down-regulation of tumor suppressor a kinase anchor protein 12 in human hepatocarcinogenesis by...
TRANSCRIPT

Down-Regulation of Tumor Suppressor A KinaseAnchor Protein 12 in Human Hepatocarcinogenesis by
Epigenetic MechanismsBenjamin Goeppert,1 Peter Schmezer,2 Celine Dutruel,2 Christopher Oakes,2 Marcus Renner,1
Marco Breinig,1 Arne Warth,1 Monika Nadja Vogel,3 Michel Mittelbronn,4 Arianeb Mehrabi,5 Georg Gdynia,1
Roland Penzel,1 Thomas Longerich,1 Kai Breuhahn,1 Odilia Popanda,2 Christoph Plass,2
Peter Schirmacher,1 and Michael Andre Kern1
The A kinase anchor protein 12 (AKAP12) is a central mediator of protein kinase A andprotein kinase C signaling. Although AKAP12 has been described to act as a tumor sup-pressor and its expression is frequently down-regulated in several human malignancies, theunderlying molecular mechanisms responsible for the AKAP12 reduction are poorlyunderstood. We therefore analyzed the expression of AKAP12 and its genetic and epige-netic regulatory mechanisms in human hepatocarcinogenesis. Based on tissue microarrayanalyses (n 5 388) and western immunoblotting, we observed a significant reduction ofAKAP12 in cirrhotic liver (CL), premalignant lesions (DN), and hepatocellular carcino-mas (HCCs) compared to histologically normal liver specimens (NL). Analyses of arraycomparative genomic hybridization data (aCGH) from human HCCs revealed chromo-somal losses of AKAP12 in 36% of cases but suggested additional mechanisms underlyingthe observed reduction of AKAP12 expression in hepatocarcinogenesis. Quantitative meth-ylation analysis by MassARRAY of NL, CL, DN, and HCC tissues, as well as of varioustumorigenic and nontumorigenic liver cell lines revealed specific hypermethylation of theAKAP12a promoter but not of the AKAP12b promoter in HCC specimens and in HCCcell lines. Consequently, restoration experiments performed with 5-aza-20deoxycytidinedrastically increased AKAP12a mRNA levels in a HCC cell line (AKN1) paralleled byAKAP12a promoter demethylation. As hypermethylation is not observed in CL and DN,we investigated microRNA-mediated posttranscriptional regulation as an additional mech-anism to explain reduced AKAP12 expression. We found that miR-183 and miR-186 areup-regulated in CL and DN and are able to target AKAP12. Conclusion: In addition togenetic alterations, epigenetic mechanisms are responsible for the reduction of the tumorsuppressor gene AKAP12 in human hepatocarcinogenesis. (HEPATOLOGY 2010;52:2023-2034)
Akinase anchor proteins (AKAPs) are a diversegroup of functionally related scaffolding pro-teins that target protein kinase A (PKA) and
other enzymes, thereby coordinating a range of signal-ing events.1 Human AKAP12 (synonymous: Gravin/AKAP250) is a large protein up-regulated in contact-inhibited cells and down-regulated by Src, Ras, and
PKC.2 Interestingly, AKAP12 is able to modulate bothprotein kinase A and C, indicating that this protein isinvolved in the regulation of several signaling path-ways. Other effects of AKAP12 are direct sequestrationof cyclin D1, inhibition of ERK2 activation, and actincytoskeleton interaction.3 Cyclin D1 overexpression isa frequent event in hepatocarcinogenesis and has been
Abbreviations: aCGH, array-based comparative genomic hybridization; AKAP12, a kinase anchor protein 12; 5-aza-dC, 5-aza-20deoxycytidine; CL, cirrhoticliver; DN, dysplastic nodule; FFPE, formalin-fixed paraffin-embedded; HCC, hepatocellular carcinoma; HBV, hepatitis B virus; HCV, hepatitis C virus; miRNA,microRNA; NL, normal liver; PCR, polymerase chain reaction; PHH, primary human hepatocytes; PT, noncirrhotic peritumorous tissue; SSeCKS, Src-suppressed Ckinase substrate; TMA, tissue microarray; UTR, untranslated region.From the 1Institute of Pathology and 5Department of General, Visceral, and Transplantation Surgery, University Hospital Heidelberg, Heidelberg, Germany;
2Division of Epigenomics and Cancer Risk Factors, German Cancer Research Center (DKFZ), Heidelberg, Germany; 3Department of Diagnostic Radiology, UniversityHospital Tubingen, Germany; 4Edinger Institute, University Hospital Frankfurt am Main, Frankfurt am Main, Germany.Received March 25, 2010; accepted August 17, 2010.Supported by the Deutsche Krebshilfe grants Ke107685 to M.A.K. and P.S.; the Deutsche Forschungsgemeinschaft (DFG) grant SFB/TRR77 to M.A.K., T.L., P.S.
2023

shown to occur early in the development of hepatocel-lular carcinoma (HCC) in the mouse.4,5 SSeCKS, therodent ortholog of AKAP12, has been demonstrated toact as tumor suppressor in vitro and in vivo.6,7 Thisfunction was also described in human solid neoplasms,such as prostate and gastric cancer,7,8 but data con-cerning the role of AKAP12 in human hepatocarcino-genesis are scarce.The AKAP12 gene locus in human and rodents
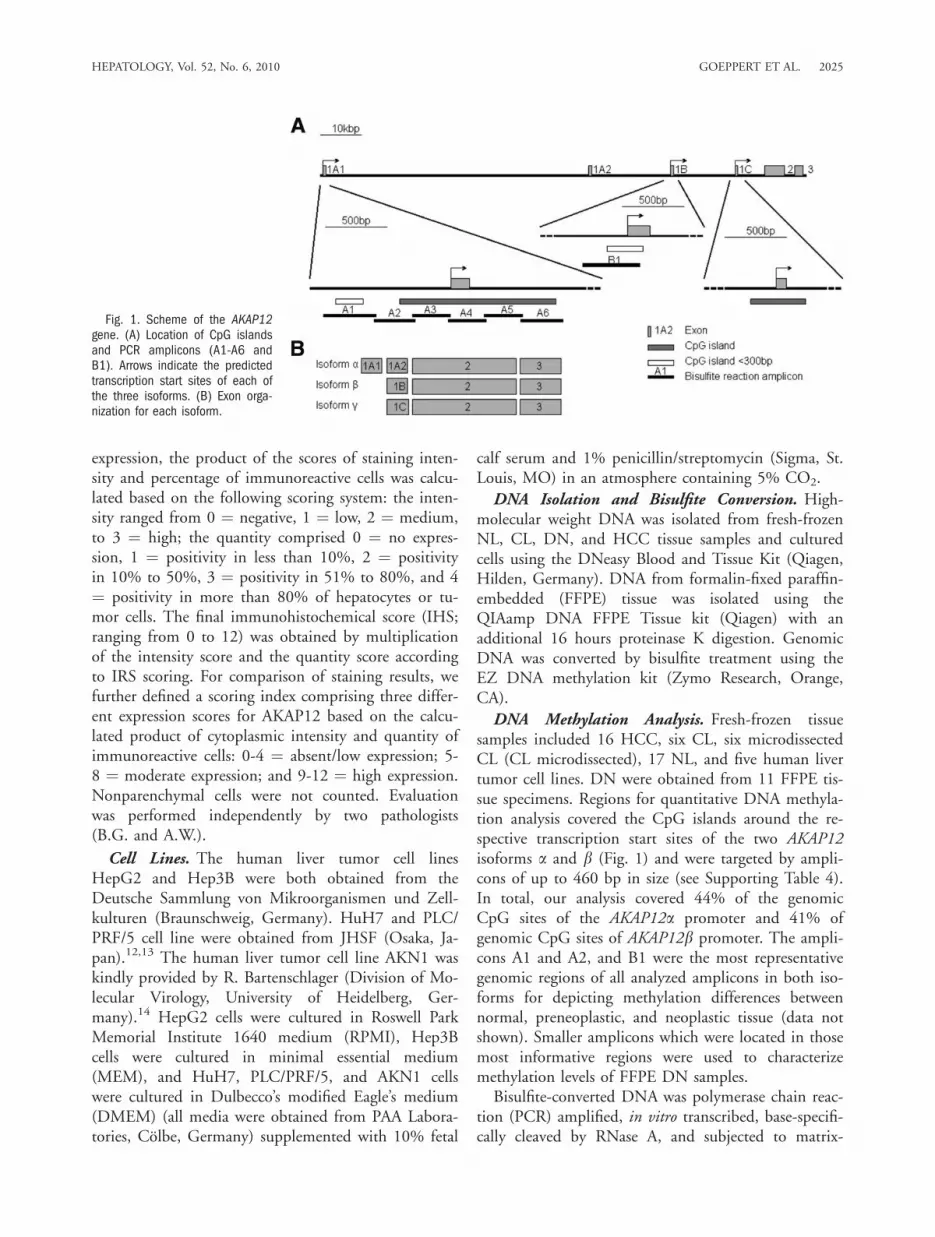
encodes three major transcripts under the control ofthree independent promoters, designated a, b, and c(Fig. 1). The two major protein isoforms of AKAP12,a and b, are expressed ubiquitously in most cell andtissue types, whereas the expression of isoform c is re-stricted to testes.9 The proteins translated from eachtranscript are encoded by a single large exon and share>95% amino acid sequence homology; however, theydiffer in their N-terminal domains. These isoforms arefrequently posttranslationally modified; for example,only the a isoform is myristoylated at the N-terminus,a modification that facilitates AKAP12a associationwith plasma membranes and vesicles of the endoplas-mic reticulum.9
In one of our recent studies analyzing 63 HCCs byaCGH, the AKAP12 gene locus on chromosome6q24-25.2 showed chromosomal losses in 36% (23/63) of all analyzed cases, whereas gains were observedonly in 5% (3/63).10 Yet this is an incomplete explana-tion of the observed AKAP12 down-regulation inHCC. Here, we show protein expression data of thetumor suppressor AKAP12 in a large series of humanliver specimens, containing typical pathohistologicalfeatures of hepatocarcinogenesis. In order to elucidatemechanisms of AKAP12 down-regulation, we have an-alyzed genetic, epigenetic, and posttranscriptionalmechanisms. In summary, we here propose three dif-ferent mechanisms of AKAP12 down-regulation inhepatocarcinogenesis: microRNA (miRNA) interfer-ence in preneoplastic lesions, genetic alterations, andAKAP12a promoter hypermethylation in HCCs.
Materials and MethodsPatients and Tissue Specimens. A total of 388
human liver tissue samples were evaluated by tissue
microarrays (TMAs). TMA#1 (n ¼ 225) containedtwo representative areas (diameter: 0.6 mm) of 14histologically normal liver (NL), 38 dysplastic nod-ule (DN), 135 hepatocellular carcinoma (HCC;grading: 29 � G1, 76 � G2, 30 � G3), 14 cir-rhotic (CL), and 24 noncirrhotic peritumorous (PT)liver tissue samples. The independently designed,processed, and evaluated TMA#2 (n ¼ 163) con-tained two representative areas (diameter: 0.6 mm)of 20 NL, 77 HCC (grading: 13 � G1, 55 �G2, 9 � G3), and 66 PT tissue samples (see Sup-porting Table 7 and Supporting Fig. 1). All speci-mens were fixed in 4% formalin (pH 7.4) and em-bedded in paraffin. Tissue specimens were obtainedfrom the tissue bank of the National Center of Tu-mor Diseases (Heidelberg, Germany). All specimenswere surgically resected at the University of Heidel-berg and histologically classified according to estab-lished criteria by three pathologists (TL, MAK, andPS). The study was approved by the institutionalethics committee (206/05). TMAs were processed aspreviously described.11
Immunohistochemistry. Immunohistochemical anal-ysis was performed according to standard protocolsusing the avidin biotin complex-method and diamino-benzidine as chromogen. AKAP12 immunohistochem-istry of TMA#1 was performed using a goat polyclonalanti-AKAP12 antibody (dilution 1:100; Santa CruzBiotechnology, Santa Cruz, CA). AKAP12 immunohis-tochemistry of TMA#2 was performed using a mousemonoclonal anti-AKAP12 antibody (dilution 1:100;Abcam, Cambridge, MA). All sections were counter-stained with hemalum. Specificity of the reaction wascontrolled by omitting the primary antibody. Immuno-histochemistry of factors used in the correlation analy-sis was performed as described.11
Immunoblotting. Western immunoblotting was per-formed using the following primary antibodies: goatpolyclonal anti-AKAP12 (dilution 1:1000; Santa CruzBiotechnology, Santa Cruz, CA) and a mouse mono-clonal anti-AKAP12 antibody (dilution 1:1000;Abcam, Cambridge, MA). For further information, seeSupporting Information.Tissue Microarray Analysis. For semiquantitative
immunohistochemical assessment of AKAP12
Address reprint requests to: Dr. Benjamin Goeppert, Institute of Pathology, University of Heidelberg, Im Neuenheimer Feld 220/221, D-69120 Heidelberg,Germany. E-mail: [email protected]; fax: 0049-6221-56-5251.CopyrightVC 2010 by the American Association for the Study of Liver Diseases.View this article online at wileyonlinelibrary.com.DOI 10.1002/hep.23939Potential conflict of interest: Nothing to report.Additional Supporting Information may be found in the online version of this article.
2024 GOEPPERT ET AL. HEPATOLOGY, December 2010
expression, the product of the scores of staining inten-sity and percentage of immunoreactive cells was calcu-lated based on the following scoring system: the inten-sity ranged from 0 ¼ negative, 1 ¼ low, 2 ¼ medium,to 3 ¼ high; the quantity comprised 0 ¼ no expres-sion, 1 ¼ positivity in less than 10%, 2 ¼ positivityin 10% to 50%, 3 ¼ positivity in 51% to 80%, and 4¼ positivity in more than 80% of hepatocytes or tu-mor cells. The final immunohistochemical score (IHS;ranging from 0 to 12) was obtained by multiplicationof the intensity score and the quantity score accordingto IRS scoring. For comparison of staining results, wefurther defined a scoring index comprising three differ-ent expression scores for AKAP12 based on the calcu-lated product of cytoplasmic intensity and quantity ofimmunoreactive cells: 0-4 ¼ absent/low expression; 5-8 ¼ moderate expression; and 9-12 ¼ high expression.Nonparenchymal cells were not counted. Evaluationwas performed independently by two pathologists(B.G. and A.W.).
Cell Lines. The human liver tumor cell linesHepG2 and Hep3B were both obtained from theDeutsche Sammlung von Mikroorganismen und Zell-kulturen (Braunschweig, Germany). HuH7 and PLC/PRF/5 cell line were obtained from JHSF (Osaka, Ja-pan).12,13 The human liver tumor cell line AKN1 waskindly provided by R. Bartenschlager (Division of Mo-lecular Virology, University of Heidelberg, Ger-many).14 HepG2 cells were cultured in Roswell ParkMemorial Institute 1640 medium (RPMI), Hep3Bcells were cultured in minimal essential medium(MEM), and HuH7, PLC/PRF/5, and AKN1 cellswere cultured in Dulbecco’s modified Eagle’s medium(DMEM) (all media were obtained from PAA Labora-tories, Colbe, Germany) supplemented with 10% fetal
calf serum and 1% penicillin/streptomycin (Sigma, St.Louis, MO) in an atmosphere containing 5% CO2.
DNA Isolation and Bisulfite Conversion. High-molecular weight DNA was isolated from fresh-frozenNL, CL, DN, and HCC tissue samples and culturedcells using the DNeasy Blood and Tissue Kit (Qiagen,Hilden, Germany). DNA from formalin-fixed paraffin-embedded (FFPE) tissue was isolated using theQIAamp DNA FFPE Tissue kit (Qiagen) with anadditional 16 hours proteinase K digestion. GenomicDNA was converted by bisulfite treatment using theEZ DNA methylation kit (Zymo Research, Orange,CA).
DNA Methylation Analysis. Fresh-frozen tissuesamples included 16 HCC, six CL, six microdissectedCL (CL microdissected), 17 NL, and five human livertumor cell lines. DN were obtained from 11 FFPE tis-sue specimens. Regions for quantitative DNA methyla-tion analysis covered the CpG islands around the re-spective transcription start sites of the two AKAP12isoforms a and b (Fig. 1) and were targeted by ampli-cons of up to 460 bp in size (see Supporting Table 4).In total, our analysis covered 44% of the genomicCpG sites of the AKAP12a promoter and 41% ofgenomic CpG sites of AKAP12b promoter. The ampli-cons A1 and A2, and B1 were the most representativegenomic regions of all analyzed amplicons in both iso-forms for depicting methylation differences betweennormal, preneoplastic, and neoplastic tissue (data notshown). Smaller amplicons which were located in thosemost informative regions were used to characterizemethylation levels of FFPE DN samples.Bisulfite-converted DNA was polymerase chain reac-
tion (PCR) amplified, in vitro transcribed, base-specifi-cally cleaved by RNase A, and subjected to matrix-
Fig. 1. Scheme of the AKAP12gene. (A) Location of CpG islandsand PCR amplicons (A1-A6 andB1). Arrows indicate the predictedtranscription start sites of each ofthe three isoforms. (B) Exon orga-nization for each isoform.
HEPATOLOGY, Vol. 52, No. 6, 2010 GOEPPERT ET AL. 2025

assisted laser desorption, ionization-time-of-flight massspectrometry (MassARRAY technique; Sequenom, SanDiego, CA) as described.15 DNA methylation stand-ards (0%, 20%, 40%, 60%, 80%, and 100% methyl-ated genomic DNA) were used for data correction.
Microdissection of CL Samples. To assure that themethylation analysis of CL samples was not contami-nated by nonparenchymal cells, microdissection wasperformed. CL tissue samples were cut into 18-lm-thick sections using a cryostat (Leica CM1850; LeicaMicrosystems, Wetzlar, Germany) and laser microdis-section was performed as described.16
5-aza-dC Treatment. Human HCC cell lines(AKN1, HepG2, and HuH7) were incubated for 72hours with 10 lmol/L 5-aza-dC (Sigma-Aldrich, St.Louis, MO) with a medium change every 24 hours.Genomic DNA was isolated from treated cells asdescribed and total RNA was purified using Trizol rea-gent (Invitrogen, Karlsruhe, Germany).
Reverse Transcription and Quantitative Real-Time PCR. Quantitative mRNA expression analysis ofAKAP12 isoforms and reference genes was performedon a LightCycler 480 (Roche Diagnostics, Mannheim,Germany) using the Absolute QPCR SYBR GreenMix (Thermo Scientific, Waltham, MA) asdescribed.17 Briefly, total RNA from the cell lines(1 lg) was reverse transcribed using SuperScript IIIreverse transcriptase (Invitrogen) and oligodT primersor random hexamers for tissue samples. (For primersand real-time PCR conditions, see Supporting Table5.) Reaction products were characterized by meltingpoint analysis and relative quantification with effi-ciency correction (LightCycler Software 4, RocheDiagnostics). Mean normalized ratios were determinedfor each sample, using HPRT1, TBP, and ACTB as ref-erence genes.
miRNA Purification and Quantification in FFPEand Fresh-Frozen Tissues. For purification of miRNAfrom FFPE tissue sections we used the miRNeasyFFPE Kit according to the manufacturers’ instructions(Qiagen). miRNAs were isolated from fresh-frozen tis-sues using Trizol (Invitrogen). For FFPE tissues,cDNA was synthesized using miScript Reverse Tran-scription kit (Qiagen), and real-time quantitative PCR(qPCR) was performed using miScript SYBR GreenPCR kit and specific human miScript assays for hsa-miR-183 and hsa-miR-186 (Qiagen) in an ABI-Prism7300 Real-time PCR system (Applied Biosystems,Darmstadt, Germany). RNU6B was used as endoge-nous reference RNA.
Functional Interaction Studies of miRNAs andAKAP12. Human miR-183 and miR-186 were clonedinto the pCMX-PL1 expression plasmid by PCRamplification of 6100 bp of the pre-miRNA sequenceas annotated in the UCSC, hg18. The conserved30end of the AKAP12 30-untranslated region (30UTR)was cloned by insertion of hybridized oligonucleotides(120 bp) into the 30end of the Firefly luciferase genein the pMirReport plasmid (Ambion, Austin, TX).AKAP12 UTR/miRNA interactions were performed byexpression in HEK293T cells using a TK-Renilla plas-mid (Promega, Madison, WI) as a transfection control.Regulation of endogenous AKAP12 mRNA was deter-mined by overexpression of miRNAs followed byRNA isolation using Trizol; cDNA synthesis, andqPCR as described.
Statistical Analyses and Software. Data are pre-sented as mean values 6 SD. The Spearman rank coeffi-cient was used as a statistical measure of association. ForTMA correlation analysis, r > 0.3 and P < 0.001 wereconsidered as biologically relevant. To account for multi-ple testing (TMA-data), the a-level was adjusted accord-ing to Bonferroni. The statistical comparison betweentwo groups was accomplished with the nonparametricMann-Whitney U test (SPSS version 11).
ResultsDown-Regulation of AKAP12 in CLs, DNs, and
in HCCs. A previous study of our group demonstratedlosses of genomic material coding for the AKAP12gene locus in 36% of human HCCs.10 As data aboutAKAP12 expression in the process of human hepato-carcinogenesis are scarce, we aimed to define altera-tions of AKAP12 protein levels in hepatocarcinogene-sis. We detected AKAP12 expression by immuno-histochemistry using TMAs containing a total numberof 388 human liver tissue samples, including NL, CL,DN, and HCC. Immunohistochemistry showed astrong cytoplasmic staining pattern in hepatocytes,with a partly granular, partly homogeneous appearance(Fig. 2A). Statistical analyses of TMA#1 revealed a sig-nificantly higher relative immunoreactivity of AKAP12in human NL compared to HCCs (P < 0.0001, Fig.2B). According to our scoring index, 72% of NL sam-ples showed a high or moderate AKAP12 expression,whereas in the HCC group only 27% (G1), 36%(G2), and 23% (G3) of samples showed a high ormoderate expression. Overall, in 68% of the HCCsamples AKAP12 expression dropped below the 25thpercentile compared to the NL group. AKAP12 expres-sion in noncirrhotic PT was comparable to NL, whereas
2026 GOEPPERT ET AL. HEPATOLOGY, December 2010

in CL and DN a significant down-regulation ofAKAP12 was observed (P < 0.01, P < 0.05; Fig. 2B).Focusing on the group of DN and HCC (all differen-tiation grades), we detected a statistically significantdown-regulation of AKAP12 correlating with thereduced differentiation grade from DN toward G3-HCC (P < 0.01; Fig. 2B). TMA#2 (n ¼ 163; con-taining NL, PT, and HCC specimens) confirmed theresults of TMA#1 (see Supporting Table 7 and Sup-porting Fig. 1).TMA results were confirmed by analyzing protein
extracts of human NL tissues and HCC samples ofvarious differentiation grades by western immunoblot.In NL specimens, AKAP12 was strongly or at leastmoderately expressed, whereas in HCC samples,AKAP12 expression was reduced or not detectable.Semiquantitative densitometry of western immunoblotsrevealed a 10-fold to 100-fold higher AKAP12 expres-sion in NL compared to HCC (Fig. 3A). In addition,we examined AKAP12 expression in various hepaticcell lines and in primary human hepatocytes (PHH).These immunoblots showed a reduced or absentAKAP12 expression in the HCC cell lines, whereas inPHH, AKAP12 expression was clearly detectable.Semiquantitative densitometry of western immunoblotsrevealed a 30-fold to 600-fold higher AKAP12 expres-sion in PHH compared to HCC cell lines (Fig. 3B).
Correlation Analysis of AKAP12 Expression WithClinical Parameters, Proliferation Rate, and OtherFactors Involved in Hepatocarcinogenesis. Correla-tion analysis for AKAP12 expression and the prolifera-tion marker Ki-67 showed a statistically significantinverse correlation (r ¼ �0.318; P < 0.001). AKAP12levels did not show any correlation at the protein levelwith other factors involved in hepatocarcinogenesis.More interestingly, no correlation of AKAP12 withcyclin D1 was detected. No significant statistical corre-lation was detected after testing etiological factors,such as chronic viral hepatitis (HBV and HCV), clini-cal parameters, tumor staging (TNM), or gender withAKAP12 levels (see Supporting Table 2 and 3).
Isoform-Specific AKAP12 mRNA Expression inTissue Samples. Because the used antibodies recognizethe C-terminal domain of both AKAP12 isoforms, weseparately examined AKAP12a and b expression at themRNA level in NL, CL, and HCC tissues (Fig. 4A,B).We found that AKAP12a was the predominant isoformexpressed in all tissues. AKAP12a mRNA expressionshowed a statistically significant decrease during hepato-carcinogenesis, correlating with TMA protein data.
Fig. 2. Down-Regulation of AKAP12 correlates with dedifferentiation inhuman hepatocarcinogenesis. (A) In NL, a strong cytoplasmic expression ofAKAP12 is observed in hepatocytes, whereas in DN and in HCC a distinctreduction of AKAP12 was detectable. Original magnification: �50 (left),�200 (right). (B) Box plot illustration of TMA analyses demonstrate AKAP12expression in NL and a significant stepwise reduction in CL, DN (CL: P <0.01; DN: P < 0.05), and further in the group of HCCs (P< 0.0001).
HEPATOLOGY, Vol. 52, No. 6, 2010 GOEPPERT ET AL. 2027

Regulation of AKAP12 Expression by DNA Meth-ylation. In a cohort of 63 HCCs recently analyzed byaCGH, the AKAP12 gene locus on chromosome 6q24-25.2 showed chromosomal losses in 36% (23/63) andgains in 5% (3/63) of cases.10 In addition, no alterationsof the AKAP12 gene locus on chromosome 6q24-25.2were detectable in all analyzed cell lines (see SupportingTable 1). However, these data do not sufficiently explainthe distinct decrease of AKAP12 protein expressionobserved in this study. As a possible suppressive mecha-nism, we investigated the DNA methylation of pro-moter-related CpG islands of both AKAP12 isoforms inNL, CL, DN, and HCC samples by quantitative Mas-sARRAY analysis (Fig. 1). In tumor samples, hypermeth-ylation was detected in the AKAP12a promoter (Fig.5A,B), but not in the AKAP12b promoter (Fig. 5C,D).Methylation analysis of the AKAP12b promoter showeda methylation value higher than 10% only for oneHCC sample. For the AKAP12a promoter, a mean
methylation of 12% for NL and CL, 15% for DN, andof 30% for HCC was observed. As high amounts offibroblasts, infiltrating immune cells, and other nonpar-enchymal cells may inflict on the genuine methylationstatus of hepatocytes in CL, microdissection of hepato-cytes was performed. However, the methylation valuesof CL specimens after microdissection (18%) did notsignificantly differ from undissected samples of the sametissues.Elevated DNA methylation levels of the AKAP12a
promoter were also detected in HCC cell lines. Thisanalysis revealed DNA methylation of 96% (AKN1),42% (HuH7), 41% (Hep3B), 24% (HepG2), and20% (PLC/PRF/5) (Fig. 5A,B). Methylation analysisof the AKAP12b promoter in cell lines showed meth-ylation values lower than 1% (Fig. 5C,D). Hypermeth-ylation of the AKAP12a promoter was confirmed byan independent method (combined bisulfite restrictionanalysis; see Supporting Fig. 2).
Fig. 3. Western blot analysis shows AKAP12 down-regulation in HCC tissue lysates and in HCC cell lines. (A) HCC tissue samples of variousdifferentiation grades (G1-G3) show a 10-fold to 100-fold reduction of AKAP12 expression when compared to NL tissue samples. (B) HCC celllines (PLC/PRF/5, HepG2, Hep3B, HuH7, AKN1) show a 30-fold to 600-fold reduction of AKAP12 signal when compared to PHH. A glioblastomacell line (U87) served as a positive control. Densitometry was performed and is indicated as density index (DI, normalized ratio).
Fig. 4. AKAP12 mRNA expression in tissue samples. (A) AKAP12a mRNA levels in fresh-frozen tissue samples of NL, CL, and HCC. Horizontalbars indicate mean values. HCC tissue samples show statistically significant lower AKAP12a mRNA levels compared to CL and NL (P < 0.05;Mann-Whitney U test). (B) AKAP12b mRNA levels in fresh-frozen tissue samples of NL, CL, and HCC. Horizontal bars indicate mean values. NL,CL, and HCC tissue samples do not show statistically significant differences in AKAP12b mRNA expression. In comparison with (A), AKAP12a isthe predominantly expressed isoform in all subgroups.
2028 GOEPPERT ET AL. HEPATOLOGY, December 2010

To verify the functional relationship between pro-moter hypermethylation and loss of AKAP12 geneexpression, methylation and mRNA expression levelsof both isoforms were compared before and after treat-ment with 5-aza-dC in cell lines AKN1, HepG2, andHuH7. Isoform-specific mRNA expression of AKAP12differed between untreated hepatic cell lines and PHHbut confirmed protein data (Fig. 3B; Fig. 6A). The 5-aza-dC treatment resulted in a decrease in AKAP12apromoter methylation in the highly methylated AKN1cell line (Fig. 6B), accompanied by a strong increase inAKAP12a mRNA expression (Fig. 6C), demonstratinga relationship between AKAP12a expression and meth-
ylation of its promoter. In HepG2 and HuH7 cells,which showed lower methylation levels than AKN1,demethylation as well as re-expression of 5-aza-dC wasmoderate. Similarly, only a marginal increase in expres-sion was detected for isoform b with its unmethylatedpromoter (Fig. 6B,C). Data were confirmed in two in-dependent experiments (see Supporting Table 6).Regulation of AKAP12 Expression by miRNAs. Alth-
ough we have demonstrated that silencing of AKAP12 isassociated with DNA hypermethylation in HCC, pro-moter methylation does not explain the loss of expressionin earlier stages of hepatocarcinogenesis. Thus, we postu-lated that a posttranscriptional mechanism may cause
Fig. 5. MassARRAY analysis ofAKAP12 promoter methylation. (A)Quantitative DNA methylation analy-sis of the AKAP12a promoterregion. Each column represents agroup of one to five CpGs analyzed;each row represents a sample. Per-centages of methylation span from0% to 100%. Gray squares indicateunavailable data. (B) The meanpercentage of methylation for eachsample represented in (A). Horizon-tal bars represent mean methyla-tion values for each group. HCCtissue samples show statisticallysignificant hypermethylation whencompared to NL (P < 0.001), CL(P < 0.01), and DN tissue sam-ples (P < 0.01; Mann-Whitney Utest). (C) Quantitative DNA methyla-tion analysis of the AKAP12b pro-moter region using MassARRAY. (D)The average percent methylation foreach sample represented in (C).
HEPATOLOGY, Vol. 52, No. 6, 2010 GOEPPERT ET AL. 2029

silencing in CL and DN. A candidate list of up-regulatedmiRNAs was established by a literature search of miRNAexpression profiles in CL and HCC,18 which was cross-referenced with miRNAs predicted to target the 30UTRof AKAP12 using miRWalk, a composite software suite ofseveral miRNA target prediction programs (http://www.ma.uni-heidelberg.de/apps/zmf/mirwalk). Thissearch strategy identified miR-183 and miR-186 aspotential regulators of AKAP12 in hepatocarcinogenesis.We then performed an expression analysis of miR-183and miR-186 in FFPE and fresh-frozen tissue samples.Expression analysis in FFPE samples showed a signifi-cant up-regulation of miR-183 (P < 0.01, all groups ver-sus NL) and a trend toward miR-186 up-regulation inDN and CL tissues (Fig. 7A). Further analysis of miR-
186 expression from fresh-frozen tissues showed an up-regulation of miR-186 in CL (see Supporting Fig. 3; P <0.05). We next determined if miR-183 and miR-186 candirectly regulate AKAP12 levels. The 30UTR ofAKAP12a/b harbors three and four putative target sitesfor miR-183 and miR-186, respectively, as predicted byTargetScan (Fig. 7B).19 The highly conserved 30 end ofthe AKAP12 30UTR contains two putative binding sitesfor each miRNA and was cloned into the 30 end of Fireflyluciferase. Nucleotide sequences were mutated to ablateeither both miR-183 or both miR-186 sites. MiR-183and miR-186 were cloned into expression plasmids andtransfected in HEK293T cells (see Supporting Fig. 4).Expression of increasing amounts of both miRNAs causeda dose-dependent increase in the mature miRNAs aboveendogenous levels (range 9 to 27 and 1.8 to 20-fold formiR-183 and miR-186, respectively). Coexpression ofmiR-183 with the wild-type (WT) and 186-mutatedUTR resulted in a measurable (1.3-fold) decrease in lucif-erase expression as compared to the 183-mutated UTR(Fig. 7C). Coexpression of miR-186 with the WT UTRresulted in a distinct decrease (two-fold) in luciferaseexpression compared to the 186-mutated UTR. Expres-sion of miR-186 in HEK293T cells resulted in a 2-foldreduction of endogenous AKAP12 mRNA levels (Fig.7D). Expression of miR-183 did not reduce endogenousAKAP12 levels. Both a and b isoforms were reduced tosimilar levels following miRNA-dependent mRNAknockdown (data not shown).
Discussion
Down-regulation and tumor suppressor activity of thescaffold protein AKAP12 has been shown in severalmalignancies,7,8,20 but, so far the role of AKAP12 inhepatocarcinogenesis is almost completely unknown.Here, we present protein expression data of AKAP12 ina large number of human liver tissue specimens (n ¼388), revealing a significant down-regulation ofAKAP12 in HCC compared to NL. Remarkably, CLand DN already showed reduced AKAP12 expression,suggesting that down-regulation of AKAP12 is an earlyevent in hepatocarcinogenesis. Indeed, recent findings inprostate cancer have led to the theory that AKAP12may act as a key player in early stages of carcinogenesis.6
TMA data demonstrate that AKAP12 expression is pro-gressively down-regulated during HCC progression.Accordingly, we propose a two-step model of AKAP12down-regulation in hepatocarcinogenesis, with a first sig-nificant down-regulation from NL to CL and DN. Thesecond step takes place during HCC progression.Because expression characteristics and functional data
Fig. 6. 5-aza-dC treatment causes DNA demethylation and reactiva-tion of the hypermethylated AKAP12a gene in AKN1 cells. (A) Isoform-specific AKAP12 mRNA expression levels in various untreated HCC celllines and PHH. (B) 5-aza-dC treatment strongly reduces promotermethylation levels of AKAP12a in AKN1 cells compared to HepG2 andHuH7 cells, which display lower baseline methylation levels. AKAP12bis unmethylated in all cell lines tested (black bars, untreated cells;white bars, cells treated with 10 lmol/L 5-aza-dC for 72 hours). (C)5-aza-dC treatment causes the re-expression of AKAP12a in AKN1cells. Only minor increases in expression of AKAP12a in HuH7 cellsand AKAP12b in all cell lines are observed. Histograms represent theaverage value of three technical replicates of the quantitative real-timePCR, with the corresponding SD.
2030 GOEPPERT ET AL. HEPATOLOGY, December 2010

obtained in prostate and gastric cancer suggest a tumorsuppressive function of AKAP12,6,8 its down-regulationin the majority of CL and DN may contribute to theincreased risk of malignant transformation.Because little is known about interaction of
AKAP12 with other factors, we correlated AKAP12expression at the protein level with the expression ofother factors involved in hepatocarcinogenesis. CyclinD1 overexpression is a common finding in hepatocar-cinogenesis, which has been shown to occur very earlyin hepatocarcinogenesis in mouse models.4,5 InNIH3T3 cells, it has been shown that SSeCKS expres-sion induces G1 arrest marked by a decrease in cyclinD1 expression.21 Interestingly, our study did not reveala statistically significant inverse correlation of AKAP12
with cyclin D1, although our TMA analysis showedincreasing cyclin D1 levels during hepatocarcinogene-sis. As expected, AKAP12 showed an inverse correla-tion with the proliferation marker Ki-67.As we previously demonstrated, AKAP12 down-regula-
tion may partly be caused by chromosomal loss of theAKAP12 gene locus (see Supporting Table 1).10 However,this finding did not sufficiently explain down-regulationof AKAP12 in most HCCs. Because aberrant methylationstatus has been identified to be of mechanistic and prog-nostic significance in human HCC,22 we tested epigeneticalterations in the AKAP12 promoter region. Our studydemonstrates hypermethylation of AKAP12a promoter inhuman HCC specimens and in various HCC cell lines.Thus, gene silencing by promoter hypermethylation may
Fig. 7. AKAP12 regulation by miR-183 andmiR-186 in hepatocarcinogenesis. (A) Expres-sion levels of miR-183 and miR-186 relativeto RNU6B in NL (n ¼ 5), CL (n ¼ 13), DN(n ¼ 5), and HCC (n ¼ 12) in FFPE tissues.Error bars represent standard errors of themean. MiR-183 is significantly higherexpressed in CL, DN, and HCC versus NL(P < 0.01; P < 0.001; Mann-Whitney Utest). (B) Schematic representation of theAKAP12 30UTR depicting the position of puta-tive miR-183 and miR-186 binding sites, theregion analyzed for functional miRNA/UTRinteractions, and the level of mammaliansequence conservation (UCSC, hg18). (C)Functional interaction assays were performedby expressing the highly conserved (30end)portion of the AKAP12 30UTR linked to the lu-ciferase gene in HEK293T cells. MiR-183,miR-186, and control (Let-7i and miR-196a)miRNAs were coexpressed. Luciferase expres-sion is displayed relative to the 30UTR wherethe corresponding predicted binding siteswere mutated. Error bars represent the SD ofthree independent experiments. (D) Endoge-nous AKAP12 mRNA expression levels follow-ing expression of increasing amounts of miR-183 and miR-186 expression plasmids inHEK293T cells. Data represent the average ofa and b isoforms. Error bars represent the SDof three technical replicates.
HEPATOLOGY, Vol. 52, No. 6, 2010 GOEPPERT ET AL. 2031

be the cause for the significant decrease of AKAP12 pro-tein levels in HCC cells. This concept of AKAP12 down-regulation is in line with studies in lung and gastric cancerwhich described the AKAP12 gene as a target for epige-netic silencing.8,23 Although existing antibodies fail to dis-tinguish between AKAP12 isoforms, data on AKAP12aand b transcripts suggest that hypermethylation of theAKAP12a promoter is predominantly responsible for epi-genetic silencing of AKAP12. This is supported by thefact that the highly methylated HCC cell line AKN1decreased AKAP12a promoter methylation after 5-aza-dCtreatment resulting in increased expression of AKAP12amRNA. The distinct hypermethylation of only theAKAP12a promoter seems to be specific for humanHCC, because data obtained in other malignancies, e.g.,gastric cancer, showed a hypermethylation of both,AKAP12a and b promoter region.8 A coordinate controlbetween the AKAP12 promoters might be involved inhepatocarcinogenesis; however, our data in human HCCdo not support this hypothesis.Interestingly, methylation analyses did not show hyper-
methylation of the AKAP12 promoter in CL and DN,although AKAP12 was down-regulated at the protein level.Because methylation analysis of CL tissue specimens wasoriginally performed without the use of microdissection,we assumed that a "dilution effect" by nonparenchymalcells (mainly fibrocytes and fibroblasts) might conceal hy-permethylation of hepatocytes in these samples. However,even improved analysis employing microdissected CL sam-ples of the same tissue specimens failed to confirm pro-moter hypermethylation as the cause of AKAP12 down-regulation in CL and DN. In search of posttranscriptionalmechanisms for AKAP12 down-regulation we detected analternative regulatory mechanism in CL and DN by miR-183 and miR-186. Both of these miRNAs are up-regu-lated in the precancerous stages where promoter hyper-methylation is absent; and, via a direct interaction with theAKAP12 transcript, both miRNAs can regulate mRNAlevels to various degrees, with miR-186 demonstrating astrong ability to regulate endogenous transcript levels.Regarding the observed genetic and epigenetic alterationsin HCC, this represents an interesting interplay betweendifferent epigenetic regulatory mechanisms in the course ofhuman hepatocarcinogenesis. A connection between epige-nome and miRNome and alteration in the balance of thiscomplicated network as a possible mechanism leading tocancer has been described recently.24
Additional mechanisms may also account forAKAP12 down-regulation. In CL, it could be shownthat a histone deacetylase inhibitor influences SSeCKSexpression.25 Apart from aberrant patterns of histonemodification, involvement of chromatin modifications
in the expression of the AKAP12a isoform was recentlyshown by its re-expression after treatment of mousefibroblasts with a histone deacetylase inhibitor.26 Differ-ent models support the hypothesis that CpG islandmethylation may follow histone modification to stablylock silenced genes.27 It is therefore conceivable that theobserved de novo DNA methylation of the AKAP12apromoter in HCCs may be also triggered by histonemodifications which are already present in CL.In summary, the data presented here demonstrate
that the tumor suppressor AKAP12 is down-regulatedduring hepatocarcinogenesis in a stepwise manner:early in cirrhosis and in premalignant lesions, and latein HCC dedifferentiation. We could identify differentepigenetic mechanisms responsible for this stepwisedown-regulation. In CL and DN, down-regulation ofAKAP12 is at least partly caused by interaction of twospecific miRNAs, whereas in HCC genetic loss and toa significant extent hypermethylation of the AKAP12apromoter are responsible for AKAP12 reduction.
Acknowledgment: We thank Peter Waas, Anna-LisaLackner, and Otto Zelezny (Division of Epigenomicsand Cancer Risk Factor, German Cancer ResearchCenter), and Eva Eiteneuer and John Moyers (Instituteof Pathology, University of Heidelberg) for their excel-lent technical assistance.
References1. Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr
Opin Cell Biol 2007;19:192-198.
2. Frisch SM. cAMP takes control. Nat Cell Biol 2000;2:E167-E168.
3. Su B, Bu Y, Engelberg D, Gelman IH. SSeCKS/Gravin/AKAP12 inhib-its cancer cell invasiveness and chemotaxis by suppressing a protein ki-nase C-Raf/MEK/ERK pathway. J Biol Chem 2010;285:4578-4586.
4. Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS, et al.Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activa-tion. Oncogene 2007;26:7103-7110.
5. Yamamoto M, Tamakawa S, Yoshie M, Yaginuma Y, Ogawa K. Neo-plastic hepatocyte growth associated with cyclin D1 redistribution fromthe cytoplasm to the nucleus in mouse hepatocarcinogenesis. Mol Car-cinog 2006;45:901-913.
6. Akakura S, Huang C, Nelson PJ, Foster B, Gelman IH. Loss of theSSeCKS/Gravin/AKAP12 gene results in prostatic hyperplasia. CancerRes 2008;68:5096-5103.
7. Su B, Zheng Q, Vaughan MM, Bu Y, Gelman IH. SSeCKS metastasis-suppressing activity in MatLyLu prostate cancer cells correlates withvascular endothelial growth factor inhibition. Cancer Res 2006;66:5599-5607.
8. Choi MC, Jong HS, Kim TY, Song SH, Lee DS, Lee JW, et al.AKAP12/Gravin is inactivated by epigenetic mechanism in human gas-tric carcinoma and shows growth suppressor activity. Oncogene 2004;23:7095-7103.
9. Streb JW, Kitchen CM, Gelman IH, Miano JM. Multiplepromoters direct expression of three AKAP12 isoforms with distinctsubcellular and tissue distribution profiles. J Biol Chem 2004;279:56014-56023.
2032 GOEPPERT ET AL. HEPATOLOGY, December 2010

10. Schlaeger C, Longerich T, Schiller C, Bewerunge P, Mehrabi A, ToedtG, et al. Etiology-dependent molecular mechanisms in human hepato-carcinogenesis. HEPATOLOGY 2008;47:511-520.
11. Singer S, Ehemann V, Brauckhoff A, Keith M, Vreden S, SchirmacherP, et al. Protumorigenic overexpression of stathmin/Op18 by gain-of-function mutation in p53 in human hepatocarcinogenesis. HEPATOLOGY
2007;46:759-768.12. Bressac B, Galvin KM, Liang TJ, Isselbacher KJ, Wands JR, Ozturk
M. Abnormal structure and expression of p53 gene in human hepato-cellular carcinoma. Proc Natl Acad Sci U S A 1990;87:1973-1977.
13. Tsuji T, Miyazaki M, Fushimi K, Mihara K, Inoue Y, Ohashi R, et al.Cyclin E overexpression responsible for growth of human hepatictumors with p21WAF1/CIP1/SDI1. Biochem Biophys Res Commun1998;242:317-321.
14. Nussler AK, Vergani G, Gollin SM, Dorko K, Morris SM Jr., DemetrisAJ, et al. Isolation and characterization of a human hepatic epithelial-like cell line (AKN-1) from a normal liver. In Vitro Cell Dev BiolAnim 1999;35:190-197.
15. Pallasch CP, Patz M, Park YJ, Hagist S, Eggle D, Claus R, et al. miRNAderegulation by epigenetic silencing disrupts suppression of the oncogenePLAG1 in chronic lymphocytic leukemia. Blood 2009;114:3255-3264.
16. Funke B, Autschbach F, Kim S, Lasitschka F, Strauch U, Rogler G,et al. Functional characterisation of decoy receptor 3 in Crohn’s disease.Gut 2009;58:483-491.
17. Wiebalk K, Schmezer P, Kropp S, Chang-Claude J, Celebi O, Debus J,et al. In vitro radiation-induced expression of XPC mRNA as a possiblebiomarker for developing adverse reactions during radiotherapy. Int JCancer 2007;121:2340-2345.
18. Wang Y, Lee AT, Ma JZ, Wang J, Ren J, Yang Y, et al. Profiling micro-RNA expression in hepatocellular carcinoma reveals microRNA-224up-regulation and apoptosis inhibitor-5 as a microRNA-224-specifictarget. J Biol Chem 2008;283:13205-13215.
19. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flankedby adenosines, indicates that thousands of human genes are microRNAtargets. Cell 2005;120:15-20.
20. Flotho C, Paulun A, Batz C, Niemeyer CM. AKAP12, a gene withtumour suppressor properties, is a target of promoter DNA methyla-tion in childhood myeloid malignancies. Br J Haematol 2007;138:644-650.
21. Lin X, Nelson P, Gelman IH. SSeCKS, a major protein kinase C sub-strate with tumor suppressor activity, regulates G(1)-->S progression bycontrolling the expression and cellular compartmentalization of cyclinD. Mol Cell Biol 2000;20:7259-7272.
22. Calvisi DF, Ladu S, Gorden A, Farina M, Lee JS, Conner EA, et al.Mechanistic and prognostic significance of aberrant methylation in themolecular pathogenesis of human hepatocellular carcinoma. J ClinInvest 2007;117:2713-2722.
23. Tessema M, Willink R, Do K, Yu YY, Yu W, Machida EO, et al. Pro-moter methylation of genes in and around the candidate lung cancersusceptibility locus 6q23-25. Cancer Res 2008;68:1707-1714.
24. Iorio MV, Piovan C, Croce CM. Interplay between microRNAs andthe epigenetic machinery: an intricate network. Biochim Biophys Acta2010 May 20. Epub ahead of print.
25. Rombouts K, Knittel T, Machesky L, Braet F, Wielant A, HellemansK, et al. Actin filament formation, reorganization and migration areimpaired in hepatic stellate cells under influence of trichostatin A, ahistone deacetylase inhibitor. J Hepatol 2002;37:788-796.
26. Bu Y, Gelman IH. v-Src-mediated down-regulation of SSeCKS metas-tasis suppressor gene promoter by the recruitment of HDAC1 into aUSF1-Sp1-Sp3 complex. J Biol Chem 2007;282:26725-26739.
27. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how thegenome integrates intrinsic and environmental signals. Nat Genet2003;33(Suppl):245-254.
HEPATOLOGY, Vol. 52, No. 6, 2010 GOEPPERT ET AL. 2033