development and characterisation of pilchard (sardinops sagax neopilchardus) cell lines derived from...
TRANSCRIPT

1. Introduction
In 1995 and again in 1998–99, mass mortalities ofpilchards (Sardinops sagax neopilchardus) occurredaround the southern coastline of Australia. In the1995 mortality, a herpesvirus was observed in thegills of all affected fish examined [10, 17]. Numerousattempts to isolate this pilchard herpesvirus onvarious poikilothermic cell lines have not been suc-cessful [10]. During the 1998/99 mortality, a her-pesvirus was also observed in affected fish but inlower numbers (Hyatt, pers. comm.). The inabilityto isolate and grow this virus in CHSE-214, RTG-2,BF-2, FHM or EPC fish cell lines has severelylimited the progress of research on this virus. Duringthe disease investigations, it became evident that apilchard cell line, susceptible to infection by thepilchard herpesvirus, would facilitate further studieswith this virus. This report describes the establish-ment and characterisation of two pilchard cell lineswhich should provide diagnostic and research toolsfor pilchard biology in general, and the study ofvarious marine viruses in particular. It is noteworthythat the worldwide growth of the marine aquacultureindustry in recent years has led to an increasedinterest in the development of marine fish cell linesespecially for use in virus isolation and growth [2,9, 14, 16]. Although methods used closely followthose developed for other vertebrates [7, 13], modi-fications are sometimes necessary and the number
of cell lines from marine fish are still relatively few[8].
2. Materials
Pilchards. During the 1998/99 pilchard mortalityevent, pilchards (Sardinops sagax neopilchardus)were collected in collaboration with staff from theMarine and Freshwater Resources Institute,Queenscliff, Victoria and a commercial fisher fromLakes Entrance, Victoria. In an attempt to obtaintissues from normal uninfected fish, juvenilepilchards approximately 10 months of age werenetted from a fishing trawler off the coast of LakesEntrance in Victoria, Australia in November 1998.The pilchards were alive when netted but died, orwere moribund, shortly after and were immediatelyplaced on ice. Seventeen juvenile fish (of similar size,measuring approximately 10 cm in fork length) wererandomly selected for cell culture, and appropriatetissues were removed aseptically while at sea andplaced into transport medium on ice. Tissues weretransported to the Australian Animal HealthLaboratory (AAHL) and further processed, approxi-mately 6 hours after collection.
Transport medium. Medium used for transport ofexcised tissue from the field was Eagle’s minimumessential medium with Earle’s salts (EMEM) (Gibco-
Methods in Cell Science 25: 105–113 (2003).© 2004 Kluwer Academic Publishers. Printed in the Netherlands.
Development and characterisation of pilchard (Sardinops sagaxneopilchardus) cell lines derived from liver and heart tissues
Lynette M. Williams, Mark St. J. Crane & Nicholas GudkovsCSIRO Livestock Industries, Australian Animal Health Laboratory, AAHL Fish Diseases Laboratory, Private Bag 24,Geelong, Victoria, 3220, Australia
Accepted in revised form 9 April 2003
Abstract. Two cell lines have been established fromjuvenile pilchards (Sardinops sagax neopilchardus)caught in waters off the Victorian coast of Australia.Following establishment of primary cultures derivedfrom different pilchard tissues, using various cellculture media, a pilchard liver (PL) cell line and apilchard heart (PH) cell line have been maintainedin Eagle’s minimal essential medium supplementedwith 10% foetal bovine serum for over four years.The cell lines have been cryopreserved in liquidnitrogen and can be recovered from storage with
Key words: Cell culture, Fish viruses, Pilchard, Sardinops sagax neopilchardus, Tissue culture
good cell viability. Stock cell cultures have beenmaintained at 20–22 °C on a continuous basis innormal atmosphere (100% air), with weekly sub-culture at a split ratio of 3:1. The origin of the cellcultures was confirmed by PCR analysis usingprimers designed to be specific for pilchard mito-chondrial DNA. In addition, the liver cell line wascloned and both the parental cell line and clonesthereof were shown to be susceptible to a broad rangeof marine and freshwater viral pathogens of fish.

BRL1 Cat. #41500-034) supplemented with 10 mMhepes (BDH2 Cat. #44285 6A) buffer, 2 mM gluta-mine (ICN Biomedicals Inc.3 Cat. #1580115), 2%(v/v) foetal bovine serum (CSL4), 50 µg/ml gen-tamycin sulphate (Sigma5 Cat. #G-3623), and 5µg/ml amphotericin B (Bristol-Myers Squibb Co.6
‘Apothecon’ Fungizone-Amphotercin B).
Washing medium. Phosphate buffered saline, PBSApH 7.4 (without Ca2+ and Mg2+ ions), supplementedwith 50 µg/ml gentamycin sulphate and 5 µg/mlamphotericin B was used to rinse the tissues toremove excess blood.
Trypsinisation medium. 0.25% (w/v) trypsin (Difco7
trypsin 1:250 Cat #0152-15) in hepes buffered saline,adjusted to pH 7.2 with 1 N sodium hydroxide andfiltered through 22 µm filter was sterility checked at37 °C prior to being stored frozen (–20 °C). Thetrypsin solution was used to treat tissues in attemptsto obtain single cell suspensions of the variouspilchard tissues for subsequent culture.
Growth medium. Initially, explanted and trypsinisedtissues were processed and cultured in 6 differentmedia:1. Eagle’s minimum essential medium with Earle’s
salts (EMEM) (Gibco-BRL) supplemented with10 mM hepes buffer.
2. Double strength EMEM supplemented with 10mM hepes buffer.
3. Medium 199 (M199) (ICN Biomedicals Inc.) supplemented with 10 mM hepes buffer.
4. Double strength M199 supplemented with 10mMhepes buffer.
5. Leibovitz (L-15) medium (Gibco-BRL) supple-mented with 1 mM hepes buffer.
6. Double strength L-15 supplemented with 1mMhepes buffer.
All media were further supplemented with 2 mM glutamine, 20% (v/v) foetal bovine serum (FBS), 50µg/ml gentamycin sulphate and 5 µg/ml amphotericinB.
Immunocytochemical reagents. Cells undergoingDNA synthesis were detected in cultures using acellular proliferation assay kit, commercially avail-able from Roche8 (Cat. #1 299 964), and then counterstained using either Mayer’s haematoxylin(Dako,9 Lillie’s modification) or Diff-Quik II counterstain (Lab Aids Pty Ltd,10 modification ofWright’s stain).
PCR reagents. DNA was extracted using thePuregene® DNA Isolation Kit (Gentra Systems11) asper the manufacturer’s instructions. Other reagentssuch as Platinum™ Taq DNA Polymerase and PCRbuffer were obtained from Life Technologies.12
3. Procedures
Preparation of tissues for primary cell cultures
Seventeen juvenile fish were netted from the trawlerand immediately placed on ice. Processing one fishat a time and using sterile instruments and aseptictechniques, the operculum was removed and gillsexcised. After the gills were removed, surface ster-ilisation was carried out by immersing the fish in80% (v/v) ethanol. Liver, heart, spleen and kidneywere removed and each placed in sterile containerscontaining an excess (20–50 ml) of transportmedium. Dissections took approximately one and onehalf hours to complete and tissues were maintainedon ice in transport medium for approximately sixhours from death while being transported to the lab-oratory for further processing.
At the laboratory and using aseptic technique, eachtissue was processed individually. Containers weresurface-disinfected with Virkon® (Antec InternationalLtd.13) and all processes carried out in a laminar flowcabinet. Tissues were washed four times in washingmedium, then, half the volume of each tissue placedin a 50 ml sterile, centrifuge tube with wash mediumand held at 4 °C for subsequent trypsin treatment.
Tissue explants
The remaining tissues, with the exception of gills,were minced in 10 ml wash medium using twoscalpel blades until a suspension of small tissue fragments was obtained. Each tissue suspension wasthen aspirated into a 10 ml pipette, placed in a 50 ml tube and centrifuged at 100 ×g for 5 minutesat approximately 4 °C. The supernatant of each tissuewas removed and the pellet resuspended in 6ml washmedium. Aliquots (1 ml) were placed into 25 cm2
(Corning) tissue culture flasks containing 5 ml ofeach type of growth medium.
Due to the volume of blood contained in the gill,after washing, half the gill tissue was treated withtrypsin and the remaining half had gill archesremoved with a scalpel blade from the remainingtissues, and the gill filaments were minced. The tissuehomogenate was aspirated and dispensed. Aliquots(2 ml) were placed in 25 cm2 (Corning) tissue cultureflasks each containing 5 ml of one of the mediumtypes. The remaining gill arch and filament tissue wascentrifuged at 100 ×g for 5 minutes at approximately4 °C. The resulting pellet was resuspended inwashing medium and 1 ml aliquots added to 25 cm2
(Corning) tissue culture flasks each containing 5 mlof one of the medium types. Cultures containingexplanted tissue were incubated at 22 °C, left undis-turbed for seven days, to allow optimum cell attach-ment, before examination by light microscopy.
106 MICS ART NO. 3003 PIPS NO. 5139801

Trypsinisation of tissues
Kidney, liver, spleen and heart tissues were slowlyagitated in 5 ml trypsinisation medium on a magneticstirrer at room temperature (22–24 °C). Trypsinisa-tion times varied between tissues (Table 1).
After initial trypsinisation, supernatants of eachtissue were removed into sterile tubes, and 5% (v/v)foetal bovine serum added to protect the cells fromthe effects of further trypsinisation. A second trypsin-isation treatment on the remaining tissues was undertaken (Table 1). Again the supernatants wereremoved. The two supernatants of each tissue werecombined and then aliquoted into six 25 cm2
(Corning) tissue culture flasks, each containing 5 mlof one medium type.
Cultures containing trypsinised tissue were incubated at 22 °C and examined twice weekly usinglight microscopy, to determine cell attachment, cellgrowth, and occurrence of any microbial contamina-tion.
Routine maintenance of cultures
Confluent cell monolayers of liver and heart cultures,maintained in single strength EMEM, have been sub-cultured using standard techniques (treatmentwith 0.25% trypsin/versene solution to obtain a singlecell suspension). A split ratio of 3:1 was used toestablish new cultures which grew to confluency in5 to 7 days at an incubation temperature of 22 °C.Within the first few months of maintenance most ofthe cultures established from other tissues or in otherculture medium did not thrive and were eventuallyterminated.
Storage of cells in liquid nitrogen
Standard methods using dimethyl sulphoxide(DMSO; Sigma D-5879) as a cryoprotectant forstorage of cells were used. Briefly, a single cell suspension was obtained from monolayer culturesusing the method described above and the cell densitydetermined. The single cell suspension was cen-trifuged at 100 ×g and the supernatant discarded. Thecell pellet was resuspended in a volume of growthmedium containing 10% (v/v) FBS and 10% (v/v)
DMSO to obtain a cell density of 3 million cells/ml.Cryotubes containing 1.5 ml aliquots were placed inan insulated container which, when placed at –80 °Cfor 24 hours, provided control of the freezing rate.Following freezing, the cryotubes were placed inliquid nitrogen storage. Viability of frozen stocks waschecked by thawing one cryotube from each batchand determining whether viable, growing culturescould be re-established.
Temperature range
Cultures were initiated at 22 °C and all subsequentcultures were maintained at this temperature. Todetermine the optimal growth temperature, replicatepilchard liver cell cultures (passage 63) were incu-bated at 15, 22, 25 and 30 °C. These cultures wereexamined daily, by light microscopy, and the relative,non-quantitative cell number and viability wasrecorded for cultures at each temperature.
Virus susceptibility
Replicate pilchard liver cell cultures at passage 56were seeded in 25 cm2 tissue culture flasks and incubated at 22 °C. Following overnight incubation,growth medium was removed from the near-confluentcultures and duplicate cultures were exposed to oneof the viral isolates (Table 2) in a minimal volumeof medium (at a multiplicity of infection < 0.01). Replicate cultures were incubated at either15 or 19 °C for one hour to allow virus adsorption.Following virus adsorption, 5 ml growth medium wasadded to each culture. Duplicate cultures inoculatedwith each virus were incubated at either 15 or 19 °C.Cultures were examined daily, by light microscopy,and any changes in culture morphology, typical ofviral cytopathic effect (CPE), were noted.
The presence of viral particles in samples fromcell cultures displaying viral-like CPE was confirmedby transmission electron microscopy (results notshown).
Cellular proliferation assay
Actively replicating cells were identified using animmunocytochemical assay, available as a commer-
MICS ART NO. 3003 PIPS NO. 5139801 107
Table 1. Summary of trypsin treatments applied to individual pilchard tissue
Tissue 1st trypsin treatment 2nd trypsin treatment Effect of treatment
Heart 04 min 07 min Undigested tissue remainingLiver 04 min 07 min Good dissociationKidney 12 min 05 min Good dissociationSpleen 19 min 13 min Good dissociationGill 13 min 15 min Undigested tissue remaining
Trypsin treatment undertaken at room temperature (22–24 °C).

cial kit, for the detection of exogenous 5-bromo-2′deoxy-uridine (BrdU) incorporation into cellularDNA using a BrdU-specific monoclonal antibodyprovided. A Roche (Cat. #1 299 964) kit was adaptedfor use on fish cell lines. Both the fixative and thetemperature of fixation were modified for use withfish cells. The fixative of choice was aceticacid:ethanol (1:3) used at room temperature (approx-imately 22 °C) for 45 minutes. Acetic acid/ethanolreproducibly yielded good results, was compatiblewith other kit reagents, and was used with otherfinfish cell lines such as the rainbow trout gonad cellline, RTG-2 (ATCC CCL-55) and the epitheliomapapulosum cyprini (EPC) cell line [6]. Apart frommodifications to the fixation of cell cultures, theassay was conducted according to the manufacturer’sinstructions.
On completion of the assay, microscopic exami-nation revealed that unlabelled cells were difficultto visualize. Thus two commercially available counterstains were evaluated for use:• Mayer’s haematoxylin (Dako, Lillie’s modifica-
tion)• Diff-Quik II counterstain (Lab Aids Pty Ltd,
modification of Wright’s stain)A number of experiments were conducted usingduplicate BrdU-labelled cell cultures, which werethen stained with either one of the two counterstainsto determine the most appropriate counterstain foreach cell type. Counterstains were applied to cellcultures after the immunoperoxidase reaction andwashing steps were completed. Cell cultures weretreated with Mayer’s haematoxylin for 1 minute,washed with tap water, and then a blueing agent,
Scott’s tap water, was added for 1 minute thenwashed off using tap water. Diff Quik was addeddirectly to cell cultures, drained and washed off intap water. After drying, coverslips were applied usingan aqueous mounting medium (Gurr2 Aquamountimproved).
PCR for confirmation of pilchard origin of celllines
A polymerase chain reaction (PCR) specific forSardinops spp., in combination with sequenceanalysis of the PCR product was used to determineif the established cell lines were derived frompilchard tissue. PCR primers were designed from theconsensus sequence of 4 DNA mitochondrial controlregions from Sardinops neopilchardus previouslylodged with GenBank (Accession Numbers U95929to U95932).
Genomic DNA isolated from the spleen of a frozenpilchard (Sardinops neopilchardus) was used as apositive control; sterile distilled water served as the negative control. DNA was extracted fromactively growing pilchard cell cultures or from frozenspleen using the Puregene® DNA Isolation Kit(Gentra Systems) as per the manufacturer’s instruc-tions. The concentration of DNA used for PCR wasquantified using a GeneQuant II® DNA calculator(Invitrogen).
PCRs were performed in 0.2 ml thin walled PCRtubes (Quantum Scientific Pty Ltd)14 in a GeneAmp®
9600 thermal cycler (Perkin Elmer-Cetus).15 Each 25 µl reaction contained 1–10 ng of template DNA,0.25 U of Platinum™ Taq DNA polymerase, PCR
108 MICS ART NO. 3003 PIPS NO. 5139801
Table 2. Listing of the sources of the fish viruses used in this study
Virus Source
Infectious pancreatic necrosis virus (IPNV) CEFAS, Weymouth laboratory, Weymouth, United Kingdom[Sp serotype] www.cefas.co.uk/weymouthlab.htm
Infectious haematopoietic necrosis virus (IHNV) Western Fisheries Research Centre, Seattle, Washington, USA[WRAC strain]
Viral haemorrhagic septicaemia virus (VHSV) National Institute for Agricultural Research (INRA), France[serotype 23.75] www.inra.fr/ENG/
Oncorhynchus masou virus (OMV) [serotype P-17] Faculty of Fisheries Hokkaido University, Japanwww.hokudai.ac.jp
Epizootic haematopoietic necrosis virus (EHNV) Regional Veterinary Laboratory, Benalla, Australia
Pilchard orthomyxo-like virus AFDL, AAHL, Geelong, Australiawww.csiro.au
Spring viraemia of carp virus (SVCV) CEFAS, Weymouth laboratory, Weymouth, United Kingdom[S-30 reference strain] www.cefas.co.uk/weymouthlab.htm
Infectious salmon anaemia virus (ISAV) Department of Fisheries and Oceans, New Brunswick, Canada[Canadian reference strain] www.glf.dfo-mpo.gc.ca
Atlantic salmon reovirus AFDL, AAHL, Geelong, Australiawww.csiro.au

buffer (Life Technologies), 6 mM MgCl2, 8 pmoleseach of the primers 5′-ATCTTATCATTCATGATAT-GCACGC-3′ (Pil-1) and 5′-TTATACTACAC-CGCGCGTCG-3′ (Pil-2), and all four deoxy-nucleotide triphosphates at 0.2 mM each. Reactionmixes were held for 2 min at 94 °C then amplifiedfor 25 cycles, with denaturation for 30 sec at 94 °C,annealing for 30 sec at 56 °C, and elongation for 1 min at 72 °C. A final extension was performed at 72 °C for 3 min. The expected PCR product size was472 bp. The primary PCR products were purifiedusing a Qiagen™ PCR product purification kit andboth DNA strands (typically 50 ng DNA) weresequenced with the ABI Prism® BigDye® TerminatorCycle Sequencing Ready Reaction Kit (AppliedBiosystems™)16 with the same primers used for theprimary PCR. Sequencing reaction products wereanalysed with an Applied Biosystems™ Model 377DNA sequencer. The resulting sequence data werethen subjected to blastn analysis [1]. All bioinfor-matics analyses were conducted on BioNavigator.comas provided by Entigen Corporation (http://www.entigen.com).
Cell cloning
A monolayer culture of PL cells was treated with0.25% trypsin/versene solution and individual cellswere cloned by the limiting dilution technique [11].Increasing dilutions of the cell suspension wereplaced in wells of 96-well culture plates and incu-bated at 22 °C. When cultures derived from singlecells reached confluency in wells, they were sub-cultured and expanded until sufficient numbers ofcells were obtained to allow cryopreservation androutine sub-culturing at weekly intervals.
To determine if the clones varied in virus suscep-tibility compared to the original parent PL cell line,cultures from three clones, in 25 cm2 flasks, wereinoculated with virus isolates (Table 2) and incubatedat 15 °C. Virus-inoculated cultures were examined bylight microscopy for the development of viral CPE.
4. Results and discussion
Primary cell cultures
After 2 days incubation, it was evident that cells incultures derived from trypsinised tissues in thedouble strength media were not attaching to the substrate. Nevertheless, cultures in double strengthmedia which remained uncontaminated with bacteriaand/or fungi, were retained for relatively prolongedperiods (up to 50 days) to ensure that any slowgrowing cells were not overlooked. Eventually, it wasclear that these cultures were not thriving and theywere discarded. The use of double-strength mediumto compensate for the marine habitat of these finfishappears to be unnecessary and, indeed, detrimentalto the primary cell cultures. Cell cultures have beengrown in 10% FBS and maintained for longer periodsin 5% FBS. Cells were initially cultured in mediacontaining 20% FBS and this was reduced to 10%as the cells did not appear to be growing and thereduction in FBS appeared to be beneficial to cellgrowth, indicating that 20% FBS may be excessiveand supra optimal. These observations are similar toprevious reports on the establishment of cell linesfrom marine finfish [5, 13, 16].
It is noted that the pilchard herpesvirus, the occurrence of which prompted this study, grows inthe gills of affected pilchards [10] and, for thisreason, it would have been desirable to obtain gillcell lines. Unfortunately, the primary gill cell culturesdid not survive. Contamination was the most signif-icant problem when attempting to culture gill tissue.Despite extensive washing of the excised tissue, usingantibiotic-supplemented media, cultures wereovercome by bacterial contamination. It is interestingto note that despite the cultures being heavily con-taminated, fibroblastic cells in 1× L-15 medium wereattached and appeared to be growing under the layerof contamination. For larger fish, a technique toperfuse gills with saline containing antibiotics andanticoagulant, while the fish was under anaesthetic,could be used, in an attempt to obtain tissue depletedof blood cells and bacteria which would be moresuitable for cell culture. However, the small size ofthe pilchards in this instance, was a limiting factor.
Cultures derived from kidney and spleen tissues,depending on treatments, demonstrated variabledegrees of cell attachment and spreading. Some ofthe cultures were sub-cultured once but then failed toshow any evidence of cellular replication and were
MICS ART NO. 3003 PIPS NO. 5139801 109
Table 3. Results summary: susceptibility of PL cell lineto fish viruses
Virus Time post inoculation andtemperature at which viralCPE was well developed
IPNV serotype Sp 7 days at 15 and 19 °C
IHNV (WRAC strain) 7 days at 15 °C; CPE did not develop at 19 °C
VHSV serotype 23.75 7 days at 15 and 19 °C
OMV serotype P-17 No CPE after 36 days at 15 and 19 °C.
EHNV 7 days at 15 and 19 °C
Pilchard orthomyxo-like virus 7 days at 15 and 19 °C
SVCV S-30 reference strain 7 days at 15 and 19 °C
ISAV Canadian ref. strain No CPE after 36 days at 15 and 19 °C
Atlantic salmon reovirus 7 days at 15 and 19 °C

eventually discarded. While cultures derived fromsplenic tissue appeared to have cells attached andsurvived in culture for 55 days, cellular replicationwas not evident and long-term cultures did not eventuate. Whether this inability to thrive was dueto the relatively small amounts of tissue initiallycultured or to the use of unsuitable culture medium,is not clear. The cells appeared to be ‘healthy’throughout the incubation period and thus it wouldseem worthwhile to attempt initiation of further cellcultures using larger amounts of splenic tissue.
Cell cultures derived from explanted heart tissueappeared to have sufficient numbers of cells but thesedid not attach to the substrate and all cultures werediscarded after 13 days incubation. Similar observa-tions were made on cultures derived from trypsinisedheart tissue, with one exception. Trypsinised hearttissues maintained in single strength EMEM havebeen subcultured to passage number 35. Interestingly,up to passage #10 the cells had a fibroblastic mor-phology which has since changed to a stable epithe-lial-like morphology. It is unclear why the cellularmorphology in the heart cultures ‘changed’ after thetenth passage from a fibroblastic-like appearance to an epithelial-like morphology. Whether a sub-population of epithelial-like cells outgrew the fibroblasts or whether a morphological transforma-tion [3] occurred cannot be determined. Nevertheless,following this transformation, the epithelial appear-ance of the heart cells appeared to be a stable characteristic of this cell line and no further mor-phological changes have occurred with continuouspassage.
Trypsinised liver and heart were the only tissueswhich have produced long-term cultures. Trypsinisa-tion, rather than the use of tissue explants, appearedto be the more successful technique. Cultures derivedfrom explants of liver tissue did not thrive in anymedium and were eventually discarded. Of the livercell cultures derived from trypsinised tissue, thosecultures maintained in single strength EMEM demonstrated morphological characteristics of epithelial cells and appeared to thrive better thanthose cultures maintained with L-15 or M199 media.After 44 days in primary culture with two mediumchanges, cells appeared to be overgrowing and weresubcultured. The foetal bovine serum concentrationwas reduced from 20% to 10% (v/v) in the secondary(passage #1) cell cultures after 48 days. Currently,cultures are subcultured routinely every 7 to 9 days,at a split ratio of 3:1. Stocks of PL cells have beencryopreserved in liquid nitrogen and successfullyrecovered at various passage numbers.
In summary, confluent monolayers of liver andheart tissue were obtained after approximately 5weeks incubation. After initial sub-culturing andincubation for various time periods, monolayercultures could be sub-cultured on a weekly basis,using trypsin-versene solution to obtain a single cell
suspension. A split ratio of 3:1 was used to establishnew cultures which grew to confluency in 5 to 7 daysat an incubation temperature of 22 °C. Cultures ofPL cells were treated with trypsin-versene at roomtemperature (22–24 °C) for approximately 5 minutes,while, initially, PH cells were treated for 15–25minutes. For later passages the exposure time wasreduced to less than 8 minutes.
From a range of tissues derived from juvenilepilchards and using various conditions of incubation,two pilchard cell lines have been established. The PL cell line has been sub-cultured in excess of 80passages over a 4-year period, while the PH cell linehas been sub-cultured in excess of 40 passages overthe same 4-year period. Cultures have been grownin a standard cell culture medium (EMEM) supple-mented with 10% FBS, 2 mM glutamine and 10 mMhepes. Although M-199 and L-15 media were usedon initial cultures, further trials using these and othermedium types were not pursued.
Storage of cells in liquid nitrogen
PL cells were cryopreserved in liquid nitrogen andrecovered successfully at passage numbers 8, 13, 15,16, 29, 48, 53 and 64. Heart cell cultures have beencryopreserved and recovered at passage numbers 9,12 and 14. Three clones were sub-cultured 12 timesprior to cryopreservation in liquid nitrogen and havebeen successfully recovered.
Cell proliferation assay
The Roche kit was relatively simple to apply to fishcell cultures. Following some minor modifications,the kit was shown to be a useful indicator of cell proliferation for fish cell cultures. Counterstainingthe cultures made the immunocytochemical labellingeasier to interpret. Mayer’s haematoxylin appeared tobe the better counterstain for the RTG-2 cell linewhile ‘Diff-Quik’ appeared better for the PL cell line(Figure 3). It is interesting to note that the ratio of
110 MICS ART NO. 3003 PIPS NO. 5139801
Figure 1. Photomicrograph of cloned PL cell line atpassage number 58 (Bar = 100 µm).

labelled to non-labelled cells is greater in the longerestablished fish cell line, RTG-2, than the PL cellline. It was noted that the longer the pilchard celllines were maintained in culture, the better theyseemed to adapt to in vitro culture resulting in celllines with less variable features, for example cellularmorphology became more uniform and the periodbetween subcultures became more consistent.
While cell proliferation assays have been devel-oped for use with mammalian tissues or cell cultures,usually incubated at 37 °C, this work has shown thatassays can be adapted for use with poikilothermic celllines from finfish. Similarly, other such assays havebeen used in the characterisation of primary cellcultures from invertebrates [12].
Incubation temperature
Cultures from both liver and heart tissue have beengrown on a routine basis at 22 °C. To determine theeffect of temperature on cell cultures, PL cells weregrown at 15, 22, 25 and 30 °C. All cultures wereincubated and observed for a period of 4 weeks.Incubation temperature had no effect on cellular morphology. Cultures at 15 °C reached confluency ata slower rate than at other incubation temperaturesbut cell cultures at the higher incubation temperatures(25–30 °C) showed some deterioration (abnormalcellular morphology) towards the end of the incuba-tion period.
Stock cell cultures are grown at 22 °C, the temperature at which the primary cultures were established and which is well within the normal temperature range for pilchards. It is likely thatduring this initial culture period, cells best suited tothis temperature were selected and it is not surprisingthat this temperature appears to be the optimum forcell growth. As with other fish cell lines as well asother vertebrate cell lines, the pilchard cell linesdemonstrate a permissive temperature range [5, 8, 13,16].
Virus susceptibility
The mass pilchard mortality events of 1995 and1998/9 associated with a herpesvirus infection [10,17] were the primary prompts for this study. It wasrecognised that an ability to grow the pilchard herpesvirus in cell cultures would greatly facilitatefurther research on this virus and the developmentof diagnostic procedures for the detection and iden-tification of the virus.
PL cells were susceptible to infection by IPNV (Spstrain), IHNV (WRAC strain), VHSV (strain 23.75),EHNV, pilchard orthomyxo-like virus (Crane et al.,manuscript in preparation), SVCV (strain S-30), andAtlantic salmon reovirus [4]. Cultures inoculatedwith these viruses demonstrated viral CPE (Figure 3). Growth of IHNV appeared to be temper-ature dependent. At 15 °C the virus appeared to growwell but IHNV did not appear to grow in culturesincubated at 19 °C. The PL line was refractory toinfection with OMV and ISAV. These results wereconfirmed by electron microscopic examination ofcell culture material (results not shown). The threePL cell clones, incubated at 15 °C only, did not
MICS ART NO. 3003 PIPS NO. 5139801 111
Figure 2. Photomicrograph of PH cell line at passagenumber 8 (Bar = 100 µm).
Figure 3. Photomicrographs of (a) PL cell line at passage 60 and (b) RTG-2 cell line at passage 99, stained using theRoche immunocytochemical cell proliferation kit. Note the darkly stained nuclei which were undergoing active DNAsynthesis at the time of fixation (Bar = 100 µm).
demonstrate any variation in virus susceptibility tothe non-cloned parental PL cell line.
Attempts to grow the pilchard herpesvirus on thesecell lines have not been successful to date. Due to thesub-optimal collection and storage conditions usedfor the herpesvirus during the disease outbreaks, itis likely that the available frozen stocks contain non-viable virus. Thus the susceptibility of these pilchardcell lines to the pilchard herpes virus cannot be determined. Nevertheless, it has been demonstratedthat the PL cell line is susceptible to a broad rangeof fish viruses and will be evaluated as a host cellline for the pilchard herpesvirus if fresh virusbecomes available.
PCR analysis
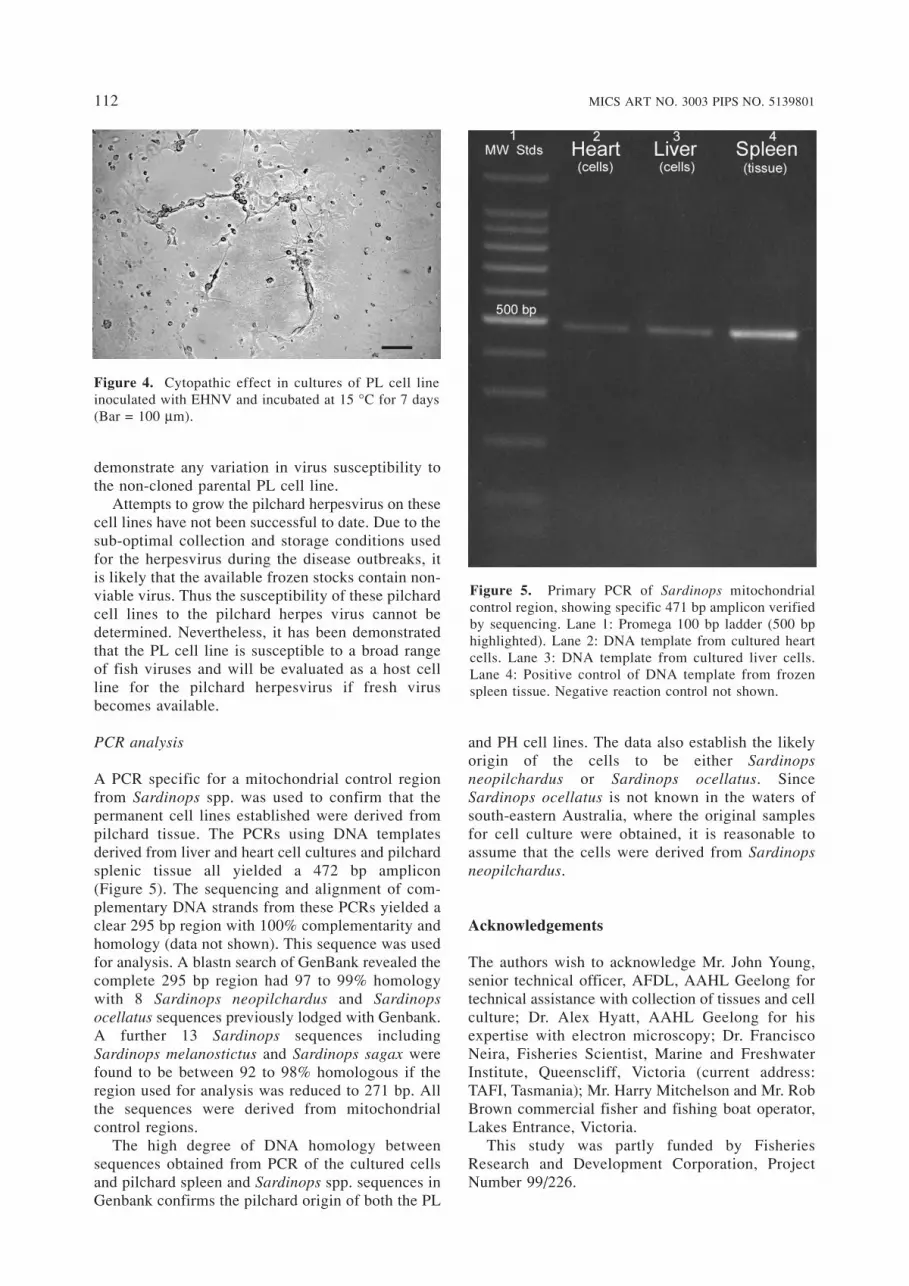
A PCR specific for a mitochondrial control regionfrom Sardinops spp. was used to confirm that the permanent cell lines established were derived frompilchard tissue. The PCRs using DNA templatesderived from liver and heart cell cultures and pilchardsplenic tissue all yielded a 472 bp amplicon (Figure 5). The sequencing and alignment of com-plementary DNA strands from these PCRs yielded aclear 295 bp region with 100% complementarity andhomology (data not shown). This sequence was usedfor analysis. A blastn search of GenBank revealed thecomplete 295 bp region had 97 to 99% homologywith 8 Sardinops neopilchardus and Sardinops ocellatus sequences previously lodged with Genbank.A further 13 Sardinops sequences includingSardinops melanostictus and Sardinops sagax werefound to be between 92 to 98% homologous if theregion used for analysis was reduced to 271 bp. Allthe sequences were derived from mitochondrialcontrol regions.
The high degree of DNA homology betweensequences obtained from PCR of the cultured cellsand pilchard spleen and Sardinops spp. sequences inGenbank confirms the pilchard origin of both the PL
and PH cell lines. The data also establish the likelyorigin of the cells to be either Sardinopsneopilchardus or Sardinops ocellatus. SinceSardinops ocellatus is not known in the waters ofsouth-eastern Australia, where the original samplesfor cell culture were obtained, it is reasonable toassume that the cells were derived from Sardinopsneopilchardus.
Acknowledgements
The authors wish to acknowledge Mr. John Young,senior technical officer, AFDL, AAHL Geelong fortechnical assistance with collection of tissues and cellculture; Dr. Alex Hyatt, AAHL Geelong for his expertise with electron microscopy; Dr. FranciscoNeira, Fisheries Scientist, Marine and FreshwaterInstitute, Queenscliff, Victoria (current address:TAFI, Tasmania); Mr. Harry Mitchelson and Mr. RobBrown commercial fisher and fishing boat operator,Lakes Entrance, Victoria.
This study was partly funded by FisheriesResearch and Development Corporation, ProjectNumber 99/226.
112 MICS ART NO. 3003 PIPS NO. 5139801
Figure 4. Cytopathic effect in cultures of PL cell lineinoculated with EHNV and incubated at 15 °C for 7 days(Bar = 100 µm).
Figure 5. Primary PCR of Sardinops mitochondrialcontrol region, showing specific 471 bp amplicon verifiedby sequencing. Lane 1: Promega 100 bp ladder (500 bphighlighted). Lane 2: DNA template from cultured heartcells. Lane 3: DNA template from cultured liver cells.Lane 4: Positive control of DNA template from frozenspleen tissue. Negative reaction control not shown.

Notes on suppliers
01. Gibco-BRL Invitrogen Australia Pty Limited, 122/45Gilby Road, Mt. Waverly, Victoria, 3149, Australia
02. BDH – supplied by Merck Pty Ltd, 207 ColchesterRoad, Kilsyth, Victoria, 3137, Australia
03. ICN Biomedicals Inc, 1263 South Chillicothe Road,Aurora, OH 44202, USA
04. CSL Ltd. 45 Poplar Road, Parkville, Victoria, 3052,Australia
05. Sigma-Aldrich Pty Ltd, Unit 2, 14 Anella Avenue,Castle Hill, New South Wales, 3154, Australia
06. Bristol-Myers Squibb Co, 556 Princess Highway,Noble Park, Victoria, 3174, Australia
07. Difco Laboratories, Detroit, MI, USA08. Roche Diagnostics Australia Pty Ltd, 31 Victoria
Avenue, Castle Hill, New South Wales, 3154,Australia
09. Dako Corporation, Carpinteria, CA 93013, USA10. Lab Aids Pty Ltd, Unit 3, 3 Gondola Road, Narrabeen,
New South Wales, 2101, Australia11. Gentra Systems, 13355 10th Avenue N, Suite 120,
Minneapolis, MN 55441, USA12. Life Technologies. Invitrogen Australia Pty Limited,
122/45 Gilby Road, Mt. Waverly, Victoria, 3149,Australia
13. Antec International Ltd, Chilton Industrial Estate,Sudbury, Suffolk, CO102XD, UK
14. Quantum Scientific Pty Ltd, 8/200 Turner Street, PortMelbourne, Victoria, 3207, Australia
15. Perkin-Elmer Instruments, PO Box 600, Knoxfield,Victoria, 3180, Australia
16. Applied Biosystems, 52 Rocco Drive, Scoresby,Victoria, 3179, Australia
References
01. Altschul SF, Madden TL, Schäffer AA, Zhang J,Zhang Z, Miller W, Lipman, DJ (1997). GappedBLAST and PSI-BLAST: a new generation of proteindatabase search programs. Nucl Acids Res 25:3389–3402.
02. Chang SF, Ngoh GH, Kueh LFS, Qin QW, Chen CL,Lam TJ, Sin YM (2001). Development of a tropicalmarine fish cell line from Asian seabass (Lates calcarifer) for virus isolation. Aquaculture 192:133–145.
03. Crane MStJ (1999). Mutagenesis and cell transfor-mation in cell culture. Meth Cell Sci 21: 245–253.
04. Crane, MStJ, Hardy-Smith P, Williams LM, HyattAD, Eaton LM, Gould A, Handlinger J, Kattenbelt J,Gudkovs N (2000). First isolation of an aquatic birnavirus from farmed and wild fish species inAustralia. Dis Aquat Org 43: 1–14.
05. Fernandez RD, Yoshimizu M, Kimura T, Exura Y(1993). Characterisation of three continuous cell linesfrom marine fish. J Aquat Animal Hlth 5: 127–136.
06. Fijan N, Sulimanovic D, Bearzotti M, Muzinic D,
Zwillenberg LO, Chilmonczyk S, Vautherot JF, deKinkelin P (1983). Some properties of the EpitheliomaPapulosum Cyprini (EPC) cell line from carp(Cyprinus carpio). Ann Virol (Inst Pasteur) 134E:207–220.
07. Freshney RI (1994). Culture of Animal Cells – aManual of Basic Techniques (3rd ed.) Wiley-Liss,New York.
08. Fryer JL, Lannan CN (1994) Three decades of fishcell culture: current listing of cell lines derived fromfishes. J Tissue culture Methods 16: 87–94.
09. Ganassin RC, Sanders SM, Kennedy CJ, Joyce EM,Bols NC (1999). Development and characterization ofa cell line from Pacific herring, Clupea harenguspallasi, sensitive to both naphthalene cytotoxicity andinfection by viral hemorrhagic septicemia virus. CellBiol Toxicol 15: 299–309.
10. Hyatt AD, Hine PM, Jones B, Whittington R, KearnsC, Wise TG, Crane MS, Williams LM (1997).Epizootic mortality in the pilchard Sardinops sagaxneopilchardus in Australia and New Zealand in 1995.II. Identification of a herpesvirus within the gillepithelium. Dis Aquat Org 28: 17–29.
11. McFarland DC (2000). Preparation of pure cellcultures by cloning. Meth Cell Sci 22: 63–66.
12. Mulford AL, Lyng F, Mothersill C, Austin B (2001).Development and characterisation of primary cellcultures from the hematopoietic tissues of the DublinBay prawn, Nephrops norvegicus. Meth Cell Sci 22:265–275.
13. Nicholson BL (1985). Techniques in fish cell culture.In: Techniques in the Life Sciences, C1. Setting Upand Maintenance of Tissue and Cell Cultures, C105:1–16.
14. Perez-Prieto SI, Rodriguez-Saint-Jean S, Garcia-Rosado E, Castro D, Alvarez MC, Borrego JJ (1999).Virus susceptibility of the fish cell line SAF-1 derivedfrom gilt-head seabream. Dis Aquat Org 35: 149–153.
15. Qin QW, Lam TJ, Sin YM, Shen H, Chang SF, NgohGH, Chen CL (2001). Electron microscopic observa-tions of the marine fish iridovirus isolated frombrown-spotted grouper, Epinephelus tauvina. J VirolMeth 98: 17–24.
16. Watanabe Y, Hanada H, Usiyama M (1981).Monolayer cell cultures from marine fishes. FishPathol 15: 201–205.
17. Whittington RJ, Jones JB, Hine PM, Hyatt AD (1997).Epizootic mortality in the pilchard Sardinops sagaxneopilchardus in Australia and New Zealand in 1995.I. Pathology and epizootiology. Dis Aquat Org 28:1–15.
Address for correspondence: Lynette M. Williams, CSIROLivestock Industries, Australian Animal HealthLaboratory, AAHL Fish Diseases Laboratory, Private Bag24, Geelong, Victoria, 3220, AustraliaPhone: +61 3 5227 5000; Fax: +61 3 5227 5555E-mail: [email protected]
MICS ART NO. 3003 PIPS NO. 5139801 113