cell biology of gingival wound healing
TRANSCRIPT

Periodontology 2000, Vol. 24, 2000, 127–152 Copyright C Munksgaard 2000Printed in Denmark ¡ All rights reserved
PERIODONTOLOGY 2000ISSN 0906-6713
Cell biology ofgingival wound healingLARI HAKKINEN, VELI-JUKKA UITTO & HANNU LARJAVA
An overview
Wound healing is a critical process for the organism.The attempt of this chapter is to critically summarizethe recent advancements in the wound-healing pro-cess. Because of the excessive amount of infor-mation (search with the key words wound healingproduced 2052 citations within the last 12 months),we will focus on two major events in wound healing,namely re-epithelialization and granulation tissueformation, leaving out many other key events suchas clot formation and angiogenesis. At the end, wewill also discuss the special features of wound heal-ing in oral cavity. It is commonly stated that oralwounds heal better than other wounds such as der-mal wounds. We will explore evidence supportingthis assumption.
Most of the data presented in this chapter refer tostudies with gingival and dermal full-thicknesswounds and no special attempts have been madeto focus on the healing in tooth-gingiva interphasebecause only a few studies are available to reportspecial molecular events in humans. In addition,several previous articles have addressed periodontalwound healing and regeneration in great detail (114,276). It is reasonable to assume, however, that thebasic cell biological events of wound healing will fol-low the same principles at the tooth-gingiva inter-phase, at least at the supracrestal locations. We willalso try to make several back-and-forth citations be-tween in vivo and in vitro studies. This is necessarybecause functional studies have been difficult toperform with animal models of wound healing in amanner that would allow meaningful conclusions tobe drawn. In vitro studies with cultured keratino-cytes and fibroblasts are still the only way to explorefunctions of various molecules. Recent molecular bi-ology techniques have made it possible to eliminatethe expression of molecules of interest and investi-gate how the function is changed consequently. Invivo studies have dealt with immunolocalization of
127
extracellular molecules and their expression using insitu hybridization. In addition, transgenic animalsprovide information how wound healing is altered inthe absence or following overexpression of a knownmolecule (153). Many of these studies have providednew critical information but others have resulted inconfusing results, such as showing no alterationseven though a molecule has been thought to be criti-cally important for wound healing. It is believed thatin these cases there are other related proteins thatcan provide similar functions and compensate forthe absence of the studied molecule. Wound healinghas become well protected during evolution becauseof its critical importance. Numerous molecules ap-pear to overlap each other’s functions. Duringwound healing, several molecules that are usuallypresent only during embryonal development arefound in the granulation tissue. Epithelial cells startto express extracellular matrix receptors that are notnormally present in the resting epithelium. Fibro-blastic cells with a special phenotype are found inthe healing granulation tissue. Where they comefrom is still largery unknown but possible sourceswill be discussed in this chapter. Expression of vari-ous proteins by the resident cells is also influencedby the new environment. For example, in fibroblaststhe expression of hundreds of genes is altered by theexposure to serum alone. Proteolytic enzymes re-lease growth factors from the wounded basementmembrane and connective tissue matrix. Matrixdegradation produces biologically active peptidesfrom the matrix proteins that could have specificfunctions in tissue repair. Clarification of the com-plex interplay between new matrix, growth factors,matrix degradation products and cells during woundhealing will be a challenging task. This review fo-cuses on re-epithelialization and granulation tissueformation with the special emphasis on the inte-grins, since cell adhesion serves as a foundation forcell migration, matrix turnover and differentiationduring wound healing.

Hakkinen et al.
Integrins
Integrins play key roles in re-epithelialization andgranulation tissue formation during wound healingthrough their function in cell adhesion and sig-naling. A brief overview is, therefore, presented de-scribing the structure and function of integrins thatwe hope will help the reader to better understandhow they participate in the regulation of woundhealing. Integrins are cell surface–associated dimericglycoproteins that function as cell-to-extracellularmatrix adhesion receptors (4, 16, 160). Throughbinding to the extracellular matrix proteins integrinsmediate information transfer from the extracellularmatrix to the cell interior leading to alterations incell functions and ultimately in cell behaviour (100,160). Integrins are known to play an important rolein regulating a wide range of cell functions duringgrowth, development, differentiation, and immuneresponse (4). Integrins are composed of a single aand a single b subunit that are non-covalently linkedto each other. At least 17 different a and 8 b subunitsare currently known. These subunits can variouslycombine to form more than 23 different cell surfacereceptors that have distinct ligand-binding speci-ficities. Both a and b subunits are transmembraneglycoproteins that cooperate in integrin binding toligands. Ligand binding causes clustering of the re-ceptors, which leads to cytoskeletal organization andsignaling (Fig. 1) (160). Integrin-binding sites are tar-gets for therapeutic applications aiming at blockingintegrin function. Domain crystal structures are
Fig. 1. Integrins and cell signalling. Integrins mediate celladhesion to extracellular matrix proteins (ECM) leadingto organization of cytoskeleton and signaling. Integrinsalso collaborate with growth factors in regulation of cellgrowth. Some integrins can also directly activate growthfactors such as transforming growth factor b1 (TGFb1).Proteolytic enzymes are also found in cell adhesion sitesallowing cells to detach and subsequently migrate.
128
being used for designing new molecules that havehigh affinity to the binding site but are irrelevant tothe original peptide sequence. Commercial productsare already available for cardiovascular applications(146). Several other compounds are in clinical trialsand many other similar products will follow and maybe used for periodontal applications in the future.
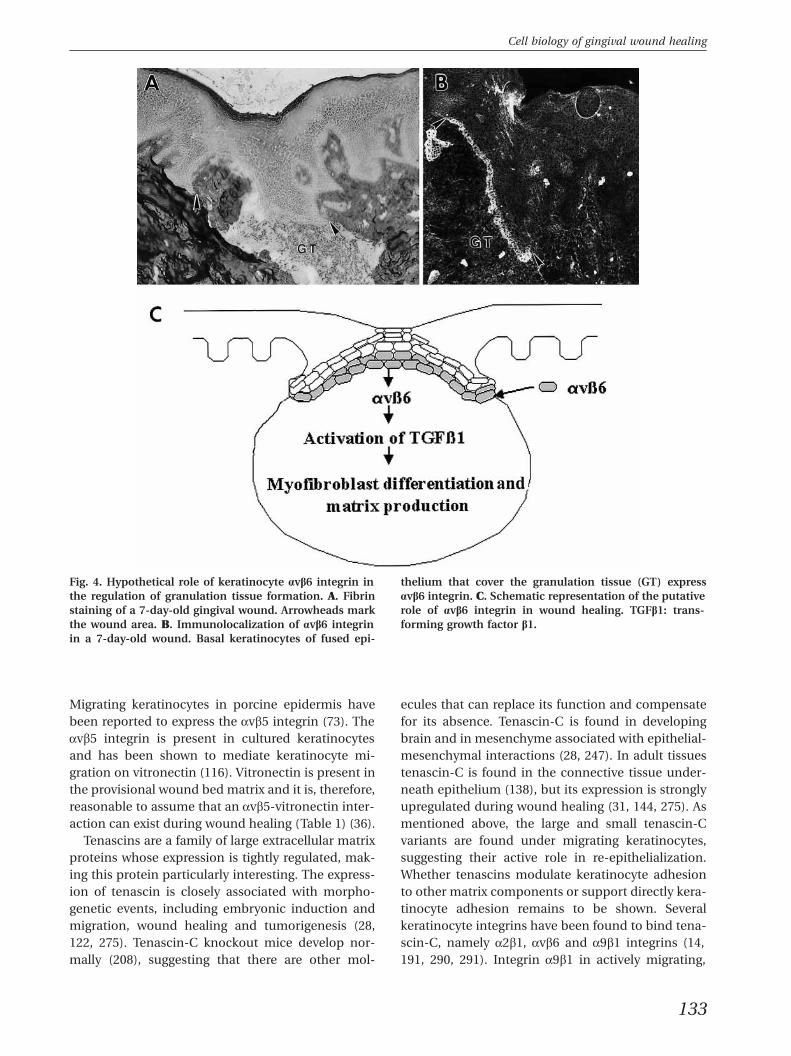
The number of proteins known to associate withintegrins is rapidly increasing (96). Integrin associ-ated protein, transmembrane-4 superfamily, growthfactor receptors and urokinase-type plasminogen ac-tivator receptor appear to have a regulatory functionon integrins. Growth factor receptors accumulate inthe same structures as integrins and regulate inte-grin functions (Fig. 1) (161). In the context of woundhealing, the synergy between integrin and growthfactor receptors is probably a key process in theregulation of cell proliferation (100). Integrins avb1and avb6 can also bind growth factors such as trans-forming growth factor b1 through its latency-acti-vated peptide that contains an Arg-Gly-Asp (RGD)peptide sequence (166, 167). It has recently beenshown that epithelial cells can bind and also activatetransforming growth factor b1 through avb6 integrin(Fig. 1); this mechanism may play an important rolein the connective tissue bridge formation under-neath the epithelium that has covered the wound. Itis evident that the epithelium has a much more ac-tive role in the regulation of connective tissue forma-tion than previously assumed.
Epithelial wound healing
Re-epithelialization
Wound healing is a complex phenomenon that in-volves series of controlled events including the for-mation of a provisional extracellular matrix that ismainly composed of fibrin, fibronectin and vitronec-tin and the migration of epithelial cells from the edg-es of the wound (33, 108). After the epithelium hasbeen disrupted by tissue injury, re-epithelializationmust occur as rapidly as possible in order to re-es-tablish tissue integrity. Keratinocytes start movinginto the defect about 24 hours after the injury (277).Epithelial cells from residual epithelial structuresdissolve their hemidesmosomal connections and de-tach from basement membrane and move quicklyacross the wound defect. Later, as the re-epithelializ-ation proceeds, the proliferation of keratinocytesthat feed the advancing epithelial edge becomesmore important. It is still somewhat unclear whichcells in the epithelium first move into the wound.

Cell biology of gingival wound healing
There is some evidence that the suprabasal keratino-cytes are the first migratory cells sliding over thebasal keratinocytes. Epithelial cell migration on theexposed connective tissue matrix underneath thefibrin-fibronectin clot has been commonly de-scribed. In small gingival wounds (Fig. 2), however,the keratinocytes cut their way directly through theclot and may not interact with the connective tissuematrix at all (132). Migrating keratinocytes are highlyphagocytic, allowing them to penetrate throughtissue debris or the clot (277). Degradation of the fi-brin clot appears to be critical for wound healing,since wounds in animals that lack the plasminogengene do not re-epithelialize (199). It seems likely thatintegrins play a role in the fibrin clot removal. Mi-grating cells must be able to focalize the proteolysisinto the leading edge of epithelium (227). This couldbe done by activation of proteolytic enzymes at spe-cific sites at the cell membrane. It has been foundthat urokinase type plasminogen activator receptoris able to associate with integrins (283). This is anexample how a cell is able to focalize fibrinolysis byplasmin and promote subsequent migration by inte-grins. Modulation of other matrix componets may
Fig. 2. Stages of healing of full thickness gingival wounds (about 3 mm deep and 2 mm wide). FC: fibrin clot; GT:granulation tissue.
129
also be necessary. Migrating keratinocytes expressmatrix metalloprotease-9 (type IV collagenase), ma-trix metalloprotease-1 (interstitial collagenase) andmatrix metalloprotease-10 (stromelysin), which allmay be required if the cells encounter the exposedmatrix (207, 210, 266). Blocking matrix metalloprote-ase activity prevents keratinocyte migration into thewounds in cell culture (147). This proteolytic modu-lation of the matrix underneath migrating cells mustbe well controlled, since overexpression of matrixmetalloproteases is a common finding in nonhealingchronic wounds (187). In chronic inflammation, ma-trix metalloproteases such as matrix metalloprotea-se-9 produced by keratinocytes may bind to the cellsurface and into the extracellular matrix resulting ina delayed clearance (147). Thus, regulation of matrixmetalloproteases and their inhibitors must be wellbalanced for normal wound healing (266).
During wound healing, keratinocytes function torapidly cover the exposed connective tissue (236,277). This process depends upon a variety of interac-tions between cells and the extracellular matrix. Asthe basal keratinocytes at the wound margin are ex-posed to the new provisional matrix, the phenotype
Hakkinen et al.
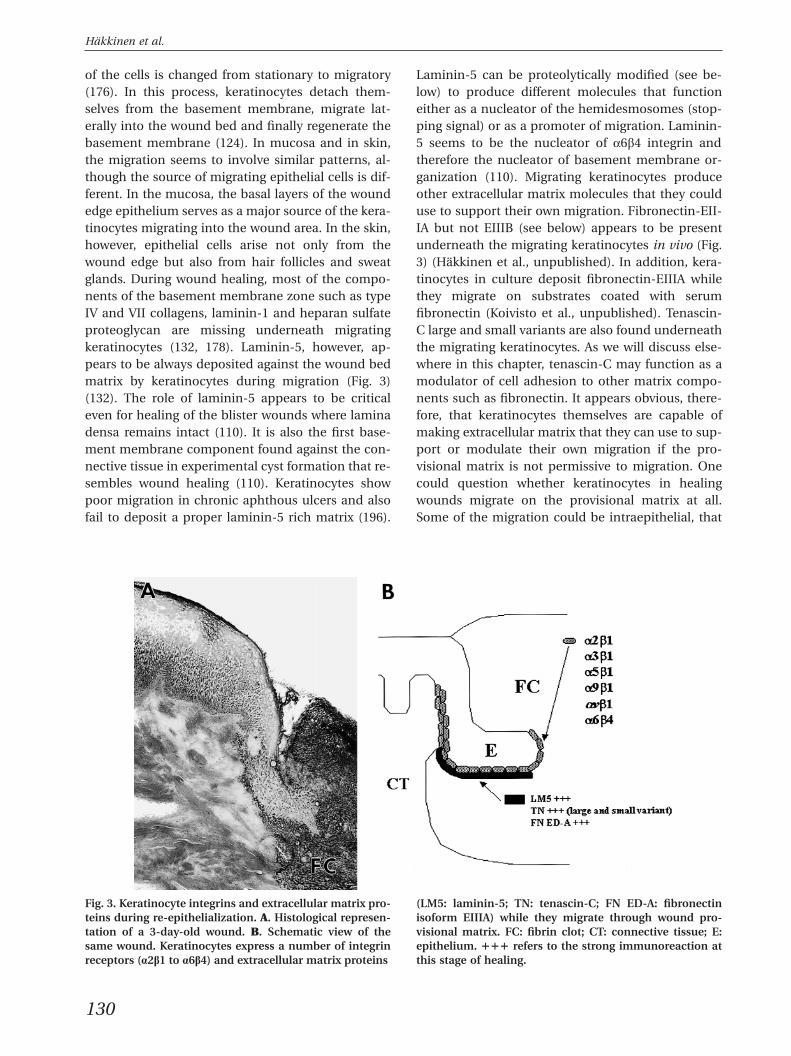
of the cells is changed from stationary to migratory(176). In this process, keratinocytes detach them-selves from the basement membrane, migrate lat-erally into the wound bed and finally regenerate thebasement membrane (124). In mucosa and in skin,the migration seems to involve similar patterns, al-though the source of migrating epithelial cells is dif-ferent. In the mucosa, the basal layers of the woundedge epithelium serves as a major source of the kera-tinocytes migrating into the wound area. In the skin,however, epithelial cells arise not only from thewound edge but also from hair follicles and sweatglands. During wound healing, most of the compo-nents of the basement membrane zone such as typeIV and VII collagens, laminin-1 and heparan sulfateproteoglycan are missing underneath migratingkeratinocytes (132, 178). Laminin-5, however, ap-pears to be always deposited against the wound bedmatrix by keratinocytes during migration (Fig. 3)(132). The role of laminin-5 appears to be criticaleven for healing of the blister wounds where laminadensa remains intact (110). It is also the first base-ment membrane component found against the con-nective tissue in experimental cyst formation that re-sembles wound healing (110). Keratinocytes showpoor migration in chronic aphthous ulcers and alsofail to deposit a proper laminin-5 rich matrix (196).
Fig. 3. Keratinocyte integrins and extracellular matrix pro- (LM5: laminin-5; TN: tenascin-C; FN ED-A: fibronectinteins during re-epithelialization. A. Histological represen- isoform EIIIA) while they migrate through wound pro-tation of a 3-day-old wound. B. Schematic view of the visional matrix. FC: fibrin clot; CT: connective tissue; E:same wound. Keratinocytes express a number of integrin epithelium. πππ refers to the strong immunoreaction atreceptors (a2b1 to a6b4) and extracellular matrix proteins this stage of healing.
130
Laminin-5 can be proteolytically modified (see be-low) to produce different molecules that functioneither as a nucleator of the hemidesmosomes (stop-ping signal) or as a promoter of migration. Laminin-5 seems to be the nucleator of a6b4 integrin andtherefore the nucleator of basement membrane or-ganization (110). Migrating keratinocytes produceother extracellular matrix molecules that they coulduse to support their own migration. Fibronectin-EII-IA but not EIIIB (see below) appears to be presentunderneath the migrating keratinocytes in vivo (Fig.3) (Hakkinen et al., unpublished). In addition, kera-tinocytes in culture deposit fibronectin-EIIIA whilethey migrate on substrates coated with serumfibronectin (Koivisto et al., unpublished). Tenascin-C large and small variants are also found underneaththe migrating keratinocytes. As we will discuss else-where in this chapter, tenascin-C may function as amodulator of cell adhesion to other matrix compo-nents such as fibronectin. It appears obvious, there-fore, that keratinocytes themselves are capable ofmaking extracellular matrix that they can use to sup-port or modulate their own migration if the pro-visional matrix is not permissive to migration. Onecould question whether keratinocytes in healingwounds migrate on the provisional matrix at all.Some of the migration could be intraepithelial, that

Cell biology of gingival wound healing
Table 1. Epithelial integrins and their ligands during wound healing
Integrin Ligand Ligand source
a2b1 Collagen Connective tissueTenascin (?) Keratinocytes, granulation tissue
a3b1, a6b4 Laminin-5 Keratinocytes
a5b1, avb1 Serum fibronectin SerumFibronectin EIII/A Keratinocytes
a9b1 Tenascin-C Keratinocytes, granulation tissue
avb5 Vitronectin Serum, connective tissue
avb6 Fibronectin EIIIA, fibronectin EIIIB Keratinocytes, granulation tissueTransforming growth factor b1 Keratinocytes, macrophages, connective tissue cells
is, suprabasal keratinocytes sliding on basal cells atthe leading edge. After reaching the provisional ma-trix, migrating cells could stop moving and becomebasal stationary keratinocytes. Two models for epi-thelial sheet migration in wounds have been pro-posed (236). In the sliding model the cells at themargin are active and pull the cells behind them. Ina leap frog model, the migrating cells become non-motile when they adhere to the provisional woundmatrix. Then the cells behind the leading edge crawlover the newly attached keratinocytes. The exactmechanism by which multilayered epithelium mi-grates remains to be discovered, but it is possiblethat all three mechanisms described above couldfunction depending on the defect and the state ofthe healing. Embryonic re-epithelialization appearsto occur through a different mechanism. Epidermalcells do not crawl by extending lamellopodia into thewound as they do during, for example, gingivalwound healing (132). They rather use the actinfilament cables to pull wound edges together (154).
In small gingival wounds (2–3 mm distance be-tween the wound edges), the formation of new base-ment membrane starts when the migrating epithelialsheets have reached each other and fused (132). Thenucleation of the basement membrane appears tooccur at multiple sites at the same time. It has beenreported that in epidermal wounds the deposition ofbasement membrane starts from the wound marginin a zipper-like fashion (35). Whether there is a realdifference in the sequence of basement membraneorganization between gingival and epidermal heal-ing has not been shown. The reorganization of thebasement membrane is complete at 4 weeks atwhich time the localization of all basement mem-brane components such as type IV and VII collagens,laminin-1 and heparan sulfate proteoglycan appearnormal (Hakkinen et al., unpublished). Keratinocyteshave been shown to synthesize these main compo-
131
nents of the basement membrane zone (236). Sig-nificant portion of the basement membrane compo-nents are, however, synthesized by the woundfibroblasts (65). It is obvious that the cross-talk be-tween the wound keratinocytes and fibroblasts iscrucial during the reorganization of the basementmembrane zone.
Extracellular matrix interactions of keratinocytesduring re-epithelialization
As the epithelial cells undergo major environmentalchanges during the wound healing, they have to ad-just their cell surface receptors for the new demands.During wound healing keratinocytes have to migrateon the provisional matrix different from theirstationary matrix. This requires a change of express-ion levels and distribution of old integrins and ex-pression of some totally new integrins (Table 1, Fig.3) (131). As discussed throughout this chapter,fibronectin is a critical early component of the clotand the forming granulation tissue. Initially, theblood clot contains plasma fibronectin, which islater replaced by cellular fibronectin produced bykeratinocytes, fibroblasts and macrophages (33).Fibronectin can vary structurally in a tissue specificmanner by alternative splicing of three regions,namely EIIIA, EIIIB and V(IIICS) (284). Expression ofthe alternatively spliced isoforms of cellularfibronectin containing either the EIIIA or EIIIB mod-ules, which are omitted from the plasma fibronectin,are upregulated specifically during embryonic devel-opment and upon wounding (Hakkinen et al., un-published). As discussed above, EIIIA fibronectin isexpressed by migrating keratinocytes in wounds(Hakkinen et al., unpublished). The EIIIB fibronectinthat is expressed in embryonal tissues but not nor-mally in adult connective tissue is strongly upregu-lated during granulation tissue formation (Hakkinen

Hakkinen et al.
et al., unpublished, see below). Plasma fibronectinand the isoforms containing the EIIIA or EIIIB mod-ules may have different properties in cell adhesionand migration during wound repair. Human woundkeratinocytes express two major receptors that areable to bind to fibronectins, namely a5b1 and avb6integrins (Table 1) (91, 132). The classical fibronectinbinding receptor is a5b1 integrin that binds to theRGD sequence of the fibronectin molecule (1, 229).Integrin a5b1 is considered to be a specialized recep-tor for fibronectin and it mediates fibronectin-matrixassembly, cell adhesion and migration on fibronec-tin (1). Another fibronectin RGD site binding recep-tor in epithelial cells is the avb6 integrin (17). Inte-grin avb6 is expressed exclusively in epithelial cells,and it functions as a receptor for the extracellularmatrix proteins fibronectin (17, 270) and tenascin(191). Neither of the main fibronectin receptors,a5b1 or avb6 integrin, appears to be present in kera-tinocytes residing in the resting epithelium, but theexpression of both of these integrins can be inducedby wounding or placing epithelial cells in cell culture(Fig. 3). These two fibronectin receptors appear inthe wound keratinocytes at different times, a5b1during early migration and avb6 during reorganiza-tion of the basement membrane zone suggestingthat their function may be different (91, Hakkinenet al., unpublished). As mentioned earlier, epithelialintegrin avb6 may play a central role in the reorgan-ization of the basement membrane zone (Fig. 4) as itis expressed by wound keratinocytes after epithelialsheets have joined and because it is able to activatetransforming growth factor b (167). In culture, kera-tinocytes can switch between avb6 and a5b1 inte-grins in fibronectin binding if one receptor is un-functional (118).
Laminin-5 is found in the basement membraneof skin and other epithelial tissues and serves as acomponent of anchoring filaments that spanthrough the basement membrane (19, 203). Theextracellular domain of a6b4 integrin interacts withlaminin-5 (173, 203). This integrin is known to linkthe basal keratinocytes to the underlying basementmembrane, and this link mainly relies on the inter-action with laminin-5 in the anchoring filaments.The functional importance of a6b4 integrin is sub-stantiated by experiments in knockout mice that lackeither a6 or b4 integrin and cannot form hemides-mosomes, which results in blistering of skin. Lamin-in-5 is also recognized by a3b1 integrin (19). Trans-genic mice lacking a3 integrin also develop localizedblistering of the skin (49). In mice and man, a3b1integrin is occasionally found at the basal surface of
132
the basal keratinocytes, suggesting that a3b1 inte-grin collaborates with a6b4 integrin in laminin-5binding and they may be functionally interchange-able in some circumstances. Integrin a6b4 is presentin migratory epithelial cells although hemidesmo-somes are absent (132). One possible scenario couldbe connected to the functions of laminin-5 duringwound healing. Intact laminin-5 appears to be a mo-tility factor for keratinocytes, but when proteo-lytically processed it starts to function as the nu-cleator of hemidesmosomes and therefore promotesthe formation of basement membrane structures(78). This is a good example demonstrating howcomplex the wound-healing process is, since simpleproteolytic cleavage can totally reverse the functionof a protein important for the cell migration process.
Expression of b1 integrins is strongly upregulatedin keratinocytes during wound healing (20, 73, 97,108, 132). Both a2b1 and a3b1 integrins seem to beequally expressed. Integrin a3b1 is known to be ableto bind both fibronectin and laminin-5, both ofwhich are present in the provisional matrix (19, 132)although its role as a fibronection receptor in kera-tinocytes is doubtful. Integrin a2b1 is known to beable to bind many types of collagens (Table 1), suchas type I, III, V and VI that serve as a substrate formigrating keratinocytes. Direct interactions of kera-tinocytes with type IV collagen are rare but may oc-cur, for example, during healing of blister wounds,in which case keratinocytes migrate on the compo-nents of lamina densa. Keratinocytes are more likelyto interact with fibrillar collagens, such as types I,III and V collagens during healing of full-thicknesswounds. In this case, interaction of keratinocyteswith collagens is mediated via a2b1 integrin. Inte-grins are also involved in the regulation of extracellu-lar matrix degradation. Cells in the tissue adapt totheir changing environment and sense alterations inthe matrix through integrins on the cell surface. Rec-ognition of an altered matrix by integrins leadchanges in gene expression. For example, epithelialand connective tissue cells attach to collagen bya2b1 integrin which process induces the expressionof collagenase (matrix metalloproteinase-1) that cancleave the exposed collagen matrix and facilitate themigration of wound keratinocytes (186, 187, 278).
Vitronectin is an abundant protein present inblood plasma, extracellular matrices and fibrin clots.Vitronectin is known to be an active cell adhesionmediator as well as playing a role in cell migrationand invasion (190). The avb1, avb3, avIIb3 and avb5integrins are all vitronectin receptors binding to theRGD cell attachment sequence of the protein (24).
Cell biology of gingival wound healing
Fig. 4. Hypothetical role of keratinocyte avb6 integrin in thelium that cover the granulation tissue (GT) expressthe regulation of granulation tissue formation. A. Fibrin avb6 integrin. C. Schematic representation of the putativestaining of a 7-day-old gingival wound. Arrowheads mark role of avb6 integrin in wound healing. TGFb1: trans-the wound area. B. Immunolocalization of avb6 integrin forming growth factor b1.in a 7-day-old wound. Basal keratinocytes of fused epi-
Migrating keratinocytes in porcine epidermis havebeen reported to express the avb5 integrin (73). Theavb5 integrin is present in cultured keratinocytesand has been shown to mediate keratinocyte mi-gration on vitronectin (116). Vitronectin is present inthe provisional wound bed matrix and it is, therefore,reasonable to assume that an avb5-vitronectin inter-action can exist during wound healing (Table 1) (36).
Tenascins are a family of large extracellular matrixproteins whose expression is tightly regulated, mak-ing this protein particularly interesting. The express-ion of tenascin is closely associated with morpho-genetic events, including embryonic induction andmigration, wound healing and tumorigenesis (28,122, 275). Tenascin-C knockout mice develop nor-mally (208), suggesting that there are other mol-
133
ecules that can replace its function and compensatefor its absence. Tenascin-C is found in developingbrain and in mesenchyme associated with epithelial-mesenchymal interactions (28, 247). In adult tissuestenascin-C is found in the connective tissue under-neath epithelium (138), but its expression is stronglyupregulated during wound healing (31, 144, 275). Asmentioned above, the large and small tenascin-Cvariants are found under migrating keratinocytes,suggesting their active role in re-epithelialization.Whether tenascins modulate keratinocyte adhesionto other matrix components or support directly kera-tinocyte adhesion remains to be shown. Severalkeratinocyte integrins have been found to bind tena-scin-C, namely a2b1, avb6 and a9b1 integrins (14,191, 290, 291). Integrin a9b1 in actively migrating,

Hakkinen et al.
and avb6 integrin in nonmigrating wound keratino-cytes, colocalize in the same area as tenascins (Table1) (Hakkinen et al., unpublished). These two epi-thelial integrins may, therefore, have a different rolein cell signaling caused by tenascin-C variants. Up-regulation of a9b1 and tenascin-C is also observedduring corneal wound healing (239).
Transforming growth factor b andre-epithelialization
In wounds, specific regulatory signals are requiredfor normal repair. Currently it is believed that trans-forming growth factors-b have a central role in thewound healing process. Transforming growth factorsb are a family of polypeptides that have multipleregulatory actions in cell growth, differentiation, anddevelopmental processes (155, 234, 244). Threehighly homologous transforming growth factor-bgenes have been identified in mammals, represent-ing transforming growth factor b1, transforminggrowth factor b2 and transforming growth factor b3polypeptides. Human keratinocytes of intact skin ex-press transforming growth factor b3 (216). Thisgrowth factor seems to play an important role in epi-dermal maintenance. In animal studies, only smallamounts of transforming growth factor b2 and trans-forming growth factor b3 messenger RNA have beenfound in keratinocytes of intact dermis (215). Alltransforming growth factor b isoforms are found inhealing wounds of animals (111, 137). However,Schmid et al. (216) did not find any detectable levelsof transforming growth factor b2 messenger RNA inhuman wound keratinocytes. Wound fibroblasts andmacrophages are known to express both trans-forming growth factor b1 and transforming growthfactor b2 in cells adjacent to the wound (175, 267).Transforming growth factor b1 is the major isoformin wound keratinocytes, and induction of trans-forming growth factor b1 in migrating keratinocytesis crucial for the successful re-epithelialization ofskin wounds. In vitro studies have shown that trans-forming growth factor b3 inhibits the growth of pri-mary human keratinocytes, while transforminggrowth factor b1 seems to stimulate keratinocytemotility by switching the cells from the differentiat-ing to regenerative phenotype (152) and by inducingtheir production of fibronectin (172) and laminin-5(110). In cultured human keratinocytes, trans-forming growth factor b1 has also been shown to in-crease the levels of messenger RNA for some inte-grins, such as a5, av and b5, that may facilitate mi-gration of wound keratinocytes (73, 292). In cultured
134
HaCaT keratinocytes, transforming growth factor b1and 2 specifically stimulate the expression of avb6integrin, and promote both haptotactic and epi-thelial sheet migration (118). As we have discussedearlier, the most important role for avb6 integrinmay be the activation of transforming growth factorb1 that then promotes the formation of the connec-tive tissue bridge formation under the joined woundepithelium. The first collagen fibrils are, indeed, laiddown under the epithelium in areas where the ex-pression of avb6 is strong and transforming growthfactor b1 is present in active form. Transforminggrowth factor b1 may also stimulate the proliferationof wound keratinocytes at the wound margins in-directly by inducing the expression of other polypep-tide growth factors, such as platelet-derived growthfactor (3, 22).
In addition to transforming growth factors b,many other growth factors regulate wound healing.This review will not try to list all possible factors in-volved in re-epithelialization and the readers are ad-vised to review other sources for the roles of platelet-derived growth factor, epidermal growth factor, kera-tinocyte growth factor, hepatocyte growth factor andothers in wound healing.
Connective tissue repair
Activation of fibroblasts
The resolution of a tissue defect after wounding re-quires not only re-epithelialization but also prolifer-ation and migration of fibroblasts into the woundbed where they participate in the formation ofgranulation tissue and synthesize, deposit and reor-ganize various extracellular matrix molecules.Wound repair involves phenotypic change of fibro-blasts from quiescent to proliferating cells, and sub-sequently to migratory, and then to stationary matrixproducing and contractile cells. In normal connec-tive tissue, fibroblasts reside in a quiescent state andhave a slow proliferation rate and metabolic activity.Upon wounding, fibroblasts become activated andchange their gene expression. This is supported by arecent finding with quiescent fibroblasts that show arapid change in expression of hundreds of geneswhen stimulated by serum. Interestingly, many ofthe serum-activated genes are known to be involvedin the physiology of wound repair, including controlof cell cycle and proliferation, coagulation andhemostasis, inflammation, angiogenesis, tissue re-modeling, cytoskeletal reorganization and re-epi-thelialization (107). This indicates that exposure of

Cell biology of gingival wound healing
fibroblasts to serum that is present in the blood clotin the wound initiates not only a rapid generalstimulus for cell proliferation but also a more speci-fic gene expression that controls function of othercells involved in inflammation, angiogenesis and re-epithelialization. Therefore, it is likely that fibro-blasts play a more important role in the physiologyof wound repair than has been previously realized(107).
In addition to serum stimulation, cell adhesion todifferent matrix molecules can also alter gene ex-pression and protein synthesis (4, 120, 121, 143). Celladhesion also modulates signaling initiated bygrowth factors (109, 163). During tissue repair,fibroblasts are exposed to a variety of extracellularmatrix molecules and soluble growth factors that canexert multiple signals at the same time. Additionally,cells experience tensional forces from the extracellu-lar matrix during cell migration and reorganizationof the wound matrix, which can modulate signalinginside the cell (27, 205, 228). The function of extra-cellular matrix proteins may be further modulatedby proteolytic modification (13, 143, 227), and byconformational changes induced by tensional forces(295). Therefore, the regulation of fibroblast gene ex-pression upon wounding is likely to be very complexand dependent on the balance between different ac-tivated signaling pathways (41).
Origin of wound fibroblasts
The multiple functions of wound fibroblasts raisethe question of whether all of the tasks are per-formed by a single cell type or multiple differentphenotypes are involved (Table 2). If the latter caseis correct, does the heterogeneity result from mi-gration of phenotypically different cells into woundor does it result from differentiation of the fibro-blasts that have populated the wound? These ques-tions are still largely unanswered. There is evidence,however, that connective tissue fibroblasts are het-erogeneous in several properties, including, respon-siveness to growth factors and in the ability to pro-duce specific extracellular matrix proteins (93, 94,
Table 2. Origin of wound fibroblasts
Possible sources of wound fibroblasts Steps involved
Surrounding connective tissue Migration, differentiation
Pericytes Proliferation, migration
Bone marrow Systemic control, homing, differentiation
135
185, 188, 219, 225) suggesting that signals initiatedby wounding may stimulate certain subpopulationsto enter the wound space. On the other hand, thereis also evidence that some of the fibroblasts thathave migrated into wound actually change pheno-type and differentiate into myofibroblasts (seebelow).
Progenitor fibroblast populations have been iden-tified in wounds for decades using autoradiographiclabeling techniques. Based on these studies, themain source of wound fibroblasts seems to be thesurrounding connective tissue. Typically, 2 or 3 daysafter wounding, the label-retaining cells are locatedat the subepithelial connective tissue underlying thewound edge and at the perivascular location. Itseems, however, that only a subpopulation of the lo-cal fibroblasts proliferate in response to tissue injury(225), suggesting that certain fibroblast subsets aremore responsive to growth stimulation than others.
Local fibroblasts are not the only source of woundfibroblasts. After wounding, pericytes that are resi-dents of the surroundings of the vascular endo-thelium of capillaries and venules are also inducedto proliferate and migrate into the wound (79, 106,158). Although fibroblasts have multiple functions inwound repair, their hallmark characteristic is theproperty to produce collagen and other structuralextracellular matrix proteins that replace the bloodclot. This property separates wound fibroblasts frominflammatory and vascular endothelial cells that arealso involved in the formation of granulation tissue.Accordingly, upon wounding, the pattern of gene ex-pression in pericytes is reprogrammed from a con-tractile to a migratory collagen-producing cell, sug-gesting that these cells are able to function as ma-trix-producing wound fibroblasts.
Could some of the wound fibroblasts also arisefrom bone marrow stem cells? Interestingly, earlywork by Cohnheim in 1867 suggested that some ofthe wound fibroblasts may originate from bone mar-row. This was further supported by recent findingsshowing that genetically marked bone marrowstromal cells serve as a source of progenitor cells forvarious mesenchymal tissues. Most importantly,

Hakkinen et al.
these cells apparently translocate to the target tissuewhere they acquire the phenotype of the residentcells (183, 193). These ‘‘peripheral blood fibrocytes’’accumulate in the wound already in the early acutephase but are also present in the scar tissue (15).Fibrocytes function as antigen presenting cells andcan prime naive T cells (25) and secrete a number ofcytokines involved in immune response, hematopo-esis and extracellular matrix synthesis (25, 26), sug-gesting that fibrocytes may play a role in the regula-tion of the acute inflammatory reaction as well as inthe matrix deposition in wounds.
Taken together, the matrix-producing cells thatpopulate wound granulation tissue originate fromdiverse tissue sources and, depending on their ori-gin, they may serve different functions.
Integrin expression and function in fibroblastsduring wound repair
In the connective tissue, fibroblasts are surroundedby a matrix that contains collagen and cellularfibronectin as the major components. Consequently,quiescent fibroblasts express collagen receptorsa1b1 and a2b1 and the major fibronectin receptora5b1 integrin which they use for adhesion to the ma-trix (74, 271, 281). Fibroblasts express a3b1 (281) andintegrin heterodimers of the av subfamily (Hakkinenet al., unpublished) that can also be used to bindfibronectin (4). Although cultured fibroblasts areable to express avb1, avb3 and avb5 integrins (70,73, 92), it is not completely clear with which b sub-units av actually combines to form integrin hetero-dimers in quiescent fibroblasts in vivo. In cell cul-ture, fibroblasts also express a4b1 integrin that bindsto fibronectin (72), but it is unclear whether thisintegrin is actually expressed by fibroblasts in vivo.
The b1 integrins in quiescent fibroblasts are par-tially in an inactive state and become activated afterwounding in fibroblasts that are located near the de-fect (Hakkinen et al., unpublished). Several lines ofevidence show that integrin activation and integrin-mediated cell adhesion to extracellular matrix is im-portant for induction of DNA synthesis (179). Speci-fic integrin-ligand binding is required to commenceDNA synthesis of cells. Cell adhesion to extracellularmatrix via integrins strongly influences the ability ofnormal cells to respond to soluble mitogens (43, 76,109, 221). Activated integrins can physically complexwith growth factor receptors (see above) which pro-vides a mechanism of how different signaling mol-ecules may be targeted to cell-matrix interactionsites inside the cell (161, 218). It is, therefore, prob-
136
able that the decision of the fibroblast to enter thecell cycle depends on the repertoire of integrins thatactively interact with specific extracellular matrixproteins and on the combination of growth factorsthat the cells are exposed to at the particular phaseof wound repair.
Cell migration is needed for fibroblasts to enterthe wound provisional matrix. Fibroblast migrationinto the blood clot occurs only after a lag period of3 to 5 days, the time required for cells to proliferate(32, 157). The lag phase may also be needed for thecells to be activated from quiescent state to be fullyactive migratory cells and to be recruited from theiroriginal location in the tissue or bone marrow to thewound site. It is believed that, in order to detachfrom a collagen-rich matrix, fibroblasts have todownregulate their expression of collagen receptorsand upregulate integrins that bind fibronectin, fi-brin, fibrinogen and vitronectin in the provisionalwound matrix. Accordingly, about 3 to 5 days afterinjury, fibroblasts that migrate into the blood clot ex-press the primary fibronectin receptor a5b1 integrinand a3b1 integrin while they show downregulationof collagen-binding a1 and a2 integrin expression atthe wound margin (74, 281). Platelet-derived growthfactor, which is abundantly expressed during earlywound repair, stimulates cell migration towardsfibronectin by increasing the synthesis of a5 and a3integrins (74, 117). On the other hand, platelet-de-rived growth factor downregulates a1 (74) while itstimulates a2 integrin expression by fibroblasts inculture. Therefore, the regulation of a2 integrin ex-pression in vivo and in vitro is different, possibly be-cause of the multiple signals in addition to platelet-derived growth factor that modulate integrin ex-pression in the tissue. One of the factors that modu-lates integrin expression during wound repair is thecomposition of the extracellular matrix. If fibroblastsare cultured in fibrin and fibronectin-rich matricesthat resemble the early wound matrix, they showselective upregulation of a3 and a5 integrin mRNAas opposed to cells that are in collagen-rich matrix,which show upregulation of a2 integrin (281). Thissuggests that signals initiated by growth factors andextracellular matrix molecules collaborate to induceappropriate integrin expression to support cell mi-gration on fibrin-fibronectin matrix in the earlywound repair and to allow cell adhesion on collagenlater during scar formation.
In the early wound, fibroblast migration seems tobe primarily mediated by fibronectin becausefibroblast migration from a three-dimensional colla-gen matrix into a fibrin clot depends on the presence

Cell biology of gingival wound healing
of fibronectin in the matrix and can be blocked withantibodies against a5b1 and avb3 integrins in vitro(84). Fibroblasts can also use avb3 and avb5 inte-grins for cell adhesion on vitronectin present in theclot (70) and avb3 for cell adhesion and migrationon fibrin and fibrinogen, respectively (71, 84). Cellmigration is regulated by the structural organizationof the fibronectin matrix. In the normal connectivetissue, cells deposit and arrange fibronectin into afibrillar network between the cells and other matrixmolecules using primarily a5b1 and possibly avb3integrins (279, 280, 287). In contrast, in the bloodclot, plasma fibronectin is cross-linked with fibrin toform a three-dimensional scaffold (224). The vari-ation in the architecture of the fibronectin networkcan regulate multiple cell functions, suggesting thatfibronectin in the connective tissue and in the bloodclot may elicit different signals (224, 233). Duringwound repair, cells are induced to deposit alterna-tively spliced isoforms of cellular fibronectin con-taining either the EIIIA or EIIIB modules (see above).The EIIIA or EIIIB modules have different propertiesto support cell adhesion, migration, and prolifer-ation (95, 150, 151), suggesting that they may havedifferent functions in wound repair.
Wounding induces deposition of extracellular ma-trix proteins that modulate cell adhesion. Amongthese molecules, tenascin-C, thrombospondin andsecreted protein, rich in cysteine/osteonectin(SPARC) are multifunctional molecules that canserve as ligands for multiple cell surface receptors(83). Typically, the expression of tenascin-C andthrombospondin is potently stimulated early duringgranulation tissue formation, while SPARC is ex-pressed from middle to late stages of repair. Express-ion of tenascin-C and SPARC remains elevated dur-ing tissue reorganization while the expression ofthrombospondin is only transient (50, 144). Interest-ingly, wounding specifically induces the expressionof the large tenascin-C splice isoform (144, Hakkinenet al., unpublished) that is normally expressed pre-dominantly during embryonic development (156,192). Tenascin-C contains both adhesive andcounteradhesive domains, although mostly it is be-lieved to reduce the adhesion of cells to fibronectinand therefore enhance cell migration (253). The ef-fect of tenascin-C on cell adhesion to fibronectin canalso further modulate gene expression by the cells(253). Interestingly, the large tenascin-C splice iso-form functions differently in regulation of cell ad-hesion and in binding fibronectin as compared withthe more ubiquitously expressed small isoform (29,63, 64, 113, 169, 184). Tenascin-C can serve as ligand
137
for several cell surface receptors, including avb3integrin in fibroblasts that binds to the RGD se-quence found in the third fibronectin type repeat ofthe tenascin molecule. Ligand binding by integrinsto this repeat is modulated by other counteradhesivedomains in the molecule (40).
Thrombospondin can also, like tenascin-C, modu-late cell adhesion, migration and proliferation (83).It contains an RGD sequence that is recognized byavb3 integrin expressed by fibroblasts (240, 255), butit also contains, like tenascin-C, additional bindingsites for other cell surface receptors (240). SPARC isalso able to interfere with integrin-mediated cell ad-hesion on extracellular matrix, and this is believedto occur via interaction with other cell surface mol-ecules that are able to bind SPARC and not withsteric disruption of integrin–extracellular matrix in-teraction (168). Because of their different temporalexpression pattern during wound repair, it is poss-ible that tenascin-C, thrompospondin and SPARCregulate different cellular functions. Indeed, geneknockout animal studies have indicated that throm-bospondin plays a role in regulation of the activityof transforming growth factor-b, collagen fiber or-ganization and vascularization in the connectivetissue (39, 126), which are all important processes inthe physiology of wound repair.
Wound contraction
One of the tasks of fibroblasts in granulation tissueis to bring the wound margins closer together toallow rapid wound closure. It is not completely clearhow wound contraction actually happens in vivo.The most widely held concepts suggest that woundclosure follows when central granulation tissue iscontracted either by myofibroblasts pulling the newcollagen matrix (69) and/or by fibroblasts changingthe organization of the wound matrix by migratingthrough it (54, 67). On the other hand, experimentalremoval of central granulation tissue does not seemto prevent wound contraction, suggesting an alter-native mechanism in which polarized coordinatedmigration of a rim of densely packed fibroblasts pullforward the wound edges (89).
Myofibroblasts are likely to play a role in woundcontraction and matrix deposition. This is supportedby findings that myofibroblasts are typically presentin tissues that are under mechanical stress but theyare only occasionally found in normal tissue (46).Myofibroblasts become especially prominent inpathological conditions including wound repair,tissue fibrosis and tumor stroma (42, 46, 201). These

Hakkinen et al.
cells are characterized by presence of abundantcytoplasmic microfilament bundles and a-smoothmuscle actin. They differ from normal fibroblastsmorphologically, in growth potential, expression ofproteoglycans and collagen, responsiveness to trans-forming growth factor b, organization of focal ad-hesions, and in their ability to organize cellularfibronectin in culture (51, 93, 94, 212). Fibroblastscultured from wound granulation tissue are rich inmyofibroblasts and express a similar repertoire ofintegrins as normal connective tissue fibroblasts.They are, however, phenotypically unique in thatthey show reduced cell spreading on fibronectin asa result of altered interaction of a5b1 integrin andfibronectin (92).
The differentiation of myofibroblasts occurs be-tween 6 and 15 days after wounding (42, 217). After15 days, about 70% of fibroblasts in granulationtissue express a-smooth muscle actin. Differen-tiation of myofibroblasts coincides with wound con-traction, which has led to the idea that these cellsare actually involved in this process. The differen-tiation of myofibroblasts is induced by transforminggrowth factor b in vivo and in cell culture but notwith other profibrotic growth factors and cytokines(47, 204). Importantly, myofibroblasts differentiationis controlled by integrin mediated mechanisms anddepends on the composition of the extracellular ma-trix and the tensional forces mediated from the ma-trix. Recent studies have shown that the induction ofmyofibroblast differentiation by transforminggrowth factor-b requires deposition of cellular EIIIAfibronectin in the pericellular matrix (226). In tissue,differentiation of wound fibroblasts into myofibro-blasts follows induction of EIIIA fibronectin express-ion (226). The induction of the expression of a-smooth muscle actin in fibroblasts by transforminggrowth factor b is also regulated by the changes inthe collagen matrix around the cells and on the de-velopment of intracellular tension (5, 171). This is inturn regulated by interaction between a2b1 integrinsand collagen (171). The induction of a-smoothmuscle actin expression during wound repair isthought to provide a mechanism to stop fibroblastmigration when cells reach their destination. Ter-mination of migration and formation of prominentfocal adhesions may be needed to promote stable(200) cell adhesion to matrix, which is required formyofibroblasts to acquire a matrix-synthesizing andcontractile phenotype.
In ideal wound repair, scar tissue is remodeled andreorganized to form structurally and functionally nor-mal connective tissue. When contraction stops, the
138
myofibroblasts disappear because of apoptosis andthe scar becomes less cellular and new fibroblastswith properties typical to normal connective tissuefibroblasts emerge (42, 48, 217). Apoptosis of myo-fibroblasts begins at day 12, peaks at day 20 and re-solves by day 60 after wounding (48). Factors that in-hibit a-smooth muscle actin expression and differen-tiation of myofibroblasts are not well characterized. Ithas been noted that interferon-g has an antifibroticeffect, which probably results from its ability to inhibitfibroblast differentiation into myofibroblasts (217).Additionally, myofibroblasts are more sensitive tobasic fibroblast growth factor–induced apoptosisthan normal connective tissue fibroblasts (68), whichmay provide a mechanism to reduce the amount ofmyofibroblasts in the tissue when they are no longerneeded. Interestingly, myofibroblasts persist in cer-tain pathological conditions, including hyperthroph-ic and fibrotic lesions of many organs, suggesting thatthey are involved in the accumulation of extracellularmatrix that is characteristic for these lesions (45, 217).Therefore, for normal wound repair it is importantthat the myofibroblasts undergo apoptosis and are re-placed by normal fibroblasts when wound contrac-tion is completed.
An interesting question is where the fibroblasts thatreplace myofibroblasts and that eventually maintainthe connective tissue structure during tissue homeo-stasis originate from. Because these cells have pheno-typic properties characteristic to normal connectivetissue fibroblasts, it is possible that they are derivedfrom the connective tissue next to the wound or, alter-natively, they differentiate from more primitivefibroblasts that initially populate the wound. Regard-less of their origin, it seems that replacement of thewound fibroblasts with cells of normal connectivetissue fibroblast phenotype is necessary for the com-plete regeneration of the tissue.
Mechanical tension
The interactions between cells and extracellular ma-trix during granulation tissue formation can bestudied in cell culture using cell-populated three-di-mensional fibrin or collagen gels (86, 256). In prin-ciple, the fibrin gels correspond to the situation inearly wound repair when fibroblasts have migratedinto the fibrin containing provisional matrix whilethe collagen gel model resembles later phase ofwound repair when granulation tissue fibroblasts re-side in a newly deposited collagen-rich matrix thatundergoes remodeling. In these models, fibroblastsattach to the proximal fibrin or collagen fibers and

Cell biology of gingival wound healing
in an attempt to migrate pull the fibers together toform a dense tissue-like structure. When fibroblastsare allowed to grow in fibrin gels they eventually re-place the fibrin matrix with newly deposited colla-gen (256). If the gel is not relaxed, fibroblasts tryingto pull the fibers experience high tension from thematrix that resembles wound granulation tissue.Contraction of three-dimensional collagen gel is me-diated by integrins and can be stimulated by serumand growth factors present in wound, including lyso-phosphatidic acid, transforming growth factor b andplatelet-derived growth factor (34, 87, 90, 164, 197,250). Currently, there is some controversy over whichspecific integrin-ligand interactions actually mediatecollagen contraction. Binding between fibroblastsand collagen fibrils is evidently the first and mostimportant step in the collagen gel contraction. Mostdata show that in human connective tissue fibro-blasts, collagen gel contraction is mediated by thecollagen receptor a2b1 integrin (214, 238). The rateof contraction depends on the amount of a2b1 inte-grins expressed by the cells (197). This is in accord-ance with simultaneous induction of a2 integrin ex-pression by wound fibroblasts with formation of col-lagenous scar and beginning of wound contractionin vivo (282).
Role of integrins in regulation of cell proliferationand survival in granulation tissue
During the formation of granulation tissue, fibro-blasts that emerge into the provisional wound matrixstop migrating and undergo cell proliferation. Ac-cordingly, about 7 to 10 days after wounding, thegranulation tissue becomes hypercellular containingabundant fibroblasts and endothelial cells (157, 271).After that, during tissue maturation, the cell numberin the granulation tissue gradually decreases (48,157, 271). It appears that the organization of collagenand mechanical tension mediated from the extracel-lular matrix are important factors that modulateDNA synthesis and cell survival. This is evidenced bythe finding that fibroblasts cultured under mechan-ical tension proliferate actively, but after stress relax-ation the DNA synthesis is rapidly downregulatedand cells start to undergo apoptosis (66, 87, 139,162). Stress relaxation also downregulates autophos-phorylation of growth factor receptors, which con-tributes to the growth reduction (139, 159, 241). In amechanically stressed collagen gel, cells develop anorganized cell surface fibronectin network (162, 251)that disappears after stress relaxation. Since a5b1integrin can probably serve as a mechanoreceptor
139
and mediate signals that promote cell growth (269,294), differences in fibronectin receptor engagementmay be a mechanism of how cell proliferation isregulated by mechanical signals. Therefore, reduc-tion of mechanical strain during tissue maturationmay provide a mechanism to downregulate cellgrowth and induce apoptosis to normalize the cellu-larity in the tissue. Additionally, in the later phasesof wound repair the activity of growth factors isgradually downregulated (231, 288), which willfurther reduce the number of growth stimulatory sig-nals for fibroblasts. In the regulation of cell growthand apoptosis in the granulation tissue, integrinsprobably play an important role because they me-diate the signals initiated by mechanical tension andcollaborate with growth factor signaling and mediatesignals that regulate cell survival (109, 205, 206, 228).Interestingly, fibroblasts undergo apoptosis in con-tractile collagen gels but not in fibrin gels (66), sug-gesting that specific types of integrin–extracellularmatrix interactions are needed for initiation ofapoptosis. This may provide a mechanism to preventfibroblasts from undergoing apoptosis in the earlyfibrin-rich provisional wound matrix and to allowtheir proliferation, which is needed to populate thegranulation tissue with fibroblasts.
An additional mechanism that is likely to regulatecell growth in the granulation tissue is the structuralorganization of the extracellular matrix. Duringgranulation tissue formation, fibroblasts first syn-thesize fibronectin and then type I collagen that isgradually organized from monomeric molecules toform a fibrillar collagen matrix (125). Monomericcollagen through a2b1 integrin promotes cell pro-liferation while fibrillar collagen downregulates it(123). Additionally, studies using fibronectin de-ficient cells have shown that assembly of fibronectinmatrix by the cells is required for fibronectin to pro-mote cell growth on different extracellular matrixmolecules (233). Therefore, it is probable that the de-position and organization of extracellular matrix byfibroblasts provides a feedback mechanism to regu-late cell growth during wound repair.
Regulation of protein synthesis and matrixdegradation in the granulation tissue
Remodeling of tissue during wound repair requirescontrolled synthesis and degradation of extracellularmatrix. The most important factors that regulatesynthesis and secretion of these molecules includegrowth factors and signals from the extracellular ma-trix. There is evidence that integrin binding to its

Hakkinen et al.
ligand can directly induce gene expression (102, 254,272). Most importantly, there seems to be a specificintegrin-ligand interaction that is needed for stimu-lation of specific type of protein synthesis (254, 272).In this process, the signals mediated by integrins arefurther modulated by growth factors, the structuralarchitecture of the extracellular matrix as well as bytensional forces (109, 202, 228).
During granulation tissue formation, fibroblastsare stimulated by transforming growth factor-b todeposit new extracellular matrix proteins. Interest-ingly, early matrix deposition seems to occur first atabout day 7 after injury in the granulation tissue im-mediately under the newly formed epithelium (178).This coincides with the peak in activation of latenttransforming growth factor-b from storages in theblood clot provisonal matrix and in the matrix of thegranulation tissue (81, 288) and with induction ofepithelial avb6 integrin which may regulate the acti-vation of latent transforming growth factor-b (seeabove, 91, Hakkinen et al., unpublished). Once acti-vated, transforming growth factor-b can induce itsown production by fibroblasts (139), which may be amechanism to maintain high activity of transforminggrowth factor-b in the granulation tissue.
As discussed earlier, a potent modulator of proteinsynthesis is mechanical tension. Generally, whencells are grown in collagen gels under tension theyshow a relatively high protein synthesis rate, whileafter stress relaxation the protein synthesis is down-regulated (162). The signals initiated by tension ina three-dimensional collagen matrix require specificintegrin-collagen interactions and may lead to speci-fic gene activation. Additionally, platelet-derivedgrowth factor–induced upregulation of a2 integrinexpression is stimulated if fibroblasts are cultured incollagen gel while expression of a5 and a3 integrinsis attenuated (281). Transforming growth factor-bcan also potently stimulate expression of a2 integrinin fibroblasts that interact with collagen, and this de-pends on the tensional forces that the cells experi-ence (5). It seems that tension mediated from thematrix can effectively regulate the balance betweenmatrix production and degradation. For example,upon stress relaxation the expression of type I colla-gen is downregulated while that of matrix metallo-proteinase-1 and -13 is upregulated (30, 195, 197,252). The regulation of collagen and matrix metallo-proteinase genes by collagen matrix is partially regu-lated by different collagen receptors. The downregu-lation of collagen synthesis after stress relaxation ismediated by a1b1 integrin and the upregulation ofmatrix metalloproteinase-1 and matrix metallopro-
140
teinase-13 expression occurs by a2b1 and by botha1b1 and a2b1 integrins, respectively (195, 197).Integrin a1b1 seems to be able to function as a feed-back regulator of collagen synthesis in fibroblasts(75).
Gingival wound repair: similarities to scarless fetalwound repair
There has been a general observation that woundsin the oral mucosa heal faster and with less scarringthan extraoral wounds (Table 3). Scar tissue is char-acterized by excessive accumulation of disorderly ar-ranged collagen, mostly type I and III (219), proteo-glycans (274, 289) and persistent myofibroblasts (42,46, 201), which leads to aberrant function of thetissue. Surprisingly, there are only a few reports thathave quantitatively tested this hypothesis. Neverthe-less, based mostly on animal studies it seems thatwound healing in oral mucosa, indeed, is faster andresults in less scarring than in skin (223, 286). It isprobable that oral wound healing is enhanced partlybecause of factors present in the saliva and by thespecific microflora of the oral cavity (see below). Ad-ditionally, the properties of the cells involved intissue regeneration in oral mucosa are unique andshare properties of fetal cells (219). Scar formation isdevelopmentally regulated because, in early ges-tation, fetal wound repair occurs without scar forma-tion and the transition to healing with scar forma-tion occurs late in the gestation (56, 104). Interest-ingly, the initiation of scar formation parallels theinduction of myofibroblast differentiation duringwound repair at different stages of embryonal devel-opment (56), suggesting that phenotypic modulationof wound fibroblasts into myofibroblasts may be in-volved in scar formation.
As compared with adult wounds, fetal wounds arecharacterized by the absence of clot formation andinflammatory reaction. They also show unique spa-tial and temporal organization of extracellular matrixand reduced expression of transforming growth fac-tor-b (274). Several investigations have compared theproperties of adult skin and gingival fibroblasts withfetal fibroblasts. The findings indicate that, unlikeadult skin fibroblasts, adult gingival fibroblastslocated in the papillary connective tissue sharemany properties with fetal fibroblasts, includingtheir growth and migration properties, cell mor-phology, production of migration stimulating factor,and production and response to cytokines (219). Ad-ditionally, similar to fetal fibroblasts, oral mucosal orgingival fibroblasts are able to contract three-dimen-

Cell biology of gingival wound healing
Table 3. Special features of oral wound healing
Factor Mechanism
Saliva Moisture, ionic strengthGrowth factors (EGF, TGFb, FGF, IGF etc.)Unknown factors
Bacteria Stimulation of macrophage influxDirect stimulative action on keratinocytes and fibroblasts
Phenotype of cells Fetal-like fibroblasts with unique responseSpecialized epithelium and connective tissue
sional collagen matrix faster as dermal fibroblasts(105, 237). They also populate experimental woundsfaster than their dermal counterparts in culture (2).Gingival fibroblasts also differ from dermal fibro-blasts in their ability to secrete proteolytic enzymes.Matrix metalloproteinase-13, a potent collagenase(273), is expressed in the granulation tissue duringacute wound repair at 7- and 14-day-old human gin-gival wounds but not in dermal wounds (296). Ad-ditionally, when gingival fibroblasts are inoculatedinto three-dimensional fibrin matrix they are able toreorganize and degrade the matrix rapidly. This isbecause of high expression of tissue plasminogen ac-tivator. Matrix reorganization and fibrinolysis is lessadvanced in dermal fibroblast-inoculated matrices(141, 142). The regulation of fibrinolysis by gingivalfibroblasts depends on the tensional forces that thecells experience from the matrix (140). The variousfindings described above suggest that several cellfunctions important in tissue repair are shared byfetal and gingival fibroblasts and differ from dermalfibroblasts. It is possible that gingival fibroblasts arephenotypically unique cells in adult tissue that maycontribute to the rapid healing of oral wounds withminimal scarring in the gingiva.
Role of saliva and gingivalcrevicular fluid in oral woundhealing
While it is clear that the excellent healing potentialof oral mucosa results to a large extent from the in-trinsic tissue factors such as the presence of struc-tural cells with potential for tissue regeneration,dense vasculature and high turnover rate of connec-tive tissue and epithelium, it is also apparent thatsaliva provides a unique environment in the mouthconducive for rapid tissue repair (Table 3). This be-comes obvious from the studies showing delayed
141
healing of oral wounds in people with xerostomiaor sialadenectomized animals (12, 55, 103). Animalsinstinctly lick their wounds, which appears to resultin faster healing (11). There are several physico-chemical factors in saliva favoring gingival healing.These include an appropriate pH, ionic strength, andpresence of ions such as calcium and magnesiumrequired for healing (53). Saliva also has an efficientcapacity to reduce redox activity caused by tran-sitional metal ions and inhibit the production of freeradicals that may be beneficial for the healing pro-cess (170). Lubrication of oral mucosa provided bysaliva is beneficial for wound healing. The advan-tageous effects of maintaining a moist wound en-vironment include prevention of tissue dehydrationand cell death, accelerated angiogenesis and in-creased breakdown of fibrin and tissue debris.Hence, the use of hydrocolloid occlusive dressing isa useful adjunct in facilitating cutaneous woundhealing (61). The therapeutic effect of saliva on heal-ing of skin wounds has been demonstrated exper-imentally in calves. Compared to saline-treatedwounds, saliva-treated wounds had shorter in-flammatory reaction and faster epithelial coverageand connective tissue regeneration (268). It appearsthat moisture and ionic strength are not the primaryfactors in saliva that promote tissue repair. This po-tential is probably due to the presence of several fac-tors in saliva including various growth factors andbacteria (293).
Growth factors found in saliva are synthesized bysalivary glands or derived from plasma through gin-gival crevice. Epithelial cells and connective tissuecells also produce their own growth factors that acteither in a paracrine or autocrine manner. Becausemany of the growth factors are transported to salivaalong with gingival crevicular fluid, it is conceivablethat their concentrations in gingival tissue are higherthan elsewhere in the oral cavity. Therefore the peri-odontium is in a favorable position with respect totissue healing.

Hakkinen et al.
As discussed above, wound healing is a complexphenomenon involving increased proliferation, ad-hesion and migration of cells of connective tissueand epithelium, inflammatory reactions and re-modeling of extracellular matrix. All these phenom-ena are directed by growth factors (9, 10, 82). Differ-ent growth factors have specific functions and targetcells in wound healing, and their delicate balance isrequired for optimal tissue repair. The actual roles ofeach growth factor in saliva are not known. The beststudied salivary growth factor, and possibly the mostimportant one, is epidermal growth factor. In hispioneer studies, Cohen found that a protein compo-nent of mouse submandibular gland induced pre-cocious eyelid opening and incisor tooth eruption.Later it was discovered that the factor termed epider-mal growth factor has a multitude of effects on cellproliferation, cell migration and extracellular matrixmetabolism (21, 37, 220). It has now become obviousthat epidermal growth factor is needed for the nor-mal maintenance and repair of oral mucosa (174).Interestingly, salivary epidermal growth factor alsoplays a role in the maintenance of gastric and ilealmucosal integrity (194, 213). In humans and manyother animals, salivary glands are the major epider-mal growth factor–producing organs. In humans epi-dermal growth factor is synthesized by both parotidand submandibular glands. During oral wound heal-ing, the concentration of epidermal growth factor isincreased in saliva (99, 180, 181).
Another growth factor found in saliva is the vascu-lar endothelial growth factor (243). This protein isimportant in many aspects of angiogenesis and in-flammation such as endothelial growth, permeabilityand leukocyte adherence (52). A number of othergrowth factors are also present in saliva. These in-clude nerve growth factor and members of trans-forming growth factor b, fibroblast growth factor andinsulin-like growth factor families (230). As thesesubstances have specific regulatory roles in cellgrowth and extracellular matrix formation, they areimportant in maintaining health of the oral cavityand in healing of oral mucosal tissues (293).
The presence of growth factors in crevicular fluidhas received limited attention. The major focus ofthe studies has been pro-inflammatory cytokines,such as interleukin-1 and tumor necrosis factor a(177). It is important to recognize that while one ac-tion of the crevicular fluid cytokines is promotion ofinflammation, another function is to facilitate tissuerepair. Maintenance of the normal tissue turnoverresults from a delicate balance of the cytokines. Analteration in this balance results either in persistence
142
of inflammation and tissue destruction or increasedcell growth and tissue repair.
Integrin ligands such as collagen, vitronectin,fibronectins and laminins are involved in directingthe adhesion and migration of cells (261). It is there-fore possible that these molecules, when present ingingival crevicular fluid or saliva, may modulatewound healing of gingiva. Indeed, fibronectin hasbeen found both in crevicular fluid and saliva (112,129, 257). Even though not yet reported, it is reason-able to assume that other adhesion molecules maybe present in saliva.
Extracellular matrix remodeling is an essentialpart of wound repair. In this process matrix metallo-proteinases and other neutral proteinases have animportant function. During periodontal diseasesgingival crevicular fluid and saliva are rich in col-lagenases, gelatinases and elastases (133–136, 149,232, 263–265). Most of these enzymes appear to de-rive from neutrophils migrating from inflamed peri-odontium. However, salivary gland cells and acti-vated epithelial cells and fibroblasts of periodontiumproduce their own matrix metalloproteases includ-ing collagenase-1 (matrix metalloprotease-1), col-lagenase-3 (matrix metalloprotease-13) and gela-tinase A (matrix metalloprotease-2) (209, 211, 258).In concert the matrix metalloproteases and elastasesare capable of cleaving all extracellular matrix pro-teins and proteoglycans. As discussed earlier, proteo-lytic enzymes are necessary for proper wound heal-ing. These matrix metalloproteases could also re-lease cryptic bioactive domains from matrixmolecules that may regulate cell proliferation andmigration (285). For example, fibronectin and colla-gen fragments have been demonstrated in gingivalcrevicular fluid (245) and saliva (77). Practically noinformation presently exists on the possible role ofthese extracellular matrix fragments in promotinghealing of oral wounds.
Role of bacteria in oral wound healing
The oral cavity harbors considerable amounts ofbacteria. More than 500 bacterial species have beenso far identified in the oral cavity (165). Recognizingthe limitation of the present detection methods, inreality several times more species may colonize inthe mouth. It is clear that bacteria affect woundhealing in the oral cavity, and it is well establishedthat wounds colonized by pathogenic bacteria havedelayed healing (198, 249). Clinicians are aware ofthe painful complications in extraction wound repairthat result from bacterial infection (38, 57). Much

Cell biology of gingival wound healing
less attention has been given to the fact that smallconcentrations of bacteria may increase the rate ofwound healing.
In 1921, Carrel reported that wounds of dogstreated with certain concentrations of Staphylococ-cus aureus healed faster than untreated wounds.Since then several studies have confirmed that find-ing, also using other bacterial species. Many factorsmay contribute to this effect. Inflammatory reactionthat is a prerequisite for tissue repair is accentuatedby bacterial contamination. Bacteria present in awound will attract macrophages into the area andinduce their cytokine secretion. As a consequence,blood supply and granulation tissue formation areaccentuated in the wound. Proliferation of mes-enchymal cells is increased and synthesis rate ofconnective tissue components is stimulated, leadingto greater tensile strength of the contaminatedwounds in the course of healing (115, 127, 128, 136).Bacteria contain a variety of substances, somestimulating host cell proliferation and othersexerting toxic effects. In addition, the same sub-stance can be either stimulatory or inhibitory, de-pending on its concentration in tissue. Bacteria mayact directly on epithelial cells and connective tissuecells in wounds and, depending on the type andconcentration, either accelerate or delay wound re-generation. We found that proliferation of gingivalfibroblats in culture was increased by Prevotella in-termedius but decreased by the same concentrationsof Porphyromonas gingivalis (133). Interestingly,there was a great variation in this effect betweenfibroblast populations obtained from different pa-tients. These findings imply that the potential forperiodontal repair depends both on the bacterialflora and the individual cell populations of the peri-odontal wounds. Some bacterial factors have directfibroblast and epithelial cell stimulating properties.Lipopolysaccharide of both Actinobacillus actino-mycetemcomitans and P. gingivalis slightly increasescell growth in vitro (98, 189). At higher concen-trations lipopolysaccharide from different bacterialspecies inhibits cell proliferation (8, 134). Lipopoly-saccharide as well as bacterial plaque extracts in-crease hyaluronan production in cultured gingivalfibroblasts (7, 130). Hyaluronan is a high-molecular-weight polyanionic glycosaminoglycan that plays animportant role in wound healing through specific in-teraction with other matrix molecules and cells (23).S. aureus lipoteichoic acid and protein A inducefibroblast production of hepatocyte growth factor,which stimulates epithelial proliferation (6). Thereare numerous other factors present in bacteria that
143
could modulate oral wound healing. For example, A.actinomycetemcomitans GroEL-like heat-shock pro-tein and phospholipase C are able to modulate epi-thelial cell growth and cell migration (62, 80, 294).Specific mechanisms of these effects remain to beexplored.
Conclusion
Wound healing in the oral cavity is a very complexprocess. We are just starting to uncover the complexinterplay between various cell types, growth factorsand salivary components. The focus of this chapterwas to summarize the two major events of gingivalwound healing, namely re-epithelialization and theformation of granulation tissue. Because of theunique environment of oral cavity, we also reviewedthe special features of oral healing. The interplay be-tween oral cells and their extracellular matrix, bac-teria, saliva and gingival crevicular fluid involvesmyriad factors dictating the nature of the tissue re-pair process. It would be an enormous if not imposs-ible task to sort out all these factors. Some of thefactors are already relatively well understood andcould be used for practical applications. Activestudies on certain growth factors are underway inorder to provide new tools for periodontal therapy(101). Similar studies utilizing other factors benefi-cial for wound healing can be expected when morebasic research has been done elucidating their speci-fic functions. Only then we can design precisiontools to speed-up (or slow-down) re-epithelializ-ation, granulation tissue formation and scarlesswound healing. Modulation of cell–to–extracellularmatrix adhesion will be the key target for computerdesigned drugs engineered to guide optimal tissuerepair.
Acknowledgments
We thank Colin Wiebe for critical reading of themanuscript. Wound-healing studies performed inthe laboratories of the authors are supported by theMedical Research Council of Canada.
References
1. Akiyama SK, Yamada SS, Chen W-T, Yamada KM. Analysisof fibronectin receptor function with monoclonal anti-bodies: roles in cell adhesion, migration, matrix assembly,

Hakkinen et al.
and cytoskeletal organization. J Cell Biol 1989: 109: 863–875.
2. al-Khateeb T, Stephens P, Shepherd JP, Thomas DW. Aninvestigation of preferential fibroblast wound repopu-lation using a novel in vitro wound model. J Periodontol1997: 68: 1063–1069.
3. Anatoniades HN, Galanopoulos T, Neville-Golden J, Kirit-sy CP, Lynch E. Injury induces in vivo expression of plate-let-derived growth factor (PDGF) and PDGF receptormRNAs in skin epithelial cells and PDGF mRNA in con-nective tissue fibroblasts. Proc Natl Acad Sci U S A 1991:88: 565–569.
4. Aplin AE, Howe A, Alahari SK, Juliano RL. Signal transduc-tion and signal modulation by cell adhesion receptors:the role of integrins, cadherins, immunoglobulin-cell ad-hesion molecules, and selectins. Pharmacol Rev 1998: 50:197–263.
5. Arora PD, Narani N, McCulloch CA. The compliance ofcollagen gels regulates transforming growth factor-b in-duction of a-smooth muscle actin in fibroblasts. Am JPathol 1999: 154: 871–882.
6. Baroni A, Perfetto B, Ruocco E, Rossano F. Lipoteichoicacid and protein-A from Staphylococcus aureus stimulaterelease of hepatocyte growth factor (HGF) by human der-mal fibroblasts. Arch Dermatol Res 1998: 290: 211–214.
7. Bartold PM. Lipopolysaccharide stimulation of hyaluron-ate synthesis by human gingival fibroblasts in vitro. ArchOral Biol 1991: 36: 791–797.
8. Bartold PM, Narayanan AS, Page RC. Platelet-derivedgrowth factor reduces the inhibitory effects of lipopoly-saccharide on gingival fibroblast proliferation. J Peri-odontal Res 1992: 27: 499–505.
9. Bennett NT, Schultz GS. Growth factors and wound heal-ing: biochemical properties of growth factors and theirreceptors. Am J Surg 1993: 165: 728–737.
10. Bennett NT, Schultz GS. Growth factors and wound heal-ing. II. Role in normal and chronic wound healing. Am JSurg 1993: 166: 74–81.
11. Bodner L. Effect of parotid submandibular and sublingualsaliva on wound healing in rats. Comp Biochem PhysiolA 1991: 100: 887–890.
12. Bodner L, Dayan D. Epithelium and connective tissue re-generation during palatal wound healing in desalivatedrats – a comparative study. Comp Biochem Physiol A Phy-siol 1995: 111: 415–419.
13. Boudreau N, Bissell MJ. Extracellular matrix signalling: in-tegration of form and function in normal and malignantcells. Curr Opin Cell Biol 1998: 10: 640–646.
14. Bourdon MA, Ruoslahti E. Tenascin mediates cell attach-ment through an RGD-dependent receptor. J Cell Biol1989: 108: 1149–1155.
15. Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. Cir-culating fibrocytes define a new leukocyte subpopulationthat mediates tissue repair. Mol Med 1994: 1: 71–81.
16. Burke RD. Invertebrate integrins: structure, function, andevolution. Int Rev Cytol 1999: 191: 257–284.
17. Busk M, Pytela R, Sheppard D. Characterization of theintegrin avb6 as a fibronectin-binding protein. J BiolChem 1992: 267: 5790–5796.
18. Carrel A. Cicatrization of wounds. XII. Factors initiatingregeneration. J Exp Med 1921: 34: 425–432.
19. Carter WG, Ryan MC, Gahr PJ. Epiligrin, a new cell ad-hesion ligand for integrin a3b1 in epithelial basementmembranes. Cell 1991: 65: 599–610.
144
20. Cavani A, Zambruno G, Marconi A, Manca V, Marchetti M,Giannetti A. Distinctive integrin expression in the newlyforming epidermis during wound healing in humans. JInvest Dermatol 1993: 101: 600–604.
21. Chen JD, Kim JP, Zhang K, Sarret Y, Wynn KC, Kramer RH,Woodley DT. Epidermal growth factor (EGF) promoteshuman keratinocyte locomotion on collagen by increas-ing the a2 integrin subunit. Exp Cell Res 1993: 209: 216–223.
22. Chen JWC, Rogers AA, Lydon MJ. Characterization of bio-logic properties of wound fluid collection during earlystages of wound healing. J Invest Dermatol 1992: 99: 559–564.
23. Chen WY, Abatangelo G. Functions of hyaluronan inwound repair. Wound Repair Regen 1999: 7: 79–89.
24. Cherny RC, Honan MA, Thiagaran P. Site-directed mutag-enensis of the arginine-glycine-aspartic acid in vitro nec-tin abolishes cell adhesion. J Biol Chem 1993: 268: 9725–9729.
25. Chesney J, Bacher M, Bender A, Bucala R. The peripheralblood fibrocyte is a potent antigen-presenting cell cap-able of priming naive T cells in situ. Proc Natl Acad SciU S A 1997: 94: 6307–6312.
26. Chesney J, Metz C, Stavitsky AB, Bacher M, Bucala RJ.Regulated production of type I collagen and inflamma-tory cytokines by peripheral blood fibrocytes. Immu-nology 1998: 160: 419–425.
27. Chiquet M, Matthisson M, Koch M, Tannheimer M,Chiquet-Ehrismann R. Regulation of extracellular matrixsynthesis by mechanical stress. Biochem Cell Biol 1996:74: 737–744.
28. Chiquet-Ehrismann R. What distinguishes tenascin fromfibronectin? FASEB J 1990: 4: 2598–2604.
29. Chiquet-Ehrismann R, Matsuoka Y, Hofer U, Spring J,Bernasconi C, Chiquet M. Tenascin variants: differentialbinding to fibronectin and distinct distribution in cell cul-tures and tissues. Cell Regul 1991: 2: 927–938.
30. Chiquet-Ehrismann R, Tannheimer M, Koch M, BrunnerA, Spring J, Martin D, Baumgartner S, Chiquet M. Tenas-cin-C expression by fibroblasts is elevated in stressed col-lagen gels. J Cell Biol 1994: 127: 2093–2101.
31. Chuong CM, Chen HM. Enhanced expression of neuralcell adhesion molecules and tenascin (Cytotactin) duringwound healing. Am J Pathol 1991: 38: 427–440.
32. Clark RA. Regulation of fibroplasia in cutaneous woundrepair. Am J Med Sci 1993: 306: 42–48.
33. Clark RA. Overview and general considerations. In: ClarkRAF, ed. The molecular and cellular biology of wound re-pair. New York: Plenum Press, 1996: 3–50.
34. Clark RA, Folkvord JM, Hart CE, Murray MJ, McPhersonJMJ. Platelet isoforms of platelet-derived growth factorstimulate fibroblasts to contract collagen matrices. J ClinInvest 1989: 84: 1036–1040.
35. Clark RAF, Lanigan JM, DellaPelle P, Manseau E, DvorakHF, Colvin RB. Fibronectin and fibrin provide a pro-visional matrix for epidermal cell migration duringwound reepithelialization. J Invest Dermatol 1982: 70:264–269.
36. Clark RAF, Spencer J, Larjava H, Ferguson MWJ. Re-epi-thelialization of normal human excisional wounds is as-sociated with a switch from avb5 to avb6 integrins. Br JDermatol 1996: 135: 46–51.
37. Cohen S. The stimulation of epidermal proliferation by aspecific protein (EGF). Dev Biol 1965: 12: 394–407.

Cell biology of gingival wound healing
38. Colby RC. The general practitioner’s perspective of theetiology, prevention, and treatment of dry socket. GenDent 1997: 45: 461–467, 471–472.
39. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM,Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospond-in-1 is a major activator of TGF-b1 in vivo. Cell 1998: 93:1159–1170.
40. Crossin KL. Tenascin: a multifunctional extracellular ma-trix protein with a restricted distribution in developmentand disease. J Cell Biochem 1996: 61: 592–598.
41. Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA.Suppression of ceramide-mediated programmed celldeath by sphingosine-1-phosphate. Nature 1996: 381:800–803.
42. Darby I, Skalli O, Gabbiani G. a-smooth muscle actin istransiently expressed by myofibroblasts during experi-mental wound healing. Lab Invest 1990: 63: 21–29.
43. DeMali KA, Balciunaite E, Kazlauskas A. Integrins en-hance platelet-derived growth factor (PDGF)-dependentresponses by altering the signal relay enzymes that arerecruited to the PDGF b receptor. J Biol Chem 1999: 274:19551–19558.
44. DeSimone DW, Norton PA, Hynes RO. Identification andcharacterization of alternatively spliced fibronectinmRNAs expressed in early Xenopus embryos. Dev Biol1992: 149: 357–369.
45. Desmouliere A, Badid C, Bochaton-Piallat ML, GabbianiG. Apoptosis during wound healing, fibrocontractive dis-eases and vascular wall injury. Int J Biochem Cell Biol1997: 1: 19–30.
46. Desmouliere A, Gabbiani G. Modulation of fibroblasticcytoskeletal features during pathological situations: therole of extracellular matrix and cytokines. Cell Motil Cyto-skel 1994: 29: 195–203.
47. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Trans-forming growth factor-b1 induces a-smooth muscle actinexpression in granulation tissue myofibroblasts and inquiescent and growing cultured fibroblasts. J Cell Biol1993: 122: 103–111.
48. Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosismediates the decrease in cellularity during the transitionbetween granulation tissue and scar. Am J Pathol 1995:146: 56–66.
49. DiPersio CM, Hodival-Dilke KM, Jaenisch R, Kreidberg JA,Hynes RO. a3b1 Integrin is required for normal develop-ment of the epidermal basement membrane. J Cell Biol1997: 137: 729–742.
50. DiPietro LA, Nissen NN, Gamelli RL, Koch AE, Pyle JM,Polverini PJ. Thrombospondin 1 synthesis and functionin wound repair. Am J Pathol 1996: 148: 1851–1860.
51. Dugina V, Alexandrova A, Chaponnier C, Vasiliev J, Gabbi-ani G. Rat fibroblasts cultured from various organs exhibitdifferences in a-smooth muscle actin expression, cyto-skeletal pattern, and adhesive structure organization. ExpCell Res 1998: 238: 481–490.
52. Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascu-lar permeability factor/vascular endothelial growth factorand the significance of microvascular hyperpermeabilityin angiogenesis. Curr Top Microbiol Immunol 1999: 237:97–132.
53. Edgar WM. Saliva: its secretion, composition and func-tions. Br Dent J 1992: 172: 305–312.
54. Ehrlich HP, Rajaratnam JB. Cell locomotion forces versus
145
cell contraction forces for collagen lattice contraction: anin vitro model of wound contraction. Tissue Cell 1990: 22:407–417.
55. Epstein JB, Scully C. The role of saliva in oral health andthe causes and effects of xerostomia. J Can Dent Assoc1992: 58: 217–21.
56. Estes JM, Vande Berg JS, Adzick NS, MacGillivray TE, De-smouliere A, Gabbiani G. Phenotypic and functional fea-tures of myofibroblasts in sheep fetal wounds. Differen-tiation 1994: 56: 173–181.
57. Fazakerley M, Field EA. Dry socket: a painful post-extrac-tion complication. Dent Update 1991: 18: 31–34.
58. Ffrench-Constant C, Hynes RO. Patterns of fibronectingene expression and splicing during cell migration inchicken embryos. Development 1988: 104: 369–382.
59. Ffrench-Constant C, Hynes RO. Alternative splicing offibronectin is temporally and spatially regulated in thechicken embryo. Development 1989: 106: 375–388.
60. Ffrench-Constant C, Van de Water L, Dvorak HF, HynesRO. Reappearance of an embryonic pattern of fibronectinsplicing during wound healing in the adult rat. J Cell Biol1989: 109: 903–914.
61. Field FK, Kerstein MD. Overview of wound healing in amoist environment. Am J Surg 1994: 167: 2S–6S.
62. Firth JD, Putnins EE, Larjava H, Uitto VJ. Bacterial phos-pholipase C upregulates matrix metalloproteinase ex-pression by cultured epithelial cells. Infect Immun 1997:65: 4931–4936.
63. Fischer D, Brown-Ludi M, Schulthess T, Chiquet-Ehris-mann R. Concerted action of tenascin-C domains in celladhesion, anti-adhesion and promotion of neurite out-growth. J Cell Sci 1997: 10: 1513–1522.
64. Fischer D, Tucker RP, Chiquet-Ehrismann R, Adams JC.Cell-adhesive responses to tenascin-C splice variants in-volve formation of fascin microspikes. Mol Biol Cell 1997:8: 2055–2075.
65. Fleischmajer R, Perlish JS, MacDonald ED 2nd, SchechterA, Murdoch AD, Iozzo RV, Yamada Y. There is binding of col-lagen IV to b1 integrin during early skin basement mem-brane assembly. Ann N Y Acad Sci 1998: 857: 212–227.
66. Fluck J, Querfeld C, Cremer A, Niland S, Krieg T, SollbergS. Normal human primary fibroblasts undergo apoptosisin three-dimensional contractile collagen gels. J InvestDermatol 1998: 110: 153–157.
67. Friedl P, Zanker KS, Brocker EB. Cell migration strategiesin 3-D extracellular matrix: differences in morphology,cell matrix interactions, and integrin function. MicroscRes Tech 1998: 43: 369–378.
68. Funato N, Moriyama K, Shimokawa H, Kuroda T. Basicfibroblast growth factor induces apoptosis in myofibro-blastic cells isolated from rat palatal mucosa. BiochemBiophys Res Commun 1997: 240: 21–26.
69. Gabbiani G, Ryan GB, Majne G. Presence of modifiedfibroblasts in granulation tissue and their possible role inwound contraction. Experientia 1971: 27: 549–550.
70. Gailit J, Clark RA. Studies in vitro on the role of av andb1 integrins in the adhesion of human dermal fibroblaststo provisional matrix proteins fibronectin, vitronectin,and fibrinogen. J Invest Dermatol 1996: 106: 102–108.
71. Gailit J, Clarke C, Newman D, Tonnesen MG, MosessonMW, Clark RA. Human fibroblasts bind directly to fi-brinogen at RGD sites through integrin avb3. Exp Cell Res1997: 232: 118–126.

Hakkinen et al.
72. Gailit J, Pierschbacher M, Clark RA. Expression of func-tional a4b1 integrin by human dermal fibroblasts. J InvestDermatol 1993: 100: 323–328.
73. Gailit J, Welch MP, Clark RA. TGF-b1 stimulates expressionof keratinocyte integrins during re-epithelialization of cu-taneous wounds. J Invest Dermatol 1994: 103: 221–227.
74. Gailit J, Xu J, Bueller H, Clark RA. Platelet-derived growthfactor and inflammatory cytokines have differential ef-fects on the expression of integrins a1b1 and a5b1 by hu-man dermal fibroblasts in vitro. J Cell Physiol 1996: 169:281–289.
75. Gardner H, Broberg A, Pozzi A, Laato M, Heino. Absenceof integrin a1b1 in the mouse causes loss of feedbackregulation of collagen synthesis in normal and woundeddermis. J Cell Sci 1999: 112: 263–272.
76. Giancotti FG. Integrin signalling: specificity and controlof cell survival and cell cycle progression. Curr Opin CellBiol 1997: 9: 691–700.
77. Gibbons RJ, Etherden I. Fibronectin-degrading enzymesin saliva and their relation to oral cleanliness. J Peri-odontal Res 1986: 21: 386–395.
78. Goldfinger LE, Hopkinson SB, deHart GW, Collawn S, Co-uchman JR, Jones JC. The a3 laminin subunit, a6b4 anda3b1 integrin coordinately regulate wound healing in cul-tured epithelial cells and in the skin. J Cell Sci 1999: 112:2615–2629.
79. Gould TR, Melcher AH, Brunette DM. Migration and divi-sion of progenitor cell populations in periodontal liga-ment after wounding. J Periodontal Res 1980: 15: 20–42.
80. Goulhen F, Hafezi A, Uitto VJ, Hinode D, Nakamura R,Grenier D, Mayrand D. Subcellular localization and cyto-toxic activity of the GroEL-like protein isolated fromActinobacillus actinomycetemcomitans. Infect Immun1998: 66: 5307–5313.
81. Grande JP. Role of transforming growth factor-b in tissueinjury and repair. Proc Soc Exp Biol Med 1997: 214: 27–40.
82. Greenhalgh DG. The role of growth factors in wound heal-ing. J Trauma 1996: 41: 159–167.
83. Greenwood JA, Murphy-Ullrich JE. Signalling of de-ad-hesion in cellular regulation and motility. Microsc ResTech 1998: 43: 420–432.
84. Greiling D, Clark RA. Fibronectin provides a conduit forfibroblast transmigration from collagenous stroma intofibrin clot provisional matrix. J Cell Sci 1997: 110: 861–870.
85. Grenier D, Chao G, McBride BC. Characterization of so-dium dodecyl sulfate–stable Bacteroides gingivalis pro-teases by polyacrylamide gel electrophoresis. Infect Im-mun 1989: 57: 95–99.
86. Grinnell F. Fibroblasts, myofibroblasts, and wound con-traction. J Cell Biol 1994: 124: 401–404.
87. Grinnell F, Ho CH, Lin YC, Skuta G. Differences in theregulation of fibroblast contraction of floating versusstressed collagen matrices. J Biol Chem 1999: 274: 918–923.
88. Grinnell F, Zhu M, Carlson MA, Abrams JM. Release ofmechanical tension triggers apoptosis of human fibro-blasts in a model of regressing granulation tissue. Exp CellRes 1999: 248: 608–619.
89. Gross J, Farinelli W, Sadow P, Anderson R, Bruns R. On themechanism of skin wound ‘‘contraction’’: a granulationtissue ‘‘knockout’’ with a normal phenotype. Proc NatlAcad Sci U S A 1995: 92: 5982–5986.
146
90. Gullberg D, Tingstrom A, Thuresson AC, Olsson L, Terra-cio L, Borg TK, Rubin K. b1 integrin-mediated collagengel contraction is stimulated by PDGF. Exp Cell Res 1990:186: 264–272.
91. Haapasalmi K, Zhang K, Tonnesen M, Olerud J, SheppardD, Salo T, Kramer R, Clark RAF, Uitto V-J, Larjava H. Kera-tinocytes in human wounds express avb6 integrin. J In-vest Dermatol 1996: 106: 42–48.
92. Hakkinen L, Heino J, Koivisto L, Larjava H. Altered inter-action of human granulation-tissue fibroblasts withfibronectin is regulated by a5b1 integrin. Biochim Bio-phys Acta 1994: 1224: 33–42.
93. Hakkinen L, Larjava H. Characterization of fibroblastclones from periodontal granulation tissue in vitro. J DentRes 1992: 71: 1901–1907.
94. Hakkinen L, Westermarck J, Kahari VM, Larjava H. Hu-man granulation-tissue fibroblasts show enhancedproteoglycan gene expression and altered response toTGF-b1. J Dent Res 1996: 75: 1767–1778.
95. Hashimoto-Uoshima M, Yan YZ, Schneider G, Aukhil I.The alternatively spliced domains EIIIB and EIIIA of hu-man fibronectin affect cell adhesion and spreading. J CellSci 1997: 110: 2271–2280.
96. Hemler ME. Integrin associated proteins. Curr Opin CellBiol 1998: 10: 578–585.
97. Hertle MD, Kubler M-D, Leigh IM, Watt FM. Aberrantintegrin expression during epidermal wound healing andin psoriatic epidermis. J Clin Invest 1992: 89: 1892–1901.
98. Hill SJ, Ebersole JL. The effect of lipopolysaccharide ongrowth factor-induced mitogenesis in human gingivalfibroblasts. J Periodontol 1996: 67: 1274–1280.
99. Hormia M, Thesleff I, Perheentupa J, Pesonen K, Saxen L.Increased rate of salivary epidermal growth factor secre-tion in patients with juvenile periodontitis. Scand J DentRes 1993: 101: 138–144.
100. Howe A, Aplin AE, Alahari SK, Juliano RL. Integrin signal-ling and cell growth control. Curr Opin Cell Biol 1998: 10:220–231.
101. Howell TH, Martuscelli G, Oringer J. Polypeptide growthfactors for periodontal regeneration. Curr Opin Peri-odontol 1996: 3: 149–156.
102. Huhtala P, Humphries MJ, McCarthy JB, Tremble PM,Werb Z, Damsky CH. Cooperative signalling by a5b1 anda4b1 integrins regulates metalloproteinase gene express-ion in fibroblasts adhering to fibronectin. J Cell Biol 1995:129: 867–879.
103. Humphreys-Beher MG, Macauley SP, Chegini N, van Sett-en G, Purushotham K, Stewart C, Wheeler TT, Schultz GS.Characterization of the synthesis and secretion of trans-forming growth factor-a from salivary glands and saliva.Endocrinology 1994: 134: 963–970.
104. Ihara S, Motobayashi Y, Nagao E, Kistler A. Ontogenetictransition of wound healing pattern in rat skin occurringat the fetal stage. Development 1990: 110: 671–680.
105. Irwin CR, Myrillas T, Smyth M, Doogan J, Rice C, SchorSL. Regulation of fibroblast-induced collagen gel contrac-tion by interleukin-1b. J Oral Pathol Med 1998: 27: 255–259.
106. Ivarsson M, Sundberg C, Farrokhnia N, Pertoft H, RubinK, Gerdin B. Recruitment of type I collagen producingcells from the microvasculature in vitro. Exp Cell Res1996: 229: 336–349.
107. Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JCF,

Cell biology of gingival wound healing
Trent JM, Staudt LM, Hudson J Jr, Boguski MS, LashkariD, Shalon D, Botstein D, Brown PO. The transcriptionalprogram in the response of human fibroblasts to serum.Science 1999: 283: 83–87.
108. Juhasz I, Murphy GF, Yan HC, Herlyn M, Albelda SM.Regulation of extracellular matrix proteins and integrincell substratum adhesion receptors on epithelium duringcutaneous human wound healing in vivo. Am J Pathol1993: 43: 1458–1469.
109. Juliano R. Cooperation between soluble factors and inte-grin-mediated cell anchorage in the control of cell growthand differentiation. Bioessays 1996: 18: 911–917.
110. Kainulainen T, Hakkinen L, Hamidi S, Larjava K, Kallioin-en M, Peltonen J, Salo T, Larjava H, Oikarinen A. Essentialrole of laminin-5 during re-epithelialization of wounds. JHistochem Cytochem 1998: 46: 353–360.
111. Kane CJM, Hebda PA, Mansbridge JN, Hamawalt PC. Di-rect evidence for spatial and temporal regulation of trans-forming growth factor-b1 expression during cutaneouswound healing. J Cell Physiol 1991: 148: 157–173.
112. Kanehisa J, Doi S, Yamanaka T, Takeuchi H. Salivaryfibronectin in man: an immunoblotting, radioimmuno-assay and immunohistochemical study. Arch Oral Biol1991: 36:265–272.
113. Kaplony A, Zimmermann DR, Fischer RW, Imhof BA, Od-ermatt BF, Winterhalter KH, Vaughan L. Tenascin Mr
220,000 isoform expression correlates with corneal cellmigration. Development 1991: 112: 605–614.
114. Karring T, Nyman S, Gottlow J, Laurell L. Developmentof the biological concept of guided tissue regeneration –animal and human studies. Periodontol 2000 1993: 1: 26–35.
115. Kilcullen JK, Ly QP, Chang TH, Levenson SM, SteinbergJJ. Nonviable Staphylococcus aureus and its peptidoglycanstimulate macrophage recruitment, angiogenesis, fibro-plasia, and collagen accumulation in wounded rats.Wound Repair Regen 1998: 6: 149–156.
116. Kim JP, Zhang K, Chen JD, Kramer RH, Woodley DT. Vi-tronectin-driven human keratinocyte locomotion is me-diated by the avb5 integrin receptor. J Biol Chem 1994:269: 26926–26932.
117. Kirchberg K, Lange TS, Klein EC, Jungtaubl H, Heinen G,Meyer-Ingold W, Scharffetter-Kochanek K. Induction of b1integrin synthesis by recombinant platelet-derivedgrowth factor (PDGF-AB) correlates with an enhanced mi-gratory response of human dermal fibroblasts to variousextracellular matrix proteins. Exp Cell Res 1995: 220: 29–35.
118. Koivisto L, Larjava K, Hakkinen L, Uitto V-J, Heino J, Lar-java H. Different integrins mediate cell spreading, hapto-taxis and lateral migration of HaCat keratinocytes onfibronectin. Cell Adhes Commun 1999: 7: 245–257.
119. Konturek JW, Bielanski W, Konturek SJ, Bogdal J, Oleksy J.Distribution and release of epidermal growth factor inman. Gut 1989: 30: 1194–1200.
120. Koseki N, Sato H, Yoshizato K. Suppression of growth-as-sociated phosphorylation of proteins of fibroblasts by col-lagen fibrils. Cell Adhes Commun 1996: 3: 463–474.
121. Koseki N, Yoshizato K. Collagen-induced changes in thepattern of protein synthesis of fibroblasts. Cell AdhesCommun 1994: 1: 355–366.
122. Koukoulis GK, Gould VE, Bhattacharyya A, Gould JE,Howeedy AA, Virtanen I. Tenascin in normal, reactive,
147
hyperplastic, and neoplastic tissues: biologic and patho-logic implications. Hum Pathol 1991: 22: 636–643.
123. Koyama H, Raines EW, Bornfeldt KE, Roberts JM, Ross R.Fibrillar collagen inhibits arterial smooth muscle prolifer-ation through regulation of Cdk2 inhibitors. Cell 1996: 87:1069–1078.
124. Krawczyk WS, Wilgram GF. Hemidesmosome and desmo-some morphogenesis during epidermal wound healing. JUltrastruct Res 1973: 45: 93–101.
125. Kurkinen M, Vaheri A, Roberts PJ, Stenman S. Sequentialappearance of fibronectin and collagen in experimentalgranulation tissue. Lab Invest 1980: 43: 47–51.
126. Kyriakides TR, Leach KJ, Hoffman AS, Ratner BD,Bornstein P. Mice that lack the angiogenesis inhibitor,thrombospondin 2, mount an altered foreign body reac-tion characterized by increased vascularity. Proc NatlAcad Sci U S A 1999: 96: 4449–4454.
127. Laato M, Lehtonen OP, Niinikoski J. Granulation tissue for-mation in experimental wounds inoculated with Staphylo-coccus aureus. Acta Chir Scand 1985: 151: 313–318.
128. Laato M, Niinikoski J, Lundberg C, Gerdin B. Inflamma-tory reaction and blood flow in experimental wounds in-oculated with Staphylococcus aureus. Eur Surg Res 1988:20: 33–38.
129. Lamberts BL, Pederson ED, Bial JJ, Tombasco PK.Fibronectin levels of unstimulated saliva from naval re-cruits with and without chronic inflammatory peri-odontal disease. J Clin Periodontol 1989: 16: 342–346.
130. Larjava H. Metabolic change in cultured gingival fibro-blasts exposed to bacterial extracts. Stimulation of hyalu-ronic acid synthesis. J Periodontal Res 1984: 19: 230–237.
131. Larjava H, Haapasalmi K, Salo T, Wiebe C, Uitto V-J. Kera-tinocyte integrins in wound healing and chronic in-flammation of the human periodontium. Oral Dis 1996:2: 77–86.
132. Larjava H, Salo T, Haapasalmi K, Kramer RH, Heino J. Ex-pression of integrins and basement membrane compo-nents by wound keratinocytes. J Clin Invest 1993: 92:1425–1435.
133. Larjava H, Uitto VJ. Effects of extracts from Bacteroidesgingivalis, Bacteroides intermedius, and Bacteroides asac-charolyticus on the growth of fibroblast lines obtainedfrom healthy and inflamed human gingiva. Oral MicrobiolImmunol 1987: 2: 112–116.
134. Larjava H, Uitto VJ, Eerola E, Haapasalo M. Inhibition ofgingival fibroblast growth by Bacteroides gingivalis. InfectImmun 1987: 55: 201–205.
135. Lee W, Aitken S, Sodek J, McCulloch CA. Evidence of adirect relationship between neutrophil collagenase activ-ity and periodontal tissue destruction in vivo: role of ac-tive enzyme in human periodontitis. J Periodontal Res1995: 30: 23–33.
136. Levenson SM, Kan-Gruber D, Gruber C, Molnar J, SeifterE. Wound healing accelerated by Staphylococcus aureus.Arch Surg 1983: 118: 310–320.
137. Levine JH, Moses HL, gold LI, Nanney LMB. Spatial andtemporal patters of immunoreactive transforming growthfactor b1, b2 and b3 during excitional wound repair. AmJ Pathol 1993: 143: 368–380.
138. Lightner VA, Gumkowski F, Bigner DD, Erickson HP. Tena-scin/hexabrachion in human skin: biochemical identifi-cation and localization by light and electron microscopy.J Cell Biol 1989: 108: 2483–2493.

Hakkinen et al.
139. Lin YC, Grinnell F. Decreased level of PDGF-stimulatedreceptor autophosphorylation by fibroblasts in mechanic-ally relaxed collagen matrices. J Cell Biol 1993: 122: 663–672.
140. Lorimier S, Bouthors S, Droulle C, Maquin DL, MaquartFX, Gillery P, Emonard H Hornebeck W. The rate of fi-brinolysis is increased by free retraction of human gingi-val fibroblast populated fibrin lattices. Int J Biochem CellBiol 1997: 29: 181–189.
141. Lorimier S, Gillery P, Hornebeck W, Chastang F, Laurent-Maquin D, Bouthors S, Droulle C, Potron G, Maquart FX.Tissue origin and extracellular matrix control neutral pro-teinase activity in human fibroblast three-dimensionalcultures. J Cell Physiol 1996: 168: 188–198.
142. Lorimier S, Hornebeck W, Godeau G, Pellat B, Gillery P,Maquart FX, Laurent-Maquin D. Morphometric studies ofcollagen and fibrin lattices contracted by human gingivalfibroblasts; comparison with dermal fibroblasts. J DentRes 1998: 77: 1717–1729.
143. Lukashev ME, Werb Z. ECM signalling: orchestrating cellbehaviour and misbehaviour. Trends Cell Biol 1998: 8:437–441.
144. Mackie EJ, Halfter W, Liverani D. Induction of tenascin inhealing wounds. J Cell Biol 1988: 107: 2757–2767.
145. Magnuson VL, Young M, Schattenberg DG, Mancini MA,Chen DL, Steffensen B, Klebe RJ. The alternative splicingof fibronectin pre-mRNA is altered during aging and inresponse to growth factors. J Biol Chem 1991: 266: 14654–14662.
146. Mahaffey KW, Harrington RA, Simoons ML, Granger CB,Graffagnino C, Alberts MJ, Laskowitz DT, Miller JM, SloanMA, Berdan LG, MacAulay CM, Lincoff AM, Deckers J, To-pol EJ, Califf RM. Stroke in patients with acute coronarysyndromes: incidence and outcomes in the platelet glyco-protein IIb/IIIa in unstable angina. Receptor suppressionusing integrilin therapy (PURSUIT) trial. The PURSUITInvestigators. Circulation 1999: 99: 2371–2377.
147. Makela M, Larjava H, Sorsa T, Uitto V-J. Matrix metallo-proteinase 2 (gelatinase A) is related to migration of kera-tinocytes. Exp Cell Res 1999: 251: 67–78.
148. Makela M, Salo T, Larjava H. MMP-9 from TNFa-stimu-lated keratinocytes binds to cell surface and type I colla-gen. Biochem Biophys Res Commun 1998: 253: 325–335.
149. Makela M, Salo T, Uitto VJ, Larjava H. Matrix metallopro-teinases (MMP-2 and MMP-9) of the oral cavity: cellularorigin and relationship to periodontal status. J Dent Res1994: 73: 1397–1406.
150. Manabe R, Ohe N, Maeda T, Fukuda T, Sekiguchi K J.Modulation of cell-adhesive activity of fibronectin by thealternatively spliced EDA segment. J Cell Biol 1997: 139:295–307.
151. Manabe R, Ohe N, Sekiguchi K. Alternatively spliced EDAsegment regulates fibronectin-dependent cell cycle pro-gression and mitogenic signal transduction. J Biol Chem1999: 274: 5919–5924.
152. Mansbridge JN, Hanawalt PC. Role of transforminggrowth factor-b in the maturation of human epidermalkeratinocytes. J Invest Dermatol 1988: 90: 336–341.
153. Martin P. Wound healing-aiming for perfect skin re-generation. Science 1997: 276: 75–81.
154. Martin P, Lewis J. Actin cables and epidermal movementin embryonic wound healing. Nature 1992: 360: 179–183.
155. Massaque J, Cheifetz S, Laiho M, Ralph DA, Weis FM,
148
Zentella A. Transforming growth factor-b. Cancer Surv1992: 12: 81–103.
156. Matsuoka Y, Spring J, Ballmer-Hofer K, Hofer U, Chiquet-Ehrismann R. Differential expression of tenascin splicingvariants in the chick gizzard and in cell cultures. Cell Dif-fer Dev 1990: 32: 417–423.
157. McClain SA, Simon M, Jones E, Nandi A, Gailit JO,Tonnesen MG, Newman D, Clark RA Mesenchymal cellactivation is the rate-limiting step of granulation tissueinduction. Am J Pathol 1996: 149: 1257–1270.
158. McCulloch CAG. Progenitor cell populations in the peri-odontal ligament of mice. Anat Rec 1985: 211: 258–262.
159. Mio T, Adachi Y, Romberger DJ, Ertl RF, Rennard SI. Regu-lation of fibroblast proliferation in three-dimensional col-lagen gel matrix in vitro. Cell Dev Biol Anim 1996: 32:427–433.
160. Miyamoto S, Katz BZ, Lafrenie RM, Yamada KM.Fibronectin and integrins in cell adhesion, signalling, andmorphogenesis. Ann N Y Acad Sci 1998: 857: 119–129.
161. Miyamoto S, Teramoto H, Gutkind JS, Yamada KM. Inte-grins can collaborate with growth factors for phos-phorylation of receptor tyrosine kinases and MAP kinaseactivation: roles of integrin aggregation and occupancy ofreceptors. J Cell Biol 1996: 135: 1633–1642.
162. Mochitate K, Pawelek P, Grinnell F. Stress relaxation ofcontracted collagen gels: disruption of actin filamentbundles, release of cell surface fibronectin and down-regulation of DNA and protein synthesis. Exp Cell Res1991: 193: 198–207.
163. Moghal N, Sternberg PW. Multiple positive and negativeregulators of signalling by the EGF-receptor. Curr OpinCell Biol 1999: 11: 190–196.
164. Montesano R, Orci L. Transforming growth factor b stimu-lates collagen-matrixm contraction by fibroblasts: impli-cations for wound healing. Proc Natl Acad Sci U S A 1988:85: 4894–4897.
165. Moore WE, Moore LV. The bacteria of periodontal dis-eases. Periodontol 2000 1994: 5: 66–77.
166. Munger JS, Harpel JG, Giancotti FG, Rifkin DB. Interac-tions between growth factors and integrins: latent formsof transforming growth factor-b are ligands for the inte-grin avb1. Mol Biol Cell 1998: 9: 2627–2638.
167. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, DaltonSL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rif-kin DB, Sheppard D. The integrin avb6 binds and acti-vates latent TGFb1: a mechanism for regulating pulmon-ary inflammation and fibrosis. Cell 1999: 96: 319–328.
168. Murphy-Ullrich JE, Lane TF, Pallero MA, Sage EH. SPARCmediates focal adhesion disassembly in endothelial cellsthrough a follistatin-like region and the Ca(2π)-bindingEF-hand. J Cell Biochem 1995: 57: 341–350.
169. Murphy-Ullrich JE, Lightner VA, Aukhil I, Yan YZ, EricksonHP, Hook M. Focal adhesion integrity is downregulated bythe alternatively spliced domain of human tenascin. J CellBiol 1991: 115: 1127–1136.
170. Nagler RM, Kitrossky N, Chevion M. Antioxidant activityof rat parotid saliva. Arch Otolaryngol Head Neck Surg1997: 123: 989–993.
171. Narani N, Arora PD, Lew A, Luo L, Glogauer M, Ganss B,McCulloch CA. Transforming growth factor-b induction ofa-smooth muscle actin is dependent on the deformabilityof the collagen matrix. Curr Top Pathol 1999: 93: 47–60.
172. Nickoloff BJ, Mitra RS, Riser BL, Dixit VM, Varani J. Modu-

Cell biology of gingival wound healing
lation of keratinocyte motility. Correlation with produc-tion of extracellular matrix molecules in response togrowth promoting and antiproliferative factors. Am JPathol 1988: 132: 543–551.
173. Niessen CM, Hogervorst F, Jaspars LH, de Melker AA,Delwel GO, Hulsman EH, Kuikman I, Sonnenberg A. Thea6b4 integrin is a receptor for both laminin and kalinin.Exp Cell Res 1994: 211: 360–367.
174. Noguchi S, Ohba Y, Oka T. Effect of salivary epidermalgrowth factor on wound healing of tongue in mice. Am JPhysiol 1991: 260: E620–E6205.
175. Obberghen-Schilling EV, Roche NS, Flanders KC, SpornMB, Roberts AB. Transforming growth factor b1 positivelyregulates its own expression in normal and transformedcells. J Cell Chem 1988: 263: 7741–7746.
176. Odland G, Ross R. Human wound repair. I. Epidermal re-generation. J Cell Biol 1968: 39: 135–151.
177. Okada H, Murakami S. Cytokine expression in periodontalhealth and disease. Crit Rev Oral Biol Med 1998: 9: 248–266.
178. Oksala O, Salo T, Tammi R, Hakkinen L, Jalkanen M, InkiP, Larjava H. Expression of proteoglycans and hyaluronanduring wound healing. J Histochem Cytochem 1995: 43:125–135.
179. Oktay M, Wary KK, Dans M, Birge RB, Giancotti FG. Inte-grin-mediated activation of focal adhesion kinase is re-quired for signalling to jun NH2-terminal kinase and pro-gression through the G1 phase of the cell cycle. J Cell Biol1999: 145: 1461–1470.
180. Oxford GE, Jonsson R, Olofsson J, Zelles T, Humphreys-Beher MG. Elevated levels of human salivary epidermalgrowth factor after oral and juxtaoral surgery. J Oral Max-illofac Surg 1999: 57: 154–158.
181. Oxford GE, Nguyen KH, Alford CE, Tanaka Y, Humphreys-Beher MG. Elevated salivary EGF levels stimulated byperiodontal surgery. J Periodontol 1998: 69: 479–484.
182. Pagani F, Zagato L, Vergani C, Casari G, Sidoli A, BaralleFE. Tissue-specific splicing pattern of fibronectin messen-ger RNA precursor during development and aging in rat.J Cell Biol 1991: 113: 1223–1229.
183. Pereira RF, Halford KW, O’Hara MD, Leeper DB, SokolovBP, Pollard MD, Bagasra O, Prockop DJ. Cultured adherentcells from marrow can serve as long-lasting precursorcells for bone, cartilage, and lung in irradiated mice. ProcNatl Acad Sci U S A 1995: 92: 4857–4861.
184. Phillips GR, Krushel LA, Crossin KL. Domains of tenascininvolved in glioma migration. J Cell Sci 1998: 111: 1095–1104.
185. Phipps RP, Borrello MA, Blieden TM. Fibroblast heteroge-neity in the periodontium and other tissues. J PeriodontalRes 1997: 32: 159–165.
186. Pilcher BK, Dumin JA, Sudbeck BD, Krane SM, WelgusHG, Parks WC. The activity of collagenase-1 is requiredfor keratinocyte migration on a type I collagen matrix. JCell Biol 1997: 137: 1445–1457.
187. Pilcher BK, Wang M, Qin XJ, Parks WC, Senior RM, WelgusHG. Role of matrix metalloproteinases and their inhi-bition in cutaneous wound healing and allergic contacthypersensitivity. Ann N Y Acad Sci 1999: 878: 12–24.
188. Pitaru S, McCulloch CA, Narayanan SA. Cellular originsand differentiation control mechanisms during peri-odontal development and wound healing. J PeriodontalRes 1994: 29: 81–94.
149
189. Pollanen MT, Salonen JI, Grenier D, Uitto V-J. Epithelialcell response to challenge of bacterial lipoteichoic acidsand lipopolysaccharides in vitro. J Med Microbiol 2000:49: 1–8.
190. Preissner KT. Structure and biological role of vitronectin.Annu Rev Cell Biol 1991: 7: 275–310.
191. Prieto AL, Edelman GM, Crossin KL. Multiple integrinsmediate cell attachment to cytotactin/tenascin. Proc NatlAcad Sci U S A 1993: 90: 10154–10158.
192. Prieto AL, Jones FS, Cunningham BA, Crossin KL, Edel-man GM. Localization during development of alterna-tively spliced forms of cytotactin mRNA by in situ hybridi-zation. J Cell Biol 1990: 111: 685–698.
193. Prockop DJ. Marrow stromal cells as stem cells for non-hematopoietic tissues. Science 1997: 276: 71–74.
194. Rao RK, Thomas DW, Pepperl S, Porreca F. Salivary epider-mal growth factor plays a role in protection of ileal mu-cosal integrity. Dig Dis Sci 1997: 42: 2175–2181.
195. Ravanti L, Heino J, Lopez-Otin C, Kahari VM. Inductionof collagenase-3 (MMP-13) expression in human skinfibroblasts by three-dimensional collagen is mediated byp38 mitogen-activated protein kinase. J Biol Chem 1999:274: 2446–2455.
196. Richards DW, MacPhail LA, Dekker N, Greenspan D,Greenspan JS, Lozada-Nur F, Regezi JA. Expression of lam-inin 5, fibronectin, and epithelium-associated integrins inrecurrent aphthous ulcers. J Dent Res 1996: 75: 1512–1517.
197. Riikonen T, Westermarck J, Koivisto L, Broberg A, KahariVM, Heino J. Integrin a2b1 is a positive regulator of col-lagenase (MMP-1) and collagen a1(I) gene expression. JBiol Chem 1995: 270: 13548–13552.
198. Robson MC, Stenberg BD, Heggers JP. Wound healingalterations caused by infection. Clin Plast Surg 1990: 17:485–492.
199. Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL,Dano. Impaired wound healing in mice with a disruptedplasminogen gene. Nat Med 1996: 2: 287–292.
200. Ronnov-Jessen L, Petersen OW. A function for filamen-tous-smooth muscle actin: retardation of motility infibroblasts. J Cell Biol 1996: 134: 67–80.
201. Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellularchanges involved in conversion of normal to malignantbreast: importance of the stromal reaction. Physiol Rev1996: 76: 69–125.
202. Rosenfeldt H, Lee DJ, Grinnell F. Increased c-fos mRNAexpression by human fibroblasts contracting stressed col-lagen matrices. Mol Cell Biol 1998: 18: 2659–2667.
203. Rousselle P, Lunstrum GP, Keene DR, Burgeson RE. Kal-inin: an epithelium-specific basement membrane ad-hesion molecule that is a component of anchoring fila-ments. J Cell Biol 1991: 114: 567–576.
204. Rubbia-Brandt L, Sappino AP, Gabbiani G. Locally appliedGM-CSF induces the accumulation of a-smooth muscleactin containing myofibroblasts. Virchows Arch 1991: 60:73–82.
205. Ruoslahti E. Stretching is good for a cell. Science 1997:276: 1345–1346.
206. Ruoslahti E. Fibronectin and its integrin receptors in can-cer. Adv Cancer Res 1999: 76: 1–20.
207. Saarialho-Kere UK, Kovacs SO, Pentland AP, Olerud JE,Welgus HG, Parks WC. Cell-matrix interactions modulateinterstitial collagenase expression by human keratino-

Hakkinen et al.
cytes actively involved in wound healing. J Clin Invest1993: 92: 2858–2866.
208. Saga Y, Yagi T, Ikawa Y, Sakakura T, Aizawa S. Mice de-velop normally without tenascin. Genes Dev 1992: 6:1821–1831.
209. Salo T, Lyons JG, Rahemtulla F, Birkedal-Hansen H, Larja-va H. Transforming growth factor-b1 up-regulates type IVcollagenase expression in cultured human keratinocytes.J Biol Chem 1991: 266: 11436–11441.
210. Salo T, Makela M, Kylmaniemi M, Autio-Harmainen H,Larjava H. Expression of MMP-2 and MMP-9 (72 kDa and92 kDa type IV collagenases) during early human woundhealing. Lab Invest 1994: 70: 176–182.
211. Salonen J, Uitto VJ, Pan YM, Oda D. Proliferating oral epi-thelial cells in culture are capable of both extracellularand intracellular degradation of interstitial collagen. Ma-trix 1991: 11: 43–55.
212. Sappino AP, Schurch W, Gabbiani G. Differentiation reper-toire of fibroblastic cells: expression of cytoskeletal pro-teins as marker of phenotypic modulations. Lab Invest1990: 63: 144–161.
213. Sarosiek J, Bilski J, Murty VL, Slomiany A, Slomiany BL.Role of salivary epidermal growth factor in the mainten-ance of physicochemical characteristics of oral and gas-tric mucosal mucus coat. Biochem Biophys Res Commun1988: 152: 1421–1427.
214. Schiro JA, Chan BM, Roswit WT, Kassner PD, Pentland AP,Hemler ME, Eisen AZ, Kupper TS. Integrin a2b1 (VLA-2)mediates reorganization and contraction of collagen mat-rices by human cells. Cell 1991: 67: 403–410.
215. Schmid P, Cox D, Bilbe G, McMaster G, Morrison C, StahelinH, Luscher N, Seiler W. TGF-bs and TGF-b type II receptorin human epidermis: differential expression in acute andchronic skin wounds. J Pathol 1993: 171: 191–197.
216. Schmid P, Kunz S, Cerletti N, McMaster G, Cox D. Injuryinduced expression of TGF-b1 is enhanced by exoge-nously applied TGF-bs. Biochem Biophys Res Commun1993: 194: 399–406.
217. Schmitt-Graff A, Desmouliere A, Gabbiani G. Heteroge-neity of myofibroblast phenotypic features: an example offibroblastic cell plasticity. Virchows Arch 1994: 425: 3–24.
218. Schneller M, Vuori K, Ruoslahti E. avb3 integrin associ-ates with activated insulin and PDGF receptors and po-tentiates the biological activity of PDGF. EMBO J 1997: 16:5600–5607.
219. Schor SL, Ellis I, Irwin CR, Banyard J, Seneviratne K, Dol-man C, Gilbert AD, Chisholm DM. Subpopulations of fe-tal-like gingival fibroblasts: characterisation and potentialsignificance for wound healing and the progression ofperiodontal disease. Oral Dis 1996: 2: 155–166.
220. Schultz G, Rotatori DS, Clark W. EGF and TGF-b in woundhealing and repair. J Cell Biochem 1991: 45: 346–352.
221. Schwartz MA, Baron V. Interactions between mitogenicstimuli, or, a thousand and one connections. Curr OpinCell Biol 1999: 11: 197–202.
222. Schwarzbauer JE. Alternative splicing of fibronectin: threevariants, three functions. Bioessays 1991: 13: 527–533.
223. Sciubba JJ, Waterhouse JP, Meyer J. A fine structural com-parison of the healing of incisional wounds of mucosaand skin. J Oral Pathol 1978: 7: 214–227.
224. Sechler JL, Corbett SA, Wenk MB, Schwarzbauer JE.Modulation of cell-extracellular matrix interactions. AnnN Y Acad Sci 1998: 857: 143–154.
150
225. Sempowsky GD, Borello MA, Blieden TM, Barth RK,Phipps RP. Fibroblast heterogeneity in the healing wound.Wound Repair Regen 1995: 3: 120–131.
226. Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, BorsiL, Zardi L, Gabbiani G. The fibronectin domain ED-A iscrucial for myofibroblastic phenotype induction by trans-forming growth factor-b1. J Cell Biol 1998: 142: 873–881.
227. Shapiro SD. Matrix metalloproteinase degradation ofextracellular matrix: biological consequences. Curr OpinCell Biol 1998: 10: 602–608.
228. Shyy JY, Chien S. Role of integrins in cellular responses tomechanical stress and adhesion. Curr Opin Cell Biol 1997:9: 707–713.
229. Singer II, Scott S, Kawka DW, Kazazis DM, Gailit J, Ruos-lahti E. Cell surface distribution of fibronectin and vi-tronectin receptors depends on substrate composition adextracellular matrix accumulation. J Cell Biol 1988: 106:2171–2182.
230. Skaleric U, Kramar B, Petelin M, Pavlica Z, Wahl SM.Changes in TGF-1 levels in gingiva, crevicular fluid andserum associated with periodontal inflammation inhumans and dogs. Eur J Oral Sci 1997: 105: 136–142.
231. Slavin J. The role of cytokines in wound healing. J Pathol1996: 178: 5–10.
232. Sorsa T, Uitto VJ, Suomalainen K, Vauhkonen M, Lindy S.Comparison of interstitial collagenases from human gin-giva, sulcular fluid and polymorphonuclear leukocytes. JPeriodontal Res 1988: 23: 386–393.
233. Sottile J, Hocking DC, Swiatek PJ. Fibronectin matrix as-sembly enhances adhesion-dependent cell growth. J CellSci 1998: 111: 2933–2943.
234. Sporn MB, Roberts AB. Transforming growth factor-b: re-cent progress and new challenges. J Cell Biol 1993: 119:1017–1021.
235. Steffensen B, Duong AH, Milam SB, Potempa CL, Win-born WB, Magnuson VL, Chen D, Zardeneta G, Klebe RJ.Immunohistological localization of cell adhesion proteinsand integrins in the periodontium. J Periodontol 1992: 63:584–592.
236. Stenn KS, Malhotra R. Epithelialization. In: Cohe IK, Dieg-elmann RF, Lindblad WJ, ed. Wound healing. Biochem-ical & clinical aspects. Philadelphia: WB Saunders, 1992:115–127.
237. Stephens P, Davies KJ, al-Khateeb T, Shepherd JP, ThomasDW. A comparison of the ability of intra-oral and extra-oral fibroblasts to stimulate extracellular matrix reorgan-ization in a model of wound contraction. J Dent Res 1996:75: 1358–1364.
238. Stephens P, Genever PG, Wood EJ, Raxworthy MJ. Integrinreceptor involvement in actin cable formation in an invitro model of events associated with wound contraction.Int J Biochem Cell Biol 1997: 29: 121–128.
239. Stepp MA, Zhu L J. Upregulation of a9 integrin and tenas-cin during epithelial regeneration after debridement inthe cornea. J Histochem Cytochem 1997: 45: 189–201.
240. Stomski FC, Gani JS, Bates RC, Burns GF. Adhesion tothrombospondin by human embryonic fibroblasts is me-diated by multiple receptors and includes a role for glyco-protein 88 (CD36). Exp Cell Res 1992: 198: 85–92.
241. Sundberg C, Ivarsson M, Gerdin B, Rubin K. Pericytes ascollagen-producing cells in excessive dermal scarring.Lab Invest 1996: 74: 452–466.
242. Sundberg C, Rubin K. Stimulation of b1 integrins on

Cell biology of gingival wound healing
fibroblasts induces PDGF independent tyrosine phos-phorylation of PDGFb-receptors. J Cell Biol 1996: 132:741–752.
243. Taichman NS, Cruchley AT, Fletcher LM, Hagi-Pavli EP,Paleolog EM, Abrams WR, Booth V, Edwards RM, Mala-mud D. Vascular endothelial growth factor in normal hu-man salivary glands and saliva: a possible role in themaintenance of mucosal homeostasis. Lab Invest 1998:78: 869–875.
244. Taipale J, Saharinen J, Keski-Oja J. Extracellular matrix-associated transforming growth factor-b: role in cancercell growth and invasion. Adv Cancer Res 1998: 75: 87–134.
245. Talonpoika J, Heino J, Larjava H, Hakkinen L, Paunio K.Gingival crevicular fluid fibronectin degradation in peri-odontal health and disease. Scand J Dent Res 1989: 97:415–421.
246. Tenovuo J. Antimicrobial function of human saliva – howimportant is it for oral health? Acta Odontol Scand 1998:56: 250–256.
247. Thesleff I, Partanen AM, Vainio S. Epithelial-mesenchy-mal interactions in tooth morphogenesis: the roles ofextracellular matrix, growth factors, and cell surface re-ceptors. J Craniofac Genet Dev Biol 1991: 11: 229–237.
248. Thesleff I, Viinikka L, Saxen L, Lehtonen E, PerheentupaJ. The parotid gland is the main source of human salivaryepidermal growth factor. Life Sci 1988: 43: 13–18.
249. Thomson P. The microbiology of wounds. J Wound Care1998: 7: 477–478.
250. Tingstrom A, Heldin CH, Rubin K. Regulation of fibro-blast-mediated collagen gel contraction by platelet-de-rived growth factor, interleukin-1 and transforminggrowth factor-b1. J Cell Sci 1992: 102: 315–322.
251. Tomasek JJ, Akiyama SK. Fibroblast-mediated collagen gelcontraction does not require fibronectin-a5b1 integrin in-teraction. Anat Rec 1992: 234: 153–160.
252. Trachslin J, Koch M, Chiquet M. Rapid and reversibleregulation of collagen XII expression by changes in tensilestress. Exp Cell Res 1999: 247: 320–328.
253. Tremble P, Chiquet-Ehrismann R, Werb Z. The extracellu-lar matrix ligands fibronectin and tenascin collaborate inregulating collagenase gene expression in fibroblasts. MolBiol Cell 1994: 5: 439–453.
254. Tremble P, Damsky CH, Werb Z. Components of the nu-clear signalling cascade that regulate collagenase gene ex-pression in response to integrin-derived signals. Cell Biol1995: 129: 1707–1720.
255. Tsao PW, Mousa SA. Thrombospondin mediates calciummobilization in fibroblasts via its Arg-Gly-Asp and car-boxyl-terminal domains. J Biol Chem 1995: 270: 23747–23753.
256. Tuan TL, Song A, Chang S, Younai S, Nimni ME. In vitrofibroplasia: matrix contraction, cell growth, and collagenproduction of fibroblasts cultured in fibrin gels. Exp CellRes 1996: 223: 127–134.
257. Tynelius-Bratthall G. Crevicular and salivary fibronectinbefore and after gingivitis treatment. J Clin Periodontol1988: 15: 283–287.
258. Uitto VJ, Airola K, Vaalamo M, Johansson N, PutninsEE, Firth JD, Salonen J, Lopez-Otin C, Saarialho-Kere U,Kahari VM. Collagenase-3 (matrix metalloproteinase-13)expression is induced in oral mucosal epithelium dur-ing chronic inflammation. Am J Pathol 1998: 152: 1489–1499.
151
259. Uitto VJ, Grenier D, Chan EC, McBride BC. Isolation of achymotrypsinlike enzyme from Treponema denticola. In-fect Immun 1988: 56: 2717–2722.
260. Uitto VJ, Haapasalo M, Laakso T, Salo T. Degradation ofbasement membrane collagen by proteases from someanaerobic oral micro-organisms. Oral Microbiol Immunol1988: 3: 97–102.
261. Uitto VJ, Larjava H. Extracellular matrix molecules andtheir receptors: an overview with special emphasis onperiodontal tissues. Crit Rev Oral Biol Med 1991: 2: 323–354.
262. Uitto VJ, Larjava H, Heino J, Sorsa T. A protease of Bacter-oides gingivalis degrades cell surface and matrix glyco-proteins of cultured gingival fibroblasts and induces se-cretion of collagenase and plasminogen activator. InfectImmun 1989: 57: 213–218.
263. Uitto VJ, Nieminen A, Coil J, Hurttia H, Larjava H. Oralfluid elastase as an indicator of periodontal health. J ClinPeriodontol 1996: 23: 30–37.
264. Uitto VJ, Raeste AM. Activation of latent collagenase ofhuman leukocytes and gingival fluid by bacterial plaque.J Dent Res 1978: 57: 844–851.
265. Uitto VJ, Suomalainen K, Sorsa T. Salivary collagenase.Origin, characteristics and relationship to periodontalhealth. J Periodontal Res 1990: 25: 135–142.
266. Vaalamo M, Weckroth M, Puolakkainen P, Kere J, SaarinenP, Lauharanta J, Saarialho-Kere UK. Patterns of matrixmetalloproteinase and TIMP-1 expression in chronic and-normally healing human cutaneous wounds. Br J Derma-tol 1996: 135: 52–59.
267. Van Waes C. Cell adhesion and regulatory molecules in-volved in tumor formation, hemostasis, and wound heal-ing. Head Neck 1995: 17: 140–147.
268. Varshney AC, Sharma DN, Singh M, Sharma SK, NigamJM. Therapeutic value of bovine saliva in wound healing:a histomorphological study. Indian J Exp Biol 1997: 35:535–537.
269. Wary KK, Mainiero F, Isakoff SJ, Marcantonio EE, Giancot-ti FG. The adaptor protein Shc couples a class of integrinsto the control of cell cycle progression. Cell 1996: 87: 733–743.
270. Weinacker A, Chen A, Agrez M, Cone RI, Nishimura S,Wayner E, Pytela R, Sheppard D. Role of integrin avb6 incell attachment to fibronectin: heterologous expression ofintact and secreted forms of the receptor. J Biol Chem1994: 269: 6940–6948.
271. Welch MP, Odland GF, Clark RA. Temporal relationshipsof F-actin bundle formation, collagen and fibronectinmatrix assembly, and fibronectin receptor expression towound contraction. J Cell Biol 1990: 110: 133–145.
272. Werb Z, Tremble PM, Behrendtsen O, Crowley E, DamskyCH. Signal transduction through the fibronectin receptorinduces collagenase and stromelysin gene expression. JCell Biol 1989: 109: 877–889.
273. Westermarck J, Kahari VM. Regulation of matrix metallo-proteinase expression in tumor invasion. FASEB J 1999:13: 781–792.
274. Whitby DJ, Ferguson MW. The extracellular matrix of lipwounds in fetal, neonatal and adult mice. Development1991: 112: 651–668.
275. Whitby DJ, Longaker MT, Harrison MR, Adzick NS, Fergu-son MWJ. Rapid epithelialization of fetal wounds is as-sociated with the early deposition of tenascin. J Cell Sci1991: 99: 583–586.

Hakkinen et al.
276. Wikesjo UM, Selvig KA. Periodontal wound healing andregeneration. Periodontol 2000 1999: 19: 21–39.
277. Woodley DT. Reepithelialization. In: Clark RAF, ed. Themolecular and cellular biology of wound repair. New York:Plenum Press, 1996: 339–354.
278. Woodley DT, Kalebec T, Banes AJ, Link W, Prunieras M,Liotta. Adult human keratinocytes migrating over nonvi-able dermal collagen produce collagenolytic enzymes thatdegrade type I and type IV collagen. J Invest Dermatol1986: 86: 418–423.
279. Wu C, Chung AE, McDonald JA. A novel role for a3b1 inte-grins in extracellular matrix assembly. J Cell Sci 1995: 108:2511–2523.
280. Wu C, Hughes PE, Ginsberg MH, McDonald JA. Identifi-cation of a new biological function for the integrin avb3:initiation of fibronectin matrix assembly. Cell Adhes Com-mun 1996: 4: 149–158.
281. Xu J, Clark RA. Extracellular matrix alters PDGF regulationof fibroblast integrins. J Cell Biol 1996: 132: 239–249.
282. Xu J, Zutter MM, Santoro SA, Clark RA. PDGF inductionof a2 integrin gene expression is mediated by protein ki-nase C-zeta. J Cell Biol 1996: 134: 1301–1311.
283. Xue W, Mizukami I, Todd RF III, Petty HR. Urokinase-type plasminogen activator receptors associate with b1and b3 integrins of fibrosarcoma cells: dependence onextracellular matrix components. Cancer Res 1997: 57:1682–1689.
284. Yamada KM, Clark RAF. Provisional matrix. In: Clark RAF,ed. The molecular and cellular biology of wound repair.New York: Plenum Press, 1996: 51–93.
285. Yamada Y, Kleinman HK. Functional domains of cell ad-hesion molecules. Curr Opin Cell Biol 1992: 4: 819–823.
286. Yang J, Tyler LW, Donoff RB, Song B, Torio AJ, GallagherGT, Tsuji T, Elovic A, McBride J, Yung CM, Galli SJ, WellerPF, Wong DT. Salivary EGF regulates eosinophil-derivedTGF-a expression in hamster oral wounds. Am J Physiol1996: 270: 191–202.
287. Yang JT, Hynes RO. Fibronectin receptor functions in em-bryonic cells deficient in a5b1 integrin can be replacedby aV integrins. Mol Biol Cell 1996: 7: 1737–1748.
152
288. Yang L, Qiu CX, Ludlow A, Ferguson MW, Brunner G. Ac-tive transforming growth factor-beta in wound repair: de-termination using a new assay. Am J Pathol 1999: 154:105–111.
289. Yeo TK, Brown L, Dvorak HF. Alterations in proteoglycansynthesis common to healing wounds and tumors. Am JPathol 1991: 138: 1437–1450.
290. Yokosaki Y, Monis H, Chen J, Sheppard D. Differential ef-fects of the integrins a9b1, avb3, avb6 on cell proliferativeresponses to tenascin. Roles of the b subunit extracellularand cytoplasmic domains. J Biol Chem 1996: 271: 24144–24150.
291. Yokosaki Y, Palmer EL, Prieto AL, Crossin KL, BourdonMA, Pytela R, Sheppard D. The integrin a9b1 mediatescell attachment to a non-RGD site in the third fibronectintype III repeat of tenascin. J Biol Chem 1994: 269: 26691–26696.
292. Zambruno G, Marchisio PC, Marconi A, Vaschieri C, Mel-chiori A, Giannetti A, De Luca M. Transforming growthfactor-b1 modulates b1 and b5 integrin receptor and in-duces the de novo expression of the avb6 heterodimer innormal keratinocytes: Implications for wound healing. JCell Biol 1995: 129: 853–865.
293. Zelles T, Purushotham KR, Macauley SP, Oxford GE,Humphreys-Beher MG. Saliva and growth factors: thefountain of youth resides in us all. J Dent Res 1995: 74:1826–1832.
294. Zhang, L, Goulhen F, Grenier D, Mayrand D, Uitto V-J.Effects of GroEL-like heat-stress protein of Actinobacillusactinomycetemcomitans on epithelial signal transduction.J Dent Res 1999: 78: 137.
295. Zhong C, Chrzanowska-Wodnicka M, Brown J, Shaub A,Belkin AM, Burridge K. Rho-mediated contractility ex-poses a cryptic site in fibronectin and induces fibronectinmatrix assembly. J Cell Biol 1998: 141: 539–551.
296. Ravanti L, Hakkinen L, Larjava H, Saarialho-Kere U, Fos-chi M, Han J, Kahari V-M. Transforming growth factor-b induces collagenase-3 (MMP-13) expression by humangingival fibroblast via p38 mitogen-activated protein ki-nase. J Biol Chem 1999: 274: 37292–37300.