres
sed:
APT
D: c
hang
e: 2
.03
(±4.
98).
CO
NT:
ch
ange
: -4.
20 (±
15.2
3).
Stud
y in
vest
igat
es e
ffec
t of A
PTD
on
prol
actin
, ne
urop
sych
olog
ical
and
sub
ject
ive
mea
sure
sG
ood
Har
riso
n (2
002)
37Ra
nd.,
DB,
W
ithin
SS,
FH
-13
FG
ood
POM
S-de
pres
sed:
APT
D: B
L: 0
.92
(±0.
56 S
E), p
ost-
test
: 1.1
5 (±
0.55
) CO
NT:
BL:
1.61
(±0.
52),
pos
t-te
st: 1
.15 (±
0.56
).St
udy
also
repo
rts
abse
nce
of e
ffec
ts o
f ATD
and
A
PTD
on
inte
rleuk
in-6
act
ivat
ion
Goo
d
Har
rison
(200
4)38
Rand
., D
B,
With
in S
S, F
H-
13 F
Goo
dVA
MS,
no
sign
ific
ant c
hang
es o
bser
ved;
exa
ct sc
ores
not
re
port
edSt
udy
also
inve
stig
ates
mem
ory
and
cogn
itive
eff
ects
of
ATD
and
APT
D
Mod
erat
e
Leyt
on (1
999)
, Le
yton
(2
000)
39;4
0Ra
nd.,
DB,
Bet
w.
SS, F
H-
12 A
PTD
14
CO
NT
FG
ood
POM
S**: A
PTD
: cha
nge:
-2.3
9 (±
2.1 S
E). C
ON
T: c
hang
e -
1.04
(±1.
2). V
AM
S-de
pres
sed:
APT
D: B
L: 1.
3 (±
1.6 S
D),
po
st-t
est:
1.7
(±2.
7), c
hang
e: 0
.4 (±
1.6).
CO
NT:
BL:
0.4
(±
0.5)
, pos
t-te
st: 0
.4 (±
0.4)
, cha
nge:
0.1
(±0.
4).
POM
S; n
o ex
act s
core
s re
port
ed. T
hird
arm
with
ATD
in
clud
edM
oder
ate
Lyth
e (2
005)
61Ra
nd.,
DB,
W
ithin
SS,
12 M
+F
Goo
dVA
MS,
no
sign
ific
ant c
hang
es o
bser
ved;
exa
ct sc
ores
not
re
port
edSt
udy
inve
stig
ates
eff
ect o
f APT
D o
n ne
uro
psyc
ho
logi
cal a
nd s
ubje
ctiv
e m
easu
res
Mod
erat
e
McL
ean
(200
4)62
Rand
., D
B, B
etw
. SS
19+2
0M+F
Goo
d**VA
MS-
cont
ente
dnes
s: A
PTD
: BL:
10.2
(±0.
7 SE
), p
ost-
test
: 11
.5 (±
0.5)
. CO
NT:
BL:
11.8
(±0.
5), p
ost-
test
: 11.8
(±0.
5).
Stud
y in
vest
igat
es e
ffec
t of A
PTD
on
neur
o ps
ycho
lo
gica
l and
sub
ject
ive
mea
sure
s.G
ood
A2.
Hea
lthy
con
trol
s w
ith
+ve
fam
ily h
isto
ry fo
r MD
D
Gre
vet (
2002
)63Ra
nd.,
DB,
W
ithin
SS,
6/1
0 FH
+
10 M
+FIn
suff
icie
ntPO
MS-
depr
esse
d: A
PTD
: BL:
23.
40 (±
7.21
), p
ost-
test
: 23
.50
(±6.
48).
CON
T: B
L: 2
1.30
(±8.
05),
pos
t-te
st: 2
5.20
(±
4.73
).VA
MS-
cont
ente
dnes
s: A
PTD
: BL:
2.7
8 (±
1.43)
, po
st-t
est:
3.2
4 (±
1.49)
. CO
NT:
BL:
3.12
(±1.2
6), p
ost-
test
: 2.
79 (±
1.29
).
Stud
y in
vest
igat
es e
ffec
t of A
PTD
on
neur
o ps
ycho
lo
gica
l and
sub
ject
ive
mea
sure
s.M
oder
ate

Chapter6
119
Monoamine depletion in humans
B1. P
atie
nts
wit
h M
DD
pre
viou
sly,
but
cur
rent
ly in
rem
issi
on (w
itho
ut m
edic
atio
n)
McT
avis
h (2
005)
64Ra
nd.,
DB,
W
ithin
SS ≥6
m
onth
s re
mis
si
on, a
ver a
ge 4
1 m
onth
s
15 F
Rx-
free
≥6
mth
s
Goo
d O
nly
(TYR
+ PH
E)/N
AA
ratio
pr
ovid
ed
HD
RS-
6**: A
PTD
: BL:
1.7
(±0.
7 SE
), p
ost-
test
: 2.4
(±0.
9).
CON
T: B
L: 1.
0 (±
0.4)
, pos
t-te
st: 2
.7 (±
1.1).V
AS-
depr
esse
d no
t sig
nifi
cant
ly c
hang
ed b
y A
PTD
vs
CON
T; n
o ex
act
scor
es p
rovi
ded.
Stud
y in
vest
igat
es e
ffec
ts o
f ATP
D o
n su
bjec
tive
and
a sp
atia
l rec
ogni
tion
mem
ory-
task
Goo
d
Rois
er (2
005)
65Ra
nd.,
DB,
W
ithin
SS ≥6
m
onth
s re
mis
si
on
17 M
+F
Rx-fr
ee
≥6 m
ths
Insu
ffic
ient
TYR
de
plet
ion;
(TYR
+ PH
E)/N
AA
ratio
de
crea
ses
80%)
HD
RS-
19: A
PTD
: BL:
2.1
(±2.
0), p
ost-
test
: 2.2
(±2.
0). C
ON
T:
BL: 1
.4 (±
1.3)
, pos
t-te
st: 1
.1 (±
1.1).V
AS-
cont
ente
dnes
s:
APT
D: B
L: 12
.4 (±
1.5)
, pos
t-te
st: 1
2.8
(±1.
5). C
ON
T: B
L: 12
.3
(±1.8
), p
ost-
test
: 12.
8 (±
1.8).
BD-II
pat
ient
s in
clud
ed (n
=?).
Stu
dy in
vest
igat
es
effe
cts
of A
TPD
on
subj
ectiv
e an
d ne
uroc
og n
itive
m
easu
res
Goo
d
B2. P
atie
nts
wit
h M
DD
pre
viou
sly,
but
cur
rent
ly in
rem
issi
on (c
urre
ntly
usi
ng m
edic
atio
n)
- C. P
atie
nts
curr
entl
y w
ith
MD
D (w
ith
or w
itho
ut a
ntid
epre
ssan
ts)
- APT
D =
Acu
te P
heny
lala
nine
/Tyr
osin
e D
eple
tion;
NA
A =
neu
tral
am
ino
acid
s; P
HE
= ph
enyl
alan
ine;
TYR
= ty
rosi
ne† t
yros
ine
leve
ls d
eclin
ed b
y at
leas
t 60%
For o
ther
sym
bols
and
abb
revi
atio
ns s
ee fo
otno
te
in T
able
6.2
.
Tabl
e 6.
4. In
clud
ed s
tudi
es A
cute
Phe
nyla
lani
ne/T
yros
ine
Dep
letio
n (A
PTD
). (C
ontin
ued)
Aut
hor
(yea
r)*
Des
ign
N
Ade
quac
y of
de
plet
ion†
Resu
lts
moo
d sc
ores
(sca
le, s
core
s (±
SD‡ ))
Re
mar
ks
Judg
e m
ent§

120
Chap
ter
Chapter 6
6
Tabl
e 6.
5. In
clud
ed s
tudi
es A
lpha
-met
hyl-p
ara-
tyro
sine
(AM
PT).
Aut
hor
(yea
r)*
Des
ign
N
Ade
quac
y of
dep
leti
on†
Resu
lts
moo
d sc
ores
(s
cale
, sco
res
(±SD
‡ ))
Rem
arks
Judg
emen
t
A1.
Hea
lthy
con
trol
s
Krah
n (1
999)
88Ra
nd.,
DB,
With
in
SS, F
H-
10 M
+FN
ot re
port
edH
DRS
sco
res
incr
ease
d si
gnifi
cant
(cha
nge:
3 S
D
not r
epor
ted)
in A
MPT
com
pare
d to
CO
NT
(no
mor
e da
ta p
rovi
ded)
PO
MS
no s
igni
fica
nt
chan
ges
(no
mor
e da
ta p
rovi
ded)
Stud
y in
vest
igat
es e
ffec
ts o
f AM
PT o
n m
elat
onin
sec
retio
n. P
rom
etha
zine
25m
g as
CO
NT
Poor
McC
ann
(199
3)11
0Ra
nd.,
DB,
Bet
w. S
S40
MN
ot d
eter
min
ed; u
nspe
cifie
d si
gn. p
rola
ctin
rise
in A
MPT
gr
oup
VAS-
sadn
ess**
; AM
PT: B
L: 3
.2, p
ost-
test
: 13.
1. CO
NT:
BL:
11.2
, pos
t-te
st: 1
6.9.
(no
SDs
prov
ided
) (o
nly
rest
ing
cond
ition
pro
vide
d he
re) n
o si
gn.
Diff
eren
ce A
MPT
and
pla
cebo
in re
sted
con
ditio
n
Subj
ects
wer
e ra
ndom
ized
to 1.
AM
PT +
rest
, 2.
AM
PT+S
leep
Dep
r., 3
. CO
NT
+ Sl
eepD
epr.,
4.
CO
NT
+ re
st
Mod
erat
e
McC
ann
(199
5)82
Rand
., D
B, B
etw
. SS
41 M
Not
det
erm
ined
; pro
lact
in ri
se o
f 18
8% in
AM
PT g
roup
POM
S- s
adne
ss, V
AS-
sadn
ess;
no
exac
t sco
res
prov
ided
; fig
ure
for c
alm
ness
sho
wn.
AM
PT
caus
ed m
axim
al ra
tings
of t
ensi
on, w
hich
was
re
vers
ed b
y ad
ditio
n of
+ L
-dop
a/ c
arbi
dop
a.
Non
-sig
nific
ant i
ncre
ase
in s
adne
ss re
port
ed in
A
MPT
-gro
up (r
egar
dles
s L-
dopa
/ car
bi d
opa)
.
Subj
ects
wer
e ra
ndom
ized
to 1.
AM
PT +
PLA
C,
2. A
MPT
+L-d
opa/
carb
i dop
a, 3
. CO
NT
+ L-
dopa
/ car
bi d
opa,
4. C
ON
T +
PLA
C, in
ord
er to
in
vest
igat
e th
e sp
ecif
icity
of A
MPT
-cha
nges
by
eff
ects
on
cate
chol
amin
es
Mod
erat
e
Zim
mer
man
n (1
996)
89Ra
nd.,
DB,
With
in
SS, F
H-
10 M
+FSu
ffic
ient
MH
PG d
ecre
ase
in M
+FH
DRS
, PO
MS:
No
sign
ifica
nt d
iffer
ence
s. N
o ex
act s
core
s re
port
edSt
udy
inve
stig
ates
eff
ects
of A
MPT
on
prol
actin
and
mel
aton
in s
ecre
tion,
incl
udin
g ge
nder
-eff
ects
. Pro
met
hazi
ne 5
0 m
g as
CO
NT
Mod
erat
e
A2.
Hea
lthy
con
trol
s w
ith
+ve
fam
ily h
isto
ry fo
r MD
D
- B1. P
atie
nts
wit
h M
DD
pre
viou
sly,
but
cur
rent
ly in
rem
issi
on (w
itho
ut m
edic
atio
n)
Berm
an (1
999)
122
Rand
., D
B, W
ithin
SS
≥4
mon
ths
rem
issi
on, A
ver a
ge
18 m
onth
s
15M
+F R
x fr
ee ≥
3 m
onth
s
Goo
d H
DRS
-23;
AM
PT: B
L: 3
.1 (±
3.2)
, Pea
k: 2
3.9
(±12
.0).
CO
NT:
BL:
2.8
(±3.
4), p
eak:
9.0
(±9.
7).ID
S; A
MPT
: BL
: 8.2
(±7.
2). P
eak:
23.
6 (±
14.8
). CO
NT:
BL:
6.1
(±6.
6), p
eak:
10.0
(±8.
1).R
elap
se in
10/1
4 A
MPT
-pa
tient
s an
d in
1/13
CO
NT.
No
sepa
rate
dat
a fo
r n=1
2 w
ho u
nder
wen
t bo
th te
sts.
Giv
en s
core
s ar
e pe
ak s
core
s.
Post
-tes
t sco
resn
ot re
port
ed.
Mod
erat
e

Chapter6
121
Monoamine depletion in humans
B2. P
atie
nts
wit
h M
DD
pre
viou
sly,
but
cur
rent
ly in
rem
issi
on (c
urre
ntly
usi
ng m
edic
atio
n)
Brem
ner (
2003
)103
Rand
., D
B, W
ithin
SS
Aft
er re
mis
sion
18 M
+F D
ES
NO
RN
ot re
port
edH
DRS
**; n
o ex
act s
core
s an
d SD
s re
port
ed
AM
PT: W
ith A
MPT
rela
pse:
BL:
7.4
,pos
t-te
st:
24.8
. With
out r
elap
se: B
L: 11
.5,p
ost-
test
: 12.
5 CO
NT:
With
AM
PT re
laps
e: B
L: 8
.5,p
ost-
test
: 8.5
. W
ithou
t rel
apse
: BL:
6.4
,pos
t-te
st: 1
3.2
Rela
pses
: A
MPT
: 11/
18, C
ON
T: n
ot p
rovi
ded,
0/1
8 as
sum
ed.
Stud
y pr
imar
ilyin
vest
igat
es b
rain
met
abol
ic
corr
elat
es o
f AM
PT in
duce
d de
pres
siv
e re
laps
es w
ith P
ET
Mod
erat
e
Del
gado
(199
3)17
Rand
., D
B, W
ithin
SS
‘Var
ious
’ len
gth
of re
mis
sion
14 M
+F D
ES
MA
ZFLX
SE
R
Goo
d**H
DRS
-25;
No
exac
t sco
res
repo
rted
. Sig
nifi
cant
ef
fect
s of
AM
PT v
s CO
NT
in D
ES a
nd M
AZ
trea
ted
patie
nts,
not
in F
LX o
r SER
trea
ted
patie
nts.
Patie
nts
wer
e cu
rren
tly u
sing
nor
adre
nerg
ic
or s
erot
oner
gic
antid
epre
ssan
t med
icat
ion.
M
oder
ate
C. P
atie
nts
curr
entl
y w
ith
MD
D (w
ith
or w
itho
ut a
ntid
epre
ssan
ts)
Mill
er (1
996)
115
Rand
., D
B, W
ithin
SS
17 M
+FG
ood
HD
RS-2
5**; A
MPT
: BL:
29,
pos
t-te
st: 2
8.5.
CO
NT:
BL
: 30.
5, p
ost-
test
: 29.
5. S
Ds
not r
epor
ted.
VAS:
ex
act d
ata
not p
rovi
ded;
sig
n de
crea
se in
su
bsca
les
‘tire
d’ a
nd ‘e
nerg
etic
’ for
AM
PT v
s CO
NT
Patie
nts
wer
e no
t usi
ng a
ntid
epre
ssan
t m
edic
atio
n. 2
/17
patie
nts
had
bipo
lar
depr
essi
on, 6
/17
had
co-m
orbi
d an
xiet
y,
dyst
hym
ic o
r eat
ing
diso
rder
s
Mod
erat
e
AM
PT =
Alp
ha-m
ethy
l-par
a-ty
rosi
ne; I
DS
= In
vent
ory
of D
epre
ssiv
e Sy
mpt
oms;
MA
Z =
maz
indo
l; PL
AC
= ad
ditio
nal p
lace
bo, S
leep
Dep
r. =
Slee
p de
priv
atio
n† D
eclin
e in
cat
echo
lam
ine
met
abol
ites
hom
ovan
illic
aci
d (H
VA) b
y at
leas
t 60%
or 3
-met
hoxy
-4-h
ydro
xyph
enyl
glyc
ol (M
HPG
) by
at le
ast 4
0% F
or o
ther
sym
bols
and
abb
revi
atio
ns s
ee fo
otno
te in
Tab
le 6
.2.
Tabl
e 6.
5. In
clud
ed s
tudi
es A
lpha
-met
hyl-p
ara-
tyro
sine
(AM
PT).
(Con
tinue
d)
Aut
hor
(yea
r)*
Des
ign
N
Ade
quac
y of
dep
leti
on†
Resu
lts
moo
d sc
ores
(s
cale
, sco
res
(±SD
‡ ))
Rem
arks
Judg
emen
t

Chap
ter
Chapter 6
6
122
Quantitative summary (pooling) Fifty-eight percent (52/90) of the studies supplied enough data to be included in the meta-analysis. Meta-analysis was possible for ATD and APTD studies only. Table 6.6 gives an overview ofthe number of identified studies for each pre-defined population and eligibility for meta-analysis.Due to remaining heterogeneity we used random effects models for all meta-analyses.
ATDIn figure 6.1 the pooled results of ATD in healthy controls in studies with a within subjects designare presented. Overall Hedges' g (95% CI) was -0.27(-0.45 to -0.09). We stratified results by familyhistory for MDD (negative, positive or not reported in the studies). Pooled Hedges’ g for healthycontrols with a negative family history (-0.19 (-0.43 to 0.05)) was significantly different (Qres=6.59, df= 1, p= 0.01) compared to controls with a positive family history (-0.56 (-1.00 to -0.13)). Thepooled result in studies that did not report the family history status resembled the studies with anegative family history (-0.28 (-0.57 to 0.00) Qres= 1.36, df= 1, p= 0.24). In between subjects studiessimilar results were found. Pooled Hedges’ g was -0.63 (-1.95 to 0.70) for controls with a negativefamily history and -0.06 (-0.57 to 0.45) in one study that did not report family history status (Qres=0.14, df= 1, p= 0.71). The large Hedges’ g in controls with a negative family history was largelydetermined by one study.85 Leaving this study out reduced Hedges’ g to 0.16 (-0.43 to 0.76) (datanot shown).
Table 6.6. Identified studies for different types of depletion and design and eligibility for meta-analysis.
Identified (in meta-analysis)
Within Subjects Between Subjects
Acute Tryptophan Depletion
A. Healthy controls
FH- 41 (22) 10 (5)
FH+ 5 (5) 0
B. Patients with MDD in remission
Without AD 8 (6) 1
With AD 13 (12) 1
C. Patients with Current MDD
Without AD 3 (1) 0
With AD 1 (1) 0
Para-chlorophenylalanine
B. Patients with MDD in remission
With AD 2 0
Acute Phenylalanine/Tyrosine Depletion
A. Healthy controls
FH- 5 (3) 2 (2)
FH+ 1 (1) 0
B. Patients with MMD in remission
Without AD 2 (2) 0
Alpha-methyl-para-tyrosine
A. Healthy controls
FH- 2 2
B. Patients with MDD in remission
Without AD 1 0
With AD 2 0
C. Patients with Current MDD
Without AD 1 0
AD = antidepressant; FH = family history; MDD = Major Depressive Disorder

ChapterMonoamine depletion in humans
6
123
Figures 6.2A and 6.2B show the modification of Hedges’ g by gender for healthy controls(within subjects design) with a negative or positive family history. The difference in Hedges’ gbetween males and females was most prominent in healthy controls with a negative familyhistory (0.23 (-0.10 to 0.57) vs. -0.44 (-0.81 to -0.06) respectively; Qres= 11.92, df= 1, p<0.001). Incontrols with a positive family history, males experienced a larger decrease in mood after ATD inonly one study (Hedges’ g -0.98 (-1.53 to -0.42) Qres= 11.92, df= 1, p<0.001). In contrast, femaleswith a positive family history only had a slightly larger Hedges’ g (-0.56 (-1.43 to 0.31)) thanfemales with a negative family history (Qres= 0.62, df= 1, p= 0.43).
Figures 6.3A and 6.3B present the pooled Hedges’ g for patients with MDD in remissionwithout or with current AD (within subjects design). In the remitted patients without current ADwe stratified results by length of time without AD. Only 2 studies with 3-6 months without AD47;51
largely differed in Hedges’ g (-4.35 (-7.39 to -1.31) compared to 1 study directly after successfulelectroconvulsive therapy (Hedges’ g 0.04 (-0.85 to 0.94),117 and 3 studies with at least 6 monthswithout AD (pooled Hedges’ g -0.60 (-1.38 to 0.18) (Qres= 29.34, df= 2, p< 0.0001).48;93;118 Leavingone possible outlier47 out diminished the overall pooled Hedges’ g for remitted patients withoutAD from -1.90 (-3.02 to -0.78) to -1.06 (-1.83 to -0.29), but the observed effect modification bylength of time without AD remained highly significant (p< 0.001; data not shown).
Figure 6.1.
Acute Tryptophan Depletion (ATD) in healthy controls studied in a within subjects design, stratified by status of family historyfor depression (negative or positive for Major Depressive Disorder). References to studies as indicated, different subgroupsper study handled as separate studies with appropriate pooling weights. FH- = family history negative, FH+ = family historypositive, N/A = not reported.
��� T ��� ���!� � � "��#�"����� ��� ����"�� � � "��#�"����� ���������$����"��T ! ! � � "��#�"���%� �&'(��) �( �&'(��)
���* ���"���R���* $�+�����&&,����������� ���������������������� ���������������� ����� �������������� ���������������+��-��.����&&/�* $�� ������� ���������� ��� ��������������� ���� �����������������������������+�������&&0��������� ���������������������� ��������������� � ����� �����������������������������%������"����&&1����� ���������������������� ����������������� ����� ������������������������������%R�������'���������� ���������������������� ��������������� � ������ �������������������� ��������� ������������������� ���������������������� ������������� �� ������ �������������� ��������������� �"�������2��������� ���������������������� ����������������� ���� � ������������������������������3���������&&&������� ���������������������� ����������������� ������ �������������� ���� ��������!����������������4�� ���������������������� ������ ��������� ������ �����������������������������!����������������4�� ���������������������� ���������������� ���� � ������������������������������!����������������4�� ���������������������� ��������������� ������ �����������������������������!�������������/����� ������� ���������� ��� ����� �������� ����� ������������������������������!�������������1��4�� ���������������������� ���������������� ������ ������������������������������!�������������1��4�� ���������������������� ������������� � ���� � �����������������������������!�������������1��4�� ���������������������� ������� ������� ������ �������������������������������������������������� ���������������������� ����������������� ������ �������������������������������������&&,���������� ���������������������� ������� ������ � ���� ������������� ������������������S���������* $���� ���������������������� ������ ������ ��� ������ ����������������������������������������1��������� ���������������������� ���������������� ����� �������������������������������� �����U����&&0���� ���������������������� ������� ��������� ������ ������������������������������
����������&'(��)� ������ ��������� ��� ����� �������� ������������������.���������"�����T�����V�5�''6�17� .�5��&��8�9��6�����7�)V�5�1'61(�����.����R�������..�����:�5��6'���8�5��6�2�
���* ������R���* ;�+��-��.����&&/�* ;�� ���������������������� ����� ���������� ����� ������� ���������������������%������"����&&&����� ���������������������� ������������ �� ����� ������������������������������3���������&&&������� ���������������������� ���������������� ���� �����������������������������!����������������4�� ������� ���������� ��� ����������������� ���� � �����������������������������!����������������4�� ���������������������� ����� ������� � ���� � ������� ����������������������!����������������4�� �������������������� �������������� ������ ���������������������������������S���������* ;���� �������������������� ������ �������� ���� � ������������������������������
����������&'(��)� �������������������� ������ ����������������������������.���������"�����T�����V�5���6�07� .�5�1��8�5��6�1�7�)V�5�'�60(�����.����R�������..�����:�5��6'���8�5��6���
�2�* ����������� ��!4��3���������&&&������� ���������������������� ������������ �� ������ ������������������������������3W�-X�������'������� ���������������������� ����� ���������� ����� ������� ��������������������������T�������������� ���������������������� ����������������� ������ �������� ������������ ���������� �����&&/��������� ���������������������� ������������� ��� ����� ������������������������������8��-��&&/����������� ���������������������� �������������� �� ������ ����������������������������������U����&&/�������� ������� ���������� ��� ������������ ��� ����� �������� �������������������������U����&&'�������� ���������������������� ����������������� ������ ������������������������������
����������&'(��)� ���������������������� ������ ���������������������������.���������"�����T�����V�5�06007� .�5�1��8�5��6�0�7�)V�5�2�6'(�����.����R�������..�����:�5��6&/��8�5��6�'�
�������&'(��)� �������������������� ������ ��������������������� ������.���������"�����T�����V�5�026�&7� .�5�22��8�9��6������7�)V�5�1�6/(�����.����R�������..�����:�5��6&1��8�5��6��2�
�$/ �$� �� �� �/
������� ��T��� ������� ��T���!�

Chap
ter
Chapter 6
6
124
Figure 6.2A/B.
Acute Tryptophan Depletion (ATD) in healthy controls studied in a within subjects design, stratified by status of family historyfor depression and gender included in the studies. Family history negative for MDD (A) and family history positive for MDD(B). References to studies as indicated, different subgroups per study handled as separate studies with appropriate poolingweights. FH- = family history negative, FH+ = family history positive, MDD = major depressive disorder.
<�R��S� ��������������������� ���������������������������� �����������T����������������� �����T������������������������������������������������������������������������������ ������ �����T����������* $�����������������������������������������������������������������������������������
��� T � � "��#�"����� ��� ����"�� � � "��#�"����� ���������$����"��T �&'(��) �( �&'(��)
����������+��-��.����&&/�* $�� ������ ������������������ ���������+�������&&0��������� ������ ������� ����� �������������%R�������'���������� ������ ���������������������������� �"�������2��������� ������ ��������������������������������������1��������� ���� ����������� �� ������������
����������&'(��)� ������ ������������� ������������.���������"�����T�����V�5�,60&7� .�5�/��8�5��6���7�)V�5�/&62(�����.����R�������..�����:�5��620��8�5��6�,�
���*��������%������"����&&1����� ������ ����������� � ������ �������� ������������������� ������ ����������������������������!����������������4�� ������ �����������������������������!����������������4�� ������ ����� �����������������������!����������������4�� ������ ����� ���� ������ ���������������&&,���������� ������ ������� ���������������������
����������&'(��)� ������ �������������� �������������.���������"�����T�����V�5�,6/27� .�5�'��8�5��6�&�7�)V�5�2�6,(�����.����R�������..�����:�5��6�&��8�5��6���
�2��;*���+�����&&,����������� ����� ����������� �� ����� ��������3���������&&&������� ������ �����������������������������!�������������/����� ����� ����� ����������������������!�������������1��4�� ������ ����������� � �������������!�������������1��4�� ������ ����������� �������� ��������!�������������1��4�� ���� � ���������� �� ��������������������������������� ������ ��������������������������������S���������* $���� ������ ������� ����������������������� �����U����&&0���� ������ ������������� �� �����������
����������&'(��)� ������ ��������������������������.���������"�����T�����V�5��16&27� .�5�0��8�5��6���,�7�)V�5�,�62(�����.����R�������..�����:�5��61'��8�5��6���
�������&'(��)� ����� ������� �������������������.���������"�����T�����V�5�''6�17� .�5��&��8�9��6�����7�)V�5�1'61(�����.����R�������..�����:�5��6'���8�5��6�2�
�$/ �$� �� �� �/
������� ��T��� ������� ��T���!�
<�R��S� ��������������������� ���������������������������� �����������T����������������� �����T������������������������������������������������������������������������������ �2���� �����T����������* ;�����������������������������������������������������������������������������������
��� T � � "��#�"����� ��� ����"�� � � "��#�"����� ���������$����"��T �&'(��) �( �&'(��)
����������+��-��.����&&/�* ;�� �� ��� ����������� �����������������
����������&'(��)� �� ��� ����������� ����������������.���������"�����T�������������������.����R�������..�����:�5�26/1��8�5��6���'�
���*��������%������"����&&&����� �� ��� ������� ��������������������!����������������4�� � ��� ����������� � ���������������!����������������4�� ����� ����� �����������������������!����������������4�� ������ ���������������� �����������
����������&'(��)� ������ ����������� �������� ������.���������"�����T�����V�5�,6'17� .�5�2��8�5��6�1�7�)V�5�1�62(�����.����R�������..�����:�5��6�,��8�5��6���
�2��;�*���3���������&&&������� �� ��� ����������� � ������������������S���������* ;���� � � ����������� �����������������
����������&'(��)� ������ ����������� �� �������������.���������"�����T�����V�5��6�/7� .�5����8�5��6,��7�)V�5��(�����.����R�������..�����:�5��6�,��8�5��6�2�
�������&'(��)� ����� ����������� �������� �������.���������"�����T�����V�5���6�07� .�5�1��8�5��6�1�7�)V�5�'�60(�����.����R�������..�����:�5��6'���8�5��6���
�$/ �$� �� �� �/
������� ��T��� ������� ��T���!�
�
�

ChapterMonoamine depletion in humans
6
125
Figure 6.3A/B.
Acute Tryptophan Depletion (ATD) in former depressed patients in remission, studied in a within subjects design without orwith current medication. Figure 6.3A is stratified by length of time without medication; 6.3B is stratified by type ofmedication used by patients. References to studies as indicated, different subgroups per study handled as separate studieswith appropriate pooling weights. BUP = bupropion; SNRI = Serotonin Norepinephrin Reuptake Inhibitor; SSRI = SelectiveSerotonin Reuptake Inhibitor; PHZ = phenelzine.
2�R��S� ���������������������� ��������������������������� ����� �����T��������������������������S������R�� �������� �����T����������������������������������������� ������ ������R�� �������� �����T��������������S���� ���� ���������!T���"����##��� ���������������������������������
�� T ��� "��$�"����� ��� ����"�� ��� "��$�"����� ������� !%����"��T �&'(��) �( �&'(��)
�*�+�,����������� T�*&&-�������� � ��� �����������������������������
� !������&'(��)� � ��� ���������������������������#���������"�����T����������!������#����R�����##�����.�/��0*�����/��0&��
���,%1�������2� �����������3����� ���� ����������� �����������������2� �����������1�4� ����� ����������������� � ��������2� �����������1��4� � �� ������� ��������� �� �������2� �����������1��4�� � ���� ����� �����������������������
� !������&'(��)� ������ ����������������� �� ������#���������"�����T�����5�/��60,1�� #�/�,����+��0����*���)5�/�6&03(�����#����R�����##�����.�/��06*����/��0��'�
�,�7/1���������T�������3��������� � ���� ������������������������������$8��� ������3������ � ���� �������� �� � ����� ��������������*&&-���������� � ��� ����� ������� ���������������
� !������&'(��)� ������ ����������� ������� �������#���������"�����T�����5�/�60,'�� #�/������/��0�����)5�/�-10�(�����#����R�����##�����.�/�*0'�����/��0*,�
������&'(��)� ����� ����� ���������������������#���������"�����T�����5�/�110�'�� #�/�-����+��0����*���)5�/�6&03(�����#����R�����##�����.�/�,0,,����/��0���&�
�%*� �%' �� �' �*�
�������� �!T���� �������� �!T���2�
8�R��S� ���������������������� ��������������������������� ����� �����T��������������������������S������R�� �������� �����T����������������������������������������� ������ �,����R�� �������� �����T���������������!T��� ���������������������������������������������������������������
�� T ��� "��$�"����� ��� ����"�� ��� "��$�"����� ������� !%����"��T �&'(��) �( �&'(��)
�*������� ��������8)��� �������9�������*&&-�2��8�0 � �� ������������������� ��������9�������*&&-�8���� ����� ����� �������� ������ �������������*&&6���������� � ���� ������� ���������������������$8��� ������3������ � ���� ����� � ������������ ������������������*������ � ���� ����� �������� ��������������
� !������&'(��)� ������ ����������� ���������������#���������"�����T�����5�/�*30�&�� #�/�3����/��0��-���)5�/�-*01(�����#����R�����##�����.�/�*0-�����/��0�6�
��������� ��������8)4�28)��� �������9���:����'���������� � ��� ����������� �����������������
� !������&'(��)� � ��� ����������� ����������������#���������"�����T����������!������#����R�����##�����.�/��0*1����/��0�,�
�,������� ������9;���� �������<R������������������ � ��� ����������������������������
� !������&'(��)� � ��� ��������������������������#���������"�����T����������!������#����R�����##�����.�/��016����/��0'��
�3������� ��������.��� �������=�� ������,�������� ������ ������������ ���������������
� !������&'(��)� ������ ������������ �������������#���������"�����T����������!������#����R�����##�����.�/�*0*�����/��0�-�
������&'(��)� ����� �������������������� �������#���������"�����T�����5�/�*'0�*�� #�/�-����/��0�3���)5�/�',0,(�����#����R�����##�����.�/��03,����/��0�*�
�%3 �%� �� �� �3
�������� �!T���� �������� �!T���2�
�
�

Chap
ter
Chapter 6
6
126
In remitted patients previously depressed and currently using AD, ATD caused a decrease inmood (pooled Hedges’ g -0.49 (-0.89 to -0.10). Hedges’ g varied slightly depending on type of AD(Qres= 0.92, df= 3, p= 0.82). Surprisingly, the pooled Hedges’ g for SSRIs showed a moderate pointestimate, which did not reach significance (-0.60 (-1.28 to 0.08). Especially for bupropiontreatment Hedges’ g was small and not significant (-0.25 (-0.96 to 0.47). No ATD studies withother ADs without a 5-HT mechanism of action were available for this comparison.
We stratified the results of ATD studies by length of remission, which revealed significanteffect modification. In remitted patients using AD decreased mood after ATD was especially seenin the first 5 months after the achievement of remission (pooled Hedges’ g -0.55 (-0.90 to -0.21)Qres= 47.18, df= 2, p< 0.0001).48;59;94-96;119 In contrast, remitted patients without AD showed moredecrease in mood after ≥2 months of remission(Hedges’ g -1.65 (-2.60 to -0.69) Qres= 13.81, df= 2,p= 0.001)48;51;118 (figure available on request).
Relapse rates in remitted patients with AD were increased after ATD compared to controldepletion (pooled difference in relapse rate 47% (28% to 66%); Figure 6.4). This increase in relapserate was especially seen in patients using SSRIs (47% (27% to 67%)) or a SNRI (35% (14% to 56%)). TheNE acting drug desipramine showed no significant difference in relapse rate (7% (-6% to 19%)) afterATD. This effect modification by drug was statistically significant (Qres= 18.02, df= 2, p< 0.001).
In patients who were depressed at the time of ATD we found two studies for meta-analysis.These studies included patients who used,120 or did not use87 AD (Figure 6.5). The effects of ATDwere opposed: Hedges’ g was 0.32 (-0.22 to 0.86) for patients using different types of ADs, and -0.12 (-0.45 to 0.21) for patients without AD. Two studies in depressed patients without AD werenot suitable for meta-analysis. ATD did not decrease mood during depletion in these studies.86;121
Contra-intuitively, in one study mood improved the day after depletion in 16/43 patients,121 a resultwhich was also found by one study in the meta-analysis.120
Figure 6.4.
Acute Tryptophan Depletion (ATD) in former depressed patients in remission, differences of relapse rates versus controldepletion studied in a within subjects design in remitted patients stratified by current medication use. References to studiesas indicated. SNRI = Serotonin Norepinephrine Reuptake Inhibitor; SSRI = Selective Serotonin Reuptake Inhibitor.
����� �������������� ������������� �!��"#� �������������� ������������������� ����"��� �$%�&' �% �$%�&'
(����')�������($$*�������� � ��� ��������������������������������"����($$$�������� � � � �����������������������������+��������,-������ � ��� ����������������������������������#�. �������,- � ���� ��������������� ������������
����������$%�&'� ������ ������������������/��������#�����"��������&#�0�1�2�2$�����1�3����1��4���'0�1��,%/��������� ��������������5�1�-�3����6��(�
,����'7�8�')���9�,���������� � ��� ������������ ����������������
����������$%�&'� � ��� ������������ ��������/��������#�����"����������������������/��������� ��������������5�1�3�,4����1��(�
3��:�'��;�'8:���"����($$$�������� � ���� ������������������ ���������
����������$%�&'� � ���� ������������������ ��/��������#�����"����������������������/��������� ��������������5�1�(�-����1��3�
-�<�������������������������=�����"����($$(�������� � ���� �����������������������������������($$$��������� � �� ���������������� � ��������������#�($$$���������� ����� ������������ ��� � ���������
����������$%�&'� �� ��� ������������ �������/��������#�����"��������&#�0�1�2�-�����1�,����1��-���'0�1�2$�-%/��������� ��������������5�1��(����6��(�
/������$%�&'� ����� ��������������������/��������#�����"��������&#�0�1�-�3(�����1�4����6��(���'0�1�4�3%/��������� ��������������5�1�-�4����6��(�
� ( � � � �� �(
�����������������&�8/ �����������������;/�

ChapterMonoamine depletion in humans
6
127
APTDFigure 6.6A and 6.6B show results from APTD studies in healthy controls (within and betweensubjects). APTD did not decrease mood: pooled Hedges’ g were 0.10 (-0.23 to 0.43) in withinsubjects, and 0.12 (-0.43 to 0.68) in between subjects studies. However, in one study with healthycontrols with a positive family history for MDD, a moderate but non-significant effect on moodwas found (Hedges’ g -0.49 (-1.17 to 0.19)).63 In patients with MDD in remission without AD (Figure6.7), no effect of APTD was observed in 2 studies (pooled Hedges’ g -0.02 (-0.50 to 0.47).64;65 Nostudies inpatients with current MDD were identified.
Sensitivity analysis and publication biasWe examined our assumptions for intercorrelation ofbefore-after mood ratings per condition inATD studies in healthy controls without a family history of MDD. At the same examination, we alsoexamined the assumed intercorrelation between two test conditions (ATD vs. CONT). Asexpected, increasing the assumed R to 0.8 (less conservative) increased the pooled Hedges’ gfrom -0.19 (-0.43 to 0.05) to -0.31 (-0.64 to 0.02). Reducing R to 0.2 (more conservative) decreasedthe pooled Hedges’ g to -0.14 (-0.34 to 0.06). The calculated R’s were higher than 0.5 in 80% of thestudies that reported all relevant data. Therefore, we judged the imputed value of 0.5 for R asacceptable.
Inspection of the funnel-plot of the within subjects ATD studies in healthy controls without afamily history of MDD revealed asymmetry (figure available on request). More studies with adecrease in mood after ATD vs. control depletion were published. In a Galbraith-plot of studies,the intercept in the regression equation was -2.08 (SE 0.90; p= 0.030). Therefore, we concludedthat publication bias probably distorted our findings. We did not inspect funnel-plots in otherpopulations of our review due to the limited number of studies.
Discussion
In this systematic review we performed the first meta-analysis of the mood-effects in ATD andAPTD studies. The depletion of monoamine systems (both 5-HT and NE/DA) does not decreasemood in healthy controls. However, in healthy controls with a family history of MDD the resultssuggest that mood is slightly decreased, both by ATD and APTD. Additionally, healthy females aremore affected by ATD than healthy males, especially in controls without a family history of MDD.In patients who were previously depressed but in remission without AD, ATD moderatelydecreases mood, while APTD does not significantly decrease mood. ATD has comparable moodlowering effects in patients with MDD in remission who are still using ADs. The site of action of
Figure 6.5.
Acute Tryptophan Depletion (ATD) in depressed patients stratified by use of concurrent medication (within subjects design).References to studies as indicated.
�� "� ���� �� #� ���"$��%�$�����"��� ����$�� ���"$��%�$�����"������� &'����$��� # # ���"$��%�$���(� �+)*��+ �* �+)*��+
�!��������"�������,���-����)&��������� ���������������������� �������� �������� ����� ������������������������������ &������+)*��+� ���������������������� ����� ����������������������������.���������$����������������&������.����M�����..�����/�0�!1!)����0��1�)�
�������� ����"�������������!++2���������� ���������������������� ������� �������� ������ ����������������������������� &������+)*��+� ���������������������� ������ ���������������������������.���������$����������������&������.����M�����..�����/�0��13!����0��142�
������+)*��+� �������������������� ������ ����������������������������.���������$�����������5�0�!12�6�".�0�!����0��1!2�6�+5�0�4)1�*�����.����M�����..�����/�0��1������0��124�
�'4 �'� �� �� �4
��������"�&����� ��������"�&��� #�

Chap
ter
Chapter 6
6
128
these ADs (5-HT or NE/DA) predicts the occurrence of a lowering of mood or a short relapse inMDD after depletion of the corresponding monoamine. Our findings are in line with thesummaries in previous reviews.11;18-20;24
The most consistent finding from this review is the decrease of mood and relapse into adepressed state after ATD and APTD in remitted MDD patients who use AD. In remitted patientswithout AD relapses are less prominent. Because after remission medication is often continued,the difference in mood responses after depletion might be related to the duration of the achievedremission. Previous reviews discussed the relationship between the duration of the remission andthe effect of monoamine depletion, with opposite conclusions. Bell et al. concluded that effectsof ATD were more pronounced early in recovery.123 In contrast, Booij et al. concluded thatduration of remission was not associated with mood response to ATD.21
Booij et al. investigated predictors of relapse in a pooled analysis of the individual patient dataof some of the studies included in this review.21 This is often referred to as a ‘mega-analysis’. Theyfound that recurrent depressive episodes (≥2), female gender, and a history of suicidal thoughts/attempts predicted relapse. Duration of remission did not contribute to this prediction whenconfounding was considered. We found a modest relationship between relapse and the durationof the remission after ATD. We defined the duration of remission as the reported averageduration, or – if not stated – the minimum duration of remission used as inclusion criterion for thestudies. We found that especially in the first 5 months after remission, ATD caused lowering of
Figure 6.6A/B.
Acute phenylalanine/tyrosine depletion (APTD) in healthy controls (6.6A. within subjects design; 6.6B. between subjectsdesign), stratified by status of family history for depression. References to studies as indicated. FH- = family history negative,FH+ = family history positive, N/A = not reported.
<�7��8� �,������������������������ ����� �������������� ��� ������������T��������T�� �������������������T������ ������������������������������������������������������� ���������T������ ���� ���!������������������������������������������������������������������������������
��!T ����� ���"� ���!#� $�#��%�&�!� ����#� ���!#� $�#��%�&�!���� � '���#��T " " ���!#� $�#���(� �)*+��, �+ �)*+��,
���-����#��.���-�'������ �������������� ���������������������� ���������������� ������ �������������������������
�� ����)*+��,� ���������������������� ������ ������������������������ �%��������#����T����������� ��� �%����.�����%%����/�0��1�2����0��1�2�
���-���� ��.���-�3�4��.��������������� ���������������������� ������ ��������� ����� �������� ������������ ����
�� ����)*+��,� ���������������������� ����� �������� ������������ ��� �%��������#����T����������� ��� �%����.�����%%����/�0��156����0��1�*�
�6�-�����������!��"��������!������������ �������������������� ������� ��������� ������ ���������������������������������������������� ���������������������� ���������������� �� ��� ��������������������������
�� ����)*+��,� ���������������������� ������ ������������������������ �%��������#����T�����7�0��1)68�!%�0������0��165�8�,7�0��+�� �%����.�����%%����/�0��1������0��16��
�����)*+��,� ���������������������� ������ ������������������������� �%��������#����T�����7�0�519)8�!%�0�6����0��1�)�8�,7�0�6915+�� �%����.�����%%����/�0��1:�����0��1**�
�'5 �'� �� �� �5
�;����!���� �!����� �;����!���� �!���"�
<�.��=� ;������������������������� ����� �������������� ��� �5����������T��������T�� �������������������T������ ��>�=1������������������������������������������������� ���������T������ ���� ���!������������������������������������������������������������������������������
��!T ����� ���"� ���!#� $�#�����!��� ����#� ���!#� $�#�����!������ � '���#��T " " ���!#� $�#���(� �)*+��, �+ �)*+��,
���-�'?�T����)))��������� ���������������������� ������� ����� ��� ������ �������������� �����������
�� ����)*+��,� ���������������������� ������ �������������� ��������� �%��������#����T����������� ��� �%����.�����%%����/�0��1*6����0��1:��
���-��"��;�?�������5��������� ������� �������������� ����������������� ����� ���������������������������
�� ����)*+��,� ������� �������������� ����� ������������������������� �%��������#����T����������� ��� �%����.�����%%����/�0��1�6����0��1�:�
�����)*+��,� ���������������������� ������ ������������������������� �%��������#����T�����7�0��1�98�!%�0������0��1�:�8�,7�0���1�+�� �%����.�����%%����/�0��156����0��1:9�
�'5 �'� �� �� �5
�;����!���� �!����� �;����!���� �!���"�
�
�

ChapterMonoamine depletion in humans
6
129
mood in remitted patients still using AD. However, the problem with our and Bell’s comparison isthe intraindividual spread in duration within the studies. This spread is not considered at the samelevel of detail as in a ‘mega-analysis’. In addition, confounding can only be considered in ‘mega-analysis’. Therefore – although their study did not include all available studies – we think thatBooij et al. provide the best available indication of riskfactors for mood lowering effects by ATD.
Do depletion studies elucidate the pathogenesis of MDD?The absence of robust mood effects in healthy controls indicates that mood is not a directcorrelate of 5-HT or NE levels in the brain. The only healthy controls who are modestly affected bymonoamine depletion studies are healthy controls with a positive family history for MDD. Thismight be indicative of a biological vulnerability which is revealed by depletion studies. Of interestare the findings of recent studies that combined ATD with neuroimaging or geneticsampling,47;49;51;66;68-70;100-10846;51 reviewed by Fusar-Poli et al.25 The intelligent approach of combiningdepletion with imaging or genotyping appears very promising. A summary of the complex resultsof these studies goes beyond the scope of our review. However, these neuroimaging andgenotyping studies also suggest that monoamine depletion discloses rather a ‘trait’ vulnerabilitythan a pure ‘state’ dependent change due to depletion.
Additionally, a depressive relapse after monoamine depletion in remitted patients who useAD, occurs only if the target of the depletion (5-HT, NE) coincides with the working mechanism ofthe antidepressant used. This emphasizes that AD indeed specifically affect their supposed targetsystems. However, we may only conclude that an undepleted 5-HT system is required forserotonergic AD. The same holds for the NE system and norepinephrinergic AD. Delgadoproposed an alternative explanation for the decrease in mood after monoamine depletion inpatients: depletion of e.g. 5-HT may give the same effect as abrupt discontinuation of SSRIs.124
Rapid discontinuation is also associated with mood-effects, which are considered to be differentfrom a depressive relapse.125
What certainly cannot be concluded is that MDD is caused by low levels of 5-HT and/or NE/DA.This simplification, which is often used to promote the use of AD specifically affecting 5-HT or NEor both systems, represents a Catch-22 argument, and ignores the notion that serotonergic andnorepinephrinergic AD presumably act by a final common pathway. In this pathway post-synapticchanges at the cellular level are supposed to be responsible for the remission of MDD.126 Cellularchanges include up or down regulated receptors, increased neuronal interconnections andsprouting and changes in levels of neuropeptides (e.g. corticotrophin releasing hormone). Theclinical question of how to distinguish a patient who will respond to an AD with e.g. NE effects hasnot been solved. Descriptive variables at the symptom level have not yet sufficiently predictedthe response to any selective agent. Therefore, a pragmatic approach to affect this final commonpathway might be to prescribe ADs which target both 5-HT and NE. However, the effectiveness ofthis approach is still equivocal.127-129
In patients who are depressed at the time of monoamine depletion, no further decrease inmood is observed. A ceiling effect could be responsible for this finding. But, a morestraightforward conclusion is that there is no simple relation between 5-HT or NE deficiency andmood or MDD. Nevertheless, this finding is complicated by the finding of some authors120;121 thatmood is lowered or elevated the day after ATD. A delayed decrease of mood was indicative fortreatment refractoriness,121 a finding that was not yet replicated by others.86;87;120 The number of
Figure 6.7.
Acute phenylalanine/tyrosine depletion (APTD) in former depressed patients in remission without medication, studied in awithin subjects design. References to studies as indicated.
#��%� � ������ ������� ���%&��'�&����%�� �"��&�� ���%&��'�&����%�������()���&��� * * ���%&��'�&��#+� �,-.��/ �. �,-.��/
�� ��������-������� �������������������� ����������������� ����� ����������������������������0���������-��������� ���������������������� ���������������� ����� �����������������������������
�� ��,-.��/� ���������������������� ������ ���������������������� ����1���������&�����������2�3��456!�%1�3������3��4�5�!�/2�3�7-46. ����1������� ��11�����8�3��4�9����3��4,7�
�)7 �)� �� �� �7
������%�������%��� � ������%�������%��$*

Chap
ter
Chapter 6
6
130
comparable studies to date is limited. Therefore, we think no clear conclusions or explanationscan be made, except that even in MDD patients mood is not a correlate of 5-HT or NE levels in thebrain.
We agree with the conclusion of Booij et al. and Bell et al. that a relapse of depressivesymptoms in remitted patients after depletion probably reflects a biological vulnerability of the 5-HT system in remitted patients.11;21;123 This vulnerability increases their risk to become depressed.This increased risk was further demonstrated in two prospective studies that used the responseto ATD in remitted patients to predict later relapse/recurrence.130;131 However, also these resultsneed replication. Moreover, the cause of an increased vulnerability remains uncertain. Probablythe cause is a combination of genetic, environmental and otherdeterminants (e.g. ‘scarring’ thebrain after multiple depressive episodes).123
In conclusion, monoamine depletion by ATD and APTD does not elucidate a causal factor inthe pathogenesis of MDD. However, ATD and APTD remain useful models to safely and directlymanipulate 5-HT, NE and DA function in living humans, and to study the behavioral consequencesof this manipulation, especially in subgroups of humans with an apparent vulnerability.11;12;24
Still, several methodological issues need to be addressed. First, because of the competition ofaminoacids to pass the blood-brain barrier, ATD might unwillingly result in an intracerebralincrease of tyrosine/phenylalanine.24;132 Vice-versa, APTD might increase intracerebral tryptophanavailability. Levels of other amino-acids than those depleted are not provided in the studies.Second, ATD provides a specific net lowering of 5-HT. Contrary, depletion of tyrosine andphenylalanine lowers both NE and DA, which are synthesized in the same cascade (with DAthereafter transformed into NE by dopamine beta-hydroxylase). Also AMPT interferes early in thiscascade. Although evidence from animal studies points to more DA depletion by APTD and moreNE depletion by AMPT,133 it is impossible to truly distinguish between net NE and DA depletion.Third, test re-test reliability for monoamine depletion paradigms was only tested for ATD and wasrather limited, which limits the robustness of the method.134;135 Fourth, also in healthy controlssubtle cognitive effects of monoamine depletion in the brain occur: deficits in learning andmemory consolidation and improvement in focused attention and executive function.136 Theseeffects show similarity with symptoms of MDD. The question remains whether these effectsmight represent mild first symptoms of MDD or the starting symptoms of a cascade of alteredbrain functions leading to MDD. Fifth, in line with the fourth issue, MDD does not develop withinone day. Therefore, the changes by experimental monoamine depletion by ATD and APTD may betoo short to really induce a complex biological and psychological deregulation which is recognizedas MDD. Patients suffering from gastrointestinal carcinoid tumors – 5-HT producing tumors withexpected prolonged states of secondary tryptophan depletion – are generally not depressed butdo show improved focused attention.137 However, carcinoid findings were not yet related totryptophan levels over time. Sixth, the single depletion of one monoamine system by ATD orAPTD/AMPT may be too simplistic, especially because of the complex interaction of monoaminesystems. Five dual depletion studies were not included in this review.38;138-141 In healthy controlscontrasting results were found. Hughes et al. found some decrease of mood on 3 VAMS-subscales,139 which was also found in another open study.140 However, no effects were found in 2other studies.38;141 In unmedicated patients with MDD no significant increase of MDD was foundafter dual depletion.138 It would be interesting to investigate the effects of simultaneous depletionof 5-HT and NE/DA in other populations.
Limitations of the studies and the reviewSome limitations should be mentioned. First, female gender is a risk-factor for MDD, and was alsofound to be a predictor of relapse after ATD in remitted patients. Based on several studies, genderdifferences in 5-HT metabolismare probable, and hormonal factors may play a role in 5-HTfunction.21 In our results we examined the effect of gender in healthy controls. The differencebetween males and females was most prominent in healthy controls without a family history ofMDD. In the included studies hormonal status (pre or postmenopausal state) was notdistinguished in the results nor used as inclusion criterion. Second, many small differences

ChapterMonoamine depletion in humans
6
131
between the studies existed: different composition of depletion and control drinks, differentpresentation of tryptophan/tyrosine/phenylalanine or their ratios to other neutral amino acids,different presentation of free vs. total tryptophan/tyrosine/phenylalanine values, differentmeasurements, different scales. Differences between studies undoubtedly introducedheterogeneity between the studies, which may bias the results of this review. Therefore, werecommend an international consensus protocol. Third, limited presentation of the data forced usto make assumptions, that may have influenced our findings. However, the assumptionsappeared to have little influence on the results in a sensitivity analysis. Future studies need toaddress clear presentation of their data, preferably including a description of the directions of theeffects, SDs, in cross-over designs the numbers of subjects having the control or sham first, andpreferably include a SD of the pooled difference between the depletion and the control condition.For example, only 10 studies adequately reported the number of patients treated in eachsequence in the within subjects design.54;57;64-66;68;70;95;122;135 Fourth, 6 studies included BipolarPatients. Three of these studies were also included in the meta-analysis.65;94;114 This involved 6patients of the total of 265 patients (2.2%) in the concerning meta analytical comparisons.Therefore, we consider the possibility of bias by the inclusion of this etiologically differentdisorder unlikely. Fifth, the rate of agreement in the selection of studies still may rise questionsabout the clarity of our selection criteria. However, discrepancies in agreement could easily besolved between the two reviewers. Therefore, we do not think our sensitive searches missedrelevant studies. Finally, an indication of publication bias was found. If publication bias truly exists,the studies which found no effect or an increase in mood after ATD would not have beenpublished. Indeed, the mood effects of studies that could not be included in the meta-analysiswere mostly very small and non-significant. Furthermore, the exact information in studiesrequired for meta-analysis forced us to exclude many studies. It seems natural that a non-significant result will not be given this level of detailed attention (change-scores with SDs),especially when mood-effects are not the primary outcome in these studies. Without thispublication bias the pooled effect of ATD in healthy controls would probably have been lower.Because our conclusion already is that there is no apparent mood effect of ATD in healthycontrols, we conclude that the observed publication bias will not severely distort the generalconclusion. Therefore, despite these limitations, we consider the results of our review after ourstrict methods as valid.
Conclusion and future studiesWe conclude that although ATD and APTD are important in the investigation of the monoaminesystems, monoamine depletion does not directly decrease mood. Although previously themonoamine systems were considered to be responsible for the development of MDD, theavailable evidence to date does not support a direct causal relationship with MDD. There is nosimple direct correlation of 5-HT or NE levels in the brain and mood. The depletion of 5-HT by ATDand NE/DA by APTD most clearly decreases mood in vulnerable patients who are in remission fromtheir MDD, while still using AD. Furthermore, depletion affects mood in unmedicated patients inremission or healthy controls with a family history positive for MDD. Therefore, the monoaminesystems are probably important systems in the vulnerability to become depressed. The changes inbrain metabolism in remitted patients who relapse after ATD or AMPT suggest that 5-HT and NEsystems give input to a final common pathway which needs further research to be clarified.
We suggest some lines for future research. First: three or four-armed depletion studiescomparing ATD, APTD (and – if possible – their combination) versus sham depletion to investigatethe differential effects of the 5-HT and NE systems on mood in remitted patients or controls withpositive family history for MDD. Second: a further exploration of the relation between knowngenetic polymorphisms of the 5-HT, NE and DA systems (e.g. 5-HTTPR) and biological cerebralresponses to depletion paradigms, as measured by PET/fMRI (e.g. like Neumeister et al.46;51).Third: the relations between monoamine depletion indicating biological vulnerability andpsychological vulnerability for MDD (see Booij et al.)11 Fourth: Replication, further validation andstandardization of the properties of ATD and other depletion paradigms as a diagnostic test for

Chap
ter
Chapter 6
6
132
recurrences in remitted patients (see Moreno et al.130 and Neumeister et al.131). New studies willincrease our knowledge of the 5-HT and NE systems, which are important targets in the currenttreatments of MDD. This knowledge will finally improve the treatment for MDD.
Acknowledgement
The authors wish to thank Mrs Natasha Wiebe, MMath PStat, Research Associate at theUniversity of Alberta, Canada and especially Dr. Rob J.P.M. Scholten, M.D. PhD, epidemiologistand director of the Dutch Cochrane Centre at the Academic Medical Centre, Amsterdam for theirindispensable help with the statistic methods used in this review. This study was partially fundedby the program Opleiding Onderzoekers GGZ (OOG) (project number 100-002-002) by ZonMw,the Hague, the Netherlands.
Conflict of interests
None
References
1. Murray CJ, Lopez AD. Evidence-based health policy--lessons from the Global Burden of Disease Study. Science. 1996; 274:740-743.
2. Kennedy SH, Lam RW, Cohen NL, Ravindran AV, CANMAT Depression Work Group. Clinical guidelines for the treatment ofdepressive disorders. IV. Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.
3. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
4. Anderson IM, Nutt DJ, Deakin JFW. Evidence-based guidelines for treating depressive disorders with antidepressants: Arevision of the 1993 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2000; 14: 3-20.
5. Mulrow CD, Williams JW, Jr., Trivedi M, Chiquette E, Aguilar C, Cornell JE et al. Evidence report on: Treatment ofdepression - Newer pharmacotherapies. Psychopharmacol Bull. 1999; 34: 409-795.
6. Hindmarch I. Beyond the monoamine hypothesis: mechanisms, molecules and methods. Eur Psychiatry. 2002; 17 Suppl 3:294-299.
7. Owens MJ. Selectivity of antidepressants: from the monoamine hypothesis of depression to the SSRI revolution andbeyond. J Clin Psychiatry. 2004; 65 Suppl 4: 5-10.
8. Burke WJ. Selective versus multi-transmitter antidepressants: are two mechanisms better than one? J Clin Psychiatry.2004; 65 Suppl 4: 37-45.
9. Fava M, Kendler KS. Major depressive disorder. Neuron. 2000; 28: 335-341.10. Delgado PL. How antidepressants help depression: mechanisms of action and clinical response. J Clin Psychiatry. 2004; 65
Suppl 4: 25-30.11. Booij L, Van der Does AJ, Riedel WJ. Monoamine depletion in psychiatric and healthy populations: review. Mol Psychiatry.
2003; 8: 951-973.12. Hood SD, Bell CJ, Nutt DJ. Acute tryptophan depletion. Part I: Rationale and methodology. Aust N Z J Psychiatry. 2005; 39:
558-564.13. Young SN, Smith SE, Pihl RO, Ervin FR. Tryptophan depletion causes a rapid lowering of mood in normal males.
Psychopharmacol (Berl). 1985; 87: 173-177.14. Moja EA, Lucini V, Benedetti F, Lucca A. Decrease in plasma phenylalanine and tyrosine after phenylalanine-tyrosine free
amino acid solutions in man. Life Sci. 1996; 58: 2389-2395.15. Shopsin B, Gershon S, Goldstein M, Friedman E, Wilk S. Use of synthesis inhibitors in defining a role for biogenic amines
during imipramine treatment in depressed patients. Psychopharmacol Commun. 1975; 1: 239-249.16. Gratton A. Time course analysis of para-chlorophenylalanine induced suppression of self-stimulation behavior. Pharmacol
Biochem Behav. 1982; 17: 597-602.17. Delgado PL, Miller HL, Salomon RM, Licinio J, Heninger GR, Gelenberg AJ et al. Monoamines and the mechanism of
antidepressant action: effects of catecholamine depletion on mood of patients treated with antidepressants.Psychopharmacol Bull. 1993; 29: 389-396.

ChapterMonoamine depletion in humans
6
133
18. Van der Does AJ. The effects of tryptophan depletion on mood and psychiatric symptoms. J Affect Disord. 2001; 64: 107-119.
19. Bell C, Abrams J, Nutt D. Tryptophan depletion and its implications for psychiatry. Br J Psychiatry. 2001; 178: 399-405.20. Moore P, Landolt HP, Seifritz E, Clark C, Bhatti T, Kelsoe J et al. Clinical and physiological consequences of rapid
tryptophan depletion. Neuropsychopharmacology. 2000; 23: 601-622.21. Booij L, Van der DW, Benkelfat C, Bremner JD, Cowen PJ, Fava M et al. Predictors of mood response to acute tryptophan
depletion. A reanalysis. Neuropsychopharmacology. 2002; 27: 852-861.22. Van der Does AJ. The mood-lowering effect of tryptophan depletion: possible explanation for discrepant findings. Arch
Gen Psychiatry. 2001; 58: 200-202.23. Riedel WJ, Klaassen T, Schmitt JA. Tryptophan, mood, and cognitive function. Brain Behav Immun. 2002; 16: 581-589.24. Reilly JG, McTavish SF, Young AH. Rapid depletion of plasma tryptophan: a review of studies and experimental
methodology. J Psychopharmacol. 1997; 11: 381-392.25. Fusar-Poli P, Allen P, McGuire P, Placentino A, Cortesi M, Perez J. Neuroimaging and electrophysiological studies of the
effects of acute tryptophan depletion: a systematic review of the literature. Psychopharmacol (Berl). 2006; 188:131-143.26. McNair DM, Lorr M, Droppelman LF. Manual for the Profile of Mood States. San Diego: Educational and Industrial Testing
Service; 1988.27. Zuckerman M, Lubin B. Manual for the Multiple Affect Adjective Checklist. San Diego CA: Educational and testing service;
1965.28. Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol. 1974; 47: 211-218.29. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.30. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979; 134: 382-
389.31. Kawanishi C, Lundgren S, Agren H, Bertilsson L. Increased incidence of CYP2D6 gene duplication in patients with
persistent mood disorders: ultrarapid metabolism of antidepressants as a cause of nonresponse. A pilot study. Eur J ClinPharmacol. 2004; 59: 803-807.
32. Review Manager. [Version 4.2 for Windows]. 2002. Oxford, UK, The Cochrane Collaboration. 33. The Cochrane Collaboration. RevMan 4.2 User Guide, version 4.2 for Windows. 4.2 ed. Oxford, UK: The Cochrane
Collaboration; 2002.34. Cochrane Reviewers' Handbook. 4.2.2 [Updated March 2004] ed. Chichester, UK: John Wiley & Sons, Ltd; 2004.35. DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986; 7: 177-188.36. Egger M, Davey SG, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997; 315: 629-
634.37. Harrison BJ, Olver JS, Norman TR, Nathan PJ. Effects of serotonin and catecholamine depletion on interleukin-6
activation and mood in human volunteers. Hum Psychopharmacol. 2002; 17: 293-297.38. Harrison BJ, Olver JS, Norman TR, Burrows GD, Wesnes KA, Nathan PJ. Selective effects of acute serotonin and
catecholamine depletion on memory in healthy women. J Psychopharmacol. 2004; 18: 32-40.39. Leyton M, Young SN, Pihl RO, Etezadi S, Lauze C, Blier P et al. A comparison of the effects of acute tryptophan depletion
and acute phenylalanine/tyrosine depletion in healthy women. Adv Exp Med Biol. 1999; 467: 67-71.40. Leyton M, Young SN, Pihl RO, Etezadi S, Lauze C, Blier P et al. Effects on mood of acute phenylalanine/tyrosine depletion
in healthy women. Neuropsychopharmacology. 2000; 22: 52-63.41. Benkelfat C, Ellenbogen MA, Dean P, Palmour RM, Young SN. Mood-lowering effect of tryptophan depletion. Enhanced
susceptibility in young men at genetic risk for major affective disorders. Arch Gen Psychiatry. 1994; 51: 687-697.42. Delgado PL, Price LH, Miller HL, Salomon RM, Licinio J, Krystal JH et al. Rapid serotonin depletion as a provocative
challenge test for patients with major depression: relevance to antidepressant action and the neurobiology ofdepression. Psychopharmacol Bull. 1991; 27: 321-330.
43. Hayward G, Goodwin GM, Cowen PJ, Harmer CJ. Low-dose tryptophan depletion in recovered depressed patients induceschanges in cognitive processing without depressive symptoms. Biol Psychiatry. 2005; 57: 517-524.
44. Klaassen T, Riedel WJ, van Someren A, Deutz NE, Honig A, Van Praag HM. Mood effects of 24-hour tryptophan depletionin healthy first-degree relatives of patients with affective disorders. Biol Psychiatry. 1999; 46: 489-497.
45. Klaassen T, Riedel WJ, Deutz NE, Van Praag HM. Mood congruent memory bias induced by tryptophan depletion. PsycholMed. 2002; 32: 167-172.
46. Neumeister A, Konstantinidis A, Stastny J, Schwarz MJ, Vitouch O, Willeit M et al. Association between serotonintransporter gene promoter polymorphism (5HTTLPR) and behavioral responses to tryptophan depletion in healthywomen with and without family history of depression. Arch Gen Psychiatry. 2002; 59: 613-620.
47. Neumeister A, Nugent AC, Waldeck T, Geraci M, Schwarz M, Bonne O et al. Neural and behavioral responses totryptophan depletion in unmedicated patients with remitted major depressive disorder and controls. Arch Gen Psychiatry.2004; 61: 765-773.
48. O'Reardon JP, Chopra MP, Bergan A, Gallop R, DeRubeis RJ, Crits-Christoph P. Response to tryptophan depletion in majordepression treated with either cognitive therapy or selective serotonin reuptake inhibitor antidepressants. BiolPsychiatry. 2004; 55: 957-959.
49. Praschak-Rieder N, Hussey D, Wilson AA, Carella A, Lee M, Dunn E et al. Tryptophan depletion and serotonin loss inselective serotonin reuptake inhibitor-treated depression: an [18F] MPPF positron emission tomography study. BiolPsychiatry. 2004; 56: 587-591.
50. Stewart ME, Deary IJ, Ebmeier KP. Neuroticism as a predictor of mood change: the effects of tryptophan depletion. Br JPsychiatry. 2002; 181: 242-247.

Chap
ter
Chapter 6
6
134
51. Neumeister A, Hu XZ, Luckenbaugh DA, Schwarz M, Nugent AC, Bonne O et al. Differential effects of 5-HTTLPR genotypeson the behavioral and neural responses to tryptophan depletion in patients with major depression and controls. Arch GenPsychiatry. 2006; 63: 978-986.
52. Gallagher P, Massey AE, Young AH, McAllister-Williams RH. Effects of acute tryptophan depletion on executive functionin healthy male volunteers. BMC Psychiatry. 2003; 3: 10.
53. Hughes JH, Gallagher P, Stewart ME, Matthews D, Kelly TP, Young AH. The effects of acute tryptophan depletion onneuropsychological function. J Psychopharmacol. 2003; 17: 300-309.
54. Murphy FC, Smith KA, Cowen PJ, Robbins TW, Sahakian BJ. The effects of tryptophan depletion on cognitive andaffective processing in healthy volunteers. Psychopharmacol (Berl). 2002; 163: 42-53.
55. Park SB, Coull JT, McShane RH, Young AH, Sahakian BJ, Robbins TW et al. Tryptophan depletion in normal volunteersproduces selective impairments in learning and memory. Neuropharmacology. 1994; 33: 575-588.
56. Rubinsztein JS, Rogers RD, Riedel WJ, Mehta MA, Robbins TW, Sahakian BJ. Acute dietary tryptophan depletion impairsmaintenance of "affective set" and delayed visual recognition in healthy volunteers. Psychopharmacol (Berl). 2001; 154:319-326.
57. Schmitt JA, Jorissen BL, Sobczak S, van Boxtel MP, Hogervorst E, Deutz NE et al. Tryptophan depletion impairs memoryconsolidation but improves focussed attention in healthy young volunteers. J Psychopharmacol. 2000; 14: 21-29.
58. Shansis FM, Busnello JV, Quevedo J, Forster L, Young S, Izquierdo I et al. Behavioural effects of acute tryptophandepletion in healthy male volunteers. J Psychopharmacol. 2000; 14: 157-163.
59. Booij L, Van der Does AJ, Haffmans PM, Riedel WJ, Fekkes D, Blom MJ. The effects of high-dose and low-dose tryptophandepletion on mood and cognitive functions of remitted depressed patients. J Psychopharmacol. 2005; 19: 267-275.
60. Harmer CJ, McTavish SF, Clark L, Goodwin GM, Cowen PJ. Tyrosine depletion attenuates dopamine function in healthyvolunteers. Psychopharmacol (Berl). 2001; 154: 105-111.
61. Lythe KE, Anderson IM, Deakin JFW, Elliott R, Strickland PL. Lack of behavioural effects after acute tyrosine depletion inhealthy volunteers. J Psychopharmacol. 2005; 19: 5-11.
62. McLean A, Rubinsztein JS, Robbins TW, Sahakian BJ. The effects of tyrosine depletion in normal healthy volunteers:implications for unipolar depression. Psychopharmacol (Berl). 2004; 171: 286-297.
63. Grevet EH, Tietzmann MR, Shansis FM, Hastenpflugl C, Santana LC, Forster L et al. Behavioural effects of acutephenylalanine and tyrosine depletion in healthy male volunteers. J Psychopharmacol. 2002; 16: 51-55.
64. McTavish SF, Mannie ZN, Harmer CJ, Cowen PJ. Lack of effect of tyrosine depletion on mood in recovered depressedwomen. Neuropsychopharmacology. 2005; 30: 786-791.
65. Roiser JP, McLean A, Ogilvie AD, Blackwell AD, Bamber DJ, Goodyer I et al. The subjective and cognitive effects of acutephenylalanine and tyrosine depletion in patients recovered from depression. Neuropsychopharmacology. 2005; 30: 775-785.
66. Allen PP, Cleare AJ, Lee F, Fusar-Poli P, Tunstall N, Fu CHY et al. Effect of acute tryptophan depletion on pre-frontalengagement. Psychopharmacology. 2006; 187:486-497.
67. Amin Z, Gueorguieva R, Cappiello A, Czarkowski KA, Stiklus S, Anderson GM et al. Estradiol and Tryptophan DepletionInteract to Modulate Cognition in Menopausal Women. Neuropsychopharmacology. 2006;31:2489-2497.
68. Evers EA, van der Veen FM, van Deursen JA, Schmitt JA, Deutz NE, Jolles J. The effect of acute tryptophan depletion onthe BOLD response during performance monitoring and response inhibition in healthy male volunteers. Psychopharmacol(Berl). 2006; 187: 200-208.
69. van der Veen FM, Evers EA, van Deursen JA, Deutz NE, Backes WH, Schmitt JA. Acute tryptophan depletion reducesactivation in the right hippocampus during encoding in an episodic memory task. Neuroimage. 2006; 31: 1188-1196.
70. Evers EA, van der Veen FM, Jolles J, Deutz NE, Schmitt JA. Acute tryptophan depletion improves performance andmodulates the BOLD response during a Stroop task in healthy females. Neuroimage. 2006; 32: 248-255.
71. Talbot PS, Watson DR, Barrett SL, Cooper SJ. Rapid tryptophan depletion improves decision-making cognition in healthyhumans without affecting reversal learning or set shifting. Neuropsychopharmacology. 2006; 31: 19.
72. McAllister-Williams RH, Massey AE, Rugg MD. Effects of tryptophan depletion on brain potential correlates of episodicmemory retrieval. Psychopharmacol (Berl). 2002; 160: 434-442.
73. Abbott FV, Etienne P, Franklin KB, Morgan MJ, Sewitch MJ, Young SN. Acute tryptophan depletion blocks morphineanalgesia in the cold-pressor test in humans. Psychopharmacol (Berl). 1992; 108: 60-66.
74. Crean J, Richards JB, de Wit H. Effect of tryptophan depletion on impulsive behavior in men with or without a familyhistory of alcoholism. Behav Brain Res. 2002; 136: 349-357.
75. Koszycki D, Zacharko RM, Le Melledo JM, Young SN, Bradwejn J. Effect of acute tryptophan depletion on behavioral,cardiovascular, and hormonal sensitivity to cholecystokinin-tetrapeptide challenge in healthy volunteers. Biol Psychiatry.1996; 40: 648-655.
76. Miller HEJ, Deakin JFW, Anderson IM. Effect of acute tryptophan depletion on CO2-induced anxiety in patients with panicdisorder and normal volunteers. Br J Psychiatry. 2000; 176: 182-188.
77. Oldman AD, Walsh AE, Salkovskis P, Laver DA, Cowen PJ. Effect of acute tryptophan depletion on mood and appetite inhealthy female volunteers. J Psychopharmacol. 1994; 8: 8-13.
78. Richell RA, Deakin JF, Anderson IM. Effect of acute tryptophan depletion on the response to controllable anduncontrollable noise stress. Biol Psychiatry. 2005; 57: 295-300.
79. Schmeck K, Sadigorsky S, Englert E, Demisch L, Dierks T, Barta S et al. Mood changes following acute tryptophandepletion in healthy adults. Psychopathology. 2002; 35: 234-240.
80. Weltzin TE, Fernstrom MH, Fernstrom JD, Neuberger SK, Kaye WH. Acute tryptophan depletion and increased foodintake and irritability in bulimia nervosa. Am J Psychiatry. 1995; 152: 1668-1671.
81. Coupland N, Zedkova L, Sanghera G, Leyton M, Le Melledo JM. Response to pentagastrin after acute phenylalanine andtyrosine depletion in healthy men: a pilot study. J Psychiatry Neurosci. 2001; 26: 247-251.

ChapterMonoamine depletion in humans
6
135
82. McCann UD, Thorne D, Hall M, Popp K, Avery W, Sing H et al. The effects of L-dihydroxyphenylalanine on alertness andmood in alpha-methyl-para-tyrosine-treated healthy humans. Further evidence for the role of catecholamines in arousaland anxiety. Neuropsychopharmacology. 1995; 13: 41-52.
83. Booij L, Swenne CA, Brosschot JF, Haffmans PM, Thayer JF, Van der Does AJ. Tryptophan Depletion Affects Heart RateVariability and Impulsivity in Remitted Depressed Patients with a History of Suicidal Ideation. Biol Psychiatry. 2006; 60:507-514.
84. Danjou P, Hamon M, Lacomblez L, Warot D, Kecskemeti S, Puech AJ. Psychomotor, subjective and neuroendocrineeffects of acute tryptophan depletion in the healthy volunteer. Psychiatry Psychobiol. 1990; 5: 31-38.
85. Ravindran AV, Griffiths J, Merali Z, Knott VJ, Anisman H. Influence of acute tryptophan depletion on mood and immunemeasures in healthy males. Psychoneuroendocrinology. 1999; 24: 99-113.
86. Price LH, Malison RT, McDougle CJ, McCance-Katz EF, Owen KR, Heninger GR. Neurobiology of tryptophan depletion indepression: effects of m-chlorophenylpiperazine (mCPP). Neuropsychopharmacology. 1997; 17: 342-350.
87. Price LH, Malison RT, McDougle CJ, Pelton GH, Heninger GR. The neurobiology of tryptophan depletion in depression:effects of intravenous tryptophan infusion. Biol Psychiatry. 1998; 43: 339-347.
88. Krahn LE, Lin SC, Klee GG, Lu PY, Ory SJ, Zimmermann RC. The effect of presynaptic catecholamine depletion on 6-hydroxymelatonin sulfate: a double blind study of alpha-methyl-para-tyrosine. Eur Neuropsychopharmacol. 1999; 9: 61-66.
89. Zimmermann RC, Krahn L, Klee G, Lu PY, Ory SJ, Lin SC. The impact of gender on alpha-methyl-para-tyrosine mediatedchanges in prolactin secretion and 6-hydroxymelatonin sulfate excretion. Psychoneuroendocrinology. 1996; 21: 469-478.
90. Bhatti T, Gillin JC, Seifritz E, Moore P, Clark C, Golshan S et al. Effects of a tryptophan-free amino acid drink challenge onnormal human sleep electroencephalogram and mood. Biol Psychiatry. 1998; 43: 52-59.
91. Kahkonen S, Makinen V, Jaaskelainen IP, Pennanen S, Liesivuori J, Ahveninen J. Serotonergic modulation of mismatchnegativity. Psychiatry Res. 2005; 138: 61-74.
92. Knott VJ, Howson AL, Perugini M, Ravindran AV, Young SN. The effect of acute tryptophan depletion and fenfluramine onquantitative EEG and mood in healthy male subjects. Biol Psychiatry. 1999; 46: 229-238.
93. Haynes PL, McQuaid JR, Kelsoe J, Rapaport M, Gillin JC. Affective state and EEG sleep profile in response to rapidtryptophan depletion in recently recovered nonmedicated depressed individuals. J Affect Disord. 2004; 83: 253-262.
94. Evans L, Golshan S, Kelsoe J, Rapaport M, Resovsky K, Sutton L et al. Effects of rapid tryptophan depletion on sleepelectroencephalogram and mood in subjects with partially remitted depression on bupropion.Neuropsychopharmacology. 2002; 27: 1016-1026.
95. Landolt HP, Kelsoe JR, Rapaport MH, Gillin JC. Rapid tryptophan depletion reverses phenelzine-induced suppression ofREM sleep. J Sleep Res. 2003; 12: 13-18.
96. Moore P, Gillin C, Bhatti T, DeModena A, Seifritz E, Clark C et al. Rapid tryptophan depletion, sleep electroencephalogram,and mood in men with remitted depression on serotonin reuptake inhibitors. Arch Gen Psychiatry. 1998; 55: 534-539.
97. Debener S, Strobel A, Kurschner K, Kranczioch C, Hebenstreit J, Maercker A et al. Is auditory evoked potentialaugmenting/reducing affected by acute tryptophan depletion? Biol Psychol. 2002; 59: 121-133.
98. Hughes JH, Ashton CH, Matthews D, Young AH. Acute depletion of plasma tryptophan does not alter electrophysiologicalvariables in healthy males. Psychopharmacol (Berl). 2000; 152: 119-121.
99. Voderholzer U, Hornyak M, Thiel B, Huwig-Poppe C, Kiemen A, Konig A et al. Impact of experimentally induced serotonindeficiency by tryptophan depletion on sleep EEG in healthy subjects. Neuropsychopharmacology. 1998; 18: 112-124.
100. Praschak-Rieder N, Wilson AA, Hussey D, Carella A, Wei C, Ginovart N et al. Effects of tryptophan depletion on theserotonin transporter in healthy humans. Biol Psychiatry. 2005; 58: 825-830.
101. Bremner JD, Innis RB, Salomon RM, Staib LH, Ng CK, Miller HL et al. Positron emission tomography measurement ofcerebral metabolic correlates of tryptophan depletion-induced depressive relapse. Arch Gen Psychiatry. 1997; 54: 364-374.
102. Smith KA, Morris JS, Friston KJ, Cowen PJ, Dolan RJ. Brain mechanisms associated with depressive relapse and associatedcognitive impairment following acute tryptophan depletion. Br J Psychiatry. 1999; 174: 525-529.
103. Bremner JD, Vythilingam M, Ng CK, Vermetten E, Nazeer A, Oren DA et al. Regional brain metabolic correlates of alpha-methylparatyrosine-induced depressive symptoms: implications for the neural circuitry of depression. JAMA. 2003; 289:3125-3134.
104. Yatham LN, Liddle PF, Shiah IS, Lam RW, Adam MJ, Zis AP et al. Effects of rapid tryptophan depletion on brain 5-HT(2)receptors: a PET study. Br J Psychiatry. 2001; 178: 448-453.
105. Talbot PS, Cooper SJ. Anterior cingulate and subgenual prefrontal blood flow changes following tryptophan depletion inhealthy males. Neuropsychopharmacology. 2006; 31: 09.
106. Rubia K, Lee F, Cleare AJ, Tunstall N, Fu CH, Brammer M et al. Tryptophan depletion reduces right inferior prefrontalactivation during response inhibition in fast, event-related fMRI. Psychopharmacol (Berl). 2005; 179: 791-803.
107. Evers EA, Cools R, Clark L, van der Veen FM, Jolles J, Sahakian BJ et al. Serotonergic modulation of prefrontal cortexduring negative feedback in probabilistic reversal learning. Neuropsychopharmacology. 2005; 30: 1138-1147.
108. Cools R, Calder AJ, Lawrence AD, Clark L, Bullmore E, Robbins TW. Individual differences in threat sensitivity predictserotonergic modulation of amygdala response to fearful faces. Psychopharmacol (Berl). 2005; 180: 670-679.
109. Shopsin B, Friedman E, Gershon S. Parachlorophenylalanine reversal of tranylcypromine effects in depressed patients.Arch Gen Psychiatry. 1976; 33: 811-819.
110. McCann UD, Penetar DM, Shaham Y, Thorne DR, Sing HC, Thomas ML et al. Effects of catecholamine depletion onalertness and mood in rested and sleep deprived normal volunteers. Neuropsychopharmacology. 1993; 8: 345-356.
111. Aberg-Wistedt A, Hasselmark L, Stain-Malmgren R, Aperia B, Kjellman BF, Mathe AA. Serotonergic 'vulnerability' inaffective disorder: a study of the tryptophan depletion test and relationships between peripheral and central serotoninindexes in citalopram-responders. Acta Psychiatr Scand. 1998; 97: 374-380.
112. Smith SE, Pihl RO, Young SN, Ervin FR. A test of possible cognitive and environmental influences on the mood loweringeffect of tryptophan depletion in normal males. Psychopharmacol (Berl). 1987; 91: 451-457.

Chap
ter
Chapter 6
6
136
113. Weltzin TE, Fernstrom JD, McConaha C, Kaye WH. Acute tryptophan depletion in bulimia: effects on large neutral aminoacids. Biol Psychiatry. 1994; 35: 388-397.
114. Delgado PL, Miller HL, Salomon RM, Licinio J, Krystal JH, Moreno FA et al. Tryptophan-depletion challenge in depressedpatients treated with desipramine or fluoxetine: implications for the role of serotonin in the mechanism ofantidepressant action. Biol Psychiatry. 1999; 46: 212-220.
115. Miller HL, Delgado PL, Salomon RM, Heninger GR, Charney DS. Effects of alpha-methyl-para-tyrosine (AMPT) in drug-freedepressed patients. Neuropsychopharmacology. 1996; 14: 151-157.
116. Moreno FA, Gelenberg AJ, Heninger GR, Potter RL, McKnight KM, Allen J et al. Tryptophan depletion and depressivevulnerability. Biol Psychiatry. 1999; 46: 498-505.
117. Cassidy F, Murry E, Weiner RD, Carroll BJ. Lack of relapse with tryptophan depletion following successful treatment withECT. Am J Psychiatry. 1997; 154: 1151-1152.
118. Smith KA, Fairburn CG, Cowen PJ. Relapse of depression after rapid depletion of tryptophan. Lancet. 1997; 349: 915-919.119. Spillmann MK, Van der Does AJ, Rankin MA, Vuolo RD, Alpert JE, Nierenberg AA et al. Tryptophan depletion in SSRI-
recovered depressed outpatients. Psychopharmacol (Berl). 2001; 155: 123-127.120. Booij L, Van der Does AJ, Haffmans PM, Riedel WJ. Acute tryptophan depletion in depressed patients treated with a
selective serotonin-noradrenalin reuptake inhibitor: augmentation of antidepressant response? J Affect Disord. 2005; 86:305-311.
121. Delgado PL, Price LH, Miller HL, Salomon RM, Aghajanian GK, Heninger GR et al. Serotonin and the neurobiology ofdepression. Effects of tryptophan depletion in drug-free depressed patients. Arch Gen Psychiatry. 1994; 51: 865-874.
122. Berman RM, Narasimhan M, Miller HL, Anand A, Cappiello A, Oren DA et al. Transient depressive relapse induced bycatecholamine depletion: potential phenotypic vulnerability marker? Arch Gen Psychiatry. 1999; 56: 395-403.
123. Bell CJ, Hood SD, Nutt DJ. Acute tryptophan depletion. Part II: clinical effects and implications. Aust N Z J Psychiatry. 2005;39: 565-574.
124. Delgado PL. Monoamine depletion studies: implications for antidepressant discontinuation syndrome. J Clin Psychiatry.2006; 67 Suppl 4: 22-26.
125. Rosenbaum JF, Fava M, Hoog SL, Ascroft RC, Krebs WB. Selective serotonin reuptake inhibitor discontinuation syndrome:a randomized clinical trial. Biol Psychiatry. 1998; 44: 77-87.
126. Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998; 59Suppl 14: 11-14.
127. Freemantle N, Anderson IM, Young P. Predictive value of pharmacological activity for the relative efficacy ofantidepressant drugs. Meta-regression analysis. Br J Psychiatry. 2000; 177: 292-302.
128. Ferreri M, Lavergne F, Berlin I, Payan C, Puech AJ. Benefits from mianserin augmentation of fluoxetine in patients withmajor depression non-responders to fluoxetine alone. Acta Psychiatr Scand. 2001; 103: 66-72.
129. Nelson JC, Mazure CM, Jatlow PI, Bowers MB, Jr., Price LH. Combining norepinephrine and serotonin reuptake inhibitionmechanisms for treatment of depression: a double-blind, randomized study. Biol Psychiatry. 2004; 55: 296-300.
130. Moreno FA, Heninger GR, McGahuey CA, Delgado PL. Tryptophan depletion and risk of depression relapse: A prospectivestudy of tryptophan depletion as a potential predictor of depressive episodes. Biol Psychiatry. 2000; 48: 327-329.
131. Neumeister A, Habeler A, Praschak-Rieder N, Willeit M, Kasper S. Tryptophan depletion: a predictor of future depressiveepisodes in seasonal affective disorder? Int Clin Psychopharmacol. 1999; 14: 313-315.
132. Badawy AA. Acute tryptophan or tyrosine depletion test: time for reappraisal? J Psychopharmacol. 2005; 19: 429-430.133. McTavish SF, Callado L, Cowen PJ, Sharp T. Comparison of the effects of alpha-methyl-p-tyrosine and a tyrosine-free
amino acid load on extracellular noradrenaline in the rat hippocampus in vivo. J Psychopharmacol. 1999; 13: 379-384.134. Ellenbogen MA, Young SN, Dean P, Palmour RM, Benkelfat C. Mood response to acute tryptophan depletion in healthy
volunteers: sex differences and temporal stability. Neuropsychopharmacology. 1996; 15: 465-474.135. Ellenbogen MA, Young SN, Dean P, Palmour RM, Benkelfat C. Acute tryptophan depletion in healthy young women with a
family history of major affective disorder. Psychol Med. 1999; 29: 35-46.136. Riedel WJ. Cognitive changes after acute tryptophan depletion: what can they tell us? Psychol Med. 2004; 34: 3-8.137. Russo S, Nielen MMA, Boon JC, Kema IP, Willemse PHB, De Vries EGE et al. Neuropsychological investigation into the
carcinoid syndrome. Psychopharmacol (Berl). 2003; 168: 324-328.138. Berman RM, Sanacora G, Anand A, Roach LM, Fasula MK, Finkelstein CO et al. Monoamine depletion in unmedicated
depressed subjects. Biol Psychiatry. 2002; 51: 469-473.139. Hughes JM, Matrenza C, Kemp AH, Harrison BJ, Liley D, Nathan PJ. Selective effects of simultaneous monoamine
depletion on mood and emotional responsiveness. Int J Neuropsychopharmacol. 2004; 7: 9-17.140. Leyton M, Pun VK, Benkelfat C, Young SN. A new method for rapidly and simultaneously decreasing serotonin and
catecholamine synthesis in humans. J Psychiatry Neurosci. 2003; 28: 464-467.141. Salomon RM, Miller HL, Krystal JH, Heninger GR, Charney DS. Lack of behavioral effects of monoamine depletion in
healthy subjects. Biol Psychiatry. 1997; 41: 58-64.142. Curtin F, Altman DG, Elbourne D. Meta-analysis combining parallel and cross-over clinical trials. I: Continuous outcomes.
Stat Med. 2002; 21: 2131-2144.143. Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several
studies in meta-analysis. In: Egger M, Smith GD, Altman DG, eds. Systematic reviews in healthcare. Meta-analysis in context.2 ed. London, UK: BMJ Publishers Group; 2001:285-312.
144. Altman DG. Comparing groups-categorical data. In: Altman DG, ed. Practical statistics for medical research. 1st ed. London:Chapman & Hall; 1991:229-276.
145. Barr LC, Heninger GR, Goodman W, Charney DS, Price LH. Effects of fluoxetine administration on mood response totryptophan depletion in healthy subjects. Biol Psychiatry. 1997; 41: 949-954.

ChapterMonoamine depletion in humans
6
137
146. Klaassen T, Riedel WJ, Deutz NE, van Someren A, Van Praag HM. Specificity of the tryptophan depletion method.Psychopharmacol (Berl). 1999; 141: 279-286.
147. Smith KA, Clifford EM, Hockney RA, Clark DM, Cowen PJ. Effect of tryptophan depletion on mood in male and femalevolunteers: A pilot study. Hum Psychopharmacol. 1997; 12: 111-117.
148. Morris JS, Smith KA, Cowen PJ, Friston KJ, Dolan RJ. Covariation of activity in habenula and dorsal raphe nuclei followingtryptophan depletion. Neuroimage. 1999; 10: 163-172.
Supplementary appendix: statistical formulas.
Differences between intervention and control measurementsIn depletion studies changes in mood scores typically represented mean mood-scores both before(pre) and after (post) the depletion/challenge (experimental intervention) and the placebo/sham/control-intervention. Because the mood scores were not necessarily identical at the start of theexperiment and the control, we first calculated the mean change in mood-score (pooleddifference) for the experimental and control condition separately per study. Some studies alsoprovided the standard deviation (SD) of the pooled differences. When the standard deviation wasnot reported, we calculated the standard deviation of the pooled difference for paired data:
In this formula, the correlation coefficient R was calculable in four studies only40;41;50;119 andranged between 0.42and 1.00 for the experimental and between 0.34and 0.95 for the controlcondition. To be able to calculate the standard deviation of the change between pre and post testmood scores for the rest of the studies we imputed a correlation coefficient R of 0.5. This valuewas considered to be a conservative assumption.
Statistics for studies with a within subjects designThe difference in the changes of mood scores between intervention and control were expressedas difference of change scores:
For this difference the SD of the difference was calculated by again applying formula (1), withan assumed R of 0.5.
To acknowledge the different mood scales to measure change in mood, the difference inchanges between experimental and control condition were standardized by calculating Hedges’adjusted g, which is similar to Cohen’s d, but includes an adjustment for small sample bias:33
In this formula nAB and nBA represent the number of subjects randomized to intervention orcontrol as first test in the study. If the numbers for nAB and nBA were not reported, we assumedthat the sample was split half for the two sequences. For Hedges’ ga SE was calculated:142;143
)***2(22postprepostprechange SDSDRSDSDSD −+= (1).
INTCONTchanges ChangeChangeDifference −= (2).
)9)(4
31(*SD
Hedges' CONT
−+−−=
BAABchangesDifference
INT
nnChangeChangeg (3).
( )⎟⎟⎠⎞
⎜⎜⎝
⎛−+
+∗
+=94.32
'4
2
BAABBAAB
BAAB
nngHedges
nnnnSE (4).

Chap
ter
Chapter 6
6
138
Statistics for studies with a between subjects designFor between subject studies comparable statistics were used to calculate the mean change inmood-scores. Because the between-subjects design is a parallel group design, Hedges’ g wascalculated with formula (3) in which for nAB and nBA nINT and nCONT were substituted. The formulafor the SE was slightly different to acknowledge the absence of paired data:
Statistics for Relapse ratesFor relapse rates of MDD after depletion provided in a within subjects design the difference inrelapse rates was calculated as:
in which N is the total number of patients included. The standard error then is:
in which b represents the number of patients with a relapse after the intervention but not thecontrol condition and c the number of patients with a relapse after the control but not theintervention.144 If the numbers of ‘pairs’ were not extractable from the paper, a conservativeapproach was used assuming the minimal number of patients relapsing both after theintervention and the control condition (maximal c), resulting in the largest SE.
( )⎟⎟⎠⎞
⎜⎜⎝
⎛−+
+∗+=
94.32' 2
CONTINTCONTINT
CONTINT
nngHedges
nnnnSE (5).
Nn
Nn
Difference CONTlapseINTlapse _Re_Rerate-lapseRe −= (6),
Ncbcb
NSE raterelapseDifference
2
_)(1 −−+=−
(7),

CHA
PTER
SEROTONIN TRANSPORTER BINDING WITH [123I]β-CIT SPECT IN MAJOR DEPRESSIVE DISORDER
VERSUS CONTROLS: EFFECT OF SEASON AND
GENDER.
Submitted
Henricus G. Ruhé1
Jan Booij2
Johannes B. Reitsma3
Aart H. Schene1
1 Program for Mood Disorders, Department of Psychiatry, Academic Medical Center, University ofAmsterdam, Amsterdam, the Netherlands2 Department of Nuclear Medicine, Academic Medical Center, University of Amsterdam, Amsterdam,the Netherlands3 Department of Clinical Epidemiology, Biostatistics & Bioinformatics, Academic Medical Center,University of Amsterdam, Amsterdam, the Netherlands

Chap
ter
Chapter 7
7
140
Abstract
BackgroundThe serotonin system is undoubtedly involved in the pathogenesis of major depressive disorder(MDD). More specifically the serotonin transporter (SERT) serves as a major target forantidepressant drugs. There are conflicting results about SERT availability in depressed patientsversus healthy controls (HC).
AimTo measure SERT availability and study effects of age, gender and season of scanning in MDDpatients versus HC.
MethodsWe included 49 depressed outpatients (42.3 ± 8.3 [SD] years) with a Hamilton Depression RatingScale above 18, who were drug-naïve or drug-free for ≥4 weeks, and 49 age- (±2 years) and sex-matched HC. Subjects were scanned with single photon emission computed tomography (SPECT)using [123I]β-CIT. SERT availability was expressed as specific to non-specific binding ratios (BPND) inmidbrain (MID) and diencephalon (DIENC) with cerebellar binding as a reference.
ResultsIn crude comparisons between patients and HC, we found no significant differences in MID orDIENC SERT availability. In subgroup analyses, depressed males had numerically lower MID SERTavailability than HC, whereas in women SERT availability was not different (significantdiagnosis*gender interaction; p= 0.048). In DIENC we found a comparable diagnosis*gender (p=0.002) and an additional smoking*gender (p= 0.036) interaction. In MID the season of scanningshowed a significant main-effect (p= 0.018) with higher SERT availability in winter.
ConclusionsDifferences in MID and DIENC SERT availability in MDD patients compared with HC are modifiedby gender. The season of scanning is a covariate in MID. The diagnosis*gender andgender*smoking interactions on SERT availability should be considered in future studies of thepathogenesis of MDD.

Serotonin transporter availability in MDD
141
Chapter7
Introduction
Major depressive disorder (MDD) is a highly prevalent and disabling disease, often treated byselective serotonin reuptake inhibitors (SSRIs).1 SSRIs block the serotonin transporter (SERT),which lowers the reuptake of serotonin (5-HT) from the synaptic cleft and increasesneurotransmission. Despite the fact that the working-mechanism of antidepressants supports themonoamine deficiency theory, the pathogenesis of MDD remains unclear.2;3 Therefore,differences in SERT availability in patients and healthy controls (HC) have been studied previously.
Postmortem studies have shown reduced or unchanged concentrations of SERTs in MDD-patients compared with HC, but these studies may be biased by retrospective data collection,previous antidepressant use, suicidal behavior apart from MDD or non-selective ligands (reviewedby Stockmeier).4
Cerebral SERTs in humans can be quantified in-vivo with single photon emission computedtomography (SPECT) and positron emission tomography (PET). The first SERT radioligand, iodine-123-labeled 2β-carbomethoxy-3β-(4-iodophenyl)-tropane ([123I]β-CIT) binds both to SERTs anddopamine transporters (DATs).5 Diencephalon (DIENC) and brainstem (MID) [123I]β-CIT bindingpredominantly reflect SERT, while striatal [123I]β-CIT uptake reflects DAT.6
Studies comparing depressed patients with HC have reported either decreased,7-13
unchanged,14-18 or increased19 SERT availability in MDD patients (Table S7.1). A negative correlationbetween SERT availability and severity of depression (measured by Hamilton Depression RatingScale (HDRS) scores) was reported in patients with primary MDD9 or Wilson’s disease.20
Discrepant results among studies may be explained by differences in scanning techniques,analytic methods, and subject sampling, although the effects of additional variables and theirinteraction might also explain conflicting results. Staley et al. reported lower SERT availability inthe DIENC of MDD female patients versus HC,11 and suggested that this interaction accounted forthe contradictory results between studies. Furthermore, in HC significant effects on SERTavailability have been reported for gender,21 smoking behavior,21 aging22;23 and season ofscanning.24
Our objectives were to quantify SERT availability in MDD patients versus HC while accountingfor these potential covariates and possible interactions, and to correlate SERT availability withdepression severity. Therefore, we compared [123I]β-CIT SPECT scans of drug-free MDD patientswith age- and sex-matched HC.
Methods
SubjectsAfter approval by the institutional ethical committee and written informed consent, we recruiteddepressed patients from primary care, and our outpatient department (October 2003-August2006). Patients were eligible if they were 25-55 years old, had a diagnosis of MDD (diagnosed bystructured clinical interview for DSM-IV (SCID Patient Version)), had a HDRS score >18, and weredrug-free or used no more than one antidepressant (stopped for >4 weeks and ≥5 half-lives of thisantidepressant before scanning) for the present MDD-episode. Exclusion criteria were pregnancy(or desire to become pregnant), bipolar disorder, psychotic features, primary anxiety and/orsubstance abuse disorders and acute, severe suicidal ideation. We allowed secondary co-morbidanxiety and/or substance abuse.
We individually matched each patient by gender and age (±2 years). HC were in good physicalhealth and had never used psychotropic medication. Exclusion criteria were current or lifetimepsychiatric disorder(s) according to the SCID (including abuse or addiction disorders), a BeckDepression Inventory (BDI) score >9, alcohol use >4 units per day (last month) or a 1st-degreerelative with psychiatric disorder(s). We allowed HC to have incidentally used illicit drugs unless

Chap
ter
Chapter 7
7
142
criteria for a DSM-IV disorder were met, but prohibited illicit drug use the month prior to scanning.Patients and HC received €50 and €40, respectively. No restrictions were made with respect tosmoking behavior.
Procedure & SPECT-imagingWe performed all scans 230 ±18 (SD) minutes after intravenous injection of approximately 100MBq [123I]β-CIT, when the radioligand was at equilibrium for SERT binding in brain areasexpressing high densities of SERTs.22 Radiosynthesis of [123I]β-CIT and image acquisition weredescribed earlier.25 We performed SPECT imaging using a 12-detector single slice brain-dedicatedscanner (Neurofocus 810, Strichmann Medical Equipment; Cleveland, OH) with a full-width at half-maximum resolution of 6.5 mm, throughout the 20 cm field-of-view (http://www.neurophysics.com).
Image AnalysisAfter attenuation correction and reconstruction in 3D mode (http://www.neurophysics.com),
we selected regions of interest (ROIs) for midbrain (MID), DIENC and cerebellum (CER) by usingvalidated templates (see Figure 2.3).25 One examiner (HGR), blinded for diagnosis positioned allROIs in two series. Intra-class correlation coefficients were ≥0.98 for all ROIs. If the two seriesdiffered by >5%, scans were re-evaluated by a second investigator (JB). In the analyses weaveraged the counts for the two series.
We assumed activity in CER to represent non-displaceable activity (non-specific binding andfree radioactivity).26 We calculated the binding potential (BP) as the rate of specific to non-displaceable (ND) binding (BPND = ) for MID and DIENC.27 BPND is proportional totransporter number under equilibrium conditions.
StatisticsGeneral linear models were used to analyze differences in BPND in MID and DIENC betweendepressed patients and HC using the following modeling strategy.
We first compared mean BPND in MDD patients versus HC in univariable (‘crude’) models, onlycontaining the main effect of diagnosis (categorical: MDD/HC). We then fitted multivariablemodels by adding variables to the model, which in the literature have been reported to influenceBPND. These variables included: gender (categorical: male/female), age (continuous), smoking(categorical: yes/no), season of scanning (categorical: “winter” October-March/ “summer” April-September).24 In addition, a number of specific two-way interactions were examined, againbecause they have been reported as significant in previous studies, which included:diagnosis*gender, diagnosis*smoking, gender*smoking (‘full multivariable models’). Three-wayinteractions were not examined because of the relatively small sample-size (from a statisticalperspective). The Akaike’s Information Criterion (AIC) was used to judge whether the two-wayinteractions improved the model. If a two-way interaction did not improve the fit of the model, itwas removed from the model in order to facilitate the interpretation of the model (‘reducedmultivariable models’). Main effects always remained in the model, irrespective of theirsignificance, in order to report their (lack of) impact. Diencephalon and midbrain data wereanalyzed separately, but the same set of variables and interaction terms were examined using thesame modeling approach. If significant interactions were present, we performed post-hocanalyses in order to report the absolute differences in SERT availability in the involved subgroups.Differences between subgroups were analyzed within the framework of the multivariable modeland tested for significance using the residual variance estimate of the model. For explorativeanalyses see the supplementary appendix.
We examined the association of BPND with HDRS scores using linear regression models inpatients, correcting for covariates used in the multivariable models. We used SPSS (version 15.0.1)for statistical procedures (www.spss.com). We expressed all means ±SD, except in Figures 7.1 andS7.1, where ±SEM was used for legibility.
( )CER
CERROI
ActivityActivityActivity −

Serotonin transporter availability in MDD
143
Chapter7
Results
We studied 17 male and 32 female patients with MDD, versus 17 male and 32 female HC (Table 7.1).Patients smoked significantly more (n= 27; 55.1%) than HC (n= 11; 22.4%; χ2= 11.2; df= 2; p= 0.004).Among HC we included more Caucasians (n= 44; 89.8%) than among patients (n= 31; 63.3%; χ2=11.9; df= 3; p= 0.008). We scanned 67% of patients and 55% of HC in winter (χ2= 1.55, df= 1, p= 0.213).In one female patient insufficient CER was scanned as a reference, and in three patients and onecontrol MID slices were insufficient; these were omitted in the analyses. Of 15 patients who usedantidepressants during their lives, 3 used antidepressants for the current episode. One patientused mirtazapine until 4 weeks before scanning, all others stopped antidepressants 6-132 monthsbefore scanning. Illicit drug abuse mainly involved cannabis. Lifetime MDMA-use occurred in noneof the patients and in one female control (<10 tablets; last use 8 months before scanning).
SERT availability in MDD patients versus healthy controls (‘crude models’)Mean MID BPND in patients (0.62 ±0.22 [SD]) was not significantly different from HC (0.63 ±0.19;F1,94= 0.118; p= 0.733). BPND in DIENC was 1.15 (±0.24) in patients and 1.09 (±0.26) in HC (F1,97=1.209; p= 0.274).
Multivariable models of SERT availability in MDD patients versus healthycontrolsMidbrainFor MID, the full multivariable model (including diagnosis, gender, age, smoking, season ofscanning, diagnosis*gender, diagnosis*smoking, and gender*smoking) was subsequentlyreduced by removing the non-significant interaction terms diagnosis*smoking, and
Table 7.1. Baseline characteristics of MDD patients and healthy controls (stratified by gender).
MDD patients Healthy controls (HC)
Male (n= 17) Female (n= 32) Male (n= 17) Female (n= 32)
Age (years) 43.2 ±7.8 41.8 ±8.6 43.0 ±8.1 41.9 ±8.9
Current cigarette smokers; n (%) *
Alcohol use >8 Units/ week; n (%) †12 (70.6) 3 (17.6)
15 (46.9) 4 (12.5)
3 (17.6) 7 (41.2)
8 (25.0) 8 (25.0)
Race: n (%) Caucasian Creole Asian
12 (70.6) 2 (11.8) 2 (11.8)
19 (59.4) 7 (21.9) 5 (15.6)
15 (88.2) 2 (11.8) 0
29 (90.6) 3 (9.4) 0
MDD Severity‡
First episode; n (%) Drug-naïve; n (%) Melancholic; n (%) Atypical; n (%) Suicidal thoughts, plan or attempt; n (%) Duration of episode: n (%) <5 months 5 months – 2 years > 2 years Age of first episode (years)
23.2 ±4.9 9 (52.9) 12 (70.6) 12 (70.6) 0 7 (41.2)
5 (29.4) 8 (47.1) 4 (23.5) 35.9 ±10.3
25.4 ±4.7 18 (56.3) 22 (68.8) 24 (75.0) 3 (9.4) 10 (31.3)
8 (25.0) 21 (65.6) 3 (9.4) 35.8 ±10.4
1.3 ±1.4 N/A
2.0 ±2.1 N/A
Co-morbidity: n (%) Anxiety disorder Dysthymia Alcohol dependence Drug (alcohol, cannabis, benzodiazepines) abuse
4 (23.5) 1 (5.9) 1 (5.9) 4 (23.5)
7 (21.9) 1 (3.1) 3 (9.4)3 (9.4)
N/A N/A
SPECT scan in winter; n (%) 9 (52.9) 24 (75.0%) 7 (41.2) 20 (62.5)
Numbers represent means ± SD* Significantly more smoking in MDD patients than in HC (χ2 = 11.0; 1 df; p< 0.01) † Trend of lower alcohol use in MDD patients than in HC (χ2 = 3.75; 1 df; p= 0.053)‡ Severity measured by Hamilton Depression Rating Scale (17-items) in MDD patients and by Beck Depression Inventory in HC

Chap
ter
Chapter 7
7
144
gender*smoking (AIC-decrease= 2.478; Figure 7.1A). This reduced multivariable model had asignificant diagnosis*gender interaction (F1,87= 4.039; p= 0.048). In post-hoc comparisons, MDDmales showed a trend of lower BPND compared with male HC (difference -0.124; t87= -1.718; p=0.089), while female patients and HC did not differ (difference 0.047; t87= 0.898; p= 0.372).Furthermore, the main effect of season of scanning was significant (F1,87= 5.814; p= 0.018). Scansperformed in winter showed on average 18% higher BPND than scans made in summer (F2,87=3.248; p= 0.044). The main effects of age and smoking were also included in this reduced model,but were not significant (p= 0.227 and p= 0.582, respectively; Figure 7.1A).
Diencephalon For DIENC, the full multivariable model (including the same variables as MID) could not bereduced as all three interactions terms (diagnosis*smoking, diagnosis*gender andsmoking*gender) improved the fit of the model as evaluated by the AIC (Figure 2B). This fullmultivariable model showed significant diagnosis*gender (F1,88= 10.227; p= 0.002) andsmoking*gender (F1,88= 4.541; p= 0.036) interactions. The diagnosis*smoking interaction was notsignificant (p= 0.127). In post-hoc comparisons, smoking MDD males had significant lower BPNDcompared with smoking male HC (difference -0.304; t88= -2.643; p= 0.010). In non-smoking MDDmales BPND was numerically lower than in non-smoking male HC (difference -0.140; t88= -1.340; p=0.184). Contrary, non-smoking female patients had higher BPND than non-smoking female HC(difference 0.221; t88= 3.064; p= 0.003) with almost no difference in BPND between MDD and HC insmoking females (difference 0.057; t88= 0.616; p= 0.539). Furthermore, male smoking HC hadsignificantly higher BPND than non-smoking male HC (difference 0.279; t88= -2.384; p= 0.019),while in female HC BPND was not affected by smoking (difference 0.032; t88= -0.371; p= 0.712). Themain effects of season of scanning and age were also included in this model, but were notsignificant (p= 0.679 and p= 0.247, respectively; Figure 7.1B).
Relation of SERT availability and severity of MDD With linear regression models, we found no significant relation between HDRS-scores and BPND,neither in MID, nor in DIENC when accounting for gender, age, smoking and season of scanning.
Figure 7.1. BPND values for midbrain and diencephalon.
Multivariable models for BPND in MDD patients (MDD) versus HC stratified for gender. Values represent estimated means±SEM. A. Midbrain (n= 94), corrected for main effects of diagnosis (p= 0.414), gender (p= 0.128), season of scanning (F1,87= 5.814; p=0.018), smoking (p= 0.582), age (p= 0.227) and diagnosis*gender interaction (F1,87= 4.039;p= 0.048). * Post-hoc differences t87= -1.718;p= 0.089 between: MDD males versus HC males. B. Diencephalon (n= 97), corrected for main effects of diagnosis (p= 0.476), gender (p= 0.277), season of scanning (p= 0.679),smoking (p= 0.223), age (p= 0.247) and diagnosis*gender interaction (F1,88= 10.227; p= 0.002), gender*smoking interaction(F1,88= 4.541; p= 0.036) and diagnosis*smoking interaction (p= 0.127). * Post-hoc difference t88= -2.384; p= 0.019 between smoking HC males versus non-smoking HC males. ** Post-hoc differencest88> 2.643; p< 0.01 between: smoking MDD males versus smoking HC males, non-smoking MDD females versus non-smokingHC females.
��� �� ��� �����
���
���
���
���
��� ���� ����� �
�
�
����� ������ � ���������� ������
� ���
���
�������
��
�� �� �� �����
���
���
��
���
���
����� ���� �
�� ���
����� ������ � ���������� ������
� ���
���
�������
��
� �

Serotonin transporter availability in MDD
145
Chapter7
Discussion
In the present – until now largest – study of MDD patients versus HC, we aimed to quantify SERTavailability in MDD patients versus HC while accounting for covariates and interactions, and tocorrelate SERT availability with depression severity. We did not find significant differences in SERTavailability in MID or DIENC in crude comparisons. However, a significant diagnosis*genderinteraction existed in MID and DIENC, combined with a significant gender*smoking interaction inDIENC only. Depressed males, but not females, had lower MID SERT availability compared withHC. In DIENC depressed smoking males had significantly lower MID SERT availability comparedwith smoking male HC, while non-smoking female patients had higher SERT availability thanfemale HC. Furthermore, the season of scanning influenced SERT availability in MID, with higherSERT availability in winter. We found no clinically relevant correlation of HDRS-scores with SERTavailability.
Comparison with previous studiesOur results replicate earlier reports of similar SERT availability in MID and DIENC in MDD patientsand HC.14-18 Other studies reported increased19 or decreased7-13 SERT availability in MDD patientsversus HC. However, none of these studies – except two11;13 – investigated the effect of genderand no study corrected for season. Furthermore, we replicated a significant contribution ofseason of scanning on MID BPND.24
The diagnosis*gender interaction in MID and DIENC is our most important finding. Contrary toStaley and colleagues,11;21 we found a different direction of this interaction in DIENC: significantlylower BPND in MDD males in MID (-17%) and DIENC (-18%), and higher BPND in MDD females in MID(+9%, non-significant) and DIENC (+13%, significant) than in HC. Staley et al. found 1% lower DIENCSERT availability in MDD males, and 22% lower DIENC SERT availability in MDD females. We found agender*smoking interaction in DIENC (with highest BPND in smoking healthy males), while Staleyfound higher SERT availability in the brainstem (attributable to males). Our findings suggest that afailure to stratify for a diagnosis*gender interaction may obscure differences between patientsand HC.
Methodological explanations for inconsistent findingsDespite technical differences between studies (scanning protocols, radioligands, image analyses),variation in the selection of HC (e.g. having relatives with psychiatric diagnoses) or patients (fromdifferent source populations) is the most probable explanation for inconsistent findings. Previousstudies recruited patients in general psychiatric outpatient and university clinics.7;8;10;12;18;19;28 Werecruited 65% of our patients from primary care settings. We adequately diagnosed patients bySCID, and required an HDRS >18 for inclusion. Thus, we recruited severe and often melancholicpatients that were drug-free, with 69% of the patients drug-naïve. Three studies7;13;29 includedlarger proportions of drug-naïve patients. Like Parsey et al.,10 we observed (non-significant -15%)lower MID SERT availability in drug-naïve patients (results available on request). Additionally,some studies suggested that anxiety disorders influence SERT availability,30 and MDD with co-morbid anxiety may differ from ‘pure MDD’. However, this was not observed in our sample(results available on request).
Role of SERT in the pathogenesis of MDDSERTs evacuate extracellular 5-HT from the synapse. Observed differences in SERTs betweenpatients and HC may represent differences in the number of SERT containing neurons, in thenumber of SERTs per neuron or a combination of both.
Two major mechanisms for the role of SERT in MDD are hypothesized.30 First, increased SERTavailability reduces 5-HT from the synapse more easily, which might lower 5-HT transmission,possibly leading to MDD. Second, as the brain might apply compensation mechanisms to retainhomeostasis, a decreased 5-HT transmission by MDD may result in downregulation (decrease) of

Chap
ter
Chapter 7
7
146
SERT in order to increase 5-HT transmission. A sequential occurrence of these two mechanismscould also be hypothesized: an initially increased SERT availability destabilizes (with or without anadditional factor) and leads to MDD, which is followed by a decrease in SERT to compensate fordecreased 5-HT transmission.
Differential effects of MDD on SERT availability between sexes may be explained via sex-hormones. Estrogen replacement after ovariectomy increased SERT mRNA and SERT availabilityin female rats31 and in hypothalamic regions of female macaques.32 Depressed women may havesignificantly higher 24h mean levels of diurnal estradiol rhythms, and may have highertestosterone levels compared with HC.33 Testosterone may increase SERT availability byconversion to estrogen by aromatase, which is especially available in DIENC. This could explain ourfinding of increased SERT availability in DIENC in females. In depressed men, the sex steroidtestosterone is decreased,33 with 34-61% biochemical hypogonadism in depressed malescompared with 6-14% in HC.34 This lack of testosterone in MDD may reduce SERT availability byreduced conversion to estrogen. Replacement of testosterone in castrated male rats increasedSERT mRNA and SERT availability.35 Since we did not measure sex hormones, and Best et al.36
found no relation between menstrual cycle or sex hormones and SERT availability in HC, theseexplanations remain speculative and should be examined further.
We replicated a main effect of season demonstrated previously in DIENC in 12 healthywomen37 and mesencephalon in 29 HC.24 Neumeister et al. observed decreased SERT availability inwinter.37 In contrast, Buchert et al.24 found increased SERT availability in winter, which was alsofound in our study. Serotonin modulates the effects of photic input in the suprachiasmaticnucleus (SCN). The SCN imposes a circadian rhythm by affecting hormonal and autonomic output(reviewed by Buijs).38 Serotonin release in the SCN is highest during waking and activity.39 Becauseraphe neurons show regular high firing rates during waking and decreased firing during sleep, itcould be hypothesized that during winter, with decreased daylight, more serotonergic activity isneeded, which may be mediated via the raphe input into the SCN.40 Increased serotonergicactivity (increased free synaptic serotonin) may result in a compensatory increase in SERTs.Nevertheless, the small size of the SCN (~0.27 mm3) by itself cannot explain higher SERTavailability found by SPECT or PET.
Limitations of the present studyThe cerebellum (especially the vermis) contains small amounts of SERT,41;42 which could result inan underestimation of BPND in patients and HC, expected to be 7% at most.43 Although asystematic underestimation of SERT due to DAT- or NET-rich areas in MID (substantia nigra andlocus coeruleus, respectively) cannot be ruled out, we think this does not differentially affectpatients or HC.
Second, lower levels of endogenous 5-HT (e.g. in MDD) could result in less competition withradioligands, increasing the specific binding measured. This was demonstrated in rhesus monkeyswith [123I]β-CIT SPECT44 but not in humans. After tryptophan depletion (artificially reducingendogenous 5-HT) no differences in SERT availability were observed45;46 but the radioligand([11C]DASB) in that study does not bind to the 5-HT recognition/translocation site, and may not besuitable to image such changes in extracellular 5-HT.47
Third, we used previously validated ROIs25 instead of Magnetic Resonance Imaging forcoregistration. Because these templates cover larger brain areas, BPND in small regions (raphenuclei) cannot be determined. This potential measurement error (non-differential for patients andHC), might have increased variance in our measurements, despite very good intra-ratercorrelation coefficients. Additionally, we did not correct for non-uniform photon attenuation andpartial volume effects in our gender-analyses. Greater skull thickness might underestimate BPNDin males, and smaller MID and DIENC in females might suppress BPND compared with males.However, these factors are unlikely to explain the observed interactions.

Serotonin transporter availability in MDD
147
Chapter7
Fourth, the 4-week washout of antidepressants (binding to SERT) may be too short.48 Becauseall but one patient stopped antidepressants ≥6 months before scanning, we think no substantialbias of the BPND assessment was introduced by competitive binding by traces of previousantidepressants.
Fifth, we allowed previous incidental use of illicit drugs (marijuana/cannabis n= 10, or MDMAn= 1) in our HC. Because heavy use of MDMA (>50 MDMA tablets) can damage serotoninneurons,49 we performed an additional analysis in which we excluded data from the MDMA-userin the HC group. However, this exclusion did not affect our results (results available on request).Sixth, we did not check personal or family history of psychiatric illnesses in HC, nor did we test foralcohol or drug abuse.
ConclusionWe showed lower SERT availability in MID and DIENC in depressed males and higher SERTavailability in DIENC in depressed (non-smoking) females compared with HC. We replicated aseasonal influence on MID SERT availability, and found a gender*smoking interaction on DIENCSERT availability. This study points to complex effects of gender, smoking and season on theserotonergic system in the pathogenesis of MDD.
Acknowledgements
We wish to thank patients and healthy controls for their participation in the study. We thankgeneral practitioners and psychiatric residents for appropriate referrals of patients. Staffmembers and technicians of the department of Nuclear Medicine provided indispensable help inmaking the SPECT-scans. We would like to acknowledge Dr. Maartje M. de Win, MD PhD for herhelp to recruit HC. Dr. Jules Lavalaye, MD PhD helped in the initial design of this study. Mrs.Michelle L. Miller revised the text linguistically. Dr. Ruud M. Buijs, PhD commented constructivelyon an earlier version of the manuscript. Henricus G. Ruhé received a grant from the NetherlandsOrganisation for Health Research and Development (ZonMw), program Mental Health, educationof investigators in mental health (OOG; #100-002-002).
Conflicts of interest
None
References
1. Kennedy SH, Lam RW, Cohen NL, Ravindran AV, CANMAT Depression Work Group. Clinical guidelines for the treatment ofdepressive disorders. IV. Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.
2. Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans:a meta-analysis of monoamine depletion studies. Mol Psychiatry. 2007; 12: 331-359.
3. Owens MJ. Selectivity of antidepressants: from the monoamine hypothesis of depression to the SSRI revolution andbeyond. J Clin Psychiatry. 2004; 65 Suppl 4: 5-10.
4. Stockmeier CA. Involvement of serotonin in depression: evidence from postmortem and imaging studies of serotoninreceptors and the serotonin transporter. J Psychiatr Res. 2003; 37: 357-373.
5. Innis R, Baldwin R, Sybirska E, Zea Y, Laruelle M, al Tikriti M et al. Single photon emission computed tomography imagingof monoamine reuptake sites in primate brain with [123I]CIT. Eur J Pharmacol. 1991; 200: 369-370.
6. Laruelle M, Baldwin RM, Malison RT, Zea-Ponce Y, Zoghbi SS, al Tikriti MS et al. SPECT imaging of dopamine andserotonin transporters with [123I]β-CIT: pharmacological characterization of brain uptake in nonhuman primates.Synapse. 1993; 13: 295-309.

Chap
ter
Chapter 7
7
148
7. Lehto S, Tolmunen T, Joensuu M, Saarinen PI, Vanninen R, Ahola P et al. Midbrain binding of [123I]nor-β-CIT in atypicaldepression. Prog Neuropsychopharmacol Biol Psychiatry. 2006; 30: 1251-1255.
8. Malison RT, Price LH, Berman R, van Dyck CH, Pelton GH, Carpenter L et al. Reduced brain serotonin transporteravailability in major depression as measured by [123I]-2β-carbomethoxy-3β-(4-iodophenyl)tropane and single photonemission computed tomography. Biol Psychiatry. 1998; 44: 1090-1098.
9. Newberg AB, Amsterdam JD, Wintering N, Ploessl K, Swanson RL, Shults J et al. [123I]-ADAM binding to serotonintransporters in patients with major depression and healthy controls: a preliminary study. J Nucl Med. 2005; 46: 973-977.
10. Parsey RV, Hastings RS, Oquendo MA, Huang YY, Simpson N, Arcement J et al. Lower serotonin transporter bindingpotential in the human brain during major depressive episodes. Am J Psychiatry. 2006; 163: 52-58.
11. Staley JK, Sanacora G, Tamagnan G, Maciejewski PK, Malison RT, Berman RM et al. Sex differences in diencephalonserotonin transporter availability in major depression. Biol Psychiatry. 2006; 59: 40-47.
12. Willeit M, Praschak-Rieder N, Neumeister A, Pirker W, Asenbaum S, Vitouch O et al. [123I]β-CIT SPECT imaging showsreduced brain serotonin transporter availability in drug-free depressed patients with seasonal affective disorder. BiolPsychiatry. 2000; 47: 482-489.
13. Joensuu M, Tolmunen T, Saarinen PI, Tiihonen J, Kuikka J, Ahola P et al. Reduced midbrain serotonin transporteravailability in drug-naive patients with depression measured by SERT-specific [123I] nor-β-CIT SPECT imaging. PsychiatryRes. 2007; 154: 125-131.
14. Ahonen A, Heikman P, Kauppinen T, Koskela A, Bergström K. Serotonin transporter availability in drug free depressionpatients using a novel SERT ligand [Abstract #106]. Eur J Nucl Med. 2004; 31 (suppl 1): S227.
15. Catafau AM, Perez V, Plaza P, Pascual JC, Bullich S, Suarez M et al. Serotonin transporter occupancy induced byparoxetine in patients with major depression disorder: a [123I]-ADAM SPECT study. Psychopharmacology (Berl). 2006; 189:145-153.
16. Herold N, Uebelhack K, Franke L, Amthauer H, Luedemann L, Bruhn H et al. Imaging of serotonin transporters and itsblockade by citalopram in patients with major depression using a novel SPECT ligand [123I]-ADAM. J Neural Transm. 2006;113: 659-670.
17. Meyer JH, Houle S, Sagrati S, Carella A, Hussey DF, Ginovart N et al. Brain serotonin transporter binding potentialmeasured with carbon 11-labeled DASB positron emission tomography: effects of major depressive episodes and severityof dysfunctional attitudes. Arch Gen Psychiatry. 2004; 61: 1271-1279.
18. Reivich M, Amsterdam JD, Brunswick DJ, Shiue CY. PET brain imaging with [11C](+)McN5652 shows increased serotonintransporter availability in major depression. J Affect Disord. 2004; 82: 321-327.
19. Ichimiya T, Suhara T, Sudo Y, Okubo Y, Nakayama K, Nankai M et al. Serotonin transporter binding in patients with mooddisorders: a PET study with [11C]McN5652. Biol Psychiatry. 2002; 51: 715-722.
20. Eggers B, Hermann W, Barthel H, Sabri O, Wagner A, Hesse S. The degree of depression in Hamilton rating scale iscorrelated with the density of presynaptic serotonin transporters in 23 patients with Wilson's disease. J Neurol. 2003; 250:576-580.
21. Staley JK, Krishnan-Sarin S, Zoghbi S, Tamagnan G, Fujita M, Seibyl JP et al. Sex differences in [123I]β-CIT SPECT measuresof dopamine and serotonin transporter availability in healthy smokers and nonsmokers. Synapse. 2001; 41: 275-284.
22. Pirker W, Asenbaum S, Hauk M, Kandlhofer S, Tauscher J, Willeit M et al. Imaging serotonin and dopamine transporterswith [123I]β-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000; 41: 36-44.
23. van Dyck CH, Malison RT, Seibyl JP, Laruelle M, Klumpp H, Zoghbi SS et al. Age-related decline in central serotonintransporter availability with [123I]β-CIT SPECT. Neurobiol Aging. 2000; 21: 497-501.
24. Buchert R, Schulze O, Wilke F, Berding G, Thomasius R, Petersen K et al. Is correction for age necessary in SPECT or PET ofthe central serotonin transporter in young, healthy adults? J Nucl Med. 2006; 47: 38-42.
25. de Win MM, Habraken JB, Reneman L, Brink W vd, den Heeten GJ, Booij J. Validation of [123Iβ-CIT SPECT to assessserotonin transporters in vivo in humans: a double-blind, placebo-controlled, crossover study with the selective serotoninreuptake inhibitor citalopram. Neuropsychopharmacology. 2005; 30: 996-1005.
26. Laruelle M, Vanisberg MA, Maloteaux JM. Regional and subcellular localization in human brain of [3H]paroxetine binding,a marker of serotonin uptake sites. Biol Psychiatry. 1988; 24: 299-309.
27. Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN et al. Consensus nomenclature for in vivo imaging ofreversibly binding radioligands. J Cereb Blood Flow Metab. 2007; 27: 1533-1539.
28. Dahlstrom M, Ahonen A, Ebeling H, Torniainen P, Heikkila J, Moilanen I. Elevated hypothalamic/midbrain serotonin(monoamine) transporter availability in depressive drug-naive children and adolescents. Mol Psychiatry. 2000; 5: 514-522.
29. Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K et al. Occupancy of serotonin transporters by paroxetineand citalopram during treatment of depression: a [11C]DASB PET imaging study. Am J Psychiatry. 2001; 158: 1843-1849.
30. Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. JPsychiatry Neurosci. 2007; 32: 86-102.
31. Fink G, Sumner BE, McQueen JK, Wilson H, Rosie R. Sex steroid control of mood, mental state and memory. Clin ExpPharmacol Physiol. 1998; 25: 764-775.
32. Lu NZ, Eshleman AJ, Janowsky A, Bethea CL. Ovarian steroid regulation of serotonin reuptake transporter (SERT)binding, distribution, and function in female macaques. Mol Psychiatry. 2003; 8: 353-360.
33. Swaab DF, Bao AM, Lucassen PJ. The stress system in the human brain in depression and neurodegeneration. Ageing ResRev. 2005; 4: 141-194.
34. McIntyre RS, Mancini D, Eisfeld BS, Soczynska JK, Grupp L, Konarski JZ et al. Calculated bioavailable testosterone levelsand depression in middle-aged men. Psychoneuroendocrinology. 2006; 31: 1029-1035.
35. Fink G, Sumner B, Rosie R, Wilson H, McQueen J. Androgen actions on central serotonin neurotransmission: relevance formood, mental state and memory. Behav Brain Res. 1999; 105: 53-68.

Serotonin transporter availability in MDD
149
Chapter7
36. Best SE, Sarrel PM, Malison RT, Laruelle M, Zoghbi SS, Baldwin RM et al. Striatal dopamine transporter availability with[123I]β-CIT SPECT is unrelated to gender or menstrual cycle. Psychopharmacology (Berl). 2005; 183: 181-189.
37. Neumeister A, Pirker W, Willeit M, Praschak-Rieder N, Asenbaum S, Brucke T et al. Seasonal variation of availability ofserotonin transporter binding sites in healthy female subjects as measured by [123I]-2β-carbomethoxy-3β-(4-iodophenyl)tropane and single photon emission computed tomography. Biol Psychiatry. 2000; 47: 158-160.
38. Buijs RM, Scheer FA, Kreier F, Yi C, Bos N, Goncharuk VD et al. Organization of circadian functions: interaction with thebody. Prog Brain Res. 2006; 153: 341-360.
39. Pickard GE, Weber ET, Scott PA, Riberdy AF, Rea MA. 5HT1B receptor agonists inhibit light-induced phase shifts ofbehavioral circadian rhythms and expression of the immediate-early gene c-fos in the suprachiasmatic nucleus. J Neurosci.1996; 16: 8208-8220.
40. Moore RY, Speh JC. Serotonin innervation of the primate suprachiasmatic nucleus. Brain Res. 2004; 1010: 169-173.41. Kent JM, Coplan JD, Lombardo I, Hwang DR, Huang Y, Mawlawi O et al. Occupancy of brain serotonin transporters during
treatment with paroxetine in patients with social phobia: a positron emission tomography study with [11C]McN 5652.Psychopharmacology (Berl). 2002; 164: 341-348.
42. Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB et al. Acute occupancy of brain serotonintransporter by sertraline as measured by [11C]DASB and positron emission tomography. Biol Psychiatry. 2006; 59: 821-828.
43. Kish SJ, Furukawa Y, Chang LJ, Tong J, Ginovart N, Wilson A et al. Regional distribution of serotonin transporter protein inpostmortem human brain: is the cerebellum a SERT-free brain region? Nucl Med Biol. 2005; 32: 123-128.
44. Heinz A, Jones DW, Zajicek K, Gorey JG, Juckel G, Higley JD et al. Depletion and restoration of endogenous monoaminesaffects β-CIT binding to serotonin but not dopamine transporters in non-human primates. J Neural Transm Suppl. 2004;29-38.
45. Praschak-Rieder N, Wilson AA, Hussey D, Carella A, Wei C, Ginovart N et al. Effects of tryptophan depletion on theserotonin transporter in healthy humans. Biol Psychiatry. 2005; 58: 825-830.
46. Talbot PS, Frankle WG, Hwang DR, Huang Y, Suckow RF, Slifstein M et al. Effects of reduced endogenous 5-HT on the invivo binding of the serotonin transporter radioligand [11C]DASB in healthy humans. Synapse. 2005; 55: 164-175.
47. Hummerich R, Reischl G, Ehrlichmann W, Machulla HJ, Heinz A, Schloss P. DASB -in vitro binding characteristics on humanrecombinant monoamine transporters with regard to its potential as positron emission tomography (PET) tracer. JNeurochem. 2004; 90: 1218-1226.
48. Strauss WL, Layton ME, Dager SR. Brain elimination half-life of fluvoxamine measured by 19F magnetic resonancespectroscopy. Am J Psychiatry. 1998; 155: 380-384.
49. Reneman L, Booij J, de Bruin K, Reitsma JB, de Wolff FA, Gunning WB et al. Effects of dose, sex, and long-term abstentionfrom use on toxic effects of MDMA (ecstasy) on brain serotonin neurons. Lancet. 2001; 358: 1864-1869.
50. Heinz A, Ragan P, Jones DW, Hommer D, Williams W, Knable MB et al. Reduced central serotonin transporters inalcoholism. Am J Psychiatry. 1998; 155: 1544-1549.
51. Kugaya A, Sanacora G, Staley JK, Malison RT, Bozkurt A, Khan S et al. Brain serotonin transporter availability predictstreatment response to selective serotonin reuptake inhibitors. Biol Psychiatry. 2004; 56: 497-502.
52. Meyer JH, Wilson AA, Sagrati S, Hussey D, Carella A, Potter WZ et al. Serotonin Transporter Occupancy of Five SelectiveSerotonin Reuptake Inhibitors at Different Doses: An [11C]DASB Positron Emission Tomography Study. Am J Psychiatry.2004; 161: 826-835.

Chap
ter
Chapter 7
7
150
Supplementary appendix: explorative analyses
METHODSIn addition to the analyses described in the main text, in exploratory analyses we examinedwhether potentially relevant other factors (alcohol use,50 lifetime antidepressant use10 andduration of depression episode) had an impact on BPND by adding them as main effects to thefinal multivariable model. Because our exploratory models used small subgroups for assessmentof further confounding in multivariable models, we only present these analyses as hypothesisgenerating, exploratory models.
RESULTSMidbrainFor MID differentiation of patients who never took antidepressants (drug-naïve) versus thosewho did improved the reduced model (AIC-decrease= 1.399). Drug-naïve male and female patientshad non-significantly lower BPND than patients with a history of antidepressant use (data notshown).
Differentiation of the duration of the current episode, stratified as ≤2 years or >2 years(‘chronic’ depression) improved the reduced model (AIC-decrease= 7.289). Men depressed formore than 2 years had significantly higher BPND than men depressed ≤2 years (difference 0.333;t85= -3.102; p= 0.003), men depressed ≤2 years had significantly lower BPND than HC (difference -0.204; t85= -2.795; p= 0.006; Figure S7.1A). Differences between females were not significant.
Diencephalon Differentiation between patients with or without a secondary diagnosis of alcohol abuse/dependence improved the model for DIENC (AIC-decrease= 0.602). Smoking, alcohol-abusingmale patients had numerically higher BPND than smoking male patients not abusing alcohol(difference 0.717; t85= -1.175; p= 0.064). Non-smoking, female, alcohol-abusing patients hadsignificantly lower BPND than non-smoking, female patients not abusing alcohol (difference -0.388; t85= 2.182; p= 0.032; Figure S7.1B).
Figure S7.1. BPND values for midbrain and diencephalon (exploratory models)
Explorative models for BPND in MDD patients (MDD) versus HC stratified for gender. Values represent estimated means±SEM. Alc = Alcohol abuse/dependence.A. Midbrain (n= 94), corrected for main effects of duration of episode (F2,85= 4.777;p= 0.011), gender (p= 0.294), age (p=0.380), smoking (p= 0.636), season of scanning (F1,85= 6.194;p= 0.015), and duration of episode*gender interaction (F2,85=3.953;p= 0.023). ** Post-hoc differences for MDD males between: episodes ≤2 years versus >2 years (t85= 3.102; p= 0.003), episodes ≤2 yearsversus HC (t85= 2.795; p= 0.006). B. Diencephalon (n= 97), corrected for main effects of alcohol abuse/dependence (p= 0.521), gender (p= 0.489), season ofscanning (p= 0.511), age (p= 0.486), smoking (p= 0.067) and alcohol abuse/dependence*gender interaction (F2,85= 7.236;p=0.001), gender*smoking interaction (F1,85= 7.112;p= 0.009) and alcohol abuse/dependence*smoking interaction (p= 0.099). * Post-hoc differences t85= 2.182; p= 0.032 between non-smoking MDD females without, versus with alcohol abuse/dependence.
�� �
��"�
��
����
" � ��"�
��
����
" �� �
� �
� �
� �
� �
�
� �
� � ���� ����� �
��
��
��
�������#$����%#� ������&����#$����%#
�#%�&
�%��
�&��
'���
�
�� ���
�������� �
��������
�������� �
��
�
��
���
���
���
��
��
��� ����� ���� �
�
�������#$����%#� ������&����#$����%#
�#%�&
�%��
�&��
'���
��

151
Serotonin transporter availability in MDD
Chapter7
Tabl
e S7
.1. P
revi
ous
stud
ies
mea
surin
g SE
RT a
vaila
bilit
y in
MD
D p
atie
nts
vers
us h
ealth
y co
ntro
ls.
Aut
hor &
Dat
e (r
efer
ence
)Po
pula
tion
(mea
n ag
e)Se
xN
SER
T Im
agin
g an
d R
OI*
Res
ults
Rem
arks
Aho
nen
2004
14D
rug
free
M
DD
pts
(34
±?)
Cont
rols
(35
±?)
M +
F ??
10 14
[123 I]
-AD
AM
SPE
CT, 1
0min
, 5h
, 7h
Mid
Br, T
hal,
Caud
, Put
am,
Pons
, Cer
Non
sign
. Diff
eren
ces
of V
3’’ i
n M
idBr
(7%
high
er in
pat
ient
s);
larg
e in
terin
divi
dual
var
iatio
n in
V3’
’ in
Mid
Br. O
ther
RO
Is n
o si
gn. d
iffer
ence
s.
Abs
trac
t of p
relim
inar
y re
sults
onl
y; li
ttle
info
rmat
ion
prov
ided
. G
ende
r not
repo
rted
. App
aren
tly n
o m
atch
ing
of c
ontr
ols
and
patie
nts.
Rec
ruitm
ent o
f pat
ient
s un
spec
ified
.
Cata
fau
2006
15D
rug-
free
M
DD
pts
(36
±11)
Cont
rols
(36
±11)
M +
F
10 10
[123 I]
-AD
AM
SPE
CT, 4
hM
idBr
, Tha
l, St
riatu
m, C
erSl
ight
ly lo
wer
but
non
sign
. diff
eren
ces
in S
ERT
avai
labi
lity
in
Mid
Br (-
4%),
Tha
l (-11
%) in
MD
D p
atie
nts
vs c
ontr
ols.
Patie
nts
wer
e dr
ug-fr
ee fo
r >6m
.A
ge m
atch
ed h
isto
ric c
ontr
ol g
roup
, no
mat
chin
g fo
r sex
. U
nequ
al m
ale-
fem
ale
dist
ribut
ion
in M
DD
vs.
con
trol
s. N
o in
form
atio
n on
fam
ily h
isto
ry o
f con
trol
s. R
ecru
itmen
t of
patie
nts
unsp
ecif
ied.
Dah
lstr
öm
2000
28D
rug-
naiv
e M
DD
pts
(13.
5 ±2
.5)
Dru
g-na
ive
Non
-MD
D p
ts (1
2.2
±2.9
)M
+ F
31 10
[123 I]β-
CIT
SPEC
T, 1,
4, 2
4h
Stria
tum
(24h
), PF
C, T
hal
(4h)
, Mid
Br (1
h), O
cc
Mid
Br V
3’’ i
n pa
tient
s si
gn. h
ighe
r (8%
) at 1
hr c
ompa
red
to
cont
rols
(diff
eren
ce n
ot s
ign.
at 4
h, a
gain
sig
n. a
t 24h
).
Pref
ront
al a
nd T
hal d
iffer
ence
s no
t sig
n. d
iffer
ent
Cont
rol-g
roup
is d
ue to
eth
ical
con
side
ratio
ns n
ot d
epre
ssed
bu
t a p
sych
iatr
ical
ly a
ffec
ted
grou
p of
ado
lesc
ents
. MD
D
patie
nts
and
cont
rols
are
not
mat
ched
for a
ge a
nd s
ex. N
o in
form
atio
n on
fam
ily h
isto
ry o
f con
trol
s. N
o ef
fect
of a
ge,
gend
er o
r co-
mor
bidi
ty fo
und.
Rec
ruitm
ent o
f out
patie
nts
in
univ
ersi
ty c
linic
.
Her
old
2006
16D
rug-
free
M
DD
pts
(42
±12)
Cont
rols
(36
±13)
M +
F
21 12
[123 I]
-AD
AM
SPE
CT, 4
hM
idBr
, Cer
Mid
Br V
3’’ i
n pa
tient
s 21
% hi
gher
(non
sign
.). M
DD
mal
es
nons
ign.
low
er V
3’’ t
han
MD
D fe
mal
es.
No
corr
elat
ion
with
SER
T an
d M
DD
sev
erity
ratin
g.
All
patie
nts
wer
e dr
ug-fr
ee fo
r >2m
. No
mat
chin
g fo
r age
or
sex.
Une
qual
mal
e-fe
mal
e di
strib
utio
n in
MD
D v
s. c
ontr
ols.
Stud
y al
so re
port
s on
occ
upan
cy a
fter
1 w
eek
of tr
eatm
ent
with
CIT
10 m
g (n
=13)
. Rec
ruitm
ent o
f pat
ient
s un
clea
r.
Ichi
miy
a 20
0219
Dru
g-fr
ee
MD
D p
ts (4
4.1 ±
13.5
) or
BD (4
1.7 ±
8.8)
Cont
rols
(42.
3 ±1
4.5)
M
13 21
[11C]
(+)M
cN56
52 P
ETM
idBr
, Tha
l, Ce
rSi
gn. h
ighe
r (22
%) in
Tha
l BP
in c
ompl
ete
patie
nt g
roup
vs
cont
rols
(in M
DD
onl
y (2
3%),
no
chan
ge in
Mid
Br6
patie
nts
had
BD; 1
0 pa
tient
s ha
d be
en tr
eate
d w
ith
antid
epre
ssan
ts ³6
w b
efor
e sc
anni
ng (1
pat
ient
with
BD
2w
). A
ge m
atch
ed (g
roup
wis
e??)
con
trol
s, n
ot fo
r sex
. Rec
ruitm
ent
of o
utpa
tient
s in
uni
vers
ity a
nd g
ener
al p
sych
iatr
ic c
linic
s.
Joen
suu
2007
13D
rug-
naïv
e M
DD
pts
(28.
8 ±8
.6)
Cont
rols
(30.
6 ±8
.9)
M +
F
29 19
[123 I]
nor β
-CIT
SPE
CT, 6
, 24
h M
idBr
, Cer
Sign
. low
er V
3’’ i
n pa
tient
s (1
0%) i
n M
idBr
vs
cont
rols
. N
o co
rrel
atio
n w
ith S
ERT
and
MD
D s
ever
ity ra
ting.
Alth
ough
men
tione
d as
suc
h, in
com
plet
e m
atch
ing
on g
ende
r an
d ag
e. 14
/29
pts
had
HD
RS ≤
18. R
ecru
itmen
t of o
utpa
tient
s fo
r psy
chod
ynam
ic p
sych
othe
rapy
in u
nive
rsity
clin
ic. N
o si
gn.
effe
cts
of g
ende
r (in
clud
ed p
atie
nts
mos
tly F
) or s
easo
n.
Leht
o 20
067
Dru
g-na
ïve
MD
D p
ts (2
8.3
±?)
Cont
rols
(30.
6 ±9
.2)
M +
F
29 18
[123 I]
nor-β
-CIT
SPE
CT, 5
m
in, 6
h, 2
4h
Mid
Br, S
tria
tum
, Cer
Sign
. low
er S
ERT
(-10
%) in
Mid
Br in
MD
D p
atie
nts v
s con
trol
s.
No
diff
eren
ces
betw
een
mel
anch
olic
, aty
pica
l, no
ndiff
eren
tiate
d M
DD
sub
type
s. L
inea
r inv
erse
cor
rela
tion
betw
een
SER
T an
d at
ypic
al s
core
. No
corr
elat
ion
betw
een
SERT
and
HD
RS.
Patie
nts
and
cont
rols
wer
e (g
roup
wis
e??)
mat
ched
for a
ge a
nd
sex.
No
subs
tant
ial r
atio
nale
giv
en fo
r rel
atio
n be
twee
n SE
RT
avai
labi
lity
and
atyp
ical
dim
ensi
on o
f MD
D. R
ecru
itmen
t of
outp
atie
nts
in u
nive
rsity
clin
ic.

152
Chapter 7
Chap
ter
7
Mal
ison
1998
8 ; Ku
gaya
200
451D
rug-
free
M
DD
pts
(44
±10)
Cont
rols
(45
±11)
M +
F
15 15
[123 I]β-
CIT
SPEC
T, 2
4h
Brai
nste
m, S
tria
tum
, Occ
Sign
. red
uctio
n (-1
9%) i
n br
ains
tem
V3’
’ of M
DD
vs.
con
trol
s,
but n
ot (-
11%)
in s
tria
tum
.N
o co
rrel
atio
n w
ith S
ERT
and
MD
D s
ever
ity ra
tings
Gro
up-m
atch
ing
of c
ontr
ols
for a
ge a
nd s
ex. N
ine
subj
ects
had
be
en tr
eate
d w
ith a
ntid
epre
ssan
ts ³3
w-7
y be
fore
sca
nnin
gO
ccip
ital c
orte
x w
as c
hose
n as
refe
renc
e.
Kuga
ya s
tudy
mai
nly
repo
rts
on o
ccup
ancy
of S
ERT
in T
hal a
nd
Mid
Br a
fter
2*
8 da
ys tr
eatm
ent w
ith C
IT 4
0 m
g ±
BUP
200
mg
or B
UP
200
mg
in c
ontr
ols
(n=
17) a
nd P
AR
20
mg
in M
DD
pa
tient
s (n
= 10
). Re
crui
tmen
t of o
utpa
tient
s in
uni
vers
ity c
linic
.
Mey
er 2
00129
Dru
g-fr
ee
MD
D p
ts (3
7 ±8
)Co
ntro
ls (?
±?)
M +
F
13 13
[11C]
DA
SP P
ETst
riatu
m, C
erN
o si
gn d
iffer
ence
in s
tria
tum
V3’
’ of M
DD
vs
cont
rols
. Sig
n.
effe
ct o
f age
.Ex
cept
2 (t
reat
ed >
2m b
efor
e) p
atie
nts
wer
e al
l dru
g-na
ïve;
in
divi
dual
age
mat
chin
g (±
2yea
rs),
no
sex-
mat
chin
g re
port
ed;
no s
epar
ate
mea
n ag
e pr
ovid
ed fo
r con
trol
s. S
tudy
mai
nly
repo
rts o
n oc
cupa
ncy
afte
r 4w
of t
reat
men
t with
PA
R 10
-20
mg
(n=
8) a
nd C
IT 2
0 m
g (n
=4).
Rec
ruitm
ent o
f pat
ient
s un
clea
r.
Mey
er 2
004a
52D
rug-
free
M
DD
pts
(? ±
?)Co
ntro
ls (?
±?)
M +
F
37 35
[11C]
DA
SP P
ETst
riatu
m, C
erN
o si
gn. d
iffer
ence
s in
str
iatu
m V
3’’ o
f MD
D v
s co
ntro
ls.
12 s
ubje
cts
(pat
ient
s/co
ntro
ls) w
ere
also
ana
lyze
d in
Mey
er e
t al
.29
Patie
nts
wer
e dr
ug-fr
ee fo
r >1m
. No
mat
chin
gfor
age
or
sex.
Rec
ruitm
ent o
f pat
ient
s un
spec
ifie
d.
Mey
er 2
004b
17
Dru
g fr
ee
MD
D p
ts (3
5 ±1
1)Co
ntro
ls (3
5 ±1
1)M
+F
20 20
[11C]
DA
SP P
ET
Bila
tera
l ant
erom
edia
l PF
C, d
orso
late
ral P
FC,
Ant
. Cin
g, C
auda
te,
Puta
m, T
hal,
Mid
Br, C
er
No
diff
eren
ce in
SER
T av
aila
bilit
y be
twee
n M
DD
and
co
ntro
ls fo
r any
regi
on.
With
in p
atie
nts
(but
not
in c
ontr
ols)
sig
n. c
orre
latio
ns
betw
een
incr
ease
d D
ysfu
nctio
nal A
ttitu
de S
cale
(DA
S)
scor
es a
nd in
crea
sed
SER
T av
ail.
Sign
ific
ant i
ncre
ase
in S
ERT
in p
atie
nts
vs. c
ontr
ols
in a
ll re
gion
s fo
r 8 p
atie
nts
with
hig
h D
AS
scor
es (>
190)
vs
cont
rols
.
MD
D p
atie
nts
wer
e/ d
rug
free
for ≥
3m; c
ontr
ols
wer
e in
divi
dual
ly a
ge-m
atch
ed (±
3 ye
ars)
but
not
for s
ex. A
ll pa
rtic
ipan
ts w
ere
non-
smok
ers;
no
info
rmat
ion
on fa
mily
hi
stor
y of
con
trol
s. N
o ef
fect
s of
age
or s
ex in
this
sam
ple.
Re
crui
tmen
t of p
atie
nts
unsp
ecifi
ed.
New
berg
200
59D
rug-
free
M
DD
pts
(38
±?)
Cont
rols
(37
±?)
M +
F
7 6
[123 I]
-AD
AM
SPE
CT, 4
hM
idBr
, Cer
Mid
Br S
ERT
sign
. low
er (-
7%) i
n M
DD
pat
ient
s vs
. con
trol
s.
SER
T M
idBr
cor
rela
ted
sign
ific
antly
with
sco
res
on H
DR
S2
drug
-nai
ve p
atie
nts,
2 >
3w a
nd 2
>6m
dru
g-fr
ee. N
o m
atch
ing
for a
ge o
r sex
. No
info
rmat
ion
on fa
mily
his
tory
of c
ontr
ols.
Re
crui
tmen
t of p
atie
nts
unsp
ecifi
ed.
Pars
ey 2
00610
Dru
g fr
ee
MD
D p
ts (3
8.0
±13.
4)Co
ntro
ls (3
8.8
±15.
9)M
+F
25 43
[11C]
McN
5652
PET
Mid
Br, P
utam
, Am
yg,
Thal
, Hip
poc,
Ant
. Cin
g,
Cer
Sign
. low
er (-
20%)
BP’
in a
ll re
gion
s in
MD
D p
atie
nts
vs
cont
rols
. Pos
t-ho
c as
sign
ed to
am
ygda
la a
nd m
idbr
ain
regi
ons.
Dru
g-na
ive
patie
nts
had
sign
. low
er B
P’ in
am
ygda
la a
nd
mid
brai
n vs
non
dru
g-na
ive
patie
nts.
No
corr
elat
ion
with
BP
and
depr
essi
on s
ever
ity ra
tings
MD
D p
atie
nts
wer
e dr
ug-fr
ee fo
r ≥2w
, 48%
dru
g-na
ive,
56%
in
patie
nts,
72%
fem
ales
. Con
trol
s ha
d no
1st d
egre
e po
sitiv
e fa
mily
his
tory
for p
sych
opat
holo
gyCo
ntro
ls n
ot m
atch
ed fo
r sex
, mat
chin
g fo
r age
unc
lear
, no
sex-
diff
eren
ces
obse
rved
. Rec
ruitm
ent o
f inp
atie
nts
and
outp
atie
nts.
Tabl
e S7
.1. P
revi
ous
stud
ies
mea
surin
g SE
RT a
vaila
bilit
y in
MD
D p
atie
nts
vers
us h
ealth
y co
ntro
ls.
(Con
tinue
d)
Aut
hor &
Dat
e (r
efer
ence
)Po
pula
tion
(mea
n ag
e)Se
xN
SER
T Im
agin
g an
d R
OI*
Res
ults
Rem
arks

153
Serotonin transporter availability in MDD
Chapter7
Rei
vich
200
418D
rug
free
M
DD
pts
(22-
56)
Cont
rols
(23-
59)
M+F
4 4
[11C]
(+)M
cN56
52 P
ETBi
late
ral F
ront
al, C
ing,
Th
al, P
ons,
Cer
Sign
. lar
ger B
P in
left
fron
tal (
+17%
) and
righ
t cin
gula
te
(+24
%) in
pat
ient
s vs
con
trol
s. T
hala
mus
and
pon
s no
sig
n.
diff
eren
ce
MD
D p
atie
nts
wer
e dr
ug fr
ee fo
r ≥5
half-
lives
of t
he d
rug,
≥2w
fo
r MA
O-I
and ≥3
w fo
r flu
oxet
ine.
Pat
ient
s w
ere
not m
atch
ed
for a
ge o
r sex
; no
info
rmat
ion
on fa
mily
his
tory
of c
ontr
ols.
W
ide
age-
rang
e w
ithou
t pos
sibi
lity
to c
orre
ct fo
r age
-eff
ects
on
SER
T av
aila
bilit
y. L
arge
SD
s fo
r tha
lam
us o
r pon
s R
OIs
. Re
crui
tmen
t of o
utpa
tient
s.
Stal
ey 2
00611
Dru
g-fr
ee
MD
D p
ts (3
8.8
±9.7
)Co
ntro
ls (3
8.9
±10)
M+F
32 32
[123 I]β-
CIT
SPEC
T, 2
4h
Brai
nste
m, D
ienc
epha
lon,
St
riatu
m, C
er
Sign
. red
uctio
n (-1
2%) i
n di
ence
phal
ons
V3’’
of M
DD
vs.
co
ntro
ls, b
ut n
ot in
bra
inst
em.
Inte
ract
ion
MD
D b
y se
x on
die
ncep
halo
n V3
’’: w
omen
-22%
in
MD
D, m
en -1
%.
Subj
ects
wer
e (g
roup
??) m
atch
ed fo
r sex
, age
and
sm
okin
g st
atus
. No
info
rmat
ion
on fa
mily
his
tory
of c
ontr
ols.
Re
crui
tmen
t of p
atie
nts
unsp
ecifi
ed.
Will
eit 2
00012
Dru
g-fr
ee p
atie
nts
with
SA
D (3
0.5
±8)
Cont
rols
(29.
0 ±5
.5)
M +
F
11 11
[123 I]β-
CIT
SPEC
T, 4
, 24h
Th
al, H
ypoT
hal,
Mid
Br,
Pons
, Cer
Sign
. red
uctio
n (-
15%)
in T
hala
mus
and
Hyp
otha
lam
us V
3’’ o
f M
DD
vs.
con
trol
s, b
ut n
ot (-
8%) i
n M
idBr
-Pon
s.N
o co
rrel
atio
n w
ith S
ERT
and
depr
essi
on s
ever
ity ra
tings
5 su
bjec
ts h
ad b
een
trea
ted
with
ant
idep
ress
ants
³6m
bef
ore
scan
ning
. Gro
upw
ise
mat
chin
g fo
r age
and
sex
.A
ll di
ffer
ence
s in
SPE
CT a
cqui
sitio
ns 2
4h p
.i., a
t 4h
p.i.
no
sign
ific
ant d
iffer
ence
s fo
und.
Rec
ruitm
ent o
f out
patie
nts
of
univ
ersi
ty c
linic
.
* N
amin
g of
iden
tical
regi
ons
as p
rovi
ded
in th
e st
udie
s. R
efer
ence
regi
on in
ital
ics.
Abb
revi
atio
ns: A
myg
= A
myg
dala
, Ant
. Cin
g =
Ant
erio
r Cin
gula
te, B
D =
Bip
olar
Dis
orde
r, B
P =
Bind
ing
Pote
ntia
l, Ca
ud =
Cau
datu
s, C
er =
cer
ebel
lum
, CIT
= c
italo
pram
, h =
hou
r, H
ippo
c =
Hip
poca
mpu
s,
Hyp
oTha
l = H
ypot
hala
mus
, min
= m
inut
e, m
= m
onth
, MD
D =
Maj
or D
epre
ssiv
e D
isor
der,
Mid
Br =
Mid
brai
n, O
cc =
Occ
ipita
l lob
e, P
AR
= p
arox
etin
e, P
FC =
Pre
fron
tal C
orte
x, P
utam
= P
utam
en, S
AD
=
Seas
onal
Aff
ectiv
e D
isor
der,
Tha
l = T
hala
mus
, V3’
’ = B
P, w
= w
eek
Tabl
e S7
.1. P
revi
ous
stud
ies
mea
surin
g SE
RT a
vaila
bilit
y in
MD
D p
atie
nts
vers
us h
ealth
y co
ntro
ls.
(Con
tinue
d)
Aut
hor &
Dat
e (r
efer
ence
)Po
pula
tion
(mea
n ag
e)Se
xN
SER
T Im
agin
g an
d R
OI*
Res
ults
Rem
arks


NEUROBIOLOGICAL EFFECTS OF PAROXETINE IN THE TREATMENT OF MAJOR DEPRESSIVE DISORDER
PART
Neurobiological effects of paroxetine in the treatment of Major Depressive Disorder

156

CHA
PTER
SEROTONIN TRANSPORTER GENE PROMOTER
POLYMORPHISMS MODIFY THE ASSOCIATION
BETWEEN PAROXETINE SEROTONIN TRANSPORTER
OCCUPANCY AND CLINICAL RESPONSE IN MAJOR
DEPRESSIVE DISORDER
Pharmacogenetics and Genomics 2008; in press
Henricus G. Ruhé1
Wendy Ooteman1
Jan Booij2
Martin C. Michel3
Martina Moeton4
Frank Baas4
Aart H. Schene1
1 Program for Mood Disorders, Department of Psychiatry, Academic Medical Center, University ofAmsterdam, Amsterdam, the Netherlands2 Department of Nuclear Medicine, Academic Medical Center, University of Amsterdam, Amsterdam,the Netherlands 3Department of Pharmacology and Pharmacotherapy, Academic Medical Center, University ofAmsterdam, Amsterdam, the Netherlands4 Department of Neurogenetics, Academic Medical Center, University of Amsterdam, Amsterdam, theNetherlands

Chapter 8
158
Chap
ter
8
Abstract
BackgroundIn major depressive disorder (MDD), selective serotonin reuptake inhibitors (SSRIs) target theserotonin transporter (SERT). Their 30-50% response-rates are modified by SERT promotorpolymorphisms (5-HTTLPR).
AimTo quantify the relation between SERT occupancy and response, and whether 5-HTTLPR is amodifier.
MethodsDrug-free depressed outpatients (n= 49; both sexes; 25-55 years), received paroxetine 20 mg/day.We quantified SERT occupancy with [123I]β-CIT SPECT imaging at baseline and after 6 weeks; wegenotyped 5-HTTLPR (S, LG, LA). Primary outcomes: percentage decrease in Hamilton DepressionRating Scale (HDRS17) and response (≥50% decrease of HDRS17).
ResultsA significant positive relation between SERT occupancy and clinical response existed only in theLA/LA genotype (p< 0.002). Relative to paroxetine serum concentrations midbrain SERToccupancy was numerically higher for LA/LA compared with other genotypes, but this differencewas not significant (p= 0.188).
ConclusionsHigher SERT occupancy is only associated with more clinical improvement in the LA/LA-genotype.We hypothesize that the LA/LA carriers have a more dynamic serotonergic system, which appearsmore responsive to SSRIs. ISRCTN register ISRCTN44111488

ChapterSERT-occupancy, SSRI response and 5-HTTLPR
8
159
Introduction
Major depressive disorder (MDD) is often treated with antidepressants, most commonly selectiveserotonin reuptake inhibitors (SSRIs). SSRIs occupy the serotonin transporter (SERT) with highselectivity and affinity to block presynaptic serotonin reuptake. Unfortunately, clinical responserates for SSRIs are modest (30-50%) and difficult to predict.1 Unraveling the factors that modifyclinical response may contribute to more effective SSRI treatment.
SERT occupancy can be estimated in-vivo using radioligands and SPECT or PET-imaging.2 SERToccupancy reaches a plateau at relatively low SSRI serum concentrations in healthy subjects,3-6
social phobia patients,7 and MDD patients,4;8-11 explaining why SSRI-serum levels do not predictclinical response.12 Previous studies postulated that 80% SERT occupancy by SSRIs would berequired for clinical response.4;5;8 However, when SERT occupancy-rates and clinical responsewere correlated, the same studies found no clear relationship between striatal SERT occupancyand clinical response after 4 weeks of SSRI treatment in MDD patients.4;8 Contrary, Kugaya et al.found that higher pretreatment SERT availability and greater SERT occupancy in diencephalonpredicted better treatment response after 6 weeks of SSRIs.11 Recently, Zitterl et al. found acorrelation between clomipramine diencephalon SERT occupancy and severity-scores inobsessive compulsive disorder.13 However, in these two studies SERT occupancies rangedbetween 23-61%.
Polymorphisms of the SERT gene promoter region (5-HTTLPR) are associated with itstranscriptional activity and influences the rate of serotonin uptake.14 In human lymphoblasts, cellshomozygous for the long (L) allele produce higher concentrations of SERT mRNA than cellscontaining one or two copies of the short (S) allele. Furthermore, serotonin uptake by thetransporter is >2-fold higher in cells homozygous for the L-allele. In-vivo imaging inconsistentlyshowed lower midbrain SERT availability for S-allele carrying healthy subjects compared with L-allele carriers.15-18 Individuals with an S-allele have increased anxiety related traits,19;20 elevated riskof depression after stress,21;22 increased amygdala reactivity,23;24 and increased adverse eventsafter SSRI treatment,25 compared with subjects with an L-allele. An elegant, large MRI-study inhealthy volunteers showed associations of the 5-HTTLPR S/S polymorphism with unfavorablealterations in anatomy and function of the amygdala-cingulate feedback circuit.26 This studypoints to an important role of the 5-HTTLPR-polymorphism in the development and functioning ofemotional networks involved in MDD. A meta-analysis showed associations between the 5-HTTLPR-polymorphism and clinical SSRI efficacy within 4 weeks of treatment.27 Depressedpatients without S-alleles had higher response rates to SSRIs. Thus, apart from developmental,functional, and stress reactivity effects on emotional networks, the 5-HTTLPR polymorphism alsoappears to influence the response to SSRIs.
Nowadays, the 5-HTTLPR-polymorphism is considered tri-allelic.28 The L-allele is subdivided asLG and LA by a common single nucleotide polymorphism (SNP; rs25531). The LG SNP creates afunctional AP2 transcription factor binding site, behaving like an S-allele. The tri-allelic S:LA:LG ratiois approximately 4:5:1,28 and can be reclassified into a modified bi-allelic genotype.29 In-vivo studiespredominantly found higher SERT availability for LA/LA carriers in healthy and MDD subjects.29-31
Recently, a significant association between the LA/LA allele and citalopram adverse effects32 butnot with response33 was found in MDD patients.
In summary, the relation between SERT occupancy and clinical response to SSRIs is unclear, aSERT polymorphism likely influences the development and function of the serotonergic system(e.g. SERT availability) and affects clinical response to SSRIs. Since it was not investigatedwhether the relation between SERT occupancy and clinical response is modified by the 5-HTTLPR-polymorphism, we aimed to quantify (1) the relation between SERT occupancy and clinicalresponse, (2) pre-treatment SERT availability for different tri-allelic 5-HTTLPR genotypes and (3)the modification by 5-HTTLPR with respect to the relation between SSRI-occupancy and clinicalresponse.

Chap
ter
Chapter 8
8
160
We hypothesized that (1) the relation between occupancy and clinical response is nonlinearwith increased response-rates above 80% occupancy after 6 weeks of treatment, (2) we wouldfind lower pretreatment SERT availability for the tri-allelic S’/S’ 5-HTTLPR genotype and (3)because of the higher response-rates in patients with a L-allele, the association betweenoccupancy and clinical response would be distinct for tri-allelic 5-HTTLPR genotypes. We studiedthese questions in a 6 week open trial of paroxetine 20 mg/day, in subjects who participated in thefirst phase of a randomized dose-escalation trial with [123I]β-CIT SPECT assessment of SERToccupancy.34
Methods
ParticipantsFollowing approval by the institutional medical ethical committee and written informed consent,we recruited drug-free outpatients (25-55 years)35 from primary care, our outpatient department,and public psychiatric settings (October 2003 - August 2006) and included them in the studybefore antidepressants were started. Inclusion criteria were: MDD determined by the StructuredClinical Interview for DSM-IV (SCID),36 and a Hamilton Depression Rating Scale (HDRS17)37 scoreabove 18. Patients were drug-naïve or drug-free (>4 weeks and ≥ 5 half-lives of a previousantidepressant, when treated previously) and had used no more than one antidepressanttreatment (other than paroxetine) at an effective dose for ≥ 6 weeks for the present MDD-episode. Exclusion criteria were pregnancy (or wish), bipolar disorder, psychotic features,neurological cognitive impairments (i.e. dementia), primary anxiety and/or substance abusedisorders and acute, severe suicidal ideation. We allowed secondary co-morbid anxiety and/orsubstance abuse to increase applicability of the findings of our main study.34
SSRI treatmentAfter baseline assessment, patients were treated open-label with paroxetine 20 mg/day for sixweeks. When severe adverse effects occurred, dosages were reduced to 10 mg/day and againincreased to 20 mg/day after one week. We supplied paroxetine in pill-boxes to improvetreatment adherence. We checked adherence by pill-counts and medical history.38
Benzodiazepines (temazepam 10-20 mg/day or oxazepam 10-30 mg/day) were allowed ifnecessary.
Questionnaires and measurementsPrimary clinical outcomes were the percentage decrease in HDRS17-score, and the proportion ofpatients achieving clinical response (≥50% decrease in HDRS17). We administered questionnaires atstudy-entry and after 6 weeks of treatment. In addition, depressive symptoms were monitored atweek 2 and 4 using the Maier and Bech subscales of the HDRS17,39;40 and the self-rated Inventoryfor Depressive Symptomatology (IDS-SR30) scores.41 Three trained investigators whoadministered the clinician-rated questionnaires (HDRS17 and subscales) had good inter-rateragreement (intra-class correlation coefficient = 0.98).
SPECT imaging and analysisWe performed single photon emission computed tomography (SPECT) imaging for in-vivoassessment of SERT availability at study-entry and after 6 weeks between 2 to 10 pm as describedpreviously.42 The acquisition started 230 ±18 (SD) minutes after intravenous injection of 100 MBqiodine-123-labeled 2β-carbomethoxy-3β-(4-iodophenyl)-tropane ([123I]β-CIT), when the radioligandis at equilibrium for SERT binding in brain areas expressing high densities of SERTs (i.e. midbrainand diencephalon).43 To prevent thyroid uptake of [123I], all subjects received oral potassium-

ChapterSERT-occupancy, SSRI response and 5-HTTLPR
8
161
iodide solution. We performed SPECT imaging using a 12-detector single slice brain-dedicatedscanner (Neurofocus 810, Strichmann Medical Equipment; Cleveland, OH) with a full-width at half-maximum resolution of 6.5 mm, throughout the 20 cm field-of-view (www.neurophysics.com).
After attenuation correction and reconstruction in 3D mode (www.neurophysics.com), wedefined regions of interest (RoIs) for midbrain, diencephalon and cerebellum by using validatedtemplates (see Figure 2.3).34;42 One investigator, blinded for scan session (study-entry/6 weeks),positioned all RoIs in two series. Intra-class correlation coefficients between series were >0.97 forall RoIs. If the two series differed by >5%, scans were re-evaluated by a second investigator. In theanalyses the counts were averaged for the two series.
Using activity in the cerebellum (CER) as indicator of non-displaceable activity (non-specificbinding and free radioactivity),44 we estimated the non-displaceable binding potential (BPND) ofthe radioligand to SERTby calculating the ratio of specific to non-specific binding per scan asBPND = . BPND is proportional to SERT availability under equilibrium conditions.45 In adifferent study, we found high reproducibility of SERT imaging with [123I]β-CIT SPECT afterrepeated scanning of subjects, using the same camera and scanning-protocol (de Win et al.,submitted). We calculated SERT occupancy at 6 weeks relative to the untreated SERT BPND(study-entry): OCC6 weeks = .
Paroxetine serum concentrationsWe collected blood for paroxetine serum concentrations (PSC; therapeutic range 10-75 µg/L) after6 weeks, immediately before SPECT scanning. Serum was stored at -20° C until analysis. PSCs weredetermined using a validated High Pressure Liquid Chromatography-MS/MS method (available onrequest). The lower limit of quantification was 5µg/L, the lower limit of detection was 0.3 µg/L.
Genotyping Procedures and AnalysisGenomic deoxyribonucleic acid (DNA) was isolated out of blood using a filter-based method
(QIAamp DNA Mini Kit, Qiagen Ltd, United Kingdom). The length of 5-HTTLPR28 was determinedby gel electrophoresis. The region around the polymorphism was amplified by PCR using forwardprimer tgtaaaacgacggccagtgccagcacctaacccctaat and reverse primercaggaaacagctatgaccaggggagatcctgggaga (M13 primer sequence in italics). The PCR reaction wasperformed in 10µl 1.5mM MgCl2, 0.2µM forward and reverse primer, 0.1mM dNTP’s, 0.5 UnitsHotfire Polymerase (Solis Biodyne, Estonia), Buffer B (Solis Biodyne, Estonia) and 20ng genomicDNA. The lengths of the different alleles were short ≈ 250 bp and long ≥298 bp. Genotyping of thers25531 SNP was done by sequencing (Sanger) using Big Dye Terminators (Applied Biosystems).The M13 forward primer tgtaaaacgacggccagt was used for sequencing. Reactions were performedcontaining 5ng of a forward primer, 5µl PCR product, BDT mix (Applied Biosystems) and 2.5xBDTbuffer (Applied Biosystems). The length of the 5-HTTLPRpolymorphism was confirmed by lookingat the length of the sequenced PCR product. We reclassified the genotypes as S’/S’ (S/S, LG/S, LG/LG), S’/LA (S/LA, LG/LA) and LA/LA.29 In post-hoc analyses we grouped S’/S’ and S’/LA genotypes tocontrast these with the ‘high-expression’ LA/LA genotype.
Statistical AnalysisFor cross tabulations and differences between groups we used χ2 tests, Fisher’s exact test andANOVA in SPSS for Windows v15.0.1.1 (www.spss.com). To investigate the relation between SERToccupancy and PSC, we modeled SERT occupancy (OCC) after 6 weeks in an Emax model asOCC= , in which a represents maximal SERT occupancy in the model (OCCmax) and b thePSC with 50% SERT occupancy (EC50).4-7;9 We calculated a and b by fitting a nonlinear regressionmodel that minimizes the sum of squares of the residuals (GraphPad Prism v5.00,www.graphpad.com). To assess whether PSC-occupancy or occupancy-response curves wereimproved by sub-grouping (e.g. genetic subgroups or responders versus non-responders), wefitted either one curve, or separate curves and determined whether separate curves decreased
( )CER
CERROI
ActivityActivityActivity −
( )entrystudyND
weeksNDentrystudyND
BPBPBP
−
− − _6
)(*
PSCbPSCa+

Chap
ter
Chapter 8
8
162
the Akaike Information Criterion (AIC; lower is better), which expresses the -2 log-likelihood of the(nested) model penalized for the number of independent variables in the model. We furthermoreverified these analyses by regression models including interaction terms for genotype.
To predict clinical response by SERT occupancy, we performed χ2 and ANOVA tests to estimategroup differences in clinical response-rate and proportional decrease in HDRS17, respectively, withSERT occupancy stratified as poor <50%, intermediate 51-79%, and high ≥80%, and linear andlogistic regression analyses for continuous outcomes (proportional decrease in HDRS17) anddichotomous outcomes (response). In these models occupancy was entered as a continuous anddummy-coded group variable (stratified SERT occupancy), while the models were corrected forage, baseline midbrain /diencephalon SERT availability and sex.
One responder and one non-responder were potentially non-adherent after 6 weeks (PSC<5µg/L), but were included in the analyses for three reasons. First, this estimation of medicationadherence was only based on a single measurement of PSC over a 6-week treatment period,second, patients had empty pill-boxes and had claimed adherence, and third, potential non-adherence in other patients before the measurement of PSC could not be excluded either.
Table 8.1. Characteristics of patients, stratified by clinical response after 6 weeks of paroxetine 20 mg/day.
Responders*(n= 10) Non-responders*(n= 32) p†
Age at baseline (years) 43.5 ±10.8 41.5 ±7.6 0.51
Female sex - n (%) 6 (60.0) 21 (65.6) 1.00
Current smoking - n (%) Alcohol use: n (%) ≤ 7 Units/week >7 Units/ week
5 (50.0)
7 (70.0) 3 (30.0)
17 (53.1)
30 (93.8) 2 (6.2)
1.000.08
MDD HDRS17 at baseline IDS-SR30 baseline First episode - n (%) No of episodes Drug-naïve - n (%) Used AD in current episode - n (%) Melancholic - n (%) Duration of episode: n (%) <5 months duration 5 months – 2 years duration ≥ 2 years Age of first episode (years)
23.8 ±3.7 41.0 ±7.2 4 (40.0) 2.0 ±1.1 7 (70.0) 0 (0.0) 7 (87.5)
3 (30.0) 6 (60.0) 1 (10.0) 33.3 ±11.2
25.0 ±4.7 44.7 ±9.118 (58.1)1.9 ±1.8 22 (68.8) 3 (9.4) 24 (96.0)
10 (31.3)18 (56.3) 4 (12.5) 36.4 ±9.6
0.470.280.480.921.001.000.430.97
0.40
Co-morbidity - n (%) Anxiety disorder Dysthymia Drug abuse / dependence‡
Alcohol abuse / dependence
3 (30.0) 0 (0.0) 3 (30.0) 2 (20.0)
3 (9.4) 1 (3.1)1 (3.1)1 (3.1)
0.141.000.040.14
SERT genotype S’/S’ (S/S, S/LG, LG/LG) S’/L’ (S/LA, LG/LA) L’/L’ (LA/ LA)
4 (40.0) 5 (50.0) 1 (10.0)
8 (25.0)17 (53.1)7 (21.9)
0.56
SERT availability Bsl-scan (BPND) Midbrain Diencephalon
0.60 (0.24) 1.16 (0.29)
0.61 (0.18) 1.15 (0.23 )
0.930.89
* Responders defined as patients with ≥50% decrease in baseline HDRS17-score† p values for χ2 for categorical, Fisher’s exact test for dichotomous, and independent T-tests for continuous data‡ cannabis, benzodiazepines

ChapterSERT-occupancy, SSRI response and 5-HTTLPR
8
163
Results
PatientsFrom 177 patients assessed, 53 patients were excluded and 73 refused participation. Of the 51patients included in the study, 7 patients dropped out due to paroxetine adverse effects (n= 5) orrefusal of a second scan (n= 2), leaving 44 patients who were scanned at baseline and after sixweeks of treatment. Of these 44 patients, 12 patients were treatment-responder after 6 weeks. Ofresponders, two SPECT scans could not be used for analysis due to technical reasons, leaving 42patients for the final analysis. Benzodiazepines were prescribed in 11/42 (26%) patients.
At study-entry, no significant differences were found between responders and non-responders except fordrug (cannabis/benzodiazepine) abuse or dependence with higherprevalence in responders compared with non-responders (Fisher’s Exact; p= 0.036; Table 8.1).None of the patients reported lifetime use of 3,4-methylenedioxy methamphe tamine (MDMA).
SERT occupancy and clinical response When SERT occupancy was stratified as poor (<50%), intermediate (51-79%) and high (≥80%), wefound no significant relation between SERT occupancy and response-rate or the percentagedecrease in HDRS17 after 6 weeks of paroxetine treatment, neither in midbrain nor indiencephalon (p>0.05) (Table 8.2). Neither in linear nor in logistic regression models occupancysignificantly predicted clinical response (p>0.05). The minimum occupancy rate at which responseoccurred in at least one patient was 29% in midbrain and 49% in diencephalon. We found noincrease in response-rate for SERT occupancy >80%. Only midbrain SERT-occupancy wasassociated with PSC (p=0.02). PSC significantly predicted response (OR= 1.04 (95% CI= 1.00-1.09).After exclusion of 2 potential non-adherent patients, the association of PSC with responsebecame non-significant, other results did not change.
Modification of the relation between PSC, occupancy and response bygenotype
PSC and SERT occupancy PSC-occupancy curves in both the midbrain and diencephalon were curvilinear, with significantlydifferent (non-stratified) curves for midbrain and diencephalon (determined by a decrease in AIC;figure available on request). The curves for responders versus non-responders were notsignificantly different in both brain regions. Stratification of the PSC-occupancy curves by SERTgenotype indicated numerically higher OCCmax and lower EC50 in the LA/LA genotype (n= 8/42;19%) compared with the S’/S’ (n= 12/42; 29%) and S’/LA genotypes (n= 22/42; 52%), especially inmidbrain (OCCmax for LA/LA=99.6; p=0.188) but not in diencephalon (OCCmax for LA/LA=79.3; p=1.0;
Table 8.2. SERT occupancy by paroxetine and clinical response after 6 weeks of paroxetine 20 mg/day.
Occupancy after 6 weeks paroxetine
<50% 51-79.9% ≥80% p*
Diencephalon n= 9 25 8
Mean occupancy (%) Mean PSC (µg/L) Responders n (%) % decrease HDRS17
†
28.9 ±7.43 22.2 ±10.823 (33.3) 23.9 ±10.72
65.4 ±1.72 46.4 ±7.25 4 (16.0) 27.5 ±4.96
85.0 ±1.29 46.0 ±8.83 3 (37.5) 37.7 ±8.38
0.176 0.347 0.530
Midbrain n= 9 12 18
Mean occupancy (%) Mean PSC (µg/L) Responders n (%) % decrease HDRS17
†
30.4 ±5.40 18.2 ±6.27 4 (44.4) 30.2 ±10.89
68.6 ±2.46 38.5 ±7.90 2 (16.7) 26.1 ±6.88
93.2 ±2.18 56.3 ±9.18 3 (16.7) 30.6 ±4.29
0.020 0.149 0.896
Values represent means (±SEM) unless indicated different. N= 42, including 2 non-adherent patients. PSC = Paroxetine serum concentration* χ2 for linear trend for response rates; ANOVA for % decrease† after 6 weeks of treatment

Chap
ter
Chapter 8
8
164
Figures 8.1A and B). The difference in AIC between one or separate curves indicated no significantimprovement by including genotype in the models, both in midbrain (AIC increase 2.359) anddiencephalon (AIC increase 4.037).
SERT availability and SERT occupancy by genotypeMean pretreatment SERT availability and mean SERT occupancies after 6 weeks in midbrain ordiencephalon did not significantly differ between genotypes (Table 8.3). Six of 8 patients (75%)with the LA/LA genotype reached midbrain occupancies ≥ 80% after 6 weeks compared with 12 of31 patients (39%) with other genotypes (Fisher’s exact; p= 0.112).
After exclusion of 2 potentially non-adherent patients, mean SERT occupancies in midbrainwere significantly different between genotypes (S’/S’ 80.9% ±5.80, S’/LA 65.8% ±5.17, LA/LA 91.6%±6.07; p= 0.017). Moreover, 6 of 7 patients (86%) with the LA/LA genotype reached midbrainoccupancies ≥80% after 6 weekscompared with 12 of 30 patients (40%) with the other genotypes(Fisher’s exact; p= 0.042).
Figure 8.1. Paroxetine serum concentration and SERT occupancy by paroxetine, stratified by SERT genotype.
Panels represent PSC-occupancy curves for S’/S’ vs. S’/LAvs. LA/LA in midbrain (A) and diencephalon (B). Equotation fitted: OCC 6 weeks = . Differences between curves were not significant (lower AIC for 1 fitted curve vs. three fitted curves)PSC= Paroxetine Serum Concentration, OCC6 weeks = SERT occupancy after 6 weeks of paroxetine treatment.
Table 8.3. Ethnicity, clinical response and SERT occupancy by paroxetine stratified for SERT genotype.
SERT genotype
S/S, S/LG, LG/LG S/LA, LG/LA LA/ LA p
Ethnicity - n (%) Caucasian Creole Asian
7 (24.1) 3 (42.9) 2 (33.3)
15 (51.7) 3 (42.9) 4 (66.7)
7 (24.1) 1 (14.3) 0
0.605
Clinical n= 12 n= 22 n= 8
Week 6 % decrease HDRS17 39.4±8.18 22.2±5.34 30.4±7.97 0.183
Occupancy Diencephalon n= 12 n= 22 n= 8
Baseline BPND Mean occupancy (%) Occupancy ≥80%n (%)
101.7±3.94 58.3±6.94 2 (16.7)
116.2±6.0162.7±4.25 4 (18.2)
122.9±8.8462.0±9.76 2 (25.0)
0.353 0.857 0.888
Occupancy Midbrain n= 11 n= 20 n= 8
Baseline BPND Mean occupancy (%) Occupancy ≥80%n (%)
60.1±5.20 73.6±9.04 5 (45.5)
61.9±5.0365.8±5.17 7 (35.0)
57.8±4.52 81.3±11.56 6 (75.0)
0.879 0.381 0.159*
Continuous values represent means (±SEM). N= 42, including 2 non-adherent patients.* Post-hoc LA/LAvs.S/S, S/LG, LG/LG, S/LA, LG/LA Fisher’s exact p= 0.112
�����������
� �
�
�
�
�
��
��������������������������������
����� �����
�����
����(��������������������������� �����
���� ���)
��!�
"#$%&�#�
� �
�
�
�
�
��
��������������������������������
�����
�����
�����
����(��������������������������� �����
���� ���)
��!�
� %
)(*
PSCbPSCa+

ChapterSERT-occupancy, SSRI response and 5-HTTLPR
8
165
SERT occupancy and clinical response by genotype Despite no significant differences in mean proportional HDRS17-decrease across genotypes (Table8.3), we found a significant genotype*SERT occupancy interaction in diencephalon and midbrain(Figure 8.2). Higher diencephalon SERT occupancy was associated with larger proportionalHDRS17-decreases in LA/LA carriers (ANOVA; F2,5= 27.64; p= 0.002) (Figure 8.2B; corrected for age),and LA/LA and LA/S’ carriers grouped together (ANOVA; F1,28= 8.380; p= 0.007) (Figure 8.2D). Thegenotype*SERT occupancy interaction in diencephalon was significant (ANOVA; F2,36= 3.273; p=0.049, with correction for age) (Figure 2B). Higher midbrain SERT occupancy in LA/LA carriers wasassociated with larger proportional HDRS17-decreases when corrected for age (Figure 8.2C;ANOVA; F2,5= 56.918; p< 0.001). Introduction of baseline SERT availability in diencephalon (ormidbrain) showed no significant improvement of the association between proportional HDRS17decrease and SERT occupancy (p>0.05). These results remained unchanged with absolutedecrease in HDRS17-scores instead of proportional HDRS17-decreases. Performing the sameanalyses with the bi-allelic 5-HTTLPR classification found identical directions of the effects, butexplained less of the variances (available on request).
Figure 8.2. SERT occupancy by paroxetine and proportional decrease in HDRS17, stratified by SERT genotype.
Panels A and C: Midbrain, panels B and D: Diencephalon. In panel A and C (midbrain) a significant relation between SERT occupancy and % decrease in HDRS17 (determined by theslopes of regression lines) existed for the LA/LA genotype when confounding by age was accounted for (F2,5= 56.92; p<0.001).In panels B (diencephalon) this relation was significant for the LA/LA genotype (F1,6= 15.36; p= 0.008), and confounded by age(F2,5= 27.64; p= 0.002); for panel D this relation was significant for the S/LA, LG/LA, LA/LA genotypes (F1,28= 8.380; p= 0.007).In panel A and C there was no significant interaction between the different genotype groups. In panel B and D a significantinteraction existed between the genotype groups (F2,36=3.273; p= 0.049 and F1,38= 6.293; p= 0.017 respectively).
����� ��� �������������
�� � � �� ���
�
��
�
��
�
�
��
���
�������� ��������������������������
����� �������
�����
������'������
���
����
����
�'��
��
���
������������ � ���������
�� � � �� ��� ���
�
��
�
��
�
�
��
���
�������� ��������������������������
�����
������������ �
������'������
���
����
����
�'��
��
���
� !"#!$����"
�� � � �� ���
�
��
�
��
�
�
��
���
��������������������������������
�����
�����
�����
������'������
���
����
����
�'��
��
���
% �&�� "
�� � � �� ��� ���
�
��
�
��
�
�
��
���
�������� ������������������������
�����
�����
�����
������'������
���
����
����
�'��
��
���
� &
# �

Chap
ter
Chapter 8
8
166
Discussion
We quantified the relation between SERT occupancy in midbrain and diencephalon and clinicalresponse after 6 weeks of paroxetine 20 mg/day. We found no significant relation between theseoccupancies and clinical response. Nor did we find significant differences in pretreatment SERTavailability in patients with different 5-HTTLPR-genotypes. We found a significant modifying effectof the 5-HTTLPR genotype with respect to the association between SERT occupancy and thedecrease in HDRS17-score (proportional and absolute). In diencephalon, increased SERToccupancy was associated with larger decreases in HDRS17 for LA/LA and S’/LA carriers. Inmidbrain, increased SERT occupancy was associated with larger proportional decreases in HDRS17for LA/LA carriers only. Age was a covariate in these associations. Unexpectedly, we found a trendof a modifying effect of the 5-HTTLPR genotype with respect to the association betweenparoxetine serum concentrations and occupancy.
Critique of methodsSome limitations might be addressed first. We observed a low 6 week response rate (24%), whichmay be attributable to 1) the inclusion of some patients with secondary co-morbid anxiety and/orsubstance abuse, who might show less and slower recovery,46 and 2) the relatively highpretreatment initial HDRS-scores. Nevertheless, the recent Sequential Treatment Alternatives torelieve Depression trial measured only 30% response rate after 6 weeks of citalopram as well.1
Second, we included patients who were ethnically heterogeneous (Table 8.3). This raises thepossibility of spurious results secondary to population stratification, which can be a problemwhen analyzing non-functional markers which are in linkage disequilibrium with the functionalvariants. Since we have analyzed the relation between a functional DNA sequence variant andtransporter occupancy we do not expect effects by the heterogeneity of our sample.
Third, as recruitment of MDD patients willing to undergo two SPECT scans is difficult, wecould only successfully scan 42 patients twice. This led to small genotype-subgroups, andreplication in larger samples will be necessary. Nevertheless, these 42 patients scanned twicewhile treated with the same antidepressant (and dose) is the largest patient sample to date, andallowed us to address the relation between SERT occupancy and clinical response, and itsmodification by the 5-HTTLPR-genotype for the first time. Furthermore, we had low dropout ratesand good adherence.
Fourth, in this open label study we did not control for placebo response (estimated to be 30%).Placebo-responses may potentially obscure the relation between occupancy and response.Nevertheless, our major finding of a significant relation between SERT occupancy andproportional response in LA/LA-carriers was found despite such potential placebo-effects.Additionally, a placebo comparator would have estimated test-retest variability in SERTavailability, but this variability is known to be much lower than the observed 60-70% SERToccupancy.
Fifth, we used [123I]β-CIT for SPECT imaging, which is a non-selective radioligand, and also bindsto dopamine transporters (DAT; e.g. midbrain substantia nigra).47 Nevertheless, uptake inmidbrain and diencephalon is considered to reflect predominantly SERT,48 as these structures arerich of SERT relative to DAT. Therefore, although some additional DAT-binding might haveconcealed SERT occupancy, we think the change in [123I]β-CIT-binding in diencephalon andmidbrain mainly reflects SERT occupancy. Nevertheless, it would be challenging to replicate ourstudy with selective radioligands like [11C]DASB or [123I]ADAM.
Finally, we did not further explore whether 5-HTTLPR (or other polymorphisms) alsoinfluenced transporter site kinetics. Rausch et al. showed that initial serotonin kinetic conditionsof SERT in platelets could predict treatment response in MDD patients, which was modified bySERT polymorphism.49 Patients with an L-allele were more responsive to fluoxetine than patientswith an S-allele, with the initial affinity constant Km and dose being significant covariates fortreatment outcome. It is unlikely that Km is directly affected by the SERT promoter polymorphismthat primarily regulates expression. Because the S and L variants actually represent 10 subtypes,50

ChapterSERT-occupancy, SSRI response and 5-HTTLPR
8
167
other subtypes or an unknown other polymorphism could very well be associated withdifferences in Km, which may better explain the inter-individual variation between PSC and SERToccupancy.49 Recently, Smeraldi et al.51 investigated the relation between clinical response tofluvoxamine and different L and S subtypes (identified by Nakamura et al.50). They confirmedbetter responses in depressed patients bearing the L-allele, but also found significant differencesin response among L-allele carriers according to the subtype of the L-allele, accounting for 0.6% ofthe variance. Because these subtypes are relatively uncommon, it would be very interesting tospecifically study these L-allele subtypes in future neuroimaging studies using a case-controldesign.
SERT occupancy and clinical responsePrevious studies postulated that a SERT occupancy of at least 80% would be associated withclinical response to SSRIs.4;5;8 Our data did not show significant associations betweenpretreatment SERT availability or SERT occupancy and clinical response to paroxetine, and neithersuggested an 80% SERT occupancy threshold with increased response rates. We could notreplicate a relation between SERT occupancy and decrease of symptoms.11;13 Receiver operatingcharacteristic curves relating SERT occupancy to response (available on request) showed thatSERT occupancy had no diagnostic value in the prediction of response.
The relation between SERT occupancy and clinical response is modified by 5-HTTLPR genotypeWe found that 5-HTTLPR-polymorphisms modify the relation between SERT occupancy andclinical response. Previous clinical studies and meta-analyses found superior treatment effects inL-allele carriers. Despite a low response rate in our sample, we found that in LA/LA carriers higherSERT occupancy was associated with more improvement. We put forward two possibleexplanations for this finding.
First, the LA/LA genotype is associated with a more than two-fold higher serotonin uptake inhuman lymphoblasts.14 If we assume that serotonergic cells maintain a certain tonus inneurotransmission, in LA/LA carriers more serotonin must be released as this is more effectivelyevacuated. When SERTs are blocked with paroxetine, this may increase serotoninneurotransmission more in LA/LA carriers than in other genotypes, which may result in largerpostsynaptic effects.
Second, 5-HTTLPR also modifies the development and synaptic plasticity of neural networkscritically involved in MDD.14 Our findings might thus point to differences in postsynaptic effects(evoked by increased serotonin neurotransmission) in neuronal networks that have beendeveloped differently as a result of different SERT genotypes. Pezawas et al. showed that healthysubjects with an S/S polymorphism demonstrated relative uncoupling of the amygdala from theanterior perigenual cingulate.26 S/S carriers were also associated with increased anxiety traits,increased amygdala reactivity,23 decreased mood after tryptophan depletion52 and increased riskfor MDD.22 In summary, the limbic-cortical network and the serotonergic innervations appear tobe more flexible in non S/S carriers. Our results may be supportive of the hypothesis that in LA/LAcarriers the significant association between higher SERT occupancy and increased reduction ofsymptoms could be indicative for a broader range for regulation of the serotonergic system.Higher SERT occupancy might then result in more effects of serotonergic antidepressants in LA/LAcarriers.
Interestingly, Pollock et al. found quicker response in L/L carriers when treated withparoxetine for 12 weeks, but no difference when treated with the noradrenergic antidepressantnortriptyline.53 Additionally better treatment outcomes and fewer adverse events in S/Sgenotypes were found when treated with mirtazapine (a postsynaptic 5-HT2A, 5-HT2Cantagonistand noradrenergic agonist),54;55 which were contrary to the effects of paroxetine.55 Futurepharmacogenomic studies should investigate whether non S’/S’ carriers will benefit more fromserotonergic drugs while S’/S’ carriers may benefit from noradrenergic antidepressants.Furthermore, since the mechanisms of action of antidepressants may be influenced bypolymorphisms of other genes, future research should investigate additive effects of multiplecandidate gene polymorphism combinations.56

Chap
ter
Chapter 8
8
168
Are paroxetine serum concentrations and SERT occupancy modified by 5-HTTLPR genotype?Six of 8 patients (75%) with the LA/LA genotypereached midbrain occupancies ≥80% after 6weekscompared with 12 of 31 patients (39%) with other genotypes, although this was statisticallynot significant (p= 0.112, Fisher’s exact). Which factor determines the maximum SERT occupancyremains unclear. 5-HTTLPR-polymorphisms primarily affect gene transcription, and as aconsequence affect SERT expression. Therefore, we a priori did not expect differences in SERToccupancy for different genotypes, as occupancy is expected to be independent of availableSERTs. As such, our results could represent an epiphenomenon: e.g. a different SNP in the SERTgene, in linkage disequilibrium with the 5-HTTLPR-polymorphism (e.g. rs2228673;www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=2228673) may have affected SERT occupancy.Contrary, the suggested modifying properties of the 5-HTTLPR-genotype for the relation betweenPSC and occupancy may also lack power, since the PSC-occupancy curves in diencephalon show aclear trend of modification by 5-HTTLPR. Therefore, this needs further exploration in largersamples.
A rationale for a modifying role of the 5-HTTLPR polymorphism in the occupancy of the SERTby paroxetine and clinical response could be postulated as follows. SERT imaging assesses the in-vivo binding to SERTs. After 6 weeks of treatment with paroxetine, the availability of SERTs tobind to the radiotracer is lower, presumably due to SERT blockade by the SSRI. However, it is notpossible to assess whether in addition to blockade, secondary effects occur such as down-regulation of the SERT. This might also result in lowering of the availability of SERTs to bind withthe radiotracer. Indeed, in rats down-regulation of SERTs after prolonged exposure to paroxetinehas been reported.57;58 Said differently, we are unable to discriminate between direct (i.e.,blockade of SERTs by paroxetine) and potential indirect pharmacological effects (i.e., down-regulation). Importantly, Benmansour et al., suggested that SSRI-induced down-regulation of theSERT may be a key component for the clinical response to SSRIs.57;58 Down-regulation of SERTs inrats was not caused by decreased gene transcription, but is presumably caused by increased(posttranslational) internalization of SERTs.57 Although speculative, the level of down-regulationof SERTs might be diverse for different 5-HTTLPR-genotypes, with faster or greater down-regulation in LA/LA versus other genotypes. This hypothesis could be studied in future imagingstudies with antidepressant-exposed primates (having 5-HTTLPR-polymorphisms) in which abruptdiscontinuation is possible.
ConclusionIn conclusion, we find that SERT genotype modifies the relation between SERT occupancy andclinical response, with more improvement at higher midbrain and diencephalon SERT occupancyin LA/LA carriers. Although this SERT promoter-polymorphism presumably does not influenceSERT occupancy at given PSCs, our data may point to a more flexible serotonergic system in LA/LAcarriers, which is more easily influenced by serotonergic antidepressants. This hypothesis couldserve as a starting point for future pharmacogenetic studies. This might provide the necessarydatato guide the choice of antidepressant treatment for individual patients, reducing thepatient's suffering, and lowering healthcare costs.
Acknowledgements
The authors wish to thank the patients in this study for their participation, and willingness toparticipate in the SPECT-study. We thank all participating general practitioners in the area ofAmsterdam Oost and Zuidoost, Hoofddorp, Nieuw Vennep, and Abcoude for their inclusions andreferrals for the study. Mrs. E. Miedema M.D. and Mrs. M.C. ten Doesschate M.D. wereindispensable for their help in rating questionnaires. This study was financed by a grant from theNetherlands Organisation for Health Research and Development (ZonMw), program MentalHealth, education of investigators in mental health to H.G. Ruhé (OOG; #100-002-002).

ChapterSERT-occupancy, SSRI response and 5-HTTLPR
8
169
Conflicts of interest
None
References
1. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram fordepression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006; 163: 28-40.
2. Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. JPsychiatry Neurosci. 2007; 32: 86-102.
3. Erlandsson K, Sivananthan T, Lui D, Spezzi A, Townsend CE, Mu S et al. Measuring SSRI occupancy of SERT using the noveltracer [123I]ADAM: a SPECT validation study. Eur J Nucl Med Mol Imaging. 2005; 32: 1329-1336.
4. Meyer JH, Wilson AA, Sagrati S, Hussey D, Carella A, Potter WZ et al. Serotonin Transporter Occupancy of Five SelectiveSerotonin Reuptake Inhibitors at Different Doses: An [11C]DASB Positron Emission Tomography Study. Am J Psychiatry.2004; 161: 826-835.
5. Suhara T, Takano A, Sudo Y, Ichimiya T, Inoue M, Yasuno F et al. High levels of serotonin transporter occupancy with low-dose clomipramine in comparative occupancy study with fluvoxamine using positron emission tomography. Arch GenPsychiatry. 2003; 60: 386-391.
6. Takano A, Suzuki K, Kosaka J, Ota M, Nozaki S, Ikoma Y et al. A dose-finding study of duloxetine based on serotonintransporter occupancy. Psychopharmacology (Berl). 2006; 185: 395-399.
7. Kent JM, Coplan JD, Lombardo I, Hwang DR, Huang Y, Mawlawi O et al. Occupancy of brain serotonin transporters duringtreatment with paroxetine in patients with social phobia: a positron emission tomography study with [11C]McN 5652.Psychopharmacology (Berl). 2002; 164: 341-348.
8. Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K et al. Occupancy of serotonin transporters by paroxetineand citalopram during treatment of depression: a [11C]DASB PET imaging study. Am J Psychiatry. 2001; 158: 1843-1849.
9. Catafau AM, Perez V, Plaza P, Pascual JC, Bullich S, Suarez M et al. Serotonin transporter occupancy induced byparoxetine in patients with major depression disorder: a [123I]-ADAM SPECT study. Psychopharmacology (Berl). 2006; 189:145-153.
10. Kugaya A, Seneca NM, Snyder PJ, Williams SA, Malison RT, Baldwin RM et al. Changes in human in vivo serotonin anddopamine transporter availabilities during chronic antidepressant administration. Neuropsychopharmacology. 2003; 28:413-420.
11. Kugaya A, Sanacora G, Staley JK, Malison RT, Bozkurt A, Khan S et al. Brain serotonin transporter availability predictstreatment response to selective serotonin reuptake inhibitors. Biol Psychiatry. 2004; 56: 497-502.
12. Mitchell PB. Therapeutic drug monitoring of non-tricyclic antidepressant drugs. Clin Chem Lab Med. 2004; 42: 1212-1218.13. Zitterl W, Aigner M, Stompe T, Zitterl-Eglseer K, Gutierrez-Lobos K, Wenzel T et al. Changes in Thalamus-Hypothalamus
serotonin transporter availability during clomipramine administration in patients with Obsessive-Compulsive Disorder.Neuropsychopharmacology. 2008; E-publication doi:10.1038/npp.2008.35.
14. Lesch KP, Gutknecht L. Pharmacogenetics of the serotonin transporter. Prog Neuropsychopharmacol Biol Psychiatry.2005; 29: 1062-1073.
15. Heinz A, Jones DW, Mazzanti C, Goldman D, Ragan P, Hommer D et al. A relationship between serotonin transportergenotype and in vivo protein expression and alcohol neurotoxicity. Biol Psychiatry. 2000; 47: 643-649.
16. Shioe K, Ichimiya T, Suhara T, Takano A, Sudo Y, Yasuno F et al. No association between genotype of the promoter regionof serotonin transporter gene and serotonin transporter binding in human brain measured by PET. Synapse. 2003; 48:184-188.
17. Van Dyck CH, Malison RT, Staley JK, Jacobsen LK, Seibyl JP, Laruelle M et al. Central serotonin transporter availabilitymeasured with [123I]β-CIT SPECT in relation to serotonin transporter genotype. Am J Psychiatry. 2004; 161: 525-531.
18. Willeit M, Stastny J, Pirker W, Praschak-Rieder N, Neumeister A, Asenbaum S et al. No evidence for in vivo regulation ofmidbrain serotonin transporter availability by serotonin transporter promoter gene polymorphism. Biol Psychiatry. 2001;50: 8-12.
19. Schinka JA, Busch RM, Robichaux-Keene N. A meta-analysis of the association between the serotonin transporter genepolymorphism (5-HTTLPR) and trait anxiety. Mol Psychiatry. 2004; 9: 197-202.
20. Sen S, Burmeister M, Ghosh D. Meta-analysis of the association between a serotonin transporter promoterpolymorphism (5-HTTLPR) and anxiety-related personality traits. Am J Med Genet B Neuropsychiatr Genet. 2004; 127: 85-89.
21. Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H et al. Influence of life stress on depression: moderation bya polymorphism in the 5-HTT gene. Science. 2003; 301: 386-389.
22. Lotrich FE, Pollock BG. Meta-analysis of serotonin transporter polymorphisms and affective disorders. Psychiatr Genet.2004; 14: 121-129.
23. Hariri AR, Mattay VS, Tessitore A, Kolachana B, Fera F, Goldman D et al. Serotonin transporter genetic variation and theresponse of the human amygdala. Science. 2002; 297: 400-403.
24. Hariri AR, Drabant EM, Munoz KE, Kolachana BS, Mattay VS, Egan MF et al. A susceptibility gene for affective disordersand the response of the human amygdala. Arch Gen Psychiatry. 2005; 62: 146-152.

Chap
ter
Chapter 8
8
170
25. Murphy DL, Lerner A, Rudnick G, Lesch KP. Serotonin transporter: gene, genetic disorders, and pharmacogenetics. MolInterv. 2004; 4: 109-123.
26. Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS et al. 5-HTTLPR polymorphismimpacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8: 828-834.
27. Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry. 2007; 12:247-257.
28. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD et al. Serotonin transporter promoter gain-of-functiongenotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. 2006; 78: 815-826.
29. Parsey RV, Hastings RS, Oquendo MA, Hu X, Goldman D, Huang YY et al. Effect of a triallelic functional polymorphism ofthe serotonin-transporter-linked promoter region on expression of serotonin transporter in the human brain. Am JPsychiatry. 2006; 163: 48-51.
30. Praschak-Rieder N, Kennedy J, Wilson AA, Hussey D, Boovariwala A, Willeit M et al. Novel 5-HTTLPR allele associates withhigher serotonin transporter binding in putamen: a [11C] DASB positron emission tomography study. Biol Psychiatry. 2007;62: 327-331.
31. Reimold M, Smolka MN, Schumann G, Zimmer A, Wrase J, Mann K et al. Midbrain serotonin transporter binding potentialmeasured with [11C]DASB is affected by serotonin transporter genotype. J Neural Transm. 2007; 114: 635-639.
32. Hu XZ, Rush AJ, Charney D, Wilson AF, Sorant AJ, Papanicolaou GJ et al. Association between a functional serotonintransporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Arch GenPsychiatry. 2007; 64: 783-792.
33. Kraft JB, Peters EJ, Slager SL, Jenkins GD, Reinalda MS, McGrath PJ et al. Analysis of association between the serotonintransporter and antidepressant response in a large clinical sample. Biol Psychiatry. 2007; 61: 734-742.
34. Ruhe HG, Booij J, Weert HC van, Reitsma JB, Fransen EJF, Michel MC et al. Evidence why dose-escalation of paroxetine inmajor depressive disorder is not effective: A 6-week, randomized-controlled trial with assessment of serotonintransporter occupancy. Neuropsychopharmacology. 2008. [In press].
35. van Dyck CH, Malison RT, Seibyl JP, Laruelle M, Klumpp H, Zoghbi SS et al. Age-related decline in central serotonintransporter availability with [123I]β -CIT SPECT. Neurobiol Aging. 2000; 21: 497-501.
36. First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders Patient Edition(SCID-I/P version 2.0). Translated in Dutch by. Groenestijn MAC, Akkerhuis GW, Kupka RW, Schneider N, and NolenWA.1999. Lisse, the Netherlands, Swets & Zeitlinger B.V.
37. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.38. Saunders K, Simon G, Bush T, Grothaus L. Assessing the feasibility of using computerized pharmacy refill data to monitor
antidepressant treatment on a population basis: a comparison of automated and self-report data. J Clin Epidemiol. 1998;51: 883-890.
39. Faries D, Herrera J, Rayamajhi J, DeBrota D, Demitrack M, Potter WZ. The responsiveness of the Hamilton DepressionRating Scale. J Psychiatr Res. 2000; 34: 3-10.
40. Ruhe HG, Dekker JJ, Peen J, Holman R, de Jonghe F. Clinical use of the Hamilton Depression Rating Scale: is increasedefficiency possible? A post hoc comparison of Hamilton Depression Rating Scale, Maier and Bech subscales, ClinicalGlobal Impression, and Symptom Checklist-90 scores. Compr Psychiatry. 2005; 46: 417-427.
41. Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The Inventory of Depressive Symptomatology (IDS):psychometric properties. Psychol Med. 1996; 26: 477-486.
42. de Win MM, Habraken JB, Reneman L, van den Brink W, den Heeten GJ, Booij J. Validation of [123I]β-CIT SPECT to assessserotonin transporters in vivo in humans: a double-blind, placebo-controlled, crossover study with the selective serotoninreuptake inhibitor citalopram. Neuropsychopharmacology. 2005; 30: 996-1005.
43. Pirker W, Asenbaum S, Hauk M, Kandlhofer S, Tauscher J, Willeit M et al. Imaging serotonin and dopamine transporterswith [123I]β-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000; 41: 36-44.
44. Laruelle M, Vanisberg MA, Maloteaux JM. Regional and subcellular localization in human brain of [3H]paroxetine binding,a marker of serotonin uptake sites. Biol Psychiatry. 1988; 24: 299-309.
45. Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN et al. Consensus nomenclature for in vivo imaging ofreversibly binding radioligands. J Cereb Blood Flow Metab. 2007; 27: 1533-1539.
46. Fava M, Rush AJ, Alpert JE, Balasubramani GK, Wisniewski SR, Carmin CN et al. Difference in Treatment Outcome inOutpatients With Anxious Versus Nonanxious Depression: A STAR*D Report. Am J Psychiatry. 2008; 165: 342-351.
47. Innis R, Baldwin R, Sybirska E, Zea Y, Laruelle M, al Tikriti M et al. Single photon emission computed tomography imagingof monoamine reuptake sites in primate brain with [123I]CIT. Eur J Pharmacol. 1991; 200: 369-370.
48. Laruelle M, Baldwin RM, Malison RT, Zea-Ponce Y, Zoghbi SS, al Tikriti MS et al. SPECT imaging of dopamine andserotonin transporters with [123I]β-CIT: pharmacological characterization of brain uptake in nonhuman primates.Synapse. 1993; 13: 295-309.
49. Rausch JL, Johnson ME, Fei YJ, Li JQ, Shendarkar N, Hobby HM et al. Initial conditions of serotonin transporter kineticsand genotype: influence on SSRI treatment trial outcome. Biol Psychiatry. 2002; 51: 723-732.
50. Nakamura M, Ueno S, Sano A, Tanabe H. The human serotonin transporter gene linked polymorphism (5-HTTLPR) showsten novel allelic variants. Mol Psychiatry. 2000; 5: 32-38.
51. Smeraldi E, Serretti A, Artioli P, Lorenzi C, Catalano M. Serotonin transporter gene-linked polymorphic region: possiblepharmacogenetic implications of rare variants. Psychiatr Genet. 2006; 16: 153-158.
52. Neumeister A, Hu XZ, Luckenbaugh DA, Schwarz M, Nugent AC, Bonne O et al. Differential effects of 5-HTTLPR genotypeson the behavioral and neural responses to tryptophan depletion in patients with major depression and controls. Arch GenPsychiatry. 2006; 63: 978-986.

ChapterSERT-occupancy, SSRI response and 5-HTTLPR
8
171
53. Pollock BG, Ferrell RE, Mulsant BH, Mazumdar S, Miller M, Sweet RA et al. Allelic variation in the serotonin transporterpromoter affects onset of paroxetine treatment response in late-life depression. Neuropsychopharmacology. 2000; 23:587-590.
54. Kang RH, Wong ML, Choi MJ, Paik JW, Lee MS. Association study of the serotonin transporter promoter polymorphismand mirtazapine antidepressant response in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31: 1317-1321.
55. Murphy GM, Jr., Hollander SB, Rodrigues HE, Kremer C, Schatzberg AF. Effects of the serotonin transporter genepromoter polymorphism on mirtazapine and paroxetine efficacy and adverse events in geriatric major depression. ArchGen Psychiatry. 2004; 61: 1163-1169.
56. Serretti A, Benedetti F, Zanardi R, Smeraldi E. The influence of Serotonin Transporter Promoter Polymorphism (SERTPR)and other polymorphisms of the serotonin pathway on the efficacy of antidepressant treatments. ProgNeuropsychopharmacol Biol Psychiatry. 2005; 29: 1074-1084.
57. Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG et al. Effects of chronic antidepressanttreatments on serotonin transporter function, density, and mRNA level. J Neurosci. 1999; 19: 10494-10501.
58. Benmansour S, Owens WA, Cecchi M, Morilak DA, Frazer A. Serotonin clearance in vivo is altered to a greater extent byantidepressant-induced downregulation of the serotonin transporter than by acute blockade of this transporter.JNeurosci. 2002; 22: 6766-6772.

Chap
ter
Chapter 8
8
172

CHA
PTER
EVIDENCE WHY PAROXETINE DOSE-ESCALATION
IS NOT EFFECTIVE IN MAJOR DEPRESSIVE
DISORDER: A RANDOMIZED-CONTROLLED TRIAL
WITH ASSESSMENT OF SEROTONIN TRANSPORTER
OCCUPANCY
Neuropsychopharmacology 2008; in press
Henricus G. Ruhé1
Jan Booij2
Henk C. v Weert3
Johannes B. Reitsma4
Eric J.F. Fransen5
Martin C. Michel6
Aart H. Schene1
1 Program for Mood Disorders, Department of Psychiatry, Academic Medical Center, University ofAmsterdam, Amsterdam, the Netherlands2 Department of Nuclear Medicine, Academic Medical Center, University of Amsterdam, Amsterdam,the Netherlands 3 Department of General Practice, Academic Medical Center, University of Amsterdam, Amsterdam,the Netherlands 4 Department of Clinical Epidemiology, Biostatistics & Bioinformatics, Academic Medical Center,University of Amsterdam, Amsterdam, the Netherlands 5 Department of Pharmacy and Clinical Pharmacology, Onze Lieve Vrouwe Gasthuis, Amsterdam, theNetherlands6 Department of Pharmacology and Pharmacotherapy, Academic Medical Center, University ofAmsterdam, Amsterdam, the Netherlands

Chap
ter
Chapter 9
9
174
Abstract
BackgroundDose-escalation is often used in depressed patients who fail to respond to standard doses ofSSRIs, but clinical efficacy is equivocal.
AimTo reassess the efficacy of paroxetine dose-escalation and quantify whether paroxetine dose-escalation increases occupancy of the serotonin transporter (SERT) more than placebo dose-escalation, in a randomized controlled trial.
MethodsWe recruited 107 non-psychotic, unipolar depressed outpatients (18-70 yrs; Hamilton DepressionRating Scale (HDRS17) >18) from primary care and psychiatric outpatient departments. After 6weeks open-label paroxetine 20 mg/day (T0), non-responding patients (HDRS17 decrease <50%; n=60) were randomized to double-blind paroxetine (30-50 mg/day as tolerable) or placebo dose-escalation (paroxetine 20 mg/day + placebo). Patients were followed until 6 weeks afterrandomization (T1). Forty-nine patients, drug free at study-entry, underwent SPECT-scanningbefore treatment and were scanned repeatedly at T0 and T1. Paroxetine serum concentrationsand SERT occupancy were determined at T0 and T1 (n= 32).
ResultsWe terminated the dose-escalation trial after an interim analysis. Thirty non-responding patientswere randomized to paroxetine (46.7 ±5.5 mg/day), 27 to placebo dose-escalation. Response-rates were 10/30 (33.3%) and 10/27 (37.0%), respectively. Repeated measurement analyses showedno significant effect for treatment (p= 0.88, exceeding a priori stopping rules for futility [p>0.5]).Overall dropout was higher for placebo (26.7%) than paroxetine (3.3%; p= 0.03). Paroxetine dose-escalation increased paroxetine serum concentrations (p<0.001). SPECT measurements (12patients randomized to paroxetine (46.9 ±4.8 mg) and 14 to placebo dose-escalation) showed nosignificant increase of midbrain SERT occupancy (2.5 ±26.4%, paroxetine; 3.1 ±25.8% placebo; p=0.687) nor in diencephalon (p= 0.529).
ConclusionsParoxetine dose-escalation in depressed patients has no clinical benefit over placebo dose-escalation. This is explained by the absence of significant increases of SERT occupancy byparoxetine dose-escalation, despite increased paroxetine serum concentrations. (ISRCTN register nr. ISRCTN44111488)

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
175
Introduction
Major depressive disorder (MDD) is often treated with antidepressants, particularly selectiveserotonin reuptake inhibitors (SSRIs). Unfortunately, response and remission rates are modest(30-50%), which require additional strategies to gain remission.1;2 Switching3 and augmentation4
have recently been evaluated. A third, and frequently applied option is dose-escalation,recommended in treatment guidelines1;2 and frequently used preceding other strategies. Only therecent NICE guideline is more reluctant in recommending dose-escalation.5 Although an individualpatient may improve after dose-escalation, this could also represent a delayed drug response orreflect the natural course of the disease. Prolonged (up to 10 weeks), unaltered treatment withfluoxetine 20 mg/day improved the response rates of initial week 6 non-responders.6
Theoretically, the concept of dose-escalation assumes linear dose-response relationships whichhave not been proven for SSRIs.7;8 Therefore, the efficacy of dose-escalation of SSRIs has beenquestioned.5;8-10
Previous studies did not show improved clinical effectiveness of dose-escalation, but hadserious methodological weaknesses.8-17 All previous studies increased dosages probably too early(mostly after 3-4 weeks) and too abruptly, which may have obscured true dose-escalation effectsby delayed effect of the standard doses and selective early drop-out of patients receiving truedose-escalation.9;10 Moreover, no study provided a rationale why dose-escalation was ineffective.
The primary molecular target of SSRIs is the serotonin transporter (SERT). Imaging techniquessuch as positron emission tomography (PET) and single photon emission computed tomography(SPECT) allow in-vivo labeling of SERT in the brain, which can be used to study their occupancy. Todate, several imaging studies measured SERT occupancy after short or prolonged treatment withSSRIs (Table S9.1).18-31 Particularly, Meyer et al.20 showed 60-80% SERT occupancy after standardclinical doses of SSRIs, and demonstrated curvilinear dose-response relationships for SERToccupancy by SSRIs. However, high doses of SSRIs were rarely studied,30 and dose-escalation wasnever studied.
Taking into account previous methodological criticisms, and considering the molecular targetof SSRIs, we have tested whether paroxetine dose-escalation increases SERT occupancy, andimproves depressive symptoms more than placebo dose-escalation. We performed a 6 week,multicenter, randomized study in depressed patients not responding to 6 weeks of paroxetine at20 mg/day. As a novel extension to previous clinical trials, and in order to elucidate theneurobiological basis for an expected lack of benefit of dose-escalation, we included a SPECTimaging approach. Herewith, we quantified whether paroxetine dose-escalation increased SERToccupancy more than placebo dose-escalation. This enabled us to relate clinical findings to theneurobiological correlate of SERT occupancy.
Methods
ParticipantsFollowing approval by the institutional ethical committee and written informed consent, werecruited outpatients (18-70 years) from primary care, our outpatient department, and publicpsychiatric settings between October 2003 and February 2007. Inclusion criteria were: MDDdetermined by the structured clinical interview for DSM-IV (SCID),32 and a Hamilton DepressionRating Scale (17 items; HDRS17)33 score above 18. All participants were drug-free or had undergoneno more than one antidepressant treatment (other than paroxetine) at an effective dose for ≥ 6weeks for the present MDD-episode. By the latter criterion, we avoided treatment resistance aspotential bias for inefficacy of dose-escalation. Exclusion criteria, apart from pregnancy (or wish),were bipolar disorder, psychotic features, neurological cognitive impairments (i.e. dementia),

Chap
ter
Chapter 9
9
176
primary anxiety and/or substance abuse disorders and acute, severe suicidal ideation. Contrary,we allowed secondary co-morbid anxiety and/or substance abuse to increase applicability of ourfindings.
InterventionsPatients were treated by their referring physician or were referred to our outpatient department.After assessment at study-entry, all patients were treated open-label with paroxetine 20 mg/dayfor 6 weeks (see Figure 2.1). When severe adverse effects occurred, dosages were reduced to 10mg/day and again increased to 20 mg/day after one week. We randomized all patients who did notachieve ≥50% decrease in HDRS17-score after 6 weeks, relative to study entry. They received a trueparoxetine or a placebo dose-escalation added to paroxetine 20 mg/day. Dose-escalation wasprovided in blue capsules containing 10 mg paroxetine or placebo. Randomization was stratifiedfor treatment setting (SPECT-group, outpatient department AMC, primary care, publicpsychiatry), gender and age. Within strata, we applied a minimization method to achieve abalanced distribution. We concealed allocation by using an independently operated computerprogram.
Dose-escalation consisted of incremental steps of one capsule every 5 days towards amaximum of 50 mg/day (20 mg + 3 capsules). Patients were allowed to increase at a slower pace(e.g. by 7 days) or stop further escalation (e.g. 20 mg + 2 capsules) according to adverse effects.34
No dosage adjustments were allowed during the last 3 weeks of the study. We checkedadherence by pill-counts and anamnesis.35
Outcomes and measurementsPrimary clinical outcomes were HDRS17-scores, and the proportion of patients achieving response(≥50% decrease in HDRS17) or remission (HDRS17 ≤7). Secondary outcomes were total and specific(adverse effects / inefficacy) dropout rates, the Maier and Bech 6 item subscales of theHDRS17,36;37 the Inventory for Depressive Symptomatology self-rated (IDS-SR30)38 scores, theoccurrence of adverse effects and health-related quality of life (MOS-SF36; physical and mentalcomponent scales standardized to a general Dutch population).39
We administered questionnaires at study-entry, randomization (T0), and 6 weeks afterrandomization (T1). Depressive symptoms were also monitored at week 1, 2 and 4 using the Maierand Bech subscales and IDS-SR30 (see Figure 2.1). Three trained investigators administeredclinician-rated questionnaires. Agreement between raters was good (intraclass correlationcoefficient = 0.98). Raters and patients were blinded for treatment.
Subgroup for SPECT imagingFrom all patients who entered the trial, we recruited patients who were drug-free (>4 weeks and≥5 half-lives of a previous antidepressant) as potential candidates for SPECT imaging. Thesepatients were asked to participate in the SPECT sub-study if their age was between 25-55 years toreduce variability in SERT measurements by age.40 Forty-nine patients could thus be recruited fora first SPECT scan. None of these patients reported past or present use of 3,4-methylenedioxymethamphetamine. We made a second scan in those patients who completed 6weeks of paroxetine treatment (n= 44; including 12 responders), while only randomized non-responders (n= 32) were invited for a third scan at the end of the study. We treated SPECTpatients at the AMC outpatient department. Medication was supplied in pillboxes.
SPECT imaging and analysisWe performed SPECT imaging at study-entry (baseline-scan), T0 and T1 (see Figure 2.1) between 2to 10 pm according to previously described procedures.41 We made all scans 230 ±18 (SD) minutesafter intravenous injection of approximately 100 MBq iodine-123-labeled 2β-carbomethoxy-3β-(4-iodophenyl)-tropane ([123I]β-CIT), when the radioligand is at equilibrium for SERT binding in brainareas expressing high densities of SERTs.42 To prevent thyroid uptake of [123I], all subjects received

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
177
oral potassium-iodide solution. We performed SPECT imaging using a 12-detector single slicebrain-dedicated scanner (Neurofocus 810, Strichmann Medical Equipment; Cleveland, OH) with afull-width at half-maximum resolution of 6.5 mm, throughout the 20 cm field-of-view (http://www.neurophysics.com). Blood for paroxetine serum concentrations (PSC) was collected at T0and T1 immediately before scanning. Serum was stored at -20° C until analysis. PSC weredetermined in May 2007 using a validated HPLC-MS/MS method (therapeutic range 10-75 µg/L; seeappendix). The lower limit of quantification was 5 µg/L, the lower limit of detection was 0.3 µg/L.
After attenuation correction and reconstruction in 3D mode (http://www.neurophysics.com),we defined regions of interest (RoIs) for midbrain, diencephalon and cerebellum by usingvalidated templates (see Figure 2.3).41 One examiner, blinded for scan session (baseline-scan/T0/T1), positioned all RoIs in two series. Intraclass correlation coefficients were >0.97 for all RoIs. Ifthe two series differed by >5%, scans were re-evaluated by a second investigator. In the analyseswe averaged the counts for the two series.
Using activity in cerebellum as indicator of non-displaceable activity (non-specific binding andfree radioactivity),43 we calculated specific to non-specific binding ratios per scan as BPND = . BPND is proportional to transporter number under equilibriumconditions.44 In a different study, we found high reproducibility of SERT imaging with [123I]β-CITSPECT after repeated scanning of subjects, using the same camera and scanning-protocol (de Winet al., submitted). As primary outcomes, we calculated SERT occupancies at T0 or T1 relative tountreated baseline-scan SERT availability: OCCT0 or T1 = .
Power and interim analysis We performed a-priori power-calculations for two co-primary endpoints: (a) To detect a differenceof ≥5-points in HDRS17 scores between paroxetine and placebo dose-escalation, while assuming acommon standard deviation of 7 and using a one-tailed α = 0.025 and β = 0.05, sample sizes of 60per group were required; (b) For response rates (assumed to be 50% and 30% for paroxetine vs.placebo dose-escalation) a two-tailed α = 0.05 and β = 0.20 required 110 participants per group.Because previous dose-escalation studies indicated no benefits relative to placebo dose-escalation, we planned an interim analysis after SPECT data for had been collected on at least 30randomized patients in the SPECT subgroup. Stopping criteria, using the most informativecontinuous scores in a mixed model, were predetermined using the O'Brien and Fleming45
approach, were undisclosed while performing the interim analysis, and were p<0.0026 in case ofsuperiority and p>0.50 for futility.
Data analysesAnalyses were performed while blinded for treatment allocation. We based endpoint analyses onintention to treat (ITT), with last-observation carried forward (LOCF). To examine theeffectiveness of paroxetine vs. placebo dose-escalation, we compared the proportion of patientswith response, remission and drop-outs at the end of study using χ2 or Fisher’s exact test. Weexamined differences in mean continuous endpoints by ANCOVA with treatment as factor andvalue at randomization (T0) as covariate.
We used linear mixed models to assess differences in trends over time between groups inMaier, Bech and IDS-SR30 scores. Mean scores for these questionnaires were modeled as afunction of the randomized group (paroxetine vs. placebo dose-escalation), score atrandomization, and time since randomization (categorical, four levels). The interaction betweentime*group was added to the model to test whether trends over time were different between thetwo treatment groups. We used the Akaike Information Criteria to choose the best fittingvariance/covariance structure (unstructured, compound symmetry or first-order auto-regressive)for each outcome parameter.
To examine changes in SERT occupancy between T0 and T1, we used ANCOVAs with treatmentas factor, and SERT occupancy at T0 and age as covariates. In order to obtain maximuminformation of dose-escalation in these analyses, we excluded patients that were likely non-adherent to paroxetine at T0 or T1 (PSC < 5 μg/l). Thereafter, we plotted SERT occupancy against
CER
CERROI
ActivityActivityActivity −
( )BslND
TorTNDBslND
BPBPBP 10−

Chap
ter
Chapter 9
9
178
dose and PSC. We modeled dose-response in an Emax model as OCC= , in which arepresents maximal SERT occupancy and b the PSC with 50% SERT occupancy.18;20-22;27;28 Wecalculated a and b by fitting a nonlinear regression model that minimizes the sum of squares ofthe residuals. For quantification of differences in SERT occupancy between final responders andnon-responders, we used ANCOVA models corrected for differences at T0, age and baseline-scanSERT availability in diencephalon.46 We performed all analyses in SPSS v15.0.1.1 (www.spss.com ).
Results
Patient dispositionOne-hundred and seven patients (mean age 43.8 ±9.8) started open-label paroxetine (Figure 9.1).The response-rate in the open phase was 27/107 (25.2%), and 60 non-responding patients wererandomized for the double-blind phase. Randomization over the 2 treatment-arms resulted incomparable groups (Table 9.1). Fifty-one patients completed the 6 week randomization phaseincluding 31 from the SPECT study. We obtained at least 1 post-randomization HDRS17-score for 57patients.
HDRS17 scores at study-entry (-6 weeks) were comparable in T0 responders vs. non-responders (ANOVA, F1,85= 1.972, p= 0.164). At T0, HDRS17 scores (±SD) were 7.8 ±3.62 (66.6 ±14.3%decrease) in responders vs. 20.5 ±6.25 (17.7 ±20.3% decrease) in non-responders.
Figure 9.1. Recruitment and flow of participants.
* Three patients who refused dose-escalation after randomization, never ingested study drugs and refused furtherquestionnaires, were excluded for endpoint analysis.† One SPECT patient dropped out early due to inefficacy, but for all SPECT-patients clinical data could be obtained. For SPECTanalyses, 6 patients were excluded: 1 patient missed the T1 scan (placebo dose-escalation), 3 patients were likely non-adherent at T0 (paroxetine serum concentration <5μg/l; all paroxetine dose-escalation), and 2 were likely non-adherent at T1(1 paroxetine dose-escalation, 1 placebo dose-escalation).
)(*
PSCbPSCa+
��� ��� �������� �������������
5������������ 5���� ����� ���
'�!�� ��� "�� �" 5������
'������� �� ��������5������
#!����� ������ �� ���������� 5���$��
����� �� � ������ 5���+�
%����������������� ���� �� 5������
�� ���&����� ���'�������� �� #���� �� 5���(�
3������ ������������ ���� �� 5�����
��������� 5�����
� !�� ����� 5������
�� � ������ 5���)�
"�� ��"� ���� �������5�����
3������ ��� ��* �� 5�����
��������� 5���+�
� !�� ����� 5���$�
�� � ������ 5�����
�����&��� �������� ����5����� ��� �� ���.��� ����������
� ���������� ���.�������� ��
����������
���� ��
����� ��������
��������
�� �����
���������� �������� �� ���������� �� ���.��� ��������+�
� ���������� ���.����������
������ ��� �� ��� ���� ��� �������� ���.�� !�"��! �����#�
�������������
$� ����� ��������� ��� ��"%��������&�
3���! ����'�(� � ������
���))����� ������
��*�� � �))�� ������
� (�� ��� �� ������
���������� ��� ���� ��� �������� ���.�� !�"��! �����#�

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
179
Table 9.1. Characteristics of non-responding MDD patients after 6 weeks of open treatment with paroxetine 20 mg/day (trial population).
All patients (n= 60) SPECT subgroup (n= 32)
Paroxetine DE (n= 30) Placebo DE (n= 30) Paroxetine DE (n= 16) Placebo DE (n= 16)
Age at baseline (years) 41.9 ±9.1 42.9 ±10.3 42.5 ±7.7 40.4 ±7.7
Female sex - n (%) 21 (70.0) 19 (63.3) 11 (68.8) 10 (62.5)
Marital status - n (%) Single (never married) Married Divorced WidowedEducational level - n (%) Low Intermediate HighUnemployed - n (%) Income €/month (median; 25 & 75 quartiles)
15 (51.7) 8 (27.6) 4 (13.8) 2 (6.9)
6 (20.0) 19 (63.3) 5 (16.7) 6 (20.0) 1485 (875-1769)
12 (40.0) 6 (20.0) 11 (36.7) 1 (3.3)
11 (36.7) 15 (50.0) 4 (13.3) 11 (36.7) 1197 (767-1820)
6 (37.5) 7 (43.8) 2 (12.5) 1 (6.3)
2 (12.5) 10 (62.5) 4 (25.0) 4 (25.0) 1177 (715-1530)
5 (31.3) 4 (25.0) 7 (43.8) 0
3 (18.8) 10 (62.5) 3 (18.8) 4 (25.0) 1185 (610-2277)
Current smoking - n (%) Alcohol use: n (%) ≤ 2 Units/week 3-7 Units/ week 8-21 Units/ week >22 Units/ week
13 (43.3)
20 (66.7) 6 (20.0) 2 (6.7) 2 (6.7)
16 (53.3)
21 (70.0) 7 (23.3) 1 (3.3) 1 (3.3)
6 (37.5)
10 (62.5) 4 (25.0) 1 (6.3) 1 (6.3)
11 (68.8)
11 (68.8) 5 (31.3) 0 0
Race - n (%) Caucasian Creole Asian Other
17 (56.7) 4 (13.3) 6 (20.0) 3 (10.0)
19 (63.3) 7 (23.3) 1 (3.3) 3 (10.0)
9 (56.2) 2 (12.5) 3 (18.8) 2 (12.5)
13 (81.2) 3 (18.8) 0 0
MDD HDRS17 at study-entry (-6 weeks) HDRS17 (T0)*
Maier at (T0)*
Bech (T0)*
IDS-SR30 (T0)*
First episode - n (%) No of episodes Drug-naïve - n (%) Used AD in current episode - n (%)Melancholic - n (%) Duration of episode: n (%) <5 months duration 5months – 2 years duration > 2 yearsAge of first episode (years)
24.5 ±4.7 20.1 ±6.6 10.0 ±3.0 10.5 ±2.9 38.1 ±12.3 17 (56.7) 1.6 ±0.8 24 (80.0) 3 (10.0) 23 (76.7)
9 (30.0) 19 (63.3) 2 (6.7) 37.1 ±9.1
25.5 ±5.0 21.0 ±5.9 10.5 ±3.1 11.3 ±3.0 40.8 ±12.2 21 (70.0) 1.7 ±1.8 19 (63.3) 5 (16.7) 19 (63.3)
5 (16.7) 22 (73.3) 3 (10.0) 38.1 ±11.8
25.6 ±5.0 21.3 ±7.4 10.4 ±3.6 11.0 ±3.4 40.3 ±14.6 9 (56.3) 1.6 ±0.8 14 (87.5)†
1 (6.3) 12 (75.0)
7 (43.8) 7 (43.8) 2 (12.5) 38.4 ±8.8
24.4 ±4.6 19.3 ±5.1 10.1 ±2.9 10.8 ±3.1 39.9 ±11.8 9 (56.3) 2.3 ±2.4 8 (50.0)†
2 (12.5) 12 (75.0)
3 (18.8) 11 (68.8) 2 (12.5) 34.4 ±10.2
Co-morbidity – n (%) Anxiety disorder Dysthymia Drug (alcohol, cannabis, benzodiazepines) abuse / dependence
5 (16.7) 2 (6.7) 2 (6.7)
7 (23.3) 0 1 (3.3)
0 1 (6.3) 1 (6.3)
4 (25.0) 0 0
MOS-SF36 Physical*
Mental*41.2 ±9.0 26.1 ±8.4
40.3 ±10.7 25.7 ±10.2
41.2 ±8.6 25.3 ±5.9
43.9 ±9.9 22.3 ±6.7
SERT availability baseline-scan (BPND) Midbrain DiencephalonSERT occupancy (% of BPND in Bsl-scan)* ‡
Midbrain Diencephalon
N/A N/A
N/A N/A
N/A N/A
N/A N/A
0.553 ±0.119 1.157 ±0.226
73.7±17.1 63.8 ±15.4
0.657 ±0.217 1.134 ±0.247
82.2 ±18.5 70.3 ±12.1
Numbers represent means (±standard deviation) unless specified otherwise.* at randomization (T0).† sign. difference between SPECT patients randomized to conditions (Fisher’s exact p = 0.026).‡ n = 29; excluding three patients who were likely nonadherent at T0 (PSC <5μg/l). BPND = Binding Potential (non-displaceable), DE = dose-escalation, MDD = Major Depressive Disorder, PSC = Paroxetine serum concentration

Chap
ter
Chapter 9
9
180
Clinical effectiveness of paroxetine vs. placebo dose-escalationDuring dose-escalation (T0-T1), 1, 8 and 21 patients reached final doses of 30, 40 and 50 mg/dayrespectively. The placebo group escalated to a comparable number of capsules (χ2= 0.895, df= 2,p= 0.639). Adherence based upon pill-counts was comparable between both groups (Fisher’sexact, p= 0.492).
Paroxetine dose-escalation did not yield better outcomes in depression severity and health-related quality of life compared to placebo dose-escalation (ITT; Table 9.2). The robustness of thisfinding was confirmed in the longitudinal analysis (mixed model). Changes over time in the Maiersubscale and IDS-SR30-scores (Figure 9.2), and Bech subscale-scores (available on request), werecomparable between the two groups. Overall dropout was higher with placebo (26.7%) than withparoxetine dose-escalation (3.3%; p= 0.03). Paroxetine dose-escalation had significantly moreadverse effects than placebo dose-escalation, but this did not result in higher discontinuationrates due to adverse effects (Table S9.2). Instead, adverse effects by paroxetine dose-escalationmoderately decreased over time, suggestive of habituation.
Table 9.2. Depression and health-related quality of life scores after 6 weeks paroxetine vs. placebo dose-escalation (T1); all patients and SPECT subgroup.
All patients (n = 57) SPECT subgroup (n = 32)
Paroxetine DE (n = 30) Placebo DE (n = 27) p Paroxetine DE (n = 16) Placebo DE (n = 16) p
Mean dosage mg/day
46.7 ±1.00 NA - 46.9 ±1.20 NA -
HDRS17 16.1 ±1.22 15.3 ±1.28 0.650 15.8 ±1.39 14.5 ±1.39 0.519
Maier subscale 7.5 ±0.61 7.5 ±0.64 1.000 7.6 ±0.69 7.4 ±0.69 0.868
Bech subscale 8.1 ±0.63 8.1 ±0.66 0.996 8.2 ±0.75 8.0 ±0.75 0.832
Response* - n (%) 10 (33.3) 10 (37.0) 0.788 4 (25.0) 8 (50%) 0.273
Remission† - n (%) 4 (13.3) 2 (7.4) 0.673 1 (6.3) 2 (12.5) 1.000
IDS-SR30‡ 34.8 ±1.83 32.5 ±2.05 0.406 39.2 ±2.38 32.0 ±2.38 0.042
MOS-SF36§
Physical 41.8 ±0.93 42.4 ±1.06 0.679 42.5 ±1.33 44.1 ±1.33 0.399
Mental 29.6 ±1.37 27.3 ±1.57 0.264 27.2 ±1.78 25.9 ±1.78 0.620
Scores at endpoint of the study are based on intention to treat, with last observation carried forward for early drop-outs. Values are means ±standard error, corrected for (mean) scores at randomization (T0) (ANCOVA).* ≥50% decrease in HDRS17 with baseline score (-6 weeks) as reference; Fisher’s exact test.† HDRS17 ≤7; Fisher’s exact test.‡ Due to missing values: n = 29 paroxetine DE and n = 23 placebo DE for all patients.§ Due to missing values: n = 29 paroxetine DE and n = 22 placebo DE for all patients. DE = dose-escalation.
Figure 9.2. Changes over time in Maier and IDS-SR scores after randomization.
Points represent mean Maier (A) and IDS-SR (B) scores (±SEM) adjusted for scores at randomization (T0) for paroxetine (n=30) and placebo (n= 27) dose-escalation. Mixed model analysis (Maier: n= 57, IDS-SR: n= 53): overall difference betweenparoxetine vs. placebo dose-escalation for Maier scores F4,101,400 = 0.295, p= 0.880, and for IDS-SR F4,45,519= 1,516; p= 0.213. * Mean IDS-SR score at week 4 differed significantly in favor of placebo dose-escalation (t49,161= 2.11; p= 0.040).DE = dose-escalation.
� � ���
��
��
��
��
��
�
�
���� ������� ������ �
����������
��� ��
� � � ��
�
�
��
����������������� ��
����� ������� ������ �
��������
��� ��
� �

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
181
SERT occupancy and clinical responseOf 32 randomized patients in the SPECT subgroup, only 3 (9%) previously used mirtazapine orfluoxetine in the current episode (1 patient stopped mirtazapine 4 weeks prior to scanning, othersstopped <2 months before scanning). One patient missed the T1 scan. Based upon PSC atrandomization or T1, five patients in the SPECT-subgroup (4 with paroxetine and 1 with placebodose-escalation; Fisher’s Exact: p= 0.172; see Figure 9.1) with PSC <5 μg/l were considered non-adherent, despite adherence according to pill-counts. We excluded these five patients foranalyses of changes in SERT occupancy after dose-escalation, leaving 26 T1 scans analyzable fordiencephalon occupancies after true or placebo dose-escalation. Paroxetine dose-escalationincreased mean PSC from 36.2 to 154.3 μg/l (paired t-test; p<0.001), while mean PSC in placebodose-escalation remained unchanged (Table S9.3). At randomization (T0), mean SERToccupancies (±SEM) for the paroxetine dose-escalation group were 76.2 ±4.70% in midbrain and64.3 ±4.60% in diencephalon. For the placebo dose-escalation group these were 84.6 ±4.95% and72.2 ±3.08%, respectively. Neither paroxetine nor placebo dose-escalation significantly increasedSERT occupancy further (Figure 9.3A). Plotting PSC vs. SERT occupancy showed that PSCs >50 μg/l were not associated with further increases of SERT occupancy in midbrain or diencephalon(Figure 9.3BC). Furthermore, individual changes in PSC (T0 to T1) were not significantly associatedwith changes in occupancy in midbrain (F1,24= 0.101; p= 0.754) and diencephalon (F1,27= 1.332; p=0.259; Figure S9.1).
Figure 9.3. SERT occupancy during randomized dose-escalation of paroxetine. Changes over time and relation with paroxetine serum concentration.
A. Mean SERT occupancy (±SEM) for paroxetine dose-escalation and placebo dose-escalation at randomization (T0) and after6 weeks of dose-escalation (T1). SERT occupancy was calculated as percentage of initial available SERTs (expressed as BPND)at baseline (-6 weeks) scans (see text). Changes in SERT occupancy between T0 and T1 for paroxetine dose-escalation andplacebo dose-escalation were non-significant in ANCOVA models correcting for age and differences in T0 SERT occupancy(MIDBRAIN: F1,21= 0.167; p= 0.687; DIENCEPHALON: F1,22 = 0.409; p= 0.529). For 1 patient insufficient midbrain was scanned atstudy entry to compute subsequent SERT occupancies.B & C. Data for randomized patients used from both T0 (open circles) and T1 (diamonds). Dose - occupancy relationships aremodeled as OCC= . For Midbrain (B): a = 86.0 ±4.03 (SE), b= 2.65 ±1.39 (n= 30 at T0 and n= 27 at T1); for Diencephalon(C) a= 73.3 ±2.28, b = 3.97±1.07 (n= 32 at T0 and n= 29 at T1). Dashed lines represent 95% confidence interval of fitting.DE = dose-escalation, OCC = occupancy, PAR = paroxetine, PLAC = placebo. PSC = paroxetine serum concentration.
������ ������� ������ ��������
�
�
��
��
�� �������������� �����������
����� ����� ����������
�������������� ��������������
������'�*
�������+����
������'�����'
�� !
� "� �� "� �� "� #���
�
�
��
��
��
�
�������������� �����������
����,���'��$���&���'��'������'�� �%&�!
������'�*
�� !
� "� �� "� �� "� #���
�
�
��
��
��
�
�������������� �����������
����,���'��$���&���'��'������'�� �%&�!
������'�*
�� !
�
� �
)(*
PSCbPSCa+

Chap
ter
Chapter 9
9
182
We explored whether SERT occupancy was related to clinical response at T1 irrespective ofparoxetine or placebo dose-escalation. Responders at T1 (n= 12) had numerically higher SERToccupancy (±SEM) in midbrain (91.2 ±5.8%) and diencephalon (69.2 ±2.8%) than non-responders(n= 14; 77.8 ±5.1% and 63.8 ±2.6%, respectively; ANCOVA: p= 0.107 and p= 0.178). These models usedbaseline-scan diencephalon SERT availability as covariate as this accounted for a major part ofvariance (F1,21= 4.831, p= 0.039 and F1,22= 10.407, p= 0.004 respectively). Contrary, T1 SERToccupancy in midbrain or diencephalon did not significantly predict the percentage decrease inHDRS17 in linear regression, or response status in logistic regression (neither when corrected forbaseline-scan SERT availability in diencephalon or age).
Discussion
In this randomized trial we examined clinical effectiveness of dose-escalation in MDD patients,who were non-responders to 6 weeks of 20 mg/day paroxetine, and explored potential underlyingmechanisms. Despite markedly increased drug exposure, paroxetine dose-escalation to 30-50 mg/day did not improve depressive symptoms more than placebo dose-escalation, but wasassociated with more adverse effects. Concomitantly, increased paroxetine serum concentrationswere not associated with substantially greater SERT occupancy, indicating that standardparoxetine doses (20 mg/day) already resulted in maximum SERT occupancy.
Clinical outcomes of dose-escalation in non-respondersThe dose-response relationship for paroxetine was previously examined in fixed dose, parallelgroup designs. Twenty mg/day and higher paroxetine doses yielded similar clinicalimprovements.7 Similar findings were reported from parallel-group, fixed dose studies of otherSSRIs.8 Accordingly, the usefulness of dose-escalation in non-responders to paroxetine and otherSSRIs has been questioned.8-10 However, the underlying studies had methodological short-comings, and dose-escalation remains a recommended standard approach for non-responders.1;2
If dose-escalation is applied too early (before week 6), randomization of ‘late responders’ willlikely dilute the difference between true and placebo dose-escalation, resulting in potentially falsenegative findings. While one study reported randomized dose-escalation of sertraline after 6weeks of treatment, this was compromised by a non-randomized dose-increase 2 weeks prior torandomization.17
The benefits of dose-escalation might be under-estimated if actively-treated patients drop-outearly due to adverse effects34 or become more non-adherent. Our schedule for dose-escalationdid not increase drop-out. Hypothetically, patients receiving a paroxetine dose-escalation mighthave interpreted the increased level of adverse effects as subjective clue of greater drug effects,encouraging them to persevere in the trial. At first sight a misbalance in adherence is suggestedwith 4 patients with paroxetine vs. 1 patient with placebo dose-escalation having low PSCs.However, this was not due to dose-escalation. As mentioned in Figure 9.1, in three patients, lowPSCs at T0 already classified them as likely non-adherent, with only 1 paroxetine vs 1 placebo dose-escalation patient becoming likely non-adherent during dose-escalation (T0-T1). Therefore, wethink that neither adverse effects nor non-adherence account for the observed inefficacy of dose-escalation.
Thus, the present study overcomes methodological limitations of previous SSRI dose-escalation studies by using a randomized, placebo-controlled, double-blind dose-escalation innon-responders to 6 weeks treatment with a standard dose of paroxetine. We also avoidedtreatment resistance as a factor for inefficacy of dose-escalation by inclusion of patients whoreceived no more than one effective antidepressant trial for the current episode. Under theseconditions paroxetine was not superior to placebo in dose-escalation. Moreover, our study was

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
183
more inclusive than most previous ones and thus may be more applicable to ‘real-world’ first-lineantidepressant treatment. This may also explain why we observed lower response and remissionrates than previous studies.
Neurobiological effects of dose-escalationOur pharmacokinetic and imaging measurements were designed to explore why paroxetine dose-escalation would or would not improve treatment outcomes. Our imaging of SERT occupancybypasses potential bias by inclusion of patients with ultrarapid drug-metabolism, which is oftencausally linked to non-response. Hypothetically the clinical selection of non-responders eligible fordose-escalation, might represent a selection of patients not reaching high levels of SERToccupancy.
Comparing SERT occupancies of different SSRI doses faces several methodologicalchallenges. Firstly, the assessment of occupancy requires knowledge on the available number ofSERTs. Due to inter-individual differences in available SERTs, only assessments with individualdrug-free baseline-scans yield reliable data. Secondly, a given drug dose may yield a range ofserum concentrations due to inter-patient pharmacokinetic differences. Hence, associationsbased upon serum concentrations are more reliable than those based upon administered dose.Finally, intra-individual comparisons of occupancy changes following dose-escalation are morepowerful than those with historic data.
Against this background, Voineskos et al.30 recently reported high SERT occupancies instriatum (~85%), thalamus (~79%), and midbrain (~98%) in 12 depressed patients exposed to > 4weeks of venlafaxine 225-450 mg/day, sertraline 150-200 mg/day or citalopram 60-80 mg/day in a[11C]DASB PET study. They concluded that high doses significantly increased occupancy comparedto an average of 80% SERT occupancy determined in previous studies with standard SSRIdoses,20;47 which would favor the concept of dose-escalation. However, they did not determineoccupancy relative to baseline scans of the same patients without medication, nor at standarddoses. We performed drug-free study-entry scans, in addition, >90% of patients did not useantidepressants for the current episode of MDD. Furthermore, low therapeutic dosages of severalSSRIs also yielded high SERT occupancy in most studies.18;19;21-29;48 The present study resolves thiscontroversy by showing that 4-fold increases of PSC upon paroxetine dose-escalation did notsignificantly increase SERT occupancy. This offers an explanation for our findings: SERToccupancy is limited by a ceiling effect. A PSC achieved with a 20 mg/day paroxetine dose issufficient to yield maximum SERT occupancy (Figures 9.3B,C). If low doses already yield maximumSERT occupancy, dose-escalation cannot be expected to increase treatment efficacy, which is inline with our clinical findings. Our results do not necessarily challenge the relationships betweendose, SERT occupancy and clinical response but rather suggest that these relationships existmainly at low and sub-therapeutic doses. Furthermore, the relationship between SERT occupancyand response might be confounded by other factors such as SERT gene polymorphisms.
In a recent study Owens et al.49 showed increased SERT occupancy with increasing paroxetineCR doses (12.5-75 mg/day) in an ex-vivo model using human transporter transfected cells, whichmight be at odds with our findings. However, validation of this ex-vivo method (in cultured cells)with concomitant in-vivo SPECT or PET SERT occupancy (the gold standard) is not yet available.Additionally, Zitterl et al.50 found a significant relation between SERT occupancy and clinicalresponse in obsessive compulsive disorder treated with clomipramine (150 mg/day), but did notstudy the effects of dose-escalation in their study. Therefore, our study optimally quantifies theneurobiological effects of dose-escalation of antidepressants in patients.
Critique of methodsFor logistic reasons, we used [123I]β-CIT for SPECT imaging, which is a non-selective radioligand,and also binds to dopamine transporters (DAT; e.g. substantia nigra) and norepinephrinetransporter (NET; e.g. locus coeruleus).51-53 Furthermore, imaging studies indicated increasedstriatal DATbinding after treatment with paroxetine,48;54 especially when the occipital cortex wasused as a reference.54 Nevertheless, uptake in midbrain and diencephalon is considered to reflect

Chap
ter
Chapter 9
9
184
predominantly SERT,55 as these structures are rich of SERT relative to DAT and NET. Therefore,although this non-selectivity might have concealed changes in SERT occupancies due to additionalDAT or NETbinding, we think our findings in diencephalon and midbrain mainly reflect SERToccupancy. Nevertheless, it would be challenging to replicate our study using a selective ligandfor SERT like [11C]DASB for PET or [123I]ADAM for SPECT imaging.
Our study did not investigate secondary effects of paroxetine. Many adaptive pre- and post-synaptic effects of chronic administration of SSRIs have been documented, includingneuroadaptive alterations in serotonin receptors and intracellular signalling pathways,56-61 as wellas time-dependent effects on neurogenesis.62-64 Thesehypothetical additional effects of dose-escalation remain to be investigated. Nevertheless, neither the results of our trial, nor the findingsin previous randomized controlled trials indicate that dose-escalation is an efficacious strategy forSSRI non-responders in MDD.8-10
The fourfold increase of PSC after dose-escalation from 20 to 50 mg/day may questionadherence of patients in the open phase of the study. However, paroxetine inhibits thecytochrome P450 enzyme 2D6, also responsible for the metabolism of paroxetine. Thereforenonlinear increases of PSC reflect normal paroxetine pharmacokinetics.65
Our study was discontinued after an interim analysis with relatively small patient numbers.However, the criteria for premature trial termination regarding futility had been pre-specified,making it highly unlikely that we overlooked clinically relevant differences. Moreover, theneurobiological parts of our study provide a rationale why even with much larger patient numbersno substantially different outcome can be expected. On the other hand, premature stoppingreduced the power to examine whether subgroups of patients were more responsive to dose-escalation.
The present study was not designed to test the efficacy of paroxetine per se, as this is wellestablished in patients with severe MDD (HDRS17>18).1;2;5 Therefore, we did not include a pureplacebo arm. Rather we investigated dose-escalation, and accordingly only included placeboduring dose-escalation. This approach is similar to e.g. the STAR*D project, which interestinglyreported similar response rates for open treatment with citalopram.66
ConclusionPrevious studies had failed to demonstrate a clinical benefit of dose-escalation by SSRIs, but hadmethodological limitations.8-10 Addressing those limitations, our trial replicates that dose-escalation of paroxetine above the 20 mg/day standard dose has no additional clinical benefit. Asa novel extension, we revealed the underlying neurobiological mechanism for this inefficacy:maximum SERT occupancy was already reached with the standard dose. Similarly high SERToccupancies reported with low doses of other SSRIs suggest that our conclusion may beapplicable to the entire drug class. However, this does not exclude that dose-escalation hasclinical benefits for antidepressants with additional molecular targets, e.g. the norepinephrinetransporter, such as venlafaxine. This drug has shown dose-dependency of the clinical response infixed-dose studies.67;68
If dose-escalation is not promising for paroxetine and presumably other SSRIs, two clinicaloptions remain for the treatment of non-responders to standard doses. These are eithercontinuation of treatment until 10 weeks while waiting for a potential delayed response, or achange to a different and potentially more effective treatment strategy. Both strategies willfurther improve response-rates, but studies directly comparing these strategies have not yet beenperformed.

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
185
Acknowledgements
The authors wish to thank the patients in this study for their participation, and especially thankthe patients that were willing to participate in the SPECT-study. We thank all participating generalpractitioners in the area of Amsterdam Oost and Zuidoost, Hoofddorp, Nieuw Vennep, andAbcoude for their inclusions and referrals for the study. Mrs. E. Miedema M.D. and Mrs. M.C. tenDoesschate M.D. were indispensable for their help in rating questionnaires. Mrs M. Haagesmanaged randomization and maintained blinding. This study was financed by a grant from theNetherlands Organisation for Health Research and Development (ZonMw), program MentalHealth, education of investigators in mental health to H.G. Ruhé (OOG; #100-002-002). Weespecially thank Professor M.E. Thase M.D. for his constructive comments on a previous versionof the manuscript.
Conflicts of interest
All authors reported no biomedical financial interests or potential conflicts of interest.
References
1. Kennedy SH, Lam RW, Cohen NL, Ravindran AV. Clinical guidelines for the treatment of depressive disorders. IV.Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.
2. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
3. Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006; 354: 1231-1242.
4. Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D et al. Medication augmentation after the failure ofSSRIs for depression. N Engl J Med. 2006; 354: 1243-1252.
5. NICE. Clinical Guideline 23. Depression: management of depression in primary and secondary care. December 2004. 23, 1-63. 2004. London, National Institute for Clinical Excellence.
6. Quitkin FM, Petkova E, McGrath PJ, Taylor B, Beasley C, Stewart J et al. When should a trial of fluoxetine for majordepression be declared failed? Am J Psychiatry. 2003; 160: 734-740.
7. Dunner DL, Dunbar GC. Optimal dose regimen for paroxetine. J Clin Psychiatry. 1992; 53 Suppl: 21-26.8. Adli M, Baethge C, Heinz A, Langlitz N, Bauer M. Is dose escalation of antidepressants a rational strategy after a medium-
dose treatment has failed? A systematic review. Eur Arch Psychiatry Clin Neurosci. 2005; 255: 387-400.9. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard-dose selective
serotonin reuptake inhibitors in major depressive disorder: Systematic review. Br J Psychiatry. 2006; 189: 309-316.10. Baker CB, Tweedie R, Duval S, Woods SW. Evidence that the SSRI dose response in treating major depression should be
reassessed: a meta-analysis. Depress Anxiety. 2003; 17: 1-9.11. Dornseif BE, Dunlop SR, Potvin JH, Wernicke JF. Effect of dose escalation after low-dose fluoxetine therapy.
Psychopharmacol Bull. 1989; 25: 71-79.12. Schweizer E, Rickels K, Amsterdam JD, Fox I, Puzzuoli G, Weise C. What constitutes an adequate antidepressant trial for
fluoxetine? J Clin Psychiatry. 1990; 51: 8-11.13. Fava M, Rosenbaum JF, McGrath PJ, Stewart JW, Amsterdam JD, Quitkin et al. Lithium and tricyclic augmentation of
fluoxetine treatment for resistant major depression: a double-blind, controlled study. Am J Psychiatry. 1994; 151: 1372-1374.
14. Benkert O, Szegedi A, Wetzel H, Staab HJ, Meister W, Philipp M. Dose escalation vs. continued doses of paroxetine andmaprotiline: a prospective study in depressed out-patients with inadequate treatment response. Acta Psychiatr Scand.1997; 95: 288-296.
15. Schweizer E, Rynn M, Mandos LA, DeMartinis N, Garcia-Espana F, Rickels K. The antidepressant effect of sertraline is notenhanced by dose titration: results from an outpatient clinical trial. Int Clin Psychopharmacol. 2001; 16: 137-143.
16. Fava M, Alpert J, Nierenberg A, Lagomasino I, Sonawalla S, Tedlow J et al. Double-blind study of high-dose fluoxetineversus lithium or desipramine augmentation of fluoxetine in partial responders and nonresponders to fluoxetine. Journalof Clinical Psychopharmacology. 2002; 22: 379-387.
17. Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first-line sertralinetreatment: A randomised study of extended duration of treatment, dose increase or mianserin augmentation.Psychopharmacology (Berl). 2002; 161: 143-151.

Chap
ter
Chapter 9
9
186
18. Kent JM, Coplan JD, Lombardo I, Hwang DR, Huang Y, Mawlawi O et al. Occupancy of brain serotonin transporters duringtreatment with paroxetine in patients with social phobia: a positron emission tomography study with [11C]McN 5652.Psychopharmacology (Berl). 2002; 164: 341-348.
19. Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K et al. Occupancy of serotonin transporters by paroxetineand citalopram during treatment of depression: a [11C]DASB PET imaging study. Am J Psychiatry. 2001; 158: 1843-1849.
20. Meyer JH, Wilson AA, Sagrati S, Hussey D, Carella A, Potter WZ et al. Serotonin Transporter Occupancy of Five SelectiveSerotonin Reuptake Inhibitors at Different Doses: An [11C]DASB Positron Emission Tomography Study. American Journalof Psychiatry. 2004; 161: 826-835.
21. Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB et al. Acute occupancy of brain serotonintransporter by sertraline as measured by [11C]DASB and positron emission tomography. Biol Psychiatry. 2006; 59: 821-828.
22. Suhara T, Takano A, Sudo Y, Ichimiya T, Inoue M, Yasuno F et al. High levels of serotonin transporter occupancy with low-dose clomipramine in comparative occupancy study with fluvoxamine using positron emission tomography. Arch GenPsychiatry. 2003; 60: 386-391.
23. Erlandsson K, Sivananthan T, Lui D, Spezzi A, Townsend CE, Mu S et al. Measuring SSRI occupancy of SERT using the noveltracer [123I]ADAM: a SPECT validation study. Eur J Nucl Med Mol Imaging. 2005; 32: 1329-1336.
24. Hiltunen J, Akerman KK, Kuikka JT, Bergstrom KA, Halldin C, Nikula T et al. Iodine-123 labeled nor-β-CIT as a potentialtracer for serotonin transporter imaging in the human brain with single-photon emission tomography. Eur J Nucl Med.1998; 25: 19-23.
25. Klein N, Sacher J, Geiss-Granadia T, Mossaheb N, Attarbaschi T, Lanzenberger R et al. Higher serotonin transporteroccupancy after multiple dose administration of escitalopram compared to citalopram: an [123I]ADAM SPECT study.Psychopharmacology (Berl). 2007; 191: 333-339.
26. Klein N, Sacher J, Geiss-Granadia T, Attarbaschi T, Mossaheb N, Lanzenberger R et al. In vivo imaging of serotonintransporter occupancy by means of SPECT and [123I]ADAM in healthy subjects administered different doses ofescitalopram or citalopram. Psychopharmacology (Berl). 2006; 188: 263-272.
27. Takano A, Suzuki K, Kosaka J, Ota M, Nozaki S, Ikoma Y et al. A dose-finding study of duloxetine based on serotonintransporter occupancy. Psychopharmacology (Berl). 2006; 185: 395-399.
28. Catafau AM, Perez V, Plaza P, Pascual JC, Bullich S, Suarez M et al. Serotonin transporter occupancy induced byparoxetine in patients with major depression disorder: a [123I]-ADAM SPECT study. Psychopharmacology (Berl). 2006; 189:145-153.
29. Herold N, Uebelhack K, Franke L, Amthauer H, Luedemann L, Bruhn H et al. Imaging of serotonin transporters and itsblockade by citalopram in patients with major depression using a novel SPECT ligand [123I]-ADAM. J Neural Transm. 2006;113: 659-670.
30. Voineskos AN, Wilson AA, Boovariwala A, Sagrati S, Houle S, Rusjan P et al. Serotonin transporter occupancy of high-doseselective serotonin reuptake inhibitors during major depressive disorder measured with [11C]DASB positron emissiontomography. Psychopharmacology (Berl). 2007; 193: 539-545.
31. Cavanagh J, Patterson J, Pimlott S, Dewar D, Eersels J, Dempsey MF et al. Serotonin transporter residual availabilityduring long-term antidepressant therapy does not differentiate responder and nonresponder unipolar patients. BiolPsychiatry. 2006; 59: 301-308.
32. First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders Patient Edition(SCID-I/P version 2.0). Translated in Dutch by Groenestijn MAC, Akkerhuis GW, Kupka RW, Schneider N, and Nolen WA.1999. Lisse, the Netherlands, Swets & Zeitlinger B.V.
33. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.34. Baker CB, Woods SW. Is there a SSRI dose response in treating major depression? The case for re-analysis of current data
and for enhancing future study design. Depress Anxiety. 2003; 17: 10-18.35. Saunders K, Simon G, Bush T, Grothaus L. Assessing the feasibility of using computerized pharmacy refill data to monitor
antidepressant treatment on a population basis: a comparison of automated and self-report data. J Clin Epidemiol. 1998;51: 883-890.
36. Faries D, Herrera J, Rayamajhi J, DeBrota D, Demitrack M, Potter WZ. The responsiveness of the Hamilton DepressionRating Scale. J Psychiatr Res. 2000; 34: 3-10.
37. Ruhe HG, Dekker JJ, Peen J, Holman R, de Jonghe F. Clinical use of the Hamilton Depression Rating Scale: is increasedefficiency possible? A post hoc comparison of Hamilton Depression Rating Scale, Maier and Bech subscales, ClinicalGlobal Impression, and Symptom Checklist-90 scores. Compr Psychiatry. 2005; 46: 417-427.
38. Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The Inventory of Depressive Symptomatology (IDS):psychometric properties. Psychol Med. 1996; 26: 477-486.
39. Aaronson NK, Muller M, Cohen PD, Essink-Bot ML, Fekkes M, Sanderman R et al. Translation, validation, and norming ofthe Dutch language version of the SF-36 Health Survey in community and chronic disease populations. J Clin Epidemiol.1998; 51: 1055-1068.
40. van Dyck CH, Malison RT, Seibyl JP, Laruelle M, Klumpp H, Zoghbi SS et al. Age-related decline in central serotonintransporter availability with [123I]β-CIT SPECT. Neurobiol Aging. 2000; 21: 497-501.
41. de Win MM, Habraken JB, Reneman L, van den Brink W, den Heeten GJ, Booij J. Validation of [123I]β-CIT SPECT to assessserotonin transporters in vivo in humans: a double-blind, placebo-controlled, crossover study with the selective serotoninreuptake inhibitor citalopram. Neuropsychopharmacology. 2005; 30: 996-1005.
42. Pirker W, Asenbaum S, Hauk M, Kandlhofer S, Tauscher J, Willeit M et al. Imaging serotonin and dopamine transporterswith [123I]β-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000; 41: 36-44.
43. Laruelle M, Vanisberg MA, Maloteaux JM. Regional and subcellular localization in human brain of [3H]paroxetine binding,a marker of serotonin uptake sites. Biol Psychiatry. 1988; 24: 299-309.
44. Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN et al. Consensus nomenclature for in vivo imaging ofreversibly binding radioligands. J Cereb Blood Flow Metab. 2007; 27: 1533-1539.

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
187
45. O'Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics. 1979; 35: 549-556.46. Kugaya A, Sanacora G, Staley JK, Malison RT, Bozkurt A, Khan S et al. Brain serotonin transporter availability predicts
treatment response to selective serotonin reuptake inhibitors. Biol Psychiatry. 2004; 56: 497-502.47. Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. J
Psychiatry Neurosci. 2007; 32: 86-102.48. Kugaya A, Seneca NM, Snyder PJ, Williams SA, Malison RT, Baldwin RM et al. Changes in human in vivo serotonin and
dopamine transporter availabilities during chronic antidepressant administration. Neuropsychopharmacology. 2003; 28:413-420.
49. Owens MJ, Krulewicz S, Simon JS, Sheehan DV, Thase ME, Carpenter DJ et al. Estimates of Serotonin and NorepinephrineTransporter Inhibition in Depressed Patients Treated with Paroxetine or Venlafaxine. Neuropsychopharmacology. 2008;Advance online publication; doi:10.1038/npp.2008.47.
50. Zitterl W, Aigner M, Stompe T, Zitterl-Eglseer K, Gutierrez-Lobos K, Wenzel T et al. Changes in Thalamus-HypothalamusSerotonin Transporter Availability during Clomipramine Administration in Patients with Obsessive-Compulsive Disorder.Neuropsychopharmacology. 2008; Advance online publication; doi:10.1038/npp.2008.35.
51. Innis R, Baldwin R, Sybirska E, Zea Y, Laruelle M, al Tikriti M et al. Single photon emission computed tomography imagingof monoamine reuptake sites in primate brain with [123I]CIT. Eur J Pharmacol. 1991; 200: 369-370.
52. Neumeyer JL, Wang SY, Milius RA, Baldwin RM, Zea-Ponce Y, Hoffer PB et al. [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane: high-affinity SPECT radiotracer of monoamine reuptake sites in brain. J Med Chem. 1991; 34: 3144-3146.
53. Neumeyer JL, Tamagnan G, Wang S, Gao Y, Milius RA, Kula NS et al. N-substituted analogs of 2 beta-carbomethoxy-3 beta-(4'-iodophenyl)tropane (beta-CIT) with selective affinity to dopamine or serotonin transporters in rat forebrain. J MedChem. 1996; 39: 543-548.
54. Booij J, de Jong J, de Bruin K, Knol R, de Win MM, van Eck-Smit BL. Quantification of striatal dopamine transporters with[123I]-FP-CIT SPECT is influenced by the selective serotonin reuptake inhibitor paroxetine: a double-blind, placebo-controlled, crossover study in healthy control subjects. J Nucl Med. 2007; 48: 359-366.
55. Laruelle M, Baldwin RM, Malison RT, Zea-Ponce Y, Zoghbi SS, al Tikriti MS et al. SPECT imaging of dopamine andserotonin transporters with [123I]β-CIT: pharmacological characterization of brain uptake in nonhuman primates.Synapse. 1993; 13: 295-309.
56. Meyer JH, Kapur S, Eisfeld B, Brown GM, Houle S, DaSilva J et al. The effect of paroxetine on 5-HT2A receptors indepression: an [18F]setoperone PET imaging study. Am J Psychiatry. 2001; 158: 78-85.
57. Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG et al. Effects of chronic antidepressanttreatments on serotonin transporter function, density, and mRNA level. J Neurosci. 1999; 19: 10494-10501.
58. Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006; 59: 1144-1150.59. Blier P, De Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci. 1994; 15: 220-
226.60. Davidson C, Stamford JA. Effect of chronic paroxetine treatment on 5-HT1B and 5-HT1D autoreceptors in rat dorsal raphe
nucleus. Neurochem Int. 2000; 36: 91-96.61. Kim SW, Park SY, Hwang O. Up-regulation of tryptophan hydroxylase expression and serotonin synthesis by sertraline.
Mol Pharmacol. 2002; 61: 778-785.62. Djavadian RL. Serotonin and neurogenesis in the hippocampal dentate gyrus of adult mammals. Acta Neurobiol Exp
(Wars). 2004; 64: 189-200.63. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat
hippocampus. J Neurosci. 2000; 20: 9104-9110.64. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;
33: 73-83.65. Paxil® Prescribing information and medication guide PXL:47PI (available at http://us.gsk.com/products/assets/
us_paxil.pdf). 1-44. 2008. Research Triangle Park, North Carolina, GlaxoSmithKline. 66. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram for
depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006; 163: 28-40.67. Thase ME, Shelton RC, Khan A. Treatment with venlafaxine extended release after SSRI nonresponse or intolerance: a
randomized comparison of standard- and higher-dosing strategies. J Clin Psychopharmacol. 2006; 26: 250-258.68. Rudolph RL, Fabre LF, Feighner JP, Rickels K, Entsuah R, Derivan AT. A randomized, placebo-controlled, dose-response
trial of venlafaxine hydrochloride in the treatment of major depression. J Clin Psychiatry. 1998; 59: 116-122.69. Parsey RV, Kegeles LS, Hwang DR, Simpson N, Abi-Dargham A, Mawlawi O et al. In vivo quantification of brain serotonin
transporters in humans using [11C]McN 5652. J Nucl Med. 2000; 41: 1465-1477.70. Laasonen-Balk T, Viinamaki H, Kuikka JT, Husso-Saastamoinen M, Lehtonen J, Tiihonen J. 123I-beta-CIT binding and
recovery from depression. A six-month follow-up study. Eur Arch Psychiatry Clin Neurosci. 2004; 254: 152-155.71. Pirker W, Asenbaum S, Kasper S, Walter H, Angelberger P, Koch G et al. β-CIT SPECT demonstrates blockade of 5HT-
uptake sites by citalopram in the human brain in vivo. J Neural Transm Gen Sect. 1995; 100: 247-256.72. Shang Y, Gibbs MA, Marek GJ, Stiger T, Burstein AH, Marek K et al. Displacement of serotonin and dopamine transporters
by venlafaxine extended release capsule at steady state: a [123I]2β-carbomethoxy-3β-(4-iodophenyl)-tropane singlephoton emission computed tomography imaging study. J Clin Psychopharmacol. 2007; 27: 71-75.
73. Takano A, Suhara T, Ichimiya T, Yasuno F, Suzuki K. Time course of in vivo 5-HTT transporter occupancy by fluvoxamine. JClin Psychopharmacol. 2006; 26: 188-191.
74. Tauscher J, Pirker W, de Zwaan M, Asenbaum S, Brucke T, Kasper S. In vivo visualization of serotonin transporters in thehuman brain during fluoxetine treatment. Eur Neuropsychopharmacol. 1999; 9: 177-179.
75. Viinamäki H, Kuikka JT, Tiihonen J, Lehtonen J. Change in monoamine transporter density related to clinical recovery: acase-control study. Nord J Psychiatry. 1998; 52: 39-44.

Chap
ter
Chapter 9
9
188
Supplementary appendix: methods of paroxetine serum concentrationsand results.
Paroxetine serum concentrationsPSC were determined in May 2007 using a previously validated HPLC-MS/MS method.A ThermoFinnigan (San Jose, CA, USA) Surveyor HPLC system was used, equipped with a Surveyorautosampler. Separation was carried out on a Agilent Eclipse XDB-CN column, 100 x 2.1 mm,particle size 3,5 µm (Agilent, Amstelveen, The Netherlands). A mixture of 60% (v/v) 2 mMammonium acetate (pH 3.2) and 40% (v/v) acetonitrile was used as mobile phase.Paroxetine wasextracted from serumby liquid-liquid extraction. Briefly, 500μL of serum samples were mixed withsodium carbonate solution (0.5M). An internal standard solution (disopyramide 20 µg/l) wasadded and the mixture was vortexed for 5 seconds. Thereafter, 1-chlorobutane/ethylacetatemixture (60/40; v/v) was added and vortexed for 10 minutes. After centrifugation for 10 minutes(3000g),the upper layer was transferred to a second vial, and evaporated under a nitrogenatmosphere at 40 °C. After addition of methanol the solution was vortexed and transferred ontothe HPLC-MS/MS system.Selected reaction monitoring (SRM) was used for drug quantification(SRM paroxetine 330.1/192.0 and SRM disopyramide 340.2/239.0).Calibration curves wereconstructed by adding known amounts of paroxetine to blank serum (5-75 µg/L range). The lowerlimit of quantification was 5 µg/L, which was 50% below the lower end of the therapeutic range ofparoxetine in serum (10-75 µg/L range). The lower limit of detection was 0.3 µg/L.

189
ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
Tabl
e S9
.1. S
tudi
es o
f SER
T oc
cupa
ncy
afte
r tre
atm
ent w
ith s
erot
oner
gic
antid
epre
ssan
ts
Aut
hor &
Dat
e (r
efer
ence
) Re
sults
Popu
latio
n (m
ean
age)
Sex
N
Follo
w-u
p D
esig
n In
terv
entio
n / c
ontr
ol
SER
T Im
ag
ing
and
RoI
Resu
ltsRe
mar
ks
Cata
fau
2006
28D
rug-
free
M
DD
pts
(36
±11)
M
+ F
10Bs
l, 4-
6 w
Pa
roxe
tine
20 m
g [12
3 I]-A
DA
M
SPEC
T, 4
h M
ID, T
hal,
Stria
tum
, Ce
r
In 9
pat
ient
s a
2nd s
can
was
mad
e.
Occ
upan
cies
wer
e: M
ID 6
6.4
±9.5
%, T
hal
63.0
±9.
6%, S
tria
t 61.3
±10
.8%
Cons
ider
able
var
iatio
n in
occ
upan
cy o
bser
ved.
A
utho
rs s
ugge
st th
at S
ERT
occu
panc
y is
pro
babl
y no
t rel
ated
to S
SRI r
espo
nse.
Cava
nagh
20
0631
MD
D p
ts tr
eate
d 24
N
/A m
ean
AD
use
26
.3-12
.8m
Varie
ty o
f dru
gs u
sed:
mon
othe
rapy
(n=
17):
ven
lafa
xine
(75-
300
mg)
, SSR
Is (2
0-60
mg)
, tric
yclic
(150
mg)
, Mirt
azap
ine
(30
mg)
; com
bina
tion(
n= 7
) co
mbi
natio
ns o
f 2 a
ntid
epre
ssan
ts,
addi
tion
of li
thiu
m, v
alpr
oate
, ca
rnba
maz
epin
e, T
3 or
ant
ipsy
chot
ics
; do
sage
s un
chan
ged
for ≥
2wee
ks
[123 I]β-
CIT
SPEC
T, 3
.3h
Br
St/D
ienc
, O
cc
No
occu
panc
y pe
rcen
tage
s av
aila
ble.
No
sign
ific
ant d
ifere
nce
in S
ERT
resi
dual
ac
tivity
bet
wee
n re
spon
ders
and
non
-re
spon
ders
. Wid
e ra
nge
of S
ERT
avai
labi
lity
Patie
nts
wer
e no
t sca
nned
bef
ore
trea
tmen
t (bs
l),
inst
ead
a co
mpa
rison
of b
indi
ng p
oten
tials
was
m
ade.
Hig
hly
hete
roge
neou
s gr
oup
cons
ider
ing
drug
trea
tmen
t.
Erla
ndss
on
2005
23H
ealth
y vo
lunt
eers
(3
2; 2
5-52
) M
16
Bsl,
2-7d
Ci
talo
pram
at d
iffer
ent d
osag
es (1
0-60
m
g) fo
r diff
eren
t dur
atio
ns (2
-7 d
ays)
. Vo
lunt
eers
wer
e ra
ndom
ized
to o
ne o
f 7
diff
eren
t dos
ing
regi
men
s
[123 I]
-AD
AM
SP
ECT,
4h
MID
, Tha
l, St
riat,
Cer
No
mea
n oc
cupa
ncy
for s
epar
ate
dosi
ng
regi
men
s gi
ven.
Max
imum
MID
oc
cupa
ncy
84%
Stud
y ai
ms
at fi
ndin
g th
e be
st ti
me
fram
e (s
igle
sc
an p
roto
col)
for s
cann
ing
with
[123 I]
-AD
AM
. Co
nsid
erab
le v
aria
tion
in re
latio
n oc
cupa
ncy
and
bloo
d-le
vels
of c
italo
pram
obs
erve
d.
Her
old
2006
29D
rug-
free
M
DD
pts
(42
±12)
M
+ F
21
Bsl,
7d
Cita
lopr
am 10
mg,
sca
ns w
ere
mad
e 6-
7 ho
urs
afte
r las
t app
licat
ion
of o
ral d
ose
[123 I]
-AD
AM
SP
ECT,
4h
MID
, Cer
In 13
pat
ient
s a
2nd s
can
was
mad
e.M
ean
MID
occ
upan
cy w
as 6
1% (r
ange
37-
88%)
Co
nsid
erab
le v
aria
tion
in o
ccup
ancy
obs
erve
d. N
o co
rrel
atio
n of
occ
upan
cy a
nd d
ecre
ase
in
depr
essi
on s
ever
ity.
Hilt
unen
1998
24H
ealth
y vo
lunt
eers
(3
0; 2
5-35
) M
+ F
5 Bs
l 1 p
atie
nt re
ceiv
ed c
italo
pram
30
mg
3hr
prio
r to
inje
ctio
n; 1
patie
nt re
ceiv
ed
cita
lopr
am 2
0 m
g, 1
patie
nt v
enla
faxi
ne
37.5
mg1
hr a
fter
inje
ctio
n.
[123 I]
nor-β
-CI
T SP
ECT,
0-
24h
a.o
. aC
G, M
ID,
Thal
, Ba
sGan
g,
Cer
Aft
er c
italo
pram
30
mg
3hr p
rior t
o in
ject
ion
spec
ific
bind
ing
in th
e m
idbr
ain
was
52%
less
than
in u
ntre
ated
sub
ject
s.
For v
enla
faxi
ne a
nd c
italo
pram
20
mg
no
data
giv
en.
Stud
y ai
ms
at e
stab
lishi
ng tr
acer
-pro
pert
ies
of
[123 I]
nor-β-
CIT.
Spe
cific
bin
ding
of [
123 I]
nor-β
-CIT
is
33%
high
er c
ompa
red
to [12
3 I]β-
CIT
(in o
ther
su
bjec
ts).
Kent
200
218Pa
tient
s w
ith
Soci
al F
obia
(3
5 ±1
1)
M +
F
5 Bs
l, 3-
6m
Paro
xetin
e 20
-40
mg
(titr
ated
by
clin
ical
re
spon
se) (
3-6m
) [11
C]M
cN56
52 P
ET M
ID,
Thal
, Str
iat,
H
ip, A
myg
, Ci
ngA
, Cer
Occ
by
paro
xetin
e: M
ID 9
8±3%
, Tha
l 81
±6%,
Str
iat 7
5±7%
, Hip
92±
12%,
Am
yg
94±7
%, C
ingA
81±
22%
Hig
her o
ccup
ancy
of S
ERT
than
aft
er a
cute
ch
alle
nge
with
hig
her d
oses
(see
69).
Une
xpec
ted
decr
ease
in V
T in
Cer
ebel
lum
and
whi
te m
atte
r,
indi
cativ
e of
dec
reas
e in
non
-spe
cifi
c di
strib
utio
n vo
lum
e of
liga
nd a
fter
chr
onic
trea
tmen
t with
pa
roxe
tine

190
Chapter 9
Chap
ter
9
Klei
n 20
0626
Hea
lthy
volu
ntee
rs
(26.
8 ±4
.3)
M
25
Bsl,
6h
Cita
lopr
am (1
0 m
g or
20
mg)
, es
cita
lopr
am (5
mg,
10 m
g, 2
0 m
g).
Volu
ntee
rs w
ere
rand
omiz
ed to
one
of
5 di
ffer
ent d
rug/
dosi
ng re
gim
ens
[123 I]
-AD
AM
SP
ECT,
4h
MID
/Tha
l, Ce
r
Occ
upan
cy o
f 60
±6%,
64
±6%
and
75 ±
5%
for 5
, 10
and
20 m
g es
cita
lopr
am. F
or 10
an
d 20
mg
cita
lopr
am o
ccup
ancy
65
±10%
an
d 70
±6%
Stud
y ai
ms
to d
eter
min
e ac
ute
occu
panc
y of
SER
T af
ter s
ingl
e do
se d
rug
adm
inis
trat
ion
of c
itaol
pram
or
esc
italo
pram
Klei
n 20
0725
Hea
lthy
volu
ntee
rs(2
8 ±7
) M
15
Bsl,
10d
Cita
lopr
am 2
0 m
g, e
scita
lopr
am 10
mg.
Vo
lunt
eers
wer
e ra
ndom
ized
to o
ne o
f th
e do
sing
gro
ups
[123 I]
-AD
AM
SP
ECT,
4h,
52
h M
ID/
Thal
, Cer
Occ
upan
cy e
scita
lopr
am 8
1.5 ±
5.4%
, ci
talo
pram
64.
0 ±1
2.7%
In
crea
sed
occu
panc
y ob
serv
ed fo
r esc
itaol
pram
, al
so re
pres
ente
d in
Em
ax c
urve
s. E
max
cur
ves
cons
truc
ted
with
dat
a fo
r 4h
and
52h
post
inje
ctio
n sc
ans
in 1
mod
el
Kuga
ya 2
003,
20
0446
;48
1. Vo
lunt
eers
(3
7.4
±14.
3)
M +
F
2. P
atie
nts
with
M
DD
(4
6.4
±7.6
) M
+ F
9 10
Bsl,
8d,
16d
Bsl,
1-3w
, 6w
Cita
lopr
am 4
0 m
g (8
d), C
ita lo
pra
m 4
0 m
g +
Bupr
o pi
on 10
0 m
g (8
-16d)
Pa
roxe
tine
20 m
g (6
w)
[123 I]β-
CIT
SPEC
T, 2
4h
BrSt
, Die
nc,
Cer
Occ
by
cita
lopr
am a
t 8d:
BrS
t 51.
4%, D
ienc
39
.4%;
no
sign
. cha
nge
ther
eaft
er O
cc b
y pa
roxe
tine
at 1-
3w: B
rSt 3
6.5%
;, D
ienc
29
.1% &
at 6
w B
rSt 3
2.6%
, Die
nc 2
3.4%
Incr
ease
d D
AT-b
indi
ng in
Str
iatu
m d
urin
g pr
olon
ged
SSR
I tre
atm
ent.
Bup
ropr
ion
(100
-200
m
g) o
nly
has
no in
fluen
ce o
n D
AT-b
indi
ng in
st
riatu
m In
stud
y 2
high
er B
sl D
ienc
SER
T av
aila
bilit
y pr
edic
ted
bett
er tr
eatm
ent r
espo
nse
at w
eek
6.
Laas
onen
-Bal
k 20
0470
Patie
nts
with
M
DD
(4
2.7
±?)
M +
F
18
Bsl,
6m
(≥1m
dru
g-fr
ee)
Ope
n tr
eatm
ent w
ith v
ario
us
antid
epre
ssan
ts (n
= 3)
and
/or
benz
odia
zepi
nes
or s
uppo
rtiv
e co
unse
lling
[123 I]β-
CIT
SPEC
T, 1h
&
21-2
4h M
ID,
Cer
Bind
ing
in M
ID w
as s
ign.
incr
ease
d in
re
spon
ders
to tr
eatm
ent (
corr
ecte
d fo
r ag
e an
d se
x)
Low
dep
ress
ion
seve
rity
at B
sl (H
DRS
13.9
±6.
7);
only
thre
e pa
tient
s re
ceiv
ed a
ntid
epre
ssan
ts, a
nd
six
benz
odia
zepi
nes o
nly.
Incr
ease
d SE
RT b
indi
ng in
re
pond
ers
sugg
ests
dec
reas
ed m
idbr
ain
dens
ity o
f SE
RT d
urin
g M
DD
Mey
er 2
00119
1. Pa
tient
s w
ith
MD
D ,
HD
RS ≥1
6 R
x-fr
ee (3
2 ±8
) M
+ F
2.
Hea
lthy
volu
ntee
rs
(age
-mat
ched
) M
+ F
12
17
Bsl,
4w
Bsl,
4w
1. O
pen
trea
tmen
t with
par
oxet
ine
20
mg
(n=
7), 1
0 m
g (n
= 1)
or c
italo
pram
20
mg
(n=
4) 2
. No
trea
tmen
t
[11C]
DA
SB
PET,
0-1
½ h
St
riat,
Tha
l, Ci
ngA
, PFC
, Ce
r
1. O
cc in
Str
iat a
fter
par
oxet
ine
20 m
g:
83%
±5, a
nd a
fter
cita
lopr
am 2
0 m
g: 7
7%
±10.
Occ
in T
hal a
fter
par
oxet
ine
20 m
g:
75-7
8% ±
8, a
nd a
fter
cita
lopr
am 2
0 m
g:
65-7
0% ±
7-15
. O
cc in
Cin
gA a
fter
pa
roxe
tine
20 m
g: 7
6-77
% ±1
5, a
nd a
fter
ci
talo
pram
20
mg:
77-
79%
±29.
No
rela
tions
hip
was
foun
d be
twee
n H
DRS
-sco
re
and
occu
panc
y le
vel i
n an
y R
oI.
Stria
tal O
cc
incr
ease
d w
ith h
ighe
r ser
um-le
vels
of p
arox
etin
e,
with
app
. 85%
occ
at s
erum
leve
ls o
f 28 μg
/l.
Ref
eren
ces
to o
ther
stu
dies
that
des
crib
e a
hype
rbol
ic re
latio
nshi
p of
ser
um le
vel a
nd
occu
panc
y. T
est-
rete
st in
hea
lthy
subj
ects
show
ed a
m
ean
diff
eren
ce o
f -3.
7% ±
3.7
(ran
ge -8
– 2
%) o
ver 4
w
eeks
. Mea
n ab
solu
te d
iffer
ence
was
10.9
% ±2
.9.
Tabl
e S9
.1. S
tudi
es o
f SER
T oc
cupa
ncy
afte
r tre
atm
ent w
ith s
erot
oner
gic
antid
epre
ssan
ts (C
ontin
ued)
Aut
hor &
Dat
e (r
efer
ence
) Re
sults
Popu
latio
n (m
ean
age)
Sex
N
Follo
w-u
p D
esig
n In
terv
entio
n / c
ontr
ol
SER
T Im
ag
ing
and
RoI
Resu
ltsRe
mar
ks
191
ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
Mey
er 2
00420
1. Pa
tient
s w
ith
MD
D
2. P
atie
nts
with
M
DD
and
co-
mor
bid
anxi
ety
diso
rder
3.
Hea
lthy
subj
ects
(2
0-50
) M
+ F
29
16
37
Bsl,
4w
MD
D: c
italo
pram
20-
40 m
g, fl
uoxe
tine
20 m
g, s
ertr
alin
e 50
-100
mg,
par
oxet
ine
20 m
g, v
enla
faxi
ne 7
5 m
g
MD
D+a
nxie
ty: c
italo
pram
40-
60 m
g,
fluox
etin
e 40
-60
mg,
ser
tral
ine
150-
200
mg,
par
oxet
ine
40-6
0 m
g, v
enla
faxi
ne
150-
225
mg
Hea
lthy
subj
ects
: cita
lopr
am
1-10
mg,
fluo
xetin
e 1-1
0 m
g, s
ertr
alin
e 5-
25 m
g, p
arox
etin
e 5-
10 m
g, v
enla
faxi
ne
2.4-
37.5
mg
[11C]
DA
SB
PET,
0-1
½ h
St
riat,
Tha
l, Ci
ngA
, PFC
, Cu
neus
, M
ID, C
er
Mea
n st
riata
l occ
was
81.
4 ±7
.2%
for
cita
lopr
am (2
0-40
mg)
,76.
2 ±7
.5 fo
r flu
oxet
ine
(20
mg)
, 85.
0 ±7
.0%
for
sert
ralin
e (5
0-10
0), 8
4.5
±6.0
% fo
r pa
roxe
tine
and
83.7
±2.
4 fo
r ven
lafa
xine
(7
5 m
g).
Occ
for T
hal:
72.3
±7.
6%, 6
9.1
±9.4
%, 7
6.8
±4.3
%, 7
4.7
±15.
7% a
nd 7
1.3
±10.
2% re
sp. O
cc fo
r MID
: 87.
5 ±7
.7%,
82.
3 ±9
.3%,
91.8
±4.
0%, 9
3.4
±7.7
% an
d 91
.0
±7.7
% re
sp.
12 p
atie
nst a
lso
desc
ribed
in M
eyer
et a
l. 20
0119
2
MD
D-p
atie
nts
rece
ived
sub
ther
apeu
tical
dos
es in
‘H
ealth
y’ g
roup
; in
5 pa
tient
s of
MD
D g
roup
do
sage
s w
ere
seco
ndar
ily in
crea
sed
and
also
an
alys
ed (t
wic
e) in
MD
D +
anx
iety
gro
up. N
o re
latio
n be
twee
n st
riata
l occ
upan
cy a
nd c
linic
al
rem
issi
on o
r per
cent
age
chan
ge in
Ham
ilton
de
pres
sion
sco
res.
Pars
ey 2
00069
Hea
lthy
volu
ntee
rs
(38
±12)
M
2 Bs
l 2
of 6
vol
unte
ers
rece
ived
par
oxet
ine
60m
or8
0 m
g pr
ior t
o sc
anni
ng
(blo
ckin
g st
udy)
; blo
ckin
g co
mpa
red
to
PET
befo
re u
sing
par
oxet
ine
[11C]
McN
5652
PET
, 0-2
½
h M
ID, T
hal,
Hip
, Am
yg,
Cing
A, m
TL,
Occ
by
60 m
g pa
roxe
tine
‘pre
trea
tmen
t’
(n=
1): A
myg
64.
8%, H
ip 4
6.0%
, Tha
l 38.
4%,
Mid
br 8
3.9%
, Cin
gA 2
6.4%
Mai
nstu
dy (n
= 6)
aim
s to
qua
ntify
cha
ract
eris
tics
and
prot
ocol
for [
11C]
McN
5652
.
Pars
ey 2
00621
Hea
lthy
volu
ntee
rs17
Bs
l, 4-
6d
Sert
ralin
e 25
mg,
50
mg
and
100
mg
(4
days
at d
esig
nate
d do
sage
) [11
C]D
ASB
PE
T, 0
-1½
h
Stria
t, T
hal,
Cing
, DL/
O/
M P
FC,
Am
yg,H
ip,
Insu
la,
MID
,Occ
, Ce
r
Aver
age
acro
ss 15
RoI
s: m
ax o
ccup
ancy
10
6.8
±8.3
% ra
nge
126.
9 ±3
5.9
in O
PFC
to
79.3
±10
.4%
in T
hal
Wid
e ra
nge
of R
oIs
take
n. S
tudy
als
o ex
amin
es
SERT
ava
ilabi
lity
in c
ereb
ellu
m a
nd e
xam
ines
its
role
as
refe
renc
e. S
peci
fic S
ERT
bind
ing
was
foun
d hi
gher
in th
e ce
rebe
llar v
erm
is c
ompa
red
to th
e ce
rebe
llar g
rey.
In c
oncl
usio
n no
idea
l ref
eren
ce
regi
on fo
r in
vivo
SER
T st
udie
s ca
n be
re
com
men
ded.
Pirk
er 19
9571
1. Pa
tient
s w
ith
MD
D
(44
26-7
1)
M +
F2.
‘Hea
lthy’
co
ntro
ls
(42.
3 24
-70)
M
+ F
13
11
1 sca
n so
me
whe
re in
tr
eatm
ent
Cita
lopr
am 2
0 m
g (n
= 5)
, 40
mg
(n=
6)
and
60 m
g (n
= 1)
for a
t lea
st o
ne w
eek;
on
e un
trea
ted
patie
nt.
[123 I]β-
CIT
SPEC
T, 2
, 4,
16, 2
0, 2
4h
Thal
, hy
poTh
al,
MID
, Pon
s,
Cer
No
diff
eren
ce in
bin
ding
in S
tria
t be
twee
n pa
tient
s an
d co
ntro
ls.
Cita
lopr
am p
atie
nts
show
ed s
ign.
de
crea
se in
Tha
l, hy
poTh
al, M
ID
com
pare
d to
con
trol
s. N
o di
ffer
ence
in
bind
ing
betw
een
patie
nts
with
ci
talo
pram
20
mg
or 4
0 m
g.
Stud
y in
clud
ed p
atie
nts
with
bip
olar
dis
orde
r (n=
1)
and
conv
ersi
on d
isor
der (
n= 1)
. Con
trol
gro
up
incl
uded
4 p
atie
nts
with
per
iphe
ral n
euro
logi
cal
dise
ase.
Cita
lopr
am-d
osin
g w
as n
ot b
linde
d an
d hi
gher
dos
es m
ay b
e th
e re
sult
of a
n un
clea
r se
lect
ion
proc
ess.
Spe
cific
bou
nd in
Tha
l, hy
poTh
al
and
Mid
br re
gion
pea
ked
at 4
h; in
Str
iat t
his
was
re
ache
d af
ter 1
6-24
h.
Suha
ra 2
00322
1. H
ealth
y vo
lunt
eers
(2
2.0
±2.3
) M
2.
Pat
ient
s w
ith
MD
D(3
5.7
±12.
1)
27
10
1 sca
n 1.
Clo
mip
ram
ine
5-50
mg,
fluv
oxam
ine
12.5
-50
mg
as s
ingl
e ad
min
istr
atio
n 2.
Cl
omip
ram
ine
20-2
50 m
g, fl
uvox
amin
e 25
-200
mg
as lo
ng-t
erm
adm
inis
trat
ion
[11C]
McN
5652
PET
, 0-1
½
h Th
al
1. Cl
omip
ram
ine
occ ≥8
3.9-
100%
at
dosa
ges
5-50
mg;
fluv
oxam
ine
occ ≥7
.7-
87.7
% at
dos
ages
12.5
-50
mg.
2.
Clom
ipra
min
e oc
c ≥6
1.3-10
0% a
t dos
ages
20
-250
mg;
fluv
oxam
ine
occ ≥7
6.6-
93.6
% at
dos
ages
25-
200
mg.
Hyp
erbo
lic re
latio
nshi
p be
twee
n or
al d
ose,
pla
sma
conc
entr
atio
n an
d oc
c. fo
r clo
mip
ram
ine
and
fluvo
xam
ine.
Pat
ient
dat
a su
gges
t tha
t the
du
ratio
n of
trea
tmen
t with
fluv
oxam
ine
may
be
corr
elat
ed w
ith o
cc. T
his
does
not
hol
d fo
r cl
omip
ram
ine.
Tabl
e S9
.1. S
tudi
es o
f SER
T oc
cupa
ncy
afte
r tre
atm
ent w
ith s
erot
oner
gic
antid
epre
ssan
ts (C
ontin
ued)
Aut
hor &
Dat
e (r
efer
ence
) Re
sults
Popu
latio
n (m
ean
age)
Sex
N
Follo
w-u
p D
esig
n In
terv
entio
n / c
ontr
ol
SER
T Im
ag
ing
and
RoI
Resu
ltsRe
mar
ks

192
Chapter 9
Chap
ter
9
Shan
g 20
0772
Hea
lthy
cont
rols
(2
3.6
± 6.
3)
M +
F ?
8 Bs
l, 9d
Ve
nlaf
axin
e 15
0 m
g (4
day
s st
able
dos
e)
[123 I]β-
CIT
SPEC
T, 2
3h
Die
nc, M
ID,
Stria
t, C
er
Occ
in D
ienc
: 52.
5 ±
4.7%
, in
MID
: 55.
7 ±4
.5%
Stud
y al
so in
vest
igat
es in
fluen
ce o
f ven
lafa
xine
on
stria
tal D
AT a
vaila
bilit
y, w
hich
incr
ease
s 10
.7 ±
3.0
%
Taka
no 2
006a
73H
ealth
y co
ntro
ls
(24.
3 ±4
.8)
M
6 Bs
l, 5h
, 26
h, 5
3h
Fluv
oxam
ine
50 m
g on
ce
[11C]
DA
SB
PET,
0-1
½ h
PF
C, T
hal,
Stria
t,
Am
yg, H
ip,
Cer
Mea
n oc
c of
the
5 re
gion
s of
6 s
ubje
cts
wer
e 72
.9%
± 4.
9% a
t 5 h
ours
, 50.
3% ±
11.0
% at
26
hour
s, a
nd 2
4.7%
± 15
.3%
at 5
3 ho
urs.
5h
occ
: Tha
l 71.8
±3.
5%, A
myg
71.
6 ±1
2.8%
; 53
h oc
c: T
hal 2
4.2
±12.
3%, A
myg
27.
4 ±1
8.2%
Stud
y in
vest
igat
es ti
me-
cour
se o
f occ
upan
cy o
f SE
RT
afte
r one
(low
) dos
e of
fluv
oxam
ine.
In E
max
m
odel
dep
ende
nt m
easu
rem
ents
at 3
tim
epoi
nts
are
used
as
inde
pend
ent d
ata-
poin
ts
Taka
no 2
006b
27H
ealth
y co
ntro
ls
(24.
1 ±2.
4)
M
15
Bsl,
1. 6
h,
25h,
53h
2.
7d, 9
d, 10
d
1. si
ngle
dul
oxet
ine
dose
(5, 2
0, 4
0, 6
0 m
g) (n
= 12
) 2.
dul
oxet
ine
60 m
g fo
r 7 d
ays
then
st
oppe
d (n
= 3)
[11C]
DA
SB
PET,
0-1
½ h
Th
al, C
er
1. Th
al o
cc 3
5.3-
86.5
% at
incr
easi
ng
dosa
ges:
at 6
hrs
occ:
43.
6±8.
8% (5
mg)
, 71
.3±5
.3%
(20
mg)
, 80.
6±4.
8% (4
0 m
g),
81.8
±4.3
% (6
0 m
g) 2
. Tha
l occ
: 84.
3+2.
8%
(7d)
, 71.9
+2.6
% (9
d), 4
7.1+
3.7%
(11d
)
Stud
y co
nclu
des
that
mor
e th
an 4
0 m
g is
requ
ired
to a
ttai
n 80
% oc
cupa
ncy.
Inst
ead
of p
lasm
a co
ncen
trat
ions
that
dec
reas
e in
a o
ne-e
xpon
entia
l w
ay a
fter
stop
ping
, occ
upan
cy a
ppea
rs to
dec
reas
e lin
early
and
slo
wer
than
pla
sma
conc
entr
atio
ns.
Taus
cher
1999
74Pa
tient
with
BN
an
d M
DD
(1
8)
F
1 1 s
can
Fluo
xetin
e 60
mg.
Unt
il 1 w
eek
befo
re
scan
add
ition
with
dox
epin
e 75
mg.
[12
3 I]β-
CIT
SPEC
T, 4
, 24
h Th
al,
hypo
Thal
, St
riat
Com
pare
d to
con
trol
s an
occ
in T
hal a
nd
hypo
Thal
of a
pp. 4
1% w
as e
xpec
ted
by
fluox
etin
e. H
ighe
r Str
iat D
AT th
an in
co
ntro
ls; i
n ac
cord
ance
with
oth
er M
DD
-pa
tient
s
Met
hodo
logi
cally
poo
r. R
esul
ts c
ompa
red
to a
hi
stor
ic c
ontr
ol g
roup
(n=
13, M
+ F
; age
46
±20;
age
ra
nge
24-7
1).
Viin
amäk
i 19
9875
1. Pa
tient
s w
ith
Pers
Dis
(25
&
34) M
2.
Hea
lthy
con
trol
s (2
5±3
& 3
4±1)
M
2 10
Bsl,
1-1.5
y Ps
ycho
dyna
mic
psy
chot
hera
py o
r no
trea
tmen
t
No
trea
tmen
t
[123 I]β-
CIT
SPEC
T, 1h
&
21h
mPF
C, O
C,
MID
, Tha
l, St
riat
(Suc
cesf
ully
) tre
ated
pat
ient
1: in
crea
se in
bi
ndin
g in
MID
: 34%
, mPF
C 43
%, T
hal 3
1%;
with
no
chan
ges
in u
ntre
ated
pat
ient
2;
leve
ls o
f bin
ding
pos
t-tr
eatm
ent
appr
oach
leve
ls o
f con
trol
s
Patie
nts
wer
e bo
th a
busi
ng a
lcoh
ol; p
atie
nt 1
abst
aine
d du
ring
trea
tmen
t; p
atie
nt 2
did
not
. Pa
tient
1 ha
d lo
w, a
nd p
atie
nt 2
had
hig
h H
DRS
-sc
ores
at b
asel
ine
and
follo
w-u
p. C
ontr
ols
used
al
coho
l occ
asio
nally
, no
data
on
HD
RS
are
give
n.
Met
hodo
logi
cally
poo
r, s
tudy
is d
escr
ibed
poo
rly.
Voin
esko
s 20
0730
Patie
nts
with
M
DD
(36
±9)
Hea
lthy
cont
rols
(3
2 ±9
) M+F
12
>4w
Bsl
Ve
nlaf
axin
e 22
5 m
g, 4
50 m
g, s
ertr
alin
e 15
0 m
g, 2
00 m
g, c
italo
pram
60
mg,
80
mg
[11C]
DA
SB
PET,
0-1
½ h
St
riat,
MID
, Th
al,
PFC,
ACg
, Ce
r
Occ
Str
iat:
85.
8 ±3
.4%
(ven
lafa
xine
), 8
5.8
±2.3
(ser
tral
ine)
and
85.
4±2.
0 (c
italo
pram
) Occ
MID
: 99.
5 ±4
.1%
(ven
lafa
xine
), 9
8.2
±3.2
(ser
tral
ine)
and
95
.7±1
.4 (c
italo
pram
) Occ
Tha
l: 77
.6 ±
4.7%
(v
enla
faxi
ne),
76.
3 ±7
.9 (s
ertr
alin
e) a
nd
82.2
±3.
6 (c
italo
pram
)
No
base
line
scan
s w
ere
perf
orm
ed in
pat
ient
s.
Inst
ead
as re
fere
nce
base
line
scan
s of
con
trol
s w
ere
used
to d
eter
min
e oc
cupa
ncy.
Abb
revi
atio
ns: A
myg
= A
myg
dala
, aCG
= a
nter
ior C
ingu
late
Gyr
us, B
asG
ang
= Ba
sal G
angl
ia, B
N =
Bul
imia
Ner
vosa
, BrS
t = B
rain
Stem
, Bsl
= B
asel
ine,
Cin
gA =
Cin
gula
te A
nter
ior,
d =
day
s, D
AT =
Dop
amin
e Tr
ansp
orte
r, D
ienc
= D
ienc
epha
lon,
h =
hou
rs, H
ip =
Hip
poca
mpu
s, H
DRS
= H
amilt
on D
epre
ssio
n R
atin
g Sc
ale,
hyp
oTha
l = H
ypot
hala
mus
, m =
mon
ths,
MD
D =
Maj
or d
epre
ssiv
e di
sord
er, m
TL =
med
ial
tem
pora
l lob
e, M
ID =
Mid
brai
n, m
PFC
= m
edia
l Pre
Fron
tal C
orte
x, O
C =
Occ
ipita
l Cor
tex,
Occ
= O
ccup
ancy
,Per
sDis
= P
erso
nalit
y D
isor
der,
PFC=
Pre
Fron
tal C
orte
x, R
oI =
Reg
ions
of I
nter
est,
Str
iat =
St
riatu
m, T
hal =
Tha
lam
us, w
= w
eeks
, y =
yea
rs.* m
ost s
tudi
es u
sed
antid
epre
ssan
tsSE
RT
occ.
= S
ERT
occu
panc
y re
lativ
e to
non
-spe
cifi
c bi
ndin
g (c
ereb
ellu
m)
Tabl
e S9
.1. S
tudi
es o
f SER
T oc
cupa
ncy
afte
r tre
atm
ent w
ith s
erot
oner
gic
antid
epre
ssan
ts (C
ontin
ued)
Aut
hor &
Dat
e (r
efer
ence
) Re
sults
Popu
latio
n (m
ean
age)
Sex
N
Follo
w-u
p D
esig
n In
terv
entio
n / c
ontr
ol
SER
T Im
ag
ing
and
RoI
Resu
ltsRe
mar
ks

ChapterSERT occupancy by paroxetine dose-escalation in MDD
9
193
Table S9.2. Drop-outs and emerging adverse effects reported more than once between T0 and T1 (ITT).
Paroxetine DE (n = 30) Placebo DE (n = 30) p
Drop-outs
Overall 1/30 (3.3%) 8/30 (26.7%) 0.026
Inefficacy 0/30 (0.0%) 3/30 (10.0%) 0.237
Adverse effects 0/30 (0.0%) 4/30 (13.3%) 0.112
Adverse effect rates Paroxetine DE (n = 30) Placebo DE (n = 27)
% all* T0 ≥2x RP End RP Rec. AE† T0 ≥2x RP End RP Rec. AE†
Headache 69.0 46.7 70.0 46.7 5/10 (50.0) 50.0 67.9 50.0 5/6 (83.3)
Sleepiness 67.2 63.3 70.0 56.7 6/7 (85.7) 46.4 64.3 46.4 7/8 (87.5)
Sweating 63.8 56.7 80.0‡ 63.3 3/5 (60.0) 39.3 46.4‡ 42.9 3/5 (60.0)
Dry mouth 63.8 66.7 63.4 50 1/5 (20.0) 64.3 64.3 46.4 2/4 (50.0)
Orthostatic hypotension 46.6 50.0 53.3 50.0 3/5 (60.0) 35.7 39.3 28.6 6/8 (75.0)
Constipation 44.8 36.7 50.0 43.3 2/3 (66.7) 28.6 39.3 21.4 4/7 (57.1)
Libido 41.4 36.7 56.7‡ 56.7 2/2 (100.0) 28.6 25.0‡ 39.3 1/4 (25.0)
Agitation 41.4 33.3 50.0 33.3 6/9 (66.7) 21.4 32.1 17.9 5/10 (50.0
Appetite 39.7 46.7 46.7 43.3‡ 3/7 (42.9) 35.7 32.1 14.3‡ 3/8 (37.5)
Blurry vision 37.9 40.0 43.3 43.3 0/4 (0.0) 14.3 32.1 25.0 2/4 (50.0)
Tremor 36.2 36.7 46.7 40.0‡ 4/7 (57.1) 32.1 25.0 14.3‡ 3/5 (60.0)
Dizziness 34.5 36.7 46.7 26.7 8/11 (72.7) 28.6 21.4 17.9 2/7 (28.6)
Concentration 34.5 26.7 43.3 30.0 5/8 (62.5) 28.6 25.0 25.0 4/5 (80.0)
Paraesthesia 32.8 23.3 36.7 36.7 4/5 (80.0) 32.1 28.6 25.0 2/3 (66.7)
Sleep disturbances 31.0 23.3 43.3‡ 43.3§ 8/9 (88.9) 35.7 17.9‡ 10.7§ 3/8 (37.5)
Nausea 29.3 33.3 33.3 33.3 5/11 (45.5) 28.6 25.0 14.3 4/7 (57.1)
Weakness 29.3 16.7 36.7 26.7 3/6 (50.0) 17.9 21.4 17.9 2/7 (28.6)
Diarrhea 24.1 16.7 23.3 13.3 4/5 (80.0) 25.0 25.0 17.9 3/4 (75.0)
Urinary retention 24.1 13.3 36.7‡ 23.3 2/3 (66.7) 14.3 10.7‡ 7.1 0/1 (0.0)
Other sexual dysfunction
22.8 16.7 26.7 30.0 3/3 (100) 17.9 18.5 17.9 0/0
Palpitations 22.4 16.7 26.7 16.7 3/7 (42.9) 10.7 17.9 17.9 4/9 (44.4)
Rash 10.3 6.7 6.7 3.3 1/4 (25.0) 7.1 14.3 10.7 0/2 (0.0) * Adverse effects are listed in order of overall frequency (mentioned more than once in Randomization Phase).† Recurrence of adverse effect was determined if an adverse effect was initially present during the open phase, disappeared at the end of the open phase, and reemerged during the randomization phase. Columns represent reemerging cases (nominater) of all subjects with the adverse effect initially present, but in whom it disappeared at end of the open phase (denominater). Values in brackets represent this rate as percentages.‡ Sign. difference paroxetine DE vs. placebo DE (p<0.05; Fisher’s exact test).§ Sign. difference paroxetine DE vs. placebo DE (p<0.01; Fisher’s exact test). DE = dose-escalation, T0 = time of randomization at end of open phase, ≥2x RP = mentioned more than once in randomization phase, Rec. AE = Recurrence of adverse effect.
Table S9.3.Changes in paroxetine serum concentrations (μg/l) in the SPECT subgroup due to paroxetine or placebo dose-escalation.
T0 T1 p*
Paroxetine DE (n= 11) 36.2 (22.7-52.8) 154.3 (112.4-202.7) <0.001
Placebo DE (n= 15) 60.3 (41.0-83.3) 52.2 (32.9-75.8) 0.268
Values represent means with 95% confidence intervals, computed on square-root transformed paroxetine serum concentrations. Probable adherentpatients only; in 1 patient (paroxetine dose-escalation) the measurement of paroxetine concentration was missing. The change in paroxetine serum concentrations after paroxetine dose-escalation vs. placebo dose-escalation was significant (ANCOVA correcting for paroxetine serum concentration at T0: F1,23= 59.938; p <0.001). *significance of difference between T0 and T1; paired T-test. DE = dose-escalation.

Chap
ter
Chapter 9
9
194
Figure S9.1. Changes in paroxetine serum concentration versus changes in occupancy (randomization to endpoint).
Regression lines show linear relationship (with 95% CI) between increase in paroxetine serum concentration (PSC) andchange in occupancy of midbrain (circles; beta= 0.03 ±0.08; F1,24= 0.101; p= 0.754) and diencephalon (diamonds; beta= 0.07±0.06; F1,27= 1.332; p= 0.259)
��� �� ��� ��� ��� ���
���
���
���
���
��
��
��
��
���
�+�,������������������������ ������������������������
��� ������������������������������� �����
� �
������
������
���
�����
�����
������������
�����!
�

CHA
PTER
SUCCESSFUL PHARMACOLOGICAL TREATMENT OF
MAJOR DEPRESSIVE DISORDER ATTENUATES
AMYGDALA ACTIVATION TO NEGATIVE FACIAL
EXPRESSIONS. AN FMRI STUDY
Submitted
Henricus G. Ruhé1*
Jan Booij2
Dick J.Veltman3,4
Martin C. Michel5
Aart H. Schene1
1 Program for Mood Disorders, Department of Psychiatry, Academic Medical Center, University ofAmsterdam, Amsterdam, the Netherlands2 Department of Nuclear Medicine, Academic Medical Center, University of Amsterdam, Amsterdam,the Netherlands 3 Department of Addiction Research, Academic Medical Center, University of Amsterdam,Amsterdam, the Netherlands 4 Department of Psychiatry, Vrije Universiteit, Amsterdam, the Netherlands 5Department of Pharmacology and Pharmacotherapy, Academic Medical Center, University ofAmsterdam, Amsterdam, the Netherlands

Chap
ter
Chapter 10
10
196
Abstract
BackgroundDespite modest response and remission rates to selective serotonin reuptake inhibitors (SSRIs),underlying neurobiological mechanisms of pharmacological treatment of major depressivedisorder (MDD) are scarcely investigated in vivo. Amygdala function might be a biomarker forMDD treatment-response.
Aimto quantify the changes in activation by emotional faces in amygdala and other limbic-subcortical-prefrontal structures after paroxetine treatment in MDD.
MethodsWe performed an event-related functional magnetic resonance imaging study in SCID-positive,non-psychotic, unipolar depressed patients (n= 22; M+F; 25-55 years, HDRS17>18) who werescanned at study-entry, and at 6 weeks (T0) and 12 weeks (T1) of paroxetine treatment. After 6weeks open-label paroxetine 20mg/day (T0), 15 non-responding patients (HDRS17 decrease <50%)were randomized to double-blind paroxetine (30-50mg/day) or placebo dose-escalation(paroxetine 20mg/day + placebo). The paradigm consisted of sex-judgments of fearful, angry,happy and neutral faces, relative to scrambled (baseline) faces. Twenty-one age- (±2.5 yrs) andsex-matched healthy controls (HC; without 1st degree relative with, or lifetime psychiatricdisorder) were scanned once as reference.
ResultsAt study-entry, MDD-patients showed increased ventral/limbic (amygdala and insula) anddecreased dorsal cortical activations relative to HC. At T0 and T1, 5/20 and 13/20 patients wereresponders, respectively. Treatment response was associated with decreased activations inamygdala, right insula and orbitofrontal cortex (OFC), and increased activation in rightdorsolateral prefrontal cortex (DLPFC) and nucleus accumbens. Amygdala activation wasinversely correlated with increased pregenual cingulate and DLPFC activation as well as withimprovement using the HDRS17. Paroxetine dose-escalation was associated with decreasedactivity in ventral (limbic) and dorsal cortical regions, whereas activity of right hippocampus andleft subgenual cingulate increased relative to placebo dose-escalation.
ConclusionsDecreased amygdala activation by negative faces is a biomarker for clinical response toantidepressants, but needs further exploration in other MDD-treatments. Paroxetine response-effects appear to be mediated by increased fronto-limbic control.(ISRCTN register nr. ISRCTN44111488)

Chapter10
197
Amygdala deactivation in responders to paroxetine
Introduction
Major depressive disorder (MDD) is a highly prevalent and disabling disease,1 often treated withantidepressants, particularly selective serotonin reuptake inhibitors (SSRIs). Unfortunately,response and remission rates are only modest (30-50%). An additional strategy to achieveremission2-4 is dose-escalation2;3 of which the underlying neurobiological mechanism has scarcelybeen investigated in vivo. The development of non-invasive neuroimaging techniques may aid inclarifying this issue. For example, we recently showed that a true dose-escalation of paroxetinedid not increase serotonin transporter occupancy (the molecular target of SSRIs) more thanplacebo-dose-escalation.5
Previous studies of the etiopathogenesis of MDD have provided evidence for a dysfunctionallimbic-subcortical-prefrontal network in MDD.6;7 A processing bias to perceive negative emotionswas associated with increased activation of the ‘ventral’ compartment of this network (consistingof amygdala, insula, ventral striatum, ventral anterior cingulate gyrus and prefrontal cortex),responsible for the identification of the emotional significance of stimuli and the production ofaffective states.7 Activation of the (left) amygdala to negative facial expressions (sad, angry orfearful) has been consistently found increased in MDD compared with healthy subjects, and maybe considered a biomarker for MDD.8-11
Treatment with SSRIs may not fully reverse these differences between MDD and healthysubjects. Four studies have investigated changes in brain activation after 8 weeks of treatmentwith antidepressants in 8-19 patients per study.10;12-14 Another study in patients who remitted had aduration of 22 weeks (Table S10.1).15 These studies found an increase in activation in neocorticalregions, including the cingulate gyrus (dorsal and anterior parts) after treatment withantidepressants.10;13 A decrease in amygdala activation, as originally found by Sheline et al.,12 wasreplicated in one study with sertraline (sad faces task),10 and in one study with bupropion(emotional oddball task).14 In addition, two randomized placebo-controlled studies in healthycontrols found decreased activation in right16 or bilateral17 amygdala after 7 days of treatmentwith reboxetine and citalopram, respectively. However, as most of these treatment studiesreported high response rates and one study explicitly excluded non-responders,13 it is unclearwhether the observed changes over time are drug effects, or due to clinical response. This couldbe addressed in analyses of responders vs. non-responders. Furthermore, although dose-escalation is a recommended strategy for non-responders, studies employing fMRI to investigatedose-response relationships have not yet been published.
Therefore, the aim of this study was to evaluate changes within the limbic-subcortical-prefrontal network, including the amygdala, to (negative) facial expressions after paroxetinetreatment of MDD. We specifically aimed to investigate clinicalresponse versus drug effects,including dose-escalation effects. To this end, we performed a functional MRI study in 22depressed patients who were scanned at study-entry, 6 weeks and 12 weeks of paroxetinetreatment,and 22 matched controls scanned once.
We expected to observe increased activation of the amygdala in patients versus healthycontrols at baseline, followed by an attenuation of amygdala hyperactivity together with anincrease of activation in the neocortical areas (anterior cingulate and dorsomedial and -lateralprefrontal cortex (DMPFC/DLPFC)) after 12 weeks of treatment. We hypothesized that thesechanges would correlate with clinical response, but not with dose-escalation of paroxetinecompared with placebo.

Chap
ter
Chapter 10
10
198
Methods
ParticipantsFollowing approval by the institutional ethical committee and written informed consent, werecruited 22 outpatients (25-55 years) from our outpatient department (August 2005 - August2006).5 Inclusion criteria were: MDD determined by the structured clinical interview for DSM-IV(SCID18), and a Hamilton Depression Rating Scale (17 items; HDRS17)19 score above 18. Patientswere drug-free (>4 weeks and ≥5 half-lives of a previous antidepressant) and had not used morethan one antidepressant treatment (other than paroxetine) at an effective dose for ≥6 weeks forthe present MDD-episode. Exclusion criteria, apart from pregnancy (or wish) and standard fMRIcontraindications, were bipolar disorder, psychotic features, neurological impairments, primaryanxiety and/or substance abuse disorders and acute, severe suicidal ideation.
We individually matched each patient by gender and age (±2.5 years) with a healthy control(HC), who had good physical health and had never used psychotropic medication. Exclusioncriteria for HC were any lifetime psychiatric disorder according to the SCID (including abuse oraddiction disorders), a Beck Depression Inventory (BDI)20 score >9, average alcohol use >4 unitsper day (preceding month) and a 1st-degree relative with psychiatric disorder(s).
Treatment scheduleAfter assessment at study-entry patients were treated with paroxetine 20 mg/day (supplied in pill-boxes) for 6 weeks. All patients who did not achieve ≥50% decrease in HDRS17-score after 6 weeks(T0; see Figure 2.1) were randomized (stratified for gender and age). Patients received a trueparoxetine or a placebo dose-escalation added to paroxetine 20 mg/day. Methods of dose-escalation were described previously,5 we applied incremental steps of one capsule every 5 days(reaching 30-50 mg/day according to adverse effects).21 Dosages remained unchanged the last 3weeks of the study. Adherence was checked by self-report,22 pill-counts and measured paroxetineserum concentrations.5
MeasurementsPrimary clinical outcome was the proportion of patients achieving response (≥50% decrease inHDRS17). Depression severity was measured at study-entry, randomization (T0), and 6 weeks afterrandomization (T1), also using the Inventory for Depressive Symptomatology self-rated (IDS-SR30).23 At these time-points we planned fMRI-sessions of 50 minutes, each including a cognitivetask (reported elsewhere), a structural scan and the facial expression task reported here.Agreement between trained raters was good (intra-class correlation coefficient = 0.98). Ratersand patients were blinded for the randomized dose-escalation.
Facial expression task paradigmWe used an event-related emotional faces paradigm, which reliably activates the anterior medialtemporal lobe including the amygdala.24;25 We presented four human face stimuli: angry, fearful,happy, and neutral human faces,26 and scrambled faces (with two arrows in the centre of thescreen) as baseline condition. Each face stimulus condition consisted of 10 pictures; each picturewas presented three times. Stimuli were randomized once and presented in identical order to allsubjects, using the same task for each session. Stimuli were displayed for 2500ms with a variableinterstimulus interval (400-600ms), to increase experimental power and to decrease expectancyeffects. To control for overflow effects, we displayed a baseline stimulus after each one or twoface pictures. Subjects were instructed to make gender judgements during presentation of facestimuli to control for attention differences. To familiarize participants, the task was first explainedoutside the scanner. No feedback was provided during the task.

Chapter10
199
Amygdala deactivation in responders to paroxetine
fMRI imaging We acquired fMRI scans in the afternoon/early evening using a 3Tesla Intera MRI scanner (Philips,Eindhoven, Netherlands). A 6-channel head-coil was used for radiofrequency reception, with thehead fixated in the coil by foam pads to prevent movement-artefacts. Stimuli were generated by aPentium PC and projected on a screen at the patient’s feet, visible through a mirror mounted onthe headcoil. Stimulus onset was triggered by a pulse from the scanner. Two magnet compatibleresponse boxes were used to record subject’s performance and reaction times (RTs).
At each session, we obtained a volumetric T1-weighted coronal scan (TE/TR= 4.6/9.63 msec,field of view= 24×24 cm, flip angle= 8°, number of excitations= 1, matrix= 256×256, 182 slices, slicethickness= 1.2 mm, interslice gap= 0 mm, scan time= 7 min) covering the entire brain volume,and260 T2*-weighted axial echoplanar imaging (EPI) images sensitive to blood oxygen leveldependent (BOLD) contrast (TE/TR= 35/2530.4 msec, field of view= 24×24 cm, flip angle= 90°,number of excitations= 1, matrix= 128×128, 36 ascending slices, slice thickness= 3 mm, interslicegap= 0.3 mm, scan time=10 min).
fMRI data analysis
Individual analysisFor all fMRI data-analyses we used SPM5 (Statistical Parametric Mapping; Wellcome Departmentof Cognitive Neurology, London, UK; http://www.fil.ion.ucl.ac.uk/spm/), operated under Matlabversion 7.3.0.267 (2006b; the Mathworks,Natick, Massachusetts, USA). Standard preprocessing ofscans consisted of correcting for slice-timing differences, head movements, coregistration to thestructural scan, normalization to SPM/MNI standard space, and smoothing (8 mm full-width half-maximum Gaussian filter). Next, BOLD responses were modeled to affective facial expressionsand baseline conditions for each voxel. For each subject, weighted contrasts were computed forsimple main effects across all stimulus types combined (angry/anxious, happy, and neutral facesvs. baseline = ‘all faces’), and within stimulus type contrasts (angry/anxious vs. baseline =‘negative faces’; happy vs. baseline = ‘happy faces’).
Group analysisThe contrast images obtained in individual analyses were entered into second level (randomeffects) analyses for between-group and within subject (i.e. T0-T1) comparisons (withinterdependent measures and assuming equal variances).Main effects were identified at p <0.01and FDR-corrected for multiple comparisons, with an extent threshold of three voxels.27 Plannedcontrasts were changes over time (study-entry – T0 – T1), responders versus non-responders, andeffect of paroxetine dosage. Interactive effects of time/response*stimulus type (all faces,negative faces, happy faces; masked with the relevant main effect) were identified at p<0.001 (z>3.09), uncorrected, with an extent threshold of three voxels, but in the amygdala region atp<0.005 (z>2.57).
Statistics We compared study-entry characteristics of patients and controls with independent samples t-tests for continuous and χ2 or Fisher’s exact test for categorical variables. We used linear mixedmodels (compound symmetry variance/covariance structure) to assess differences in scanperformance (reaction times and errors); between patients and healthy controls, and in patientsin trend over time. Parameter estimates were extracted for bilateral amygdala from SPM5, andthese values were used for graphical representation. With regression models, we quantified theassociation of parameter estimates with HDRS17-scores. We performed additional analyses inSPSS v15.0.1.1 (www.spss.com) and GraphPad Prism v5.00 (www.graphpad.com).

Chap
ter
Chapter 10
10
200
Results
Patients, controls and behavioural dataTable 10.1 displays demographic data and MDD ratings for patients and HC. HC had significantlyhigher education-levels. MDD-patients reported significantly higher state and trait anxiety thancontrols (p<0.001), and showed slower RTs p= 0.045) and more gender-judgement errors (p=0.032). Three male patients withdrew from the study after the study-entry scan; 2 withdrewcompletely. One female HC did not attend her visit and could not be replaced. Due to excessivemovements, two study-entry scans (one male, one female patient), two T0 scans (one male, onefemale patient) and three T1 scans (one male, two female patients) could not be analyzed. Thus,41 study-entry scans (20 patients, 21 controls), 17 T0 and 16 T1 scans were available for analyses.
Twenty patients completed the treatment protocol, 5/20 (25%) and 13/20 (65%) had a clinicalresponse at week 6 and 12 respectively. No week 6 responders deteriorated afterwards (Table10.2). A randomized dose-escalation was provided to 15 non-responding patients after 6 weeks ofparoxetine treatment; for 11 of these repeated scans were analyzable. Clinical outcomes wereneither numerically, nor statistically significantly different between true and placebo dose-escalation. RTs and errors did not change over time, and were not significantly different forresponders versus non-responders nor associated with HDRS17-scores.
Main effects at study-entry in patients and healthy controlsCombining the study-entry scans of patients and HCs (all faces contrast) showed robust activationof bilateral amygdala, fusiform gyrus, DLPFC, (anterior) insula, occipital cortices, and rightorbitofrontal cortex (OFC; extending into the right anterior insula), parietal cortex and DMPFC.These effects were also found for negative faces, except for the right amygdala, left insula, andleft DLPFC, which were not activated above threshold. With the happy faces contrast, we foundmain effects for bilateral fusiform gyrus, insula, occipital cortices, and right DLPFC, OFC (extendedfrom insula), thalamus and parietal cortex (Table S10.2).
Table 10.1. Characteristics of study-sample.
Patients (n= 22) Healthy Controls (n= 22) p
Age ±SD 43.3 ±7.93 43.7 ±7.99
Males n (%) 14 (63.6) 14 (63.6)
Handedness R/L 20/2 21/0* 0.488
Educational level n (%) Low Intermediate High
5 (22.7) 12 (54.5) 5 (22.7)
0 6 (27.3) 16 (72.7)
0.002
MDD-severity HDRS17 IDS-SR30 STAI-I STAI-II
23.1 ±3.61 42.9 ±7.69 58.9 ±8.84 62.7 ±9.25
N/A 4.6 ±3.87* 29.4 ±7.74* 29.8 ±11.14*
<0.001 <0.001 <0.001
Co-morbidity n (%) Anxiety Alcohol dependance Cannabis dependance
3 (13.6) 2 (9.1) 1 (4.5)
N/A
Scan task performance Reaction Time (ms) Correct response (%)
954.7 ±238.3 95.5 ±3.24
* 830.0 ±143.3 97.5 ±2.66
0.045 0.032
* 1 female HC did not attend; no questionnaire and scan performance data available

Chapter10
201
Amygdala deactivation in responders to paroxetine
Activations in the amygdala in MDD-patients versus HCThe all faces contrast showed higher activations in bilateral (extended) amygdala in MDD-patientsthan in HC. Higher (right) amygdala activities in patients were also found when we contrastednegative faces, but not when contrasting happy faces (Table 10.3A). In post-hoc analyses, baselineamygdala activations were not different between final treatment responders and non-responders(all contrasts).
Other activations of the limbic-subcortical-prefrontal network in MDD-patientsversus HCContrasting all faces versus baseline showed higher activations in the left insula in MDD-patientscompared with HC (Table 10.3A). For happy faces MDD-patients showed higher activation in theleft subthalamic nucleus.
With the all faces contrast, we found lower activations in MDD-patients relative to HC inbilateral ventrolateral prefrontal cortex (VLPFC), posterior and medial dorsal cingulate, leftDMPFC, bilateral DLPFC and fusiform gyrus (Table 10.3B). For negative faces, we found loweractivations in MDD-patients in bilateral VLPFC, left posterior cingulate gyrus, left DMFPC, rightDLPFC, and bilateral fusiform gyrus. With happy faces, we found lower activations in right VLPFC,right premotor cortex, and left fusiform gyrus in MDD-patients relative to HC.
In post-hoc analyses, final treatment responders showed higher activations at study-entry inthe right pregenual (rostral) cingulate (MNI 14, 44, 3; k= 4; Z= 2.77; p= 0.003; negative faces),relative to final non-responders. Instead, non-responders showed higher study-entry activationsin the subgenual cingulate (MNI 0, 26, -3; k= 11; Z= 3.90; p<0.001; negative faces).
Table 10.2. Clinical response and task performance of patients during follow-up (n= 20).
T0 (Randomization) T1 (Endpoint)
HDRS17All (n= 20) T0-Responders (n= 5) True DE (n= 8) Placebo DE (n= 7)
15.7 ±5.14 9.8 ±3.11 18.0 ±5.13 17.3 ±2.75
11.1 ±5.31 7.2 ±1.48 13.5 ±6.70 11.0 ±3.87
% decrease in HDRS17All (n= 20) T0-Responders (n= 5) True DE (n= 8) Placebo DE (n= 7)
30.9 ±26.67 59.9 ±9.53 20.5 ±26.36 22.0 ±21.01
51.6 ±25.66 70.5 ±2.24 39.4 ±34.07 52.0 ±15.09
Responders (HDRS17 ≥50%)All (n= 20) T0-Responders (n= 5) True DE (n= 8) Placebo DE (n= 7)
5 (25%) 5 (100%) - -
13 (65%) 5 (100%) 4 (50%) 4 (57%)
Remission (HDRS17 ≤7)All (n= 20) T0-Responders (n= 5) True DE (n= 8) Placebo DE (n= 7)
2 (10%) 2 (40%) - -
5 (25%) 3 (60%) 1 (13%) 1 (14%)
IDS-SR30All (n= 20) T0-Responders (n= 5) True DE (n= 8) Placebo DE (n= 7)
30.6 ±8.80 19.8 ±6.46 35.3 ±6.71 33.0 ±5.66
27.2 ±11.46 21.6 ±4.72 33.5 ±14.92 23.9 ±7.03
Scan task performance* Reaction Time (ms) Correct response (%)
936.8 ±242.8 96.3 ±3.74
921.7 ±252.2 95.6 ±6.26
Values represent means ±SD. DE= dose-escalation. All differences between True DE and Placebo DE non-significant (ANOVA; p>0.14) * n= 18 at T0 and n= 17 at T1

Chap
ter
Chapter 10
10
202
Changes in amygdala activations after 6 and 12 weeks of paroxetine treatmentAfter 6 weeks of treatment (T0), for the all faces and negative faces contrasts, the activations inbilateral amygdala regions had not significantly decreased relative to the study-entry scan (TableS10.3A). In contrast, the right lateral amygdala showed a significant increase in activation after 6weeks of treatment (all faces, negative faces and happy faces). Further exploration showed thatweek 6 treatment non-responders (n= 12) had increased amygdala activations for all faces (left z=2.33; p= 0.01; right z= 2.52; p= 0.006) and negative faces (left z= 3.22; p= 0.001; right z= 2.87; p=0.002) relative to week 6 responders.
After 12 weeks of treatment (T1), when we contrasted all faces, the right amygdala showeddecreased activations (although subtreshold), relative to study-entry (Table 10.4A). Othercontrasts did not reveal amygdala deactivations over time.
Table 10.3. Study-entry scans: MDD-patients versus HC.
Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p
A. MDD > HC
All Faces Amygdala R 18 2 -9 12 3.04 0.001
L -16 -4 -9 39 2.63 0.004
Insula L -26 4 12 30 3.13 0.001
Neg. Faces Amygdala R 18 2 -9 13 2.99 0.001
L -16 -4 -9 16 2.46 0.007
Insula L -28 6 15 32 3.26 0.001
Hap. Faces Subthalamic nucleus L -12 -6 -6 14 3.35 <0.001
B. HC > MDD
All Faces VLPFC R 50 20 -6 222 3.95 <0.001
L -34 22 -6 23 3.20 <0.001
DLPFC R 42 16 27 61 3.73 <0.001
R 48 -2 51 16 3.31 <0.001
L -54 16 0 25 3.48 <0.001
DMFPC L -4 10 63 76 3.49 <0.001
Fusiform gyrus L -42 -54 -21 61 3.61 <0.001
Cingulate, medial L -8 26 42 58 3.36 <0.001
posterior L -6 -20 51 23 3.59 <0.001
Neg. Faces DMFPC L -4 10 63 383 4.37 <0.001
VLPFC R 50 20 -6 241 3.74 <0.001
L -34 22 -6 41 3.09 0.001
DLPFC R 42 16 27 76 4.25 <0.001
Fusiform gyrus L -42 -54 -21 67 3.78 <0.001
R 46 -40 -24 49 3.10 0.001
Cerebellum L -16 -38 -21 13 3.28 0.001
Sup. Temporal gyrus R 58 -6 -12 37 3.45 <0.001
Cingulate, posterior L -12 -22 48 39 3.41 <0.001
Hap. Faces VLPFC R 54 32 3 81 3.51 <0.001
Premotor cortex R 44 2 48 27 3.37 <0.001
Sup. Temporal gyrus R 54 -48 12 34 3.51 <0.001
R 48 -36 6 27 3.20 0.001
Fusiform gyrus L -40 -54 -18 50 3.13 0.001
DLPFC= dorsolateral prefrontal cortex; HC = healthy control; MDD= major depressive disorder; MNI= Montreal neurological institute; VLPFC= ventrolateral prefrontal cortex.
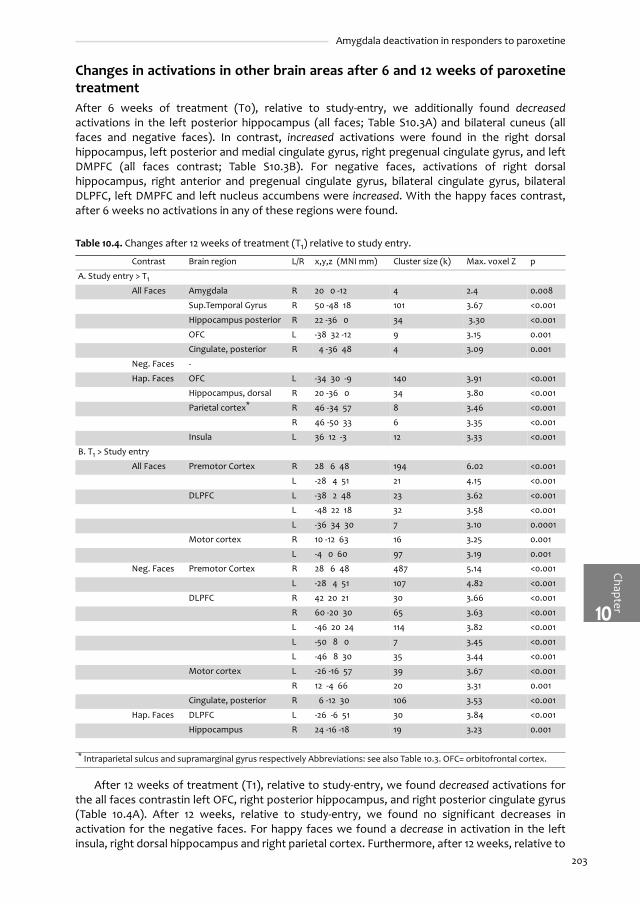
Chapter10
203
Amygdala deactivation in responders to paroxetine
Changes in activations in other brain areas after 6 and 12 weeks of paroxetinetreatmentAfter 6 weeks of treatment (T0), relative to study-entry, we additionally found decreasedactivations in the left posterior hippocampus (all faces; Table S10.3A) and bilateral cuneus (allfaces and negative faces). In contrast, increased activations were found in the right dorsalhippocampus, left posterior and medial cingulate gyrus, right pregenual cingulate gyrus, and leftDMPFC (all faces contrast; Table S10.3B). For negative faces, activations of right dorsalhippocampus, right anterior and pregenual cingulate gyrus, bilateral cingulate gyrus, bilateralDLPFC, left DMPFC and left nucleus accumbens were increased. With the happy faces contrast,after 6 weeks no activations in any of these regions were found.
After 12 weeks of treatment (T1), relative to study-entry, we found decreased activations forthe all faces contrastin left OFC, right posterior hippocampus, and right posterior cingulate gyrus(Table 10.4A). After 12 weeks, relative to study-entry, we found no significant decreases inactivation for the negative faces. For happy faces we found a decrease in activation in the leftinsula, right dorsal hippocampus and right parietal cortex. Furthermore, after 12 weeks, relative to
Table 10.4. Changes after 12 weeks of treatment (T1) relative to study entry.
Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p
A. Study entry > T1
All Faces Amygdala R 20 0 -12 4 2.4 0.008
Sup.Temporal Gyrus R 50 -48 18 101 3.67 <0.001
Hippocampus posterior R 22 -36 0 34 3.30 <0.001
OFC L -38 32 -12 9 3.15 0.001
Cingulate, posterior R 4 -36 48 4 3.09 0.001
Neg. Faces -
Hap. Faces OFC L -34 30 -9 140 3.91 <0.001
Hippocampus, dorsal R 20 -36 0 34 3.80 <0.001
Parietal cortex* R 46 -34 57 8 3.46 <0.001
R 46 -50 33 6 3.35 <0.001
Insula L 36 12 -3 12 3.33 <0.001
B. T1 > Study entry
All Faces Premotor Cortex R 28 6 48 194 6.02 <0.001
L -28 4 51 21 4.15 <0.001
DLPFC L -38 2 48 23 3.62 <0.001
L -48 22 18 32 3.58 <0.001
L -36 34 30 7 3.10 0.0001
Motor cortex R 10 -12 63 16 3.25 0.001
L -4 0 60 97 3.19 0.001
Neg. Faces Premotor Cortex R 28 6 48 487 5.14 <0.001
L -28 4 51 107 4.82 <0.001
DLPFC R 42 20 21 30 3.66 <0.001
R 60 -20 30 65 3.63 <0.001
L -46 20 24 114 3.82 <0.001
L -50 8 0 7 3.45 <0.001
L -46 8 30 35 3.44 <0.001
Motor cortex L -26 -16 57 39 3.67 <0.001
R 12 -4 66 20 3.31 0.001
Cingulate, posterior R 6 -12 30 106 3.53 <0.001
Hap. Faces DLPFC L -26 -6 51 30 3.84 <0.001
Hippocampus R 24 -16 -18 19 3.23 0.001
* Intraparietal sulcus and supramarginal gyrus respectively Abbreviations: see also Table 10.3. OFC= orbitofrontal cortex.

Chap
ter
Chapter 10
10
204
study-entry, we found increased activations in left DLPFC, and bilateral premotor and motorcortices for the all faces and negative faces contrasts, and in the right hippocampus and leftDLFPC for the happy faces contrast (Table 10.4B).
Changes in amygdala activations in relation to clinical responseWhen we compared responders and non-responders after 6 weeks and 12 weeks (full factorialmodel), we found higher right bilateral amygdala activations in non-responders relative toresponders for the all faces contrast (Table 10.5A). With the negative faces contrast, higheractivations in non-responders were significant in bilateral amygdala (p= 0.001; Figure 10.1).Moreover, the amygdala negative faces contrast estimates were significantly associated withHDRS17-scores, with higher activations in more depressed patients. No differential effectsbetween left and right amygdala were found.
In post-hoc analyses, left amygdala activations correlated positively with the contralateralamygdala, bilateral OFC and right subgenual anterior cingulate (Table S10.3). Right amygdalaactivation correlated positively with the contralateral amygdala, bilateral OFC and right DLPFC.Left amygdala signal was inversely correlated with the pregenual anterior cingulate, rightamygdala signal was inversely correlated with the left DLPFC and the left nucleus accumbens.
Table 10.5. Full-factorial model of 6-weeks and 12-weeks responders versus non-responders.
Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p
A. Non-Responders > Responders
All Faces Amygdala R 18 -2 -18 30 3.24 0.001
L -18 -10 -21 24 2.22 0.013
OFC lateral R 28 16 -18 98 3.80 <0.001
posterior R 10 12 -18 98 3.03 0.001
R 40 38 -12 39 3.56 <0.001
Insula R 42 -10 3 60 3.62 <0.001
Brainstem (L. coeruleus) R 6 -16 -12 30 3.45 <0.001
Neg. Faces Amygdala R 18 -2 -18 30 3.20 0.001
L -22 2 -18 24 3.11 0.001
OFC R 40 38 -12 33 3.48 <0.001
lateral R 26 16 -18 46* 3.01 0.001
posterior R 10 12 -18 * 2.90 0.002
Insula R 42 -10 3 49 3.33 <0.001
Hap. Faces -
B. Responders > Non-Responders
All Faces DLPFC R 20 20 63 5 3.71 <0.001
N.Accumbens L -10 10 -9 22 3.21 0.001
Neg. Faces N.Accumbens L -8 12 -9 40 3.69 <0.001
Parietal cortex L -46 -52 45 21 3.49 <0.001
Hap. Faces Cerebellum R 26 -66 -24 11 3.67 <0.001
R 8 -74 -15 78 3.64 <0.001
Hippocampus, dorsal L -24 -32 -6 15 3.38 <0.001
Cingulate, medial LR 4 16 42 41 3.29 0.001
Insula L -34 24 6 30 3.17 0.001
Hypothalamus R 8 -10 3 15 3.11 0.001 * in one OFC cluster. Abbreviations: see Table 10.3 and 10.4.

Chapter10
205
Amygdala deactivation in responders to paroxetine
Changes in activations in other brain areas in relation to clinical response Non-responders also showed significant (p≤0.001) higher activations in right OFC and right insula(all faces and negative faces), brainstem (all faces), relative to non-responders after 6 and 12weeks of treatment (Table 10.5A). In contrast, treatment responders showed higher activations inright DLPFC, and left nucleus accumbens (all faces and negative faces; Table 10.5B). Furthermore,with the happy faces contrast, responders had higher activations in the left dorsal hippocampus,bilateral medial cingulate gyrus, left insula and right hypothalamus.
Effects of true dose-escalation versus placebo dose-escalationWe investigated the interaction of randomized dose-escalation by time in full factorial models.Relative to placebo dose-escalation, 6 weeks of true dose-escalation decreased activations in leftOFC, bilateral insula, right VMPFC, left ventrolateral thalamus, right anterior cingulate, left DMPFCand motor cortex, right parietal cortex, right putamen and left globus pallidus (all faces; TableS10.5A). With negative faces decreased activations after dose-escalation were found in rightinsula, bilateral OFC, left ventrolateral thalamus, right anterior cingulate, left DMPFC and bilateral
Figure 10.1. Amygdala response relative to clinical response (HDRS17 ≥50% decrease) after 6 and 12 weeks of treatment with paroxetine.
Negative faces contrast. Week 6 & 12 scans and response status were combined in a full factorial model. For color figure see page 284A. SPM-results showing design matrix, contrast and activation in right amygdala (MNI 18,-2,-18).B. Activations of left (MNI -22,2,-18; dark) and right (MNI 18,-2,-18; white) amygdala stratified for non-response and responseat 6 and 12 weeks. Significant difference between non-responders and responders (F1,32= 4.591; p<0.0001); no significantlateralization (F1,32= 0.1517; p= 0.70).C. Plot of contrast estimate by HDRS17-score (centralized to mean). Significant positive correlations for left (0.33 ±0.12 [SE];F1,31= 7.076; p= 0.0123; r2= 0.19; line) and right (0.38 ±0.14 [SE]; F1,31= 6.929; p= 0.0131; r2= 0.18; dashed line) amygdala.Regression lines for left and right amygdala are not significantly different (F1,62= 0.0683; p= 0.79).
�� �� ��
���
��
�
�
��
��
������������������������������
������ ���
��������
���������
��
��
�
�
�
�
������������������������������
���� ���� ���� ����
���� ����������� ��
��������
���������
� �
�

Chap
ter
Chapter 10
10
206
motor cortex. With happy faces decreased activations were found in right insula, bilateral OFC,right VMPFC, bilateral anterior cingulate, left pregenual cingulate, bilateral parietal cortex andright putamen.
Relative to placebo dose-escalation, 6 weeks of true dose-escalation increased activations inright hippocampus, left subgenual cingulate, bilateral medial temporal gyrus, left parietal andDLPFC and right cerebellum (all faces; Table S10.5B). With the negative faces contrast, dose-escalation increased activations in right hippocampus, left parahippocampal gyrus, left subgenualcingulate, left medial temporal gyrus and bilateral parietal cortex and cerebellum. With happyfaces 6 weeks of dose-escalation increased activations in left parahippocampal gyrus, left medialtemporal gyrus and left DLPFC.
Discussion
This fMRI study evaluated the changes in amygdala activation to (negative) facial expressionsafter paroxetine treatment of MDD, as well as changes in other brain regions within the limbic-subcortical-prefrontal network. Depressed patients were scanned at study-entry, 6 weeks and 12weeks of paroxetine treatment. To our knowledge, this is the first fMRI study investigating SSRI-treatment with a randomized, placebo-controlled dose-escalation.
MDD-patients showed higher ventral (amygdala and insula) and decreased dorsal activationsrelative to HC at study entry. After 6 weeks of paroxetine treatment, when only 5/20 patientswere classified as responders, we found increased ventral (right amygdala) and dorsal activations(posterior, medial and pregenual cingulate cortex, left DMPFC and DLPFC). In contrast, after 12weeks of treatment (with 13/20 responders), we found decreased activations in ventral (left OFC)and increased activations in the dorsal compartment, all relative to study-entry. Treatmentresponse was associated with lower activations in amygdala, right OFC and insula, and higheractivations in right DLPFC and nucleus accumbens. The placebo-controlled randomized dose-escalation resulted in decreased activations in a number of ventral regions, but not amygdala,while right hippocampus and left subgenual cingulate activations increased, relative to placebodose-escalation. These results suggest that the attenuation of amygdala activation during SSRI-treatment is not a direct pharmacological response, but appears to be a biomarker for treatmentresponse.
Brain activations in MDD and changes following antidepressant treatmentOur study replicates an increased reactivity of the ventral ‘limbic’ structures in MDD-patients (e.g.amygdala and insula), with decreased activations in dorsal prefrontalareas (PFC and cingulate).8-11
Furthermore, like previous treatment studies10;13-15 paroxetine treatment over 12 weeks increaseddorsal cortical function, and reduced activations in the ventral structures. However, these effectswere more prominent when we distinguished treatment responders and non-responders.
In the present study, we investigated the role of the amygdala as a biomarker forantidepressant treatment. Previous reports suggested that the SSRIs sertraline and fluoxetinedecreased abnormal amygdala activations in response to negative stimuli in MDD-patients,10;12 aswas found for bupropion.14 Two studies in healthy volunteers, treated for 7 days with citalopramand the noradrenaline uptake inhibitor reboxetine also found an attenuation of amygdalaresponse to negative faces.16;17 Although we replicate this attenuation, our data suggest animportant difference regarding the interpretation previous findings. Specifically, our resultsindicate that abnormal amygdala activation is decreased in association with treatment response,but is not a direct pharmacological effect, as was also indicated by the absence of amygdala-effects after dose-escalation. Because the response-rates in previous MDD fMRI-studies werehigh; 10/11 (91%),12 13/19 (68%),10;28 and 6/8 (75%)14 this response-related effect may have beenmissed in previous group analyses.

Chapter10
207
Amygdala deactivation in responders to paroxetine
As expected, in our study amygdala activation at week 6 and 12 was correlated with otherventral structures (contralateral amygdala, OFC, subgenual cingulate) and inversely correlated topregenual (rostral) cingulate cortex and DLPFCactivity. This highlights an inhibitive connectionbetween the amygdala and the pregenual cingulate, a region that plays a crucial role inintegrating the function of dorsal and ventral prefrontal compartments.6;29-31 Previous pre-treatment, resting state, fluorodeoxyglucose positron emission tomography (PET) studies haveshown that increased metabolism in the pregenual cingulate predicted better treatmentresponse (reviewed by Dougherty and Rauch).29 This finding has been replicated in two fMRIstudies with negative faces/pictures,10;13;31 and in our post-hoc comparisons of study-entry scans.Moreover, we also found increased activations in the subgenual cingulate in final treatment non-responders,32;33 suggesting that activity of pregenual and subgenual cingulate areas may serve asadditional biomarkers for (future) treatment response.
The present study also included a paroxetine versus placebo dose-escalation comparison in asecondary randomization of treatment non-responders after 6 weeks. Relative to placebo, higherparoxetine doses was associated with decreased activity in several regions, including left OFC,bilateral insula, right VMPFC and left ventrolateral thalamus. In contrast, dose-escalation vs.placebo resulted in increased activity of the (para)hippocampal cortex and, unexpectedly, thesubgenual cingulate cortex. Two explanations can be put forward to account for these results.First, higher doses of paroxetine may further decrease ventral (limbic) hyperactivation andincrease dorsal prefrontal activation, in line with the effects seen at standard doses.10;12;14
However, the observed increased activity of the subgenual cingulate after dose-escalation is atodds with this hypothesis, because persistent or higher activation in the subgenual cingulate waspreviously associated with non-response.32;33 Also within this hypothesis, the increased activationsin the hippocampus might represent increased hippocampal neurogenesis after prolongedantidepressant administration, as has been found in rodents,34;35 and which is presumablyassociated with second-messenger effects of SSRIs resulting in increased levels of brain derivedneurotrophic factor (BDNF)36 In contrast, Mayberg et al. previously reported decreasedhippocampal metabolism in fluoxetine-responders, and no change in non-responders.33 However,these studies used fluorodeoxyglucose resting-state PET, comparing changes between week 1and week 6, so that direct comparisons are problematic.
An alternative explanation might be that dose-escalation does not lead to furtherimprovement MDD in patients, nor affect its neurobiological substrate. This would be in line withthe absence of unequivocal improvement after dose-escalation found in previous clinicaltrials.5;37-39 Following this line of argument, the increased activations in the subgenual cingulatemay be interpreted as supportive to the finding of Licht et al.,40 who reported that patients withincreased sertraline doses (200 mg/day) had a poorer response-rate relative to placebo dose-escalation (sertraline 100 mg/day).
In our study, correlation analyses showed that decreased amygdala activity in responders wasassociated with increased rostral cingulate and DLPFC activity, suggesting that antidepressantresponse is associated with increased cognitive control by the dorsal compartment as well as areduction of the hyperactivation in the ventral compartment. Recently, Chen et al. investigatedfunctional connectivity of the amygdala, and reported increased functional coupling with thefrontal and (pregenual) cingulate cortex (and striatum and thalamus) after 8 weeks of sertralinetreatment.31 Although assessed differently, we also found associations of the amygdala withthese regions. Therefore, it may be hypothesized that SSRI-exposure over time improves dorsalprefrontal regulation of abnormal limbic activity.31;41;42 However, the SSRI-effects on pregenualcingulate-amygdala coupling remain controversial,31;41 and may be modified by patient selectionand other variables (e.g. polymorphisms of the serotonin transporter promoter region andBDNF).31;43;44
In treatment responders, we found increased activation not only of dorsal prefrontal areasbut also of the left nucleus accumbens. Activation of the nucleus accumbens is associated withanticipation of reward.45 In contrast, expected loss might deactivate the nucleus accumbens.Although a recent pilot-study did not reveal differences in nucleus accumbens activation betweenMDD patients and HC during a monetary reward task,46 it might still be that during MDD either the

Chap
ter
Chapter 10
10
208
anticipation of reward is reduced, or the responses to loss are increased compared with a lessdepressed state. This might hypothetically explain the increased activations (or reduceddeactivations) seen in the nucleus accumbens in patients who showed a clinical response. Thisfinding merits further investigation of reward-processing in MDD.
LimitationsSimilar to previous studies,10;12;13 we refrained from a placebo-treated group, but instead included arandomized placebo-controlled dose-escalation. A full placebo-group, as in previous PET studies,47
might have enabled a clearer differentiation between true drug-effects and response effects.Therefore, as proposed recently, future fMRI-studies should preferably include a full placebo-group.31
Second, we scanned healthy controls once, so that differential habituation effects could notbe assessed.10;13 Nevertheless, our patient sample showed a biphasic response, the initial increasein amygdala activity being associated with a high non-response ratio at T0. In addition, we foundan overall clear distinction between treatment responders and non-responders. Both findingssuggest that changes in responsiveness during our fMRI paradigm are related to clinical staterather than time-effects. We therefore consider our results valid.
Third, the findings for the randomized dose-escalation in our modest sample size need to beinterpreted with caution, given the small sample size. Therefore, replications in larger samples arewarranted.
Fourth, the inclusion of patients with co-morbid secondary anxiety disorders (3/22) orsubstance abuse disorders (3/22), might theoretically have influenced our findings. We considerthis unlikely; first, one patient with co-morbid anxiety and alcohol dependence could not beincluded in the analyses because of excessive movements in the scans, and second, the alcoholand cannabis use was stopped during the paroxetine treatment in the remaining 2 subjects.
ConclusionIn this fMRI-study in MDD patients, we investigated changes in amygdala activation and otherbrain regions within the limbic-subcortical-cortical network after paroxetine treatment, andapplied a secondary randomized, placebo-controlled dose-escalation in non-responders after 6weeks of paroxetine at 20mg/day. We found that amygdala activation by negative faces washigher in depressed patients than in HC and that bilateral amygdala activation decreased intreatment responders only. Decreased amygdala activations were correlated with increased rightpregenual cingulate and DLPFC activations. These results support the use of amygdala activitymeasured by fMRI as a biomarker for treatment response, and indicate increased fronto-limbiccontrol as a mechanism of paroxetine drug-response effects. Future studies need to clarifywhether spontaneous, psychotherapy and/or placebo responders show similar attenuation inamygdala activation by negative faces.
Acknowledgements
The authors wish to thank the patients who participated in this fMRI-study. Mrs. E. Miedema M.D.was indispensable for clinical treatment and assisted in scanning. Mr. A. Nederveen, PhD, and Mr.M. de Ruiter, PhD and Mrs. T. Dekker are acknowledged for their contributions to scanning andanalyses. Mrs. M. Haages managed randomization and maintained blinding. This study wasfinanced by a grant from the Netherlands Organisation for Health Research and Development(ZonMw), program Mental Health, education of investigators in mental health (OOG; #100-002-002) to H.G. Ruhé, and a grant from the Dutch Brain Foundation (14F06.45).

Chapter10
209
Amygdala deactivation in responders to paroxetine
Conflicts of interest
None
References
1. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006; 3: e442.2. Kennedy SH, Lam RW, Cohen NL, Ravindran AV. Clinical guidelines for the treatment of depressive disorders. IV.
Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.3. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder
(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.4. NICE. Clinical Guideline 23. Depression: management of depression in primary and secondary care. December 2004. 23, 1-
63. 2004. London, National Institute for Clinical Excellence. 5. Ruhe HG, Booij J, Weert HCv, Reitsma JB, Fransen EJF, Michel MC et al. Evidence why dose-escalation of paroxetine in
major depressive disorder is not effective: A 6-week, randomized-controlled trial with assessment of serotonintransporter occupancy. Neuropsychopharmacology. 2008; [In Press].
6. Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-basedalgorithms for diagnosis and optimised treatment. Br Med Bull. 2003; 65: 193-207.
7. Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatricdisorders. Biol Psychiatry. 2003; 54: 515-528.
8. Abler B, Erk S, Herwig U, Walter H. Anticipation of aversive stimuli activates extended amygdala in unipolar depression. JPsychiatr Res. 2007; 41: 511-522.
9. Anand A, Li Y, Wang Y, Wu J, Gao S, Bukhari L et al. Activity and connectivity of brain mood regulating circuit indepression: a functional magnetic resonance study. Biol Psychiatry. 2005; 57: 1079-1088.
10. Fu CH, Williams SC, Cleare AJ, Brammer MJ, Walsh ND, Kim J et al. Attenuation of the neural response to sad faces inmajor depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imagingstudy. Arch Gen Psychiatry. 2004; 61: 877-889.
11. Surguladze S, Brammer MJ, Keedwell P, Giampietro V, Young AW, Travis MJ et al. A differential pattern of neuralresponse toward sad versus happy facial expressions in major depressive disorder. Biol Psychiatry. 2005; 57: 201-209.
12. Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA. Increased amygdala response to maskedemotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 2001; 50:651-658.
13. Davidson RJ, Irwin W, Anderle MJ, Kalin NH. The neural substrates of affective processing in depressed patients treatedwith venlafaxine. Am J Psychiatry. 2003; 160: 64-75.
14. Robertson B, Wang L, Diaz MT, Aiello M, Gersing K, Beyer J et al. Effect of bupropion extended release on negativeemotion processing in major depressive disorder: a pilot functional magnetic resonance imaging study. J Clin Psychiatry.2007; 68: 261-267.
15. Schaefer HS, Putnam KM, Benca RM, Davidson RJ. Event-related functional magnetic resonance imaging measures ofneural activity to positive social stimuli in pre- and post-treatment depression. Biol Psychiatry. 2006; 60: 974-986.
16. Norbury R, Mackay CE, Cowen PJ, Goodwin GM, Harmer CJ. Short-term antidepressant treatment and facial processing.Functional magnetic resonance imaging study. Br J Psychiatry. 2007; 190: 531-532.
17. Harmer CJ, Mackay CE, Reid CB, Cowen PJ, Goodwin GM. Antidepressant drug treatment modifies the neural processingof nonconscious threat cues. Biol Psychiatry. 2006; 59: 816-820.
18. First MB, Spitzer RL, Gibbon M, Williams JBW. Structured Clinical Interview for DSM-IV Axis I Disorders Patient Edition(SCID-I/P version 2.0). Translated in Dutch by. Groenestijn MAC, Akkerhuis GW, Kupka RW, Schneider N, and NolenWA.1999. Lisse, the Netherlands, Swets & Zeitlinger B.V.
19. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56-61.20. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry. 1961; 4:
561-571.21. Baker CB, Woods SW. Is there a SSRI dose response in treating major depression? The case for re-analysis of current data
and for enhancing future study design. Depress Anxiety. 2003; 17: 10-18.22. Saunders K, Simon G, Bush T, Grothaus L. Assessing the feasibility of using computerized pharmacy refill data to monitor
antidepressant treatment on a population basis: a comparison of automated and self-report data. J Clin Epidemiol. 1998;51: 883-890.
23. Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The Inventory of Depressive Symptomatology (IDS):psychometric properties. Psychol Med. 1996; 26: 477-486.
24. Hariri AR, Bookheimer SY, Mazziotta JC. Modulating emotional responses: effects of a neocortical network on the limbicsystem. Neuroreport. 2000; 11: 43-48.
25. Wolfensberger SPA, Veltman DJ, Hoogendijk WJG, Boomsma DI, de Geus EJC. Amygdala responses to emotional faces intwins discordant or concordant for the risk for anxiety and depression. Neuroimage. 2008; E-pub: doi: 10.1016/j.neuroimage.2008.01.053.
26. Ekman P, Friesen W. Pictures of facial affect. Palo Alto, CA: Consulting Psychologists; 1976.

Chap
ter
Chapter 10
10
210
27. Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discoveryrate. Neuroimage. 2002; 15: 870-878.
28. Walsh ND, Williams SC, Brammer MJ, Bullmore ET, Kim J, Suckling J et al. A longitudinal functional magnetic resonanceimaging study of verbal working memory in depression after antidepressant therapy. Biol Psychiatry. 2007; 62: 1236-1243.
29. Dougherty DD, Rauch SL. Brain correlates of antidepressant treatment outcome from neuroimaging studies indepression. Psychiatr Clin North Am. 2007; 30: 91-103.
30. Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL et al. Cingulate function in depression: apotential predictor of treatment response. Neuroreport. 1997; 8: 1057-1061.
31. Chen CH, Suckling J, Ooi C, Fu CH, Williams SC, Walsh ND et al. Functional Coupling of the Amygdala in Depressed PatientsTreated with Antidepressant Medication. Neuropsychopharmacology. 2008; 33: 1909-1918.
32. Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA et al. Reciprocal limbic-cortical function andnegative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999; 156: 675-682.
33. Mayberg HS, Brannan SK, Tekell JL, Silva JA, Mahurin RK, McGinnis S et al. Regional metabolic effects of fluoxetine inmajor depression: serial changes and relationship to clinical response. Biol Psychiatry. 2000; 48: 830-843.
34. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rathippocampus. J Neurosci. 2000; 20: 9104-9110.
35. Djavadian RL. Serotonin and neurogenesis in the hippocampal dentate gyrus of adult mammals. Acta Neurobiol Exp(Wars). 2004; 64: 189-200.
36. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;33: 73-83.
37. Adli M, Baethge C, Heinz A, Langlitz N, Bauer M. Is dose escalation of antidepressants a rational strategy after a medium-dose treatment has failed? A systematic review. Eur Arch Psychiatry Clin Neurosci. 2005; 255: 387-400.
38. Baker CB, Tweedie R, Duval S, Woods SW. Evidence that the SSRI dose response in treating major depression should bereassessed: a meta-analysis. Depress Anxiety. 2003; 17: 1-9.
39. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard-dose selectiveserotonin reuptake inhibitors in major depressive disorder: Systematic review. Br J Psychiatry. 2006; 189: 309-316.
40. Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first-line sertralinetreatment: A randomised study of extended duration of treatment, dose increase or mianserin augmentation.Psychopharmacology (Berl). 2002; 161: 143-151.
41. Anand A, Li Y, Wang Y, Wu J, Gao S, Bukhari L et al. Antidepressant effect on connectivity of the mood-regulating circuit:an FMRI study. Neuropsychopharmacology. 2005; 30: 1334-1344.
42. Mayberg HS. Modulating limbic-cortical circuits in depression: targets of antidepressant treatments. Semin ClinNeuropsychiatry. 2002; 7: 255-268.
43. Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS et al. 5-HTTLPR polymorphismimpacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8: 828-834.
44. Pezawas L, Meyer-Lindenberg A, Goldman AL, Verchinski BA, Chen G, Kolachana BS et al. Evidence of biologic epistasisbetween BDNF and SLC6A4 and implications for depression. Mol Psychiatry. 2008; 13: 709-716.
45. Knutson B, Adams CM, Fong GW, Hommer D. Anticipation of increasing monetary reward selectively recruits nucleusaccumbens. J Neurosci. 2001; 21: RC159.
46. Knutson B, Bhanji JP, Cooney RE, Atlas LY, Gotlib IH. Neural responses to monetary incentives in major depression. BiolPsychiatry. 2008; 63: 686-692.
47. Mayberg HS, Silva JA, Brannan SK, Tekell JL, Mahurin RK, McGinnis S et al. The functional neuroanatomy of the placeboeffect. Am J Psychiatry. 2002; 159: 728-737.
48. Fu CH, Williams SC, Brammer MJ, Suckling J, Kim J, Cleare AJ et al. Neural responses to happy facial expressions in majordepression following antidepressant treatment. Am J Psychiatry. 2007; 164: 599-607.

Chapter10
211
Amygdala deactivation in responders to paroxetine
Tabl
e S1
0.1
Prev
ious
fMRI
stu
dies
of a
ffec
tive
stim
uli (
face
s/IA
PS) i
n M
DD
dur
ing
trea
tmen
t with
ant
idep
ress
ants
Aut
hor &
Dat
e (r
efer
ence
) Re
sult
s
Popu
latio
n (m
ean
age)
Sex
N
Des
ign
fMRI
par
adig
mR
oIs
stud
ied
/re
port
edRe
sults
Rem
arks
Dav
idso
n et
al.
(200
3)13
MD
D P
ts R
x-fr
ee ≥
? W
eeks
, HD
RS ≥
? (3
8.2
±9.3
) H
C (2
7.8
±10.
4)
M+F
12
5
8 w
eek,
VLX
225
mg
(ope
n),
3 sc
ans
Bsl,
2wk,
8w
k; 1.
5T
Neg
ativ
e, p
ositi
ve, n
eutr
al
IAPS
evo
king
hig
h ar
ousa
l.
L.In
s, L
.CG
(A),
Vi
sual
Cor
tex,
Am
yg
Act
ivat
ion
in L
.Ins
decr
ease
d in
Pts
vs
HC
(neg
. vs
neut
ral);
aft
er 2
wee
ks P
ts =
HC;
aft
er 8
wee
ks P
ts
incr
ease
d ac
tivat
ion
vs H
C (n
.s.)
Act
ivat
ion
L.CG
(A) d
ecre
ased
in P
ts v
s H
C (n
eg. v
s ne
urtr
al);
af
ter 8
wee
ks P
ts in
crea
sed
activ
atio
n vs
HC
(n.s
.)
Bsl A
myg
act
ivity
in P
ts ≈
HC;
Bsl
DLP
FC a
ctiv
ity in
Pt
s <
HC
(L) a
nd P
ts ≈
HC
(R).
Recr
uitm
ent b
y ad
vert
isem
ent,
ass
essm
ent
not s
peci
fied
. Hig
her p
re-t
reat
men
t CG
(A)
activ
atio
n pr
edic
ted
impr
ovem
ent.
Sta
rt
stim
ulus
and
sca
nnin
g m
anua
lly. N
o pl
aceb
o.
Res
pond
ers
only
: of o
rigin
ally
14 M
DD
Pts
2
non-
resp
onde
rs w
ere
excl
uded
.
Fu e
t al.
(200
4)10
MD
D P
ts R
x-fr
ee ≥
4 w
ks, H
DRS
≥18
(4
2.8
±6.7
) M
atch
ed H
C (4
3.2
±8.8
) M
+F
19
19
8 w
eekF
LX 2
0 m
g (o
pen)
, 2
scan
s Bs
l, 8w
k; 1.
5T S
ad
Ekm
an F
aces
of v
aria
ble
inte
nsity
: stu
dy o
f act
ivat
ion
and
dyna
mic
rang
e
HCM
, PH
C, A
myg
, H
ypot
hal,
Thal
, Ins
, CG
(A+D
), VS
tria
t N
C, In
fPC,
CER
Act
ivat
ion
in a
ll re
gion
s in
crea
sed
in p
ts v
s. H
C at
bs
l. Li
mbi
c an
d su
bcor
tical
act
ivat
ions
dec
reas
ed,
dyna
mic
rang
e in
neo
cort
ical
regi
ons
incr
ease
d,
in p
ts a
fter
8 w
eeks
. Dec
reas
e in
HD
RS
~ re
duct
ion
dyna
mic
rang
e in
CG
(A) a
nd C
ER.
Patie
nts
recr
uite
d by
adv
ertis
emen
ts, a
sses
s ed
by
SCID
, no
axis
1 co
-mor
bidi
ty. N
o pl
aceb
o,
13/1
9 w
eek
8 re
spon
ders
; 9 /1
9 re
mis
sion
; HD
RS
resp
onde
rs 5
.8 ±
2.7;
non
-res
pond
ers1
5.0
±4.9
28
Fu e
t al.
(200
7)48
MD
D P
ts R
x-fr
ee ≥
4 w
ks, H
DRS
≥18
(4
2.8
±6.7
) M
atch
ed H
C (4
3.2
±8.8
) M
+F
19
19
8 w
eekF
LX 2
0 m
g (o
pen)
, 2
scan
s Bs
l, 8w
k; 1.
5T H
appy
Ek
man
Fac
es o
f var
iabl
e in
tens
ity: s
tudy
of a
ctiv
atio
n an
d dy
nam
ic ra
nge
extr
astr
iata
l cor
tex,
Li
ngG
, CER
, Cun
, CG
(P),
Put
, Tha
l, H
CM, P
reC
Act
ivat
ion
in e
xtra
stria
tal c
orte
x, C
ER, L
ingG
, Cun
, CG
(P) l
ower
in p
ts v
s H
C at
bsl
. A
ctiv
atio
n in
crea
sed
in p
ts a
fter
8w
ks. P
ts h
ave
smal
ler
dyna
mic
rang
e in
Put
, Tha
l, Pr
eC, C
G(P
) vs
HC
at
base
line
and
wk
8.
See
unde
r Fu
et a
l. (2
004)
Har
mer
et a
l. (2
006)
17H
C w
ithou
t (lif
etim
e)
psyc
hiat
ric d
isor
der
24
7 da
ys d
oubl
e bl
ind
rand
omis
ed: P
L or
CIT
20
mg
(mat
ched
on
gend
er, a
ge a
nd
IQ),
1 sc
an a
t 7 d
ays;
1.5T
M
aske
d (1
7ms
+ 16
7ms
neut
ral)
fear
ful,
happ
y or
ne
utra
l Ekm
an fa
ces
Am
yg (L
+R)
Am
yg(L
+R) a
nd M
ed.P
FC a
ctiv
ity s
igni
fica
ntly
lo
wer
aft
er fe
arfu
l fac
es u
nder
CIT
vs
PL. N
o si
gn.
effe
cts
of C
IT v
s PL
on
happ
y fa
ces.
Recr
uitm
ent n
ot s
peci
fied,
ass
essm
ent n
ot
spec
ified
. No
base
line
scan
s. N
o pa
tient
s.
Dec
reas
ed M
ed.P
FC a
ctiv
ity m
ay b
e co
ntra
dict
ory
to d
ecre
ased
Med
.PFC
act
ivity
in
MD
D. E
xpla
ined
as
decr
ease
in H
C du
e to
CIT
in
hibi
tion
of A
myg
Nor
bury
et a
l. (2
007)
16H
C w
ithou
t (lif
etim
e)
psyc
hiat
ric d
isor
der
24
7 da
ys d
oubl
e bl
ind
rand
omis
ed: P
L or
REB
8m
g,
1 sca
n at
7 d
ays;
1.5T
Mas
ked
(17m
s +
183m
s ne
utra
l) an
d ov
ert (
200m
s) fe
arfu
l, ha
ppy
or n
eutr
al E
kman
face
s
Am
yg (L
+R)
Mas
ked
Am
yg(R
) act
ivity
sig
nifi
cant
ly lo
wer
aft
er
fear
ful f
aces
und
er R
EB v
s PL
; no
sign
diff
eren
ces
in o
vert
stim
ulus
pre
sent
atio
n or
Am
yg(L
).
Incr
ease
d ac
tivity
und
er R
EB v
s PL
in F
G(R
) aft
er
happ
y fa
ces
Recr
uitm
ent n
ot s
peci
fied,
ass
essm
ent n
ot
spec
ified
. No
base
line
scan
s. N
o pa
tient
s.
Resp
onse
to m
aske
d fa
ces
mig
ht re
pres
ent
effe
ct o
f REB
on
auto
mat
ic a
spec
ts o
f pr
oces
sing
.

212
Chap
ter
Chapter 10
10
Robe
rtso
n et
al.
(200
7)14
MD
D P
ts R
x-fr
ee ≥
? W
ks, H
DRS
≥15
(4
1.4 ±
7)
M+F
108
wee
k BU
P 15
0-45
0 m
g (o
pen)
, 2 sc
ans B
sl, 8
wk;
1.5
T N
eg. I
APS
(war
fare
/vio
lenc
e)
in E
mot
iona
l Odd
ball
Task
OFC
, VM
PFC,
CG
(A.P
), In
fPFC
, FG
, D
MPF
C, A
myg
, HC
Redu
ced
activ
atio
n in
OFC
(R),
VM
PFC(
R),
CG(A
.R),
InfP
FC(R
), H
C+A
myg
dala
(R),
NC(
R),
FG(R
), D
MPF
C(L)
, CG
(P.L
) aft
er 8
wee
ks o
f BU
P In
crea
sed
activ
atio
n in
InfP
FC(L
), F
G(L
). D
ecre
ase
in a
ctiv
atio
n in
OFC
(R),
InfP
FC(R
), FG
(R) a
nd
Am
yg (L
) cor
rela
ted
with
impr
ovem
ent i
n H
DR
S
Recr
uitm
ent n
ot s
peci
fied.
Sca
ns o
f 8 s
ubje
cts
com
plet
ing
follo
w-u
p us
ed fo
r ana
lyse
s. N
o pl
aceb
o, n
o co
ntro
ls. A
fter
BU
P, 2
/8 o
f sub
ject
s sh
owed
no
impr
ovem
ent,
4/8
reac
hed
HD
RS
≤7. 3
/8 p
ts w
ithdr
ew b
efor
e 8
wee
ks, s
cans
m
ade
at w
ithdr
awal
.
Scha
efer
et a
l. (2
006)
15M
DD
or d
ysth
ymia
Pts
R
x-fr
ee ≥
4 w
ks, H
DR
S ≥2
2 (3
5.9
±?)
HC
with
out 1
st d
egre
e re
lativ
es w
ith p
sych
. D
isor
d. (2
8.2
±?)
M+F
9 14
Aver
age
22 w
eek
VLX
150-
300
mg
(ope
n), 2
sca
ns B
sl, a
fter
re
mis
sion
(4 w
eeks
HD
RS
≤10)
; 1.5
T Po
s. IA
PS (s
ocia
l in
tera
ctio
n, s
ingl
e pe
rson
, hu
man
face
s, p
eopl
e, e
rotic
a,
nonh
uman
app
etiv
e) in
pa
ssiv
e vi
ewin
g
Inf.,
Med
., Su
p.PF
C,
Ins,
Med
.DN
T,
FG(R
)
For F
aces
vs
peop
le/n
onhu
man
app
etiti
ve: I
n pa
tient
s si
gn in
crea
sed
activ
ity a
t 2nd
sca
n in
In
f.,M
ed.,S
up.P
FC(L
), In
s(L,
R),
Med
.DN
T(R)
, FG
(R),
whi
le in
con
trol
s th
ese
regi
ons
show
ed
sign
, inc
reas
ed a
ctiv
ity a
t Bsl
sca
n on
ly.
Recr
uitm
ent b
y ad
vert
isem
ent,
ass
essm
ent b
y SC
ID. 2
nd s
can
afte
r rem
issi
on o
nly.
6/9
pa
tient
s re
ache
d re
mis
sion
. Tw
o ot
her
cont
rast
s: s
ocia
l int
erac
tion
and
erot
ica
also
pr
esen
ted
in m
anus
crip
t. In
exp
erim
ents
for
face
s ex
cept
for I
ns a
nd F
G(R
) a s
igni
fica
nt
late
ralit
y w
as o
bser
ved.
No
inte
ract
ion
with
cl
inic
al im
prov
emen
t or V
LX d
ose
foun
d.
Shel
ine
et a
l. (2
001)
12M
DD
Pts
Rx-
free
≥4
wks
, HD
RS ≥
18
(40.
3 ±
?)
Mat
ched
HC
HD
RS <
8 (3
9.8
±?)
M +
F
11
11
8 w
eek
SER
~10
0 m
g (o
pen)
, 2
scan
s Bs
l, 8w
k; 1.
5T M
aske
d (4
0ms)
fear
ful,
neut
ral o
r ha
ppy
follo
wed
by
neut
ral
(160
ms)
Ekm
an F
aces
Am
yg (L
+R)
Bsl:
L.A
myg
act
ivat
ion
Pts
> H
C; R
.Am
yg P
ts≥H
C (n
.s.)
(fea
rful
/hap
py v
s ne
utra
l) Pt
s: a
fter
SER
, L+
R A
myg
act
ivat
ion
afte
r all
or fe
arfu
l fac
es s
ign.
D
ecre
ased
. Aft
er S
ER L
+R A
myg
act
ivity
in P
ts =
H
C
Recr
uitm
ent b
y ad
vert
isem
ent,
ass
essm
ent b
y D
SM-IV
crit
eria
. Aft
er S
ER m
ean
HD
RS
decr
ease
d fo
rm 2
3.3
to 9
.7. O
nly
1 non
-re
spon
der a
nd 2
par
tial r
espo
nder
s. N
o co
rrel
atio
n be
twee
n L.
Am
yg s
igna
l int
ensi
ty
chan
ge a
nd H
DR
S or
anx
iety
Abb
revi
atio
ns: A
myg
= A
myg
dala
, BU
P= b
upro
pion
, CER
= Ce
rebe
llum
, CG
= Ci
ngul
ate
gyru
s (A
-ant
erio
r, D
-dor
sal,P
-pos
terio
r), C
IT=
Cita
lopr
am, C
un=
Cune
us, D
MPF
C= D
orsa
l Med
ial P
refr
onta
l Cor
tex,
FLX
= flu
oxet
ine,
FG
= Fu
sifo
rm G
yrus
, HC=
Hea
lthy
Cont
rol,
HCM
= H
ippo
cam
pus,
HD
RS=
Ham
ilton
Dep
ress
ion
Rat
ing
Scal
e, H
ypot
hal=
Hyp
otha
lam
us, I
nfFC
= In
fFro
ntal
Cor
tex,
InfP
C= In
ferio
r Par
ieta
l Cor
tex,
In
s= In
sula
, L=
left
, Lin
gG=
Ling
ual g
yrus
, MD
D=
Maj
or D
epre
ssiv
e D
isor
der,
Med
.DN
T= M
edia
l Dor
sal N
ucle
us o
f Tha
lam
us, N
C= N
ucle
us C
auda
tus,
PFC
= Pr
efro
ntal
Cor
tex,
PH
C= P
arah
ippo
cam
pal g
yrus
, PL
= pl
aceb
o, P
reC=
Pre
cune
us, P
ts=
patie
nts,
Put
= Pu
tam
en, R
= rig
ht, R
EB=
Reb
oxet
ine,
SER
= se
rtra
line,
Tha
l= T
hala
mus
, VLX
= ve
nlaf
axin
e, V
Stria
t= V
entr
al s
tria
tum
.
Tabl
e S1
0.1
Prev
ious
fMRI
stu
dies
of a
ffec
tive
stim
uli (
face
s/IA
PS) i
n M
DD
dur
ing
trea
tmen
t with
ant
idep
ress
ants
(Con
tinue
d)
Aut
hor &
Dat
e (r
efer
ence
) Re
sult
s
Popu
latio
n (m
ean
age)
Sex
N
Des
ign
fMRI
par
adig
mR
oIs
stud
ied
/re
port
edRe
sults
Rem
arks

Chapter10
213
Amygdala deactivation in responders to paroxetine
Table S10.2 Main effects in study-entry scans: MDD-patients and HC combined.
Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p
MDD + HC
All Faces Amygdala R 20 -4 -12 12 3.73 <0.001
L -18 -4 -15 8 4.00 <0.001
Fusiform gyrus R 40 -52 -21 160 6.01 <0.001
L -42 -56 -24 99 5.34 <0.001
DLPFC R 46 16 30 405 5.94 <0.001
L -40 24 21 15 3.61 <0.001
Insula (Ant.) R 36 30 -3 249 5.21 <0.001
L -34 30 -6 119 4.49 <0.001
OFC R 32 34 -12 * 4.77 <0.001
Occipital R 12 -94 6 88 4.86 <0.001
L -14 -98 15 64 4.99 <0.001
Parietal R 48 -50 6 126 4.72 <0.001
R 48 -68 12 34 4.03 <0.001
DMPFC R 4 22 45 35 3.91 <0.001
Cuneus R 4 -64 33 15 3.84 <0.001
Neg. Faces Amygdala L -18 -4 -15 8 3.95 <0.001
Fusiform gyrus R 40 -52 -21 165 6.28 <0.001
L -42 -56 -24 82 4.61 <0.001
DLPFC R 42 16 27 339 5.65 <0.001
OFC R 34 36 -12 107 4.37 <0.001
Insula (Ant.) R 36 30 -3 * 4.35 <0.001
Occipital R 14 -96 9 45 4.31 <0.001
L -12 -98 15 56 5.24 <0.001
Parietal R 48 -50 6 61 4.25 <0.001
R 50 -68 12 9 3.73 <0.001
DMPFC R 4 22 45 11 3.66 <0.001
Cuneus R 4 -60 33 10 3.93 <0.001
Hap. Faces DLPFC R 46 16 30 173 5.60 <0.001
Fusiform gyrus R 38 -56 -21 90 5.36 <0.001
L -44 -56 -24 61 4.62 <0.001
Insula (Ant.) R 36 22 3 163 5.01 <0.001
L -34 24 0 72 4.44 <0.001
OFC R 32 34 -12 * 4.70 <0.001
Occipital R 16 -94 6 38 4.41 <0.001
L -10 -96 9 17 3.98 <0.001
Thalamus R 4 -14 6 20 4.04 <0.001
Parietal R 52 -50 6 8 3.80 <0.001
Clusters are FDR (p<0.05)-corrected with extend threshold of 3 voxels.* cluster size not reported, one region extended from right anterior insula to right OFC Abbreviations: see also Table 10.3 and 10.4. DMPFC= dorsomedial prefrontal cortex

Chap
ter
Chapter 10
10
214
Table S10.3 Changes after 6 weeks of treatment (T0) compared with study-entry.Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p
A. Study-entry > T0
All Faces Amygdala R 14 4 -12 11 1.92 0.027
L -16 0 -15 12 1.97 0.024
Cuneus LR 0 -66 12 67 4.27 <0.001
Hippocampus, posterior L -18 -48 9 7 3.60 <0.001
R 28 -36 0 31 3.27 0.001
Neg. Faces Amygdala L -14 2 -12 11 2.25 0.012
Cuneus L -2 -66 12 51 3.97 <0.001
Hap. Faces Sup.Temporal Sulcus L -48 -46 6 11 3.48 <0.001
DLPFC L -50 0 15 6 3.37 <0.001
Insula L -36 16 -18 31 3.36 <0.001
B. T0 > study-entry
All Faces Amygdala R 28 -2 -15 28 3.22 0.001
Cingulate, posterior L -12 -18 48 21 4.13 <0.001
medial L 2 4 33 70 3.37 <0.001
pregenual R 4 36 3 15 3.41 <0.001
Sup.Temporal gyrus R 62 -8 -9 8 3.79 <0.001
Hippocampus, dorsal R 28 -28 -12 31 3.74 <0.001
DMFPC L -10 22 60 32 3.39 <0.001
Inf. Temporal Gyrus L -44 -4 -39 14 3.28 0.001
Neg. Faces Amygdala R 28 0 -15 47 3.47 <0.001
DMFPC L -4 22 60 288 4.56 <0.001
Cingulate, anterior R 2 30 33 133 3.21 0.001
medial LR 2 2 36 69 3.27 0.001
pregenual R 6 34 3 5 3.43 <0.001
VLPFC L -24 58 30 82 3.89 <0.001
DLPFC L -50 26 -3 16 3.65 <0.001
L -28 44 42 55 3.51 <0.001
L -30 22 54 14 3.28 0.001
R 32 -2 54 12 3.22 0.001
R 24 42 48 136 3.09 0.001
Hippocampus, dorsal R 30 -28 -9 48 3.53 <0.001
Cerebellum L -22 -34 -27 15 3.21 0.001
Sup. Temporal gyrus R 62 -8 -9 7 3.17 0.001
N.Accumbens L -4 8 -12 9 3.13 0.001
Hap. Faces Cerebellum L -24 -32 -24 23 3.78 <0.001
Sup.Temporal gyrus R 62 -8 -9 3 3.57 <0.001
Amygdala R 22 -12 -18 8 3.22 0.001
L -18 -10 -18 3 2.47 0.007
Abbreviations: see Table 10.3, 10.4 and S10.2.

Chapter10
215
Amygdala deactivation in responders to paroxetine
Table S10.4 Positive and negative correlations with amygdala activation (negative faces).
Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p
A. Left Amygdala (MNI -22, 2, -18) activation
Positive* Hippocampus R 26 -18 -12 102 4.24 <0.001
Amygdala R 22 4 -18 127 5.09 <0.001
OFC R 24 14 -21 † 4.34 <0.001
L -38 18 -21 33 3.77 <0.001
Parahippocampal gyrus L -30 -34 -18 98 4.25 <0.001
Insula R 38 -6 12 18 3.60 <0.001
Cingulate, subgenual R 6 14 -12 10 3.59 <0.001
Parietal R 52 -54 -15 37 3.88 <0.001
Negative‡ Precuneus R 14 -58 21 4 2.78 0.003
Cingulate, pregenual R 4 50 0 14 2.73 0.001
B. Right Amygdala (MNI 18, -2, -18) activation
Positive* Hippocampus R 24 -22 -12 † 4.56 <0.001
R 30 -32 -6 17 4.51 <0.001
L -16 -30 -6 57 5.07 <0.001
Amygdala L -18 -4 -21 65 4.49 <0.001
L -18 -10 -12 42 4.20 <0.001
OFC L -38 16 -21 40 4.01 <0.001
L -34 34 -12 18 3.77 <0.001
R 40 14 -21 13 3.48 <0.001
DLPFC R 58 14 36 14 3.70 <0.001
Negative‡ DLPFC L -22 16 57 6 3.16 0.001
R 22 18 63 5 2.87 0.002
Accumbens L -10 12 -9 3 2.68 0.004 * activations for p<0.001 with extend voxel size 10† in one cluster with right amygdala ‡ activations for p<0.01 with extend voxel size 3 Abbreviations: see Table 10.3, 10.4 and S10.2.
Table S10.5 Changes between T0 and T1 for true dose-escalation (paroxetine 30-50 mg) relative to placebo dose-escalation (paroxetine 20 mg/day).
Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p
A. Decrease in activation by DE relative to placebo
All Faces DMPFC L -6 14 51 333 4.27 <0.001
DLPFC L -24 16 51 96 4.31 <0.001
R 26 14 63 7 3.10 0.001
Thalamus, ventrolateral L -8 -18 3 96 4.26 <0.001
Motor cortex R 26 -2 63 138 4.19 <0.001
L -4 -6 66 56 3.97 <0.001
OFC L -38 28 -3 226 3.93 <0.001
Insula R 44 -6 6 100 3.91 <0.001
R 36 22 3 110 3.71 <0.001
L -42 -10 3 42 3.65 <0.001
Putamen R 28 0 -3 24 3.84 <0.001
Parietal R 62 -36 39 86 3.69 <0.001
R 66 -32 18 60 3.64 <0.001
R 56 -52 39 99 3.48 <0.001
Cingulate, anterior R 14 32 18 36 3.30 <0.001
Precuneus R 12 -60 21 42 3.42 <0.001
VMPFC R 16 58 33 37 3.28 0.001
Sup.Temporal Gyrus L -42 -32 12 19 3.15 0.001
G. Pallidus L -10 0 -3 10 3.14 0.001

Chap
ter
Chapter 10
10
216
Neg. Faces Motor cortex LR -4 -6 66 62 4.42 <0.001
LR -8 -14 48 85 3.24 0.001
DLPFC L -18 6 57 101 4.22 <0.001
L -24 16 51 77 3.60 <0.001
DMFPC L -6 14 51 * 3.72 <0.001
Thalamus, ventrolateral L -6 -18 0 60 3.80 <0.001
Insula R 36 22 3 72 3.74 <0.001
R 44 -6 6 41 3.62 <0.001
OFC R 38 34 6 † 3.46 <0.001
L -36 30 -3 61 3.55 <0.001
Cingulate, anterior R 12 30 21 14 3.28 0.001
Precuneus R 12 -60 21 9 3.24 0.001
Hap. Faces Insula R 36 12 0 207 4.21 <0.001
R 32 16 -21 40 3.67 <0.001
Putamen R 22 12 3 134 3.20 0.001
Parietal L -60 -36 18 109 4.08 <0.001
R 62 -28 42 166 3.10 0.001
R 26 -10 60 10 3.55 <0.001
R 22 -26 66 67 3.21 0.001
Cingulate, anterior L -6 32 18 218 3.17 0.001
R 12 42 24 180‡ 3.59 <0.001
pregenual L -8 46 -6 127 3.90 <0.001
VMPFC R 22 58 18 180 3.91 <0.001
OFC L -28 42 -15 ¶ 3.00 0.001
R 22 46 -15 136 3.43 <0.001
R 16 16 -15 23 3.23 0.001
Premotor Cortx R 4 0 54 14 3.21 0.001
DLPFC R 26 38 45 19 3.15 0.001
L -24 20 54 67 3.10 0.001
B. Increase in activation by DE relative to placebo
All Faces Med.Temporal Gyrus L -52 -6 -12 32 4.02 <0.001
R 60 -34 -3 19 3.44 <0.001
Hippocampus R 24 -14 -18 69 3.85 <0.001
Cingulate, subgenual L -2 22 -6 48 3.65 <0.001
Parietal L -60 -42 -6 25 3.63 <0.001
DLPFC L -54 24 15 45 3.16 0.001
Cerebellum R 12 -46 -15 23 3.14 0.001
Neg. Faces Cingulate, subgenual L -2 22 -6 66 4.16 <0.001
Med.Temporal Gyrus L -52 -6 -12 65 4.12 <0.001
Hippocampus R 20 -24 -21 147 3.85 <0.001
Parietal R 8 -34 57 6 3.4 <0.001
L -40 -34 66 9 3.19 0.001
Cerebellum LR -6 -42 -15 79 3.39 <0.001
Parahippocampal gyrus L -26 -32 -21 19 3.10 0.001
Hap. Faces Med.Temporal Gyrus L -60 -42 -6 43 4.51 <0.001
Parahippocapal gyrus L -28 -32 -12 12 3.55 <0.001
DLPFC L -58 20 21 43 3.01 <0.001 * in one cluster with left DLPFC † in one cluster with right insula‡ in one cluster with right VMPFC ¶ in one cluster with left pregenual cingulate Abbreviations: see also Table 10.3, 10.4 and S10.2. VMPFC= ventromedial prefrontal cortex.
Table S10.5 Changes between T0 and T1 for true dose-escalation (paroxetine 30-50 mg) relative to placebo dose-escalation (paroxetine 20 mg/day). (Continued)
Contrast Brain region L/R x,y,z (MNI mm) Cluster size (k) Max. voxel Z p

CHA
PTER
EFFECT OF THE SELECTIVE SEROTONIN REUPTAKE
INHIBITOR PAROXETINE ON PLATELET FUNCTION IS MODIFIED BY A SLC6A4 SEROTONIN
TRANSPORTER POLYMORPHISM
Journal of Thrombosis and Haemostasis 2008; in press
Nadia Abdelmalik1,2*
Henricus G. Ruhé1*
Kurdo Barwari2
Erik-Jan van den Dool3
Joost C.M. Meijers2
Saskia Middeldorp2,4,5
Harry R. Büller2
Aart H. Schene1
Pieter W. Kamphuisen2
* Both authors contributed equally to the manuscript1 Program for Mood Disorders, Department of Psychiatry, Academic Medical Center,University ofAmsterdam, Amsterdam, the Netherlands2 Department of Vascular Medicine, Academic Medical Center, University of Amsterdam, Amsterdam,the Netherlands 3 Department of Clinical Chemistry, Academic Medical Center, University of Amsterdam, Amsterdam,the Netherlands4 Department of Clinical Epidemiology, Leiden University Medical Center, Leiden, the Netherlands 5 Department of General Internal Medicine and Endocrinology, Leiden University Medical Center,Leiden, the Netherlands

Chap
ter
Chapter 11
11
218
Abstract
BackgroundSelective serotonin reuptake inhibitors (SSRIs) have been associated with an increased bleedingtendency.
AimTo prospectively quantify the dose-response effects of paroxetine and the influence of theserotonin transporter gene (SLC6A4) promoter polymorphism (5-HTTLPR) on platelet function.
MethodsNineteen drug-free psychiatric outpatients (44.5 ±10.8 years), were tested before and after 6weeks of paroxetine treatment (20 mg/day). Based on clinical symptoms, paroxetine dosageswere increased (40-50 mg/day) for 6 more weeks in 11 patients. Parameters related to plateletfunction were assessed by bleeding time, platelet function analyzer (PFA), platelet serotonin,platelet factor 4 (PF4), β-thromboglobulin (β-TG), and aggregation tests.
ResultsParoxetine 20 mg/day increased mean bleeding time by 1.2 minutes (95% confidence interval (95%CI) -0.2-2.7) and reduced median platelet serotonin level (463 ng/109 platelets; Inter QuartileRange (IQR) 361-666), and platelet ß-TG concentration (3.1 IU/106 platelets; IQR 0.3-6.0). Otherplatelet parameters did not change significantly. Serial platelet aggregation tests did not becomeabnormal. Paroxetine dose-escalation did not further influence platelet function. However, 5-HTTLPR polymorphisms modified these effects: in LA/LA-carriers, bleeding times did not change (-0.2 minutes; (95% CI -0.6 to 0.9)), while bleeding times significantly increased in <2LA-allele carriers(2.3 minutes (95% CI 0.5 to 4.07); p= 0.032). Platelet serotonin decreases were larger in patientswithout LA-alleles (868 ng/109 platelets; (IQR 585 to 1213)) than in ≥1 LA-allele carriers (457 ng/109
platelets; (IQR 392 to 598); p= 0.035). PFA closure time and PF4 increased significantly in patientswithout LA-alleles.
ConclusionsParoxetine 20 mg/day does not increase overall bleeding time, but impairs platelet function bydecreasing the levels of platelet serotonin and platelet ß-TG. These paroxetine effects appear tobe mediated by 5-HTTLPR, with most pronounced effects in patients without LA-alleles.

ChapterParoxetine induced platelet function changes
11
219
Introduction
Selective serotonin reuptake inhibitors (SSRIs) belong to the most widely prescribed classes ofdrugs and are often used for the treatment of depression and anxiety disorders.1 However,because of their extensive use, the impact of relatively infrequent side effects is of potentialclinical importance. One of these harmful side effects is an increased bleeding tendency.
Over the years, numerous case reports2-6 and case control studies7-11 have suggested that SSRIuse is associated with an increased risk of bleeding. The bleeding pattern varied from minorbleeding to spontaneous gastro-intestinal bleeding and excessive perioperative hemorrhage. Onestudy showed that SSRI users who underwent orthopedic surgery had a nearly four-fold increasedrisk to require blood transfusion.2 In addition, large population based case-control studies haveconsistently shown an increased incidence of upper gastrointestinal bleeding associated withSSRI use,12 especially when co-medicated with nonsteroidal anti-inflammatory drugs, aspirin8;11 orcoumarins.13
The underlying pathophysiological mechanisms of the increased bleeding tendency of SSRIswere previously studied, and significant changes in platelet function related to SSRI use wereconsistently reported. These changes included a decreased aggregation with epinephrine14;15 andcollagen,15;16 a prolongation of the PFA closure time,17 and a decrease in plasma beta-thromboglobulin (β-TG)18 and plasma platelet factor 4 (PF4).18;19
Furthermore, SSRIs block serotonin transporters (SERTs), which increases serotonergictransmission between neurons and results in antidepressant effects. However, SSRIs also blockthe SERTs of blood platelets, causing decreased platelet uptake of serotonin.20 Serotoninmediates vasoconstriction, platelet aggregation, and platelet activation after vessel injury, andsince platelets cannot synthesize serotonin, treatment with an SSRI could lead to depletion ofplatelet serotonin and impair hemostasis.20
Polymorphisms of the SERT gene (SLC6A4) promoter region (5-HTTLPR) are associated withthe transcriptional activity of the SERT gene and the rate of serotonin uptake.21 The 5-HTTLPRpolymorphism is located approximately 1 kb upstream of the transcription initiation site and iscomposed of 16 repeat elements. Human lymphoblasts homozygous for the long (L) 5-HTTLPRallele produce higher concentrations of SERT mRNA than cells containing one or two copies of theshort (S) allele. Furthermore, the rate of serotonin uptake by the transporter is more than twofoldhigher in cells homozygous for the L allele.21 In Caucasians the L allele is found more frequently(57%) than the S allele (43%), with a 5-HTTLPR genotype distribution of 32% LL, 49% LS and 19% SS.However, other populations show different prevalences, especially Asians, who have more thantwo-fold higher SS genotype frequencies.22 Nowadays, the 5-HTTLPR polymorphism is consideredtri-allelic.23 The L allele can be subdivided in an LG and an LA variant by a common SNP (rs25531),which creates a functional transcription factor binding site (LG), behaving like a short allele. Thistri-allelic classification is probably more reliable to find associations between 5-HTTLPRpolymorphisms and phenotypes (e.g. SSRI adverse effects).24 In Caucasians the S:LA:LG ratio isapproximately 8:10:2, while in black patients this is 5:10:5.23
Although an association between SSRI use and platelet dysfunction is undoubted,14;17;18;25;26 itremains uncertain whether a dose-response relationship exists between SSRI use and plateletfunction.20 In addition, since functional polymorphisms of the serotonin transporter influence therate of serotonin uptake, they may also mediate the effects on hemostasis.27
In this study, we evaluated the effect of standard and increasing dosages of the SSRIparoxetine on the platelet function of SSRI free patients. In addition, we assessed whether thiseffect is influenced by the 5-HTTLPR/rs25531 polymorphism.

Chap
ter
Chapter 11
11
220
Methods
ParticipantsAfter approval by the local ethics committee and written informed consent, we recruited 19 adultpsychiatric outpatients (18-75 years) from February 2006 until February 2007. The inclusioncriterion was indication of SSRI use (for mood or anxiety disorders) as determined by the treatingphysician. We excluded patients who used aspirin, NSAIDs (in the preceding 48 hours) orantidepressants (less than 4 weeks prior to inclusion), and patients who had a history of increasedbleeding tendency.
Treatment and timing of measurementsAfter baseline assessment, patients were treated with paroxetine 20 mg/day for 6 weeks. If at 6weeks the clinical symptoms of the patients had not improved by 50%, physicians could increasethe dose (dose-escalation) to a maximum of 50 mg/day over the next 6 weeks, as recommendedby current psychiatric treatment guidelines.1 As a measure to detect increased bleeding tendency,in all patients the bleeding severity score (Tosetto et al.)28 was assessed at baseline, after 6 weeks,and after 12 weeks of treatment (if a dose-escalation was prescribed). Platelet parameters wereassessed at the same time points by technicians that were unaware of the treatment regimen.
Measurements of platelet parametersBlood samplesBlood was collected in open 10 mL tubes containing 1 ml 3.2% sodium citrate by venapuncturefrom the antecubital vein using a 19-gauge needle. Within 60 minutes after collection, platelet richplasma (PRP) was prepared by centrifugation at 190 g at room temperature for 10 minuteswithout braking. PRP was separated from the packed cells; after 30 minutes aggregation studieswere started. PECT tubes for 4 ml blood containing 0.4 ml of a mixture of 94 nmol/L ProstaglandinE1, 90 mmol/L EDTA, 0.63 mmol/L sodium carbonate and 10 mmol/L theophilline were used for thedetermination of ß-TG and PF4 in plasma. Blood samples were collected in PECT tubes to preventin vitro release of ß-TG and PF4 from platelets. K3EDTA 7.5% anti-coagulant tubes were used forthe determination of platelet count.
Platelet parametersBleeding time was measured with a standard incision using the Surgicutt device (Surgicutt Adult,ITC Edison, USA). Platelet function was measured with the PFA-100 (Siemens HealthcareDiagnostics, Marburg Germany) using epinephrine (EPI) and ADP containing cartridges. Plateletaggregation in platelet-rich-plasma with ADP, ristocetin, collagen and arachidonic acid wasanalyzed qualitatively with a standardized aggregometer (model 540-vs, Chrono-Log Corporation,Havertown, USA).
The amount of ß-TG and PF4 in platelets and plasma was measured using enzyme-linkedimmunosorbent assays (Asserachrom β-TG and PF4, Diagnostica Stago, Roche). ß-TG and PF4 inplasma were determined in PECT samples. ß-TG and PF4 in platelets were determined afterpelleting the platelets from PRP. The platelets were then destroyed by a combination of Triton (2%Triton X-100) and sonication for 15 seconds on ice (microtip, Branson, amplitude 50%). After 5minutes of centrifugation at 13,000 rpm, the supernatant was used for analysis.
SerotoninIn the sonicated supernatant, the amount of serotonin was measured in an acidified (HClO4)environment by fluorimetry (Fluostar Galaxy, BMG Offenburg, Germany).

ChapterParoxetine induced platelet function changes
11
221
Paroxetine serum concentrationsWe collected blood samples for measurement of paroxetine serum concentrations (PSC) after 6and 12 weeks of paroxetine treatment from 19 and 11 patients, respectively. PSC were measuredusing a validated HPLC-MS/MS method (details available on request). The lower limit ofquantification was 5 µg/L, which was at the lower end of the therapeutic range of paroxetine inserum (5-75 µg/L). The lower limit of detection was 0.3 µg/L.
Genotyping procedures and analysisGenotyping was performed as described earlier.23 In brief, genomic deoxyribonucleic acid (DNA)was isolated and the length of the 5-HTTLPR polymorphism was determined by gelelectrophoresis. The region around the polymorphism was amplified by PCR. Genotyping of thers25531 SNP was done by sequencing. The length of the 5-HTTLPR polymorphism was confirmedby looking at the length of the sequenced PCR product. Taking into account this SNP, wereclassified the tri-allelic genotypes into a modified bi-allelic classification: S’/S’ (S/S, LG/S, LG/LG =non LA), S’/LA (S/LA, LG/LA = 1 LA) and LA/LA (= 2 LA). Because it is unknown which allele is dominant(heterozygosity), we either grouped S’/S’ and S’/ LA as <2 LA alleles versus 2 LA,or contrasted ≥1 LAvs. non LA genotypes in our analyses.
Statistical analyses We first computed means for the platelet parameters at baseline, after 6 and 12 weeks ofparoxetine administration. We compared patients who received a further dose-escalation fromweek 6 onwards with patients who remained at 20 mg/day with χ2 tests for categorical data andindependent T-tests for continuous data. We compared the changes in platelet parameters overtime with paired T-tests (baseline vs. 6 weeks and 6 vs. 12 weeks). In case of a non-normaldistribution (assessed by histograms and Shapiro Wilks’ test) we present medians withinterquartile ranges (IQR) and used nonparametric Wilcoxon signed ranks tests for paired data.For the platelet parameters that appeared to change significantly after 6 weeks of treatment, wefurther explored the changes in linear mixed models;29 we examined the effects of paroxetinedosage by the significance of a dose*time interaction.
In addition, we investigated the relation of PSC with changes in relevant platelet parametersin linear regression models using all observations (after 6 and 12 weeks) at once. We introducedthe 5-HTTLPR polymorphism to explore the genetic influence on these relations. Because of oursample size and non-normal distribution of bleeding times and platelet parameters, we testeddifferences between genotypes with nonparametric Mann-Whitney tests. All analyses wereperformed in SPSS v15.0.1.
Results
PatientsNineteen subjects who were diagnosed with either a mood disorder (n= 13), an anxiety disorder(n= 2) or both (n= 4) participated in this study. The mean age was 44.5 years (SD 10.8) and 11 (58%)were female. Co-medication consisted of depakine (for epilepsy; n= 1), levothyroxine (n= 1) andomeprazol (n= 1).
All subjects received a standard dose of paroxetine 20 mg/day for 6 weeks(mean PSC 25.6 µg/L (95% CI 14.3 to 36.9)). In 11 subjects who failed to show sufficient clinical improvement after thisperiod, paroxetine dosages were increased to 40 (n= 4) or 50 mg/day (n= 7) for another 6 weeks(mean PSC 107.2 µg/L (95% CI 42.2 to 172.2)). The group that only received standard dosage wascomparable with the increased dosage group in type of psychiatric disorder, age and sex (Table11.1).

Chap
ter
Chapter 11
11
222
Bleeding severity scoresAt baseline, the bleeding severity score was low (<3) in all subjects. Six weeks at paroxetine 20mg/day, or a subsequent dose-escalation for 6 weeks thereafter did not affect the bleeding score.
Platelet parametersSix weeks of treatment with paroxetine non-significantly increased the mean bleeding time by 1.2minutes (95% CI -0.2 to 2.7; Paired t-test; p= 0.083; Table 11.2). Furthermore, paroxetine resulted ina significant reduction of median platelet serotonin level of 463 ng/109 platelets (IQR 361 to 666;Wilcoxon signed ranks test; p<0.001), and median platelet ß-TG level of 3.1 international units (IU)/106 platelets (IQR 0.3 to 6.0; Wilcoxon signed ranks test; p= 0.016).Other changes in plateletparameters were not statistically significant, nor did the aggregation tests reveal qualitativeabnormalities.
Dose-escalation of paroxetine to 40-50 mg/day for another 6 weeks did not lead to furtherreduction of platelet serotonin or platelet ß-TG levels or to a further increase of the bleeding time(Table 11.3). The absence of further changes in platelet ß-TG and serotonin levels were confirmedby non-significant dose*time interactions in mixed models.
There was no correlation between plasma paroxetine levels and changes in platelet functiontests, including the change in platelet serotonin and platelet ß-TG levels. Platelet aggregationtests with ADP, ristocetin, collagen and arachidonic acid were not influenced by paroxetineadministration (data not shown).
Table 11.1. Baseline characteristics of patients starting treatment with paroxetine 20 mg/day.
All (n= 19)
Dose not increased after 6 weeks (n= 8)
Dose-escalation 40-50 mg after 6 weeks (n= 11)
Age ±SD 44.5 ±10.8 45.6 ± 13.2 43.6 ± 9.2
M/F ratio 8/11 4/4 4/7
Psychiatric diagnosisMood disorder*
Anxiety disorder†
Mood + anxiety disorder Substance abuse‡
Eating disorder
n (%) 13 (68.4%) 2 (10.5%) 4 (21.1%) 4 (21.1%) 1 (5.3%)
n (%) 5 (62.5%) 1 (12.5%) 2 (25.0%) 2 (25%) 1 (12.5%)
n (%) 8 (72.7%) 1 (9.1%) 2 (18.2%) 2 (18.2%) -
* Unipolar depression (n= 16; 89.5%) and/or dysthymia (n= 3; 15.8%)† Panic disorder with or without agoraphobia (n= 4; 21.1%), Post traumatic stress disorder (n= 2; 10.5%)‡ Cannabis abuse (n= 1; 5.3%), Alcohol and cannabis abuse (n= 2; 10.5%), Benzodiazepine abuse (n= 1; 5.3%)
Table 11.2. Changes in platelet function after 6 weeks of treatment with paroxetine 20 mg/day (total n= 19).
Baseline (mean ±SD)
6 weeks (mean ±SD)
n* Difference baseline vs. 6 weeks (mean 95% CI)
p†
Platelets (x109/l) 252 ±58 249 ±59 19 -3.2 (-14.0 – 7.6) 0.547
Bleeding time (min) 4.3 ±1.1 5.3 ±2.0 13 1.2 (-0.2 – 2.7) 0.083
PFA-ADP (sec) 85.7 ±18.4 88.9 ±18.9 19 3.2 (-7.2 – 13.5) 0.530
PFA-epinephrine (sec) 112.4 ±26.4 119.5 ±33.9 19 7.1 (-7.8 – 22.0) 0.329
(median IQR) (median IQR) (median IQR)
PF4 platelets‡ (IU/106 platelets) 15.9 (14.7 – 16.7) 15.9 (13.2 – 17.9) 17 -0.2 (-2.3 – 2.7) 0.925
PF4 plasma‡ (IU/mL) 6.9 (4.0 – 12.1) 5.8 (3.4 – 17.6) 18 -0.7 (-6.8 – 8.9) 0.983
ß-TG platelets‡(IU/106 platelets) 33.5 (30.4 – 36.4) 30.7 (28.1 – 34.2) 17 -3.1 (-6.0 - -0.3) 0.016
ß-TG plasma‡ (IU/mL) 35.8 (22.4 -39.7) 28.0 (20.3 – 64.7) 18 -2.2 (-16.4 – 22.0) 0.879
Platelet serotonin‡(ng/109 platelets) 667 (489 – 758) 128 (80 – 141) 16 -463 (-666 - -361) <0.001 * n for pairs without missing values† p-values for paired t-tests for normally distributed data and paired Wilcoxon signed ranks test for non-normally distributed data‡ due to non-normal distribution medians and interquartile ranges (IQR) are shown.

ChapterParoxetine induced platelet function changes
11
223
Effects of the 5-HTTLPR polymorphismGenotype was analyzed in eighteen patients, of whom 3 had the S’/S’, 9 the S’/LA, and 6 the LA/ LAgenotype, respectively. For these 18 patients, 26 observations after 6 and 12 weeks were availablewhen combining data from these time points. In patients with the 2LA genotype, paroxetinetreatment did not increase bleeding time (-0.2 minutes; 95% CI -0.6 to 0.9), while in patientscarrying the <2LA genotype, bleeding time increased by 2.3 minutes (95%CI 0.5 to 4.1, Mann-Whitney test; p= 0.032). In a linear regression model the <2LA genotype significantly predicted thechange in bleeding time (p<0.001). This difference in bleeding time between the two genotypegroups was not explained by a decrease in platelet serotonin, dosage or PSC (linear regression;pchange> 0.37).
The median decrease in platelet serotonin levels of patients with paroxetine treatment wasnot different between the <2LA and 2LA genotypes (<2LA: 587 ng/109 platelets; IQR 393 to 841; 2LA:457 ng/109 platelets; IQR 373 to 582; Mann-Whitney test; p= 0.244). However, in patients withoutLA alleles, we found significant decreases in serotonin, and increases in PFA-ADP, PFA-EPI andplatelet PF4 after 6 and 12 weeks of paroxetine treatment (Mann-Whitney test; Table 11.4; Figure11.1).
Table 11.3. Changes in platelet parameters in patients who received a secondary dose-escalation (40-50 mg) paroxetine after 6 weeks (n= 11).
Difference baseline vs. 6 weeks(mean 95% CI)
6 weeks (mean ±SD)
12 weeks (mean ±SD)
n* Difference 6 vs.12 weeks(mean 95% CI)
p†
Platelets (x109/l) -9.9 (-23.5 – 3.7) 242 ±61 249 ±53 11 7.0 (-13.8 – 27.8) 0.470
Bleeding time (min) 1.3 (-0.1 – 3.1) 5.6 ±2.3 5.8 ±2.3 9 0.4 (-1.1 – 2.0) 0.523
PFA-ADP (sec) -1.4 (-15.8 – 13.1) 88.0 ±16.4 91.7 ±15.3 11 3.7 (-8.2 – 15.6) 0.502
PFA-epinephrine (sec) 9.5 (-14.7 – 33.6) 122.6 ±39.7 124.5 ±30.0 10 0.9 (-19.5 – 21.3) 0.923
(median IQR) (median IQR) (median IQR) (median IQR)
PF4 platelets‡ (IU/106 platelets)
-0.2 (-2.3 – 2.7) 16.0 (12.5 – 18.4) 15.0 (14.3 – 17.0) 10 -0.8 (-2.2 – 2.2) 0.799
PF4 plasma‡ (IU/mL) 0.1 (-6.8 – 8.9) 6.4 (4.3 – 18.2) 5.2 (3.7 – 12.2) 10 -2.6 (-13.4 – 9.9) 0.575
ß-TG platelets‡(IU/106 platelets)
-3.7 (-9.4 - -0.3)§ 31.2 (27.4 – 35.3) 32.8 (28.6 – 35.4) 10 1. 0 (-2.2 – 2.5) 0.575
ß-TG plasma‡ (IU/mL) 0.9 (-15.5 – 22.0) 28.0 (20.9 – 65.2) 22.7 (19.3 – 38.9) 10 -5.6 (-39.6 - -0.1) 0.074
Platelet serotonin‡
(ng/109 platelets) -499 (-780 - -366)¶ 118 (74 – 143) 100 (86 – 145) 10 1.50 (-23.50 – 14.00) 0.799
* n for pairs week 6 – week 12 without missing values† p-values for paired t-tests for normally distributed data and paired Wilcoxon signed ranks test for non-normally distributed data ‡ due to non-normal distribution medians and interquartile ranges (IQR) are shown. § Wilcoxon signed ranks test p= 0.051 ¶ Wilcoxon signed ranks test p= 0.008
Table 11.4. Changes in platelet parameters after 6 and 12 weeks of paroxetine treatment, relative to baseline, stratified for genotype.
No LA/LA ≥1 LA p*
Platelet serotonin(ng/109 platelets) -868 (-1213 – -585) -457 (-598 – -392) 0.035
PFA-ADP (sec) 23.0 (17.0 – 48.0) -0.5 (-15.5 – 5.0) 0.001
PFA-epinephrine (sec) 47.0 (26.5 – 63.5) 4 (-16.0 – 19.0) 0.001
PF4 plateletsc (IU/106 platelets) 3.2 (0.7 – 4.7) -0.4 (-1.5 – 1.7) 0.003
Values represent changes of platelet parameters of combined observations after 6 and 12 weeks of paroxetine treatment (relative to baseline; 29 observations for 18 patients). Negative numbers indicate a decrease of the parameter. * Mann-Whitney non-parametric test

Chap
ter
Chapter 11
11
224
Discussion
This study confirms that treatment with the SSRI paroxetine at 20 mg/day decreases plateletserotonin and platelet β-TG levels, and increases bleeding time. Paroxetine treatment did notaffect other platelet parameters or bleeding tendency. Dose-escalation of paroxetine to 40-50mg/day did not further change platelet parameters. Additionally, the 5-HTTLPR polymorphism ofthe patients strongly affected the effect of paroxetine on bleeding time, platelet serotonin andPF4, PFA-ADP and PFA-EPI. In patients with the LA/LA genotype, paroxetine did not influencebleeding time, whereas patients with non-LA/LA genotypes had an increased bleeding time.Especially patients with the S’/S’ genotype had a greater reduction in platelet serotonin level,andincreasedPFA-ADP, PFA-EPI and platelet PF4.
Previous studies did not investigate dose-escalation. Because of the large inter-individualdifference of plasma SSRI levels at stable doses, proper investigation of a dose-response relationrequire quantification of within-subject changes, instead of comparisons of (small) groups.14;16 Bydoing so, paroxetine at 20 mg/day already maximally prolonged bleeding time and decreasedplatelet serotonin, which was not altered by consecutive dose-escalation (to 40-50 mg/day). Thisis in accordance with the recent finding that SERT occupancy in the brain is not increased by dose-escalation.30
Of interest are two in vitro studies that reported dose-response relations for sertraline17 andvenlafaxine.31 Serebruany et al. reported a significantly prolonged in vitro PFA-ADP and PFA-epinephrine with sertraline concentrations that were claimed to mimic the plasma levelsobserved in patients using sertraline 50, 100 or 200 mg/day.17 Increased platelet aggregation withADP, arachidonic acid, epinephrine and collagen was found in vitro, but with venlafaxine
Figure 11.1. Reduction of platelet serotonin levels after paroxetine exposure stratified by SERT genotype (n= 18).
Boxplots show mean (+), median (-), interquartile range, and minimum-maximum reductions in serotonin levels after 6 and 12weeks of paroxetine treatment (compared with baseline). For 18 patients observations after 6 weeks were available, for 8patients (secondary receiving higher doses) observations after 12 weeks were available. Data are stratified for genotype: S’/S’(S/S, S/LG, LG/LG; n= 3; 3 observations after 6, and 2 after 12 weeks), S’/LA (S/LA, LG/LA; n= 9; 9 observations after 6, and 1 after12 weeks) and LA/LA (n= 6; 6 observations after 6, and 5 after 12 weeks). There was a significant difference between thegenotype group S’/S’ vs. S’/LA + LA/LA (Mann-Whitney; p= 0.035).
���� ����� ������
���
���
��
��
����
����
����
� ����������
�� �������
�� ����� ���
���� ��� �������
� ����� � ���

ChapterParoxetine induced platelet function changes
11
225
concentrations 1000-fold higher than usual plasma concentrations of treated patients.31 However,these in vitro studies incubated samples with antidepressants immediately before assessingplatelet function. Normally platelet serotonin levels decrease in vivo only after >7 days, since thecirculating platelet pool must be renewed before effects of SSRIs on platelet function aremeasurable.31 This raises concern about the validity of in vitroexperiments.
The effect of the SERT polymorphism on platelet functionThe effect of paroxetine on platelet function highly depended on the SERT polymorphism of thepatients. In mice the influence of the serotonin transporter can be studied in strains without(knockout Slc6a4-/-), with 1 (Slc6a4+/-) or with 2 alleles (Slc6a4+/+) of the SERT gene. In knockoutmice the serotonin level in peripheral tissues (e.g. platelets) is <10% compared with non-knockouts. In Slc6a4+/--mice (best resembling humans with S/S genotypes), peripheral serotoninlevels were unchanged compared with Slc6a4+/+ mice. Thus, in Slc6a4+/--mice overall serotoninhomeostasis can be retained, despite a 4 to 5 fold decrease in serotonin reuptake.32 Furthermore,a recent platelet aggregation study in Slc6a4-/--mice showed 80% reduction in ADP-inducedaggregation, not observed in Slc6a4+/--mice, while results of this study also indicated a moreprominent role of the requirement of ongoing SERT transport of serotonin in maintaining plateletaggregation.33 In other words, apart from the direct effect of SSRIs reducing serotonin granules inplatelets, blockade of the SERT will also impair the functioning of the SERT at the platelet surface,which is required for the aggregation process itself. This might explain our findings: despitecompromised serotonin reuptake in S’/S’ subjects, platelets manage to achieve adequate, normalserotonin levels under normal circumstances. However, blocking the SERT by paroxetine mightaffect this compromised transporter system more than in patients with the most effective LA/LAgenotype, resulting in larger decreases of serotonin, reduced aggregation capacity (increasedPFA-ADP and PFA-EPI closure times), and longer bleeding times.
A recent study failed to find an effect of 5-HTTLPR polymorphisms on PFA-closure time.27
Consistent with our findings, this study reported no significant differences in bleeding tendency,but unfortunately platelet serotonin levels were not measured. Furthermore, in this study the bi-allelic variant of 5-HTTLPR was genotyped, while the reclassification of the ‘tri-allelic’ A to G SNP inthe long allele as an S’-variant is probably more precise to detect differences.
LimitationsDue to the small number of patients, the results of dose-escalation and genotype must beinterpreted with some restraint. However, despite the modest sample size of our study, theeffects of the genetic variants on platelet serotonin and bleeding time were demonstrated inconservative nonparametric tests. We furthermore consider our findings as valid, because ourbleeding time abnormalities and platelet serotonin findings for subgroups of SERT genotypesclearly coincide. We did not test changes in platelet parameters over time in control patientswithout SSRI, nor did we investigate dose-escalation in a randomized design. Additionally, oursample size did not allow further exploration of large versus small changes in platelet parameterswithin genotype groups.34
Clinical relevanceTwo potential clinical implications emerge. First, dose-escalation of paroxetine above 20 mg/daydoes not further impair platelet function, as it is already maximally impaired at a standard dose of20 mg/day. In other words, increased bleeding tendency associated with SSRI use will occurirrespective of the administered dose. Second, and most important, the impairment of plateletfunction appears to depend on the common functional SERT promoter polymorphism. Althoughwe only investigated paroxetine, we think this finding, if replicated, can be extrapolated to otherSSRIs.35
The absence of an increased bleeding tendency or abnormal bleeding severity score mayrepresent low sensitivity of the bleeding severity score to detect changes. However, this may alsosuggest that the observed decrease in platelet function is not clinically relevant in patients

Chap
ter
Chapter 11
11
226
without trauma or surgery, unless a preexisting platelet abnormality is present.20 However,because of their enhanced antiplatelet response to SSRIs, patients with a <2LA genotype may beat increased risk of bleeding complications during surgery or while using concomitantanticoagulants.
With a low prevalence of clinically important bleeding complications associated with SSRI use,determination of the SERT polymorphism in all patients who start with an SSRI will not be cost-effective. However, for patients with a previous severe bleeding episode (either with or withoutSSRI use) genotyping may be considered. When an S’/S’ genotype is found, switching to another,non-serotonergic antidepressant might be advisable. Generally, non-serotonergic antidepressants(i.e. mirtazapine, maprotiline, doxepin or bupropion) are associated with fewer hospitalizationsdue to bleeding complications than intermediate serotonin reuptake inhibitors (e.g. venlafaxineand amitriptyline; Odds ratio 1.9, 95% CI 1.1 to 3.5), and high degree serotonin reuptake inhibitors(fluoxetine, sertraline, clomipramine and paroxetine; Odds ratio 2.6, 95% CI 1.4-4.8), althoughcalculated odds ratios differed for individual antidepressants.9 The decision to switch must thendepend on the risks (e.g. oncoming surgery) and benefits (clinical response) for individualpatients.
ConclusionThis study shows that the SSRI paroxetine already maximally impairs platelet function at 20 mg/day, with no overall effect on bleeding time, but significant decreases in platelet serotonin and β-TG levels. Dose-escalation does not further influence hemostasis. In addition, the effect ofparoxetine on platelet function appears to be largely mediated by the common functional 5-HTTLPR SERT polymorphism, with patients with the S’/S’ polymorphism having the largesthemostatic impairment with larger decreases in serotonin, and increases in PFA closure time andplatelet PF4.
Acknowledgements
We wish to thank patients for their participation in the study. We thank general practitioners andpsychiatric residents for appropriate referrals of patients.
Conflicts of interests
All authors reported no biomedical financial interests or potential conflicts of interest. This studywas partly funded by a grant from the Netherlands Organization for Health Research andDevelopment (ZonMw), program Mental Health, education of investigators in mental health(OOG; #100-002-002) to Henricus G. Ruhé.

ChapterParoxetine induced platelet function changes
11
227
References
1. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
2. Movig KL, Janssen MW, de Waal MJ, Kabel PJ, Leufkens HG, Egberts AC. Relationship of serotonergic antidepressantsand need for blood transfusion in orthopedic surgical patients. Arch Intern Med. 2003; 163: 2354-2358.
3. Sewnath ME, van HR, Koopman MM, Levi MM, Gouma DJ. [Increased perioperative blood loss during treatment withparoxetine]. Ned Tijdschr Geneeskd. 2002; 146: 1800-1802.
4. Ceylan ME, psan-Omay MH. Bleeding induced by SSRIs. Eur Psychiatry. 2005; 20: 570-571.5. Linnebur SA, Saseen JJ, Pace WD. Venlafaxine-associated vaginal bleeding. Pharmacotherapy. 2002; 22: 652-655.6. Lake MB, Birmaher B, Wassick S, Mathos K, Yelovich AK. Bleeding and selective serotonin reuptake inhibitors in childhood
and adolescence. J Child Adolesc Psychopharmacol. 2000; 10: 35-38.7. de Abajo FJ, Montero D, Rodriguez LA, Madurga M. Antidepressants and risk of upper gastrointestinal bleeding. Basic
Clin Pharmacol Toxicol. 2006; 98: 304-310.8. Wessinger S, Kaplan M, Choi L, Williams M, Lau C, Sharp L et al. Increased use of selective serotonin reuptake inhibitors in
patients admitted with gastrointestinal haemorrhage: a multicentre retrospective analysis. Aliment Pharmacol Ther.2006; 23: 937-944.
9. Meijer WE, Heerdink ER, Nolen WA, Herings RM, Leufkens HG, Egberts AC. Association of risk of abnormal bleeding withdegree of serotonin reuptake inhibition by antidepressants. Arch Intern Med. 2004; 164: 2367-2370.
10. Dalton SO, Johansen C, Mellemkjaer L, Norgard B, Sorensen HT, Olsen JH. Use of selective serotonin reuptake inhibitorsand risk of upper gastrointestinal tract bleeding: a population-based cohort study. Arch Intern Med. 2003; 163: 59-64.
11. de Abajo FJ, Rodriguez LA, Montero D. Association between selective serotonin reuptake inhibitors and uppergastrointestinal bleeding: population based case-control study. BMJ. 1999; 319: 1106-1109.
12. Serebruany VL. Selective serotonin reuptake inhibitors and increased bleeding risk: are we missing something? Am J Med.2006; 119: 113-116.
13. Schalekamp T, Klungel OH, Souverein PC, de BA. Increased bleeding risk with concurrent use of selective serotoninreuptake inhibitors and coumarins. Arch Intern Med. 2008; 168: 180-185.
14. Laine-Cessac P, Shoaay I, Garre JB, Glaud V, Turcant A, Allain P. Study of haemostasis in depressive patients treated withfluoxetine. Pharmacoepidemiol Drug Saf. 1998; 7 Suppl 1: S54-S57.
15. Hergovich N, Aigner M, Eichler HG, Entlicher J, Drucker C, Jilma B. Paroxetine decreases platelet serotonin storage andplatelet function in human beings. Clin Pharmacol Ther. 2000; 68: 435-442.
16. Markovitz JH, Shuster JL, Chitwood WS, May RS, Tolbert LC. Platelet activation in depression and effects of sertralinetreatment: An open-label study. Am J Psychiatry. 2000; 157: 1006-1008.
17. Serebruany VL, Gurbel PA, O'Connor CM. Platelet inhibition by sertraline and N-desmethylsertraline: a possible missinglink between depression, coronary events, and mortality benefits of selective serotonin reuptake inhibitors. PharmacolRes. 2001; 43: 453-462.
18. Pollock BG, Laghrissi-Thode F, Wagner WR. Evaluation of platelet activation in depressed patients with ischemic heartdisease after paroxetine or nortriptyline treatment. J Clin Psychopharmacol. 2000; 20: 137-140.
19. Musselman DL, Marzec UM, Manatunga A, Penna S, Reemsnyder A, Knight BT et al. Platelet reactivity in depressedpatients treated with paroxetine: preliminary findings. Arch Gen Psychiatry. 2000; 57: 875-882.
20. Skop BP, Brown TM. Potential vascular and bleeding complications of treatment with selective serotonin reuptakeinhibitors. Psychosomatics. 1996; 37: 12-16.
21. Lesch KP, Gutknecht L. Pharmacogenetics of the serotonin transporter. Prog Neuropsychopharmacol Biol Psychiatry.2005; 29: 1062-1073.
22. Murphy DL, Lerner A, Rudnick G, Lesch KP. Serotonin transporter: gene, genetic disorders, and pharmacogenetics. MolInterv. 2004; 4: 109-123.
23. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD et al. Serotonin transporter promoter gain-of-functiongenotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. 2006; 78: 815-826.
24. Hu XZ, Rush AJ, Charney D, Wilson AF, Sorant AJ, Papanicolaou GJ et al. Association between a functional serotonintransporter promoter polymorphism and citalopram treatment in adult outpatients with major depression. Arch GenPsychiatry. 2007; 64: 783-792.
25. Alderman CP, Seshadri P, Ben-Tovim DI. Effects of serotonin reuptake inhibitors on hemostasis. Ann Pharmacother. 1996;30: 1232-1234.
26. McCloskey DJ, Postolache TT, Vittone BJ, Nghiem KL, Monsale JL, Wesley RA et al. Selective serotonin reuptakeinhibitors: measurement of effect on platelet function. Transl Res. 2008; 151: 168-172.
27. Hougardy DM, Egberts TC, van der GF, Brenninkmeijer VJ, Derijks LJ. Serotonin transporter polymorphism and bleedingtime during SSRI therapy. Br J Clin Pharmacol. 2008; 65: 761-766.
28. Tosetto A, Rodeghiero F, Castaman G, Goodeve A, Federici AB, Batlle J et al. A quantitative analysis of bleeding symptomsin type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost. 2006;4: 766-773.
29. Dean CB, Nielsen JD. Generalized linear mixed models: a review and some extensions. Lifetime Data Anal. 2007.30. Ruhe HG, Booij J, Weert HCv, Reitsma JB, Fransen EJF, Michel MC et al. Evidence why dose-escalation of paroxetine in
major depressive disorder is not effective: A 6-week, randomized-controlled trial with assessment of serotonintransporter occupancy. Neuropsychopharmacology. [In Press].

Chap
ter
Chapter 11
11
228
31. Sarma A, Horne MK, III. Venlafaxine-induced ecchymoses and impaired platelet aggregation. Eur J Haematol. 2006; 77:533-537.
32. Murphy DL, Lesch KP. Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci.2008; 9: 85-96.
33. Carneiro AM, Cook EH, Murphy DL, Blakely RD. Interactions between integrin alphaIIbbeta3 and the serotonintransporter regulate serotonin transport and platelet aggregation in mice and humans. J Clin Invest. 2008; 118: 1544-1552.
34. Schreijer AJ, Cannegieter SC, Meijers JC, Middeldorp S, Buller HR, Rosendaal FR. Activation of coagulation system duringair travel: a crossover study. Lancet. 2006; 367: 832-838.
35. Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile ofantidepressants and their metabolites. J Pharmacol Exp Ther. 1997; 283: 1305-1322.

ChapterParoxetine induced platelet function changes
11
229


SUMMARY AND GENERAL DISCUSSION
PART
Summary and general discussion

232

CHA
PTER
SUMMARY, CONCLUSIONS AND GENERAL DISCUSSION

234

ChapterSummary and general discussion
12
235
Summary
In this chapter, a summary of the conclusions of our studies and the discussion of these findingswill be presented. Possible limitations and strengths of the thesis will be discussed and directionsfor future research will be presented.
Summary of this thesis and conclusions
This thesis addressed several questions:
1 Is a short, easy to use clinician rated questionnaires as effective and precise as the routineHamilton depression rating scale (HDRS)?In chapter 3, we reanalyzed the treatment-outcomes of two antidepressant-psychotherapytrials.1 In line with previous reports,2-5 we found that the effect-sizes of two 6-item sub-scalesof the HDRS (17-items) – the Maier6 and Bech7 sub-scales – were comparable to the originalHDRS in the measurement of depression severity, and the sensitivity to measure changes.Furthermore, this comparability was stable across the full range of response to treatment,across both pharmacotherapy and psychotherapy, and for patients with different baselineseverities of their depression. With an item response theory approach, we calculated aconversion table linking HDRS-scores and Maier and Bech scores, and determined cut-offpoints for remission for these subscales compared with conventional HDRS definitions.8-10
With these subscales clinicians can measure depression severity and clinical response moreefficient than with the original HDRS.
2 What is the evidence for dose-escalation as a strategy for non-response to a first SSRI?In chapter 4, we presented a systematic review of the evidence for the dose responserelationship of selective serotonin reuptake inhibitors (SSRIs) in major depressive disorder(MDD).11 In our literature-search, we identified 8 dose-escalation studies that increaseddosages after at least 3 weeks of a standard dosage.12-19 Furthermore, 3 systematic reviewswere published by then, which included three20;21 or four22 of the eight identified dose-escalation studies. Only one of the dose-escalation studies approached stringentmethodological criteria.19 We found no evidence for increased efficacy of dose-escalationwithin the first 4 weeks of treatment. Dose-escalation after 6 weeks appeared less effectivethan continuing the same dose. We found some, but limited evidence for efficacy of dose-escalation after 8 weeks, particularly in partial responders. This effect was seen within 4weeks after dose-escalation. Irrespective of efficacy, dose-escalation unequivocally increasedside-effects, but effects on drop-out rates due to side effects were less straightforward. Wetherefore concluded that the available evidence for dose-escalation neither unequivocallyconfirmed its efficacy, nor deemed it ineffective. This conclusion was the starting point for theDELPHI-study, described in the chapters 2 and 7 to 10.
3 What is the evidence for switching antidepressants as a strategy for non-response to a firstSSRI?In chapter 5, we presented a systematic review of the evidence for switching after failure of afirst SSRI in MDD.23 In addition to our literature-search we included four studies released afterthese searches, of which three studies from the Sequenced Treatment Alternatives to RelieveDepression (STAR*D) trial.24-27 We identified eight randomized controlled trials (RCTs),24-31 and23 open switch studies. Studies were of variable methodological quality and carried out inheterogeneous populations. The STAR*D results largely increased the amount, and quality ofthe available evidence, but did not show differential class effects to guide switching. Threerandomized switch-studies investigating the switch from SSRI to another SSRI or SNRI(venlafaxine) were eligible for a meta-analysis.24;25;28 This meta-analysis showed that in themost favorable analysis, venlafaxine was slightly more effective as a second antidepressant,

Chap
ter
Chapter 12
12
236
compared with other SSRIs (number needed to treat (NNT) = 10 (95% confidence interval (95%CI) 6.3-33.3). We therefore concluded that after the failure of a first SSRI switching is open toall antidepressant classes (except irreversible MAO-inhibitors), without clearrecommendations other than those that apply for the selection of initial treatment.
4 Does the depletion of monoamine (5-HT and NA/DA) systems lower mood in humans, and isthis lowering of mood different across different populations?In chapter 6, we presented a systematic review of monoamine depletion studies reportingmood effects of depletion.32 As an extension of previous systematic reviews of monoaminedepletion studies,33-38 we pooled the results of 53 small-sized depletion studies with anadapted pooling technique (modified from conventional meta-analyses of RCTs to handle thestatistically paired cross-over designs, and including an adjustment for small sample bias).Pooling is important because it quantifies the balance of positive versus negative studies.
We pooled 45 acute tryptophan depletion (ATD) studies and 8 acute phenylalanine/tyrosine depletion (APTD) studies, acutely lowering serotonergic and dopaminergic/norepinephrinergic neurotransmission, respectively. Serotonin or dopamine/norepinephrinedepletion did not decrease mood in healthy controls, but slightly lowered mood in healthycontrols with a family history of MDD. In drug-free patients with MDD in remission, amoderate mood decrease was found for ATD, without an effect of APTD. ATD induced relapsein patients with MDD in remission who used serotonergic antidepressants. MDD patients didnot experience mood deterioration after ATD while depressed.
We concluded that simple, direct correlations of serotonin or dopamine/norepinephrinelevels in the brain and mood do not exist. Because the serotonin or dopamine/norepinephrinedepletion induced by ATD or APTD most clearly decreases mood in vulnerable individuals(patients who are in remission from MDD and healthy controls with a positive family historyfor MDD), we concluded that the monoamine systems are important systems in thevulnerability to become depressed. Moreover, the changes in brain metabolism in remittedpatients who relapse after ATD or AMPT suggest that the serotonergic andnorepinephrinergic systems give input to a final common pathway which needs furtherresearch to be clarified.
5 Do MDD-patients and healthy controls differ in the number of central serotonin transporters,and is the amount of available SERTs correlated with depression severity?In chapter 7,we measured the availabilities of the central serotonin transporters (SERTs) inbaseline SPECT scans of the DELPHI-SPECT participants, and compared these with SERTavailabilities in age- and sex-matched healthy controls.39 Because of earlier reports, weinvestigated potential covariates as smoking behaviour,40 age,41;42 season of scanning43 andpossible interactions with gender40;44 in multivariate models. We found interaction effects ofsmoking and diagnosis with gender for diencephalon SERT availability. For midbrain SERTavailability, an interaction of gender with diagnosis was found, while season of scanning was acovariate. These interaction effects were expressed as lower SERT availability in midbrain anddiencephalon in depressed males, and higher SERT availability in diencephalon in depressed(non-smoking) females compared with healthy controls. These findings point to complexeffects of gender, smoking and season of scanning on the serotonergic system in healthycontrols and patients with MDD, which must be considered in future studies comparing SERTavailabilities in MDD patients versus healthy controls.
6 Does a common genetic polymorphism of the promoter region of the serotonin transportergene (SLC6A4) modify the association between the SERToccupancy by paroxetine and theclinical response?In chapter 8, we studied the relationship between clinical response after 6 weeks ofparoxetine treatment and SERT occupancy (which is the change in SERT availability after 6weeks of treatment relative to the pre-treatment SERT availability).45 Because clinicalresponse to SSRIs is likely associated with polymorphisms of the serotonin transporter genepromoter region (5-HTTLPR),46;47 we also aimed to investigate whether this relation of

ChapterSummary and general discussion
12
237
response and SERT occupancy was modified by the 5-HTTLPR genotype. We obtained study-entry and week 6 SPECTscans for 44 patients treated with paroxetine 20 mg/day, of which 42scans were analyzable (10 responders and 32 non-responders).
We found that for all patients, SERT occupancy was not associated with clinical response(expressed as the proportional decrease in HDRS relative to study entry). However, when wegrouped patients by 5-HTTLPR polymorphism, we found that in patients with the(favorable)46;47 LA/LA variant a significant relation between SERT occupancy and clinicalresponse (absolute and proportional change in HDRS) existed. A previous study found abetter amygdala-cingulate coupling in subjects with an L-allele relative to the unfavorable S/Svariant,48 probably reflecting differences in the development of the serotonergic systeminduced by 5-HTTLPR.49 As an explanation of our findings, we therefore hypothesized thatpatients with the 5-HTTLPR LA/LA genotype have a more flexible serotonergic system, which ismore easily influenced by serotonergic antidepressants.
7 Is dose-escalation of paroxetine an effective clinical strategy for non-response in MDD?In chapter 9, we studied the strategy of dose-escalation which was found equivocallyefficacious in the systematic review,11 as described in chapter 4. In this study, we addressedthe methodological flaws found in previous dose-escalation trials,50 but moreover, we alsomeasured whether paroxetine dose-escalation increased SERT occupancy more than placebodose-escalation.51 After inclusion of 49 SPECT patients (with successful follow-up of 31patients participating in the randomized placebo-controlled dose-escalation SPECT study), weperformed an interim analysis including all randomized patients (n= 57), and testeddifferences between placebo and true dose-escalation with pre-defined cut-off values forfutility and superiority. We found no clinical benefits of true dose-escalation compared withplacebo, and so the trial was stopped because of futility.
Most important, our data also showed that true dose-escalation of paroxetine did notincrease SERT occupancy more than placebo dose-escalation, probably because a plateau wasreached at standard doses already. We concluded, that in line with previous, though moreequivocal evidence,52;53 dose-escalation is not beneficial in MDD. Therefore, two clinicaloptions remain for the treatment of MDD-patients who do not respond to standard doses:either continuation of treatment until 10 weeks while waiting for a potential delayedresponse, or a switch to a different and potentially more effective treatment strategy (e.g.another SSRI or SNRI or TCA, or psychotherapy, or a combination).
8 Does treatment with paroxetine normalize amygdala hyperactivation in MDD?In chapter 10, we describe the (negative) faces task of the DELPHI-fMRI sub-study.54 Westudied the effects of paroxetine treatment on the hyperactivation of the amygdala (andother brain areas) during a depressive episode. We specifically investigated: 1) whetheramygdala-activation by (negative) facial expressions differed from healthy controls, 2)whether amygdala activation changed after 6 and 12 weeks of treatment, 3) whether theamygdala activation merely changed by paroxetine treatment or in relation with clinicalresponse, and 4) whether dose-escalation of paroxetine in week 6 non-responders affectedactivations, compared with placebo-dose-escalation. Apart from the amygdala, we alsoinvestigated these questions for other brain areas. We performed fMRI scans in 22 MDDpatients and 21 age- and sex-matched healthy controls (controls were scanned once).
Compared with healthy controls, we found increased activations in bilateral (extended)amygdala and left insula in MDD-patients. In contrast with previous studies,55-57 we found anincrease in right amygdala activation after 6 weeks of treatment, which was attributed to thehigh number (12/20) of treatment non-responders. This increased amygdala-activation wasreduced thereafter until week 12. When we analyzed the differences in week 6 and 12treatment responders versus week 6 and 12 non-responders, bilateral amygdala activationwas reduced in responders versus non-responders. Amygdala activations in week 6 and 12were associated with HDRS-scores. Furthermore, non-responders had higher activations in

Chap
ter
Chapter 12
12
238
right orbitofrontal cortex (OFC) and insula, while treatment responders showed higheractivation in right dorsolateral prefrontal cortex (DLPFC) and left nucleus accumbens. Thesedifferences were non-existent at study-entry.
Although randomized groups became small, changes over time in the true dose-escalationgroup showed decreased activations in ventral and dorsal regions, while activations of theright hippocampus and left subgenual cingulate increased,when compared with changesobserved after placebo dose-escalation. We concluded that in line with previous treatmentstudies56-59 paroxetine treatment over 12 weeks reduced activations in the amygdala and other(ventral) emotion-generating structures, and increased cortical function (in the dorsalregulatory structures; see chapter 1, pages 18 and 21), especially in treatment responders.These findings may point to increased fronto-limbic control as a mechanism of paroxetinedrug-response effects, which was supported by findings of other groups.60;61
9 What are the changes in hemostasis and blood platelet parameters when patients are treatedwith paroxetine, and are these changes modified by dose-escalation or a geneticpolymorphism of the promoter region of the serotonin transporter gene?In chapter 11, we present the results of a study investigating an infrequent, but dangerousadverse effect of SSRIs: increased bleeding tendency.62 In this study, we measured plateletparameters and coagulation, while we applied a secondary (non-randomized) dose-escalationin paroxetine treatment non-responders. We additionally investigated whether 5-HTTLPRpolymorphisms modified these effects on platelet parameters. We found that a standard doseof paroxetine (20 mg/day) already significantly decreased platelet serotonin levels andplatelet β-thromboglobulin (β-TG), without further decreases after dose-escalation.Moreover, we found that 5-HTTLPR polymorphisms modified these effects: compared withLA/LA-carriers, bleeding time significantly increased in <2LA-allele carriers, and plateletserotonin decrease was larger in patients without LA-alleles. Furthermore, the plateletfunction analyzer closure time significantly increased in patients without LA-alleles. Althoughthe observed bleeding tendency presumably is not clinically relevant in patients withouttrauma or surgery (unless a preexisting platelet abnormality is present),63 these findings areapplicable to patients with a previous severe bleeding episode. For them genotyping may beconsidered; when an S’/S’ genotype is found, switching to another, non-serotonergicantidepressant might be advisable.
Clinical relevance
Antidepressants have comparable efficacy, however only 50% of the MDD-patients respond to thefirst antidepressant trial given, while fewer achieve full remission of symptoms.64;65 When themiracle doesn’t happen, there are 5 strategies for non-response: prolongation of the initial trial,dose-escalation, switching to another drug, augmentation with another drugora combination ofantidepressants. This thesis addresses switching strategies23 and dose-escalation.11;51
SwitchingDespite a small benefit of a switch to venlafaxine compared with a second SSRI (in the mostfavorable analysis only), we concluded that switching-options after a first SSRI are open to allantidepressant classes, without clear recommendations other than those that apply for theselection of initial treatment. One more RCT comparing venlafaxine and citalopram,66 and threeopen studies, one with venlafaxine (with randomization to different doses)67, one with avenlafaxine-mirtazapine combination strategy,68 and one with duloxetine69 appeared thereafter.
In addition, another meta-analysis was published, comparing a non-SSRI switch (venlafaxine(3 studies) or mirtazapine (1 study) or buproprion (1 study)) versus a second SSRI.70 This meta-analysis included 2 different studies (abstracts from symposia)71;72 compared with our meta-analysis, and excluded one study with questionable methodology, like we did in our most

ChapterSummary and general discussion
12
239
favorable analysis.24 Their conclusion was that the NNT for remission was 22 (confidence intervalnot given), modestly in favor of a non-SSRI switch, which is far below the standard for clinicalrelevance (NNT ≤10) as suggested by the United Kingdom’s National Institute of ClinicalExcellence.73;74 When they considered response as an outcome, they found no significant benefit,comparable to our results.70 In the final manuscript of the previous abstract,71 Lenox-Smith andJiang reported increased efficacy of venlafaxine over citalopram (flexible doses) in a subgroup ofpatients with severe MDD (HDRS21>31), who participated in a multicenter RCT of SSRI non-responders.66 However, this difference was only significant for the continuous scores of theHDRS21 (difference approximately 4.7 points; SD not given), and not for the remission rates.Therefore, we think that only at the level of public health, the difference between switching-strategies might be interesting, but not at the level of individual patients.
Dose-escalationIn our systematic review of dose-escalation it was concluded that the available evidence neitherunequivocally confirmed its efficacy, nor deemed it ineffective. This was in agreement withanother review, performed by an independent group, and published just before ours.52 The resultsof the dose-escalation RCT in this thesis first of all addressed the methodological shortcomings ofprevious studies, and was stopped for futility after an interim analysis.51 Even though the numbersof patients randomized to placebo or true dose-escalation were modest (n= 27 and n= 30,respectively), the changes in HDRS-scores and subscales were far from significantly different,while the level of this significance was far above the a prioricut-off for futility(which wasundisclosed at the time of analysis). This finding aligns with the dose-escalation study by Licht etal.19 Unfortunately, the premature termination of the trial precluded planned secondary analysesto distinguish subgroups who still might benefit form dose-escalation (e.g. partial responders;defined as 25-50% reduction of HDRS).15;18 In our sample the subgroup of partial respondersconsisted of 24 patients (from 60 randomized; 40%), and did not show any indication of beneficialtreatment-effects for true dose-escalation. Nevertheless this observation cannot replace truesubgroup analyses in larger samples.
Our sub-study of the changes in SERT occupancy during the randomized, placebo-controlleddose-escalation phase further corroborates our clinical findings. We therefore conclude that oursystematic review, our dose-escalation study and our imaging study provide substantial evidencefor clinicians, that dose-escalation is not an effective strategy for depressed patients who do notrespond to 6 weeks of a first SSRI at a standard dose.
In a recent meta-analysis of 9 placebo-controlled, fixed-dose, dose-finding studies of SSRIs,75
Papakostas et al. found a modest but statistically significant difference of 4% higher responserates in patients who started with higher than standard doses of citalopram, escitalopram,fluoxetine, fluvoxamine, paroxetineand sertraline (p= 0.04). This 4% difference in response ratescorresponds with a NNT of 25, which is clinically not impressive, nor relevant. In their meta-analysis, Papakostas et al. could not investigate whether this difference was attributable to asingle SSRI. However, when inspecting their data, all paroxetine trials did not show benefits ofhigher paroxetine doses compared with standard 20 mg/day doses, with relative risks of 1.00 (95%CI 0.67-1.50), 1.02 (95% CI 0.69-1.52) and 0.94 (95% CI 0.65-1.35). In contrast, the rate ofdiscontinuation due to adverse events was also higher in the patients treated in the abovestandard doses group (16.5% compared with 9.8%). Therefore, to our opinion, it remains alsoquestionable whether higher doses of SSRIs at the initiation of treatment indeed areadvantageous to MDD-patients. Instead, they could give patients more adverse effects andpreclude prolonged exposure to tolerable standard doses. Finally, but most important for thisthesis, these findings presumably hardly apply to paroxetine.
Other findings of clinical relevanceBesides the switching and dose-escalation strategies, two other relevant points from this thesisemerge. First, we showed that the Maier and Bech subscales of the HDRS17 are equivalent to thefull HDRS17.1 Others even suggested that these subscales might measure the core-symptoms and

Chap
ter
Chapter 12
12
240
severity of MDD more specifically, and their use might even be more efficient,2;3 because of thereduction of noise by less responsive items.76 We therefore propose that the Maier or Bech-subscales should be used in the clinical assessment of (treatment of) MDD patients, instead of, orbesides self-rating scales like the Inventory for depressive symptoms self-rated (IDS-SR) or Beckdepression inventory (BDI). This will be of great importance in patients who cannot reliably fill outself-rated questionnaires, like in MDD with psychotic features or MDD with severe cognitivedysfunction. Of course, providing a shorter, easy to use clinician rated scale will not translate indirect implementation of one of these subscales, but may at least reduce the time burden ofrating scales for physicians.
Second, our findings regarding the increased bleeding tendency in patients merit attention.62
Increased bleeding tendency, especially in combination with non-steroidal anti-inflammatorydrugs (NSAIDs) was observed before.77-82 This even led to the recommendation to use TCAs whenconcomitant NSAIDs should be used in the guideline for Dutch general practitioners.83 However,the serotonergic TCA clomipramine, presumably has comparable effects. Because of the relativelylow incidence of dangerous gastro-intestinal bleeding complications in general (4.3/1000 SSRItreatment years; 14.5/1000 SSRI+NSAID/aspirin treatment years; relative to 1.2/1000/year inuntreated humans),80 the increased bleeding tendency by SSRIs is of no clinicalrelevance inpatients without trauma, surgery or a previous severe bleeding episode.63 However, in patientswith a previous severe bleeding episode, or in patients who need elective surgery, and maybe alsoin patients regularly using NSAIDs, genotyping of the 5-HTTLPR may be considered. When an S’/S’genotype is found, switching to another, non-serotonergic antidepressant might be advisable. Forwhich of the aforementioned subgroups this genotyping will be cost-effective, remains to beinvestigated.
Neurobiology of the treatment of major depressive disorder
With our studies we also aimed to investigate the neurobiology of paroxetine treatment effects inMDD in vivo. The enhancement of serotonergic neurotransmission by paroxetine is undoubted,caused by a reduced serotonin uptake after SERT occupancy as the primary mechanism.Therefore, the most important question which we addressed was whether dose-escalation ofparoxetine would increase SERT occupancy compared with placebo.
SERT occupancy after short or prolonged treatment with SSRIs was investigated before,84-96
even at variable and higher doses.86;96 However, without a pre-assessment of SERT availabilitybefore treatment96 and without randomization to a placebo-controlled dose-escalation,86 one cannever exclude the possibility that non-responders after 6 weeks of a standard dose consist of aselected sample who had lower SERT occupancy. This hypothetically lower SERT occupancy couldthen be considered explanatory for non-response, and might increase by subsequent dose-escalation compared with placebo dose-escalation.
Our SPECT measurements in the subgroup of the DELPHI study showed no overall relationbetween SERT occupancy and clinical response in the open phase of treatment with paroxetineover 6 weeks. Furthermore, we found no evidence for the postulated 80% SERT occupancy as arequisite for clinical response.45 In addition, the second, randomized, placebo-controlled dose-escalation phase of DELPHI showed no differences in changes in SERT occupancy betweenplacebo and true dose-escalation for 6 more weeks.51 With this latter finding, we are the firstgroup that studied sequential dose-escalation in non-responders, controlling for placebo-effectsand measurement-bias by repeated SPECT scans. Interestingly, we did find an associationbetween SERT occupancy and clinical response in the subgroup of patients with the (tri-allelic) LA/LA polymorphism of the 5-HTTLPR. Because the tri-allelic LA:LG ratio is approximately 5:1,97 ourfinding aligns with data from previous treatment studies which found increased response rates insubjects with the bi-allelic L/L 5-HTTLPR polymorphism.46;47

ChapterSummary and general discussion
12
241
However, the mechanism why increased SERT occupancy is associated with increased clinicalresponse in LA/LA carrying patients, cannot be explained by higher SERT occupancies in LA/LApatients.45 Probably, the explanation must be sought in differences in the function of theserotonergic system (and its development) mediated by 5-HTTLPR polymorphisms. From previouslines of research, it may be concluded that the S-allele predisposes to a more reactive arousalsystem, and that the L-allele accounts for more flexibility and/or the possibility to react and adaptbetter to external changes imposed on the subject.98 Furthermore an elegant fMRI-study showedthat both the anatomy and functional connections between limbic, subcortical and corticalregions are mediated by 5-HTTLPR polymorphisms, with S/S carriers showing relative uncouplingof the amygdala-pregenual cingulate connectivity.48
The biochemical consequences of chronic administration of SSRIs are thought to include anincrease in extracellular levels of serotonin followed by neuroadaptive alterations in serotoninreceptors and postsynaptic intracellular signalling pathways (see figure 1.2, page 20), as well astime-dependent effects on neurogenesis.99;100 Not surprisingly, animal studies demonstrated thatin mice knocked out for the murine Slc6a4 SERT transporter (Slc6a4-/-), fluoxetine andfluvoxamine are ineffective on behavioural and other measures, while mice with at least one copyof the allele (Slc6a4+/- or Slc6a4+/+) showed the typical antidepressant effects.99 This is pivotalevidence for the working mechanism of SSRIs, for which SERTs are apparently required to bepresent. The Slc6a4+/- mice have ~50% less expression and transportfunction and can be viewed asa model for humans with the S/S genotype.101 Therefore, the findings of almost identical effectsof SSRIs in Slc6a4+/- or Slc6a4+/+ mice102 do not support the findings of differential effects for 5-HTTLPR polymorphisms from meta-analyses of human studies.46;47 However, the 5-HTTLPRpolymorphism is only present in humans and higher order primates (e.g. macaques). To ourknowledge, no studies in primates investigated the mediating effects of 5-HTTLPR polymorphismson the relation between SERT occupancy and behavioural effects. Furthermore, we are unawareof studies investigating dose-response relationships of SSRIs in animals. Finally, although a robustincrease in anxiety-like behaviourwas found in Slc6a4-/-mice compared with other genotypes, sofar no studies were performed to investigate the differences in brain development and functionalconnections for mice with a Slc6a4-/-, Slc6a4+/- or Slc6a4+/+ genotype.103
Serotonin has an important role in the regulation of neuronal information processing byconstraining (instead of inhibiting) the reactivity of the brain to internal and external stressors.104
In general, serotonin appears 1) to prevent an overshoot of other dynamic systems (e.g.constraining the release of dopamine after rewarding stimuli), and 2) to control the sensitivity ofthe system to perturbation by new elements entering the system (e.g. constraining a naturalpropensity to switch to alternate behaviours; this can be provoked by serotonin depletion). Thisconstraint can be experimentally challenged by acute tryptophan depletion (ATD), which resultsin increased perception of aversive stimuli, increased food intake, increased sexual, aggressive,depression-like and anxiety-like behaviours, but also decreased sensitivity to cues of punishment,and less flexibility to change behaviour in rats.104
Besides the cognitive effects,98 our meta-analysis quantified the mild mood lowering effectsby ATD in humans, which were especially seen in healthy controls with depressed relatives, andpatients who had remitted from a previous MDD-episode.32 Because mood-effects can beconsidered as more complex behaviour, these may be more difficult to provoke in relatively short-term ATD studies compared with longer-lasting studies in animals. Nevertheless, the large body ofresearch on the serotonergic system has now changed the categorical model –decreasedserotonin is specific for depression – into a model in which alterations in the serotonergicneurotransmission can be seen as a biological risk-factor, which interacts with innate and/orexternal factors. This so-called serotonergic vulnerability is now seen as neither a sufficient, nornecessary factor to develop and maintain MDD, but in combination with other factors increasesthe risk for MDD.98

Chap
ter
Chapter 12
12
242
Our fMRI-study,54 corroborates the hypothesis that paroxetine treatment interferes with the(maladaptive) functioning of various interconnected regions in the limbic-subcortical-corticalnetwork.105;106 Like others, we found increased ventral* activations (e.g. in the amygdala andinsula), and decreased dorsal* activations (anterior cingulate, DMPFC, DLPFC) in MDD-patientsrelative to healthy controls. We furthermore found that the ventral hyperactivation and dorsalhypoactivation was normalized in treatment responders, but not in non-responders. Recently,Chen et al. demonstrated increased functional coupling of the amygdala with the frontal and(pregenual) cingulate cortex (and striatum and thalamus) after 8 weeks of sertraline treatment.61
Although we did not investigate this coupling per se, we found that the amygdala responses in ourpatients were correlated with pregenual cingulate and frontal regions.
Furthermore, in responders we found increased activation in the nucleus accumbens, pointingto the involvement of the limbic-cortico-striato-pallido-thalamic network, which indirectlysuggests the involvement of changes in dopaminergic neurotransmission after paroxetinetreatment.107 Two coinciding explanations can be given for this increased activation of the nucleusaccumbens. First, in rodents, chronic treatment with antidepressants (among others SSRIs)potentiated dopamine transmission, due to an increased sensitivity of postsynaptic dopaminereceptors and possibly also due to a decreased sensitivity of presynaptic (inhibitive) dopamineautoreceptors, preferentially in the limbic system.108 Second, again in rodents, decreases inserotonin are associated with a release of normally inhibited behaviour, including impairedsuppression of behaviour inducing punishment.104 Activation of the nucleus accumbens isassociated with anticipation of reward,109 while the nucleus accumbens is deactivated by loss orpunishment. Therefore, it might be postulated that the increase in serotonergicneurotransmission by SSRIs, reduces a lack of constraint on dopaminergic neurotransmission inthe nucleus accumbens, with subsequently less punishment perceived. This would then result inless deactivation of the nucleus accumbens. The fact that this was demonstrated in treatmentresponders relative to non-responders might suggest that in non-responders this enhanceddopaminergic constraint might not be achieved.
As interesting, but contradictive finding, we did not find any indication that dose-escalationincreased SERT occupancy, but at the other hand, we found indications that a true dose-escalation decreased hyperactivations in the ventral compartment (OFC, insula, VMPFC,ventrolateral thalamus), and increased activation in the dorsal compartment (hippocampus,DLPFC, and parietal cortex). Also we counter intuitively found an increased activation in thesubgenual cingulate after dose-escalation.54 As explained, we could unfortunately not disentangleresponse- and drug-effects. If the decreases in the ventral, and the increases in the dorsalcompartment could have been attributed to responders to dose-escalation, while the increasedactivation in the subgenual cingulate was associated with non-responders (despite dose-escalation), this would have made our findings less surprising.
Limitations
Limitations of the studies in this thesis were discussed in each chapter. However, some must berecapitulated in this general discussion.
At the moment of publication of this thesis, the systematic reviews presented in the chapters4, 5, and 6 might have become outdated, since the literature searches were performed untilOctober 200632 or February 2005.11;23 Although we did not repeat the searches until July 2008, forthe dose-escalation and switching reviews, we commented on additional studies that weencountered thereafter in this general discussion. We did not review additional monoaminedepletion studies, as this should have resulted in new meta-analyses.
* For explanation of the ventral and dorsal compartments of the limbic-subcortical-cortical network, see chapter 1, page 18.

ChapterSummary and general discussion
12
243
Furthermore, in contrast with other authors,53;75 we did not perform a meta-analysis for thedose-escalation studies, and only for the venlafaxine versus second SSRI switch studies. At thispoint, we considered the dose-escalation studies with various moments of randomization afterthe initiation of treatment too heterogeneous to pool. For switching, we decided not to pool (bythen unpublished) data on various dual acting antidepressants versus second SSRIs.
In the SPECT-studies, we used [123I]β-CIT for SPECT imaging, which is a non-selectiveradioligand, and also binds to the dopamine transporter (DAT; e.g. substantia nigra) andnorepinephrine transporter (NET; e.g. locus coeruleus).41;110-112 Nowadays, selective SERT ligandslike [11C]DASB for PET or [123I]ADAM for SPECT are available. However, previous imaging study inprimates, showed that [123I]β-CIT uptake in midbrain and diencephalon predominantly reflectsSERT,113 as these structures are rich of SERT relative to DAT and NET. Although this non-selectivitymight have slightly concealed changes in SERT occupancies due to additional DAT- or NET-binding,we think our findings in diencephalon and midbrain mainly reflect SERT occupancy. Furthermore,if one would consider non-selectivity as relevant, the use of SPECT-scans at randomization as areference for changes in occupancy will further reduce the magnitude of this non-selectivity bias,because non-selectivity will be measured systematically between T0 and T1. Despite this rationale,in future studies, a selective ligand for SERT ([11C]DASB or [123I]ADAM) should be considered.Furthermore, it would be a challenge to replicate our study using one of these selective ligands.
Although the region-of-interest (RoI) approach to determine non-displaceable bindingpotential (BPND) and SERT occupancies is a valid and widely accepted method,114 we did not co-register individual SPECT images with individual structural MRI scans. This might have reducedsignal-to-noise variance in our measurements. However, because we applied a randomized,double-blind dose-escalation, and analyzed the SPECT scans while blinded for the moment intreatment (T0 or T1) and intervention, we think that this RoI approach only affectedmeasurement-bias, and did not introduce differential bias between the intervention groups.
Scientific studies are powered to investigate major questions, but often additional questionswill be explored. In the case-control study of SERT availability of MDD-patients versus controls,only small subgroups remained for effect-modifying variables, which might have increased thechance of detecting spurious results. We therefore presented these differences as exploratoryanalyses only. For the SERT occupancy-response relation by genotype interaction, we alsocompared small groups, but nevertheless found significant results, which we consider valid.Nevertheless, replication in a larger sample would strengthen these results. Because of theinterim-analysis, we stopped our trial for futility. Thereafter, our modest sample size of therandomized dose-escalation for example precluded proper subgroup analyses. Our fMRI-study,which was relatively large for imaging studies found robust effects. However, the comparison ofthe dose-escalation versus placebo dose-escalation in our fMRI study was hindered by a modestnumber of patients in the true dose-escalation group. This precluded the investigation of the 3-way interaction intervention by time by response, which could have properly answered whicheffects were drug-induced. Therefore, as indicated, these results must be considered with somerestraint. Finally, in the hemostasis study, we studied 19 patients, but did so in a within-subjectdesign, which has more power to detect changes. Again the number of subjects receiving a dose-escalation was modest, which might have precluded changes in other platelet parameters withsmaller effect-sizes.
A general limitation is that our neurobiological investigations did not investigateadaptiveneuronal effects caused by paroxetine. We either investigated the pre-synaptic effects of dose-escalation on SERT occupancy, or the changes in the final common pathway: the changes inactivations of the limbic-subcortical-cortical network. Many adaptive pre- and post-synapticeffects of SSRIs have been documented,100;115-123 mostly in animal studies, but also in humans.124 Wedid not investigate any of these specific secondary effects of SSRIs. The radiation burden forscientific research did not allow additional radioligand scans of other receptors (e.g. 5-HT2A or5-HT1A) or other monoamines (e.g. endogenous dopamine release). Furthermore, additionalchallenges (e.g. gepirone to assess 5-HT1A receptor sensitivity125) were considered tooburdensome in these patients. We did not perform animal studies to investigate dose-escalation,

Chap
ter
Chapter 12
12
244
for example in microdialysis experiments or studies investigating SERT occupancy, SERT down-regulation, or other secondary effects of serotonergic neurotransmission on receptors and/orother pathways.
Nevertheless these limitations, one should bear in mind that our, and previous clinical studiesdid not support dose-escalation as a rational strategy for SSRI non-responders in MDD. Therefore,the merits of additional dose-escalation studies would be to provide fundamental insights inworking mechanisms.
Finally, this thesis might also make clinicians wonder why initial, high doses of SSRI have beenfound effective in (some) fixed-dose, dose-finding studies in anxiety disorders,126-129 and especiallyin obsessive compulsive disorder (OCD).130-134 Of course, anxiety disorders are beyond the scope ofthis thesis, but this question remains intriguing and might warrant an approach as we did forMDD.
Strengths
First of all, a major strength of this thesis is its clinical starting point. All psychiatrists recognize theproblem of non-response to a first SSRI, and most of them increase the dose as a first strategy,followed by switching.135-139 As such, this thesis covers an important clinical topic, which could bevery relevant for future treatment guidelines.
Our systematic reviews were conducted in accordance with the stringent guidelines of theCochrane Collaboration.140 We performed ‘sensitive’ literature searches in several databases,decreasing the chance of missing valuable studies. All studies were critically appraised andabstracted. We only performed meta-analyses when clinical heterogeneity allowed pooling, amethodological requirement which is often violated by referring to a random effects approach.141
In the obvious heterogenous populations investigated in the monoamine depletion review, weachieved better homogeneity by stratification. In this latter meta-analyses, we acknowledged thestatistical problems of pooling crossover studies (having dependent measurements) and adaptedthe standard statistical approach to solve this problem.32
The empirical studies in this thesis were designed carefully and the methodology of the dose-escalation trial was based on a thorough review of the previous dose-escalation studies.11;50 Wetherefore randomized patients after sufficient time to expect not many further delayed clinicalresponses. Also, we gradually increased doses which obviously prevented early drop-out due tointolerable adverse effects. Most importantly, our SERT occupancy SPECT protocol was unique,and provided the rationale why dose-escalation was ineffective.
With the fMRI-study, which could be started after a second grant was obtained, we lookedbeyond SERT occupancy to investigate the effects of paroxetine treatment on emotion-regulation. Although the dose-escalation results of this study are not unambiguous, this study isonly the sixth fMRI-study investigating the treatment-effects of SSRIs. The fact that we scannedpatients three times, and that many patients initially were treatment non-responders allowed usto investigate the distinction in brain activations between drug-effects and clinical response,which is an important addition to the existing literature.
Finally, we also looked at peripheral effects of SSRIs, far beyond the brain, but alsoconsidering the inhibition of SERTs by paroxetine in association with hemostasis. Increasedbleeding tendency is an infrequent but potentially dangerous adverse effect. We showed thateven standard doses of paroxetine might affect hemostasis, and suggested subgroups who areeligible for secondary prevention by genetic screening of the 5-HTTLPR.

ChapterSummary and general discussion
12
245
Future research
Future studies with data from DELPHISeveral lines of research on data from the DELPHI-study still need to be worked out. We collectedsalivary samples at study-entry, T0 and T1 (see chapter 2, page 36). We will investigate thechanges in cortisol by treatment over time, and might be able to associate these changes withclinical response or other clinical or imaging (e.g. SERT) variables. We therefore will collaboratewith the group of Kirschbaum and Rohleder.142;143 In addition, we collected blood-samples atstudy-entry, T0 and T1, to determine ex-vivo SERT and norepinephrine inhibition byparoxetine.144;145 Finally, we determined omega-3 and omega-6 polyunsaturated fatty acids,146
again at study-entry, T0 and T1, which will be analyzed in relation with clinical response toparoxetine, and additionally in relation with SERT availability and occupancy.147
Furthermore, we are currently working on a sub-study which investigates the association ofmidbrain and amygdala SERT occupancy with the changes in amygdala-activation after (negative)facial expressions in the patients who underwent both SPECT and fMRI scans.
Future clinical studiesThe findings presented in this thesis may be the starting point of new research.- For better evidence-based recommendations about when to choose between switching,
augmentation, combination, or psychotherapeutic strategies as a next step, comparisons ofthese strategies relative to each other are necessary. This will require future algorithm-basedswitch studies, including psychotherapy and augmentation or combination strategies toimprove our knowledge to guide treatment for SSRI non-responders. In these studies, one armshould be to continue treatment (e.g. for another 6-8 weeks, depending on the timing ofswitching) as a ‘placebo’ control. Of course the somehow disappointing results fromSTAR*D148;149 should be reconsidered when such a major endeavor is set up again.
- As a smaller enterprise, the relative benefits of switching after 6 weeks versus a continuationof the same treatment for another 6 weeks would provide important guidance to clinicians.Quitkin et al. showed that MDD-patients treated with fluoxetine 20 mg/day who were non-responders at week 6, showed remission rates at week 12 between 31%-41%, indicative of asubstantial delayed remission.150 However studies directly comparing such prolongedtreatment versus switching have not yet been performed in MDD.
- The controversial evidence advocating initial, high doses of SSRI in panic disorder,126;128 socialfobia,151 posttraumatic stress disorder,152 OCD130;131;134 and other anxiety disorders127 necessitatesa well-performed meta-analysis of fixed-dose, dose-finding studies in anxiety disorders,stratified by disorder and drug. In addition, dose-escalation studies in these disorders would beinteresting, especially when combined with the investigation of neurobiological mechanismsby neuroimaging. For example, the effects of a randomized, placebo-controlled dose-escalation on dopaminergic D2-like receptors, or dopamine transporters in OCD would beinteresting.
Future neurobiological studiesIn order to develop more successful treatment approaches, we need to understand better whicheffects of treatment are required for recovery, and which biomarkers are associated withtreatment response. Therefore, more fundamental, neurobiological studies should aim at severalgaps in the evidence.- In order to elucidate SSRI working-mechanisms, the increased coupling between amygadala
and frontal and cingulate cortices61 must be investigated with other SSRIs. Secondly, it shouldbe investigated whether 5-HTTLPR polymorphisms mediate this coupling sec,48 or the increaseafter SSRIs, or both. If the magnitude of coupling would be mediated by 5-HTTLPRpolymorphisms, this would also corroborate our findings for the association between SERToccupancy with clinical response in LA/LA carriers.45

Chap
ter
Chapter 12
12
246
- In order to study secondary effects of SSRI treatment, neuroimaging approaches to quantifythe effects on 5-HT1A receptors during treatment and after dose-escalation are warranted inpatients. Although a desensitization of 5-HT1A was demonstrated for chronic SSRI treatment inanimals,118;153 the question remains whether this desensitization is also associated with 5-HT1Adownregulation and/or whether dose-response effects occur. This study should ideally beperformed in combination with gepirone-challenges at different moments during treatment toassess 5-HT1A receptor sensitivity.125
- In order to investigate secondary effects of serotonin, e.g. on endogenous dopamine releasein the nucleus accumbens and other brain regions, microdialysis experiments in animals,chronically treated with SSRIs, will further investigate dose-escalation and time dependentchanges in SSRI treatment.
- The hypothesis of the involvement of dopamine in mood and MDD must be investigated.107
Serotonergic antidepressants indirectly influence the dopaminergic system. Responders toamitriptyline showed decreased [123I]IBZM SPECT binding to striatal dopamine D2 receptors,relative to pretreatment.154 This decrease correlated with a decrease in HDRS-score. This mightrepresent an increased tonic dopamine release after amitriptyline, but may also be attributedto improved psychomotor activity in responders. Nevertheless, a failure to achieve alterationsin the dopaminergic system by antidepressants, might explain non-response or residualsymptoms (sleep disturbances, diminished pleasure, loss of interest, fatigue, and decreasedmotivation), which are often observed in clinical practice.107 Therefore, investigation of thedopaminergic effects of SSRIs, in combination with or versus behavioural approaches (re-activation) in combined fMRI and SPECT/PET studies in (retarded) MDD-patients are necessary.
- The effects of SSRIs on neurogenesis also merit further research. After prolongedantidepressant administration in rodents, increased hippocampal neurogenesis was found.121;123
The second-messenger effects of SSRIs upregulate cAMP responsive element binding protein(CREB) in the neuron’s nucleus, which consequently regulates CREB-directed genetranscription.117;155 To date, it is unclear which specific genes transcription are affected by CREB.Therefore the exact role of second messenger systems (and CREB in particular) in response toSSRIs remain to be elucidated.117 One of the genes that is (positively) influenced by CREB is thebrain derived neurotrophic factor (BDNF) gene. BDNF is a plethoric factor, which regulatesneuronal survival, migration, phenotypic differentiation, axonal and dendritic growth andsynapse formation. As such BDNF has a role in cognitive functions (e.g. in memory andhippocampal plasticity).100 BDNF and serotonin are closely interlinked: BDNF promotesproduction of serotonin, upregulates serotonin uptake/release, and modifies firing rates ofneurons in the raphe nuclei, while serotonin upregulates BDNF. This stimulating feedbackmechanism may be involved in the selection of useful synapses, which could be hypothesizedto be a requisite for clinical response.119 Few studies investigated the long-term changes inBDNF measured in venous blood of MDD patients treated with antidepressants.156-158 Althoughdifferent effects for different drugs were seen, generally an increase in BDNF after 6 monthswas found, significantly associated with a decrease in HDRS. Nevertheless, the role of BDNF asbiomarker for antidepressant response is equivocal. Furthermore, due to dilution, peripheralmeasures may not be adequate, so sampling from an internal jugular vein might be better.159
Therefore, this line of research might need development of the methods to measure BDNF(and CREB) more locally in the brain of humans.

ChapterSummary and general discussion
12
247
Conclusion
This thesis addresses the pharmacological treatment of Major Depressive Disorder (MDD), andfocuses on what to do when patients do not respond to a standard dose of an antidepressant. Intwo systematic reviews, we summarized the evidence for various switch strategies and dose-escalation, and concluded that for both strategies insufficient evidence existed to unambiguouslyguide clinicians. We thereafter performed a randomized controlled trial in which we studied dose-escalation in depressed patients who had not responded to 6 weeks of paroxetine (20 mg/day),and combined clinical measurements with SPECT and fMRI neuroimaging in subgroups of theparticipants (DELPHI-study). With the DELPHI-study, we show that dose-escalation of paroxetinehas no clinical benefit over placebo dose-escalation, and furthermore quantify the absence of apharmacological effect on the target of paroxetine: the SERT.
Another systematic review and four studies in this thesis, investigate the role of monoaminesand SERT in the etiology of MDD and the neurobiological effects of pharmacological treatmentwith paroxetine. From these studies we conclude that MDD is not necessarily caused by amalfunction of the serotonergic system perse. MDD may better be considered as a disturbance ofthe emotional and cognitive functioning in the limbic-subcortical-cortical network. The increase ofserotonergic neurotransmission by paroxetine (and likely other antidepressants), appears toimprove the constraint of neuronal information processing, with less emotional activation andmore cognitive control in treatment responders relative to non-responders. The findings of thisthesis can be the starting point for further investigation of biomarkers for treatment response, inorder to develop better understanding of MDD and a more effective treatment approaches.
References
1. Ruhe HG, Dekker JJ, Peen J, Holman R, de Jonghe F. Clinical use of the Hamilton Depression Rating Scale: is increasedefficiency possible? A post hoc comparison of Hamilton Depression Rating Scale, Maier and Bech subscales, ClinicalGlobal Impression, and Symptom Checklist-90 scores. Compr Psychiatry. 2005; 46: 417-427.
2. Faries D, Herrera J, Rayamajhi J, DeBrota D, Demitrack M, Potter WZ. The responsiveness of the Hamilton DepressionRating Scale. J Psychiatr Res. 2000; 34: 3-10.
3. Entsuah R, Shaffer M, Zhang J. A critical examination of the sensitivity of unidimensional subscales derived from theHamilton Depression Rating Scale to antidepressant drug effects. J Psychiatr Res. 2002; 36: 437-448.
4. O'Sullivan RL, Fava M, Agustin C, Baer L, Rosenbaum JF. Sensitivity of the six-item Hamilton Depression Rating Scale. ActaPsychiatr Scand. 1997; 95: 379-384.
5. Hooper CL, Bakish D. An examination of the sensitivity of the six-item Hamilton Rating Scale for Depression in a sample ofpatients suffering from major depressive disorder. J Psychiatry Neurosci. 2000; 25: 178-184.
6. Maier W, Philipp M. Improving the assessment of severity of depressive states: a reduction of the Hamilton DepressionScale. Pharmacopsychiatry. 1985; 18: 114-115.
7. Bech P, Gram LF, Dein E, Jacobsen O, Vitger J, Bolwig TG. Quantitative rating of depressive states. Acta Psychiatr Scand.1975; 51: 161-170.
8. Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW et al. Conceptualization and rationale for consensusdefinitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48: 851-855.
9. Prien RF, Carpenter LL, Kupfer DJ. The definition and operational criteria for treatment outcome of major depressivedisorder. A review of the current research literature. Arch Gen Psychiatry. 1991; 48: 796-800.
10. Ballenger JC. Clinical guidelines for establishing remission in patients with depression and anxiety. J Clin Psychiatry. 1999;60 Suppl 22: 29-34.
11. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard-dose selectiveserotonin reuptake inhibitors in major depressive disorder: Systematic review. Br J Psychiatry. 2006; 189: 309-316.
12. Dornseif BE, Dunlop SR, Potvin JH, Wernicke JF. Effect of dose escalation after low-dose fluoxetine therapy.Psychopharmacol Bull. 1989; 25: 71-79.
13. Schweizer E, Rickels K, Amsterdam JD, Fox I, Puzzuoli G, Weise C. What constitutes an adequate antidepressant trial forfluoxetine? J Clin Psychiatry. 1990; 51: 8-11.
14. Fava M, Cohen L, Rosenbaum JF, Reiter S, et a. High-dose fluoxetine in the treatment of depressed patients notresponsive to a standard dose of fluoxetine. J Affect Disord. 1992; 25: 229-234.
15. Fava M, Rosenbaum JF, McGrath PJ, Stewart JW, Amsterdam JD, Quitkin et al. Lithium and tricyclic augmentation offluoxetine treatment for resistant major depression: a double-blind, controlled study. Am J Psychiatry. 1994; 151: 1372-1374.

Chap
ter
Chapter 12
12
248
16. Benkert O, Szegedi A, Wetzel H, Staab HJ, Meister W, Philipp M. Dose escalation vs. continued doses of paroxetine andmaprotiline: a prospective study in depressed out-patients with inadequate treatment response. Acta Psychiatr Scand.1997; 95: 288-296.
17. Schweizer E, Rynn M, Mandos LA, DeMartinis N, Garcia-Espana F, Rickels K. The antidepressant effect of sertraline is notenhanced by dose titration: results from an outpatient clinical trial. Int Clin Psychopharmacol. 2001; 16: 137-143.
18. Fava M, Alpert J, Nierenberg A, Lagomasino I, Sonawalla S, Tedlow J et al. Double-blind study of high-dose fluoxetineversus lithium or desipramine augmentation of fluoxetine in partial responders and nonresponders to fluoxetine. JClinPsychopharmacol. 2002; 22: 379-387.
19. Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first-line sertralinetreatment: A randomised study of extended duration of treatment, dose increase or mianserin augmentation.Psychopharmacology (Berl). 2002; 161: 143-151.
20. Bollini P, Pampallona S, Tibaldi G, Kupelnick B, Munizza C. Effectiveness of antidepressants. Meta-analysis of dose-effectrelationships in randomised clinical trials. Br J Psychiatry. 1999; 174: 297-303.
21. Corruble E, Guelfi JD. Does increasing dose improve efficacy in patients with poor antidepressant response: a review.Acta Psychiatr Scand. 2000; 101: 343-348.
22. Baker CB, Tweedie R, Duval S, Woods SW. Evidence that the SSRI dose response in treating major depression should bereassessed: A meta-analysis. Depress Anxiety. 2003; 17: 1-9.
23. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Switching antidepressants after a first selective serotonin reuptake inhibitorin major depressive disorder: a systematic review. J Clin Psychiatry. 2006; 67: 1836-1855.
24. Baldomero EB, Ubago JG, Cercos CL, Ruiloba JV, Calvo CG, Lopez RP. Venlafaxine extended release versus conventionalantidepressants in the remission of depressive disorders after previous antidepressant failure: ARGOS study. DepressAnxiety. 2005; 22: 68-76.
25. Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006; 354: 1231-1242.
26. Fava M, Rush AJ, Wisniewski SR, Nierenberg AA, Alpert JE, McGrath PJ et al. A comparison of mirtazapine andnortriptyline following two consecutive failed medication treatments for depressed outpatients: a STAR*D report. Am JPsychiatry. 2006; 163: 1161-1172.
27. McGrath PJ, Stewart JW, Fava M, Trivedi MH, Wisniewski SR, Nierenberg AA et al. Tranylcypromine versus venlafaxineplus mirtazapine following three failed antidepressant medication trials for depression: a STAR*D report. Am J Psychiatry.2006; 163: 1531-1541.
28. Poirier MF, Boyer P. Venlafaxine and paroxetine in treatment-resistant depression. Double-blind, randomisedcomparison. Br J Psychiatry. 1999; 175: 12-16.
29. Ferreri M, Lavergne F, Berlin I, Payan C, Puech AJ. Benefits from mianserin augmentation of fluoxetine in patients withmajor depression non-responders to fluoxetine alone. Acta Psychiatr Scand. 2001; 103: 66-72.
30. Nolen WA, van de Putte JJ, Dijken WA, Kamp JS. L-5HTP in depression resistant to re-uptake inhibitors. An opencomparative study with tranylcypromine. Br J Psychiatry. 1985; 147: 16-22.
31. Nolen WA, van de Putte JJ, Dijken WA, Kamp JS, et a. Treatment strategy in depression: II. MAO inhibitors in depressionresistant to cyclic antidepressants: Two controlled crossover studies with tranylcypromine versus L-5-hydroxytryptophanand nomifensine. Acta Psychiatr Scand. 1988; 78: 676-683.
32. Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans:a meta-analysis of monoamine depletion studies. Mol Psychiatry. 2007; 12: 331-359.
33. Bell C, Abrams J, Nutt D. Tryptophan depletion and its implications for psychiatry. Br J Psychiatry. 2001; 178: 399-405.34. Booij L, Van der Does AJ, Riedel WJ. Monoamine depletion in psychiatric and healthy populations: review. Mol Psychiatry.
2003; 8: 951-973.35. Van der Does AJ. The effects of tryptophan depletion on mood and psychiatric symptoms. J Affect Disord. 2001; 64: 107-
119.36. Moore P, Landolt HP, Seifritz E, Clark C, Bhatti T, Kelsoe J et al. Clinical and physiological consequences of rapid
tryptophan depletion. Neuropsychopharmacology. 2000; 23: 601-622.37. Booij L, Van der DW, Benkelfat C, Bremner JD, Cowen PJ, Fava M et al. Predictors of mood response to acute tryptophan
depletion. A reanalysis. Neuropsychopharmacology. 2002; 27: 852-861.38. Van der Does AJ. The mood-lowering effect of tryptophan depletion: possible explanation for discrepant findings. Arch
Gen Psychiatry. 2001; 58: 200-202.39. Ruhe HG, Booij J, Reitsma JB, Schene AH. Serotonin transporter binding with [123I]ß-CIT SPECT in major depressive
disorder versus controls: effect of season and gender. [Submitted]. 40. Staley JK, Krishnan-Sarin S, Zoghbi S, Tamagnan G, Fujita M, Seibyl JP et al. Sex differences in [123I]β-CIT SPECT measures
of dopamine and serotonin transporter availability in healthy smokers and nonsmokers. Synapse. 2001; 41: 275-284.41. Pirker W, Asenbaum S, Hauk M, Kandlhofer S, Tauscher J, Willeit M et al. Imaging serotonin and dopamine transporters
with [123I]β-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000; 41: 36-44.42. van Dyck CH, Malison RT, Seibyl JP, Laruelle M, Klumpp H, Zoghbi SS et al. Age-related decline in central serotonin
transporter availability with [123I]β-CIT SPECT. Neurobiol Aging. 2000; 21: 497-501.43. Buchert R, Schulze O, Wilke F, Berding G, Thomasius R, Petersen K et al. Is correction for age necessary in SPECT or PET of
the central serotonin transporter in young, healthy adults? J Nucl Med. 2006; 47: 38-42.44. Staley JK, Sanacora G, Tamagnan G, Maciejewski PK, Malison RT, Berman RM et al. Sex differences in diencephalon
serotonin transporter availability in major depression. Biol Psychiatry. 2006; 59: 40-47.45. Ruhé HG, Ooteman W, Booij J, Michel MC, Moeton M, Baas F et al. Serotonin transporter gene promoter polymorphisms
modify the association between paroxetine serotonin transporter occupancy and clinical response in major depressivedisorder. Pharmacogenetics and Genomics. 2008; [In press].

ChapterSummary and general discussion
12
249
46. Smits KM, Smits LJ, Schouten JS, Stelma FF, Nelemans P, Prins MH. Influence of SERTPR and STin2 in the serotonintransporter gene on the effect of selective serotonin reuptake inhibitors in depression: a systematic review. MolPsychiatry. 2004; 9: 433-441.
47. Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry. 2007; 12:247-257.
48. Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS et al. 5-HTTLPR polymorphismimpacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8: 828-834.
49. Lesch KP, Gutknecht L. Pharmacogenetics of the serotonin transporter. Prog Neuropsychopharmacol Biol Psychiatry.2005; 29: 1062-1073.
50. Baker CB, Woods SW. Is there a SSRI dose response in treating major depression? The case for re-analysis of current dataand for enhancing future study design. Depress Anxiety. 2003; 17: 10-18.
51. Ruhe HG, Booij J, Weert HC van, Reitsma JB, Fransen EJF, Michel MC et al. Evidence why dose-escalation of paroxetine inmajor depressive disorder is not effective: A 6-week, randomized-controlled trial with assessment of serotonintransporter occupancy. Neuropsychpharmacology.[In Press].
52. Adli M, Baethge C, Heinz A, Langlitz N, Bauer M. Is dose escalation of antidepressants a rational strategy after a medium-dose treatment has failed? A systematic review. Eur Arch Psychiatry Clin Neurosci. 2005; 255: 387-400.
53. Baker CB, Tweedie R, Duval S, Woods SW. Evidence that the SSRI dose response in treating major depression should bereassessed: a meta-analysis. Depress Anxiety. 2003; 17: 1-9.
54. Ruhe HG, Booij J, Veltman DJ, Michel MC, Schene AH. Successful treatment of major depressive disorder by paroxetineattenuates amygdala activation to negative facial expressions. An fMRI study. [Submitted].
55. Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA. Increased amygdala response to maskedemotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry. 2001; 50:651-658.
56. Fu CH, Williams SC, Cleare AJ, Brammer MJ, Walsh ND, Kim J et al. Attenuation of the neural response to sad faces inmajor depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imagingstudy. Arch Gen Psychiatry. 2004; 61: 877-889.
57. Robertson B, Wang L, Diaz MT, Aiello M, Gersing K, Beyer J et al. Effect of bupropion extended release on negativeemotion processing in major depressive disorder: a pilot functional magnetic resonance imaging study. J Clin Psychiatry.2007; 68: 261-267.
58. Davidson RJ, Irwin W, Anderle MJ, Kalin NH. The neural substrates of affective processing in depressed patients treatedwith venlafaxine. Am J Psychiatry. 2003; 160: 64-75.
59. Schaefer HS, Putnam KM, Benca RM, Davidson RJ. Event-related functional magnetic resonance imaging measures ofneural activity to positive social stimuli in pre- and post-treatment depression. Biol Psychiatry. 2006; 60: 974-986.
60. Anand A, Li Y, Wang Y, Wu J, Gao S, Bukhari L et al. Antidepressant effect on connectivity of the mood-regulating circuit:an FMRI study. Neuropsychopharmacology. 2005; 30: 1334-1344.
61. Chen CH, Suckling J, Ooi C, Fu CH, Williams SC, Walsh ND et al. Functional Coupling of the Amygdala in Depressed PatientsTreated with Antidepressant Medication. Neuropsychopharmacology. 2008; 33: 1909-1918.
62. Abdelmalik N, Ruhé HG, Barwari K, Dool E-Jvd, Meijers JCM, Middeldorp S et al. The effect of the selective serotoninreuptake inhibitor paroxetine on platelet function is modified by a SLC6A4 serotonin transporter polymorphism. JThromb Haemost. 2008; [In Press].
63. Skop BP, Brown TM. Potential vascular and bleeding complications of treatment with selective serotonin reuptakeinhibitors. Psychosomatics. 1996; 37: 12-16.
64. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram fordepression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006; 163: 28-40.
65. Thase ME. Evaluating antidepressant therapies: remission as the optimal outcome. J Clin Psychiatry. 2003; 64 Suppl 13: 18-25.
66. Lenox-Smith AJ, Jiang Q. Venlafaxine extended release versus citalopram in patients with depression unresponsive to aselective serotonin reuptake inhibitor. Int Clin Psychopharmacol. 2008; 23: 113-119.
67. Thase ME, Lu Y, Joliat MJ, Detke MJ. Remission in placebocontrolled trials of duloxetine with an SSRI comparator.Presented atthe annual meeting of the American Psychiatric Association, May 2003, San Francisco, CA . 2003.
68. Malhi GS, Ng F, Berk M. Dual-dual action? Combining venlafaxine and mirtazapine in the treatment of depression. Aust N ZJ Psychiatry. 2008; 42: 346-349.
69. Perahia DG, Quail D, Desaiah D, Corruble E, Fava M. Switching to duloxetine from selective serotonin reuptake inhibitorantidepressants: a multicenter trial comparing 2 switching techniques. J Clin Psychiatry. 2008; 69: 95-105.
70. Papakostas GI, Fava M, Thase ME. Treatment of SSRI-resistant depression: a meta-analysis comparing within- versusacross-class switches. Biol Psychiatry. 2008; 63: 699-704.
71. Lenox-Smith AJ, Schaeffer P, Reynolds A, Williard L. A double blind trial of venlafaxine XR versus citalopram in patientswith severe depression. J Psychopharmacol. 2001; 15 Suppl 3: A11.
72. Thase ME, Kremer C, Rodrigues H. Mirtazapine versus sertraline after SSRI nonresponse [Abstract]. American PsychiatricAssociation.2001 annual meeting: New Orleans LA. 2001.
73. Cipriani A, Barbui C, Brambilla P, Furukawa TA, Hotopf M, Geddes JR. Are all antidepressants really the same? The case offluoxetine: a systematic review. J Clin Psychiatry. 2006; 67: 850-864.
74. NICE. Clinical Guideline 23. Depression: management of depression in primary and secondary care. December 2004. 23, 1-63. 2004. London, National Institute for Clinical Excellence.

Chap
ter
Chapter 12
12
250
75. Papakostas GI, Charles D, Fava M. Are typical starting doses of the selective serotonin reuptake inhibitors sub-optimal? Ameta-analysis of randomized, double-blind, placebo-controlled, dose-finding studies in major depressive disorder. WorldJ Biol Psychiatry. 2007; 1-8.
76. Santor DA, Coyne JC. Examining symptom expression as a function of symptom severity: Item performance on theHamilton Rating Scale for Depression. Psychol Assess. 2001; 13: 127-139.
77. de Abajo FJ, Montero D, Rodriguez LA, Madurga M. Antidepressants and risk of upper gastrointestinal bleeding. Basic ClinPharmacol Toxicol. 2006; 98: 304-310.
78. Wessinger S, Kaplan M, Choi L, Williams M, Lau C, Sharp L et al. Increased use of selective serotonin reuptake inhibitors inpatients admitted with gastrointestinal haemorrhage: a multicentre retrospective analysis. Aliment Pharmacol Ther.2006; 23: 937-944.
79. Meijer WE, Heerdink ER, Nolen WA, Herings RM, Leufkens HG, Egberts AC. Association of risk of abnormal bleeding withdegree of serotonin reuptake inhibition by antidepressants. Arch Intern Med. 2004; 164: 2367-2370.
80. Dalton SO, Johansen C, Mellemkjaer L, Norgard B, Sorensen HT, Olsen JH. Use of selective serotonin reuptake inhibitorsand risk of upper gastrointestinal tract bleeding: a population-based cohort study. Arch Intern Med. 2003; 163: 59-64.
81. van Walraven C, Mamdani MM, Wells PS, Williams JI. Inhibition of serotonin reuptake by antidepressants and uppergastrointestinal bleeding in elderly patients: retrospective cohort study. BMJ. 2001; 323: 655-658.
82. Serebruany VL. Selective serotonin reuptake inhibitors and increased bleeding risk: are we missing something? Am J Med.2006; 119: 113-116.
83. van Marwijk HW, Grundmeijer HG, Bijl D, van Gelderen MG, de Haan M, van Weel-Baumgarten EM et al. NHG-StandaardDepressieve stoornis (depressie) (eerste herziening). Huisarts Wet. 2003; 46: 614-623.
84. Kent JM, Coplan JD, Lombardo I, Hwang DR, Huang Y, Mawlawi O et al. Occupancy of brain serotonin transporters duringtreatment with paroxetine in patients with social phobia: a positron emission tomography study with [11C]McN 5652.Psychopharmacology (Berl). 2002; 164: 341-348.
85. Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K et al. Occupancy of serotonin transporters by paroxetineand citalopram during treatment of depression: a [11C]DASB PET imaging study. Am J Psychiatry. 2001; 158: 1843-1849.
86. Meyer JH, Wilson AA, Sagrati S, Hussey D, Carella A, Potter WZ et al. Serotonin Transporter Occupancy of Five SelectiveSerotonin Reuptake Inhibitors at Different Doses: An [11C]DASB Positron Emission Tomography Study. AmJPsychiatry.2004; 161: 826-835.
87. Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB et al. Acute occupancy of brain serotonintransporter by sertraline as measured by [11C]DASB and positron emission tomography. Biol Psychiatry. 2006; 59: 821-828.
88. Suhara T, Takano A, Sudo Y, Ichimiya T, Inoue M, Yasuno F et al. High levels of serotonin transporter occupancy with low-dose clomipramine in comparative occupancy study with fluvoxamine using positron emission tomography. Arch GenPsychiatry. 2003; 60: 386-391.
89. Erlandsson K, Sivananthan T, Lui D, Spezzi A, Townsend CE, Mu S et al. Measuring SSRI occupancy of SERT using the noveltracer [123I]ADAM: a SPECT validation study. Eur J Nucl Med Mol Imaging. 2005; 32: 1329-1336.
90. Hiltunen J, Akerman KK, Kuikka JT, Bergstrom KA, Halldin C, Nikula T et al. Iodine-123 labeled nor-beta-CIT as a potentialtracer for serotonin transporter imaging in the human brain with single-photon emission tomography. Eur J Nucl Med.1998; 25: 19-23.
91. Klein N, Sacher J, Geiss-Granadia T, Mossaheb N, Attarbaschi T, Lanzenberger R et al. Higher serotonin transporteroccupancy after multiple dose administration of escitalopram compared to citalopram: an [123I]ADAM SPECT study.Psychopharmacology (Berl). 2007; 191: 333-339.
92. Klein N, Sacher J, Geiss-Granadia T, Attarbaschi T, Mossaheb N, Lanzenberger R et al. In vivo imaging of serotonintransporter occupancy by means of SPECT and [123I]ADAM in healthy subjects administered different doses ofescitalopram or citalopram. Psychopharmacology (Berl). 2006; 188: 263-272.
93. Takano A, Suzuki K, Kosaka J, Ota M, Nozaki S, Ikoma Y et al. A dose-finding study of duloxetine based on serotonintransporter occupancy. Psychopharmacology (Berl). 2006; 185: 395-399.
94. Catafau AM, Perez V, Plaza P, Pascual JC, Bullich S, Suarez M et al. Serotonin transporter occupancy induced byparoxetine in patients with major depression disorder: a [123I]-ADAM SPECT study. Psychopharmacology (Berl). 2006; 189:145-153.
95. Herold N, Uebelhack K, Franke L, Amthauer H, Luedemann L, Bruhn H et al. Imaging of serotonin transporters and itsblockade by citalopram in patients with major depression using a novel SPECT ligand [123I]-ADAM. J Neural Transm. 2006;113: 659-670.
96. Voineskos AN, Wilson AA, Boovariwala A, Sagrati S, Houle S, Rusjan P et al. Serotonin transporter occupancy of high-doseselective serotonin reuptake inhibitors during major depressive disorder measured with [11C]DASB positron emissiontomography. Psychopharmacology (Berl). 2007; 193: 539-545.
97. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD et al. Serotonin transporter promoter gain-of-functiongenotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. 2006; 78: 815-826.
98. Jans LA, Riedel WJ, Markus CR, Blokland A. Serotonergic vulnerability and depression: assumptions, experimentalevidence and implications. Mol Psychiatry. 2007; 12: 522-543.
99. Fox MA, Andrews AM, Wendland JR, Lesch KP, Holmes A, Murphy DL. A pharmacological analysis of mice with a targeteddisruption of the serotonin transporter. Psychopharmacology (Berl). 2007; 195: 147-166.
100. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;33: 73-83.
101. Murphy DL, Lesch KP. Targeting the murine serotonin transporter: insights into human neurobiology. Nat Rev Neurosci.2008; 9: 85-96.
102. Holmes A, Yang RJ, Murphy DL, Crawley JN. Evaluation of antidepressant-related behavioral responses in mice lackingthe serotonin transporter. Neuropsychopharmacology. 2002; 27: 914-923.

ChapterSummary and general discussion
12
251
103. Holmes A, Murphy DL, Crawley JN. Abnormal behavioral phenotypes of serotonin transporter knockout mice: parallelswith human anxiety and depression. Biol Psychiatry. 2003; 54: 953-959.
104. Spoont MR. Modulatory role of serotonin in neural information processing: implications for human psychopathology.Psychol Bull. 1992; 112: 330-350.
105. Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-basedalgorithms for diagnosis and optimised treatment. Br Med Bull. 2003; 65: 193-207.
106. Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatricdisorders. Biol Psychiatry. 2003; 54: 515-528.
107. Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry. 2007; 64: 327-337.
108. D'Aquila PS, Collu M, Gessa GL, Serra G. The role of dopamine in the mechanism of action of antidepressant drugs. Eur JPharmacol. 2000; 405: 365-373.
109. Knutson B, Adams CM, Fong GW, Hommer D. Anticipation of increasing monetary reward selectively recruits nucleusaccumbens. J Neurosci. 2001; 21: RC159.
110. Innis R, Baldwin R, Sybirska E, Zea Y, Laruelle M, al Tikriti M et al. Single photon emission computed tomography imagingof monoamine reuptake sites in primate brain with [123I]CIT. Eur J Pharmacol. 1991; 200: 369-370.
111. Neumeyer JL, Wang SY, Milius RA, Baldwin RM, Zea-Ponce Y, Hoffer PB et al. [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane: high-affinity SPECT radiotracer of monoamine reuptake sites in brain. J Med Chem. 1991; 34: 3144-3146.
112. Neumeyer JL, Tamagnan G, Wang S, Gao Y, Milius RA, Kula NS et al. N-substituted analogs of 2 beta-carbomethoxy-3 beta-(4'-iodophenyl)tropane (β-CIT) with selective affinity to dopamine or serotonin transporters in rat forebrain. J Med Chem.1996; 39: 543-548.
113. Laruelle M, Baldwin RM, Malison RT, Zea-Ponce Y, Zoghbi SS, al Tikriti MS et al. SPECT imaging of dopamine andserotonin transporters with [123I]β-CIT: pharmacological characterization of brain uptake in nonhuman primates.Synapse. 1993; 13: 295-309.
114. de Win MM, Habraken JB, Reneman L, Brink Wvan den, den Heeten GJ, Booij J. Validation of [123I]β-CIT SPECT to assessserotonin transporters in vivo in humans: a double-blind, placebo-controlled, crossover study with the selective serotoninreuptake inhibitor citalopram. Neuropsychopharmacology. 2005; 30: 996-1005.
115. Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG et al. Effects of chronic antidepressanttreatments on serotonin transporter function, density, and mRNA level. J Neurosci. 1999; 19: 10494-10501.
116. Benmansour S, Owens WA, Cecchi M, Morilak DA, Frazer A. Serotonin clearance in vivo is altered to a greater extent byantidepressant-induced downregulation of the serotonin transporter than by acute blockade of this transporter.JNeurosci. 2002; 22: 6766-6772.
117. Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006; 59: 1144-1150.118. Blier P, De Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci. 1994; 15: 220-
226.119. Castren E. Is mood chemistry? Nat Rev Neurosci. 2005; 6: 241-246.120. Davidson C, Stamford JA. Effect of chronic paroxetine treatment on 5-HT1B and 5-HT1D autoreceptors in rat dorsal raphe
nucleus. Neurochem Int. 2000; 36: 91-96.121. Djavadian RL. Serotonin and neurogenesis in the hippocampal dentate gyrus of adult mammals. Acta Neurobiol Exp
(Wars). 2004; 64: 189-200.122. Kim SW, Park SY, Hwang O. Up-regulation of tryptophan hydroxylase expression and serotonin synthesis by sertraline.
Mol Pharmacol. 2002; 61: 778-785.123. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat
hippocampus. J Neurosci. 2000; 20: 9104-9110.124. Meyer JH, Kapur S, Eisfeld B, Brown GM, Houle S, DaSilva J et al. The effect of paroxetine on 5-HT2A receptors in
depression: an [18F]setoperone PET imaging study. Am J Psychiatry. 2001; 158: 78-85.125. Sargent P, Williamson DJ, Pearson G, Odontiadis J, Cowen PJ. Effect of paroxetine and nefazodone on 5-HT1A receptor
sensitivity. Psychopharmacology (Berl). 1997; 132: 296-302.126. Mavissakalian MR, Ryan MT. Rational treatment of panic disorder with antidepressants. Ann Clin Psychiatry. 1998; 10: 185-
195.127. Rickels K, Zaninelli R, McCafferty J, Bellew K, Iyengar M, Sheehan D. Paroxetine Treatment of Generalized Anxiety
Disorder: A Double-Blind, Placebo-Controlled Study. Am J Psychiatry. 2003; 160: 749-756.128. Practice guideline for the treatment of patients with panic disorder. Work Group on Panic Disorder. American Psychiatric
Association. Am J Psychiatry. 1998; 155: 1-34.129. Richtlijn Angststoornissen. Multidisciplinaire richtlijn Angststoornissen. Richtlijn voor de diagnostiek en behandeling van
volwassen cliënten met een angststoornis. Velde Vvd.2003. Utrecht, Trimbos-instituut, in opdracht van de LandelijkeStuurgroep Multidisciplinaire Richtlijnontwikkeling in de GGZ.
130. Denys D. Pharmacotherapy of obsessive-compulsive disorder and obsessive-compulsive spectrum disorders. PsychiatrClin North Am. 2006; 29: 553-584.
131. Figee M, Denys D. New pharmacotherapeutical approaches to obsessive compulsive disorder. CNS Spectr. 2008; [Inpress].
132. Ninan PT, Koran LM, Kiev A, Davidson JR, Rasmussen SA, Zajecka JM et al. High-dose sertraline strategy fornonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry.2006; 67: 15-22.
133. Dougherty DD, Rauch SL, Jenike MA. Pharmacotherapy for obsessive-compulsive disorder. J Clin Psychol. 2004; 60: 1195-1202.

Chap
ter
Chapter 12
12
252
134. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder.Available online at http//www.psych.org/psych_pract/treatg/pg/prac_guide.cfmArlington, VA: American PsychiatricAssociation; 2007:1-96.
135. Wittkampf KA, Ruhé HG, Huyser J, Schene AH. De Creatieve Psychiater? Een onderzoek onder Nederlandse psychiatersnaar de behandeling van depressieve stoornissen bij het falen van de eerste SSRI.2001. Amsterdam, ZorgprogrammaStemmingsstoornissen MFO Psychiatrie AMC/ De Meren.
136. Byrne S, Rothschild AJ. Psychiatrists' responses to failure of maintenance therapy with antidepressants. Psychiatr Serv.1997; 48: 835-837.
137. Fredman SJ, Fava M, Kienke AS, White CN, Nierenberg AA, Rosenbaum JF. Partial response, nonresponse, and relapsewith selective serotonin reuptake inhibitors in major depression: a survey of current "next-step" practices. J ClinPsychiatry. 2000; 61: 403-408.
138. Mischoulon D, Nierenberg AA, Kizilbash L, Rosenbaum JF, Fava M. Strategies for managing depression refractory toselective serotonin reuptake inhibitor treatment: a survey of clinicians. Can J Psychiatry. 2000; 45: 476-481.
139. Shergill SS, Katona CL. Pharmacological choices after one antidepressant fails: a survey of UK psychiatrists. J AffectDisord. 1997; 43: 19-25.
140. Cochrane Collaboration. Preparing, maintaining and promoting the accessibility of systematic reviews of the effects ofhealth care interventions. Internet: http://www.cochrane.org/. 2001. Cochrane Collaboration.
141. DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986; 7: 177-188.142. Rohleder N, Nater UM, Wolf JM, Ehlert U, Kirschbaum C. Psychosocial stress-induced activation of salivary alpha-amylase:
an indicator of sympathetic activity? Ann N Y Acad Sci. 2004; 1032: 258-263.143. Schmidt-Reinwald A, Pruessner JC, Hellhammer DH, Federenko I, Rohleder N, Schurmeyer TH et al. The cortisol response
to awakening in relation to different challenge tests and a 12-hour cortisol rhythm. Life Sci. 1999; 64: 1653-1660.144. Owens MJ, Krulewicz S, Simon JS, Sheehan DV, Thase ME, Carpenter DJ et al. Estimates of Serotonin and Norepinephrine
Transporter Inhibition in Depressed Patients Treated with Paroxetine or Venlafaxine. Neuropsychopharmacology. 2008.145. Gilmor ML, Owens MJ, Nemeroff CB. Inhibition of norepinephrine uptake in patients with major depression treated with
paroxetine. Am J Psychiatry. 2002; 159: 1702-1710.146. Parker G, Gibson NA, Brotchie H, Heruc G, Rees AM, Hadzi-Pavlovic D. Omega-3 fatty acids and mood disorders. Am J
Psychiatry. 2006; 163: 969-978.147. du Bois TM, Deng C, Bell W, Huang XF. Fatty acids differentially affect serotonin receptor and transporter binding in the
rat brain. Neuroscience. 2006; 139: 1397-1403.148. Rush AJ, Fava M, Wisniewski SR, Lavori PW, Trivedi MH, Sackeim HA et al. Sequenced treatment alternatives to relieve
depression (STAR*D): rationale and design. Control Clin Trials. 2004; 25: 119-142.149. Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D et al. Acute and longer-term outcomes in
depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006; 163: 1905-1917.150. Quitkin FM, Petkova E, McGrath PJ, Taylor B, Beasley C, Stewart J et al. When should a trial of fluoxetine for major
depression be declared failed? Am J Psychiatry. 2003; 160: 734-740.151. Stein DJ, Ipser JC, Balkom AJv. Pharmacotherapy for social anxiety disorder. Cochrane Database of Systematic Reviews.
2000; Issue 4.: Art. No.: CD001206. DOI: 10.1002/14651858.CD001206.pub2.152. Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database of Systematic
Reviews. 2006; Issue 1: Art. No.: CD002795. DOI: 10.1002/14651858.CD002795.pub2.153. Blier P, De Montigny C. Serotonin and drug-induced therapeutic responses in major depression, obsessive-compulsive
and panic disorders. Neuropsychopharmacology. 1999; 21: 91S-98S.154. Ebert D, Feistel H, Loew T, Pirner A. Dopamine and depression--striatal dopamine D2 receptor SPECT before and after
antidepressant therapy. Psychopharmacology (Berl). 1996; 126: 91-94.155. Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008; 358: 55-68.156. Aydemir O, Deveci A, Taneli F. The effect of chronic antidepressant treatment on serum brain-derived neurotrophic factor
levels in depressed patients: a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry. 2005; 29: 261-265.157. Matrisciano F, Bonaccorso S, Ricciardi A, Scaccianoce S, Panaccione I, Wang L et al. Changes in BDNF serum levels in
patients with major depression disorder (MDD) after 6 months treatment with sertraline, escitalopram, or venlafaxine. JPsychiatr Res. 2008.
158. Piccinni A, Marazziti D, Catena M, Domenici L, Del Debbio A, Bianchi C et al. Plasma and serum brain-derived neurotrophicfactor (BDNF) in depressed patients during 1 year of antidepressant treatments. J Affect Disord. 2008; 105: 279-283.
159. Barton DA, Esler MD, Dawood T, Lambert EA, Haikerwal D, Brenchley C et al. Elevated brain serotonin turnover in patientswith depression: effect of genotype and therapy. Arch Gen Psychiatry. 2008; 65: 38-46.

CHA
PTER
SAMENVATTING EN CONCLUSIES

254

ChapterSamenvatting en conclusies
13
255
Samenvatting en conclusies
In deze Nederlandse samenvatting wordt een overzicht gegeven van een aantal basiskenmerkenen een aantal problemen in de farmacologische behandeling van depressie. Deze problemenstonden aan de basis van de studies beschreven in dit proefschrift. Hiervoor is eenonderzoeksproject opgezet met verschillende substudies (beschreven in hoofdstuk 2). Ditonderzoek had de overkoepelende naam DELPHI-studie, wat staat voor ‘Dose-escalationLegitimate? Pharmacology and Imaging studies in depression’. In het Nederlands: ‘Isdosisverhoging legitiem? Farmacologische en neuroimaging studies bij depressie.
Na de achtergrond wordt een samenvatting gegeven van de belangrijkste resultaten van destudies, de klinische relevantie ervan, gevolgd door een afsluitende conclusie.
Achtergrond en problemen in de behandeling van depressie
Major Depressive Disorder (MDD), in het Nederlands depressieve stoornis, verder aangeduid metdepressie, is een veelvoorkomende en invaliderende ziekte die in potentie recidiveert of verwordttot een chronische aandoening. Depressie staat momenteel wereldwijd op de tweede plaats vaninvaliderende ziekten en komt per jaar bij ±5.5% van de volwassenen voor. Ongeveer 12-14% vanalle mannen en 22-24% van alle vrouwen wordt gedurende hun leven getroffen door depressie.1;2
Depressie gaat gepaard met hoge directe kosten betreffende de behandeling en indirecte kostenals gevolg van verminderde arbeidsproductiviteit en het verlies van kwaliteit van leven. Naastverschillende vormen van psychotherapie wordt farmacotherapie met antidepressiva vaakaangewend als behandeling.3-8
Antidepressiva kunnen worden ingedeeld in vier groepen of ‘klassen’: Selectieve serotonineheropname remmers (SSRIs), tricyclische antidepressiva (TCAs), monoamine oxidase remmers(MAOIs) en een restgroep, waaronder de zogenaamde ‘dual action’ antidepressiva. SSRIs, 'dual-action' antidepressiva en TCAs worden het meest gebruikt.9 De meeste antidepressivabeïnvloeden de balans van verschillende boodschapperstoffen in het brein, de‘neurotransmitters’ serotonine (5-HT) en noradrenaline (zogenaamde monoaminen). Deboodschapperstoffen zijn noodzakelijk voor de communicatie tussen zenuwcellen. Communicatievindt plaats door het uitscheiden van de boodschapperstoffen in de ruimte tussen dezenuwcellen: de synaps. De meeste antidepressiva blokkeren een belangrijk onderdeeltje van dezenuwcel: de serotonine (of noradrenaline) heropname transporter (respectievelijk SERT of NAT),waardoor de hoeveelheid serotonine en/of noradrenaline in de synaps wordt verhoogd. Op dezemanier verhogen de meeste antidepressiva de serotonerge en/of noradrenerge signaaloverdrachtof neurotransmissie. Als dit echter nader wordt bekeken, hebben verschillende groepenantidepressiva ook specifieke, aanvullende effecten op de zenuwcellen; zogenaamde pre- enpost-synaptische effecten (zie hoofdstuk 1, pagina 19).10;11 In reactie op de verhoogde serotonergeen/of noradrenerge signaaloverdracht worden ‘second messenger’ systemen geactiveerd, diemen op dit moment slechts gedeeltelijk begrijpt.12
Het brein is ook een systeem van met elkaar verbonden kernen of schakelcentra, ook welneuronale netwerken genoemd. Behandeling met antidepressiva blijkt aanzienlijke veranderingenin de functie van dergelijke neuronale netwerken te veroorzaken (zie hoofdstuk 1, pagina 21).13-15
Tot op heden is echter onvoldoende duidelijk welke veranderingen noodzakelijk en specifiek zijnom een depressie te genezen. Het lijkt erop dat de effecten van een toegenomensignaaloverdracht gemedieerd worden door de genetische eigenschappen van patiënten(bijvoorbeeld het 5-HTTLPR polymorphisme16) (zie hoofdstuk 1, pagina 22). Er is namelijk eenverband tussen genetische eigenschappen van patiënten en de effecten van behandeling metantidepressiva.17 Ook blijken de ontwikkeling van het brein en verbindingen in de neuronalenetwerken genetisch bepaald te zijn.18

Chap
ter
Chapter 13
13
256
Antidepressiva hebben een vergelijkbare effectiviteit; ongeveer 50% van de depressievepatiënten reageert op het eerste voorgeschreven antidepressivum. Bij slechts een kleinerpercentage van de patiënten wordt volledige remissie van de depressieve klachten bereikt.19;20
Daarom is onvoldoende verbetering op een eerste antidepressivum een groot klinisch probleem.Ten eerste wordt een behandelaar geconfronteerd met de vraag hoe de verandering van
symptomen goed en efficiënt te meten. Om op juiste gronden klinische beslissingen te nemen, isde beschikbaarheid en implementatie van valide, korte en eenvoudige meetinstrumentenbelangrijk. Ten tweede is het de vraag wat het optimale moment is om de gestarteantidepressieve behandeling te evalueren en zonodig te wijzigen: aangezien antidepressiva enigeweken tijd nodig hebben om een verbetering te bewerkstelligen, moet dit niet te vroeg, maar ookniet te laat plaatsvinden.21
Als een patiënt geen verbetering (veelal gedefinieerd als 50% verbetering van de klachten;‘respons’) ervaart op de gestarte SSRI, zijn er vijf farmacologische strategieën: 1) nog enkeleweken ongewijzigd voortzetten van de aanvankelijke behandeling, 2) dosisverhoging, 3) hetveranderen of ‘switchen’ van het antidepressivum, 4) het toevoegen van een middel dat dewerking van het antidepressivum versterkt (augmentatie) en 5) het combineren van verschillendeantidepressiva. Over al deze strategieën is de wetenschappelijke kennis beperkt. Om goede‘evidence-based’ behandelalgoritmen te ontwikkelen zijn goede systematischeliteratuuroverzichten noodzakelijk. Naast het overzicht over de effectiviteit van behandelingen,verduidelijken deze overzichten ook waar zich lacunes in de kennis over de behandeling metantidepressiva bevinden. Deze lacunes kunnen in nieuwe onderzoeksprojecten wordenbestudeerd.
De verlate start van de werking van antidepressiva, de beperkte werkzaamheid vanantidepressiva en de verscheidenheid aan farmacologische effecten in het brein, roepen de meerfundamentele vraag op wat het pathofysiologische model voor depressie is. Meer specifiek kanmen zich afvragen: wat is precies de onderbouwing van het uitgangspunt van de werking van demeeste antidepressiva: de monoamine hypothese en de betrokkenheid van het serotonergesysteem als een oorzakelijke verklaring van het ontstaan van depressie?
Samenvatting en conclusies van de studies in dit proefschrift
Dit proefschrift behandelt verschillende vragen:
1 Is een korte, door de behandelaar in te vullen vragenlijst even effectief en precies als degebruikelijke Hamilton depressieschaal (HDRS)?In hoofdstuk 3 heranalyseren wij de behandeluitkomsten van twee antidepressiva/psychotherapie onderzoeken met als doel de evaluatie van de uitkomsten bepaald doorverschillende subschalen van de oorspronkelijke HDRS.22 Conform andere studies,23-26 vondenwij dat de twee 6-items bevattende subschalen van Maier27 en Bech28 overeenkomen met deoriginele 17-items bevattende HDRS. Dit gold voor het meten van de ernst van depressie en degevoeligheid om veranderingen in de tijd te meten. Wij berekenden een conversietabel vanoorspronkelijke HDRS-scores naar de Maier en Bech scores (en vice versa). Ook bepaalden weafkappunten voor remissie bij deze subschalen.29-31 Wij concluderen dat met deze subschalenclinici de ernst en respons van patiënten efficiënter kunnen meten dan met de oorspronkelijkeHDRS.
2 Wat is het wetenschappelijke bewijs voor dosisverhoging als strategie voor patiënten die nietverbeterden op een eerste SSRI?In hoofdstuk 4 beschrijven wij een systematisch literatuuroverzicht van het wetenschappelijkebewijs voor een dosis-respons relatie voor SSRIs bij de behandeling van depressie.32 Wijconcludeerden dat er géén wetenschappelijk bewijs bestaat dat dosisverhoging van SSRIseffectief is in de eerste 4 weken van de behandeling met een standaarddosering. Verder lijktdosisverhoging na 6 weken behandeling met een standaarddosering zelfs minder effectief

ChapterSamenvatting en conclusies
13
257
dan het ongewijzigd doorgebruiken van de oorspronkelijke dosering. Dosisverhoging gaatgepaard met een toename van de bijwerkingen zonder dat patiënten de behandeling stakenvanwege deze bijwerkingen. Vanwege methodologische tekortkomingen concludeerden wijdesondanks dat het wetenschappelijke bewijs voor dosisverhoging noch een duidelijktoegenomen werkzaamheid, noch een duidelijke onwerkzaamheid aantoont. Deze conclusievormde het startpunt voor de DELPHI-studie, beschreven in de hoofdstukken 2 en 7 tot enmet 10.
3 Wat is het wetenschappelijke bewijs voor het ‘switchen’ van antidepressiva als strategie voorpatiënten die niet verbeterden op een eerste SSRI?In hoofdstuk 5 beschrijven wij een systematisch literatuuroverzicht waarin hetwetenschappelijke bewijs voor het wisselen (‘switchen’) van antidepressiva in het geval dat eronvoldoende verbetering van de depressieve klachten optreedt bij het eersteantidepressivum.33 Voor drie gerandomiseerde switch-studies die het switchen naar een 2e
SSRI vergeleken met een switch naar een SNRI (venlafaxine) konden wij een meta-analyseuitvoeren.34-36 Deze meta-analyse liet alleen in de meest rooskleurige analyse een beperktemeerwaarde van het switchen naar venlafaxine als tweede antidepressivum zien. Wijconcluderen dat bij onvoldoende respons op een eerste SSRI, het ‘switchen’ naar elk andermiddel kan plaatsvinden, zonder duidelijke verschillen in werkzaamheid en dus vergelijkbaar ismet de keuze van het eerste middel.
4 Verlaagt de depletie van monoaminerge systemen de stemming van mensen en is dezestemmingsdaling verschillend voor verschillende subgroepen van mensen?In hoofdstuk 6 beschrijven wij een systematisch literatuuroverzicht van hetwetenschappelijke bewijs dat zogenaamde monoamine depletie studies de stemming vanonderzochte proefpersonen verminderen.37 Hierbij voerden wij een meta-analyse uit, zodat deresultaten van 53 kleine depletie studies konden worden samengevat (‘gepooled’). Wijvonden dat serotonerge, noch noradrenerge/dopaminerge depletie een stemmingsdalinggeeft in gezonde controlepersonen, terwijl depletie de stemming van gezondecontrolepersonen met een eerstegraads familielid dat depressief is geweest enigszinsverminderd. Bij medicatievrije patiënten die na een depressie een volledige remissie van hunklachten hebben bereikt vonden we een matige stemmingsdaling. Monoamine depletieveroorzaakt een terugval bij patiënten die na een depressie een volledige remissie van hunklachten hebben bereikt, echter alleen als deze patiënten een serotonerg werkendantidepressivum gebruiken. Bij depressieve patiënten vindt geen verdere stemmingsdalingplaats na depletie.
Wij concluderen dat stemming niet direct is gecorreleerd aan de hoeveelheid serotonine,noradrenaline of dopamine in het brein. Wel lijken monoaminerge systemen een belangrijkerol te spelen in de kwetsbaarheid om depressief te worden.
5 Verschillen depressieve patiënten en gezonde controlepersonen in het aantal serotoninetransporters en is het aantal serotonine transporters gecorreleerd met de ernst van dedepressie?In hoofdstuk 7 onderzoeken we het aantal serotonine transporters (SERTs) in het brein vandepressieve patiënten, zoals gemeten in de eerste (baseline) SPECT scans van de deelnemersaan de DELPHI-SPECT studie. Wij vonden diverse interactie-effecten tussen geslacht enziektestatus met betrekking tot het aantal SERTs in zowel het diencephalon ((hypo)-thalamusregio) als het midbrain (de hersenstam), met daarnaast interactie-effecten tussenroken en ziektestatus (alleen in het diencephalon). Deze interactie-effecten resulteerden ineen lager aantal SERTs bij depressieve mannen maar juist meer SERTs bij depressieve vrouwen(ten opzichte van gezonde controlepersonen). Daarnaast bleek het seizoen waarin de scangemaakt wordt een duidelijke verandering van het aantal SERTs in het midbrain te geven.Deze bevindingen wijzen op complexe effecten van geslacht, roken en het seizoen waarin descan gemaakt wordt op het aantal SERTs bij depressieve patiënten en gezonde controles.Deze effecten moeten in toekomstig onderzoek van het aantal SERTs bij depressievepatiënten versus gezonde controlepersonen worden meegewogen in de analyses.

Chap
ter
Chapter 13
13
258
6 Verandert een veelvoorkomend genetisch polymorfisme van de promotor regio van het genvoor de serotoninereceptor (SLC6A4) de associatie tussen de SERT bezettingsgraad doorparoxetine en de klinische respons?In hoofdstuk 8 onderzoeken wij de relatie tussen klinische respons na 6 weken paroxetinegebruik en de mate van bezetting van het farmacologische doelwit van paroxetine: deserotonine transporter. De mate van bezetting, ofwel bezettingsgraad van de SERT is hierinde verandering van de hoeveelheid gemeten SERTs na 6 weken behandeling ten opzichte vande hoeveelheid gemeten SERTs vóór behandeling (baseline).38 Daarbij onderzoeken we of derelatie tussen klinische respons en bezettingsgraad wordt beïnvloedt door genetischevarianten in het SERT-gen, de 5-HTTLPR polymorfismen.
Wij vonden dat voor alle patiënten samen, de SERT bezettingsgraad niet geassocieerd ismet de klinische respons. Echter, nadat we de patiënten onderverdeelden op basis van hun 5-HTTLPR polymorfisme, vonden we alleen bij patiënten met het (gunstige)17;39 LA/LA genotypeeen significante relatie tussen SERT bezettingsgraad en klinische respons. Wij verklaren onzebevindingen met de hypothese dat patiënten met het 5-HTTLPR LA/LA genotype mogelijk eenmeer flexibel serotonerg systeem bezitten,18 dat beter beïnvloed kan worden doorserotonerge antidepressiva.
7 Is dosisverhoging van paroxetine een effectieve klinische strategie bij onvoldoende responsbij de behandeling van depressie?In hoofdstuk 9 onderzoeken wij dosisverhoging als strategie voor patiënten die onvoldoendeverbeteren op een standaarddosering paroxetine gedurende 6 weken in eengerandomiseerde klinische studie (RCT). Dit onderzoek was een gevolg van de twijfels over deeffectiviteit van dosisverhoging (zie ook hoofdstuk 4).32 Als meest belangrijke aanvullingonderzoeken we in een subgroep van de patiënten of een echte dosisverhoging vanparoxetine de SERT bezettingsgraad meer doet toenemen dan een placebo-verhoging.40
In een interim analyse analyseerden wij 57 gerandomiseerde patiënten en vonden dat ergeen verschil in werkzaamheid bestaat tussen een echte en een placebo dosisverhoging vanparoxetine. De studie werd hierop afgebroken in verband met futiliteit. Als belangrijkstebevinding laat ons onderzoek zien dat een echte dosisverhoging van paroxetine geen groteretoename van de SERT bezettingsgraad geeft dan een placebo dosisverhoging. Dit is teverklaren doordat waarschijnlijk al bij een standaarddosering een plateau van SERT bezettingwordt bereikt. We concluderen, in overeenstemming met eerdere, echter meer twijfelachtigegegevens,41;42 dat dosisverhoging geen meerwaarde heeft in de behandeling van depressie.
8 Wordt de hyperactivatie van de amygdala bij depressie door een behandeling met paroxetinegenormaliseerd?In hoofdstuk 10 onderzoeken we de emotionele respons van het brein op een (negatieve)gezichtentaak in de DELPHI-fMRI substudie.43 We onderzoeken hierin de effecten vanbehandeling met paroxetine op de vaak vastgestelde hyperactivatie van de amygdala* tijdenseen depressie. Ten opzichte van gezonde controlepersonen vonden we bij depressievepatiënten een toegenomen activiteit in beide amygdala’s en de daaraan grenzende regio. Wijvonden aanvankelijk een toename van de activatie van de rechter amygdala na 6 wekenbehandeling, die kon worden toegeschreven aan de patiënten (12/20) die onvoldoendeverbeterden (non-responders). Deze toegenomen amygdala-activatie nam af in de periode totweek 12. Vooral bij de patiënten die in week 6 en/of week 12 ≥50% verbetering van hunklachten ervaren, nam de amygdala-activatie beiderzijds af, terwijl de amygdala-activatie inweek 6 en 12 geassocieerd was met de HDRS-score. Patiënten met onvoldoende responshadden meer activatie in andere emotie ‘genererende’ (ventrale; zie hoofdstuk 1, pagina 18)gebieden, terwijl responderende patiënten meer activatie toonde in emotie ‘regulerende’(dorsale; zie hoofdstuk 1, pagina 18) gebieden. We concluderen dat, in overeenstemming meteerdere studies,44-47 12 weken behandeling met paroxetine de amygdala en andere emotie
* De amygdala is een amandelvormig onderdeel van de hersenen, dat zich zowel in de linker als rechter hersenhelft bevindt,en onder andere betrokken is bij automatische reacties op angstige situaties.

ChapterSamenvatting en conclusies
13
259
genererende gebieden minder actief doet zijn en dat de corticale, regulerende functieverbeterd. Dit gebeurt vooral bij patiënten die een klinische respons ervaren. Dezebevindingen wijzen op een toegenomen frontaal-limbische controle als werkingsmechanismevoor paroxetine geïnduceerde behandeleffecten (zie hoofdstuk 1, pagina 21).
9 Hoe veranderen de haemostase en stollingsparameters in bloedplaatjes gedurende eenbehandeling met paroxetine en worden deze veranderingen gemedieerd doordosisverhoging of een genetisch polymorfisme van de promotor regio van het serotoninetransporter gen?In hoofdstuk 11 beschrijven we de resultaten van een weinig voorkomende, maar potentieelgevaarlijke bijwerking van SSRIs: toegenomen bloedingsneiging.48 We vonden dat eenstandaard dosis paroxetine (20 mg/dag) al een significante daling van serotonine en β-thromboglobuline (β-TG) in de plaatjes veroorzaakt, zonder dat dit verder afneemt bijdosisverhoging. Bovendien vonden we dat het 5-HTTLPR polymorfisme deze effectenmedieert: ten opzichte van LA/LA-dragers, neemt de bloedingstijd significant toe in patiëntenmet <2LA-allelen en hadden patiënten zonder een LA-allel, sterkere veranderingen in plaatjesaggregatie gerelateerde parameters. Deze bevindingen zijn van belang voor patiënten meteen eerdere ernstige bloeding; bij hen kan genotypering overwogen worden, waarbij bij eenS’/S’ genotype het starten van, of het switchen naar een niet-serotonerg antidepressivummoet worden overwogen.
Klinische relevantie
Switchen Ondanks een klein voordeel van een eventuele switch naar venlafaxine in plaats van een tweedeSSRI (slechts in de meest gunstige analyse), concluderen wij dat de switch-opties na een eersteSSRI identiek zijn aan de keuze voor het eerste middel. Op basis van de literatuur kunnen geenduidelijke aanbevelingen worden gegeven voor een keuze na een eerste SSRI. Sinds onssystematische literatuuroverzicht verschenen er nog één nieuwe RCT die switchen naarvenlafaxine vergeleek met switchen naar citalopram,49 en drie open-label studies.50-52
Recent werd er een andere meta-analyse gepubliceerd, waarin een niet-SSRI switch(venlafaxine, mirtazapine of buproprion) werd vergeleken met switchen naar een tweede SSRI.53
In deze meta-analyse includeerdende auteurs 2 extra studies (abstracts van symposia)54;55 enexcludeerdenzij (net als wij in onze meest rooskleurige analyse34) een methodologischtwijfelachtige studie. Deze meta-analyse concludeert dat er bij switch naar venlafaxine eenstatistisch significant, doch klinisch niet relevant voordeel bestaat in het aantal patiënten dat eenremissie van de klachten bereikt.5;56 Voor het uitkomstcriterium respons werd geen significantverschil vastgesteld.53 Onze conclusie blijft met deze aanvullende bevindingen ongewijzigd; er lijktvoor individuele patiënten geen duidelijk verschil tussen de verschillende switch-opties tebestaan.
Dosisverhoging Na ons systematische literatuuroverzicht concluderen wij dat er geen overtuigend bewijs tenfaveure of tegen dosisverhoging bestaat. Een onafhankelijke onderzoeksgroep concludeerdehetzelfde op basis van een ander overzichtsartikel.41 De resultaten van onzedosisverhogingsstudie (hoofdstuk 9) verdisconteerde de methodologische beperkingen vaneerdere studies en werd vanwege futiliteit afgebroken na een interim-analyse.40 Hoewel hetaantal gerandomiseerde patiënten beperkt was, waren de verschillen tussen echte en placebodosisverhoging zodanig klein (en niet significant) dat de studie voortijdig moest wordenbeëindigd vanwege futiliteit. Dit houdt in dat het gevonden verschil in HDRS-scores kleiner wasdan een vooraf berekend noodzakelijk verschil om nog enige kans op een daadwerkelijk verschilte verwachten, indien het onderzoek voltooid zou worden conform de oorspronkelijke planning.

Chap
ter
Chapter 13
13
260
Zodoende beschermde deze interim-analysepatiënten om blootgesteld te worden aan eeninterventie waarvan op dat moment bijna 100% zeker was dat er uiteindelijk geen meerwaarde vanzou worden vastgesteld. Deze uitkomst is in overeenstemming met het eerdere onderzoek vanLicht et al.57 Helaas maakt het voortijdig staken van onze studie het onmogelijk om eventuelesubgroepen te identificeren waarin dosisverhoging wellicht toch effectief is (bijvoorbeeld depatiënten die gedeeltelijk (partieel) verbeterden).58;59
De substudie waarin wij de verandering in de SERT bezettingsgraad bepaalden bij patiëntendie een dosisverhoging kregen, versterkt de hierboven beschreven klinische bevindingen. Wijconcluderen op basis van ons systematisch literatuuroverzicht, onze RCT en onze imaging studie,dat dosisverhoging van SSRIs geen effectieve strategie is voor depressieve patiënten die na 6weken onvoldoende verbetering ervaren op een standaarddosering.
Een recente meta-analyse van 9 placebo-gecontroleerde, ‘fixed-dose’ studies, gericht op hetvinden van de optimale dosering (‘dose-finding’) van SSRIs,60 concludeerde dat er een gering,maar statistisch significant voordeel was van hogere doseringen van citalopram, escitalopram,fluoxetine, fluvoxamine, paroxetine en/of sertraline, gegeven vanaf het begin van de behandeling(4% meer patiënten met een respons; p= 0.04). Het kleine verschil wordt echter als klinisch nietrelevant beoordeeld. De auteurs konden niet aangeven dat de significantie van de resultaten aanéén van de SSRIs kon worden toegeschreven. Een nadere inspectie van hun gegevens laat in iedergeval geen enkel voordeel van hogere doseringen paroxetine zien, maar wel een duidelijk grotereuitval vanwege bijwerkingen (16.5% ten opzichte van 9.8% bij standaarddoseringen). Daarom blijfthet volgens ons dubieus of hogere doseringen SSRIs vanaf het begin van de behandeling zinvolzijn bij depressieve patiënten. Het zou zelfs zo kunnen zijn dat hogere doseringen mensen eerderdoen stoppen vanwege bijwerkingen, zodat een adequate, langerdurende blootstelling aan (quabijwerkingen acceptabele) standaarddoseringen voorkomen wordt.
Andere klinisch relevante resultaten Twee andere relevante bevindingen komen in dit proefschrift naar voren. Ten eerste blijkt dat deMaier en Bech subschalen resultaten geven die overeenkomen met de volledige HDRS17.22 Andereauteurs suggereren dat deze subschalen beter de kernsymptomen van depressie meten en dathet gebruik van subschalen zelfs efficiëntere metingen geeft23;24 vanwege de afname invariantie.61 De Maier of Bech subschalen kunnen dus gebruikt worden in de klinische beoordelingvan (de behandeling van) depressieve patiënten, naast zelfinvullijsten als de ‘Inventory fordepressive symptomatolgy self-rated’ (IDS-SR) of de ‘Beck depression inventory’ (BDI). Dit isvooral belangrijk bij mensen die zelfinvullijsten niet betrouwbaar kunnen invullen (bijvoorbeeld bijpsychotische kenmerken of bij ernstige cognitieve beperkingen).
Ten tweede verdient de verhoogde bloedingsneiging bij SSRI-gebruik meer aandacht.48 Ditwas al bekend vooral in combinatie met NSAIDs.62-67 De standaard van het Nederlands HuisartsGenootschap adviseert een TCA als de patiënt tevens NSAIDs gebruikt,4 hetgeen volgens ons nietgeheel correct is, aangezien een serotonerg werkend TCA als clomipramine vermoedelijkvergelijkbare problemen geeft. Vanwege de relatief lage incidentie van potentieellevensbedreigende gastro-intestinale bloedingen (4.3/1000 SSRI behandeljaren; 14.5/1000SSRI+NSAID/aspirine behandeljaren; ten opzichte van 1.2/1000/jaar in onbehandeldevolwassenen),65 lijkt de toegenomen bloedingsneiging bij SSRIs irrelevant bij patiënten die geeneerdere ernstige bloedingsincidenten hebben doorgemaakt, of een trauma of operatiemeemaken. Echter, bij patiënten met anamnestisch een eerder ernstig bloedingsincident, of bijpatiënten die een electieve operatie zullen ondergaan en wellicht ook bij patiënten die regelmatigNSAIDs gebruiken, is genotypering van het 5-HTTLPR een overweging. Bij het vaststellen van hetS’/S’ genotype zou dan een switch naar een niet-serotonerg werkend antidepressivum kunnenworden geadviseerd. De (kosten-) effectiviteit van deze benaderingswijze dient nog wel teworden onderzocht.

ChapterSamenvatting en conclusies
13
261
Neurobiologie van de behandeling van depressie
Met ons onderzoek onderzochten we ook de neurobiologie van depressiebehandeling metparoxetine in vivo. De algemene beschouwing omtrent dit deel van het proefschrift vraagtaanzienlijke uitleg en enige voorkennis, welke gedetailleerder uiteen wordt gezet in deEngelstalige ‘general discussion’.
Met de SPECT-scans gedurende de gerandomiseerde, dubbelblinde dosisverhogings fase40
laten wij als eersten ondubbelzinnig zien dat er bij dosisverhoging geen sprake is van een stijgingvan de SERT-bezettingsgraad. Dit kon op basis van eventuele vertekening in eerder onderzoekniet zo eenduidig worden geconcludeerd.68-80 Bovendien wijzen onze gegevens op een matigerelatie tussen bezettingsgraad en klinische respons, in tegenstelling tot een eerder gepostuleerde80% bezettingsgraad die hiervoor vereist zou zijn.38;81 De feiten dat de relatie tussen bezetting enklinische respons alleen lijkt te bestaan voor dragers van het LA/LA genotype van het 5-HTTLPRgen en dat datzelfde genotype de anatomie en connectiviteit van emotiegerelateerde netwerkenbeïnvloedt,18 wijzen erop dat de (heilzame) effecten van een verhoogde serotonergesignaaloverdracht waarschijnlijk afhankelijk zijn van de ontwikkeling van het serotonergesysteem.
Een toename van serotonine geeft in het algemeen een beter controle of beteugeling van deinformatieverwerking in het brein,82 met 1) een vermindering van de kans op doorschieten in eenreactie van andere systemen (bijvoorbeeld het dopaminesysteem) en 2) een verminderdegevoeligheid voor verstorende in- en externe invloeden op het brein. Deze effecten worden metdepletie van serotonine omgekeerd beïnvloedt. Daarnaast beïnvloedt serotonine de aanmaak van‘brain derived neurotrophic factor’ (BDNF),83 een eiwit dat betrokken is bij neuroplasticiteit. Onzeen andere fMRI-studies kunnen in dat licht worden geïnterpreteerd. Bij responders verbeterde decognitieve controle en nam de emotionele hyperactivatie af.43 Uit een eerder onderzoek bleek datbehandeling met sertraline een toename veroorzaakte in de functionele koppeling tussen deamygdala en de frontale en (pregenuale) cingulaire cortex,84 respectievelijk emotionele encontrolerende, cognitieve onderdelen van het limbische-subcortico-corticale netwerk. Mogelelijkis deze toegenomen functionele koppeling een gevolg van de verhoogde serotonergeneurotransmissie, mogelijk het gevolg van een toename van BDNF, of beide. Dit dient nog verderonderzocht te worden.
Beperkingen van het onderzoek
In ieder hoofdstuk en de Engelstalige ‘general discussion’ zijn de mogelijke beperkingen van destudies van dit proefschrift afzonderlijk beschreven. De belangrijkste hiervan worden in dezeNederlandse samenvatting aangestipt.
Voor de SPECT-studies gebruikten wijeen niet-selectieve radioligand ([123I]β-CIT), die ook bindtaan dopamine transporters (bijvoorbeeld in de substantia nigra) en noradrenaline transporters(bijvoorbeeld in de locus coeruleus).85-88 Inmiddels zijn er selectieve SERT liganden zoals [11C]DASBvoor PET of [123I]ADAM voor SPECT beschikbaar. Eerdere beeldvormende onderzoeken in apenlieten zien dat de opname van [123I]β-CIT in het midbrain en diencephalon voornamelijk bindingaan SERTs vertegenwoordigt.89 Wij menen daarom dat onze bevindingen in het diencephalon enmidbrain vooral de SERT-bezettingsgraad weergeven. Tevens reduceert het gebruik van deSPECT-scans ten tijde van randomisatie (als een uitgangspunt voor de veranderingen doordosisverhoging) een mogelijke vertekening door de niet-selectieve ligand. Uiteraard zou intoekomstig onderzoek het gebruik van selectieve liganden ([11C]DASB of [123I]ADAM) overwogenmoeten worden en is het in meerdere opzichten een uitdaging om ons onderzoek met dergelijkeliganden te herhalen.

Chap
ter
Chapter 13
13
262
In het case-control onderzoek naar de beschikbaarheid van SERTs bij depressieve patiëntenversus gezonde proefpersonen bleven kleine subgroepen over bij het onderzoeken van effect-mediërende variabelen. Daarom hebben wij deze verschillen als exploratieve analysesgepresenteerd. Ten aanzien van de associatie tussen SERT bezettingsgraad en klinische responsvergeleken wij ook kleine groepen, maar vonden wij statistisch sterk significante verbanden diewij daarom als valide beschouwen. Om toevallige bevindingen uit te sluiten is replicatie van dezebevindingen wenselijk. Na de interim-analyse staakten wij onze vergelijkende studie vanwegefutiliteit, wat verdere analyse van subgroepen niet toeliet. De fMRI-studie was een voorneuroimaging vrij grote studie, waarin wij robuuste effecten vonden. Echter ook hierbij was hetaantal patiënten in de dosisverhogingsgroep beperkt voor het onderzoeken van drie-weginteracties tussen dosisverhoging, tijd en respons.
Ons neurobiologische onderzoek onderzocht geen van de specifieke secundaire effecten vanSSRIs. Er zijn vele adaptieve pre- en post-synaptische effecten van SSRIs bekend,10;83;90-97 dezewerden meestal bij knaagdieren onderzocht.98 De maximale stralingsbelasting voorwetenschappelijk onderzoek liet geen ander scanonderzoek met radioliganden toe, evenminonderzochten wij de effecten van dosisverhoging in diermodellen. Vooropgesteld moet echterblijven, dat de klinische effecten zowel in ons, als in eerder onderzoek geen effectiviteit vandosisverhoging laten zien. Aanvullend dierexperimenteel onderzoek zou daarom vooral meerfundamentele kennis over het werkingsmechanisme van SSRIs en de neurobiologie vandosisverhoging geven.
Tenslotte kunnen clinici zich bij het lezen van het onderzoek in dit proefschrift af gaan vragenwaarom initieel hogere doseringen van SSRIs effectief zijn gebleken in (sommige) ‘fixed-dose,dose-finding’ studies bij patiënten met een angststoornis,99-102 en vooral bij de obsessiefcompulsieve stoornis (OCD).103-107 Dit vormt een intrigerend vraagstuk dat buiten het kader van ditproefschrift valt, maar een vergelijkbare aanpak als in ons onderzoek rechtvaardigt.
Methodologisch sterke kenmerken van het onderzoek
Dit proefschrift is gebaseerd op een relevant klinisch vraagstuk. Alle behandelaren kennen hetprobleem dat patiënten onvoldoende verbeteren op een eerste SSRI en dosisverhoging is bijnaaltijd de eerste stap, gevolgd door een switch van antidepressivum.108-112 Dit proefschrift kandaarom relevant zijn voor toekomstige richtlijnen.
Onze systematische literatuuroverzichten werden uitgevoerd conform de strenge richtlijnenvan de Cochrane Collaboration.113 Wij voerden ‘sensitieve’ zoekacties van de literatuur, zodat dekans op het missen van waardevolle studies geminimaliseerd werd. Bovendien werden allegevonden studies kritisch beoordeeld en samengevat, waarbij wij alleen een meta-analyseuitvoerden als de klinische homogeniteit dit toeliet. In onze meta-analyse van de monoaminedepletiestudies hanteerden wij bovendien het statistische probleem van het ‘poolen’ van ‘cross-over’ studies.37
De empirische studies uit dit proefschrift werden zorgvuldig opgezet, waarbij demethodologische problemen van eerdere dosisverhogingsstudies werden voorkomen.32;114
Hiertoe randomiseerden wij patiënten pas na 6 weken behandeling, zodat de kans op een verlaterespons werd verminderd. Ook leidde ons dosisverhogingsschema niet tot een toename van deuitval (door bijwerkingen) in de dosisverhogingsgroep. Tenslotte was onze SPECT-studie die heteffect op de SERT bezettingsgraad kwantificeerde uniek en gaf dit een duidelijke rationalewaarom dosisverhoging niet werkzaam is.
Met het uitvoeren van de fMRI-studieonderzochten we de effecten op de emotieregulatie alsgevolg was van de behandeling met paroxetine. Dit was de zesde fMRI studie die debehandeleffecten van SSRIs bij depressie onderzocht. Het feit dat wij 3 scans maakten en dat veelpatiënten initieel niet voldoende verbeterden gaf ons de mogelijkheid om het verschil inhersenactivatie tussen responders en non-responders bij paroxetinebehandeling te onderzoeken.Dit is een belangrijke aanvulling op de bestaande literatuur.

ChapterSamenvatting en conclusies
13
263
Verder keken wij ook naar de perifere effecten van paroxetine, buiten het brein, maar methetzelfde mechanisme: de blokkade van de opname van serotonine in de plaatjes en de effectendaarvan op de bloedstolling. Toegenomen bloedingsneiging is een infrequente, maar potentieelgevaarlijk complicatie. Wij lieten zien, dat ook de standaarddosering van paroxetine invloed kanhebben op de haemostase, vooral in de subgroep van patiënten die drager zijn van het S’/S’polymorfisme van het 5-HTTLPR gen. Deze groep is mogelijk ook gebaat bij secundaire preventievan toekomstige bloedingsproblemen bij het gaan of blijven gebruiken van SSRIs.
Toekomstig onderzoek
De studies in dit proefschrift stimuleren om nieuwe studies en systematischeliteratuuroverzichten te starten. Een aantal onderdelen van de DELPHI-studie dienen noguitgewerkt te worden, terwijl andere vraagstukken met deze studies onbeantwoord zullenblijven. In de Engelstalige ‘general discussion’ wordt een aanzet gegeven voor toekomstigestudies, die zich met name zullen moeten richten op de vergelijking van verschillende strategiëenvoor onvoldoende respons, en het meer fundamentele begrip van de dwarsverbanden tussenverschillende neurobiologische systemen. Hierbij valt te denken aan een RCT waarin switchen na6 weken wordt vergeleken met 6 weken doorbehandelen, onderzoek bij depressieve patiëntennaar de beïnvloeding van het dopaminerge systeem door SSRIs, en de effecten van antidepressivaop de neurogenese.
Conclusie
Dit proefschrift heeft betrekking op de behandeling van depressie met antidepressiva en vooralop het vraagstuk wat te doen als patiënten niet verbeteren op een standaarddosering. Op basisvan twee systematische literatuuroverzichten concluderen wij dat er onvoldoendewetenschappelijk bewijs bestaat voor een duidelijke aanbeveling over het switchen naar eenander antidepressivum en het verhogen van de dosering. In de hierop volgende gerandomiseerdeplacebo-gecontroleerde studie (de DELPHI-studie) onderzochten wij de klinische werkzaamheidvan dosisverhogingen bij depressieve patiënten die onvoldoende waren verbeterd na 6 wekenparoxetine gebruik (20 mg/dag). Daarnaast onderzochten we in een subgroep deneurobiologische effecten van dosisverhoging in SPECT en fMRI neuroimaging studies. Met deDELPHI-studie tonen we aan dat dosisverhoging bij paroxetine geen meerwaarde heeft tenopzichte van een placebo-verhoging. Bovendien kwantificeren we dat dosisverhoging deblokkade van het farmacologische doel van paroxetine, de SERT, niet doet toenemen.
Eveneens onderzochten wij in dit proefschrift de rol van monoaminen en de SERT in deetiologie van depressie en de neurobiologische effecten van behandeling met paroxetine. Opbasis van deze studies concluderen wij dat depressie niet noodzakelijkerwijs veroorzaakt wordtdoor een slecht werkend serotonerg systeem. Depressie kan beter worden gezien als eenverstoring van de balans tussen het emotionele en cognitieve functioneren van het limbisch-subcortico-corticale netwerk. De toename van de serotonergeneurotransmissie als gevolg vanbehandeling met paroxetine (en waarschijnlijk andere SSRIs) lijkt te liggen in een beterebeteugeling van de neuronale verwerking van informatie. Patiënten die verbeteren na debehandeling laten een afname van de emotionele activiteit (bijv. in de amygdala) en een toenamevan de cognitieve controle zien. De bevindingen van dit onderzoek kunnen dienen alsuitgangspunt voor het verder ontwikkelen van biomarkers voor behandel-uitkomsten, met alsdoel het ontwikkelen van een beter begrip van het ziektebeeld depressie en effectieverebehandelingen.

Chap
ter
Chapter 13
13
264
Referenties
1. Bijl RV, Zessen Gv, Ravelli A. Psychiatrische morbiditeit onder volwassenen in Nederland: het NEMESIS-onderzoek. II.Prevalentie van psychiatrische stoornissen. NedTijdschr Geneesk. 1997; 141: 2453-2460.
2. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006; 3: e442.3. Richtlijn Depressie. Multidisciplinaire Richtlijn Depressie. Richtlijn voor de diagnostiek en behandeling van volwassen
cliënten met een depressie. Brandt-Dominicus JC.2005. Utrecht, Trimbos-instituut, in opdracht van de LandelijkeStuurgroep Multidisciplinaire Richtlijnontwikkeling in de GGZ.
4. van Marwijk HW, Grundmeijer HG, Bijl D, van Gelderen MG, de Haan M, van Weel-Baumgarten EM et al. NHG-StandaardDepressieve stoornis (depressie) (eerste herziening). Huisarts Wet. 2003; 46: 614-623.
5. NICE. Clinical Guideline 23. Depression: management of depression in primary and secondary care. December 2004. 23, 1-63. 2004. London, National Institute for Clinical Excellence.
6. Kennedy SH, Lam RW, Cohen NL, Ravindran AV, CANMAT Depression Work Group. Clinical guidelines for the treatment ofdepressive disorders. IV. Medications and other biological treatments. Can J Psychiatry. 2001; 46 Suppl 1: 38S-58S.
7. American Psychiatric Association. Practice guideline for the treatment of patients with major depressive disorder(revision). American Psychiatric Association. Am J Psychiatry. 2000; 157: 1-45.
8. Anderson IM, Nutt DJ, Deakin JFW. Evidence-based guidelines for treating depressive disorders with antidepressants: Arevision of the 1993 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2000; 14: 3-20.
9. Ruhe HG, Rijswijk Ev, Verkes RJ. Biologische interventies bij depressie. In: Huyser J, Schene AH, Sabbe B, Spinhoven Ph,eds. Handboek depressieve stoornissen. Utrecht: De Tijdstroom; 2008:201-218.
10. Blier P, De Montigny C. Current advances and trends in the treatment of depression. Trends Pharmacol Sci. 1994; 15: 220-226.
11. Nemeroff CB, Owens MJ. Neuropharmacology of paroxetine. Psychopharmacol Bull. 2003; 37 Suppl 1: 8-18.12. Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008; 358: 55-68.13. Malhi GS, Lagopoulos J. Making sense of neuroimaging in psychiatry. Acta Psychiatr Scand. 2008; 117: 100-117.14. Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based
algorithms for diagnosis and optimised treatment. Br Med Bull. 2003; 65: 193-207.15. Phillips ML, Drevets WC, Rauch SL, Lane R. Neurobiology of emotion perception II: Implications for major psychiatric
disorders. Biol Psychiatry. 2003; 54: 515-528.16. Hu XZ, Lipsky RH, Zhu G, Akhtar LA, Taubman J, Greenberg BD et al. Serotonin transporter promoter gain-of-function
genotypes are linked to obsessive-compulsive disorder. Am J Hum Genet. 2006; 78: 815-826.17. Serretti A, Kato M, De Ronchi D, Kinoshita T. Meta-analysis of serotonin transporter gene promoter polymorphism (5-
HTTLPR) association with selective serotonin reuptake inhibitor efficacy in depressed patients. Mol Psychiatry. 2007; 12:247-257.
18. Pezawas L, Meyer-Lindenberg A, Drabant EM, Verchinski BA, Munoz KE, Kolachana BS et al. 5-HTTLPR polymorphismimpacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat Neurosci. 2005;8: 828-834.
19. Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L et al. Evaluation of outcomes with citalopram fordepression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006; 163: 28-40.
20. Thase ME. Evaluating antidepressant therapies: remission as the optimal outcome. J Clin Psychiatry. 2003; 64 Suppl 13: 18-25.
21. Quitkin FM, Petkova E, McGrath PJ, Taylor B, Beasley C, Stewart J et al. When should a trial of fluoxetine for majordepression be declared failed? Am J Psychiatry. 2003; 160: 734-740.
22. Ruhe HG, Dekker JJ, Peen J, Holman R, de Jonghe F. Clinical use of the Hamilton Depression Rating Scale: is increasedefficiency possible? A post hoc comparison of Hamilton Depression Rating Scale, Maier and Bech subscales, ClinicalGlobal Impression, and Symptom Checklist-90 scores. Compr Psychiatry. 2005; 46: 417-427.
23. Faries D, Herrera J, Rayamajhi J, DeBrota D, Demitrack M, Potter WZ. The responsiveness of the Hamilton DepressionRating Scale. J Psychiatr Res. 2000; 34: 3-10.
24. Entsuah R, Shaffer M, Zhang J. A critical examination of the sensitivity of unidimensional subscales derived from theHamilton Depression Rating Scale to antidepressant drug effects. J Psychiatr Res. 2002; 36: 437-448.
25. O'Sullivan RL, Fava M, Agustin C, Baer L, Rosenbaum JF. Sensitivity of the six-item Hamilton Depression Rating Scale. ActaPsychiatr Scand. 1997; 95: 379-384.
26. Hooper CL, Bakish D. An examination of the sensitivity of the six-item Hamilton Rating Scale for Depression in a sample ofpatients suffering from major depressive disorder. J Psychiatry Neurosci. 2000; 25: 178-184.
27. Maier W, Philipp M. Improving the assessment of severity of depressive states: a reduction of the Hamilton DepressionScale. Pharmacopsychiatry. 1985; 18: 114-115.
28. Bech P, Gram LF, Dein E, Jacobsen O, Vitger J, Bolwig TG. Quantitative rating of depressive states. Acta Psychiatr Scand.1975; 51: 161-170.
29. Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW et al. Conceptualization and rationale for consensusdefinitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48: 851-855.
30. Prien RF, Carpenter LL, Kupfer DJ. The definition and operational criteria for treatment outcome of major depressivedisorder. A review of the current research literature. Arch Gen Psychiatry. 1991; 48: 796-800.
31. Ballenger JC. Clinical guidelines for establishing remission in patients with depression and anxiety. J Clin Psychiatry. 1999;60 Suppl 22: 29-34.

ChapterSamenvatting en conclusies
13
265
32. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard-dose selectiveserotonin reuptake inhibitors in major depressive disorder: Systematic review. Br J Psychiatry. 2006; 189: 309-316.
33. Ruhe HG, Huyser J, Swinkels JA, Schene AH. Switching antidepressants after a first selective serotonin reuptake inhibitorin major depressive disorder: a systematic review. J Clin Psychiatry. 2006; 67: 1836-1855.
34. Baldomero EB, Ubago JG, Cercos CL, Ruiloba JV, Calvo CG, Lopez RP. Venlafaxine extended release versus conventionalantidepressants in the remission of depressive disorders after previous antidepressant failure: ARGOS study. DepressAnxiety. 2005; 22: 68-76.
35. Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME et al. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006; 354: 1231-1242.
36. Poirier MF, Boyer P. Venlafaxine and paroxetine in treatment-resistant depression. Double-blind, randomisedcomparison. Br J Psychiatry. 1999; 175: 12-16.
37. Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans:a meta-analysis of monoamine depletion studies. Mol Psychiatry. 2007; 12: 331-359.
38. Ruhé HG, Ooteman W, Booij J, Michel MC, Moeton M, Baas F et al. Serotonin transporter gene promoter polymorphismsmodify the association between paroxetine serotonin transporter occupancy and clinical response in major depressivedisorder. Pharmacogenetics and Genomics. 2008; [In press].
39. Smits KM, Smits LJ, Schouten JS, Stelma FF, Nelemans P, Prins MH. Influence of SERTPR and STin2 in the serotonintransporter gene on the effect of selective serotonin reuptake inhibitors in depression: a systematic review. MolPsychiatry. 2004; 9: 433-441.
40. Ruhe HG, Booij J, Weert HCv, Reitsma JB, Fransen EJF, Michel MC et al. Evidence why dose-escalation of paroxetine inmajor depressive disorder is not effective: A 6-week, randomized-controlled trial with assessment of serotonintransporter occupancy. Neuropsychopharmacology. 2008; [In Press].
41. Adli M, Baethge C, Heinz A, Langlitz N, Bauer M. Is dose escalation of antidepressants a rational strategy after a medium-dose treatment has failed? A systematic review. Eur Arch Psychiatry Clin Neurosci. 2005; 255: 387-400.
42. Baker CB, Tweedie R, Duval S, Woods SW. Evidence that the SSRI dose response in treating major depression should bereassessed: a meta-analysis. Depress Anxiety. 2003; 17: 1-9.
43. Ruhe HG, Booij J, Veltman DJ, Michel MC, Schene AH. Successful treatment of major depressive disorder by paroxetineattenuates amygdala activation to negative facial expressions. An fMRI study. 2008; [Submitted].
44. Davidson RJ, Irwin W, Anderle MJ, Kalin NH. The neural substrates of affective processing in depressed patients treatedwith venlafaxine. Am J Psychiatry. 2003; 160: 64-75.
45. Fu CH, Williams SC, Cleare AJ, Brammer MJ, Walsh ND, Kim J et al. Attenuation of the neural response to sad faces inmajor depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imagingstudy. Arch Gen Psychiatry. 2004; 61: 877-889.
46. Schaefer HS, Putnam KM, Benca RM, Davidson RJ. Event-related functional magnetic resonance imaging measures ofneural activity to positive social stimuli in pre- and post-treatment depression. Biol Psychiatry. 2006; 60: 974-986.
47. Robertson B, Wang L, Diaz MT, Aiello M, Gersing K, Beyer J et al. Effect of bupropion extended release on negativeemotion processing in major depressive disorder: a pilot functional magnetic resonance imaging study. J Clin Psychiatry.2007; 68: 261-267.
48. Abdelmalik N, Ruhé HG, Barwari K, Dool E-Jvd, Meijers JCM, Middeldorp S et al. The effect of the selective serotoninreuptake inhibitor paroxetine on platelet function is modified by a SLC6A4 serotonin transporter polymorphism. JThromb Haemost. 2008; [In Press].
49. Lenox-Smith AJ, Jiang Q. Venlafaxine extended release versus citalopram in patients with depression unresponsive to aselective serotonin reuptake inhibitor. Int Clin Psychopharmacol. 2008; 23: 113-119.
50. Thase ME, Lu Y, Joliat MJ, Detke MJ. Remission in placebocontrolled trials of duloxetine with an SSRI comparator.Presented atthe annual meeting of the American Psychiatric Association, May 2003, San Francisco, CA . 2003.
51. Malhi GS, Ng F, Berk M. Dual-dual action? Combining venlafaxine and mirtazapine in the treatment of depression. Aust N ZJ Psychiatry. 2008; 42: 346-349.
52. Perahia DG, Quail D, Desaiah D, Corruble E, Fava M. Switching to duloxetine from selective serotonin reuptake inhibitorantidepressants: a multicenter trial comparing 2 switching techniques. J Clin Psychiatry. 2008; 69: 95-105.
53. Papakostas GI, Fava M, Thase ME. Treatment of SSRI-resistant depression: a meta-analysis comparing within- versusacross-class switches. Biol Psychiatry. 2008; 63: 699-704.
54. Lenox-Smith AJ, Schaeffer P, Reynolds A, Williard L. A double blind trial of venlafaxine XR versus citalopram in patientswith severe depression. J Psychopharmacol. 2001; 15 Suppl 3: A11.
55. Thase ME, Kremer C, Rodrigues H. Mirtazapine versus sertraline after SSRI nonresponse [Abstract]. American PsychiatricAssociation.2001 annual meeting: New Orleans LA . 2001.
56. Cipriani A, Barbui C, Brambilla P, Furukawa TA, Hotopf M, Geddes JR. Are all antidepressants really the same? The case offluoxetine: a systematic review. J Clin Psychiatry. 2006; 67: 850-864.
57. Licht RW, Qvitzau S. Treatment strategies in patients with major depression not responding to first-line sertralinetreatment: A randomised study of extended duration of treatment, dose increase or mianserin augmentation.Psychopharmacology (Berl). 2002; 161: 143-151.
58. Fava M, Rosenbaum JF, McGrath PJ, Stewart JW, Amsterdam JD, Quitkin et al. Lithium and tricyclic augmentation offluoxetine treatment for resistant major depression: a double-blind, controlled study. Am J Psychiatry. 1994; 151: 1372-1374.
59. Fava M, Alpert J, Nierenberg A, Lagomasino I, Sonawalla S, Tedlow J et al. Double-blind study of high-dose fluoxetineversus lithium or desipramine augmentation of fluoxetine in partial responders and nonresponders to fluoxetine. J ClinPsychopharmacol. 2002; 22: 379-387.

Chap
ter
Chapter 13
13
266
60. Papakostas GI, Charles D, Fava M. Are typical starting doses of the selective serotonin reuptake inhibitors sub-optimal? Ameta-analysis of randomized, double-blind, placebo-controlled, dose-finding studies in major depressive disorder. WorldJ Biol Psychiatry. 2007; 1-8.
61. Santor DA, Coyne JC. Examining symptom expression as a function of symptom severity: Item performance on theHamilton Rating Scale for Depression. Psychol Assess. 2001; 13: 127-139.
62. de Abajo FJ, Montero D, Rodriguez LA, Madurga M. Antidepressants and risk of upper gastrointestinal bleeding. Basic ClinPharmacol Toxicol. 2006; 98: 304-310.
63. Wessinger S, Kaplan M, Choi L, Williams M, Lau C, Sharp L et al. Increased use of selective serotonin reuptake inhibitors inpatients admitted with gastrointestinal haemorrhage: a multicentre retrospective analysis. Aliment Pharmacol Ther.2006; 23: 937-944.
64. Meijer WE, Heerdink ER, Nolen WA, Herings RM, Leufkens HG, Egberts AC. Association of risk of abnormal bleeding withdegree of serotonin reuptake inhibition by antidepressants. Arch Intern Med. 2004; 164: 2367-2370.
65. Dalton SO, Johansen C, Mellemkjaer L, Norgard B, Sorensen HT, Olsen JH. Use of selective serotonin reuptake inhibitorsand risk of upper gastrointestinal tract bleeding: a population-based cohort study. Arch Intern Med. 2003; 163: 59-64.
66. van Walraven C, Mamdani MM, Wells PS, Williams JI. Inhibition of serotonin reuptake by antidepressants and uppergastrointestinal bleeding in elderly patients: retrospective cohort study. BMJ. 2001; 323: 655-658.
67. Serebruany VL. Selective serotonin reuptake inhibitors and increased bleeding risk: are we missing something? Am J Med.2006; 119: 113-116.
68. Kent JM, Coplan JD, Lombardo I, Hwang DR, Huang Y, Mawlawi O et al. Occupancy of brain serotonin transporters duringtreatment with paroxetine in patients with social phobia: a positron emission tomography study with [11C]McN 5652.Psychopharmacology (Berl). 2002; 164: 341-348.
69. Meyer JH, Wilson AA, Ginovart N, Goulding V, Hussey D, Hood K et al. Occupancy of serotonin transporters by paroxetineand citalopram during treatment of depression: a [11C]DASB PET imaging study. Am J Psychiatry. 2001; 158: 1843-1849.
70. Meyer JH, Wilson AA, Sagrati S, Hussey D, Carella A, Potter WZ et al. Serotonin Transporter Occupancy of Five SelectiveSerotonin Reuptake Inhibitors at Different Doses: An [11C]DASB Positron Emission Tomography Study. Am J Psychiatry.2004; 161: 826-835.
71. Parsey RV, Kent JM, Oquendo MA, Richards MC, Pratap M, Cooper TB et al. Acute occupancy of brain serotonintransporter by sertraline as measured by [11C]DASB and positron emission tomography. Biol Psychiatry. 2006; 59: 821-828.
72. Suhara T, Takano A, Sudo Y, Ichimiya T, Inoue M, Yasuno F et al. High levels of serotonin transporter occupancy with low-dose clomipramine in comparative occupancy study with fluvoxamine using positron emission tomography. Arch GenPsychiatry. 2003; 60: 386-391.
73. Erlandsson K, Sivananthan T, Lui D, Spezzi A, Townsend CE, Mu S et al. Measuring SSRI occupancy of SERT using the noveltracer [123I]ADAM: a SPECT validation study. Eur J Nucl Med Mol Imaging. 2005; 32: 1329-1336.
74. Hiltunen J, Akerman KK, Kuikka JT, Bergstrom KA, Halldin C, Nikula T et al. Iodine-123 labeled nor-β-CIT as a potentialtracer for serotonin transporter imaging in the human brain with single-photon emission tomography. Eur J Nucl Med.1998; 25: 19-23.
75. Klein N, Sacher J, Geiss-Granadia T, Mossaheb N, Attarbaschi T, Lanzenberger R et al. Higher serotonin transporteroccupancy after multiple dose administration of escitalopram compared to citalopram: an [123I]ADAM SPECT study.Psychopharmacology (Berl). 2007; 191: 333-339.
76. Klein N, Sacher J, Geiss-Granadia T, Attarbaschi T, Mossaheb N, Lanzenberger R et al. In vivo imaging of serotonintransporter occupancy by means of SPECT and [123I]ADAM in healthy subjects administered different doses ofescitalopram or citalopram. Psychopharmacology (Berl). 2006; 188: 263-272.
77. Takano A, Suzuki K, Kosaka J, Ota M, Nozaki S, Ikoma Y et al. A dose-finding study of duloxetine based on serotonintransporter occupancy. Psychopharmacology (Berl). 2006; 185: 395-399.
78. Catafau AM, Perez V, Plaza P, Pascual JC, Bullich S, Suarez M et al. Serotonin transporter occupancy induced byparoxetine in patients with major depression disorder: a [123I]-ADAM SPECT study. Psychopharmacology (Berl). 2006; 189:145-153.
79. Herold N, Uebelhack K, Franke L, Amthauer H, Luedemann L, Bruhn H et al. Imaging of serotonin transporters and itsblockade by citalopram in patients with major depression using a novel SPECT ligand [123I]-ADAM. J Neural Transm. 2006;113: 659-670.
80. Voineskos AN, Wilson AA, Boovariwala A, Sagrati S, Houle S, Rusjan P et al. Serotonin transporter occupancy of high-doseselective serotonin reuptake inhibitors during major depressive disorder measured with [11C]DASB positron emissiontomography. Psychopharmacology (Berl). 2007; 193: 539-545.
81. Meyer JH. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. JPsychiatry Neurosci. 2007; 32: 86-102.
82. Spoont MR. Modulatory role of serotonin in neural information processing: implications for human psychopathology.Psychol Bull. 1992; 112: 330-350.
83. Martinowich K, Lu B. Interaction between BDNF and serotonin: role in mood disorders. Neuropsychopharmacology. 2008;33: 73-83.
84. Chen CH, Suckling J, Ooi C, Fu CH, Williams SC, Walsh ND et al. Functional Coupling of the Amygdala in Depressed PatientsTreated with Antidepressant Medication. Neuropsychopharmacology. 2008; 33: 1909-1918.
85. Pirker W, Asenbaum S, Hauk M, Kandlhofer S, Tauscher J, Willeit M et al. Imaging serotonin and dopamine transporterswith [123I]β-CIT SPECT: binding kinetics and effects of normal aging. J Nucl Med. 2000; 41: 36-44.
86. Innis R, Baldwin R, Sybirska E, Zea Y, Laruelle M, al Tikriti M et al. Single photon emission computed tomography imagingof monoamine reuptake sites in primate brain with [123I]CIT. Eur J Pharmacol. 1991; 200: 369-370.

ChapterSamenvatting en conclusies
13
267
87. Neumeyer JL, Wang SY, Milius RA, Baldwin RM, Zea-Ponce Y, Hoffer PB et al. [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane: high-affinity SPECT radiotracer of monoamine reuptake sites in brain. J Med Chem. 1991; 34: 3144-3146.
88. Neumeyer JL, Tamagnan G, Wang S, Gao Y, Milius RA, Kula NS et al. N-substituted analogs of 2 beta-carbomethoxy-3 beta-(4'-iodophenyl)tropane (β-CIT) with selective affinity to dopamine or serotonin transporters in rat forebrain. J Med Chem.1996; 39: 543-548.
89. Laruelle M, Baldwin RM, Malison RT, Zea-Ponce Y, Zoghbi SS, al Tikriti MS et al. SPECT imaging of dopamine andserotonin transporters with [123I]β-CIT: pharmacological characterization of brain uptake in nonhuman primates.Synapse. 1993; 13: 295-309.
90. Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG et al. Effects of chronic antidepressanttreatments on serotonin transporter function, density, and mRNA level. J Neurosci. 1999; 19: 10494-10501.
91. Benmansour S, Owens WA, Cecchi M, Morilak DA, Frazer A. Serotonin clearance in vivo is altered to a greater extent byantidepressant-induced downregulation of the serotonin transporter than by acute blockade of this transporter. JNeurosci. 2002; 22: 6766-6772.
92. Blendy JA. The role of CREB in depression and antidepressant treatment. Biol Psychiatry. 2006; 59: 1144-1150.93. Castren E. Is mood chemistry? Nat Rev Neurosci. 2005; 6: 241-246.94. Davidson C, Stamford JA. Effect of chronic paroxetine treatment on 5-HT1B and 5-HT1D autoreceptors in rat dorsal raphe
nucleus. Neurochem Int. 2000; 36: 91-96.95. Djavadian RL. Serotonin and neurogenesis in the hippocampal dentate gyrus of adult mammals. Acta Neurobiol Exp
(Wars). 2004; 64: 189-200.96. Kim SW, Park SY, Hwang O. Up-regulation of tryptophan hydroxylase expression and serotonin synthesis by sertraline.
Mol Pharmacol. 2002; 61: 778-785.97. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat
hippocampus. J Neurosci. 2000; 20: 9104-9110.98. Meyer JH, Kapur S, Eisfeld B, Brown GM, Houle S, DaSilva J et al. The effect of paroxetine on 5-HT(2A) receptors in
depression: an [18F]setoperone PET imaging study. Am J Psychiatry. 2001; 158: 78-85.99. Mavissakalian MR, Ryan MT. Rational treatment of panic disorder with antidepressants. Ann Clin Psychiatry. 1998; 10: 185-
195.100. Rickels K, Zaninelli R, McCafferty J, Bellew K, Iyengar M, Sheehan D. Paroxetine Treatment of Generalized Anxiety
Disorder: A Double-Blind, Placebo-Controlled Study. Am J Psychiatry. 2003; 160: 749-756.101. Practice guideline for the treatment of patients with panic disorder. Work Group on Panic Disorder. American Psychiatric
Association. Am J Psychiatry. 1998; 155: 1-34.102. Richtlijn Angststoornissen. Multidisciplinaire richtlijn Angststoornissen. Richtlijn voor de diagnostiek en behandeling van
volwassen cliënten met een angststoornis. Velde V vd. 2003. Utrecht, Trimbos-instituut, in opdracht van de LandelijkeStuurgroep Multidisciplinaire Richtlijnontwikkeling in de GGZ.
103. Denys D. Pharmacotherapy of obsessive-compulsive disorder and obsessive-compulsive spectrum disorders. PsychiatrClin North Am. 2006; 29: 553-84.
104. Figee M, Denys D. New pharmacotherapeutical approaches to obsessive compulsive disorder. CNS Spectr. 2008; [Inpress].
105. Ninan PT, Koran LM, Kiev A, Davidson JR, Rasmussen SA, Zajecka JM et al. High-dose sertraline strategy fornonresponders to acute treatment for obsessive-compulsive disorder: a multicenter double-blind trial. J Clin Psychiatry.2006; 67: 15-22.
106. Dougherty DD, Rauch SL, Jenike MA. Pharmacotherapy for obsessive-compulsive disorder. J Clin Psychol. 2004; 60: 1195-1202.
107. American Psychiatric Association. Practice guideline for the treatment of patients with obsessive-compulsive disorder.Available online at http//www.psych.org/psych_pract/treatg/pg/prac_ guide.cfm ed. Arlington, VA: American PsychiatricAssociation; 2007:1-96.
108. Wittkampf KA, Ruhé HG, Huyser J, Schene AH. De Creatieve Psychiater? Een onderzoek onder Nederlandse psychiatersnaar de behandeling van depressieve stoornissen bij het falen van de eerste SSRI.2001. Amsterdam, ZorgprogrammaStemmingsstoornissen MFO Psychiatrie AMC/ De Meren.
109. Byrne S, Rothschild AJ. Psychiatrists' responses to failure of maintenance therapy with antidepressants. Psychiatr Serv.1997; 48: 835-837.
110. Fredman SJ, Fava M, Kienke AS, White CN, Nierenberg AA, Rosenbaum JF. Partial response, nonresponse, and relapsewith selective serotonin reuptake inhibitors in major depression: a survey of current "next-step" practices. J ClinPsychiatry. 2000; 61: 403-408.
111. Mischoulon D, Nierenberg AA, Kizilbash L, Rosenbaum JF, Fava M. Strategies for managing depression refractory toselective serotonin reuptake inhibitor treatment: a survey of clinicians. Can J Psychiatry. 2000; 45: 476-481.
112. Shergill SS, Katona CL. Pharmacological choices after one antidepressant fails: a survey of UK psychiatrists. J AffectDisord. 1997; 43: 19-25.
113. Cochrane Collaboration. Preparing, maintaining and promoting the accessibility of systematic reviews of the effects ofhealth care interventions. Internet: http://www.cochrane.org/. 2001. Cochrane Collaboration.
114. Baker CB, Woods SW. Is there a SSRI dose response in treating major depression? The case for re-analysis of current dataand for enhancing future study design. Depress Anxiety. 2003; 17: 10-18.


ABBREVIATIONS, COLOR FIGURES, PUBLICATIONS, ACKNOWLEDGEMENT AND CURRICULUM VITAE
PART
Abbreviations, color figures, publications, acknowledgement and curriculum vitae

270

ABBREVIATIONS

272

List of abbreviations
273
List of abbreviations
[123I]ß-CIT [123I]methyl 3ß-(4-iodophenyl) tropane-2 ß –carboxylate 5-HT serotonin 5-HTTLPR Serotonin transporter promoter polymorphism95% CI 95% confidence interval Β-TG β-thromboglobulinAHCPR Agency of healthcare policy and researchAIC Akaike’s information criterionAMC Academic Medical Center AMPT Alpha-methyl-para-tyrosine ANOVA Analysis of variance model ATD Acute tryptophan depletion APTD Acute phenylalanine/tyrosine depletion AUC Area under the curve BDNF Brain derived neurotrophic factor BDI Beck depression inventoryBOLD Blood oxygen level dependentBPND Binding potential (non-displacable) cAMP Cyclic adenosine monophosphateCCDAN Cochrane collaboration depression anxiety and neurosis groupCER CerebellumCGI-I/S Clinical global impression scale (improvement/severity) COMT Catechol-O-methyltransferase CI95 95% Confidence interval CREB cAMP response element binding protein CYP P450 Cytochrome P450DA Dopamine DAT Dopamine transporter DE Dose-escalationDELPHI Dose-escalation legitimate? Pharmacology and imaging studies in depressionDIENC DiencephalonDLPFC Dorsolateral prefrontal cortexDMPFC Dorsomedial prefrontal cortexDNA Deoxyribonucleic acidDLPFC Dorsolateral prefrontal cortex DSM-IV Diagnostic and statistical manual, 4th edition E-S Effect sizeEEG Electric encephalogramEMEA European agency for the evaluation of medicinal productsEPI Echoplanar imageFDA Food and drug administrationfMRI functional Magnetic Resonance Imaging HC Healthy controlsHDRS Hamilton depression rating scale IDS-C/SR Inventory for depressive symptoms – clinician / self-rated IQR Inter Quartile RangeIRT Item response theoryITT Intention to treatIU International unitsLOCF Last observation was carried forward LoE Level of evidenceMAACL Multiple affect adjective checklistMADRS Montgomery Åsberg depression rating scale MAO(-I) Monoamine oxidase (inhibitor)MHRA (British) Medicines and healthcare products regulatory agencyMID Midbrain MDD Major depressive disorder

List of abbreviations
274
MDMA 3,4-methylenedioxy methamphe tamine (XTC)MNI Montreal Neurological Institute MOS-SF36 Medical outcome study short form (measuring health related quality of life)MRI, sMRI/fMRI Magnetic resonance imaging, structural/functional N/A Not appropriateNA/NE Noradrenaline (=norepinephrine)NAT/NET Nordrenaline transporter NERI Norepineprine reuptake inhbitorsNNT/NNH Number needed to treat/ harm NSAID Non-steroidal anti-inflammatory drugOCC Occupancy OCD Obsessive compulsive disorderOFC Orbitofrontal cortex PCPA Para-chlorophenylalaninePCR Polymerase chain reactionPET Positron emission tomography PFA Platelet function analyzerPF4 Platelet factor 4PFC Prefrontal cortexPOMS Profile of Mood StatesPRP Platelet rich plasmaPSC Paroxetine serum concentrationsPUFAs Poly unsaturated fatty acidsQIDS-SR Quick-inventory for depressive symptoms RCT Randomized controlled trial RIMA Reversible inhibitor of monoamine oxidase A ROC Receiver operator characteristic (curve)RoI Regions of interestRR Relative risk RT Reaction timeSCID Structured clinical interview for DSM-IVSCL Symptoms check list (90 items)SCN Suprachiasmatic nucleusSD Standard deviation SE(M) Standard error (of the mean)SERT Serotonin transporter SLC6A4 Serotonin transporter geneSNP Single nucleotide polymorphismSNRI Serotonin and norepinephrine reuptake inhibitor SPECT Single photon emission computed tomography SPM5 Statistical Parametric Mapping version 5SPSP Short psychodynamic supportive psychotherapySSRI Selective serotonin reuptake inhibitor STAR*D Sequenced treatment alternatives to relieve depressionTCA Tricyclic antidepressant TE Echo time TR Repetition time TRD Treatment-resistant depressionVA(M)S Visual analogue (mood)scalesVLPFC Ventrolateral prefrontal cortex VMPFC Ventromedial prefrontal cortex

COLOR FIGURES

276

Color Figures
277
Figure 1.2. Transporters, receptors and second messenger systems involved in the effects of antidepressants.

Color Figures
278
Figure adapted from Belmaker and Agam.79 The left half of the presynaptic neuron representsa serotonergic neuron, the right half a norepinephrinergic neuron.
In the presynaptic neuron, serotonin is synthesized from tryptophan by tryptophanhydroxylase and stored in vesicles. Likewise, norepinephrine is synthesized from tyrosine bytyrosine hydroxylase. These vesicles merge with the cell membrane when the neuron isdepolarized, thereby releasing their contents into the synaptic cleft.
After release, serotonin and norepinephrine are transported back into the presynaptic neuronby serotonine and norerepinephrine transporters. Furthermore, serotonin and norepinephrineare catabolized by the monoamine-oxidase A (MAO-A) enzyme.
In the synaptic cleft, serotonin and norepinephrine affect both the pre-and post-synapticneuron. The pre-synaptic 5-HT1A and 5-HT1B auto-receptors decrease serotonin release byinhibitory feedback; the α2-adrenergic receptor does the same for the release of norepinephrine.
Post-synaptically, serotonin and norepinephrine bind to G-protein-coupled monoaminereceptors (MARs): the cyclicAMP (cAMP)-coupled receptor, which activates protein kinase A(PKA), and the Phosphatidylinositol (PI)-coupled receptor, which activates phospholipase C (PLC)which thereafter form inositol triphosphate (IP3) and diacylglycerol (DAG). IP3 and DAG activateprotein kinase C (PKC). Both PKA and PKC finally activate cAMP responsive element binding(CREB) protein, which stimulates DNA transcription. For example, this might result in theproduction of brain derived neurotrophic factor (BDNF).

Color Figures
279
DELPHI-SPECT and DELPHI-fMRI represent sub-studies nested within the total study. For these2 sub-studies drug-free patients were recruited and treated in the Academic Medical Center.

Color Figures
280
Figure 2.1. Design of the DELPHI-study.
������������� ����
������ �
������������������
�� ����� �� ����� !" �����
����������������
� �#�$����% #& #� ' &��� �'��(")
�������������
����� ����� ��
����� *���(�+�,���(� ���"������"����(�
�� -.�� �� -.�� !" -.��

Color Figures
281
Figure 2.3. Regions of Interest (RoI) for Midbrain, Cerebellum and Diencephalon.

Color Figures
282
Example of SPECT images after 3D reconstruction. Templates with fixed RoIs are shown ingreen.
A. Midbrain (circle) and cerebellum. B. Striatum (for demarcation midbrain-diencephalon) anddiencephalon (circle). RoIs were positioned by hand and situated based on anatomy andmaximum concentration of activity/ml in the RoI.

Color Figures
283
Negative faces contrast. Week 6 and 12 scans and response status were combined in a fullfactorial model.
A. SPM-results showing design matrix, contrast and activation in right amygdala (MNI 18,-2,-18).
B. Activations of left (MNI -22,2,-18; blue) and right (MNI 18,-2,-18; red) amygdala stratified fornon-response and response at 6 and 12 weeks. Significant difference between non-respondersand responders (F1,32= 4.591; p<0.0001); no significant lateralization (F1,32= 0.1517; p= 0.70).
C. Plot of contrast estimate by HDRS17-score (centralized to mean). Significant positivecorrelations for left (0.33 ±0.12 [SE]; F1,31= 7.076; p= 0.0123; r2= 0.19; shown in blue) and right (0.38±0.14 [SE]; F1,31= 6.929; p= 0.0131; r2= 0.18; shown in red) amygdala. Regression lines for left andright amygdala are not significantly different (F1,62= 0.0683; p= 0.79).

Color Figures
284
Figure 10.1. Amygdala response relative to clinical response (HDRS17 ≥50% decrease) after 6 and 12 weeks of treatment with paroxetine.
����
��
�����������
���������������
���
������������
������ ���
������������������������
���������������
���
������������
����
����
����
����
���� ��������
�����
�����������������
�� �

PUBLICATIONS

286

Publications
287
Papers and chapters
Ruhe HG, Booij J, Weert HCv, Reitsma JB, Fransen EJF, Michel MC, Schene AH. Evidence why dose-escalation of paroxetine in major depressive disorder is not effective: A 6-week, randomized-controlled trial with assessment of serotonin transporter occupancy. Neuropsychopharmacology.2008; [In Press].
Ruhe HG, Booij J, Reitsma JB, Schene AH. Serotonin transporter binding with [123I]ß-CIT SPECT inmajor depressive disorder versus controls: effect of season and gender. 2008; [Submitted].
Abdelmalik N, Ruhé HG, Barwari K, Dool E-Jvd, Meijers JCM, Middeldorp S, Büller HR, Schene AH,Kamphuisen PW. The effect of the selective serotonin reuptake inhibitor paroxetine on plateletfunction is modified by a SLC6A4 serotonin transporter polymorphism. J Thromb Haemost. 2008;[In Press].
Visser I, Wekking EM, de Boer AGEM, van Hout MSE, van Dorsselaar S, Ruhé HG, Huyser J, van derLaan G, van Dijk FJH, Schene AH. Mental disorders in chronic solvent induced encephalopathy:prevalence and symptom profiles. 2008; [Submitted].
Baas K, Wittkampf KA, Weert HC van, Lucassen P, Huyser J, Hoogen H van den, Lisdonk, E van de,Bindels P, Bockting CL, Ruhé HG, Schene AH. Screening on depression in high risk groups ingeneral practice is not useful: results from a prospective cohort study in general practice. Br JPsychiatry. 2008; [In press].
Ruhe HG, Rijswijk Ev, Verkes RJ. Biologische interventies bij depressie. In: Huyser J, Schene AH,Sabbe B, Spinhoven Ph, eds. Handboek depressieve stoornissen. Utrecht: De Tijdstroom; 2008:201-218.
Ruhe HG, Booij J, Veltman DJ, Michel MC, Schene AH. Successful treatment of major depressivedisorder by paroxetine attenuates amygdala activation to negative facial expressions. An fMRIstudy. 2008; [Submitted].
Ruhé HG, Ooteman W, Booij J, Michel MC, Moeton M, Baas F, Schene AH. Serotonin transportergene promoter polymorphisms modify the association between paroxetine serotonin transporteroccupancy and clinical response in major depressive disorder. Pharmacogenetics and Genomics.2008; [In press].
Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine anddopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol Psychiatry.2007; 12: 331-359.
Baas KD, Ruhé HG. Gedragsactivatie effectief in de behandeling van depressie. Tijdschr vPsychiatrie.2007; 49: 411.
Ruhé HG. Polymorfisme van het serotoninetransporteiwit niet gerelateerd aan respons, maar welaan bijwerkingen bij citalopramgebruik. Ned Tijdschr Geneeskd. 2007;151: 2641.
Ruhe HG, Huyser J, Swinkels JA, Schene AH. Dose escalation for insufficient response to standard-dose selective serotonin reuptake inhibitors in major depressive disorder: Systematic review. Br JPsychiatry. 2006; 189: 309-316.
Ruhe HG, Huyser J, Swinkels JA, Schene AH. Switching antidepressants after a first selectiveserotonin reuptake inhibitor in major depressive disorder: a systematic review. J Clin Psychiatry.2006; 67: 1836-1855.

Publications
288
Ruhé HG. Suïciderisico door antidepressiva opnieuw onder de loep genomen. Tijdschr vPsychiatrie.2006;48: 494-495.
Ruhe HG, Dekker JJ, Peen J, Holman R, de Jonghe F. Clinical use of the Hamilton DepressionRating Scale: is increased efficiency possible? A post hoc comparison of Hamilton DepressionRating Scale, Maier and Bech subscales, Clinical Global Impression, and Symptom Checklist-90scores. Compr Psychiatry. 2005; 46: 417-427.
Ruhe HG, Swinkels JA, Schene AH. Vervolgstrategieën bij onvoldoende respons op een eersteSSRI. Pharmaceutisch Weekblad. 2005; 44: 1382-1385.
Schene AH, Huyser J, Ruhe HG. Validiteit van de diagnose depressie. In: Koerselman F, VergouwenAC, eds. De gecompliceerde kliniek 2005: over de geldigheid van diagnoses. Amsterdam: Benecke;2005.
Ruhé HG. Farmacotherapie bij unipolaire depressie. Bijblijven. 2003;19:12-25.
Ruhe HG. Review: Low dose tricyclic antidepressants may be effective for adults with acutedepressive disorder. Evid Based Ment Health. 2003;6:46.
Ruhé HG, Huyser J, Swinkels JA, Schene AH. Wat te doen als de eerste pil niet werkt? EenEvidence-Based richtlijn voor de biologische behandeling van depressie bij onvoldoende responsop een standaarddosering van een eerste SSRI. 2002. Amsterdam, ZorgprogrammaStemmingsstoornissen MFO Psychiatrie AMC/ De Meren.
Ruhé HG, Becker HE, Jessurun P, Marees CH, Heeringa M, Vermeulen HD. Agranulocytosis andgranulocytopenia associated with quetiapine. Acta Psychiatr Scand. 2001;104:311-3.
Swinkels JA, Schene AH, Ruhé HG. Evidence Based Mental Health: inhoud en methode. In: ScheneAH, Boer F, Heeren TJ, Henselmans HWJ, Trijsburg RW, Vandereycken W, Velden K van der, eds.Jaarboek voor psychiatrie en psychotherapie 2001-2002. Houten/Diegem: Bohn Stafleu van Loghum.2001: 298-313.
de Vegt F, Dekker JM, Ruhe HG, Stehouwer CD, Nijpels G, Bouter LM, Heine RJ. Hyperglycaemia isassociated with all-cause and cardiovascular mortality in the Hoorn population: the Hoorn Study.Diabetologia. 1999;42:926-31.
Jager A, Kostense PJ, Ruhe HG, Heine RJ, Nijpels G, Dekker JM, Bouter LM, Stehouwer CD.Microalbuminuria and peripheral arterial disease are independent predictors of cardiovascularand all-cause mortality, especially among hypertensive subjects: five-year follow-up of the HoornStudy. Arterioscler Thromb Vasc Biol. 1999;19:617-24.
Dekker J, Ruhé E, Nijpels G, Heine R. Diabetes en cardiovasculaire complicaties. In: Broese vanGroenau M, Deeg DJH, Knipscheer CPM, Ligthart GJ, eds. VU-visies op verandering. Amsterdam:Thela-Thesis 1998: 21-24.

Publications
289
Published abstracts
Ruhé HG, Booij J, v Weert HC, Reitsma J, Michel MC, Schene AH. Randomized dose escalation ofparoxetine in depressed patients visualized by [123I]β-CIT SPECT. [Abstract P1.22]. J AffectDisord.2008;107 Suppl 1:S77.
RuhéHG, Booij J, ScheneAH. Season and gender modify in-vivo serotonin transporter binding indepressed patients and healthy controls. [Abstract P1.23]. J Affect Disord.2008;107 Suppl 1:S78.
Ruhé HG, Booij J, VeltmanDJ, Reitsma J, MichelMC, ScheneAH. The effect of paroxetine onamygdala reactivity after emotional faces measured with fMRI. [Abstract P1.24]. J AffectDisord.2008;107 Suppl 1:S78.
Schene AH, Ruhé HG, Huyser J, Speckens A, Nolen WA. Wat leert STAR*D ons over de behandelingvan depressie, nu en in de toekomst? [Abstract S-39].Tijdschr v Psychiatrie.2008;50 Suppl 1: S106-109.
Ruhé HG, Ooteman W, Visser I. Etiologie en neuroimaging van depressie. [Abstract PO-63].Tijdschrv Psychiatrie.2008;50 Suppl 1: S185-188.
Ruhé HG, Booij J, v Weert HC, Michel MC, Schene AH. Randomized dose escalation of paroxetinein depressed patients visualized by [123I]β-CIT SPECT [Abstract 344]. Eur J Nucl Med Mol Imag.2007;34 Suppl 2:S195.
Ruhé HG, Booij J, Schene AH. Effect of season and gender on in-vivo serotonin transporter bindingin depressed patients versus healthy controls. A [123I]β-CIT SPECT study [Abstract 345].Eur J NuclMed Mol Imag.2007;34 Suppl 2:S195.
Ruhé HG, Huyser J, Swinkels JA, Schene AH. Failure to respond to the first SSRI in majordepressive disorder: an evidence-based guideline for timing of treatment changes, increasing thedose and switching. [Abstract]. J Affect Disord.2004;78 Suppl 1:S89.

Publications
290

DANKWOORD

292

Dankwoord
293
Dankwoord
Het beste bewaar je tot het laatste. In de afgelopen 5 jaar heb ik met veel plezier met vele mensensamengewerkt zonder wie dit proefschrift onmogelijk zou zijn geweest. In mijn gedachten heb ikdit dankwoord al vele malen geschreven.
In de eerste plaats is deze studie alleen mogelijk geworden dankzij heel veel patiënten, dieondanks hun depressie bereid waren verwezen te worden naar het AMC. Ik ben u allenbuitengewoon erkentelijk. Zonder deze deelname was het onderzoek nooit geslaagd. Allepatiënten die naast de ‘gewone’ dosisverhogingsstudie ook nog meededen aan het SPECT en/offMRI onderzoek wil ik dubbel bedanken. U kwam op moeilijke tijden, soms meerdere keren perweek terug voor deze scans, soms ook als de somberheid nog niet zo was verbeterd.
Ook de gezonde proefpersonen die bereid waren SPECT en fMRI scans te ondergaan wil ikhartelijk bedanken. Ik hoop uw interesse in wetenschappelijk onderzoek te hebben gestimuleerdmet mijn uitleg over wat we onderzochten. Dankzij uw bereidheid is het mogelijk resultaten tevergelijken met een niet-zieke toestand.
Graag wil ik ook alle huisartsen, psychiaters en sociaal psychiatrisch verpleegkundigenbedanken die in de periode dat wij op zoek waren naar deelnemers voor ons onderzoek trouwbleven verwijzen. Wellicht dat wij in de toekomst opnieuw met jullie samen kunnen werken.
Prof.dr. Aart Schene, mijn promotor, jou ben ik veel dank verschuldigd. Om te beginnenrealiseerde jij mijn aanstelling als kersverse psychiater bij het zorgprogrammaStemmingsstoornissen, waarmee jij direct jouw vertrouwen aangaf. Vervolgens je keuze omondanks onvolledige financiering – tegen jouw gewoonte in – toch een aanvang te maken met deprojecten waar ik mijn zinnen op had gezet. Jouw no-nonsense houding bij het bespreken vanzakelijke, technische, en organisatorische aspecten van onderzoek en ondertussen ookmanagement zijn prijzenswaardig en leren mij veel. Bovendien kan ik het erg goed met je vinden.Het veel samenwerken aan projecten, op de gekste tijden bereikbaar zijn, snel reageren opgeschriften en jouw positieve benadering zijn kenmerkend en geven mij inspiratie om hier noglang mee door te gaan.
Dr. Jan Booij, mijn co-promotor, dank ook voor jouw denkwerk, enthousiasme engelijkwaardige begeleiding tijdens het uitvoeren van dit onderzoek. Jij regelde de infrastructuur inde samenwerking met de XTC-studie, de scan-faciliteiten, de uitleg over de analyses van de scansen mijn verdere scholing op het gebied van de nucleaire geneeskunde. Met jou is het altijd prettigsamenwerken geweest, laten we dit vooral voortzetten.
De leden van de promotiecommissie hebben als eersten het product van mijn werk mogenlezen en beoordelen: Prof.dr. Damiaan Denys, dank dat je in de commissie wilde plaatsnemen, endank voor de interessante wetenschappelijke discussies die we sinds jouw komst alskernhoogleraar al hebben gevoerd. Ik verheug me op de toekomst, waarin onze interessessamenkomen in nieuwe onderzoeksprojecten, hetgeen ongetwijfeld tot meer discours zal leiden.
Prof.dr. Rudi Dierckx, ook dank aan jou voor het beoordelen van het manuscript. Hopelijk gaatons andere project door, zodat we in de toekomst aan gezamenlijke studies kunnen werken.
Prof.dr. Berthe van Eck-Smit, dank voor de faciliteiten die door de afdeling Nucleairegeneeskunde werden geschapen om dit onderzoek te kunnen doen. Dank voor het beoordelenvan het manuscript, maar ook mijn dank voor de interesse in mijn onderzoek, als wij elkaar op F2ontmoetten.
Prof.dr. C.G. Kruse, mijn hartelijke dank voor uw beoordeling van mijn manuscript. Ik waardeerhet enorm dat u ondanks uw drukke reisschema aanwezig kan zijn bij de verdediging.
Prof.dr. Willem Nolen, biologische psychiater van het eerste uur en daarbij ervaren clinicus ophet gebied van de stemmingsstoornissen. Hartelijk dank voor jouw interesse in mijn activiteiten,waar je al sinds 2001 bij betrokken bent. Dat jij voor mijn commissie zou worden gevraagd wasvoor mij vanzelfsprekend, ik vind het een eer dat je mijn manuscript zo spoedig van een oordeelvoorzag. Het proefschrift is af, de studies zijn (bijna) gepubliceerd, en we moeten voort, op zoeknaar nieuwe strategieën in de behandeling van depressie.

Dankwoord
294
Dr. Robbert-Jan Verkes, ook jou wil ik hartelijk danken voor je bereidheid vragen te stellen bijmijn verdediging. Dank voor de plezierige samenwerking tijdens het schrijven van het hoofdstukin het Handboek Depressie, dat smaakte naar meer.
Jochanan Huyser, tot drie keer toe was jij op de jou toevertrouwde wijze - strategisch aan dezijlijn - een beïnvloeder van formaat. Je nam mij eind 1996 aan als AGNIO. Vervolgens bracht jij mede beginselen van depressiebehandeling bij. Je kwam, kort na een jaar zoeken naar een‘kristallisatiepunt’, met het aanbod om het richtlijnproject te gaan doen. Ik heb genoten van jestimulerende enthousiasme tijdens dit project, en denk met bewondering terug aan jouw kundein het voorzitten van de commissievergaderingen. Toen we collegae psychiaters waren, heb jijmijn keuze om de neuroimaging van de DELPHI-studie te gaan doen met een kleine suggestierichting gegeven. Dat ik, nu jij het AMC hebt verlaten, de dagelijkse leiding van ‘jouw’behandelcentrum Stemmingsstoornissen over heb mogen nemen beschouw ik als een grote eer.
Voor alle praktische en logistieke support was een telefoontje naar Marianne Haages meestalvoldoende. Marianne, fantastisch hoe jij al sinds het richtlijnproject enthousiast hebtmeegewerkt, en daarvoor ben ik je duizendmaal dank verschuldigd. Met jouw invulling krijgt deprofielschets voor een ervaren secretaris/secretaresse een onbereikbare dimensie.
Mijn collegae van de afdeling stemmingsstoornissen, Guido Nabarro, Prof.dr. Jan Swinkels,Mirjam van Zon, alle verpleegkundigen, assistenten in opleiding, ergotherapeuten,maatschappelijk werkers, psychomotore therapeuten en psychologen, jullie hebben allemaalbijgedragen aan het tot stand komen van dit boek. Ook de ex-collegae Nicole Eussen, ArjenRonner, Nicole de Boer en Pierre Delfgauw horen in dit rijtje thuis. Dank voor jullie interesse,waarneming, vragen, samenwerking op het gebied van onderzoek en ingewikkelde casuïstiek, hetverdragen van mijn afwezigheid, maar vooral jullie degelijke vakkennis en enthousiasme in debehandeling van onze depressieve patiënten. Dagelijks laten jullie zien waar het bij behandelenvan depressieve, zieke mensen echt om gaat: betrokkenheid met gepaste distantie, veelvuldigproberen en herhalen, gedoseerde activatie, soms toegeeflijk zijn maar zonder hierbij de eigengrenzen te overschrijden, gecombineerd met een professionele aandacht voor stagnatie enalarmsignalen. Ik vind het, nogmaals, een eer om met jullie te werken en hoop daar mee door tekunnen gaan.
Een speciaal woord van dank gaat uit naar Mascha ten Doesschate en Elizabeth Miedema, dieveel bijdroegen aan de opzet en de dataverzameling voor de DELPHI-studie. Mascha, veel succesmet de afronding van jouw promotie. Elizabeth, gezamenlijk werkten wij anderhalf jaar aan deinclusie. Dank voor je toewijding en bereidheid om logistieke zaken omtrent de patiëntenzorg vanmij over te nemen. Andere leden van onze onderzoeksgroep: Karin, Kim, Ieke, Anja, Wendy,Hanneke, Maarten, Monique, Judith en sinds kort Maaike en Mathilda, dank voor de prettigesamenwerking, jullie bijdragen aan het onderzoekersoverleg, de gezamenlijke projecten die zijnafgerond, nog lopen of gaan komen. Ik verheug me nu al op een volgend bezoek aan ISAD. Nada,Michiel, Koen en Max, dank voor jullie input bij mijn projecten. Maaike, sinds kort toegetreden alsAGIKO, jij startte jouw promotie in een tijd dat ik nog druk was/ben met de afronding van ditproefschrift. Ik bewonder je degelijkheid waarmee je complexe zaken uitdiept en kijk uit naar deverdere ontwikkeling van onze biomarker plannen.
Alle collegae van andere afdelingen ben ik dankbaar voor de prettige contacten, expertise entoegankelijkheid. Dr. Henk van Weert, met jou heb ik veel werk verzet om ons verwijzersnetwerkop te bouwen. Dank voor je betrokkenheid, je inzet en je hulp om DELPHI tot een succes temaken. Prof.dr. Dick Veltman, jij leerde mij in meerdere middagen één-op-één onderwijs hetwerken met SPM5. Het lijkt me goed hiermee door te gaan en vooral samen nieuwe fMRI studieste gaan doen. Prof.dr. Martin Michel, jij besteedde als co-auteur bijzonder veel aandacht aan mijnmanuscripten. Dank voor je kritische commentaren, het wordt er inderdaad alleen maar betervan. Dr. Hans Reitsma, “gelijk hebben en krijgen is niet altijd hetzelfde”, dank voor al jouw hulp bijook voor een gewone epidemioloog ingewikkelde statistiek; ik heb veel van je geleerd. Prof.dr.Koos Zwinderman, dat geldt ook voor jou. Dr. Maartje de Win, aanvankelijk kon ik voor de SPECT-scans meeliften op jouw XTC-studie. Dank hiervoor en voor jouw ordentelijke planning waar altijdop te bouwen was. De medewerkers van de afdeling Nucleaire geneeskunde, dank voor hetverdragen van al mijn ‘geregel’ om de scans op de juiste momenten te kunnen maken, en dank

Dankwoord
295
voor jullie hulp bij het scannen in de avonduren. Dr. Rob Scholten, ik ken je vanaf mijn eersteserieuze onderzoeksinspanningen op het EMGO. Ondanks dat ik door jou gedegen aan de tandwerd gevoeld tijdens mijn epidemiolgie-examen moet ik je nog steeds lastig vallen voor meta-analytische problemen, waarvan jij zelfs verzuchtte dat ze zo ingewikkeld zijn. Dank je voor jeprettige, relativerende en snelle hulp. Ook Dr. Astrid Goossens van de KEB wil ik bedanken voorhaar enthousiasme tijdens het richtlijnproject. Dr. Aart Nederveen, dank voor je rustige, gedegenhulp bij het instellen van de MRI-scanner en alle antwoorden op andere vragen die ik je hebgesteld. Raschel en Sandra, dank voor de eerste hulp bij MRI-problemen. Tessa, jij was nooit teberoerd om extra te komen scannen als MRI-piloot. Michiel de Ruiter, veel dank voor het delenvan jouw kennis van SPM en fMRI, ook als ik je op vakantie of in de supermarkt belde. Zsuzsika,dank voor je expertise bij het voorbewerken van de ToL, en succes met je eigen studies. EricFranssen, dank voor de snelle bepalingen van de paroxetine-spiegels en je commentaren opeerdere versies van het artikel. Remco Knol, dank voor je prachtige illustratie (figuur 1.2) en veelsucces de dag na mij. Peter Remijnse, we hadden een goede inclusie luchtbrug, moeten we vakerdoen. Ik ben zeer benieuwd naar jouw vorderingen. Prof.dr. Frank Baas, Marja Jakobs en MartinaMoeton, dank voor jullie hulp bij de genetische bepalingen, en de interpretatie ervan. Nadia,Pieter-Willem, Saskia, Joost, Eric-Jan, Kurdo en Prof.dr. Harry Büller, wat een mooi project is onzestollings-studie geworden, dank voor de samenwerking. Bep de Lange, sinds anderhalf jaar mijnbuurvrouw, dank voor je zorg, je interesse en natuurlijk de schutbladen op momenten dat hetecht nodig is. Prof.dr. Frans de Jonghe, Prof.dr. Jack Dekker en Jaap Peen van de MentrumDepressie groep, dank voor het gezamenlijk werken aan het HDRS-artikel, meten is weten, enefficiënt meten is sneller weten.
Eugenie Oosterhuis, u verdient een aparte vermelding in dit dankwoord. Het bereiken van eenplateau na een stevige beklimming wil niet zeggen dat je er niet alsnog af kan vallen; wel weet jebeter waar de gevaarlijke randen zitten. Dank dus opnieuw.
Leden van de intervisiegroep, Ethy, Agnes, Joop, Saskia en Hiske, gelukkig dat ik van jullie‘mag blijven’ als ik gepromoveerd ben. Dank voor jullie begripvolle, constructieve doch stevigeprobleemanalyses, waaruit nieuwe oplossingen opborrelden. Gelukkig is het ook erg gezellig ombij elkaar te komen.
De afgelopen jaren heeft het proefschrift een steeds groter deel van mijn tijd in beslaggenomen. Vele vrienden zal dat opgevallen zijn. Wouter en Jet, Mark, Cees en Dorte, Henk enMary, Mary en Bob, Miriam, Tamar en Ilan, Laetitia en Erik, Lilian en Frank, Ellen en Edwin, Stephanen Ellen, Natascha en Thijs, Michiel en Mathieu, Jef en Sylvia: het is af. Laten we dit vieren met eengroot feest. Graag nodig ik jullie deze winter uit om lekker bij de open haard te zitten, te eten ente kletsen.
Ook mijn (schoon-)familie wil ik bedanken voor de support van de laatste maanden ennatuurlijk de jaren ervoor. Pap en mam, van jullie heb ik heel veel meegekregen, maar absoluutook een zeker fanatisme, dat onontbeerlijk is voor een promotie. Dank daarvoor, en voor deinvesteringen die jullie in de afgelopen 40 jaar op alle terreinen in mij gedaan hebben. Dat de wegeigenlijk belangrijker is dan het doel, staat soms in schril contrast met ons fanatisme; laten we dusnog maar lang onderweg zijn, opdat we dat beter leren. Toine en Quinten, wat bof ik met jullie alsbroers, en dat gevoel wordt alleen maar sterker. Jullie hebben allebei een geweldige vrouw inonze familie geïntroduceerd. Toine en Andrea, geweldig dat jullie voor deze gelegenheid hierheenwillen komen. Quinten en Miriam, bij jullie is het altijd gezellig. Laten we elkaar vaak zien, ondanksalle opleiding, promotie en andere drukte. Miriam, heel veel succes ook met jouw laatste loodjes.Toon en Johanna, dank voor alle support bij dit proefschrift, praktisch, logistiek en geestelijk.Jullie voelden op de een of andere manier vaak goed aan wanneer een handje hulp gewenst was.Het is heerlijk dat ik jullie met Katelijne heb leren kennen.
Jan Willem, beste vriend sinds de Bhagwan-discotheek in 1986. Wat hebben wij al veel metelkaar meegemaakt en wat ben ik blij dat jij bij deze gelegenheid mijn paranimf bent. Quinten, jeblijft mijn broertje, ook jij bent mijn ideale paranimf. Ik wil jullie beiden speciaal bedanken voor hetovernemen van een aantal belangrijke klussen rondom de promotie, waarbij jullie ook nog evende redactie van de laatste hoofdstukken deden. Dat heeft mij veel ruimte gegeven om me op delaatste loodjes te concentreren.

Dankwoord
296
Het beste bewaar je tot het laatste. Laura en Merle, wat een feest dat jullie bij ons zijngeboren; we zouden nooit meer zonder jullie kunnen. Ik weet zeker dat ik het meest trots op julliezal zijn, als jullie op woensdag op de eerste rij zitten. Dank voor de tientallen tekeningen, dezaterdagochtenden dat we met de lego speelden (en nog gaan spelen), jullie verhalen enongelofelijke creativiteit. Hiermee hebben jullie mij geholpen om te blijven zien dat je somsgewoon even moet gaan spelen. Laura, jouw zelfvertrouwen is een lust om te zien, evenals je lachals je weer iets grappigs herkent. Merle, ik zou nog wel honderd jaar met jouw gezang wakkerwillen worden. En…. gelukkig zijn jullie veel sneller dan ik in het maken van boeken!
Katelijne, dank voor de mogelijkheid die jij biedt om mij op de wetenschap te richten en al hetandere dat je voor me betekent. Hoewel je mij ook op gezette tijden, op meer of minderdiplomatiek wijze, naar het nut van mijn exercities vraagt, heb ook jij de afgelopen jaren veel in ditproefschrift geïnvesteerd. Ik realiseer me maar al te goed dat jij mij op meerdere momentenbehoedt voor doordraven, wat vermoedelijk in mijn genen zit, maar niet minder mijn eigenverantwoordelijkheid is. Ik ga je niet beloven dat het allemaal anders gaat worden na 5 November,daarvoor ben ik te nieuwsgierig, heb ik teveel plezier in dit werk en hebben we niets aan lozebeloftes. Het vinden van een juiste, iets evenwichtiger balans moet wel weer kunnen. Gelukkig zalik maar eenmaal promoveren. Kus.
Eric Ruhe
Amstelveen, September 2008

CURRICULUM VITAE

298

Curriculum Vitae
299
Curriculum Vitae
Eric Ruhé werd geboren op 18 November 1968 in Amsterdam, als oudste van drie zoons. Toen hijvijf was verhuisde het gezin naar Overijssel, waar zij tien jaar op het platteland woonden. Hiernaverhuisden zij naar Den Haag, waar hij in 1986 het eindexamen VWO behaalde.
Vanaf 1986 studeerde hij geneeskunde aan de Universiteit van Amsterdam, waar hij in 1991 hetdoctoraal examen behaalde. Hierna vond een reis naar Kenia en Tanzania plaats waarbij hij zijneerste kennismaking met wetenschappelijk onderzoek had bij het uitvoeren van deresistentiebepaling van de malaria parasiet voor de middelen chloroquine en amodiaquine via deAfrikaanse flying docters (AMREF).
In 1992 startte hij zijn co-schappen en behaalde in Juli 1995 het artsexamen, na opnieuw 6maanden in Tanzania te hebben doorgebracht tijdens een tropen co-schap. Als een der laatste inNederland vervulde hij vanaf 1995 zijn vervangende militaire dienst bij het instituut voorExtramuraal Geneeskundig Onderzoek (EMGO) van de Vrije Universiteit, waar hij de toenmaligecross-sectionele gegevens van de Hoorn Studie voorzag van een mortaliteits- enmorbiditeitsregistratie. Deze mortaliteits- en morbiditeitsregistratie is tot op heden operationeel.In deze periode volgde hij de postacademische opleiding tot klinisch-epidemioloog.
Vanaf 1997 startte hij zijn carrière in de psychiatrie met een baan als AGNIO bij hetzorgprogramma Stemmingsstoornissen van het Academisch Medisch Centrum (AMC). Van 1998tot 2002 was hij in het AMC in opleiding tot psychiater (opleider Prof.dr. B.P.R. Gersons),aangevuld met twee keuzestages bij de stichting Mentrum. Tijdens de opleiding psychiatrie konzijn interesse in onderzoek bij stemmingsstoornissen verder worden ontwikkeld door eensubsidie vanuit het AMC. Met deze subsidie werd in 2001-2002 een richtlijn voor het klinischhandelen bij onvoldoende respons op het eerste antidepressivum opgesteld, waarvan enkelepublicaties in dit proefschrift zijn opgenomen. Deze AMC-richtlijn werd eveneens gebruikt bij desamenstelling van de multidisciplinaire richtlijn Depressie die in deze periode werd ontwikkeld.Deze richtlijn vormde tevens het uitgangspunt voor subsidieaanvragen en de andere studies die indit proefschrift staan beschreven. Sinds zijn inschrijving in het specialistenregister in Juni 2003,werkt hij als stafpsychiater bij het zorgprogramma Stemmingsstoornissen van het AMC, en heefthij zich verder gespecialiseerd in de behandeling van uni- en bipolaire depressies.
Eric Ruhé leeft sinds 1994 samen met Katelijne Schmeink, met wie hij in 1998 is getrouwd. Zijhebben samen twee prachtige dochters: Laura (2000) en Merle (2002).

300
Curriculum Vitae
Eric Ruhé was born in Amsterdam on November 18th, 1968. He is the eldest of three sons. At theage of five, the family moved to Overijssel, a rural part of the Netherlands, where they lived forten years. The family then moved to The Hague, where he completed his secondary school (VWO).
From 1986 until 1991, he studied medicine at the University of Amsterdam. His first encounterwith scientific research was in Kenya and Tanzania, where he participated in the determination ofresistance of plasmodium Falciparum against chloroquine and amodiaquine, a project conductedfor the African flying doctors service (AMREF).
He started his internships in 1992 and received his medical degree in 1995, after spendinganother 6 months in Tanzania, performing a special internship in tropical medicine. He thereafterserved his civil service at the Vrije Universiteit, at the institute for extramural medicine (EMGO).He developed a mortality and morbidity registration for the Hoorn Studie, which was until then across-sectional study. This mortality and morbidity registration is still operational until now.During this civil service, he received his masters in clinical epidemiology.
Since 1997, he has been working in psychiatry, first as a non-resident (AGNIO) at the programfor Mood Disorders of the Academic Medical Center (AMC). From 1998 until 2002, he specializedin psychiatry at the AMC and Mentrum. During his residency he combined his epidemiological andpsychiatric knowledge, and performed a clinical practice guideline project, funded by the AMC.This local guideline was also incorporated in the Dutch multidisciplinary guideline for depressionwhich was developed at the same time. Three publications of this thesis result form the guidelineproject. Thereafter two grant applications were written and awarded, which enabled the otherstudies of this thesis. Since his registration as a psychiatrist in 2003, he has been working as aspecialized clinical psychiatrist at theprogram for Mood Disorders at the AMC.
He shares his life with Katelijne Schmeink, they were married in 1998. Together they have twowonderful daughters: Laura (2000) and Merle (2002).