characterisation of macrophage capping protein as a novel
TRANSCRIPT

i
Characterisation of macrophage
capping protein as a novel
inflammatory mediator
Patrick Heng
B.Biomed.Sc., B.Sc. (Hons.)
Submitted in total fulfilment of the requirements of the degree
of Doctor of Philosophy
August 2017
Department of Pharmacology and Therapeutics
The University of Melbourne
Supervisors
Dr. Graham Mackay
Professor Mark Hogarth

i
Abstract
Immune cells such as mast cells and macrophages play important roles in
initiating, perpetuating, and resolving inflammation. These cells release
soluble factors that mediate the common features of inflammation in both
health and disease. Whilst many mediators have been identified and
characterised, the function of other released mediators remains unclear.
Preliminary studies in our laboratory have identified macrophage capping
protein (CapG) as a novel factor released from mast cells. Intracellular CapG
is known to be a regulator of actin polymerisation. However, extracellular
CapG is also known to be constitutively secreted from resting macrophages
and found to be elevated in inflammatory disorders such as rheumatoid
arthritis. However, the function of this protein at the extracellular level
remains unclear. We hypothesised that CapG is a novel inflammatory
mediator that contributes to inflammation.
The main findings of this thesis are as below:
1. CapG is predominantly expressed intracellularly in immune cells in
both primary and immortalised macrophages and mast cell lines.
2. CapG is released from activated mast cells and macrophages,
including microglia. Furthermore, release of CapG from LPS-
stimulated macrophages is mediated through TLR4 and is modulated
by the anti-inflammatory glucocorticoid dexamethasone.
3. Messenger RNA levels of CapG are downregulated in LPS-stimulated
macrophages. However, CapG message levels are elevated in tissue
samples obtained from mouse models of inflammation, as well as in
human brain samples obtained from post-mortem Alzheimer’s
disease sufferers.
4. To facilitate an examination of the extracellular role of CapG, we have
developed a human CapG mammalian expression system that was
functionally validated using actin polymerisation assays.
5. Recombinant CapG was shown to significantly induce pro-
inflammatory cytokine release from a variety of different cell types.

ii
In summary, we have shown CapG is released following immune cell
activation and is able to trigger pro-inflammatory cytokine release from
other cells. Combined, the studies in this thesis reveal extracellular CapG as
a novel pro-inflammatory mediator. The regulation of CapG at the gene
level also points to a role in ongoing inflammatory diseases. This thesis sets
the foundation for further analysis of the role of CapG in inflammatory
diseases through the use of mice with knockout of the CapG gene or with
CapG neutralising antibodies. Such studies will identify if CapG is indeed a
novel therapeutic target to alleviate the burden of chronic inflammatory
diseases.

iii
Declaration
I, Patrick Heng declare that the following Thesis entitled
“Characterisation of macrophage capping protein as a novel
inflammatory mediator” and the work presented in it are my
own. I confirm that:
This work is completed wholly while in candidature for a
Doctorate of Philosophy in Pharmacology while at the
University of Melbourne.
Where I have quoted from the work of others, the source is
always given. With the exception of such quotations, this
Thesis is entirely my own work.
Where the thesis is based on work completed by myself jointly
with others, I have made clear exactly what was done by others
and what I have contributed myself.
Parts of this Thesis have been presented at scientific
conferences
The thesis is less than 100,000 words.
__________________
Patrick Heng

iv
Conference Abstracts
Heng P, Xia YC, Harris T, Wines B, Hogarth PM, Stewart AG, Mackay
GA (2014) “Identification and Characterisation of the Biological Roles
of Novel Mast Cell Mediators.”
In:
American Thoracic Society Conference. San Diego, USA.
Airway Inflammation and Remodelling Conference. Melbourne,
Australia.

v
Acknowledgements
I would like to thank a few people who have helped me in
immeasurable ways and have helped made my journey a memorable
one. Firstly, I would like to thank Dr. Graham Mackay, my primary
supervisor who has taught me most of what I know about science and
research. Thank you for being patient with me, teaching and guiding
me, and having confidence in me at times when I was doubting myself.
Thank you also for letting me play music in the laboratory, even
though there were times when even I thought my taste in music was
questionable.
Secondly, I would like to thank my co-supervisor Prof. Mark Hogarth
and members of his laboratory, including Bruce and May Lin who
helped me during my time at the Burnet Institute. Thank you for
looking after me and making me feel comfortable during my time
there. I would also like to thank Prof. Alastair Stewart, who provided
me with helpful advice and kind words during my time at the
Department. Thank you for your guidance and advice throughout my
time in the department.
To the past and present members of the Mackay and Stewart
Laboratory–Amanda, Christine, Connie, Danica, Ebony, Meina, Sai,
Shenna, Tippy, Trudi and others whom I missed – I thank you for
teaching me and making my time in the laboratory fun and also
tolerating my moody music.
I would also like to thank members of the Department of
Pharmacology and Therapeutics – Danny, Christine, Peter, Jimmy,
Michael and Tony in particular – for supporting me throughout my
time in the department. I would also like to extend my gratitude to
my fellow Ph.D./Masters colleagues –Ash, Dalia, Khammy, Marianna,
Meaghan, Myles, Nat, Zach– thank you for the company in and out of

vi
the Department – I wish you nothing but the best in your future
endeavours and hope we cross paths in the future.
Finally, I would like to thank my family and friends. To my parents, I
thank you for your constant support and patience for me throughout
my life – I am not the person I am now without your guidance. Words
cannot express how grateful I am and how proud I am to call you my
parents. To my sister, thank you for looking out for me even though
you didn’t need to or when you were busy. To my friends – the “geng
kartu”, the badminton gang, and my high school friends – your
company, the outings, dinners/lunches, board games, and online
games were much needed stress-relief sessions that I will never forget.

vii
Table of Contents
Abstract i
Declaration iii
Conference Abstracts iv
Acknowledgements v
List of Figures xvi
Chapter 1
General Introduction 1
1.1 The immune system 2
1.2 The mast cell: its place in the immune system 2
1.3 The macrophage: another key player of the immune system 4
1.4 The innate immune system: the first line of defense 5
1.5 The involvement of mast cells and macrophages in the adaptive
immune system 7
1.6 Mast cell and macrophage activation through IgE 8
1.7 Mast cell and macrophage activation through PRRs 13
1.8 Mediator release from activated mast cells and macrophages 15
1.9 Mast cells and macrophages in disease 18 1.9.1 Type I hypersensitivity-associated diseases 19
1.9.1.1 Allergic Asthma 20 1.9.2 Type III Hypersensitivity 25
1.9.2.1 Rheumatoid Arthritis 25 1.9.3 Neuroinflammation 27

viii
1.10 A brief overview of current treatments of inflammatory disorders
28 1.10.1 β2-adrenoceptor agonists 29 1.10.2 Glucocorticoids 30 1.10.3 Omalizumab 31 1.10.4 Mast cell stabilisers 32 1.10.5 Anti-cytokine therapy 33 1.10.6 Treatment of Asthma and Rheumatoid Arthritis: Not one size
fits all 35
1.11 Identification of macrophage capping protein as a putative novel
mast cell mediator 36
1.12 Gelsolin superfamily as regulators of actin polymerisation 37
1.13 Gelsolin and CapG 39
1.14 Well established intracellular roles of Gelsolin and CapG 42
1.15 An established role for extracellular gelsolin 43
1.16 An emerging role for extracellular CapG 44
1.17 Aims of this thesis 46
Chapter 2
General Methods 49
2.1 Cell Culture 50 2.1.1 Human mast cell-1 (HMC-1) cells transfected with the α-subunit
of the human FcεRI (HMCα cells) 50 2.1.1.1 HMCα cell stimulation 50
2.1.2 Laboratory of Allergic Diseases (LAD2) cells 51 2.1.2.1 LAD2 cell stimulation 51
2.1.3 Rat Basophil Leukaemia (RBL) cells 52 2.1.4 THP-1 cells 52
2.1.4.1 THP-1 cell stimulation 52 2.1.5 BV2 cells 53
2.1.5.1 BV2 cell stimulation 54

ix
2.1.6 Mouse bone marrow derived-mast cells 54 2.1.7 Human airway smooth muscle (hASM) cells 54
2.1.7.1 hASM cell stimulation 55 2.1.8 BEAS2B cells 55
2.1.8.1 BEAS2B cell stimulation 55 2.1.9 Human Embryonic Kidney-293 (HEK293) cells 56
2.1.9.1 Flp-InTM-293 cells 56 2.1.9.2 293-EBNA or HEK293E cells 56
2.1.10 COS-7 cells 57 2.1.11 SW982 cells 57
2.1.11.1 SW982 cell stimulation 57
2.2 Rat peritoneal cell (RPC) collection and rat peritoneal mast cell
(RPMC) purification 57 2.2.1 Rat peritoneal macrophage isolation and stimulation 59
2.3 Flow cytometry (FACS) analysis for intracellular staining of CapG 59
2.4 Measurement of mast cell degranulation via β-hexosaminidase
release 60
2.5 Immunofluorescence Microscopy 61 2.5.1 THP-1 cells 61
2.6 Measurement of cytokine levels using enzyme-linked
immunosorbent assays (ELISA) 62
2.7 Protein extraction, sample preparation and Bradford protein assay
64 2.7.1 Bradford Assay 64
2.8 SDS-PAGE Gel Electrophoresis 65 2.8.1 Western Blotting 65 2.8.2 Coomassie staining of SDS-PAGE gels 65
2.9 mRNA extraction, cDNA synthesis and quantitative PCR (qPCR) 66 2.9.1 Sample collection 66
2.9.1.1 Cell samples 66 2.9.1.2 Mouse models 67
2.9.1.2.1 LPS and Respiratory Syncytial Virus (RSV) models 67 2.9.1.2.2 APPSWE/PS-1ΔE9 (APP/PS-1) model 67
2.9.1.3 Human monocytes 67

x
2.9.2 mRNA extraction 68 2.9.3 cDNA synthesis 68 2.9.4 Quantitative real-time PCR (qPCR) 68
2.10 Recombinant expression of CapG in HEK and COS cells 70 2.10.1 Flp-InTM-293 and COS cells 70 2.10.2 EBNA293 cells 70
2.11 Purification of CapG 72
2.12 Concentration and dialysis of purified CapG 72
2.13 Statistical analysis 72
Chapter 3
Cellular expression and release of Macrophage Capping
Protein (CapG) - a potential pro-inflammatory mediator?
75
3.1 Introduction 76
3.2 Specific Methods 85 3.2.1 Cell culture 85 3.2.2 Rat peritoneal cell (RPC) collection and isolation of rat peritoneal
macrophages and mast cell (RPMC) 85 3.2.3 mRNA extraction, cDNA synthesis and qPCR 85 3.2.4 Western blot analysis 85
3.2.4.1 Intracellular CapG expression in different cell types 85 3.2.4.2 Measuring CapG release from stimulated cells 88
3.2.4.2.1 Supernatants 88 3.2.4.2.2 Cell pellets 88
3.2.5 Immunofluorescence 88 3.2.6 Flow Cytometry analysis (FACS) for intracellular staining of CapG
89 3.2.7 Degranulation Assay 89 3.2.8 Measurement of cytokine levels using enzyme-linked
immunosorbent assays (ELISA) 89

xi
3.2.9 Statistical analysis 90
3.3 Results 92 3.3.1 CapG gene expression in a variety of cell types 92 3.3.2 Cell distribution of CapG in rodent and human cell lines 93 3.3.3 Purification of RPMC and expression of CapG from different rat
peritoneal cell populations 97 3.3.4 CapG is released from LAD2 cells following IgE/FcεRI activation,
but not HMCα cells 100 3.3.5 LPS induces CapG release from THP-1 cells in a concentration-
dependent manner 107 3.3.6 CapG release from LPS-stimulated THP-1 cells is inhibited by
dexamethasone and the TLR4 blocking antibody HTA-125 112 3.3.7 CapG released is also enhanced by LPS in BV2 cells, but not
affected by dexamethasone 120
3.4 Discussion 122
Chapter 4
Characterisation of CapG gene expression in vitro and in
vivo models of peripheral and central inflammatory
diseases 136
4.1 Introduction 137
4.2 Specific Methods 144 4.2.1 Animal models 144
4.2.1.1 LPS and RSV models 144 4.2.1.2 APPSWE/PS-1ΔE9 (APP/PS-1) model 144
4.2.2 Human post-mortem brain tissues 145 4.2.3 Cell stimulation 146
4.2.3.1 Human monocytes 146 4.2.4 mRNA extraction and qPCR 146
4.2.4.1 mRNA extraction and cDNA synthesis 146 4.2.4.2 qPCR 147
4.2.5 Statistical analysis 147

xii
4.3 Results 148 4.3.1 CapG mRNA expression is differentially expressed in
macrophages following LPS stimulation 148 4.3.2 CapG mRNA expression is elevated in lungs of RSV and LPS-
treated mice, but not in total BAL cells 153 4.3.3 CapG expression is not affected by LPS stimulation in BV2 cells
159 4.3.4 CapG expression is upregulated in Alzheimer’s disease patients
161
4.4 Discussion 164
Chapter 5
Generation and purification of human CapG using a
mammalian expression system 173
5.1 Introduction 174
5.2 Specific Methods 179 5.2.1 Cloning and plasmid expansion 179
5.2.1.1 pcDNA5/FRT/TO vector 179 5.2.1.2 pCEP-Pu vector 179
5.2.2 Cell culture and transfection 182 5.2.2.1 pcDNA5/FRT/TO vector – Flp-InTM-293 and COS-7 cells 182
5.2.2.1.1 Analysis of human recombinant CapG production 183 5.2.2.2 pCEP-Pu vector – EBNA-293 cells 183
5.2.2.2.1 Optimisation of CapG production by EBNA-293 cells 183 5.2.3 Western Blotting 184 5.2.4 Purification of recombinant human CapG 185
5.2.4.1 Strep-Tactin® column 185 5.2.4.2 HisTALON™ column 186
5.2.5 Protein concentration, dialysis and analysis by Coomassie Blue-
staining 187 5.2.5.1 Protein concentration 187 5.2.5.2 Dialysis 189

xiii
5.2.5.3 Coomassie Blue staining 189 5.2.6 Mass Spectrometry 189
5.2.6.1 In-gel digestion 189 5.2.7 Actin polymerization assay 190
5.3 Results 192 5.3.1 CapG is expressed in Flp-In™ 293 cells following transient
transfection 192 5.3.2 Flp-In™ 293 cell-derived released CapG is likely associated with
cell death 195 5.3.3 CapG expression was detected in transiently transfected COS-7
197 5.3.4 Transfected EBNA-293 cells secrete CapG protein 199 5.3.5 Purification using a HisTALON™ column yields higher quantities
of recombinant CapG compared to purification using a Strep-Tactin®
column 202 5.3.6 Optimisation of EBNA-293 growth in different culture conditions
204 5.3.7 Examining the degree of purity of purified CapG and the
identification of protein bands from purified samples 207 5.3.8 His-CapG reduces the rate of pyrene-actin polymerisation 211
5.4 Discussion 216
Chapter 6
Functional characterisation of the role of extracellular
CapG 226
6.1 Introduction 227
6.2 Specific methods 233 6.2.1 Cell culture and stimulation 233
6.2.1.1 Human airway smooth muscle (hASM) cells 233 6.2.1.2 THP-1 cells 233 6.2.1.3 BEAS2B cells 233 6.2.1.4 SW982 cells 234

xiv
6.2.2 Cell viability measurement 234 6.2.3 Measurement of cytokine levels using enzyme-linked
immunosorbent assays (ELISA) 235 6.2.3.1 IL-8 235 6.2.3.2 CCL2 235
6.2.4 Statistical analysis 235
6.3 Results 236 6.3.1 Bacterially-expressed recombinant CapG trigger IL-8 and IL-6
release from primary human airway smooth muscle cells. 236 6.3.2 bac-CapG induces IL-8 release from THP-1 cells 240 6.3.3 Polymyxin B dampens the biological activity of bac-CapG on IL-8
release from THP-1 and SW982 cells 245 6.3.4 His-CapG triggers IL-6 and IL-8 release from hASM cells 251 6.3.5 His-CapG triggers IL-8, but not CCL2 release from THP-1 cells 254
6.4 Discussion 259
Chapter 7
General Discussion 266
7.1 CapG is primarily expressed in haematopoietic immune cells 271
7.2 CapG is released from mast cells 271
7.3 Regulation of CapG expression in inflammatory conditions 273
7.4 CapG – a role in neuroinflammation? 276
7.5 Recombinant CapG (both commercial and in-house generated)
triggered cytokine release from a variety of different cell types 278
7.6 Future directions 282
7.7 Concluding remarks 284
References 287

xv
List of Tables
Table 1.1. Mediators released from FcεRI-activated human mast cells and its
effects in asthma pathogenesis. 21-22
Table 1.2 Potential dual roles of macrophages in allergic asthma. 24
Table 2.1. List of antibodies used in flow cytometry analysis. 60
Table 2.2. List of working dilution concentrations used in ELISA experiments.
63
Table 2.3. List of antibodies used in Western blotting analysis. 66
Table 2.4 List of TaqMan® and KicQStart® SYBR® Green primers used in this
study.
71
Table 3.1. List of cells studied for CapG expression. 86-87
Table 3.2. List of antibodies used in experiments. 91
Table 3.3. Threshold cycle numbers of human and rat CapG in different cell
types. 92
Table 5.1. Proteins identified by Mass Spectrometry. 210

xvi
List of Figures
Figure 1.1. Mast cell activation. 11
Figure 1.2. Actin treadmilling. 38
Figure 1.3 Regulation of actin polymerisation by gelsolin and CapG. 41
Figure 1.4. Could CapG be an important inflammatory mediator? 47
Figure 3.1. CapG is released from mast cells and macrophages. 83
Figure 3.2. CapG protein is highly conserved between species. 84
Figure 3.3. CapG expression in human and rodent mast cells and macrophages.
95
Figure 3.4. CapG expression in primary cells. 96
Figure 3.5. Identification of mast cells and analysis of CapG expression in distinct
rat peritoneal cell subpopulations. 98
Figure 3.6. LAD2 cells degranulate, as measured by β-hexosaminidase release,
following stimulation with various stimuli. 102
Figure 3.7. CapG release is only enhanced in antigen stimulated early passaged
LAD2 cells. 103
Figure 3.8. HMCα cells did not consistently release CapG following antigen
stimulation. 105
Figure 3.9. Comparison of IL-8 cytokine release from stimulated HMCα cells
between current and previous studies. 106
Figure 3.10. THP-1 cells release CapG following LPS stimulation. 108
Figure 3.11. THP-1 cells release IL-8 following LPS stimulation. 109
Figure 3.12. CapG is released from THP-1 cells in response to LPS in a time-
dependent manner. 110
Figure 3.13. IL-8 release from LPS-stimulated THP-1 cells was significantly
reduced following dexamethasone pre-treatment. 114
Figure 3.14. Dexamethasone reduces CapG release from LPS-stimulated THP-1
cells. 115

xvii
Figure 3.15. CapG is released from THP-1 cells upon LPS stimulation, with
release inhibited by dexamethasone. 117
Figure 3.16. Inhibition of CapG release from LPS-stimulated THP-1 cells pre-
treated with HTA-125. 118
Figure 3.17 LPS induces CapG release from BV2 cells and this is unaffected by
dexamethasone. 121
Figure 3.18. Summary of Chapter 3. 134
Figure 4.1. Overview of the APP/PS-1 mouse phenotype. 145
Figure 4.2. Expression of CapG mRNA is decreased in THP-1 cells upon LPS
stimulation. 150
Figure 4.3. CapG gene expression is decreased in primary GM-CSF differentiated human macrophages following LPS stimulation, but not in undifferentiated human monocytes. 151
Figure 4.4. Expression of CapG mRNA is decreased upon LPS stimulation of rat peritoneal macrophages cells at both 4 and 24 hours. 152
Figure 4.5. Differential CapG gene expression in BAL cells and lung extracts
obtained from LPS treated mice. 155
Figure 4.6. CapG gene is differentially expressed in BAL cells and lung extracts
obtained from RSV infected mice. 157
Figure 4.7. LPS does not modulate expression of CapG and KC in the mouse
microglial-like BV2 cell line. 160
Figure 4.8. CapG message levels are significantly elevated in Alzheimer’s disease
patients. 162
Figure 4.9. Expression of CapG message increases over time in APP/PS-1 mice.
163
Figure 5.1. Schematic diagram of the two vectors used in this study. 181
Figure 5.2. The amino acid sequence and the calculated extinction coefficient of
CapG. 188
Figure 5.3. Transient and stably-transfected Flp-In™ 293 cells express CapG in
cell pellets and supernatants. 193
Figure 5.4. The presence of CapG in transfected Flp-In™ 293 cell supernatants is
likely related to release following cell death. 196

xviii
Figure 5.5. Transiently transfected COS-7 cells express and secrete CapG after
tetracycline induction. 198
Figure 5.6. Recombinant CapG is expressed in supernatants of transfected EBNA-293 cells. 201
Figure 5.7. Purification of CapG from EBNA-293 supernatants using HisTALON™
and Strep-Tactin® resins. 203
Figure 5.8. Optimisation of EBNA-293 cell growth to maximise CapG production.
206
Figure 5.9. Concentration of CapG and analysis of purity of the concentrated
material. 209
Figure 5.10. His-CapG reduces the rate of pyrene-actin polymerisation. 213
Figure 5.11. CapG slows the rate of pyrene actin polymerisation. 214
Figure 6.1. Outline of chapter 6. 232
Figure 6.2. IL-8 and IL-6 is released from hASM cells following bac-CapG
stimulation. 238
Figure 6.3. THP-1 cells release IL-8 when stimulated with CapG. 241
Figure 6.4. Recombinant bac-CapG triggers IL-8 release from the airway
epithelial cell lines BEAS2B. 243
Figure 6.5. Recombinant bac-CapG and LPS triggers IL-8 release from the
synovial fibroblast cell lines SW982. 244
Figure 6.6. Polymyxin B significantly reduces IL-8 release from THP-1 cells
stimulated with LPS and bac-CapG. 247
Figure 6.7. Polymyxin B significantly reduces recombinant IL-8 release from
SW982 cells stimulated with LPS but not bac-CapG. 249
Figure 6.8. Polymyxin B did not affect bac-CapG-mediated IL-8 release from
hASM cells. 250
Figure 6.9. IL-8 and IL-6 is released from hASM cells following His-CapG
stimulation. 252
Figure 6.10. His-CapG triggers IL-8 cytokine release from THP-1 cells. 255
Figure 6.11. CapG does not trigger CCL2 release from THP-1 cells. 257

xix
Figure 6.12. Polymyxin B does not affect IL-8 release from His-CapG stimulated
THP-1 cells. 258
Figure 7.1 Outcomes of this thesis. 270

xx
List of Abbreviations and Glossary
AD Alzheimer’s disease
AM Alveolar macrophages
APC Antigen presenting cell
ASM Airway smooth muscle
ANOVA Analysis of variance
AP-1 Activator protein-1
AV/PE Strepavidin conjugated Phycoerythrin
BCR B-cell receptor
BCL-2 B-cell lymphoma 2
BCL-XL B-cell lymphoma-extra large
BEAS2B Human bronchial epithelial cell line
bFGF Basic fibroblast growth factor
BMMCs Bone marrow-derived mast cells
BSA Bovine serum albumin
BV2 Murine microglia cell line
c-Src Proto-oncogene tyrosine-protein kinase Src
Ca2+ Calcium
CaCl2 Calcium chloride
CapG Macrophage capping protein
CCL C-C motif chemokine ligand
CD Cluster of differentiation
cKitR c-kit, stem cell factor receptor

xxi
Clec9a C-type lectin domain family 9 member A
CLR C-type lectin receptors
CO2 Carbon dioxide
COS-7 Monkey kidney-derived fibroblast-like cell line
CRISPR Clustered regularly interspaced short palindromic repeats
DAMP Damage associated molecular patterns
dNTP Deoxynucleotide triphosphate
DMARD Disease-modifying antirheumatic drugs
DMSO Dimethyl sulfoxide
DSCG Disodium cromoglycate
DTT Dithiothreitol
ECACC European Collection of Authenticated Cell Cultures
EBNA Epstein Barr nuclear antigen
EBNA293 Epstein Barr nuclear antigen-expressing HEK293 cells
ELISA Enzyme linked immunosorbent assay
ET-1 Endothelin-1
FACS Fluorescence-activated cell sorting
FBS Fetal bovine serum
FcεRI High affinity IgE binding Fc receptor
FcεRII Low affinity IgE binding Fc receptor
FcγRIIa Low affinity IgG binding Fc receptor
Flp-In™-293 Flp-In™-expressing HEK293 cells
FITC Fluorescein isothiocyanate

xxii
FSC Forward-side scatter
G domain Gelsolin domain
GC Glucocorticoid
GILZ Glucocorticoid-induced leucine zipper
GM-CSF Granulocyte macrophage colony-stimulating factor
GR Glucocorticoid receptor
GR-α Glucocorticoid receptor-alpha
GR-β Glucocorticoid receptor-beta
GR-γ Glucocorticoid receptor-gamma
HAGG Heat-activated gamma globulin
hASM Human airway smooth muscle
HBSS Hank’s buffered salt saline
HEK293 Human embryonic kidney-293 cell line
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
hIgE Human IgE
HMC-1 Human mast cell line-1
HMCα FcεRIα transfected HMC-1 mast cell line
HMGB1 High mobility group box 1 protein
HRP Horseradish peroxidase
HSP Heat shock proteins
ICAM-3 Intercellular adhesion molecule 3
IFN-γ Interferon gamma
Ig Immunoglobulin

xxiii
IgA Immunoglobulin A
IgD Immunoglobulin D
IgE Immunoglobulin E
IgG Immunoglobulin G
IgM Immunoglobulin M
IL Interleukin
IMDM Iscove's Modified Dulbecco's Medium
IVIG Intravenous immunoglobulin
JW8 Anti-NIP antibody-producing cell line
KC Chemokine (C-X-C motif) ligand 1
LABA Long-acting beta-agonists
LAD2 Laboratory of allergic diseases-2 cells
LPS Lipopolysaccharide
LRC-TriCEPS Ligand-receptor capture
LTC4 Leukotriene C4
LTD4 Leukotriene D4
M1 macrophage Classically activated macrophages
M2 macrophage Alternatively activated macrophages
mABs Monoclonal antibodies
MBL Mannose-binding lectin
MCT Tryptase positive mast cells
MCTC Tryptase and chymase positive mast cells
MgSO4 Magnesium sulfate

xxiv
MIP-1α Macrophage inflammatory protein 1-alpha
MIP-1β Macrophage inflammatory protein 1-beta
MS4A2 Membrane spanning 4-domains A2
NaOH Sodium hydroxide
NECA 5'-N-Ethylcarboxamidoadenosine
NFκB Nuclear factor kappa-light-chain-enhancer of activated B cells
NIP 4-Hydroxy-3-iodo-5-nitrophenylacetyl
NIP-BSA NIP-hapten conjugated to BSA protein
NIP-IgE NIP-specific IgE
NOD Nucleotide-binding oligomerization domain
NOD-1 NOD-containing protein 1
PAMP Pathogen-associated molecular pattern
PBS Phosphate Buffered Saline
PD Parkinson’s disease
PGD2 Prostaglandin E2
PGE2 Prostaglandin E2
PNAG p-nitrophenyl N-acetyl-β-D-glucosaminide
PRR Pathogen recognition receptor
PVDF Polyvinylidene difluoride
qPCR Quantitative real time polymerase chain reaction
RA Rheumatoid Arthritis
RBL-2H3 Rat basophilic leaukaemia-2H3 cell line
RIG-1 Retinoic acid-inducible gene 1

xxv
RPC Rat peritoneal cells
RPMI Roswell Park Memorial Institute medium
RPMC Rat peritoneal mast cells
SABA Short-acting beta-agonists
SAv Strepavidin
SDS Sodium dodecyl sulfate
SDS-PAGE SDS-polyacrylamide gel electrophoresis
SipA Salmonella invasion protein A
SSC Size-side scatter
SW982 Human synovial fibroblast cell line
Syk Spleen tyrosine kinase
TBS Tris-buffered saline
TBST TBS with 0.05% (v v-1) Tween20
Th1 T helper type 1 cells
Th2 T helper type 2 cells
TLR Toll-like receptor
TMB substrate 3,3’5,5’-tetramethylbenzidine
TNF-α Tumour necrosis factor-alpha
TNP-BSA 2,4,6-trinitrophenol conjugated to BSA
WEHI-3BD IL-3 secreting cell line

xxvi

1
Chapter 1
General Introduction

2
1.1 The immune system
The immune system involves communication between cells, tissues and
organs that work as an intricate network maintaining homeostasis and protecting
the body from invading pathogens. The immune system can be categorised into
two broad and overlapping categories: the innate and adaptive immune systems.
The innate immune system provides a first line of defence against invading
pathogens which includes physical barriers such as the skin and mucosa, as well
as secreting antimicrobial molecules. Another major role of the innate immune
system is to identify, process and present foreign pathogens to activate the adaptive
immune system. Acquired immunity generated by the adaptive immune system,
relies on a high degree of specificity and immunological memory so that a long-
lasting protection is provided for the host. This ensures that an appropriate and
efficient immune response is mounted upon re-encountering the same pathogen.
Another key feature of the adaptive immune system is immunological tolerance,
whereby the immune system is able to distinguish between self and non-self
(foreign) antigens. Thus, both the adaptive and innate immune systems must work
in concert and in a tightly regulated manner to ensure that the host remains healthy.
Despite the protective features of the immune system, sometimes it acts aberrantly
and mounts a response against its own cells or tissues, resulting in disorders such
as autoimmune-diseases and allergy (Dempsey et al, 2003).
While the immune system is composed of a range of many different effector
cells, this thesis will focus primarily on mast cells and macrophages, both key
innate immune cells that interface between innate and adaptive immunity with
critical roles in inflammation.
1.2 The mast cell: its place in the immune system
Since their discovery over a century ago by Paul Ehrlich, studies on mast
cells have expanded from them being just the primary source of histamine to
broader roles in both innate and adaptive immune responses (Galli et al, 2005).

3
Mast cells are localised in tissues that are in close contact with the environment
including the skin, lung, and gut, thus allowing these cells to act as a first line of
defense in host immunity (Urb & Sheppard, 2012). Histologically, these cells often
vary in shape and size, but they are best recognised as cells containing a round
nuclei, surrounded by an abundance of intracellular granules containing heparin
and histamine, which can be visualised using metachromatic dyes such as toluidine
blue (Leclere et al, 2006).
Mast cells originate from pluripotent haematopoietic stem cells in the bone
marrow, but they migrate to peripheral tissue sites such as the skin and lung, where
under the influence of growth factors and cytokines such as interleukins (IL), these
cells complete their differentiation and end their migration (Galli & Tsai, 2012;
Marshall & Bienenstock, 1994; Yong, 1997). For example, mast cells express the
c-KIT receptor (CD117), which is crucial for cell maturation. This receptor binds
to stem cell factor (SCF) expressed on the surface of and released by fibroblasts,
endothelial and stromal cells (Stone et al, 2010). In addition, the phenotype and
behaviour of mast cells are tightly regulated by a range of cytokine molecules. For
example, interferon-gamma (IFNγ) and IL-4 induce apoptosis in developing mast
cells (Bailey et al, 2004; Mann-Chandler et al, 2005), whilst IL-5, IL-6 and IL-9
promote mast cell proliferation, maturation and recruitment respectively (Conti et
al, 2002; Eller et al, 2011; Stone et al, 2010). Mast cells are also long-lived cells
that can re-enter the cell cycle and proliferate following appropriate stimulation.
Indeed, this plastic phenotype is crucial for mast cells during certain conditions
such as helminth infections, prolonged immune responses or in tissue remodelling
(Galli et al, 2005).
It is interesting to note that the tissue microenvironment in which the mast
cell resides also affects the phenotype of the cell, hence the concept of mast cell
heterogeneity. As a result, the morphology, biochemistry and function of mast cells
may differ depending on tissue location. For example, mast cells from the
gastrointestinal tract in various species are of smaller size compared to other sites

4
(Welle, 1997). Mast cells can also be distinguished by the presence of tryptase or
chymase proteases stored in granules. Tryptases are negatively charged, trypsin-
like proteases that are present in a tetrameric form in mast cell granules, whereas
chymases are chymotrypsin-like proteases stored in mast cells as basic-charged
monomers (Welle, 1997). Mast cell tryptase-only positive cells (MCT) cells are
predominantly found in the lung, small intestinal mucosa, whereas tryptase and
chymase positive cells (MCTC) are located primarily in the skin, small intestinal
submucosa and tonsils (Irani et al, 1989; Welle, 1997). The expression of different
proteases in mast cells also affect cell reactivity to different pharmacological
agents. For example, MCTC degranulate to a range of stimuli including high-
affinity IgE receptor (FcεRI) cross-linking, complement proteins such as C5a,
polybasic compounds such as compound 48/80, and the neuropeptide substance P.
However MCT seem to only respond largely to FcεRI cross-linking (Oskeritzian et
al, 2005). Furthermore, these mast cell subtypes can be distinguished by cytokine
content: IL-4 is detected in both subtypes but is predominantly found in MCTC,
whilst IL-5 and IL-6 are expressed more specifically in MCT (Bradding et al,
1995). The degree of mast cell heterogeneity was further explored in more detail
in a recent study comparing the functions and mediators released from IgE-
activated mast cells derived from the bone marrow and peritoneum (Shubin et al,
2016).
1.3 The macrophage: another key player of the immune system
Similar to mast cells, macrophages also play key roles in both the innate
and adaptive immune systems. Like mast cells, macrophages with distinct
phenotypes are distributed in various tissues such as bone (osteoclasts), brain
(microglia), liver (Kupffer cells), and lung (alveolar macrophages). Despite the
different names and phenotypes, they share related features and functions (Murray
& Wynn, 2011a).

5
Macrophages originate from a committed progenitor cell in the bone
marrow that is responsible for generating the mononuclear phagocyte system
(Doulatov et al, 2010). The mononuclear phagocyte system is defined as a family
of cells consisting of blood monocytes and tissue macrophages (Hume, 2006).
Monocytes circulate in the blood for up to 2 days, after which they undergo cell
death and are removed (Italiani & Boraschi, 2014). However, in inflammatory
conditions, they can be recruited to damaged tissues, and can differentiate into
macrophages, where they exhibit a longer life span and play a role in maintaining
the inflammatory response (Parihar et al, 2010; Yang et al, 2014).
Macrophages can also be distinguished based on their functional activities.
Classically activated M1 macrophages mediate host defense and immunity, whilst
the alternatively activated M2 macrophages are involved in suppressing host
immunity and regulating wound healing. Other macrophage types include
regulatory macrophages (that secrete IL-10, which dampens the immune response
and limit immunopathology), as well as tumour-associated macrophages and
monocytic subset of myeloid-derived suppressor cells, both of which are
associated with regulating tumour immunity (Hutchinson et al, 2011; Laoui et al,
2011; Mills, 2012).
Functionally, macrophages maintain tissue homeostasis and like mast cells,
act as sentinels, where they have proteolytic and catabolytic activities and are able
to engulf pathogens by phagocytosis, removing dead cells and debris, and also
contribute to tissue remodelling following injury (Gordon & Taylor, 2005; Wynn
& Barron, 2010).
1.4 The innate immune system: the first line of defense
In vertebrates, anatomical barriers provide the first line of defence against
pathogen infections. This includes physical barriers such as the skin and chemical
barriers such as mucus and sweat. For example, the portion of the cell exposed to
the lumen (apical surface) contains a mesh of transmembrane proteins known as

6
tight junctions that function to link neighbouring cells as well as regulate the
passage of ions and molecules through the extracellular space between cells
(Guttman & Finlay, 2009). In addition, tight junctions also restrict the penetration
of invading pathogens from the lumen into tissues (Urb & Sheppard, 2012).
Although these barriers serve as an effective first line of defence against
infections, several pathogens such as Helicobacter pylori and rotavirus have
developed effective strategies to bypass the mucosal lining and epithelial barrier
and are thus able to penetrate through and invade the host (Amieva et al, 2003;
Nava et al, 2004). However, the innate immune system is able to detect invading
pathogens by recognising signature foreign molecular patterns which are tightly
conserved in specific microbial classes (Akira et al, 2006). Collectively, these are
termed as pathogen-associated molecular patterns (PAMPs) and include microbial
lipid membranes, peptidoglycan cell walls, proteins and DNA (Mogensen et al,
2008). The innate immune system recognises PAMPs through germline-encoded
pattern-recognition receptors (PRRs). PRRs can be distinguished into three
groups: membrane-bound PRRs such as Toll-like receptors and C-type lectin
receptors; cytoplasmic PRR such as NOD-Like receptors; and secreted PRR such
as receptors of the complement system (Akira et al, 2006; Gomez et al, 2009;
Meylan et al, 2006).
In addition to PAMPs, PRRs are also able to detect damage-associated
molecular patterns (DAMPs). As the name suggests, DAMPs are endogenous
danger signals derived from cells during trauma, stress, and tissue damage (Tang
et al, 2012). DAMPs can be localised within the nucleus (chromatin-associated
protein HMGB1) or cytoplasm (heat shock proteins), or present in the extracellular
matrix (hyaluronic acid) and in plasma (complement proteins C3a, C4a, and C5a),
as well as non-protein molecules including DNA, RNA, ATP, and uric acid (Tang
et al, 2012). Indeed, mast cells and macrophages express different types of PRRs
that are key in detection and clearance of DAMPs and PAMPs, thus maintaining
tissue homeostasis, which will be discussed later.

7
1.5 The involvement of mast cells and macrophages in the adaptive immune
system
Although more commonly associated with a role in the innate immune
system, both mast cells and macrophages are also actively involved in the adaptive
immune system. They are in close physical proximity to other immune cells such
as T cells, where they act as antigen presenting cells (APC) by presenting
processed antigen from engulfed pathogens to T cells, resulting in the activation
of the immune response against the specific pathogen (Banovac et al, 1989;
Stelekati et al, 2009; Underhill et al, 1999). In addition, both mast cells and
macrophages release mediators that influence the activation and recruitment of T
cells. T cells play a crucial role in the adaptive immune system where they are
involved in host defense against a variety of pathogens (Akbari & Umetsu, 2005).
Mast cells can release a range of cytokines (IL-4 and IL-6) and chemotactic factors
(tumour necrosis factor-alpha (TNFα)), and express cell surface adhesion
molecules that facilitate T cell migration, polarisation, activation and cytokine
production (Mekori & Metcalfe, 1999; Nakae et al, 2005). In turn, T cells can also
influence mast cell recruitment and activation. For example, a specific subset of T
regulatory cells was found to secrete IL-9, a mast cell growth and activation factor,
that allows mast cells to release mediators that can lead to beneficial effects such
as allograft tolerance, but also detrimental effects such as allergy (Lora et al, 2003;
Lu et al, 2006). Similarly, T cells in contact with antigen-presenting macrophages
release interferon (IFN)-γ. Along with this, a second co-stimulatory signal is
initiated between both cells. This costimulatory signal is composed of CD40
(expressed on macrophages) and CD40L (expressed on T cells) that eventually
leads to the activation of macrophages (Buhtoiarov et al, 2005; Kennedy et al,
1996).
In addition to T cells, mast cells and macrophages interact with other cells
of the adaptive immune system such as B cells. Mature B cells (or plasma cells)
are known for their ability to produce antibodies against a specific antigen. B cells

8
express B cell-receptors (BCR) that allow the cells to bind to a foreign pathogen,
internalise and process the antigen into fragments. These fragments are later
presented to T helper cells. During this interaction, a costimulatory signal CD40
(expressed on B cells) and CD40L (expressed on T cells) is required for B cell
activation, and thus leading to the production of antibodies targeting the specific
antigen (Parker, 1993). Interestingly, mast cells also express CD40L, hence
allowing these cells to activate B cells in the absence of T cells (Hong et al, 2013).
In addition, distinct mast cell populations can release mediators such as IL-4, IL-
5, IL-6 and IL-13 that influences B cell development (Merluzzi et al, 2010).
Although B cells are able to initiate an immune response independently of
APCs, studies have shown that B cells are able to acquire antigens and directly
transfer the antigen to other APCs including mast cells and macrophages, thus
allowing the B cells to “focus” the immune system towards the rapid recognition
and clearance of pathogens (Harvey et al, 2007). Interestingly, a study has reported
that pre-B cells contain macrophage transcription factors such as CCAAT-
enhancer-binding proteins (C/EBPα) that enables them to be reprogrammed to
functional macrophages under the right stimulus, suggesting that this rapid process
can be induced under pathological conditions such as infection (Rapino et al, 2013;
Xie et al, 2004).
1.6 Mast cell and macrophage activation through IgE
Although mast cells and macrophages can be activated through many
different stimuli including PAMPs such as LPS, DAMPs such as ATP, cytokines
such as IL-4 and 5 and immunoglobulin (Ig)-E and G (IgG) (Gilfillan & Tkaczyk,
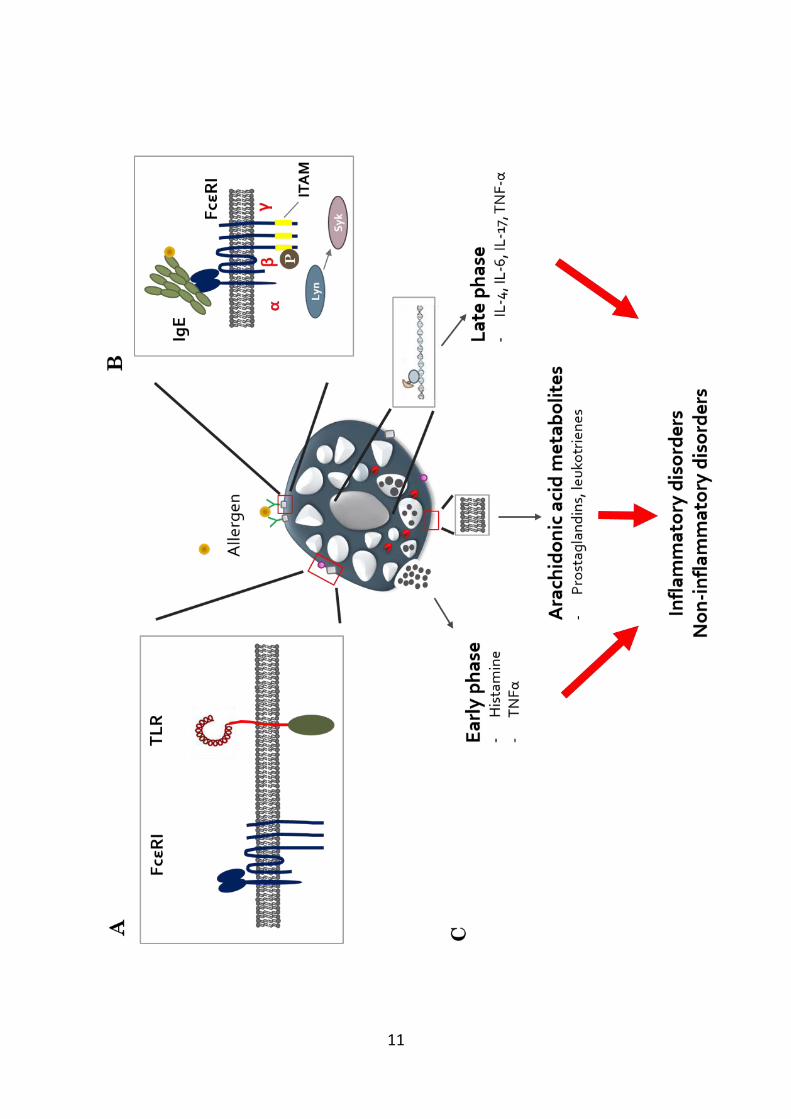
2006), this section focuses primarily on cell activation through IgE (Figure 1.1a).
There are five distinct isotypes of antibodies found in humans: IgA, IgD,
IgG, IgM, and IgE. Of the five different immunoglobulin subtypes, IgE is least
abundant in serum, with concentrations ranging between 50-300 ng/mL in healthy
individuals, compared to approximately 10 mg/mL of serum IgG (Sutton & Gould,

9
1993). Circulating IgE has a shorter half-life compared to the other antibody
subclasses (approximately 12 hours). However, receptor-bound IgE has a much
longer half-life, ranging from weeks to months (Stone et al, 2010). Interestingly,
serum IgE concentrations are elevated in patients with atopic diseases such as
atopic dermatitis and atopic asthma, as well as other disorders such as parasitic and
non-parasitic infections, inflammatory diseases such Kawasaki’s disease, and
cystic fibrosis (Stone et al, 2010). Mature B cells are the primary source of IgE,
where the IgE antibodies are initially generated against specific antigens presented
by dendritic cells, B cells or other APCs. This then leads to the antigen-specific
IgE binding to IgE receptors on the surface of mast cells or basophils, hence
“sensitising” the cells so that when the antigen subsequently invades, the cells will
activate readily and mount an allergic type immune response to eliminate the
pathogen, such as helminth infections in the gastrointestinal tract. In turn, the
activated mast cells release mediators that promote mucus hypersecretion and
increased motility in the gastrointestinal tract that in turn facilitates in helminth
expulsion by peristalsis, as well as recruiting other inflammatory cells which are
important for preventing subsequent reinfections. (Al-Qaoud et al, 2000; Bancroft
et al, 1998; Madden et al, 2002; Quinnell et al, 2004).
Mast cells are best known to be activated by IgE through antigen-induced
crosslinking of specific IgE pre-bound to the high affinity IgE receptor FcεRI
present on the mast cell surface (Sibilano et al, 2014). Studies have also shown
that monomeric IgE has several effects on mast cells including promoting survival
by inducing the pro-survival protein Bcl-XL, promoting mast cell maturation in
vitro, and stabilising and upregulating the expression of FcεRI on mast cell surface
(Kalesnikoff et al, 2001; Kashiwakura et al, 2008; Stone et al, 2010).
The FcεRI receptor expressed on mast cells consists of an IgE-binding α-
subunit, a tetraspanin β-subunit (MS4A2) which functions as a signal amplifier
and receptor stabiliser, and two identical γ-subunits linked by disulphide bonds
that act as the primary signal transducer (Sibilano et al, 2014). The cytoplasmic

10
tails of the β and γ-subunits contain immunoreceptor tyrosine-based activation
motifs (ITAMs) that are associated with the tyrosine-protein kinase Lyn (Gilfillan
& Tkaczyk, 2006). Following the crosslinking by antigen of IgE bound to FcεRI,
Lyn phosphorylates the tyrosine residues in the ITAM motifs of the β and γ
subunits, recruiting spleen tyrosine kinase (Syk). Syk activates a number of
downstream signalling pathways that result in changes in cell morphology and
transcriptional activities with subsequent release of a vast array of mediators that
initiate and propagate immune responses (Figure 1.1b) (Gilfillan & Tkaczyk,
2006; Stone et al, 2010).
In addition to FcεRI, IgE has also been shown to bind to different receptors
including FcεRII and galectin-3, a member of the lectin family. Galectin-3 is
expressed in cultured primary mast cells, tissue mast cells and mast cell lines (Chen
et al, 2006; Frigeri & Liu, 1992). Galectin-3 has been previously shown to be
involved in mast cell biology by potentiating the IgE-FcεRI-mediated mast cell
effects, as mast cells derived from galectin-3 knockout mice showed a reduction
in histamine and IL-4 release following IgE/FcεRI activation (Chen et al, 2006).
Although not as well-studied as in mast cells, both FcεRI and FcεRII are also
expressed on macrophages and studies have shown the involvement of IgE-
mediated activity of macrophages, along with other inflammatory cells, including
mast cells, in the pathogenesis of aortic aneurysms and allergen reactions in atopic
subjects (Wang et al, 2014a; Ying et al, 1998). Activation of alveolar macrophages
through the FcεRII receptor leads to the release of both pro- and anti-inflammatory
cytokines. In patients with allergic asthma, there is an upregulation of FcεRII
receptor expression on the surface of macrophages, thus implicating a contribution
of IgE-mediated macrophage activities in disease progression (Balhara & Gounni,
2012; Gosset et al, 1999; Vecchiarelli et al, 1994).
11

12
Figure 1.1. Mast cell activation. (A) Mast cells express a range of receptors
including FcεRI and TLRs that allows recognition of foreign antigens and
pathogens. (B) Mast cells are most commonly activated by antigen crosslinking
IgE bound to the high-affinity IgE receptor FcεRI. The FcεRI consists of the IgE-
binding α-subunit, and the β and two γ-subunits that contain immunoreceptor
tyrosine-based activation motifs (ITAMs) that are phosphorylated by the tyrosine-
protein kinase (Lyn) following receptor activation. Lyn-phopshorylated ITAMs
recruits spleen tyrosine kinase (Syk), which in turn leads to a downstream
signalling pathway. (C) Activated mast cells release mediators through different
pathways. Preformed mediators stored in granules such as histamine and TNFα are
released rapidly (early phase). In addition arachidonic acid, which is the
polyunsaturated fatty acid present in the phospholipids of the cell membrane is
metabolised, resulting in the synthesis of different classes of mediators including
prostaglandins and leukotrienes, which are released minutes following cell
activation. Finally, activated mast cells initiate the synthesis of many pro-
inflammatory cytokines that are subsequently released hours after cell activation
(late phase). Many of these mediators contribute to the symptoms commonly
observed in inflammatory disorders including asthma and rheumatoid arthritis.
However, there is a growing appreciation for the possible involvement of mast cell
mediators in non-inflammatory disorders such as cancer.

13
1.7 Mast cell and macrophage activation through PRRs
As discussed previously, mast cells and macrophages are strategically
located around the body, which allows these cells to act as sentinels of the immune
system. As first line defenders against pathogens, they are able to initiate an
immune response that adequately and effectively contain and remove the invading
pathogen. As such, these inflammatory cells express a range of different receptors
including different types of PRRs such as Toll-like receptors (TLRs) and C-type
lectin receptors (CLRs) on their cell surfaces which allows them to detect bacterial,
viral and fungal PAMPs, as well as host DAMPs.
TLRs are type 1-membrane glycoproteins containing leucine-rich motifs in
their extracellular domains and an IL-1 receptor-like cytoplasmic signalling
domain (TIR) (Bowie & O'Neill, 2000). To date, there are 12 members of the TLR
family and each recognise different types of PAMPs: TLR1, TLR2, and TLR6
recognises pathogen lipid components, whilst TLR7-9 recognises signature
pathogen nucleic acids. Other TLRs such as TLR4 recognise different ligands with
different structures such as the bacterial endotoxin component lipopolysaccharide
(LPS), virus envelop proteins, the plant diterpine paclitaxel, and proteins such as
fibronectin and heat-shock proteins (Akira et al, 2006). TLRs are expressed on
immune cells including macrophages, mast cells and dendritic cells and can be
expressed both intracellularly in the lysosome or endosome membranes (TLRs 3,
7-9) and on cell surface (TLRs 1, 2, 4-6) (Akira et al, 2006; Sandig & Bulfone-
Paus, 2012). In addition, TLRs can also detect DAMPs released from damaged
cells such as heat shock proteins and high mobility group box 1 protein (HMGB1)
(Asea, 2008; Park et al, 2004).
In recent years, there is a growing appreciation about the importance of the
membrane-bound PRR family CLRs and their role in antimicrobial defense. CLRs
are present on different inflammatory cells such as macrophages, mast cells,
neutrophils, and dendritic cells (Deng et al, 2015; Diebold, 2009; Vukman et al,
2013). Like TLRs, CLRs are able to detect bacterial, fungal and virus infections

14
and mediate host immunity against these pathogens. For example, Dectin-1 and
Dectin-2 are involved in mediating defense against fungal pathogens including C.
albicans, A. fumigatus, and P. carinii (Drummond & Brown, 2011; Saijo et al,
2010). Other examples of CLRs involved in mediating host defense include mincle
(mycobacteria) (Marakalala et al, 2010), mannose receptor (gram-negative
bacteria) (Vukman et al, 2013), and dendritic cell-specific ICAM-3 grabbing-
nonintegrin (virus) (Boily-Larouche et al, 2012). In addition, other CLRs such as
Clec9a have been reported to recognise DAMPs such as actin (Zhang et al, 2012).
Whilst some TLRs and CLRs sense pathogens at the cell surface, others are
capable of detecting invading pathogens in the cell cytosol. These cytoplasmic
PRRs are generally classified as NOD-Like Receptors (NLRs) or RIG-I-Like
Receptors (RLRs), which are involved in recognising bacterial peptidoglycan
motifs, fungal and viral components (Franchi et al, 2009; Meylan et al, 2006).
TLR2 and the NLR receptor subtype Nod-1 work in concert to recognise
peptidoglycan, which triggers cell activation (Feng et al, 2007). The RIG-I
receptor is known to be a virus sensor as it has been shown to recognise Dengue
virus, Sendai virus and Influenza A virus (Graham et al, 2013; Lappalainen et al,
2013; St John et al, 2011).
In addition, some PRRs can also be present extracellularly. These soluble
secreted PRRs function to detect, bind and initiate an effective response to
eliminate the pathogen often through complement activation in the extracellular
space (Dempsey et al, 2003). An example of secreted PRR is mannose-binding
lectin (MBL), which is initially synthesised in the liver and then circulates in the
bloodstream. It is able to recognise PAMPs and then initiates clearance by
activating the complement system and through the promotion of cell phagocytosis
(Ip et al, 2009).
Activation of PRRs following engagement with microbial components or
damaged cell signature molecules typically results in downstream signalling
pathways that leads to gene transcription and cytokine production (Akira et al,

15
2006; Hardison & Brown, 2012). Depending on the pathogen, mast cell activation
by PRRs result in several outcomes including degranulation, release of proteases
and mediators that promotes enhanced vascular permeability and also increased
inflammatory cellular recruitment to the site of infection (Abraham & St John,
2010). In addition, activation of mast cells by TLRs further sensitises mast cell
responses including enhancing IgE-mediated degranulation (Saluja et al, 2012).
Similarly, depending on the ligand and engagement to its corresponding PRR,
activated macrophages can polarise to different macrophage subtypes, where it
releases either pro- and anti-inflammatory cytokines (Zhou et al, 2015).
The importance of mast cell and macrophage in pathogen recognition,
clearance and resolution is highlighted using mouse knock-out studies. Several
studies utilising mast cell-deficient mice showed an increase susceptibility to
infection and mortality primarily due to poorer pathogen clearance and this was
restored by the re-introduction of mast cells from wild-type mice (Aoki et al, 2013;
Echtenacher et al, 1996; Lawrence et al, 2004; Malaviya et al, 1996). In addition,
TLR4 was found to be crucial in mast cell-mediated enterobacterial clearance as
mast cell-deficient mice reconstituted with mast cells expressing a mutant TLR-4
had significantly higher mortality due to poor neutrophil recruitment and defective
pro-inflammatory cytokine production (Supajatura et al, 2001). Similarly, studies
utilising macrophage-depleted mice resulted in reduced bacterial clearance,
increased bacterial growth and impaired tissue repair following injury (Burnett et
al, 2004; Goren et al, 2009). A key feature linked to the spectrum of activity of
activated mast cells and macrophages is their ability to release a vast array of
mediators. The types of cytokines and mediators released from mast cells and
macrophages are discussed next.
1.8 Mediator release from activated mast cells and macrophages
As described above, recognition of invading pathogens through different
PRR families, whether expressed on cell surface or intracellularly, or by other

16
pathways such as IgE, leads to cell activation, and in some cases results in mediator
release from activated cells (Figure 1.1c).
Mediators released from mast cells can be generally classified into three
groups: preformed mediators that are stored in granules and released following cell
activation, such as histamine, mediators such as TNFα and proteases that lead to
inflammatory processes including enhanced vascular permeability and leukocyte
recruitment (Bradding et al, 1994; St John & Abraham, 2013). Other granular
mediators include antimicrobial peptides such as the beta-defensin family and
cathelicidin released from both human and murine mast cells, have been shown to
be protective against microbial agents such as Group A Streptococcus infection in
the skin, and further studies on cathelicidin has also been shown to promote
neutrophil recruitment to the site of infection (Di Nardo et al, 2008). These
proteases and peptides released from mast cells can also negate the damaging
effects of certain toxins and endogenous mediators such as endothelin-1 (ET-1).
ET-1 is derived from vascular endothelial cells with potent vasoconstrictor
activity. However, ET-1-mediated vascular changes in pathological processes such
as sepsis can result in fatal consequences. Chymase and carboxypeptidase A
released from mast cells were shown to limit the morbidity and mortality
associated with ET-1 administration in mice (Maurer et al, 2004; Metsarinne et al,
2002). Similarly, sarafotoxin which shares structural similarity with ET-1 (Kloog
et al, 1988), is a cardiotoxic peptide derived from snake venom was also shown to
have its pathological effects heightened in mast-cell deficient mice, thus
highlighting the important of mast cells in attenuating the toxicity of certain
substances (Metz et al, 2006).
A second class of mediators are rapidly synthesised and released from
activated mast cells minutes after cell activation. These mediators are derived from
arachidonic acid, which are polyunsaturated fatty acids present on the cell
membrane (Moncada & Vane, 1979). The metabolism of this fatty acid by
enzymes such as cyclooxygenase and lipoxygenase gives rise to mediators such as

17
prostaglandins (PG) such as PGD2 and PGE2 and leukotrienes (LT) such as LTC4,
that contribute to many symptoms associated with inflammation including fever,
increased vascular permeability and pain sensitivity (Burd et al, 1989; Matsushima
et al, 2004; Peters et al, 1984; Wakahara et al, 2001).
The third class of mediators are released from mast cells typically hours
after cell activation. Activation of mast cells initiates a downstream signalling
cascade, resulting in the gene transcription, synthesis and release of pro-
inflammatory cytokines such as IL-1, IL-3, IL-4, IL-5, IL-6, IL-8, TNFα and
chemokines such as IL-16, CCL1 (also known as eotaxin), CCL2 (also known as
MCP-1), CCL3 (also known as MIP-1α), and CCL4 (also known as MIP-1β)
(Collington et al, 2010; Gonzalo et al, 2007; Wang et al, 1998). Together, the
cytokines and chemokines released from activated mast cells play an important
role in mediating key inflammatory features.
Similar to mast cells, activated macrophages release both pro and anti-
inflammatory mediators. The microenvironment provides diverse signals that
leads to polarisation to different macrophage phenotypes (Arango Duque &
Descoteaux, 2014). Exposure of naïve monocytes or recruited macrophages to T-
helper 1 (Th1) cytokines (TNFα and IFN-γ) drive macrophages towards the
classically activated M1 macrophages, which are involved in host-defense against
pathogens by exhibiting increased microbicidal and tumouricidal capacity (Mosser
& Edwards, 2008). Activated M1 macrophages release pro-inflammatory
cytokines such as IL-1, IL-6, IL-12, IL-23 and TNFα (Bromander et al, 1991;
Chomarat et al, 2000; Verreck et al, 2004; Xing et al, 2000). In contrast, the
cytokine profile from alternatively activated M2 macrophages are different
compared to other macrophage populations (Loke et al, 2002). During the wound
healing process, it is thought that the T-helper 2 (Th2) cytokines IL-4, IL-13 and
IL-21 are key in polarising macrophages towards the M2 phenotype (Hofmann et
al, 2014; Li et al, 2013; Salmon-Ehr et al, 2000). These cytokines stimulate
arginase activity in macrophages, which converts arginine to urea and ornithine.

18
In turn, ornithine is converted to proline and polyamines which are important in
would healing and cell proliferation (Kreider et al, 2007).
Finally, macrophages can also differentiate into another subpopulation of
anti-inflammatory macrophages known as regulatory macrophages.
Reprogramming of these macrophages requires two co-stimulatory signals, for
example the first signal being a ligand such as histamine, prostaglandin, adenosine,
and dopamine; and the second signal being a TLR ligand (Edwards et al, 2006;
Hasko et al, 2007; Hasko et al, 2002; Sirois et al, 2000; Strassmann et al, 1994).
Regulatory macrophages produce IL-10, which exerts a range of autocrine and
paracrine anti-inflammatory effects including inhibition of pro-inflammatory
cytokine release from macrophages, promoting macrophage accumulation and
differentiation in damaged tissues, and also acting as APCs to inhibit IFNγ
production from T-helper 1 cells (Fiorentino et al, 1991a; Fiorentino et al, 1991b;
Gazzinelli et al, 1992; Wang et al, 2001).
Combined, the vast array of cytokines and chemokines released from mast
cells and macrophage demonstrates their importance in modulating the immune
response in the event of pathogen infections or injuries, and their crucial role in
maintaining homeostasis. However, inappropriate activation of these cells can
have detrimental effects and lead to numerous inflammatory disorders.
1.9 Mast cells and macrophages in disease
In response to pathogens, noxious substances or signals from damaged
cells, mast cells and macrophages initiate an appropriate course of inflammation
and repair at the site of insult. However, in some instances inappropriate activation
of the immune system involving these cells can lead to undesirable and damaging
effects, termed hypersensitivity (Warrington et al, 2011). Hypersensitivity
reactions are classically defined into 4 types, however only Type I and Type III
hypersensitivity reactions and the involvement of mast cells and macrophages in

19
these processes are discussed here. In addition, the involvement of these cells in
the brain and how they may contribute to neuronal degeneration is also considered.
1.9.1 Type I hypersensitivity-associated diseases
Type I hypersensitivity, more commonly known as an ‘atopy’ or ‘allergy’,
is an inflammatory disease where IgE has been shown to be the key effector.
Allergic diseases occur due to an over-activation of the immune system to a
particular substance that is usually otherwise harmless. The reaction can lead to
responses ranging from a mild irritation to fatal consequences such as anaphylaxis
(Kim & Fischer, 2011). An allergic reaction is typically a two-step process:
i. Production of allergen specific-IgE and the sensitisation of mast
cells
This process occurs when an individual is exposed to an allergen, which is
recognised by dendritic cells that mature and migrate to the lymph nodes. Here,
they process and present the allergen to T cells that leads to T cell maturation to
Th2 cells. Matured Th2 cells in turn interact with B-cells and in an IL-4 or IL-13
dependent manner, trigger B-cell maturation to plasma cells that secrete antigen-
specific IgE (Galli & Tsai, 2012). This antigen-specific IgE binds to multiple cell
types through various IgE receptors such as FcεRI on mast cells and basophils
(Fuller et al, 1986; Stone et al, 2010).
ii. Re-exposure and binding of allergen resulting in activation of mast
cells.
Subsequent re-exposure of the specific antigen results in binding to cell-
fixed IgE and the crosslinking of FcεRI receptors on cell surface and hence cell
activation, leading to activation of downstream signalling pathways that results in
degranulation, de novo synthesis and secretion of inflammatory mediators. In Type
I hypersensitivity disorders, this exaggerated mast cell response leads to elevated
pro-inflammatory mediator release such as histamine and prostaglandins that can

20
contribute to disease symptoms such as bronchospasm and mucus hypersecretion,
as observed in allergic asthma.
1.9.1.1 Allergic Asthma
Asthma is one of the most common lung diseases worldwide, and is the
most prevalent in industrialised countries (Kim & Bernstein, 2009). It is estimated
that approximately 300 million individuals worldwide suffer from asthma
(Pawankar, 2014). The most common type of asthma is allergic asthma. Patients
with allergic asthma have higher serum IgE concentrations compared to non-
allergic asthmatics (Sandeep et al, 2010). One of the hallmark cellular features of
allergic asthma is the infiltration of mast cells into the airway smooth muscle
bundles (Brightling et al, 2002). Asthma is characterised by pathological features
such as airway inflammation, reversible airway obstruction, mucus hypersecretion,
increased airway hyperreactivity and airway remodelling (Kraneveld et al, 2012;
Reuter et al, 2010; Yu et al, 2006). These symptoms manifest due to the function
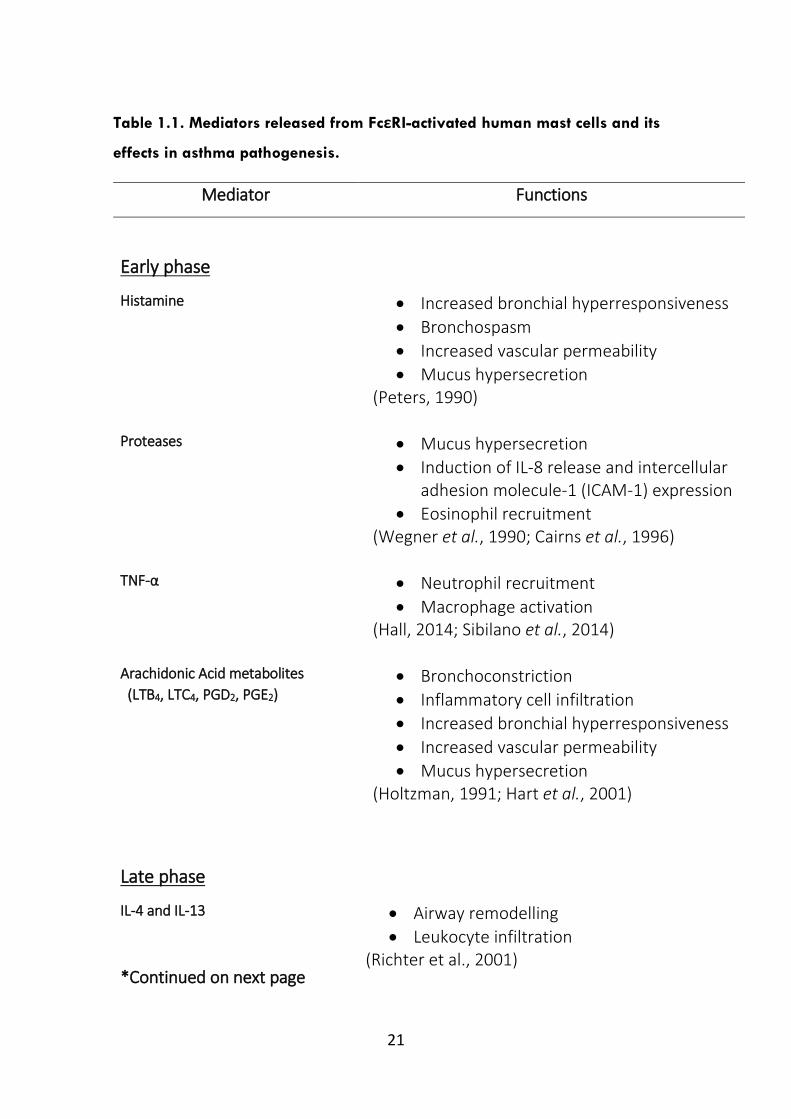
of mediators released from activated mast cells (Table 1.1). Studies using mice
deficient in FcεRI and other studies targeting IgE-binding to FcεRI show reduction
in allergic airway inflammation and airway hyperresponsiveness, thus
demonstrating the important role for mast cells and IgE in disease pathology
(D'Amato et al, 2014; Mayr et al, 2002).
21
Table 1.1. Mediators released from FcεRI-activated human mast cells and its
effects in asthma pathogenesis.
Mediator Functions
Early phase
Histamine Increased bronchial hyperresponsiveness
Bronchospasm
Increased vascular permeability
Mucus hypersecretion (Peters, 1990)
Proteases Mucus hypersecretion
Induction of IL-8 release and intercellular adhesion molecule-1 (ICAM-1) expression
Eosinophil recruitment (Wegner et al., 1990; Cairns et al., 1996)
TNF-α
Neutrophil recruitment
Macrophage activation (Hall, 2014; Sibilano et al., 2014)
Arachidonic Acid metabolites
(LTB4, LTC4, PGD2, PGE2)
Bronchoconstriction
Inflammatory cell infiltration
Increased bronchial hyperresponsiveness
Increased vascular permeability
Mucus hypersecretion (Holtzman, 1991; Hart et al., 2001)
Late phase
IL-4 and IL-13
*Continued on next page
Airway remodelling
Leukocyte infiltration (Richter et al., 2001)

22
IL-5 and GM-CSF
Eosinophil differentiation, activation, survival
(Gregory et al., 2003)
IL-6 and IL-13
Increased mucus secretion and IgE synthesis
(Rincon et al., 2012; Neveu et al., 2006)
IL-8
Neutrophil recruitment (Ordonez et al., 2000)
IL-17 Macrophage activation (Song et al., 2008)
IL-33 Inflammatory cell recruitment and activation
(Borish et al., 2011)
Chemokines (Eotaxin, CCL2, MIP-1α)
Leukocyte infiltration (Collington et al., 2010)
Alveolar macrophages (AM) also play an extensive role in allergic asthma.
They are the most abundant resident immune effector cells in the alveolar space
and play a critical role in regulating pulmonary immune responses (Guth et al,
2009; Peters-Golden, 2004). AMs have been shown to exhibit both pro-and anti-
inflammatory roles in asthma that exacerbate or resolve allergic asthma,
respectively (Table 1.2). In allergic asthma, mast cells release mediators that are
able to induce IL-17A production from AMs (Song et al, 2008). IL-17A promotes
inflammatory cell recruitment and pro-inflammatory cytokine production such as
IL-4, IL-5, and further IL-17A (Song et al, 2008). In addition, activated AMs
release other cytokines (such as TNFα and IL-8), reactive oxygen species and
arachidonic acid metabolites that promote airway inflammation (Gosset et al,
1999; Lohmann-Matthes et al, 1994).

23
Macrophages also exhibit anti-inflammatory roles in asthma to modulate
inflammation (Bang et al, 2011). It is suggested that the anti-inflammatory
cytokines IL-10 and IL-12 play important roles in regulating asthmatic
inflammation as they regulate the synthesis and activity of pro-inflammatory
cytokines (Chung, 2001). In several murine experiments, the adoptive transfer of
AMs from non-sensitised mice to macrophage-depleted and allergen-sensitised
mice resulted in several anti-inflammatory effects including more effective
phagocytosis of apoptotic cells to remove inflammatory signals released from
damaged cells, and suppression of APC activity and T cell activation (Careau et
al, 2006; Holt et al, 1993). This anti-inflammatory role of AMs has been
implicated in regulating bronchial hyperresponsiveness in rats (Careau &
Bissonnette, 2004). In addition, macrophage function such as phagocytosis was
impaired in children with poorly controlled asthma (Fitzpatrick et al, 2008).
Indeed, several studies have shown that anti-inflammatory cytokines such as IL-
10, IL-12, IFN-γ are downregulated in asthmatics (Chung, 2001; Gosset et al,
1999; Tomita et al, 2002). These studies highlighting both the pro and anti-
inflammatory roles of macrophages have provided insight into the dual importance
of macrophages in asthma.

24
Table 1.2 Potential dual roles of macrophages in allergic asthma.
Mediator Functions
Pro-inflammatory
TNFα and IL-17 Increased airway hyperreactivity
Airway inflammation
IL-8 Neutrophil recruitment
CCL2, MIP-1α, IL-1β Airway inflammation
Arachidonic acid metabolites Airway inflammation and modulation of smooth muscle tone
Reactive oxygen species (Superoxide anion O2-, hydrogen peroxide H2O2)
Antimicrobial defense and chronic lung injury
Anti-inflammatory
IL-10 and IL-12 Suppression of inflammatory response
Nitric Oxide Suppression of pro-inflammatory cytokine production
Table adapted from Balhara et al., 2012.

25
1.9.2 Type III Hypersensitivity
In Type III hypersensitivity diseases, the antibodies IgG or IgM are
generated against soluble self-antigens that leads to the formation of immune
complexes. Although macrophages are able to phagocytose larger immune
complexes, they are unable to clear smaller immune complexes in the blood
stream. As a result, the smaller complexes are deposited in blood vessels, lungs,
kidneys, joints and skin. These complexes are able to initiate inflammatory
responses by complement activation, resulting in several inflammation processes
including mast cell, macrophage, and basophil activation and neutrophil influx,
eventually leading to oedema, haemorrhage and tissue damage (Jin et al, 2012;
Nigrovic et al, 2010; Skokowa et al, 2005; Warrington et al, 2011). An example
of type III hypersensitivity disease is Rheumatoid Arthritis, an inflammatory
disorder that manifests in part from the deposition of immune complexes in the
joints.
1.9.2.1 Rheumatoid Arthritis
Rheumatoid Arthritis (RA) is an autoimmune disorder characterised by
chronic inflammation in many tissues and organs, but primarily occurs at synovial
joints. It is a condition that affects approximately 1% of the world’s population
(Gibofsky, 2012). Inflammation of the synovial membrane lining the joint, termed
synovitis, leads to swelling, due to the accumulation of synovial fluids, and pain
when moving. If untreated, the inflammation can lead to erosion of the joint surface
and eventually causes loss of movement and function of the joints (Majithia &
Geraci, 2007). Although the exact cause of RA is not fully understood, genetic and
environmental factors have been implicated. Synovial mast cells have also been
shown to be involved in the pathogenesis of RA. Although they account for
approximately 3% of total cell population of a normal synovial membrane, their
numbers are elevated in RA patients (Crisp, 1984; Nigrovic & Lee, 2005). Mast
cells have been previously reported to be a primary source of IL-17, a key mediator

26
in RA pathogenesis (Hueber et al, 2010). IL-17 is able to bind to its respective
receptor present on synoviocytes and promotes their proliferation, survival and
migration and matrix destruction. (Lee et al, 2013b; Moran et al, 2009).
In RA, macrophages are also heavily implicated in disease progression.
Synovial macrophage numbers show a positive correlation with disease severity
(Mulherin et al, 1996). Macrophages can be activated by a range of different
stimuli including cytokines such as mast-cell derived IL-17, chemokines, immune
complexes, lipid metabolites and hormones (Kinne et al, 2007). Activated
macrophages are able to perpetuate disease progression by releasing pro-
inflammatory cytokines (TNFα and IL-1) that activate neighbouring cells, release
chemokines (CXCL12) that promote inflammatory cell infiltration, release tissue-
degrading enzymes and reactive oxygen species that destroy cartilage, tendon and
bone (Burrage et al, 2006; Hot & Miossec, 2011; Jovanovic et al, 1998; Kinne et
al, 2000; Mirshafiey & Mohsenzadegan, 2008).
Amongst the different cytokines involved in RA, TNFα released from both
activated macrophages and mast cells has been shown to play a central role in
disease pathogenesis (Lee et al, 2013a; Parameswaran & Patial, 2010). TNFα
induces pro-inflammatory cytokine release from synovial fibroblasts, promotes
angiogenesis, and also promotes matrix degradation through stimulating resident
chondrocytes to release matrix metalloproteinases (Nigrovic & Lee, 2005). The
importance of TNFα in RA is supported by several key observations: TNFα alone
or in concert with IL-1 drives synovitis (van den Berg et al, 1999), and transgenic
mice expressing human TNFα develop chronic inflammatory polyarthritis (Keffer
et al, 1991). Results from these findings suggests targeting TNFα is a viable
treatment option for RA and indeed, anti-TNFα antibodies adalimumab and
infliximab are clinically approved and has been shown to reduce signs and
symptoms of RA (Navarro-Sarabia et al, 2006; Tarp et al, 2016). However, as will
be discussed later, TNFα remains a critical inflammatory cytokine of the immune
system and whilst neutralisation of its activity has been shown to be therapeutically

27
beneficial, it can also have rare but serious side effects including increased risk of
infection and tumour malignancy (Bongartz et al, 2006). Moreover, a subgroup of
RA sufferers do not respond to anti-TNFα treatment (Wu et al, 2016). The lack of
efficacy of these agents can sometimes be explained by the generation of host
antibodies against the drug treatments (van Schouwenburg et al, 2013). However,
in other cases this is not the cause and this suggests that whilst TNFα is a key
player in RA pathogenesis, there are other mediators or factors that contribute to
the pathogenesis of RA. Identifying these new targets and generating therapeutic
treatments with different mechanisms of action therefore are of importance
(Rubbert-Roth & Finckh, 2009).
1.9.3 Neuroinflammation
Neuroinflammation is classically defined as inflammation in the nervous
system that may result in neurodegenerative events. In the brain, microglia are
often considered as the brain macrophages, and these cells are the predominant cell
population of the innate immune system of the central nervous system (Streit et al,
2004). During embryonic development, cells from the myeloid lineage in the bone
marrow give rise to the microglial population in the CNS (Alliot et al, 1999). Like
macrophages, microglia maintain brain homeostasis as they respond, activate and
clear foreign pathogens or injury. However, chronic activation of microglia may
cause neuronal damage through the release of pro-inflammatory cytokines,
reactive oxygen species and proteinases (Dheen et al, 2007). Microglia are
implicated in having both causative and perpetuating roles in common
neurodegenerative disorders including Alzheimer’s disease and Parkinson’s
disease (Lull & Block, 2010). One of the hallmark feature of Alzheimer’s disease
is the formation of β-amyloid plaques that are usually cleared by microglia.
However, in Alzheimer’s disease the accumulation of these plaques leads to
neuronal death. Furthermore, the accumulation and activation of microglia leads
to further neuronal loss. Similarly in Parkinson’s disease, accumulation of
activated microglia in the substantia nigra is a prominent feature of this disease.

28
Cytokines such as TNFα secreted from activated microglia are implicated in
neuronal death in Parkinson’s disease (Nagatsu et al, 2000).
Microglia also respond to pro-inflammatory signals released from other
cells, including mast cells (Skaper et al, 2012). Like other mast cells in the body,
the function of brain mast cells is no different. They act as sentinels in the brain
against foreign pathogen, and are able to release pro-inflammatory mediators and
proteinases that disrupt the blood brain barrier. This leads to recruitment of
inflammatory cells such as mast cells and monocytes from the blood to the
damaged site in the brain, allowing the cells to communicate with neurons,
astrocytes and microglia (Khalil et al, 2007; Silverman et al, 2000; Theriault et al,
2015). The involvement of mast cells in neurodegenerative disorders remains
controversial. In Alzheimer’s disease, one study reports that the β-amyloid triggers
mast cell activation (Niederhoffer et al, 2009). Interestingly, vasoactive intestinal
peptide-activated mast cells was found to have a neuroprotective role in a rat
Parkinson’s disease model (Tuncel et al, 2005).
Combined, there is evidence showing the involvement of mast cells and
microglial/macrophages in many different immune-related disorders as discussed
in Section 1.9. Many of the pro-inflammatory processes mediated by these cells
arise from the mediators released following cell activation. Thus, targeting these
cells, or the specific mediators they release, as therapeutic treatments has resulted
in improved signs and symptoms, as discussed below.
1.10 A brief overview of current treatments of inflammatory disorders
There are currently many treatment options available for managing asthma
and RA. However, none of these treatments cure the disease, rather they facilitate
an improvement in disease symptoms and quality of life. In asthma, β2-
adrenoceptor agonists such as salbutamol whilst not classically anti-inflammatory,
relieve bronchoconstriction by relaxing airway smooth muscle cells and are
currently recommended as a combination therapy alongside glucocorticoids in

29
asthma management (Chung et al, 2009; Sears & Lotvall, 2005). Glucocorticoids
(GCs) such as prednisolone have potent anti-inflammatory effects and are
prescribed to both asthma and RA patients (Da Silva et al, 2006; Donohue & Ohar,
2004). In relation to mast cell and macrophages, GCs, along with other agents
including omalizumab and disodium cromoglycate (DSCG) can modulate cell
activity. In addition to this, monoclonal antibody therapy targeting specific
cytokines are release from these cells also therapeutically beneficial to a degree,
as discussed below.
1.10.1 β2-adrenoceptor agonists
The β2-adrenoceptor agonists can be classified as short acting (SABA) such
as salbutamol or long acting (LABA) such as salmeterol. SABAs are typically fast-
acting, and are used as rescue medications to relieve acute asthma attacks. In
contrast, LABAs are used for long-term control of asthma symptoms (Cazzola et
al, 2012). In the airways, β2-agonists target the β2-adrenoceptor on airway smooth
muscle cells. Activation of these receptors leads to inhibition of contractile
signalling in the smooth muscle cells, thus resulting in bronchodilation (Pera &
Penn, 2016). Additionally, β2-adrenoceptor are also expressed on other resident
airway cells including alveolar macrophages, airway epithelial cells and
circulating inflammatory cells, where the receptor triggers anti-inflammatory
actions, including stabilising mast cells and downregulating of pro-inflammatory
cytokine production (Hanania & Moore, 2004). However, β2-agonists
monotherapy is not recommended in asthma as there is a low but increased risk of
death in patients (Nelson et al, 2006). In addition, prolonged use of β2-agonists can
lead to β2-adrenoceptor desensitisation uncoupling of the receptor and the
signalling proteins, or the internalisation and downregulation of β2-adrenoceptor
from cell surface. As a result, the β2-adrenoceptor-mediated bronchodilatory effect
is no longer effective (Pera & Penn, 2016). To limit these side effects, the Global
Initiative for Asthma (GINA) recommends the usage of inhaled SABA on demand
as the first choice of therapy. More commonly, β2-adrenoceptor agonists are used

30
alongside low dose inhaled glucocorticoids as a combination therapy
recommended for controlling the disease symptoms (Horak et al, 2016)
1.10.2 Glucocorticoids
As previously mentioned, GCs remain one of the most effective treatment
for a range of different inflammatory disorders including asthma and RA. They are
able to passively diffuse into cells and bind to the glucocorticoid receptors (GR)
localised in the cytoplasm. There are three commonly known GR receptor
isoforms: GR-α, GR-β and GR-γ (Oakley & Cidlowski, 2013). However, GR-α is
the predominant receptor known to mediate the effects of glucocorticoids and its
actions will be discussed here. GRs normally exists as part of a complex that
includes chaperone proteins such as heat-shock proteins (HSP) 90 and 70,
members of the FK506 immunophillin family and non-receptor tyrosine kinases
including c-Src (Cain & Cidlowski, 2015). Binding of GCs to GRs result in a
conformational change that allows the dissociation of the receptor from the
multiprotein complex. This allows the GC-GR complex to translocate into the
nucleus and exert its effect by either downregulating (transrepression) pro-
inflammatory gene products such as IL-1β and IL-8 or upregulating
(transactivation) of gene products such as the mitogen activated protein-kinase
phosphatase-1 (MKP-1) and glucocorticoid-induced leucine zipper (GILZ) genes,
which have been reported to exert anti-inflammatory effects (Newton & Holden,
2007). Several studies have reported that GCs are able to modulate mast cell
activity by reducing mast cell numbers and surface FcεRI expression, as well as
downregulating signalling molecules commonly associated with activated-mast
cell signalling pathways (Andrade et al, 2004; Finotto et al, 1997; Yamaguchi et
al, 2001). In macrophages, GCs exerts their anti-inflammatory role by
downregulating gene expression of chemokines (CXCL10 and MIP-1) and
cytokines (IL-1β), as well as antimicrobial peptide genes and the generation of
reactive oxygen species (Berkman et al, 1995; Di Rosa et al, 1990; Kulkarni et al,
2016). In addition, GCs also promote the expression of anti-inflammatory genes

31
(GILZ) and the cytokine IL-10 from macrophages following LPS and IL-1β
stimulation (Berrebi et al, 2003; John et al, 1998b).
Although the therapeutic onset of GCs-mediated genomic action usually
occurs between 4-24 hours following treatment, several studies have shown that
anti-inflammatory effects exerted by GCs on mast cell activation can be observed
as early as 5 minutes (Alangari, 2010; Zhou et al, 2008). There is a growing
appreciation of the anti-inflammatory properties exerted by GCs acting through
non-genomic pathways. However, the exact mechanism of this effect is not as well
characterised through its genomic actions. It is hypothesised that the non-genomic
effects of GCs are mediated through the components liberated from the GR-
multiprotein complex that are able to integrate into signalling pathways, although
this requires further investigation (Cain & Cidlowski, 2015).
In RA, there is strong evidence that administration of low doses of GCs can
delay joint erosion progression (Andrade et al, 2004). GCs are also given as a
“bridge therapy” when initiating treatment with disease-modifying anti-rheumatic
drugs (DMARDs), which can have a relatively slow onset of effect (Kavanaugh &
Wells, 2014). However, long term use of both local and more notably systemic
GCs are associated with several adverse effects including oral candidiasis,
cataracts, increased skin fragility, weight gain, osteoporosis, mood swings and
increased risk of infections (Kim & Mazza, 2011).
1.10.3 Omalizumab
Alongside GCs and β2-adrenoceptor agonists, the humanised monoclonal
antibody omalizumab is also used in treating a subset (high IgE) of patients with
allergic asthma (Holgate et al, 2004). Omalizumab competes with FcεRI for
binding to the Cε3 domain of IgE. As a result, the effector functions of IgE are
inhibited as there is less IgE available to bind to the FcεRI present on the mast cell
and basophil surface. By reducing free IgE levels in serum, omalizumab indirectly
downregulates FcεRI receptor expression, reducing early and late phase allergic

32
reactions in response to allergen and as well as reducing airway thickness and
eosinophil infiltration (D'Amato, 2002; Fahy, 2000; Riccio et al, 2012). Although
omalizumab is relatively safe and well tolerated, several factors including cost and
compliance have limited its use (McNicholl & Heaney, 2008). Currently,
omalizumab is used as an add-on therapy with corticosteroids and β2-adrenoceptor
agonists in patients suffering from poorly controlled severe asthma. This
combination therapy produces a significant reduction in asthma exacerbation and
hospital visits compared to patients receiving placebo, and also reduces the usage
of GCs without compromising symptom control (Holgate et al, 2004; Humbert et
al, 2005; Lanier et al, 2003; Pace et al, 2011).
1.10.4 Mast cell stabilisers
Although their molecular mechanisms of action remains controversial, mast
cell “stabilisers” are considered as an alternative therapeutic approach as they
inhibit the release of mediators from activated mast cells. Originally derived from
plants, these compounds have demonstrated anti-allergic activity in both in vitro
and in vivo screening assays (Finn & Walsh, 2013). Some of these compounds
have other biological activities. For example, ketotifen and rupatadine are
primarily known as histamine (H1) receptor antagonists, however both agents have
been reported to also block mast cell activation (Bradford, 1976; Queralt et al,
2000). One of these “stabilisers”, disodium cromoglycate (DSCG) was inspired
from Khellin, a compound extracted from seeds of the Amni visnaga plant
(Edwards, 2014; Finn & Walsh, 2013). Khellin was used as a diuretic and smooth
muscle relaxant, however reports of its bronchodilatory activity lead to the
development and synthesis of DSCG (Edwards, 2014; Kennedy & Stock, 1952).
Alongside DSCG, other mast cell “stabilisers” such as nedocromil sodium (NS)
were also discovered. NS acts similarly as DSCG as both agents were previously
reported to have anti-inflammatory and anti-allergic properties (Okayama et al,
1992; Valletta & Boner, 1994). However, several factors including weak efficacy,
difference in therapeutic activity due to mast cell heterogeneity and a lack of

33
understanding of their mechanisms of action have limited the suitability of these
compounds as therapeutic agents for allergic and inflammatory diseases (Finn &
Walsh, 2013; Horak et al, 2016; Okayama et al, 1992).
Recent studies however have identified DSCG and NS as agonists at the G-
protein coupled receptor 35 (GPR35) receptor (Yang et al, 2010). GPR35 has been
linked to a wide range of diseases such as heart failure, atherosclerosis and
inflammation (Mackenzie et al, 2011). GPR35 is present on many different types
of inflammatory cells including mast cells, monocytes, macrophages and
neutrophils. Studies utilising GPR35 agonists demonstrated an attenuation on
LPS-mediated TNFα release from human blood mononuclear cells (Wang et al,
2006). In mast cells and basophils, GPR35 mRNA was found to be upregulated
following IgE-dependent activation (Yang et al, 2010). In addition, GPR35 was
also demonstrated to be involved in monocyte and neutrophil adhesion (Barth et
al, 2009). These findings suggest a role for GPR35 in immune regulation and that
drug discovery programs targeting GPR35 as a novel therapeutic approach for
allergic treatment are enticing.
Despite being “de-orphanised” over a decade ago, the lack of potent GPR35
agonists and antagonists and species selectivity issues have delayed the progress
of elucidating the pharmacology and roles of this receptor and its relation to disease
pathology. However, several recently-developed in vitro assays have identified
more potent ligands which will likely facilitate a better understanding of the role
of this receptor (Mackenzie et al, 2011; Milligan, 2011).
1.10.5 Anti-cytokine therapy
As with omalizumab, monoclonal antibodies (mAbs) have revolutionised
the treatment of inflammatory disorders. Most mAbs target cytokines and
chemokines that are known to be elevated in inflammatory diseases including
asthma to RA, and the roles of these cytokines in these diseases have been
previously discussed here and by others (Hansbro et al, 2011; Siebert et al, 2015).

34
Thus, targeting specific molecules for therapy have become an enticing avenue for
therapy. In severe eosinophilic asthma patients, mepoluzimab (a mAb neutralising
IL-5) shows a reduction in asthma exacerbations and improved asthma control (Bel
et al, 2014; Ortega et al, 2014). The anti-TNFα mAb infliximab was reported to
be more efficacious in patients with refractory asthma, where membrane bound
TNFα is elevated compared to patients with moderate asthma. However, due to the
heterogeneity associated with the disease, it is likely that only a small subset of
asthmatics benefit from anti-TNFα therapy (Brightling et al, 2008; Desai &
Brightling, 2010; Erin et al, 2006; Morjaria et al, 2008).
In RA, targeting and subsequent blockade of TNFα-mediated signalling has
shown significant therapeutic benefits in patients (Tarp et al, 2016; Yamanaka,
2015). Interestingly, the neutralisation of TNFα in RA patients also resulted in
decreased risk of Alzheimer’s disease progression, thus highlighting a potential
role for TNFα in both RA and Alzheimer’s disease (Fuggle et al, 2014). Despite
the effectiveness of targeting TNFα for achieving beneficial outcomes, there are
rare but severe adverse effects associated with anti-TNFα therapy including
increased total cholesterol levels, increased risk of infection, glomulonephritis and
sarcoidosis (Feldmann & Maini, 2001; Kroesen et al, 2003; Scott, 2014; Seriolo et
al, 2006; Toussirot et al, 2008). Furthermore, the cost of anti-TNFα treatment,
similar to other biologics such as omalizumab, is an economic burden to the
patients and the healthcare systems, and is hence often considered as not cost-
effective (Siebert et al, 2015; Sullivan & Turk, 2008).
In contrast, other trials investigating various mAb therapies showed
promising signs in in vitro assays and animal models, but did not demonstrate
clinical efficacy in asthma treatment such as MEDI-528 (anti-IL-9 mAb) and
AMG-317 (anti-IL-4 receptor-α antagonist) (Corren et al, 2010; Oh et al, 2013).
In some cases, the lack of efficacy of certain treatments can be related to patient-
acquired resistance to the drug (van Schouwenburg et al, 2013). In addition, there
are numerous adverse effects associated with use of mAbs that range from

35
infections to cancer and to autoimmune diseases depending on the target of the
mAb and thus the benefits and limitations of mAbs as a therapeutic treatment needs
to be carefully considered (Hansel et al, 2010).
1.10.6 Treatment of Asthma and Rheumatoid Arthritis: Not one size fits all
Asthma is clinically characterised as variable airways obstruction and
bronchial hyperresponsiveness. However, different subsets of asthmatic patients
experience varying degrees of symptom relief to a specific course of treatment,
such as mepoluzimab in relieving severe eosinophilic asthmatics (Menzella et al,
2015). In addition, a small subset of asthmatic patients are unable to adequately
control the symptoms despite taking medications. As a result, these patients are
unable to lead a high quality life, and account for approximately 50% of the total
health care cost for asthma, and thus highlight an unmet medical need for
managing this disorder (Morjaria & Polosa, 2010; Wang et al, 2010). An
explanation as to why the efficacy of treatment differs on an individual basis is
likely due to the pathophysiological heterogeneity displayed in asthma. Asthma
can be currently considered as a “symptom”, which consists of different
mechanistic phenotypes producing airway obstruction. Therefore, a better patient-
to-patient classification of disease phenotype is necessary, as this allows
physicians to identify and establish a “personalised medication” regimen for
symptom relief (Corren, 2013; Lotvall et al, 2011). Nevertheless, treatment options
remain challenging for clinicians and the search for novel effective drugs is
ongoing and novel anti-cytokine therapy co-administered with conventional
treatments may lead to significant improvements in disease management (Gallelli
et al, 2013; Zidek et al, 2009).
Similarly, understanding the clinical and molecular properties of individual
patients is also critical for RA sufferers due to the heterogeneous nature of the
disease (Taylor et al, 2016). In particular, differences in the dominant cytokine
mediating disease pathogenesis can account for lack of clinical efficacy, as well as

36
other factors including the development of drug resistance, intolerance and safety
reasons (Emery, 2012). Nevertheless, understanding the heterogenic nature of this
disease permits personalised medication for these patients (de Jong et al, 2014).
Taken together, there is a need for further treatment advances in asthma and RA
that address these domains of contemporary unmet need.
1.11 Identification of macrophage capping protein as a putative novel mast cell
mediator
There is still an unmet medical need in asthma and RA, and these patients
account for a large proportion of the economic burden of the disease. Research is
ongoing to identify other molecular targets that can offer new treatment
alternatives to current therapy. In mast cells, there are still mediators released from
these complex cells in which the function is still not fully understood. For example,
activated mast cells release β-hexosaminidase, an enzyme located in the mast cell
granules and often used as a marker for degranulation in in vitro studies (Stone et
al, 2010; Xiong & Rodgers, 1997). However, a recent study has demonstrated the
importance β-hexosaminidase in mediating degradation of bacterial cell wall
components, thus playing a role in host defense against infection (Fukuishi et al,
2014). Thus, although the characteristics of most pro and anti-inflammatory
mediators have been well established, there is still much to learn of the mediator
repertoire released from inflammatory cells, and of interest to us, in mast cells and
macrophages.
In a recent study, we demonstrated that the supernatant from a stably
transfected human mast cell line-1 expressing the α-subunit of FcεRI (HMCα) was
able to trigger cytokine release from human airway smooth muscle (hASM) cells
following antigen challenge (Xia et al, 2013b). Interestingly, cytokine release from
the hASM cells was not due to the known mediators released from HMCα. Thus,
an activity-based proteomics analysis lead to the identification of seven novel
protein mediators. These putative mediators were then purchased with preliminary

37
studies identifying macrophage capping protein (CapG) as the novel mediator
triggering cytokine release from hASM cells. CapG is a member of the gelsolin
superfamily of members that are known for their role in regulating actin
polymerisation. Whilst its intracellular role is well defined, to date little is known
about the role of extracellular CapG.
1.12 Gelsolin superfamily as regulators of actin polymerisation
Cytoskeletal rearrangement is important to many different cellular
processes including cell morphological changes and motility. These processes are
tightly regulated by a wide variety of proteins. One of the key proteins involved in
this process is actin. During actin assembly the monomeric actin, G-actin
(globular-actin), polymerizes with other monomers, creating a long helical double-
stranded polymer known as F-actin (filamentous-actin) (Figure 1.2). Actin
polymerization is a thermodynamically unfavourable process, which begins with
a lag phase, where G-actin aggregates into short, unstable oligomers. Once the
oligomers reach a certain length it acts as a nucleus, and this initiates a rapid
elongation phase by which actin monomers polymerise at both ends of the
filament, known as the plus (barbed) and minus end. However, polymerisation
preferentially occurs at the plus end over the minus end. The difference in
polymerisation rates results in a net loss of actin monomers at the minus end and
a net gain of monomers at the plus (barbed) end. At a particular intermediate
subunit concentration, the filament achieves an equilibrium state where the actin
subunits cycle rapidly between the free and filamentous state while the filament
length remains unchanged. This phenomenon is termed as actin treadmilling
(Blanchoin et al, 2014).
Regulation of actin polymerisation is tightly controlled by a range of
different proteins, including members of the gelsolin superfamily. There are 7
known mammalian members in this superfamily: gelsolin, CapG, adseverin, villin,
advillin, supervillin and flightless I (Silacci et al, 2004). Although most members
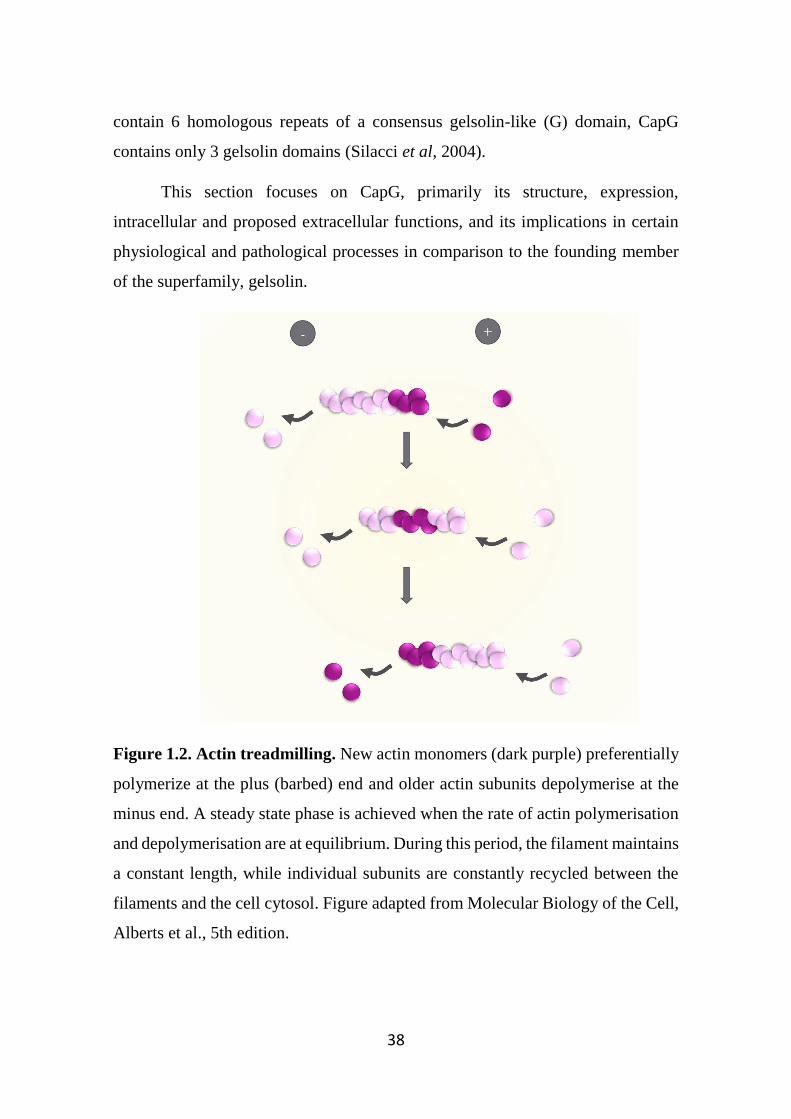
38
contain 6 homologous repeats of a consensus gelsolin-like (G) domain, CapG
contains only 3 gelsolin domains (Silacci et al, 2004).
This section focuses on CapG, primarily its structure, expression,
intracellular and proposed extracellular functions, and its implications in certain
physiological and pathological processes in comparison to the founding member
of the superfamily, gelsolin.
Figure 1.2. Actin treadmilling. New actin monomers (dark purple) preferentially
polymerize at the plus (barbed) end and older actin subunits depolymerise at the
minus end. A steady state phase is achieved when the rate of actin polymerisation
and depolymerisation are at equilibrium. During this period, the filament maintains
a constant length, while individual subunits are constantly recycled between the
filaments and the cell cytosol. Figure adapted from Molecular Biology of the Cell,
Alberts et al., 5th edition.

39
1.13 Gelsolin and CapG
Gelsolin is widely expressed in a variety of cell types, and is important in
regulating cell shape, motility and apoptosis (Koya et al, 2000; Sun et al, 1999).
As previously mentioned, gelsolin contains 6 G domains and the function of each
domain was elucidated through a combination of several techniques including X-
ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, X-ray
footprinting, electron microscopy and molecular modelling, which have all
provided useful insights on the structure and molecular function of gelsolin
(McGough et al, 2003). The G1-G3 and G4-G6 domains are homologous and
connected by a linker (Sun et al, 1999). When resting intracellular Ca2+ levels are
elevated from submicromolar to millimolar concentrations, Ca2+ binds to the G5
and G6 domains, which results in a conformational change that leads to gelsolin
activation (Ashish et al, 2007; Kolappan et al, 2003). The G2 domain binds to actin
subunits, and brings the G1 domain to its own actin binding site. Binding of the
G1 domain to the ‘barbed’ end of actin and coordinates a “pincer” movement that
causes steric strain hence severing the filaments (Burtnick et al, 2004; Sun et al,
1999). The G3 domain was recently discovered to modulate the severing and
depolymerising activity of the protein (Qian et al, 2015). Finally, structural
analysis of the G4-G6 domain showed that the G4 domain exhibits similar actin-
binding and severing capacity as G1 and G2 (Kolappan et al, 2003). Once the actin
filament is severed, gelsolin caps the barbed ends to prevent the addition of
monomers to the filament (McGough et al, 2003) (Figure 1.3a). Gelsolin
dissociates from actin only when there is: (1) an increase of phosphatidylinositol
3,4 or 4,5-bisphosphate (PIP2) and (2) there is a decrease in intracellular calcium
(Kumar & Khurana, 2004; Silacci et al, 2004). Once these two conditions are met,
gelsolin disassociates from actin and this leads to the generation of multiple
polymerization-competent, short filaments which are then able to rebuild the
cytoskeleton (Deaton et al, 1992; Sun et al, 1999). Although gelsolin is important
in mediating routine cell processes, pathogens are also able to take advantage of

40
this actin polymerisation process. Salmonella bacteria force their own
internalisation into cells and release proteins such as salmonella invasion protein
A (SipA) to ensure its survival by promoting actin polymerisation by inhibiting
gelsolin-mediated actin severing and also promoting the re-annealing of gelsolin-
severed actin filaments (McGhie et al, 2004).
Even though CapG (also known as gCap39 and Mbh-1) and gelsolin are
members of the same family, there are several distinct features that exist between
the two proteins. CapG does not have the G4-G6 domains and thus contains only
3 G (G1-G3) domains. Similar to gelsolin, CapG binds and caps actin filaments,
but it doesn’t sever the filaments (Figure 1.3b). Mutagenesis studies performed on
CapG has shown that it lacks specific amino acid sequences and domains essential
for actin severing (Zhang et al, 2006). Indeed, chimeric studies replacing these
amino acid sequences with the corresponding gelsolin amino acid sequences
resulted in a gain in actin-severing function by CapG, highlighting these regions
as being critical for actin severing (Zhang et al, 2006).
The expression profiles of CapG and gelsolin also differ: Whilst the cellular
expression of CapG and gelsolin are similar in most cell types, CapG is highly
expressed in macrophages and dendritic cells, where it represents approximately
1% of total cytoplasmic protein. However, CapG expression is undetectable in
platelets which are however rich in gelsolin (Witke et al, 2001). Unlike gelsolin,
CapG can be present in the cell nucleus, where its nuclear transport is regulated by
several proteins including the guanosine triphosphate hydrolase enzyme Ran,
nuclear transport factor 2 (NTF2), and importin-β (De Corte et al, 2004; Van Impe
et al, 2008). In addition to regulating nuclear actin, other studies have implicated
nuclear CapG tumour cell metastasis by modulating gene expression (De Corte et
al, 2004; Yu et al, 1990). Similar to gelsolin, CapG activity is also regulated by
PIP2 and intracellular calcium. However, unlike gelsolin, it readily disassociates
from actin filaments either upon decreasing calcium concentrations or increased
PIP2 levels independent of calcium concentration (Yu et al, 1990).
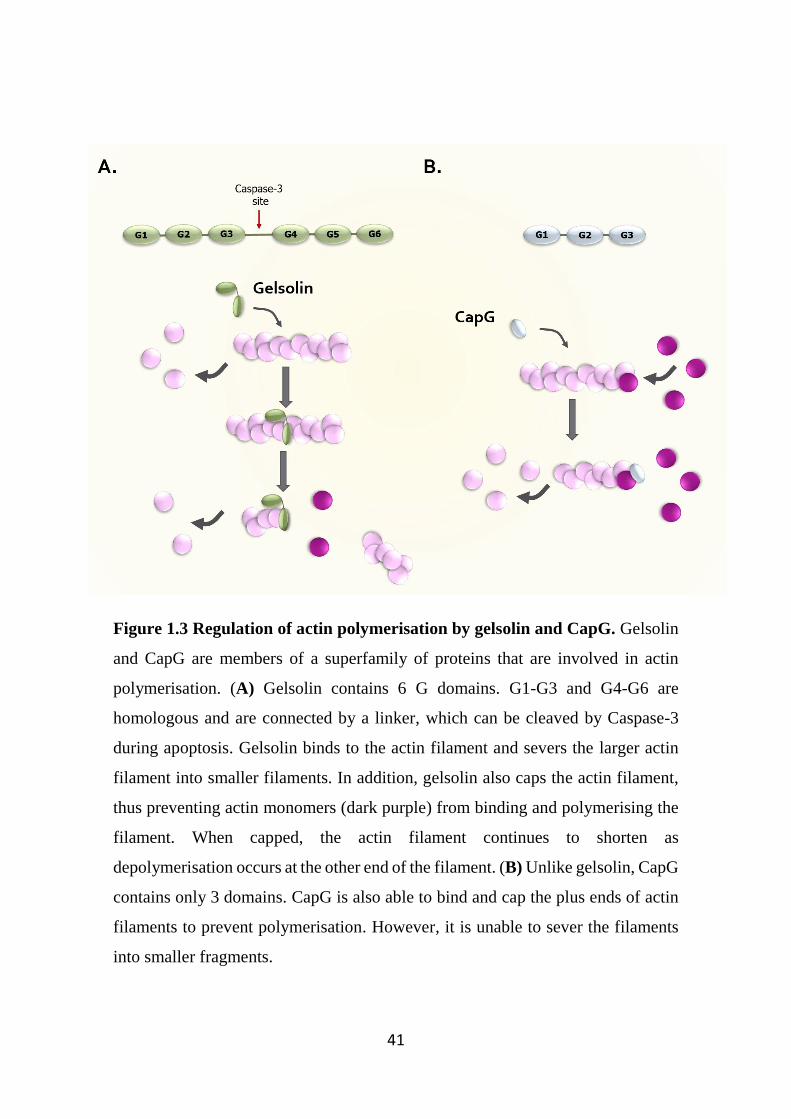
41
Figure 1.3 Regulation of actin polymerisation by gelsolin and CapG. Gelsolin
and CapG are members of a superfamily of proteins that are involved in actin
polymerisation. (A) Gelsolin contains 6 G domains. G1-G3 and G4-G6 are
homologous and are connected by a linker, which can be cleaved by Caspase-3
during apoptosis. Gelsolin binds to the actin filament and severs the larger actin
filament into smaller filaments. In addition, gelsolin also caps the actin filament,
thus preventing actin monomers (dark purple) from binding and polymerising the
filament. When capped, the actin filament continues to shorten as
depolymerisation occurs at the other end of the filament. (B) Unlike gelsolin, CapG
contains only 3 domains. CapG is also able to bind and cap the plus ends of actin
filaments to prevent polymerisation. However, it is unable to sever the filaments
into smaller fragments.

42
1.14 Well established intracellular roles of Gelsolin and CapG
Studies utilising gelsolin knockout mice (Gsn-/-) showed that these mice do
not exhibit gross phenotypic defects, and are viable and fertile (Cantu et al, 2012).
However, the loss of gelsolin resulted in impairments of cell-related functions,
thus, highlighting the importance of gelsolin in actin-related cell processes. For
example, fibroblasts derived from these mice displayed reduced motility and
migration in response to serum or epidermal growth factor (EGF) (Azuma et al,
1998). In addition, the absence of gelsolin in platelets resulted in a prolonged
bleeding time due to diminished gelsolin-mediated actin severing in platelets,
corresponding to decreased shape change and hence a reduced clotting rate.
Similarly, recruitment of neutrophils to thioglycolate-induced local inflammation
was reduced in these mice (Witke et al, 1995). A separate study performed on
human gingival fibroblasts microinjected with anti-gelsolin antibody resulted in a
reduction in cell migration (Arora & McCulloch, 1996). Combined, these studies
highlight the importance of gelsolin in regulation of the actin cytoskeleton
network.
In addition, gelsolin also plays a dual role in apoptosis. The intact gelsolin
protein is anti-apoptotic as it stabilizes the membrane potential of mitochondria
thus preventing mitochondria dysfunction events including the release of
cytochrome C, a key initiator of the apoptotic process in an actin-independent
manner (Koya et al, 2000). However, during apoptosis gelsolin is cleaved by the
protease caspase-3 (Cryns & Yuan, 1998). Specifically, caspase-3 cleaves the
linker connecting the G1-G3 domains to the G4-G6 domains (Figure 1.3). The
resulting cleavage product is pro-apoptotic as it severs actin filaments independent
of calcium regulation (Kothakota et al, 1997). Expression of this fragment in live
vascular smooth muscle cells led to the collapse of the actin cytoskeleton, which
is a key characteristic of apoptosis (Geng et al, 1998).
Like its family members, binding and modulating actin assembly allows
CapG to control actin-based cell motility. Loss of CapG activity results in

43
defective cell motility as seen in neutrophils derived from CapG knockout mice,
which exhibited impaired cell recruitment in response to the chemoattractant N-
formyl-methionyl-leucyl-phenylalanine (FMLP) (Renz et al, 2008). In addition,
macrophages derived from the bone marrow of CapG knockout mice showed
reduced complement and IgG-mediated phagocytosis compared to wild type
macrophages (Parikh et al, 2003). In keeping with its role in innate immune
functions, these knockout mice were also found to be more susceptible to certain
bacterial infections (Parikh et al, 2003).
1.15 An established role for extracellular gelsolin
Although the gelsolin superfamily are best known for regulating actin
assembly and thus mediating a variety of intracellular activities, both gelsolin and
CapG are also present in plasma. Cytoplasmic and plasma gelsolin are derived
from a single gene by alternative splicing. The secreted form differs from the
cytoplasmic form by the presence of a 27-amino acid signalling peptide and a
further 24-amino acid sequence present at the N-terminus (Nag et al, 2013).
Plasma gelsolin is present in substantial quantities (190-300 µg/mL) and is
thought to be secreted largely by skeletal muscle (Kwiatkowski et al, 1988; Lee &
Galbraith, 1992). Calcium ions are present at millimolar concentrations in the
plasma, thus allowing plasma gelsolin to exist in a fully activated state (Nag et al,
2013). Plasma gelsolin is thought to be essentially identical to its intracellular
isoform: binding and severing actin filaments but in this scenario the actin being
in plasma. During normal cell turnover or tissue injury, a variety of cytoplasmic
proteins are released including actin (Lind et al, 1986). If uncleared, actin may
increase the viscosity of extracellular fluids such as plasma and hence impair tissue
perfusion (Dahl et al, 1999). The actin-scavenger system, which consists of
gelsolin and a group of Vitamin D binding proteins (also known as ‘group-specific
component proteins’) work in concert to bind, sever and clear actin from the blood
through the liver (Erukhimov et al, 2000).

44
In certain conditions such as fulminant hepatic necrosis, adult respiratory
distress syndrome, septic shock and complicated pregnancies, plasma actin is
present at high concentrations (Erukhimov et al, 2000). As a result, the actin-
scavenger system is saturated. This can have serious consequences. Rats injected
with a high dose of G-actin showed rapid death due to pulmonary venous
obstruction by actin filaments and endothelial injury (Haddad et al, 1990). The
concentrations of plasma gelsolin have also been considered as a prognostic
biomarker for acute diseases and infections (Lee et al, 2006; Lee et al, 2007). The
lower the concentration of circulating gelsolin, the less favourable the prognosis
becomes. In addition, rats administered with exogenous gelsolin had reduced
morbidity from sepsis by reducing cytokine expression and tissue injury (Cohen et
al, 2011). Although the mechanism of action of gelsolin mediating this protective
effect is unclear, it is plausible that gelsolin can neutralise the high concentration
of plasma actin in these septic rats, thus reducing morbidity. Recent studies have
identified actin as a DAMP molecule, it being a ligand for the PRR Clec9a on
dendritic cells (Ahrens et al, 2012; Zhang et al, 2012). Activated dendritic cells
through the Clec9a receptor has the potential to activate the immune system in
response to intracellular infections or it may potentially exacerbate autoimmune
diseases such as systemic lupus erythematosus (Zhang et al, 2012). By clearing
actin, these findings therefore suggests a potential anti-inflammatory role mediated
by plasma gelsolin.
1.16 An emerging role for extracellular CapG
Similar to gelsolin, CapG can also be detected extracellularly. The protein
was initially discovered to be secreted constitutively from macrophages. Through
Western blot analysis, CapG was shown to be secreted from macrophage-like cell
lines such as the mastocytoma cell line P815 and histiocytic lymphoma U937 cells,
but not from non-macrophage cell lines (Dabiri et al, 1992; Johnston et al, 1990).
In addition, transfection of COS cells (fibroblast-like kidney cell lines) with CapG
resulted in large amounts of the protein being secreted compared to non-

45
transfected COS cells (Johnston et al, 1990). CapG has also been shown to be
present in human plasma, although the concentrations in plasma (0.3-0.5 µg/mL)
are considerably lower than gelsolin (Johnston et al, 1990). However, unlike
gelsolin, there is no noticeable molecular size difference between the secreted
CapG and intracellular CapG isoforms (Johnston et al, 1990). The absence of a
well-defined signal peptide suggests that CapG is secreted through a non-canonical
pathway similar to IL-1β, basic fibroblast growth factor (bFGF) and certain
mediators that act as DAMPs (Carta et al, 2009; Johnston et al, 1990).
Interestingly, the release of these mediators can be triggered by cell injury, and are
associated with inflammatory responses (Dinarello, 2009; Srikrishna & Freeze,
2009; Zittermann & Issekutz, 2006). In keeping with this, it has also been
suggested that in response to cell injury, regardless of physiological or pathological
origin, CapG might also be secreted from macrophages at high concentrations and
was proposed as a potential pro-inflammatory mediator (Johnston et al, 1990).
In summary, CapG and gelsolin are members of the gelsolin superfamily
and both are important in modulating actin assembly and regulation of consequent
cell motility and morphology. There is however evidence to show that both
proteins are also involved in other cellular processes. Whilst the intracellular roles
of gelsolin and CapG as well as the extracellular role of gelsolin have been well
established, the role of secreted CapG is not well understood. As mentioned earlier,
preliminary studies have identified CapG as a previously undescribed mediator
released from IgE/antigen activated mast cells that triggered cytokines release
from human airway smooth muscle cells. This finding suggests CapG acts as a
potential novel pro-inflammatory mediator. However, this novel finding requires
further investigation. A clearer understanding of the actions of CapG will provide
valuable insights into its function in inflammatory disease and whether the protein
serves as a novel target for anti-inflammatory therapy.

46
1.17 Aims of this thesis
CapG is a member of the gelsolin superfamily, which are best known for
their roles as regulators of actin polymerisation and involvement in cellular
processes such as cell motility. CapG is highly expressed in inflammatory cells
such as macrophages, where it has also been demonstrated to be involved in key
macrophage functions such as phagocytosis. CapG has also been reported to be
present in the extracellular space. However, since this discovery over two decades
ago, there has been limited understanding of its extracellular role. It is known that
macrophages constitutively release CapG and low levels of CapG have been
detected in plasma. Beyond this, the basic characteristics of extracellular CapG is
limited – it is unclear if the release of CapG can be regulated following cell
activation (Figure 1.4). Moreover, preliminary studies have implicated CapG as
playing a role in triggering pro-inflammatory mediator release from the human
airway smooth muscle cells, and that CapG is also elevated in RA patients
(Balakrishnan et al, 2014). These findings are suggestive of CapG acting as a novel
pro-inflammatory mediator.
Therefore, the aims of this project were to:
1) Characterise the expression of CapG in a range of different cell types, and
determine changes in the expression of CapG following cell stimulation.
2) Determine changes in the CapG mRNA levels from activated macrophages
in response to various stimuli, as well as in several disease settings,
including infection and neuroinflammation.
3) Generate and optimise a mammalian expression system to produce a
significant quantity of human recombinant CapG to facilitate
characterisation of its biological role.
4) Identify the role of extracellular CapG as a regulator of pro-inflammatory
mediator release in a range of different cell types.
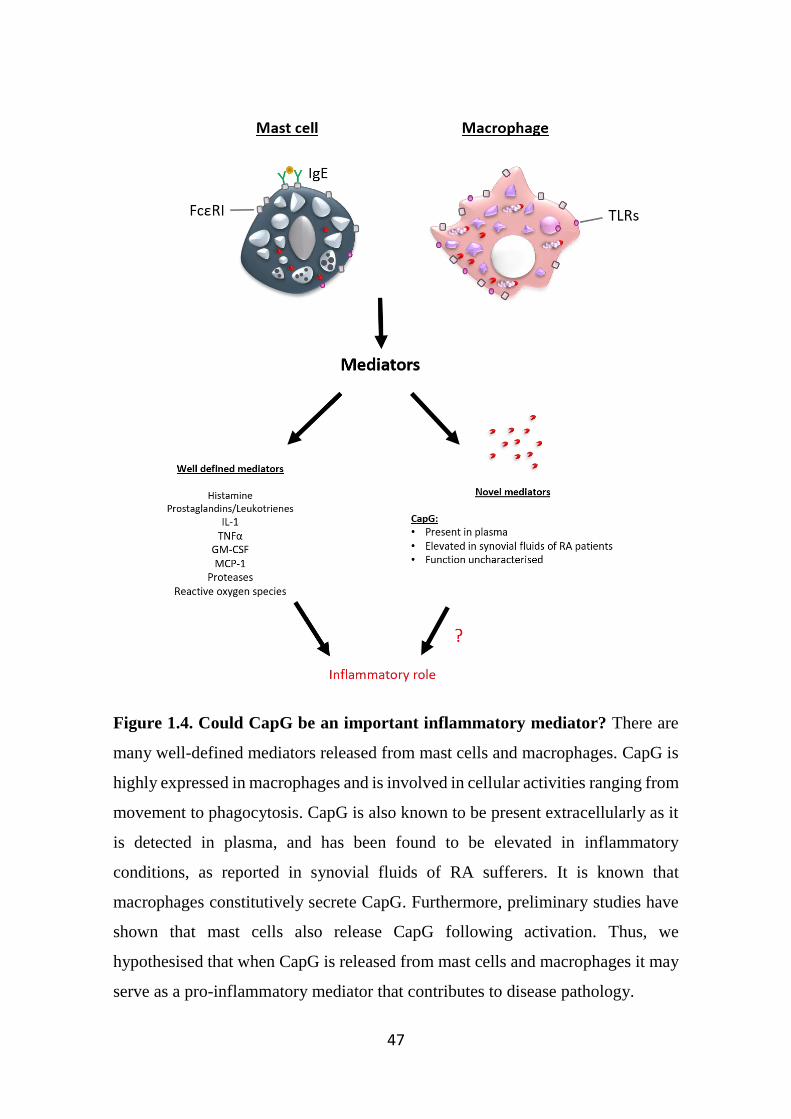
47
Figure 1.4. Could CapG be an important inflammatory mediator? There are
many well-defined mediators released from mast cells and macrophages. CapG is
highly expressed in macrophages and is involved in cellular activities ranging from
movement to phagocytosis. CapG is also known to be present extracellularly as it
is detected in plasma, and has been found to be elevated in inflammatory
conditions, as reported in synovial fluids of RA sufferers. It is known that
macrophages constitutively secrete CapG. Furthermore, preliminary studies have
shown that mast cells also release CapG following activation. Thus, we
hypothesised that when CapG is released from mast cells and macrophages it may
serve as a pro-inflammatory mediator that contributes to disease pathology.

48

49
Chapter 2
General Methods

50
The description of the general methods in this chapter pertains to common
experimental protocols conducted in the following results chapters. Where
necessary, a more detailed description of experimental protocols performed in each
specific chapter is described in their respective chapters. Throughout this project,
fetal bovine serum (FBS), used as a supplement in cell culture growth medium,
was purchased from various vendors (most commonly Lonza and Sigma-Aldrich,
both from Victoria, Australia). Regardless, the contents of this serum is still of
highest quality, as guaranteed by the vendors and no noticeable differences in
cultured cell characteristics between both serum types was observed.
2.1 Cell Culture
2.1.1 Human mast cell-1 (HMC-1) cells transfected with the α-subunit of the
human FcεRI (HMCα cells)
The HMC-1 cell line (kindly provided by Dr. Joseph Butterfield, Mayo
Clinic) was initially derived from a patient diagnosed with mast cell leukaemia
(Butterfield et al, 1988; Nilsson et al, 1994). HMC-1 cells were cultured in
Iscove’s modified Dubecco’s medium (IMDM) (Life Technologies),
supplemented with FBS (10% v v-1), alpha-thioglycerol (0.01% v v-1; Sigma-
Aldrich), glutamax (1% v v-1), penicillin (100 U/mL) and streptomycin (100
μg/mL) (all from Life Technologies).
The procedure for generation of HMCα cells, where HMC-1 cells were
transfected with the IgE-binding α subunit of FcεRI, has been described previously
(Xia et al, 2011). HMCα cells were routinely cultured as above, but in the presence
of G418 (125 μg/mL; Geneticin®, Life Technologies).
2.1.1.1 HMCα cell stimulation
HMCα cells were sensitised for 2-3 days with human IgE (hIgE; 1:100
dilution) from conditioned media derived from JW8 cells (ECACC), a cell line
known to secrete 4-hydroxy-3-iodo-5-nitrophenylacetyl (NIP) specific IgE (NIP-

51
IgE) (Bruggemann et al, 1987). The cells were then harvested by pipetting up and
down to break cell clumps and then centrifuged (300 g, 5 mins) and washed once
in serum-free IMDM. Cells were then resuspended in serum-free IMDM (as above
but without FBS), and then seeded at a density of 1 million cells/well in 24-well
plates (Corning, Victoria, Australia). Cells were stimulated with various
concentrations of NIP-conjugated BSA (NIP-BSA; Biosearch Technologies,
Novato, CA, USA) (antigen), or the stable adenosine receptor agonist 5’-(N-
ethylcarboxamido)-adenosine (NECA; Sigma-Aldrich) or the calcium ionophore
ionomycin (Abcam, Cambridge, MA, USA). Cell supernatants were harvested four
hours after stimulation, centrifuged (300 g, 5 mins) and stored at -80ºC prior to
analysis.
2.1.2 Laboratory of Allergic Diseases (LAD2) cells
LAD2 cells (kindly provided by Dr. Arnold Kirshenbaum, NIH, Bethesda,
MD, USA) are a mast cell line initially derived from a patient with mast cell
sarcoma/leukaemia (Kirshenbaum et al, 2003). Unlike other mast cell lines, the
LAD2 cells maintain many of the key characteristics of primary mast cells such as
highly abundant granules and ability to degranulate and release mediators such as
TNFα in response to IgE/antigen challenge (Zhang et al, 2011). Hence this cell
line has been extensively utilised in mast cell-related studies. The LAD2 cells were
cultured in StemPro 34 media, supplemented with StemPro 34 nutrient supplement
(2.6% v v-1) (Life Technologies, Victoria, Australia), glutamax (1% v v-1),
penicillin (100 U/mL) and streptomycin (100 μg/mL) and rhSCF (50-100 ng/mL,
Peprotech, Rocky Hills, CT, USA). Prior to cell-culture use, the complete media
was sterilised by filtration using a 0.2 µm filter (Millipore, Victoria, Australia).
2.1.2.1 LAD2 cell stimulation
The protocol for LAD2 cell stimulation is similar to HMCα cells, with the
following exceptions: LAD2 cells were incubated with hIgE for 1 day in complete
StemPro34 media supplemented with StemPro34 Nutrient Supplement and rhSCF.

52
LAD2 cells were then seeded at a density of 1x106 cells/mL on a 24-well plate,
and then stimulated with various concentrations of antigen (NIP-BSA), substance
P (Sigma-Aldrich), compound 48/80 (Sigma-Aldrich), NECA or ionomycin. Cell
supernatants were harvested four hours after stimulation, centrifuged (300 g, 5
mins) and stored at -80ºC.
2.1.3 Rat Basophil Leukaemia (RBL) cells
RBL cells (kindly provided by Prof. Hannah Gould, Kings College London,
United Kingdom) were cultured in minimum essential medium (MEM) (Life
Technologies), supplemented with FBS (5% v v-1), glutamax (1% v v-1), penicillin
(100 U/mL) and streptomycin (100 μL/mL).
2.1.4 THP-1 cells
THP-1 cells (kindly provided by A/Prof Steven Bozinovski, Royal
Melbourne Institute of Technology University, Victoria, Australia) are a
monocytic cell line that were initially obtained from a patient with acute monocytic
leukaemia (Tsuchiya et al, 1980). The cells were cultured in Roswell Park
Memorial Institute (RPMI) medium, supplemented with FBS (10% v v-1),
glutamax (1% v v-1), penicillin (100 U/mL), streptomycin (100 μg/mL) and 2-beta-
mercaptoethanol (11 μM; Life Technologies).
2.1.4.1 THP-1 cell stimulation
In the presence of phorbol 12-myristate 13-acetate (PMA), THP-1 cells can
be differentiated into macrophage-like cells (Auwerx, 1991). Differentiated THP-
1 cells are known to release pro-inflammatory cytokines such as IL-8 in response
to LPS, as well as other stimuli including IL-17 and IgG (Alonso et al, 2000; Tamai
et al, 2003; Tsuchiya et al, 1980; Turner-Brannen et al, 2011). Therefore, unless
otherwise stated, in experiments involving THP-1 cells, the cells were first treated
with PMA (100 nM) for 2 days before use.

53
THP-1 cells were seeded at a density of 500,000 cells/well in 24-well plate
and incubated with PMA (100 nM; Sigma-Aldrich) in serum-complete RPMI and
were allowed to attach and differentiate for 2 days. Cells were then serum-starved
for a further 24 hrs in incomplete RPMI supplemented with bovine serum albumin
(BSA, 0.25% v v-1; Life Technologies). Cells were then washed and fresh serum-
free RPMI added. Cells were then stimulated with various stimuli including LPS,
IL-17, and IgG in both monomeric and heat-aggregated form (gift from Prof. Mark
Hogarth, Burnet Institute, Victoria, Australia) for 24 hrs. For kinetic studies
examining the release of CapG, cells were stimulated with LPS for 1, 2, 4 and 24
hrs.
To study the regulation of CapG release from LPS-stimulated THP-1 cells
in the presence of an anti-TLR antibody (HTA-125) or the glucocorticoid
dexamethasone, the cells were pre-treated with either HTA-125 (1 µg/mL; Santa
Cruz Biotechnology, Dallas, TX, USA) or dexamethasone (100 nM; Sigma-
Aldrich) for 30 min prior to LPS stimulation.
After the respective time points, supernatants were collected and
centrifuged (300 g, 5 mins). In addition, THP-1 cells were also harvested by
trypsinisation, cell viability assessed using trypan blue (Sigma-Aldrich) exclusion
and both supernatants and cell pellets were stored at -80ºC for downstream
purposes.
2.1.5 BV2 cells
The murine microglial cell line BV2 (kindly provided by Dr. Peter Crack,
The University of Melbourne) were maintained in DMEM medium supplemented
with FBS (5% v v-1), glutamax (1% v v-1), penicillin (100 U/mL) and streptomycin
(100 µg/mL).

54
2.1.5.1 BV2 cell stimulation
One hundred thousand BV2 cells were seeded in 24-well plates in DMEM
supplemented with FBS (5% v v-1). On the following day, the media was removed
and replaced with DMEM supplemented with FBS (2% v v-1). Cells were
stimulated with LPS (1, 10 and 100 ng/mL) for 24 hours. Following stimulation,
supernatants were removed and centrifuged (300 g, 5 mins). Cells were also
harvested by trypsinisation and stored at -80ºC for downstream purposes.
2.1.6 Mouse bone marrow derived-mast cells
Three C57/BL6 mice (as approved by the Burnet Institute animal ethics
committee) were killed by CO2 asphyxiation. Bone marrow cells were flushed
from the femurs of these mice using a 1 mL syringe and 20 gauge needle (both
Terumo). Cells were cultured independently in RPMI medium, further
supplemented with FBS (10% v v-1), glutamax (1% v v-1), penicillin (100 U/mL)
and streptomycin (100 μg/mL), β-mercaptoethanol (55 nM) (all Life
Technologies), sodium pyruvate (1 mM), non-essential amino acids (1 x), HEPES
(5 mM) (all Hyclone), and WEHI-3BD cell conditioned media (50% v v-1), as a
source of IL-3. Cells were cultured in a CO2 humidified atmosphere at 37°C. Every
week the suspension cells were removed from the flask and retained, replenished
with new media and cultured in fresh flasks. After 5 weeks of culture, cells were
phenotypically characterised via FACS analysis and histological examination.
Prior to all experiments, cell viability was assessed using trypan blue exclusion.
Cells from week 5 to 12 in cultures with >90% viability were used in experiments.
2.1.7 Human airway smooth muscle (hASM) cells
Human airway smooth muscle was obtained through dissection of
macroscopically normal bronchi (0.5-2 cm diameter) obtained from lung transplant
specimens (kindly provided by the Alfred Hospital, Victoria, Australia) as
approved by the University of Melbourne human ethics committee. hASM cells

55
were prepared as previously described (Fernandes et al, 1999), and were cultured
in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with FBS (10%
v v-1), L-glutamine (2 mM), penicillin (100 U/mL), streptomycin (100 µg/mL),
HEPES (16 mM), sodium bicarbonate (0.2% v v-1), sodium pyruvate (1 mM) and
non-essential amino acids (1% v v-1). Cells at passages between 9-13 were used,
during which the proliferative response to FBS and growth factors remained
unchanged (Hirst et al, 1992). hASM cells were, unless stated, serum starved for
24 hours (medium as above, but with BSA (0.25% v v-1) before use.
2.1.7.1 hASM cell stimulation
hASM cells were seeded at a density of 10,000 cells/well in 96-well flat
bottom plates (Corning) in serum-complete DMEM. On the following day, cells
were washed and serum-free DMEM supplemented with BSA (0.25% v v-1) was
added to the cells. The cells were stimulated with a range of different stimuli on
the following day including LPS, recombinant CapG, and TNFα (Sigma-Aldrich).
After 24 hours, supernatants were harvested, centrifuged (300 g, 5 mins) and stored
at -80ºC.
2.1.8 BEAS2B cells
BEAS2B cells (ATCC) are an immortalised bronchial epithelial cell line
derived from normal human bronchial epithelial cells using the AD12-SV40 virus
(Reddel et al, 1988). Cells were maintained in LHC-9 medium (Life Technologies)
supplemented with FBS (2% v v-1), L-glutamine (2 mM), penicillin (100 U/mL)
and streptomycin (100 µg/mL).
2.1.8.1 BEAS2B cell stimulation
Cells were seeded at a density of 50,000 cells/well in 96-well flat bottom
plates for 2 days in serum-complete DMEM media. Cells were then serum-starved
for a further 24 hrs in incomplete DMEM supplemented with BSA (0.25% v v-1).

56
Cells were then stimulated for 24 hrs, after which the supernatants were harvested,
centrifuged (300 g, 5 mins) and stored at -80ºC.
2.1.9 Human Embryonic Kidney-293 (HEK293) cells
The HEK293 cell line was originally derived from primary cultures of
human embryonic kidney (HEK) cells transformed with sheared adenovirus DNA
(Graham et al, 1977) and has been used extensively in transfection studies (Shaw
et al, 2002) as well as a tool for expressing recombinant proteins (Thomas &
Smart, 2005). For high level protein expression, two 293 cell lines were examined:
the Flp-InTM-293 cell line and the 293-Ebstein-Barr Nuclear Antigen (EBNA293)
cell line.
2.1.9.1 Flp-InTM-293 cells
The Flp-InTM-293 cells (kindly provided by Prof Peter McIntyre, RMIT
University, Melbourne) contain a Flp-Recombination Target (FRT) site, which
allows homologous recombination between the cell genome and a vector
expressing the identical FRT-site such as the pcDNA5 vector (Craig, 1988). This
therefore allows for the generation of stable cell-lines that ensure homogenous
expression of the desired protein. The cells were maintained in high-glucose
DMEM medium, supplemented with FBS (10% v v-1), glutamax (1% v v-1),
penicillin (100 U/mL) and streptomycin (100 μg/mL).
2.1.9.2 293-EBNA or HEK293E cells
The EBNA293 cell line is another adapted HEK cell line stably expressing
the Epstein-Barr virus nuclear antigen (EBNA1), allowing the cells to produce
high quantities of recombinant protein after appropriate transfection (Baldi et al,
2007). EBNA293 cells (kindly provided by Dr Amanda Gavin, Burnet Institute,
Melbourne) were maintained in RPMI medium, supplemented with FBS (5% v v-
1), glutamax (1% v v-1), penicillin (100 U/mL) and streptomycin (100 μg/mL).

57
2.1.10 COS-7 cells
The COS-7 cells are an African green monkey kidney fibroblast-like cell
line that are also commonly used in transfection experiments, and were previously
found to secrete CapG following gene transfection (Gluzman, 1981; Johnston et
al, 1990). COS-7 cells were kindly provided by Dr. Mark Hullett (La Trobe
University, Melbourne) and were maintained in RPMI medium supplemented with
FCS (10% v v-1), glutamax (1% v v-1), penicillin (100 U/mL) and streptomycin
(100 μg/mL).
2.1.11 SW982 cells
The synovial fibroblasts cell line SW982 (kindly provided by Prof Jia Lin
Yang, Prince of Wales Hospital, New South Wales (NSW), Australia) were
maintained in DMEM, FCS (5% v v-1), glutamax (1% v v-1), penicillin (100 U/mL)
and streptomycin (100 μg/mL).
2.1.11.1 SW982 cell stimulation
SW982 cells were seeded at a density of 50,000 cells/well in a 96-well flat
bottom plate for 2 days in serum-complete DMEM media. Cells were then serum-
starved for a further 24 hrs in incomplete DMEM supplemented with BSA
(0.25%). Cells were then stimulated for 24 hrs, after which the supernatants were
harvested, centrifuged (300 g, 5 mins) and stored at -80ºC.
2.2 Rat peritoneal cell (RPC) collection and rat peritoneal mast cell (RPMC)
purification
Sprague Dawley rats for use in undergraduate practical classes (Department
of Pharmacology and Therapeutics, University of Melbourne) were isofluorane-
anaesthetised and decapitated using a guillotine. All animal work was approved by
the University of Melbourne animal ethics committee (Ethics code: 1513746).
Following primary use, wash buffer (ice cold phosphate buffered saline (PBS),

58
supplemented with BSA (0.25% w v-1)) was injected into the peritoneal cavity of
the rats using a 10 mL syringe and 20 gauge needle. The peritoneum was gently
massaged prior to incision and collection of lavage fluid made using a transfer
pipette. The mixed peritoneal cells were stored on ice, and any washings heavily
contaminated with red blood cells were discarded.
The cells were spun down at (300 g, 5 mins, 4ºC) and washed twice with
wash buffer prior to mast cell isolation using a Percoll density gradient as described
by others (Mackay & Pearce, 1996). Briefly, an isotonic Percoll solution was
prepared by mixing 9 parts Percoll with one part of a 10 fold concentrated Hanks
buffered salt solution (HBSS) containing HEPES (5 µM, pH 7.3). The solution
was then gently mixed with the cells (previously resuspended in 1 mL of wash
buffer) and overlayed with wash buffer (1 mL) to create an interface. The cells
were then centrifuged (140 g, 25 min, 4ºC) and the supernatant and interface cells
were aspirated and discarded, leaving a mast cell pellet, which was then removed
in a minimal volume and washed twice. For flow cytometry analysis, cells were
then resuspended in wash buffer and left to stand on ice until use. For downstream
gene and protein analysis, cells were pelleted and stored in -80ºC.
To assess purity of cells, approximately 100,000 non-purified rat peritoneal
cells as well as purified mast cells were washed with RPMI-1640 media
supplemented with BSA (0.25% v v-1) before they were spun (300 rpm, 10 mins)
onto glass slides using a Shandon cytospin (Thermo Scientific, Victoria,
Australia), and left to air dry overnight. Slides were stained with Wright-Giemsa
stain (0.4% w v-1 in methanol, Sigma-Aldrich) for 1 min, rinsed with distilled water
for 5 min twice and air dried overnight before microscopy (Leica Microsystems
GmbH, Germany). Pictures were taken using an Olympus DP80 microscope
(Olympus, Victoria, Australia) and viewed using Image-Pro Plus software
(MediaCybernetics, Rockville, MD). Several independent fields of view were
imaged to count cells and assess purity and representative images were then
captured.

59
2.2.1 Rat peritoneal macrophage isolation and stimulation
To isolate rat peritoneal macrophages, peritoneal cells obtained from lavage
were centrifuged (300g, 8 mins) and resuspended in pre-warmed medium
(incomplete DMEM supplemented with BSA (0.25% v v-1)). Cells were counted
and approximately 500,000 cells/well were plated onto a 24-well plate. Cells were
incubated for 2 hours (37ºC, 5% CO2) to allow macrophages to adhere to plastic
and non-adherent cells were gently washed off with PBS. Macrophages were then
stimulated with LPS for 4 and 24 hrs, and cell pellets were harvested through
trypsinisation and stored in -80ºC for downstream purposes.
2.3 Flow cytometry (FACS) analysis for intracellular staining of CapG
Intracellular expression of CapG was measured by flow cytometric analysis.
Cells were fixed and permeabilised prior to antibody incubation using a BD
Cytofix/Cytoperm™ Fixation/Permeabilisation solution kit (BD Bioscience,
NSW, Australia). Briefly, cells were fixed and permeabilised in the BD
Cytofix/Cytoperm™ solution (20 mins, 4ºC). The cells were then washed, and
permeabilised with BD Perm/Wash™ buffer. Two hundred thousand cells were
transferred to FACS-compatible tubes (BD Bioscience) and then labelled with
primary antibodies (1 hour, 4ºC), followed by incubation with secondary
biotinylated antibody (1 hour, 4ºC), followed by incubation of streptavidin
conjugated with APC-Cy7™ fluorophore (45 mins, 4ºC) (Table 2.1). All samples
were washed twice with BD Perm/Wash™ buffer between each step.
All FACS analysis was analysed using a BD LSRFortessa™ (BD
Bioscience). For data acquisition, a typical forward scatter (FSC) versus side
scatter (SSC) gate was set to exclude cell debris and aggregates. 2 x 104 cells in a
defined gate were collected and visualised by histogram (log fluorescence vs cell
number). FlowJo software (Tree Star, OR) was used for data analysis.
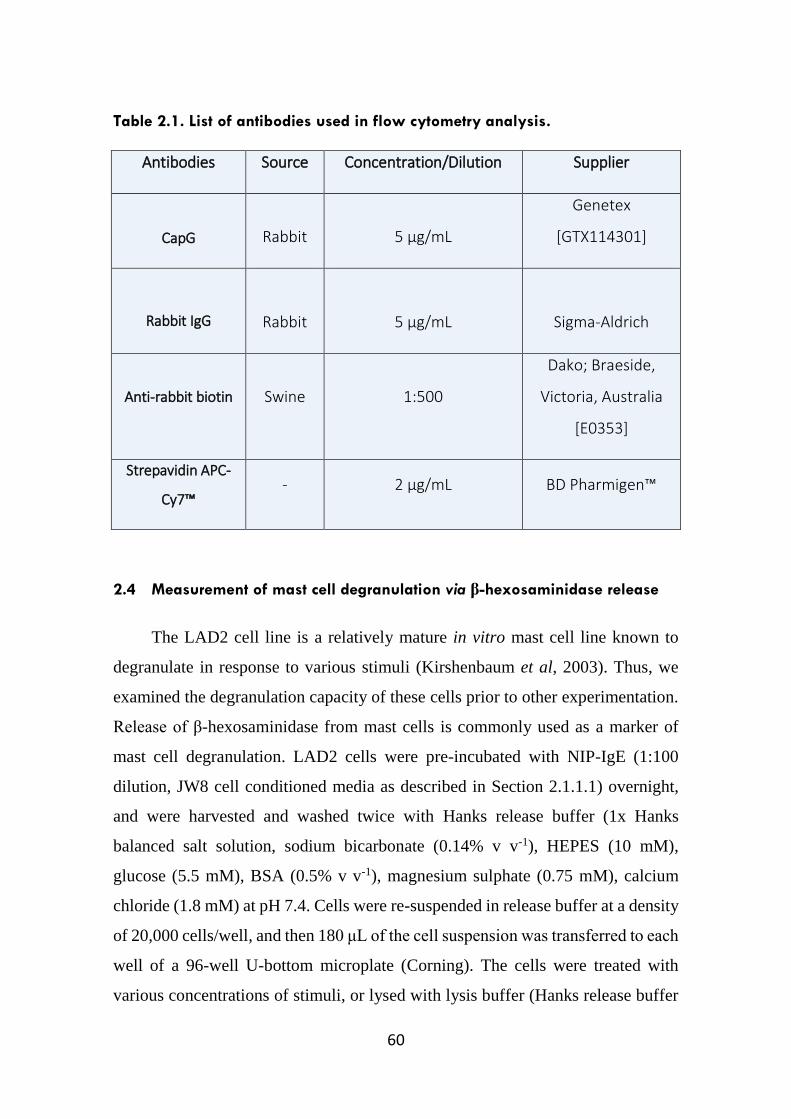
60
Table 2.1. List of antibodies used in flow cytometry analysis.
Antibodies Source Concentration/Dilution Supplier
CapG Rabbit 5 µg/mL
Genetex
[GTX114301]
Rabbit IgG
Rabbit
5 µg/mL
Sigma-Aldrich
Anti-rabbit biotin Swine 1:500
Dako; Braeside,
Victoria, Australia
[E0353]
Strepavidin APC-
Cy7™ - 2 µg/mL BD Pharmigen™
2.4 Measurement of mast cell degranulation via β-hexosaminidase release
The LAD2 cell line is a relatively mature in vitro mast cell line known to
degranulate in response to various stimuli (Kirshenbaum et al, 2003). Thus, we
examined the degranulation capacity of these cells prior to other experimentation.
Release of β-hexosaminidase from mast cells is commonly used as a marker of
mast cell degranulation. LAD2 cells were pre-incubated with NIP-IgE (1:100
dilution, JW8 cell conditioned media as described in Section 2.1.1.1) overnight,
and were harvested and washed twice with Hanks release buffer (1x Hanks
balanced salt solution, sodium bicarbonate (0.14% v v-1), HEPES (10 mM),
glucose (5.5 mM), BSA (0.5% v v-1), magnesium sulphate (0.75 mM), calcium
chloride (1.8 mM) at pH 7.4. Cells were re-suspended in release buffer at a density
of 20,000 cells/well, and then 180 μL of the cell suspension was transferred to each
well of a 96-well U-bottom microplate (Corning). The cells were treated with
various concentrations of stimuli, or lysed with lysis buffer (Hanks release buffer

61
containing 0.1% Triton X (Sigma-Aldrich) in duplicate wells (at 37°C, 45 min)
with gentle mixing (80 rpm), and the plate was then centrifuged (220 g, 5 min) to
sediment cells. 50 μL of supernatants were transferred to a flat bottom 96-well
microplate, and released β-hexosaminidase in supernatants measured by the
addition of 50 μL of the substrate p-nitrophenyl N-acetyl-β-D-glucosaminide
(pNAG, 4 mM made up in citrate-phosphate buffer; pH 4.5; Sigma-Aldrich). After
incubation with substrate (90 mins), the reaction was stopped by the addition of
100 μL of glycine (0.4 M; pH 10.7) to each well. 4-nitrophenol, which is the
product of pNAG cleavage, was detected by absorbance at 405 nm using a
microplate-reader (Multiskan Ascent®; Thermo Scientific). The non-stimulated
samples were assessed to quantify spontaneous release of β-hexosaminidase, and
the total amount of cellular β-hexosaminidase enzyme was obtained from cell lysis
of control wells. After subtracting the values of the spontaneous release from all
samples, the extent of degranulation was calculated as a percentage of the total
amount of cellular β-hexosaminidase.
2.5 Immunofluorescence Microscopy
2.5.1 THP-1 cells
Fifty-thousand THP-1 cells were seeded in an 8-well chamber slide
(LabTek, Brendale, Australia) for 48 hours in the presence of PMA (100 nM). The
cells were then serum-starved overnight prior to LPS-stimulation on the following
day. In addition, some cells were pre-incubated with dexamethasone (100 nM) for
30 minutes prior to LPS stimulation. After 24 hours, cells were fixed in neutral
buffered formalin (10% v v-1) for 15 min and then washed in PBS (3 times), and
then permeabilised with Triton-X (0.1% v v-1) for 15 mins. Following this, the cells
were incubated with blocking buffer (BSA (1% v v-1), Tween-20 (0.1% v v-1;
Sigma-Aldrich) in PBS) for 30 min prior to the addition of primary antibodies,
which were then left overnight at 4ºC. On the following day, the cells were washed
with Tween-20 (0.1% v v-1) in PBS (3 x 5 mins) prior to incubation with a

62
fluorophore-conjugated secondary antibody for 2 hours. After this, cells were then
washed in PBS-containing Tween-20 (3 x 5 mins) and cells were incubated with
DAPI (10 mins). Cells were again washed in PBS prior to the slides being
coverslipped with the aid of a mounting medium (Dako). The slides were then
dried overnight at 4ºC prior to fluoromicroscopy imaging using the Axio Observer
Z1 Microscope (Carl Zeiss AG, Germany). Images were captured using the Zeiss
Microscope imaging software ZEN 2 Pro.
2.6 Measurement of cytokine levels using enzyme-linked immunosorbent
assays (ELISA)
Cytokine levels from harvested supernatants were assayed using
commercially available sandwich ELISA kits (OptEIA; BD Bioscience) in
accordance with the manufacturer’s instructions but with some modifications.
Briefly, capture antibodies were diluted to the recommended concentrations in
coating buffer (carbonate-bicarbonate buffer; pH 9.6). 96-well high binding
ELISA microplates (Corning) were coated with diluted capture antibody overnight
at 4°C. On the following day, the wells were washed three times with wash buffer
(PBS with Tween 20 (0.05% v v-1)), prior to the addition of blocking buffer (PBS
with FBS (10% v v-1)) to block non-specific sites (1 hr, room temperature). The
wells were then washed three times with wash buffer. When human IL-8 or CCL2
cytokine levels were measured, 50 μL of sample (suitably diluted where necessary)
or cytokine standard in corresponding culture medium was added and incubated (3
hr, room temperature or overnight, 4°C). Following incubation, plates were
washed five times with wash buffer and then incubated with a detecting mix
(detection antibody with HRP-conjugated streptavidin, diluted in blocking buffer
at recommended concentrations) for a further hour at room temperature. The
protocol for quantifying human IL-6 cytokine levels was different, in that the
detecting antibody was added alongside the supernatant samples and co-incubated
for 3 hours, and then plates were washed 5 times prior to the 1 hr incubation of
HRP-conjugated streptavidin alone. Following this, plates were washed seven
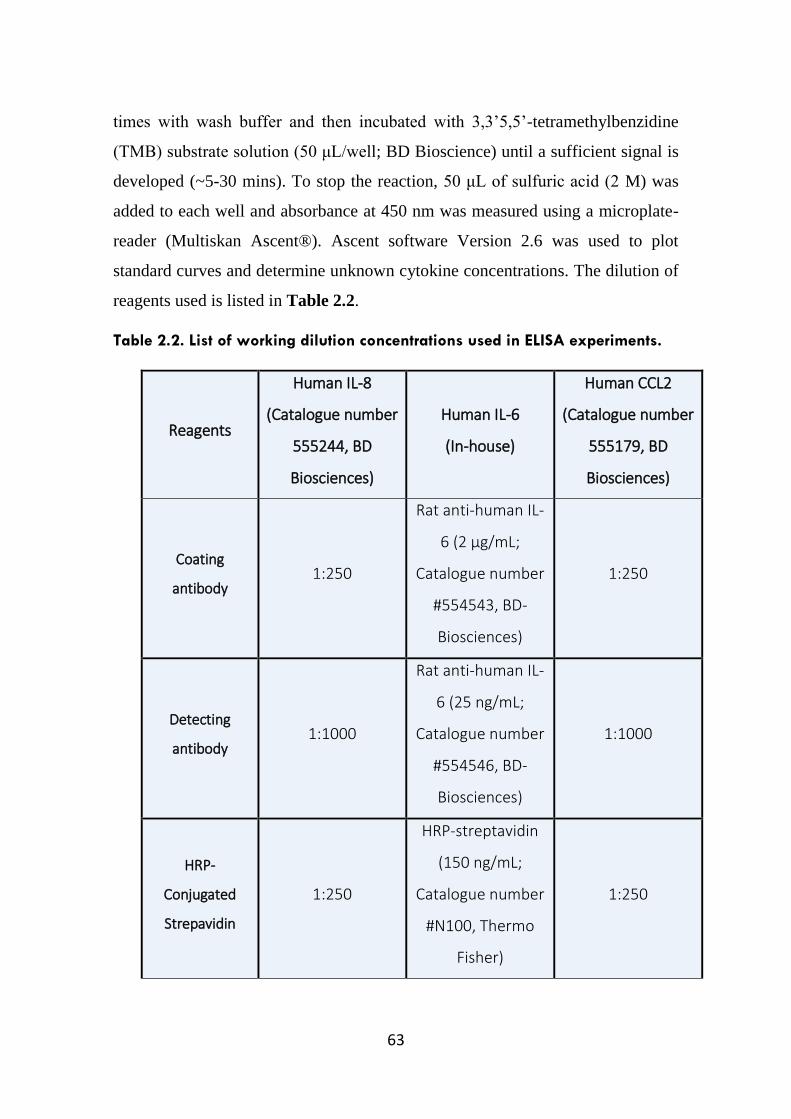
63
times with wash buffer and then incubated with 3,3’5,5’-tetramethylbenzidine
(TMB) substrate solution (50 μL/well; BD Bioscience) until a sufficient signal is
developed (~5-30 mins). To stop the reaction, 50 μL of sulfuric acid (2 M) was
added to each well and absorbance at 450 nm was measured using a microplate-
reader (Multiskan Ascent®). Ascent software Version 2.6 was used to plot
standard curves and determine unknown cytokine concentrations. The dilution of
reagents used is listed in Table 2.2.
Table 2.2. List of working dilution concentrations used in ELISA experiments.
Reagents
Human IL-8
(Catalogue number
555244, BD
Biosciences)
Human IL-6
(In-house)
Human CCL2
(Catalogue number
555179, BD
Biosciences)
Coating
antibody 1:250
Rat anti-human IL-
6 (2 µg/mL;
Catalogue number
#554543, BD-
Biosciences)
1:250
Detecting
antibody 1:1000
Rat anti-human IL-
6 (25 ng/mL;
Catalogue number
#554546, BD-
Biosciences)
1:1000
HRP-
Conjugated
Strepavidin
1:250
HRP-streptavidin
(150 ng/mL;
Catalogue number
#N100, Thermo
Fisher)
1:250

64
2.7 Protein extraction, sample preparation and Bradford protein assay
Cells were harvested and washed twice with ice-cold PBS followed by cell
lysis in reduced SDS buffer (Tris-HCL (0.05M), SDS (0.01% v v-1), glycerol
(0.01% v v-1), dithiothreitol (DTT; 50 mM)). Lysates were passed through a 20-
gauge needle to facilitate shearing of DNA and the lysates were then heated for 3
min in boiling water.
Alternatively, cells were washed twice in ice-cold PBS, and then lysed by
the addition of lysis buffer (Brij 80 (1% v v-1), NaCl (0.15 M), Tris (0.1 M), sodium
orthovanadate (1 mM) and cOmplete, mini EDTA-free protease inhibitor tablet
(0.03% w v-1 in Milli-Q water; Roche) for 15 min on ice. The lysates were collected
and clarified by centrifugation (10,000g for 10 min at 4°C). Protein concentrations
were determined by Bradford Protein Assay (Section 2.7.1). Protein lysates were
prepared for SDS-PAGE gel electrophoresis by addition of an appropriately
concentrated reducing SDS sample buffer (as above) and heated for 3 min in
boiling water. Lysates that were not processed immediately were stored at -20°C.
2.7.1 Bradford Assay
The Bradford protein assay was used to quantify total protein levels from cell
lysates as described above (Bradford, 1976). A protein standard curve was
constructed using BSA, these being prepared in duplicate from a stock solution of
1 mg/mL BSA dissolved in PBS. Protein samples were made up to 100 μL with
PBS and a NaOH solution (0.2 M, 100 µL) was then added to both unknown
samples and standards. Both samples and standards were mixed and proteins were
solubilised for 15 min prior to the addition of Milli-Q H2O (600 µL). Bio-Rad
protein assay dye reagent (200 μL; Bio-Rad Laboratories, NSW, Australia) was
then added to all the samples. The samples were mixed and gently vortexed before
aliquots of 200 μL were transferred to a 96-well microplate prior to measuring the
absorbance at 595 nm using a microplate-reader (Multiskan Ascent®). Ascent

65
software version 2.6 was used to plot standard curves and determine the unknown
sample concentrations.
2.8 SDS-PAGE Gel Electrophoresis
2.8.1 Western Blotting
Protein samples prepared in sample buffer were separated by standard SDS-
polyacrylamide gel electrophoresis (SDS-PAGE; 10%-12% acrylamide) under
reducing and denaturing conditions. The resolved proteins were then transferred
onto either nitrocellulose (Bio-Rad Laboratories) or Hybond™ polyvinylidene
difluoride (PVDF) membranes (Thermo Scientific). The transfer process was
performed in transfer buffer (methanol (30% v v-1), Tris (25 mM), glycine (192
mM)) for 1 hour. Following wet transfer, the nitrocellulose or PVDF membranes
were stained with Ponceau-S dye to detect protein bands on membranes. The
membranes were then washed in Tris-buffered saline (TBS) with Tween20 (0.05%
v v-1; TBST) to remove the dye, and then blocked in skimmed milk protein (5% w
v-1) dissolved in TBST for 1 hr at room temperature. Following washing (3 x 5
min) with TBST the membranes were probed with various primary antibodies
overnight at 4⁰C (Table 2.3). Subsequently, the membranes were washed (4 x 5
min) in TBST prior to incubation with the appropriate horseradish peroxidase
(HRP)–conjugated secondary antibody at room temperature for 2 hrs. Following
washing (4 x 5 min), immunoreactive proteins were visualised by enhanced
chemiluminescence (Clarity™ Western ECL; Bio-Rad Laboratories) using a
ChemiDoc™ MP System (Bio-Rad Laboratories). Images were captured using the
Bio-Rad Image Lab™ Software.
2.8.2 Coomassie staining of SDS-PAGE gels
To visualise protein bands following gel electrophoresis, gels were stained
overnight in Coomassie Blue dye (Coomassie Blue R-250 (0.25% w v-1) in
methanol (50% v v-1), Milli-Q water (40% v v-1) and glacial acetic acid (10% v v-
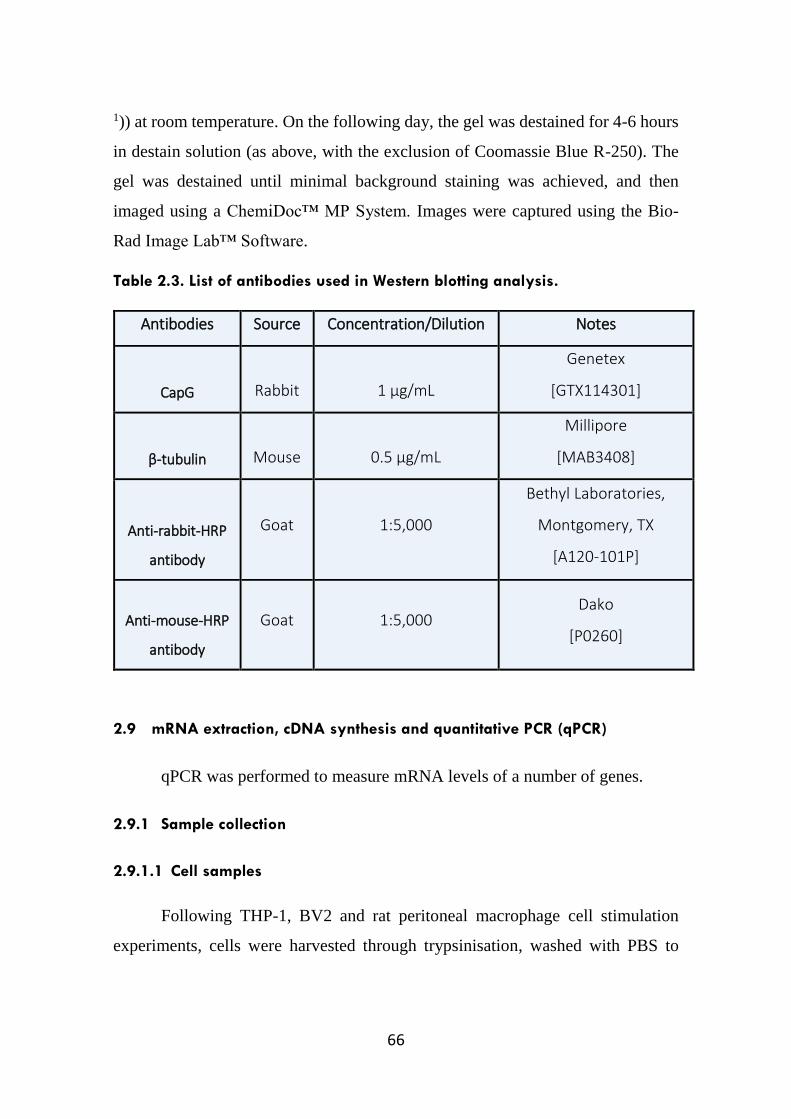
66
1)) at room temperature. On the following day, the gel was destained for 4-6 hours
in destain solution (as above, with the exclusion of Coomassie Blue R-250). The
gel was destained until minimal background staining was achieved, and then
imaged using a ChemiDoc™ MP System. Images were captured using the Bio-
Rad Image Lab™ Software.
Table 2.3. List of antibodies used in Western blotting analysis.
Antibodies Source Concentration/Dilution Notes
CapG
Rabbit
1 µg/mL
Genetex
[GTX114301]
β-tubulin
Mouse
0.5 µg/mL
Millipore
[MAB3408]
Anti-rabbit-HRP
antibody
Goat 1:5,000
Bethyl Laboratories,
Montgomery, TX
[A120-101P]
Anti-mouse-HRP
antibody
Goat 1:5,000 Dako
[P0260]
2.9 mRNA extraction, cDNA synthesis and quantitative PCR (qPCR)
qPCR was performed to measure mRNA levels of a number of genes.
2.9.1 Sample collection
2.9.1.1 Cell samples
Following THP-1, BV2 and rat peritoneal macrophage cell stimulation
experiments, cells were harvested through trypsinisation, washed with PBS to

67
remove residual media and then pelleted (300 g, 5 mins, 4ºC) prior to storage in -
80ºC for downstream gene analysis.
2.9.1.2 Mouse models
2.9.1.2.1 LPS and Respiratory Syncytial Virus (RSV) models
C57BL/6 mice (used with the approval of the University of Melbourne
animal ethics committee, ethics codes: 1312919 and 1212356) were intranasally
inoculated with LPS (10 μg/kg) or RSV (Strain A2, ATCC; 2 x 106 virions/mouse)
under isoflurane anaesthesia. 24 hours after LPS treatment, or 5 days after RSV
treatment, mice were killed by pentobarbitone (150 mg/kg, Provet, Australia). The
harvest of lung and bronchoalveolar lavage fluid (BALF) was performed by Ms.
Shenna Langenbach (Department of Pharmacology and Therapeutics, University
of Melbourne).
2.9.1.2.2 APPSWE/PS-1ΔE9 (APP/PS-1) model
C57BL/6 APP/PS-1 mice (used with the approval of the University of
Melourne animal ethics committee, ethics code: 1312746) were aged for up to 13
months before experimental use. Mice were fed ad-libitum on a standard dry-chow
diet and had open access to water. The APP/PS-1 mouse is commonly used as a
model of amyloidosis in AD, expressing mutations that provide relevance to the
human disease setting (Liu et al, 2008). Mice were killed by cervical dislocation
and brain tissue was harvested and snap frozen in liquid nitrogen. This process was
conducted by Dr. Myles Minter (Department of Pharmacology and Therapeutics,
University of Melbourne).
2.9.1.3 Human monocytes
mRNA samples of stimulated human blood monocytes in the presence and
absence of macrophage-colony stimulating factor (M-CSF) were kindly provided
by Prof. Alastair Stewart (Department of Pharmacology and Therapeutics,

68
University of Melbourne). Briefly, human monocytes were separated from
peripheral blood mononuclear cells by adherence. Monocytes were differentiated
to a macrophage phenotype by culturing in M-CSF (10 ng/mL) for 7 days. M-CSF
treated or non-treated cells were then activated with various stimuli either
individually, or in combination for 24 hours. Cell samples were stored at -80ºC for
downstream gene analysis.
2.9.2 mRNA extraction
mRNA was extracted from samples using an ISOLATE II RNA Mini Kit
according to manufacturer’s protocol (Bioline, NSW, Australia). Messenger RNA
was quantified and purity assessed using a NanoDrop™ 2000 spectrophotometer
with measurements made at 260/280 nm (Thermo Scientific).
2.9.3 cDNA synthesis
The purified total RNA was used as a template to generate first-strand
cDNA by reverse transcription (RT) using a High-Capacity RNA-to-cDNA kit
(Life Technologies). A reaction mix was generated containing RNA (0.2 µg),
reverse transcription buffer mix (2x, 2.5 µL), RT enzyme mix (20x, 0.25 µL) (Life
Technologies) in a final volume of 5 µL. Reverse transcription was performed
using a thermocycler (Mastercycler® Pro, Eppendorf) using the following
conditions: 37⁰C/60 min, 95⁰C/5 min, 4⁰C/hold until collection. The resulting
cDNA was diluted in 1:20 with sterile RNAse free water and stored at -20⁰C until
use.
2.9.4 Quantitative real-time PCR (qPCR)
For measuring the expression of human and rat genes, TaqMan® primer
sequences were purchased as recommended from Life Technologies. For the
measurement of mouse genes, gene specific forward and reverse KicQStart®
SYBR® Green primers were purchased from Sigma-Aldrich (Table 2.4).

69
An ABI Prism 7900HT sequence detection system (Life Technologies) was
used to quantitatively analyse the level of gene expression in the samples tested.
All reactions were conducted in 384 well plates (Life Technologies). For
measuring gene expression levels in human and rat samples, each reaction (5 µL)
contains diluted cDNA (2 µL); TaqMan® Fast Advanced Master Mix (3 µL;
Invitrogen) that included gene-specific TaqMan® Gene Expression Assays
primers (0.2 µM; Invitrogen). The amplification conditions for real time PCR
were: 50⁰C/2 min for uracil-N-glycosylase (UNG) incubation to prevent carryover
contamination between reactions, 95⁰C/20 seconds to activate the DNA
polymerase, which was then followed by 40 cycles of denaturation at 95⁰C/1
second and annealing/extension at 60⁰C/20 seconds.
In experiments measuring gene expression from mouse samples, each
reaction (5 μL) contained diluted cDNA (1.5 μL); SYBR Green Master Mix (3.5
μL; Invitrogen) including gene-specific forward and reverse primers (10 μM;
Sigma). The amplification conditions for real time PCR using the SYBR® Green
Master Mix were: 50⁰C/2 min for uracil-N-glycosylase (UNG) incubation,
95⁰C/10 min to activate the DNA polymerase, which was then followed by 40
cycles of denaturation at 95⁰C/15 secs and annealing/extension at 60⁰C/1 min.
In all gene expression studies, a suitable housekeeping gene (usually
ubiquitin C (UBC)) was used as a control gene for measuring fold change of the
gene of interest in different treatment groups.
The software SDS 2.1 (Applied Biosystems) was used to generate threshold
cycle (Ct) values. Messenger RNA expression levels were expressed as a relative
expression of the target gene against the housekeeper gene such as Ubiquitin C
(UBC). Firstly, ΔCt was calculated by subtracting the Ct value of target gene from
Ct value of UBC. ΔΔCt was calculated as the Ct change relative to the non-treated
control group, where the ΔCt value of treated group is subtracted from ΔCt of the
control group. The relative fold expression of the target genes was then calculated
as 2-ΔΔCt.

70
2.10 Recombinant expression of CapG in HEK and COS cells
2.10.1 Flp-InTM-293 and COS cells
Flp-InTM-293 and COS cells were transfected with a codon-optimised full
length cDNA of CapG cloned into pcDNA5 vector (GenScript, Piscataway, NJ).
Stable transfections were performed by the co-transfection of CapG and Flp-
recombinase (pOG44) expression vectors using FuGENE® HD (Promega,
Victoria, Australia). Transfected cells were selected and maintained in DMEM
(HEK Flp-In 293 cells) or RPMI (COS-7 cells with no Flp-In sites) containing FCS
(10% v v-1) and hygromycin (100 µg/mL; Life Technologies). A detailed protocol
for transfection, protein expression and supernatant harvest is described in Section
5.2.2.1.
2.10.2 EBNA293 cells
EBNA-293 cells were transfected with the full length cDNA of the CapG
sequence cloned into pCEP-Pu vector (kindly provided by Dr. Amanda Gavin,
(The Scripps Institute, San Diego, CA)) (Van Craenenbroeck et al, 2000). Cells
were transfected using Lipofectamine® transfection reagent (Life Technologies)
in accordance with the manufacturer’s instruction. A detailed protocol for
transfection, protein expression and supernatant harvest is described in Section
5.2.2.2.
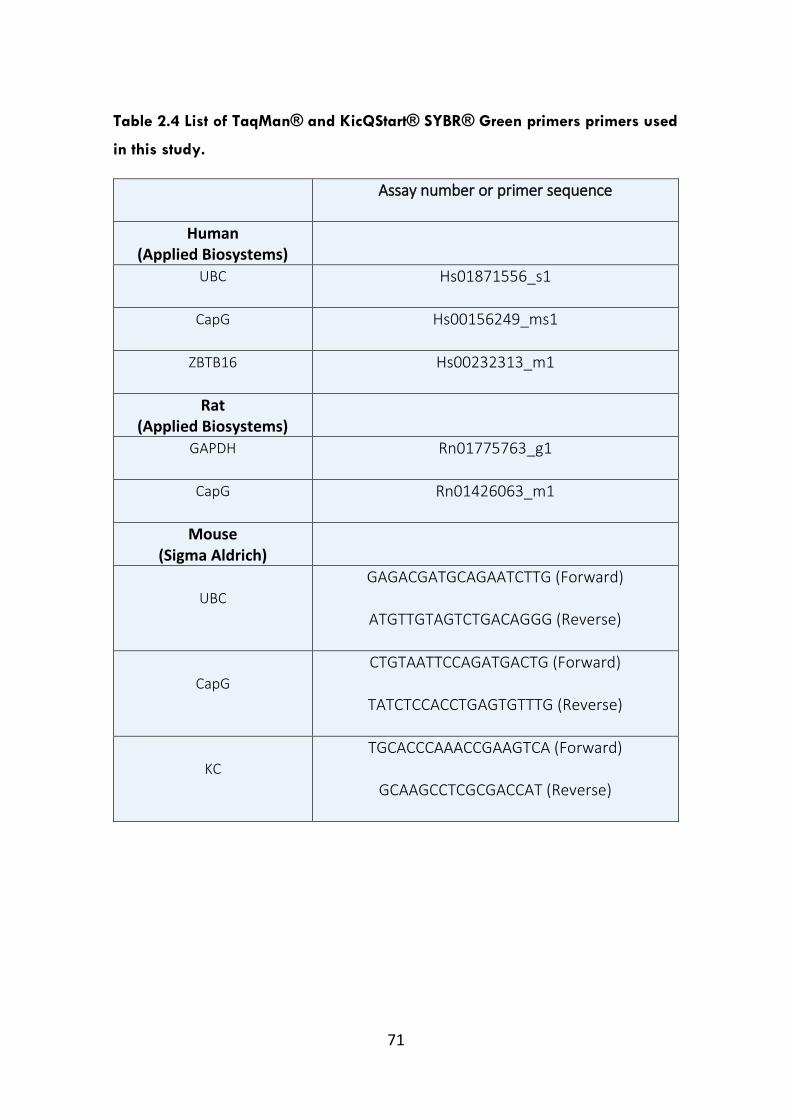
71
Table 2.4 List of TaqMan® and KicQStart® SYBR® Green primers primers used
in this study.
Assay number or primer sequence
Human (Applied Biosystems)
UBC Hs01871556_s1
CapG Hs00156249_ms1
ZBTB16 Hs00232313_m1
Rat (Applied Biosystems)
GAPDH Rn01775763_g1
CapG Rn01426063_m1
Mouse (Sigma Aldrich)
UBC
GAGACGATGCAGAATCTTG (Forward)
ATGTTGTAGTCTGACAGGG (Reverse)
CapG
CTGTAATTCCAGATGACTG (Forward)
TATCTCCACCTGAGTGTTTG (Reverse)
KC TGCACCCAAACCGAAGTCA (Forward)
GCAAGCCTCGCGACCAT (Reverse)

72
2.11 Purification of CapG
The transfection experiments performed on the Flp-InTM-293 cells results in
recombinant CapG protein expressing a Strep-tag at the C-terminus. In contrast,
transfection studies performed on EBNA293 cells will yield an alternate
recombinant CapG expressing both a His-tag and a Strep tag at the N and C-
terminus respectively. The presence of these tags allows expressed CapG to be
purified using two different purification systems: Strep-Tactin® resin (IBA,
Göttingen, Germany) that binds to the Strep-tag section of CapG, and the
HisTALON™ cobalt resin (Clontech, Mountain View, CA) that binds to the His-
tag sequence on CapG. Both Strep-Tactin® and HisTALON™ purification
methods are described in more detail in Sections 5.2.4.1 and 5.2.4.2, respectively.
The concentration of all eluted fractions were determined using a NanoDrop™
2000 spectrophotometer with protein absorbance measurements made at 280 nm.
2.12 Concentration and dialysis of purified CapG
Eluted fractions containing recombinant CapG, as determined by
NanoDrop™ 2000 spectrophotometer measurements, were pooled and then
concentrated approximately 10 fold using a 10 kDa cut-off centrifugal concentrator
(Amicon® Ultra 4 mL filters, Millipore), and was quantified and later transferred
into dialysis tubing (Spectrum Laboratories, Victoria, Australia) as described in
Section 5.2.5.1 and 5.2.5.2. Finally, the recombinant CapG was visualised by
Coomassie Blue staining following gel electrophoresis, as described in Section
2.8.2.
2.13 Statistical analysis
In Chapters 3 and 6, where applicable, data from ELISA and Western
blotting analysis are expressed as the means ± standard error of mean (SEM),
where n represents the number of independent primary cell cultures used or
numbers of experiments repeated using cell line. Differences between treatments

73
were determined by analysis of variance (ANOVA) followed by Dunnett’s post-
hoc test for comparisons. One-way repeated measures ANOVA was used for
comparison between three or more groups. Two way ANOVA followed by
Bonferroni’s post-hoc test was used to analyse data when responses were
influenced by two categorised factors of interest.
In Chapter 4, data from qPCR analysis were expressed as means ± SEM,
where n represents the number of independent primary cell cultures, mouse or
patient samples, or numbers of independent experiments repeated using cell line.
A one-sampled t-test was applied to compare significance of fold change between
control and treatment groups.
Results shown were plotted using Graphpad Prism software (version 6.01).
If a statistical significance was obtained, then * denotes p<0.05, ** denotes p<0.01,
and *** denotes p<0.001.

74

75
Chapter 3
Cellular expression and release of Macrophage Capping Protein
(CapG) - a potential pro-inflammatory mediator?

76
3.1 Introduction
Mast cells are one of the key inflammatory cells of the immune system.
These cells can be activated by a range of diverse receptors including
immunoreceptors such as the high affinity IgE-receptor (FcεRI), pathogen
recognition receptors toll-like receptors (TLRs) and nod-like receptors (NLRs) (St
John & Abraham, 2013). Mast cell activation by antigen crosslinking of IgE/FcεRI
is well established and plays a key role in diseases such as Type I hypersensitivity
reactions including allergic asthma (Sibilano et al, 2014). Asthma is a
multifactorial airway disease that is usually characterised by mucus
hypersecretion, airway obstruction and hyperreactivity and remodeling. This
chronic airway inflammation is thought to be orchestrated by a host of different
cytokines and chemokines secreted by mast cells and other inflammatory
leukocytes such as T cells, eosinophils, basophils and macrophages as reviewed in
Chapter 1. In addition, studies have also shown that airway structural cells such as
airway epithelial and smooth muscle cells release pro-inflammatory mediators that
exacerbate and may indeed trigger the disease condition (Balhara & Gounni, 2012;
John et al, 1998a; Lloyd & Hessel, 2010; Reibman et al, 2003).
A hallmark feature of allergic asthma is the infiltration of mast cells into
the airway smooth muscle bundles (Bradding & Brightling, 2007). When
activated, mast cells release a variety of mediators that bind to their respective
receptors present on neighbouring cells, initiating an inflammatory cascade as
described in Chapter 1. In addition, alveolar macrophages also play a role in
asthma pathology as these cells are able to release mediators that can further
exacerbate the disease pathology (Balhara & Gounni, 2012). These pro-
inflammatory cytokines promote airway remodelling and hyperresponsiveness
whilst chemokines promote the recruitment of inflammatory cells to the airways
(Hart, 2001). In addition, anti-inflammatory cytokines such as IL-10, IL-12 and
IFNγ are also reportedly decreased in asthmatic patients (Chung, 2001).

77
Current mainstay therapies in asthma such as glucocorticoids and β2-
adrenoceptor agonists are relatively successful in alleviating asthma symptoms.
However, they possess several undesirable side effects (Dahl, 2006). Although
glucocorticoids are an effective treatment due to their anti-inflammatory
properties, prolonged use of high-dose levels have serious systemic side effects
including impaired growth in children, decreased bone-mineral density, skin
thining and bruising (Dahl, 2006). Short acting and long acting β-adrenoceptor
agonists such as albuterol and salmeterol, respectively, are known to provide
immediate symptom relief. However, it has been shown that prolonged use of these
treatments, in the absence of glucocorticoids, can lead to loss of asthma control
such as increased airway-hyperresponsiveness and in some cases be potentially
fatal (Xia et al, 2013a).
Given the strong evidence of cytokine-driven pathologies in asthma, there
is growing interest in the generation of monoclonal antibodies (mAbs) targeting
specific cytokines or receptors as an alternative treatment to current mainstay
therapies. To date, the most established monoclonal antibody therapy used in
asthma treatment is omalizumab, which binds to the FcεRI binding site of plasma
IgE thereby preventing IgE binding to FcεRI present on mast cell and basophils.
As a result, the IgE/FcεRI interaction is neutralised and prevents mast cell
activation. In addition, omalizumab, by reducing circulating free IgE, also
downregulates FcεRI expression on basophils and mast cells (Hamilton et al, 2005;
Prussin et al, 2003). Indeed, omalizumab provides both short and long term
benefits for severe allergic asthma patients as it reduces the clinical symptoms
associated with asthma, such as unprovoked exacerbations and patient quality of
life is hence improved (Abraham et al, 2016; Humbert et al, 2005). Currently, the
GINA guideline recommends omalizumab as an add-on therapy alongside β-
adrenoceptor agonists and inhaled glucorticosteroids for treating severe asthma
(Horak et al, 2016). Most recently, omalizumab was also approved as a therapeutic
agent for patients with chronic urticaria, where it targets mast cell activation and

78
thus prevents mediator release, which has been long associated with urticaria
pathogenesis (Maurer et al, 2013; Wu & Jabbar-Lopez, 2015).
Following on from the success of omalizumab as an additive therapy in
allergic asthma, several other mAbs have also been trialled clinically as an
alternate treatment to relieve asthmatic symptoms. However, the therapeutic
effects in clinical trials have been mixed: The anti-IL-5 humanised mAb
mepolizumab has been shown to be clinically effective primarily in patients with
severe eosinophilic asthma (Menzella et al, 2015; Ortega et al, 2014). In addition,
the anti-IL-4 receptor monoclonal antibody dupilumab was found to improve
clinical responses in patients with moderate to severe asthma (Beck et al, 2014;
Thaci et al, 2016; Wenzel et al, 2013). In contrast, other trials investigating other
mAb therapies showed promising signs in in vitro assays and animal models but
did not demonstrate clinical efficacy such as MEDI-528 (anti-IL-9 mAb) and
AMG-317 (anti-IL-4 receptor antagonist) (Corren et al, 2010; Oh et al, 2013).
A likely explanation to the limited clinical efficacies of monoclonal
antibodies in asthma treatment is due to the complexity and spectrum of the
disease. Genetic and environmental factors influence the disease pathology
(Holgate et al, 2007). As a result these different factors contribute to different
“endotypes” of asthma, a terminology used to define “a subtype of a condition,
which is defined by a distinct functional or pathophysiological mechanism”
(Lotvall et al, 2011). Among these asthma endotypes are allergic
bronchopulmonary mycosis, eosinophil or neutrophil-driven asthma, aspirin-
exacerbated respiratory disease and many more (Lotvall et al, 2011; Wesolowska-
Andersen & Seibold, 2015). The ability to distinguish these distinct asthma
endotypes is important in terms of more effective and personalised treatments for
patients in the future (Skloot, 2016). Currently, there is a still an unmet medical
need in asthma and thus novel therapeutic agents that target mediators involved in
the underlying immune dysfunction in asthma are still keenly sought after.

79
Although many mediators have been identified to play a role in asthma,
novel mediators are also being reported to be released from activated mast cells
with roles yet to be elucidated (Xia et al, 2013b). In this study, it was discovered
that antigen/IgE mediated activation of the FcεRIα-subunit transfected human
mast cell (HMCα) cells produced cellular secretion of soluble factor/factors that in
turn induced pro-inflammatory cytokine release from human airway smooth
muscle cells (hASM). This effect was not attributed to the two predominant
cytokines released from HMCα cells (CCL2 and MIP-1β) as determined by a 17-
human inflammatory cytokine Bio-Plex assay. Therefore, an activity based-
proteomics approach was performed to identify novel factors secreted from HMCα
upon stimulation. Through mass spectrometry analysis, six soluble proteins were
identified and later examined for their ability to recapitulate the initial findings. Of
these, only macrophage capping protein (CapG) was shown to stimulate IL-8 and
IL-6 release from hASM cells (Xia et al, unpublished) (Figure 3.1a).
CapG (also known as gCap39) is a member of the gelsolin family, known
for regulating actin polymerisation. Cytoskeletal rearrangement is crucial in many
intracellular processes including cell division and motility. During actin assembly,
the monomeric actin, G-actin (globular-actin) polymerizes with other monomers,
creating a long helical double-stranded polymer known as F-actin (filamentous-
actin) (Figure 3.1b). The gelsolin superfamily of proteins are key regulators of
this polymerization process. All members of the gelsolin superfamily are able to
bind and cap actin filaments. However, CapG is the only member that does not
sever the filaments. This is likely explained by the divergence of CapG from other
family members resulting in a loss of amino acid sequences (84-91 and 124-128).
Indeed, restoration of these amino acids using chimeric approaches resulted in a
gain in actin-severing function by CapG, thus highlighting the importance of this
region for actin severing (Zhang et al, 2006). CapG is also highly conserved
between species, as humans share a 93% sequence identity with rats and 91%
sequence similarity with mouse (Figure 3.2).

80
CapG is highly expressed in macrophages as it represents 1% of total
cytoplasmic protein (Witke et al, 2001). Macrophages are originally derived from
monocytes that originate from the bone marrow. The monocytes enter the
bloodstream and can migrate to peripheral tissues, where under the influence of
local growth factors and pro-inflammatory mediators, they differentiate into
macrophages (Italiani & Boraschi, 2014; Shi & Pamer, 2011). Macrophages, or
related cells, reside in every tissue of the body, in different guises including
microglia (brain), Kupffer cells (liver) and osteoclasts (bone). These cells can be
distinguished into different subtypes depending on mediator or pathogen
influences: Interferon-γ (IFN-γ) or lipopolysaccharide (LPS) drive the
macrophages towards the classically activated M1 phenotype which mediate
defence against foreign pathogens, whilst the interleukins (IL)-4 and 13 polarise
the macrophages towards the alternatively activated M2 phenotype which have
anti-inflammatory functions and regulate wound healing (Murray & Wynn,
2011b). Other subsets of macrophages include regulatory macrophages, tumour-
associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs)
(Murray & Wynn, 2011b).
The strategic location of mature macrophages in tissues allow these cells to
perform important immune surveillance activities which include phagocytosis and
antigen presentation. Phagocytosis is defined as the process that is used by cells to
internalize large particles including debris, apoptotic cells and pathogens (Murray
& Wynn, 2011b). This enables macrophages to process the foreign material and
present this processed antigen to neighbouring T cells, which then initiates an
immune response to target the removal of the specific antigen (Murray & Wynn,
2011b). There are distinct modes of phagocytosis employed by macrophages to
remove different pathogens. For example, the surface proteins internalins present
on Listeria monocytogenes bacteria binds to receptors on macrophages, resulting
in tyrosine phosphorylation that induces cytoskeleton rearrangement in
macrophages to phagocytose the bacteria (Cossart & Lecuit, 1998). In contrast,

81
contact between the Salmonella enterica bacteria and macrophage induces
cytoskeleton rearrangement in macrophages that internalises the bacteria in a non-
specific mechanism, which is similar to macropinocytosis (Hayward & Koronakis,
1999). The differences in pathogen clearance and the role of CapG in phagocytosis
were evident in CapG-knockout mice, as macrophages from these animals showed
an impaired clearance of Listeria bacteria but not Salmonella bacteria (Parikh et
al, 2003). In addition, loss of CapG in macrophages also resulted in impairments
of IgG and complement-mediated phagocytosis and vesicle rocketing (Witke et al,
2001).
CapG is also expressed in other immune cells such as neutrophils, where it
was demonstrated to be involved in neutrophil chemotaxis as neutrophils in CapG-
knockout mice showed slower migration towards a chemotactic gradient, which in
turn may impair its ability to clear Listeria pathogens in the liver of knockout mice
(Parikh et al, 2003).
Aside from macrophages, CapG is also reportedly overexpressed in the
nucleus and cytoplasm of several types of cancer, including ovarian, hepatocellular
and lung adenocarcinomas (Glaser et al, 2014; Kimura et al, 2013; Shao et al,
2011). Others have found that high CapG expression is related to increased tumour
size and targeting CapG by nanobody injection, or siRNA mediated knockdown
studies resulted in reduced cancer cell motility and invasion (Glaser et al, 2014;
Thompson et al, 2007; Van Impe et al, 2013). In addition, a study shows an
association between CapG and non-small cell lung cancer, as well as a likely role
in cigarette smoke-induced carcinogenesis (Zhu et al, 2012).
In contrast to its intracellular roles described above, the possibility of CapG
release occurring and its significance has been considered less. Like the founding
member gelsolin, CapG can be detected in the plasma (Johnston et al, 1990).
Although most proteins targeted for secretion typically contains a signal peptide
sequence on the N-terminus of the precursor protein (Martoglio, 2003), this was

82
not found in the primary structure of CapG and both secreted and intracellular
CapG were of identical size (Johnston et al, 1990).
Plasma gelsolin has long been established as a part of the actin-scavenger
system that is crucial in clearing plasma actin to prevent clotting by
depolymerising actin filaments and promoting its clearance from the circulation.
It is possible that extracellular CapG may function to clear actin from the
extracellular environment through a non-actin severing mechanism (Johnston et
al, 1990). However, the non-canonical pathway targeting CapG for release is
reminiscent of DAMPs such as IL-1β and together with our own earlier data
suggest it may potentially have a pro-inflammatory role.
This chapter describes the investigation of CapG as a potential pro-
inflammatory mediator, examining its expression and release from mast cells and
macrophages in vitro.
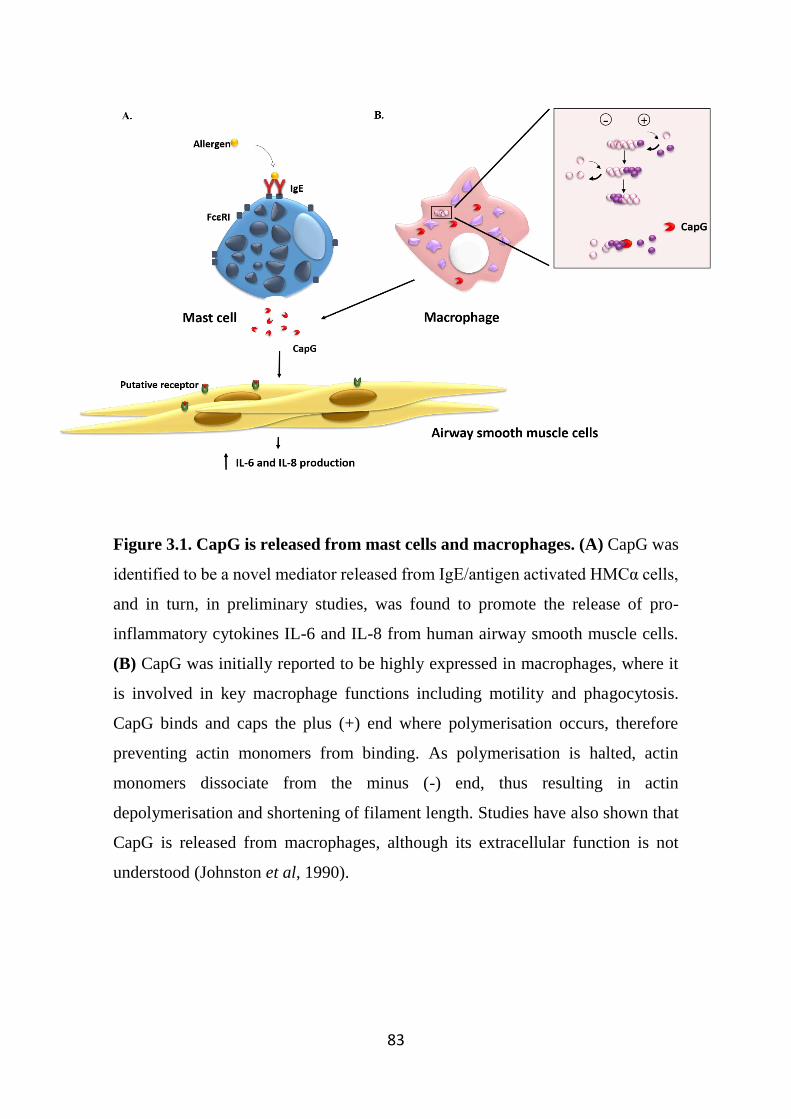
83
Figure 3.1. CapG is released from mast cells and macrophages. (A) CapG was
identified to be a novel mediator released from IgE/antigen activated HMCα cells,
and in turn, in preliminary studies, was found to promote the release of pro-
inflammatory cytokines IL-6 and IL-8 from human airway smooth muscle cells.
(B) CapG was initially reported to be highly expressed in macrophages, where it
is involved in key macrophage functions including motility and phagocytosis.
CapG binds and caps the plus (+) end where polymerisation occurs, therefore
preventing actin monomers from binding. As polymerisation is halted, actin
monomers dissociate from the minus (-) end, thus resulting in actin
depolymerisation and shortening of filament length. Studies have also shown that
CapG is released from macrophages, although its extracellular function is not
understood (Johnston et al, 1990).

84
Figure 3.2. CapG protein is highly conserved between species. CapG sequences
from human, rat and mouse were compared and aligned using the Basic Local
Alignment Search Tool (BLAST) accessed from the National Centre for
Biotechnology Information (NCBI). There is a 93% sequence identity shared
between humans and rat, and a 91% sequence identity between humans and mouse.

85
3.2 Specific Methods
3.2.1 Cell culture
The cells utilised for stimulation studies included the human mast cell lines
HMCα (HMC-1 cells transfected with the α-subunit of FcεRI) and LAD2, and the
human monocytic cell line THP-1. The origins and culture of these cells were
described in Section 2.1.
3.2.2 Rat peritoneal cell (RPC) collection and isolation of rat peritoneal
macrophages and mast cell (RPMC)
All animal work was approved by the University of Melbourne animal
ethics committee (Ethics code: 1513746). Rat peritoneal cell harvest and
collection, as well as isolation of rat peritoneal macrophages and mast cells, was
as described in Section 2.2.
3.2.3 mRNA extraction, cDNA synthesis and qPCR
Cell pellets were lysed and RNA extracted (Section 2.9.1). The extracted
RNA was reverse transcribed into cDNA (Section 2.9.2). Briefly, cDNA was
synthesised from 200 ng of total RNA with the high-capacity RNA-to-cDNA kit
(Life Technologies) according to the manufacturer’s instructions. The cDNA
synthesised was used for gene expression analysis. qPCR analysis of CapG gene
expression was performed on the cDNA using the methods described in Section
2.9.3 (see Table 2.3 for the primers utilised).
3.2.4 Western blot analysis
3.2.4.1 Intracellular CapG expression in different cell types
A range of cell types were assessed for intracellular CapG expression
(Table 3.1). Cell pellets were lysed in lysis buffer and the protein concentration
determined by a Bradford protein assay as described in Section 2.7. The samples
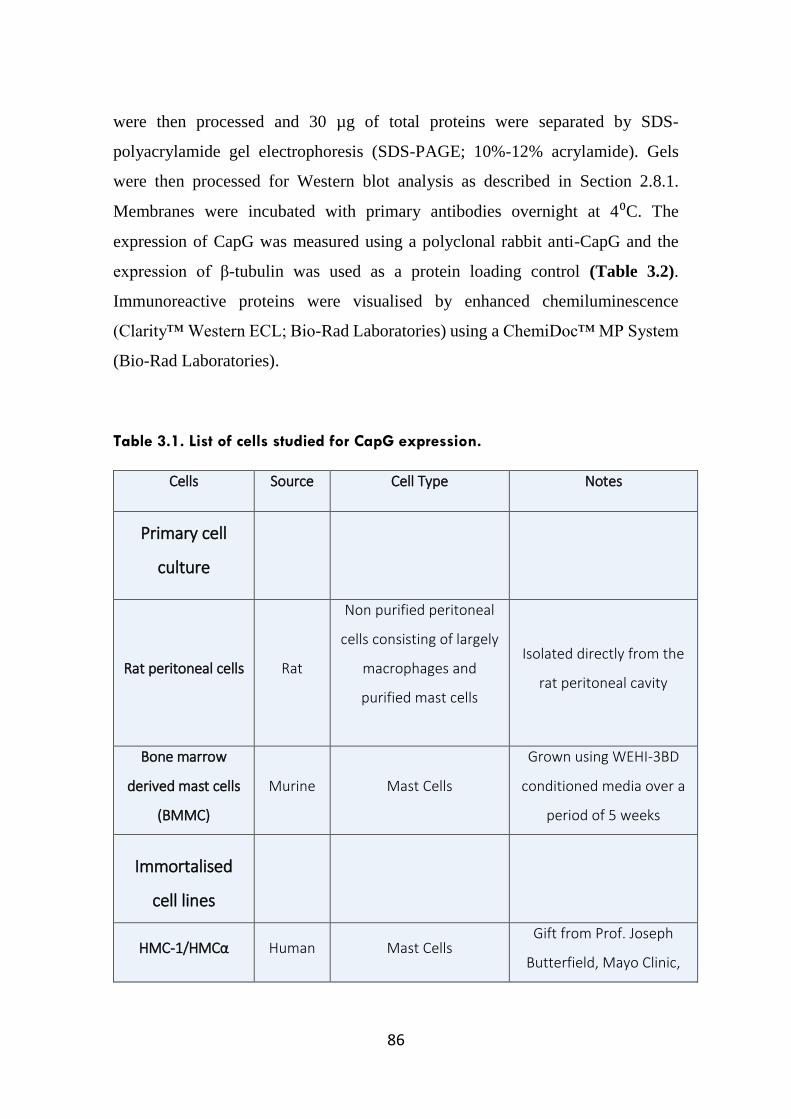
86
were then processed and 30 µg of total proteins were separated by SDS-
polyacrylamide gel electrophoresis (SDS-PAGE; 10%-12% acrylamide). Gels
were then processed for Western blot analysis as described in Section 2.8.1.
Membranes were incubated with primary antibodies overnight at 4⁰C. The
expression of CapG was measured using a polyclonal rabbit anti-CapG and the
expression of β-tubulin was used as a protein loading control (Table 3.2).
Immunoreactive proteins were visualised by enhanced chemiluminescence
(Clarity™ Western ECL; Bio-Rad Laboratories) using a ChemiDoc™ MP System
(Bio-Rad Laboratories).
Table 3.1. List of cells studied for CapG expression.
Cells Source Cell Type Notes
Primary cell
culture
Rat peritoneal cells Rat
Non purified peritoneal
cells consisting of largely
macrophages and
purified mast cells
Isolated directly from the
rat peritoneal cavity
Bone marrow
derived mast cells
(BMMC)
Murine Mast Cells
Grown using WEHI-3BD
conditioned media over a
period of 5 weeks
Immortalised
cell lines
HMC-1/HMCα Human Mast Cells Gift from Prof. Joseph
Butterfield, Mayo Clinic,
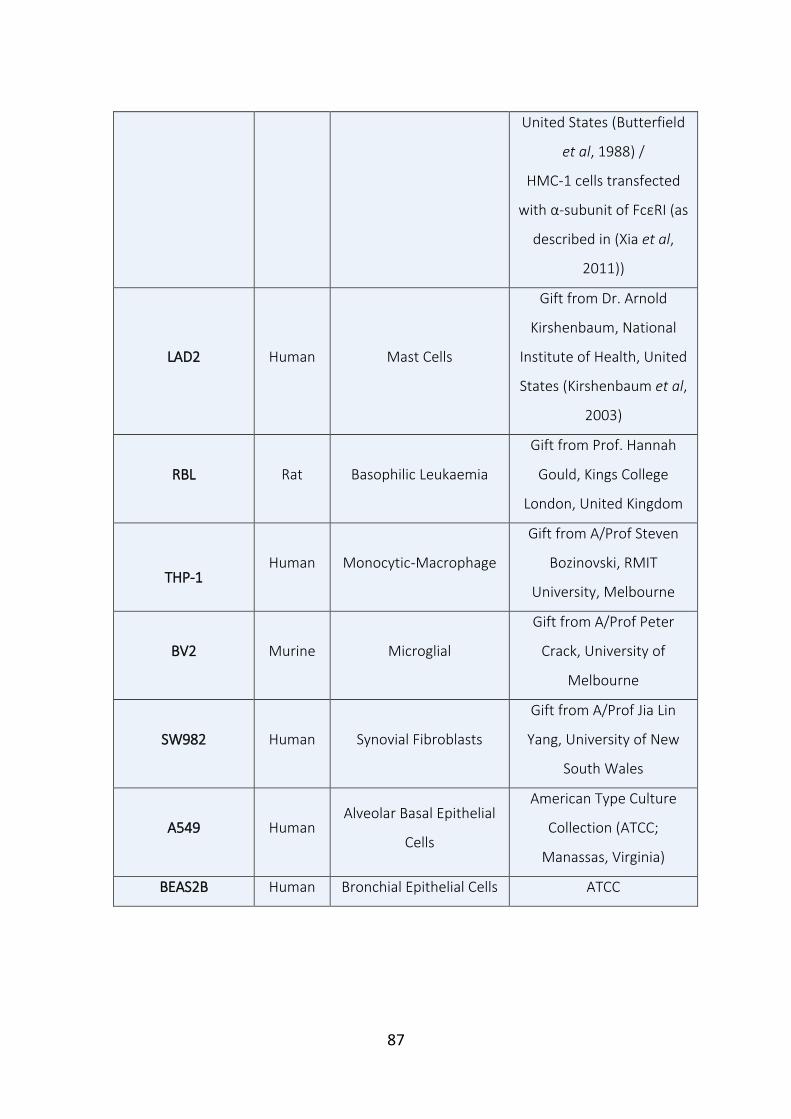
87
United States (Butterfield
et al, 1988) /
HMC-1 cells transfected
with α-subunit of FcεRI (as
described in (Xia et al,
2011))
LAD2 Human Mast Cells
Gift from Dr. Arnold
Kirshenbaum, National
Institute of Health, United
States (Kirshenbaum et al,
2003)
RBL Rat Basophilic Leukaemia
Gift from Prof. Hannah
Gould, Kings College
London, United Kingdom
THP-1 Human Monocytic-Macrophage
Gift from A/Prof Steven
Bozinovski, RMIT
University, Melbourne
BV2 Murine Microglial
Gift from A/Prof Peter
Crack, University of
Melbourne
SW982 Human Synovial Fibroblasts
Gift from A/Prof Jia Lin
Yang, University of New
South Wales
A549 Human Alveolar Basal Epithelial
Cells
American Type Culture
Collection (ATCC;
Manassas, Virginia)
BEAS2B Human Bronchial Epithelial Cells ATCC

88
3.2.4.2 Measuring CapG release from stimulated cells
3.2.4.2.1 Supernatants
Supernatants from stimulated and non-stimulated HMCα, LAD2 and THP-
1 cells (Sections 2.1.1.1, 2.1.2.1 and 2.1.4.1, respectively) were harvested by
centrifugation (300 g, 5 min). Samples were processed for Western blot analysis
as described in Section 2.8.1.
Western blot band intensities were quantified by densitometry using the Fiji
imaging software (National Institutes of Health, Bethesda, MD). The data were
then expressed as a percentage of CapG release as compared to non-stimulated
cells.
3.2.4.2.2 Cell pellets
Pellets from THP-1 stimulated cells were lysed in lysis buffer and the
protein concentration determined by a Bradford protein assay as described in
Section 2.7. The samples were then processed for Western blot analysis as
described in Section 2.8.1. Both polyclonal rabbit anti-CapG and anti β-tubulin
antibodies were used. Immunoreactive proteins were visualised by enhanced
chemiluminescence using a ChemiDoc™ MP System. Band intensities were
quantified using the Fiji imaging software, and were normalised to their respective
β-tubulin expression, and data expressed as a percentage of non-stimulated cell
CapG levels.
3.2.5 Immunofluorescence
The protocol for immunofluorescence studies visualising intracellular
CapG from THP-1 cells following LPS stimulation was as described in Section
2.5.

89
3.2.6 Flow Cytometry analysis (FACS) for intracellular staining of CapG
To study the intracellular expression of CapG in mast cells and
macrophages isolated from rat peritoneal lavage, cells were fixed and
permeabilised using the Cytofix/Cytoperm™ Fixation/Permeabilisation solution
kit (BDBioscience; NSW, Australia), and then incubated with anti-CapG antibody
or an isotype control antibody and samples processed as described in Section 2.3.
Cells were analysed by flow cytometry (BD LSRFortessa™,
BDBioscience). For data acquisition, a forward scatter (FSC) versus side scatter
(SSC) gate was set to identify cell subpopulations based on size and granularity.
50,000 gated macrophage and mast cells were collected and visualised on a
histogram (log fluorescence vs cell number). A rightward shift of the histogram,
compared to the isotype control indicated positive CapG expression. FlowJo
version 10.0.8 software (Tree Star, OR) was used for data analysis.
3.2.7 Degranulation Assay
Release of β-hexosaminidase from mast cells is commonly used as a
measure of mast cell degranulation activity. Because the HMCα cells are a
relatively immature mast cell line, as demonstrated by the paucity in granules, they
seem unable to degranulate upon stimulation through the IgE-antigen pathway
(Xia et al, 2011). In contrast, the LAD2 cell lines have been known to degranulate
in response to various stimuli and were also used in this study (Kirshenbaum et al,
2003). The process of IgE-sensitisation, stimulation and measurements of β-
hexosaminidase was described in Section 2.4.
3.2.8 Measurement of cytokine levels using enzyme-linked immunosorbent
assays (ELISA)
Stimulated and non-stimulated HMCα, LAD2 and THP-1 supernatants
were harvested by centrifugation (300 g, 5 mins) and assayed for IL-8 levels using
commercially available ELISA Kits (OptEIA; BDBioscience, North Ryde, NSW,

90
Australia) as described in Section 2.6. The absorbance was measured at 450 nm
using a plate-reader (Multiskan Ascent, Thermo Scientific) and Ascent software
version 2.6 was used to plot standard curves used to determine unknown IL-8
cytokine concentrations.
3.2.9 Statistical analysis
Data from ELISA and Western blotting analysis were expressed as the
means ± standard error of mean (SEM), where n represents the number of
independent primary cell cultures used or numbers of experiments conducted on
immortalised cell lines. If applicable, an appropriate statistical analysis test was
performed (refer to Section 2.13).
Results shown were plotted using Graphpad Prism software (version 6.01).
If a statistical significance was obtained, then * denotes p<0.05, ** denotes p<0.01,
and *** denotes p<0.001.
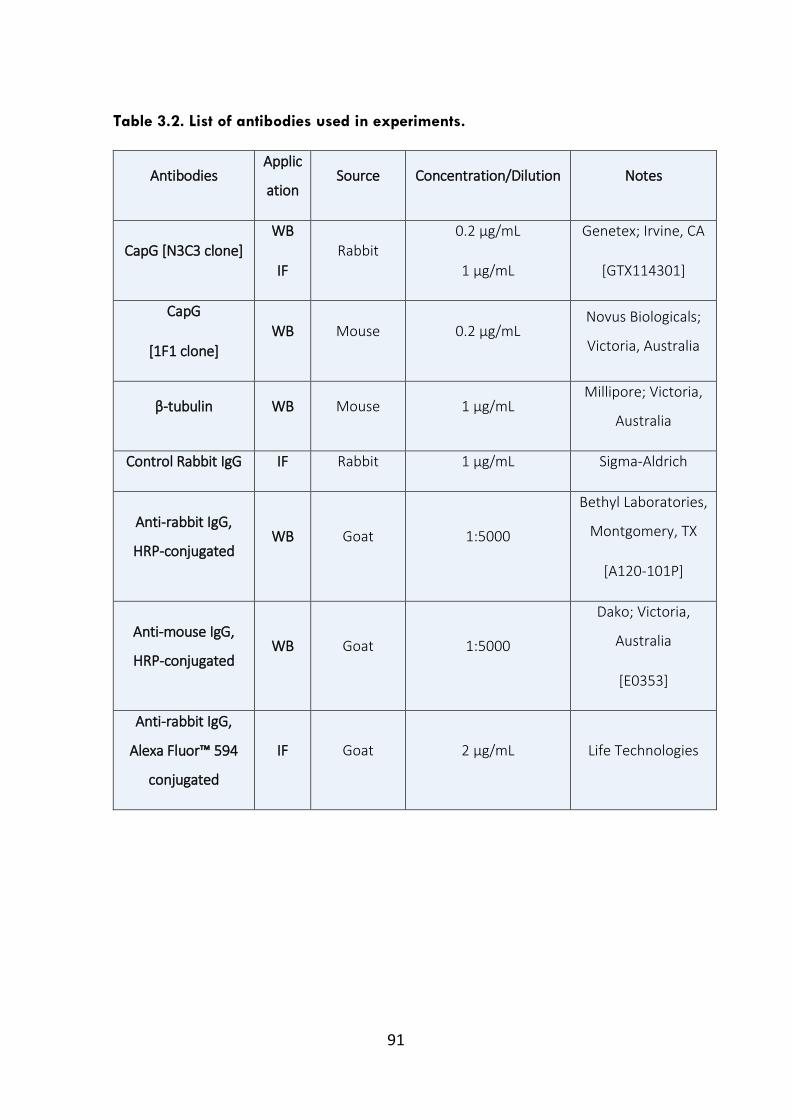
91
Table 3.2. List of antibodies used in experiments.
Antibodies Applic
ation Source Concentration/Dilution Notes
CapG [N3C3 clone] WB
IF Rabbit
0.2 µg/mL
1 µg/mL
Genetex; Irvine, CA
[GTX114301]
CapG
[1F1 clone] WB Mouse 0.2 µg/mL
Novus Biologicals;
Victoria, Australia
β-tubulin WB Mouse 1 µg/mL Millipore; Victoria,
Australia
Control Rabbit IgG IF Rabbit 1 µg/mL Sigma-Aldrich
Anti-rabbit IgG,
HRP-conjugated WB Goat 1:5000
Bethyl Laboratories,
Montgomery, TX
[A120-101P]
Anti-mouse IgG,
HRP-conjugated WB Goat 1:5000
Dako; Victoria,
Australia
[E0353]
Anti-rabbit IgG,
Alexa Fluor™ 594
conjugated
IF Goat 2 µg/mL Life Technologies

92
3.3 Results
3.3.1 CapG gene expression in a variety of cell types
CapG expression was measured in equal concentrations of total RNA
isolated from classically defined “immune” immortalised cell lines such as mast
cells (HMCα, LAD2), monocytes (THP in both undifferentiated and PMA-
differentiated cells) and the “non-immune” bronchial epithelial cell line BEAS2B.
In addition, CapG gene expression was also measured in primary rat peritoneal
macrophages and mast cells. CapG mRNA levels are higher in the HMCα, LAD2,
THP-1 cells compared to BEAS-2B, as indicated by the lower cycle number in
these immune cells (Table 3.3). CapG was also highly expressed in RBL cell line
and rat peritoneal macrophages. Although CapG gene expression was detectable
in purified rat peritoneal mast cells (RPMC), expression was lower when compared
to macrophages.
Table 3.3. Threshold cycle numbers of human and rat CapG in different cell
types.

93
3.3.2 Cell distribution of CapG in rodent and human cell lines
The cellular distribution of CapG was investigated in cells from humans,
rats and mice by Western blotting (Figure 3.3). CapG expression was first
investigated in immortalised cell lines with Western blot of cell lysates. CapG
expression was detected in immune cell lines including LAD2 and HMCα (mast
cells), THP-1 (monocytic), and BV2 (microglial). Interestingly, although CapG
message levels were detected in the RBL cells, CapG protein was not detected in
the rat basophilic leukaemia RBL cells, another commonly used mast cell model
in in vitro studies. However, CapG protein was weakly detected in the non-
haemapoeitic cell lines tested such as SW982 (fibroblast) and BEAS2B (bronchial
epithelial) and A549 (alveolar basal epithelial) (Figure 3.3A). This was also
confirmed by densitometry analysis (Figure 3.3B). Blotting for β-tubulin indicated
that approximately equal quantities of total cell lysates had been loaded.
We sought to determine whether CapG was also expressed in vivo and in
vitro primary mast cells. Cultured BMMCs are a commonly used mast cell model
in in vitro studies as they provide reasonably high yields of homogenous cells and
demonstrate mast cell-like characteristics such as expression of different
immunoglobulin (Ig) receptors and degranulation. However, these cells are
relatively immature and lack certain mast cell characteristics (Malbec et al, 2007).
Thus, we sought to examine CapG expression in mature in vivo differentiated mast
cells. However, an important limitation associated with this is the dependency of
fresh tissue and the limited yield of purified mast cells (Bischoff, 2007).
A readily available source of mature in vivo differentiated mast cells can be
obtained by peritoneal lavage of rats. Mast cells represent only 1-4% of total
peritoneal cells, which are predominantly composed of macrophages (95%) (Allen
et al, 1980). Sedimentation techniques using a Percoll® density gradient have been
proven useful for yielding reasonably pure mast cells however this results in poor
yields (Arock et al, 2008). Following purification, non-purified and purified cells
were cyto-spun onto slides and mast cells were differentially stained with a

94
commonly used metachromatic stain, Wright-Giemsa (Leclere et al, 2006).
Following Percoll® purification, mast cells represented around 95% of the cell
population (Figure 3.4A).
CapG protein expression was measured in cultured BMMCs, rat peritoneal
cells and purified peritoneal mast cells (Figure 3.4B), and densitometry analysis
performed (Figure 3.4C). CapG was readily detected in BMMCs. Similarly, CapG
expression was also detected in rat peritoneal cells. However, this expression was
likely due to expression in macrophages where CapG represents approximately 1%
of total protein content (Witke et al, 2001). However, CapG expression was not
detected in purified mast cells, although this was likely due to a low yield of cells
following Percoll purification. This was further confirmed by the lack of β-tubulin
detected in RPMCs, despite our attempts to load equal amounts of total cell lysates
for each sample. It is possible that measurements of total cell proteins in RPMC
lysate was skewed by the high abundance of granular proteins during the
quantification of lysate protein resulting in loading of a relatively low amount of
other cellular proteins.
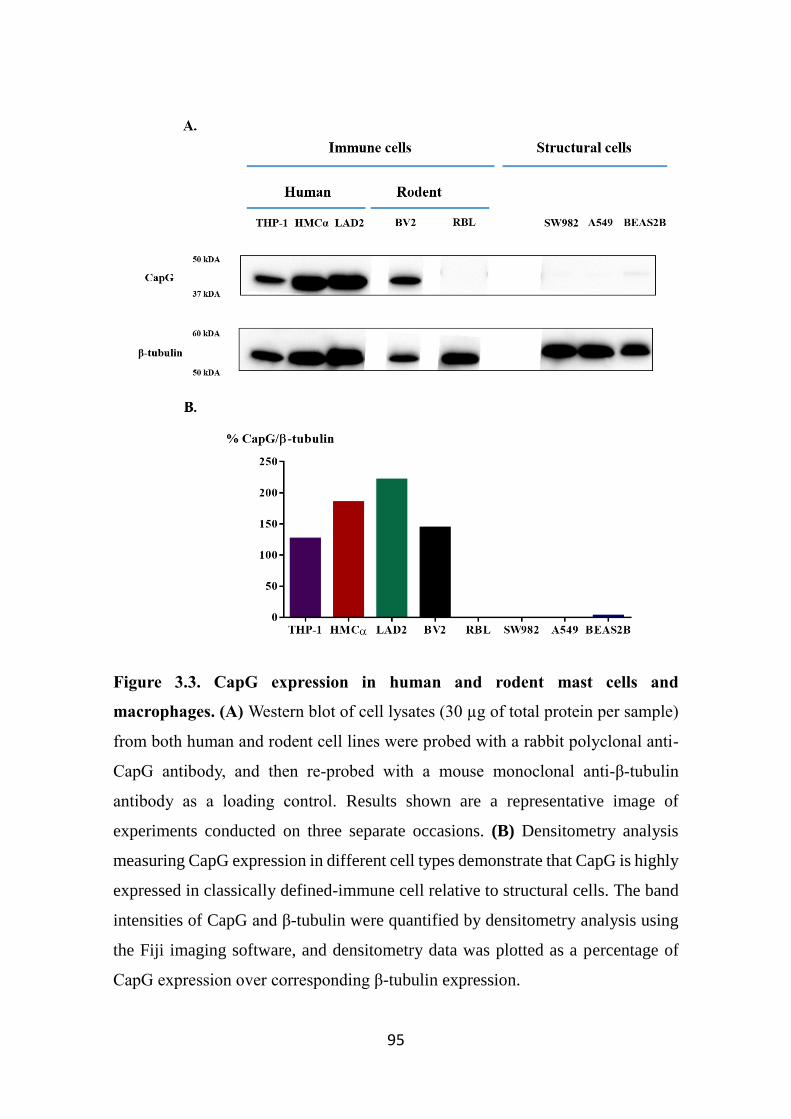
95
Figure 3.3. CapG expression in human and rodent mast cells and
macrophages. (A) Western blot of cell lysates (30 µg of total protein per sample)
from both human and rodent cell lines were probed with a rabbit polyclonal anti-
CapG antibody, and then re-probed with a mouse monoclonal anti-β-tubulin
antibody as a loading control. Results shown are a representative image of
experiments conducted on three separate occasions. (B) Densitometry analysis
measuring CapG expression in different cell types demonstrate that CapG is highly
expressed in classically defined-immune cell relative to structural cells. The band
intensities of CapG and β-tubulin were quantified by densitometry analysis using
the Fiji imaging software, and densitometry data was plotted as a percentage of
CapG expression over corresponding β-tubulin expression.
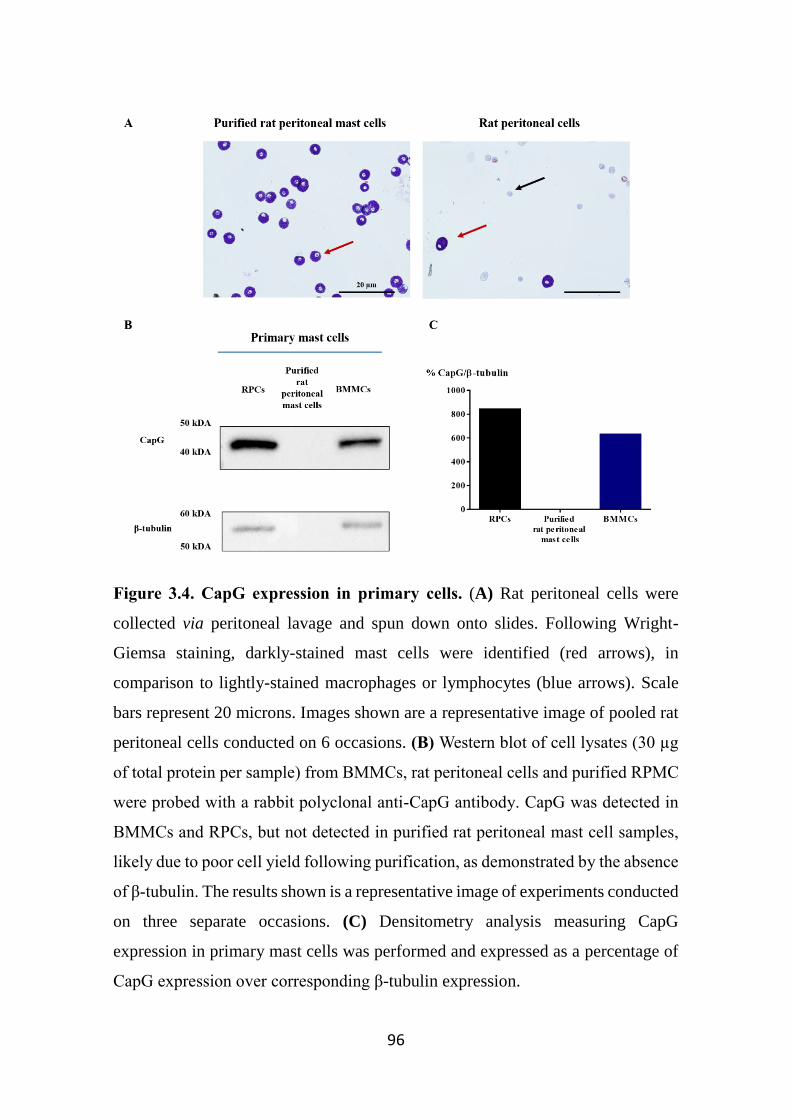
96
Figure 3.4. CapG expression in primary cells. (A) Rat peritoneal cells were
collected via peritoneal lavage and spun down onto slides. Following Wright-
Giemsa staining, darkly-stained mast cells were identified (red arrows), in
comparison to lightly-stained macrophages or lymphocytes (blue arrows). Scale
bars represent 20 microns. Images shown are a representative image of pooled rat
peritoneal cells conducted on 6 occasions. (B) Western blot of cell lysates (30 µg
of total protein per sample) from BMMCs, rat peritoneal cells and purified RPMC
were probed with a rabbit polyclonal anti-CapG antibody. CapG was detected in
BMMCs and RPCs, but not detected in purified rat peritoneal mast cell samples,
likely due to poor cell yield following purification, as demonstrated by the absence
of β-tubulin. The results shown is a representative image of experiments conducted
on three separate occasions. (C) Densitometry analysis measuring CapG
expression in primary mast cells was performed and expressed as a percentage of
CapG expression over corresponding β-tubulin expression.

97
3.3.3 Purification of RPMC and expression of CapG from different rat
peritoneal cell populations
Since CapG expression by Western blotting analysis was challenging in
RPMCs (Figure 3.4B), we used flow cytometry as an alternative method for
examining protein expression. Both rat peritoneal cells and purified RPMCs were
fixed and permeabilised prior to intracellular staining for CapG expression. The
purified mast cell populations were used in flow cytometric analysis to distinguish
mast cells over other cell types in subsequent analysis using non-purified samples
(Figure 3.5B). Using flow cytometry, cell populations in the peritoneal lavage
preparation can be distinguished on the basis of their forward scatter (cell size) and
side scatter (cell granularity) and stained for intracellular CapG expression
following permeabilisation. Three distinct cell subpopulations were identified, the
largest population likely being macrophages (80-90%). A second subpopulation of
cells above the macrophages was defined as mast cells population as their forward
and size scatter profiles matched to that of the purified mast cell samples (Figure
3.5A). Finally, a third subpopulation characterised by smaller cell size in
comparison to macrophages was identified as likely lymphocytes. Following
intracellular staining for CapG, all three cell subpopulations expressed the protein,
as indicated by a rightward shift compared to cells incubated with an isotype
control antibody (Figure 3.5B). There was a greater rightward shift in the
macrophage samples, indicative of a stronger expression of CapG in these cells,
which was expected.
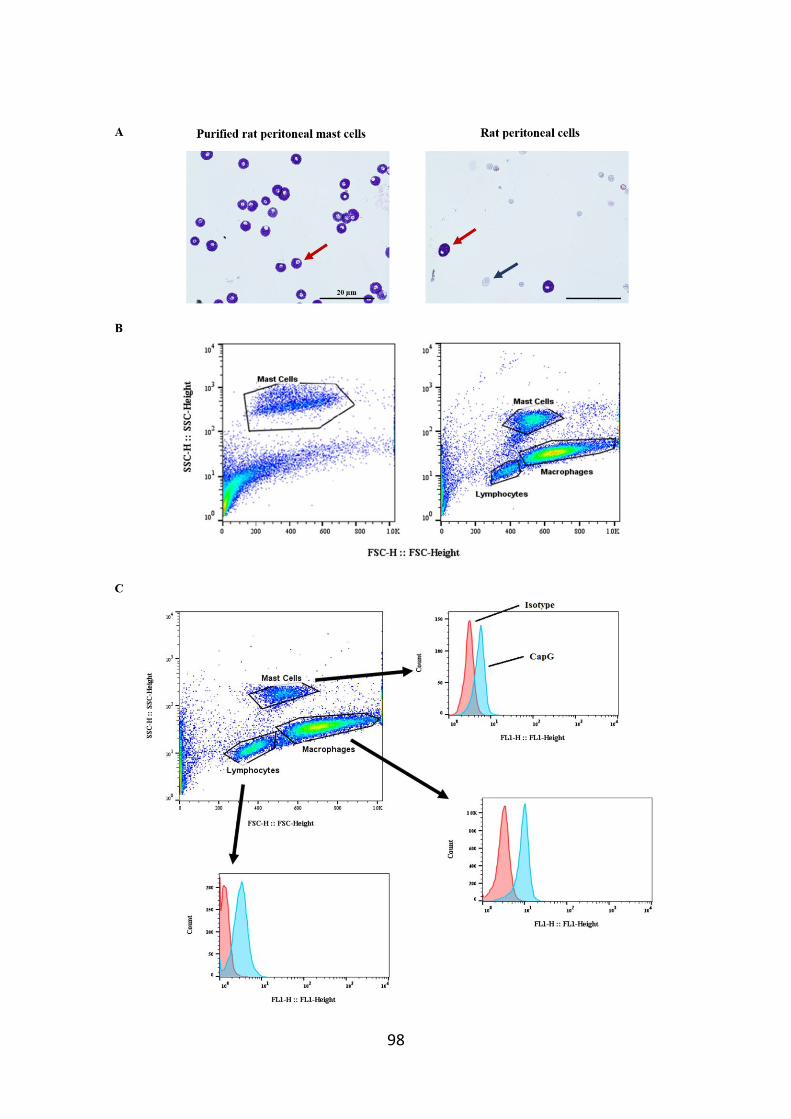
98

99
Figure 3.5. Identification of mast cells and analysis of CapG expression in
distinct rat peritoneal cell subpopulations. Using flow cytometric analysis,
purified mast cells (A) and non-purified rat peritoneal cells (B) samples were
separated by cell granularity (side scatter, y-axis) and size (forward scatter, x-axis).
A population of mast cells were identified through Percoll® purification and this
gate was used as a reference for the mast cell population in non-purified samples.
(C) The intracellular expression of CapG detected by flow cytometry in rat
peritoneal cell populations. Mast cells, macrophage and lymphocyte populations
were distinguished by forward and side scatter profiles. Rat peritoneal cells were
fixed and stained for CapG expression. In addition, some cells were stained with
an isotype control antibody. All three cell types expressed CapG as indicated by a
rightward shift (blue) compared to isotype control antibody (red). Flow cytometry
results shown are representative of experiments conducted on 4 pooled samples,
each containing rat peritoneal cells harvested from three rats.

100
3.3.4 CapG is released from LAD2 cells following IgE/FcεRI activation, but not
HMCα cells
The leukaemia-derived LAD2 mast cell line are commonly used in mast
cell research (Kirshenbaum et al, 2003). Since they express intracellular CapG, we
sought to determine whether CapG is released from these cells and if this is
affected by cell stimulation. Prior to this, the functional capacity of LAD2 cells
was first assessed by degranulation, as measured by β-hexosaminidase release
(Figure 3.6). Following one day of IgE sensitisation (NIP-IgE), LAD2 cells were
challenged with a range of different stimuli such as antigen (NIP-BSA), the
neuropeptide substance P, and the calcium ionophore ionomycin. β-
hexosaminidase release from stimulated cells was measured as a percentage of
total enzyme, as determined by β-hexosaminidase release from lysed cells. β-
hexosaminidase release increased from antigen concentrations ranging from 0.1
ng/mL to 30 ng/mL. At higher concentrations, β-hexosaminidase release from
LAD2 cells plateaued at approximately 60% of total β-hexosaminidase. Similarly,
LAD2 cells challenged with either substance P or ionomycin both released β-
hexosaminidase at a concentration-dependent manner, with β-hexosaminidase
release plateauing at approximately 90% of total β-hexosaminidase.
LAD2 cells were next examined for CapG release in response to various
stimuli by Western blotting. LAD2 cells basally released CapG, but in response to
antigen, CapG was further released in an apparent bell-shaped pattern, with release
peaking at 1 ng/mL (Figure 3.7A). This release pattern is different compared to
antigen-induced β-hexosaminidase release as antigen release was maximal at
higher antigen concentrations (Figure 3.6). It should be noted that in later
experiments, there was noticeable variation in CapG release from antigen-
stimulated LAD2 cells. In earlier experiments, there was enhanced CapG released
from LAD2 cells upon antigen stimulation, whereas in experiments utilising LAD2
cells at higher passage, CapG release was not affected by antigen stimulation
(Figure 3.7B). In these experiments, degranulation assays were also performed

101
and there were noticeable reductions in β-hexosaminidase release in response to
various stimuli (data not shown), indicating that high passage number was a likely
explanation in the diminished functional capacity of LAD2 cells.
Despite producing an extensive degranulation response, there was no
difference in CapG release between basal CapG release and substance P-induced
CapG release. Although there was an increase in CapG release from cells
stimulated with ionomycin, this was likely associated with cell death, as
determined by trypan blue viability staining, with consequent non-regulated
release of CapG (Figure 3.7C).
In addition to LAD2 cells, CapG release from stimulated HMCα cells was
also examined (Figures 3.8). Similar to LAD2 cells, HMCα cells were found to
basally release CapG. There was no consistent concentration-dependent trend
suggestive of CapG release from antigen-stimulated HMCα cells. CapG was also
not released from HMCα cells stimulated with the metabolically stable adenosine
analog NECA. In addition, ionomycin also triggered CapG release from cells
although release was again likely due to cell death as determined by trypan blue
staining. As our previous preliminary data had shown CapG release from antigen-
activated HMCα cells, we sought to establish whether these cells still responded
to IgE/antigen activation by secreting IL-8 (Xia et al, 2011). In comparison to prior
studies, IL-8 release from HMCα cells in this study were considerably lower and
although we attempted to optimise the experimental conditions, such as through
using freshly thawed cells and newly made stimuli, we were unable to recapitulate
preliminary data from our previous experiments (Figure 3.9). This suggests that
the HMCα model was not optimal for detailing CapG release.
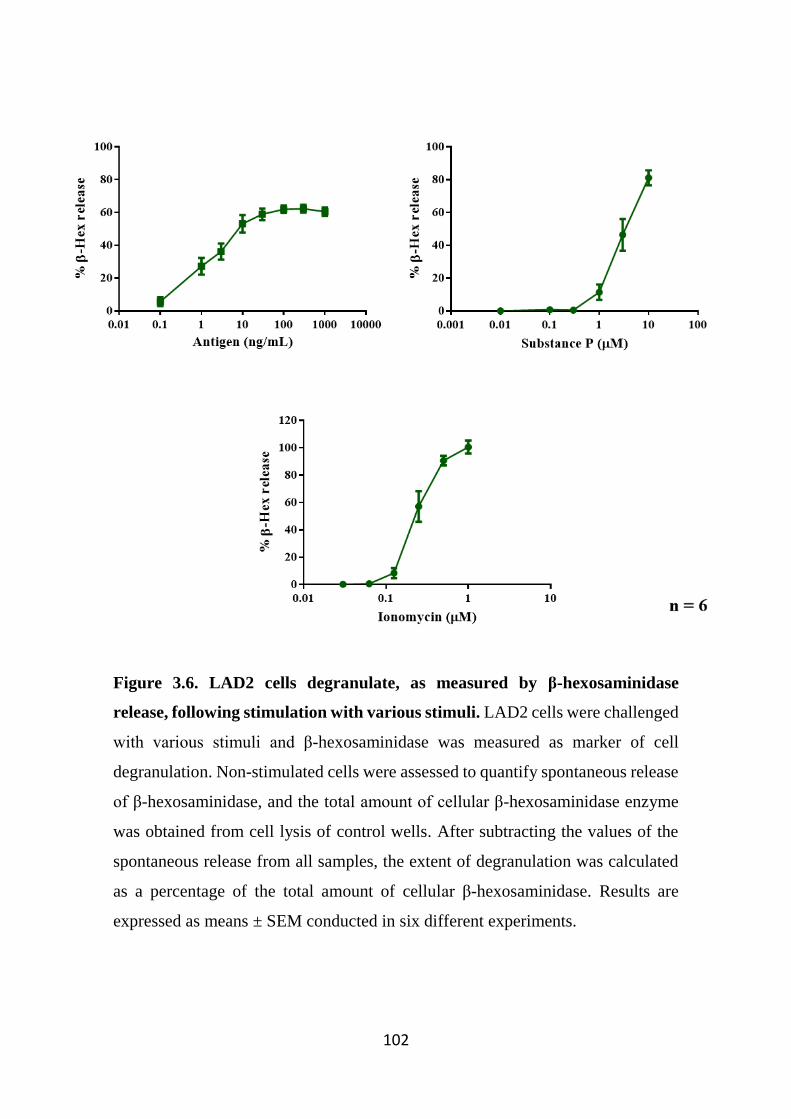
102
Figure 3.6. LAD2 cells degranulate, as measured by β-hexosaminidase
release, following stimulation with various stimuli. LAD2 cells were challenged
with various stimuli and β-hexosaminidase was measured as marker of cell
degranulation. Non-stimulated cells were assessed to quantify spontaneous release
of β-hexosaminidase, and the total amount of cellular β-hexosaminidase enzyme
was obtained from cell lysis of control wells. After subtracting the values of the
spontaneous release from all samples, the extent of degranulation was calculated
as a percentage of the total amount of cellular β-hexosaminidase. Results are
expressed as means ± SEM conducted in six different experiments.
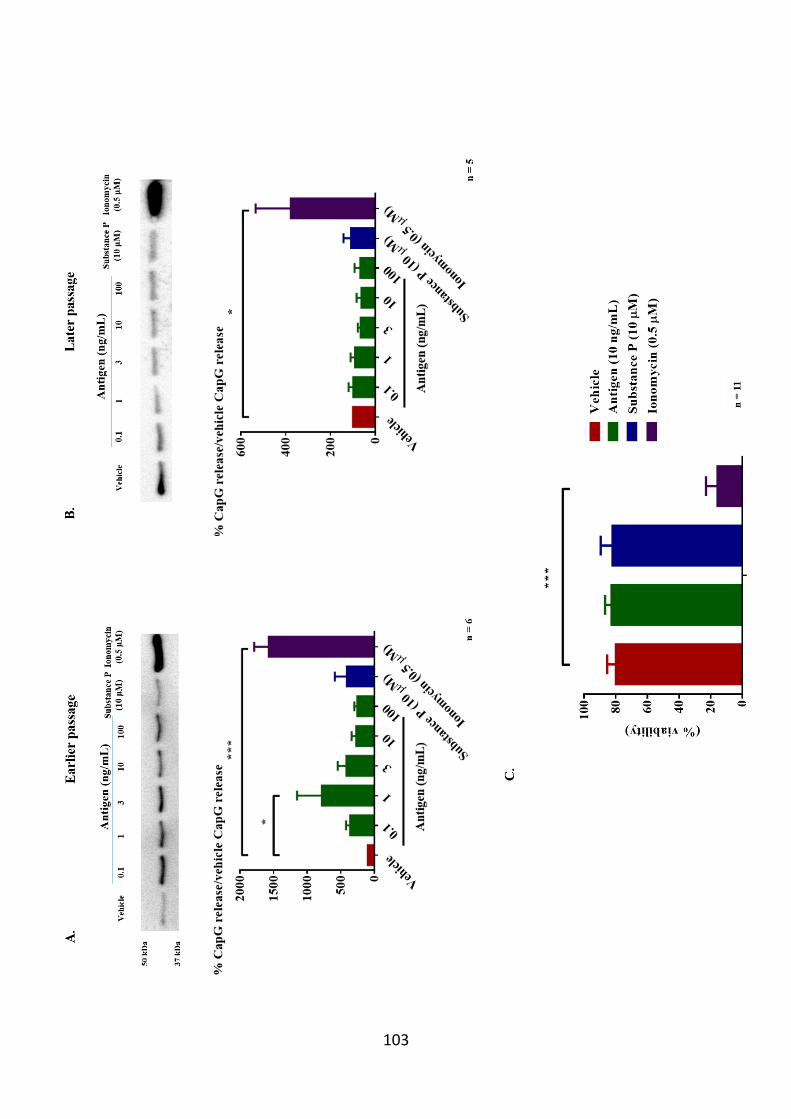
103

104
Figure 3.7. CapG release is only enhanced in antigen stimulated early
passaged LAD2 cells. CapG release was measured from LAD2 cells following 4
hour stimulation by Western blotting and quantitated by densitometry analysis.
There was a noticable change in CapG release pattern from antigen-stimulated
LAD2 cells between (A) early passage cells and (B) older passaged cells. Band
intensities of secreted CapG from cells stimulated with antigen was expressed as a
percentage of basal CapG release ± SEM of 6 or 5 experiments, respectively. A
one-way ANOVA followed by Bonferroni’s post-hoc test was applied for
comparing CapG release from stimulated LAD2 cells compared to vehicle control
(Vehicle). *p<0.05, and ***p<0.001 compared to vehicle group. (C) Following
cell stimulation, cell viability was measured by trypan blue exclusion. Cell
viability data were expressed as a percentage of viable cells over total cells counted
for each condition.
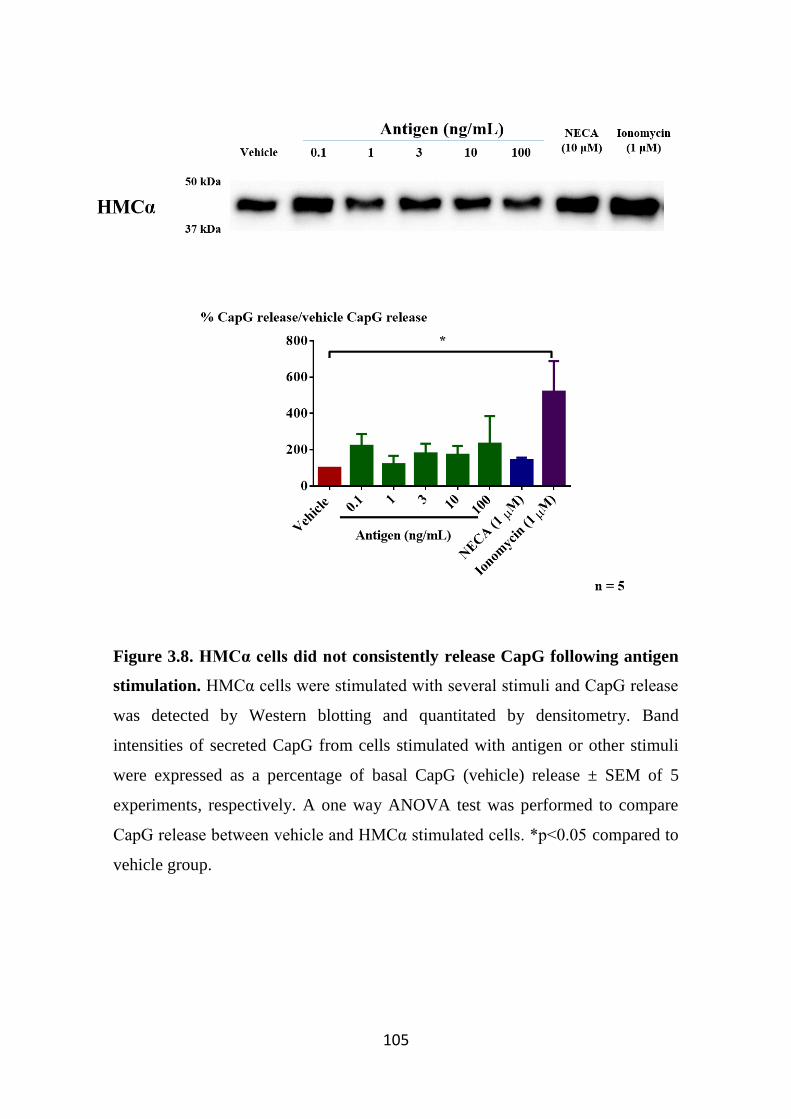
105
Figure 3.8. HMCα cells did not consistently release CapG following antigen
stimulation. HMCα cells were stimulated with several stimuli and CapG release
was detected by Western blotting and quantitated by densitometry. Band
intensities of secreted CapG from cells stimulated with antigen or other stimuli
were expressed as a percentage of basal CapG (vehicle) release ± SEM of 5
experiments, respectively. A one way ANOVA test was performed to compare
CapG release between vehicle and HMCα stimulated cells. *p<0.05 compared to
vehicle group.
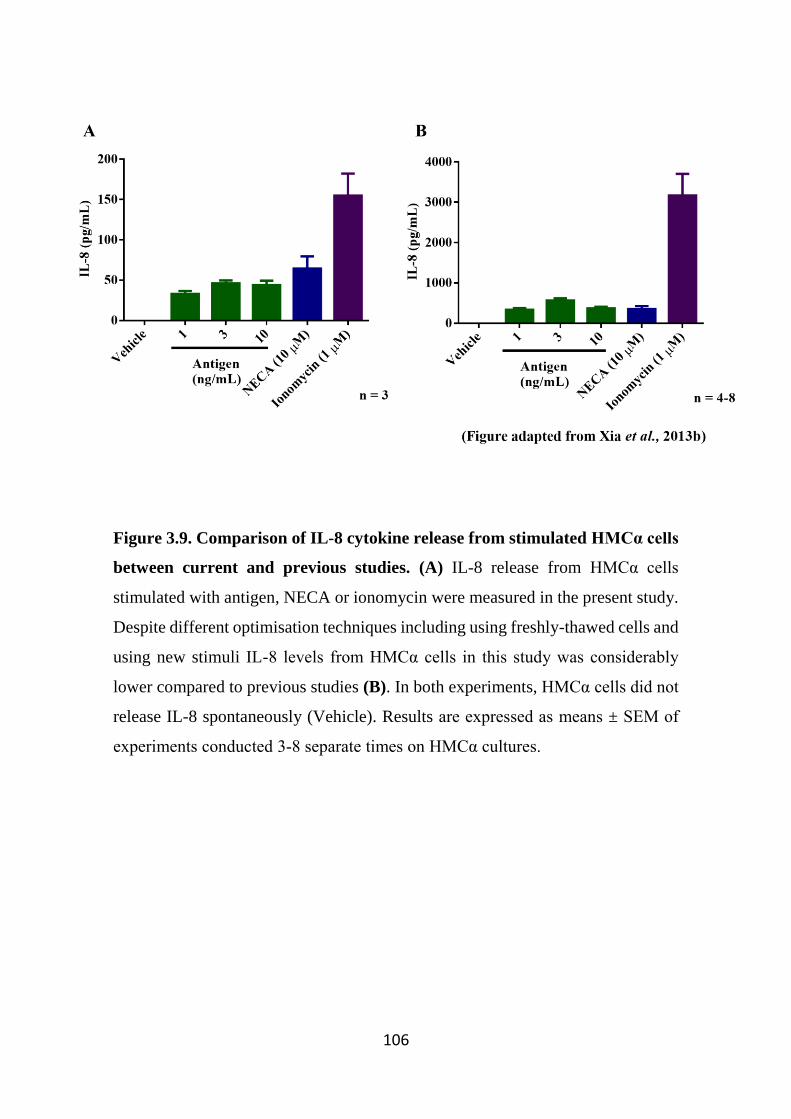
106
Figure 3.9. Comparison of IL-8 cytokine release from stimulated HMCα cells
between current and previous studies. (A) IL-8 release from HMCα cells
stimulated with antigen, NECA or ionomycin were measured in the present study.
Despite different optimisation techniques including using freshly-thawed cells and
using new stimuli IL-8 levels from HMCα cells in this study was considerably
lower compared to previous studies (B). In both experiments, HMCα cells did not
release IL-8 spontaneously (Vehicle). Results are expressed as means ± SEM of
experiments conducted 3-8 separate times on HMCα cultures.

107
3.3.5 LPS induces CapG release from THP-1 cells in a concentration-dependent
manner
Although CapG is known to be released constitutively from macrophages,
it is not known if its release can be regulated by stimuli. PMA-differentiated THP-
1 cells are a commonly used model for macrophage studies and hence used in this
study. We first sought to characterise the release of CapG from THP-1 cells in
response to various stimuli that have been previously shown to trigger THP-1 cell
activation. Of these, there was a concentration-dependent release in CapG from
THP-1 cells stimulated with 0.01 to 10 ng/mL of LPS. However, there was no
difference in CapG levels released from cells stimulated with IL-17 (50 ng/mL) or
between monomeric or heat-aggregated IgG (HAGG) (1 µg/mL) (Figure 3.10),
indicating that CapG release from activated THP-1 cells is ligand-dependent.
To further investigate the effects of LPS on CapG release, the concentration
of LPS was increased in the following experiments. In addition, we also sought to
determine the kinetics of CapG release from stimulated cells. Hence, after 2 days
of PMA treatment, THP-1 cells were stimulated with LPS (1, 10 and 100 ng/mL)
for 1, 2, 4 and 24 hours. Both supernatants and cell pellets were harvested.
Measured IL-8 release was used as a positive control for LPS activity with cytokine
levels increasing over time particularly between 4 and 24 hours, and also in a
concentration-dependent manner (Figure 3.11A). In addition, THP-1 cells
maintained high viability following stimulation, as assessed by trypan blue
staining (Figure 3.11B). Thus, release of CapG from THP-1 cells is not associated
with cell death.
Compared to the vehicle treated THP-1 cells, CapG was released from
THP-1 cells when stimulated with LPS (100 ng/mL) at 4 and 24 hours (Figure
3.12A). Interestingly, the intracellular expression of CapG was not reduced over
time and was unaffected by LPS, likely relating to high concentrations of CapG in
these cells. β-tubulin was also analysed as a loading control and was consistent
across all samples (Figure 3.12B).
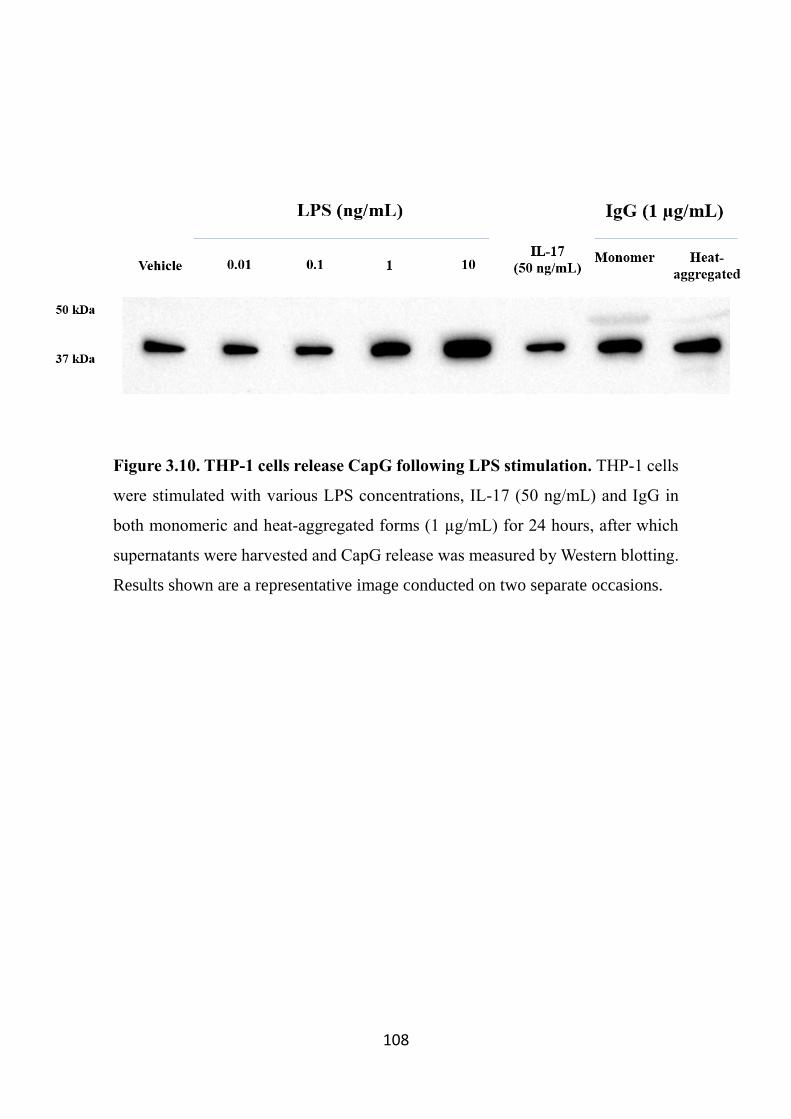
108
Figure 3.10. THP-1 cells release CapG following LPS stimulation. THP-1 cells
were stimulated with various LPS concentrations, IL-17 (50 ng/mL) and IgG in
both monomeric and heat-aggregated forms (1 µg/mL) for 24 hours, after which
supernatants were harvested and CapG release was measured by Western blotting.
Results shown are a representative image conducted on two separate occasions.
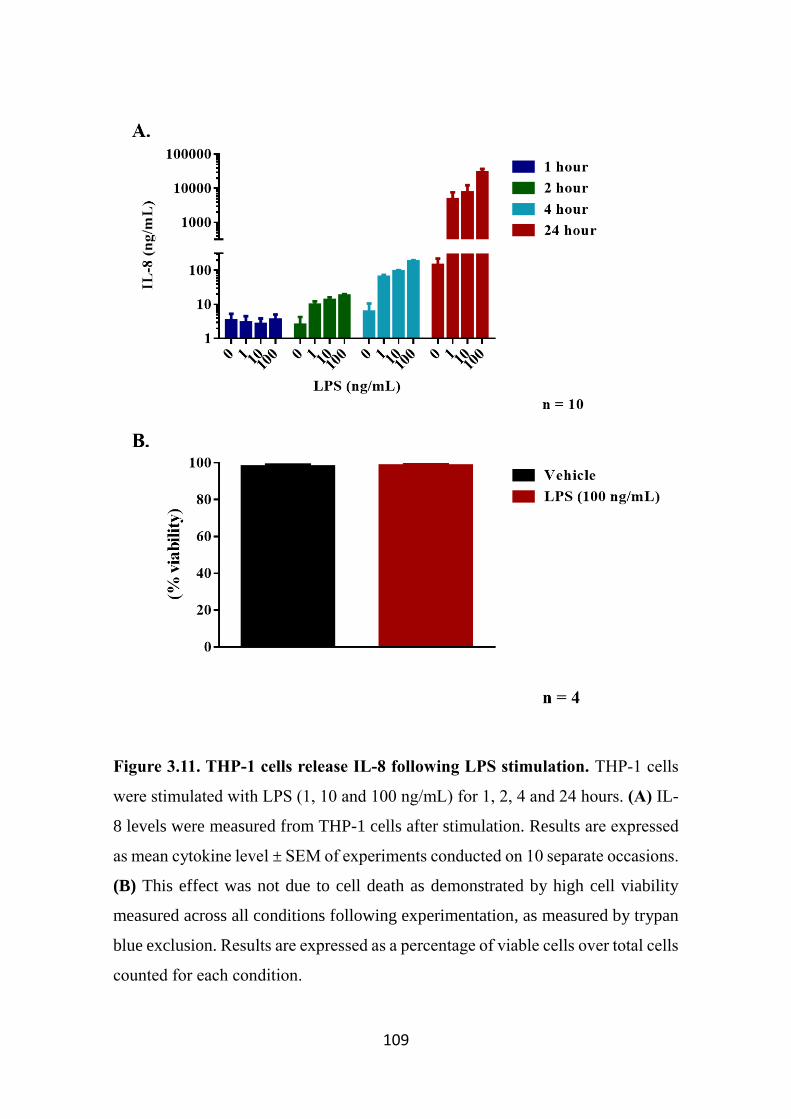
109
Figure 3.11. THP-1 cells release IL-8 following LPS stimulation. THP-1 cells
were stimulated with LPS (1, 10 and 100 ng/mL) for 1, 2, 4 and 24 hours. (A) IL-
8 levels were measured from THP-1 cells after stimulation. Results are expressed
as mean cytokine level ± SEM of experiments conducted on 10 separate occasions.
(B) This effect was not due to cell death as demonstrated by high cell viability
measured across all conditions following experimentation, as measured by trypan
blue exclusion. Results are expressed as a percentage of viable cells over total cells
counted for each condition.
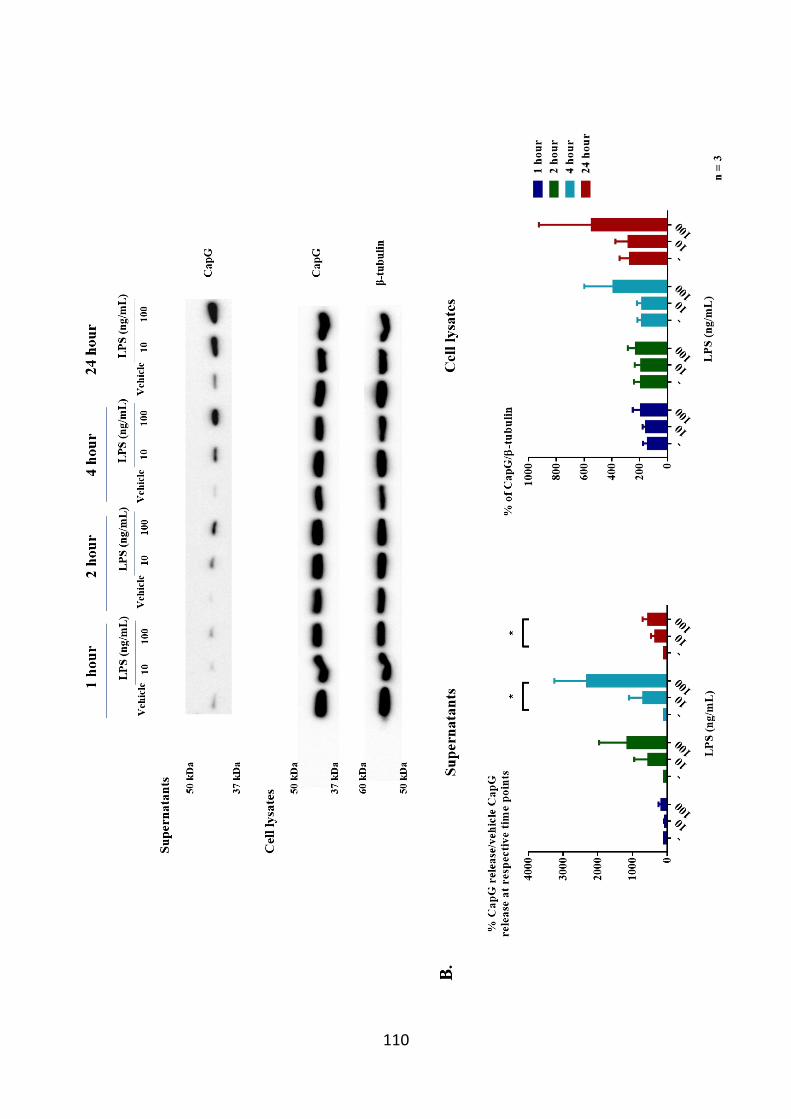
110

111
Figure 3.12. CapG is released from THP-1 cells in response to LPS in a time-
dependent manner. THP-1 cells were treated with PMA for 2 days prior to LPS
stimulation. Cells were stimulated with varying concentrations of LPS for 1, 2, 4
and 24 hours and supernatants and pellets were harvested. (A) Western blot of
supernatants and cell lysates were examined for CapG expression, and β-tubulin
measured as a loading control (in all lysates). (B) Densitometry analysis measuring
CapG release from THP-1 cells demonstrated a significant increase in CapG
release from cells stimulated with LPS (100 ng/mL) for 4 and 24 hrs. The band
intensities of CapG measured in supernatants and pellets were quantified by
densitometry analysis using the Fiji imaging software. Release of CapG from LPS-
stimulated THP-1 cells was normalised to basal CapG release (Vehicle) at the
respective time points and the data were then expressed as a percentage of CapG
release/basal release ± SEM of all 3 experiments. One-way repeated measures
ANOVA followed by Bonferroni’s post-hoc test was applied for multiple
comparisons. *p<0.05 compared to basal release at matching time points. In
contrast, band intensities of CapG in cell pellets were normalised to matching β-
tubulin and the data were expressed as a percentage of CapG expression/β-tubulin
± SEM of all 3 experiments.

112
3.3.6 CapG release from LPS-stimulated THP-1 cells is inhibited by
dexamethasone and the TLR4 blocking antibody HTA-125
Since glucocorticoids were previously reported to inhibit LPS-mediated IL-
8 release from THP-1 cells (Mogensen et al, 2008), we sought to examine whether
these compounds would also affect CapG release from THP-1 cells following LPS
stimulation. THP-1 cells were pre-treated with the anti-inflammatory
glucocorticoid dexamethasone for 30 minutes prior to 24 hour-LPS stimulation
and both cell pellets and supernatants were harvested. As expected, IL-8 release
from LPS-induced THP-1 cells was significantly reduced in dexamethasone
treated cells compared to untreated cells (Figure 3.13A). This was not influenced
by cell death as all conditions had high cell viability (Figure 3.13B). Thus, like
LPS, dexamethasone did not affect cell viability during THP-1 cell stimulation.
Interestingly, dexamethasone also significantly reduced CapG release from LPS-
stimulated cells THP-1 cells. However, intracellular CapG protein expression was
unaffected by the presence of dexamethasone (Figure 3.14).
The expression of intracellular CapG in THP-1 cells was also visualised by
immunofluorescence microscopy 24 hours after LPS stimulation (Figure 3.15).
Cytoplasmic CapG (red) was detected in un-treated THP-1 cells. However, CapG
staining was decreased when cells were stimulated with LPS as indicated by a
reduction in red staining. Furthermore, dexamethasone inhibited CapG release
from THP-1 cells in both resting and LPS-stimulated cells as indicated by a more
intense red stain in cells pre-treated with dexamethasone compared to non-
dexamethasone treated conditions. Isotype control antibody used in this study
showed no staining. It should be noted here that the observed reduction in
intracellular CapG in response to LPS is not in keeping with Western blot analysis,
which will be discussed later.
Although LPS is known to exert its effects through a variety of signalling
pathways, activation of cells is primarily mediated by the engagement of LPS to
TLR4 (Hoshino et al, 1999a; Lu et al, 2008). Previous studies have also

113
demonstrated that LPS-mediated activity on THP-1 cells can be inhibited by
targeting the TLR4 receptor using a mouse monoclonal anti-TLR4 antibody (HTA-
125) (Su et al, 2003). Thus, we examined whether inhibition of LPS/TLR4
signalling would modulate CapG release from THP-1 cells following LPS
stimulation. Cells were pre-incubated with HTA-125 for 30 minutes prior to LPS
stimulation for 24 hrs. A monoclonal mouse isotype antibody (mIgG) was also
included in this study as a negative control. Although the antibodies alone did not
affect CapG release from cells, there was a significant decrease in CapG release in
THP-1 cells pre-treated with HTA-125 before LPS stimulation (10 and 100 ng/mL)
(Figure 3.16A). Interestingly, HTA-125 or the isotype antibody did not affect IL-
8 release from LPS-stimulated THP-1 cells (Figure 3.16C).
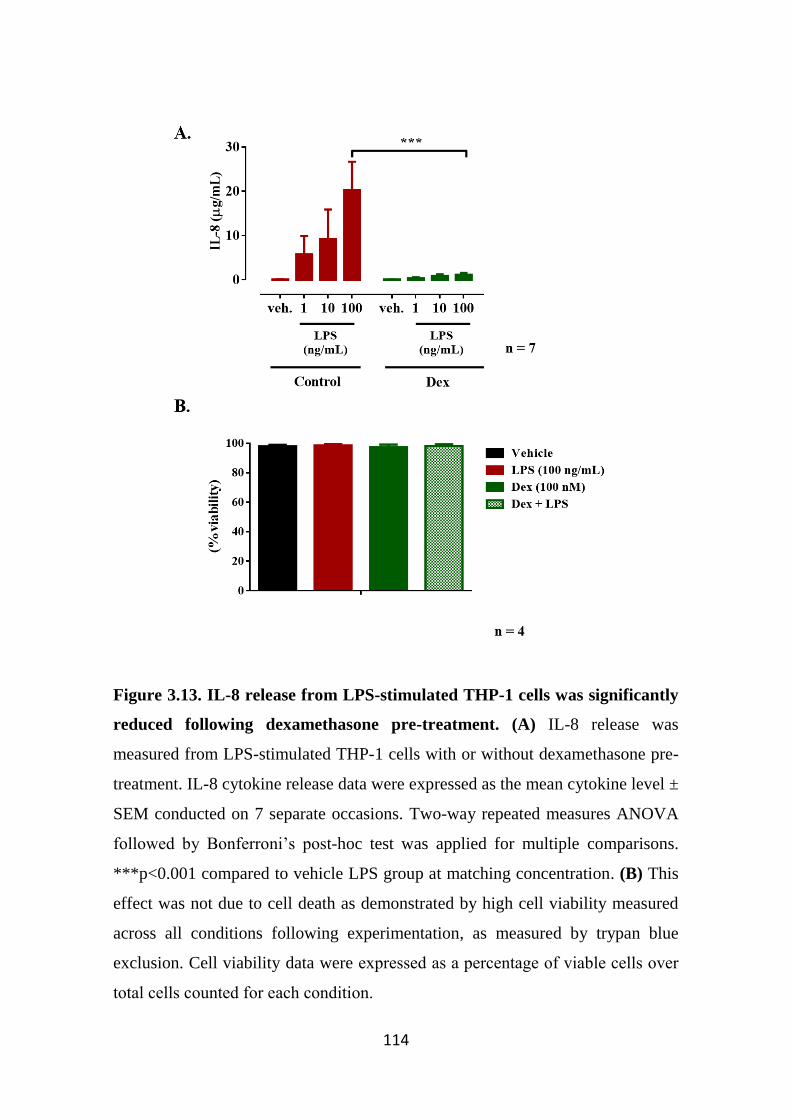
114
Figure 3.13. IL-8 release from LPS-stimulated THP-1 cells was significantly
reduced following dexamethasone pre-treatment. (A) IL-8 release was
measured from LPS-stimulated THP-1 cells with or without dexamethasone pre-
treatment. IL-8 cytokine release data were expressed as the mean cytokine level ±
SEM conducted on 7 separate occasions. Two-way repeated measures ANOVA
followed by Bonferroni’s post-hoc test was applied for multiple comparisons.
***p<0.001 compared to vehicle LPS group at matching concentration. (B) This
effect was not due to cell death as demonstrated by high cell viability measured
across all conditions following experimentation, as measured by trypan blue
exclusion. Cell viability data were expressed as a percentage of viable cells over
total cells counted for each condition.
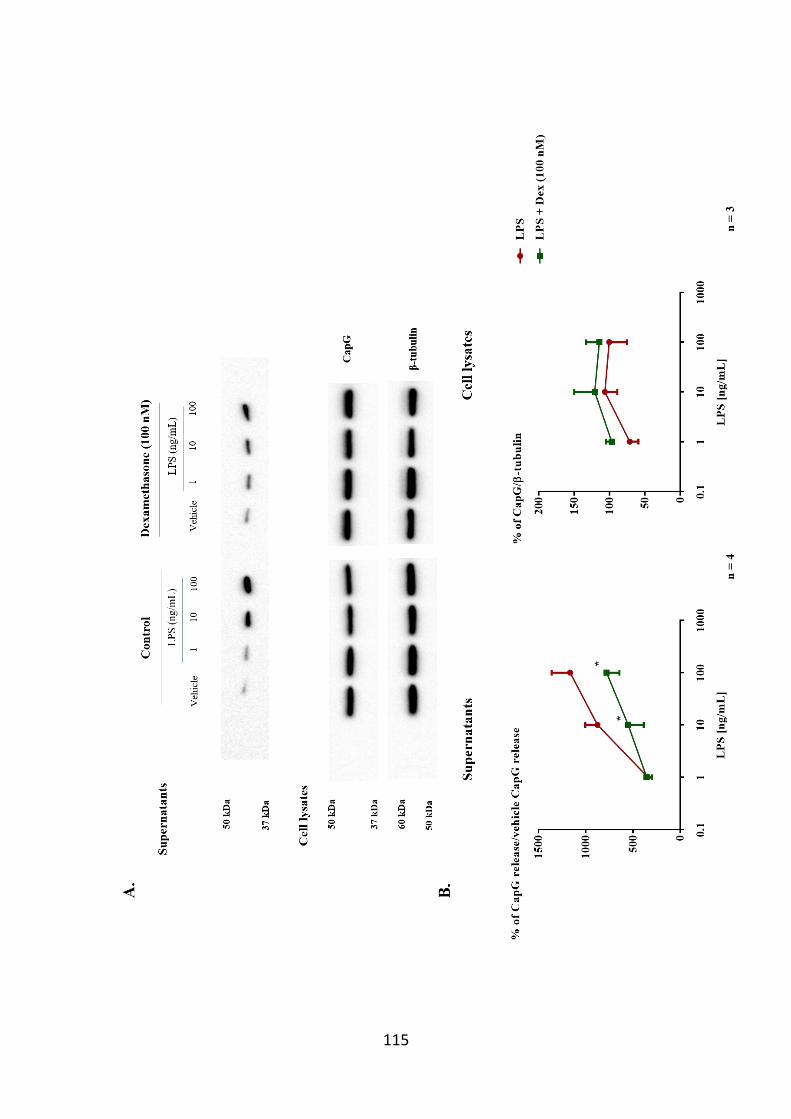
115

116
Figure 3.14. Dexamethasone reduces CapG release from LPS-stimulated
THP-1 cells. (A) THP-1 cells were pre-treated with dexamethasone (100 nM) for
30 min and then treated with LPS. After 24 hours, supernatants and pellets were
harvested and assayed for CapG levels. Blots shown are representative of
experiments conducted on 4 (supernatants) and 3 (lysates) separate occasions. (B)
Densitometry analysis measuring CapG release from THP-1 cells demonstrated a
significant decrease in CapG release from cells treated with dexamethasone prior
to LPS treatment (10 and 100 ng/mL). The band intensities of CapG measured in
supernatants were quantified by densitometry using the Fiji imaging software.
Release of CapG from LPS-stimulated THP-1 cells was normalised to basal CapG
release (Vehicle) in the presence and absence of dexamethasone. The data were
then expressed as a percentage of CapG release/basal release ± SEM of all 3
experiments. Two-way repeated measures ANOVA followed by Bonferroni’s
post-hoc test was applied for multiple comparisons between CapG release from
THP-1 cells in the presence and absence of dexamethasone. *p<0.05 compared to
LPS group at respective concentrations. Band intensities of CapG in cell pellet
samples were normalised to matching β-tubulin and the data were expressed as a
percentage of CapG expression/β-tubulin ± SEM of all 3 experiments.
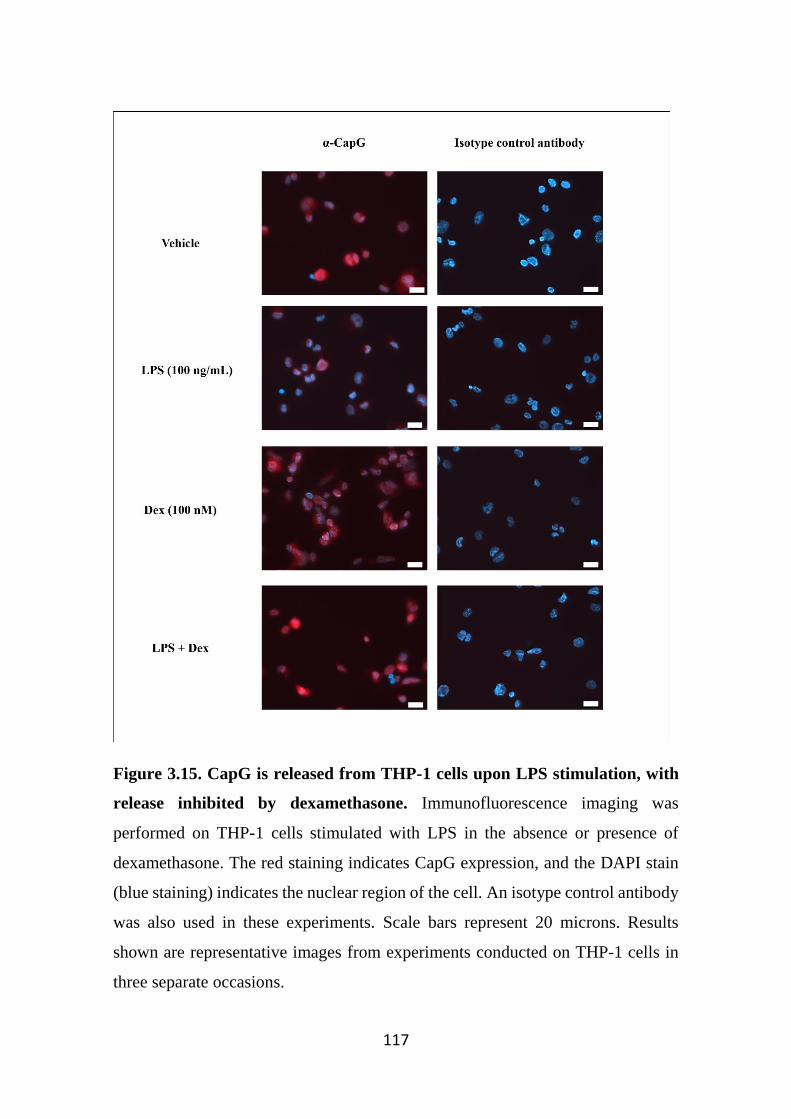
117
Figure 3.15. CapG is released from THP-1 cells upon LPS stimulation, with
release inhibited by dexamethasone. Immunofluorescence imaging was
performed on THP-1 cells stimulated with LPS in the absence or presence of
dexamethasone. The red staining indicates CapG expression, and the DAPI stain
(blue staining) indicates the nuclear region of the cell. An isotype control antibody
was also used in these experiments. Scale bars represent 20 microns. Results
shown are representative images from experiments conducted on THP-1 cells in
three separate occasions.
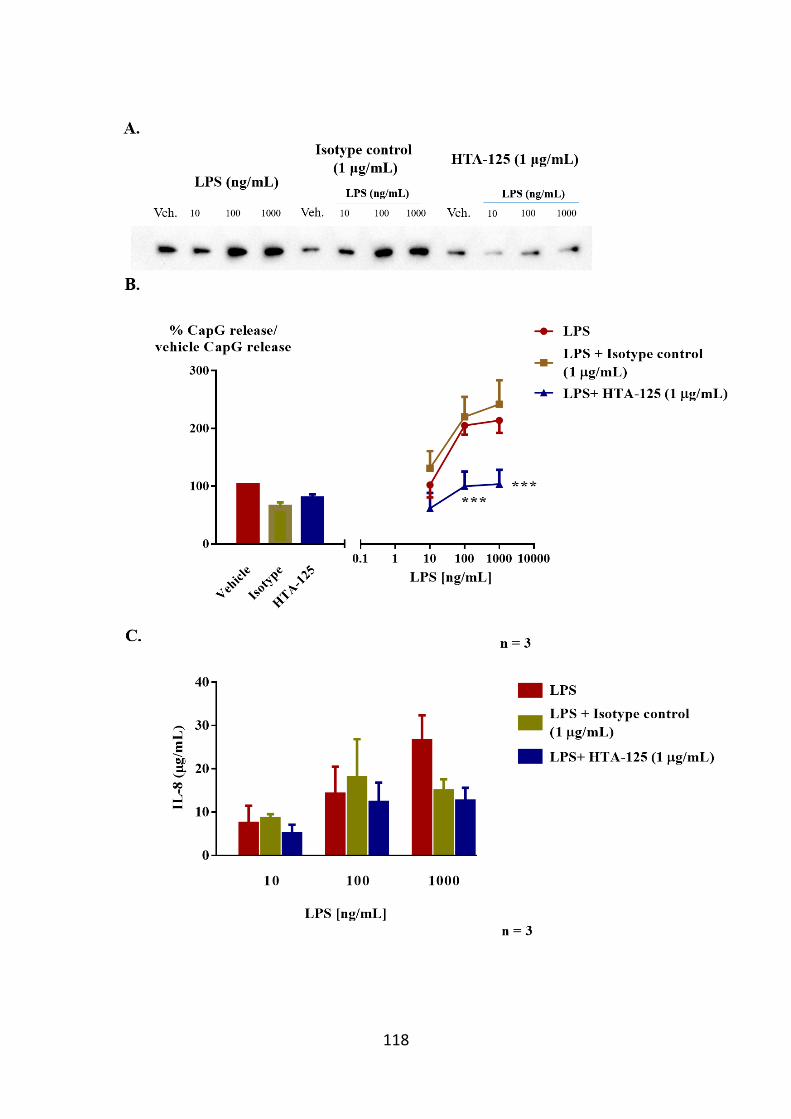
118

119
Figure 3.16. Inhibition of CapG release from LPS-stimulated THP-1 cells pre-
treated with HTA-125. (A) CapG release from LPS-stimulated THP-1 cells is
inhibited by HTA-125. (B) HTA-125 significantly reduced CapG release from
THP-1 cells stimulated with LPS (100 ng/mL and 1000 ng/mL). The band
intensities of CapG released from supernatants were quantified by densitometry
analysis using the Fiji imaging software. Secreted CapG from cells stimulated with
LPS ± HTA-125 or isotype control were normalised to basal CapG release from
THP-1 cells. The data were then expressed as a percentage of basal CapG (Veh.)
release ± SEM of all 3 of these experiments. Two-way repeated measures ANOVA
followed by Bonferroni’s post-hoc test was applied for multiple comparisons.
***p<0.001 compared to LPS group at respective concentrations. (C) In addition,
IL-8 cytokine levels from LPS-stimulated THP-1 cells treated with HTA-125 were
also quantified by ELISA. However, HTA-125 did not inhibit IL-8 release from
LPS-stimulated THP-1 cells. In addition, an isotype control antibody was also
included in these studies. Results are expressed as the mean cytokine level ± SEM
conducted on 3 separate occasions.

120
3.3.7 CapG released is also enhanced by LPS in BV2 cells, but not affected by
dexamethasone
The murine microglial cell line BV2 is frequently used as a substitute for
primary microglia in in vitro studies of neurological disorders and inflammation
(Henn et al, 2009). Microglia are often considered as the macrophages of the brain
and act as one of the main forms of immune defense in the central nervous system
(Perry & Teeling, 2013). Several studies have characterised the activation of BV2
cells (Dilshara et al, 2014; Olajide et al, 2013). We have previously shown that
BV2 cells express CapG (Figure 3.3). Thus, we sought to examine if CapG can
also be released following LPS stimulation like the THP-1 cells. BV2 cells
released CapG in response to LPS in a concentration-dependent manner, reaching
statistical significance at higher concentrations (100 ng/mL). There was a trend for
dexamethasone alone to trigger CapG release in BV2 cells. However, this did not
reach statistical significance over 4 independent experiments. In addition,
dexamethasone did not inhibit CapG release from LPS-stimulated BV2 cells
(Figure 3.17), indicating very different mechanisms for LPS stimulated release
from PMA differentiated THP-1 macrophage like cells and the microglial line
BV2.
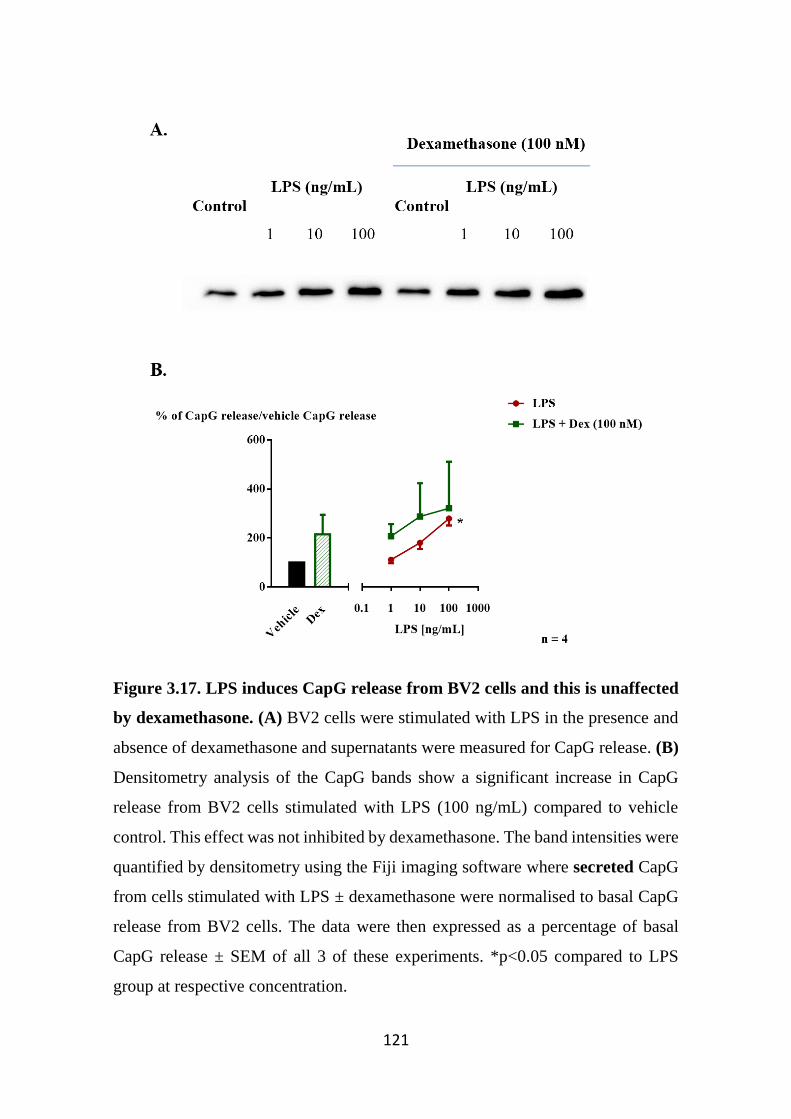
121
Figure 3.17. LPS induces CapG release from BV2 cells and this is unaffected
by dexamethasone. (A) BV2 cells were stimulated with LPS in the presence and
absence of dexamethasone and supernatants were measured for CapG release. (B)
Densitometry analysis of the CapG bands show a significant increase in CapG
release from BV2 cells stimulated with LPS (100 ng/mL) compared to vehicle
control. This effect was not inhibited by dexamethasone. The band intensities were
quantified by densitometry using the Fiji imaging software where secreted CapG
from cells stimulated with LPS ± dexamethasone were normalised to basal CapG
release from BV2 cells. The data were then expressed as a percentage of basal
CapG release ± SEM of all 3 of these experiments. *p<0.05 compared to LPS
group at respective concentration.

122
3.4 Discussion
Macrophage capping protein (CapG) is a member of the gelsolin protein
family, that is best known for its regulatory role in actin polymerization and thus
plays an important role in maintaining cell structure, integrity and cell mobility
(Mishra et al, 1994). As the name suggests, CapG is highly expressed in
macrophages, where it is estimated to represent 1% of total cytoplasmic protein,
and is important in key macrophage functions such as membrane ruffling
(formation of thin cell surface membrane protrusions) and phagocytosis (Witke et
al, 2001). The gelsolin superfamily are best known for regulating these largely
intracellular processes. However, both gelsolin and CapG can be detected in
plasma and whilst the role of plasma gelsolin is relatively well established (as
described in Chapter 1), the biological role of secreted CapG is still poorly
understood.
Cytoplasmic and plasma gelsolin are derived from a single gene by
alternative splicing. The secreted form differs from the cytoplasmic form by the
presence of a 25-amino acid signalling peptide and the presence of disulfide bonds
between the cysteine residues 188 and 201 (McGough et al, 2003). Plasma gelsolin
is present in substantial quantities in the blood (190-300 µg/mL) with its function
proposed to be identical to its intracellular role, binding and severing actin
filaments, but in this scenario the actin being in the plasma. During normal cell
turnover or tissue injury, a variety of cytoplasmic proteins are released including
actin (Lind et al, 1986). If not cleared, actin may increase the viscosity of
extracellular fluids such as plasma and hence impair tissue perfusion (Dahl et al,
1999). The actin-scavenger system, which includes gelsolin works in concert to
bind, sever and clear actin from the blood through the reticuloendothelial system
in the liver (Erukhimov et al, 2000).
Similar to gelsolin, CapG has also been shown to be secreted from cells,
especially from macrophages. Through Western blot analysis, CapG was shown to
be secreted from the murine macrophage cell lines P388D1 and J774, but not from

123
a number of non-macrophage cell lines (Johnston et al, 1990). In addition,
transfection of CapG into COS cells (fibroblast-like kidney cell lines) resulted in
CapG secretion compared to non-transfected COS cells (Johnston et al, 1990).
CapG was also shown to be present in human plasma, although the concentrations
in plasma (0.3-0.5 µg/mL) are considerably lower than gelsolin (Johnston et al,
1990). Unlike gelsolin, there is no noticeable molecular size difference between
the secreted CapG and intracellular CapG isoforms (Johnston et al, 1990). The
absence of a well-defined signal peptide suggests that CapG is secreted through a
non-canonical pathway similar to other mediators such as IL-1β and basic
fibroblast growth factor (bFGF) (Carta et al, 2009; Johnston et al, 1990).
Interestingly, the release of these mediators can be triggered by cell injury, and are
associated with inflammatory responses (Dinarello, 2009; Srikrishna & Freeze,
2009; Zittermann & Issekutz, 2006). In keeping with this, it has been suggested
that in response to cell injury, regardless of physiological or pathological origin,
CapG might also be released from macrophages at high concentrations and might
act as a potential pro-inflammatory mediator (Johnston et al, 1990).
A recent quantitative proteomics analysis approach followed by validation
via Western blotting analysis showed that CapG was found to be upregulated in
the synovial fluid of rheumatoid arthritis patients where mast cells and
macrophages are known to play a role (Balakrishnan et al, 2014; Ma & Pope, 2005;
Suurmond et al, 2011).
Recent studies in our laboratory identified several previously undescribed
mediators released from the activated human mast cell line HMCα using an
activity based proteomics approach. Of these, CapG was identified and subsequent
preliminary studies showed that CapG was able to stimulate the release of pro-
inflammatory cytokine IL-8 from human airway smooth muscle (hASM) cells in
vitro. IL-8 has previously been reported to promote the recruitment and activation
of neutrophils into the airways (John et al, 1998a). Combined, these findings
support the notion that CapG when released from cells acts as a pro-inflammatory

124
mediator. The major focus of this chapter was hence to examine and characterise
the expression and release of CapG from mast cells and macrophages.
Since CapG is known to be expressed in inflammatory cells such as
macrophages and neutrophils, we sought to examine the expression of CapG gene
and protein in several primary cells and cell lines of different cell origin from both
haematopoietic and structural cells. There was strong intracellular CapG
expression in several immortalised human mast cell lines such as HMCα and
LAD2 cells. As expected, CapG was also strongly expressed in the
monocytic/macrophage cell line THP-1 cells (Dabiri et al, 1992; Onoda et al,
1993). In comparison, although CapG gene expression was detectable in cell lines
of both haematopoietic and structural cell types, CapG protein was undetected or
at best weakly detected in the non-haematopoietic cells A549, BEAS2B and
SW982. This finding is in keeping with data obtained from the Human Protein
Atlas, where many cells express message for CapG, however protein expression is
low (CapG_Cell_Line_Atlas, 2016). It is however interesting to note that CapG
protein expression is upregulated in several tumour cells of epithelial origins with
metastatic properties, and that targeting CapG resulted in a reduction in this
metastatic property (Li et al, 2015; Van Impe et al, 2013). Thus, it is likely that
under normal circumstances, CapG protein expression in structural cells is very
low and tightly regulated. However, loss of control in tumorigenesis results in
enhanced CapG protein production and expression, leading to aberrant cell activity
such as metastasis. Indeed, a key feature associated with tumour cell metastasis is
the dynamic reorganisation of the actin-cytoskeleton network that contributes to
altered cell motility (Fife et al, 2014).
CapG expression in mast cells of different species such as RBLs (rat) and
primary bone marrow mast cells (BMMCs) was also examined. Interestingly,
CapG expression was undetected in the RBL cell line. It is unclear why CapG
expression is absent in RBL cells as mRNA for CapG is comparable to the other
immune cells examined. This is unlikely attributed to species specificity as the

125
antibody readily detected CapG in rat peritoneal cells. Furthermore, analysis of
CapG sequences in human, rat and mouse CapG shows high sequence homology
between all species (Figure 3.2). Although these cells are commonly used for mast
cell studies in vitro, these immortalised cell line originate from basophils and it is
possible that CapG is not expressed in these inflammatory cell types (Passante &
Frankish, 2009).
In contrast, CapG was readily detected in the primary murine bone marrow
derived mast cells (BMMCs). However, these cells are derived from bone marrow
cells and cultured in vitro, and are relatively immature and lack certain mast cell
characteristics such as poor response to IgG immune complexes (Malbec et al,
2007). Thus, we sought to examine CapG expression in mature in vivo
differentiated mast cells. Hence, CapG expression was analysed in mature, in vivo
differentiated rat macrophages and mast cells that were obtained by peritoneal
lavage of rats. Although there was detectable CapG expression in non-purified rat
peritoneal cells, this expression is likely due to macrophage population, which
account for 85-90% of total cells obtained from lavage (Allen et al, 1980).
However, expression of CapG in purified rat peritoneal mast cell was not detected.
A likely explanation for the lack of CapG protein undetected in the purified RPMC
is likely attributed to the low cell numbers following purification as confirmed by
the absence of β-tubulin in this sample. Although the amount of protein loaded in
these experiments was based on Bradford protein assay measurements, the protein
quantity measured in these samples may have consisted of primarily of granular
proteins, resulting in underestimation of total non-granular protein load.
An alternative approach to examine intracellular CapG in the primary cells
was through intracellular staining and measuring CapG expression by flow
cytometry analysis. This approach also can distinguish different cell subsets in
heterogeneous cell populations (such as rat peritoneal cells) and thus would be
useful for identifying CapG expression in different subpopulations in the rat
peritoneal cells (Krutzik et al, 2004). The forward and size scatter profile of the

126
purified mast cells was identified and used as an indicator of the mast cell
subpopulation in non-purified samples. There was a positive CapG expression in
macrophages, mast cells and lymphocytes, which was previously confirmed in
proteomics studies conducted by others who have documented these findings
online (CapG_Cell_Line_Atlas, 2016).
Taken together, these data suggest that CapG is expressed in human and
mouse mast cells lines, but is not expressed in the rat basophilic leukaemic cell
line RBL. CapG was also detected in primary mouse BMMCs and rat peritoneal
macrophages. Whilst by Western blot CapG was not detected in RPMC, this was
likely due to technical issues as the protein was detected using flow cytometry. It
is more likely that CapG was undetected in Western blot due to low cell numbers,
as demonstrated by the absence of β-tubulin in relevant blots. Nevertheless, we
have shown that CapG is expressed in both in vitro and in vivo-derived mast cells.
As previously mentioned, preliminary studies identified CapG as a novel
mediator secreted from the human mast cell line HMCα cells upon IgE/antigen
stimulation. Therefore, we sought to further characterise CapG and its relation to
mast cell biology, in particular how this protein is released from activated mast
cells. In these earlier studies, the HMC-1 cells used were stably-transfected with
the FcεRIα-subunit, thus permitting IgE-dependent activation (Xia et al, 2011).
However, when these studies were repeated, we were unable to detect differences
in CapG release from IgE/antigen activated HMCα cells compared to non-
stimulated controls. In addition, IL-8 cytokine was also measured in this study, and
interestingly, there was a noticeable reduction in cytokine release from these cells
in response to antigen compared to previous studies. Furthermore, these cells also
responded poorly to NECA, another stimuli that was previously shown by us and
others to trigger high levels of IL-8 cytokine release from stimulated cells (Xia et
al, 2013b). Although we have attempted different methods including establishing
new cultures from cryovials and using newly made stimuli, IL-8 release from these
cells remains considerably less than previous studies. Therefore, data

127
interpretation using this cell line is difficult due to the general loss of cell reactivity
to various stimuli.
Another commonly used mast cell line in recent years is the LAD2 cell line,
which are a more mature mast cell line compared to HMC-1 cells. To assess cell
functionality, we first analysed LAD2 cell degranulation in response to various
known stimuli such as IgE/antigen, substance P and ionomycin as previously
analysed by others (Kirshenbaum et al, 2003). In our hands, we observed the β-
hexosaminidase degranulation from these cells is similar to that previously
reported.
When LAD2 cells were investigated for CapG release upon stimulation, we
observed a concentration-dependent release of CapG at low concentrations of
antigen (0.1 – 3 ng/mL), with statistical significance achieved at 1 ng/mL.
However, at higher antigen concentrations, CapG release from LAD2 cells was
diminished, resulting in a bell-shaped release pattern. This pattern of mediator
release from IgE-FcεRI activated mast cells is consistent with reports that higher
antigen concentrations engage signalling molecules (Huber, 2013; Magro &
Alexander, 1974), that in turn down-regulates the mast cell activity involved in
CapG release.
There are several differences between CapG and β-hexosaminidase release
from activated LAD2 cells. Like CapG, β-hexosaminidase release from antigen-
stimulated LAD2 cells was also in a concentration-dependent manner. However,
peak β-hexosaminidase release was observed at higher antigen concentrations (30
ng/mL or higher), where this concentration range did not trigger significant CapG
release from LAD2 cells. Thus, this suggests that CapG release from LAD2 cells
is likely mediated through a pathway distinct to degranulation.
However, it should be noted that although the Western blot data shown in
Figure 3.7 were a representative image of several independent experiments, there
is variability in the results, where in some experiments CapG release was not

128
affected by antigen-stimulation. Most Western blotting experiments showing
LAD2 cells release of CapG in a bell-shaped trend were performed on cells from
earlier passages. Whilst subsequent repeat experiments performed on older
passage cells showed no effect of antigen on CapG release. Likewise, antigen
stimulated β-hexosaminidase release was found to be lost in the later passages of
the LAD2 cells. Whilst experiments should ideally be conducted in cells in earlier
passage, the LAD2 cells are difficult to propagate following cryopreservation and
have a generally slow growth rate (Radinger et al, 2010). Therefore, while CapG
is released from antigen-stimulated LAD2 cells, further data obtained with early
passage cells will be necessary to consolidate this conclusion. Given the
inconsistency of the LAD2 cultures in future studies it would also be ideal to
examine CapG release from in vitro differentiated primary mast cells.
Although the neuropeptide substance P induced strong LAD2 cell
degranulation between 0.1 µM to 10 µM, CapG was not be released from substance
P stimulated LAD2 cells at a high concentration (3 µM). This suggests that CapG
release is independent of the substance P and its receptors neurokinin 1 and Mas-
related G protein coupled receptor X1 (MRGPRX1) (McNeil et al, 2015; O'Connor
et al, 2004). Ionomycin was used as a positive control in this study, and although
there was a noticeable increase in CapG release from ionomycin-stimulated cells,
this was most likely due to the calcium ionophore inducing cell death as indicated
by trypan-blue exclusion results.
In summary of mast cell data, this study has shown that CapG is released
from antigen, but not substance P stimulated LAD2 cells. Moreover, this selective
release is unlikely associated with the degranulation pathway. Further
investigation is required to better understand the regulated release of this protein
from mast cells. In particular, studies to elucidate the signalling cascade and
release pathways of CapG are important as knowledge of these mechanisms may
provide novel therapeutic strategies for targeting CapG and its pro-inflammatory
actions (as discussed in Chapter 6) as an alternative to current inflammatory

129
disease treatments. However, studies were not straightforward to interpret due to
variabilities in results likely associated with cell passage and difficulties in
culturing human mast cells.
Since CapG is known to be released from macrophages, we sought to
examine the regulation of CapG release from this cell type. THP-1 cells are a
commonly used cell line studying monocyte/macrophage function in vitro. These
non-adherent cells were initially derived from a patient diagnosed with acute
monocytic leukaemia and have been shown to exhibit monocyte features such as
morphology, gene expression and expression of membrane antigens (Reyes et al,
1999; Tsuchiya et al, 1980). In addition, THP-1 monocytes can also be
differentiated into macrophages following treatment with PMA as well as other
stimuli (Daigneault et al, 2010), where the cells undergo a morphological change
and become strongly adherent to tissue-culture plastics. These cells also express
classical macrophage markers and have increased production of several secretory
products, thus making this a useful cell line for monocyte/macrophage studies
(Daigneault et al, 2010). Thus, we investigated whether THP-1 cells release CapG
and whether this process can be regulated by cell stimulation. In our hands, we
observed basal CapG release from THP-1 cells. Interestingly, when we examined
CapG release from THP-1 cells is response to various stimuli, only LPS was able
to trigger CapG release in a concentration and time-dependent manner. This clearly
shows that CapG can be released in a regulated fashion following CapG
stimulation.
The release pattern of CapG from activated mast cells and macrophages is
thought to be reminiscent to that of IL-1β, a pro-inflammatory cytokine where its
secretion mechanism is not well understood. Similar to CapG, it is known that IL-
1β is not targeted for release through the conventional signal-peptide mechanism.
However, it is proposed that IL-1β is secreted in continuum, and the strength and
extent of secretion is dependent on the type of inflammatory stimulus (Lopez-
Castejon & Brough, 2011). This secretion pattern is in keeping with the

130
observations in this present study, where CapG is basally secreted from mast cells
and macrophages and secretion levels are heightened from stimulated cells.
A limitation associated with quantifying CapG released in supernatants by
Western blotting is the lack of an appropriate loading control between each
samples to normalise the data. Following gel electrophoresis, the nitrocellulose or
PVDF membranes were stained with Ponceau-S dye to detect protein bands on
membranes. However, protein bands stained consisted primarily of proteins
present in BSA, and large albumin bands present in samples (65 kDa) prevented
accurate densitometry analysis. An alternative method to detect and quantify CapG
levels in supernatants is by ELISA. Although there are commercial CapG ELISA
kits available, this was considered not cost effective due to the cost of the kit and
the limited samples permitted to be analysed. Attempts at generating an in-house
ELISA assay that allows quantification of CapG levels in supernatants were
unfortunately not fruitful. This may have been due to the commercially available
anti-CapG antibodies having dominant and overlapping epitopes which prohibited
the development of an assay with these reagents.
To further examine the regulation of CapG release from inflammatory cells,
we studied the effects of the glucocorticoid dexamethasone on CapG release from
THP-1 cell following LPS stimulation. Previous studies have shown
dexamethasone effectively reduces expression of pro-inflammatory cytokines such
as TNFα and IL-8 (Mogensen et al, 2008; Steer et al, 2000). Here, dexamethasone
treatment reduced CapG release from LPS-stimulated THP-1 cells. This was also
confirmed in immunofluorescence studies. It is should be noted here that although
the immunofluorescence study shows a reduction in intracellular CapG staining in
LPS-stimulated THP-1 cells, this finding is dissimilar to Western blotting
experiments performed on THP-1 cell pellets following LPS stimulation, where
CapG expression was not affected by LPS stimulation in the presence or absence
of dexamethasone. This difference is likely due to the experimental set up focusing
on detecting small amounts of released CapG while the intracellular CapG remains

131
abundant, leading to signal oversaturation in Western blotting analysis, thus
masking more substantial but partial loss of intracellular CapG in some treatments.
In sort, the superior quantitation and individual cell data from fluorescence
microscopy demonstrates LPS treatment results in CapG release, and also
substantially depletes intracellular CapG.
Although LPS is known to activate THP-1 cells primarily through the
TLR4-pathway, there have been studies demonstrating that LPS stimulation of
THP-1 cells can lead to activation of several other pathways (Kayagaki et al,
2013). To determine whether CapG release from THP-1 cells is mediated through
the LPS-TLR4 pathway, the anti-TLR4 antibody (HTA-125) was used to inhibit
the LPS-TLR4 pathway interaction. Indeed, inhibition of the TLR4 signalling
pathway resulted in reduction in CapG release from LPS-stimulated THP-1 cells.
TLR4 signalling can be separated into two pathways resulting in the transcription
and translation of different mediators. For example, TNFα, IL-1β and macrophage
chemoattractant protein-1 (CCL2) production is mediated through the myeloid
differentiation primary response gene 88 (MyD88) pathway. In contrast, IFNβ and
nitric oxide is produced through a MyD88-independent pathway (Zughaier et al,
2005). It would therefore be necessary to identify the pathway that mediates CapG
release from THP-1 cells upon stimulation in subsequent studies. Interestingly in
this study, IL-8 release from THP-1 cells was not inhibited by HTA-125 pre-
treatment, suggesting that LPS may be able to activate THP-1 cells through a TLR-
4 independent pathway. Indeed, a recent study showed that LPS was able to
activate non-canonical inflammation independent of TLR4 signalling, which
triggers the release of the pro-inflammatory cytokine IL-1β, which in turn
promotes IL-8 secretion (Carmi et al, 2009; Kayagaki et al, 2013).
In addition to THP-1 cells, we sought to examine whether CapG can also
be released from other macrophage-like cell lines. The BV2 cell line shares similar
characteristics to primary microglia, thus making this cell line a useful model for
studying brain-related disorders (Henn et al, 2009). Previous studies have shown

132
that LPS induces pro-inflammatory mediator release from BV2 cells (Dilshara et
al, 2014). Hence, we examined whether CapG is also released from BV2 cells
following LPS stimulation. BV2 cells released CapG in concentration-dependent
at which statistically significance was achieved at high LPS concentrations. Since
dexamethasone has been previously reported to inhibit LPS-mediated reactive
oxygen species production in BV2 cells (Huo et al, 2011), we sought to examine
whether dexamethasone would inhibit CapG release from these cells. However,
dexamethasone did not attenuate CapG release from BV2 cells following LPS
stimulation. This suggests that dexamethasone does not affect signalling pathways
associated with CapG release in BV2 cells. An explanation to the differences in
dexamethasone effect on CapG release from LPS-stimulated THP-1 and BV2 cells
could be related to macrophage heterogeneity. Others have reported that
macrophages derived from different tissues have distinct functional and secretory
properties (Blasi et al, 1994). In this study, microglia were found to be poorly
responsive to the H. candida fungus, whilst other macrophage cell lines responded
strongly to this stimulus. Furthermore, a recent study found several markers that
were distinct to both macrophages and microglia, further highlighting the
heterogeneity observed in these cells (Hickman et al, 2013), and likely
contributing to the differences in cellular responses observed in this present study.
Another likely explanation for the different responses could also be due to
heterogeneity associated with the glucocorticoid receptor, where factors including
alternative splicings, translational isoforms and post translational modifications
can account for the likely differences in glucocorticoid signalling between these
cells (Oakley & Cidlowski, 2013).
In summary, we have shown that mast cells along with macrophages
express CapG. In addition, CapG was found to be basally released from mast cells.
Our data showing CapG release from mast cells following IgE/FcεRI activation
require further consolidation with early passage LAD2 cells (Figure 3.18A).
Although it was previously reported by others that CapG is basally released from

133
macrophages, we also found for the first time that release is heightened in LPS-
stimulated THP-1 and BV2 cells (Figure 3.17B). CapG release form THP-1 cells
is inhibited in the presence of the anti-inflammatory glucocorticoid agent
dexamethasone and the anti-TLR4 receptor antibody HTA-125. This suggests that
CapG in its secreted form shows the properties of a novel mediator that potentially
modulates the inflammatory microenvironment, which may exacerbate disease
pathology. As most of this chapter focuses primarily on in vitro data, it would be
therefore interesting to analyse CapG expression, in particular its mRNA level, in
in vitro and in vivo studies. This is described in Chapter 4. In addition, the role of
extracellular CapG and its potential to serve as a pro-inflammatory mediator is
examined in Chapter 6.
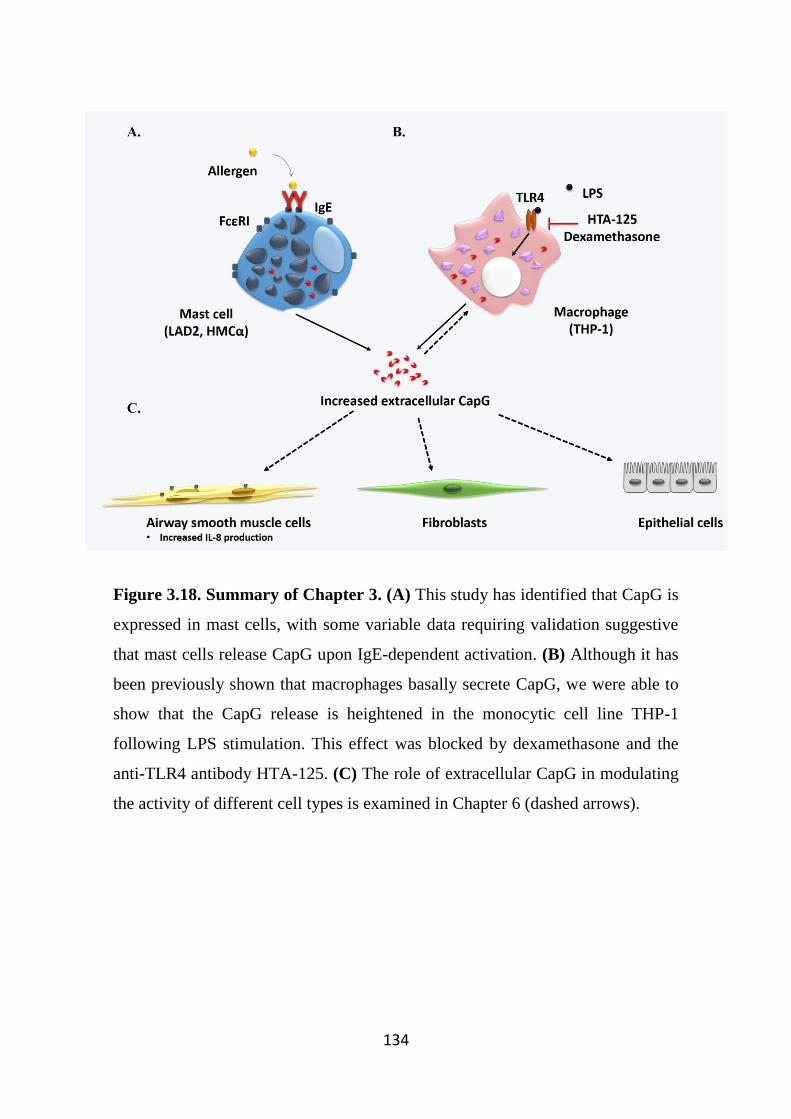
134
Figure 3.18. Summary of Chapter 3. (A) This study has identified that CapG is
expressed in mast cells, with some variable data requiring validation suggestive
that mast cells release CapG upon IgE-dependent activation. (B) Although it has
been previously shown that macrophages basally secrete CapG, we were able to
show that the CapG release is heightened in the monocytic cell line THP-1
following LPS stimulation. This effect was blocked by dexamethasone and the
anti-TLR4 antibody HTA-125. (C) The role of extracellular CapG in modulating
the activity of different cell types is examined in Chapter 6 (dashed arrows).

135

136
Chapter 4
Characterisation of CapG gene expression in vitro and in vivo
models of peripheral and central inflammatory diseases

137
4.1 Introduction
Mast cells and macrophages are immune cells that are distributed in most
tissues throughout the body where they play a key role the initiation, perpetuation
and the resolution of inflammation (Fujiwara & Kobayashi, 2005; Theoharides et
al, 2012). These cells can be activated by a range of stimuli such as cytokines,
antigens and pathogens as discussed earlier (Fujiwara & Kobayashi, 2005;
Marshall, 2004). Activated cells often result in the release of a wide variety of
mediators (Arango Duque & Descoteaux, 2014; Hart, 2001; Merluzzi et al, 2010).
Although the functions of many of these cytokines have been well described, mast
cells secrete other poorly characterised potential mediators (Xia et al, 2013b). One
of the proteins identified, macrophage capping protein (CapG) was also shown to
be released basally from macrophages (Johnston et al, 1990).
As a member of the gelsolin superfamily, CapG is best known as an
intracellular protein involved in regulating the cell cytoskeletal network by binding
and capping the barbed ends of actin to modulate actin polymerisation. In addition,
we and others have shown that other inflammatory cells such as mast cells and
neutrophil can also express this protein intracellularly, suggesting a likely
importance in inflammation (Parikh et al, 2003). However, the role of CapG in
inflammation remains poorly understood. In particular, not much is known of the
extracellular role of CapG. Previous studies by others have demonstrated that
CapG levels are elevated in the synovial fluid of rheumatoid arthritis patients
(Balakrishnan et al, 2014). In addition, our further studies, described in Chapter 3,
also demonstrated that CapG release is enhanced from IgE-activated mast cells and
LPS-stimulated macrophages. Combined, this indicates that elevated levels of
extracellular CapG during inflammatory conditions suggests a potential pro-
inflammatory role for this protein. During inflammation, many inflammatory-
associated genes are transcriptionally regulated (Medzhitov & Horng, 2009). Thus,
we sought to ascertain whether CapG gene expression would be modulated in
inflammatory conditions. As the role of CapG is better characterised in

138
macrophages, all in vitro studies examining CapG message levels were performed
in macrophages.
LPS is a major lipid and polysaccharide component of the outer wall of
most Gram-negative bacteria and elicits a strong innate immune response in a
diverse range of eukaryotic species ranging from insects to humans (De Castro et
al, 2012). LPS is a commonly used and well-characterised stimulus to recapitulate
clinical aspects of lung inflammation (Conti et al, 2010; Hakansson et al, 2012).
LPS activates resident immune cells including macrophages (Meng & Lowell,
1997), mast cells (Vosskuhl et al, 2010), T cells (Eisenbarth et al, 2002),
neutrophils (Soler-Rodriguez et al, 2000), as well as cells that are not classically
defined as immune cells such as epithelial cells that are important early sentinels
of infection (Schulz et al, 2002).
The host immune system typically recognises LPS via Toll-like receptor-4
(TLR4) and this triggers a cascade of signalling pathways that initiates host
defense and also primes the acquired immune response (Hoshino et al, 1999b;
Ravasi et al, 2002). LPS is able to drive macrophages towards the classically
activated (M1) phenotype, where it induces a dramatic change in expression of
different genes in macrophages, where gene transcription and subsequent release
of pro-inflammatory cytokines such as tumour necrosis factor-α (TNFα),
interleukins (IL-) 1, 6, 8, 10, 12 and 15 (Ravasi et al, 2002; Rossol et al, 2011;
Wang et al, 2014b). As previously demonstrated in Chapter 3, LPS also triggered
the release of CapG from macrophages, and this effect was reduced by the
inhibition of the TLR4 receptor. However, the gene regulation of CapG by LPS
has not been explored.
Macrophages also express other TLRs that enables these cells to recognise
other pathogens, including respiratory syncytial virus (RSV). RSV infection
triggers severe exacerbation of asthma, worsens disease symptoms and also
impairs lung function (Zomer-Kooijker et al, 2014). RSV infections are also the
leading cause of infant hospitalisation, accounting for more than 70% of

139
bronchiolitis-associated hospitalisation cases in the developed world (Henrickson
et al, 2004). Furthermore, asthma is also associated with increased susceptibility
for severe RSV disease (Stensballe et al, 2009). Inflammatory cells such as
macrophages express TLR2 that recognise RSV, and this also polarises the cells
towards the M1 macrophage phenotype as they release pro-inflammatory
mediators including TNFα and IL-6, as well as promoting neutrophil recruitment
and activation of dendritic cells in the lung (Murawski et al, 2009). Interestingly,
RSV can also promote tissue resolution by inducing the release of IL-4 and IL-13
from macrophages that in turn promotes circulating monocytes to differentiate into
the alternatively-activated (M2) phenotype (Shirey et al, 2010). Moreover, RSV is
also able infect and hijack macrophages, where several macrophage functions
including phagocytosis and the release of pro-inflammatory cytokines are
diminished (Franke-Ullmann et al, 1995; Rivera-Toledo & Gomez, 2012; Senft et
al, 2010). This compromised macrophage function leaves the host susceptible to
subsequent secondary infections (Franke-Ullmann et al, 1995). As CapG plays a
prominent role in several macrophage functions including phagocytosis, it is
important to examine its gene expression profile and how this might be modulated
by RSV.
As previously mentioned, macrophages are located in various tissues
including bone, liver and brain. Microglia are often described as the resident
macrophages of the brain and represent approximately 10% of cells in the human
brain (Cucchiarini et al, 2003). Microglia, like macrophages, arise from the
myeloid cell lineage that initially migrate into embryonic brain during
development. Resting microglia and macrophages can often be differentiated by
specific criteria such as morphological appearance, immunological/molecular
marker expression and functional characteristics. For example, resting microglia
are morphologically characterised by a small soma and branching (ramified
processes) as opposed to the oval, rounded or amoeboid shape of macrophages
(Gate et al, 2010). However, both cells share similar characteristics (ElAli &

140
Rivest, 2015; Gensel et al, 2009; Qian & Flood, 2008). Activated microglia are
able to undergo morphological changes similar to macrophages and are able to
express several cell surface markers following activation (Gate et al, 2010).
Microglia are the major inflammatory cell type in the brain, and become activated
following tissue damage or pathogen invasion should the tightly-regulated
environment controlled by the blood-brain barrier be compromised (Daneman,
2012). Microglia are involved in phagocytosis to remove foreign pathogens or
substances and damaged neurons, as well as secreting inflammatory mediators
such as prostaglandins, TNFα, IL-1, and free radicals such as nitric oxide and
superoxide (Qian & Flood, 2008). In addition, microglia are also important in
inflammation resolution and tissue repair (Ginhoux et al, 2013). However,
undesirable microglia activation and inflammation is observed in a range of brain
pathologies, where microglia exert detrimental effects on neurons, and contribute
to the pathologies of common neurological disorders such as Alzheimer’s and
Parkinson’s disease (Kingwell, 2012; Perry et al, 2010).
Alzheimer’s and Parkinson’s diseases are two of the most common
neurodegenerative diseases (Nussbaum & Ellis, 2003). Both diseases share several
similarities, where neurons are damaged and die over the course of the disease.
Both disorders can ultimately lead to dementia, with Alzheimer’s disease
constituting two thirds of overall dementia cases, whilst Parkinson’s disease
accounts for a smaller portion of cases (Nussbaum & Ellis, 2003). Despite some
similarities, the pathology and neurophysiological differences associated with both
disorders differ. In Alzheimer’s disease (AD), the aggregation of misfolded β-
amyloid oligomers and tau protein is heavily implicated in neuronal death and
leads to brain atrophy due to the shrinkage of the cerebral cortex and hippocampus
(Crespo-Biel et al, 2012; Double et al, 1996). This often results in patients
suffering cognitive deficits and altered behaviour (Reitz & Mayeux, 2014).
Clinical work and animal studies suggests a strong role for microglia involvement
preceding amyloid plaque formation (Heneka et al, 2005). Microglia aggregate in

141
close proximity to the amyloid plaques and the plaques are able to directly activate
microglia through a range of receptors including antibody (Fc) receptors, Toll-like
receptors, and complement receptors, thus implicating several different pathways
in Alzheimer’s disease pathogenesis (Doens & Fernandez, 2014). In addition,
microglia can also be activated by DAMPs released from damaged neurons
(Bolmont et al, 2008; ElAli & Rivest, 2015). Whilst early microglia activation is
thought to be beneficial in clearing the toxic plaques from the brain, sustained
activation of these inflammatory cells likely has detrimental effects that lead to
exacerbation of inflammation, enhanced amyloid plaque deposition and the
progression of neurodegeneration (Hickman et al, 2008).
In contrast, the aggregation of the α-synuclein protein is believed to
contribute to chronic inflammation-induced neurodegeneration and subsequent
death of dopamine-producing neurons in the substantia nigra and striatum of
patients suffering from Parkinson’s disease (PD) (Qian & Flood, 2008). Whilst this
14 kDa monomeric protein is produced by healthy neurons, in PD patients the
protein forms oligomers called Lewy bodies found in neurons, which is a hallmark
feature of PD (McKeith, 2004). PD patients often suffer from resting tremors,
posture instability and rigidity (Dauer & Przedborski, 2003). Post-mortem analysis
of the substantia nigra in PD brains showed evidence of microglial activation in
the regions where the degeneration of dopamine-producing neurons is highly
prominent (McGeer et al, 1988). In addition, elevated levels of pro-inflammatory
mediators such as IL-1β, TNFα, eicosanoids, as well as increased free radicals,
within the substantia nigra suggests a strong association between microglial
activation and PD disease progression (Hunot et al, 1996; Mogi et al, 1994).
An interesting feature commonly associated with neuroinflammatory
disorders such as AD and PD, is the infiltration of systemic macrophages into the
brain (Stoll & Jander, 1999). Macrophages are important in the clearance of
amyloid plaques in AD, as diminished macrophage recruitment is associated with
increased plaque load and mortality in mice (El Khoury et al, 2007). However, in

142
AD patients, infiltrated macrophages are poorly differentiated and are unable to
clear amyloid plaques, often resulting in cell apoptosis, as opposed to the mature
macrophages of healthy control patients which are able to phagocytose the amyloid
plaques effectively (Fiala et al, 2005). In addition, brain samples of AD patients
also have higher levels of pro-inflammatory cytokines released from microglia
compared to control (Wang et al, 2015). This results in an exacerbated
inflammatory response, likely as a compensatory mechanism initiated by the
adaptive immune system to aid macrophages in plaque clearance (Fiala et al,
2005). In PD, the role of macrophages in disease pathology is not as clear as AD.
Aggregated misfolded α-synuclein that is commonly observed in PD pathology is
usually stored intracellularly in neurons and likely does not activate macrophages
as strongly as amyloid plaques in AD (Pey et al, 2014). However, release of these
protein from damaged neurons in the substantial nigra results in activation and
release of mediators that exacerbates inflammation and subsequent neuronal
degeneration at the nigra (Zhang et al, 2005).
A recent study showed that in the brain of AD patient brain samples
exhibited a higher expression of the marker CD163, a member of the scavenger
receptor cysteine-rich superfamily group B, which is expressed selectively on
circulating monocytes and tissue macrophages (Pey et al, 2014). This study
highlights the infiltration and likely involvement of systemic macrophages in AD
pathology. Although samples from PD patients also showed increased CD163
expression compared to control, the expression was less pronounced, suggesting
macrophages are not as heavily involved in PD pathology in comparison to AD
(Pey et al, 2014). Nevertheless, there is strong evidence of systemic macrophages
and their involvement in the pathogenesis of neuroinflammatory disorders.
In relation to CapG, it is known that this protein acts as a regulator of actin
polymerisation (Silacci et al, 2004). In addition, previous results (Chapter 3)
suggest that CapG is a novel pro-inflammatory mediator that can be released from
activated immune cells including macrophages and microglia. To our knowledge,

143
the gene expression profile of CapG in inflammatory disease models is not
understood. Thus, we sought to examine and compare gene expression of CapG in
several inflammatory settings. In particular, we examined CapG gene expression
between resting and activated macrophages in vitro as well as compared gene
expression between brain samples obtained from control, AD and PD patients.
In addition, we were also interested in examining CapG expression in
different mouse models of inflammation. This includes utilising an APP/PS-1
transgenic mouse model, which express mutations that provide relevance to the
human disease setting, and thus a commonly used model for AD (Liu et al, 2008).
In addition, since LPS was found to be a strong stimulus for CapG release in
macrophages, we sought to examine whether CapG gene expression would differ
in LPS-mediated lung inflammation in mouse model. Finally, we also examined
CapG gene expression in a RSV model of lung inflammation. Since both LPS and
RSV were administered into mice intranasally, CapG gene expression was
primarily focused on the lungs and cells of the bronchoalveolar space. Knowledge
of CapG gene expression in these inflammatory conditions is important as they
might serve as potential therapeutic targets for treating inflammatory-related
disorders, or may serve as a marker of disease progression.

144
4.2 Specific Methods
4.2.1 Animal models
4.2.1.1 LPS and RSV models
C57BL/6 mice (used following approval of the University of Melbourne
animal ethics committee; Ethics codes: 1312919 and 1212356) were intranasally
inoculated with LPS (10 μg/kg) or RSV (Strain A2, ATCC; 2 x 106 virions/mouse)
as described in Section 2.9.1.2.1. After the treatment period, mice were killed by
pentobarbitone (150 mg/kg, Provet, Australia). The harvest of lung and
bronchoalveolar (BAL) fluid via lavage was performed by Ms. Shenna
Langenbach (Department of Pharmacology and Therapeutics, University of
Melbourne).
Mouse BAL cells were collected by bronchoalveolar lavage and the lungs
harvested and stored in liquid nitrogen. Differential cell counts were performed to
identify cell subpopulations using Diff-Quik staining as per the manufacturer’s
protocol (Kwik Diff Stain Kit, Thermo Scientific). The lungs were crushed with
liquid nitrogen and stored in -80 Cº for future processing.
4.2.1.2 APPSWE/PS-1ΔE9 (APP/PS-1) model
The APP/PS-1 transgenic mouse express the amyloid precursor protein
carrying the Swedish mutation (APPSWE) and mutant human presenilin 1 (PS-1 ΔE9)
both directed to the CNS neuron (Jankowsky et al, 2004). The APPSWE transgene
promotes secretion of human β-amyloid protein at high levels. The PS-1 ΔE9 protein
is found in early-onset familial AD, and plays a crucial role in regulating the
secretion of β-amyloid protein. As a result, these mice display an early onset of
Alzheimer’s disease. Hence, this mouse model is commonly used to study AD
disease pathogenesis and developing new therapies (Liu et al, 2008). Several
characteristics of the APP/PS-1 mouse model are described in Figure 4.1. Mice
were aged for 9 and 13 months before experimental use. Mice (used following

145
approval of the University of Melbourne animal ethics committee; Ethics code:
1312746) were killed by cervical dislocation and brain cortex tissue harvested and
snap frozen in liquid nitrogen as previously described (Minter et al, 2016).
Figure 4.1. Overview of the APP/PS-1 mouse phenotype. The APP/PS-1
transgenic mouse model is a commonly used model for studying Alzheimer’s
disease as these mice progressively develop β-amyloid plaques and exhibit many
symptoms associated with Alzheimer’s disease.
4.2.2 Human post-mortem brain tissues
mRNA samples of the human cortical brain from four patients diagnosed
with either Alzheimer’s or Parkinson’s diseases at death, were kindly provided by

146
Dr Tony Frugier (Victorian Brain Bank Network, Australia). In addition, aged
matched post-mortem brain cDNA samples were also provided as a control in this
study.
4.2.3 Cell stimulation
Cell pellets from stimulated and non-stimulated THP-1, BV2 and rat
peritoneal macrophages (Sections 2.1.4, 2.1.5 and 2.2.1) were harvested by
centrifugation (300 g, 5 min). Samples were lysed and stored in -80ºC for
downstream purposes.
4.2.3.1 Human monocytes
The cDNA samples of stimulated human blood monocytes kindly provided
by Prof. Alastair Stewart (University of Melbourne) were as described in Section
2.9.1.3. In the presence of GM-CSF, monocytes can undergo differentiation into
macrophages in vitro (Bender et al, 2004). Following differentiation of human
monocytes to macrophages using GM-CSF, cells were stimulated with a range of
stimuli including those known to polarise macrophage to M1 or M2 phenotypes.
After 24 hours of stimulation, cells were lysed in TRIzol reagent (Invitrogen) and
RNA extracted (in accordance with the manufacturer’s protocol) and reverse
transcribed into cDNA as per the manufacturer’s protocol.
4.2.4 mRNA extraction and qPCR
4.2.4.1 mRNA extraction and cDNA synthesis
Following cell stimulation, THP-1, BV2 and rat peritoneal macrophage cell
pellets were harvested and cells were lysed and mRNA extracted. Messenger RNA
extraction experiments were performed using the Qiagen RNAEasy® Plus Kit
(Qiagen, Mortlake, NSW, Australia) in accordance with the manufacturer’s
protocol.

147
The collection of LPS or RSV-infected mouse lung samples and BAL cells
was performed by Ms. Shenna Langenbach (Department of Pharmacology and
Therapeutics, University of Melbourne). All samples were stored at -80ºC prior to
use. Messenger RNA extraction of these samples was performed using the Qiagen
RNAEasy® Plus Kit in accordance with the manufacturer’s protocol.
mRNA from APP/PS-1 mice were extracted by phenol-chloroform
separation using TRIzol® reagent (Chomczynski & Sacchi, 2006) as performed
by Dr. Myles Minter (Department of Pharmacology and Therapeutics, University
of Melbourne).
Following all mRNA extraction procedures, the purified RNA was used to
generate first-strand cDNA by reverse transcription, as described in Section 2.9.3.
4.2.4.2 qPCR
qPCR gene expression analysis was performed using the methods described
in Section 2.9.4 (refer to Table 2.3 for the primers utilised).
4.2.5 Statistical analysis
Data from qPCR analysis were expressed as means ± standard error of mean
(SEM), where n represents the number of independent primary cell cultures, mouse
or patient samples, or numbers of experiments repeated using cell line. If
applicable, an appropriate statistical analysis test was performed (refer to Section
2.13).
Results shown were plotted using Graphpad Prism software (version 6.01).
If a statistical significance was obtained, then * denotes p<0.05, ** denotes p<0.01,
and *** denotes p<0.001.

148
4.3 Results
4.3.1 CapG mRNA expression is differentially expressed in macrophages
following LPS stimulation
Since studies from Chapter 3 showed that CapG and IL-8 release from THP-
1 cells can be modulated by LPS and dexamethasone, we sought to examine both
CapG and IL-8 gene regulation by LPS in the presence and absence of
dexamethasone. In addition, Zinc Finger and BTB Domain Containing 16
(ZBTB16), a gene known to be strongly induced by glucocorticoids was also
measured. IL-8 gene expression levels were significantly increased in LPS-
stimulated THP-1 cells, indicating robust LPS-mediated activation had occurred
(Figure 4.2A). However, CapG gene expression levels in LPS-stimulated THP-1
cells was decreased compared to untreated THP-1 cells (Figure 4.2B).
Interestingly, although dexamethasone inhibited IL-8 and CapG release from THP-
1 cells, dexamethasone did not modulate either of these genes in LPS-stimulated
cells compared to non-dexamethasone treated cells. As expected the gene
expression levels of ZBTB16 were higher in cells treated with dexamethasone,
demonstrating induction of GC-regulated genes. However, LPS did not affect
ZBTB16 gene expression in THP-1 cells or affect dexamethasone-mediated
expression of this gene.
CapG gene expression was also examined in primary human monocytes and
macrophages derived from peripheral blood mononuclear cells through GM-CSF
treatment following stimulation with a range of mediators that are known to
polarise macrophages towards the M1 (IFNγ, TNFα, and LPS) or M2 (IL-4 and
IL-10) phenotype (Figure 4.3). Although not statistically significant, there was a
trend for CapG expression in non-differentiated monocytes to be upregulated
when treated with TNFα and LPS compared to resting cells. IFNγ alone did not
affect CapG gene expression, and co-treatment of IFNγ with LPS or TNFα also
did not affect CapG gene expression. In contrast, the M2-polarising cytokines IL-

149
4 and IL-10 did not appear to have an effect on CapG expression in monocytes
(Figure 4.3A). Interestingly, in GM-CSF differentiated macrophages, only cells
stimulated with LPS showed significant reduction in CapG gene expression in
macrophages, a similar pattern observed in the THP-1 studies (Figure 4.3B).
Furthermore, the LPS-induced reduction in CapG gene expression was not affected
by co-stimulation with IFNγ.
In addition, regulation of CapG gene expression in rat peritoneal
macrophages stimulated with LPS for 4 hours and 24 hours was examined. Similar
to THP-1 cells and GM-CSF differentiated human macrophages, there was a
significant decrease in CapG gene expression at both time points in LPS-treated
rat macrophages (Figure 4.4). Although gene expression appeared to be lower at
4 hrs compared to 24 hrs stimulation, this did not reach statistical significance.
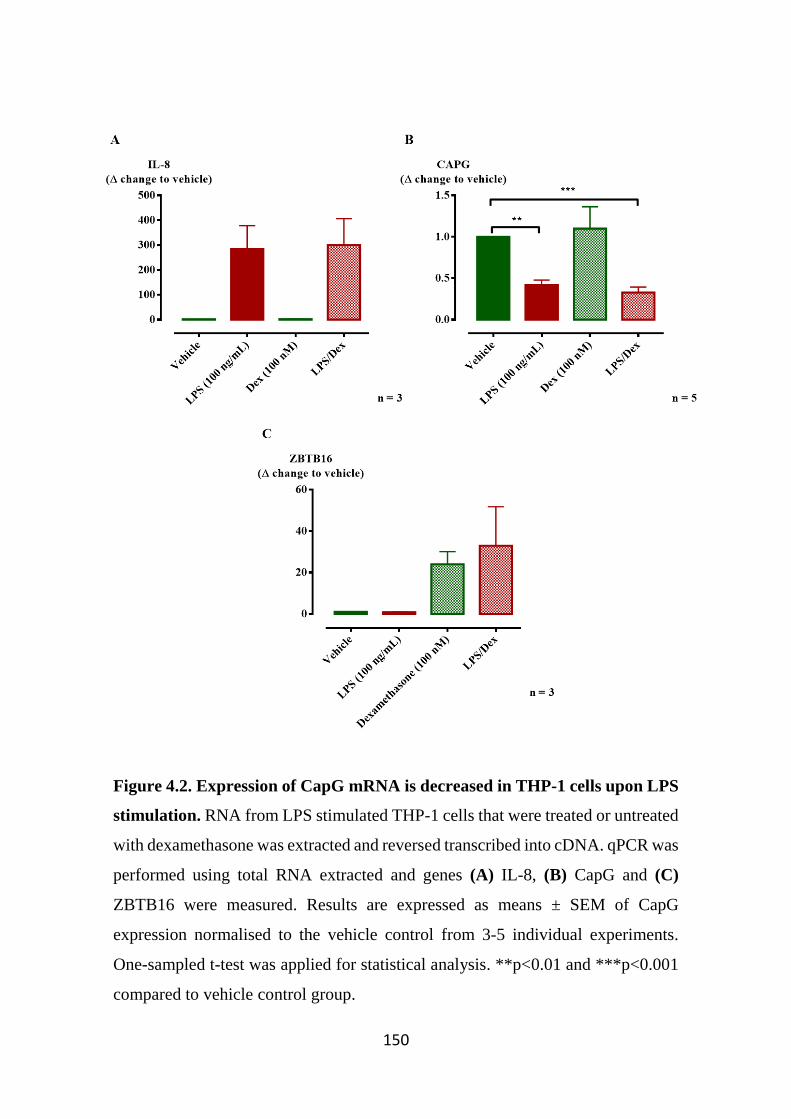
150
Figure 4.2. Expression of CapG mRNA is decreased in THP-1 cells upon LPS
stimulation. RNA from LPS stimulated THP-1 cells that were treated or untreated
with dexamethasone was extracted and reversed transcribed into cDNA. qPCR was
performed using total RNA extracted and genes (A) IL-8, (B) CapG and (C)
ZBTB16 were measured. Results are expressed as means ± SEM of CapG
expression normalised to the vehicle control from 3-5 individual experiments.
One-sampled t-test was applied for statistical analysis. **p<0.01 and ***p<0.001
compared to vehicle control group.
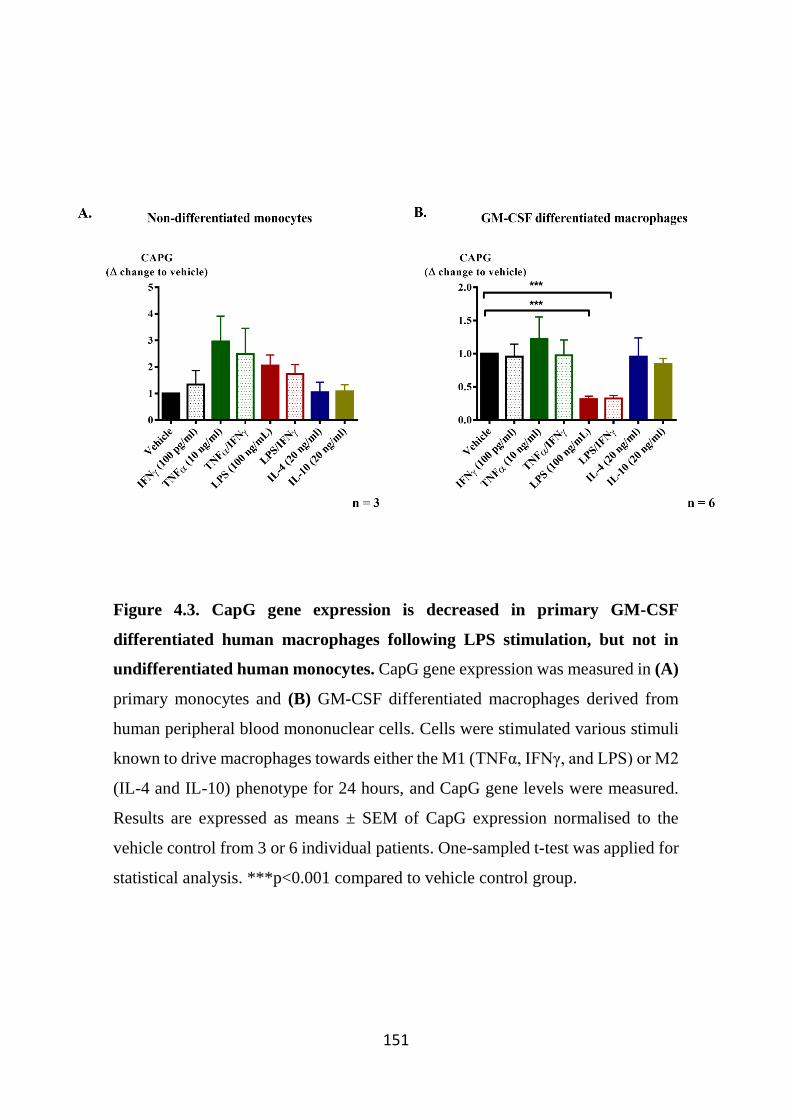
151
Figure 4.3. CapG gene expression is decreased in primary GM-CSF
differentiated human macrophages following LPS stimulation, but not in
undifferentiated human monocytes. CapG gene expression was measured in (A)
primary monocytes and (B) GM-CSF differentiated macrophages derived from
human peripheral blood mononuclear cells. Cells were stimulated various stimuli
known to drive macrophages towards either the M1 (TNFα, IFNγ, and LPS) or M2
(IL-4 and IL-10) phenotype for 24 hours, and CapG gene levels were measured.
Results are expressed as means ± SEM of CapG expression normalised to the
vehicle control from 3 or 6 individual patients. One-sampled t-test was applied for
statistical analysis. ***p<0.001 compared to vehicle control group.
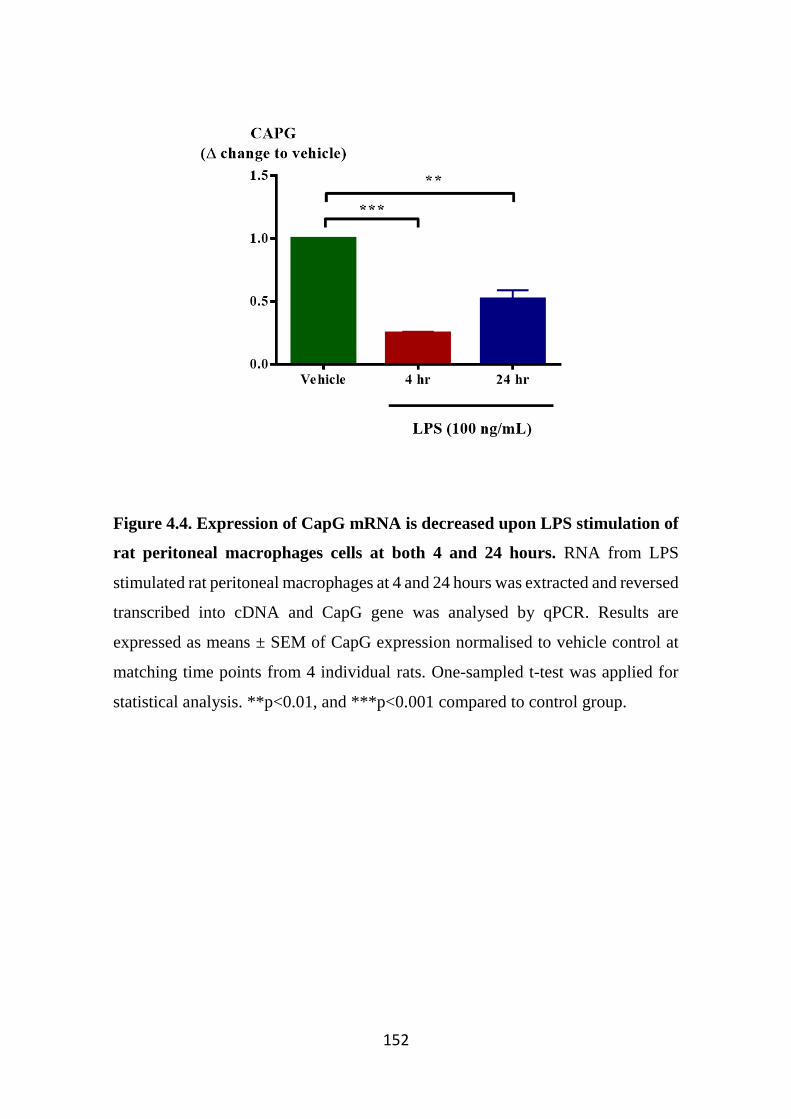
152
Figure 4.4. Expression of CapG mRNA is decreased upon LPS stimulation of
rat peritoneal macrophages cells at both 4 and 24 hours. RNA from LPS
stimulated rat peritoneal macrophages at 4 and 24 hours was extracted and reversed
transcribed into cDNA and CapG gene was analysed by qPCR. Results are
expressed as means ± SEM of CapG expression normalised to vehicle control at
matching time points from 4 individual rats. One-sampled t-test was applied for
statistical analysis. **p<0.01, and ***p<0.001 compared to control group.

153
4.3.2 CapG mRNA expression is elevated in lungs of RSV and LPS-treated
mice, but not in total BAL cells
Next, we investigated CapG cellular gene expression in samples collected
from mice treated with either LPS or RSV as two different models of lung
inflammation. Mice treated with saline were used as vehicle controls in this study.
In both studies, differential cell counts were performed and BAL cells obtained by
bronchoalveolar lavage and lung extracts were measured for both CapG and KC
gene levels. KC was used as a measure of induced lung inflammation in this study
as it has been previously shown to be upregulated in response to LPS, but not RSV
(Miller et al, 2004; Ohmori et al, 1995).
Compared to vehicle control, total BAL cell counts were significantly
increased in LPS treated mice, predominantly due to elevated neutrophil levels
(Figure 4.5A). Compared to vehicle control, CapG gene expression was
significantly downregulated in the BAL cells obtained from LPS-treated mice.
However, there was a significant increase in CapG gene expression in the total
lung extract from LPS-treated mice. As expected, KC gene expression was
significantly upregulated in both BAL cells and lung extracts of LPS-treated mice
compared to vehicle (Figure 4.5B).
In the RSV studies, there was an increase in total BAL cell count in RSV-
treated mice compared to vehicle control due largely to the increased monocytes
and lymphocytes, as previously observed (Collins et al, 2005). However, due to a
small sample size and variability in the RSV-treated animals this did not reach
statistical significance (Figure 4.6A). Although there was a significant decrease in
CapG gene expression in BAL cells, gene expression was upregulated in lung
extracts. Compared to the vehicle control, there was no significant differences in
KC gene expression between BALF cells of vehicle control and RSV-treated mice
(Figure 4.6B), which as previously observed (Miller et al, 2004). This is likely
because the KC chemokine is a neutrophil chemoattractant, and it has been
previously thought these cells lack the innate signalling system that enables RSV

154
recognition, (Bataki et al, 2005). Furthermore, we and others (Collins et al, 2005)
have shown no noticeable differences in neutrophil numbers in both treatment
groups. However, KC expression was significantly upregulated in the lungs of the
RSV-treated mice, likely due to increased inflammatory cell infiltration in the
lungs as well as increased gene expression from airway epithelial cells (Miller et
al, 2004).
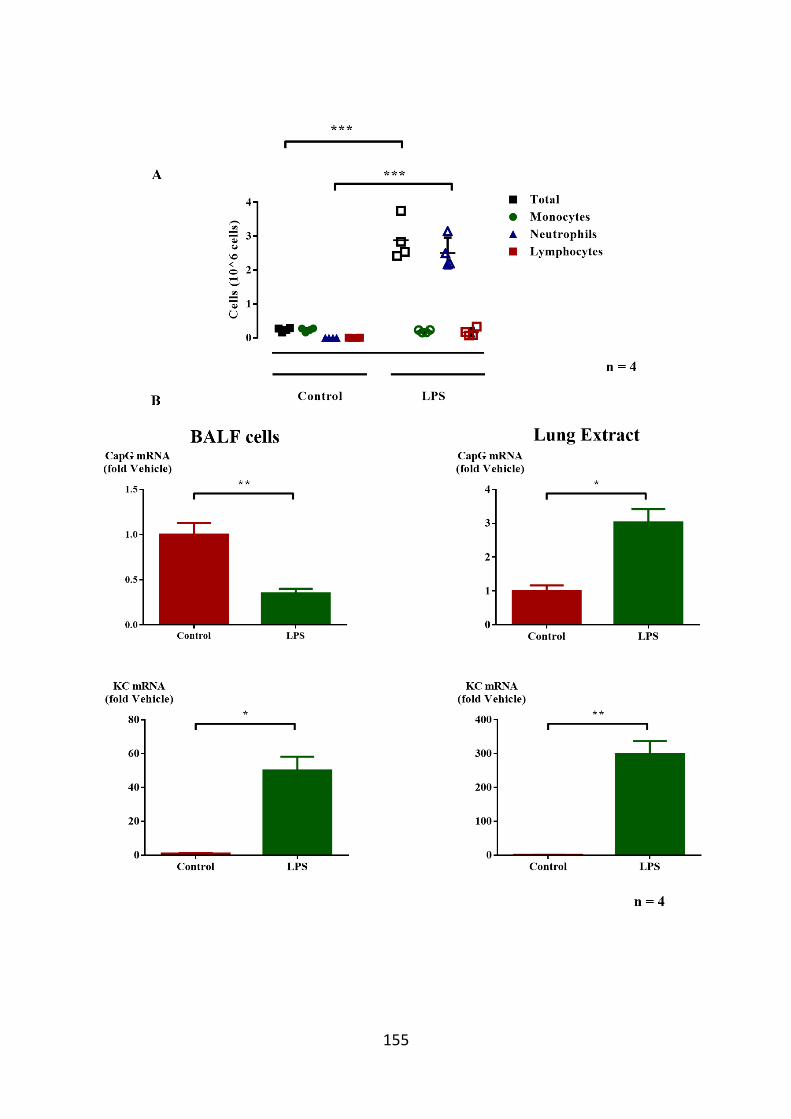
155

156
Figure 4.5. Differential CapG gene expression in BAL cells and lung extracts
obtained from LPS treated mice. Lung extracts and BAL cells were collected
from LPS-treated mice (10 μg/kg) and matching vehicle controls. (A) Differential
cell counts performed on the BAL fluid obtained from mice in different treatment
groups. There was an increase in total BAL cells in LPS-treated mice,
predominantly due to neutrophil infiltration. BAL cell enumeration is expressed as
individual cell counts, corresponding to cell types obtained from 4 individual mice
in each treatment group. An ordinary one-way ANOVA statistical analysis was
applied for comparing the cell numbers of each cell type between vehicle control
and treatment group. (B) The genes CapG and KC were measured in the BAL cells
and lung extracts. Gene expression results are expressed as means ± SEM of CapG
expression normalised to the house-keeper gene UBC from 4 individual mice in
each treatment group. One-sampled t-test was applied for statistical analysis.
*p<0.05 and **p<0.01 compared to control group.
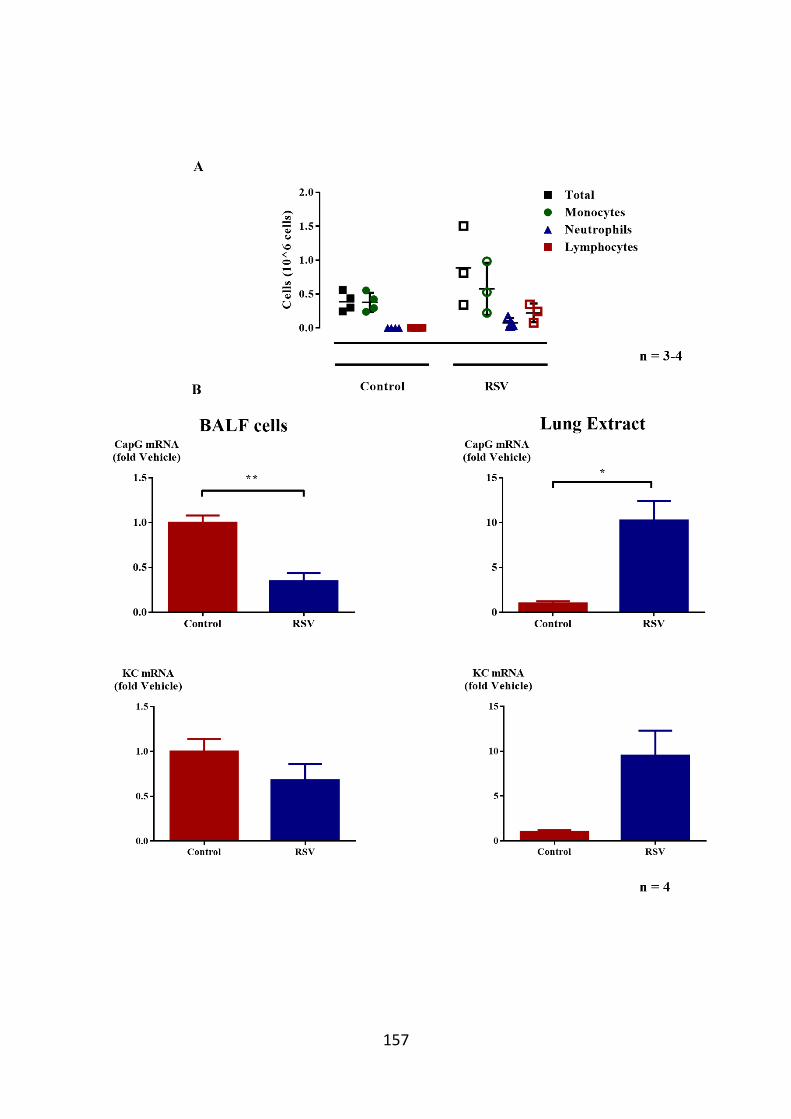
157

158
Figure 4.6. CapG gene is differentially expressed in BAL cells and lung
extracts obtained from RSV infected mice. Lung extracts and BAL fluids were
collected from RSV-infected mice (Strain A2; 2 x 106 virions/mouse) and
matching vehicle control. (A) Differential cell counts were performed on BAL
cells obtained from mice. BAL cells obtained from RSV-treated mice showed an
increased monocyte population compared to vehicle control group. Results are
expressed in a scatter-plot of individual differential cell counts measured in 3-4
mice in each treatment group. BAL cell enumeration was expressed as individual
cell counts, corresponding to cell types obtained from 3-4 individual mice in each
treatment group. (B) The genes CapG and KC were measured in the BAL cells and
lung extracts. Gene expression results are expressed as means ± SEM of CapG
expression conducted on 4 mice in each treatment group and normalised to the
control. A one-sampled t-test was applied for statistical analysis. *p<0.05 and
**p<0.01 compared to control group.

159
4.3.3 CapG expression is not affected by LPS stimulation in BV2 cells
CapG gene expression was also examined in the murine microglia cell line
BV2. Gene expression of CapG and KC were measured in BV2 cells stimulated
with LPS for 24 hours in the presence and absence of dexamethasone. In contrast
to gene expression studies in human and rat macrophages, LPS had no effect on
CapG and KC gene expression in BV2 cells (Figure 4.7). Although
dexamethasone did not affect CapG gene expression in the presence and absence
of LPS, KC expression was elevated although this was not statistically significant.
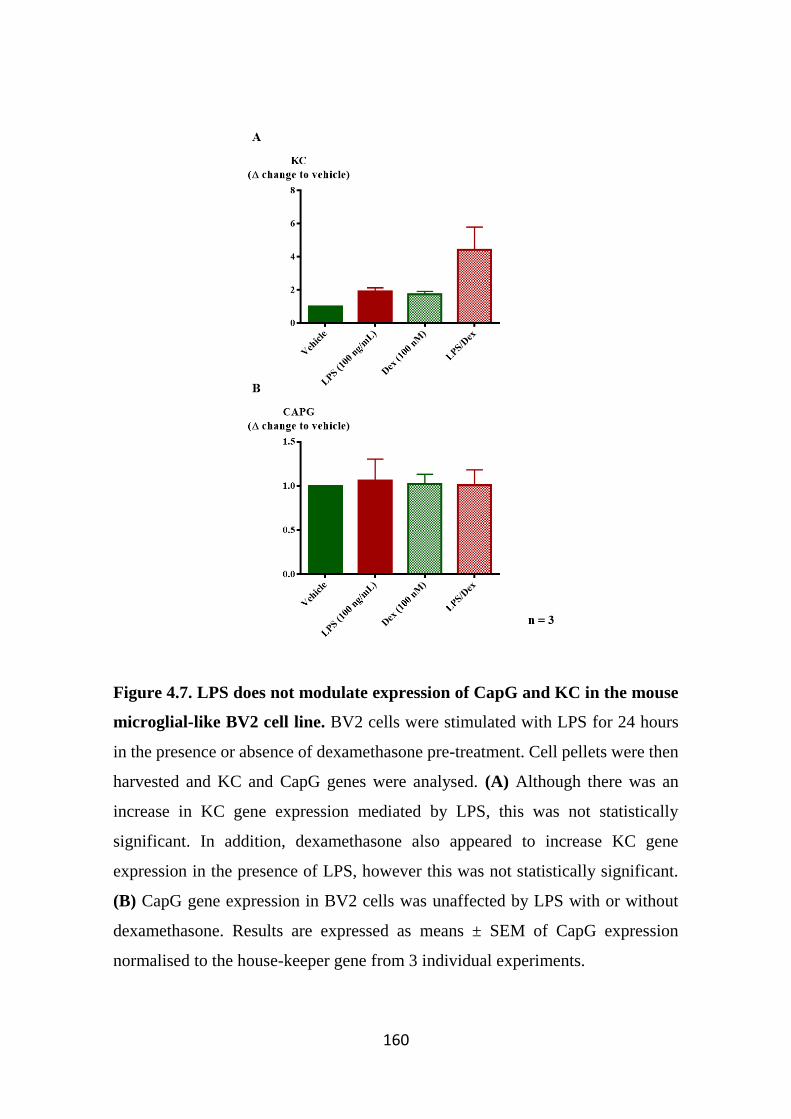
160
Figure 4.7. LPS does not modulate expression of CapG and KC in the mouse
microglial-like BV2 cell line. BV2 cells were stimulated with LPS for 24 hours
in the presence or absence of dexamethasone pre-treatment. Cell pellets were then
harvested and KC and CapG genes were analysed. (A) Although there was an
increase in KC gene expression mediated by LPS, this was not statistically
significant. In addition, dexamethasone also appeared to increase KC gene
expression in the presence of LPS, however this was not statistically significant.
(B) CapG gene expression in BV2 cells was unaffected by LPS with or without
dexamethasone. Results are expressed as means ± SEM of CapG expression
normalised to the house-keeper gene from 3 individual experiments.

161
4.3.4 CapG expression is upregulated in Alzheimer’s disease patients
Since microglia play a crucial role in the pathogenesis of neurological
disorders such as AD and PD, we sought to examine whether CapG message levels
were differentially expressed in patients diagnosed with these disorders. Post-
mortem brain samples from four AD and PD were examined. In addition, brain
samples from healthy control patients were included as a control. In comparison
to control patients, CapG gene expression was significantly upregulated in AD
patients. CapG gene expression was however unchanged in PD patients (Figure
4.8).
The APP/PS-1 transgenic mice are a commonly used mouse model to study
AD as these mice are characterised by the deposition of β-amyloid plaques in the
hippocampus and cortex (Jankowsky et al, 2004). These mice also exhibit several
symptoms of AD including social recognition memory impairments and abnormal
anxiety levels (Cheng et al, 2013). Thus, we examined CapG expression in brain
of these mice. Mice aged 9 and 13 months with age-matched wild-type controls
were killed and the cortex tissue was isolated for gene analysis. Although CapG
gene expression was unchanged in mice aged for 9 months, there was a significant
increase in CapG expression in 13-month aged mice compared to control (Figure
4.9).
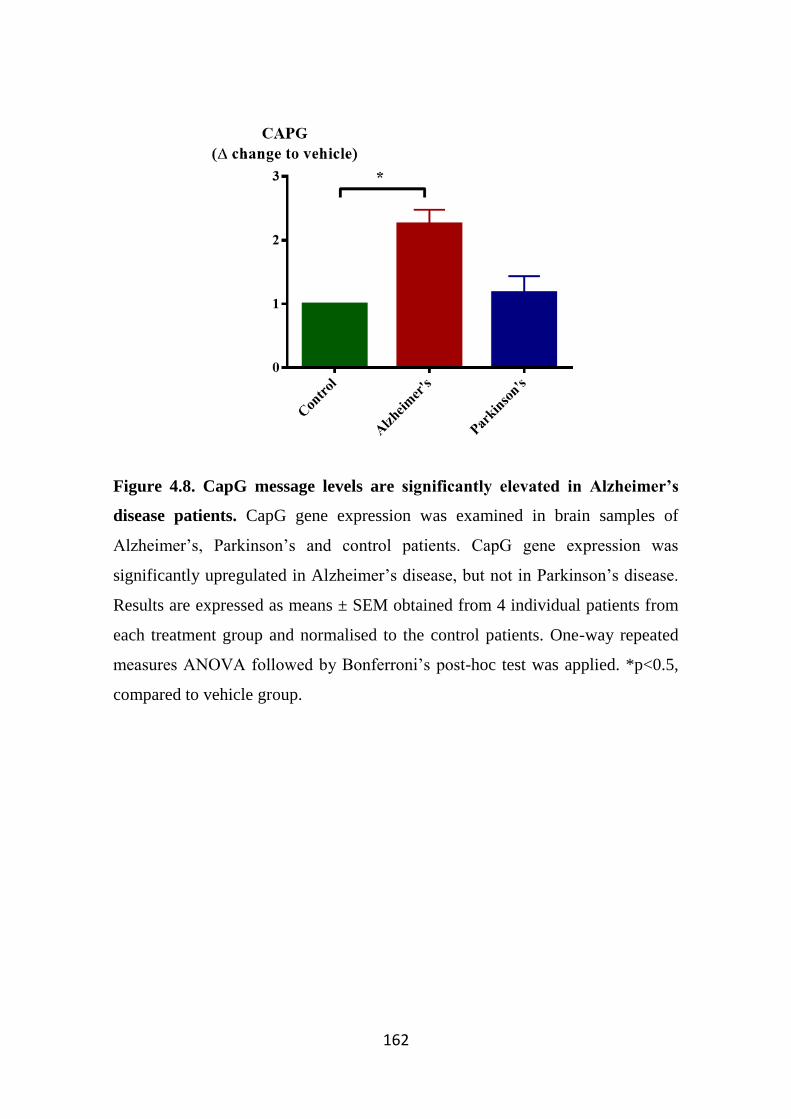
162
Figure 4.8. CapG message levels are significantly elevated in Alzheimer’s
disease patients. CapG gene expression was examined in brain samples of
Alzheimer’s, Parkinson’s and control patients. CapG gene expression was
significantly upregulated in Alzheimer’s disease, but not in Parkinson’s disease.
Results are expressed as means ± SEM obtained from 4 individual patients from
each treatment group and normalised to the control patients. One-way repeated
measures ANOVA followed by Bonferroni’s post-hoc test was applied. *p<0.5,
compared to vehicle group.
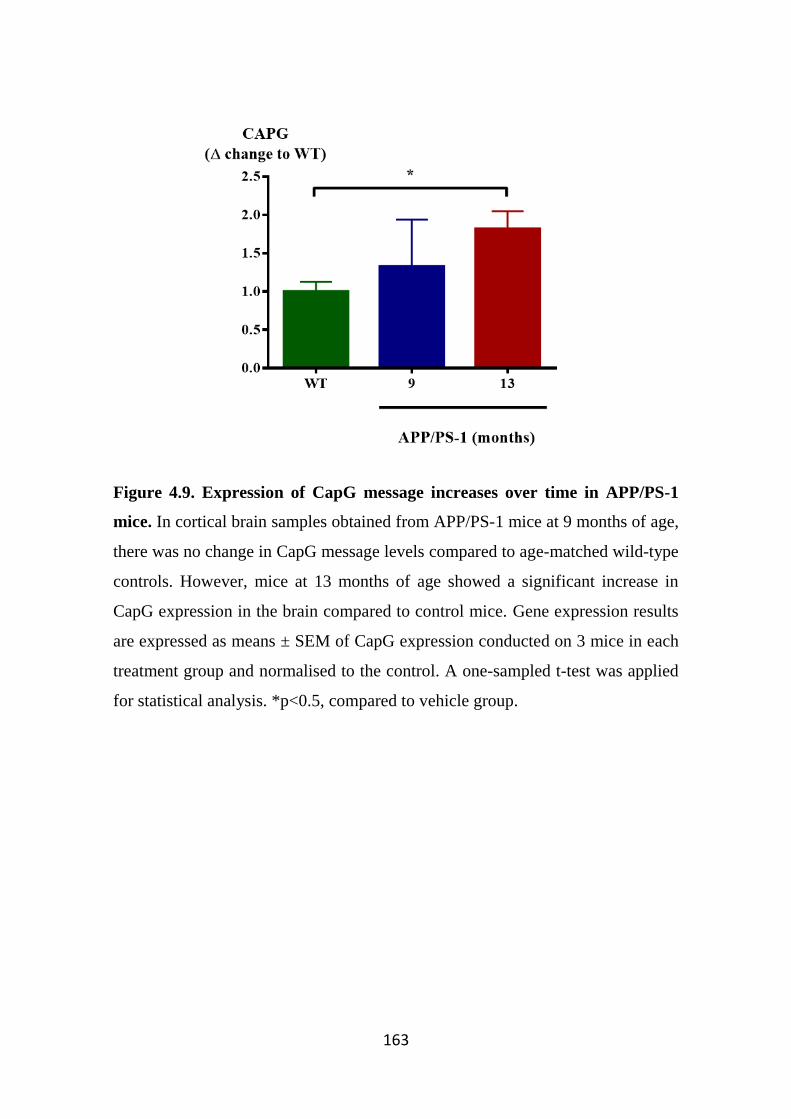
163
Figure 4.9. Expression of CapG message increases over time in APP/PS-1
mice. In cortical brain samples obtained from APP/PS-1 mice at 9 months of age,
there was no change in CapG message levels compared to age-matched wild-type
controls. However, mice at 13 months of age showed a significant increase in
CapG expression in the brain compared to control mice. Gene expression results
are expressed as means ± SEM of CapG expression conducted on 3 mice in each
treatment group and normalised to the control. A one-sampled t-test was applied
for statistical analysis. *p<0.5, compared to vehicle group.

164
4.4 Discussion
The role of CapG as a regulator of actin polymerisation is well documented
and characterised (Hubert et al, 2008; Hubert et al, 2009; Mishra et al, 1994;
Silacci et al, 2004). In addition, several recent studies have shown that intracellular
CapG overexpression is implicated in various types of carcinoma (Glaser et al,
2014; Ichikawa et al, 2013; Kimura et al, 2013; Morofuji et al, 2012; Shao et al,
2011; Zhu et al, 2012). However, the role of CapG in inflammation is still poorly
understood. It was previously reported that a loss of CapG expression results in
host defense impairment to specific infections (Parikh et al, 2003). In addition, the
protein is constitutively secreted from macrophages, and as we have shown, in
response to the inflammogen LPS. This indicates a potential pro-inflammatory role
for CapG in the inflammatory response. However, to our knowledge, specific gene
expression of CapG during inflammation has not been examined. In this chapter,
we were interested in characterising and examining the gene expression of CapG
under normal and inflammatory conditions in macrophages in vitro, and whether
this would also be translated in vivo by examining gene expression in two different
models of lung inflammation, where mice were treated with either LPS or RSV. In
addition, as macrophages are found in different tissues, we sought to examine
CapG gene expression in the brain, by determining CapG message levels in the
murine microglia BV2 cells stimulated with LPS. Finally, expression of CapG was
also measured in the cortical tissue of brain samples obtained from Alzheimer’s
and Parkinson’s disease sufferers and compared with donors who died of other
causes.
CapG message levels were downregulated in the GM-CSF differentiated
macrophages following LPS stimulation. Along with LPS, the differentiated
macrophages were also stimulated with mediators known to drive macrophages
towards either the classically-activated M1 (TNFα, LPS, IFNγ) or alternatively-
activated M2 (IL-4, IL-10) phenotypes. However, in response to these treatments
CapG gene expression levels were unchanged compared to control, suggesting that

165
CapG is unlikely to be differentially expressed in M1 or M2 macrophages. In
addition, IFNγ did not modulate LPS mediated downregulating of CapG
expression. To examine whether CapG mRNA downregulation by LPS was
affected only in differentiated macrophages, undifferentiated monocytes were also
stimulated with LPS, along with the M1 and M2 driving cytokines. However, in
monocytes, CapG gene expression remained unchanged in response to these
cytokines, as well as LPS. A possible explanation for the differences in CapG
expression between LPS-stimulated macrophages and monocytes could be related
to the sensitivity of the cells to LPS stimulation, as it has been previously shown
that macrophages are more sensitive to LPS compared to monocytes, and also
result in greater pro-inflammatory cytokine production in macrophages (Gessani
et al, 1993; Takashiba et al, 1999). Hence, the increased sensitivity to LPS is
reflected in macrophages, but not in monocytes. In addition, the downregulation
of CapG mRNA mediated by LPS was also observed in experiments conducted on
mature rat peritoneal macrophages and THP-1 cells.
Combined with data from Chapter 3, where it was observed that LPS-
stimulated CapG was released from THP-1 cells, this suggests that both reduced
CapG message expression and protein release contribute to diminished
intracellular CapG in LPS-stimulated macrophages. The reduction of intracellular
CapG would be predicted to result in an impairment in regular macrophage activity
such as phagocytosis, as previously reported (Witke et al, 2001). In addition, others
have also reported that LPS induces suppression of phagocytosis and diminishing
macrophage infiltration through disruption of the cytoskeletal network
(Wonderling et al, 1996; Zhou et al, 1999). Furthermore, others have also shown
that LPS inhibits macrophage phagocytosis of apoptotic neutrophils (Feng et al,
2011). Whilst this particular study demonstrated that LPS inhibits phagocytosis by
regulating TNFα and growth arrest-specific gene 6, it is possible that LPS can also
downregulate CapG expression and so function, thus limiting macrophage activity.
Although not statistically significant, CapG gene expression appeared to be lower

166
in rat macrophages stimulated at 4 hours compared to 24 hours, suggesting a
recovery period as a response by the macrophages to restore CapG message back
to basal levels following initial loss of expression mediated by LPS.
To further examine CapG expression during inflammation in vivo, two
different murine inflammatory lung models were used. Mice were intranasally
administered with LPS or RSV and after the inoculation period, CapG expression
was measured in lungs obtained from infected and control mice. Differential cell
counts showed in both treatment groups that BAL cell counts were higher
compared to matching vehicle control, indicating inflammatory cell infiltration to
the inflamed lung. The predominant cells infiltrating the lungs of LPS-treated mice
were neutrophils. These differential cell count data were in keeping with previous
studies reported by others (Asti et al, 2000). When CapG gene levels were
measured, there was a noticeable reduction in CapG message in BAL cells
obtained from treated mice. The downregulation of CapG in LPS-treated mice is
in keeping with our in vitro studies, where LPS supresses CapG expression, thus
impairing macrophage activity. However, downregulation of message CapG could
also be due to the increased neutrophils in total BAL cells, where neutrophils are
known to express CapG, but likely not as highly expressed as macrophages (Witke
et al, 2001). Thus, this might have contributed to lowered CapG message levels in
total RNA measured in BAL cells.
Although there appeared to be elevated BAL cell counts in the RSV-treated
mice, this was not statistically significant, likely due to the small sample size.
However, there was a trend for an increase in monocyte and lymphocyte
population in response to RSV. Despite this, CapG gene expression was also
downregulated in the BAL cells of RSV-treated mice. It has been previously
reported that RSV can attenuate macrophage activity, where it alters gene
expression profiles, and induces release of the anti-inflammatory cytokine IL-10
from macrophages, as well as supressing the release of immunoregulatory
cytokines that are important for virus clearance (Panuska et al, 1995; Rivera-

167
Toledo & Gomez, 2012; Senft et al, 2010). Thus, it is possible that RSV can
downregulate CapG message which in turn results in impairment of macrophage-
mediated inflammatory activity.
Whilst downregulation of CapG could be mediated by pathogens to escape
from host immune system, it is also important to consider that downregulation of
CapG in macrophages may also be a defence mechanism employed by the host
immune system to eliminate any foreign pathogens. Whilst it was observed that
CapG knockout macrophages did show increased susceptibility to certain
infections, these cells were able to phagocytose other types of bacteria,
highlighting that CapG plays an important, but not crucial role in phagocytosis
(Parikh et al, 2003). More importantly, the key observation of this study highlights
the impaired motility in cells derived from the CapG knockout mice. Furthermore,
it has been previously observed that LPS-activated macrophages have reduced
migratory properties (Vogel et al, 2014). Thus, it is plausible to expect that the loss
of CapG gene and CapG protein in LPS-stimulated macrophages results in loss of
cell movement. This in turn leads to macrophages remaining confined to the site
of activation and thus participate in pathogen clearance. Similarly, monocytes can
recognise RSV through the TLR2 receptor, resulting in monocyte activation and
maturation to macrophages (Murawski et al, 2009). Thus, like LPS, it is possible
that activated macrophages downregulate CapG to limit cellular movement and
prioritise in pathogen clearance. In addition, the time course in vitro experiment
performed on rat peritoneal macrophages suggest a likely recovery of CapG
message levels back to basal levels may suggest that CapG is re-upregulated to
allow macrophages to re-establish CapG to facilitate pathogen clearance.
In contrast, there was a significant increase in CapG message in lung
extracts obtained from both LPS and RSV-treated mice. The upregulated
expression of CapG in these tissues could be associated with an increase in
macrophage population due to LPS or RSV-mediated monocyte differentiation
into macrophages (Jones et al, 2006; Tsuji et al, 2000). Alternatively, this could

168
also be explained by the increase in inflammatory cell infiltration to the lungs
following treatment. However, it is also worth considering that CapG expression
is also upregulated in the classically defined “non-immune” cell types. We have
previously demonstrated that message CapG can be expressed in classically
defined non-haematopoietic cell types, including bronchial epithelial cells,
although at lower levels compared to inflammatory cells (Table 3.3), a finding that
was also previously described (Dabiri et al, 1992). However, several studies have
shown that CapG expression can be upregulated in non-haematopoietic cell types
such as epithelial cells, where these cells have increased cell metastasis capacity
(Renz et al, 2008). Thus, our findings suggests that expression of CapG message
in these “non-immune” cells such as bronchial epithelial cells could potentially be
regulated by RSV and thus requires further investigation.
Since previous studies (Chapter 3) have shown that the murine microglial
cell line BV2 also secrete CapG following LPS stimulation, we sought to examine
whether the CapG gene expression would also be modulated by LPS. In contrast
to the THP-1 cells and the primary human and rat macrophage gene expression
study, CapG (and KC) genes in microglia was found to be not affected by LPS
stimulation. Thus, whilst both macrophages and microglia release CapG following
LPS stimulation, the gene regulation in these cell types may be different. This
highlights the divergent responses observed in different macrophage populations,
which could explain differences in gene regulation following cell activation. It is
also important to consider that this study examines CapG (and KC) gene
expression of activated microglia in vitro, where is it has been previously shown
that the gene expression profiles is different between microglia cultured in vivo
and in vitro (Schmid et al, 2009). Therefore, it would be interesting to obtain adult
microglia from mice injected with LPS at the CNS region and compare gene
expression results from this present study. Similar to THP-1 studies,
dexamethasone did not affect CapG gene expression in microglia, thus

169
demonstrating that in a macrophage setting CapG is not a dexamethasone-
inducible gene.
Interestingly, the CapG gene expression is significantly enhanced in the
cortical region of Alzheimer’s disease sufferers, where microglia are known to
accumulate and are key players in disease pathogenesis (Hickman et al, 2008).
Thus, total CapG message in the brain samples of AD patients could be due to
increased microglial activity. However, perivascular macrophages are known to
also be recruited to the injury site and facilitate resolution of local immune
response by modulating activated microglia activity (Shechter et al, 2009). In
addition, there is also evidence to show that they can have detrimental effects on
pathology independently of microglia (Fiala et al, 2005). Further studies
examining CapG in the brain of a commonly used Alzheimer’s disease mouse
model APP/PS-1 also showed upregulated expression. Thus, elevated CapG gene
expression in the brain could also be associated with the increased infiltration of
perivascular macrophages into the brain during neuroinflammation, which results
in macrophage-mediated detrimental roles in Alzheimer’s disease pathogenesis.
When we compared CapG message levels between brain samples obtained
from Parkinson’s disease sufferers and healthy controls, there was no difference in
CapG expression between both groups. It has been previously reported that
perivascular macrophages have a greater role in disease progression in AD over
PD (Pey et al, 2014). As macrophages highly express CapG, infiltration of these
inflammatory cells into the cortical region of AD sufferers could explain the
elevated CapG gene expression. However, neuroinflammation in PD primarily
occurs at the substantia nigra region of the brain (Bender et al, 2006; Liu & Hong,
2003; Saggu et al, 1989). Whilst our data might suggest no difference in CapG
message levels between Parkinson’s disease patients and normal healthy
individuals, it is likely that this is not an accurate comparison of CapG expression
in the brain as these samples provided to us are obtained from the cortical region,
where PD disease pathology is not as pronounced as AD (Rocha et al, 2015;

170
Shepherd et al, 2000). Ideally, the expression of CapG in the substantia nigra
between both healthy and PD sufferers should be compared.
In summary, CapG message levels are downregulated in LPS-activated
macrophages. Furthermore, CapG expression level is also downregulated in BAL
cells obtained from both LPS and RSV-treated mice. Interestingly, when message
levels were examined in lung tissues, CapG gene was found to be elevated both
LPS and RSV-treated mice. However, differences in BAL cell enumeration and
the magnitude of CapG message upregulation between these inflammatory models
suggests that these pathogenic agents mediate different signalling pathways. As
LPS and RSV can also interact with other cell types such as epithelial cells, we
hypothesise that interaction between these inflammogens and cells can lead to
differential CapG expression in different cell types. However, this requires further
investigation. In addition to lung inflammation, we also examined the effects of
LPS in other inflammatory settings, such as the brain. LPS however, did not affect
CapG gene expression in microglia, which are the resident brain macrophages. The
difference in CapG message levels between macrophages and microglia highlights
a degree of macrophage heterogeneity and that engagement of LPS to these cells
triggers different signalling pathways. Finally, CapG expression was found to be
upregulated in brain samples obtained from Alzheimer’s disease sufferers,
suggesting a likely role for CapG in AD disease pathology. This result was also
observed in brain samples of APP/PS-1 mice, which is an established mouse model
of Alzheimer’s disease. Taken together, results from in vivo studies show that
CapG gene expression is elevated during certain inflammatory conditions.
However, our in vitro macrophage data show that CapG gene expression is
downregulated in LPS-stimulated macrophages. Combined, these studies suggest
that infiltration of macrophages to the site of inflammation may account for the
elevated CapG message levels in whole tissue. It is also possible that once
macrophages reach the site of inflammation, CapG message levels are reduced as
these cells employ a “fight, not flight” defense mechanism, which may explain the

171
reduction in CapG message levels in macrophages in in vitro studies. However, it
is also important to consider whether CapG message levels in other cell types is
also differentially expressed during inflammation. Knowledge and understanding
of CapG expression may provide a better understanding of its involvement in
certain inflammatory diseases and evaluate its potential as a possible novel
biomarker. Combined with earlier studies in Chapter 3, the differential expression
of CapG message and protein levels in inflammation is consistent with our initial
hypothesis that CapG plays a likely role in inflammation, and its extracellular and
putative pro-inflammatory role is later examined and discussed in Chapter 6.

172

173
Chapter 5
Generation and purification of human CapG using a
mammalian expression system

174
5.1 Introduction
CapG is a well-defined actin regulatory protein that is highly expressed in
macrophages where it has been shown to be involved in multiple macrophage
functions (Witke et al, 2001). It belongs to a group of actin-regulatory proteins
known as the gelsolin family. However, CapG contains only three gelsolin domain
repeats unlike the conventional six. In addition, this protein is able to bind and cap
but not sever actin filaments (Dabiri et al, 1992). In addition to macrophages, we
and others have shown that CapG is expressed in other inflammatory cells such as
mast cells, microglia and dendritic cells. CapG is important in cellular motility as
demonstrated by the reduced motility in neutrophils and reduced ruffling activity
in dendritic cells derived from CapG knockout mice when exposed to a
chemoattractant (Witke et al, 2001).
Certain members of the gelsolin family have also been previously reported
to be present extracellularly. Extracellular gelsolin is part of the extracellular actin-
scavenger system, where it and another protein Gc-globulin/Vitamin D-binding
protein (VDP), are crucial in clearing extracellular actin to prevent actin
polymerisation in the extracellular space, which if left uncontrolled leads to
undesirable effects such as vascular occlusion (Lee & Galbraith, 1992). In
addition, extracellular gelsolin has also been reported to bind to the bacterial
endotoxin LPS and lipoteichoic acid, reducing cellular responses to these
components such as inhibiting LPS or LTA-induced IL-8 release human
neutrophils (Bucki et al, 2008). The structures and sizes of cytoplasmic and
extracellular gelsolin are different as extracellular gelsolin is approximately 85
kDa, whereas cytoplasmic gelsolin is approximately 80 kDa. The key difference
between these two isoforms is the presence of a 25 amino acid leader peptide
sequence that targets the protein towards cellular secretion pathways
(Kwiatkowski et al, 1986; Martoglio, 2003), and is cleaved prior to release (Pottiez
et al, 2010).

175
The presence of CapG in the extracellular space and in plasma has also been
previously reported (Johnston et al, 1990). Unlike gelsolin, cytoplasmic and
extracellular CapG are identical in mass. However, the exact extracellular role of
this protein remains to be elucidated. Work on examining CapG secretion from
macrophages has shown that its release is mediated through an unknown non-
canonical pathway that differs from the classical signal peptide-targeted release of
gelsolin (Johnston et al, 1990). As shown in Chapter 3, CapG was found to be
constitutively released from the monocytic cell line, THP-1. This finding is in
keeping with studies reported by others studying different macrophage cell lines
(Johnston et al, 1990). Interestingly, we observed that CapG release is enhanced
following LPS-stimulation, and this effect was blocked by the anti-inflammatory
glucocorticoid dexamethasone and by TLR4 blockade. We have also found that
CapG is also released from the human mast cell line LAD2 following antigen/IgE
activation. In addition, CapG expression has also been shown to be increased in
the synovial fluid of rheumatoid arthritis patients (Balakrishnan et al, 2014),
further highlighting a possible inflammatory role for CapG. Therefore, our
hypothesis is that CapG when secreted is a potential pro-inflammatory mediator
that is capable of stimulating perhaps multiple pro-inflammatory pathways.
In order to study and understand the biological role of secreted CapG, a
substantial amount of protein is desirable. Commercially available material is of
high cost and produced using a bacterial expression system, increasing the
likelihood of bacterial contaminants present that could confound data
interpretation. This is especially true for studies proposed on the monocytic cell
line THP-1, which have been shown to respond to the bacterial endotoxin LPS at
nanogram per millilitre concentrations (Chanput et al, 2013; Park et al, 2007). To
overcome this limitation, we sought to generate an in-house mammalian
expression system that permits the generation of high yields of recombinant CapG,
which can then be used for downstream studies to elucidate its potential role as a
pro-inflammatory mediator.

176
Three mammalian systems were examined:
1. Flp-In™ 293 cells are a specialised HEK293 cell line that have been
previously shown to secrete recombinant proteins in large quantities
following transfection (Waldner et al, 2011). The pcDNA5/FRT/TO
vector utilised in these transfection studies is a tetracycline-inducible
expression vector designed for use with the Flp-In™ cell lines (Figure
5.1A). The Flp-In™ cell line has an Flp-Recombination Target (FRT)
site. The FRT site, in the presence of Flp recombinase, catalyses a
homologous recombination between the cellular and plasmid FRT sites,
thus allowing stable genome integration of the vector into Flp-In™ cell
lines and therefore allowing the generation of isogenic stable cell lines
(Craig, 1988; Sauer, 1994). This system was chosen as it allowed future
opportunities to examine the activity of CapG mutants. Other features
of this vector include a hygromycin resistance gene to allow for
selection of stable cell lines. A hybrid human cytomegalovirus
(CMV)/TetO2 promoter is also included in the vector to promote high
level, tetracycline-regulated expression of the gene of interest. This
system does not make use of a signal peptide to encourage protein
secretion. As CapG has been shown to be secreted from cells through
the non-canonical secretory pathway, this cell model also facilitates
studying the mechanism of secretion upon tetracycline induction. The
ability to regulate CapG production by tetracycline induction would be
useful for future studies examining the intracellular role of CapG.
An 8 amino acid Strep-tag sequence was introduced to the C-terminus
end of the CapG sequence to facilitate downstream purification
processes.
2. In addition to the modified HEK 293 cell lines, we also examined COS-
7 cells as an alternative expression system. This cell line was derived
from the green kidney monkey culture, CV-1 that were transformed with

177
the polyomavirus SV40 that produces large T antigens, that in turn
permits replication of plasmids (Gluzman, 1981). Hence, plasmids
containing the cDNA or genomic insert of interest will result in
generation of the protein at relatively high levels over a short period of
time (Aruffo, 2002). In addition, CapG was previously shown to be
secreted from these cells following transfection (Johnston et al, 1990).
3. An alternative expression system used was the EBNA-293 cell line. In
recent years, transient gene expression systems have been the forefront
method for generating large quantities of recombinant protein in a high-
throughput manner (Baldi et al, 2007; Durocher et al, 2002; Meissner et
al, 2001). Similar to the pcDNA5 vector, the pCEP-Pu vector uses the
CMV promoter to promote high recombinant protein expression
(Figure 5.1B). In addition, the vector also incorporated a BM40 signal
peptide at the N-terminus of the CapG protein. The BM40 signal peptide
is derived from osteonectin and targets protein for secretion (Holden et
al, 2005). A key advantage of utilising TGE and EBNA-293 cells over
the Flp-In™ 293 cell lines is the ability of the EBNA-293 cells to
produce recombinant proteins of interest at high yields shortly after
transfection (Baldi et al, 2007). The pCEP-Pu vector is an episomal
vector that contains the EBNA-1 (Epstein-Barr nuclear antigen 1) gene,
which encodes a viral DNA binding protein (EBNA-1) that is essential
for the extra chromosomal existence of the plasmid. The EBNA-1
protein binds and interacts with oriP (latent origin of replication)-
containing episomal vectors, and promotes the tethering of the foreign
vector to chromosomes during cell mitosis (Hung et al, 2001). This
allows for more plasmid copies to persist in the transfected cells
throughout the production phase, hence maintaining the expression
levels of the recombinant protein (Van Craenenbroeck et al, 2000). In
addition, EBNA-1 also recruits DNA replication proteins to the oriP site
and enhances transcription through the binding of an EBNA-1-

178
dependent transcriptional enhancer (FR) located at the oriP site
(Kohfeldt et al, 1997). The EBNA-293 cell line has been commonly
used in expression studies and has been shown to be a powerful tool for
protein production, with recombinant protein yields up to milligram/litre
levels (Meissner et al, 2001). Another key advantage of utilising the
EBNA-293 cells is its relative flexibility and ease of use including its
capacity to propagate in suspension culture and in low serum conditions,
which limits the amounts of potential protein contaminants for later
purification process (Meissner et al, 2001).
Following expression, CapG must be purified prior to functional assays to
validate CapG as a novel pro-inflammatory mediator. In this study, we compared
the effectiveness of CapG purification using two different methods: Strep-Tactin®
Sepharose resins (Strep-Tag) or HisTALON™ metal affinity chromatography
resin (His-Tag). Once purified, the material was then assessed for its functional
integrity using an actin polymerisation assay.
This chapter aimed to generate a mammalian CapG expression and
purification system that would permit the later analysis of CapG as a putative pro-
inflammatory mediator.

179
5.2 Specific Methods
5.2.1 Cloning and plasmid expansion
5.2.1.1 pcDNA5/FRT/TO vector
The pcDNA5/FRT/TO vector containing the full length cDNA of human
CapG with a C-terminus Strep-tag (WSHPQFEK) was purchased from GenScript
(Piscataway, NJ, USA). The sequence was codon optimized, with the introduction
of synonymous mutations that favours protein expression. The sequence was
cloned between the BamHI and XhoI site (Figure 5.1a). The vector was
transformed in DH5α-competent E.coli cells and grown on a Luria Broth (LB) agar
plate in the presence of ampicillin (100 µg/mL) at 37ºC overnight. On the
following day, colonies were selected and grown in LB for another 18 hours at
37ºC in a shaking incubator (225 rpm). On the following day, the bacterial culture
was harvested and cells were spun down (3000 rpm, 15 mins) and plasmid DNA
isolated from bacterial cells using a Zyppy™ Plasmid Maxiprep Kit (Zymo
Research, Irvine, CA) in accordance with the manufacturer instructions. The
plasmid DNA was then quantified using a NanoDrop™ 2000 spectrophotometer
(Thermo Scientific), and the plasmid DNA was sequenced and validated by the
Centre for Translational Pathology (The University of Melbourne).
5.2.1.2 pCEP-Pu vector
The validated CapG sequence including strep-tag was excised from the
pcDNA5 vector using the restriction enzymes BamHI and XhoI (New England
Biolabs, Ipswich, MA) and ligated into compatible NheI and NotI sites of the
pCEP-Pu vectors which was kindly provided by Dr. Amanda Gavin (The Scripps
Institute, San Diego, CA). The vector contains the signal peptide sequence of the
human extracellular matrix protein BM-40 as well as a puromycin-resistance gene
as a selection marker (Kohfeldt et al, 1997). Two pCEP-Pu vectors were used in
this study, with and without a polyhistidine-tag. In the first vector, the

180
polyhistidine-tag is located following the signal peptide sequence, and thus located
on the N-terminus of CapG (HIS+). In contrast, the second vector does not contain
a polyhistidine-tag (HIS-). Following this, the vectors were transformed into
TOP10-competent cells and grown in similar conditions as above and the plasmid
DNA isolated as above.
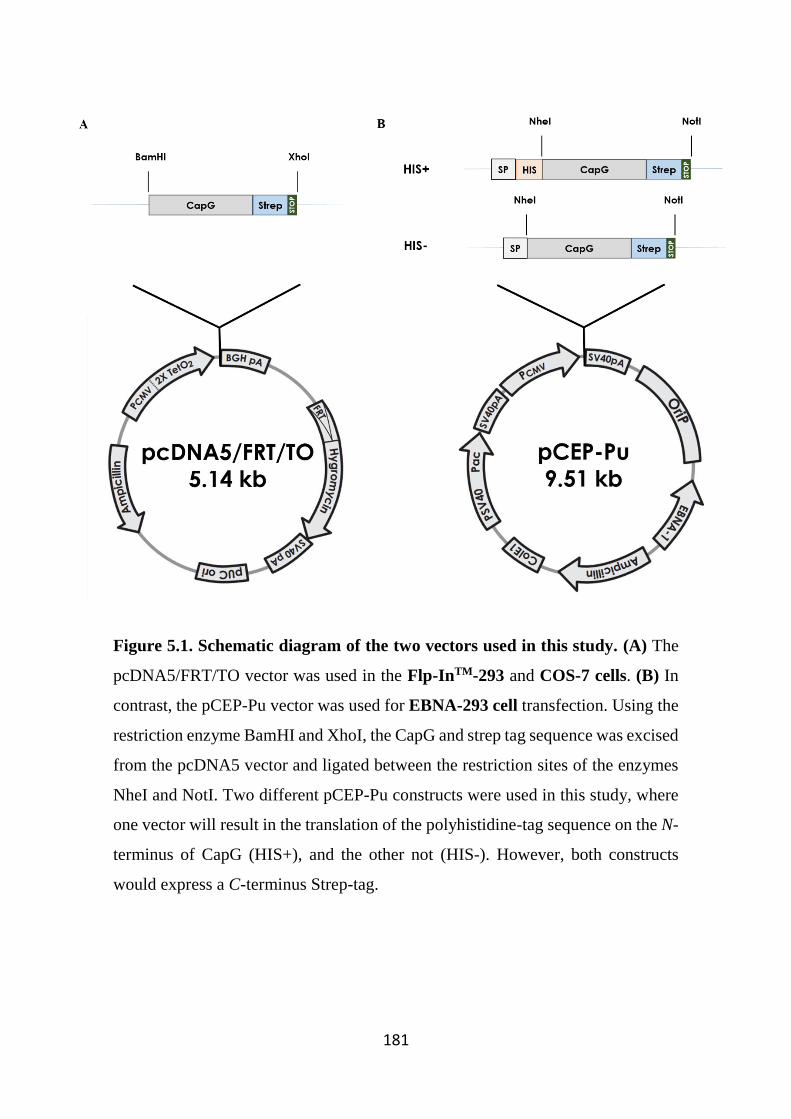
181
Figure 5.1. Schematic diagram of the two vectors used in this study. (A) The
pcDNA5/FRT/TO vector was used in the Flp-InTM-293 and COS-7 cells. (B) In
contrast, the pCEP-Pu vector was used for EBNA-293 cell transfection. Using the
restriction enzyme BamHI and XhoI, the CapG and strep tag sequence was excised
from the pcDNA5 vector and ligated between the restriction sites of the enzymes
NheI and NotI. Two different pCEP-Pu constructs were used in this study, where
one vector will result in the translation of the polyhistidine-tag sequence on the N-
terminus of CapG (HIS+), and the other not (HIS-). However, both constructs
would express a C-terminus Strep-tag.

182
5.2.2 Cell culture and transfection
5.2.2.1 pcDNA5/FRT/TO vector – Flp-InTM-293 and COS-7 cells
The growth and maintenance of Flp-InTM-293 cells and COS-7 cells is
previously described in Sections 2.1.9.1 and Section 2.1.10, respectively.
Transient transfections used to inform stable transfection studies were
performed by co-transfection of CapG and GFP expression vectors using
FuGENE®6 HD (Promega) or Lipofectamine 2000 (Life Technologies). Cells
transfected with an empty vector or no vector were used as controls in this study.
On the following day, cells were assessed for transfection efficiency by
immunofluorescence imaging using a Leica microscope and images were captured
using the Leica LAS image analysis software (Solms, Germany). Following this,
cells were treated with tetracycline (1 µg/mL; Sigma-Aldrich, Victoria, Australia)
and supernatants and cell pellets harvested after 48 hours.
For stable transfections, the cells were co-transfected with the CapG vector
and the Flp recombinase vector, pOG44 (Lyznik et al, 1996). The addition of
pOG44 in this transfection results in a targeted integration of the CapG gene to the
same locus in every cell, therefore ensuring homogenous levels of CapG
expression. The Flp-In™ 293 cells were transfected with FuGENE®6 HD
transfection reagent (Promega) in accordance to the manufacturer’s instruction.
Twenty four hours following transfection, cells were selected in culture media,
above supplemented with hygromycin B (250 μg/mL, Life Technologies) to select
for transfected cells expressing the pCDNA5/CapG vector, and blasticidin (5
μg/mL, Life Technologies) to select for pOG44-expression. Antibiotic-resistant
cells were expanded and cultured over a period of 1 month. To induce CapG
expression, the cells were treated with tetracycline (1 μg/mL, Sigma-Aldrich) and
after 3 days the pellets and supernatants were harvested.

183
5.2.2.1.1 Analysis of human recombinant CapG production
Following tetracycline induction, supernatants were collected and spun
down (300 g, 5 min) using a table-top centrifuge (Heraeus Multifuge 3SR Plus,
Thermo Scientific). Cells were then trypsinised and pelleted by centrifugation (300
g, 5 min). All samples were stored in -20ºC for later experiments examining CapG
expression.
5.2.2.2 pCEP-Pu vector – EBNA-293 cells
EBNA-293 cells (kindly provided by Dr Amanda Gavin) were maintained
in RPMI medium, supplemented with FBS (10% v v-1) (Lonza, Victoria,
Australia), glutamax (1% v v-1), penicillin (100 U/mL) and streptomycin (100
μg/mL) (Life Technologies). Transfections were performed with Lipofectamine®
2000, in accordance with the manufacturer’s protocol. 6 hours following
transfection, transfection reagent was removed from the cells and replaced with
fresh media. On the following day, the cells were selected with puromycin (2
μg/mL, Life Technologies). Puromycin-resistant cells were expanded and cultured
over a period of 3 months. When confluent, cells were passaged and the
supernatants were harvested, centrifuged (300 g, 5 min) and stored at -20ºC for
analysis of CapG expression.
5.2.2.2.1 Optimisation of CapG production by EBNA-293 cells
Optimisation and scale-up experiments were performed to increase
production of recombinant CapG from EBNA-293 cells. The cells were cultured
in different types of culture medium to establish a protocol which would provide
optimal yield. Cells were cultured in either RPMI or IMDM, and maintained in
different concentrations of FBS to identify the optimal conditions for propagating
the cells. Cells were grown in multi-tiered 175 cm2 flasks (BDBioscience) in the
presence of puromycin. A week later, the supernatant was then removed and cells
were washed twice in PBS prior to trypsinisation. The harvested supernatant was

184
spun down by centrifugation (300 g, 15 min, 4ºC) and stored at 4ºC prior to
purification. Cells were assessed for viability by trypan blue staining, and then re-
seeded and replenished with fresh media in the multi-tiered flasks. Cells were left
to grow until confluent, where supernatants were then once again harvested for
purification. This process was repeated until cells reached a high passage, where
the cells were replaced by new cultures that were brought up freshly from
cryopreservation.
In addition, EBNA-293 cells were also grown in suspension in the
chemically defined, protein-free medium CD293 (Life Technologies) in a roller
bottle flask. The flask was gassed with 5% CO2 in air for 15 min daily and then
placed in a shaking incubator (37ºC, 60 rpm). A week after seeding, supernatants
were harvested, centrifuged (300 g, 15 min, 4ºC) and stored at 4ºC prior to
purification.
5.2.3 Western Blotting
Protein samples prepared in sample buffer were separated by SDS-
polyacrylamide gel electrophoresis (SDS-PAGE; 10%-12% v v-1) as described in
Section 2.8.1, with the following exception: After membranes were probed with
anti-CapG antibody, membranes were then stripped for re-probing with other
antibodies. Briefly, membranes were incubated with mild stripping buffer (glycine
(15 mg/mL), sodium dodecyl sulfate (1 mg/mL), Tween 20 (1% v v-1, pH 2.2) for
10 minutes, then washed in PBS (2 x 10 mins), followed by washes in
TBS/Tween20, and then blocked in skimmed milk (5% in TBS/Tween 20) for 1
hour prior to re-probing with anti-His-tag antibody (0.5 μg/mL, Aviva Systems
Biology Corporation, San Diego, CA). The membrane was then striped and re-
probed with an anti-Strep-tag II antibody (20 ng/mL; IBA Life Sciences,
Göttingen, Germany). Wherever applicable, commercially purchased recombinant
CapG (bac-CapG, 30 ng; ab95385, Abcam, Cambridge, MA) was used as a loading

185
control for CapG staining, as well as a calibrator for determining the amount of
CapG present in cell pellets and supernatants.
5.2.4 Purification of recombinant human CapG
Since the recombinant human CapG produced in EBNA-293 cells
expressed both His-tag and Strep tag at the N and C-terminus respectively, the
supernatants were purified using two different purification systems: Strep-Tactin®
resin (IBA Life Sciences) and a HisTALON™ column (Clontech, Mountain View,
CA). Prior to passing the supernatants through the column, all supernatants were
filter steriled (0.2 micron). The concentration of all eluted fractions was
determined using a NanoDrop™ 2000 spectrophotometer with protein absorbance
measurements made at 280 nm.
5.2.4.1 Strep-Tactin® column
The presence of the Strep-tag on the C-terminus of the CapG protein was
designed to serve as a purification tag where the Strep-tag protein binds with high
affinity to an engineered streptavidin derivative known as Strep-Tactin®. This
process is reported as a fast and simple one-step purification and thus in theory a
useful method for efficient purification of protein (Junttila et al, 2005).
The Strep-Tactin® Sepharose® kit was purchased from IBA (Göttingen,
Germany). The resins were packed in an Econo-Column (BioRad) with 2 mL resin
bed. The conditioned medium was then passed through the column at a flow rate
of 1 mL/min (4ºC, overnight). On the following day, the column was washed with
a proprietary wash buffer (Buffer W), and proteins were eluted with a proprietary
elution buffer (Buffer E) that contained D-desthiobiotin, which is a derivative of
biotin that competes with the Strep-tagged CapG for the biotin binding pockets on
Strep-Tactin. The column was regenerated by passing through a regeneration
buffer (Buffer R) which contains HABA (2- [4’-hydroxy-benzeneazo] benzoic
acid) to displace D-desthiobiotin from the binding pocket. HABA was later

186
removed by washing the column with Buffer W. The elution process was
performed at room temperature.
5.2.4.2 HisTALON™ column
His-tagged proteins are commonly purified and detected via the ability of
the string of histidine residues to bind different types of immobilised charged metal
ions such as nickel and cobalt (Porath, 1992). Cobalt charged resins have a higher
degree of binding specificity for his-tagged proteins compared to nickel resin, thus
limiting possible co-purification of contaminants. In addition, the nickel resins are
reported to have higher metal ion leakage, resulting in a reduction in the number
of reactive sites available for binding, and therefore reducing subsequent yields of
purified protein. In addition, free metal ions can also have a detrimental effect on
protein activity, and also form salt bridges leading to protein precipitation, which
is known to be toxic to cells and tissues (Hochuli et al, 1987; Porath, 1988). Thus,
cobalt-charged resins were used in this study. TALON His-Tag Purification Resin
(2 mL; Clontech Laboratories, Mountain View, CA) initially stored in ethanol
(70%) was packed into a Poly-Prep column (Bio-Rad) and extensively washed in
PBS prior to the addition of His-CapG containing supernatants. To reduce non-
specific binding to the column, imidazole (5 mM; ChemSupply, SA, Australia)
was added to the supernatants prior to purification. The supernatant was
recirculated through the column overnight (4⁰C) at a rate of 1 mL/min and on the
following day, the column was washed extensively with PBS containing imidazole
(5 mM). Bound protein was then eluted from the column using a high
concentration of imidazole (100 mM). Eluted fractions (1 mL) were collected and
protein concentration was determined using a NanoDrop™ 2000
spectrophotometer with measurements made at 280 nm (Thermo Scientific). To
regenerate the columns for subsequent runs, bound imidazole was removed from
the TALON column by passing through 2-(N-Morpholino) ethanesulfonic acid
hydrate, 4-Morpholineethanesulfonic acid solution (MES-Hydrate; 20 mM, pH
5.0; Sigma-Aldrich) containing NaCl (100 mM; ChemSupply). PBS was then

187
passed through the column to remove the MES-hydrate solution and the column
stored in 70% ethanol at 4ºC until the next purification run. When necessary, the
column was regenerated by passing cobalt chloride (50 mM) through the resin,
followed by extensive washing prior to the next purification run.
CapG protein expression from eluted fractions was visualised by Western
blotting analysis.
5.2.5 Protein concentration, dialysis and analysis by Coomassie Blue-staining
5.2.5.1 Protein concentration
The eluents containing measurable concentrations of purified protein were
pooled and then concentrated 10 fold in a 10 kDa centrifugal concentrator (10
minutes, 1200 g, 4ºC; Millipore, Victoria, Australia). The final protein
concentration was again determined using a NanoDrop™ 2000, with the
absorbance measured at 280 nm. Using the absorbance measurement and the
predicted CapG extinction coefficient of 1.32 (as determined by ProtParam tool
available on the ExPASy (Expert Protein Analysis System) server) (Wilkins et al,
1999) (Figure 5.2), the concentration of CapG was determined using the following
formula:
𝐶𝑎𝑝𝐺 𝑐𝑜𝑛𝑐𝑒𝑛𝑡𝑟𝑎𝑡𝑖𝑜𝑛 (𝑚𝑔/𝑚𝐿) = 𝐴𝑏𝑠𝑜𝑟𝑏𝑎𝑛𝑐𝑒 𝑎𝑡 280 𝑛𝑚
1.32
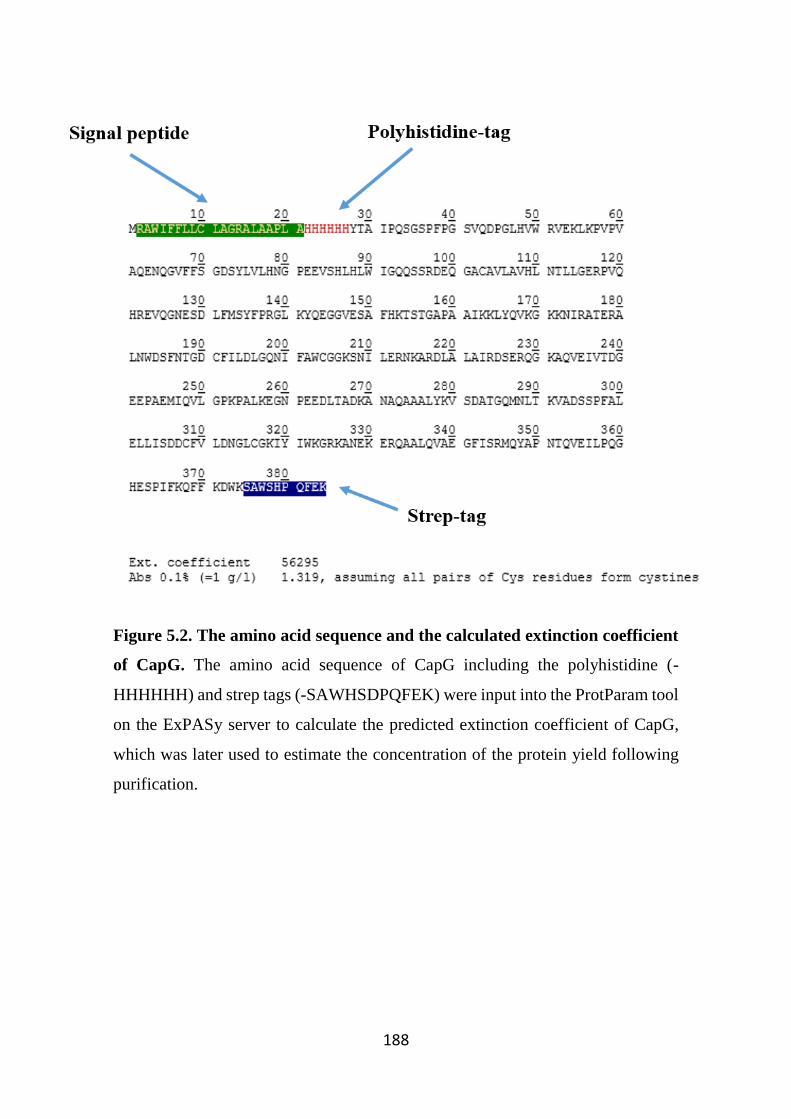
188
Figure 5.2. The amino acid sequence and the calculated extinction coefficient
of CapG. The amino acid sequence of CapG including the polyhistidine (-
HHHHHH) and strep tags (-SAWHSDPQFEK) were input into the ProtParam tool
on the ExPASy server to calculate the predicted extinction coefficient of CapG,
which was later used to estimate the concentration of the protein yield following
purification.

189
5.2.5.2 Dialysis
Imidazole was removed from the concentrated material by dialysis
(Spectrum Laboratories, Victoria, Australia). After three rounds of dialysis into
PBS (1:1000 dilution on each occasion, pH 7.4, 4ºC), the protein solution was
collected and protein concentration determined again using the NanoDrop™ 2000
spectrophotometer.
5.2.5.3 Coomassie Blue staining
Following dialysis, the purified, concentrated recombinant CapG was
visualised by Coomassie Blue staining. Following gel electrophoresis (as
described in section 2.8.2), the gels were stained overnight in Coomassie Blue dye
(0.25% Coomassie Blue R-250 (w v-1) in 50% methanol (v v-1), 40% Milli-Q water
(v v-1) and 10% glacial acetic acid (v v-1)) at room temperature to visualize protein
bands in samples. On the following day, the gel was destained for 4-6 hours in
destain solution (as above, with the exclusion of 0.25% Coomassie Blue R-250 (w
v-1)). Once the gel has been destained until minimal background staining was
achieved, the gel was imaged using ChemiDoc™ MP System (Bio-Rad
Laboratories, Hercules, CA).
5.2.6 Mass Spectrometry
5.2.6.1 In-gel digestion
Putative purified CapG and unknown bands were analysed by mass
spectrometry. Coomassie Blue stained gel bands were excised and washed twice
in Milli-Q water (2 x 15 mins) and then washed with 50% acetonitrile/50 mM
ammonium bicarbonate to remove all Coomassie stain from gel bands (2 x 15
mins). The gel bands were then dehydrated in acetonitrile until the gel plugs turned
opaque, and subsequently dried using a vacuum centrifuge (10 mins; Maxi dry lyo,
Dynavac, MA). Following this, the bands were incubated in sequence grade,
modified trypsin (100 ng/μL; Roche Diagnostics Gmbh, NSW, Australia) for

190
approximately 20 minutes to allow the trypsin to infuse into the bands for “in-gel”
digestion. Excess trypsin was removed using a pipette and the bands were
incubated with ammonium bicarbonate (25 mM) overnight in 37ºC to ensure
proper hydration. On the following day, the digestion process was stopped with
the addition of formic acid (10% v v-1). Supernatants were recovered, stored at -
20ºC and used for peptide mass mapping. The samples were sent to the La Trobe
Institute for Molecular Science (LIMS, Melbourne, Australia), where mass
spectrometry analysis was performed to identify the proteins in samples.
5.2.7 Actin polymerization assay
Pyrene-labelled actin (where a pyrenyl group is covalently linked to the
cysteine 374 of the C-terminus of the globular actin (G-actin) molecule) is
commonly used in assays measuring actin polymerization (Crosbie et al, 1994). It
has been previously shown that the emitted fluorescence intensity of pyrene
increases 7-12 fold upon polymerization (Cooper et al, 1983). In the
polymerization assay, lyophilised pyrene-labelled rabbit smooth muscle actin
(10% pyrene actin mixed with 90% unlabelled actin, Hypermol, Germany) was
reconstituted in sterile deionised water. Subsequently, pyrene actin was diluted to
0.45 mg/mL in general actin buffer (Tris-HCl (5 mM, pH 8.0) and CaCl2 (0.2 mM)
supplemented with ATP (0.2 mM) (all Sigma)) and left on ice for 1 hour to allow
for depolymerisation of any actin oligomers. Actin was subsequently centrifuged
at 13,000 rpm (4ºC, 30 mins) to remove residual nucleating centres that might
trigger spontaneous polymerisation. The actin polymerization assay was
performed in a 384-well white plates (Packard Bio Science Culturplate; Arvada,
Colorado, USA) in a total reaction volume of 20 µL. This was made up of pyrene
actin (0.45 mg/mL), test material (2 µL) and 10x actin polymerization buffer (KCl
(500 mM), MgCl2 (20 mM), and ATP (10 mM), 2 µL (all Sigma)). Pyrene actin
and test compounds were added into wells and the plate was spun down (400 g, 1
min). Actin polymerisation fluorescence was measured by increasing fluorescence
using the FlexStation II spectrophotometer (Molecular Devices, Sunnyvale, CA)

191
at an excitation wavelength of 360 nm and emission wavelength at 405 nm. The
plate was read for 3 minutes to establish a baseline and to determine if the test
compounds were intrinsically capable of initiating actin polymerization. After 3
minutes, the polymerisation buffer was added into each well and fluorescence
intensity was measured. The polymerisation assay was conducted for 30 mins, with
fluorescence readings taken at 30 second intervals. The fluorescence
measurements at each time point were consolidated and plotted using Graphpad
Prism software (version 6.01).

192
5.3 Results
5.3.1 CapG is expressed in Flp-In™ 293 cells following transient transfection
Flp-In™ 293 cells were transfected with the pcDNA5/FRT/TO expression
vector containing a CapG sequence with an N-terminus Strep-tag to permit
downstream purification. This study also included cells transfected with an empty
vector control, or cells that were mock transfected. Cells were also co-transfected
with a GFP construct as an indicator of transfection efficiency. Twenty four hours
after transfection, immunofluorescence imaging was performed on the Flp-In™
293 cells and there was an approximate 60-70% transfection efficiency as
indicated by the positive GFP expression in the transfected cells (Figure 5.3A). 72
hours after tetracycline induction, supernatants and pellets were harvested and
samples resolved by Western blotting to determine CapG expression. CapG was
only detected in cell lysates and supernatants of cells transfected with the CapG
vector following tetracycline induction. In contrast, CapG was not detected in cell
lysates and supernatants of non-transfected Flp-In™ 293 cells and the empty-
vector transfected cells, demonstrating that the Flp-In™ 293 cells do not natively
express CapG (Figure 5.3B). In stable transfection experiments, transfected cells
were selected by hygromycin treatment and allowed to grow for a period of time
before the cells were examined for CapG expression again following tetracycline
treatment for 72 hours. Similar to the transient transfection studies, CapG was only
detected in pellets and supernatants of cells stably transfected with the CapG vector
(Figure 5.3C). In addition, bac-CapG (30 ng) was included as a loading control,
which permitted densitometry analysis to estimate the concentration of CapG
present in the supernatants of transfected cells, which was approximately 2.5
µg/mL.
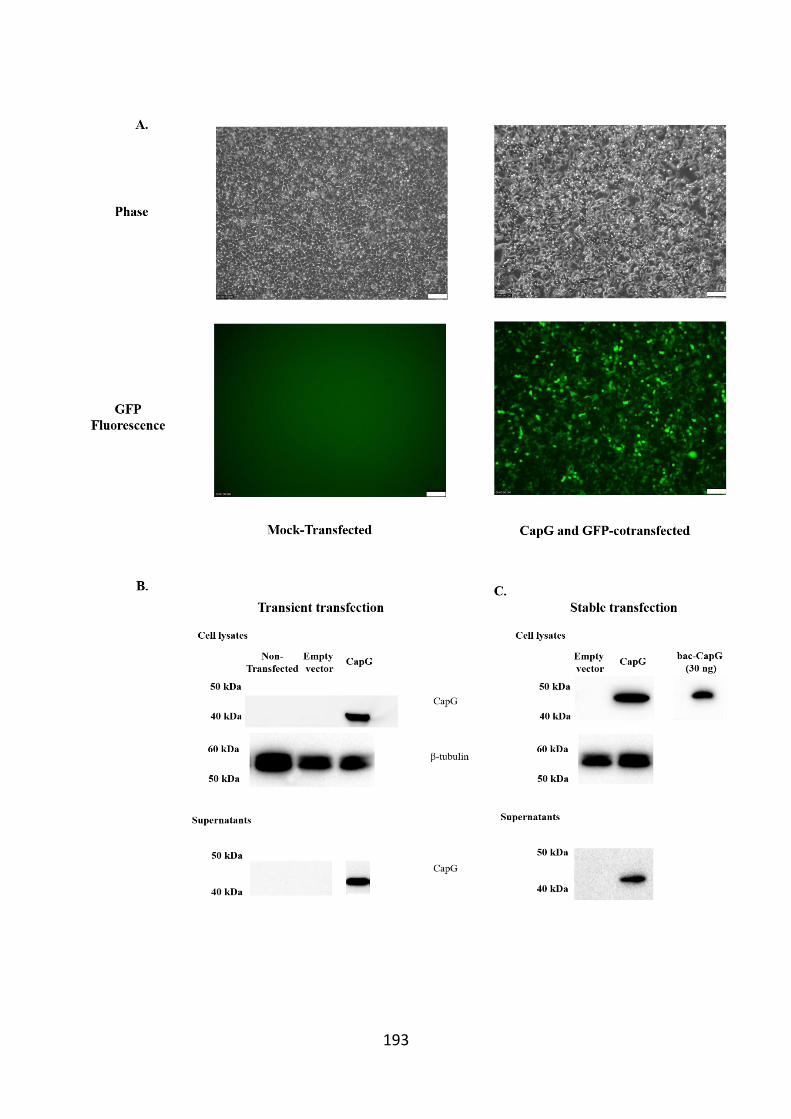
193

194
Figure 5.3. Transient and stably-transfected Flp-In™ 293 cells express CapG
in cell pellets and supernatants. In transfection experiments, Flp-In™ 293 cells
were transfected with an empty vector control or a pcDNA5-CapG vector. In
addition, a non-transfected control was also used in the transient transfection
experiments. (A) Transiently-transfected cells were co-transfected with a GFP
vector to measure transfection efficiency and GFP was visualised the day
following transfection. Phase microscopy images of the transfected cells were also
visualised to assess cell viability and morphology. Scale bars (in white) represent
100 microns. (B) Cells were treated with tetracycline for 72 hours and both cell
lysates and supernatants analysed for CapG expression. (C) In stable transfections,
cells were selected with hygromycin following transfection and resistant cells were
cultured and also examined for CapG expression following 72 hour tetracycline
induction. Immunofluorescence and Western blotting results shown are a
representative of 3 separate transfection studies.

195
5.3.2 Flp-In™ 293 cell-derived released CapG is likely associated with cell
death
Since CapG was expressed in both transient and stably-transfected Flp-In™
293 cells and also detected in supernatants, we sought to examine the kinetics of
CapG release from stably transfected cells, which would also determine the ideal
time point to harvest supernatants. Cells were seeded in serum-free DMEM media
to limit the amount of potential extraneous protein that might make CapG
purification more challenging. In addition, cells were treated with tetracycline to
induce CapG expression. CapG was readily detected in cell lysates 24 hour
following induction and expression increased over 5 days (Figure 5.4A). In
contrast, CapG was only detected in the supernatants 3 days after tetracycline
induction, but similarly CapG levels in supernatants increased over time. In
addition, commercially purchased recombinant CapG (bac-CapG, 30 ng) was
visualised and used as a loading control in this study. Cell viability was also
examined in this study and after the third day total cell viability was observed to
decline to approximately 80% and this continued, as indicated by an approximate
50% reduction in cell viability by the end of the time course experiment (Figure
5.4B). The differences in CapG expression in cell pellets and supernatants suggests
that whilst the transfected cells are able to produce CapG, they are however unable
to secrete this protein. Taken together, data suggest that the detected CapG in
supernatants was likely due to release from these dead/dying cells.
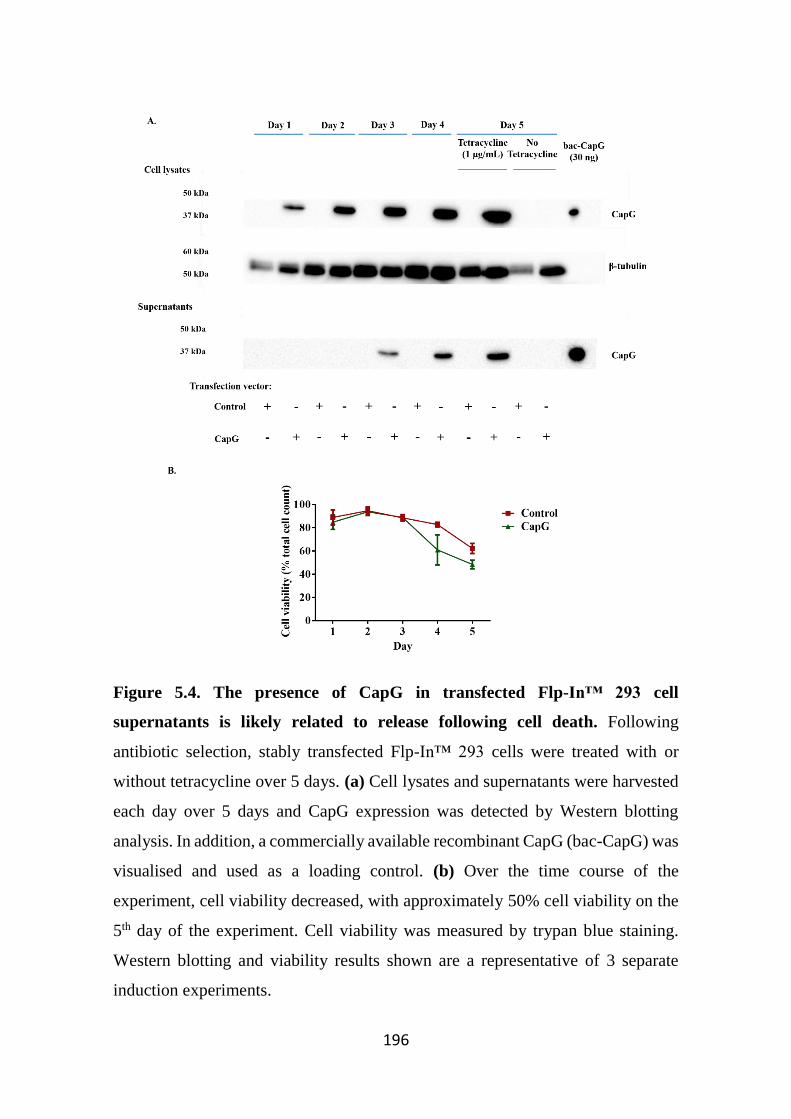
196
Figure 5.4. The presence of CapG in transfected Flp-In™ 293 cell
supernatants is likely related to release following cell death. Following
antibiotic selection, stably transfected Flp-In™ 293 cells were treated with or
without tetracycline over 5 days. (a) Cell lysates and supernatants were harvested
each day over 5 days and CapG expression was detected by Western blotting
analysis. In addition, a commercially available recombinant CapG (bac-CapG) was
visualised and used as a loading control. (b) Over the time course of the
experiment, cell viability decreased, with approximately 50% cell viability on the
5th day of the experiment. Cell viability was measured by trypan blue staining.
Western blotting and viability results shown are a representative of 3 separate
induction experiments.

197
5.3.3 CapG expression was detected in transiently transfected COS-7
Since the presence of CapG in supernatant from transfected-Flp-In™ 293
cells was likely due to cell death and the expulsion of intracellular proteins was
not ideal for downstream purification, we sought to identify other expression
system candidates. We examined monkey kidney fibroblast cell line COS-7 as
these cells were previously shown to secrete CapG following transfection
(Johnston et al, 1990). In similar transfection conditions to the Flp-In™ 293 cells,
there was an approximate 70-80% transfection efficiency through visualisation of
GFP following transfection (Figure 5.5A). Following tetracycline treatment for 72
hours, cell lysates and supernatants were harvested and CapG expression detected
by Western blotting. Similar to the Flp-In 293 cell lines, intracellular CapG is not
natively expressed in COS-7 cells and was only detected in cells transfected with
the CapG vector. Furthermore, CapG was also detected in supernatants of CapG-
transfected cells (Figure 5.5B). Similar to the Flp-In™ 293 studies, bac-CapG (30
ng) was utilised as a loading control in this study. The estimated amount of CapG
present in transfected COS-7 supernatant was approximately 2 µg/mL, as
determined by ImageJ densitometry analysis. Release of CapG from transfected
COS-7 cells was not due to cell death, as determined by high cell viability
measured after supernatant harvest (Figure 5.5C). Whilst COS-7 cells were able
to release CapG unlike the Flp-In™ cells, this cell line was not an ideal expression
system for CapG production due to low protein yield. In addition, because these
cells were transiently transfected, the transfected CapG gene is not stably
integrated into the cell genome, and can results in the loss of CapG gene in
transfected cells over time due to environmental factors and cell division (Kim &
Eberwine, 2010).
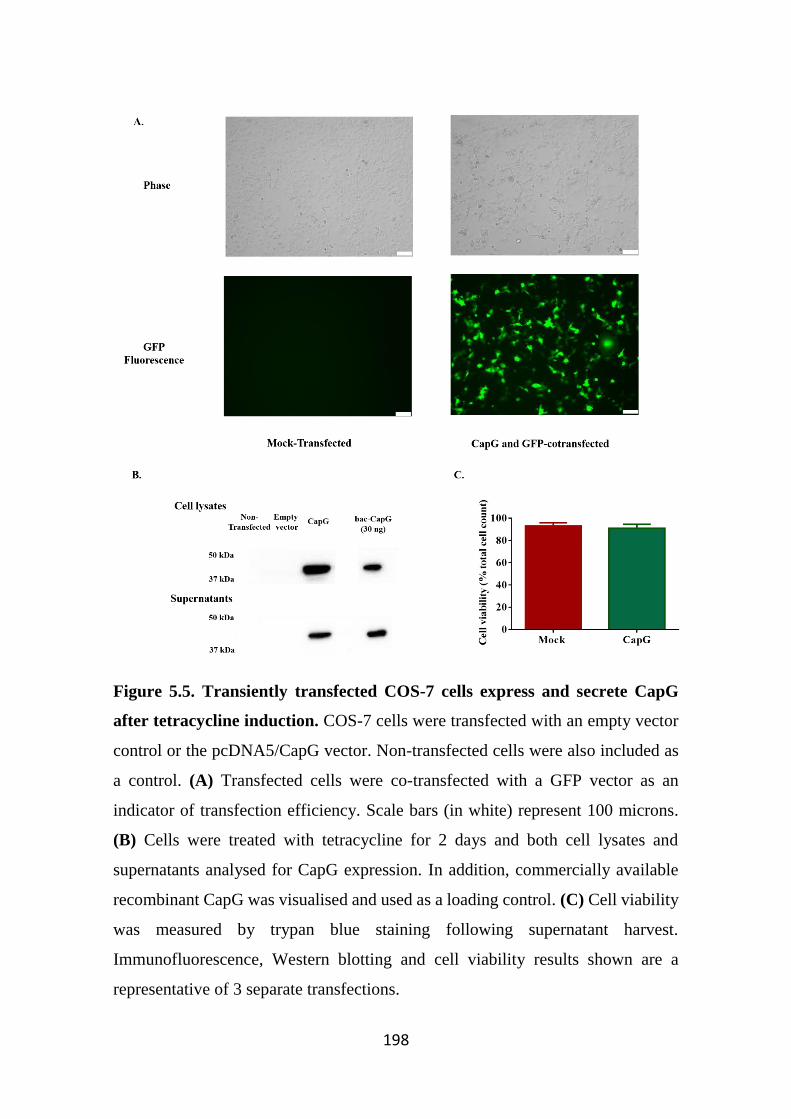
198
Figure 5.5. Transiently transfected COS-7 cells express and secrete CapG
after tetracycline induction. COS-7 cells were transfected with an empty vector
control or the pcDNA5/CapG vector. Non-transfected cells were also included as
a control. (A) Transfected cells were co-transfected with a GFP vector as an
indicator of transfection efficiency. Scale bars (in white) represent 100 microns.
(B) Cells were treated with tetracycline for 2 days and both cell lysates and
supernatants analysed for CapG expression. In addition, commercially available
recombinant CapG was visualised and used as a loading control. (C) Cell viability
was measured by trypan blue staining following supernatant harvest.
Immunofluorescence, Western blotting and cell viability results shown are a
representative of 3 separate transfections.

199
5.3.4 Transfected EBNA-293 cells secrete CapG protein
Since the Flp-In™ 293 and COS-7 cells were shown to not be ideal systems
for generating mammalian CapG, we examined another expression system that
would be able to secrete and efficiently produce CapG at higher yields. Previous
studies performed on the Epstein-Barr Virus Nuclear Antigen (EBNA)-293 cells
demonstrated that this cell line was able to produce high concentrations of
recombinant proteins in a short period (Meissner et al, 2001). The pCEP-Pu/CapG
vector used in this study was generated through the excision of the CapG-Strep-
tagged sequence from the pcDNA5 vector and re-ligating the sequence into a
pCEP-Pu vector (as described in Section 5.2.1). To prevent the likelihood of the
EBNA-293 cells being unable to natively secrete CapG, as observed in the Flp-
In™ 293 cells, a signal peptide sequence was included in the vector to target the
protein for secretion. Two different vectors were used in this study, the first vector
results in the translation of CapG protein identical to previous experiments with
only a Strep-tag (HIS-), and the second containing a hexa-histidine-tag (HIS+)
located at the N-terminus of the protein to be used as an alternative method of
purification (HIS+) (Figure 5.6A).
Cells were transfected with either (HIS-) or (HIS+) vectors and selected
with the antibiotic puromycin. The puromycin-resistant cells were cultured over a
period of time and conditioned media was harvested and assessed for CapG protein
(Figure 5.6B). CapG was not detected in supernatants of mock-transfected cells.
Utilising the bac-CapG control, we were able to estimate an approximate
concentration of CapG present in both supernatant samples (approximately 2.8
µg/mL (HIS+) and 3.3 µg/mL (HIS-), as determined by ImageJ densitometry
analysis). These cells released CapG at higher quantities compared to previous
transfections performed on Flp-In™ 293 and COS-7 cells. When we examined the
expression of His-tag, as expected only the cells transfected with the HIS+ vector
showed positive staining. (Figure 5.6C). The membrane was then stripped to
remove residual antibodies, and then re-probed for Strep-tag presence. Both

200
recombinant proteins expressed the Strep tag, as evident by Western blotting
analysis. The presence of CapG in the supernatants was not due to cell death
(unlike the Flp-In™ 293 cells) as determined by trypan blue staining (Figure
5.6D). It should be noted that the native molecular weight of CapG is
approximately 42 kDa, however the recombinant CapG produced from the EBNA-
293 cells ran at approximately 47 kDa likely due to the presence of His and Strep
tags present at both N and C terminus, respectively.
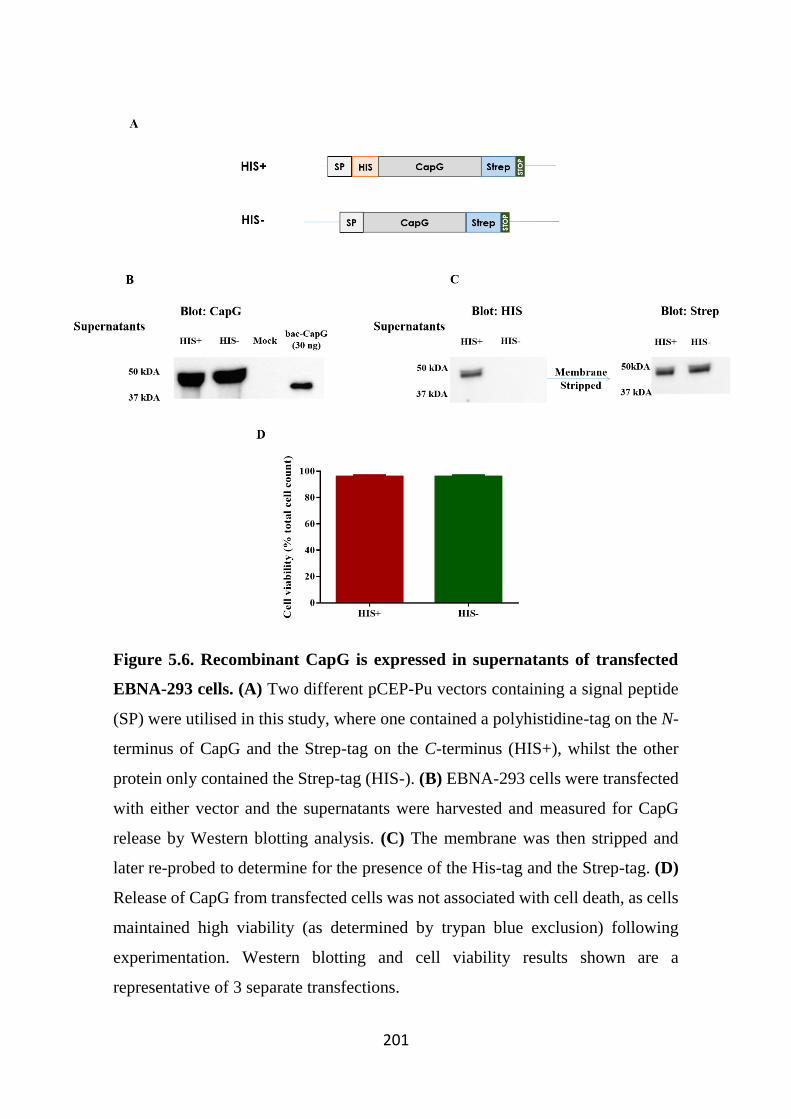
201
Figure 5.6. Recombinant CapG is expressed in supernatants of transfected
EBNA-293 cells. (A) Two different pCEP-Pu vectors containing a signal peptide
(SP) were utilised in this study, where one contained a polyhistidine-tag on the N-
terminus of CapG and the Strep-tag on the C-terminus (HIS+), whilst the other
protein only contained the Strep-tag (HIS-). (B) EBNA-293 cells were transfected
with either vector and the supernatants were harvested and measured for CapG
release by Western blotting analysis. (C) The membrane was then stripped and
later re-probed to determine for the presence of the His-tag and the Strep-tag. (D)
Release of CapG from transfected cells was not associated with cell death, as cells
maintained high viability (as determined by trypan blue exclusion) following
experimentation. Western blotting and cell viability results shown are a
representative of 3 separate transfections.

202
5.3.5 Purification using a HisTALON™ column yields higher quantities of
recombinant CapG compared to purification using a Strep-Tactin®
column
Once supernatants were harvested from EBNA-293 cells, the conditioned
medium was passed through either a HisTALON™ column or a Strep-Tactin®
column and the eluted material analysed for CapG protein by Western Blotting.
The His-tagged CapG protein bound to the HisTALON™ column was eluted
largely in fractions 2 and 3 with efficient clearance of CapG from all culture
supernatants (Figure 5.7). In contrast, there was a much lower efficiency of CapG
purification from supernatants using a Strep-Tactin® column. Modest quantities of
purified CapG were observed in Fraction 3 with minimal differences in CapG
levels in supernatants before and after passing over the column. It should be noted
in the Western blot image in Figure 5.7, that there was also CapG detected in
Fraction 1 eluted from the Strep-Tactin® column, likely a result of poor washing
of the column following the last purification run. Since the HisTALON™ column
was a more efficient method for CapG protein purification, we chose the His-
tagged CapG expressing cell line to generate CapG for all downstream studies.

203
Figure 5.7. Purification of CapG from EBNA-293 supernatants using
HisTALON™ and Strep-Tactin® resins. The EBNA-293 supernatants were
harvested and passed through either a HisTALON™ cobalt resin column or Strep-
Tactin® resin column and fractions collected. Samples of each fractions were
resolved by gel electrophoresis and Western blotting, and the membrane probed
for CapG expression. There was a substantial amount of CapG detected in fractions
2-5 collected from supernatants that passed through the HisTALON™ column. In
contrast, there was a modest yield of recombinant CapG from supernatants passing
through the Strep-Tactin® resin column. Western blots shown are representative
of experiments conducted on 3 separate occasions.

204
5.3.6 Optimisation of EBNA-293 growth in different culture conditions
Several optimisation processes were performed to identify the ideal
conditions for the production and purification of CapG from EBNA-293 cells.
EBNA-293 cells were cultured in different types of media including RPMI, IMDM
and the chemically defined serum free media CD293. There were no noticeable
differences in cell growth rate and morphology between cells cultured in RPMI or
IMDM media. However, IMDM was preferred over RPMI as this was a more cost
effective option for large scale cell culture. In addition, cells were also grown in
media supplemented with different concentrations of FBS to identify a serum
concentration that was ideal for cell growth whilst limiting the presence of serum
proteins that might interfere with protein purification. The EBNA-293 cells
cultured in medium supplemented with two different FBS concentrations (1% and
10%) maintained high viability by the end of the experimental time point. CapG
was present at higher concentrations from cells cultured in 10% FBS in comparison
to cells cultured in 1% FBS (Figure 5.8A), likely due to the faster rate of growth
and hence increased amount of cells. However, at these higher FBS concentrations,
other proteins including serum albumin were also present in higher amounts
potentially affecting downstream purification of CapG. To avoid this
complication, cells were cultured in lower concentrations of FBS (1%).
It has been previously shown by others that the EBNA-293 cells can be
cultured in suspension (Meissner et al, 2001). Thus, we examined CapG
production from EBNA-293 cells when grown in suspension in roller flasks. In
addition, adherent EBNA-293 cells were cultured in a 5-layered (5 x 175 cm2)
multi-tiered flask, as an alternate method from standard culture flasks to improve
protein yield (Abraham et al, 2011). Supernatants from both culture vessels were
harvested a week after initial seeding and CapG expression was compared to CapG
production from EBNA-293 cells cultured in a standard 175 cm2 culture flask. Of
these, CapG production from cells cultured in the multi-tiered flasks showed
greater CapG production. Cells grown in suspension had a slower growth rate and

205
measuring CapG quantities in supernatants were similar to CapG levels obtained
from conditioned media from adherent EBNA-293 cells grown in standard T175
flasks (Figure 5.8B). Thus, the cells were propagated in IMDM, 1% FBS in a
multi-tiered flask as this was most efficient for CapG expression.
In addition, we examined whether the transfected cells would secrete CapG
consistently over numerous passages. Cells were passaged when 90-100%
confluency was reached and supernatants were collected at the end of 5, 10 and 15
weeks in culture. CapG expression was detected in cells grown in culture over 3
months, demonstrating that the expression of CapG in transfected cells was very
stable (Figure 5.8C).
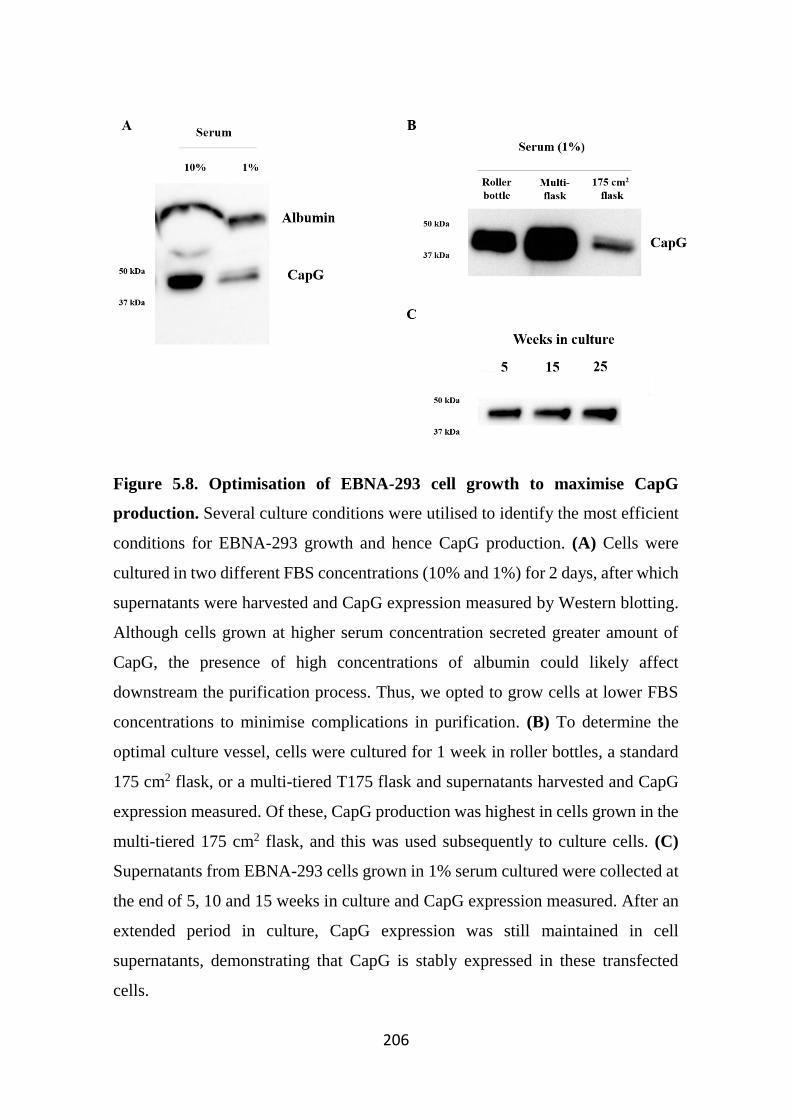
206
Figure 5.8. Optimisation of EBNA-293 cell growth to maximise CapG
production. Several culture conditions were utilised to identify the most efficient
conditions for EBNA-293 growth and hence CapG production. (A) Cells were
cultured in two different FBS concentrations (10% and 1%) for 2 days, after which
supernatants were harvested and CapG expression measured by Western blotting.
Although cells grown at higher serum concentration secreted greater amount of
CapG, the presence of high concentrations of albumin could likely affect
downstream the purification process. Thus, we opted to grow cells at lower FBS
concentrations to minimise complications in purification. (B) To determine the
optimal culture vessel, cells were cultured for 1 week in roller bottles, a standard
175 cm2 flask, or a multi-tiered T175 flask and supernatants harvested and CapG
expression measured. Of these, CapG production was highest in cells grown in the
multi-tiered 175 cm2 flask, and this was used subsequently to culture cells. (C)
Supernatants from EBNA-293 cells grown in 1% serum cultured were collected at
the end of 5, 10 and 15 weeks in culture and CapG expression measured. After an
extended period in culture, CapG expression was still maintained in cell
supernatants, demonstrating that CapG is stably expressed in these transfected
cells.

207
5.3.7 Examining the degree of purity of purified CapG and the identification of
protein bands from purified samples
HisTALON™ purified eluent was concentrated and protein concentration
determined using a spectrophotometer. Taking the predicted extinction coefficient
of the protein (1.32 as calculated by ProtParam tool, ExPASy) into account, the
protein concentration was estimated to be approximately 200 µg/mL. The
concentrated and non-concentrated material was visualised by Western blotting
(Figure 5.9A). To assess the purity of the sample, the purified protein was loaded
onto a SDS-polyacrylamide gel under reducing conditions. The proteins were
resolved by electrophoresis and the gel stained with Coomassie Blue to identify
proteins present (Figure 5.9B).
A dominant band at 47 kDa coincided with the CapG Western blot was
observed, as predicted. However, additional higher molecular weight bands were
also detected. All three bands were excised and analysed by mass spectrometry.
The quantitative analysis of proteins identified is shown in Table 5.1. Results were
attained using the ‘spectral count’ method, which is the number of spectra
matching peptides from a protein and is used as a surrogate measure of protein
abundance (Choi et al, 2008; Zhu et al, 2010). In addition, complementary
densitometry analysis was performed to quantify the abundance of each protein
band relative to total protein (Figure 5.9C). As expected, the most abundant
protein at approximately 47 kDa band was CapG (53%). The most abundant
proteins identified in the samples taken from the two contaminating bands were
epididymis luminal proteins 213 and 214 (140 kDa; 34%) and cDNA FLJ61580
(110 kDa; 13%). It is unclear why these protein bands are present. However,
similar sized contaminant bands were also observed when purifying unrelated His-
tagged proteins (His-sFcεRIα), suggesting an interaction with the HisTALON™
matrix that is independent of CapG. Whilst these contaminating bands could likely
be removed by further purification processes such as size-based exclusion
chromatography, it was likely that this process would have resulted in potentially

208
significant loss of His-CapG, thus limiting downstream functional studies and so
no further purification was conducted.
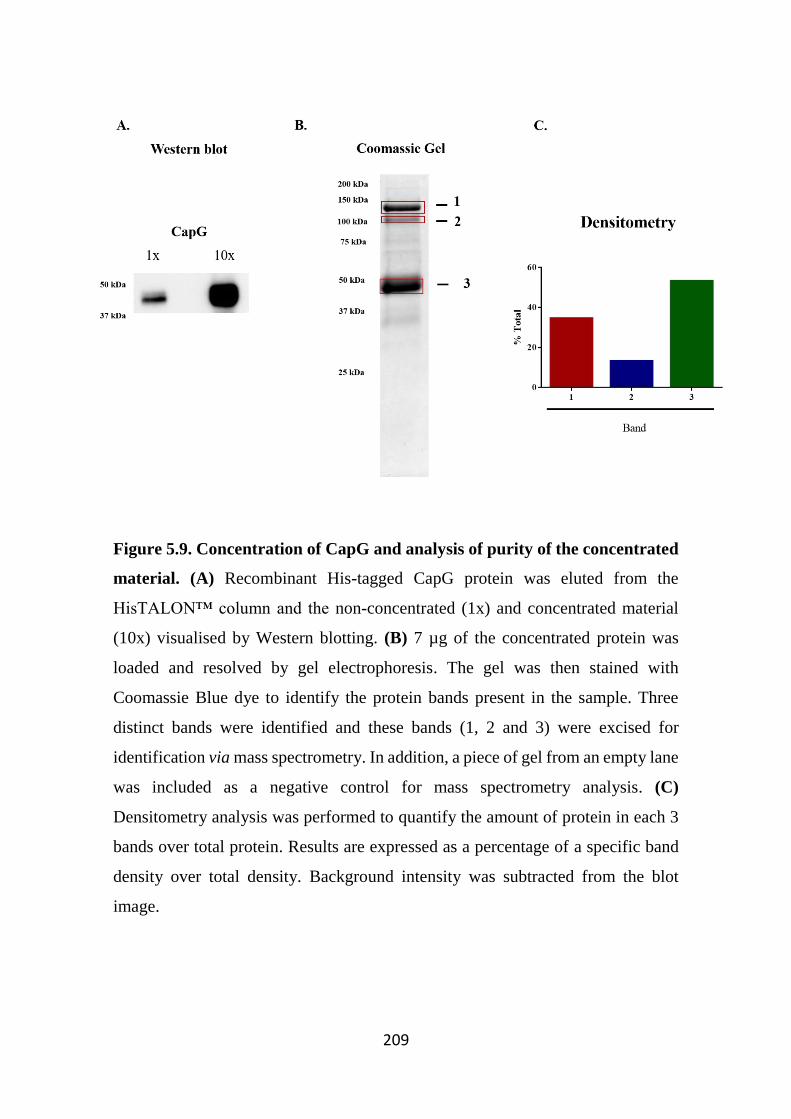
209
Figure 5.9. Concentration of CapG and analysis of purity of the concentrated
material. (A) Recombinant His-tagged CapG protein was eluted from the
HisTALON™ column and the non-concentrated (1x) and concentrated material
(10x) visualised by Western blotting. (B) 7 µg of the concentrated protein was
loaded and resolved by gel electrophoresis. The gel was then stained with
Coomassie Blue dye to identify the protein bands present in the sample. Three
distinct bands were identified and these bands (1, 2 and 3) were excised for
identification via mass spectrometry. In addition, a piece of gel from an empty lane
was included as a negative control for mass spectrometry analysis. (C)
Densitometry analysis was performed to quantify the amount of protein in each 3
bands over total protein. Results are expressed as a percentage of a specific band
density over total density. Background intensity was subtracted from the blot
image.

210
Table 5.1. Proteins identified by mass spectrometry. The top most abundant
proteins identified from bands 1, 2 and 3 are listed. Proteins identified from the
blank lane such as keratin were discounted in all samples. Each protein’s name,
accession number and abundance in spectral counts are listed.

211
5.3.8 His-CapG reduces the rate of pyrene-actin polymerisation
Prior to examining the potential novel functions of the in-house generated
recombinant CapG (His-CapG), we first sought to determine whether the
recombinant protein was biologically active in a ‘traditional’ assay of its activity.
Since CapG is best known for its role in binding to actin filaments and inhibiting
actin polymerisation, we examined the function of His-CapG in this setting.
Pyrene-actin assays are commonly used in vitro for studying actin polymerisation
(Cooper et al, 1983). In its monomeric form, pyrene actin is weakly fluorescent.
As actin monomers polymerise following the addition of a polymerisation buffer,
the fluorescence signal increases. This polymerisation process is easily measurable
and readily quantified.
In our hands, we observed this expected increase in fluorescence intensity
following the addition of the polymerisation buffer, with the fluorescence intensity
plateauing at approximately 1,000 secs (Refer to PBS trace in Figure 5.10).
In the presence of His-CapG, whilst there was no change in peak
fluorescence intensity, the rate of pyrene-actin polymerisation was slower, as
evidenced by longer time to establish peak fluorescence compared to buffer only
control (Figure 5.10 and 5.11A). Similarly, the commercially-purchased
recombinant CapG (bac-CapG) also decreased the rate of actin polymerisation.
However, this was only performed once due to limited availability of material. To
examine whether the contaminating bands observed in Figure 5.9B would affect
actin polymerisation, we examined the effects of His-sFcεRIα on actin
polymerisation. This protein also had similar sized contaminating bands observed
in Coomassie staining. However, the His-sFcεRIα protein did not affect actin
polymerisation, demonstrating that the contaminating bands did not affect actin
polymerisation (Figure 5.10). To estimate the effects of His-CapG on the rate of
actin polymerisation, linear regression analysis was performed to compare the
slopes of the buffer only control and His-CapG (Figure 5.11B). Analysis was
performed between the start of addition of actin polymerisation buffer (t = 240 s)

212
and the time where submaximal fluorescence activity was reached for both buffer
only (t = 840 s) and His-CapG (t = 1350 s). In the presence of CapG, the slope of
the curve was flatter compared to control, indicative that CapG impaired actin
polymerisation.
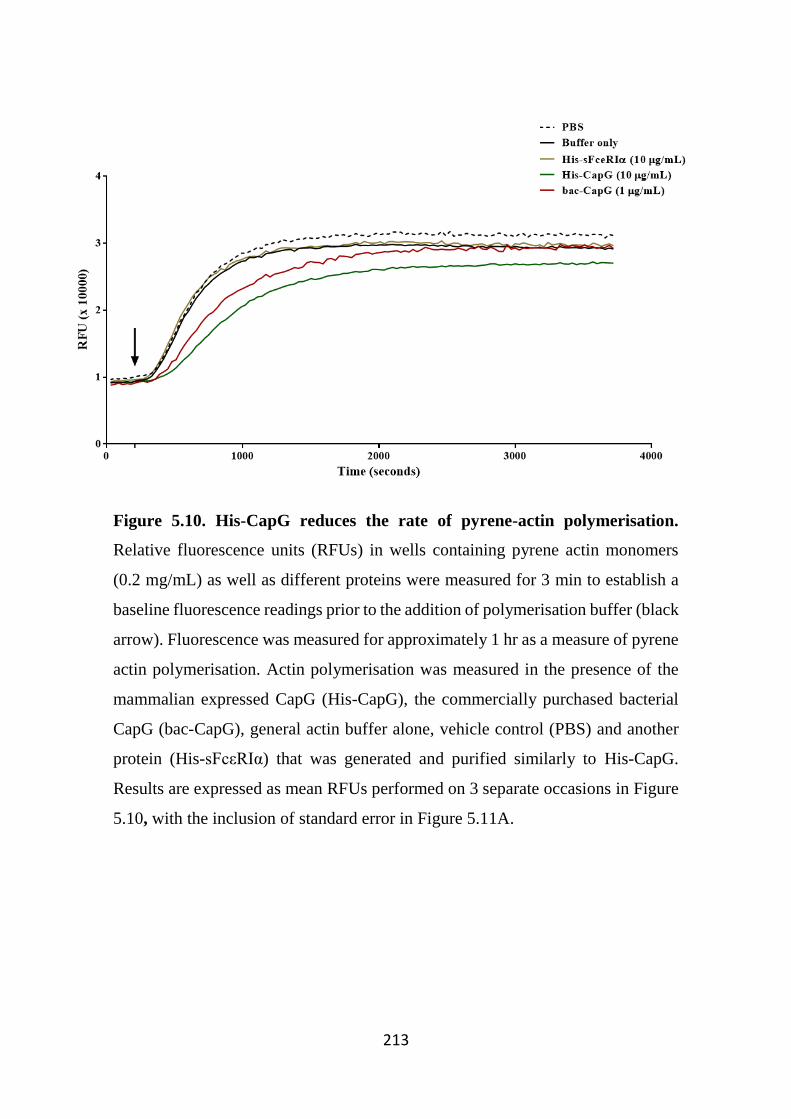
213
Figure 5.10. His-CapG reduces the rate of pyrene-actin polymerisation.
Relative fluorescence units (RFUs) in wells containing pyrene actin monomers
(0.2 mg/mL) as well as different proteins were measured for 3 min to establish a
baseline fluorescence readings prior to the addition of polymerisation buffer (black
arrow). Fluorescence was measured for approximately 1 hr as a measure of pyrene
actin polymerisation. Actin polymerisation was measured in the presence of the
mammalian expressed CapG (His-CapG), the commercially purchased bacterial
CapG (bac-CapG), general actin buffer alone, vehicle control (PBS) and another
protein (His-sFcεRIα) that was generated and purified similarly to His-CapG.
Results are expressed as mean RFUs performed on 3 separate occasions in Figure
5.10, with the inclusion of standard error in Figure 5.11A.
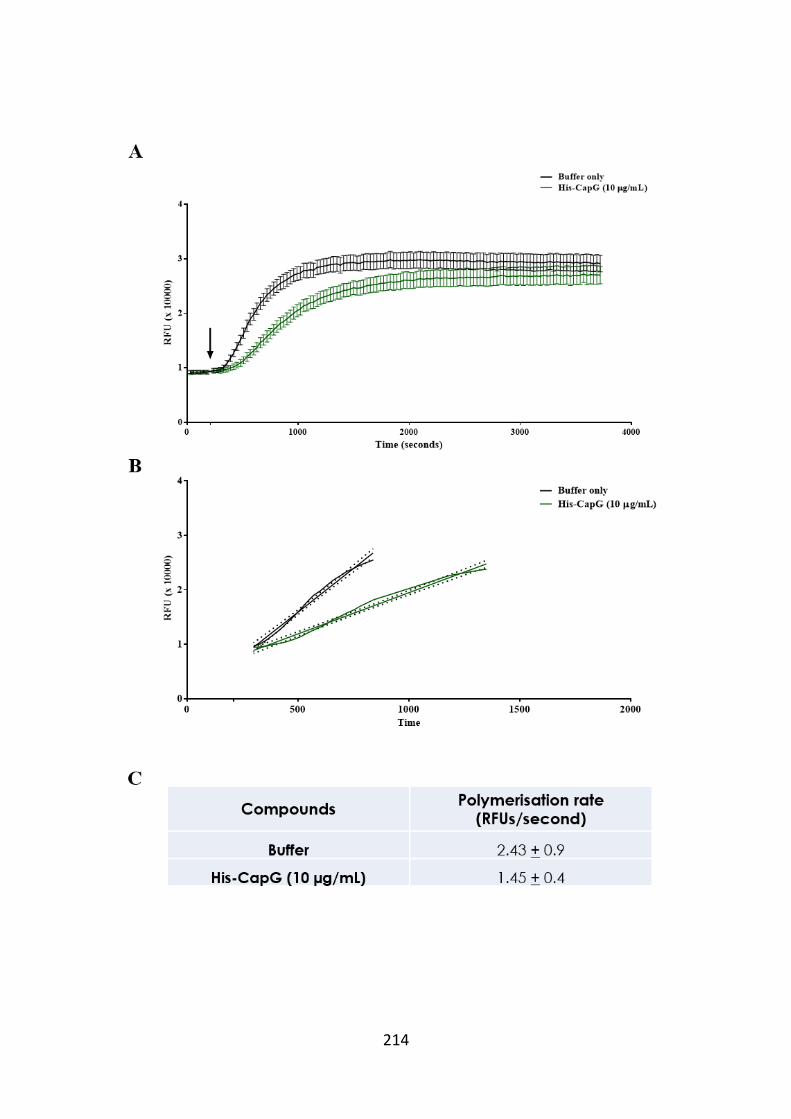
214

215
Figure 5.11. CapG slows the rate of pyrene actin polymerisation. (A) The
fluorescence intensity of actin polymerisation in the presence of buffer only
(control) or His-CapG was compared. Results are plotted as mean fluorescence
intensity ± SEM of relative fluorescence units performed on three separate
occasions. (B) Linear interpolation analysis was applied at the data points of the
elongation phase, which is defined by the time of the addition of actin
polymerisation buffer (t = 240 s) towards the time at which fluorescence activity
has peaked (t = 840 or 1350 for buffer control and His-CapG, respectively). (C)
The rate of actin polymerisation was also calculated, showing that His-CapG
decreased the rate of pyrene actin polymerisation.

216
5.4 Discussion
The aim of this chapter was to generate and optimize a mammalian
expression system that produces high quantities of recombinant human CapG for
downstream functional studies to test the hypothesis that extracellular CapG is a
potential pro-inflammatory mediator. Human recombinant CapG is available
commercially. However, this protein is generated in a bacterial expression system
and not validated for cellular assays and thus likely could contain bacterial
contaminants that might affect data interpretation. In addition, this material is of
high cost when considering the requirements of this material for cellular assays.
Several advantages of utilising mammalian expression systems for recombinant
proteins includes the ability to produce proper protein folding and post
translational modifications similar to its native cellular state, which are often
important for biological activity (Khan, 2013). In addition, the use of the
mammalian expression system might help understand the CapG secretory pathway
as well as facilitate in other studies examining both the intracellular and
extracellular roles of CapG.
Three different CapG mammalian expression systems were examined in
this study. First, the Flp-In™ 293 cells were transfected with a pcDNA5/CapG
vector and initially appeared to be an ideal mammalian expression system. This
was determined by high transfection efficiency and inducible protein present in
both pellets and supernatants of transfected cells following drug induction. The
Flp-In™ 293 cells do not natively express CapG as this protein was detectable only
in the CapG-transfected cells. In our hands, intracellular CapG was readily
detected in transfected cells. In contrast, CapG was only detectable in the
supernatants 3 days following treatment, which was not consistent with previous
studies by others demonstrating that this system readily produces and secretes
proteins that can often be detected in supernatants as early as 24 hours post-
tetracycline treatment (Lu et al, 2011). The delay in CapG release from transfected
cells could likely be attributed to these cells lacking the machinery required for

217
non-canonical secretory pathway for CapG release. It was previously observed that
CapG in macrophages was secreted through a non-canonical signal peptide-
independent pathway (Johnston et al, 1990). Therefore, despite the successful
transfections of the Flp-In™ 293 cells with the pcDNA5/CapG vector, the cells
may intrinsically lack the appropriate secretory machinery that targets CapG for
secretion. To date, knowledge of the pathway of CapG secretion is still not well
understood. Cellular transfection utilising this vector was also designed to
elucidate the native secretory pathway of CapG. However, under our conditions,
the presence of CapG in supernatants was most likely attributed to release from
dying or lysed cells. More importantly, the amount of CapG detected in
supernatants was also at relatively low concentrations that was not ideal for
purification purposes. Thus, utilising this system for generating CapG under these
conditions is not ideal for purifying recombinant CapG, as many proteins released
from dead/dying cells are DAMPs which are also capable of activating cells and
therefore affecting downstream experiments (Bianchi, 2007).
Although the Flp-In™ 293 cell lines may not be an ideal expression system
for recombinant CapG production, these cells lines could be advantageous to study
and better understand the intracellular roles of CapG as this cell line does not
natively express this protein. Moreover, the tetracycline-regulated expression of
CapG could also serve as a useful tool for studying the biological characteristics
of intracellular CapG, including extension mutagenesis studies. In recent years,
there is increasing evidence implicating CapG as a promoter of tumour invasion
(Glaser et al, 2014; Ichikawa et al, 2013; Kimura et al, 2013; Morofuji et al, 2012;
Shao et al, 2011). Previous studies have utilised HEK293 cells in cell migration
and wound closure experiments such as scratch-wound assays (McParland et al,
2011). Thus, this transfected cell line could be useful in characterising the role of
CapG in a variety of cell biological functions including tumour cell invasion and
metastasis.

218
Previous studies have shown that COS-7 cells transfected with CapG were
able to secrete the protein and this was not attributed to cell death (Johnston et al,
1990). This finding was consistent with the present study, as evidenced by the
release of CapG in transiently transfected-COS-7 cells following tetracycline
treatment. However, the CapG yield in cell supernatants was relatively low for
downstream purposes. In addition, these cells do not the express the FRT site
required by the vector to generate stably-transfected cell lines. Thus, whilst
confirming earlier studies, these cells were not progressed for CapG production.
One of the limitations associated with utilising the Flp-In™ 293 or COS-7
cells was the low amount of secreted CapG produced from these cells. To address
this, we utilised an alternative transfection method that relies on episomal
replication instead of the conventional vector-genome integration. There are
several advantages in utilising episomal vectors: Firstly, the inserted gene of
interest will not be interrupted or constrained which can sometimes occur with
integration of the vector to cellular DNA. Secondly, the presence of the gene of
interest will not affect the cell’s own genomic material. Thirdly, episomal vectors
exist in multiple copies in the nucleus, which allows for amplication in the gene of
interest. Finally, the use of episomal vectors often results in higher transfection
efficiency compared to genome-integrating plasmids (Van Craenenbroeck et al,
2000). As mentioned previously, this episomal vector encodes for the EBNA-1
protein which is crucial for interaction with the oriP sequence to maintain vector
stability and episomal replication in EBNA-293 cells (Cachianes et al, 1993).
Studies utilising EBNA-293 cells have shown to produce recombinant
proteins in large quantities within a reasonable timeframe (Meissner et al, 2001).
In the earlier transfection studies, the Flp-In™ 293 and COS-7 cells were
transfected with the pcDNA5/CapG vector, which did not include a signal peptide
to target the protein for release. As mentioned previously, the rationale for initially
excluding the signal peptide sequence was to study and elucidate the native
secretory pathway of this protein. However, in Flp-In™ 293 cells, the protein did

219
not appear to be targeted for secretion. Since the EBNA-293 cells also shared the
same parental cell lines with Flp-In™ 293 cells, it was likely that these cells would
also lack the appropriate secretory machinery to target CapG towards secretion.
Therefore, a new vector with the inclusion of a BM40 signal peptide was
constructed to encourage secretion of CapG. This signal peptide was initially
derived from osteonectin sequence and was included at the N-terminus of the CapG
protein (Holden et al, 2005). The CapG-Strep tag sequence from the initial
pcDNA5 vector was excised and ligated into a pCEP-Pu vector, which was in turn
used in transfecting EBNA-293 cells. Compared to the previous results seen in
Flp-In™ 293 cells and COS-7 cells, the EBNA-293 cells produced and secreted
higher concentrations of CapG which was not associated with cell death.
The conditions of culturing EBNA-293 cells were further optimised to
generate sufficient quantities for downstream studies. Following several
comparisons, including different culture media type and various serum
concentrations, cells were cultured in 1% serum in IMDM as this was the most
cost-efficient option and provided an equilibrium between cell growth, CapG
production and reduced additional protein burden. In addition, the cells were
cultured in 5-layered multi-tiered flask for maximising protein yield and
generating more concentrated material, which would be advantageous for
downstream purification. Adherent EBNA-293 cells grown in the 5-layered multi-
tiered flask grew rapidly, even in low serum media. Every week, supernatants were
harvested and cells were reseeded back in the flask and allowed to propagate and
supernatants collected on the following week. Throughout the entire culture
process, the transfected EBNA-293 cells maintained CapG expression
intracellularly and, more importantly, there was consistently high concentrations
of CapG secreted from the cells.
The recombinant material was also examined for the presence of His and
Strep-tags, respectively, with both tags being detected by Western blotting. These
tags were crucial for downstream purification purposes to obtain high purity

220
material. Since the recombinant CapG expressed both His and Strep tags, the
protein could be subjected to two different purification strategies: 1. A Strep-
Tacin® affinity-chromatography system containing specialised resins that binds
specifically to the Strep-tag portion of the material, or 2. A HisTALON™ metal
affinity column utilising cobalt-charged resins that binds to His-tagged proteins.
The supernatants were subjected to purification through both systems and when
compared, the recombinant CapG yield through the Strep-Tacin® column was
poor. This low protein yield could be overcome by including a repeating Strep-tag
on CapG, as it has been previously shown to increase protein affinity to the Strep-
Tacin® matrices (Schmidt et al, 2013). Another recommended approach to
increase protein yield is by eluting the protein with sodium dodecyl sulfate (SDS).
However, this method is undesirable as it can lead to sample contamination with
Strep-Tacin® released from the resin itself, and SDS can also affect downstream
cellular assays (Ivanov et al, 2014). Thus, purification of the recombinant protein
with Strep-Tacin® resin was not ideal for our purposes.
In contrast, there were high yields of recombinant CapG purified using the
HisTALON™ column and this method was hence used for subsequent purification
processes. This column was able to bind to most of the His-tag recombinant CapG
after passing the supernatants through the column over a period of time, and this
was confirmed by Western blotting analysis, where CapG expression was reduced
in supernatants that had passed through the column, and CapG was strongly
detected in several eluted fractions.
These fractions were pooled and concentrated approximately 10-fold prior
to gel electrophoresis separation under reducing conditions. The protein bands
were stained by Coomassie Blue dye and three strongly-stained bands were
excised to identify their identity by mass spectrometry. A label-free quantitative
proteomics approach was used for protein identification involving a method known
as spectral count, which is defined by the total number of spectra identified for a
protein as a measure of protein abundance in a sample (Lundgren et al, 2010).

221
Distinct proteins were identified in the three bands as listed in Table 5.1. The most
abundant protein present in the sample was sized at approximately 45-47 kDa, and
as expected, was identified as CapG. In addition, two other high molecular weight
proteins at approximately 140 kDa and 110 kDa were also identified. The 140 kDa
band was identified as epididymis luminal proteins 214 (52 kDa) and 213 (26 kDa).
Both proteins contain immunoglobulin domains which might form protein-protein
interactions and create large complexes. However, it should be noted that proteins
were resolved in SDS-PAGE under reducing conditions, thus the presence of these
two low molecular weight proteins at 140 kDa suggests strong chemical bonds
forming stable complexes between the proteins. The function of these proteins
however, remains poorly understood in the literature.
A protein known as cDNA FLJ61580 was detected only in band 2 (110
kDa). This protein is 108 kDa sized and thus correlates with the approximate size
of band 2. cDNA FLJ61580 is highly similar to calsyntenin-1, a type 1
transmembrane protein that belongs to the cadherin superfamily. This protein was
previously reported to be found in the postsynaptic membrane, where its
cytoplasmic domain was reported to bind to intracellular calcium (Vogt et al,
2001). Indeed, cDNA FLJ61580 and calsyntenin-1 share a 98.1% sequence
identity, where there is a 19 amino acid sequence present only in cDNA FLJ61580.
Similar to the proteins identified in band 1, little is known of the function of cDNA
FLJ61580. However, it is likely that this protein shares a similar function to
calsynthenin-1 due to high sequence similarities. Although calsynthenin-1 is more
commonly associated with a role in maintaining the postsynaptic densities in
neurons, this protein has been previously reported to be secreted from α cells in
the pancreatic islets of Langerhans, thus suggestive of a possible endocrine role
for this protein (Rindler et al, 2008). Whilst the function of cDNA FLJ61580
remains poorly understood, the protein could likely be intrinsically secreted from
these EBNA-293 cells, which to our knowledge has not been described in the
literature.

222
Whilst purification of highly expressed polyhistidine recombinant proteins
can generate high degree of pure protein, it is known that purification from
mammalian cells has an increased likelihood of background binding compared to
bacterial cells as mammalian proteins contain a higher percentage of His residues
(Kimple et al, 2013). However, analysis of the amino acid sequences of the
epididymis luminal proteins 213, 214 and cDNA FLJ61580 proteins show that
these proteins do not contain rich strings of histidine residues. In addition, these
protein bands were also absent in Western blot analysis probing for CapG
expression, indicative that these high molecular weight proteins are unlikely to be
associated with CapG. Thus, the source of these contaminating bands is unclear.
Although a range of different imidazole elution conditions was used, we were
unable to remove these contaminating bands. It should also be noted these high
molecular weight bands were also observed during purification of another
unrelated His-tagged protein (His-sFcεRIα). Hence, it is likely that these proteins
may have distinct properties that permit binding with relatively high affinity to the
column, and displacement of this protein during the elution phase results in co-
purification with CapG.
Although CapG was able to be purified from EBNA293 conditioned
medium using the HisTALON™ column, the high molecular weight
contaminating bands could be removed by an additional purification step such as
sized-based gel filtration chromatography that separates proteins on the basis of
their molecular weight. Since the two most prominent bands have a significantly
higher molecular weight under denaturing conditions compared to CapG, this
method could be ideal for purifying CapG further. However, further purification
steps would lead to a reduction in CapG yield. Nevertheless, despite the presence
of the contaminating bands, CapG was the most abundant protein present in the
solution. Thus, we continued with this material to assess the biological activity of
CapG.

223
To validate whether the purified CapG was biologically active, we
examined its ability to modulate actin polymerisation, as CapG is known to bind
to actin filaments and impede polymerisation. Pyrene-labelled actin has been one
of the most commonly used tools for studying actin polymerisation. In its
monomeric form, pyrene-actin is weakly fluorescent. However, in the presence of
actin polymerisation buffer (as described in Section 5.2.7), the actin monomers
polymerise into filaments, resulting in an enhanced fluorescent signal that is easily
measurable (Cooper et al, 1983). In addition, pyrene-actin polymerisation
characteristics has been shown to be comparable to that of native actin as they both
share similar elongation rates, polymerisation kinetics and Ostwald viscosity,
which is a measurement of both polymer length and weight concentration (Cooper
et al, 1983).
Previous experiments characterising the role of intracellular CapG showed
that CapG caps actin filaments at the polymerising end, thus during actin
polymerisation, CapG is able to prolong the lag period (the phase prior to actin
elongation where G-actin aggregates into short, unstable oligomers) (Van Impe et
al, 2013; Young et al, 1990). Similarly, in our hands, there was an increased lag
time in actin polymerisation and a slower rate of actin polymerisation in the
presence of CapG. Over time, CapG did not affect the maximal actin fluorescence
signal as there was no difference in peak fluorescence in presence or absence of
CapG. This is likely because of CapG binding and capping actin filaments
reversibly (Van Impe et al, 2013). The bacterial recombinant CapG was also
included as a positive control and this material was also able to reduce the rate of
pyrene actin polymerisation. In addition, the control His-sFcεRIα protein purified
in the same manner as His-CapG did not have an effect on actin polymerisation,
indicating that the contaminating bands present in both proteins did not affect actin
polymerisation.
In summary, several different cell types were utilised as a mammalian
expression system for generating recombinant human CapG, as an alternative to

224
the commercially available recombinant CapG that is produced through a bacterial
system. Of these, the EBNA-293 cell line was determined as the most effective
system for generating high yields of recombinant CapG. The presence of the His-
tag sequence at the N-terminus of the recombinant protein facilitated in a
successful purification through the use of a HisTALON™ column. Western blot
imaging and mass spectrometry analysis confirmed that the eluted material was
CapG, and the actin polymerisation studies showed that the in-house material was
able to reduce the rate of actin polymerisation, indicating that the recombinant
CapG generated is functionally active. Chapter 6 examines the activity of
mammalian-expressed and purified CapG and its ability to regulate cellular
activity. In some studies, this material is compared to a commercially available,
bacterially expressed recombinant CapG.

225

226
Chapter 6
Functional characterisation of the role of extracellular CapG

227
6.1 Introduction
Inflammation is a key immune process that is activated by any stimulus that
may disrupt tissue homeostasis (Maskrey et al, 2011). This complex biological
process involves a range of different inflammatory cells including resident immune
cells such as macrophages and mast cells, and other immune cells such as
neutrophils, eosinophils and basophils that are recruited to the inflammatory site.
Together, these cells mediate host defense against pathogens such as bacterial,
fungal and viral infections. Along with pathogen clearance, tissue repair and
resolution is also a crucial endpoint of an inflammatory process. The restoration of
resident tissue mononuclear cells (macrophages and lymphocytes) to basal
numbers and the apoptosis and removal of infiltrating inflammatory cells is
necessary as these cells can do harm to the original site of injury if left uncleared
(Maskrey et al, 2011; Serhan et al, 2007).
Many different soluble mediators orchestrate the initiation, infiltration and
the resolution processes. For example, interleukin (IL)-6 and IL-8 promote the
recruitment of leukocytes such as neutrophils (Kaplanski et al, 2003; Miyamoto et
al, 2003), whilst other mediators such as histamine and prostaglandins contribute
to other inflammatory processes including vascular leak, swelling and pain
(Claesson-Welsh, 2015; Ricciotti & FitzGerald, 2011). However, some mediators
are also responsible for the resolution of inflammation. Lipid-derived mediators
such as lipoxins exert their anti-inflammatory effects on many cell types including
monocytes, where they reduce pro-inflammatory cytokine release, and neutrophils
and eosinophils, where they inhibit cell migration and chemotaxis (Serhan et al,
2008). Other mediators such as IL-10 have been shown to limit inflammatory
processes by inhibiting release of inflammatory mediators from cells such as
epithelial (Yilma et al, 2012), as well as promoting neutrophil apoptosis (Cox,
1996).
Many of the symptoms of asthma manifest as a result of mediators released
from activated mast cells (Hall & Agrawal, 2014; Hart, 2001; Holtzman, 1991;

228
Neveu et al, 2009; Nunomura et al, 2005; Sibilano et al, 2014). These mediators
are able to modulate the actions of different cell types such as hASMs, epithelial
cells and fibroblasts. hASM is considered the major contractile element in the
lungs, where it induces and modulates both structural and functional responses of
the airway (Prakash, 2013). hASM cells actively participate in airway responses
to injury, inflammation and infection. In asthma, the hASM layer is markedly
increased in mass and is hyperresponsive (Brightling et al, 2002; Woodman et al,
2008). In addition, these cells release pro and anti-inflammatory mediators that
modulates the local inflammatory environment, drive the proliferation, migration
and apoptosis of other cells, and are also able to release extracellular matrix
proteins that regulates structural changes (Prakash, 2013). In addition to hASM
cells, mast cell mediators can also activate many other cell types. For example,
activated mast cells release tryptase, which is a mitogen for epithelial cells and
also promotes IL-8 release and increases adhesion molecule expression on
epithelial cells (Cairns & Walls, 1996).
Alongside mast cells, hASM and epithelial cells, macrophages are also
known to play a role in allergic asthma pathophysiology. Macrophages are one of
the most abundant leukocytes found in the human lungs and are distinguished into
three classes depending on their location: bronchial, alveolar and interstitial. Of
these, alveolar macrophages (AMs) have in particular been shown to be involved
in asthma pathology. Under normal conditions, AMs are considered as an immune-
suppressing population (Snelgrove et al, 2008). Regulation of AMs is mediated
through cell-cell and soluble mediator interactions, which creates a tightly
regulated environment that limits unwanted inflammatory response. Studies have
shown that cells such as bronchial and alveolar epithelial cells release mediators
such as transforming growth factor-β (TGF-β) (Sacco et al, 1992), and IL-10
(Bonfield et al, 1995) that keeps the activity of AMs in check. However, in the
inflamed lungs, where the airway epithelium is damaged, these negative regulatory
signals are lost, and coupled with the increased presence of pro-inflammatory

229
mediators in the environment, drives AMs to a pro-inflammatory phenotype
(Hussell & Bell, 2014). In addition, tryptase activated epithelial cells release
monocyte chemotactic protein-1 (CCL2) that recruits further AMs to the site of
inflammation (Lee et al, 2015). In airway disorders, AMs have been reported to be
involved in increased airway inflammation, hyperreactivity and inflammatory cell
recruitment (de Nadai et al, 2006; Heaton et al, 2005; Lukacs et al, 1995; Yang et
al, 2009).
AMs can be activated by several mechanisms including IgE-FcεRII, and
through PRRs that enables them to recognise damage-associated molecular
patterns (DAMPs) as well as pathogen-associated molecular patterns (PAMPs)
(Bianchi, 2007; Gosset et al, 1999). In addition, activated mast cells release
mediators that can also activate AMs (Song et al, 2008).
Following cell activation, AMs release a range of different pro-
inflammatory cytokines including TNF, IL-1β, IL-8, and IL-17 (Gosset et al,
1999). The release of these pro-inflammatory cytokines results in the progression
of the severity of asthma pathophysiology features including increased airway
smooth muscle tone and mucus hypersecretion (Berry et al, 2007; Nakae et al,
2002; Whelan et al, 2004). In addition, like mast cells, AMs release CCL2 which
in turn recruits circulating monocytes to the inflamed tissue, thus further
exacerbating the inflammatory process (Brieland et al, 1992; Jiang et al, 1992).
The interaction between immune cells and non-haematopoietic cells is also
observed in other inflammatory disorders not just in asthma. In rheumatoid arthritis
(RA), which is a condition characterised by chronic inflammation of the joints,
both macrophages, and to a lesser extent mast cells, are known to contribute to
disease pathology. These cells release pro-inflammatory cytokines that can lead to
fibroblast proliferation and collagen synthesis, as well as initiating signalling
cascades that lead to further cytokine release, an increased expression of adhesion
molecules and induction of matrix-degrading enzymes that destroys the cartilage,
tendon and bone (Huber et al, 2006; McInnes & Schett, 2007).

230
In summary, haematopoietic immune cells such as mast cells and
macrophages exert many of its pro-inflammatory roles by releasing cytokines and
chemokines that in turn activate resident cells or recruit other immune cells to the
site of inflammation. Whilst many of these mediators have been identified and
well-studied, there are still likely other novel mediators that might be of
importance to disease pathology and serve as novel targets for drug therapy for
inflammatory diseases.
As previously mentioned, unpublished studies in our laboratory have
identified several previously undescribed proteins released from activated mast
cells that may act as inflammatory mediators (Xia et al, 2013b). Of particular
interest to us was macrophage capping protein (CapG), which was found to be
released from activated mast cells and macrophages, as described in Chapter 3. It
has been previously reported that CapG is released constitutively from
macrophages and that it is present in plasma (Johnston et al, 1990). However,
knowledge in regards to CapG function as a released mediator is unclear. CapG
may be released from dead or dying cells and act potentially as a damage
associated molecular pattern (DAMP). DAMPs are cell-derived molecules that are
localised within the cell nucleus and cytoplasm, but can also consist of components
of the extracellular matrix and serum (Tang et al, 2012). These molecules are
normally sequestered but can be actively secreted from cells or passively released
from dying cells or damaged extracellular matrix (Land, 2015). DAMPs bind to
PRRs expressed on immune cells. This in turn results in the initiation of protective
mechanisms including inflammation and apoptosis to remove the dying or dead
cells (Land, 2015).
On the other hand, extracellular CapG may act similarly to its family
member gelsolin in the extracellular space. Extracellular gelsolin is involved in
clearing extracellular actin to prevent extracellular polymerisation and thus flow
occlusion, which can have serious consequences if left uncleared (Lee et al, 2007).

231
Therefore, it is plausible that extracellular CapG may also play a role in regulating
extracellular actin polymerisation.
Whilst it has been previously reported that CapG is constitutively released
from resting macrophages, results from Chapter 3 showed the regulated release of
CapG from LPS-stimulated macrophages. In addition, IgE/FcεRI-activated LAD2
mast cell lines also released CapG (summarised in Figure 6.1A). Furthermore,
CapG levels were reported to be elevated in the synovial fluids of RA patients
(Balakrishnan et al, 2014). Taken together, we hypothesised that CapG released
from activated mast cells and macrophages acts as a pro-inflammatory mediator
that is able to exacerbate the inflammatory state.
As discussed in Chapter 5, commercially available recombinant human
CapG is generated using a bacterial expression system. However, this material is
costly and because it is not intended for cell stimulation experiments the
purification of this recombinant protein is not as stringent as for instance
recombinant cytokines. Therefore, we generated recombinant human CapG in-
house using a mammalian expression system, as described in Chapter 5. Functional
assays validated the activity of the in-house recombinant CapG. In this chapter we
sought to examine whether CapG is able to act as a novel inflammatory mediator,
triggering cytokine release from different cell types, by comparing the activity of
the mammalian His-tagged CapG (His-CapG) versus the commercial bacterial
recombinant CapG (bac-CapG) (Figure 6.1B).
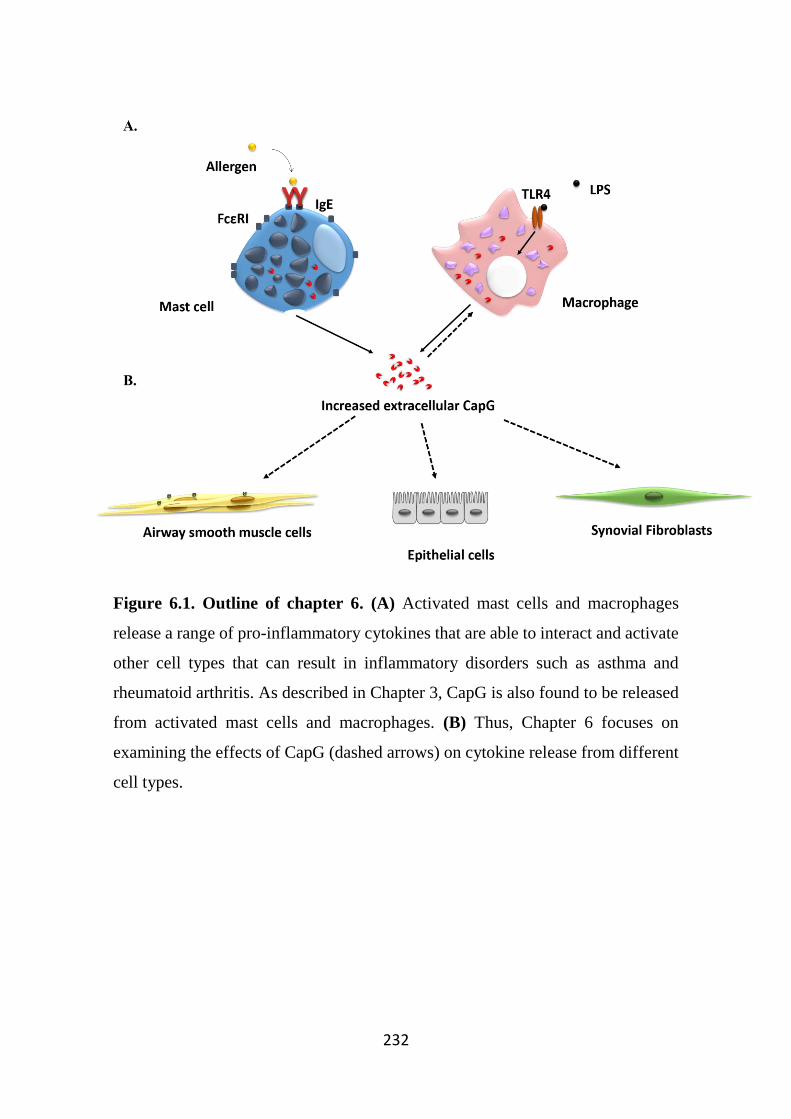
232
Figure 6.1. Outline of chapter 6. (A) Activated mast cells and macrophages
release a range of pro-inflammatory cytokines that are able to interact and activate
other cell types that can result in inflammatory disorders such as asthma and
rheumatoid arthritis. As described in Chapter 3, CapG is also found to be released
from activated mast cells and macrophages. (B) Thus, Chapter 6 focuses on
examining the effects of CapG (dashed arrows) on cytokine release from different
cell types.

233
6.2 Specific methods
6.2.1 Cell culture and stimulation
6.2.1.1 Human airway smooth muscle (hASM) cells
The protocol for obtaining, culturing and stimulating hASM cells was
described earlier in Section 2.1.7. Cells were stimulated with lipopolysaccharide
(LPS; Sigma-Aldrich), TNFα (Sigma-Aldrich), and recombinant CapG that was
either purchased (bac-CapG, ab95385, Abcam, Cambridge, MA) or generated in-
house (His-CapG). In addition, cells were also challenged with recombinant CapG
or LPS that were either treated with the antibiotic polymyxin B (PMXB; 10 µg/mL,
Sigma-Aldrich), which has been shown to inhibit the effects of LPS through
physical binding (Cardoso et al, 2007), or heat treated at 97ºC for 30 min prior to
exposure to cells.
6.2.1.2 THP-1 cells
The cultivation and maintenance of THP-1 cells was described earlier in
Section 2.1.4. The experimental protocol for cell stimulation is essentially as
described in Section 2.1.4.1. However, cells were seeded in 96-well plates
(Corning) at a density of 100,000 cells/well to conserve CapG material.
Similar to hASM cells, THP-1 cells were also stimulated with LPS and
recombinant CapG pre-treated with PMXB or heat-treated for 30 minutes. After
24 hours, supernatants were harvested for cytokine analysis.
6.2.1.3 BEAS2B cells
The bronchial epithelial cell line BEAS2B is commonly used in in vitro
studies of airway inflammation (Ohtoshi et al, 1998). The cells were cultivated and
maintained as described earlier in Section 2.1.8. In cell stimulation experiments,
cells were seeded at a density of 70,000 cells/well in a 96 well plate in serum-
complete RPMI. The following day, cells were replenished with fresh serum-free

234
RPMI for 24 hours. The cells were then stimulated with CapG for a further 24
hours.
6.2.1.4 SW982 cells
The synovial fibroblast cell line SW982 was maintained as described earlier
in Section 2.1.11. Prior to stimulation, cells were seeded at a density of 50,000
cells/well in a 96-well flat bottom plate for 2 days in serum-complete DMEM
media. Cells were then serum-starved for a further 24 hrs in incomplete DMEM
supplemented with BSA (0.25%). Cells were then stimulated under conditions
similar to THP-1 cells as described in Section 6.2.1.2.
6.2.2 Cell viability measurement
Following stimulation, cell viability was assessed in order to examine if
stimuli had any cytotoxic actions on cells. In these studies, resazurin dye was used.
This dye is blue and weakly-fluorescent that in the presence of reducing enzymes
undergoes an irreversible chemical reduction into the highly fluorescent pink
resorufin. Several mitochondrial enzymes such as mononucleotide dehydrogenase,
flavin adenine dinucleotide dehydrogenase and cytochromes converts resazurin to
resorufin (O'Brien et al, 2000). The pink resorufin is then secreted from cells,
which can then be quantified fluorometrically. In this study, after harvesting
supernatants for cytokine analysis, cells were incubated with resazurin dye
(Sigma-Aldrich; diluted 1:10 in stimulation medium) and incubated at 37ºC, 5%
CO2. An empty well containing no cells was also included to quantify any
spontaneous reduction of resazurin. Fluorometric measurements were measured
using a FlexStation® II machine, with the excitation and emission wavelengths set
at 570 nm and 585 nm, respectively.

235
6.2.3 Measurement of cytokine levels using enzyme-linked immunosorbent
assays (ELISA)
6.2.3.1 IL-8
The pro-inflammatory cytokine IL-8 is known to be released from hASM
and THP-1 cells in response to a range of stimuli and was thus measured in this
study (John et al, 1998a; Tamai et al, 2003). IL-8 levels were measured using a
commercially available IL-8 ELISA kit as described earlier (Section 2.6).
6.2.3.2 CCL2
LPS-activated THP-1 cells have also been previously reported to release
CCL2, a chemokine that promotes recruitment and infiltration of monocyte and
macrophages (Deshmane et al, 2009; Harrison et al, 2005). CCL2 cytokine levels
released from THP-1 cells were measured using a commercially available CCL2
ELISA kit as described earlier (Section 2.6).
6.2.4 Statistical analysis
Data from ELISA analysis were expressed as the means ± standard error of
mean (SEM), where n represents the number of independent primary cell cultures
used or numbers of experiments repeated using cell line. If applicable, an
appropriate statistical analysis test was performed (refer to Section 2.13).
Results shown were plotted using Graphpad Prism software (version 6.01).
If a statistical significance was obtained, then * denotes p<0.05, ** denotes p<0.01,
and *** denotes p<0.001.

236
6.3 Results
6.3.1 Bacterially-expressed recombinant CapG trigger IL-8 and IL-6 release
from primary human airway smooth muscle cells.
Whilst the generation of the mammalian recombinant CapG was ongoing,
we sought to examine whether the commercially purchased bacterial recombinant
CapG was able to trigger cytokine release from different cell types. Primary hASM
cultures obtained from 4-5 donors were stimulated with bacterial recombinant
CapG (bac-CapG) for 24 hrs and supernatants were harvested. bac-CapG triggered
IL-8 production from hASM cells in a concentration-dependent manner, with
statistical significance reached at higher bac-CapG (5 and 10 µg/mL). Cells were
also stimulated with other stimuli including LPS (1 µg/mL) and TNFα (10 ng/mL).
TNFα was used as a positive control, as demonstrated in other studies (John et al,
1998a), and this was also observed across all cultures. In contrast, IL-8 release
from hASM cells stimulated with LPS at 1 µg/mL was similar to basal IL-8 release
(Figure 6.2A), indicating that LPS is a weak stimulator of hASM cells, which has
been previously described (Morris et al, 2005).
In addition to IL-8, IL-6 cytokine level was also measured from bac-CapG-
stimulated hASM cells. IL-6 release was elevated from cells stimulated at higher
bac-CapG concentrations, however this did not reach statistical significance at the
concentrations tested (Figure 6.2B). However, due to limited quantities of the bac-
CapG available, cytokine release from hASM cells stimulated at higher bac-CapG
concentrations was not investigated. In response to TNFα, there was also a
significant increase in IL-6 cytokine production from hASM cells. Similar to IL-8
cytokine results, we and others have shown that LPS did not induce IL-6 release
from hASM cells (Shan et al, 2006).
Cytokine release from cells was not due to cell death as indicated by there
being no statistical significance in cell viability (as measured by resazurin)

237
between stimulated cells compared to vehicle control, although there appeared to
be a trend for lowered viability in cells treated with LPS (Figure 6.2C).
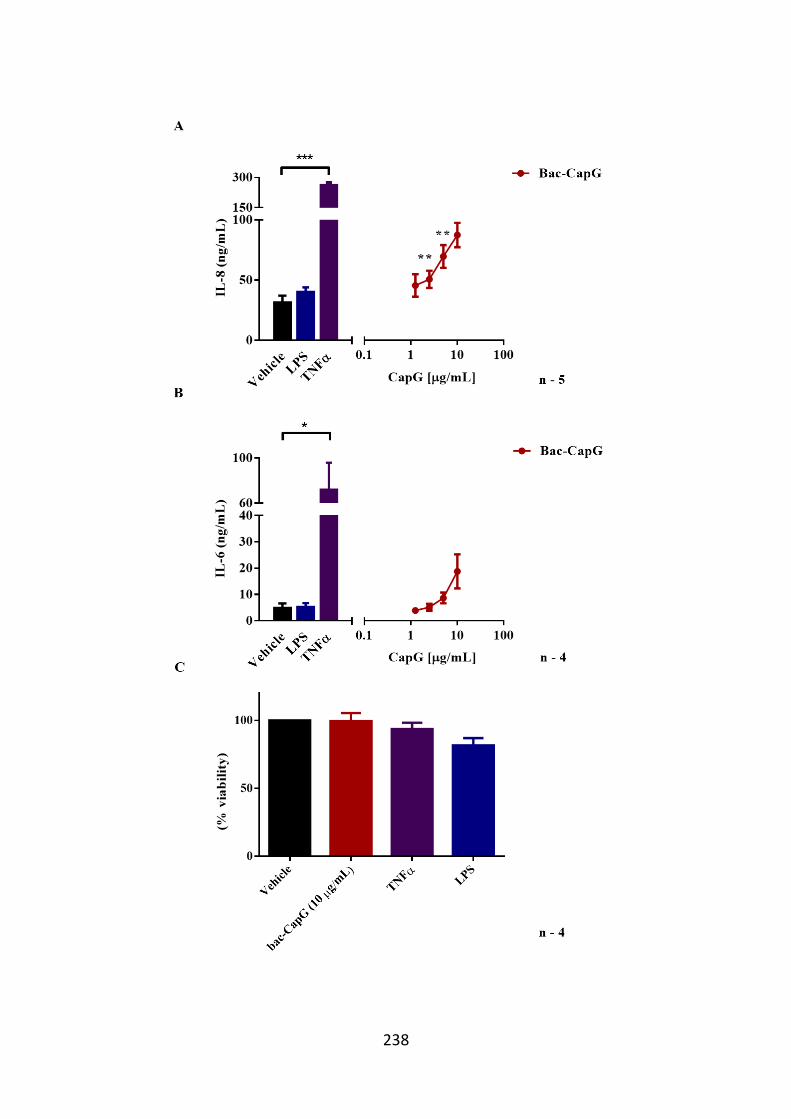
238

239
Figure 6.2. IL-8 and IL-6 is released from hASM cells following bac-CapG
stimulation. Primary hASM cells were serum starved for 24 hours, and then
treated with human recombinant CapG expressed in E. coli (bac-CapG). Cytokine
release from hASM cells stimulated with TNFα (100 ng/mL) and LPS (1 µg/mL)
was also measured. The vehicle control group in this study was non-treated hASM
cells (black bar). hASM supernatants were harvested after 24 hour stimulation and
cytokine levels were measured by immunoassay. (A) Compared to vehicle control,
there was a concentration-dependent increase in IL-8 release in response to higher
CapG concentrations. (B) However, bac-CapG did not trigger significant IL-6
cytokine release from hASM cells. In contrast, TNFα induced high levels of IL-8
and IL-6 from hASM cells. Results are expressed as mean cytokine level ± SEM
conducted on 4-5 individual primary hASM cultures. One-way repeated measures
ANOVA followed by the Dunnett’s post-hoc test was applied for comparison of
each condition to vehicle; *p<0.05, and ***p<0.001 for comparison with the
vehicle group. (C) Following cell stimulation, cell viability was assessed using the
resazurin dye. Fluorometric measurements were measured over time and compared
between different treatments and vehicle control. Results are expressed as the ratio
± SEM between the fluorescence readings of each condition over the vehicle
control conducted on the primary hASM cultures. For clarity, the fluorescence
measurements of only the highest concentration of bac-CapG, LPS and TNFα are
displayed. However, it should be noted that the cell viability assay was performed
across all treatment conditions and cell viability measurements were similar to
control.

240
6.3.2 bac-CapG induces IL-8 release from THP-1 cells
Although CapG is known to be released from activated THP-1 cells as
described in Chapter 3, we were also interested in determining whether CapG is
able induce autocrine cell activation. Hence, we examined IL-8 cytokine release
from stimulated THP-1 cells. Stimulation with bac-CapG (Figure 6.3A) induced
a concentration dependent increase in IL-8 release from THP-1 cells. In addition,
cells were also stimulated with LPS as a positive control. As expected, LPS
triggered a very large concentration-dependent IL-8 release from cells (Figure
6.3B). The release of IL-8 from CapG or LPS was not due to cell death as indicated
by similar resazurin readings to vehicle control (Figure 6.3C).
In addition, we also investigated the effects of CapG on cytokine release
from other cell types known to interact with and be regulated by mast cells and
macrophages such as epithelial cells and fibroblasts. The effects of bac-CapG on
the bronchial epithelial cell line BEAS2B was examined. Cells were stimulated
overnight and supernatants were harvested and assayed for IL-8 levels by ELISA.
In response to bac-CapG, the BEAS2B cells released IL-8 (Figure 6.4).
As previously discussed, macrophages and to a lesser extent mast cells have
also been implicated in RA pathology, and previous studies have reported elevated
CapG levels in the synovial fluids of RA patients. Thus, we also examined the
effects of bac-CapG on the synovial fibroblasts cell line SW982. Similar to other
cell types, there was a concentration-dependent IL-8 release from CapG stimulated
SW982 cells (Figure 6.5A). In addition to CapG, cells were also stimulated with
LPS as a positive control. As expected, there was a concentration-dependent
increase in IL-8 release from SW982 cells stimulated with LPS (Figure 6.5B).
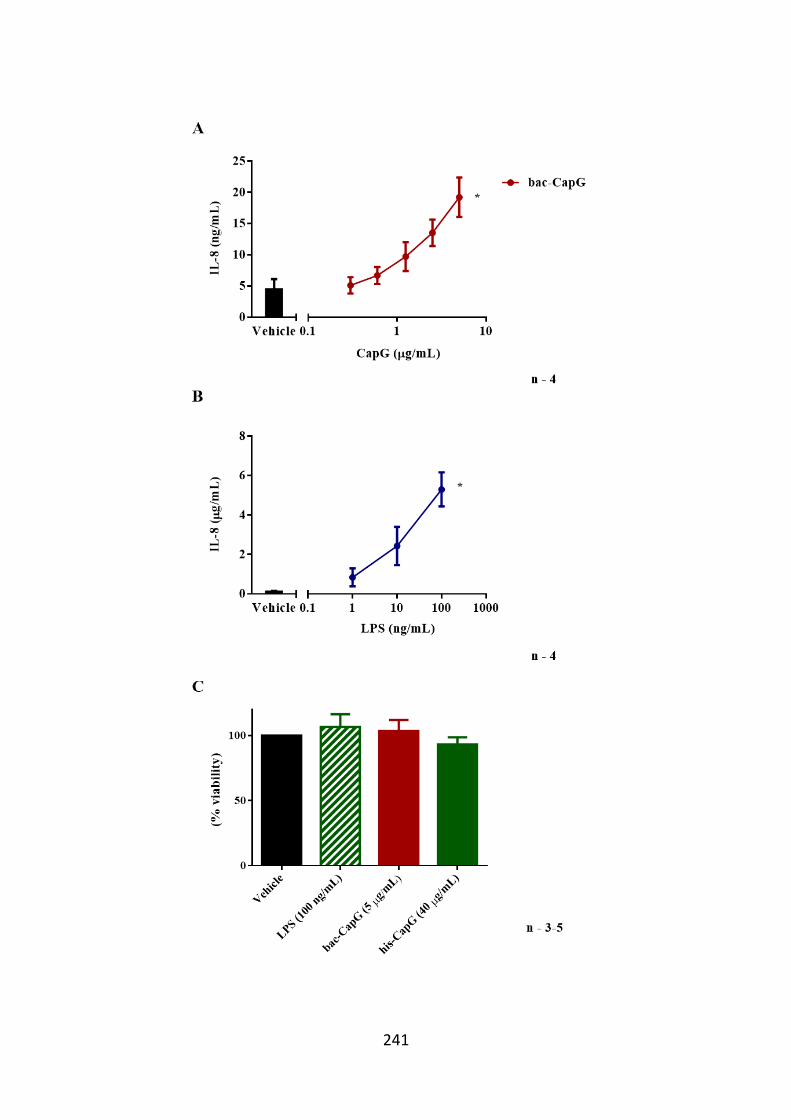
241

242
Figure 6.3. THP-1 cells release IL-8 when stimulated with CapG. THP-1 cells
were PMA-treated for 48 hours, and further serum-starved for 24 hours prior to
stimulation with a range of concentrations of (A) bac-CapG for 24 hours and IL-8
levels measured by immunoassay. Compared to basal IL-8 levels (black bar), there
was a significant increase in IL-8 release in response to bac-CapG. (B) Cells were
also stimulated with LPS as a positive control in this study. IL-8 cytokine results
are expressed as mean percentage relative to vehicle control ± SEM, conducted on
THP-1 cells on 3-5 separate occasions. One-way repeated measures ANOVA
followed by the Dunnett’s post-hoc test were applied for multiple comparisons;
*p<0.05 for comparison with the vehicle group. (C) Following cell stimulation,
cell viability was measured by incubation with resazurin dye and fluorometric
readings were measured over time. There were no differences in readings between
treated cells and vehicle control. Results are expressed as the ratio ± SEM between
the fluorescence readings of each condition over the vehicle control conducted on
THP-1 cells. For clarity, the fluorescence measurements of only the highest
concentration of His-CapG, bac-CapG and LPS were displayed. However, it
should be noted that the cell viability assay was performed across all treatment
conditions and cell viability data were similar to control.
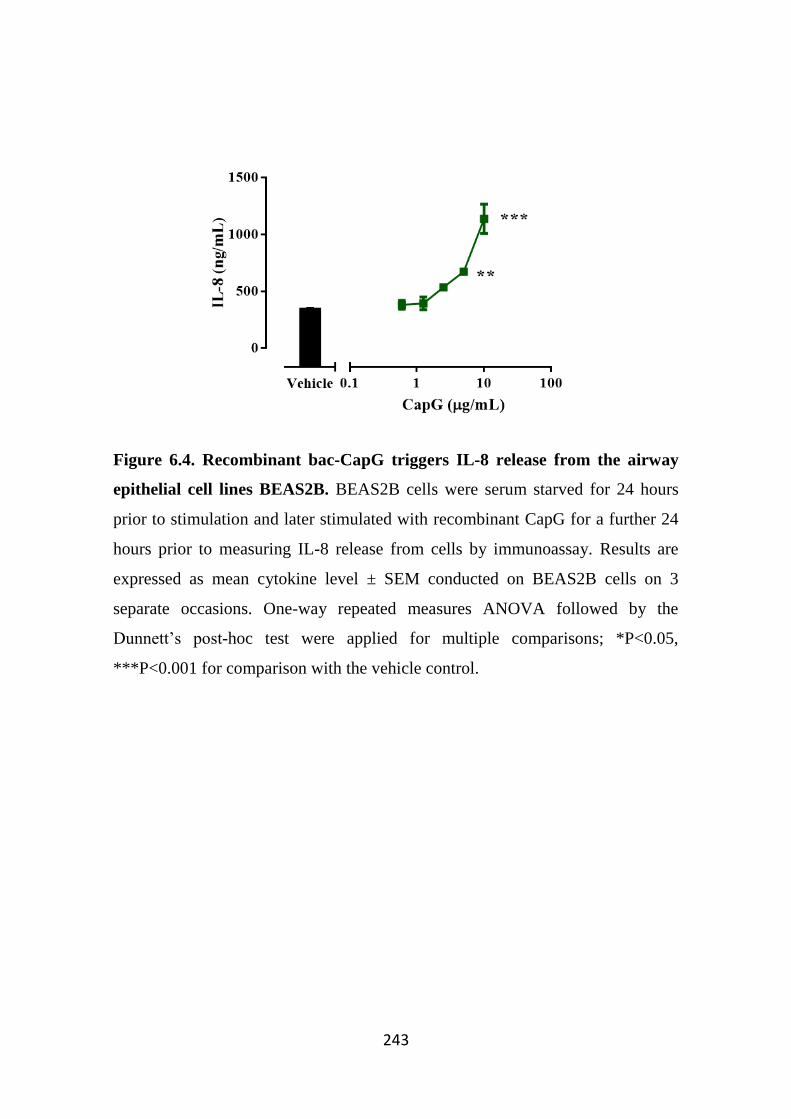
243
Figure 6.4. Recombinant bac-CapG triggers IL-8 release from the airway
epithelial cell lines BEAS2B. BEAS2B cells were serum starved for 24 hours
prior to stimulation and later stimulated with recombinant CapG for a further 24
hours prior to measuring IL-8 release from cells by immunoassay. Results are
expressed as mean cytokine level ± SEM conducted on BEAS2B cells on 3
separate occasions. One-way repeated measures ANOVA followed by the
Dunnett’s post-hoc test were applied for multiple comparisons; *P<0.05,
***P<0.001 for comparison with the vehicle control.
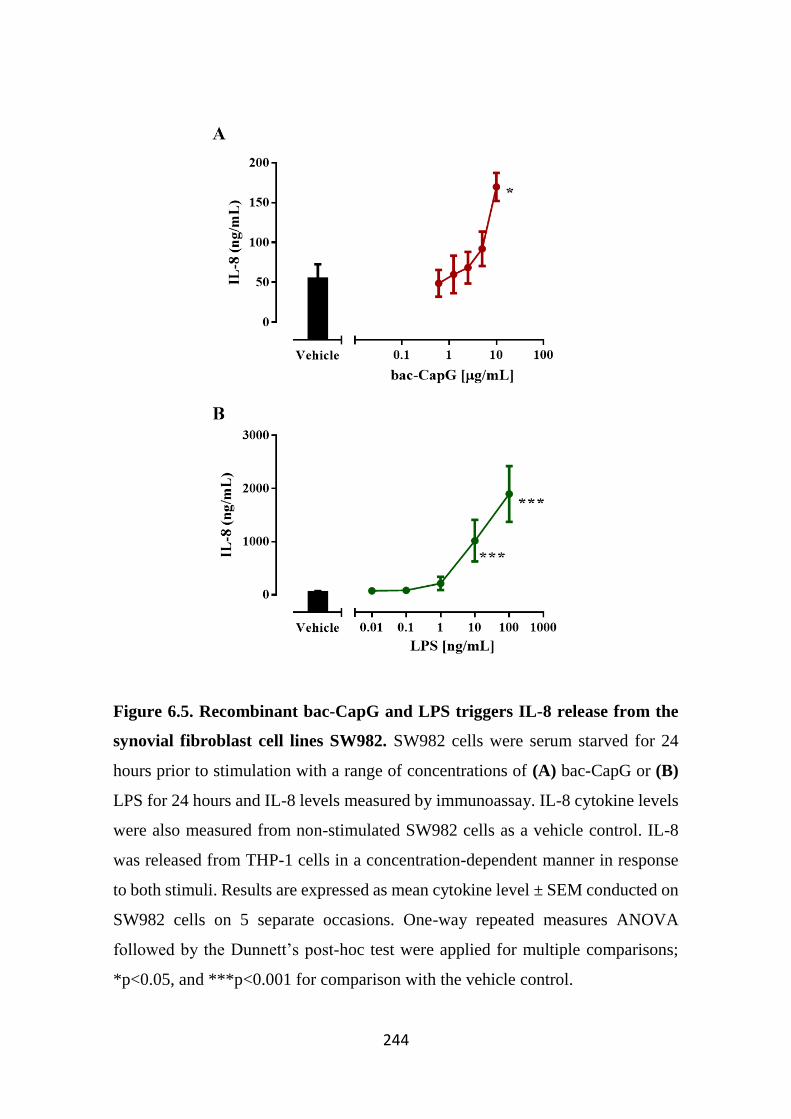
244
Figure 6.5. Recombinant bac-CapG and LPS triggers IL-8 release from the
synovial fibroblast cell lines SW982. SW982 cells were serum starved for 24
hours prior to stimulation with a range of concentrations of (A) bac-CapG or (B)
LPS for 24 hours and IL-8 levels measured by immunoassay. IL-8 cytokine levels
were also measured from non-stimulated SW982 cells as a vehicle control. IL-8
was released from THP-1 cells in a concentration-dependent manner in response
to both stimuli. Results are expressed as mean cytokine level ± SEM conducted on
SW982 cells on 5 separate occasions. One-way repeated measures ANOVA
followed by the Dunnett’s post-hoc test were applied for multiple comparisons;
*p<0.05, and ***p<0.001 for comparison with the vehicle control.

245
6.3.3 Polymyxin B dampens the biological activity of bac-CapG on IL-8 release
from THP-1 and SW982 cells
Since the purchased recombinant bac-CapG was generated in a bacterial
expression system, and the material was not as necessarily highly purified as are
recombinant cytokines used for cell treatments, we sought to examine whether the
presence of additional factors, particularly as endotoxins like LPS, that could affect
cytokine release from stimulated cells. Coomassie staining of bac-CapG showed a
strong staining at approximately 40 kDa, which correlates to the size of CapG
(Figure 6.6A). However, there were also several faint but noticeable lower
molecular weight proteins present, indicative of other proteins present. In addition,
the likely presence of other non-protein molecules such as bacterial sugars
including LPS could also potentially confound data interpretation.
We and others have shown that THP-1 cells respond strongly to even low
concentrations of bacterial products like LPS. To examine whether the presence of
bacterial components (such as LPS) could affect cell stimulation, both LPS and
bac-CapG were pre-treated with polymyxin B (1 µg/mL), which is known
neutralise LPS activity by directly binding to LPS (Tsuzuki et al, 2001). As
expected, LPS alone significantly induced IL-8 release from THP-1 cells.
However, in the presence of polymyxin B, IL-8 release from THP-1 cells
stimulated with LPS was significantly reduced (Figure 6.6B). It is interesting to
note that Polymyxin B did not completely inhibit IL-8 release from LPS-stimulated
THP-1 cells. This may suggest that (1) that the concentration of polymyxin B used
was too low to completely inhibit THP-1 response to LPS, or (2) that THP-1 cells
are very sensitive to LPS that only low concentrations of non-polymyxin-B-
neutralised LPS is required to trigger THP-1 response.
In addition, we examined whether polymyxin B was able to affect THP-1
cells stimulated with a high concentration of bac-CapG (5 µg/mL) known to trigger
IL-8 release (refer to Figure 6.3A). Interestingly, cytokine release was reduced
when cells were stimulated with polymyxin-B treated CapG (Figure 6.6C).

246
Despite this, bac-CapG was still able to significantly increase IL-8 cytokine release
from THP-1 cells. Therefore, this data suggest that IL-8 release from THP-1 cells
stimulated with bac-CapG is only partly due to CapG itself. The presence of
bacterial components such as LPS also triggered IL-8 release from cells, thus
partly confounding our results but also validating the necessity of alternative
expression systems to study CapG.
Since both bac-CapG and LPS also triggered a strong IL-8 release from
SW982 cells, we sought to determine whether polymyxin B treatment could also
affect IL-8 release from these cells in response to these stimuli. Similar to THP-1
results, polymyxin B significantly reduced LPS-mediated IL-8 release from
SW982 cells (Figure 6.7A). However, polymyxin B did not affect IL-8 release
from bac-CapG stimulated SW982 cells (Figure 6.7B).
The data obtained from THP-1 and SW982 cells suggest that the presence
of bacterial contaminants in bac-CapG, likely LPS, can indeed confound data
interpretation of bac-CapG. However, these experiments utilising polymyxin B has
demonstrated that the concentration of these contaminants are too low that only
certain cell lines very sensitive to LPS such as THP-1 will be affected. In contrast,
other cell lines less sensitive to LPS such as SW982 cells, are not affected by these
contaminants, as demonstrated by the lack of effect of polymyxin B on cell
response to bac-CapG.
When hASM cultures were stimulated with polymyxin B-treated bac-
CapG, IL-8 cytokine levels were similar to bac-CapG responses in the absence of
polymyxin B (Figure 6.8). This result was not surprising, as unlike the THP-1 and
SW982 cells, hASM cells respond weakly to LPS as observed in Figure 6.1. This
finding demonstrates that polymyxin B does not affect the signalling pathways
mediated by CapG. Combined, this finding demonstrates that whilst bac-CapG is
able to trigger cytokine release from cells, the presence of bacterial components
such as LPS can confound results and data interpretation.
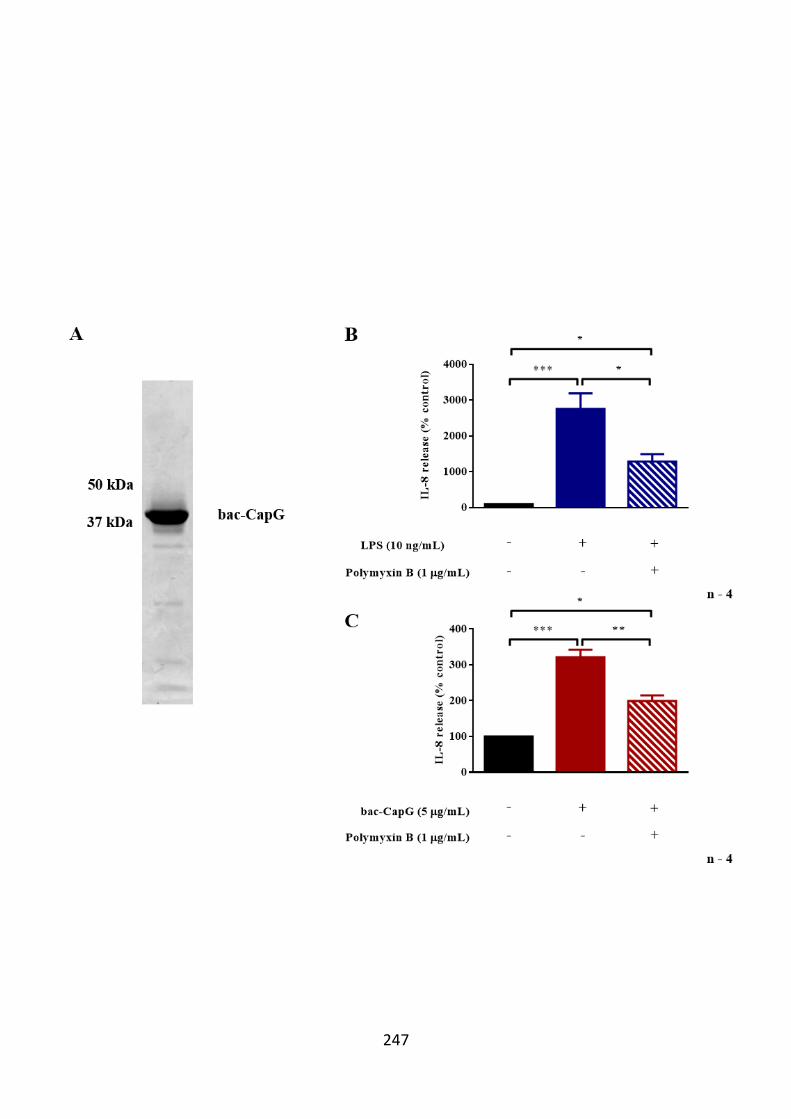
247

248
Figure 6.6. Polymyxin B significantly reduces IL-8 release from THP-1 cells
stimulated with LPS and bac-CapG. (A) The recombinant bac-CapG was
resolved by gel electrophoresis and the gel was then stained with Coomassie Blue
dye to visualise protein bands. A strongly-stained band was detected at
approximately 40 kDa, which is the predicted molecular weight of CapG.
However, other faint low molecular weight bands were also present in the
recombinant bac-CapG protein. (B) IL-8 release from THP-1 cells stimulated with
LPS pre-treated with polymyxin B (30 mins) or untreated LPS was quantified by
immunoassay following 24 hour stimulation. Compared to untreated recombinant
LPS, there was a significant reduction in IL-8 release from THP-1 cells stimulated
with polymyxin B-treated LPS. (C) Similarly, IL-8 release from THP-1 cells
stimulated with polymyxin B-treated bac-CapG was also significantly reduced
compared to bac-CapG alone. Results are expressed as a percentage of IL-8
cytokine release ± SEM normalised to basal IL-8 release from THP-1 cells
conducted on 4 separate occasions. One-way repeated measures ANOVA followed
by the Dunnett’s post-hoc test were applied for multiple comparisons. *p>0.05,
**p>0.01 and ***p>0.001 for comparing differences in IL-8 release between LPS
or bac-CapG-stimulated THP-1 cells in the presence or absence of polymyxin B.
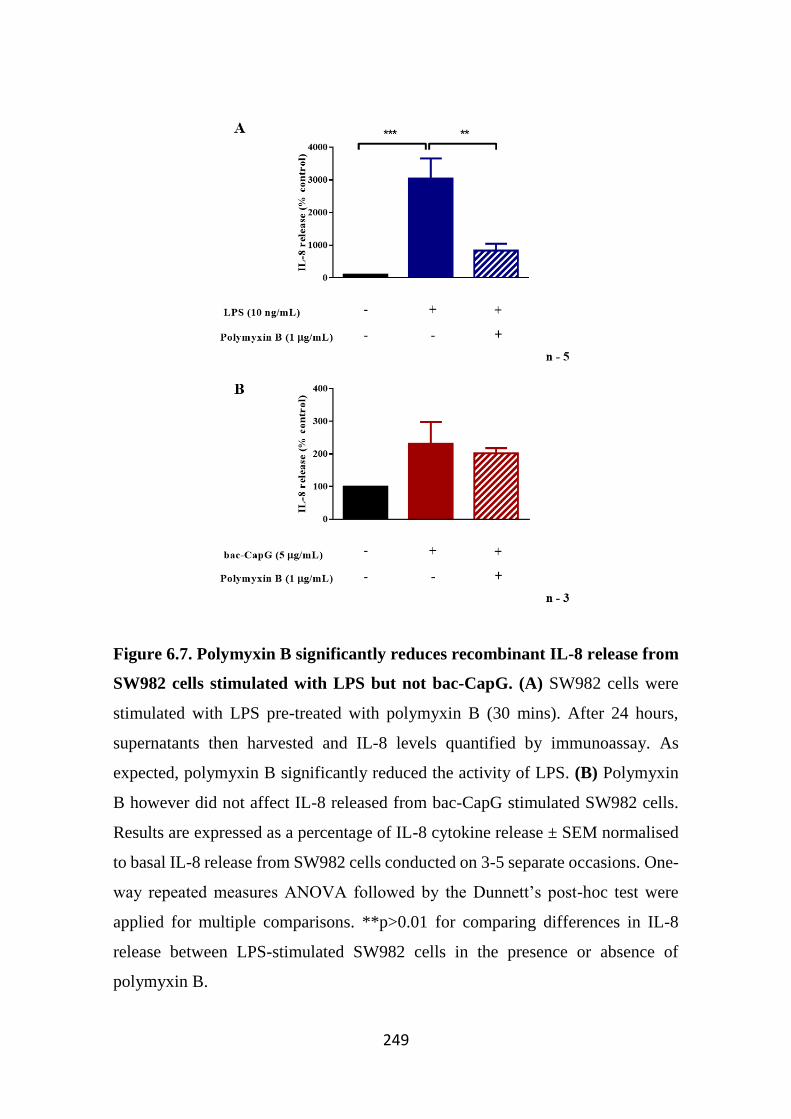
249
Figure 6.7. Polymyxin B significantly reduces recombinant IL-8 release from
SW982 cells stimulated with LPS but not bac-CapG. (A) SW982 cells were
stimulated with LPS pre-treated with polymyxin B (30 mins). After 24 hours,
supernatants then harvested and IL-8 levels quantified by immunoassay. As
expected, polymyxin B significantly reduced the activity of LPS. (B) Polymyxin
B however did not affect IL-8 released from bac-CapG stimulated SW982 cells.
Results are expressed as a percentage of IL-8 cytokine release ± SEM normalised
to basal IL-8 release from SW982 cells conducted on 3-5 separate occasions. One-
way repeated measures ANOVA followed by the Dunnett’s post-hoc test were
applied for multiple comparisons. **p>0.01 for comparing differences in IL-8
release between LPS-stimulated SW982 cells in the presence or absence of
polymyxin B.
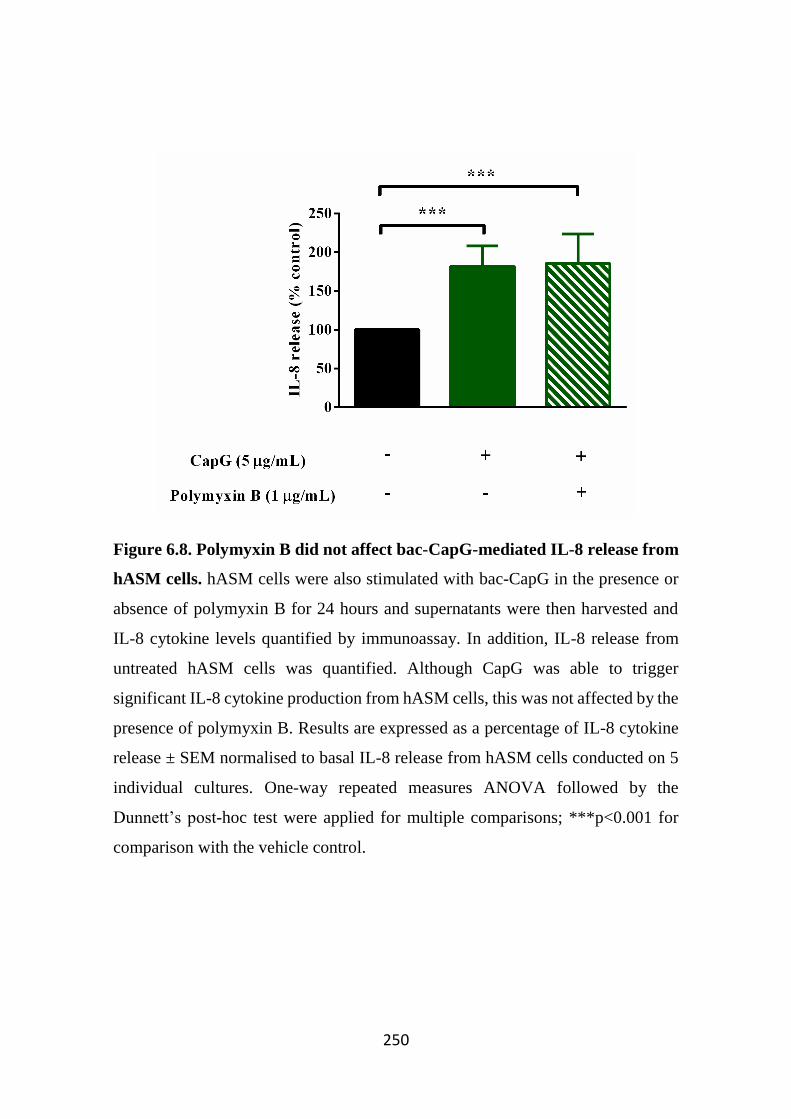
250
Figure 6.8. Polymyxin B did not affect bac-CapG-mediated IL-8 release from
hASM cells. hASM cells were also stimulated with bac-CapG in the presence or
absence of polymyxin B for 24 hours and supernatants were then harvested and
IL-8 cytokine levels quantified by immunoassay. In addition, IL-8 release from
untreated hASM cells was quantified. Although CapG was able to trigger
significant IL-8 cytokine production from hASM cells, this was not affected by the
presence of polymyxin B. Results are expressed as a percentage of IL-8 cytokine
release ± SEM normalised to basal IL-8 release from hASM cells conducted on 5
individual cultures. One-way repeated measures ANOVA followed by the
Dunnett’s post-hoc test were applied for multiple comparisons; ***p<0.001 for
comparison with the vehicle control.

251
6.3.4 His-CapG triggers IL-6 and IL-8 release from hASM cells
Following the generation and functional validation of mammalian
expressed His-CapG in Chapter 5, we examined whether cell stimulation with His-
CapG could recapitulate previous studies utilising bac-CapG by examining
cytokine release from hASM cells. Cells were stimulated with a range of His-CapG
concentrations for 24 hrs. Supernatants were harvested and IL-8 levels measured.
His-CapG triggered IL-8 release from hASM cells in a concentration-dependent
manner (Figure 6.9A). Similarly, His-CapG also triggered IL-6 release from
hASM cells in a concentration-dependent manner (Figure 6.9B). Cytokine release
mediated by His-CapG was not due to cell death, as determined by high cell
viability measured following cell stimulation (Figure 6.9C). It should be noted
that His-sFcεRIα was also used as a stimulus in this study, however, this stimulus
did not trigger cytokine release from cells (data not shown).
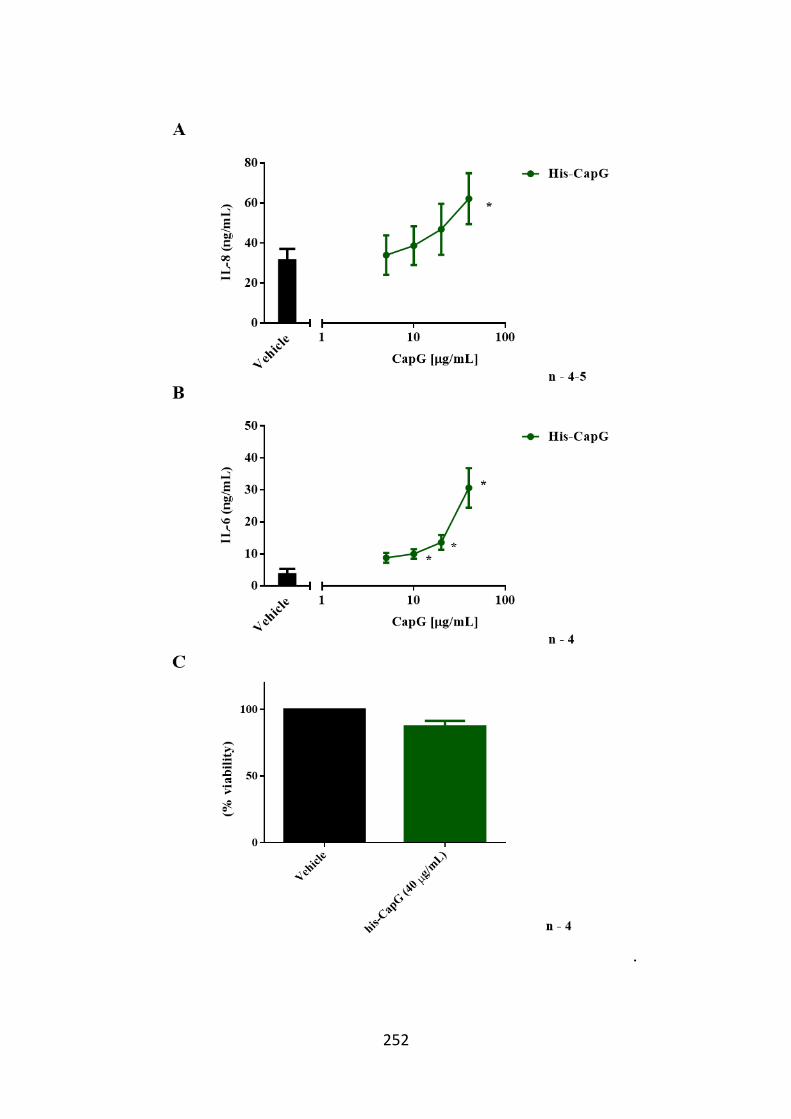
252
.

253
Figure 6.9. IL-8 and IL-6 is released from hASM cells following His-CapG
stimulation. We investigated whether the mammalian expressed recombinant His-
CapG could trigger cytokine release from primary hASM cells. hASM
supernatants cells were stimulated with His-CapG and supernatants were harvested
after 24 hour stimulation and cytokine levels were measured by immunoassay. (A)
Compared to vehicle control, there was a concentration-dependent increase in IL-
8 release in response to higher CapG concentrations. The vehicle control group in
this study was non-treated hASM cells (black bar). (B) Similarly, His-CapG also
triggered significant IL-6 cytokine release from hASM cells. Results are expressed
as mean cytokine level ± SEM conducted on 4-5 individual primary hASM
cultures. One-way repeated measures ANOVA followed by the Dunnett’s post-
hoc test was applied for comparison of each condition to vehicle; *p<0.05
compared to vehicle control group. (C) Following cell stimulation, cell viability
was assessed using the resazurin dye. Fluorometric measurements were measured
over time and compared between different treatments and vehicle control. Results
are expressed as the ratio ± SEM between the fluorescence readings of each
condition over the vehicle control conducted on the primary hASM cultures. For
clarity, the fluorescence measurements of only the highest concentration of His-
CapG is displayed. However, the cell viability assay was performed across all
treatment conditions and cell viability measurements were similar to control.

254
6.3.5 His-CapG triggers IL-8, but not CCL2 release from THP-1 cells
Similar to hASM cells, THP-1 cells were also stimulated with a range of
His-CapG concentrations. Similar to bac-CapG, His-CapG was also able to trigger
IL-8 release from THP-1 cells (Figure 6.10A). IL-8 release from THP-1 cells in
response to His-CapG was in a concentration-dependent manner, with statistical
significance reached at the highest His-CapG concentration (40 µg/mL). Release
of IL-8 from stimulated cells was not due to cell death, as determined by high cell
viability measured following stimulation (Figure 6.20B).
Previous studies have shown that LPS is able to trigger CCL2 release from
THP-1 cells (Harrison et al, 2005). Thus we were interested in determining
whether CapG was able to similarly trigger CCL2 release from these cells. As
expected, LPS induced CCL2 release from THP-1 cells in a concentration-
dependent manner, with statistical significance reached at the highest LPS
concentration (100 ng/mL) (Figure 6.11A). However, His-CapG did not trigger
CCL2 release from THP-1 cells (Figure 6.11B). This is an interesting observation
as it demonstrates that THP-1 cell activation by His-CapG triggers different
signalling pathways that leads to IL-8 but not CCL2 cytokine release.
Since polymyxin B reduced bac-CapG mediated IL-8 release from LPS-
stimulated THP-1 cells, we sought to investigate whether this effect would also be
observed in THP-1 cells stimulated with His-CapG. However, treatment of
polymyxin B of His-CapG did not affect IL-8 release from THP-1 cells (Figure
6.12).
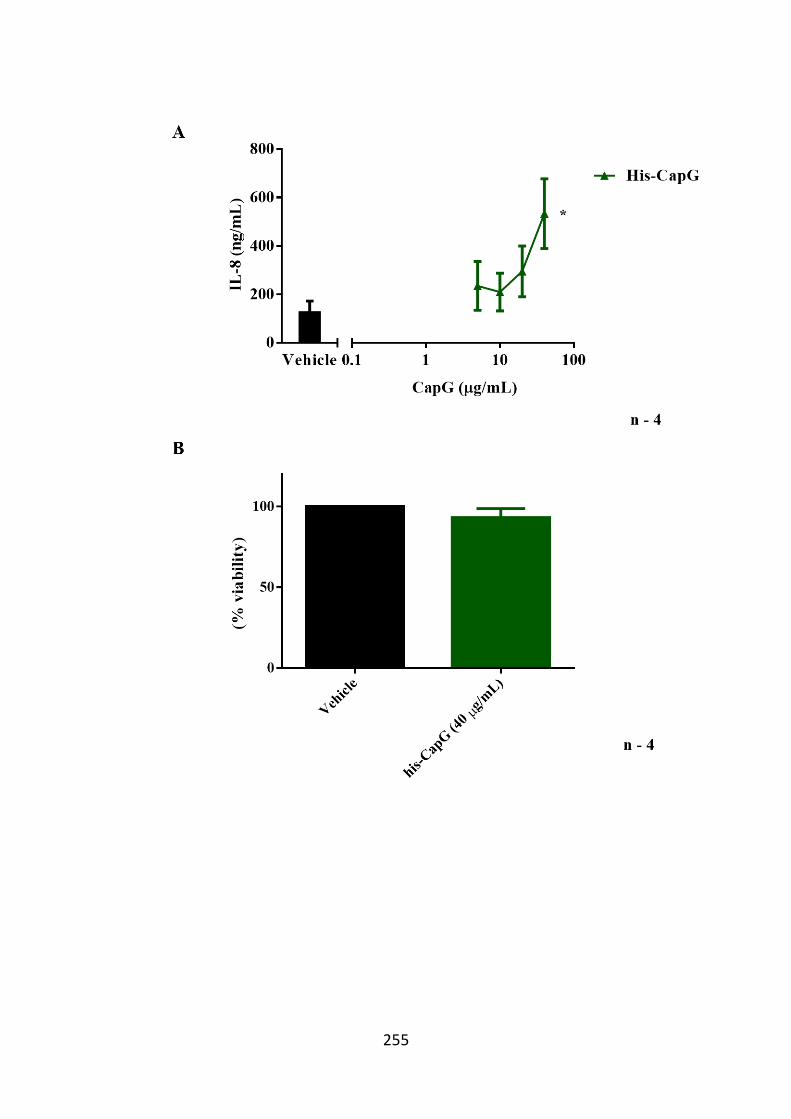
255

256
Figure 6.10. His-CapG triggers IL-8 cytokine release from THP-1 cells. THP-
1 cells were stimulated with His-CapG and IL-8 cytokine release was examined.
(A) Compared to vehicle control, there was a concentration-dependent increase in
IL-8 release in response to higher CapG concentrations. Results are expressed as
mean cytokine level ± SEM conducted on 4 separate occasions. One-way repeated
measures ANOVA followed by the Dunnett’s post-hoc test was applied for
comparison of each condition to vehicle; *p<0.05 compared to vehicle control
group. (B) Following cell stimulation, cell viability was assessed using the
resazurin dye. Fluorometric measurements were measured over time and compared
between different treatments and vehicle control. Results are expressed as the ratio
± SEM between the fluorescence readings of each condition over the vehicle
control conducted on the THP-1 cells. For clarity, the fluorescence measurements
of only the highest concentration of His-CapG is displayed. However, the cell
viability assay was performed across all treatment conditions and cell viability
measurements were similar to control.
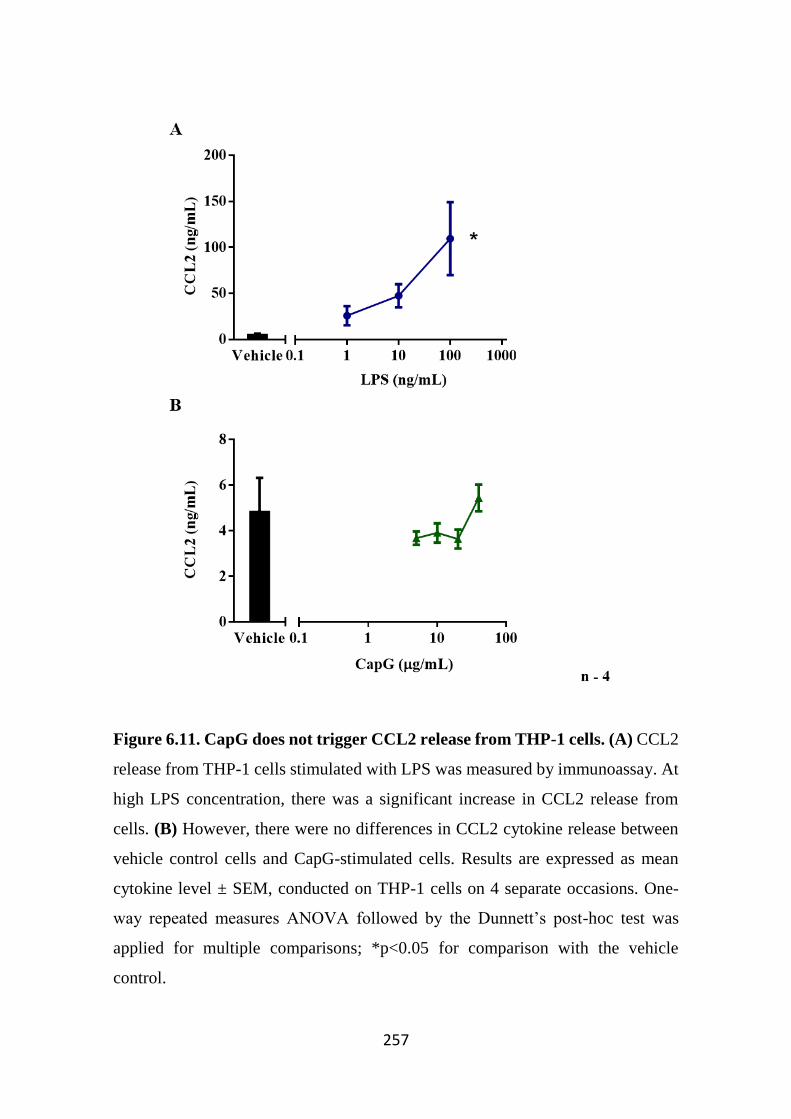
257
Figure 6.11. CapG does not trigger CCL2 release from THP-1 cells. (A) CCL2
release from THP-1 cells stimulated with LPS was measured by immunoassay. At
high LPS concentration, there was a significant increase in CCL2 release from
cells. (B) However, there were no differences in CCL2 cytokine release between
vehicle control cells and CapG-stimulated cells. Results are expressed as mean
cytokine level ± SEM, conducted on THP-1 cells on 4 separate occasions. One-
way repeated measures ANOVA followed by the Dunnett’s post-hoc test was
applied for multiple comparisons; *p<0.05 for comparison with the vehicle
control.
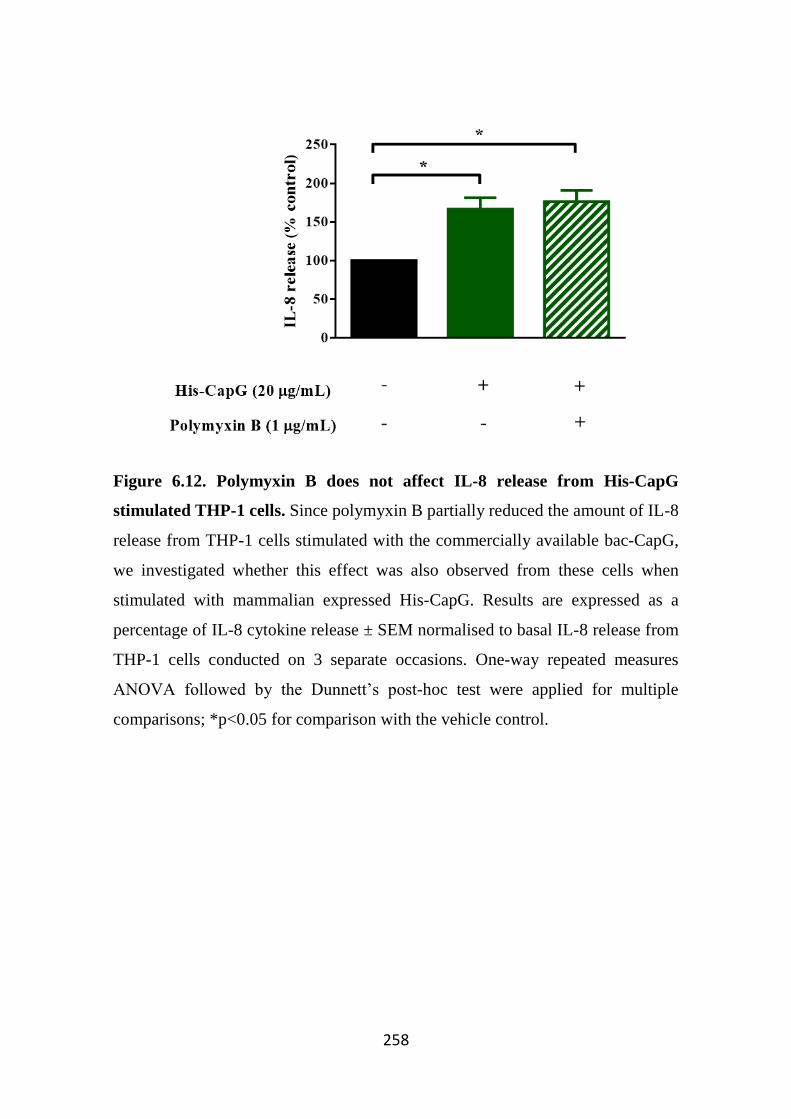
258
Figure 6.12. Polymyxin B does not affect IL-8 release from His-CapG
stimulated THP-1 cells. Since polymyxin B partially reduced the amount of IL-8
release from THP-1 cells stimulated with the commercially available bac-CapG,
we investigated whether this effect was also observed from these cells when
stimulated with mammalian expressed His-CapG. Results are expressed as a
percentage of IL-8 cytokine release ± SEM normalised to basal IL-8 release from
THP-1 cells conducted on 3 separate occasions. One-way repeated measures
ANOVA followed by the Dunnett’s post-hoc test were applied for multiple
comparisons; *p<0.05 for comparison with the vehicle control.

259
6.4 Discussion
Although extracellular gelsolin has a well-established role in clearing
extracellular actin and thus preventing extracellular actin polymerisation and
possible obstruction of vascular flow, the extracellular role of one of its other
family members CapG remains to be elucidated. CapG is constitutively secreted
from macrophages (Johnston et al, 1990) and in keeping with this, we have also
shown that the PMA-differentiated monocytic cell line THP-1 cells also basally
release CapG, and levels are further enhanced following LPS stimulation (Chapter
3). However, CapG release is not restricted to macrophages as the mast cell line
LAD2 cells also release CapG upon antigen/FcεRI activation. As CapG natively
lacks the ability to sever actin filaments, it is likely that this protein may possess
other roles unrelated to actin clearance (Zhang et al, 2006). It has been previously
reported that in healthy individuals, extracellular CapG is detectable in plasma.
However, levels of CapG is comparatively low (0.2 – 0.4 µg/mL) compared to
gelsolin (200 – 300 µg/mL). A recent proteomic study showed that CapG is
upregulated in the synovial fluid obtained from rheumatoid arthritis patients,
suggestive of a potential detrimental role in disease pathology (Balakrishnan et al,
2014). Therefore, whilst CapG is present extracellularly at low concentrations
relative to gelsolin, its plasma concentrations could be increased due to elevated
release from macrophages and mast cells, which may in turn lead to pro-
inflammatory effects.
To examine the role of extracellular CapG as a pro-inflammatory mediator,
the effects of recombinantly expressed CapG on cytokine release from different
cell types was investigated. When cells were stimulated with CapG at low
concentrations, there was no significant cytokine release from cells. However, at
higher CapG concentrations, hASM cells released IL-8 and IL-6. IL-8 cytokine
release was also observed in CapG stimulated macrophages and bronchial
epithelial cells. Furthermore, CapG was also observed to trigger IL-8 release in a
concentration-dependent manner from the synovial fibroblasts cell line SW982.

260
Interestingly, studies have also reported upregulated CapG expression in the
synovium of RA patients (Balakrishnan et al, 2014), suggesting that CapG may be
involved in pro-inflammatory cytokine release from synovial fibroblasts in this
setting.
In inflammatory disorders such as RA or asthma and chronic obstructive
pulmonary disease where macrophages and mast cells play a key role in disease
pathogenesis (Ma & Pope, 2005; Maruotti et al, 2007), these pro-inflammatory
cells release a range of cytokines and mediators that binds to their respective
receptors present on neighbouring cells (de Boer et al, 1998; Scott et al, 1997). We
propose here that increased CapG secretion from these activated cells can lead to
a pro-inflammatory environment. It is known that CapG is present in plasma at
lower concentrations (less than 0.5 µg/mL). This is likely due to the constitutive
secretion of this protein from resting macrophages and mast cells, where this
concentration range is unlikely to trigger cell activation. However, activated
macrophages and mast cells can likely trigger enhanced CapG production, where
the elevated CapG levels (greater than 5 µg/mL) may promote a pro-inflammatory
environment by inducing release of other pro-inflammatory cytokines such as IL-
8 and IL-6, as we have demonstrated. This release pattern has been previously
observed for the pro-inflammatory cytokine IL-1β, where it is proposed that IL-1β
is constitutively from “non-activated” cells but release can be heightened
following cell activation (Lopez-Castejon & Brough, 2011). It is also interesting
to note that the secretion of both CapG and IL-1β are independent of signal
peptides, suggesting that both cytokines may be released in a similar manner,
although this requires further investigation.
In addition to hASMs, epithelial cells and fibroblasts, we observed that
CapG can also trigger IL-8 release from macrophages. This finding suggests that
macrophages not only release CapG that activates neighbouring cells, but likely
express putative receptors for CapG that leads to IL-8 release, serving in an
autocrinic positive feedback loop that potentially leads to further exacerbation of

261
the inflammatory environment. This autocrine signalling is also observed with
other pro-inflammatory cytokines such as interferon-γ, IL-1β and IL-15, where
their release from cells regulates the release of further other pro-inflammatory
cytokines (Alleva et al, 1997; Attur et al, 1998; Held et al, 1999; Munder et al,
1998). Taken together, our findings suggests that the putative CapG receptor is
expressed on different cell types, and that binding of CapG to its receptor leads to
the secretion of pro-inflammatory mediators following cell activation.
Thus, identification of the putative CapG receptor and characterisation of
the signalling pathways associated with the CapG-receptor is necessary.
Identification of this putative receptor could be performed using the ligand-
receptor capture (LRC) technology. This approach relies on the labelling of a
ligand, in this case CapG to a labelled crosslinker known as LRC-TriCEPS™, and
this complex can then be incubated with cells. Binding of this complex to the
receptor can then be isolated, purified and then analysed by mass spectrometry.
Indeed, this approach was recently used to identify the receptor for the novel
adipokine CTRP3 (Li et al, 2016).
It should be noted here that the purchased recombinant bac-CapG used in
these initial studies was of bacterial origin. Thus, bacterial contaminants such as
LPS could influence or potentially dominate the observed activity of the bac-
CapG. This effect was particularly evident in THP-1 cells, where the cells were
sensitive to low concentrations of LPS. Experiments utilising the antibiotic
polymyxin B demonstrated that whilst cells released IL-8 in response to bac-CapG,
only part of the cytokine release was due to the presence of conatminants such as
LPS. However, it is likely that the contaminants were present at low concentrations
that only certain cell lines sensitive to these endotoxins such as THP-1 cells would
be affected. Indeed, not all cell types examined were affected by the presence of
these contaminants. For example, whilst polymyxin B reduced IL-8 release from
LPS-stimulated SW982 cell, this antibiotic did not inhibit bac-CapG-mediated
cytokine release from these cells. In addition, hASM cells are known to be weak

262
responders to LPS, but here we still observed release of IL-8 in response to bac-
CapG, confirming that LPS was not contributing to these cell activation. It has
been previously shown that the BEAS2B cells also respond poorly to LPS (Schulz
et al, 2002). Whilst we did not examine the effects of polymyxin B on IL-8 release
from activated BEAS2B cells, we predict that the LPS presence in bac-CapG is
unlikely to affect IL-8 release from these cells.
Taken together, the data presented in this chapter suggest that bac-CapG is
able to induce IL-8 release from different cell types such as monocytes (THP-1),
airway smooth muscle cells (hASM), bronchial epithelial cells (BEAS2B), and
synovial fibroblasts (SW982). However, data interpretation is partly confounded
by the presence of likely bacterial contaminants that are able to trigger IL-8 release
in some cells sensitive to activation by LPS. Nevertheless, clearly in LPS
unresponsive cells, such as hASM and BEAS2B, cell activation dependent on
CapG was demonstrated with the bac-CapG preparations. Also with the LPS
sensitive THP-1 cells, cell activation was demonstrated with polymyxin B treated
bac-CapG, strongly indicating cell activation by CapG itself. Thus, this effect is
dependent on cell type and sensitivity to the contaminants as not all cell types
responded to LPS or had effects modulated by polymyxin B.
To overcome this limitation, a mammalian expressed recombinant human
CapG (His-CapG) was generated in house. This process, along with functional
validation experiments were described in Chapter 5. Thus, the present chapter also
aimed at ascertaining whether the in-house material was also able to induce
cytokine release from cells following stimulation. When examined, His-CapG was
able to trigger cytokines IL-6 and IL-8 from hASM cells, and IL-8 from THP-1
cells. It should be noted here that there is a differences in potency between
recombinant CapGs as higher concentrations of His-CapG were required to trigger
cytokine release from cells compared to bac-CapG. This could be related to the
changes in protein structure and folding, due to the presence of purification tags
present on both N and C terminus of His-CapG, as described in Chapter 5. In

263
addition, the His-CapG concentrations used in these studies were determined based
on calculated absorbance measurements. Although CapG was the most abundant
protein in the sample, other high molecular weight contaminating proteins were
also identified (Figure 5.9). These protein contaminants were also present in
another his-tagged protein (His-sFcεRIα) that was generated for other purposes
unrelated to His-CapG. When examining the function of His-CapG, the His-
sFcεRIα protein was used as a control and this protein did not trigger cytokine
release in cell stimulation studies (data not shown). Nevertheless, these studies
utilising His-CapG reinforces previous studies utilising the bac-CapG,
demonstrating that CapG is likely a novel pro-inflammatory protein that triggers
the release of other pro-inflammatory mediators. It should be noted here that due
to limited yield of His-CapG following protein purification, stimulation
experiments were not performed on BEAS2B and SW982 cells, but should be
conducted in future studies.
Interestingly, when the release of the chemokine CCL2 from THP-1 cells
was examined, His-CapG did not induce cytokine release from cells. In our hands,
LPS triggered CCL2 cytokine release from THP-1 cells in a concentration-
dependent manner and this finding has been confirmed by others (Schecter et al,
1997). The difference in CCL2 and IL-8 release from THP-1 cells following CapG
stimulation suggests that CapG acts on signalling pathway involved in IL-8
release, but not CCL2. Whilst there are similarities in the pathways involved in the
production of IL-8 and CCL2, previous studies have documented distinct pathways
involved in the production of these chemokines (Bauermeister et al, 1998). In
stimulated human retinal pigment epithelial cells, the transcription factors activator
protein 1 (AP-1) and nuclear factor κB (NF-κB) leads to the production of both
CCL2 and IL-8, respectively (Bian et al, 2004). However, inhibition of AP-1
resulted in loss of production of CCL2 but not IL-8, indicating that AP-1 is an
important regulator for CCL2 (Bian et al, 2004). Whilst both NF-κB and AP-1 can
be simultaneously activated by many stimuli, NF-κB is able to regulate AP-1

264
transcriptional activity, hence regulating gene expression in response to different
stimuli (Fujioka et al, 2004). Hence, we propose here that activation of cells by
CapG likely leads to activation of NF-κB but not AP-1, thus resulting in production
of IL-8, but not CCL2. To investigate this, THP-1 cells could be incubated with
benznidazole, which selectively inhibits the NF-κB-dependent, but not AP-1
activity in macrophages (Manarin et al, 2010). Thus, examining IL-8 and CCL2
cytokine release from His-CapG and/or various concentrations of LPS stimulated
cells in the presence and absence of benznidazole could provide further insights
on the signalling pathways associated with CapG.
In summary, we have been able to demonstrate that CapG is able to induce
pro-inflammatory cytokine release from hASM cells and a range of other cell
types. Taken together, this suggests that CapG is a novel pro-inflammatory
mediator that can be constitutively secreted from cells, but release is elevated
during pro-inflammatory conditions. This in turn leads to the release of other pro-
inflammatory cytokines. However, more studies are required to gain further
insights into the role of CapG in inflammatory disorders. This is discussed more
fully in Chapter 7.

265

266
Chapter 7
General Discussion

267
The immune system plays an important role in maintaining homeostasis. It
is composed of cells, tissues and organs that recognises and responds to foreign
substances and microorganisms in order to maintain the host in a healthy state. A
key feature of the immune system is inflammation, which is a process where a
localised region of the body becomes reddened, swollen, hot and hyperalgesic
(Majno & Joris, 2004). These physiological responses are regulated by a variety
of mediators that can be released from immune cells such as mast cells and
macrophages. These cells express receptors that allow them to recognise antigens,
invading pathogens or damaged cells. When activated, mast cells and macrophages
release cytokines, proteases and arachidonic acid metabolites that play roles in
driving inflammatory symptoms (Aderem, 2003; Moon et al, 2014). In addition,
these cells release agents that promotes the recruitment and infiltration of other
inflammatory cells to the site of tissue damage. Finally, other mediators that have
both anti-inflammatory and pro-resolution properties emerge to restore tissue
homeostasis (Serhan et al, 2008).
In certain cases, the immune system can act aberrantly triggering sustained
inflammation which can then lead to targeting and damage of the host’s own cells
and tissues (Warrington et al, 2011). Both mast cells and macrophages have been
implicated in these peripheral hypersensitivity-related disorders such as asthma
and rheumatoid arthritis (Bradding & Arthur, 2016; Hueber et al, 2010; Ma &
Pope, 2005; Peters-Golden, 2004). In addition, both cell types are also implicated
in neuroinflammation, which plays a major role in many neurodegenerative
disorders (Zhang et al, 2016). In many of these inflammatory-related disorders, the
mediators released from activated mast cells and macrophages contribute to the
pathophysiological features associated with the disease. Thus, targeting and
preventing actions of these mediators is a potential avenue for both existing and
new therapeutics (Astrakhantseva et al, 2014).
Whilst there has been various degrees of success in utilising anti-cytokine
agents (Brightling et al, 2008; Erin et al, 2006), other treatments have shown

268
disappointing clinical benefits despite promising outcomes in pre-clinical work
and initial clinical trials (Corren et al, 2010; Oh et al, 2013). In many inflammatory
diseases, a significant percentage of patients are poor responders towards current
available treatments and thus there is still an unmet medical need for effective anti-
inflammatory drugs.
Although many of the mediators released from mast cells and macrophages
are well established, there are likely other novel mediators where their functions
remains to be elucidated (Xia et al, 2013b). Of particular interest to us is
macrophage capping protein (CapG), which was shown to be secreted from
activated human mast cells. CapG belongs to the gelsolin superfamily of proteins,
which are involved in regulating actin polymerisation. This protein is highly
expressed in macrophages, and studies have shown its involvement in
macrophage-related activities including phagocytosis and cell motility (Parikh et
al, 2003). In addition, others have also reported the presence of CapG in the
extracellular space. However, its extracellular function remains poorly understood.
Macrophages constitutively secrete this protein through a non-canonical pathway
that is independent of signal peptide-targeted release (Johnston et al, 1990). In
addition, CapG is also found to be elevated in the synovial fluid of rheumatoid
arthritis sufferers although its role in disease pathology remains to be elucidated
(Balakrishnan et al, 2014).
The expression, characterisation and function of extracellular CapG was
investigated in this Thesis. This provided new knowledge in regards to the ability
of CapG to act as a novel inflammatory mediator.
The main outcomes of this project (as shown in Figure 7.1) are that:
1. CapG is primarily expressed in haematopoietic immune cells;
2. CapG is released from antigen-stimulated LAD2 mast cells;
3. CapG is released from LPS-stimulated THP-1 cells, which can be
modulated by pharmacological agents;

269
4. CapG gene expression is downregulated in activated macrophages,
but upregulated in in vivo models of inflammation; and
5. Using a mammalian expression system to generate recombinant
CapG, we have shown that this protein is able to trigger cytokine
release from a variety of cell types.
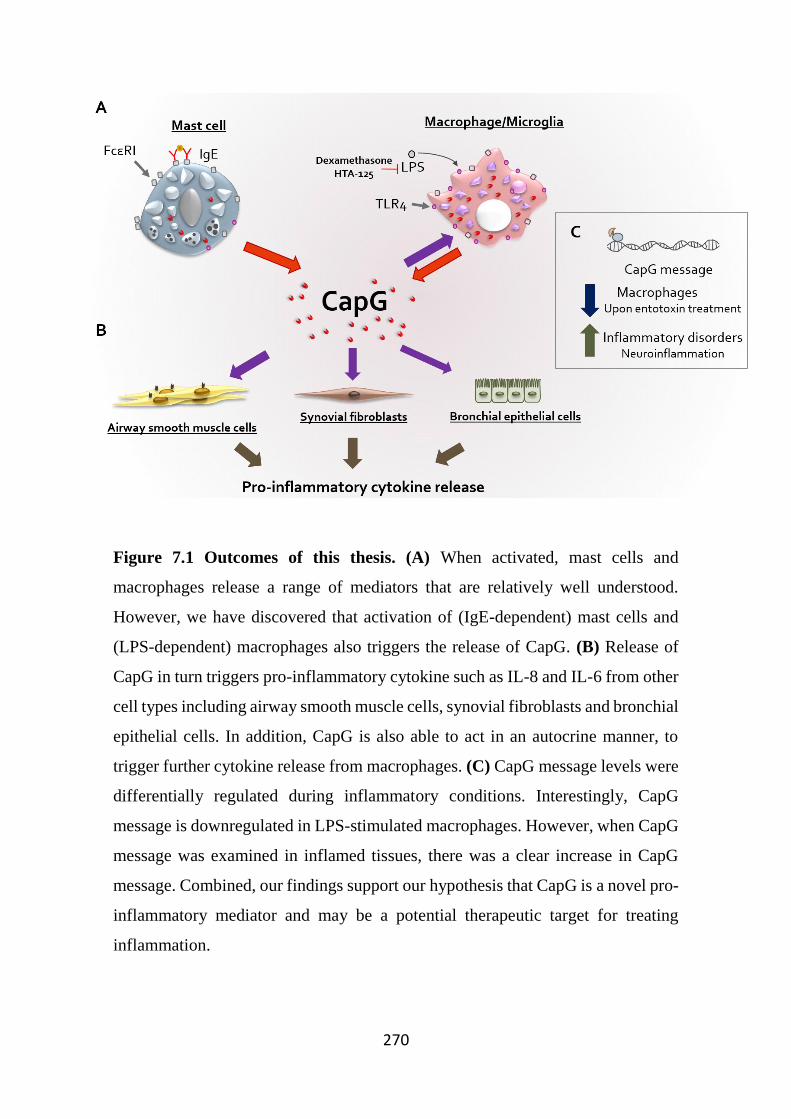
270
Figure 7.1 Outcomes of this thesis. (A) When activated, mast cells and
macrophages release a range of mediators that are relatively well understood.
However, we have discovered that activation of (IgE-dependent) mast cells and
(LPS-dependent) macrophages also triggers the release of CapG. (B) Release of
CapG in turn triggers pro-inflammatory cytokine such as IL-8 and IL-6 from other
cell types including airway smooth muscle cells, synovial fibroblasts and bronchial
epithelial cells. In addition, CapG is also able to act in an autocrine manner, to
trigger further cytokine release from macrophages. (C) CapG message levels were
differentially regulated during inflammatory conditions. Interestingly, CapG
message is downregulated in LPS-stimulated macrophages. However, when CapG
message was examined in inflamed tissues, there was a clear increase in CapG
message. Combined, our findings support our hypothesis that CapG is a novel pro-
inflammatory mediator and may be a potential therapeutic target for treating
inflammation.

271
7.1 CapG is primarily expressed in haematopoietic immune cells
It has been previously demonstrated that CapG protein is found largely in
inflammatory cells such as macrophages and neutrophils (Witke et al, 2001). In
our hands, we too observed strong CapG protein expression in macrophages and
mast cells. Although CapG is also detected in non-haematopoietic cells (eg. airway
epithelium), expression is weaker compared to mast cells and macrophages.
Examination of CapG gene expression also correlates to higher CapG message in
immune cells compared to these structural cells. This finding is in keeping with
data obtained from The Human Protein Atlas (CapG_Cell_Line_Atlas, 2016).
Others have recently shown the involvement of CapG in cell metastasis of several
cancerous cell lines including breast epithelial cells (Westbrook et al, 2016; Zhu
et al, 2012), suggesting that expression of this protein may be tightly regulated in
structural cells, and that dysregulation of this can perhaps lead to aberrant cell
activity such as metastasis.
It is interesting to note that amongst all the haematopoietic cell lines
examined, CapG expression was absent in the rat basophilic leukaemia (RBL) cell
line. Although this immortalised cell line has some features of blood basophils,
they are commonly used in vitro as a mast cell-like model (Passante & Frankish,
2009). It is unclear why CapG was undetected in this rat cell line as expression
was detectible in mature rat peritoneal macrophages and mast cells. In addition,
the sequence homology between human and rat CapG is highly conserved and the
antibody used in this study detects a specific sequence that is conserved in human,
rat and mouse protein. It is possible that CapG may not be expressed in basophils,
although it is found in neutrophil granulocytes. Examination of CapG expression
in purified primary basophils should be performed to determine this.
7.2 CapG is released from mast cells
Preliminary data from our laboratory on antigen-activated HMCα cells
identified CapG as a potential novel mediator that was capable of inducing pro-

272
inflammatory cytokine release from hASM cells. However, our data were
inconsistent with previous findings as we did not observe any changes in CapG
release between stimulated and non-stimulated HMCα cells. In addition to CapG,
IL-8 release from these antigen-stimulated HMCα cells was also significantly
lower compared to previous studies (Xia et al, 2013b), suggesting that the cells
utilised in this present study may have lost reactivity to the antigen/IgE signalling
pathway. Although we performed experiments on new cells taken from
cryostorage, we were unable to recapitulate previous findings. Thus, to confirm
previous preliminary studies, it is desirable to perform these experiments on newly
FcεRIα-subunit transfected HMC cells.
Although the HMCα cells were transfected with the α-subunit of FcεRI, it
should be noted that whilst the levels of FcεRIγ subunits are high, FcεRIβ levels
are barely detectable at the mRNA level (Guhl et al, 2010). This is of potential
significance as FcεRIγ and FcεRIβ subunits contain ITAM motifs that are crucial
for downstream signalling pathway. This suggests that the presence of FcεRIβ
could be important in propagating and amplifying signalling pathways to promote
CapG release. Moreover, the HMC-1 cell line is relatively immature, has sparse
granules, and do not classically degranulate upon stimulation (Butterfield et al,
1988; Nilsson et al, 1994).
In contrast to HMC-1 cells, the LAD2 mast cell line share several
characteristics with more mature mast cells, such as constitutively expressing the
high-affinity IgE receptor FcεRI and the ability to degranulate (Radinger et al,
2010). Thus, we examined CapG release from antigen-stimulated LAD2 cells.
CapG release from LAD2 cells appeared to be FcεRI-specific as other stimuli
(such as substance P) did not produce its release. Interestingly, CapG release from
LAD2 cells at higher antigen concentrations was lower compared to lower antigen
concentrations. This bell-shaped release trend has been previously observed by
others, where it has been described that higher antigen concentrations engages

273
inhibitory molecules that in turn down-regulates mast cell activity (Huber, 2013;
Magro & Alexander, 1974).
How is CapG released? Could it be through classical degranulation? This
seems unlikely due to differences in degranulation (as measured though β-
hexosaminidase release) and CapG release from substance P-stimulated LAD2
cells. In these studies, LAD2 cells readily released β-hexosaminidase in response
to substance P but even at high concentrations, substance P did not trigger CapG
release from these cells. Our findings here demonstrates that CapG can be released
from mast cells constitutively, and when activated, these cells are capable of
releasing greater quantities of CapG independent of the degranulation pathway.
7.3 Regulation of CapG expression in inflammatory conditions
Despite knowledge of its release from macrophages, not much is known in
regards to if CapG release from these cells is regulated (Johnston et al, 1990).
Thus, we examined CapG release from the monocytic cell line THP-1 in response
to different stimuli. Of these relatively limited stimuli, only LPS was found to
induce CapG release from these cells. Furthermore, release of CapG was
concentration and time-dependent, and its release was modulated by the anti-
inflammatory glucocorticoid dexamethasone. However, whilst the other stimuli
examined (such as aggregated IgG and IL-17) did not trigger CapG release, it is
possible that CapG release can also be differentially regulated by other stimuli
including other TLR ligands, and this should be further investigated.
Gene expression characterisation of CapG and its regulation was also
examined in this Thesis. Interestingly, CapG message levels were downregulated
in all LPS-stimulated macrophage models including THP-1 cells, rat peritoneal
macrophages and GM-CSF-differentiated human peripheral blood macrophages.
Taken with the CapG release data from THP-1 cells, this suggests that intracellular
CapG expression appears to be diminished in macrophages following LPS
stimulation. This observation was also found at the protein level in

274
immunohistocytochemistry data (Chapter 3). In macrophages, several genes have
previously been reported to be downregulated by LPS, where these genes are
commonly associated with pro-apoptosis and redox homeostasis (Mikita et al,
2001; Sharif et al, 2007). However, the well-established roles of CapG are not
commonly associated with these downregulated genes. As CapG is known to be
associated with macrophage-related activity such as cell motility, it is likely that
the loss of intracellular CapG prevent macrophages migrating away from site of
cell activation, resulting in a “macrophage stasis” phenotype. In turn, this allows
macrophages to prioritise in mediating pathogen clearance. In the literature, it has
been shown that inhibition of CapG in tumour cells results in loss of cell metastasis
(Li et al, 2015; Van Impe et al, 2013), thus supporting the notion that loss of CapG
prevents macrophages from migrating. However, CapG is also important in
macrophage functions such as phagocytosis and others have also demonstrated that
LPS is able to suppress phagocytosis activity through the disruption of cytoskeletal
network (Wonderling et al, 1996). Whilst CapG was not highlighted as a target
protein for LPS-mediated suppression, this finding combined with our results,
suggests that CapG could also be downregulated as an avoidance strategy by Gram
positive bacteria (via LPS). Thus, the downregulation of CapG in macrophages
could be due to (1) a host response to better facilitate bacterial clearance, or (2) a
survival mechanism by the bacteria to avoid host defense.
Previous studies on CapG knockout mice have demonstrated that these mice
were susceptible to specific strains of bacterial infections, depending on how these
pathogens were cleared by macrophages (Parikh et al, 2003). In the study, the
authors demonstrated that loss of CapG resulted in a reduction in the rate of
phagocytosis activity. Despite this, the cells were still able to phagocytose these
pathogens, demonstrating that CapG is important, but not crucial for macrophage
phagocytosis. In addition, the loss of CapG did not affect other macrophage
functions such as micropinocytosis (a form of endocytosis that mediates the uptake
of molecules, antigens and nutrients, (Lim & Gleeson, 2011)). Thus, this suggests

275
that although CapG is important for several macrophage functions, these cells are
well-equipped with other mechanisms to promote pathogen clearance that are
independent of CapG. Taking these findings into consideration, the loss of CapG
expression in macrophages following LPS-activation seen in our studies suggests
that this is likely then a mechanism employed by macrophages to promote
pathogen clearance.
CapG gene expression was also examined in two different in vivo mouse
models of inflammation (Chapter 4). Inflammatory cells in the bronchoalveolar
fluid (BALF) were obtained and CapG mRNA levels examined in both the
lipopolysaccharide (LPS) and respiratory syncytial virus (RSV) inflammation
models. In both inflammatory models, CapG expression in BALF cells was
downregulated. This concurs with that observed in in vitro macrophage studies
described above, where CapG message was downregulated in inflammatory cells
upon LPS stimulation. It is also worth considering that CapG represents
approximately 1% of the total macrophage protein (Witke et al, 2001). As
discussed earlier, CapG in macrophages is key in several intracellular macrophage
functions. However, there is also a likelihood that the abundance of CapG permits
it to act like a preformed mediator that is readily released from macrophages upon
activation, and that the amount of intracellular CapG remaining is sufficient for
other macrophage activity such as phagocytosis.
Although CapG message is downregulated in the BALF cells of both
inflammatory models, there are several differences observed in these models. In
particular, neutrophil accumulation is commonly observed in the BALFs following
LPS treatment (Asti et al, 2000). Whilst neutrophils have been previously
described to exhibit CapG-dependent activities, CapG is not known to be present
in abundance in neutrophils in comparison with macrophages. Thus, it is also
possible that the downregulation of CapG in BALF cells of LPS-treated mice could
at least be partly due to increased neutrophil infiltration reducing the relative

276
percentage of macrophages nad thus resulting in a reduction in total CapG message
in samples.
In contrast, it has been shown that monocyte and lymphocyte cell counts
are elevated in RSV inflammatory models (Collins et al, 2005). Thus, the
downregulation of CapG message observed is more likely in keeping with earlier
in vitro macrophage studies, where the decreased CapG message in these
inflammatory cells is associated with facilitating viral clearance. As RSV is known
to activate macrophages through the TLR2 receptor, examining if CapG message
levels are downregulated in THP-1 cells, as well as whether CapG is released from
these cells following RSV activation in vitro should be performed. This would
provide further insights into CapG regulation during viral infections.
In addition, CapG message levels was also examined in whole lung tissue
of LPS and RSV-treated mice. Interestingly, CapG message was upregulated in
the lungs of both inflammatory models. This significant increase in CapG message
could be attributed to increase in macrophages derived from monocyte
differentiation, or due to increased inflammatory cell infiltration to the lungs.
However, these data might also suggest that CapG gene may potentially be
upregulated in structural cells in the lung during inflammation, where, as
previously discussed, CapG message and protein was found to be upregulated in
several non-haematopoietic cells in cancer (De Corte et al, 2004; Westbrook et al,
2016). Thus, in vitro studies on the effects of LPS or RSV on CapG expression in
structural cells is necessary to provide a better understanding of the in vivo results.
7.4 CapG – a role in neuroinflammation?
Macrophages and macrophage-like cells are found in different tissues,
including the brain where the resident macrophages, known as microglia, are the
primary immune cells involved in maintaining brain homeostasis. Investigation of
the effects of LPS on the murine microglia BV2 cell line revealed similarities to
THP-1, where there was an upregulation in CapG release following endotoxin

277
stimulation. However, dexamethasone did not affect LPS-mediated CapG release
from these cells, although it has been previously demonstrated that dexamethasone
can attenuate LPS activity in BV2 cells (Huo et al, 2011). In addition, CapG
message levels were not affected by LPS in BV2 stimulated cells. The differences
observed in CapG gene and protein regulation in these two cell lines could be
related to macrophage heterogeneity, likely due to the origins of these cell lines,
as previously described.
A potentially clinically intriguing outcome of the work was that CapG
mRNA was upregulated in the cortical region of the brain of Alzheimer’s disease
(AD) sufferers. The increase in CapG message might be due to its increased
expression in activated microglia in AD sufferers. However, perivascular
macrophages are also thought to be heavily involved in clearance of β-amyloid
plaques (Pey et al, 2014; Theriault et al, 2015). Whilst microglia express both
CapG message and protein, the infiltration of perivascular macrophages to the
inflamed cortical region could also account for the observed upregulated
expression of CapG message in AD sufferers. Similarly, CapG mRNA was also
found to be upregulated in the cortex of the APP/PS-1 mice, a commonly used
mouse model for studying AD pathology. Thus, in vivo studies are necessary to
determine whether the upregulated CapG message observed is due to activation of
microglia or increased influx of perivascular macrophages. Interestingly, CapG
message was only significantly elevated in 12-month aged mice, suggesting that
CapG may only be involved in late-stages of the disease pathology. Since the
clearance of plaques is central to AD pathology, it will be of importance to
determine if the increased CapG expression is reflective of its involvement as a
protective mechanism that is simply overwhelmed in AD progression or is simply
a marker of pathology. If the former, CapG and the plaque removal process may
be an opportunity for therapeutic intervention and if the latter CapG expression
may still prove to be a useful diagnostic marker.

278
Our findings demonstrate a potential role for CapG in AD as indicated by
the increased CapG gene expression in AD sufferers. However, CapG gene
expression was not elevated in Parkinson’s disease (PD) sufferers. This could be
explained by several factors. Firstly, it has been suggested that perivascular
macrophages infiltration is more prominent and plays a more significant role in
AD progression compared to PD (Pey et al, 2014), thus accounting for a greater
CapG gene expression in AD. However, it should be noted that the brain samples
obtained in this study were from the cortical region of the patient brains. Whilst
inflammation and neuroinflammation in the cerebral cortex is the hallmark of AD,
PD pathology manifests largely from neurodegeneration of dopaminergic neurons
in the substantia nigra (Bender et al, 2006; Saggu et al, 1989). Thus, these data
might not provide an accurate depiction of CapG gene expression in the brains of
PD sufferers, and that investigating CapG message levels in the substantial nigra
of PD patients would be more informative as to whether CapG plays a role in PD
pathogenesis. In addition, in vitro studies examining the effects of CapG message
regulation and release from BV2 following stimulation with β-amyloid or α-
synuclein peptides could be performed as this provides further information in
regards to the possible role of CapG in these neurodegenerative disorders.
7.5 Recombinant CapG (both commercial and in-house generated) triggered
cytokine release from a variety of different cell types
Whilst Chapter 3 examined the regulated release of CapG from activated
mast cells and macrophages, the logical extension to this was to examine the
function of extracellular CapG and its relevance to inflammation. Recombinant
human CapG can be purchased commercially and is generally used as a control
protein for proteomics analysis. However, for our purposes, the purification of this
protein is likely to be not as stringent as those recombinant proteins used for in
vitro cell stimulation studies. Indeed, Coomassie staining of this protein revealed
several contaminating protein bands. In addition, the cost of the recombinant
material was limiting with regards to our proposed experiments. Hence, we

279
established an in-house mammalian expression system for generating recombinant
CapG (as described in Chapter 5). CapG production from different cell lines was
compared. Of these, the EBNA-293 cell line was found to be best suited for
generating CapG as this system permitted high yields of recombinant CapG
(approximately 3 µg/mL) that would facilitate numerous downstream experiments,
including: using material for in vitro cell stimulation assays and future experiments
including observing the effects of CapG in in vivo animal studies. After several
growth optimisation experiments, we were able to establish effective culture
conditions to permit CapG production.
Prior to its use in downstream studies, the recombinant CapG produced
from EBNA-293 cells was purified using a HisTALON™ metal affinity column,
and then the protein sample was resolved with gel electrophoresis and the protein
bands stained. As expected, the most abundant protein was CapG. However, two
other high molecular weight protein bands were also visualised. Mass
spectrometry analysis identified these proteins as epididymis luminal proteins 213
and 214 (140 kDa) and cDNA FLJ61580 (110 kDa). To our knowledge, the
function of these proteins are not well understood. It should also be noted these
high molecular weight bands were also observed during purification of another
unrelated His-tagged protein (His-sFcεRIα). Thus, it is likely that these proteins
have distinct properties that permit binding with high affinity to the column, as a
range of imidazole elution conditions could not remove these proteins from the
eluted CapG. Since these bands have a significantly higher molecular weight than
CapG, subsequent purification via gel filtration chromatography should provide
highly-purified CapG.
The established biological activity of CapG is its effect on the initiation and
kinetics of actin polymerisation (Van Impe et al, 2013; Young et al, 1990). A
standard assay for actin polymerisation is the pyrene actin-polymerisation assay,
and was hence used to test the biological activity of the mammalian His-CapG.
The His-CapG was active in prolonging the start of actin polymerisation and the

280
rate of polymerisation. These activities were also apparent with the commercial
bac-CapG, indicating that the His-CapG secreted from the EBNA-293 cells was
functional. Thus, the His-CapG generated is functionally active and hence suitable
for downstream experiments examining its role as a possible mediator of
inflammation.
Whilst generation of His-CapG was ongoing, we initiated our studies by
examining the effects of the commercially purchased bac-CapG on different cell
types. bac-CapG triggered IL-8 release from a range of cell types testing including
human airway smooth muscle (hASM) cells, macrophages (THP-1), airway
epithelial cells (BEAS2B) and synovial fibroblasts (SW982). In addition, IL-6
cytokine levels were also elevated in CapG-stimulated hASM cells. It should be
noted that bacterial endotoxin contaminants present in bac-CapG was also able to
trigger cytokine release from macrophages and the synovial fibroblast cell line
SW982. Thus, although bac-CapG triggered cytokine release from cells, this
material did not allow us to unequivocally define the pro-inflammatory role of
CapG. However, the inhibition of LPS by using polymyxin B and the LPS
insensitivity of some of the cell lines powerfully demonstrated a role for CapG
stimulating pro-inflammatory cytokine secretion by macrophages, fibroblasts and
hASM cells.
Following the generation and validation of His-CapG, we were able to
complete our characterisation of the pro-inflammatory activity of CapG that began
with bac-CapG. Similar to bac-CapG, His-CapG also significantly increased
release of IL-6 and IL-8 from hASM cells as well as IL-8 from THP-1 cells.
Interestingly, His-CapG did not induce CCL2 release from THP-1 cells, suggesting
that CapG activates a signalling pathway discriminatory between IL-6/IL-8 and
CCL2. This demonstrates a unique signalling pathway for CapG that is distinct
from LPS which stimulates production of both IL-6, IL-8 and CCL2. A previous
study demonstrated inhibition of the phosphoinositide 3-kinase (PI3K) and the
protein kinase B (Akt) pathway resulted in inhibition of CCL2, but not IL-8 (Bian

281
et al, 2004). Thus, this suggests a divergence in signalling pathways leading to the
secretion of these cytokines, and that CapG may only be involved in one of these
signalling pathway.
Whilst both bac-CapG and mammalian His-CapG were able to trigger
cytokine release from THP-1 and hASM cells, it should be noted that the bac-
CapG was more potent that His-CapG, despite the higher inhibitory activity of the
mammalian His-CapG in the pyrene-actin polymerisation assay. The difference in
potencies could be attributed to different factors. Firstly, the presence of either or
both purification Strep and His-tags could have altered CapG protein folding and
structure although gross changes are argued against by the activity of mammalian
His-CapG in the polymerisation assay. Secondly, it is likely that the measured
protein concentration may not be an accurate depict the actual concentration of
His-CapG. As stated in Chapter 5, protein concentration was calculated using a
formula involving the absorbance measurement of the protein solution and the
predicted CapG extinction coefficient. However, Coomassie staining of the His-
CapG in solution revealed other high molecular weight contaminating bands.
Although His-CapG is the predominant protein present in solution, the presence of
these contaminating band would contribute to incorrect absorbance measurements,
thus overestimating the actual concentration of His-CapG.
Another possibility with the His-CapG preparation is that the minor
contaminating bands may also affect cytokine release from cells. However, when
cells were also stimulated with a His-tagged control protein that contained similar
levels of these high molecular weight contaminants, there was no significant
cytokine release. To avoid this, in future studies His-CapG should be purified to
homogeneity by sized-based exclusion chromatography. Nevertheless, results
utilising the mammalian His-CapG preparation confirmed our findings in earlier
bac-CapG studies.
In this present study, we found that mast cells and macrophages release
CapG constitutively. It is thus conceivable that this manner of CapG release

282
contributes to the low CapG concentrations in plasma (0.3 – 0.5 µg/mL). At this
concentration, we find that CapG does not trigger cytokine release from other cell
types. However, it is possible that activated mast cells and macrophages release
sufficient amounts of CapG that at a local level produce a threshold concentration
(10 µg/mL and above) that triggers inflammatory responses such as cytokine
release from neighbouring cells, as we observed. This concentration vs. response
relationship further suggests CapG as a possible novel pro-inflammatory mediator.
7.6 Future directions
Characterisation of the release and expression of CapG from mast cells and
macrophages and its activities on different cell types is novel and intriguing.
Although this present study has shown that extracellular CapG is a potential novel
pro-inflammatory mediator, there are numerous experiments that should be
performed to further characterize the nature and effects of CapG in both normal
and disease conditions. Because of the novelty of this project, the experimental
tools required for further studies also need to be further optimised, as outlined
below.
Although the primary focus of this Thesis was on extracellular CapG, there
is also much to learn and understand about the characteristics of intracellular
CapG. In particular, recent studies suggesting CapG as a promoter of tumour
metastasis could be of therapeutic importance (Ichikawa et al, 2013; Kimura et al,
2013; Li et al, 2015; Shao et al, 2011). Although the stably CapG-transfected
HEK293 cell lines were not an ideal expression system (as discussed in Chapter
5), the readily-controllable CapG expression permits this cell line to be a useful
tool for understanding CapG inside the cell and its involvement in tumour
metastasis.
Our results suggest that CapG may be of importance in Alzheimer’s disease
pathology. Thus, examining behavioural studies and brain morphological changes
in APP/PS-1 mice in the absence of CapG would allow us to determine the

283
importance of this protein. This could be performed by using CapG gene-silencing
techniques including siRNA transfections or clustered regularly interspaced short
palindromic repeats (CRISPR) techniques, or by using nanobodies that target
CapG protein. Alternatively, studies could also be performed on mice generated
by crossing both APP/PS-1 and CapG knockout animals. It has been previously
shown that CapG knockout mice do not exhibit any gross morphological changes
(Parikh et al, 2003). Thus, generation of APP/PS-1/CapG-knockout mice would
also allow us to examine whether AD pathology is promoted or reduced in these
mice.
Whilst we have used His-CapG to study the role of CapG as a pro-
inflammatory mediator in an in vitro setting, this recombinant protein can also be
used in experiments to better understand its role in vivo. This could be performed
by intradermal injection of CapG at a localised region and examining if symptoms
associated with inflammation emerge (for example, Evans’ Blue dye
extravasation). Knowledge of the function of CapG in these in vivo studies will
provide us with clearer insights into its inflammatory actions and help determine
if targeting CapG may be of therapeutic benefit.
Whilst it is known that CapG is important in macrophage functions such as
phagocytosis, our results demonstrate that CapG, when released, has the ability to
further stimulate macrophages. Thus, it would be interesting to characterise LPS-
stimulated macrophages in the absence of CapG to determine its autocrinic
activities. However, macrophages are known to be difficult to transfect through
conventional methods, as they express several degrading enzymes that can disrupt
the transfection process (Zhang et al, 2009). However, recent advances in gene-
editing technology such as the CRISPR method have been shown to be relatively
successful in macrophages (Jing et al, 2015). Alternatively, CapG activity can also
be targeted using anti-CapG nanobodies, that have been shown to impair breast
cancer metastasis (Van Impe et al, 2013).

284
As previously discussed, CapG is known to be present at low concentrations
in plasma. We hypothesise that during inflammatory conditions, activated mast
cells and macrophages release pro-inflammatory mediators including CapG that
exacerbates inflammatory conditions including triggering cytokine release from
other cells. Thus, comparison of CapG concentration in plasma between healthy
individuals and mastocytosis patients, a condition caused by the presence of high
mast cell numbers, or other systemic inflammatory diseases would be insightful.
It is also worth comparing CapG gene and protein levels in classically defined non-
haematopoietic structural cells between normal and inflammatory conditions to
ascertain whether these cells may also contribute to elevated CapG expression in
inflammation. These studies would provide further insight to whether CapG can
be potentially used as a marker of certain inflammatory disorders.
Although data from this Thesis demonstrate that CapG is able to trigger
certain pro-inflammatory cytokine release from different cell types, future studies
are also necessary to identify the putative receptor for CapG on the cell surface
and the signalling pathways involved. It is interesting to note that CapG was able
to trigger cytokine release from many different cell types, suggesting that this
receptor might be ubiquitously expressed and that CapG may serve as a DAMP
that is released from damaged cells. Thus, understanding this receptor provides a
better insight of the involvement of CapG and its relevance in diseases. Targeting
the putative receptor may be achieved with small molecules, thus expanding the
possible therapeutic modulation of CapG activity.
7.7 Concluding remarks
This study presents novel insights into the role of extracellular CapG.
Firstly, CapG protein is expressed primarily in haematopoietic immune cells such
as mast cells and macrophages. In contrast, CapG protein is little expressed in
classically defined non-immune cells. However, it is likely that under some
circumstances such as inflammation and cancer, CapG protein expression is

285
elevated in these cells. Secondly, IgE-activated mast cells and LPS-activated
macrophages release CapG into the surrounding environment, which in turn can
be regulated by pharmacological agents including the anti-inflammatory
corticosteroid dexamethasone. However, macrophage heterogeneity was observed
in this regard. This Thesis also describes the generation of several useful tools to
better understand CapG, including generation of CapG from a mammalian
expression system, as an alternative to a bacterial expression system where
endotoxins were shown to confound data interpretation. Both CapG sources were
able to triggers release of pro-inflammatory cytokine such as IL-6 and IL-8 release
from a range of cell types including airway smooth muscle and fibroblast cells.
This finding supports our hypothesis that CapG is a novel pro-inflammatory
mediator. Finally, CapG mRNA levels were found to be differentially regulated in
inflammatory conditions. In particular, CapG expression was downregulated in
LPS-activated macrophages. Combined with CapG release, the loss of intracellular
CapG in these cells suggests a “macrophage stasis” phenotype. We propose that
this “stasis” phenotype mediated by the loss of CapG, prevents cell movement,
hence allowing macrophages to prioritise pathogen killing and perhaps the
resolution of inflammation. Thus, this Thesis demonstrates that CapG can be likely
added to the list of inflammatory mediators although more information is required
to determine its importance and the possibility for its modulation to treat
inflammatory diseases.

286

287
References

288
A
Abraham EJ, Slater KA, Sanyal S, Linehan K, Flaherty PM, Qian S (2011) Scale-up of mammalian cell culture using a new multilayered flask. Journal of Visualized Experiments 58
Abraham I, Alhossan A, Lee CS, Kutbi H, MacDonald K (2016) 'Real-life' effectiveness studies of omalizumab in adult patients with severe allergic asthma: systematic review. Allergy 71(5): 593-610
Abraham SN, St John AL (2010) Mast cell-orchestrated immunity to pathogens. Nature Reviews Immunology 10(6): 440-52
Aderem A (2003) Phagocytosis and the inflammatory response. Journal of Infectious Diseases 187 Suppl 2: S340-5
Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, Fletcher G, Durkin C, Postigo A, Skehel M, Batista F, Thompson B, Way M, Reis e Sousa C, Schulz O (2012) F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36(4): 635-45
Akbari O, Umetsu DT (2005) Role of regulatory dendritic cells in allergy and asthma. Current Allergy and Asthma Reports 5(1): 56-61
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124(4): 783-801
Al-Qaoud KM, Pearlman E, Hartung T, Klukowski J, Fleischer B, Hoerauf A (2000) A new mechanism for IL-5-dependent helminth control: neutrophil accumulation and neutrophil-mediated worm encapsulation in murine filariasis are abolished in the absence of IL-5. International Immunology 12(6): 899-908
Alangari AA (2010) Genomic and non-genomic actions of glucocorticoids in asthma. Annals of Thoracic Medicine 5(3): 133-9
Allen RC, Sannes PL, Spicer SS, Hong CC (1980) Comparisons of alveolar and peritoneal macrophages: soluble protein, esterase, dipeptidyl aminopeptidase II, and proteinase inhibitor. Journal of Histochemistry and Cytochemistry 28(9): 947-52
Alleva DG, Kaser SB, Monroy MA, Fenton MJ, Beller DI (1997) IL-15 functions as a potent autocrine regulator of macrophage proinflammatory cytokine production: evidence for differential receptor subunit utilization associated with stimulation or inhibition. Journal of Immunology 159(6): 2941-51

289
Alliot F, Godin I, Pessac B (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Research Developmental Brain Research 117(2): 145-52
Alonso A, Bayon Y, Renedo M, Crespo MS (2000) Stimulation of Fc gamma R receptors induces monocyte chemoattractant protein-1 in the human monocytic cell line THP-1 by a mechanism involving I kappa B-alpha degradation and formation of p50/p65 NF-kappa B/Rel complexes. International Immunology 12(4): 547-54
Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S (2003) Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300(5624): 1430-4
Andrade MV, Hiragun T, Beaven MA (2004) Dexamethasone suppresses antigen-induced activation of phosphatidylinositol 3-kinase and downstream responses in mast cells. Journal of Immunology 172(12): 7254-62
Aoki R, Kawamura T, Goshima F, Ogawa Y, Nakae S, Nakao A, Moriishi K, Nishiyama Y, Shimada S (2013) Mast cells play a key role in host defense against herpes simplex virus infection through TNF-alpha and IL-6 production. Journal of Investigative Dermatology 133(9): 2170-9
Arango Duque G, Descoteaux A (2014) Macrophage cytokines: involvement in immunity and infectious diseases. Frontiers in Immunology 5: 491
Arock M, Le Nours A, Malbec O, Daeron M (2008) Ex vivo and in vitro primary mast cells. Methods in Molecular Biology 415: 241-54
Arora PD, McCulloch CA (1996) Dependence of fibroblast migration on actin severing activity of gelsolin. The Journal of Biological Chemistry 271(34): 20516-23
Aruffo A (2002) Transient expression of proteins using COS cells. Current Protocols in Molecular Biology Chapter 16: Unit 16 12
Asea A (2008) Heat shock proteins and toll-like receptors. Handbook of Experimental Pharmacology(183): 111-27
Ashish, Paine MS, Perryman PB, Yang L, Yin HL, Krueger JK (2007) Global structure changes associated with Ca2+ activation of full-length human plasma gelsolin. Journal of Biological Chemistry 282(35): 25884-92
Asti C, Ruggieri V, Porzio S, Chiusaroli R, Melillo G, Caselli GF (2000) Lipopolysaccharide-induced lung injury in mice. I. Concomitant evaluation of inflammatory cells and haemorrhagic lung damage. Pulmonary Pharmacology & Therapeutics 13(2): 61-9

290
Astrakhantseva IV, Efimov GA, Drutskaya MS, Kruglov AA, Nedospasov SA (2014) Modern anti-cytokine therapy of autoimmune diseases. Biochemistry (Moscow) 79(12): 1308-21
Attur MG, Patel IR, Patel RN, Abramson SB, Amin AR (1998) Autocrine production of IL-1 beta by human osteoarthritis-affected cartilage and differential regulation of endogenous nitric oxide, IL-6, prostaglandin E2, and IL-8. Proceedings of the Association of American Physicians 110(1): 65-72
Auwerx J (1991) The human leukemia cell line, THP-1: a multifacetted model for the study of monocyte-macrophage differentiation. Experientia 47(1): 22-31
Azuma T, Witke W, Stossel TP, Hartwig JH, Kwiatkowski DJ (1998) Gelsolin is a downstream effector of rac for fibroblast motility. EMBO Journal 17(5): 1362-70
B
Bailey DP, Kashyap M, Mirmonsef P, Bouton LA, Domen J, Zhu J, Dessypris EN, Ryan JJ (2004) Interleukin-4 elicits apoptosis of developing mast cells via a Stat6-dependent mitochondrial pathway. Experimental Hematology 32(1): 52-9
Balakrishnan L, Bhattacharjee M, Ahmad S, Nirujogi RS, Renuse S, Subbannayya Y, Marimuthu A, Srikanth SM, Raju R, Dhillon M, Kaur N, Jois R, Vasudev V, Ramachandra Y, Sahasrabuddhe NA, Prasad Ts K, Mohan S, Gowda H, Shankar S, Pandey A (2014) Differential proteomic analysis of synovial fluid from rheumatoid arthritis and osteoarthritis patients. Clinical Proteomics 11(1): 1
Baldi L, Hacker DL, Adam M, Wurm FM (2007) Recombinant protein production by large-scale transient gene expression in mammalian cells: state of the art and future perspectives. Biotechnology Letters 29(5): 677-84
Balhara J, Gounni AS (2012) The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunology 5(6): 605-9
Bancroft AJ, McKenzie AN, Grencis RK (1998) A critical role for IL-13 in resistance to intestinal nematode infection. Journal of Immunology 160(7): 3453-61
Bang BR, Chun E, Shim EJ, Lee HS, Lee SY, Cho SH, Min KU, Kim YY, Park HW (2011) Alveolar macrophages modulate allergic inflammation in a murine model of asthma. Experimental and Molecular Medicine 43(5): 275-80

291
Banovac K, Neylan D, Leone J, Ghandur-Mnaymneh L, Rabinovitch A (1989) Are the mast cells antigen presenting cells? Immunological Investigations 18(7): 901-6
Barth MC, Ahluwalia N, Anderson TJ, Hardy GJ, Sinha S, Alvarez-Cardona JA, Pruitt IE, Rhee EP, Colvin RA, Gerszten RE (2009) Kynurenic acid triggers firm arrest of leukocytes to vascular endothelium under flow conditions. Journal of Biological Chemistry 284(29): 19189-95
Bataki EL, Evans GS, Everard ML (2005) Respiratory syncytial virus and neutrophil activation. Clinical and Experimental Immunology 140(3): 470-7
Bauermeister K, Burger M, Almanasreh N, Knopf HP, Schumann RR, Schollmeyer P, Dobos GJ (1998) Distinct regulation of IL-8 and MCP-1 by LPS and interferon-gamma-treated human peritoneal macrophages. Nephrology, Dialysis, Transplantation 13(6): 1412-9
Beck LA, Thaci D, Hamilton JD, Graham NM, Bieber T, Rocklin R, Ming JE, Ren H, Kao R, Simpson E, Ardeleanu M, Weinstein SP, Pirozzi G, Guttman-Yassky E, Suarez-Farinas M, Hager MD, Stahl N, Yancopoulos GD, Radin AR (2014) Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. New England Journal of Medicine 371(2): 130-9
Bel EH, Wenzel SE, Thompson PJ, Prazma CM, Keene ON, Yancey SW, Ortega HG, Pavord ID, the SI (2014) Oral Glucocorticoid-Sparing Effect of Mepolizumab in Eosinophilic Asthma. New England Journal of Medicine
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature Genetics 38(5): 515-7
Bender AT, Ostenson CL, Giordano D, Beavo JA (2004) Differentiation of human monocytes in vitro with granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor produces distinct changes in cGMP phosphodiesterase expression. Cellular Signalling 16(3): 365-74
Berkman N, Jose PJ, Williams TJ, Schall TJ, Barnes PJ, Chung KF (1995) Corticosteroid inhibition of macrophage inflammatory protein-1 alpha in human monocytes and alveolar macrophages. American Journal of Physiology 269(4 Pt 1): L443-52
Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, Maillot MC, Portier A, Couderc J, Galanaud P, Peuchmaur M, Riccardi C, Emilie D (2003) Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood 101(2): 729-38
Berry M, Brightling C, Pavord I, Wardlaw A (2007) TNF-alpha in asthma. Current Opinion in Pharmacology 7(3): 279-82

292
Bian ZM, Elner SG, Yoshida A, Elner VM (2004) Differential involvement of phosphoinositide 3-kinase/Akt in human RPE MCP-1 and IL-8 expression. Investigative Ophthalmology and Visual Science 45(6): 1887-96
Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of Leukocyte Biology 81(1): 1-5
Bischoff SC (2007) Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nature Reviews Immunology 7(2): 93-104
Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J (2014) Actin dynamics, architecture, and mechanics in cell motility. Physiological Reviews 94(1): 235-63
Blasi E, Puliti M, Pitzurra L, Barluzzi R, Mazzolla R, Adami C, Cox GW, Bistoni F (1994) Comparative studies on functional and secretory properties of macrophage cell lines derived from different anatomical sites. FEMS Immunology and Medical Microbiology 9(3): 207-15
Boily-Larouche G, Milev MP, Zijenah LS, Labbe AC, Zannou DM, Humphrey JH, Ward BJ, Poudrier J, Mouland AJ, Cohen EA, Roger M (2012) Naturally-occurring genetic variants in human DC-SIGN increase HIV-1 capture, cell-transfer and risk of mother-to-child transmission. PLoS One 7(7): e40706
Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. Journal of Neuroscience 28(16): 4283-92
Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hilliard JB, Berger M (1995) Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. American Journal of Respiratory Cell and Molecular Biology 13(3): 257-61
Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V (2006) Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 295(19): 2275-85
Borish L, Steinke JW (2011) Interleukin-33 in asthma: how big of a role does it play? Current Allergy and Asthma Reports 11(1): 7-11
Bowie A, O'Neill LA (2000) The interleukin-1 receptor/Toll-like receptor superfamily: signal generators for pro-inflammatory interleukins and microbial products. Journal of Leukocyte Biology 67(4): 508-14

293
Bradding P, Arthur G (2016) Mast cells in asthma--state of the art. Clinical and Experimental Allergy 46(2): 194-263
Bradding P, Brightling C (2007) Mast cell infiltration of airway smooth muscle in asthma. Respiratory Medicine 101(5): 1045; author reply 1046-7
Bradding P, Okayama Y, Howarth PH, Church MK, Holgate ST (1995) Heterogeneity of human mast cells based on cytokine content. Journal of Immunology 155(1): 297-307
Bradding P, Roberts JA, Britten KM, Montefort S, Djukanovic R, Mueller R, Heusser CH, Howarth PH, Holgate ST (1994) Interleukin-4, -5, and -6 and tumor necrosis factor-alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. American Journal of Respiratory Cell and Molecular Biology 10(5): 471-80
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72: 248-54
Brieland JK, Jones ML, Clarke SJ, Baker JB, Warren JS, Fantone JC (1992) Effect of acute inflammatory lung injury on the expression of monocyte chemoattractant protein-1 (MCP-1) in rat pulmonary alveolar macrophages. American Journal of Respiratory Cell and Molecular Biology 7(2): 134-9
Brightling C, Berry M, Amrani Y (2008) Targeting TNF-alpha: a novel therapeutic approach for asthma. Journal of Allergy and Clinical Immunology 121(1): 5-10
Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID (2002) Mast-cell infiltration of airway smooth muscle in asthma. New England Journal of Medicine 346(22): 1699-705
Bromander A, Holmgren J, Lycke N (1991) Cholera toxin stimulates IL-1 production and enhances antigen presentation by macrophages in vitro. Journal of Immunology 146(9): 2908-14
Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, Waldmann H, Neuberger MS (1987) Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. Journal of Experimental Medicine 166(5): 1351-61
Bucki R, Byfield FJ, Kulakowska A, McCormick ME, Drozdowski W, Namiot Z, Hartung T, Janmey PA (2008) Extracellular gelsolin binds lipoteichoic acid and modulates cellular response to proinflammatory bacterial wall components. Journal of Immunology 181(7): 4936-44

294
Buhtoiarov IN, Lum H, Berke G, Paulnock DM, Sondel PM, Rakhmilevich AL (2005) CD40 ligation activates murine macrophages via an IFN-gamma-dependent mechanism resulting in tumor cell destruction in vitro. Journal of Immunology 174(10): 6013-22
Burd PR, Rogers HW, Gordon JR, Martin CA, Jayaraman S, Wilson SD, Dvorak AM, Galli SJ, Dorf ME (1989) Interleukin 3-dependent and -independent mast cells stimulated with IgE and antigen express multiple cytokines. Journal of Experimental Medicine 170(1): 245-57
Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM, Cohen DA (2004) Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. Journal of Leukocyte Biology 75(4): 612-23
Burrage PS, Mix KS, Brinckerhoff CE (2006) Matrix metalloproteinases: role in arthritis. Frontiers in Bioscience 11: 529-43
Burtnick LD, Urosev D, Irobi E, Narayan K, Robinson RC (2004) Structure of the N-terminal half of gelsolin bound to actin: roles in severing, apoptosis and FAF. EMBO Journal 23(14): 2713-22
Butterfield JH, Weiler D, Dewald G, Gleich GJ (1988) Establishment of an immature mast cell line from a patient with mast cell leukemia. Leukemia Research 12(4): 345-55
C
Cachianes G, Ho C, Weber RF, Williams SR, Goeddel DV, Leung DW (1993) Epstein-Barr virus-derived vectors for transient and stable expression of recombinant proteins. Biotechniques 15(2): 255-9
Cain DW, Cidlowski JA (2015) Specificity and sensitivity of glucocorticoid signaling in health and disease. Best Practice & Research Clinical Endocrinology & Metabolism 29(4): 545-56
Cairns JA, Walls AF (1996) Mast cell tryptase is a mitogen for epithelial cells. Stimulation of IL-8 production and intercellular adhesion molecule-1 expression. Journal of Immunology 156(1): 275-83
Cantu C, Bose F, Bianchi P, Reali E, Colzani MT, Cantu I, Barbarani G, Ottolenghi S, Witke W, Spinardi L, Ronchi AE (2012) Defective erythroid maturation in gelsolin mutant mice. Haematologica 97(7): 980-8
CapG_Cell_Line_Atlas (2016) Available at: http://www.proteinatlas.org/ENSG00000042493-CAPG/cell/HPA019092 (accessed 21 June 2015).

295
Cardoso LS, Araujo MI, Goes AM, Pacifico LG, Oliveira RR, Oliveira SC (2007) Polymyxin B as inhibitor of LPS contamination of Schistosoma mansoni recombinant proteins in human cytokine analysis. Microbial Cell Factories 6: 1
Careau E, Bissonnette EY (2004) Adoptive transfer of alveolar macrophages abrogates bronchial hyperresponsiveness. American Journal of Respiratory Cell and Molecular Biology 31(1): 22-7
Careau E, Proulx LI, Pouliot P, Spahr A, Turmel V, Bissonnette EY (2006) Antigen sensitization modulates alveolar macrophage functions in an asthma model. American Journal of Physiology Lung Cellular and Molecular Physiology 290(5): 871-9
Carmi Y, Voronov E, Dotan S, Lahat N, Rahat MA, Fogel M, Huszar M, White MR, Dinarello CA, Apte RN (2009) The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis. Journal of Immunology 183(7): 4705-14
Carta S, Castellani P, Delfino L, Tassi S, Vene R, Rubartelli A (2009) DAMPs and inflammatory processes: the role of redox in the different outcomes. Journal of Leukocyte Biology 86(3): 549-55
Catley MC, Coote J, Bari M, Tomlinson KL (2011) Monoclonal antibodies for the treatment of asthma. Pharmacology & Therapeutics 132(3): 333-51
Cazzola M, Page CP, Calzetta L, Matera MG (2012) Pharmacology and therapeutics of bronchodilators. Pharmacology Reviews 64(3): 450-504
Chanput W, Mes JJ, Savelkoul HF, Wichers HJ (2013) Characterization of polarized THP-1 macrophages and polarizing ability of LPS and food compounds. Food and Function 4(2): 266-76
Chen HY, Sharma BB, Yu L, Zuberi R, Weng IC, Kawakami Y, Kawakami T, Hsu DK, Liu FT (2006) Role of galectin-3 in mast cell functions: galectin-3-deficient mast cells exhibit impaired mediator release and defective JNK expression. Journal of Immunology 177(8): 4991-7
Cheng D, Logge W, Low JK, Garner B, Karl T (2013) Novel behavioural characteristics of the APP(Swe)/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Behavioural Brain Research 245: 120-7
Choi H, Fermin D, Nesvizhskii AI (2008) Significance analysis of spectral count data in label-free shotgun proteomics. Molecular & Cellular Proteomics 7(12): 2373-85
Chomarat P, Banchereau J, Davoust J, Palucka AK (2000) IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nature Immunology 1(6): 510-4

296
Chomczynski P, Sacchi N (2006) The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nature Protocols 1(2): 581-5
Chung F (2001) Anti-inflammatory cytokines in asthma and allergy: interleukin-10, interleukin-12, interferon-gamma. Mediators of Inflammation 10(2): 51-9
Chung KF, Caramori G, Adcock IM (2009) Inhaled corticosteroids as combination therapy with beta-adrenergic agonists in airways disease: present and future. European Journal of Clinical Pharmacology 65(9): 853-71
Claesson-Welsh L (2015) Vascular permeability--the essentials. Upsala Journal of Medical Sciences 120(3): 135-43
Cohen TS, Bucki R, Byfield FJ, Ciccarelli NJ, Rosenberg B, DiNubile MJ, Janmey PA, Margulies SS (2011) Therapeutic potential of plasma gelsolin administration in a rat model of sepsis. Cytokine 54(3): 235-8
Collington SJ, Hallgren J, Pease JE, Jones TG, Rollins BJ, Westwick J, Austen KF, Williams TJ, Gurish MF, Weller CL (2010) The role of the CCL2/CCR2 axis in mouse mast cell migration in vitro and in vivo. Journal of Immunology 184(11): 6114-23
Collins RA, Gualano RC, Zosky GR, Atkins CL, Turner DJ, Colasurdo GN, Sly PD (2005) Hyperresponsiveness to inhaled but not intravenous methacholine during acute respiratory syncytial virus infection in mice. Respiratory Research 6: 142
Conti G, Tambalo S, Villetti G, Catinella S, Carnini C, Bassani F, Sonato N, Sbarbati A, Marzola P (2010) Evaluation of lung inflammation induced by intratracheal administration of LPS in mice: comparison between MRI and histology. Magnetic Resonance Materials in Physics, Biology and Medicine 23(2): 93-101
Conti P, Kempuraj D, Di Gioacchino M, Boucher W, Letourneau R, Kandere K, Barbacane RC, Reale M, Felaco M, Frydas S, Theoharides TC (2002) Interleukin-6 and mast cells. Allergy and Asthma Proceedings 23(5): 331-5
Cooper JA, Walker SB, Pollard TD (1983) Pyrene actin: documentation of the validity of a sensitive assay for actin polymerization. Journal of Muscle Research and Cell Motility 4(2): 253-62
Corren J (2013) Asthma phenotypes and endotypes: an evolving paradigm for classification. Discov Med 15(83): 243-9

297
Corren J, Busse W, Meltzer EO, Mansfield L, Bensch G, Fahrenholz J, Wenzel SE, Chon Y, Dunn M, Weng HH, Lin SL (2010) A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. American Journal of Respiratory and Critical Care Medicine 181(8): 788-96
Cossart P, Lecuit M (1998) Interactions of Listeria monocytogenes with mammalian cells during entry and actin-based movement: bacterial factors, cellular ligands and signaling. EMBO Journal 17(14): 3797-806
Cox G (1996) IL-10 enhances resolution of pulmonary inflammation in vivo by promoting apoptosis of neutrophils. American Journal of Physiology 271(4 Pt 1): 566-71
Craig NL (1988) The mechanism of conservative site-specific recombination. Annual Review of Genetics 22: 77-105
Crespo-Biel N, Theunis C, Van Leuven F (2012) Protein tau: prime cause of synaptic and neuronal degeneration in Alzheimer's disease. International Journal of Alzheimer's Disease 2012: 251426
Crisp AJ (1984) Mast cells in rheumatoid arthritis. Journal of the Royal Society of Medicine 77(6): 450-1
Crosbie RH, Miller C, Cheung P, Goodnight T, Muhlrad A, Reisler E (1994) Structural connectivity in actin: effect of C-terminal modifications on the properties of actin. Biophysical Journal 67(5): 1957-64
Cryns V, Yuan J (1998) Proteases to die for. Genes & Development 12(11): 1551-70
Cucchiarini M, Ren XL, Perides G, Terwilliger EF (2003) Selective gene expression in brain microglia mediated via adeno-associated virus type 2 and type 5 vectors. Gene Therapy 10(8): 657-67
D
D'Amato G (2002) Treating atopic asthma with the anti-IgE monoclonal antibody. Monaldi Archives for Chest Disease 57(2): 117-9
D'Amato G, Stanziola A, Sanduzzi A, Liccardi G, Salzillo A, Vitale C, Molino A, Vatrella A, D'Amato M (2014) Treating severe allergic asthma with anti-IgE monoclonal antibody (omalizumab): a review. Multidisciplinary Respiratory Medicine 9(1): 23

298
Da Silva JA, Jacobs JW, Kirwan JR, Boers M, Saag KG, Ines LB, de Koning EJ, Buttgereit F, Cutolo M, Capell H, Rau R, Bijlsma JW (2006) Safety of low dose glucocorticoid treatment in rheumatoid arthritis: published evidence and prospective trial data. Annals of the Rheumatic Diseases 65(3): 285-93
Dabiri GA, Young CL, Rosenbloom J, Southwick FS (1992) Molecular cloning of human macrophage capping protein cDNA. A unique member of the gelsolin/villin family expressed primarily in macrophages. Journal of Biological Chemistry 267(23): 16545-52
Dahl B, Schiodt FV, Ott P, Gvozdenovic R, Yin HL, Lee WM (1999) Plasma gelsolin is reduced in trauma patients. Shock 12(2): 102-4
Dahl R (2006) Systemic side effects of inhaled corticosteroids in patients with asthma. Respiratory Medicine 100(8): 1307-17
Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH (2010) The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS One 5(1): e8668
Daneman R (2012) The blood-brain barrier in health and disease. Annals of Neurology 72(5): 648-72
Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39(6): 889-909
de Boer WI, van Schadewijk A, Sont JK, Sharma HS, Stolk J, Hiemstra PS, van Krieken JH (1998) Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. American Journal of Respiratory and Critical Care Medicine 158(6): 1951-7
De Castro C, Holst O, Lanzetta R, Parrilli M, Molinaro A (2012) Bacterial lipopolysaccharides in plant and mammalian innate immunity. Protein and Peptide Letters 19(10): 1040-4
De Corte V, Van Impe K, Bruyneel E, Boucherie C, Mareel M, Vandekerckhove J, Gettemans J (2004) Increased importin-beta-dependent nuclear import of the actin modulating protein CapG promotes cell invasion. Journal of Cell Science 117(Pt 22): 5283-92
de Jong TD, Vosslamber S, Verweij CL (2014) Moving towards personalized medicine in rheumatoid arthritis. Arthritis Research & Therapy 16(3): 110
de Nadai P, Charbonnier AS, Chenivesse C, Senechal S, Fournier C, Gilet J, Vorng H, Chang Y, Gosset P, Wallaert B, Tonnel AB, Lassalle P, Tsicopoulos A (2006) Involvement of CCL18 in allergic asthma. Journal of Immunology 176(10): 6286-93

299
Deaton JD, Guerrero T, Howard TH (1992) Role of gelsolin interaction with actin in regulation and creation of actin nuclei in chemotactic peptide activated polymorphonuclear neutrophils. Molecular Biology of the Cell 3(12): 1427-35
Dempsey PW, Vaidya SA, Cheng G (2003) The art of war: Innate and adaptive immune responses. Cellular and Molecular Life Sciences 60(12): 2604-21
Deng Z, Ma S, Zhou H, Zang A, Fang Y, Li T, Shi H, Liu M, Du M, Taylor PR, Zhu HH, Chen J, Meng G, Li F, Chen C, Zhang Y, Jia XM, Lin X, Zhang X, Pearlman E, Li X, Feng GS, Xiao H (2015) Tyrosine phosphatase SHP-2 mediates C-type lectin receptor-induced activation of the kinase Syk and anti-fungal TH17 responses. Nature Immunology 16(6): 642-52
Desai D, Brightling C (2010) TNF-alpha antagonism in severe asthma? Recent Patents on Inflammation & Allergy Drug Discovery 4(3): 193-200
Deshmane SL, Kremlev S, Amini S, Sawaya BE (2009) Monocyte chemoattractant protein-1 (MCP-1): an overview. Journal of Interferon and Cytokine Research 29(6): 313-26
Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Current Medicinal Chemistry 14(11): 1189-97
Di Nardo A, Yamasaki K, Dorschner RA, Lai Y, Gallo RL (2008) Mast cell cathelicidin antimicrobial peptide prevents invasive group A Streptococcus infection of the skin. Journal of Immunology 180(11): 7565-73
Di Rosa M, Radomski M, Carnuccio R, Moncada S (1990) Glucocorticoids inhibit the induction of nitric oxide synthase in macrophages. Biochemical and Biophysical Research Communications 172(3): 1246-52
Diebold SS (2009) Activation of dendritic cells by toll-like receptors and C-type lectins. Handbook of Experimental Pharmacology(188): 3-30
Dilshara MG, Lee KT, Jayasooriya RG, Kang CH, Park SR, Choi YH, Choi IW, Hyun JW, Chang WY, Kim YS, Lee HJ, Kim GY (2014) Downregulation of NO and PGE2 in LPS-stimulated BV2 microglial cells by trans-isoferulic acid via suppression of PI3K/Akt-dependent NF-kappaB and activation of Nrf2-mediated HO-1. International Immunopharmacology 18(1): 203-11
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annual Review of Immunology 27: 519-50
Doens D, Fernandez PL (2014) Microglia receptors and their implications in the response to amyloid beta for Alzheimer's disease pathogenesis. Journal of Neuroinflammation 11: 48

300
Donohue JF, Ohar JA (2004) Effects of corticosteroids on lung function in asthma and chronic obstructive pulmonary disease. Proceedings of the American Thoracic Society 1(3): 152-60
Double KL, Halliday GM, Kril JJ, Harasty JA, Cullen K, Brooks WS, Creasey H, Broe GA (1996) Topography of brain atrophy during normal aging and Alzheimer's disease. Neurobiology of Aging 17(4): 513-21
Doulatov S, Notta F, Eppert K, Nguyen LT, Ohashi PS, Dick JE (2010) Revised map of the human progenitor hierarchy shows the origin of macrophages and dendritic cells in early lymphoid development. Nature Immunology 11(7): 585-93
Drummond RA, Brown GD (2011) The role of Dectin-1 in the host defence against fungal infections. Current Opinion in Microbiology 14(4): 392-9
Durocher Y, Perret S, Kamen A (2002) High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Research 30(2): 9
E
Echtenacher B, Mannel DN, Hultner L (1996) Critical protective role of mast cells in a model of acute septic peritonitis. Nature 381(6577): 75-7
Edwards AM (2014) Chromones. Chemical Immunology and Allergy 100: 317-22
Edwards JP, Zhang X, Frauwirth KA, Mosser DM (2006) Biochemical and functional characterization of three activated macrophage populations. Journal of Leukocyte Biology 80(6): 1298-307
Eisenbarth SC, Piggott DA, Huleatt JW, Visintin I, Herrick CA, Bottomly K (2002) Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. Journal of Experimental Medicine 196(12): 1645-51
El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature Medicine 13(4): 432-8
ElAli A, Rivest S (2015) Microglia in Alzheimer's disease: A multifaceted relationship. Brain, Behaviour, and Immunity: 138–150

301
Eller K, Wolf D, Huber JM, Metz M, Mayer G, McKenzie AN, Maurer M, Rosenkranz AR, Wolf AM (2011) IL-9 production by regulatory T cells recruits mast cells that are essential for regulatory T cell-induced immune suppression. Journal of Immunology 186(1): 83-91
Emery P (2012) Optimizing outcomes in patients with rheumatoid arthritis and an inadequate response to anti-TNF treatment. Rheumatology (Oxford) 51 Suppl 5: 22-30
Erin EM, Leaker BR, Nicholson GC, Tan AJ, Green LM, Neighbour H, Zacharasiewicz AS, Turner J, Barnathan ES, Kon OM, Barnes PJ, Hansel TT (2006) The effects of a monoclonal antibody directed against tumor necrosis factor-alpha in asthma. American Journal of Respiratory and Critical Care Medicine 174(7): 753-62
Erukhimov JA, Tang ZL, Johnson BA, Donahoe MP, Razzack JA, Gibson KF, Lee WM, Wasserloos KJ, Watkins SA, Pitt BR (2000) Actin-containing sera from patients with adult respiratory distress syndrome are toxic to sheep pulmonary endothelial cells. American Journal of Respiratory and Critical Care Medicine 162(1): 288-94
F
Fahy JV (2000) Reducing IgE levels as a strategy for the treatment of asthma. Clinical and Experimental Allergy 30 Suppl 1: 16-21
Feldmann M, Maini RN (2001) Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annual Review of Immunology 19: 163-96
Feng BS, He SH, Zheng PY, Wu L, Yang PC (2007) Mast cells play a crucial role in Staphylococcus aureus peptidoglycan-induced diarrhea. American Journal of Pathology 171(2): 537-47
Feng X, Deng T, Zhang Y, Su S, Wei C, Han D (2011) Lipopolysaccharide inhibits macrophage phagocytosis of apoptotic neutrophils by regulating the production of tumour necrosis factor alpha and growth arrest-specific gene 6. Immunology 132(2): 287-95
Fernandes D, Guida E, Koutsoubos V, Harris T, Vadiveloo P, Wilson JW, Stewart AG (1999) Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. American Journal of Respiratory Cell and Molecular Biology 21(1): 77-88
Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, Lossinsky AS, Graves MC, Gustavson A, Sayre J, Sofroni E, Suarez T, Chiappelli F, Bernard G (2005) Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer's disease patients. Journal of Alzheimer's Disease 7(3): 221-32

302
Fife CM, McCarroll JA, Kavallaris M (2014) Movers and shakers: cell cytoskeleton in cancer metastasis. British Journal of Pharmacology 171(24): 5507-23
Finn DF, Walsh JJ (2013) Twenty-first century mast cell stabilizers. British Journal of Pharmacology 170(1): 23-37
Finotto S, Mekori YA, Metcalfe DD (1997) Glucocorticoids decrease tissue mast cell number by reducing the production of the c-kit ligand, stem cell factor, by resident cells: in vitro and in vivo evidence in murine systems. Journal of Clinical Investigation 99(7): 1721-8
Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O'Garra A (1991a) IL-10 inhibits cytokine production by activated macrophages. Journal of Immunology 147(11): 3815-22
Fiorentino DF, Zlotnik A, Vieira P, Mosmann TR, Howard M, Moore KW, O'Garra A (1991b) IL-10 acts on the antigen-presenting cell to inhibit cytokine production by Th1 cells. Journal of Immunology 146(10): 3444-51
Fitzpatrick AM, Holguin F, Teague WG, Brown LA (2008) Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. Journal of Allergy and Clinical Immunology 121(6): 1372-8
Franchi L, Warner N, Viani K, Nunez G (2009) Function of Nod-like receptors in microbial recognition and host defense. Immunological Reviews 227(1): 106-28
Franke-Ullmann G, Pfortner C, Walter P, Steinmuller C, Lohmann-Matthes ML, Kobzik L, Freihorst J (1995) Alteration of pulmonary macrophage function by respiratory syncytial virus infection in vitro. Journal of Immunology 154(1): 268-80
Frigeri LG, Liu FT (1992) Surface expression of functional IgE binding protein, an endogenous lectin, on mast cells and macrophages. Journal of Immunology 148(3): 861-7
Fuggle NR, Howe FA, Allen RL, Sofat N (2014) New insights into the impact of neuro-inflammation in rheumatoid arthritis. Frontiers in Neuroscience 8: 357
Fujioka S, Niu J, Schmidt C, Sclabas GM, Peng B, Uwagawa T, Li Z, Evans DB, Abbruzzese JL, Chiao PJ (2004) NF-kappaB and AP-1 connection: mechanism of NF-kappaB-dependent regulation of AP-1 activity. Molecular and Cellular Biology 24(17): 7806-19
Fujiwara N, Kobayashi K (2005) Macrophages in inflammation. Current Drug Targets Inflammation and Allergy 4(3): 281-6

303
Fukuishi N, Murakami S, Ohno A, Yamanaka N, Matsui N, Fukutsuji K, Yamada S, Itoh K, Akagi M (2014) Does beta-hexosaminidase function only as a degranulation indicator in mast cells? The primary role of beta-hexosaminidase in mast cell granules. Journal of Immunology 193(4): 1886-94
Fuller RW, Morris PK, Richmond R, Sykes D, Varndell IM, Kemeny DM, Cole PJ, Dollery CT, MacDermot J (1986) Immunoglobulin E-dependent stimulation of human alveolar macrophages: significance in type 1 hypersensitivity. Clinical and Experimental Immunology 65(2): 416-26
G
Gallelli L, Busceti MT, Vatrella A, Maselli R, Pelaia G (2013) Update on anticytokine treatment for asthma. BioMed Research International 2013: 104315
Galli SJ, Nakae S, Tsai M (2005) Mast cells in the development of adaptive immune responses. Nature Immunology 6(2): 135-42
Galli SJ, Tsai M (2012) IgE and mast cells in allergic disease. Nature Medicine 18(5): 693-704
Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP (2006) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiology of Disease 24(3): 516-24
Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T (2010) Macrophages in Alzheimer's disease: the blood-borne identity. Journal of Neural Transmission (Vienna) 117(8): 961-70
Gazzinelli RT, Oswald IP, James SL, Sher A (1992) IL-10 inhibits parasite killing and nitrogen oxide production by IFN-gamma-activated macrophages. Journal of Immunology 148(6): 1792-6
Geng YJ, Azuma T, Tang JX, Hartwig JH, Muszynski M, Wu Q, Libby P, Kwiatkowski DJ (1998) Caspase-3-induced gelsolin fragmentation contributes to actin cytoskeletal collapse, nucleolysis, and apoptosis of vascular smooth muscle cells exposed to proinflammatory cytokines. European Journal of Cell Biology 77(4): 294-302
Gensel JC, Nakamura S, Guan Z, van Rooijen N, Ankeny DP, Popovich PG (2009) Macrophages promote axon regeneration with concurrent neurotoxicity. Journal of Neuroscience 29(12): 3956-68
Gessani S, Testa U, Varano B, Di Marzio P, Borghi P, Conti L, Barberi T, Tritarelli E, Martucci R, Seripa D, et al. (1993) Enhanced production of LPS-induced cytokines during differentiation of

304
human monocytes to macrophages. Role of LPS receptors. Journal of Immunology 151(7): 3758-66
Gibofsky A (2012) Overview of epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis. American Journal of Managed Care 18(13 Suppl): S295-302
Gilfillan AM, Tkaczyk C (2006) Integrated signalling pathways for mast-cell activation. Nature Reviews Immunology 6(3): 218-30
Ginhoux F, Lim S, Hoeffel G, Low D, Huber T (2013) Origin and differentiation of microglia. Frontiers in Cellular Neuroscience 7: 45
Glaser J, Neumann MH, Mei Q, Betz B, Seier N, Beyer I, Fehm T, Neubauer H, Niederacher D, Fleisch MC (2014) Macrophage capping protein CapG is a putative oncogene involved in migration and invasiveness in ovarian carcinoma. BioMed Research International 2014: 379847
Gluzman Y (1981) SV40-transformed simian cells support the replication of early SV40 mutants. Cell 23(1): 175-82
Gomez SA, Arguelles CL, Guerrieri D, Tateosian NL, Amiano NO, Slimovich R, Maffia PC, Abbate E, Musella RM, Garcia VE, Chuluyan HE (2009) Secretory leukocyte protease inhibitor: a secreted pattern recognition receptor for mycobacteria. American Journal of Respiratory and Critical Care Medicine 179(3): 247-53
Gonzalo JA, Qiu Y, Lora JM, Al-Garawi A, Villeval JL, Boyce JA, Martinez AC, Marquez G, Goya I, Hamid Q, Fraser CC, Picarella D, Cote-Sierra J, Hodge MR, Gutierrez-Ramos JC, Kolbeck R, Coyle AJ (2007) Coordinated involvement of mast cells and T cells in allergic mucosal inflammation: critical role of the CC chemokine ligand 1:CCR8 axis. Journal of Immunology 179(3): 1740-50
Gordon S, Taylor PR (2005) Monocyte and macrophage heterogeneity. Nature Reviews Immunology 5(12): 953-64
Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M, Waisman A, Pfeilschifter J, Frank S (2009) A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. American Journal of Pathology 175(1): 132-47
Gosset P, Tillie-Leblond I, Oudin S, Parmentier O, Wallaert B, Joseph M, Tonnel AB (1999) Production of chemokines and proinflammatory and antiinflammatory cytokines by human alveolar macrophages activated by IgE receptors. Journal of Allergy and Clinical Immunology 103(2 Pt 1): 289-97

305
Graham AC, Hilmer KM, Zickovich JM, Obar JJ (2013) Inflammatory response of mast cells during influenza A virus infection is mediated by active infection and RIG-I signaling. Journal of Immunology 190(9): 4676-84
Graham FL, Smiley J, Russell WC, Nairn R (1977) Characteristics of a human cell line transformed by DNA from human adenovirus type 5. Journal of General Virology 36(1): 59-74
Gregory B, Kirchem A, Phipps S, Gevaert P, Pridgeon C, Rankin SM, Robinson DS (2003) Differential regulation of human eosinophil IL-3, IL-5, and GM-CSF receptor alpha-chain expression by cytokines: IL-3, IL-5, and GM-CSF down-regulate IL-5 receptor alpha expression with loss of IL-5 responsiveness, but up-regulate IL-3 receptor alpha expression. Journal of Immunology 170(11): 5359-66
Guhl S, Babina M, Neou A, Zuberbier T, Artuc M (2010) Mast cell lines HMC-1 and LAD2 in comparison with mature human skin mast cells--drastically reduced levels of tryptase and chymase in mast cell lines. Experimental Dermatology 19(9): 845-7
Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW (2009) Lung environment determines unique phenotype of alveolar macrophages. American Journal of Physiology Lung Cellular and Molecular Physiology 296(6): L936-46
Guttman JA, Finlay BB (2009) Tight junctions as targets of infectious agents. Biochimica et Biophysica Acta 1788(4): 832-41
H
Haddad JG, Harper KD, Guoth M, Pietra GG, Sanger JW (1990) Angiopathic consequences of saturating the plasma scavenger system for actin. Proceedings of the National Academy of Sciences of the United States of America 87(4): 1381-5
Hakansson HF, Smailagic A, Brunmark C, Miller-Larsson A, Lal H (2012) Altered lung function relates to inflammation in an acute LPS mouse model. Pulmonary Pharmacology & Therapeutics 25(5): 399-406
Hall S, Agrawal DK (2014) Key mediators in the immunopathogenesis of allergic asthma. International Immunopharmacology
Hamilton RG, Marcotte GV, Saini SS (2005) Immunological methods for quantifying free and total serum IgE levels in allergy patients receiving omalizumab (Xolair) therapy. Journal of Immunological Methods 303(1-2): 81-91

306
Hanania NA, Moore RH (2004) Anti-inflammatory activities of beta2-agonists. Current Drug Targets Inflammation and Allergy 3(3): 271-7
Hansbro PM, Kaiko GE, Foster PS (2011) Cytokine/anti-cytokine therapy - novel treatments for asthma? British Journal of Pharmacology 163(1): 81-95
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ (2010) The safety and side effects of monoclonal antibodies. Nature Reviews Drug Discovery 9(4): 325-38
Hardison SE, Brown GD (2012) C-type lectin receptors orchestrate antifungal immunity. Nature Immunology 13(9): 817-22
Harrison LM, van den Hoogen C, van Haaften WC, Tesh VL (2005) Chemokine expression in the monocytic cell line THP-1 in response to purified shiga toxin 1 and/or lipopolysaccharides. Infection and Immunity 73(1): 403-12
Hart PH (2001) Regulation of the inflammatory response in asthma by mast cell products. Immunology and Cell Biology 79(2): 149-53
Harvey BP, Gee RJ, Haberman AM, Shlomchik MJ, Mamula MJ (2007) Antigen presentation and transfer between B cells and macrophages. European Journal of Immunology 37(7): 1739-51
Hasko G, Pacher P, Deitch EA, Vizi ES (2007) Shaping of monocyte and macrophage function by adenosine receptors. Pharmacology & Therapeutics 113(2): 264-75
Hasko G, Szabo C, Nemeth ZH, Deitch EA (2002) Dopamine suppresses IL-12 p40 production by lipopolysaccharide-stimulated macrophages via a beta-adrenoceptor-mediated mechanism. Journal of Neuroimmunology 122(1-2): 34-9
Hayward RD, Koronakis V (1999) Direct nucleation and bundling of actin by the SipC protein of invasive Salmonella. EMBO Journal 18(18): 4926-34
Heaton T, Rowe J, Turner S, Aalberse RC, de Klerk N, Suriyaarachchi D, Serralha M, Holt BJ, Hollams E, Yerkovich S, Holt K, Sly PD, Goldblatt J, Le Souef P, Holt PG (2005) An immunoepidemiological approach to asthma: identification of in-vitro T-cell response patterns associated with different wheezing phenotypes in children. Lancet 365(9454): 142-9
Held TK, Weihua X, Yuan L, Kalvakolanu DV, Cross AS (1999) Gamma interferon augments macrophage activation by lipopolysaccharide by two distinct mechanisms, at the signal transduction level and via an autocrine mechanism involving tumor necrosis factor alpha and interleukin-1. Infection and Immunity 67(1): 206-12

307
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F (2005) Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. Journal of Neuroinflammation 2: 22
Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M (2009) The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX 26(2): 83-94
Henrickson KJ, Hoover S, Kehl KS, Hua W (2004) National disease burden of respiratory viruses detected in children by polymerase chain reaction. Pediatric Infectious Disease Journal 23(1 Suppl): S11-8
Hickman SE, Allison EK, El Khoury J (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. Journal of Neuroscience 28(33): 8354-60
Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nature Neuroscience 16(12): 1896-905
Hirst SJ, Barnes PJ, Twort CH (1992) Quantifying proliferation of cultured human and rabbit airway smooth muscle cells in response to serum and platelet-derived growth factor. American Journal of Respiratory Cell and Molecular Biology 7(6): 574-81
Hochuli E, Dobeli H, Schacher A (1987) New metal chelate adsorbent selective for proteins and peptides containing neighbouring histidine residues. Journal of Chromatography 411: 177-84
Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, Frantz S (2014) Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circulation Heart Failure 7(5): 822-30
Holden P, Keene DR, Lunstrum GP, Bachinger HP, Horton WA (2005) Secretion of cartilage oligomeric matrix protein is affected by the signal peptide. Journal of Biological Chemistry 280(17): 17172-9
Holgate ST, Chuchalin AG, Hebert J, Lotvall J, Persson GB, Chung KF, Bousquet J, Kerstjens HA, Fox H, Thirlwell J, Cioppa GD, Omalizumab 011 International Study G (2004) Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clinical and Experimental Allergy 34(4): 632-8
Holgate ST, Davies DE, Powell RM, Howarth PH, Haitchi HM, Holloway JW (2007) Local genetic and environmental factors in asthma disease pathogenesis: chronicity and persistence mechanisms. European Respiratory Journal 29(4): 793-803

308
Holt PG, Oliver J, Bilyk N, McMenamin C, McMenamin PG, Kraal G, Thepen T (1993) Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. Journal of Experimental Medicine 177(2): 397-407
Holtzman MJ (1991) Arachidonic acid metabolism. Implications of biological chemistry for lung function and disease. American Review of Respiratory Disease 143(1): 188-203
Hong GU, Park BS, Park JW, Kim SY, Ro JY (2013) IgE production in CD40/CD40L cross-talk of B and mast cells and mediator release via TGase 2 in mouse allergic asthma. Cellular Signalling 25(6): 1514-25
Horak F, Doberer D, Eber E, Horak E, Pohl W, Riedler J, Szepfalusi Z, Wantke F, Zacharasiewicz A, Studnicka M (2016) Diagnosis and management of asthma - Statement on the 2015 GINA Guidelines. Central European Journal of Medicine 128(15-16): 541-54
Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S (1999a) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. Journal of Immunology 162(7): 3749-52
Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S (1999b) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162(7): 3749-52
Hot A, Miossec P (2011) Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Annals of the Rheumatic Diseases 70(5): 727-32
Huber LC, Distler O, Tarner I, Gay RE, Gay S, Pap T (2006) Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology (Oxford) 45(6): 669-75
Huber M (2013) Activation/Inhibition of mast cells by supra-optimal antigen concentrations. Cell Communication and Signalling 11(1): 7
Hubert T, Van Impe K, Vandekerckhove J, Gettemans J (2008) The F-actin filament capping protein CapG is a bona fide nucleolar protein. Biochemical and Biophysical Research Communications 377(2): 699-704
Hubert T, Van Impe K, Vandekerckhove J, Gettemans J (2009) The actin-capping protein CapG localizes to microtubule-dependent organelles during the cell cycle. Biochemical and Biophysical Research Communications 380(1): 166-70
Hueber AJ, Asquith DL, Miller AM, Reilly J, Kerr S, Leipe J, Melendez AJ, McInnes IB (2010) Mast cells express IL-17A in rheumatoid arthritis synovium. Journal of Immunology 184(7): 3336-40

309
Humbert M, Beasley R, Ayres J, Slavin R, Hebert J, Bousquet J, Beeh KM, Ramos S, Canonica GW, Hedgecock S, Fox H, Blogg M, Surrey K (2005) Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 60(3): 309-16
Hume DA (2006) The mononuclear phagocyte system. Current Opinion in Immunology 18(1): 49-53
Hung SC, Kang MS, Kieff E (2001) Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. Proceedings of the National Academy of Sciences of the United States of America 98(4): 1865-70
Hunot S, Boissiere F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, Hirsch EC (1996) Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience 72(2): 355-63
Huo Y, Rangarajan P, Ling EA, Dheen ST (2011) Dexamethasone inhibits the Nox-dependent ROS production via suppression of MKP-1-dependent MAPK pathways in activated microglia. BMC Neuroscience 12: 49
Hussell T, Bell TJ (2014) Alveolar macrophages: plasticity in a tissue-specific context. Nature Reviews Immunology 14(2): 81-93
Hutchinson JA, Riquelme P, Geissler EK, Fandrich F (2011) Human regulatory macrophages. Methods in Molecular Biology 677: 181-92
I
Ichikawa H, Kanda T, Kosugi S, Kawachi Y, Sasaki H, Wakai T, Kondo T (2013) Laser microdissection and two-dimensional difference gel electrophoresis reveal the role of a novel macrophage-capping protein in lymph node metastasis in gastric cancer. Journal of Proteome Research 12(8): 3780-91
Ip WK, Takahashi K, Ezekowitz RA, Stuart LM (2009) Mannose-binding lectin and innate immunity. Immunological Reviews 230(1): 9-21
Irani AM, Bradford TR, Kepley CL, Schechter NM, Schwartz LB (1989) Detection of MCT and MCTC types of human mast cells by immunohistochemistry using new monoclonal anti-tryptase and anti-chymase antibodies. Journal of Histochemistry and Cytochemistry 37(10): 1509-15

310
Italiani P, Boraschi D (2014) From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Frontiers in Immunology 5: 514
Ivanov KI, Basic M, Varjosalo M, Makinen K (2014) One-step purification of twin-strep-tagged proteins and their complexes on strep-tactin resin cross-linked with bis(sulfosuccinimidyl) suberate (BS3). Journal of Visualized Experiments(86)
J
Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Human Molecular Genetics 13(2): 159-70
Jiang Y, Beller DI, Frendl G, Graves DT (1992) Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. Journal of Immunology 148(8): 2423-8
Jin G, Matsushita T, Hamaguchi Y, Le Huu D, Ishii T, Hasegawa M, Obata K, Karasuyama H, Takehara K, Fujimoto M (2012) Basophils and mast cells play critical roles for leukocyte recruitment in IgE-mediated cutaneous reverse passive Arthus reaction. Journal of Dermatological Science 67(3): 181-9
Jing W, Zhang X, Sun W, Hou X, Yao Z, Zhu Y (2015) CRISPR/CAS9-Mediated Genome Editing of miRNA-155 Inhibits Proinflammatory Cytokine Production by RAW264.7 Cells. BioMed Research International 2015: 326042
John M, Au BT, Jose PJ, Lim S, Saunders M, Barnes PJ, Mitchell JA, Belvisi MG, Chung KF (1998a) Expression and release of interleukin-8 by human airway smooth muscle cells: inhibition by Th-2 cytokines and corticosteroids. American Journal of Respiratory Cell and Molecular Biology 18(1): 84-90
John M, Lim S, Seybold J, Jose P, Robichaud A, O'Connor B, Barnes PJ, Chung KF (1998b) Inhaled corticosteroids increase interleukin-10 but reduce macrophage inflammatory protein-1alpha, granulocyte-macrophage colony-stimulating factor, and interferon-gamma release from alveolar macrophages in asthma. American Journal of Respiratory and Critical Care Medicine 157(1): 256-62
Johnston PA, Yu FX, Reynolds GA, Yin HL, Moomaw CR, Slaughter CA, Sudhof TC (1990) Purification and expression of gCap39. An intracellular and secreted Ca2(+)-dependent actin-binding protein enriched in mononuclear phagocytes. Journal of Biological Chemistry 265(29): 17946-52

311
Jones A, Morton I, Hobson L, Evans GS, Everard ML (2006) Differentiation and immune function of human dendritic cells following infection by respiratory syncytial virus. Clinical & Experimental Immunology 143(3): 513-22
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP (1998) IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. Journal of Immunology 160(7): 3513-21
Junttila MR, Saarinen S, Schmidt T, Kast J, Westermarck J (2005) Single-step Strep-tag purification for the isolation and identification of protein complexes from mammalian cells. Proteomics 5(5): 1199-203
K
Kalesnikoff J, Huber M, Lam V, Damen JE, Zhang J, Siraganian RP, Krystal G (2001) Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity 14(6): 801-11
Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C (2003) IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends in Immunology 24(1): 25-9
Kashiwakura J, Xiao W, Kitaura J, Kawakami Y, Maeda-Yamamoto M, Pfeiffer JR, Wilson BS, Blank U, Kawakami T (2008) Pivotal advance: IgE accelerates in vitro development of mast cells and modifies their phenotype. Journal of Leukocyte Biology 84(2): 357-67
Kavanaugh A, Wells AF (2014) Benefits and risks of low-dose glucocorticoid treatment in the patient with rheumatoid arthritis. Rheumatology 53(10): 1742-51
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341(6151): 1246-9
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G (1991) Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO Journal 10(13): 4025-31
Kennedy MC, Stock JP (1952) The bronchodilator action of khellin. Thorax 7(1): 43-65

312
Kennedy MK, Picha KS, Fanslow WC, Grabstein KH, Alderson MR, Clifford KN, Chin WA, Mohler KM (1996) CD40/CD40 ligand interactions are required for T cell-dependent production of interleukin-12 by mouse macrophages. European Journal of Immunology 26(2): 370-8
Khalil M, Ronda J, Weintraub M, Jain K, Silver R, Silverman AJ (2007) Brain mast cell relationship to neurovasculature during development. Brain Research 1171: 18-29
Khan KH (2013) Gene expression in Mammalian cells and its applications. Advanced Pharmaceutical Bulletin 3(2): 257-63
Kim H, Bernstein JA (2009) Air pollution and allergic disease. Current Allergy and Asthma Reports 9(2): 128-33
Kim H, Fischer D (2011) Anaphylaxis. Allergy, Asthma, and Clinical Immunology 7 Suppl 1: S6
Kim H, Mazza J (2011) Asthma. Allergy, Asthma, and Clinical Immunology 7 Suppl 1: S2
Kim TK, Eberwine JH (2010) Mammalian cell transfection: the present and the future. Analytical and Bioanalytical Chemistry 397(8): 3173-8
Kimple ME, Brill AL, Pasker RL (2013) Overview of affinity tags for protein purification. Current Protocols in Protein Science 73: Unit 9.9
Kimura K, Ojima H, Kubota D, Sakumoto M, Nakamura Y, Tomonaga T, Kosuge T, Kondo T (2013) Proteomic identification of the macrophage-capping protein as a protein contributing to the malignant features of hepatocellular carcinoma. Journal of Proteomics 78: 362-73
Kingwell K (2012) Neurodegenerative disease: Microglia in early disease stages. Nature Reviews Neurology 8(9): 475
Kinne RW, Brauer R, Stuhlmuller B, Palombo-Kinne E, Burmester GR (2000) Macrophages in rheumatoid arthritis. Arthritis Research & Therapy 2(3): 189-202
Kinne RW, Stuhlmuller B, Burmester GR (2007) Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Research & Therapy 9(6): 224
Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, Rao VK, Metcalfe DD (2003) Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leukemia Research 27(8): 677-82

313
Kloog Y, Ambar I, Sokolovsky M, Kochva E, Wollberg Z, Bdolah A (1988) Sarafotoxin, a novel vasoconstrictor peptide: phosphoinositide hydrolysis in rat heart and brain. Science 242(4876): 268-70
Kohfeldt E, Maurer P, Vannahme C, Timpl R (1997) Properties of the extracellular calcium binding module of the proteoglycan testican. FEBS Letters 414(3): 557-61
Kolappan S, Gooch JT, Weeds AG, McLaughlin PJ (2003) Gelsolin domains 4-6 in active, actin-free conformation identifies sites of regulatory calcium ions. Journal of Molecular Biology 329(1): 85-92
Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry TJ, Kirschner MW, Koths K, Kwiatkowski DJ, Williams LT (1997) Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 278(5336): 294-8
Koya RC, Fujita H, Shimizu S, Ohtsu M, Takimoto M, Tsujimoto Y, Kuzumaki N (2000) Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. Journal of Biological Chemistry 275(20): 15343-9
Kraneveld AD, Sagar S, Garssen J, Folkerts G (2012) The two faces of mast cells in food allergy and allergic asthma: the possible concept of Yin Yang. Biochimica et Biophysica Acta 1822(1): 93-9
Kreider T, Anthony RM, Urban JF, Jr., Gause WC (2007) Alternatively activated macrophages in helminth infections. Current Opinion in Immunology 19(4): 448-53
Kroesen S, Widmer AF, Tyndall A, Hasler P (2003) Serious bacterial infections in patients with rheumatoid arthritis under anti-TNF-alpha therapy. Rheumatology (Oxford) 42(5): 617-21
Krutzik PO, Irish JM, Nolan GP, Perez OD (2004) Analysis of protein phosphorylation and cellular signaling events by flow cytometry: techniques and clinical applications. Clinical Immunology 110(3): 206-21
Kulkarni NN, Gunnarsson HI, Yi Z, Gudmundsdottir S, Sigurjonsson OE, Agerberth B, Gudmundsson GH (2016) Glucocorticoid dexamethasone down-regulates basal and vitamin D3 induced cathelicidin expression in human monocytes and bronchial epithelial cell line. Immunobiology 221(2): 245-52
Kumar N, Khurana S (2004) Identification of a functional switch for actin severing by cytoskeletal proteins. Journal of Biological Chemistry 279(24): 24915-8
Kwiatkowski DJ, Mehl R, Izumo S, Nadal-Ginard B, Yin HL (1988) Muscle is the major source of plasma gelsolin. Journal of Biological Chemistry 263(17): 8239-43

314
Kwiatkowski DJ, Stossel TP, Orkin SH, Mole JE, Colten HR, Yin HL (1986) Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature 323(6087): 455-8
L
Lalonde R, Kim HD, Maxwell JA, Fukuchi K (2005) Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neuroscience Letters 390(2): 87-92
Land WG (2015) The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I - Promoting inflammation and immunity. Sultan Qaboos University Medical Journal 15(1): e9-e21
Lanier BQ, Corren J, Lumry W, Liu J, Fowler-Taylor A, Gupta N (2003) Omalizumab is effective in the long-term control of severe allergic asthma. Annals of Allergy, Asthma & Immunology 91(2): 154-9
Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, Nikolaou A, Morias Y, De Baetselier P, Van Ginderachter JA (2011) Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. International Journal of Developmental Biology 55(7-9): 861-7
Lappalainen J, Rintahaka J, Kovanen PT, Matikainen S, Eklund KK (2013) Intracellular RNA recognition pathway activates strong anti-viral response in human mast cells. Clinical and Experimental Immunology 172(1): 121-8
Lawrence CE, Paterson YY, Wright SH, Knight PA, Miller HR (2004) Mouse mast cell protease-1 is required for the enteropathy induced by gastrointestinal helminth infection in the mouse. Gastroenterology 127(1): 155-65
Leclere M, Desnoyers M, Beauchamp G, Lavoie JP (2006) Comparison of four staining methods for detection of mast cells in equine bronchoalveolar lavage fluid. Journal of Veterinary Internal Medicine / American College of Veterinary Internal Medicine 20(2): 377-81
Lee H, Kashiwakura J, Matsuda A, Watanabe Y, Sakamoto-Sasaki T, Matsumoto K, Hashimoto N, Saito S, Ohmori K, Nagaoka M, Tokuhashi Y, Ra C, Okayama Y (2013a) Activation of human synovial mast cells from rheumatoid arthritis or osteoarthritis patients in response to aggregated IgG through Fcgamma receptor I and Fcgamma receptor II. Arthritis and Rheumatism 65(1): 109-19

315
Lee PS, Drager LR, Stossel TP, Moore FD, Rogers SO (2006) Relationship of plasma gelsolin levels to outcomes in critically ill surgical patients. Annals of Surgery 243(3): 399-403
Lee PS, Waxman AB, Cotich KL, Chung SW, Perrella MA, Stossel TP (2007) Plasma gelsolin is a marker and therapeutic agent in animal sepsis. Crit Care Med 35(3): 849-55
Lee SY, Kwok SK, Son HJ, Ryu JG, Kim EK, Oh HJ, Cho ML, Ju JH, Park SH, Kim HY (2013b) IL-17-mediated Bcl-2 expression regulates survival of fibroblast-like synoviocytes in rheumatoid arthritis through STAT3 activation. Arthritis Research & Therapy 15(1): R31
Lee WM, Galbraith RM (1992) The extracellular actin-scavenger system and actin toxicity. New England Journal of Medicine 326(20): 1335-41
Lee YG, Jeong JJ, Nyenhuis S, Berdyshev E, Chung S, Ranjan R, Karpurapu M, Deng J, Qian F, Kelly EA, Jarjour NN, Ackerman SJ, Natarajan V, Christman JW, Park GY (2015) Recruited alveolar macrophages, in response to airway epithelial-derived monocyte chemoattractant protein 1/CCl2, regulate airway inflammation and remodeling in allergic asthma. American Journal of Respiratory Cell and Molecular Biology 52(6): 772-84
Li BK, Guo K, Li CY, Li HL, Zhao PP, Chen K, Liu CX (2015) Influence of suppression of CapG gene expression by siRNA on the growth and metastasis of human prostate cancer cells. Genetics and Molecular Research 14(4): 15769-78
Li SN, Wang W, Fu SP, Wang JF, Liu HM, Xie SS, Liu BR, Li Y, Lv QK, Li ZQ, Xue WJ, Huang BX, Chen W, Liu JX (2013) IL-21 modulates release of proinflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Mediators of Inflammation 2013: 548073
Li Y, Ozment T, Wright GL, Peterson JM (2016) Identification of Putative Receptors for the Novel Adipokine CTRP3 Using Ligand-Receptor Capture Technology. PLoS One 11(10): e0164593
Lim JP, Gleeson PA (2011) Macropinocytosis: an endocytic pathway for internalising large gulps. Immunology and Cell Biology 89(8): 836-43
Lind SE, Smith DB, Janmey PA, Stossel TP (1986) Role of plasma gelsolin and the vitamin D-binding protein in clearing actin from the circulation. Journal of Clinical Investigation 78(3): 736-42
Liu B, Hong JS (2003) Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. Journal of Pharmacology and Experimental Therapeutics 304(1): 1-7

316
Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, Mamounas L, Lyons WE, Blue ME, Lee MK (2008) Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. Journal of Neuroscience 28(51): 13805-14
Lloyd CM, Hessel EM (2010) Functions of T cells in asthma: more than just T(H)2 cells. Nature Reviews Immunology 10(12): 838-48
Lohmann-Matthes ML, Steinmuller C, Franke-Ullmann G (1994) Pulmonary macrophages. European Respiratory Journal 7(9): 1678-89
Loke P, Nair MG, Parkinson J, Guiliano D, Blaxter M, Allen JE (2002) IL-4 dependent alternatively-activated macrophages have a distinctive in vivo gene expression phenotype. BMC Immunology 3: 7
Lopez-Castejon G, Brough D (2011) Understanding the mechanism of IL-1beta secretion. Cytokine and Growth Factor Reviews 22(4): 189-95
Lora JM, Al-Garawi A, Pickard MD, Price KS, Bagga S, Sicoli J, Hodge MR, Gutierrez-Ramos JC, Briskin MJ, Boyce JA (2003) FcepsilonRI-dependent gene expression in human mast cells is differentially controlled by T helper type 2 cytokines. Journal of Allergy and Clinical Immunology 112(6): 1119-26
Lotvall J, Akdis CA, Bacharier LB, Bjermer L, Casale TB, Custovic A, Lemanske RF, Jr., Wardlaw AJ, Wenzel SE, Greenberger PA (2011) Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. Journal of Allergy and Clinical Immunology 127(2): 355-60
Lu H, Khurana S, Verma N, Manischewitz J, King L, Beigel JH, Golding H (2011) A rapid Flp-In system for expression of secreted H5N1 influenza hemagglutinin vaccine immunogen in mammalian cells. PLoS One 6(2): e17297
Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J, Strom TB, Zheng XX, Noelle RJ (2006) Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature 442(7106): 997-1002
Lu YC, Yeh WC, Ohashi PS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42(2): 145-51
Lukacs NW, Strieter RM, Chensue SW, Widmer M, Kunkel SL (1995) TNF-alpha mediates recruitment of neutrophils and eosinophils during airway inflammation. Journal of Immunology 154(10): 5411-7

317
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7(4): 354-65
Lundgren DH, Hwang SI, Wu L, Han DK (2010) Role of spectral counting in quantitative proteomics. Expert Review of Proteomics 7(1): 39-53
Lyznik LA, Rao KV, Hodges TK (1996) FLP-mediated recombination of FRT sites in the maize genome. Nucleic Acids Research 24(19): 3784-9
M
Ma Y, Pope RM (2005) The role of macrophages in rheumatoid arthritis. Current Pharmaceutical Design 11(5): 569-80
Mackay GA, Pearce FL (1996) Extracellular guanosine 3',5'-cyclic monophosphate and disodium cromoglycate share a similar spectrum of activity in the inhibition of histamine release from isolated mast cells and basophils. International Archives of Allergy and Immunology 109(3): 258-65
Mackenzie AE, Lappin JE, Taylor DL, Nicklin SA, Milligan G (2011) GPR35 as a Novel Therapeutic Target. Frontiers in Endocrinology 2: 68
Madden KB, Whitman L, Sullivan C, Gause WC, Urban JF, Jr., Katona IM, Finkelman FD, Shea-Donohue T (2002) Role of STAT6 and mast cells in IL-4- and IL-13-induced alterations in murine intestinal epithelial cell function. Journal of Immunology 169(8): 4417-22
Magro AM, Alexander A (1974) Histamine release: in vitro studies of the inhibitory region of the dose-response curve. Journal of Immunology 112(5): 1762-5
Majithia V, Geraci SA (2007) Rheumatoid arthritis: diagnosis and management. American Journal of Medicine 120(11): 936-9
Majno G, Joris I (2004) Cells, Tissues, and Disease. Principles of General Pathology (ed 2.). New York: Oxford University Press
Malaviya R, Ikeda T, Ross E, Abraham SN (1996) Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-alpha. Nature 381(6577): 77-80
Malbec O, Roget K, Schiffer C, Iannascoli B, Dumas AR, Arock M, Daeron M (2007) Peritoneal cell-derived mast cells: an in vitro model of mature serosal-type mouse mast cells. Journal of Immunology 178(10): 6465-75

318
Manarin R, Pascutti MF, Ruffino JP, De Las Heras B, Bosca L, Bottasso O, Revelli S, Serra E (2010) Benznidazole blocks NF-kappaB activation but not AP-1 through inhibition of IKK. Molecular Immunology 47(15): 2485-91
Mann-Chandler MN, Kashyap M, Wright HV, Norozian F, Barnstein BO, Gingras S, Parganas E, Ryan JJ (2005) IFN-gamma induces apoptosis in developing mast cells. Journal of Immunology 175(5): 3000-5
Marakalala MJ, Graham LM, Brown GD (2010) The role of Syk/CARD9-coupled C-type lectin receptors in immunity to Mycobacterium tuberculosis infections. Clinical & Developmental Immunology 2010: 567571
Marshall JS (2004) Mast-cell responses to pathogens. Nature Reviews Immunology 4(10): 787-99
Marshall JS, Bienenstock J (1994) The role of mast cells in inflammatory reactions of the airways, skin and intestine. Current Opinion in Immunology 6(6): 853-9
Martoglio B (2003) Intramembrane proteolysis and post-targeting functions of signal peptides. Biochemical Society Transactions 31(Pt 6): 1243-7
Maruotti N, Crivellato E, Cantatore FP, Vacca A, Ribatti D (2007) Mast cells in rheumatoid arthritis. Clinical Rheumatology 26(1): 1-4
Maskrey BH, Megson IL, Whitfield PD, Rossi AG (2011) Mechanisms of resolution of inflammation: a focus on cardiovascular disease. Arteriosclerosis, Thrombosis, and Vascular Biology 31(5): 1001-6
Matsushima H, Yamada N, Matsue H, Shimada S (2004) TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. Journal of Immunology 173(1): 531-41
Maurer M, Rosen K, Hsieh HJ, Saini S, Grattan C, Gimenez-Arnau A, Agarwal S, Doyle R, Canvin J, Kaplan A, Casale T (2013) Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. New England Journal of Medicine 368(10): 924-35
Maurer M, Wedemeyer J, Metz M, Piliponsky AM, Weller K, Chatterjea D, Clouthier DE, Yanagisawa MM, Tsai M, Galli SJ (2004) Mast cells promote homeostasis by limiting endothelin-1-induced toxicity. Nature 432(7016): 512-6

319
Mayr SI, Zuberi RI, Zhang M, de Sousa-Hitzler J, Ngo K, Kuwabara Y, Yu L, Fung-Leung WP, Liu FT (2002) IgE-dependent mast cell activation potentiates airway responses in murine asthma models. Journal of Immunology 169(4): 2061-8
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38(8): 1285-91
McGhie EJ, Hayward RD, Koronakis V (2004) Control of actin turnover by a salmonella invasion protein. Molecular Cell 13(4): 497-510
McGough AM, Staiger CJ, Min JK, Simonetti KD (2003) The gelsolin family of actin regulatory proteins: modular structures, versatile functions. FEBS Letters 552(2-3): 75-81
McInnes IB, Schett G (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nature Reviews Immunology 7(6): 429-42
McKeith I (2004) Dementia with Lewy bodies. Dialogues in Clinical Neuroscience 6(3): 333-41
McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, Dong X (2015) Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519(7542): 237-41
McNicholl DM, Heaney LG (2008) Omalizumab: the evidence for its place in the treatment of allergic asthma. Journal of Core Evidence 3(1): 55-66
McParland V, Varsano G, Li X, Thornton J, Baby J, Aravind A, Meyer C, Pavic K, Rios P, Kohn M (2011) The metastasis-promoting phosphatase PRL-3 shows activity toward phosphoinositides. Biochemistry 50(35): 7579-90
Medzhitov R, Horng T (2009) Transcriptional control of the inflammatory response. Nature Reviews Immunology 9(10): 692-703
Meissner P, Pick H, Kulangara A, Chatellard P, Friedrich K, Wurm FM (2001) Transient gene expression: recombinant protein production with suspension-adapted HEK293-EBNA cells. Biotechnology and Bioengineering 75(2): 197-203
Mekori YA, Metcalfe DD (1999) Mast cell-T cell interactions. Journal of Allergy and Clinical Immunology 104(3 Pt 1): 517-23
Meng F, Lowell CA (1997) Lipopolysaccharide (LPS)-induced macrophage activation and signal transduction in the absence of Src-family kinases Hck, Fgr, and Lyn. Journal of Experimental Medicine 185(9): 1661-70

320
Menzella F, Lusuardi M, Galeone C, Taddei S, Zucchi L (2015) Profile of anti-IL-5 mAb mepolizumab in the treatment of severe refractory asthma and hypereosinophilic diseases. Journal of Asthma and Allergy 8: 105-14
Merluzzi S, Frossi B, Gri G, Parusso S, Tripodo C, Pucillo C (2010) Mast cells enhance proliferation of B lymphocytes and drive their differentiation toward IgA-secreting plasma cells. Blood 115(14): 2810-7
Metsarinne KP, Vehmaan-Kreula P, Kovanen PT, Saijonmaa O, Baumann M, Wang Y, Nyman T, Fyhrquist FY, Eklund KK (2002) Activated mast cells increase the level of endothelin-1 mRNA in cocultured endothelial cells and degrade the secreted Peptide. Arteriosclerosis, Thrombosis, and Vascular Biology 22(2): 268-73
Metz M, Piliponsky AM, Chen CC, Lammel V, Abrink M, Pejler G, Tsai M, Galli SJ (2006) Mast cells can enhance resistance to snake and honeybee venoms. Science 313(5786): 526-30
Meylan E, Tschopp J, Karin M (2006) Intracellular pattern recognition receptors in the host response. Nature 442(7098): 39-44
Mikita T, Porter G, Lawn RM, Shiffman D (2001) Oxidized low density lipoprotein exposure alters the transcriptional response of macrophages to inflammatory stimulus. Journal of Biological Chemistry 276(49): 45729-39
Miller AL, Bowlin TL, Lukacs NW (2004) Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. Journal of Infectious Diseases 189(8): 1419-30
Milligan G (2011) Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends in Pharmacological Sciences 32(5): 317-25
Mills CD (2012) M1 and M2 Macrophages: Oracles of Health and Disease. Critical Reviews in Immunology 32(6): 463-88
Minter MR, Moore Z, Zhang M, Brody KM, Jones NC, Shultz SR, Taylor JM, Crack PJ (2016) Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathologica Communications 4(1): 72
Mirshafiey A, Mohsenzadegan M (2008) The role of reactive oxygen species in immunopathogenesis of rheumatoid arthritis. Iranian Journal of Allergy, Asthma, and Immunology 7(4): 195-202
Mishra VS, Henske EP, Kwiatkowski DJ, Southwick FS (1994) The human actin-regulatory protein cap G: gene structure and chromosome location. Genomics 23(3): 560-5

321
Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A (2003) Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. Journal of Immunology 170(9): 4665-72
Mogensen TH, Berg RS, Paludan SR, Ostergaard L (2008) Mechanisms of dexamethasone-mediated inhibition of Toll-like receptor signaling induced by Neisseria meningitidis and Streptococcus pneumoniae. Infection and Immunity 76(1): 189-97
Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, Nagatsu T (1994) Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neuroscience Letters 180(2): 147-50
Moncada S, Vane JR (1979) Arachidonic acid metabolites and the interactions between platelets and blood-vessel walls. New England Journal of Medicine 300(20): 1142-7
Moon TC, Befus AD, Kulka M (2014) Mast cell mediators: their differential release and the secretory pathways involved. Frontiers in Immunology 5: 569
Moran EM, Mullan R, McCormick J, Connolly M, Sullivan O, Fitzgerald O, Bresnihan B, Veale DJ, Fearon U (2009) Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-alpha, Oncostatin M and response to biologic therapies. Arthritis Research & Therapy 11(4): R113
Morjaria JB, Chauhan AJ, Babu KS, Polosa R, Davies DE, Holgate ST (2008) The role of a soluble TNFalpha receptor fusion protein (etanercept) in corticosteroid refractory asthma: a double blind, randomised, placebo controlled trial. Thorax 63(7): 584-91
Morjaria JB, Polosa R (2010) Recommendation for optimal management of severe refractory asthma. Journal of Asthma and Allergy 3: 43-56
Morofuji N, Ojima H, Onaya H, Okusaka T, Shimada K, Sakamoto Y, Esaki M, Nara S, Kosuge T, Asahina D, Ushigome M, Hiraoka N, Nagino M, Kondo T (2012) Macrophage-capping protein as a tissue biomarker for prediction of response to gemcitabine treatment and prognosis in cholangiocarcinoma. Journal of Proteomics 75(5): 1577-89
Morris GE, Whyte MK, Martin GF, Jose PJ, Dower SK, Sabroe I (2005) Agonists of toll-like receptors 2 and 4 activate airway smooth muscle via mononuclear leukocytes. American Journal of Respiratory and Critical Care Medicine 171(8): 814-22
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nature Reviews Immunology 8(12): 958-69

322
Mulherin D, Fitzgerald O, Bresnihan B (1996) Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis and Rheumatism 39(1): 115-24
Munder M, Mallo M, Eichmann K, Modolell M (1998) Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. Journal of Experimental Medicine 187(12): 2103-8
Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, Kurt-Jones EA, Finberg RW (2009) Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. Journal of Virology 83(3): 1492-500
Murray PJ, Wynn TA (2011a) Obstacles and opportunities for understanding macrophage polarization. Journal of Leukocyte Biology 89(4): 557-63
Murray PJ, Wynn TA (2011b) Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology 11(11): 723-37
N
Nag S, Larsson M, Robinson RC, Burtnick LD (2013) Gelsolin: the tail of a molecular gymnast. Cytoskeleton (Hoboken) 70(7): 360-84
Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Cytokines in Parkinson's disease. Journal of Neural Transmission Supplementum(58): 143-51
Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y (2002) Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17(3): 375-87
Nakae S, Suto H, Kakurai M, Sedgwick JD, Tsai M, Galli SJ (2005) Mast cells enhance T cell activation: Importance of mast cell-derived TNF. Proceedings of the National Academy of Sciences of the United States of America 102(18): 6467-72
Nava P, Lopez S, Arias CF, Islas S, Gonzalez-Mariscal L (2004) The rotavirus surface protein VP8 modulates the gate and fence function of tight junctions in epithelial cells. Journal of Cell Science 117(Pt 23): 5509-19
Navarro-Sarabia F, Ariza-Ariza R, Hernandez-Cruz B, Villanueva I (2006) Adalimumab for treating rheumatoid arthritis. Journal of Rheumatology 33(6): 1075-81

323
Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM, Group SS (2006) The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest 129(1): 15-26
Neveu WA, Allard JB, Dienz O, Wargo MJ, Ciliberto G, Whittaker LA, Rincon M (2009) IL-6 is required for airway mucus production induced by inhaled fungal allergens. Journal of Immunology 183(3): 1732-8
Newton R, Holden NS (2007) Separating transrepression and transactivation: a distressing divorce for the glucocorticoid receptor? Molecular Pharmacology 72(4): 799-809
Niederhoffer N, Levy R, Sick E, Andre P, Coupin G, Lombard Y, Gies JP (2009) Amyloid beta peptides trigger CD47-dependent mast cell secretory and phagocytic responses. International Journal of Immunopathology and Pharmacology 22(2): 473-83
Nigrovic PA, Lee DM (2005) Mast cells in inflammatory arthritis. Arthritis Research & Therapy 7(1): 1-11
Nigrovic PA, Malbec O, Lu B, Markiewski MM, Kepley C, Gerard N, Gerard C, Daeron M, Lee DM (2010) C5a receptor enables participation of mast cells in immune complex arthritis independently of Fcgamma receptor modulation. Arthritis and Rheumatism 62(11): 3322-33
Nilsson G, Blom T, Kusche-Gullberg M, Kjellen L, Butterfield JH, Sundstrom C, Nilsson K, Hellman L (1994) Phenotypic characterization of the human mast-cell line HMC-1. Scandinavian Journal of Immunology 39(5): 489-98
Nunomura S, Gon Y, Yoshimaru T, Suzuki Y, Nishimoto H, Kawakami T, Ra C (2005) Role of the FcepsilonRI beta-chain ITAM as a signal regulator for mast cell activation with monomeric IgE. International Immunology 17(6): 685-94
Nussbaum RL, Ellis CE (2003) Alzheimer's disease and Parkinson's disease. New England Journal of Medicine 348(14): 1356-64
O
O'Brien J, Wilson I, Orton T, Pognan F (2000) Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. European Journal of Biochemistry 267(17): 5421-6
O'Connor TM, O'Connell J, O'Brien DI, Goode T, Bredin CP, Shanahan F (2004) The role of substance P in inflammatory disease. Journal of Cellular Physiology 201(2): 167-80

324
Oakley RH, Cidlowski JA (2013) The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. Journal of Allergy and Clinical Immunology 132(5): 1033-44
Oh CK, Leigh R, McLaurin KK, Kim K, Hultquist M, Molfino NA (2013) A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respiratory Research 14: 93
Ohmori Y, Fukumoto S, Hamilton TA (1995) Two structurally distinct kappa B sequence motifs cooperatively control LPS-induced KC gene transcription in mouse macrophages. Journal of Immunology 155(7): 3593-600
Ohtoshi T, Takizawa H, Okazaki H, Kawasaki S, Takeuchi N, Ohta K, Ito K (1998) Diesel exhaust particles stimulate human airway epithelial cells to produce cytokines relevant to airway inflammation in vitro. Journal of Allergy and Clinical Immunology 101(6 Pt 1): 778-85
Okayama Y, Benyon RC, Rees PH, Lowman MA, Hillier K, Church MK (1992) Inhibition profiles of sodium cromoglycate and nedocromil sodium on mediator release from mast cells of human skin, lung, tonsil, adenoid and intestine. Clinical & Experimental Allergy 22(3): 401-9
Olajide OA, Bhatia HS, de Oliveira AC, Wright CW, Fiebich BL (2013) Inhibition of Neuroinflammation in LPS-Activated Microglia by Cryptolepine. Evidence Based Complementary and Alternative Medicine 2013: 459723
Onoda K, Yu FX, Yin HL (1993) gCap39 is a nuclear and cytoplasmic protein. Cell Motility and the Cytoskeleton 26(3): 227-38
Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, Humbert M, Katz LE, Keene ON, Yancey SW, Chanez P, the MI (2014) Mepolizumab Treatment in Patients with Severe Eosinophilic Asthma. New England Journal of Medicine
Oskeritzian CA, Zhao W, Min HK, Xia HZ, Pozez A, Kiev J, Schwartz LB (2005) Surface CD88 functionally distinguishes the MCTC from the MCT type of human lung mast cell. Journal of Allergy and Clinical Immunology 115(6): 1162-8
P
Pace E, Ferraro M, Bruno A, Chiappara G, Bousquet J, Gjomarkaj M (2011) Clinical benefits of 7 years of treatment with omalizumab in severe uncontrolled asthmatics. Journal of Asthma 48(4): 387-92

325
Panuska JR, Merolla R, Rebert NA, Hoffmann SP, Tsivitse P, Cirino NM, Silverman RH, Rankin JA (1995) Respiratory syncytial virus induces interleukin-10 by human alveolar macrophages. Suppression of early cytokine production and implications for incomplete immunity. Journal of Clinical Investigation 96(5): 2445-53
Parameswaran N, Patial S (2010) Tumor necrosis factor-alpha signaling in macrophages. Critical Reviews in Eukaryotic Gene Expression 20(2): 87-103
Parihar A, Eubank TD, Doseff AI (2010) Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. Journal of Innate Immunity 2(3): 204-15
Parikh SS, Litherland SA, Clare-Salzler MJ, Li W, Gulig PA, Southwick FS (2003) CapG(-/-) mice have specific host defense defects that render them more susceptible than CapG(+/+) mice to Listeria monocytogenes infection but not to Salmonella enterica serovar Typhimurium infection. Infection and Immunity 71(11): 6582-90
Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS (2007) Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflammation Research 56(1): 45-50
Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. Journal of Biological Chemistry 279(9): 7370-7
Parker DC (1993) T cell-dependent B cell activation. Annual Review of Immunology 11: 331-60
Passante E, Frankish N (2009) The RBL-2H3 cell line: its provenance and suitability as a model for the mast cell. Inflammation Research 58(11): 737-45
Pawankar R (2014) Allergic diseases and asthma: a global public health concern and a call to action. World Allergy Organization Journal 7(1): 12
Pera T, Penn RB (2016) Bronchoprotection and bronchorelaxation in asthma: New targets, and new ways to target the old ones. Pharmacology & Therapeutics
Perry VH, Nicoll JA, Holmes C (2010) Microglia in neurodegenerative disease. Nature Reviews Neurology 6(4): 193-201
Perry VH, Teeling J (2013) Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Seminars in Immunopathology 35(5): 601-12

326
Peters-Golden M (2004) The alveolar macrophage: the forgotten cell in asthma. American Journal of Respiratory Cell and Molecular Biology 31(1): 3-7
Peters SP (1990) Mast cells and histamine in asthma. Journal of Allergy and Clinical Immunology 86(4 Pt 2): 642-6
Peters SP, MacGlashan DW, Jr., Schulman ES, Schleimer RP, Hayes EC, Rokach J, Adkinson NF, Jr., Lichtenstein LM (1984) Arachidonic acid metabolism in purified human lung mast cells. Journal of Immunology 132(4): 1972-9
Pey P, Pearce RK, Kalaitzakis ME, Griffin WS, Gentleman SM (2014) Phenotypic profile of alternative activation marker CD163 is different in Alzheimer's and Parkinson's disease. Acta Neuropathologica Communications 2: 21
Porath J (1988) High-performance immobilized-metal-ion affinity chromatography of peptides and proteins. Journal of Chromatography 443: 3-11
Porath J (1992) Immobilized metal ion affinity chromatography. Protein Expression and Purification 3(4): 263-81
Pottiez G, Haverland N, Ciborowski P (2010) Mass spectrometric characterization of gelsolin isoforms. Rapid Communications in Mass Spectrometry 24(17): 2620-4
Prakash YS (2013) Airway smooth muscle in airway reactivity and remodeling: what have we learned? American Journal of Physiology Lung Cellular and Molecular Physiology 305(12): L912-33
Prussin C, Griffith DT, Boesel KM, Lin H, Foster B, Casale TB (2003) Omalizumab treatment downregulates dendritic cell FcepsilonRI expression. Journal of Allergy and Clinical Immunology 112(6): 1147-54
Q
Qian D, Nan Q, Yang Y, Li H, Zhou Y, Zhu J, Bai Q, Zhang P, An L, Xiang Y (2015) Gelsolin-Like Domain 3 Plays Vital Roles in Regulating the Activities of the Lily Villin/Gelsolin/Fragmin Superfamily. PLoS One 10(11): e0143174 Qian L, Flood PM (2008) Microglial cells and Parkinson's disease. Immunologic Research 41(3): 155-64

327
Queralt M, Brazis P, Merlos M, de Mora F, Puigdemont A (2000) In vitro inhibitory effect of rupatadine on histamine and TNF-alpha release from dispersed canine skin mast cells and the human mast cell line HMC-1. Inflammation Research 49(7): 355-60
Quinnell RJ, Pritchard DI, Raiko A, Brown AP, Shaw MA (2004) Immune responses in human necatoriasis: association between interleukin-5 responses and resistance to reinfection. Journal of Infectious Diseases 190(3): 430-8
R
Radinger M, Jensen BM, Kuehn HS, Kirshenbaum A, Gilfillan AM (2010) Generation, isolation, and maintenance of human mast cells and mast cell lines derived from peripheral blood or cord blood. Current Protocols in Immunology Chapter 7: Unit 7 37
Rapino F, Robles EF, Richter-Larrea JA, Kallin EM, Martinez-Climent JA, Graf T (2013) C/EBPalpha induces highly efficient macrophage transdifferentiation of B lymphoma and leukemia cell lines and impairs their tumorigenicity. Cell Reports 3(4): 1153-63
Ravasi T, Wells C, Forest A, Underhill DM, Wainwright BJ, Aderem A, Grimmond S, Hume DA (2002) Generation of diversity in the innate immune system: macrophage heterogeneity arises from gene-autonomous transcriptional probability of individual inducible genes. Journal of Immunology 168(1): 44-50
Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park JB, Rhim JS, Harris CC (1988) Transformation of human bronchial epithelial cells by infection with SV40 or adenovirus-12 SV40 hybrid virus, or transfection via strontium phosphate coprecipitation with a plasmid containing SV40 early region genes. Cancer Research 48(7): 1904-9
Reibman J, Hsu Y, Chen LC, Bleck B, Gordon T (2003) Airway epithelial cells release MIP-3alpha/CCL20 in response to cytokines and ambient particulate matter. American Journal of Respiratory Cell and Molecular Biology 28(6): 648-54
Reitz C, Mayeux R (2014) Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochemical Pharmacology 88(4): 640-51
Renz M, Betz B, Niederacher D, Bender HG, Langowski J (2008) Invasive breast cancer cells exhibit increased mobility of the actin-binding protein CapG. International Journal of Cancer 122(7): 1476-82
Reuter S, Stassen M, Taube C (2010) Mast cells in allergic asthma and beyond. Yonsei Medical Journal 51(6): 797-807

328
Reyes L, Davidson MK, Thomas LC, Davis JK (1999) Effects of Mycoplasma fermentans incognitus on differentiation of THP-1 cells. Infection and Immunity 67(7): 3188-92
Riccio AM, Dal Negro RW, Micheletto C, De Ferrari L, Folli C, Chiappori A, Canonica GW (2012) Omalizumab modulates bronchial reticular basement membrane thickness and eosinophil infiltration in severe persistent allergic asthma patients. International Journal of Immunopathology and Pharmacology 25(2): 475-84
Ricciotti E, FitzGerald GA (2011) Prostaglandins and inflammation. Arteriosclerosis, Thrombosis, and Vascular Biology 31(5): 986-1000
Richter A, Puddicombe SM, Lordan JL, Bucchieri F, Wilson SJ, Djukanovic R, Dent G, Holgate ST, Davies DE (2001) The contribution of interleukin (IL)-4 and IL-13 to the epithelial-mesenchymal trophic unit in asthma. American Journal of Respiratory Cell and Molecular Biology 25(3): 385-91
Rincon M, Irvin CG (2012) Role of IL-6 in asthma and other inflammatory pulmonary diseases. International Journal of Biological Sciences 8(9): 1281-90
Rindler MJ, Xu CF, Gumper I, Cen C, Sonderegger P, Neubert TA (2008) Calsyntenins are secretory granule proteins in anterior pituitary gland and pancreatic islet alpha cells. Journal of Histochemistry and Cytochemistry 56(4): 381-8
Rivera-Toledo E, Gomez B (2012) Respiratory syncytial virus persistence in macrophages alters the profile of cellular gene expression. Viruses 4(12): 3270-80
Rocha NP, de Miranda AS, Teixeira AL (2015) Insights into Neuroinflammation in Parkinson's Disease: From Biomarkers to Anti-Inflammatory Based Therapies. BioMed Research International 2015: 628192
Rossol M, Heine H, Meusch U, Quandt D, Klein C, Sweet MJ, Hauschildt S (2011) LPS-induced cytokine production in human monocytes and macrophages. Critical reviews in Immunology 31(5): 379-446
Rubbert-Roth A, Finckh A (2009) Treatment options in patients with rheumatoid arthritis failing initial TNF inhibitor therapy: a critical review. Arthritis Research & Therapy 11 Suppl 1: S1
S
Sacco O, Romberger D, Rizzino A, Beckmann JD, Rennard SI, Spurzem JR (1992) Spontaneous production of transforming growth factor-beta 2 by primary cultures of bronchial epithelial cells. Effects on cell behavior in vitro. Journal of Clinical Investigation 90(4): 1379-85

329
Saggu H, Cooksey J, Dexter D, Wells FR, Lees A, Jenner P, Marsden CD (1989) A selective increase in particulate superoxide dismutase activity in parkinsonian substantia nigra. Journal of Neurochemistry 53(3): 692-7
Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, Komatsu R, Miura N, Adachi Y, Ohno N, Shibuya K, Yamamoto N, Kawakami K, Yamasaki S, Saito T, Akira S, Iwakura Y (2010) Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32(5): 681-91
Salmon-Ehr V, Ramont L, Godeau G, Birembaut P, Guenounou M, Bernard P, Maquart FX (2000) Implication of interleukin-4 in wound healing. Laboratory Investigation 80(8): 1337-43
Saluja R, Delin I, Nilsson GP, Adner M (2012) FcepsilonR1-mediated mast cell reactivity is amplified through prolonged Toll-like receptor-ligand treatment. PLoS One 7(8): e43547
Sandeep T, Roopakala MS, Silvia CR, Chandrashekara S, Rao M (2010) Evaluation of serum immunoglobulin E levels in bronchial asthma. Lung India 27(3): 138-40
Sandig H, Bulfone-Paus S (2012) TLR signaling in mast cells: common and unique features. Frontiers in Immunology 3: 185
Sauer B (1994) Site-specific recombination: developments and applications. Current Opinion in Biotechnology 5(5): 521-7
Schecter AD, Rollins BJ, Zhang YJ, Charo IF, Fallon JT, Rossikhina M, Giesen PL, Nemerson Y, Taubman MB (1997) Tissue factor is induced by monocyte chemoattractant protein-1 in human aortic smooth muscle and THP-1 cells. Journal of Biological Chemistry 272(45): 28568-73
Schmid CD, Melchior B, Masek K, Puntambekar SS, Danielson PE, Lo DD, Sutcliffe JG, Carson MJ (2009) Differential gene expression in LPS/IFNgamma activated microglia and macrophages: in vitro versus in vivo. Journal of Neurochemistry 109 Suppl 1: 117-25
Schmidt TG, Batz L, Bonet L, Carl U, Holzapfel G, Kiem K, Matulewicz K, Niermeier D, Schuchardt I, Stanar K (2013) Development of the Twin-Strep-tag(R) and its application for purification of recombinant proteins from cell culture supernatants. Protein Expression and Purification 92(1): 54-61
Schulz C, Farkas L, Wolf K, Kratzel K, Eissner G, Pfeifer M (2002) Differences in LPS-induced activation of bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes (A-549). Scandinavian Journal of Immunology 56(3): 294-302

330
Scott BB, Weisbrot LM, Greenwood JD, Bogoch ER, Paige CJ, Keystone EC (1997) Rheumatoid arthritis synovial fibroblast and U937 macrophage/monocyte cell line interaction in cartilage degradation. Arthritis and Rheumatism 40(3): 490-8
Scott LJ (2014) Etanercept: a review of its use in autoimmune inflammatory diseases. Drugs 74(12): 1379-410
Sears MR, Lotvall J (2005) Past, present and future--beta2-adrenoceptor agonists in asthma management. Respiratory Medicine 99(2): 152-70
Senft AP, Taylor RH, Lei W, Campbell SA, Tipper JL, Martinez MJ, Witt TL, Clay CC, Harrod KS (2010) Respiratory syncytial virus impairs macrophage IFN-alpha/beta- and IFN-gamma-stimulated transcription by distinct mechanisms. American Journal of Respiratory Cell and Molecular Biology 42(4): 404-14
Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA, Perretti M, Rossi AG, Wallace JL (2007) Resolution of inflammation: state of the art, definitions and terms. FASEB Journal 21(2): 325-32
Serhan CN, Chiang N, Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews Immunology 8(5): 349-61
Seriolo B, Paolino S, Sulli A, Fasciolo D, Cutolo M (2006) Effects of anti-TNF-alpha treatment on lipid profile in patients with active rheumatoid arthritis. Annals of the New York Academy of Sciences 1069: 414-9
Shan X, Hu A, Veler H, Fatma S, Grunstein JS, Chuang S, Grunstein MM (2006) Regulation of Toll-like receptor 4-induced proasthmatic changes in airway smooth muscle function by opposing actions of ERK1/2 and p38 MAPK signaling. American Journal of Physiology Lung Cellular and Molecular Physiology 291(3): L324-33
Shao F, Zhang R, Don L, Ying K (2011) Overexpression of gelsolin-like actin-capping protein is associated with progression of lung adenocarcinoma. Tohoku Journal of Experimental Medicine 225(2): 95-101
Sharif O, Bolshakov VN, Raines S, Newham P, Perkins ND (2007) Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunology 8: 1
Shaw G, Morse S, Ararat M, Graham FL (2002) Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB Journal 16(8): 869-71

331
Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, Rolls A, Mack M, Pluchino S, Martino G, Jung S, Schwartz M (2009) Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Medicine 6(7): e1000113
Shepherd CE, Thiel E, McCann H, Harding AJ, Halliday GM (2000) Cortical inflammation in Alzheimer disease but not dementia with Lewy bodies. Archives of Neurology 57(6): 817-22
Shi C, Pamer EG (2011) Monocyte recruitment during infection and inflammation. Nature Reviews Immunology 11(11): 762-74
Shirey KA, Pletneva LM, Puche AC, Keegan AD, Prince GA, Blanco JC, Vogel SN (2010) Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunology 3(3): 291-300
Shubin NJ, Glukhova VA, Clauson M, Truong P, Abrink M, Pejler G, White NJ, Deutsch GH, Reeves SR, Vaisar T, James RG, Piliponsky AM (2016) Proteome analysis of mast cell releasates reveals a role for chymase in the regulation of coagulation factor XIIIA levels via proteolytic degradation. Journal of Allergy and Clinical Immunology
Sibilano R, Frossi B, Pucillo CE (2014) Mast cell activation: A complex interplay of positive and negative signaling pathways. European Journal of Immunology
Siebert S, Tsoukas A, Robertson J, McInnes I (2015) Cytokines as therapeutic targets in rheumatoid arthritis and other inflammatory diseases. Pharmacological Reviews 67(2): 280-309
Silacci P, Mazzolai L, Gauci C, Stergiopulos N, Yin HL, Hayoz D (2004) Gelsolin superfamily proteins: key regulators of cellular functions. Cellular and Molecular Life Sciences 61(19-20): 2614-23
Silverman AJ, Sutherland AK, Wilhelm M, Silver R (2000) Mast cells migrate from blood to brain. Journal of Neuroscience 20(1): 401-8
Sirois J, Menard G, Moses AS, Bissonnette EY (2000) Importance of histamine in the cytokine network in the lung through H2 and H3 receptors: stimulation of IL-10 production. Journal of Immunology 164(6): 2964-70
Skaper SD, Giusti P, Facci L (2012) Microglia and mast cells: two tracks on the road to neuroinflammation. FASEB Journal 26(8): 3103-17
Skloot GS (2016) Asthma phenotypes and endotypes: a personalized approach to treatment. Current Opinion in Pulmonary Medicine 22(1): 3-9

332
Skokowa J, Ali SR, Felda O, Kumar V, Konrad S, Shushakova N, Schmidt RE, Piekorz RP, Nurnberg B, Spicher K, Birnbaumer L, Zwirner J, Claassens JW, Verbeek JS, van Rooijen N, Kohl J, Gessner JE (2005) Macrophages induce the inflammatory response in the pulmonary Arthus reaction through G alpha i2 activation that controls C5aR and Fc receptor cooperation. Journal of Immunology 174(5): 3041-50
Snelgrove RJ, Goulding J, Didierlaurent AM, Lyonga D, Vekaria S, Edwards L, Gwyer E, Sedgwick JD, Barclay AN, Hussell T (2008) A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nature Immunology 9(9): 1074-83
Soler-Rodriguez AM, Zhang H, Lichenstein HS, Qureshi N, Niesel DW, Crowe SE, Peterson JW, Klimpel GR (2000) Neutrophil activation by bacterial lipoprotein versus lipopolysaccharide: differential requirements for serum and CD14. Journal of Immunology 164(5): 2674-83
Song C, Luo L, Lei Z, Li B, Liang Z, Liu G, Li D, Zhang G, Huang B, Feng ZH (2008) IL-17-producing alveolar macrophages mediate allergic lung inflammation related to asthma. Journal of Immunology 181(9): 6117-24
Srikrishna G, Freeze HH (2009) Endogenous damage-associated molecular pattern molecules at the crossroads of inflammation and cancer. Neoplasia 11(7): 615-28
St John AL, Abraham SN (2013) Innate immunity and its regulation by mast cells. Journal of Immunology 190(9): 4458-63
St John AL, Rathore AP, Yap H, Ng ML, Metcalfe DD, Vasudevan SG, Abraham SN (2011) Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proceedings of the National Academy of Sciences of the United States of America 108(22): 9190-5
Steer JH, Kroeger KM, Abraham LJ, Joyce DA (2000) Glucocorticoids suppress tumor necrosis factor-alpha expression by human monocytic THP-1 cells by suppressing transactivation through adjacent NF-kappa B and c-Jun-activating transcription factor-2 binding sites in the promoter. Journal of Biological Chemistry 275(24): 18432-40
Stelekati E, Bahri R, D'Orlando O, Orinska Z, Mittrucker HW, Langenhaun R, Glatzel M, Bollinger A, Paus R, Bulfone-Paus S (2009) Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity 31(4): 665-76
Stensballe LG, Simonsen JB, Thomsen SF, Larsen AM, Lysdal SH, Aaby P, Kyvik KO, Skytthe A, Backer V, Bisgaard H (2009) The causal direction in the association between respiratory syncytial virus hospitalization and asthma. Journal of Allergy and Clinical Immunology 123(1): 131-137 e1

333
Stoll G, Jander S (1999) The role of microglia and macrophages in the pathophysiology of the CNS. Progress in Neurobiology 58(3): 233-47
Stone KD, Prussin C, Metcalfe DD (2010) IgE, mast cells, basophils, and eosinophils. Journal of Allergy and Clinical Immunology 125(2 Suppl 2): S73-80
Strassmann G, Patil-Koota V, Finkelman F, Fong M, Kambayashi T (1994) Evidence for the involvement of interleukin 10 in the differential deactivation of murine peritoneal macrophages by prostaglandin E2. Journal of Experimental Medicine 180(6): 2365-70
Streit WJ, Mrak RE, Griffin WS (2004) Microglia and neuroinflammation: a pathological perspective. Journal of Neuroinflammation 1(1): 14
Su B, Ceponis PJ, Lebel S, Huynh H, Sherman PM (2003) Helicobacter pylori activates Toll-like receptor 4 expression in gastrointestinal epithelial cells. Infection and Immunity 71(6): 3496-502
Sullivan SD, Turk F (2008) An evaluation of the cost-effectiveness of omalizumab for the treatment of severe allergic asthma. Allergy 63(6): 670-84
Sun HQ, Yamamoto M, Mejillano M, Yin HL (1999) Gelsolin, a multifunctional actin regulatory protein. Journal of Biological Chemistry 274(47): 33179-82
Supajatura V, Ushio H, Nakao A, Okumura K, Ra C, Ogawa H (2001) Protective roles of mast cells against enterobacterial infection are mediated by Toll-like receptor 4. Journal of Immunology 167(4): 2250-6
Sutton BJ, Gould HJ (1993) The human IgE network. Nature 366(6454): 421-8
Suurmond J, Dorjee AL, Boon MR, Knol EF, Huizinga TW, Toes RE, Schuerwegh AJ (2011) Mast cells are the main interleukin 17-positive cells in anticitrullinated protein antibody-positive and -negative rheumatoid arthritis and osteoarthritis synovium. Arthritis Research & Therapy 13(5): R150
T
Takashiba S, Van Dyke TE, Amar S, Murayama Y, Soskolne AW, Shapira L (1999) Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kappaB. Infection and Immunity 67(11): 5573-8

334
Tamai R, Sugawara S, Takeuchi O, Akira S, Takada H (2003) Synergistic effects of lipopolysaccharide and interferon-gamma in inducing interleukin-8 production in human monocytic THP-1 cells is accompanied by up-regulation of CD14, Toll-like receptor 4, MD-2 and MyD88 expression. Journal of Endotoxin Research 9(3): 145-53
Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT (2012) PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunological Reviews 249(1): 158-75
Tarp S, Furst DE, Dossing A, Ostergaard M, Lorenzen T, Hansen MS, Singh JA, Choy EH, Boers M, Suarez-Almazor ME, Kristensen LE, Bliddal H, Christensen R (2016) Defining the optimal biological monotherapy in rheumatoid arthritis: A systematic review and meta-analysis of randomised trials. Seminars in Arthritis and Rheumatism
Taylor PC, Moore A, Vasilescu R, Alvir J, Tarallo M (2016) A structured literature review of the burden of illness and unmet needs in patients with rheumatoid arthritis: a current perspective. Rheumatology International 36(5): 685-95
Thaci D, Simpson EL, Beck LA, Bieber T, Blauvelt A, Papp K, Soong W, Worm M, Szepietowski JC, Sofen H, Kawashima M, Wu R, Weinstein SP, Graham NM, Pirozzi G, Teper A, Sutherland ER, Mastey V, Stahl N, Yancopoulos GD, Ardeleanu M (2016) Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet 387(10013): 40-52
Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, Asadi S, Vasiadi M, Weng Z, Miniati A, Kalogeromitros D (2012) Mast cells and inflammation. Biochimica et Biophysica Acta 1822(1): 21-33
Theriault P, ElAli A, Rivest S (2015) The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimer's Research & Therapy 7(1): 41
Thomas P, Smart TG (2005) HEK293 cell line: a vehicle for the expression of recombinant proteins. Journal of Pharmacological and Toxicological Methods 51(3): 187-200
Thompson CC, Ashcroft FJ, Patel S, Saraga G, Vimalachandran D, Prime W, Campbell F, Dodson A, Jenkins RE, Lemoine NR, Crnogorac-Jurcevic T, Yin HL, Costello E (2007) Pancreatic cancer cells overexpress gelsolin family-capping proteins, which contribute to their cell motility. Gut 56(1): 95-106
Tomita K, Lim S, Hanazawa T, Usmani O, Stirling R, Chung KF, Barnes PJ, Adcock IM (2002) Attenuated production of intracellular IL-10 and IL-12 in monocytes from patients with severe asthma. Clinical Immunology 102(3): 258-66

335
Toussirot E, Pertuiset E, Kantelip B, Wendling D (2008) Sarcoidosis occuring during anti-TNF-alpha treatment for inflammatory rheumatic diseases: report of two cases. Clinical & Experimental Rheumatology 26(3): 471-5
Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K (1980) Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). International Journal of Cancer 26(2): 171-6
Tsuji S, Matsumoto M, Takeuchi O, Akira S, Azuma I, Hayashi A, Toyoshima K, Seya T (2000) Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette-Guerin: involvement of toll-like receptors. Infect Immun 68(12): 6883-90
Tsuzuki H, Tani T, Ueyama H, Kodama M (2001) Lipopolysaccharide: neutralization by polymyxin B shuts down the signaling pathway of nuclear factor kappaB in peripheral blood mononuclear cells, even during activation. Journal of Surgical Research 100(1): 127-34
Tuncel N, Sener E, Cerit C, Karasu U, Gurer F, Sahinturk V, Baycu C, Ak D, Filiz Z (2005) Brain mast cells and therapeutic potential of vasoactive intestinal peptide in a Parkinson's disease model in rats: brain microdialysis, behavior, and microscopy. Peptides 26(5): 827-36
Turner-Brannen E, Choi KY, Arsenault R, El-Gabalawy H, Napper S, Mookherjee N (2011) Inflammatory cytokines IL-32 and IL-17 have common signaling intermediates despite differential dependence on TNF-receptor 1. Journal of Immunology 186(12): 7127-35
U
Underhill DM, Bassetti M, Rudensky A, Aderem A (1999) Dynamic interactions of macrophages with T cells during antigen presentation. Journal of Experimental Medicine 190(12): 1909-14
Urb M, Sheppard DC (2012) The role of mast cells in the defence against pathogens. PLoS Pathogens 8(4): e1002619
V
Valletta EA, Boner AL (1994) Cromoglycate and nedocromil: influence on airway reactivity. Mediators of Inflammation 3(7): S15-9
Van Craenenbroeck K, Vanhoenacker P, Haegeman G (2000) Episomal vectors for gene expression in mammalian cells. European Journal of Biochemistry 267(18): 5665-78

336
van den Berg WB, Joosten LA, Kollias G, van De Loo FA (1999) Role of tumour necrosis factor alpha in experimental arthritis: separate activity of interleukin 1beta in chronicity and cartilage destruction. Annals of the Rheumatic Diseases 58 Suppl 1: I40-8
Van Impe K, Bethuyne J, Cool S, Impens F, Ruano-Gallego D, De Wever O, Vanloo B, Van Troys M, Lambein K, Boucherie C, Martens E, Zwaenepoel O, Hassanzadeh-Ghassabeh G, Vandekerckhove J, Gevaert K, Fernandez LA, Sanders NN, Gettemans J (2013) A nanobody targeting the F-actin capping protein CapG restrains breast cancer metastasis. Breast Cancer Research 15(6): R116
Van Impe K, Hubert T, De Corte V, Vanloo B, Boucherie C, Vandekerckhove J, Gettemans J (2008) A new role for nuclear transport factor 2 and Ran: nuclear import of CapG. Traffic 9(5): 695-707
van Schouwenburg PA, Rispens T, Wolbink GJ (2013) Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nature Reviews Rheumatology 9(3): 164-72
Vecchiarelli A, Siracusa A, Monari C, Pietrella D, Retini C, Severini C (1994) Cytokine regulation of low-affinity IgE receptor (CD23) on monocytes from asthmatic subjects. Clinical and Experimental Immunology 97(2): 248-53
Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, Kastelein R, Kolk A, de Waal-Malefyt R, Ottenhoff TH (2004) Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proceedings of the National Academy of Sciences of the United States of America 101(13): 4560-5
Vogel DY, Heijnen PD, Breur M, de Vries HE, Tool AT, Amor S, Dijkstra CD (2014) Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. Journal of Neuroinflammation 11: 23
Vogt L, Schrimpf SP, Meskenaite V, Frischknecht R, Kinter J, Leone DP, Ziegler U, Sonderegger P (2001) Calsyntenin-1, a proteolytically processed postsynaptic membrane protein with a cytoplasmic calcium-binding domain. Molecular and Cellular Neurosciences 17(1): 151-66
Volianskis A, Kostner R, Molgaard M, Hass S, Jensen MS (2010) Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1deltaE9-deleted transgenic mice model of ss-amyloidosis. Neurobiology of Aging 31(7): 1173-87
Vosskuhl K, Greten TF, Manns MP, Korangy F, Wedemeyer J (2010) Lipopolysaccharide-mediated mast cell activation induces IFN-gamma secretion by NK cells. Journal of Immunology 185(1): 119-25

337
Vukman KV, Ravida A, Aldridge AM, O'Neill SM (2013) Mannose receptor and macrophage galactose-type lectin are involved in Bordetella pertussis mast cell interaction. Journal of Leukocyte Biology 94(3): 439-48
W
Wakahara S, Fujii Y, Nakao T, Tsuritani K, Hara T, Saito H, Ra C (2001) Gene expression profiles for Fc epsilon RI, cytokines and chemokines upon Fc epsilon RI activation in human cultured mast cells derived from peripheral blood. Cytokine 16(4): 143-52
Waldner C, Rempel O, Schutte F, Yanik M, Solomentsew N, Ryffel GU (2011) Double conditional human embryonic kidney cell line based on FLP and PhiC31 mediated transgene integration. BMC Research Notes 4: 420
Wang HW, Tedla N, Lloyd AR, Wakefield D, McNeil PH (1998) Mast cell activation and migration to lymph nodes during induction of an immune response in mice. Journal of Clinical Investigation 102(8): 1617-26
Wang J, Lindholt JS, Sukhova GK, Shi MA, Xia M, Chen H, Xiang M, He A, Wang Y, Xiong N, Libby P, Wang JA, Shi GP (2014a) IgE actions on CD4+ T cells, mast cells, and macrophages participate in the pathogenesis of experimental abdominal aortic aneurysms. EMBO Molecular Medicine 6(7): 952-69
Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, Ling L (2006) Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. Journal of Biological Chemistry 281(31): 22021-8
Wang N, Liang H, Zen K (2014b) Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Frontiers in Immunology 5: 614
Wang W, Li JJ, Foster PS, Hansbro PM, Yang M (2010) Potential therapeutic targets for steroid-resistant asthma. Current Drug Targets 11(8): 957-70
Wang WY, Tan MS, Yu JT, Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Annals of Translational Medicine 3(10): 136
Wang ZQ, Bapat AS, Rayanade RJ, Dagtas AS, Hoffmann MK (2001) Interleukin-10 induces macrophage apoptosis and expression of CD16 (FcgammaRIII) whose engagement blocks the cell death programme and facilitates differentiation. Immunology 102(3): 331-7
Warrington R, Watson W, Kim HL, Antonetti FR (2011) An introduction to immunology and immunopathology. Allergy, Asthma, and Clinical Immunology 7 Suppl 1: S1

338
Welle M (1997) Development, significance, and heterogeneity of mast cells with particular regard to the mast cell-specific proteases chymase and tryptase. Journal of Leukocyte Biology 61(3): 233-45
Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, Wang L, Kirkesseli S, Rocklin R, Bock B, Hamilton J, Ming JE, Radin A, Stahl N, Yancopoulos GD, Graham N, Pirozzi G (2013) Dupilumab in persistent asthma with elevated eosinophil levels. New England Journal of Medicine 368(26): 2455-66
Wesolowska-Andersen A, Seibold MA (2015) Airway molecular endotypes of asthma: dissecting the heterogeneity. Current Opinion in Allergy and Clinical Immunology 15(2): 163-8
Westbrook JA, Cairns DA, Peng J, Speirs V, Hanby AM, Holen I, Wood SL, Ottewell PD, Marshall H, Banks RE, Selby PJ, Coleman RE, Brown JE (2016) CAPG and GIPC1: Breast Cancer Biomarkers for Bone Metastasis Development and Treatment. Journal of the National Cancer Institute 108(4)
Whelan R, Kim C, Chen M, Leiter J, Grunstein MM, Hakonarson H (2004) Role and regulation of interleukin-1 molecules in pro-asthmatic sensitised airway smooth muscle. European Respiratory Journal 24(4): 559-67
Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, Hochstrasser DF (1999) Protein identification and analysis tools in the ExPASy server. Methods in Molecular Biology 112: 531-52
Witke W, Li W, Kwiatkowski DJ, Southwick FS (2001) Comparisons of CapG and gelsolin-null macrophages: demonstration of a unique role for CapG in receptor-mediated ruffling, phagocytosis, and vesicle rocketing. Journal of Cell Biology 154(4): 775-84
Witke W, Sharpe AH, Hartwig JH, Azuma T, Stossel TP, Kwiatkowski DJ (1995) Hemostatic, inflammatory, and fibroblast responses are blunted in mice lacking gelsolin. Cell 81(1): 41-51
Wonderling RS, Ghaffar A, Mayer EP (1996) Lipopolysaccharide-induced suppression of phagocytosis: effects on the phagocytic machinery. Immunopharmacology and Immunotoxicology 18(2): 267-89
Woodman L, Siddiqui S, Cruse G, Sutcliffe A, Saunders R, Kaur D, Bradding P, Brightling C (2008) Mast cells promote airway smooth muscle cell differentiation via autocrine up-regulation of TGF-beta 1. Journal of Immunology 181(7): 5001-7
Wu C, Wang S, Xian P, Yang L, Chen Y, Mo X (2016) Effect of Anti-TNF Antibodies on Clinical Response in Rheumatoid Arthritis Patients: A Meta-Analysis. BioMed Research International 2016: 7185708

339
Wu KC, Jabbar-Lopez ZK (2015) Omalizumab, an Anti-IgE mAb, receives approval for the treatment of chronic idiopathic/spontaneous urticaria. Journal of Investigative Dermatology 135(1): 13-5
Wynn TA, Barron L (2010) Macrophages: master regulators of inflammation and fibrosis. Seminars in Liver Disease 30(3): 245-57
X
Xia Y, Kelton CM, Xue L, Guo JJ, Bian B, Wigle PR (2013a) Safety of long-acting beta agonists and inhaled corticosteroids in children and adolescents with asthma. Therapeutic Advances in Drug Safety 4(6): 254-63
Xia YC, Harris T, Stewart AG, Mackay GA (2013b) Secreted factors from human mast cells trigger inflammatory cytokine production by human airway smooth muscle cells. International Archives of Allergy and Immunology 160(1): 75-85
Xia YC, Sun S, Kuek LE, Lopata AL, Hulett MD, Mackay GA (2011) Human mast cell line-1 (HMC-1) cells transfected with FcepsilonRIalpha are sensitive to IgE/antigen-mediated stimulation demonstrating selectivity towards cytokine production. International Immunopharmacology 11(8): 1002-11
Xie H, Ye M, Feng R, Graf T (2004) Stepwise reprogramming of B cells into macrophages. Cell 117(5): 663-76
Xing Z, Zganiacz A, Santosuosso M (2000) Role of IL-12 in macrophage activation during intracellular infection: IL-12 and mycobacteria synergistically release TNF-alpha and nitric oxide from macrophages via IFN-gamma induction. Journal of Leukocyte Biology 68(6): 897-902
Xiong S, Rodgers K (1997) Effects of malathion metabolites on degranulation of and mediator release by human and rat basophilic cells. Journal of Toxicology and Environmental Health 51(2): 159-75
Y
Yamaguchi M, Hirai K, Komiya A, Miyamasu M, Furumoto Y, Teshima R, Ohta K, Morita Y, Galli SJ, Ra C, Yamamoto K (2001) Regulation of mouse mast cell surface Fc epsilon RI expression by dexamethasone. International Immunology 13(7): 843-51

340
Yamanaka H (2015) TNF as a Target of Inflammation in Rheumatoid Arthritis. Endocrine, Metabolic & Immune Disorders Drug Targets 15(2): 129-34
Yang J, Zhang L, Yu C, Yang XF, Wang H (2014) Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomarker Research 2(1): 1
Yang M, Kumar RK, Foster PS (2009) Pathogenesis of steroid-resistant airway hyperresponsiveness: interaction between IFN-gamma and TLR4/MyD88 pathways. Journal of Immunology 182(8): 5107-15
Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, Reagan JD (2010) G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 86(1): 1-5
Yilma AN, Singh SR, Fairley SJ, Taha MA, Dennis VA (2012) The anti-inflammatory cytokine, interleukin-10, inhibits inflammatory mediators in human epithelial cells and mouse macrophages exposed to live and UV-inactivated Chlamydia trachomatis. Mediators of Inflammation 2012: 520174
Ying S, Barata LT, Meng Q, Grant JA, Barkans J, Durham SR, Kay AB (1998) High-affinity immunoglobulin E receptor (Fc epsilon RI)-bearing eosinophils, mast cells, macrophages and Langerhans' cells in allergen-induced late-phase cutaneous reactions in atopic subjects. Immunology 93(2): 281-8
Yong LC (1997) The mast cell: origin, morphology, distribution, and function. Experimental and Toxicologic Pathology 49(6): 409-24
Young CL, Southwick FS, Weber A (1990) Kinetics of the interaction of a 41-kilodalton macrophage capping protein with actin: promotion of nucleation during prolongation of the lag period. Biochemistry 29(9): 2232-40
Yu FX, Johnston PA, Sudhof TC, Yin HL (1990) gCap39, a calcium ion- and polyphosphoinositide-regulated actin capping protein. Science 250(4986): 1413-5
Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ (2006) Mast cells can promote the development of multiple features of chronic asthma in mice. Journal of Clinical Investigation 116(6): 1633-41

341
Z
Zhang B, Alysandratos KD, Angelidou A, Asadi S, Sismanopoulos N, Delivanis DA, Weng Z, Miniati A, Vasiadi M, Katsarou-Katsari A, Miao B, Leeman SE, Kalogeromitros D, Theoharides TC (2011) Human mast cell degranulation and preformed TNF secretion require mitochondrial translocation to exocytosis sites: relevance to atopic dermatitis. Journal of Allergy and Clinical Immunology 127(6): 1522-31 e8
Zhang JG, Czabotar PE, Policheni AN, Caminschi I, San Wan S, Kitsoulis S, Tullett KM, Robin AY, Brammananth R, van Delft MF, Lu J, O'Reilly LA, Josefsson EC, Kile BT, Chin WJ, Mintern JD, Olshina MA, Wong W, Baum J, Wright MD, Huang DC, Mohandas N, Coppel RL, Colman PM, Nicola NA, Shortman K, Lahoud MH (2012) The Dendritic Cell Receptor Clec9A Binds Damaged Cells via Exposed Actin Filaments. Immunity 36(4): 646-57
Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS, Zhang J (2005) Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB Journal 19(6): 533-42
Zhang X, Edwards JP, Mosser DM (2009) The expression of exogenous genes in macrophages: obstacles and opportunities. Methods in Molecular Biology 531: 123-43
Zhang X, Wang Y, Dong H, Xu Y, Zhang S (2016) Induction of Microglial Activation by Mediators Released from Mast Cells. Cellular Physiology and Biochemistry 38(4): 1520-31
Zhang Y, Vorobiev SM, Gibson BG, Hao B, Sidhu GS, Mishra VS, Yarmola EG, Bubb MR, Almo SC, Southwick FS (2006) A CapG gain-of-function mutant reveals critical structural and functional determinants for actin filament severing. EMBO Journal 25(19): 4458-67
Zhou J, Liu DF, Liu C, Kang ZM, Shen XH, Chen YZ, Xu T, Jiang CL (2008) Glucocorticoids inhibit degranulation of mast cells in allergic asthma via nongenomic mechanism. Allergy 63(9): 1177-85
Zhou L, Cao X, Fang J, Li Y, Fan M (2015) Macrophages polarization is mediated by the combination of PRR ligands and distinct inflammatory cytokines. International Journal of Clinical and Experimental Pathology 8(9): 10964-74
Zhou Y, Yang Y, Warr G, Bravo R (1999) LPS down-regulates the expression of chemokine receptor CCR2 in mice and abolishes macrophage infiltration in acute inflammation. Journal of Leukocyte Biology 65(2): 265-9
Zhu W, Smith JW, Huang CM (2010) Mass spectrometry-based label-free quantitative proteomics. Journal of Biomedicine and Biotechnology 2010: 840518

342
Zhu WY, Hunag YY, Liu XG, He JY, Chen DD, Zeng F, Zhou JH, Zhang YK (2012) Prognostic evaluation of CapG, gelsolin, P-gp, GSTP1, and Topo-II proteins in non-small cell lung cancer. Anatomical Record (Hoboken) 295(2): 208-14
Zidek Z, Anzenbacher P, Kmonickova E (2009) Current status and challenges of cytokine pharmacology. British Journal of Pharmacology 157(3): 342-61
Zittermann SI, Issekutz AC (2006) Basic fibroblast growth factor (bFGF, FGF-2) potentiates leukocyte recruitment to inflammation by enhancing endothelial adhesion molecule expression. American Journal of Pathology 168(3): 835-46
Zomer-Kooijker K, Uiterwaal CS, van der Gugten AC, Wilbrink B, Bont LJ, van der Ent CK (2014) Decreased lung function precedes severe respiratory syncytial virus infection and post-respiratory syncytial virus wheeze in term infants. European Respiratory Journal 44(3): 666-74
Zughaier SM, Zimmer SM, Datta A, Carlson RW, Stephens DS (2005) Differential induction of the toll-like receptor 4-MyD88-dependent and -independent signaling pathways by endotoxins. Infection and Immunity 73(5): 2940-50

Minerva Access is the Institutional Repository of The University of Melbourne
Author/s:Heng, Patrick
Title:Characterisation of macrophage capping protein as a novel inflammatory mediator
Date:2017
Persistent Link:http://hdl.handle.net/11343/191467
Terms and Conditions:Terms and Conditions: Copyright in works deposited in Minerva Access is retained by thecopyright owner. The work may not be altered without permission from the copyright owner.Readers may only download, print and save electronic copies of whole works for their ownpersonal non-commercial use. Any use that exceeds these limits requires permission fromthe copyright owner. Attribution is essential when quoting or paraphrasing from these works.