enzymatic properties of hepatitis c virus ns3-associated helicase
TRANSCRIPT

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
Journal of General Virology (2000), 81, 1335–1345. Printed in Great Britain. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Enzymatic properties of hepatitis C virus NS3-associatedhelicase
Chantal Paolini, Raffaele De Francesco and Paola Gallinari
Istituto di Ricerche di Biologia Molecolare ‘P. Angeletti ’ (IRBM), Via Pontina Km 30.600, 00040 Pomezia (Rome), Italy
The hepatitis C virus non-structural protein 3 (NS3) possesses a serine protease activity in the N-terminal one-third, whereas RNA-stimulated NTPase and helicase activities reside in the C-terminalportion. In this study, an N-terminal hexahistidine-tagged full-length NS3 polypeptide wasexpressed in Escherichia coli and purified to homogeneity by conventional chromatography.Detailed characterization of the helicase activity of NS3 is presented with regard to its binding andstrand release activities on different RNA substrates. On RNA double-hybrid substrates, theenzyme was shown to perform unwinding activity starting from an internal ssRNA region of at least3 nt and moving along the duplex in a 3« to 5« direction. In addition, data are presented suggestingthat binding to ATP reduces the affinity of NS3 for ssRNA and increases its affinity for duplex RNA.Furthermore, we have ascertained the capacity of NS3 to specifically interact with and resolve thestem–loop RNA structure (SL I) within the 3«-terminal 46 bases of the viral genome. Finally, ouranalysis of NS3 processive unwinding under single cycle conditions by addition of heparin in bothhelicase and RNA-stimulated ATPase assays led to two conclusions: (i) NS3-associated helicaseacts processively ; (ii) most of the NS3 RNA-stimulated ATPase activity may not be directly coupledto translocation of the enzyme along the substrate RNA molecule.
IntroductionHepatitis C virus (HCV) presents a major health problem.
The global prevalence of chronic hepatitis C is estimated toaverage 3 %. In industrialized countries, HCV accounts for20% of cases of acute hepatitis, 70% of cases of chronichepatitis, 40% of cases of end-stage cirrhosis, 60% of cases ofhepatocellular carcinoma and 30% of liver transplants (re-viewed by Houghton, 1996). HCV contains an approximately9±6 kilobase positive-sense ssRNA genome and is classified inthe Flaviviridae family of animal viruses (reviewed by Rice,1996). Its genome consists of a conserved 5« non-translatedsequence that serves as an internal ribosome entry site (5«-NTR), a single open reading frame that encodes a polyproteinof more than 3000 amino acids, and a 3« non-translated regionthat contains tracts of poly(UC)
nfollowed by a conserved
98 nt sequence (3«-NTR). Proteolytic processing of the viralpolyprotein by cellular and virus-encoded proteases generatesmature core, envelope and non-structural (NS) proteins (C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B) (reviewed by
Author for correspondence: Paola Gallinari.
Fax 39 06 91093225. e-mail Gallinari!IRBM.it
Clarke, 1997 ; Lohmann et al., 1996). Non-structural protein 3(NS3) is a bifunctional enzyme exhibiting a protease activity inthe N-terminal one-third and an RNA helicase activity in theC-terminal portion. The NS3 protease belongs to the serineprotease family and is responsible for processing the HCVpolyprotein at NS3}NS4A, NS4A}NS4B, NS4B}N5A andNS5A}NS5B junctions (reviewed by De Francesco et al.,1998 ; Kwong et al., 1998). The helicase activity, associatedwith a nucleic acid-stimulated NTPase activity (Gwack et al.,1995 ; Jin & Peterson, 1995 ; Kim et al., 1995 ; Porter, 1998 ;Preugschat et al., 1996), is presumed to be involved in thereplication and}or translation of viral RNA. The minimalrequirement for these latter functions lies in the C-terminal 465amino acids of NS3, which represent a functionally andstructurally separate domain (Kim et al., 1995). Recently, wehave demonstrated that although the N-terminal proteasedomain has little if any effect on the ATPase and helicaseactivities of NS3 (Gallinari et al., 1998), NS4A-mediatedstabilization of NS3 in the active protease conformation cannegatively affect its ability to unwind dsRNA (Gallinari et al.,1999).
Different three-dimensional structures of the isolated NS3helicase domain have been determined (Cho et al., 1998 ; Kim
0001-6757 # 2000 SGM BDDF

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
C. Paolini, R. De Francesco and P. GallinariC. Paolini, R. De Francesco and P. Gallinari
et al., 1998 ; Yao et al., 1997). The enzyme comprises threedomains termed 1, 2 and 3, with domains 1 and 2 beingstructurally similar. The ATP-binding site is situated in a cleftbetween domains 1 and 2 that is lined with conserved sequencemotifs that are the hallmarks of helicases (Gorbalenya &Koonin, 1993). NS3 is a member of a large class of helicasesthat have a 3« to 5« directionality and share a number ofstructural features (Bird et al., 1998). Other members of thisclass include the Rep, PcrA and UvrD bacterial DNA helicasesfor which much structural data became available recently(Korolev et al., 1997 ; Subramanya et al., 1996). NS3 helicasedomains 1 and 2 fold similar to domains 1A and 2A of theabove-mentioned enzymes, but with a slightly differentconnectivity (Bird et al., 1998). The third domain has nostructural similarity with Rep or PcrA but sits in a positionapproximately equivalent to that occupied by domain 1B in theRep helicase (Korolev et al., 1998). The structure of a complexof the NS3 helicase domain with a (dU)
)oligonucleotide (Kim
et al., 1998) demonstrated the nucleic acid in a channel thatseparates domain 3 from domains 1 and 2, in a positionequivalent to the binding site for ssDNA in the Rep (Korolevet al., 1997) and PcrA (Velankar et al., 1999) enzymes. Threedifferent models for the mechanism of NS3 helicase have beenproposed, one to accompany each of the three publishedcrystal structures. To get more insight on the overallmechanism of unwinding, we have analysed the effect thatbinding of ATP (or non-hydrolysable ATP analogues) to thefull-length (FL) enzyme exerts on its relative affinity for ssRNAand dsRNA.
In order for a helicase to unwind duplex nucleic acids in aprocessive manner, the enzyme should destabilize the hy-drogen bonds between the base pairs, translocate to the nextbase-paired region, and repeat the cycle without dissociatingfrom the RNA substrate. Here we have analysed the FL-NS3processive unwinding under single cycle conditions usingheparin as a trapping molecule both in helicase and ATPaseassays.
The helicase activity of NS3 was previously shown torequire substrates with a free 3« single-stranded tail (Gwack etal., 1996 ; Tai et al., 1996). In this study, we have measured NS3unwinding activity on double-hybrid substrates designed tomimic stem–loop structures, containing internal ssRNAregions of different lengths. The ability of NS3 to interactpreferentially with HCV genomic or antigenomic RNA has notbeen addressed so far. We and others have previously shownthat in the absence of ATP, NS3 binds ssRNA tightly with noparticular sequence specificity and with efficiency dependingonly on the length of ssRNA (Gallinari et al., 1998 ; Gwack etal., 1996 ; Kanai et al., 1995 ; Tai et al., 1996). Therefore, it wasof interest to assess whether NS3 shows a selective interactionwith the 3« ends of the viral genome and antigenome, thepresumed initiation sites for negative- and positive-strandRNA synthesis, respectively. We have studied the interactionand the helicase function of FL-NS3 with a series of RNA
oligonucleotides derived from the 46 nt stem–loop structure(SL I) at the 3« terminus of HCV genomic positive-strand RNA(Blight & Rice, 1997 ; Kolykhalov et al., 1996 ; Tanaka et al.,1996). The stability and structural conservation of SL Isuggested that it represents a recognition site for viral and}orcellular proteins involved in virus replication. While some ofthe protein–RNA interactions at the 3«-end of the genomehave been recently elucidated (Cheng et al., 1999 ; Ito & Lai,1997 ; Tsuchihara et al., 1997), our data represent the firstevidence of the ability of HCV NS3 to recognize and resolveRNA secondary structures within the viral 3«-NTR.
Methods+ Expression and purification of a histidine-tagged FL-NS3protein. The cDNA fragment encoding FL-NS3 (residues 1027–1657 ofthe BK strain HCV polyprotein) was excised from pT7NS3-FL vector(Gallinari et al., 1998) and cloned between the NdeI and HindIII restrictionsites of pET14b expression vector (Novagen). The recombinant proteincontaining an N-terminal hexahistidine tag was expressed in E. coli BL21(DE3) cells (Studier et al., 1998) and purified as previously described(Gallinari et al., 1998), with the following modifications. After con-centration by ammonium sulphate precipitation, soluble NS3 wasdialysed against a buffer containing 25 mM HEPES (pH 8), 20% glycerol,0±5 M NaCl, 0±1% n-octyl-β--glucopyranoside (Calbiochem), and10 mM β-mercaptoethanol and loaded on a 15 ml Ni#+-charged HiTrapmetal-chelating column (Pharmacia) equilibrated in the same buffer. Theprotein was eluted with 3 column vols of the chromatographic buffercontaining 200 mM imidazole and further purified on a Superdex 20026}60 gel filtration column (Pharmacia) equilibrated with buffer A(Gallinari et al., 1998) containing 0±2 M NaCl. The peak fractions werethen loaded on a poly(U)–Sepharose affinity column (Pharmacia) andpure NS3 was eluted in buffer A containing 0±5 M NaCl. Proteinconcentration was estimated by Bio-Rad assay and UV absorptionspectroscopy at 280 nm by using a molar extinction coefficient of64200 M−" cm−".
+ ATPase activity assays. ATPase activity was directly determinedby monitoring [γ-$#P]ATP hydrolysis by thin-layer chromatography, asdescribed by Gallinari et al. (1998). RNA titration assays were carried outby incubating 20 nM enzyme in the presence of increasing concentrationsof ssRNA or partial dsRNA (0 to 10 µM) for 30 min at 37 °C in helicaseactivity buffer [25 mM MOPS–NaOH (pH 7), 2±5 mM DTT, 2±5 URNasin (Promega), 100 µg}ml BSA, 5% glycerol, 3 mM MgCl
#]
containing 1 mM ATP and 2 µCi [γ-$#P]ATP (6000 Ci}mmol, 10 mCi}ml ; Dupont NEN) in a volume of 10 µl. For analysis under singleprocessive cycle conditions, increasing amounts of heparin were added tothe samples as described in the legend to Fig. 6. After termination with5 mM EDTA, 0±5 µl aliquots were spotted onto polyethyleneiminecellulose sheets and developed by ascending chromatography in 150 mMLiCl, 150 mM formic acid (pH 3±0). The cellulose sheets were dried andreleased [$#P]phosphoric acid was quantified with a PhosphorImagerusing ImageQuant software.
+ Helicase assays. Helicase partial double-stranded substrates wereobtained by annealing the corresponding complementary RNA (Genset)or DNA (Primm) synthetic oligonucleotides described in the legends toFigs 2, 4 and 5. Gel-purification and annealing were performed asdescribed (Gallinari et al., 1998). The release strand was 5« end-labelledwith [γ-$#P]ATP by T4 polynucleotide kinase (Pharmacia) prior to theannealing reaction. The assays were performed in 20 µl of helicase
BDDG

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
Enzymology of HCV NS3 helicaseEnzymology of HCV NS3 helicase
activity buffer (see above) containing 1±25 nM $#P-labelled RNA sub-strate and the indicated enzyme concentrations. After pre-incubationfor 15 min at 23 °C, 5 mM ATP was added to start the helicase reaction.Unless otherwise specified, this was carried out at 37 °C for 30 min andthen stopped by adding 5 µl of termination buffer (0±1 M Tris pH 7±5,20 mM EDTA, 0±5% SDS, 0±1% NP-40, 0±1% bromophenol blue, 0±1%xylene cyanol). For analysis under single processive cycle conditions,heparin was used as trapping molecule as detailed in the legend to Fig. 6.Aliquots (8 µl) were analysed on native 8–12% polyacrylamide gelscontaining 0±5¬ Tris–borate–EDTA. Strand separation was visualizedby autoradiography and the efficiency of the helicase reaction wascalculated by quantification of the radioactivity using a PhosphorImagerand ImageQuant software. Percentage of unwinding was calculated asthe ratio between the release strand-associated radioactivity and totalradioactivity of both the unwound substrate and the release strand.
+ Binding assays. Gel retardation reaction mixtures (20 µl) in helicaseactivity buffer contained 1±25 nM $#P 5« end-labelled probe and theindicated enzyme concentrations. Where specified, MgCl
#was omitted
from the reactions and either 5 mM ATP or 5 mM β,γ-methylene-ATP(AMP-PCP, Fluka) was added as indicated. Unless otherwise specified,after incubating for 30 min at 23 °C, suitable aliquots were eitherelectrophoresed on a native 6% polyacrylamide gel containing 0±25¬Tris–borate–EDTA or UV-irradiated as described in the legend to Fig. 4.Bands corresponding to the protein-bound and unbound probe werevisualized by autoradiography and quantification of the radioactivity wasperformed as above. The efficiency of the binding reaction was calculatedas the ratio between the radioactivity associated with the protein-boundprobe and the total radioactivity of both the protein-bound and unboundprobe.
ResultsPurification of the N-terminal histidine-tagged FL-NS3protein
We have expressed in E. coli and purified to homogeneitythe FL-NS3 67 kDa protein containing an N-terminal hexa-histidine tag to facilitate purification (Fig. 1). We have used aprotocol successfully devised for the production of the solubleFL-NS3 untagged protein (Gallinari et al., 1998), with themodifications described in Methods. A comparative analysis ofthe ATPase, helicase and NS4A-stimulated protease activitiesof the pure histidine-tagged protein proved it to be functionallyindistinguishable from the corresponding untagged enzyme(not shown).
NS3 helicase activity does not require a free 3«-end onthe template strand
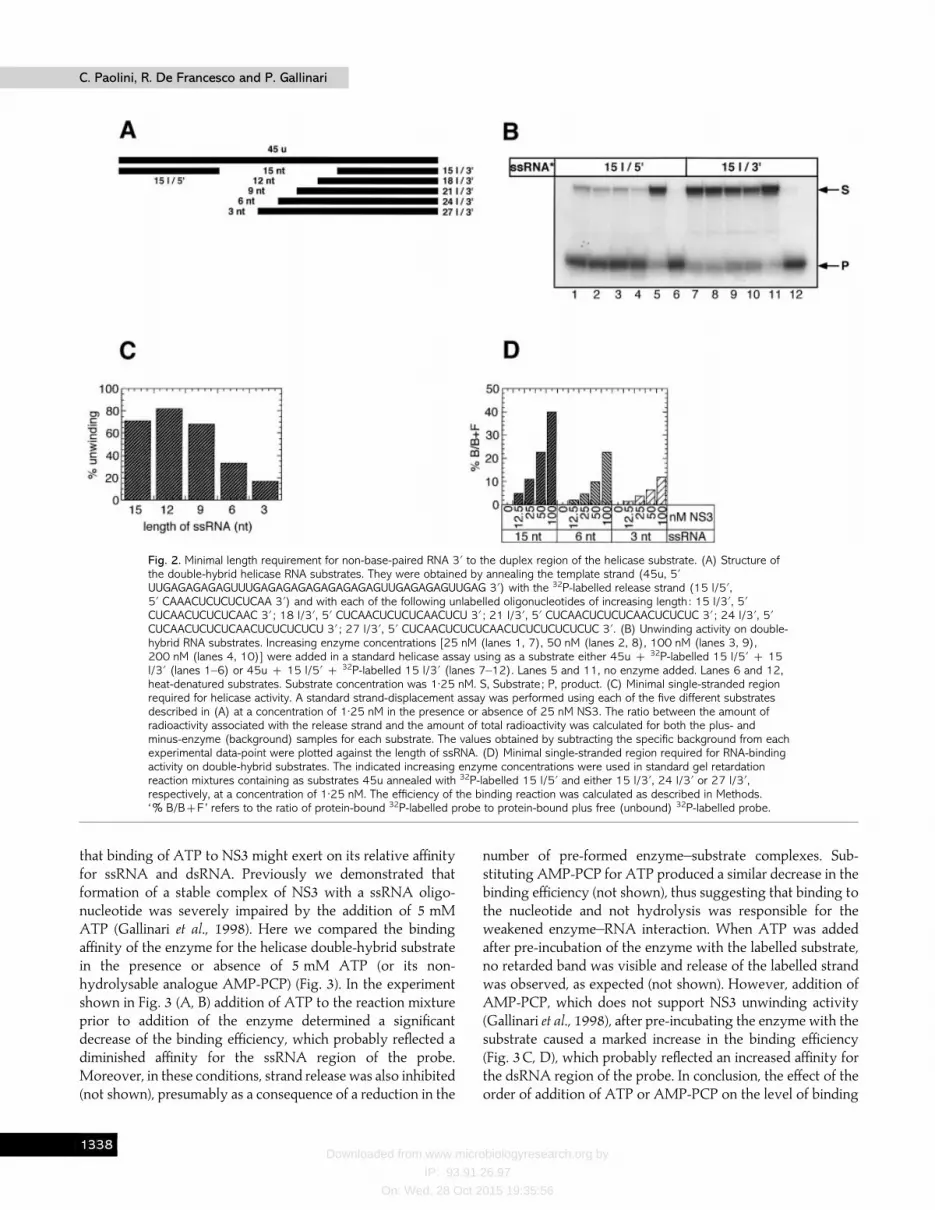
Earlier evidence showed that NS3 helicase unwinds sub-strates possessing a 3« non-base-paired region on the templatestrand (Gwack et al., 1996 ; Tai et al., 1996). To investigate theabsolute requirement for a free 3«-end, we constructed an RNAsubstrate containing two duplex regions each of 15 nt,separated by a 15 nt single-stranded region on the templatestrand (Fig. 2A; 15 l}3«-containing substrate). As shown in Fig.2 (B) (lanes 1 to 5), the 15 l}5« oligonucleotide was efficientlyreleased from the double-hybrid substrate to a level com-parable to that seen with a standard single-hybrid substrate
Fig. 1. SDS–PAGE analysis by Coomassie blue staining of histidine-taggedNS3 purification steps. Lanes : 1, non-induced lysate ; 2, induced lysate ;3, soluble fraction ; 4, insoluble fraction ; 5, 50% ammonium sulphate cut ;6 and 7, Ni2+-charged HiTrap Chelating input and pool, respectively ;8, Superdex 200 pool ; 9, poly(U)–Sepharose pool ; 10, molecular massmarkers. The purified NS3 collected in the poly(U)–Sepharose pool washomogeneous and about 10 mg was obtained per litre of bacteria.
(Gallinari et al., 1998), indicating that the enzyme is able toperform unwinding activity starting from an internal ssRNAregion. Furthermore, NS3 helicase activity showed a marked 3«to 5« directionality, since the release of 15 l}5« was significantlymore effective than that of 15 l}3« (Fig. 2B ; lanes 7 to 11). Todetermine the minimal single-stranded region required foractivity, we constructed a series of substrates containinginternal ssRNA regions of decreasing length (Fig. 2A). Thequantification of the NS3 unwinding efficiency on thesedouble-hybrid substrates (Fig. 2C) indicated that strand-releasewas not affected by shortening the non-base-paired regionfrom 15 down to 9 nt. Further reduction of the ssRNA lengthto 6 and 3 nt caused a decrease of NS3 unwinding efficiency to50% and 25% of maximal activity, respectively. The bindingaffinity of NS3 to these substrates, analysed by gel retardationassay, showed the same ssRNA length-dependent inversecorrelation demonstrated for unwinding (Fig. 2D). Theseobservations are in agreement with published data (Preugschatet al., 1996) showing, by fluorimetric measurements, that theaffinity of NS3 helicase domain for single-stranded oligo-nucleotides is maximal with more than 10 nt.
Effect of nucleotides on the affinity of NS3 for ssRNAand dsRNA
Although we have shown that NS3 is able to form stablecomplexes with double-hybrid substrates containing single-stranded regions down to 3 nt in length, no direct binding wasobserved on ssRNA molecules shorter than 18 nt in the sameexperimental conditions (unpublished results). Since NS3bound ssRNA molecules with lower affinity than partialdsRNA molecules containing ssRNA regions of equal size, acontribution to the overall binding affinity of NS3 for theselatter substrates could be given by additional interactions withthe dsRNA region. We were interested in evaluating the effect
BDDH
Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
C. Paolini, R. De Francesco and P. GallinariC. Paolini, R. De Francesco and P. Gallinari
Fig. 2. Minimal length requirement for non-base-paired RNA 3« to the duplex region of the helicase substrate. (A) Structure ofthe double-hybrid helicase RNA substrates. They were obtained by annealing the template strand (45u, 5«UUGAGAGAGAGUUUGAGAGAGAGAGAGAGAGUUGAGAGAGUUGAG 3«) with the 32P-labelled release strand (15 l/5«,5« CAAACUCUCUCUCAA 3«) and with each of the following unlabelled oligonucleotides of increasing length : 15 l/3«, 5«CUCAACUCUCUCAAC 3« ; 18 l/3«, 5« CUCAACUCUCUCAACUCU 3« ; 21 l/3«, 5« CUCAACUCUCUCAACUCUCUC 3« ; 24 l/3«, 5«CUCAACUCUCUCAACUCUCUCUCU 3« ; 27 l/3«, 5« CUCAACUCUCUCAACUCUCUCUCUCUC 3«. (B) Unwinding activity on double-hybrid RNA substrates. Increasing enzyme concentrations [25 nM (lanes 1, 7), 50 nM (lanes 2, 8), 100 nM (lanes 3, 9),200 nM (lanes 4, 10)] were added in a standard helicase assay using as a substrate either 45u 32P-labelled 15 l/5« 15l/3« (lanes 1–6) or 45u 15 l/5« 32P-labelled 15 l/3« (lanes 7–12). Lanes 5 and 11, no enzyme added. Lanes 6 and 12,heat-denatured substrates. Substrate concentration was 1±25 nM. S, Substrate ; P, product. (C) Minimal single-stranded regionrequired for helicase activity. A standard strand-displacement assay was performed using each of the five different substratesdescribed in (A) at a concentration of 1±25 nM in the presence or absence of 25 nM NS3. The ratio between the amount ofradioactivity associated with the release strand and the amount of total radioactivity was calculated for both the plus- andminus-enzyme (background) samples for each substrate. The values obtained by subtracting the specific background from eachexperimental data-point were plotted against the length of ssRNA. (D) Minimal single-stranded region required for RNA-bindingactivity on double-hybrid substrates. The indicated increasing enzyme concentrations were used in standard gel retardationreaction mixtures containing as substrates 45u annealed with 32P-labelled 15 l/5« and either 15 l/3«, 24 l/3« or 27 l/3«,respectively, at a concentration of 1±25 nM. The efficiency of the binding reaction was calculated as described in Methods.‘% B/BF’ refers to the ratio of protein-bound 32P-labelled probe to protein-bound plus free (unbound) 32P-labelled probe.
that binding of ATP to NS3 might exert on its relative affinityfor ssRNA and dsRNA. Previously we demonstrated thatformation of a stable complex of NS3 with a ssRNA oligo-nucleotide was severely impaired by the addition of 5 mMATP (Gallinari et al., 1998). Here we compared the bindingaffinity of the enzyme for the helicase double-hybrid substratein the presence or absence of 5 mM ATP (or its non-hydrolysable analogue AMP-PCP) (Fig. 3). In the experimentshown in Fig. 3 (A, B) addition of ATP to the reaction mixtureprior to addition of the enzyme determined a significantdecrease of the binding efficiency, which probably reflected adiminished affinity for the ssRNA region of the probe.Moreover, in these conditions, strand release was also inhibited(not shown), presumably as a consequence of a reduction in the
number of pre-formed enzyme–substrate complexes. Sub-stituting AMP-PCP for ATP produced a similar decrease in thebinding efficiency (not shown), thus suggesting that binding tothe nucleotide and not hydrolysis was responsible for theweakened enzyme–RNA interaction. When ATP was addedafter pre-incubation of the enzyme with the labelled substrate,no retarded band was visible and release of the labelled strandwas observed, as expected (not shown). However, addition ofAMP-PCP, which does not support NS3 unwinding activity(Gallinari et al., 1998), after pre-incubating the enzyme with thesubstrate caused a marked increase in the binding efficiency(Fig. 3C, D), which probably reflected an increased affinity forthe dsRNA region of the probe. In conclusion, the effect of theorder of addition of ATP or AMP-PCP on the level of binding
BDDI

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
Enzymology of HCV NS3 helicaseEnzymology of HCV NS3 helicase
Fig. 3. Effect of ATP and AMP-PCP on the NS3 binding affinity for ssRNA and dsRNA. (A) Increasing enzyme concentrations[25 nM (lanes 1, 4), 50 nM (lanes 2, 5) and 100 nM (lanes 3, 6)] were added in a standard gel retardation reaction mixtureusing as a probe 45u RNA annealed with 32P-labelled 15 l/5« and unlabelled 15 l/3« RNAs (see Fig. 2) at a concentration of1±25 nM. Lane 7, no enzyme added. Reactions were performed in 3 mM MgCl2 and either in the absence or in the presence of5 mM ATP. Where present, ATP was always added before the enzyme and after 15 min of pre-incubation at 23 °C, the bindingreactions were continued for an additional 30 min at 37 °C. Aliquots (8 µl) were analysed on a native 6%polyacrylamide/0±25¬ TBE gel using autoradiography. B, Protein-bound probe; F, free probe. (B) Quantification of theradioactive bands in the experiment shown in (A) was performed as described in Methods. (C) Gel retardation reactionmixtures were as in (A), but in lanes 2, 4 and 6 the non-hydrolysable ATP analogue AMP-PCP was added at the end of a15 min pre-incubation of the enzyme with the probe at 23 °C. In lanes 1, 3 and 5, no AMP-PCP was added. The bindingreactions were carried out for 30 min extra at 37 °C and samples were analysed as above. Lane 7, no enzyme added.(D) Quantification of the radioactive bands in the experiment shown in (C) was performed as described in Methods.
activity to the partial dsRNA substrate suggests that theenzyme–nucleotide complex has a lower affinity for ssRNAand a higher affinity for duplex RNA than does the non-complexed protein.
NS3 interacts with the stem–loop RNA structure (SL I)within the 3«-terminal 46 nt of the HCV genome
Analysis of the secondary structure of the X region at the3«-end of the HCV genomic positive-strand (Blight & Rice,1997) revealed a stable stem–loop structure (SL I) within the3«-terminal 46 bases containing a 6 nt single-stranded loop and2 nt representing bulges (Fig. 4A). A common role envisagedfor viral helicases is that of causing RNA strand separationduring genome replication. We assessed the ability of FL-NS3to bind a 46 nt RNA oligonucleotide representing SL I. Gelretardation and UV-cross-linking experiments (Fig. 4B, C,
respectively) indicated that NS3 is able to bind tightly and inan ATP-sensitive manner to the stable stem–loop structure,presumably interacting with the 6 ribonucleotide single-stranded loop. Furthermore, the competition experimentshown in Fig. 4 (D, E) indicated that a linear dsRNA containinga 3« ssRNA tail of 6 nt only partially inhibited the complexformation between NS3 and SL I RNA at concentrations atwhich the unlabelled SL I RNA did compete efficiently. Thisobservation suggests that the preferential binding observedmight depend not only on the presence of the 6 nt loop butalso on the presence of RNA secondary structure.
NS3 can resolve an SL I-containing stem–loop RNAstructure
Next we addressed whether the helicase activity of FL-NS3was able to resolve the SL I RNA structure by unwinding the
BDDJ

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
C. Paolini, R. De Francesco and P. GallinariC. Paolini, R. De Francesco and P. Gallinari
Fig. 4. Binding of NS3 to SL I. (A) Structure of SL I. A 46-mer RNA oligonucleotide with the sequence 5« GCAUGACUGCAGA-GAGUGCUGAUACUGGCCUCUCUGCAGAUCAUGU 3« was 32P 5«-end-labelled, denatured and renatured under the conditions foroligonucleotide annealing described in Methods. An RNA binding experiment was performed exactly as described in Fig. 3(A),using labelled SL I as a probe. At the end of the binding reaction, 8 µl aliquots were analysed by native PAGE (B) and theremaining 12 µl aliquots were UV-irradiated (0±12 J, 260 nM) for 15 min at 4 °C using a Stratalinker 2400 apparatus(Stratagene). Samples were analysed by SDS–PAGE (C). (D) Binding reactions were performed in the presence of 1±25 nMlabelled SL I probe, 20 nM enzyme and the indicated fold-excess concentrations of unlabelled specific (SL I) and non-specificcompetitor RNAs. The non-specific competitor was a partial dsRNA of unrelated sequence containing a 20 nt duplex region anda 6 nt single-stranded 3«-end tail. It was obtained by annealing the 26-mer oligonucleotide (5« GUUGAGAGAGAGAGAGUUUGA-GAGAG 3«) with the 32P-labelled 20-mer oligonucleotide (5« CAAACUCUCUCUCUCUCAAC 3«). (E) Quantification of theradioactive bands in the experiment shown in (D) was performed as described in Methods. E, Specific competitor RNA;*, non-specific competitor RNA.
stem region upon binding to the 6 nt loop. First attempts todevise an unwinding assay using optimized NS3 helicaseconditions and the 46-mer SL I RNA oligonucleotide as asubstrate failed to reveal any reaction product co-migratingwith the corresponding linearized form in a native gel (notshown). A possible explanation resides in the extremethermodynamic stability of SL I [∆G¯®26±5 kcal}mol witha melting temperature of 85 °C; Blight & Rice, 1997], whichprobably favours re-annealing of the base-paired region, thuspreventing the isolation of enzymatically melted RNAmolecules. Therefore, by annealing oligonucleotides 1 and 2schematized in Fig. 5 (A), we created a new stem–loop substrate
in which we extended both the 5«- and 3«-ends of SL I stemwith complementary tails of 10 nt and introduced a nick in themiddle of the original base-paired region. This new stem–loopsubstrate (SL I-l, Fig. 5A) allowed us to monitor strand releaserather than a melting product, with the assumption that thetwo unwinding reactions were equivalent in all other respects.To verify that the presence of the nick in the stem structurewould not destabilize the base-paired region by creating anartificial single-stranded tail which could trigger unwinding,we designed two negative control substrates. By annealingoligonucleotide 1 either with oligonucleotides 3 and 4 or 3aand 4a, blunt-ended linear dsDNA molecules were constructed
BDEA

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
Enzymology of HCV NS3 helicaseEnzymology of HCV NS3 helicase
Fig. 5. Unwinding activity on SL I-derived substrates. (A) Structures of the SL I-derived DNA substrates. SL I-l was obtained byannealing the 20-mer oligonucleotide 1 (5« TTGAGAGAGAGCATGACTGC 3«) with the 46-mer oligonucleotide 2(5« AGAGAGTGCCGATACTGGCCTCTCTGCAGATCATGCTCTCTCTCAA 3«). C1 was obtained by annealing to the 36-meroligonucleotide 3 (5« CCCACTGGCCTCTCTGCAGATCATGCTCTCTCTCAA 3«) both the oligonucleotide 1 and the 16-meroligonucleotide 4 (5« AGAGAGTGCCAGTGGG 3«). C2 was obtained by annealing to the 35-mer oligonucleotide 3a(5« CCCACTGGCCTCTCTGCAGTCATGCTCTCTCTCAA 3«) both the oligonucleotide 1 and the 15-mer oligonucleotide 4a(5« AGAGAGGCCAGTGGG 3«). In all substrates, oligonucleotide 1 was 5« end-labelled with 32P (release strand). (B) Unwindingactivity on SL I-derived substrates. Increasing enzyme concentrations [6±25 nM (lanes 1, 6, 11), 12±5 nM (lanes 2, 7, 12),25 nM (lanes 3, 8, 13), 50 nM (lanes 4, 9, 14)] were added in a standard strand-displacement assay using as a substrate SLI-l (lanes 1–5), C1 (lanes 6–10) or C2 (lanes 11–15). Lanes 5, 10, 15, no enzyme added to SL I-l, C1 or C2–containingsamples, respectively. Substrate concentration was 1±25 nM. (C) Quantification of radioactive bands in the experiment shown in(B) was performed as described in Methods and the efficiency of unwinding versus NS3 concentration determined as inFig. 2(C). E, SL I-l substrate ; *, C1 substrate ; _, C2 substrate. (D) Binding activity on SL I-derived substrates. Enzyme(6±25–50 nM) was added in standard gel retardation mixtures containing either SL I-l, C1 or C2 in the absence of MgCl2.Lane assignment was as in (B).
which no longer included the single-stranded loop but retainedthe nick-containing extended stem with (C1) or without (C2)bulges, respectively (Fig. 5A). As shown in Fig. 5 (B, C), thelabelled oligonucleotide 1 was efficiently released from the SLI-derived stem–loop structure by the NS3-associated helicaseactivity and the enzyme showed a significant preference forthis substrate compared with the negative controls. Thisdifference was reflected in a higher binding affinity for thestem–loop than for the blunt-ended substrates (Fig. 5D), thusconfirming that NS3 was able to interact with the 6 nt single-stranded loop and to unwind the double-stranded SL I stemstarting from it. The presence of the two bulges did not causeany significant additional destabilization of the base-pairedregion as judged by the two control curves in Fig. 5 (C).
Effect of heparin on ATP hydrolysis and helicaseactivities
NS3 helicase has a large intrinsic ATPase activity (kcat
¯3 s−") which is stimulated up to 30-fold by ssRNA or DNA(Preugschat et al., 1996). We have compared the stimulation ofNS3 ATPase activity in the presence of increasing con-
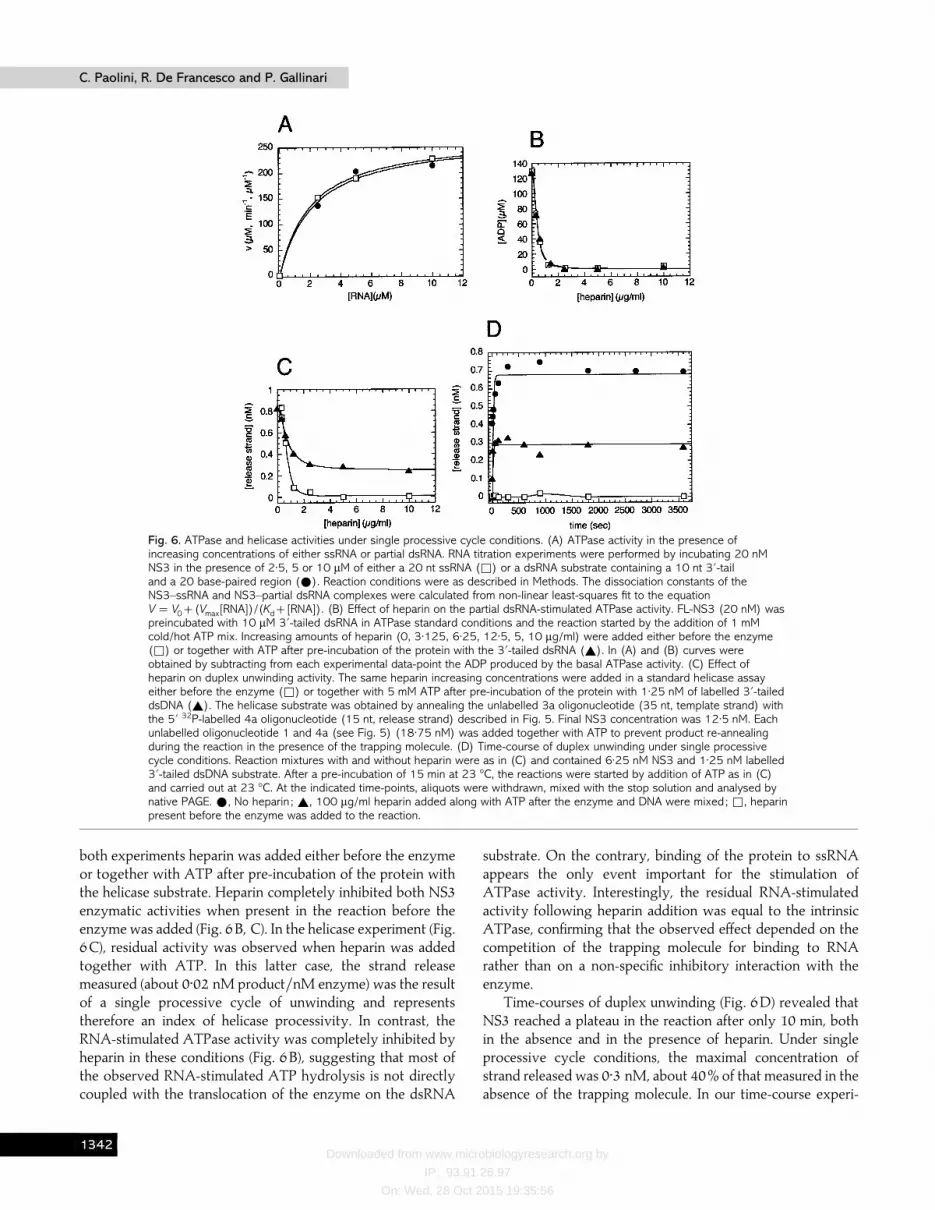
centrations of either a 20 nt ssRNA or a dsRNA substratecontaining a 10 nt 3«-tail (Gallinari et al., 1998). The twotitration curves shown in Fig. 6 (A) were very similar, indicatingthat the degree of stimulation of the ATPase activity observedwith the ssRNA and the tailed dsRNA was identical (approxi-mately 10-fold). The dissociation constant values for the twoactivator RNAs were also very similar (2 and 2±1 µM,respectively). This result suggests that the presence of a duplexregion does not influence the efficiency of the RNA-mediatedactivation of ATP hydrolysis. Although it is known that NS3unwinding activity is ATP-dependent (Tai et al., 1996), themechanism of coupling ATP hydrolysis to unwinding of theduplex is not completely understood. We reasoned that ifATPase activity in the presence of a 3«-tailed dsRNA waseffectively coupled with the unwinding reaction, we should beable to determine the enzyme processivity by measuring thehydrolysis of ATP under single processive cycle conditions.To this aim, we added increasing concentrations of heparin ina standard ATPase reaction stimulated by the addition of asaturating amount of 3«-tailed dsRNA (Fig. 6B). In parallel, weperformed the same heparin titration experiment adding thetrapping molecule in a standard helicase assay (Fig. 6C). In
BDEB
Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
C. Paolini, R. De Francesco and P. GallinariC. Paolini, R. De Francesco and P. Gallinari
Fig. 6. ATPase and helicase activities under single processive cycle conditions. (A) ATPase activity in the presence ofincreasing concentrations of either ssRNA or partial dsRNA. RNA titration experiments were performed by incubating 20 nMNS3 in the presence of 2±5, 5 or 10 µM of either a 20 nt ssRNA (*) or a dsRNA substrate containing a 10 nt 3«-tailand a 20 base-paired region (E). Reaction conditions were as described in Methods. The dissociation constants of theNS3–ssRNA and NS3–partial dsRNA complexes were calculated from non-linear least-squares fit to the equationV¯V0(Vmax[RNA])/(Kd[RNA]). (B) Effect of heparin on the partial dsRNA-stimulated ATPase activity. FL-NS3 (20 nM) waspreincubated with 10 µM 3«-tailed dsRNA in ATPase standard conditions and the reaction started by the addition of 1 mMcold/hot ATP mix. Increasing amounts of heparin (0, 3±125, 6±25, 12±5, 5, 10 µg/ml) were added either before the enzyme(*) or together with ATP after pre-incubation of the protein with the 3«-tailed dsRNA (_). In (A) and (B) curves wereobtained by subtracting from each experimental data-point the ADP produced by the basal ATPase activity. (C) Effect ofheparin on duplex unwinding activity. The same heparin increasing concentrations were added in a standard helicase assayeither before the enzyme (*) or together with 5 mM ATP after pre-incubation of the protein with 1±25 nM of labelled 3«-taileddsDNA (_). The helicase substrate was obtained by annealing the unlabelled 3a oligonucleotide (35 nt, template strand) withthe 5« 32P-labelled 4a oligonucleotide (15 nt, release strand) described in Fig. 5. Final NS3 concentration was 12±5 nM. Eachunlabelled oligonucleotide 1 and 4a (see Fig. 5) (18±75 nM) was added together with ATP to prevent product re-annealingduring the reaction in the presence of the trapping molecule. (D) Time-course of duplex unwinding under single processivecycle conditions. Reaction mixtures with and without heparin were as in (C) and contained 6±25 nM NS3 and 1±25 nM labelled3«-tailed dsDNA substrate. After a pre-incubation of 15 min at 23 °C, the reactions were started by addition of ATP as in (C)and carried out at 23 °C. At the indicated time-points, aliquots were withdrawn, mixed with the stop solution and analysed bynative PAGE. E, No heparin ; _, 100 µg/ml heparin added along with ATP after the enzyme and DNA were mixed; *, heparinpresent before the enzyme was added to the reaction.
both experiments heparin was added either before the enzymeor together with ATP after pre-incubation of the protein withthe helicase substrate. Heparin completely inhibited both NS3enzymatic activities when present in the reaction before theenzyme was added (Fig. 6B, C). In the helicase experiment (Fig.6C), residual activity was observed when heparin was addedtogether with ATP. In this latter case, the strand releasemeasured (about 0±02 nM product}nM enzyme) was the resultof a single processive cycle of unwinding and representstherefore an index of helicase processivity. In contrast, theRNA-stimulated ATPase activity was completely inhibited byheparin in these conditions (Fig. 6B), suggesting that most ofthe observed RNA-stimulated ATP hydrolysis is not directlycoupled with the translocation of the enzyme on the dsRNA
substrate. On the contrary, binding of the protein to ssRNAappears the only event important for the stimulation ofATPase activity. Interestingly, the residual RNA-stimulatedactivity following heparin addition was equal to the intrinsicATPase, confirming that the observed effect depended on thecompetition of the trapping molecule for binding to RNArather than on a non-specific inhibitory interaction with theenzyme.
Time-courses of duplex unwinding (Fig. 6D) revealed thatNS3 reached a plateau in the reaction after only 10 min, bothin the absence and in the presence of heparin. Under singleprocessive cycle conditions, the maximal concentration ofstrand released was 0±3 nM, about 40% of that measured in theabsence of the trapping molecule. In our time-course experi-
BDEC

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
Enzymology of HCV NS3 helicaseEnzymology of HCV NS3 helicase
ments, we were not able to measure with accuracy theamplitude of the rapid phase of the reaction. This valuedepends also on the length of the duplex region that isunwound by the enzyme in a single cycle, 15 base pairs in oursubstrate, and further experiments on much longer duplexsubstrates are needed to more precisely define the intrinsic rateof unwinding by NS3.
DiscussionRecent mechanistic studies on E. coli Rep helicase form the
basis for a detailed understanding of the kinetic mechanism forDNA unwinding by a helicase (Lohman & Bjornson, 1996).The alternative mechanisms proposed for the action of helicasehave been classified as ‘passive ’ or ‘ active ’ (reviewed by Birdet al., 1998). In the passive-type mechanism, the enzyme bindspreferentially to ssDNA and unwinds dsDNA by interactingwith the ssDNA that is formed transiently from the duplex asthe result of thermal fluctuations. In contrast, the active rollingmodel implies the binding of helicase alternatively to ssDNAand dsDNA. During catalysis, the alternating affinity forssDNA and dsDNA is coupled to ATP binding and hydrolysisas the enzyme rolls along the duplex. Although the originalactive model applied only to oligomeric enzymes with multipleDNA-binding sites on different subunits (Wong & Lohman,1992), a more general proposal was recently put forward fromthe comparison of two different structures of the monomericPcrA DNA helicase complexed with a single-strand-tailedDNA duplex in the presence or absence of a non-hydrolysableATP analogue (Velankar et al., 1999). In this latter model, theconformational changes that occur on binding ATP not onlydestabilize the interaction with ssDNA, allowing the sliding ofone of the helicase domains along it, but also set up the proteinsurface to bind duplex DNA, hence creating strain at the basepairing at the fork. Biochemical and structural data showedNS3 to be monomeric under a range of different conditions(Gallinari et al., 1998 ; Kim et al., 1998 ; Porter et al., 1998 ; Yaoet al., 1997). It has been demonstrated previously that ATPdecreases the affinity of the NS3 helicase domain for a (dU)
")by 95% (Preugschat et al., 1996). Our data indicate that the FLenzyme in complex with ATP or an ATP non-hydrolysableanalogue has a lower affinity for ssRNA and a higher affinityfor duplex DNA than does the non-complexed protein. Theseobservations are consistent with the ‘ inchworm’ activemechanism proposed for the monomeric PcrA helicase dis-cussed above, in which the free energy of hydrolysis of ATP isutilized for both unidirectional translocation and strandseparation. Additional crystallographic structures of NS3complexed with its substrates are, however, needed to shedmore light on the mechanistic details of its unwinding function.
It is reasonable to assume that following HCV infection, theinitiation of negative-strand RNA synthesis depends on aninitial recognition and specific binding of the replicativecomplex to the 3«-end of the viral genomic RNA. NS5B
polymerase has been recently shown to specifically interactwith conserved stem–loop structures in the 3« coding region ofthe HCV genomic RNA (Cheng et al., 1999). Since the 98 nt Xregion at the 3« terminus of the HCV genome is highlyconserved in sequence and has a stable secondary structure(Blight & Rice, 1997 ; Ito & Lai, 1997 ; Kolykhalov et al.,1996 ; Tanaka et al., 1996), it has been proposed to be involvedin viral RNA synthesis and in multiple protein–RNA inter-actions. It has been recently suggested that NS5B polymerasecan use this region as a cis-acting sequence to initiate HCVRNA synthesis in vitro (Oh et al., 1999), although previousstudies failed to demonstrate a specific interaction betweenNS5B and a viral RNA containing the 98 nt sequence (Chenget al., 1999 ;Lohmann et al., 1997). Furthermore, cellular proteinsincluding polypyrimidine tract-binding protein have beendemonstrated to bind specifically to the SL 2 and SL 3stem–loop structures of the conserved X region (Ito & Lai,1997 ; Tsuchihara et al., 1997). For efficient transcriptioninitiation at the 3«-end of the HCV positive- and negative-strands, NS3-associated unwinding activity might be requiredto remove the secondary structure on the template RNA. Ouranalysis of NS3 unwinding activity on double-hybrid sub-strates indicates that the minimal internal non-base-pairedregion for optimal strand displacement lies between 9 and 6 nt,while shortening this region down to 3 nt causes a decrease to25% of maximal activity. This would suggest that NS3 mightrequire three or more non-base-paired ribonucleotides totrigger its unwinding activity and resolve the secondarystructure elements on the template RNA. NS3 protein fromHCV-related dengue virus has been recently demonstrated tointeract with stem–loop structures in the 3« non-coding regionof the genomic RNA that plays an important role in theinitiation of the negative-strand RNA synthesis (Chen et al.,1997 ; Cui et al., 1998). Our data indicate that HCV NS3protein is able to bind tightly and with some specificity to thestable stem–loop structure SL I formed by the 3«-terminal 46bases of HCV positive-strand RNA (Blight & Rice, 1997).Furthermore, NS3-associated helicase activity is able to resolvethis kind of structure in a standard unwinding assay, pre-sumably through the initial binding to the 6 nt loop followedby the ATP-dependent translocation of the enzyme along thebase-paired stem. The specificity of the interaction mightdepend only on the presence of RNA secondary structure andnot on the primary sequence. Indeed all base changes identifiedwithin SL I in different HCV genotypes occur either in thesingle-stranded loop or, when they arise in the double-strandedstem, compensatory mutations are always present whichpreserve the secondary structure (Blight & Rice, 1997). Itwould be of interest to assess whether NS3 could interact withother stem–loop structures within the ends of the genomic andanti-genomic RNA, and the degree of binding selectivity in theabsence or presence of other replication factors.
Processivity of the NS3 helicase was inferred by its capacityto unwind a tailed RNA substrate containing a 15 nt duplex
BDED

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
C. Paolini, R. De Francesco and P. GallinariC. Paolini, R. De Francesco and P. Gallinari
region under single processive cycle conditions. We includedheparin to trap the enzyme not bound to RNA. Heparin hasbeen used as a nucleic acid analogue in studies of a number ofenzymes including DNA helicases (Korangy & Julin, 1992,1993) and affinity chromatography on a heparin column wasused in the purification protocol of native FL NS3 (Gallinari etal., 1998). The effect exhibited by the order of addition of thetrapping molecule on the amount of strand released by NS3helicase activity is consistent with a processive action of theenzyme. This implies that heparin does not bind to the enzymewhile unwinding its substrate. A similar inhibitory effect on thehelicase activity of NS3 was observed by including excessamounts of oligo(U)
")in the assay (not shown). Assuming that
the enzyme progresses on the duplex RNA by two base pairsfor every molecule of ATP hydrolysed (Porter et al., 1998),theoretically 200 nM ATP (half of the amount of base pairscontained in 20 nM of 20 nt dsRNA) should be hydrolysed inthe presence of heparin by 20 nM enzyme during a completesingle cycle of unwinding (assuming that all the enzyme iscatalytically active). On the contrary, the ATPase activitystimulated by partial dsRNA was completely inhibited byheparin, independent of the order of addition. This wouldsuggest that the extent of ATP hydrolysis measured in thisassay is not coupled, for the most part, with the translocationof the enzyme on duplex RNA but is only reflecting NS3binding to the ssRNA tail. This is consistent with theunanticipated features of a K1235E mutant in motif I of NS3helicase (Kim et al., 1997). This mutation almost completelyabolished both the intrinsic ATPase and helicase activities ofthe isolated helicase domain, although the RNA-stimulatedATPase activity was only partially reduced. Similar resultswere also reported for a different mutation at the same Kresidue in FL-NS3 (Wardell et al., 1999). It has been reportedthat, under single cycle conditions, the processivity of the NS3helicase domain is low (Porter et al., 1998). The evidencepresented here with FL-NS3 does not support or disagree withthis observation and additional experiments are required toestablish whether the isolated helicase domain and the FLenzyme are equally processive in vitro. Furthermore, theassociation to other cellular and}or viral proteins mightincrease the processivity of NS3 in vivo as has been shownfor several viral and cellular replication helicases (Boehmer,1998 ; Dong et al., 1996 ; Phillips et al., 1997).
We are especially grateful to L. Tomei and C. Steinku$ hler for manyhelpful discussions and for critically reviewing this manuscript. We thankM. Emili for artwork.
ReferencesBird, L. E., Subramanya, H. S. & Wingley, D. B. (1998). Helicases : aunifying structural theme? Current Opinion in Structural Biology 8, 14–18.
Blight, K. J. & Rice, C. M. (1997). Secondary structure determination ofthe conserved 98-base sequence at the 3« terminus of hepatitis C virusgenome RNA. Journal of Virology 71, 7345–7352.
Boehmer, P. E. (1998). The herpes simplex virus type-1 single-strandDNA-binding protein, ICP8, increases the processivity of the UL9protein DNA helicase. Journal of Biological Chemistry 273, 2676–2683.
Chen, C.-J., Kuo, M.-D., Chien, L.-J., Hsu, S.-L., Wang, Y.-M. & Lin,J.-H. (1997). RNA–protein interactions : involvement of NS3, NS5, and3« noncoding regions of Japanese encephalitis virus genomic RNA.Journal of Virology 71, 3466–3473.
Cheng, J.-C., Chang, M.-F. & Chang, S. C. (1999). Specific interactionbetween the hepatitis C virus NS5B RNA polymerase and the 3« end ofthe viral RNA. Journal of Virology 73, 7044–7049.
Cho, H.-S., Ha, N.-C., Kang, L.-W., Chung, K. M., Back, S. H., Jang,S. K. & Oh, B.-H. (1998). Crystal structure of RNA helicase fromgenotype 1b hepatitis C virus. Journal of Biological Chemistry 273,15045–15052.
Clarke, B. (1997). Molecular virology of hepatitis C virus. Journal ofGeneral Virology 78, 2397–2410.
Cui, T., Sugrue, R. J., Xu, Q., Lee, A. K. W., Chan, Y.-C. & Fu, J. (1998).Recombinant dengue virus type 1 NS3 protein exhibits specific viralRNA binding and NTPase activity regulated by the NS5 protein. Virology246, 409–417.
De Francesco, R., Pessi, A. & Steinku$ hler, C. (1998). The hepatitis Cvirus NS3 proteinase : structure and function of a zinc-containing serineproteinase. In Therapies for Viral Hepatitis, pp. 235–245. Edited by R. F.Schinazi, J.-P. Sommadossi & H. C. Thomas. London : InternationalMedical Press.
Dong, F., Weitzel, S. E. & von Hippel, P. H. (1996). A coupled complexof T4 DNA replication helicase (gp41) and polymerase (gp43) canperform rapid and processive DNA strand-displacement synthesis.Proceedings of the National Academy of Sciences, USA 93, 14456–14461.
Gallinari, P., Brennan, D., Nardi, C., Brunetti, M., Tomei, L.,Steinku$ hler, C. & De Francesco, R. (1998). Multiple enzymatic activitiesassociated with recombinant NS3 protein of hepatitis C virus. Journal ofVirology 72, 6758–6769.
Gallinari, P., Paolini, C., Brennan, D., Nardi, C., Steinku$ hler, C. & DeFrancesco, R. (1999). Modulation of hepatitis C virus NS3 protease andhelicase activities through the interaction with NS4A. Biochemistry 38,5620–5632.
Gorbalenya, A. E. & Koonin, E. V. (1993). Helicases : amino acidsequence comparison and structure–function relationship.Current Opinionin Structural Biology 3, 419–429.
Gwack, Y., Wook, D., Han, J. H. & Choe, J. (1995). NTPase activity ofhepatitis C virus NS3 protein expressed in insect cells. Molecular Cell 5,171–175.
Gwack, Y., Kim, D. W., Han, J. H. & Choe, J. (1996). Characterization ofRNA binding activity and RNA helicase activity of the hepatitis C virusNS3 protein. Biochemical and Biophysical Research Communications 225,654–659.
Houghton, M. (1996). Hepatitis C viruses. In Fields Virology, 3rd edn,pp. 1035–1058. Edited by B. N. Fields, D. M. Knipe & P. M. Howley.New York : Raven Press.
Ito, T. & Lai, M. C. (1997). Determination of the secondary structure ofand cellular protein binding to the 3«-untranslated region of the hepatitisC virus RNA genome. Journal of Virology 71, 8698–8706.
Jin, L. & Peterson, D. L. (1995). Expression, isolation, and character-ization of the hepatitis C virus ATPase}RNA helicase. Archives ofBiochemistry & Biophysics 323, 47–53.
Kanai, A., Tanabe, K. & Kohara, M. (1995). Poly(U) binding activity ofhepatitis C virus NS3 protein, a putative RNA helicase. FEBS Letters 376,221–224.
BDEE

Downloaded from www.microbiologyresearch.org by
IP: 93.91.26.97
On: Wed, 28 Oct 2015 19:35:56
Enzymology of HCV NS3 helicaseEnzymology of HCV NS3 helicase
Kim, D. W., Gwack, Y., Han, J. H. & Choe, J. (1995). C-terminal domainof the hepatitis C virus NS3 protein contains an RNA helicase activity.Biochemical and Biophysical Research Communications 215, 160–166.
Kim, D. W., Kim, J., Gwack, Y., Han, J. H. & Choe, J. (1997). Mutationalanalysis of the hepatitis C virus RNA helicase. Journal of Virology 71,9400–9409.
Kim, J. R., Morgernstern, K. A., Griffith, J. P., Dwyer, M. D., Thomson,J. A., Murcko, M. A., Lin, C. & Caron, P. R. (1998). Hepatitis C virusNS3 RNA helicase domain with a bound oligonucleotide : the crystalstructure provides insights into the mode of unwinding. Structure 6,89–100.
Kolykhalov, A. A., Feinstone, S. M. & Rice, C. M. (1996). Identificationof a highly conserved sequence element at the 3« terminus of hepatitis Cvirus genome RNA. Journal of Virology 70, 3363–3371.
Korangy, F. & Julin, D. A. (1992). A mutation in the consensus ATP-binding sequence of the RecD subunit reduces the processivity of theRecBCD enzyme from Escherichia coli. Journal of Biological Chemistry 267,3088–3095.
Korangy, F. & Julin, D. A. (1993). Kinetics and processivity of ATPhydrolysis and DNA unwinding by the RecBC enzyme from Escherichiacoli. Biochemistry 32, 4873–4880.
Korolev, S., Hsieh, J., Gauss, G. H., Lohman, T. M. & Waksman, G.(1997). Major domain swiveling revealed by the crystal structures ofcomplexes of E. coli Rep helicase bound to single-stranded DNA andADP. Cell 90, 635–647.
Korolev, S., Yao, N., Lohman, T. M., Weber, P. C. & Waksman, G.(1998). Comparisons between the structures of HCV and Rep helicasesreveal structural similarities between SF1 and SF2 super-families ofhelicases. Protein Science 7, 605–610.
Kwong, A. D., Kim, J. L., Rao, G., Lipovsek, D. & Raybuck, S. A. (1998).Hepatitis C virus NS3}4A protease. Antiviral Research 40, 1–18.
Lohman, T. M. & Bjornson, K. P. (1996). Mechanisms of helicase-catalyzed DNA unwinding. Annual Review of Biochemistry 65, 169–214.
Lohmann, V., Koch, J. O. & Bartenschlager, R. (1996). Processingpathways of the hepatitis C virus proteins. Journal of Hepatology 24,11–19.
Lohmann, V., Ko$ rner, F., Herian, U. & Bartenschlager, R. (1997).Biochemical properties of hepatitis C virus NS5B RNA-dependent RNApolymerase and identification of amino acid sequence motifs essential forenzymatic activity. Journal of Virology 71, 8416–8428.
Oh, J.-W., Ito, T. & Lai, M. M. C. (1999). A recombinant hepatitis C virusRNA-dependent RNA polymerase capable of copying the full-lengthviral RNA. Journal of Virology 73, 7694–7702.
Phillips, R. J., Hickleton, D. C., Boehmer, P. E. & Emmerson, P. T.(1997). The RecB protein of Escherichia coli translocates along single-stranded DNA in the 3« to 5« direction : a proposed ratchet mechanism.Molecular and General Genetics 254, 319–329.
Porter, D. J. T. (1998). A kinetic analysis of the oligonucleotide-modulated ATPase activity of the helicase domain of the NS3 proteinfrom hepatitis C virus. Journal of Biological Chemistry 273, 14247–14253.
Porter, D. J. T., Short, S. A., Hanlon, M. H., Preugschat, F., Wilson,J. E., Willard, D. H., Jr & Consler, T. G. (1998). Product release is themajor contributor to k
catfor the hepatitis C virus helicase-catalyzed
strand separation of short duplex DNA. Journal of Biological Chemistry273, 18906–18914.
Preugschat, F., Averett, D. R., Clarke, B. E. & Porter, D. J. T. (1996). Asteady-state and pre-steady state kinetic analysis of the NTPase activityassociated with the hepatitis C virus NS3 helicase domain. Journal ofBiological Chemistry 271, 24449–24457.
Rice, C. M. (1996). Flaviviridae : the viruses and their replication. In FieldsVirology, 3rd edn, pp. 931–960. Edited by B. N. Fields, D. M. Knipe & P.M. Howley. New York : Raven Press.
Studier, F. W., Rosenberg, A. H., Dunn, J. J. & Dubendorff, J. W.(1998). Use of the T7 RNA polymerase to direct expression of clonedgenes. Methods in Enzymology 185, 60–89.
Subramanya, H. S., Bird, L. E., Brannigan, J. A. & Wigley, D. B. (1996).Crystal structure of a DExx box helicase. Nature 384, 379–383.
Tai, C.-L., Chi, W.-K., Chen, D.-S. & Hwang, L.-H. (1996). The helicaseactivity associated with hepatitis C virus nonstructural protein 3 (NS3).Journal of Virology 70, 8477–8484.
Tanaka, T., Kato, N., Cho, M. J. & Shimotohno, K. (1996). Structure ofthe 3« terminus of the hepatitis C virus. Journal of Virology 70, 3307–3312.
Tsuchihara, K., Tanaka, T., Hijikata, M., Kuge, S., Toyoda, H., Nomoto,A., Yamamoto, N. & Shimotohno, K. (1997). Specific interaction ofpolypyrimidine tract-binding protein with the extreme 3«-terminalstructure of the hepatitis C virus genome, the 3«X. Journal of Virology 71,6720–6726.
Velankar, S. S., Soultanas, P., Dillingham, M. S., Subramanya, H. S. &Wigley, D. B. (1999). Crystal structures of complexes of PcrA DNAhelicase with a DNA substrate indicate an inchworm mechanism. Cell 97,75–84.
Wardell, A. D., Errington, W., Ciaramella, G., Merson, J. & McGarvey,M. J. (1999). Characterization and mutational analysis of the helicaseand NTPase activities of hepatitis C virus full-length NS3 protein. Journalof General Virology 80, 701–709.
Wong, I. & Lohman, T. M. (1992). Allosteric effects of nucleotidecofactors on Escherichia coli Rep helicase-DNA binding. Science 256,350–355.
Yao, N., Hesson, T., Cable, M., Hong, Z., Kwong, A. D., Le, H. V. &Weber, P. C. (1997). Structure of the hepatitis C virus RNA helicasedomain. Nature Structural Biology 4, 463–467.
Received 12 October 1999; Accepted 5 January 2000
BDEF