drug approval system in malaysia
TRANSCRIPT

1
DRUG APPROVAL SYSTEM
IN MALAYSIA
Rosilawati Ahmad
Deputy Director, Centre for Product Registration
National Pharmaceutical Regulatory Agency (NPRA), Malaysia
2019 K-Pharma Academy (KPA) for ASEAN
Sharing content
Intro-Pharmaceutical
Services Program& NPRA
Categories of Products & Registration
requirements
Registration process & timelines
Issues Identified in Korean product dossier
assessed at NPRASummary
2
Sharing content
Intro-Pharmaceutical
Services Program & NPRA
Regulatory activities
Registration requirements &
process
Challenges & Regulatory reform
Summary
3
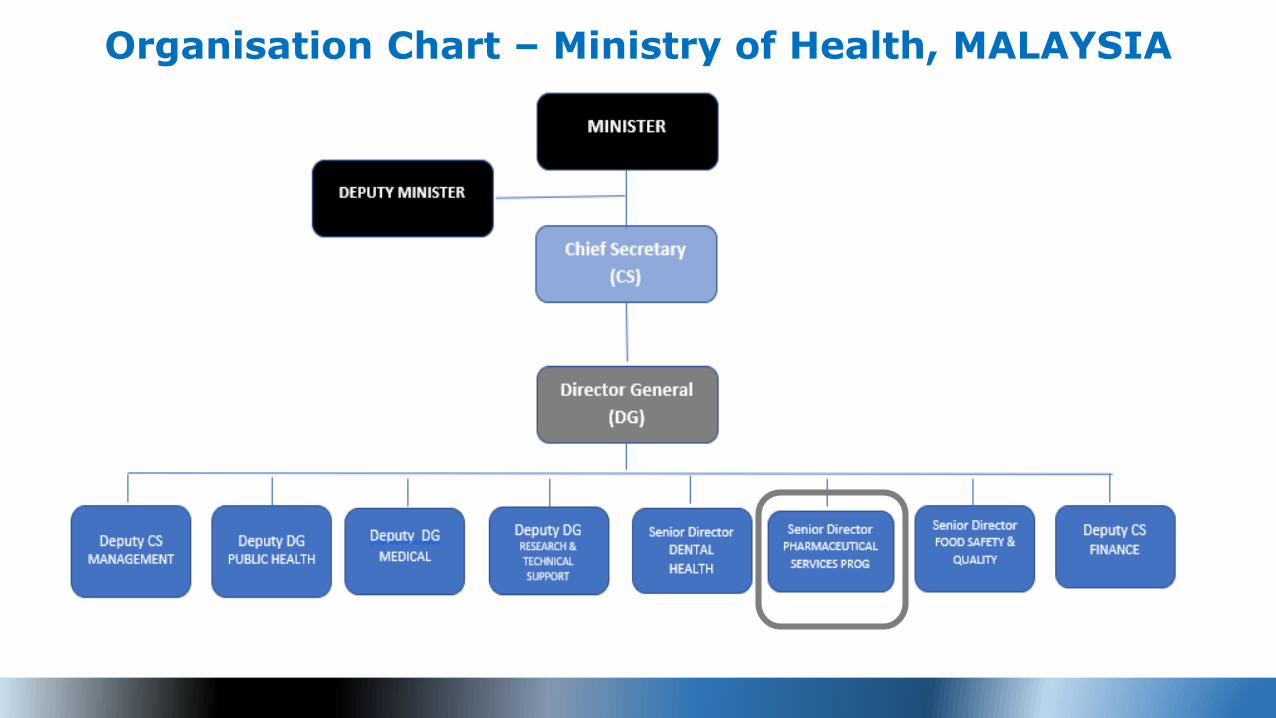
Organisation Chart – Ministry of Health, MALAYSIA
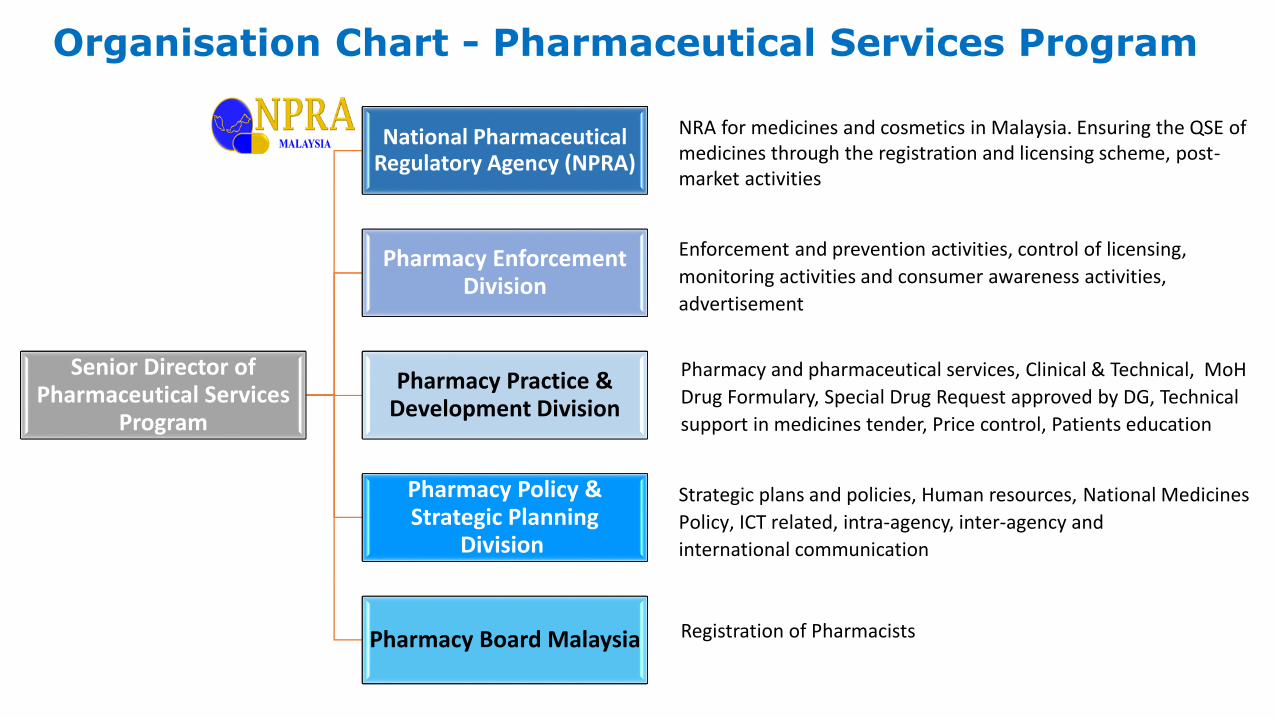
Organisation Chart - Pharmaceutical Services Program
Senior Director of Pharmaceutical Services
Program
National Pharmaceutical Regulatory Agency (NPRA)
Pharmacy Enforcement Division
Pharmacy Practice & Development Division
Pharmacy Policy & Strategic Planning
Division
Pharmacy Board Malaysia
Pharmacy and pharmaceutical services, Clinical & Technical, MoH
Drug Formulary, Special Drug Request approved by DG, Technical
support in medicines tender, Price control, Patients education
Strategic plans and policies, Human resources, National Medicines
Policy, ICT related, intra-agency, inter-agency and
international communication
Enforcement and prevention activities, control of licensing,
monitoring activities and consumer awareness activities,
advertisement
Registration of Pharmacists
NRA for medicines and cosmetics in Malaysia. Ensuring the QSE of medicines through the registration and licensing scheme, post-market activities

Poisons Act 1952 (Revised 1989)
Sales of Drugs Act 1952 (Revised 1989)
Dangerous Drugs Act 1952 (Revised 1980)
Registration of Pharmacists Act 1952 (Revised 1989)
Medicine (Advertisement & Sale) Act 1956 (Revised 1983)
Legislation

Legislation
Control of Drugs and Cosmetics Regulations CDCR 1984, Regulation 7(1) states:
• No person shall manufacture, sell, supply, import, possess or administer any product unless:
• (a) the product is a registered product, and
• (b) the person holds the appropriate license required & issued under these Regulations.
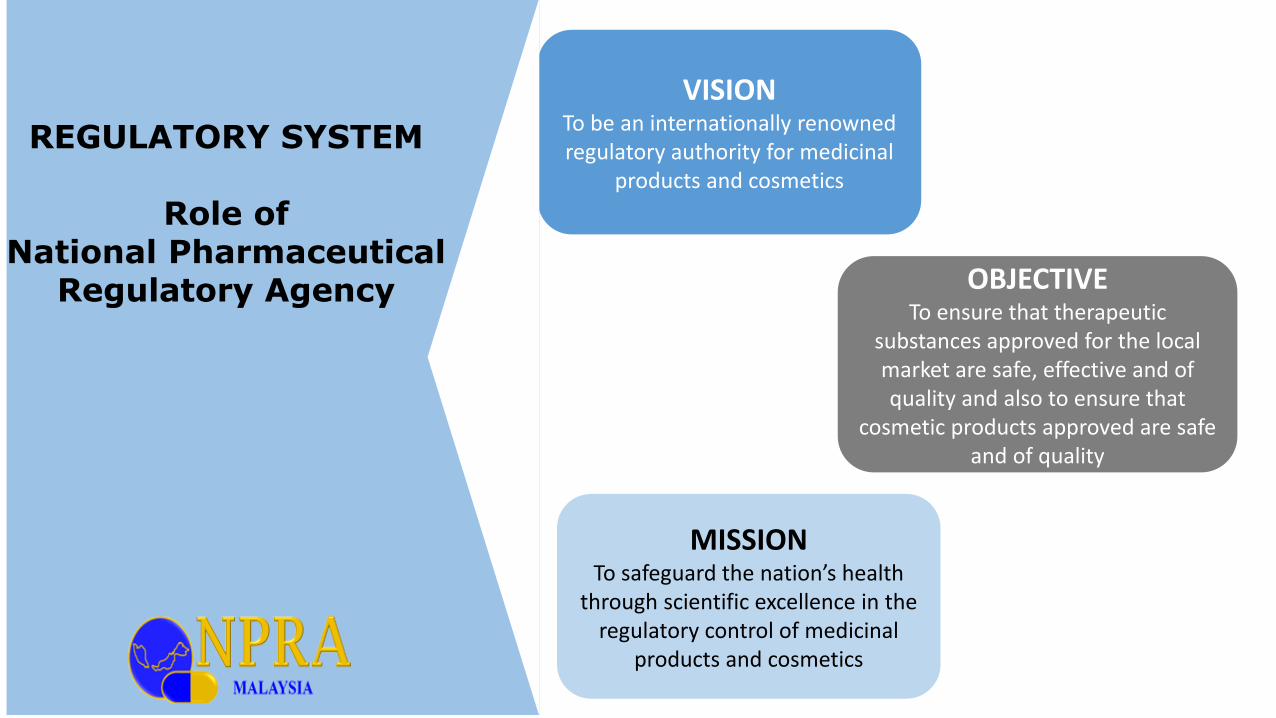
VISIONTo be an internationally renowned regulatory authority for medicinal
products and cosmetics
MISSIONTo safeguard the nation’s health
through scientific excellence in the regulatory control of medicinal
products and cosmetics
OBJECTIVETo ensure that therapeutic
substances approved for the local market are safe, effective and of quality and also to ensure that
cosmetic products approved are safe and of quality
REGULATORY SYSTEM
Role of National Pharmaceutical
Regulatory Agency
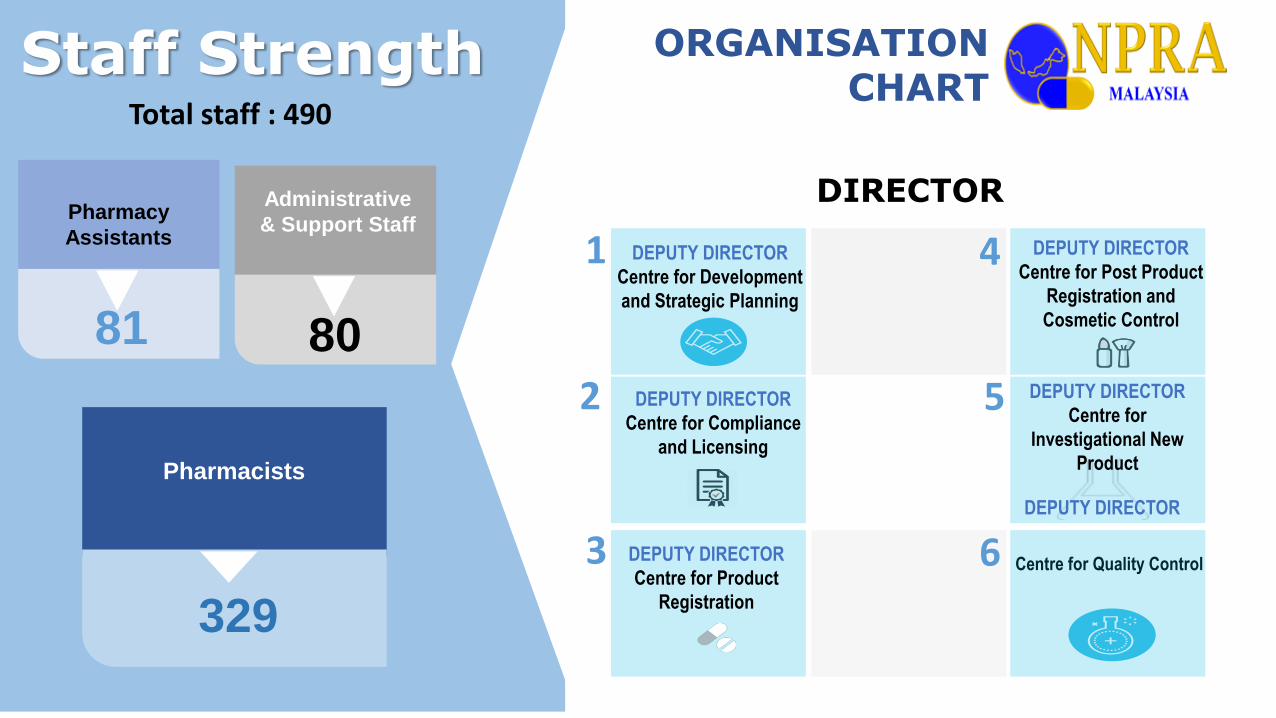
ORGANISATION CHART
81
Pharmacy
Assistants
329
Pharmacists
Total staff : 490
Staff Strength
Centre for Quality Control
DEPUTY DIRECTOR
Centre for Post Product
Registration and
Cosmetic Control
DIRECTOR
DEPUTY DIRECTOR
Centre for
Investigational New
Product
DEPUTY DIRECTOR
Centre for Product
Registration
DEPUTY DIRECTOR
Centre for Compliance
and Licensing
DEPUTY DIRECTOR
Centre for Development
and Strategic Planning
80
Administrative
& Support Staff
1
3
2
4
5
6DEPUTY DIRECTOR
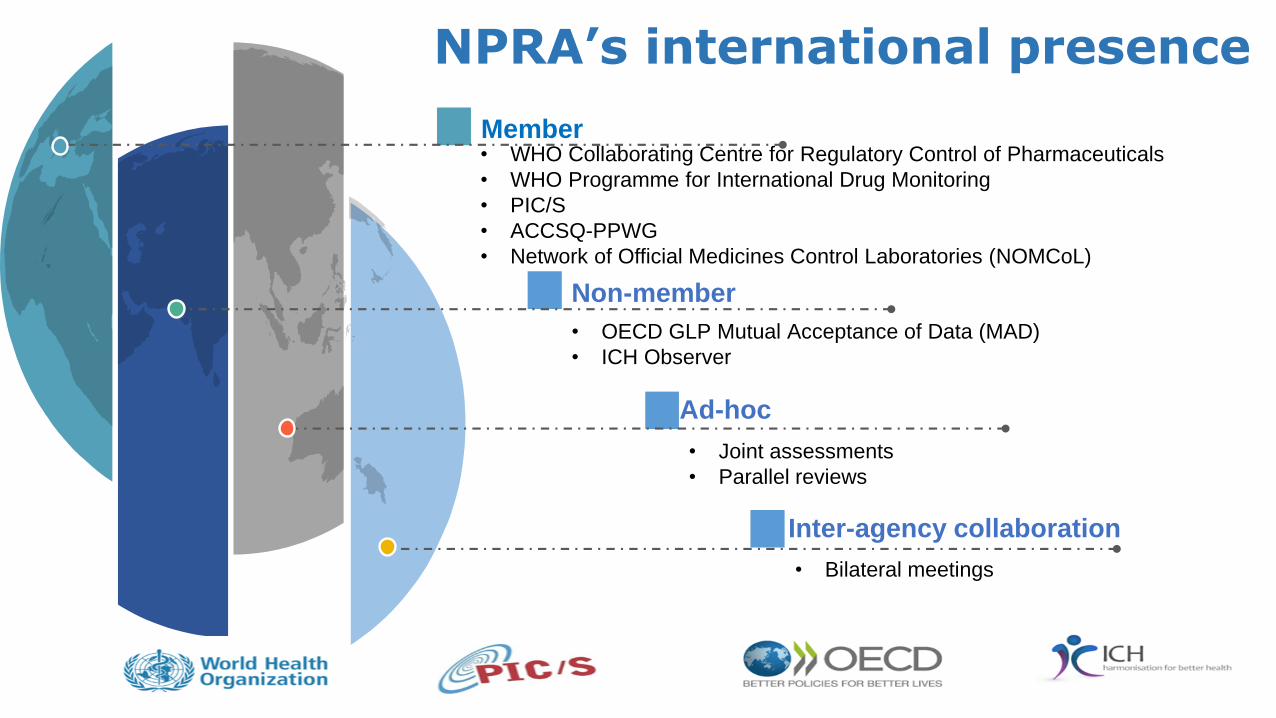
NPRA’s international presence
10
Member
Non-member
Ad-hoc
Inter-agency collaboration
• WHO Collaborating Centre for Regulatory Control of Pharmaceuticals
• WHO Programme for International Drug Monitoring
• PIC/S
• ACCSQ-PPWG
• Network of Official Medicines Control Laboratories (NOMCoL)
• OECD GLP Mutual Acceptance of Data (MAD)
• ICH Observer
• Joint assessments
• Parallel reviews
• Bilateral meetings
Sharing content
Intro-Pharmaceutical
Services Program& NPRA
Categories of Products & Registration
requirements
Registration process & timelines
Issues Identified in Korean product dossier
assessed at NPRASummary
11
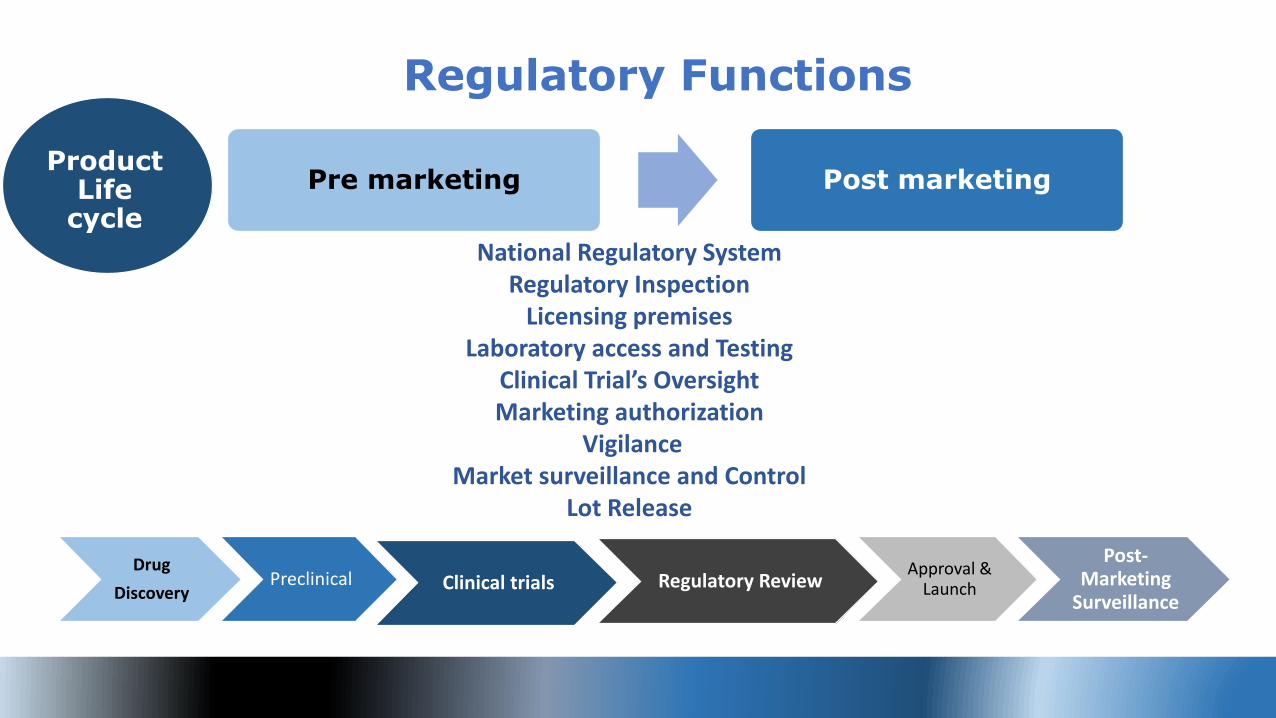
12#RegulatoryReform
Pre marketing Post marketing
Regulatory Functions
Product Life
cycle
Drug
DiscoveryPreclinical Clinical trials Regulatory Review
Approval & Launch
Post-Marketing
Surveillance
National Regulatory System Regulatory Inspection
Licensing premises Laboratory access and Testing
Clinical Trial’s Oversight Marketing authorization
Vigilance Market surveillance and Control
Lot Release
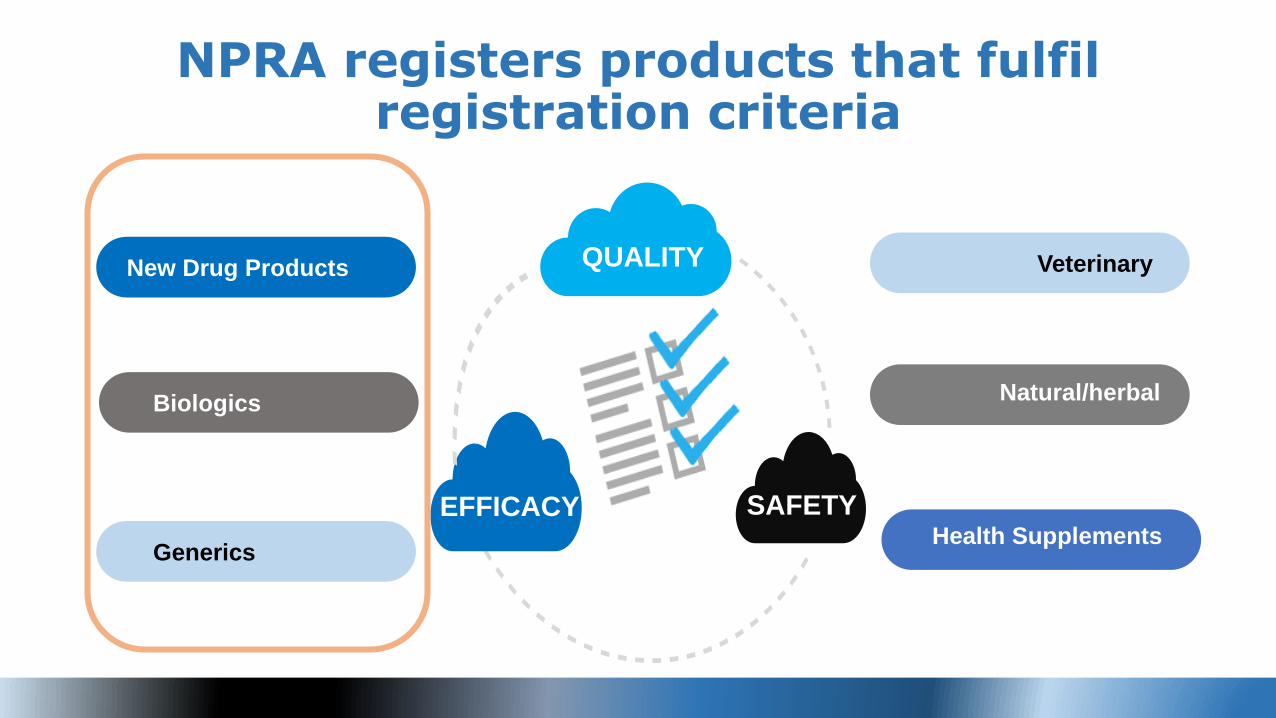
NPRA registers products that fulfil registration criteria
New Drug Products
Biologics
Health Supplements Generics
Natural/herbal
VeterinaryQUALITY
EFFICACY SAFETY
13

New Drug ProductAny pharmaceutical product that has not been previously registered in
accordance with the legal provisions in Malaysia

Types of New Drug Product
New Chemical Entities
Hybrid
15
Product containing an active moiety/ radiopharmaceutical
substance that has not been registered in any
pharmaceutical product [full product dossier required]
Other products, e.g. combination of registered NCEs, new
dosage form, new strength, new route of administration
[abbreviated product dossier allowed]

Biologics
Biologic product refers to a product whose active substance is made by or derived from a living organism (plant, human, animal or microorganism)
May be produced by biotechnology methods and other sophisticated cutting-edge technologies.
The product imitates natural biological substances in our bodies such as hormones, enzymes or antibodies.
Therapeutics made out of proteinsthat can differ in size (large) and structural complexity
E.g. Vaccines
Blood/Plasma derived medicinal products
Biotechnology products including biosimilarCell and Gene Therapy Products (CGTPs)

GenericsA generic product is a product that is essentially similar to a currently registered product in
Malaysia. Classified into two groups:
Scheduled Poison
(Known as Controlled Medicine/ Controlled Poison) Products containing poisons as listed in the First Schedule under Poisons Act 1952
Non-scheduled Poison
(Known as Non-Poison or “Over-the-Counter”, OTC) Products containing active ingredients which are not listed in the First Schedule under Poisons Act 1952; and is excluding active ingredient which is categorized under health supplements or natural products or cosmetics

PROCEDURES FOR REGISTRATION
2
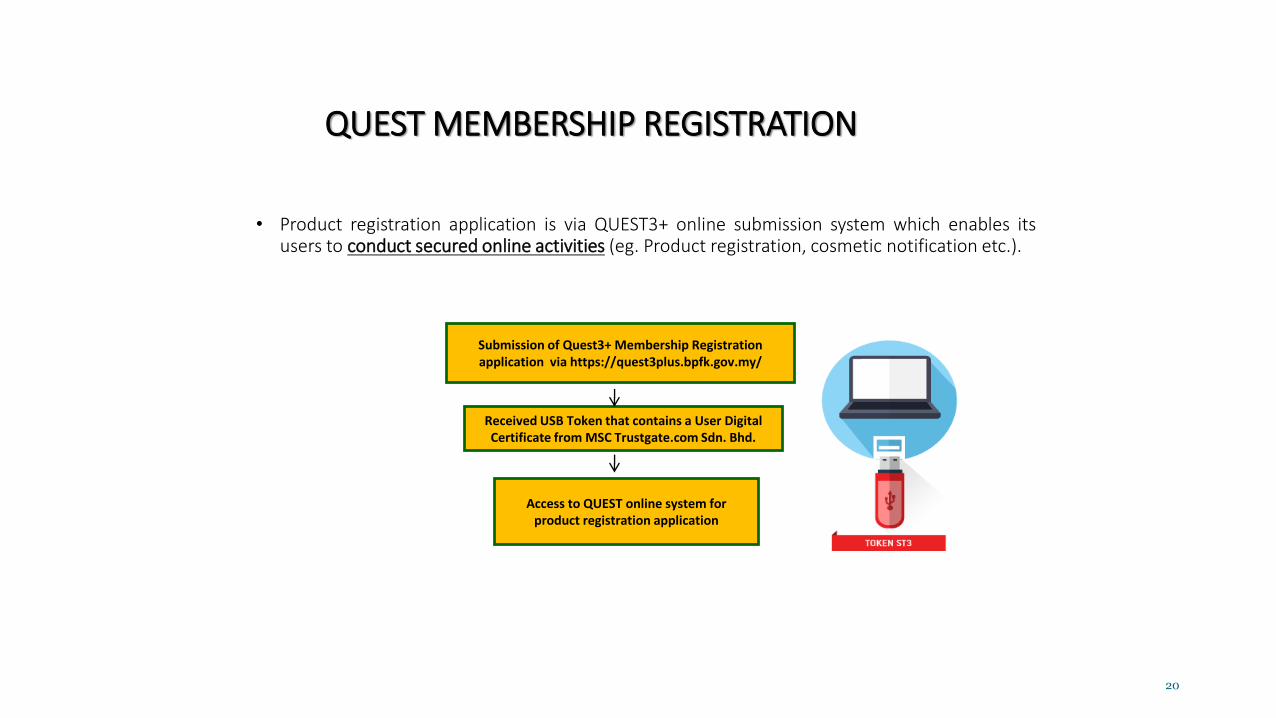
QUEST MEMBERSHIP REGISTRATION
• Product registration application is via QUEST3+ online submission system which enables itsusers to conduct secured online activities (eg. Product registration, cosmetic notification etc.).
20
Submission of Quest3+ Membership Registration application via https://quest3plus.bpfk.gov.my/
Received USB Token that contains a User Digital Certificate from MSC Trustgate.com Sdn. Bhd.
Access to QUEST online system for product registration application

REQUIREMENTS FOR REGISTRATION
21

Registration requirements for pharmaceutical products
InternationalWHO
ICH
EMA
US FDA
Regional
ASEAN Guidelines for:
Stability
Process validation
Variation
Local
Drug Registration Guidance Document
NPRA Guidelines
DCA Directives
Circulars 22

GUIDELINES
Drug Registration Guidance Document(DRGD)
ASEAN Guidelines on Process Validation
ASEAN Guidelines for the Conduct of BA/BE Studies
ASEAN Guidelines for Drug Product Stability Study
ASEAN Guidance on ACTD
Other guidilines may be applicable ) as and when needed

NPCB
MOH
Registration Application Format
ASEAN Common Technical Dossier / ASEAN
Common Technical Requirements ( ACTD/ACTR)
- adopted and adapted from ICH requirements
Implemented since July 2003

Registration Application Format
ASEAN Common Technical Dossier / ASEAN Common Technical Requirements ( ACTD/ACTR)
- adopted and adapted from ICH requirements
Implemented since July 2003

GMP Requirements
CANADA
AUSTRALIA
Malaysia -Member since 2002ARGENTINA LATVIAAUSTRALIA LIECHTENSTEINAUSTRIA LITHUANIA
BELGIUM MALAYSIACANADA MALTACYPRUS NETHERLANDSCZECH REP. (SUKL) NEW ZEALANDCZECH REP. (ISCVBM) NORWAYDENMARK POLANDESTONIA PORTUGALFINLAND ROMANIAFRANCE (ANSM) SINGAPORE FRANCE (ANSES) SLOVAK REPUBLICGERMANY SLOVENJIAGREECE SOUTH AFRICAHUNGARY SPAINICELAND SWEDENINDONESIA SWITZERLAND ISRAEL TAIWANIRELAND UKRAINEITALY UNITED KINGDOMJAPAN UNITED STATESKOREA
Pharmaceutical Inspection Cooperation Scheme
•PIC/s standards (also known as the Pharmaceuticals Inspection Cooperation
Scheme ).
•If > 1 manufacturer involved, GMP certification should be available for all
manufacturers
26
Sharing content
Intro-Pharmaceutical
Services Program& NPRA
Categories of Products & Registration
requirements
Registration process & timelines
Issues Identified in Korean product dossier
assessed at NPRASummary
27
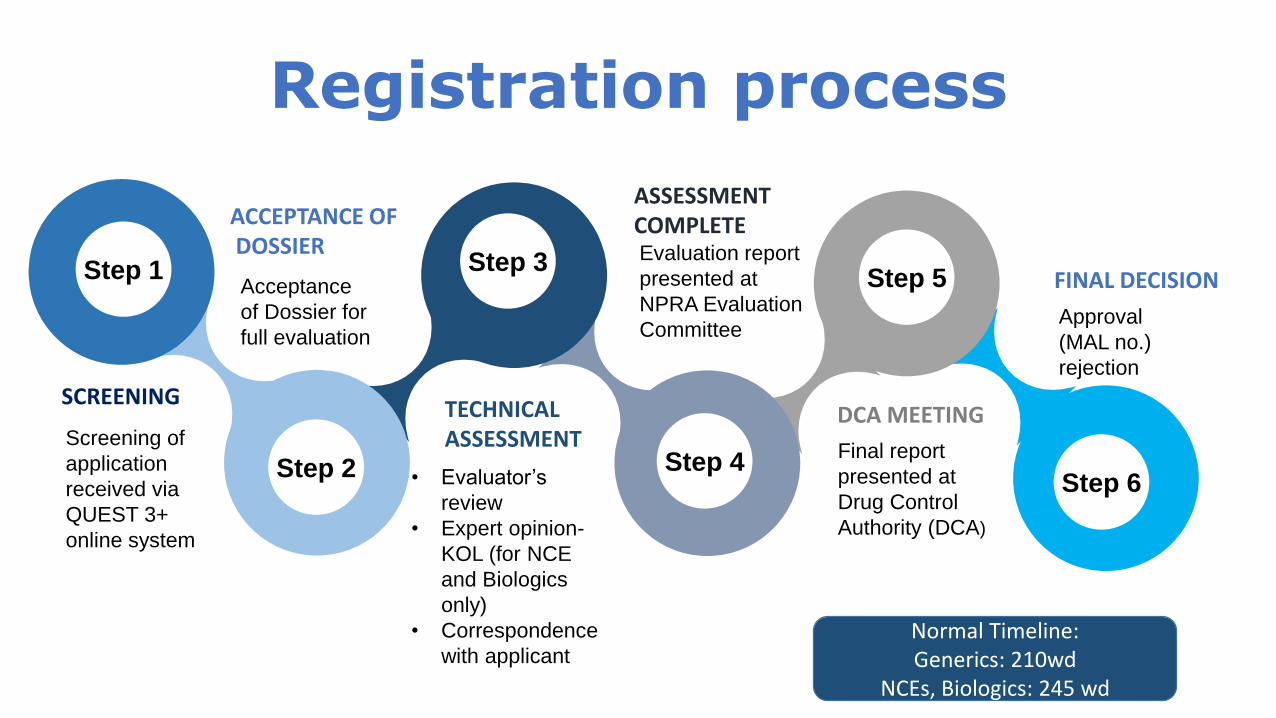
Registration process
Step 6
Step 5
Step 4
Step 3
Step 2
Step 1
TECHNICAL ASSESSMENT
• Evaluator’s
review
• Expert opinion-
KOL (for NCE
and Biologics
only)
• Correspondence
with applicant
DCA MEETING
Final report
presented at
Drug Control
Authority (DCA)
ACCEPTANCE OFDOSSIER
Acceptance
of Dossier for
full evaluation
ASSESSMENT COMPLETEEvaluation report
presented at
NPRA Evaluation
Committee
FINAL DECISION
Approval
(MAL no.)
rejection
SCREENING
Screening of
application
received via
QUEST 3+
online system
Normal Timeline: Generics: 210wd
NCEs, Biologics: 245 wd
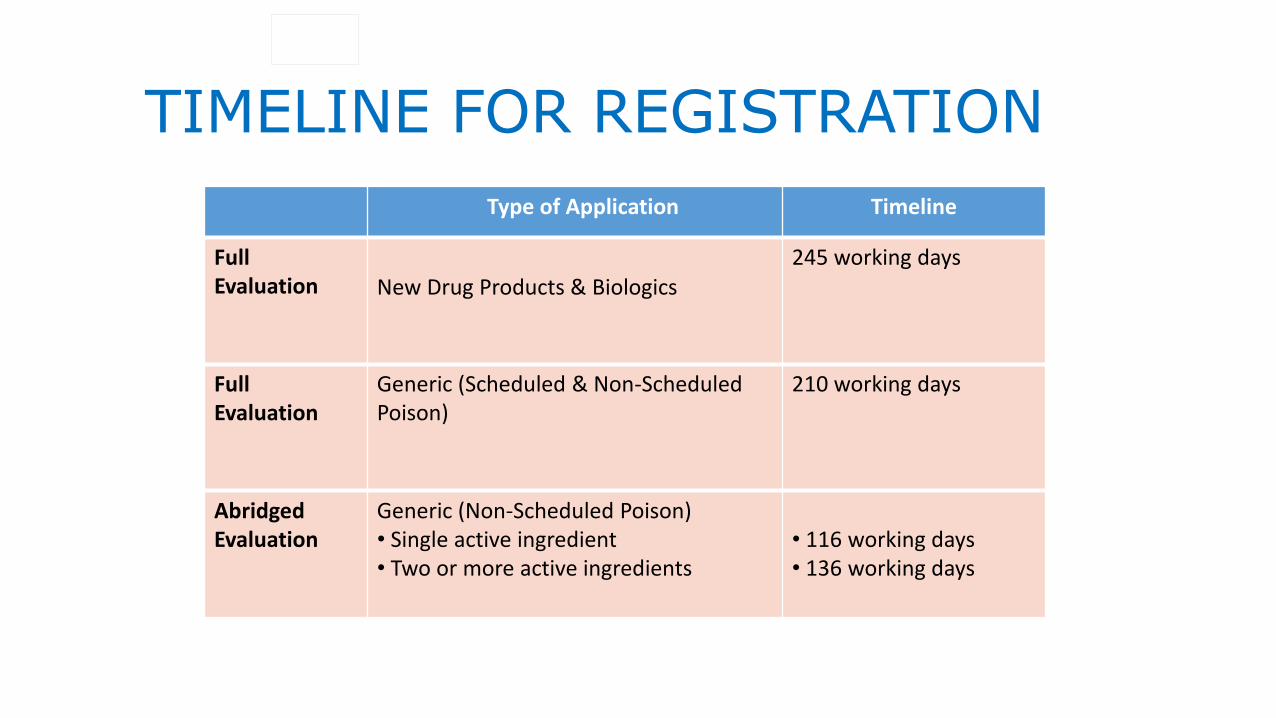
TIMELINE FOR REGISTRATION
Type of Application Timeline
Full Evaluation New Drug Products & Biologics
245 working days
Full Evaluation
Generic (Scheduled & Non-Scheduled Poison)
210 working days
Abridged Evaluation
Generic (Non-Scheduled Poison)• Single active ingredient• Two or more active ingredients
• 116 working days• 136 working days
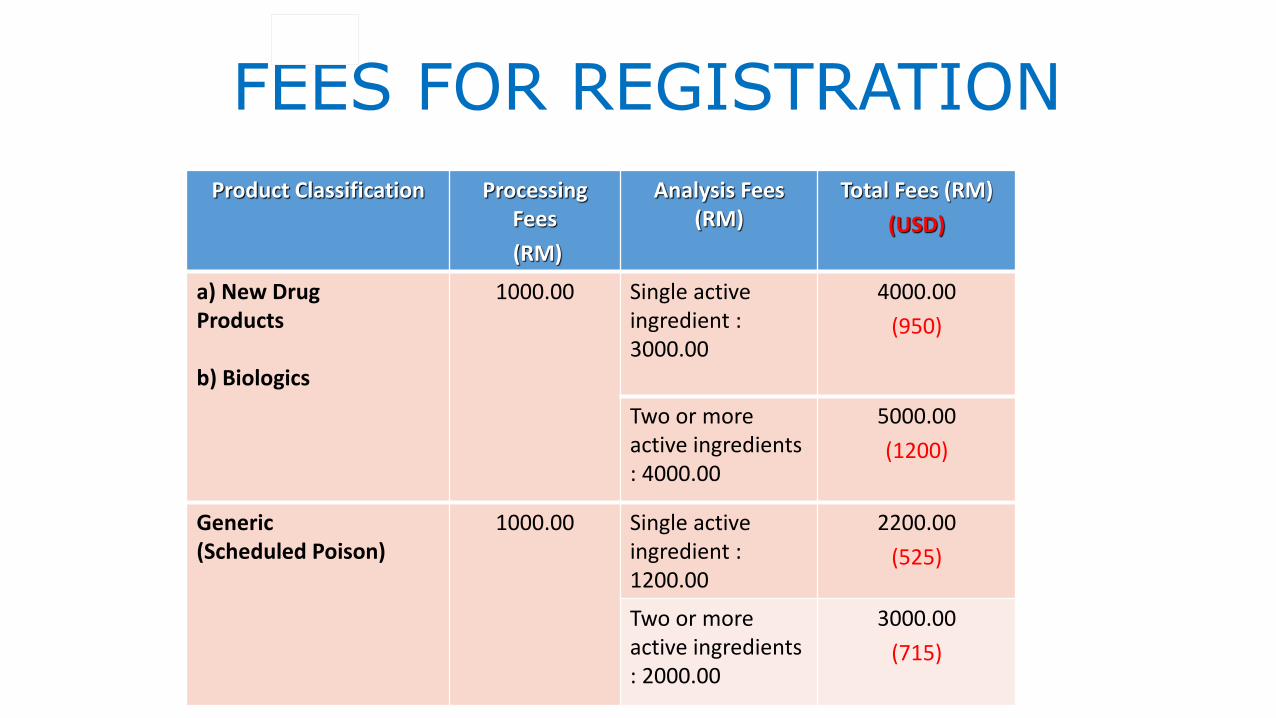
FEES FOR REGISTRATION
Product Classification Processing Fees
(RM)
Analysis Fees (RM)
Total Fees (RM)
(USD)
a) New DrugProducts
b) Biologics
1000.00 Single active ingredient : 3000.00
4000.00
(950)
Two or more active ingredients : 4000.00
5000.00
(1200)
Generic (Scheduled Poison)
1000.00 Single active ingredient : 1200.00
2200.00
(525)
Two or more active ingredients : 2000.00
3000.00
(715)

Facilitating Approval to new/innovative medicines/generics/biosimilars– alternative registration pathway
PRIORITY REVIEW
(i) Unmet medical needs with no treatment options locally available
(ii)Life-saving with no treatment options locally available
(iii)first *generic/ biosimilar product, or first locally manufactured generic/biosimilar product.
CONDITIONAL REGISTRATION
• Products for unmet need supported by early clinical data such as phase II clinical data (based on fully validated surrogate endpoints)
• Conditionally registered for 2 years period with specific conditions
FACILITATED REGISTRATION
PATHWAY
• Leveraging on a Stringent Regulatory Authority decision and information -rely on assessment report issued by the reference agencies
• Abbreviated and Verification Review

To allow promising new medicines to reach patients with unmet need earlier based on early phase data such as phase II clinical data to support the efficacy and safety.
To provide guidance on the application necessary for implementation of conditional registration
To ensure that appropriate measures are in place to manage the risks inherent as additional data are still required.
32
Guidelines on Conditional Registration for NCEs and Biologics
NPRA National Pharmaceutical Regulatory Agency
1
2
3
NPRA National Pharmaceutical Regulatory Agency
Published May 2018
NPRA National Pharmaceutical Regulatory Agency
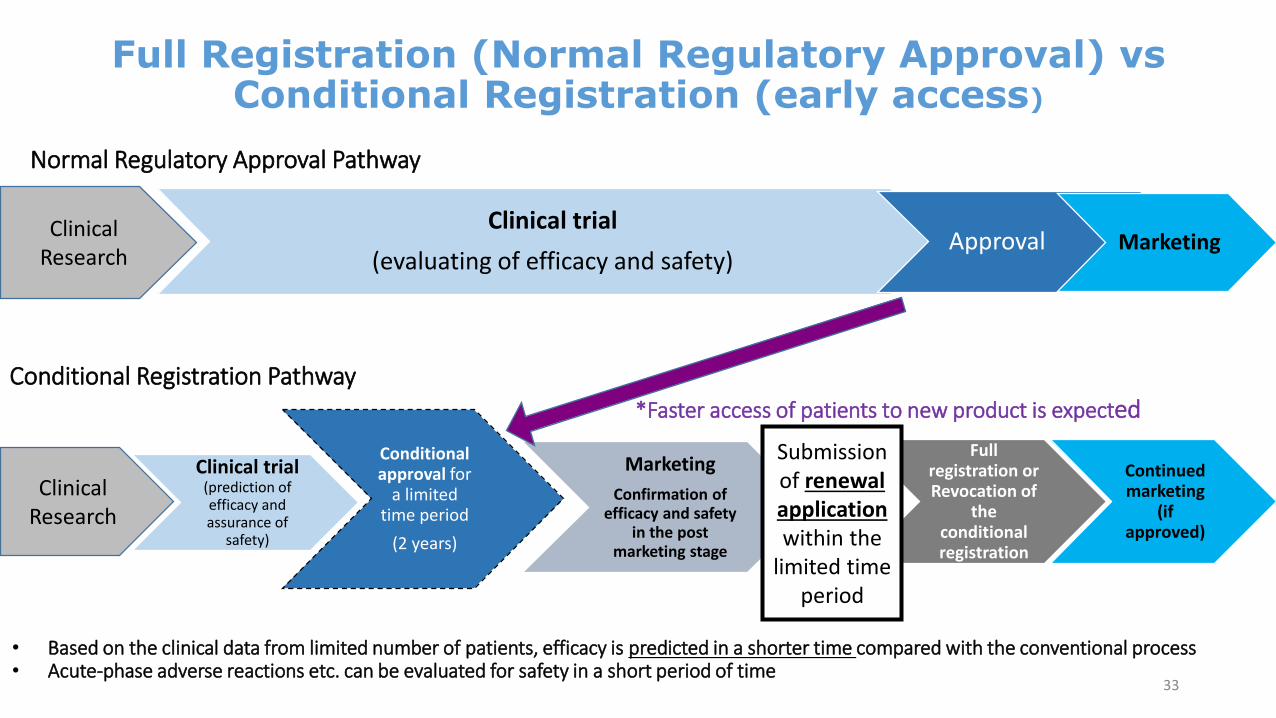
Full Registration (Normal Regulatory Approval) vs Conditional Registration (early access)
Clinical trial
(evaluating of efficacy and safety)Approval Marketing
33
Clinical Research
Clinical Research
Clinical trial (prediction of efficacy and assurance of
safety)
Conditional approval for
a limited time period
(2 years)
Marketing
Confirmation of efficacy and safety
in the post marketing stage
Full registration or Revocation of
the conditional registration
Continued marketing
(if approved)
Submission of renewal application within the
limited time period
Conditional Registration Pathway*Faster access of patients to new product is expected
• Based on the clinical data from limited number of patients, efficacy is predicted in a shorter time compared with the conventional process• Acute-phase adverse reactions etc. can be evaluated for safety in a short period of time
Normal Regulatory Approval Pathway

34
Guidelines on Facilitated Registration Pathway: Abbreviated and Verification Review
NPRA National Pharmaceutical Regulatory Agency NPRA National Pharmaceutical Regulatory Agency NPRA National Pharmaceutical Regulatory Agency
Published March 2019
ensure innovative medicines addressing
current unmet medical needs can be accessible to patients in need in a timely manner
reduce duplication, especially for products where safety and efficacy have already been confirmed by Stringent Regulatory Authorities
drive greater focus toward risk-based evaluations, focusing on what is locally critical versus what can be leveraged/relied upon from decisions made by SRAs
1
2
3
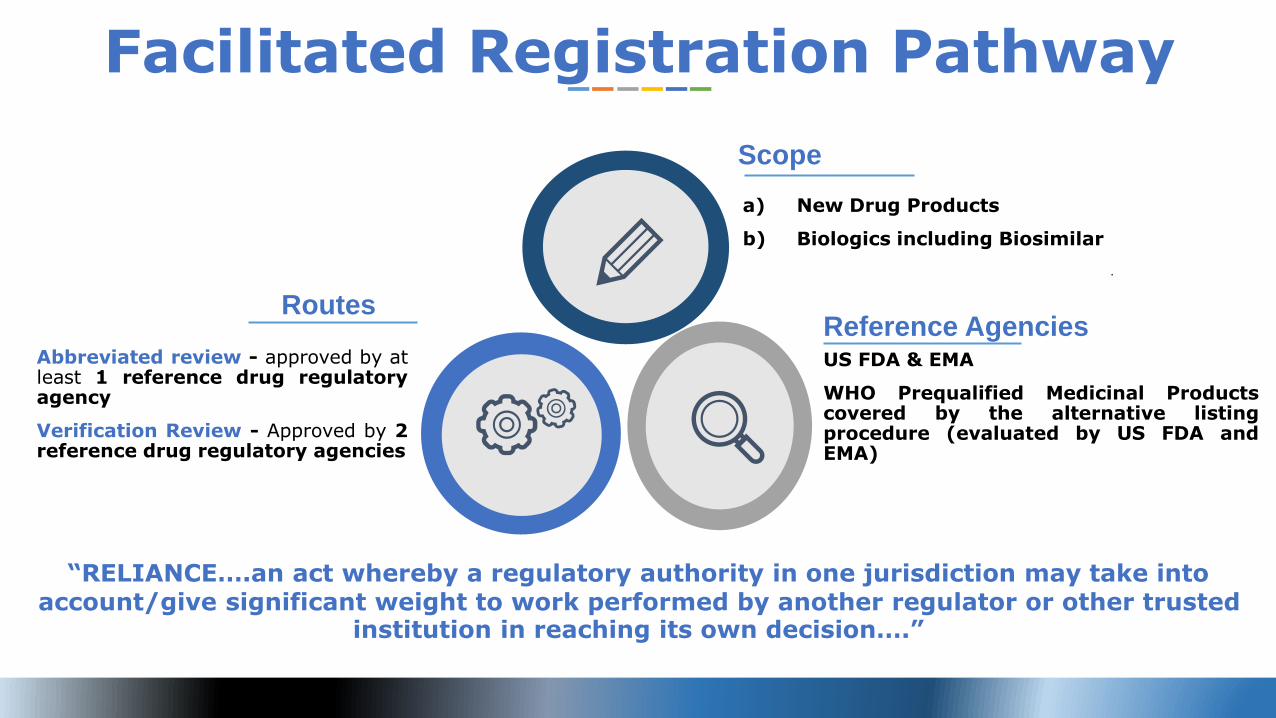
a) New Drug Products
b) Biologics including Biosimilar
.
Scope
Abbreviated review - approved by atleast 1 reference drug regulatoryagency
Verification Review - Approved by 2reference drug regulatory agencies
Routes
US FDA & EMA
WHO Prequalified Medicinal Productscovered by the alternative listingprocedure (evaluated by US FDA andEMA)
Reference Agencies
Facilitated Registration Pathway
“RELIANCE….an act whereby a regulatory authority in one jurisdiction may take into account/give significant weight to work performed by another regulator or other trusted
institution in reaching its own decision….”

Documents required
-Complete Common Technical Document-stability study complies with ASEAN stability guideline-Protocol of Analysis & analytical method validation –checklist as Appendix
FULL DOSSIER
Complete assessment report including assessment on the Q&A documents between the PRH and reference DRA and all annexes. Note: may consider accepting public assessment reports accompanied by redacted information and Q&A provided that the applicant has shown proof and effort to obtain the unredacted assessment reports
PROOF OF
APPROVALProof of approval from the chosen reference DRA-
DECLARATION LETTERRelevant declaration letter(s) issued by the product owner/PRH- all aspects of the DS & DP quality and intended direction(s) for use, including but not limited to the formulation, manufacturing site(s), release and shelf life specifications, primary packaging and active pharmaceutical ingredient(s) source are identical to that currently approved by the chosen reference drug regulatory agency at the time of submission
ASSESSMENT REPORT
1
4
3
2
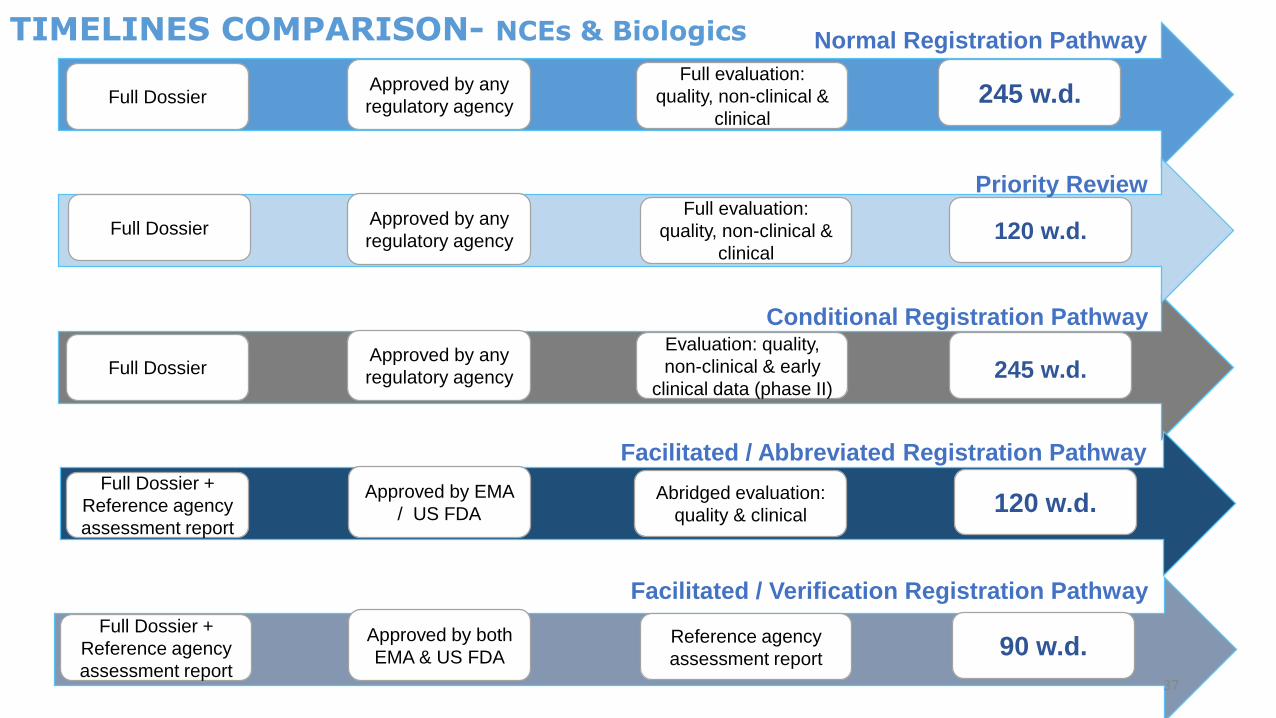
Full DossierApproved by any
regulatory agency
Full evaluation:
quality, non-clinical &
clinical
245 w.d.
Normal Registration Pathway
Facilitated / Abbreviated Registration Pathway
Facilitated / Verification Registration Pathway
Conditional Registration Pathway
Full DossierApproved by any
regulatory agency
Evaluation: quality,
non-clinical & early
clinical data (phase II)245 w.d.
Full Dossier +
Reference agency
assessment report
Approved by EMA
/ US FDAAbridged evaluation:
quality & clinical120 w.d.
Full Dossier +
Reference agency
assessment report
Approved by both
EMA & US FDAReference agency
assessment report90 w.d.
37
Full DossierApproved by any
regulatory agency
Full evaluation:
quality, non-clinical &
clinical120 w.d.
Priority Review
TIMELINES COMPARISON- NCEs & Biologics
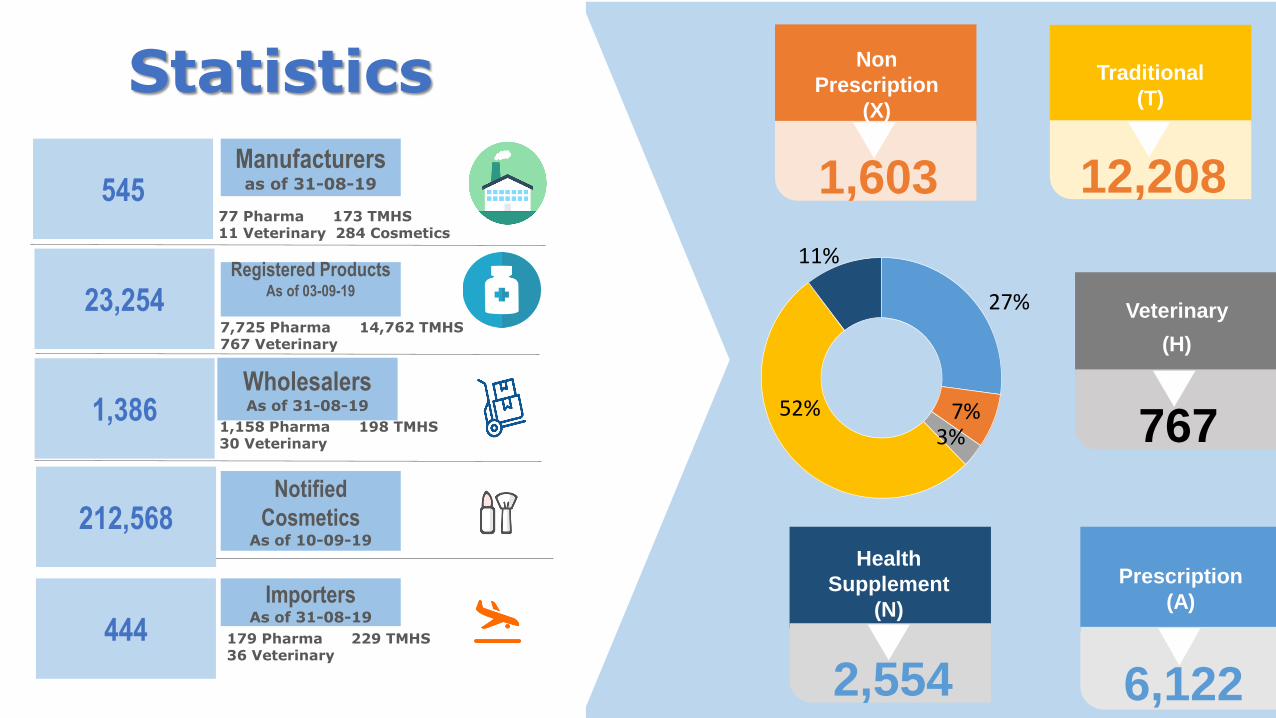
Statistics
27%
7%3%
52%
11%
212,568Notified
CosmeticsAs of 10-09-19
444
ImportersAs of 31-08-19
1,386Wholesalers As of 31-08-19
23,254Registered Products
As of 03-09-19
545Manufacturersas of 31-08-19
77 Pharma 173 TMHS11 Veterinary 284 Cosmetics
12,208
Traditional
(T)
1,603
Non
Prescription
(X)
6,122
Prescription
(A)
2,554
Health
Supplement
(N)
767
Veterinary
(H)
1,158 Pharma 198 TMHS30 Veterinary
179 Pharma 229 TMHS36 Veterinary
7,725 Pharma 14,762 TMHS767 Veterinary
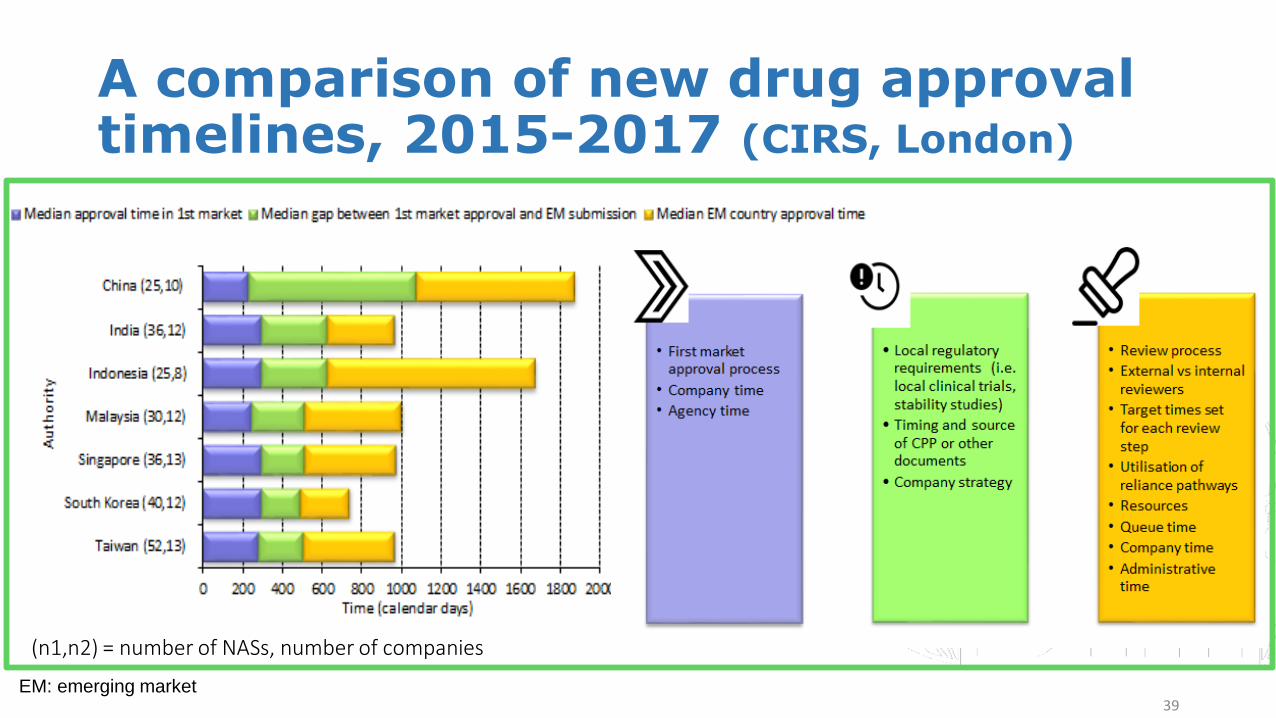
A comparison of new drug approval timelines, 2015-2017 (CIRS, London)
Trends in the Regulatory Landscapefor the Approval of New Medicinesin Asia
R&D BRIEFING 72
To address the complex challenges in the global regulatory environment and the growing demand for patient access to new medicines, regulatory agencies in Asia are actively engaging in regulatory-strengthening and capacity-building initiatives including the use of priority pathways, reliance in the prior reviews of trusted authorities and work sharing to facilitate better utilisation of resources.
This R&D Briefing focuses on the trends observed for 8 countries in the Asia region for 166 new active substances approved from 2009-2017*. The briefing explores converging review times, joint regional submissions and the positive effects of regulatory reform in China.*See page 6 for methodology
Briefing Highlights• From 2008-2017 average review times in India, Taiwan,
Singapore, South Korea and Malaysia converged to an average between 400-500 calendar days; this average increased slightly in 2017.
• From 2015-2017 median review time was longest in Indonesia (1057 days) and China (800 days).
• From 2009-2017 there was minimal variability in regulatory review times in Malaysia, Singapore, South Korea and Taiwan and review timing in South Korea was consistently under 1 year.
• Government-mandated 2015 regulatory review improvements in China have resulted in 133 new drugs being approved through a new priority review route and an application backlog decrease from 22,000 to 3,440.
• A number of countries and regions have developed facilitated regulatory review pathways. These reliance pathways are in place in Indonesia, Malaysia, Singapore and Thailand. China, Taiwan and Vietnam conduct priority review. Rolling submissions and conditional approval is possible in South Korea. These facilitated pathways contribute to a shortening of regulatory review times and increased regulatory efficiency.
Regulatory review processes and timelinesThe time to regulatory approval of new active substance (NAS) in Asia can be measured by three distinct time points: 1. time of approval in the first market, which generally is a first-wave market (USA or Europe); 2. the submission gap, (time between first-market approval and submission to a particular authority); 3. marketing authorisation (MA) time (time between submission and approval, which includes company and agency time). These time points are influenced by a number of factors, one of which is the regulatory landscape within different jurisdictions (Figure 1).
Figure 1: Overall median roll out time to Asian countries for NASs approved 2015-2017 and factors influencing their roll out.
(n1,n2) = number of NASs, number of companies
39EM: emerging market
Sharing content
Intro-Pharmaceutical
Services Program& NPRA
Categories of Products & Registration
requirements
Registration process & timelines
Issues Identified in Korean product dossier
assessed at NPRASummary
40

New Drug Products: Issues Identified in Korean product dossier assessed at NPRA
Quality
Clinical
41
• Inconsistent information on product composition and specifications among
different sections of the dossier
• No data on degradation products of active pharmaceutical ingredient (API) and
drug product (e.g. stress stability testing)
• No justification on proposed drug product quality control test specifications
• Incomplete process validation report (e.g. no sampling plan)
• Incomplete stability data (e.g. no photostability study, only 3 months’ real time
stability data with a proposed shelf life of 36 months)
• Limited clinical data from Phase III studies to support efficacy and safety of all
proposed indications – this led to DCA approval of one out of all proposed
indications
• Clinical trial did not include active comparators with established use in treating
a particular disease
• Transdermal patch product did not include studies on adhesion (including the
influence of external factor such as heat and/or under other ‘in-use conditions’),
skin irritation and sensitisation

Biologics: Issues Identified in Korean product dossier assessed at NPRA (based on limited no. of products)
General
Clinical
42
• Translation issue from Korean to English- some may not be accurate
e.g. related clinical study protocol-impact on the different conduct of
the Clinical Trial
• Translation of NPRA queries from English to Korean and vice versa –
lost in translation-the true meaning of the questions raised were not
direct effectively to the applicant. Hence, unsatisfactory response

Generics: Issues Identified in Korean product dossier assessed at NPRA
Quality
Clinical
43
• Test for degradation products is not conducted• Test parameters are not included as per ASEAN Guideline on Stability study
of Drug Product. (e.g. Microbial limit test, drug release rate for transdermal patches)
• Process validation: Media Fill report with insufficient number of containers filled, incomplete media fill incubation information, filter validation report and information for tunnel sterilization
• In-use stability data /Compatibility Data upon dilution is not provided• Final concentration of diluted product is different from innovator product
Sharing content
Intro-Pharmaceutical
Services Program& NPRA
Categories of Products & Registration
requirements
Registration process & timelines
Issues Identified in Korean product dossier
assessed at NPRASummary
44
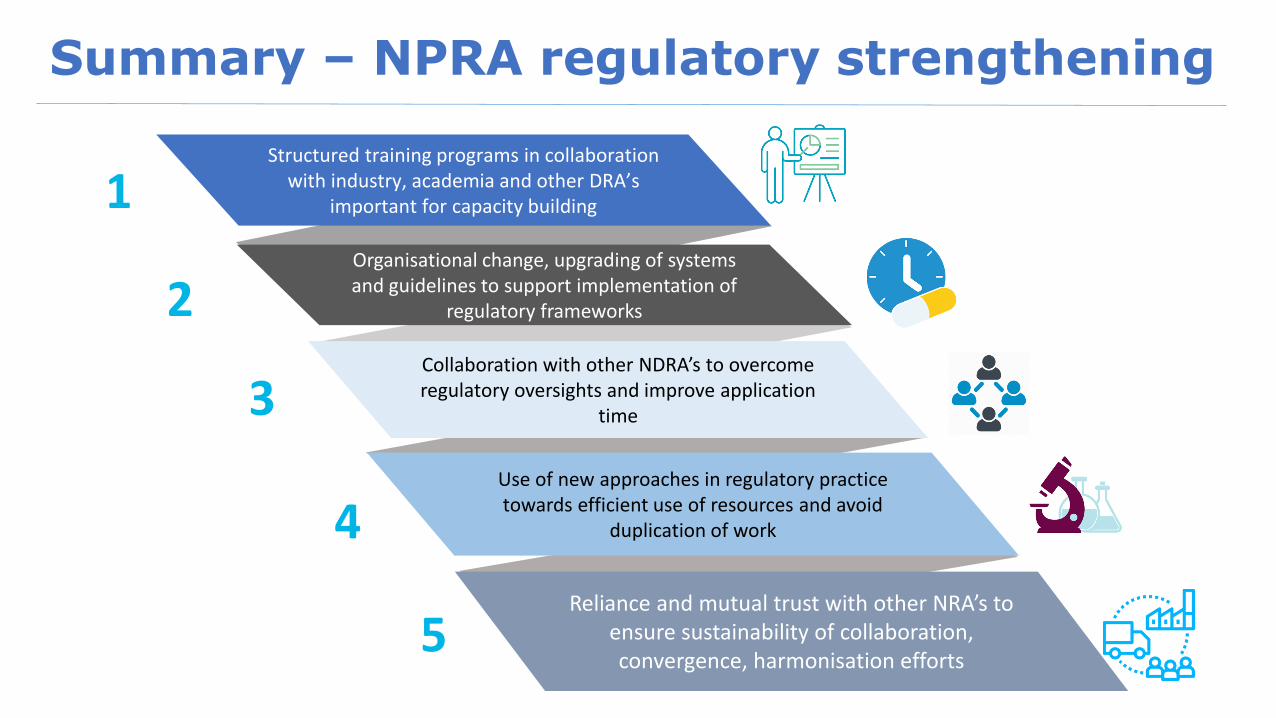
Summary – NPRA regulatory strengthening
Structured training programs in collaboration with industry, academia and other DRA’s
important for capacity building
Organisational change, upgrading of systems and guidelines to support implementation of
regulatory frameworks
Collaboration with other NDRA’s to overcome regulatory oversights and improve application
time
Use of new approaches in regulatory practice towards efficient use of resources and avoid
duplication of work
Reliance and mutual trust with other NRA’s to ensure sustainability of collaboration, convergence, harmonisation efforts
1
2
3
4
5

Further reference
• Address : Lot 36, Jalan Universiti, 46200 Petaling Jaya, Selangor, Malaysia.
• Telephone : +603-78835510
• Fax : +603-79581312
• Email: [email protected]
• Website : www.npra.gov.my

47