enhancement of charge remote fragmentation in protonated peptides by high-energy cid maldi-tof-ms...
TRANSCRIPT

ELSEVIER and la” Processes
International Journal of Mass Spectrometry and Ion Processes 169/l 70 (1997) 23 I-240
Enhancement of charge remote fragmentation in protonated peptides by high-energy CID MALDI-TOF-MS using ‘ ‘cold’ ’ matrices
E. Stimson”, 0. Truong”, W.J. Richter”, M.D. Waterfield”,h, A.L. Burlingame”.‘.*
Received 11 May 1997: revised 25 July 1997: accepted I4 August 1997
Abstract
Delayed extraction matrix-assisted laser desorption ionisalion time-of-flight mass spectrometry (DE-MALDI-TOF-MS) is employed to evaluate its potential for peptide sequencing using both post-source decay (PSD) and high-energy collision- induced dissociation (CID). This work provides evidence that complete amino-acid sequences may be obtained employing a dual approach including PSD of [M + H]+ ions using a “hot” matrix (cu-cyano-4-hydroxycinnamic acid, CHCA), followed by high-energy CID using “cold” matrices (2,5_dihydroxybenzoic acid, DHB; 2,6-dihydroxyacetophenone/di-ammonium hydro- gen citrate, DHAP/DAHC). This strategy ensures that PSD results in a rich variety of product ions derived from charge-driven processes that provide gross structural information. High-energy CID (20 keV collision energy range) of low internal energy [M + H]+ ions is then employed to reveal complementary side-chain detail (i.e. Leu/Ile distinction) in a manner highly selective for charge remote fragmentation (CRF), because PSD is largely reduced. As expected from the known behaviour of protonated peptides at IO keV collision energies, charge fixation at basic sites required for CRF is more pronounced in CID than in PSD. We have obtained spectra for a synthetic peptide that approximate the results and performance of MALDI high-energy CID obtained on sector-based instrumentation (EBE-oa-TOF). 0 I997 Elsevier Science B.V.
Kcyword.7: High-energy CID; MALDI; Peptide sequencing
1. Introduction
The recent incorporation of delayed extraction (DE) and modified detector design in MALDI- TOF mass spectrometry has brought this instrumentation into competitive performance with other existing ion optical systems for bio- macromolecular structural characterization due to its impressive mass-measurement accuracy,
* Corresponding author. Present address: Department of Pharma- ceutical Chemistry, University of California, San Francisco, CA 94143.0446, USA. Telephone: 415/476-5641, fax: 415/476-0688, email: [email protected]
high mass resolution and very high sensitivity [I 1. An application of unusually broad importance in biomedical research involves the rapid identifi- cation of two-dimensional gel-derived proteins [2,3] for correlation with differential gene expression [2,4] or discovery of gene function [5]. In the simplest terms, this involves enzymatic hydrolysis of gel-derived proteins followed by peptide mass mapping [2,3,6-141 and de novo sequence determination [2,4,1 1,151. While con- ventional MALDI mass spectra recorded in either the linear or reflector mode provide molecular- weight information for intact molecules [16], the mass spectra resulting from spontaneous
0168-I 176/97/$17.00 0 1997 Elsevier Science B.V. All rights reserved PII SO 16% I I76(97)00227-9

232 E. Stimson et al./International Journal of Mass Spectrometr?, and Ion Processes 169/l 70 (1997) 231-240
(metastable) decomposition of these species, termed post-source decay (PSD) [ 17,181, provide at least partial amino-acid composition and sequence information for identification of gel spots in tumour-cell lysates [ 19,201. Hence, such spectra are of considerable practical utility for use in protein, gene and EST database interrogation [ 19-231. Using further amino-acid compositional constraints provided by accurate molecular mass measurements (e.g. for Gln/Lys distinction), this information is sufficient to “identify” proteins (including homologous proteins) using mismatch tolerant-interrogation strategies [21-241. In general terms the nature and extent of fragmentation observed in PSD spectra is insufficient to establish complete, unambiguous peptide sequence de novo, but this can be influenced to some extent by the amount of internal energy deposited during ionisation (for example, choice of matrix, laser fluence, etc.), sample purity, amount of sample, etc. For instance, some matrices are “hotter” than others and produce markedly enhanced fragmentation patterns that comprise ion types analogous to those seen in multiply charged species in electrospray collision-induced disso- ciation [25], except they also display significant neutral losses from sequence ions as well as internal fragments [26]. However, even in these cases, the spectra do not usually provide sufficient structural information to solve more challenging problems such as the detailed structure elucidation of covalently modified proteins [27-301 and the design of oligonucleo- tide probes for gene cloning [ 15,26,30]. To date, this type of detailed information has been obtained mostly from high-energy collision- induced dissociation (CID) experiments using sector-based tandem instruments (for recent reviews, see [ 15,311). Of course, it would be highly desirable to develop less expensive techniques such as MALDI time-of-flight (TOF) mass spectrometry that would produce sequence information of comparable quality to that obtained using tandem sector and
FTMS instruments. In this study we demonstrate the importance of matrix selection in controlling the type and degree of fragmenta- tion processes observed in both PSD and high- energy CID spectra using a high-performance MALDI-TOF mass spectrometer of extended flight path [32].
2. Experimental
2. I. Materials
2. I. 1. Peptides Human transferrin was purchased from Sigma
(St Louis, MO) and des-Arg’-, des-Arg’-brady- kinin and the synthetic MHC peptide were provided by Dr C. M. John and Dr F. R. Masiarz, respectively. Approximately 50 pmol of transfenin was reduced and carboxymethylated by known procedures prior to digestion with bovine trypsin (2%, w/w; Boehringer Mannheim) in 50 mM ammonium bicarbonate buffer (pH 8.4) for 5 h at 37°C. The tryptic peptides were separated by capillary reverse-phase high-performance liquid chromatography (HPLC) (Applied Biosystems, 140D) on a C is column (150 x 0.5 mm, Brownlee) by using 0.1% trifluoroacetic acid (TFA) as solvent A and 0.08% TFA in acetonitrile as solvent B. Solvent B was increased by 0.5% per minute during the gradient elution and the column effluent was monitored at 2 10 nm. The purified peptides were analysed as described in the subsequent section.
2.2. Mass spectrometry
MALDI-PSD and -CID mass spectra were obtained on a Voyager Elite XL reflectron time-of-flight mass spectrometer (PerSeptive Biosystems, Inc., Framingham, MA) equipped with a nitrogen laser (337 nm, 3 ns pulse), delayed extraction (DE) and a collision cell for CID [32]. The delay time setting was 100 ns and the acceleration voltage was 20 kV. Spectra were
‘a
E. Stimson et al./International Journal of Mass Spectrometty and ton Processes 169/170 (1997) 231-240
_____
Y9
233
-...__,_ _ .._ ,. I I 1 .____+
200 400 600 8bO 1000 1200
!b F
Yl
8 I Q E 3
2
i
R N
- ml2 200 400 600 800 1000 1200
r ---
1’ F I
yd lM + HlJ i
260 460 660 aio 1000 do0
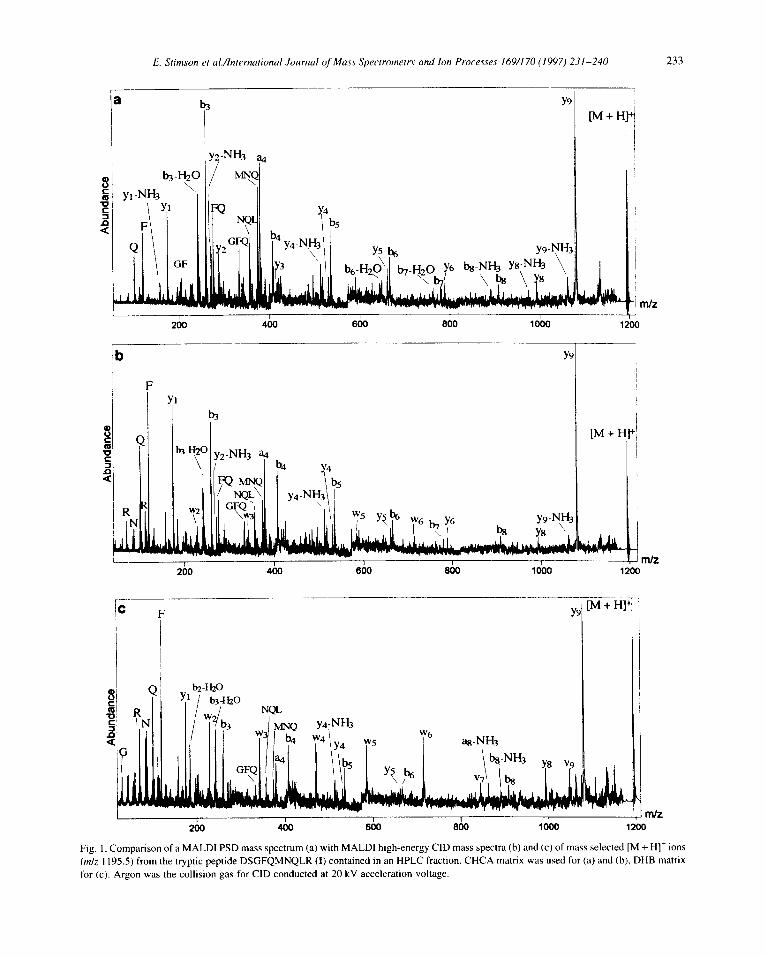
Fig. I. Comparison of a MALDI PSD mass spectrum (a) with MALDI high-energy CID mass spectra (b) and (c) of mass selected [M + H]’ ions (m/z I 195.5) from the tryptic peptide DSGFQMNQLR (I) contained in an HPLC fraction. CHCA matrix was used for (a) and (b), DHB matrix for (c). Argon was the collision gas for CID conducted at 20 kV acceleration voltage.
234 E. Stirnson et ul./Intrrrutionul Journal of Muss Spectrometq and lm Procr~srs 169/I 70 (I 997) 231-240
a
PGF
,‘3-NH3 .__
b5 I
I Y3 I yd’JH3
b7+H20 "7, 7
Y? Ilk?7 I 100 200 300 400 500 600 760 8&l m/z
C P/R F
l--n--- [M+H]-
m/z

E. Stimson et ul./Intermtional Journal of Muss Spectromet~ and Ion Processes 169/l 70 (1997) 231-240 235
produced by accumulating data from 100-200 laser shots, and recorded using a transient recorder of 2 ns channel width. For CID experiments the collision cell was filled with argon until the pressure in the source chamber reached approxi- mately 1x10” torr. To obtain complete PSD and CID spectra a series of reflectron TOF spectral segments were acquired, each optimized to focus fragment ions within different m/z ranges. Regions of each were stitched together in the usual way to produce the final PSD and CID spectra. Time-to-mass conversion was based on calibration using the monoisotopic [M + H]+ ion of angiotensin II (Sigma, St Louis, MO). For sample preparation, the peptide solutions were mixed with the chosen matrix solution to a final concentration of l-5 pmol ~1.’ and 1 ~1 of the solu- tion was placed on the sample plate and air dried. The matrices used were (a-cyano4-hydroxy- cinnamic acid (CHCA), 2,5-dihydroxybenzoic acid (DHB) (Hewlett-Packard, Palo Alto, CA) and 2,6-dihydroxyacetophenone/di-ammonium hydrogen citrate (DHAP/DAHC) (10: 1; v:v) (Aldrich, Milwaukee, WI) [33-351.
3. Results and discussion
The post-source decay (PSD) and high-energy collision-induced dissociation (CID) capabilities of the Elite XL [32] allow peptide sequencing by mass selection (i.e. flight time) of molecular ions of interest (typically [M + HI+) followed by mass analysis of the resulting fragments. Basically, two major categories of fragment classes are observed that provide information on peptide sequence and structure: (i) products of PSD such as bi, y; and internal ions resulting from charge-driven heterolytic cleavages of N-protonated CO-NH bonds and resembling, to
a certain degree, low-energy CID (eV collision energy range) [25,36]; and (ii) products of high- energy CID ( L 20 keV) such as d;, vj and Wj ions resulting from charge remote fragmentation (CRF) which require higher activation energies (in analogy to CRF at collision energies around 10 keV in sector instruments [2,11,15,36]). In the case of tryptic peptides, which have traditionally played a major role in sequencing by tandem MS [ 15,371, they contain a site of preferential protonation at their basic C-termini, and there- fore can undergo CRF reactions. In the following section a comparison is made between the PSD and CID behaviour of various arginine containing peptides using a “hot” matrix (CHCA) that favours extensive PSD, either alone or in conjunction with CID, or using a “cold” matrix (DHB or DHAP/DAHC) that minimizes PSD fragmentation as a contributor to CID.
The spectra shown in Fig. 1 a and Fig. 1 b permit direct comparison of PSD of the transfertin tryptic peptide DSGFQMNQLR (I,[M + H]+ at m/z 1195.5) with high-energy CID using the same CHCA matrix. At first glance both spectra display rather similar distributions of mainly a,, b;, and yj ions accompanied by abundant internal fragments. Charge retention at the basic site is not immediately obvious in the presence of the dominant y9 ion that arises from the well known preferential elimination of Asp [38]. This finding is not uncommon, however, since lesser extents of charge fixation at basic sites have been reported for PSD in comparison to low- and high-energy CID [36]. Except for Gln/Lys distinctions at positions 5 and 8 (identified as Gln by an independent accurate mass-measurement in reflector mode, not shown), and Leu/Ile at position 9, the complete order of amino-acids can be deduced from PSD alone (Fig. lb).
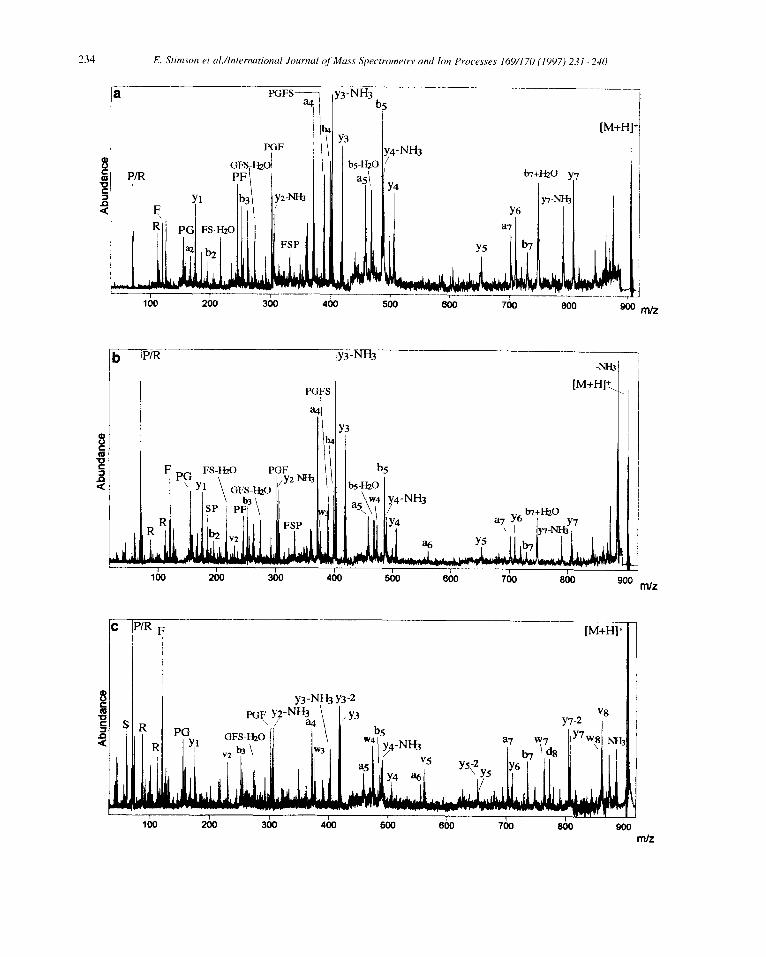
Fig. 2. Comparison of a MALDI PSD mass spectrum (a) with MALDI high-energy CID mass spectra(b) and (c) of mass selected [M + H]’ ions (4~ 905.5) from des-Arg’ bradykinin PPGFSPFR (II) using I pmol of peptide. CHCA matrix was used for (a) and (b), DHB matrix for (c). Argon was the collision gas for CID conducted at 20 kV acceleration voltage.
E. Stimson et ul./lnternutional Journal of Mass Spectrometr)~ and Ion Processes 169/170 (1997) 231-240
PGFS-Hz0
Y7 b7+Hzo
I
, -
as--
R I
R I

E. Stimson et al./lnternational Journal of Mass Spectrometry and Ion Processes 169/l 70 (1997) 231-240 231
While this example illustrates the usefulness of “hot” matrix PSD for maximizing the amount of sequence information that may be obtained for a particular sequence, often supplementary side- chain information will be required, necessitating the use of high-energy CID in additional experiments. To optimize the CID conditions with respect to the matrix, preliminary experiments were carried out using CHCA. The resulting high-energy CID spectrum (Fig. lb) now shows several CRF ions of low relative abundance, i.e. wz, w3, w5 and we, that indicate enhanced deposition of internal energy. Despite the low relative abundance of these CRF ions, w2 solves one of the ambiguities described above by permitting identification of residue 9 as Leu. In contrast to accurate molecular mass determination, the presence of w3 and w6 arising from side-chain neutral losses of C2H4N0 and C3H8N species, respectively, fail to distinguish Gln over Lys at positions 5 and 8 [39]. Overall, these CRF ions are important but could be easily overlooked because of the continuing prevalence of PSD fragmentation with this choice of matrix. The desired enhancement of this crucial side-chain information was investigated by selection of a matrix that can lower parent-ion internal energy and thus reduce excessive PSD fragmentation. Hence, use of a “cooler” matrix such as DHB [40,41] leads to markedly enriched CRF for this peptide as illustrated in Fig. lc. Except for the typical immonium ions at low mass (notably N, Q and F), the desired Wj ions together with v7 and vI) are now major contributors to fragment ion current. In particular, the predominance of ions of the Wj series in Fig. 1~ relative to the h; and vj ions indicate, in this first case, that the choice of the “cold” matrix in CID does result in a clear increase not only of charge fixation, but more importantly, of CRF over PSD.
To extend these findings, further measure- ments were performed using the same approach on the standard peptides des-Arg ‘- and des-Arg’- bradykinin (Fig. 2 and Fig. 3, respectively). These two isomers display relatively strong charge localization at either the C- or N-terminal Arg residue, respectively. The PSD spectrum (Fig. 2a, CHCA matrix) of the des-Arg ‘- isomer PPGFSPFR (II, [M + H]+ at m/z 904.5) shows a complete series of yj ions which define the complete peptide sequence without ambiguity, reflecting C-terminal charge fixation. However, some of the N-terminal bi ions such as b5 and b5-H20 are inconsistent with charge fixation at the C-terminus, but can be attributed to the preferential backbone cleavage commonly observed N-terminally to proline residues (Pro-6). High-energy CID in the same matrix (Fig. 2b) produces some wj and vi ions that are not readily recognized within the dense PSD contribution, but nevertheless indicate increased C-terminal charge fixation. Reduction of PSD in the CID spectrum obtained with DHB matrix (Fig. 2c) again enhances the expected vJ and wj ions. Indicative of Pro residues, y3-2 and y7-2 ions derived from CRF have also increased relative to their respective yj satellites that have charge-driven origin and form readily recognized doublets 2 Da apart. Although the principle of the approach chosen is confirmed, in this case the CRF ions provide only redundant sequence information.
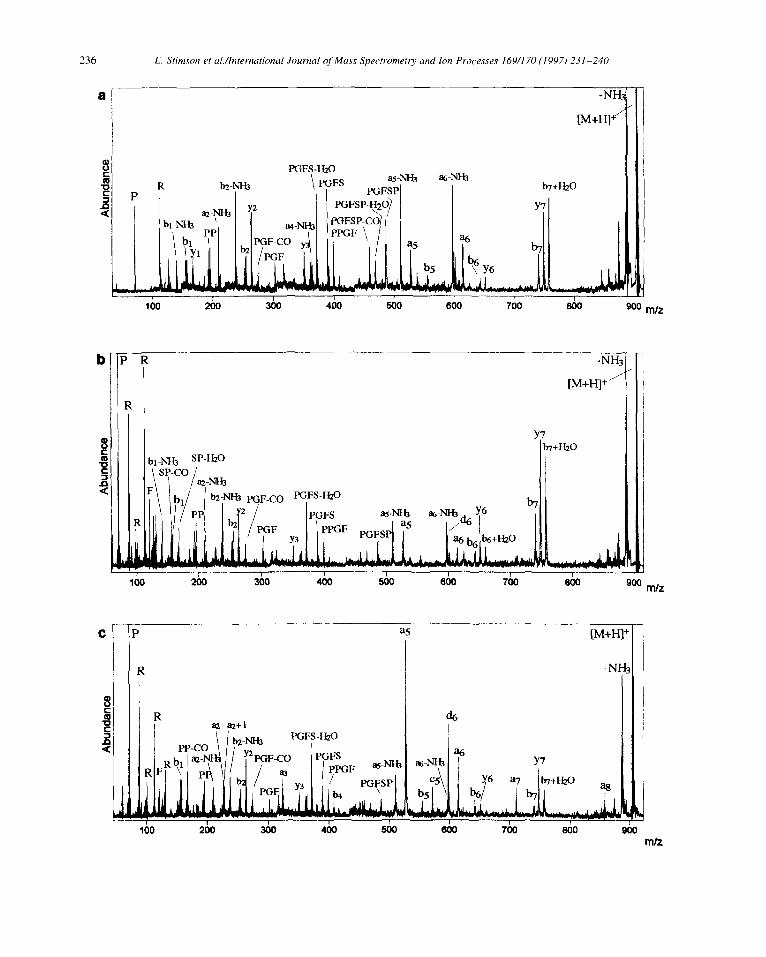
Analogous PSD and CID experiments were carried out on the des-Arg’ isomer RPPGFSPF (III, M + HI+ at m/z 904.5). This peptide is known to favour N-terminal charge fixation. Thus, in the PSD spectrum of III with CHCA matrix (Fig. 3a), an extensive series of a;-type ions and related secondary products (ai-NH3) are observed along with some C-terminal bi
Fig. 3. Comparison of a MALDI PSD mass spectrum (a) with MALDI high-energy CID mass spectra (b) and (c) of mass selected [M + H]’ ions (m/z 905.5) from des-Arg9-bradykinin RPPGFSPF (III) using 1 pmol of peptide. CHCA matrix was used for (a) and (b), DHAP/DAHC (IO: I; WV) matrix for (c). Argon was the collision gas for CID conducted at 20 kV acceleration voltage.
238 E. Stimson et al./lntermtional Journal of Muss Spectrometry and ton Processes 169/170 (1997) 231-240
b+W a
bs- PR
JH3
PRTLV-N&
I
[M+Kl’
C 1005
PRTL-NH3
PRTNH3 b-NH3 \ ds I.
do
3
It; ’ R
IJd
.H20
ds \
PR !- 1
9
LV-CO ,
i LV
jll
qH3

E. Stimson et ul./lntrrationrrl Journul of Mass Spectrometry and Ion Processes 169/170 (1997) 231-240 239
ions and derivatives thereof (e.g. b, + Hz0 [42,43]). Also, internal ions such as PPGF, PGFS and PGF are observed that arise from favourable N-terminal cleavage at proline residues. The C-terminal yj ions (notably y?, y6 and y,) are exceptions in view of strong N-terminal charge fixation, and are due to the same “proline effect”. As expected, high-energy CID using CHCA (Fig. 3b) enhances the relative abundance of immonium ions (P, R) and, to some degree, induces additional formation of the typical N-terminal CRF ion d6 that indicates Ser-6 as the only residue in III capable of direct side-chain loss. As in the previous examples, high-energy CID using a “cool” matrix (in this case DHAP/DAHC in Fig. 3c) gives enhanced CRF ions exemplified by the dominance of the d6 ion over other neighbouring peaks along with strong N-terminal charge fixation. In addition, while the a2 + 1 ion is clearly a CRF product, the increased abundancies of ai ions (cf. a5, a6 and a7) also suggest at least partial CRF origin.
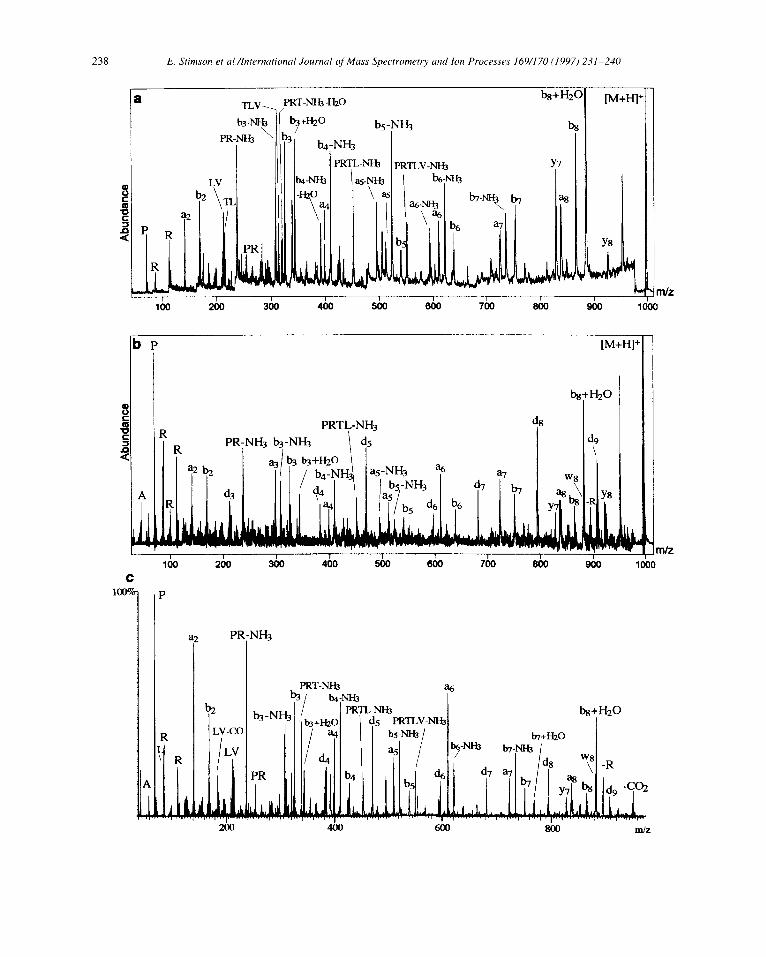
Finally, the same set of experiments were performed on a synthetic MHC peptide, APRTLVLLL (IV,[M + H]+ ion at m/z 995.6) that possesses Arg in a non-terminal position (Arg-3). This peptide still produces ions which suggest at least some degree of charge fixation close to the N-terminus. Hence, even PSD with a CHCA matrix gives a spectrum of IV (Fig. 4a) that shows mainly N-terminal a; and b, type ions along with products of addition or elimination of small neutrals (e.g. b8 + H20, b5-NH3 etc.). High-energy CID in CHCA (not shown), gives the expected spectral pattern that is still “rich” in PSD, whereas only three of the N-terminal di ions, namely Ltg, d5, and dg, gain moderate abundance. These
latter ions now constitute a complete ion series ranging from d3 to dg and are greatly enhanced throughout the entire high-energy CID spectrum acquired with the “cold” matrix DHB (Fig. 4b). Importantly, side-chain losses of C3H7 from corresponding ai + 1 precursors produce d 5, d7, d8 and dg ions that unambiguously define the residues at positions 5, 7, 8 and 9 as Leu rather than Ile. It is interesting to note that high-energy MALDI- CID performed on a sector-TOF hybrid instrument with orthogonal acceleration (EBE-ao-TOF) previously has shown almost identical CRF behaviour of this peptide (Fig. 4c, adapted with permission from [26]).
4. Conclusions
The combination of “hot” matrix DE- MALDI-TOF PSD with ‘ ‘cold’ ’ matrix high-energy CID is shown to approximate pre- sent-day performance of well-established tech- niques using relatively expensive tandem (sector-type) instrumentation for peptide- sequence determination. The use of a “cold” matrix in conjunction with high-energy CID effectively increases, relative to PSD, charge fixation at basic residues, which is an essential requirement for CRF. This is in accordance with results reported on previous CID measurements carried out at the 10 keV collision energy level [36]. However, to become truly competitive in terms of sensitivity and component selection specificity, both the timed precursor ion selection “mass resolution” currently available with the XL instrument for PSD and CID, as well as the efficiency of the CID process itself, requires further improvement.
Fig. 4. Comparison of a MALDI PSD mass spectrum (using 1 pmol peptide) (a) with MALDI high-energy CID mass spectra taken with the Elite XL using 2 pmol peptide (b) and an EBE-ao-TOF hybrid instrument (see [26]) (c) of mass selected [M + HI’ ions (m/z 995.6) from the synthetic MHC peptide APRTLVLLL (IV). CHCA matrix was used for (a), DHB matrix for(b) and (c). Argon and xenon were the collision gases for CID conducted at 20 kV or 0.8 kV acceleration voltage for (b) and (c), respectively.

240 E. Stimson et al./lntermtional Journal of Mass Spectrometg and Ion Processes 169/l 70 (I 997) 231-240
Acknowledgements
We acknowledge Rainer Cramer at the Ludwig Institute for helpful discussions. Financial support was provided by the Ludwig Institute for Cancer Research, London Branch.
References
[I] M. Vestal, Desorption ‘96, Bomholm, Denmark, 1996. [2] SC. Hall, D.M. Smith, F.R. Masiarz, V.W. Soo, H.M. Tran,
L.B. Epstein. A.L. Burlingame, Proc. Nat1 Acad. Sci. USA 90 (1993) 1927.
[3] S. D. Patterson, Biochem. Sot. Trans. (1996) 255. [4] K.R. Clauser, S.C. Hall, D.M. Smith, J.W. Webb, L.E.
Andrews, H.M. Tran, L.B. Epstein, A.L. Burlingame, Proc. Nat] Acad. Sci. USA 92 (1995) 5072.
[5] E. Lander, Science 274 (1966) 536. [6] W.J. Henzel, T.M. Billeci, J.T. Stults, S.C. Wang, C. Grimley,
C. Watanabe, Proc. Nat] Acad. Sci. USA 90 (I 993) 501 I. [7] D.J. Pappin, P. Hojrup, A.J. Bleasby, Curr. Biol. 3 (1993) 327. [8] H.H. Rasmussen, E. Mortz, M. Mann, P. Roepstorff, J.E. Celis,
Electrophoresis 15 (I 994) 406. [9] H. Ji, R.H. Whitehead, G.E. Reid, R.L. Moritz, L.D. Ward,
R.J. Simpson. Electrophoresis 15 (I 994) 391. [lo] J.T. Stults, W.J. Henzel, S.C. Wang, C. Watanabe, in: A.L.
Burlingame, S.A. Carr (Eds.), Mass Spectrometry in the Biological Sciences, Humana Press, Totowa, NJ, 1996, p. 151.
[II] SC. Hall, D.M. Smith, K.R. Clauser, L.E. Andrew& F.C. Walls, J.W. Webb, H.M. Tran, L.B. Epstein, A.L. Burlingame. in: A.L. Burlingame, S.A. Carr (Eds.), Mass Spectrometry in the Biological Sciences, Humana Press. Totowa, NJ, 1996, p. 17 1.
[ 121 M. Wilm, T. Houthaeve, G. Talbo, R. Kellner, P. Mortensen, M. Mann, in: A.L. Burlingame, S.A. Carr (Eds.), Mass Spectrometry in the Biological Sciences, Humana Press, Totowa, NJ, 1996, p. 245.
[ 131 D. Figeys, A. Ducret, J.R. Yates III, R. Aebersold, Nature Biotech. 14 (I 996) 1579.
[ 141 A. Shevchenko, O.N. Jensen, A.V. Podtelejnikov, F. Sagliocco, M. Wilm, 0. Vorm, P. Mortensen, A. Shevchenko, H. Bouch- erie, M. Mann, Proc. Natl Acad. Sci. USA 93 (1996) 14440.
[15] K.F. Medzihradszky, A.L. Burlingame, Methods: A Companion to Methods in Enzymology 6 (1994) 284.
[ 161 R.C. Beavis, B.T. Chait, Proc. Nat1 Acad. Sci. USA 84 (1990) 6873.
[ 171 B. Spengler, D. Kirsch, R. Kaufmann, Rapid Commun. Mass Spectrom. 5 (1991) 198.
[I81 R. Kaufman, D. Kirsch, B. Spengler, Int. J. Mass Spectrom. Ion Processes 131 (1994) 355.
[I91
[201
PII
WI P31 ~241
~251
WI
~271
WI
~291
1301
[311
[321
[331
[341
[351
[361
[371 [381
[391 [401
r411
r421
[431
L.B. Epstein, D.M. Smith, N.M. Matsui, H.M. Tran, C. Sullivan, 1. Raineri, A.L. Burlingame, K.R. Clauser, S.C. Hall, L.E. Andrews. Electrophoresis 17 (1996) 1655. C.M. Sullivan, D.M. Smith, N.M. Matsui, L.E. Andrews, K.R. Clauser, A. Chapeaurouge, A.L. Burlingame, L.B. Epstein, Cancer Res. 57 (1997) I 137. K.R. Clauser, P. Baker. A.L. Burlingame, Proceedings of the 44th ASMS Conference on Mass Spectrometry and Allied Topics, Portland, OR, 1996, p. 365. http://rafael.ucsf.edu. K.R. Clauser, P. Baker, A.L. Burlingame, in preparation. K.R. Clauser, R. Foulk, P. Baker, S. Fisher, A.L. Burlingame, Proceedings of the 45th ASMS Conference on Mass Spectro- metry and Allied Topics, Palm Springs. CA, 1997. D.F. Hunt, J.R. Yates, J. Shabanowitr, S. Winston, C.R. Hauer. Proc. Nat1 Acad. Sci. USA 83 (1986) 6233. K.F. Medzihradszky, R.H. Bateman. M.R. Green, G.W. Adams, A.L. Burlingame, J. Am. Sot. Mass Spectrom. 7 (1996) I. S.X. Wang, M. Mure, K.F. Medzihradszky, A.L. Burlingame, D.E. Brown, D.M. Dooley, A.J. Smith, H.M. Kagan. J.P. Klinman, Science 273 (1996) 1078. T. Nakamura, Z. Yu, M. Fainzilber. A.L. Burlingame, Protein Sci. 5 (1996) 524. Y. Qiu, L. Z. Benet, A. L. Burlingame, Drug Metab. Disp. submitted. K.F. Medzihradszky, D.A. Maltby, Y. Qiu, Z. Yu, S.C. Hall, Y. Chen, A.L. Burlingame, Int. J. Mass Spectrom. Ion Processes 160 (1997) 357. A.L. Burlingame, R.K. Boyd, S.J. Gaskell, AnalChem. 68 (1996) 599R. M.L. Vestal, P. Juhasz, S.A. Martin, Rapid Commun. Mass Spectrom. 9 (1995) 1044. J.J. Gorman, B.L. Ferguson, T.B. Nguyen, Rapid Commun. Mass Spectrom. IO (1996) 529. C.J. Pitt, J.J. Gorman, Rapid Commun. Mass Spectrom. IO (1996) 1786. U. Pieles, W. Zuercher, M. Schaer, H.E. Moser, Nucl. Acid Res. 21 (1993) 1786. E.C. Rouse, W. Yu, S.A. Martin, J. Am. Sot. Mass Spectrom. 6 (1995) 822. K. Biemann, H. Scoble, Science 237 (1987) 992. W. Yu, J.E. Vath, M.C. Huberty, S.A. Martin, Anal. Chem. 65 (1993) 3015. K. Biemann, Methods Enzymol. I93 ( 1990) 468. K. Strupat, M. Karas, F. Hillenkamp, Int. J. Mass Spectrom. Ion Proc. 1 I1 (1991) 89. B. Spengler, D. Kirsch, R. Kaufmann, J. Phys. Chem. 96 (1992) 9678. G.C. Thome, K.D. Ballard, S.J. Gaskell, J. Am. Sot. Mass Spectrom. 1 (1990) 249. K.D. Ballard, S.J. Gaskell, J. Am. Chem. Sot I I4 (1992) 64.