cardiovascular disease, single nucleotide polymorphisms; and the renin angiotensin system: is there...
TRANSCRIPT

SAGE-Hindawi Access to ResearchInternational Journal of HypertensionVolume 2010, Article ID 281692, 13 pagesdoi:10.4061/2010/281692
Review Article
Cardiovascular Disease, Single Nucleotide Polymorphisms; andthe Renin Angiotensin System: Is There a MicroRNA Connection?
Terry S. Elton,1, 2, 3 Sarah E. Sansom,1 and Mickey M. Martin1
1 Davis Heart and Lung Research Institute, The Ohio State University, DHLRI 515, Columbus, OH 43210, USA2 Division of Pharmacology, College of Pharmacy, The Ohio State University, OH 43210, USA3 Department of Medicine, College of Medicine, Division of Cardiology, The Ohio State University, OH 43210, USA
Correspondence should be addressed to Terry S. Elton, [email protected]
Received 4 May 2010; Accepted 25 June 2010
Academic Editor: Stephen B. Harrap
Copyright © 2010 Terry S. Elton et al. This is an open access article distributed under the Creative Commons Attribution License,which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Essential hypertension is a complex disorder, caused by the interplay between many genetic variants, gene-gene interactions, andenvironmental factors. Given that the renin-angiotensin system (RAS) plays an important role in blood pressure (BP) control,cardiovascular regulation, and cardiovascular remodeling, special attention has been devoted to the investigation of single-nucleotide polymorphisms (SNP) harbored in RAS genes that may be associated with hypertension and cardiovascular disease.MicroRNAs (miRNAs) are a family of small, ∼21-nucleotide long, and nonprotein-coding RNAs that recognize target mRNAsthrough partial complementary elements in the 3′-untranslated region (3′-UTR) of mRNAs and inhibit gene expression bytargeting mRNAs for translational repression or destabilization. Since miRNA SNPs (miRSNPs) can create, destroy, or modifymiRNA binding sites, this review focuses on the hypothesis that transcribed target SNPs harbored in RAS mRNAs, that altermiRNA gene regulation and consequently protein expression, may contribute to cardiovascular disease susceptibility.
1. Introduction
Identifying the genes and mutations that contribute todisease is a central aim in human genetics. Single nucleotidepolymorphisms (SNPs) are mutations that occur at genomepositions at which there are two distinct nucleotide residues(alleles) that each appear in a significant portion (i.e., aminor allele frequency greater than 1%) of the humanpopulation [1]. There are some estimated 14 million SNPs[2] in the human genome that occur at a frequency ofapproximately one in 1,200–1,500 bp [3]. SNPs can affectprotein function by changing the amino acid sequences(nonsynonymous SNP) or by perturbing their regulation(e.g., affecting promoter activity [4], splicing process [5],and DNA and pre-mRNA conformation). When SNPs occurin 3′-UTRs, they may interfere with mRNA stability andtranslation by altering polyadenylation and protein/mRNAregulatory interactions. Recently, a new layer of posttran-scriptional miRNA-mediated gene regulation has been dis-covered and shown to control the expression levels of a largeproportion of genes (reviewed in [6]). Importantly, SNPs in
microRNA (miRNA) target sites (miRSNPs) represent a spe-cific class of regulatory polymorphisms in the 3′-UTR thatmay lead to the dysregulation of posttranscriptional geneexpression. Thus, for miRNAs acting by this mechanism, themiRSNPs may lead to heritable variations in gene expression.Given that the renin angiotensin system (RAS) is intricatelyinvolved in the pathogenesis of cardiovascular disease [7–12],we review and discuss the presently available evidence formiRSNPs-mediated RAS gene regulation and its importancefor phenotypic variation and disease.
2. Current View ofthe Renin Angiotensin System
The RAS plays a critical role in regulating the physiologicalprocesses of the cardiovascular system [reviewed in [7–14]].The primary effector molecule of this system, angiotensinII (Ang II), has emerged as a critical hormone that affectsthe function of virtually all organs, including heart, kidney,vasculature, and brain, and it has both beneficial and

2 International Journal of Hypertension
ACEACE
ACE2
ACE2
Renin
Am A
Am B/N
C C
CCCC
N
NN
NN
N
Angiotensinogen
Angiotensin I
Angiotensin II
Angiotensin III Angiotensin IV
PRR MasR
VasoconstrictionHypertrophyProliferation
Pro-oxidationProfibrosis
VasodilationAntihypertrophyAntiproliferation
AntioxidationAntifibrosis
HypertensionCardiac/vascular remodeling
Atherosclerosis
AntihypertensiveAntiremodeling
Antiatherosclerosis
AT1R AT2R AT4R/IRAP
Angiotensin (1–9)
Angiotensin (1–7)
Figure 1: Summary of the RAS incorporating the Ang peptide family and physiological effects mediated via ATR subtypes. Under theclassical RAS schema, Ang II is produced, via renin and ACE, to act with equal affinity on two ATR subtypes, AT1R and AT2R (large arrows).However, it is now appreciated that a number of breakdown products of Ang II, namely, Ang (1–7), Ang III, and Ang IV exert their ownunique effects that are distinct (and often opposite) to those of Ang II. Such effects are often mediated via newly recognized receptors suchas MasR for Ang (1–7) and AT4R (also known as IRAP) for Ang IV, or additionally via AT2R stimulation. ACE2 is also a new pathway forthe formation of Ang (1–7). Newly identified Ang receptor binding proteins associated with different ATR subtypes may also modify ATRactivation. Thus, overstimulation of AT1R (and PRR) by Ang II, which can contribute to a plethora of cardiovascular disease processes, maybe counter-regulated by a number of non-AT1R mechanisms. Most notably, AT2R stimulation usually causes opposing effects to AT1R, asindicated. It is also likely that the MasR exerts a similar counter-regulatory role whereas the evidence is more preliminary and speculativefor AT4R/IRAP. In terms of mediators, Ang II itself stimulates AT2R whereas the shorter Ang peptides stimulate their cognate receptors andpossibly also AT2R.
pathological effects [7–14]. Acute stimulation with Ang IIregulates salt/water homeostasis and vasoconstriction, mod-ulating blood pressure, while chronic stimulation promoteshyperplasia and hypertrophy of vascular smooth musclecells (VSMCs). In addition, long-term exposure to Ang IIalso plays a pathophysiological role in cardiac hypertro-phy and remodeling, myocardial infarction, hypertension,atherosclerosis, in-stent restenosis, reduced fibrinolysis, andrenal fibrosis [7–14].
Ang II, an octapeptide hormone, is produced sys-temically via the classical RAS and locally via the tissueRAS [7–14]. In the classical RAS, circulating renal-derivedrenin cleaves hepatic-derived angiotensinogen to form thedecapeptide angiotensin I (Ang I), which is converted byangiotensin-converting enzyme (ACE) in the lungs to thebiologically active Ang II (Figure 1). Alternatively, a recentlyidentified carboxypeptidase, ACE2, cleaves one amino acid
from either Ang I or Ang II [15–18], decreasing Ang II levelsand increasing the metabolite Ang 1–7, which has vasodilatorproperties. Thus, the balance between ACE and ACE2 isan important factor controlling Ang II levels [15–18]. AngII is also further degraded by aminopeptidases to Ang III(Ang 2–8) and Ang IV (Ang 3–8) (Figure 1) [7]. Althoughthe RAS was originally regarded as a circulating system,many of its components are localized in tissues, includingthe heart, brain, blood vessels, adrenal, kidney, liver andreproductive organs, indicating the existence of local tissueRASs [19]. In addition to ACE-dependent pathways of AngII formation, non-ACE pathways have also been described.Chymotrypsin-like serine protease (chymase) may representan important mechanism for conversion of Ang I to AngII in the human heart, kidney, and vasculature and maybe particularly important in pathological conditions such ascoronary heart disease [20].

International Journal of Hypertension 3
The biological responses to Ang II are mediated byits interaction with two distinct high-affinity G protein-coupled receptors (GPCRs) designated AT1R and AT2R(Figure 1) [7]. Both AT1R and AT2R possess similar affinityfor Ang II [21]; however, pharmacologically, these receptorscan be distinguished according to inhibition by specificantagonists. For example, AT1R are selectively antagonizedby biphenylimidazoles such as losartan (angiotensin receptorblockers, ARB) [21] whereas tetrahydroimidazopyridinessuch as PD123319 specifically inhibit AT2R [21, 22]. Inter-estingly, all of the classical actions of Ang II, includingvasoconstriction, effects on fluid and electrolyte homeostasis,and influences on cellular growth and differentiation, havebeen shown to be due to stimulation of AT1R located on theplasma membrane of cells [7–12]. Additionally, the majorityof the pathophysiological effects (i.e., cardiac hypertrophyand remodeling, myocardial infarction, hypertension, etc.)of Ang II are also mediated via the AT1R [7–12]. In contrast,it is thought that the AT2R counter-regulates AT1R function(reviewed in [13, 14]). It is also speculated that duringcardiovascular disease, AT2R upregulation and activation byAng II, or angiotensin peptide fragments (i.e., Ang III, AngIV, and/or Ang 1–7) may limit AT1R-mediated overactivityand cardiovascular pathologies [13, 14].
Although the AT1R and AT2R have been intensivelyinvestigated it is now clear that angiotensin fragments canbind to and activate other receptor subtypes. For example,Ang 1–7 acts on the Mas GPCR (MasR) and has vasodilatoryand antiproliferative effects. This arm of the RAS is alsothought to counterbalance the effects of Ang II acting onthe AT1R (Figure 1) (reviewed in [18]). Additionally, AngIV can bind to the angiotensin II type 4 receptor (AT4R)or the membrane-bound, insulin-regulated aminopeptidase(IRAP) (Figure 1) and mediate the enhancement of cognitivefunction, modulate blood flow, increase natriuresis, inhibitcardiomyocyte hypertrophy, and improve endothelial func-tion in animal models of atherosclerosis [23, 24].
Finally, recent studies now suggest that renin, the aspartylprotease that cleaves angiotensinogen into Ang I, andprorenin, its proenzyme inactive form, can bind to what isnow designated as the (pro)renin receptor (PRR) (reviewedin [25–27]). Interestingly, the binding of renin/prorenin toPRR has been shown to have two major consequences. First,the binding of renin to its receptor increases angiotensinogenconversion to Ang I by five-fold, and prorenin, which isvirtually inactive in solution, also displays enzymatic activityfollowing receptor binding [25–27]. Second, receptor-boundrenin/prorenin activates the MAP kinases ERK1/2 andp38 pathways, which in turn, leads to the upregulationof profibrotic and cyclooxygenase-2 genes independent ofAng II generation [25–27]. Therefore, the activation andpotentiation of renin/prorenin enzymatic activity, togetherwith specific PRR-mediated signaling, could have strikingeffects on cardiovascular regulation. Taken together, thesestudies suggest that RAS is unexpectedly complex andmultilayered. New components and functions of the RASare still being unraveled and the physiological significance,and ultimately the clinical relevance, of these factors remainlargely undefined.
3. Overview of miRNA Biology
MicroRNAs (miRNAs) are endogenous, short (20–23nucleotide), and single-stranded nonprotein-coding RNAmolecules that regulate gene expression (reviewed in [28]).These molecules act by binding to their target mRNAs,preferentially to the 3′-UTR, using a partial base-pairingmechanism. In order for a miRNA to give rise to functionalconsequences, the 7-8 nucleotides (nt) at the most 5′ endmust have exact complementarity to the target mRNA,generally referred to as the “seed” region [29]. The currentmodel for inhibition of expression by a miRNA suggeststhat a miRNA either inhibits translation or induces degra-dation of its target mRNA, depending upon the overalldegree of complementarity of the binding site, number ofbinding sites, and the accessibility of those binding sites[30–32].
In mammals, computational predictions indicate thatmiRNAs may regulate 60% of all human protein codinggenes [33], and have been increasingly implicated in thecontrol of various biological processes, including cell dif-ferentiation, cell proliferation, development and apoptosis,and many pathological processes such as cancer, Alzheimer’sdisease, and cardiovascular disease [34–36]. There are esti-mated to be >1,000 miRNAs encoded by the human genome[37, 38], each of which can act on multiple target mRNAs.Conversely, individual mRNAs are commonly targeted bymultiple miRNAs, which results in a combinatorial repres-sion of gene expression more robust than the suppressionthat results from a single miRNA [39, 40]. Although miRNAsare known to mediate posttranscriptional gene silencing inthe cytoplasm, recent evidence indicates that at least somefraction of mammalian miRNAs may also activate or inhibitgene expression at the transcriptional level [41, 42]. Takentogether, these miRNA phenomena allow for enormouscombinatorial complexity and regulatory potential.
4. miRNA Biogenesis
Mature miRNAs are processed from primary miRNA tran-scripts (pri-miRNAs), which are either transcribed fromindependent miRNA genes or are portions of introns ofprotein-coding RNA polymerase II transcripts (Figure 2)[43–45]. miRNAs tend to cluster throughout the genome andmany of these clusters are likely transcribed as polycistrons[46–48]. Although little is known regarding the regulationof miRNA transcription, it is recognized that miRNAexpression is usually regulated by established transcriptionalmechanisms. Interestingly, however, it has been shown thateach miRNA located within the same genomic cluster maybe transcribed and regulated independently [49].
During the transcriptional process, pri-miRNAs foldinto hairpin structures containing imperfectly base-pairedstems and are endonucleolytically cleaved by the nuclearmicroprocessor complex formed by the RNase III typeendonuclease Drosha and the DiGeorge critical region8 (DGCR8) protein [50]. The Drosha/DGCR8 complexprocesses pri-miRNAs into ∼70-nucleotide hairpins knownas pre-miRNAs (Figure 2) [28, 51]. In animals, pre-miRNAs
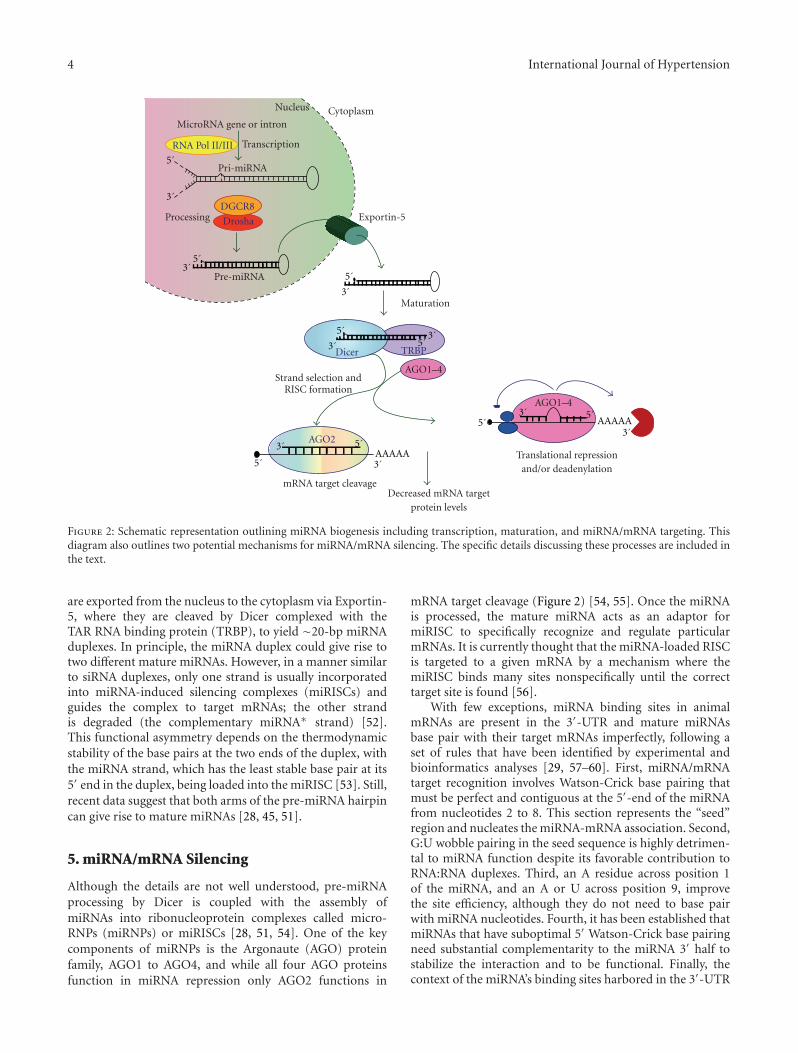
4 International Journal of Hypertension
TRBP
Drosha
DGCR8
RNA Pol II/III
Dicer
AGO2
AAAAA
AAAAA
3´
3´
3´
3´
3´
3´
3´
3´
3´
5´
5´
5´
5´
5´
5´
5´
5´
5´
CytoplasmNucleus
MicroRNA gene or intron
Transcription
Pri-miRNA
Processing
Pre-miRNA
AGO1–4
AGO1–4
Exportin-5
Maturation
Strand selection andRISC formation
mRNA target cleavage
Translational repressionand/or deadenylation
Decreased mRNA targetprotein levels
Figure 2: Schematic representation outlining miRNA biogenesis including transcription, maturation, and miRNA/mRNA targeting. Thisdiagram also outlines two potential mechanisms for miRNA/mRNA silencing. The specific details discussing these processes are included inthe text.
are exported from the nucleus to the cytoplasm via Exportin-5, where they are cleaved by Dicer complexed with theTAR RNA binding protein (TRBP), to yield ∼20-bp miRNAduplexes. In principle, the miRNA duplex could give rise totwo different mature miRNAs. However, in a manner similarto siRNA duplexes, only one strand is usually incorporatedinto miRNA-induced silencing complexes (miRISCs) andguides the complex to target mRNAs; the other strandis degraded (the complementary miRNA∗ strand) [52].This functional asymmetry depends on the thermodynamicstability of the base pairs at the two ends of the duplex, withthe miRNA strand, which has the least stable base pair at its5′ end in the duplex, being loaded into the miRISC [53]. Still,recent data suggest that both arms of the pre-miRNA hairpincan give rise to mature miRNAs [28, 45, 51].
5. miRNA/mRNA Silencing
Although the details are not well understood, pre-miRNAprocessing by Dicer is coupled with the assembly ofmiRNAs into ribonucleoprotein complexes called micro-RNPs (miRNPs) or miRISCs [28, 51, 54]. One of the keycomponents of miRNPs is the Argonaute (AGO) proteinfamily, AGO1 to AGO4, and while all four AGO proteinsfunction in miRNA repression only AGO2 functions in
mRNA target cleavage (Figure 2) [54, 55]. Once the miRNAis processed, the mature miRNA acts as an adaptor formiRISC to specifically recognize and regulate particularmRNAs. It is currently thought that the miRNA-loaded RISCis targeted to a given mRNA by a mechanism where themiRISC binds many sites nonspecifically until the correcttarget site is found [56].
With few exceptions, miRNA binding sites in animalmRNAs are present in the 3′-UTR and mature miRNAsbase pair with their target mRNAs imperfectly, following aset of rules that have been identified by experimental andbioinformatics analyses [29, 57–60]. First, miRNA/mRNAtarget recognition involves Watson-Crick base pairing thatmust be perfect and contiguous at the 5′-end of the miRNAfrom nucleotides 2 to 8. This section represents the “seed”region and nucleates the miRNA-mRNA association. Second,G:U wobble pairing in the seed sequence is highly detrimen-tal to miRNA function despite its favorable contribution toRNA:RNA duplexes. Third, an A residue across position 1of the miRNA, and an A or U across position 9, improvethe site efficiency, although they do not need to base pairwith miRNA nucleotides. Fourth, it has been established thatmiRNAs that have suboptimal 5′ Watson-Crick base pairingneed substantial complementarity to the miRNA 3′ half tostabilize the interaction and to be functional. Finally, thecontext of the miRNA’s binding sites harbored in the 3′-UTR

International Journal of Hypertension 5
of target mRNAs, also influence the functional importanceof these sites [61]. For example, miRNA site efficacy can beimproved if the site is positioned at least 15 nt downstreamfrom the stop codon, away from the center of long 3′-UTRs,and near AU-rich nucleotide regions. These factors can makethe 3′-UTR regions less structured and hence more accessibleto miRNP recognition.
When an endogenous miRISC programmed with miRNAbinds to a recognition site that is perfectly complementary,this target mRNA will be cleaved by the miRISC (Figure 2)[56, 62–65]. In contrast, miRISCs that are imperfectlymatched with target mRNAs can repress translation initia-tion at either the cap-recognition stage [66–70] or the 60Ssubunit joining stage [71]. Alternatively, binding of miRISCscan induce deadenylation and decay of target mRNAs [62].
6. Computational Algorithms toPredict miRNA/mRNA Targets
Computational miRNA/mRNA target programs remain theonly source for rapid prediction of miRNA recognition sitesharbored within the 3′-UTR of target mRNAs. Therefore,the development of reliable computational target predictionprograms is critical in advancing our understanding ofmiRNA function. Given that miRNA functionality usuallyrequires seed sequence complementarity [28, 61] the mainprediction feature used in most of these programs is thesequence alignment of the miRNA seed to the 3′-UTR of can-didate target genes. Additionally, many current algorithmsalso utilize conservation of miRNA/mRNA target sites acrossspecies as an important parameter for the identification ofbona fide targets; notably however, the conservation of amiRNA binding site harbored in a given mRNA target is nota requirement for a functional miRNA.
A recent review article [72] comparing eight of themost commonly used algorithms for miRNA target pre-diction for the human and mouse genome programsdemonstrated that the four top algorithms, DIANA-microT 3.0 (http://microrna.gr/microT) [73], TargetScan 5.0(http://www.targetscan.org/) [74], Pictar (http://pictar.org/)[75], and ElMMo (from http://www.mirz.unibas.ch) [76] allhave a precision of ∼50% with a sensitivity that ranges from6% to 12%. Of the top four performing programs, it isimportant to note that TargetScan is the most up-to-dateregarding the number of miRNAs and genes used and Pictaris least updated [72]. Most investigators assume that mRNAtargets predicted by more than one algorithm are moreaccurate than other targets thus leading to higher predictionprecision. However, Alexiou et al. [72] demonstrated thatmany of the algorithm combinations performed worse thanthe prediction of a single algorithm. These investigatorsreason that the better specificity of a combination is achievedby a higher price for the sensitivity. Taking this intoaccount, our laboratory has analyzed all of the classical andnonclassical RAS components for putative miRNA bindingsites by the TargetScan algorithm (Table 1). Importantly, thisanalysis suggests that miRNAs may play a major role inregulating the expression of RAS proteins.
Although many programs are available online for theprediction of individual mRNA targets of miRNAs (seeabove), the identification of authentic mRNA targets remainsproblematic. Mammalian miRNAs bind to the mRNA withimperfect complementarity thus how binding sites arerecognized is only partially understood. Therefore, somebioinformatically predicted targets turn out to be falseand others are entirely overlooked. Experimental validationof targets is therefore an important step in defining thefunctions of individual miRNAs (for review, see [77]).
7. Experimentally ValidatedmiRNA/RAS Targets
Although classical and nonclassical RAS components harborputative miRNA binding sites, very few of these siteshave been experimentally validated. Our laboratory hasdemonstrated that miR-155 specifically interacted withthe algorithm-predicted binding site harbored in the 3′-UTR of the human AT1R (hAT1R) mRNA (Table 1) [78,79]. Additionally, miR-155 gain-of-function experiments(i.e., cells were transfected with partially double-strandedRNAs that mimic the Dicer cleavage product and aresubsequently processed into their respective mature miR-NAs) inhibited the expression of the hAT1R and alsoattenuated Ang II-induced signaling via the hAT1R infibroblasts and vascular smooth muscle cells (VSMCs)[78, 79]. These results also demonstrated that transfectionwith miR-155 did not significantly decrease hAT1R steadystate mRNA levels, suggesting that miR-155 can decreasehAT1R expression by inhibiting translation of the mRNA,rather than targeting it for degradation. In contrast, loss-of-function experiments (i.e., cells were transfected withmiRNA inhibitors; antisense single-stranded chemically-enhanced oligonucleotides, ASO) demonstrated that trans-fection of anti-miR-155 not only increased hAT1R expres-sion but also enhanced Ang II-induced signaling via thehAT1R, indicating that miR-155 plays a physiological rolein regulating the expression of hAT1Rs in human fibroblastsand VSMCs [78, 79]. Recently, our laboratory also demon-strated that hAT1R expression can be regulated by miR-802[80].
In support of the miR-155/hAT1R studies describedabove, trisomy 21 (Ts21) mediated overexpression of miR-155 (the bic/miR-155 gene is located on human chromosome21 and is triplicated in Down syndrome [DS] individuals)[81, 82] resulted in the attenuation of hAT1R protein levelsin fibroblasts isolated from one monozygotic twin with DSwhen compared to fibroblasts isolated from the unaffectedeuploid twin [83]. Interestingly, individuals with DS havesignificantly lower systolic and diastolic blood pressures[84–87], a reduced risk of vascular anomalies [88], anda low prevalence of coronary artery disease [89–93] whencompared with the general population. Given that the over-expression of miR-155 in Ts21 results in attenuated hAT1Rprotein levels, we speculate that this may be one mechanismwhich contributes to the lack of cardiovascular diseaseobserved in individuals with DS [84–87].

6 International Journal of Hypertension
Table 1: TargetScan∗ algorithm-predicted RAS putative miRNA/mRNA target sites.
RAS Component Total Conserved miRNA Targets Total Poorly Conserved miRNA Targets Overall Total miRNA Targets
AGTR1 (AT1R) 0 56 56
AGTR2 (AT2R) 2 96 98
ACE 0 26 26
ACE2 1 57 58
AGT 1 31 32
REN 0 15 15
ATP6AP2 (PRR) 4 49 53
MAS1 (MasR) 0 12 12
LNPEP (AT4R) 5 42 47∗
Source: TargetScan (April, 2009). The number of potential targets is dramatically dependent upon the algorithm utilized.
Finally, Boettger et al. [94] demonstrated by genomic,proteomic, and transcriptional analyses that mouse ACEmRNA is a miR-145 target. The miR-143/-145 gene clusterincludes miR-143 and miR-145, which lie within a 1.7-kbhighly conserved region of mouse chromosome 18 [94].miRNA microarray hybridization experiments revealed thatmiR-143 and miR-145 are enriched in murine vascularsmooth muscle cells (VSMCs) [94, 95]. In the mouseembryo, miR-143/-145 expression is restricted to heart, vas-cular, and visceral SMCs [94, 95]. During late fetal and post-natal development, miR-143/-145 expression is downregu-lated in the heart but persists in vascular and visceral SMCs.Homozygous miR-143/-145 knockout mice were viable butexhibited thinning of the arterial tunica media (muscularlayer), with a reduction in the number of contractile VSMCsand a concomitant increase in the number of proliferativeVSMCs [94]. Physiological characterization of miR-143/-145 deficient mice and arterial segments from these animalsrevealed defects in Ang II-induced VSMC contractility andhomeostatic control of blood pressure. Consistent with thisobservation, pharmacological inhibition of ACE or the AT1Rpartially reversed vascular dysfunction and normalized geneexpression in the mutant mice [94]. Since miR-145 regulatesthe expression of ACE, when the miR-143/-145 gene clusteris mutated, the levels of membrane-bound ACE in VSMCsincrease, causing the chronic stimulation of VSMCs by AngII, which in turn results in desensitization and “angiotensinresistance” of VSMCs. Given that miR-143/145 mutant micedeveloped neointimal lesions in the absence of hyperlidemia,lipid depositions, and foam cells also highlights the potentialrole of VSMCs in the pathogenetic process leading toatherosclerosis. The enhancement of Ang II signaling due tothe increased expression of ACE would certainly contributeto this process, since increased levels of Ang II have beenshown to promote atherosclerotic lesions in apoE-deficientmice [96].
Although the hAT1R and mouse ACE are experimentallyvalidated targets of miRNAs, it is important to note thatnone of these recognition sites are conserved across species(Table 1). Therefore, although miR-155 and -802 represshAT1R expression in humans, these miRNAs will not lead tothe repression of AT1R levels in mice or rats. Likewise, miR-
145 will repress ACE expression in mice but will not regulateACE levels in humans.
8. miRSNPs
Since a large number of miRNA binding sites are harboredin the 3′-UTRs of RAS component mRNAs (Table 1), thereis a high probability that SNPs will occur within miRNAtarget sites. By definition, miRSNPs have the potential tocreate, destroy, or modify the efficiency of miRNA binding,if the SNP occurs in the seed region (i.e., the region ofbase-pairing between nucleotides 2 and 8 of the miRNA andcomplementary nucleotides in the target mRNA) [97–99].Based on this definition, there are two mechanisms by whichmiRSNPs can be functionally important: as a gain- or as aloss-of-function variation. A gain-of-function effect wouldresult if the SNP enhances the targeting of the miRNA orcreates a new target site in the 3′-UTR of the mRNA. Inthis scenario, protein expression of the target mRNA wouldbe attenuated. In contrast, a loss-of-function effect wouldresult when the SNP decreases or abolishes the interactionof the miRNA with its mRNA target, thus resulting inan augmentation of protein expression. In support of thisconclusion, miRSNPs have been implicated in Tourettesyndrome [100], papillary thyroid cancer [101], muscularityin sheep [102], hereditary spastic paraplegia type 31 [103],methotrexate resistance [104], breast cancer [105], andtumor susceptibility [106]. These examples include bothgain- and loss-of-function miRSNPs.
9. The Human AT1R Gene and miRSNPs
The hAT1R gene has been found to be highly polymorphic[107]. In particular, a SNP has been described in which thereis an A/C transversion at position +1166 (i.e., 1166 base-pairs downstream from the start codon, dsSNP# rs5186)located in the 3′-UTR of the hAT1R gene. The increasedfrequency of the +1166 C-allele has been associated withhypertension [108–115], cardiac hypertrophy [116–118],aortic stiffness [119–121], myocardial infarction [122], heartfailure [123–125], abdominal aortic aneurysms [126, 127],

International Journal of Hypertension 7
3´
3´
G G G G A U A G U G C U A A U C G U A A U U
5´
5´
U U C A C U A C C A - A A U G A G C A U U A G . .. .
miR-155
hAT1R mRNA(A-allele)
(a)
3´
3´5´
5´
. .. .
G G G G A U A G U G C U A A U C G U A A U U
U U C A C U A C C A - A A U G A G C C U U A G
miR-155
hAT1R mRNA(C-allele)
(b)
Figure 3: The human AT1R +1166 A/C SNP occurs in the miR-155-binding site. (a) Complementarity between miR-155 and the hAT1R3′-UTR site targeted (70–90 bp downstream from the human AT1R stop codon). The +1166 A/C SNP corresponds to the nucleotide86 bp downstream from the human AT1R stop codon (shown in red print). The binding of miR-155 to the hAT1R 3′-UTR target sitefulfills the requirement of a 7-bp seed sequence of complementarity at the miRNA 5′ end when the +1166 A-allele is expressed. (b)Complementarity between miR-155 and the human AT1R 3′-UTR harboring the +1166 C-allele. If the +1166 C-allele is expressed, theseed sequence requirement would not be met and, as a consequence, it would be expected that human AT1R expression would be elevated[79].
and increased oxidative stress levels in human heart failure[128]. However, the physiological relevance of this poly-morphism is uncertain because of its location within thenoncoding region of the hAT1R gene. Since our laboratoryhas previously demonstrated that miR-155 interacted with aspecific cis-response element localized in the hAT1R 3′-UTR[78], we investigated whether or not there was a correlationbetween the +1166 A/C SNP and the miR-155-binding site[79, 83]. Importantly, computer alignment revealed that the+1166 A/C SNP occurs within the cis-response element atthe site where miR-155 was shown to interact (Figure 3).The interaction between miR-155 and the hAT1R 3′-UTRharboring the A-allele fulfills the seed sequence rules [29]since there is a 7-bp region of complementarity betweenthe 5′ end of miR-155 and the hAT1R mRNA target site(Figure 3(a)). In contrast, if a hAT1R mRNA that harborsthe +1166 C-allele is expressed, the complementary seedsite is interrupted (Figure 3(b)), and the thermodynamicsof the miRNA:mRNA duplex would be significantly altered(i.e., a decrease in free energy) [79]. Therefore, the presenceof the +1166 C-allele miRSNP would decrease the abilityof miR-155 to interact with the cis-regulatory site locatedin the hAT1R 3′-UTR. As a consequence, it would beexpected that aberrantly high levels of the hAT1R would besynthesized. In support of this hypothesis we demonstratedthat when the hAT1R cis-response element harboring the C-allele was present in luciferase mRNAs, the ability of miR-155 to inhibit luciferase activity was significantly attenuated[79]. When identical experiments were performed utilizingmutant miR-155, which restored perfect Watson-Crick com-plementarity, luciferase activity was decreased to levels thatwere comparable with experiments utilizing the hAT1R cis-response element harboring the A-allele and miR-155 [79].To further demonstrate that the presence of the +1166 C-allele can influence hAT1R density, expression constructs thatproduced hAT1R mRNAs containing either the A- or C-allelewere cotransfected with miR-155 or mut-miR-155. Theseexperiments again demonstrated that when seed sequence
complementarity was not fulfilled, regardless of whethermiR-155 or mutant miR-155 was utilized, hAT1R levelswere always higher than the levels obtained when perfectcomplementarity was present between the miRNA and thehAT1R cis-response element [79]. Taken together, thesestudies provide the first feasible biochemical mechanism bywhich the +1166 A/C polymorphism (i.e., miRSNP) canlead to increased hAT1R densities and possibly cardiovasculardisease.
10. miRSNPS and RAS Component Genes
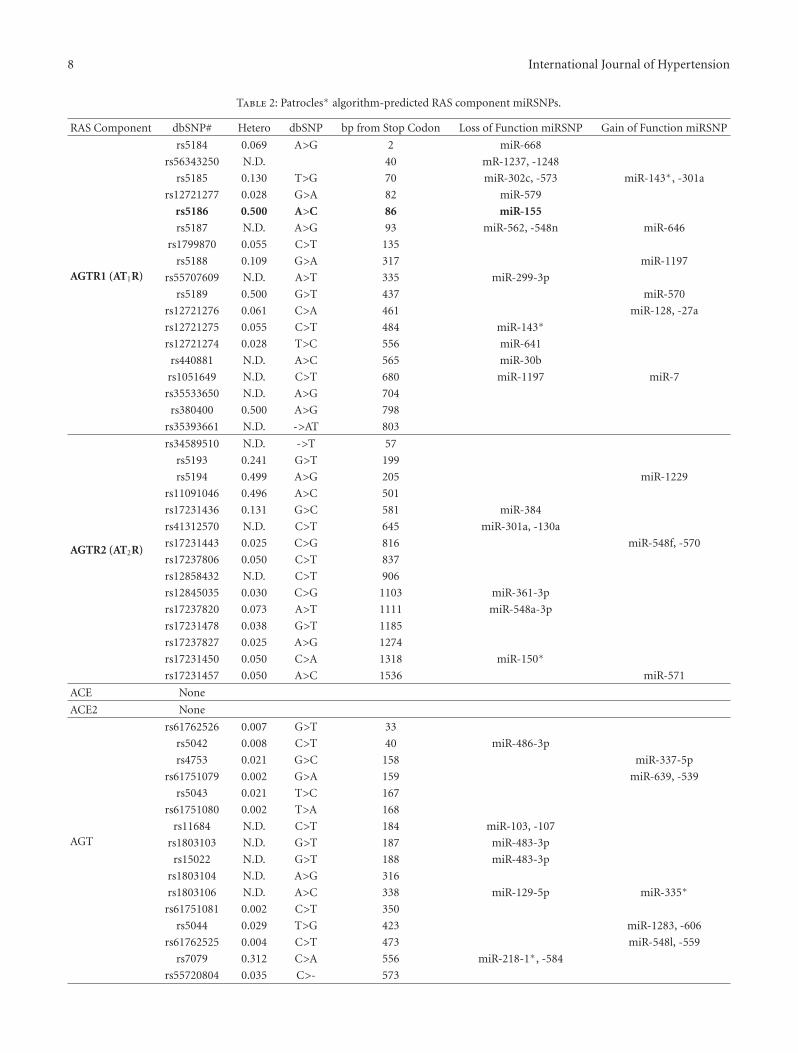
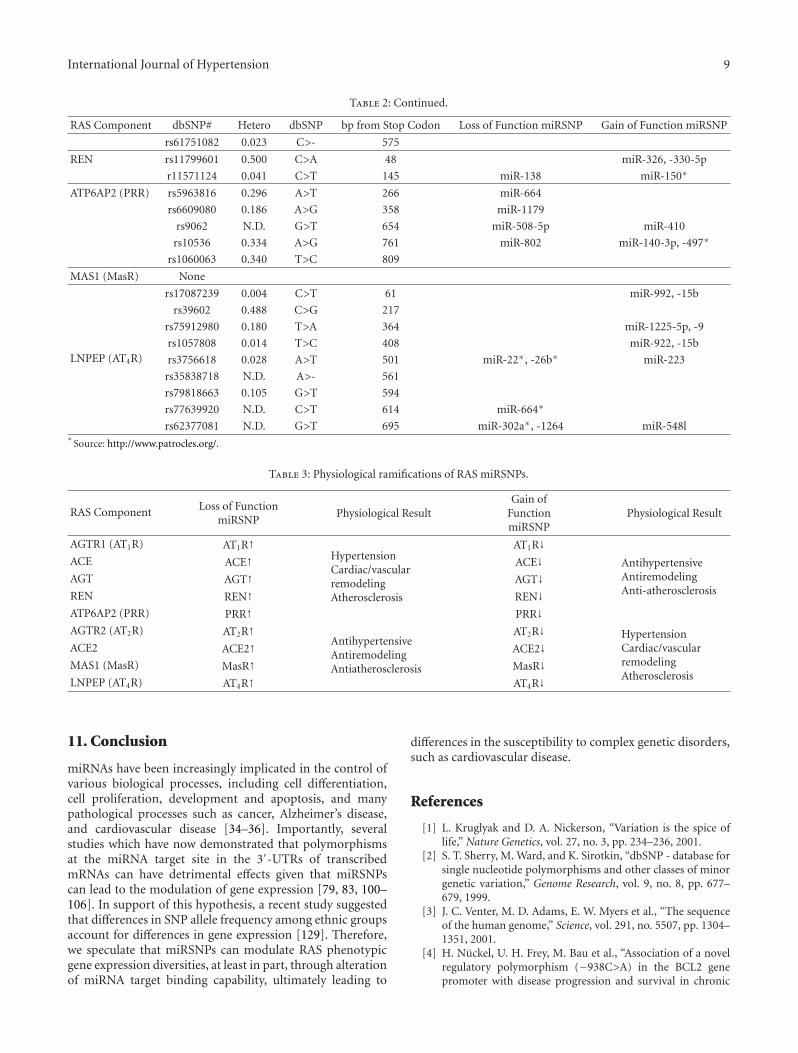
To begin to investigate whether other RAS componentsharbored SNPs in their 3′-UTRs which created miRSNPs,the SNP Geneview Report (http://www.ncbi.nlm.nih.gov/nucleotide/) for each gene was surveyed and all of the iden-tified SNPs were analyzed utilizing the Patrocles algorithm(http://www.patrocles.org/) [99]. Specifically the “PatroclesFinder” was utilized since it allows one to compare twosequences and subsequently determines the miRNA bindingsites that are different between the two sequences. Thus,a region of the transcribed 3′-UTR which harbored thecommon allele was compared with the same region contain-ing the mutated allele (select motif length: human 7 mers)and the generation of putative miRSNPs was determined(Table 2). Importantly, this analysis demonstrated that the3′-UTRs of the RAS components harbor a number ofmiRSNPs some of which alter or destroy legitimate miRNAbinding sites and others that create novel, illegitimatetarget sites. Hypothetically, if a loss-of-function miRSNPoccurred in the AGTR1, ACE, AGT, REN, or ATP6AP2 gene,an increased incidence of hypertension, cardiac/vascularremodeling, and atherosclerosis would be observed (Table 3).In contrast, if a loss-of-function miRSNP occurred inthe AGTR2, ACE2, MAS1, or LNPEP gene, a decreasedincidence of hypertension, cardiac/vascular remodeling, andatherosclerosis would be expected (Table 3).
8 International Journal of Hypertension
Table 2: Patrocles∗ algorithm-predicted RAS component miRSNPs.
RAS Component dbSNP# Hetero dbSNP bp from Stop Codon Loss of Function miRSNP Gain of Function miRSNP
AGTR1 (AT1R)
rs5184 0.069 A>G 2 miR-668
rs56343250 N.D. 40 mR-1237, -1248
rs5185 0.130 T>G 70 miR-302c, -573 miR-143∗, -301a
rs12721277 0.028 G>A 82 miR-579
rs5186 0.500 A>C 86 miR-155
rs5187 N.D. A>G 93 miR-562, -548n miR-646
rs1799870 0.055 C>T 135
rs5188 0.109 G>A 317 miR-1197
rs55707609 N.D. A>T 335 miR-299-3p
rs5189 0.500 G>T 437 miR-570
rs12721276 0.061 C>A 461 miR-128, -27a
rs12721275 0.055 C>T 484 miR-143∗
rs12721274 0.028 T>C 556 miR-641
rs440881 N.D. A>C 565 miR-30b
rs1051649 N.D. C>T 680 miR-1197 miR-7
rs35533650 N.D. A>G 704
rs380400 0.500 A>G 798
rs35393661 N.D. ->AT 803
AGTR2 (AT2R)
rs34589510 N.D. ->T 57
rs5193 0.241 G>T 199
rs5194 0.499 A>G 205 miR-1229
rs11091046 0.496 A>C 501
rs17231436 0.131 G>C 581 miR-384
rs41312570 N.D. C>T 645 miR-301a, -130a
rs17231443 0.025 C>G 816 miR-548f, -570
rs17237806 0.050 C>T 837
rs12858432 N.D. C>T 906
rs12845035 0.030 C>G 1103 miR-361-3p
rs17237820 0.073 A>T 1111 miR-548a-3p
rs17231478 0.038 G>T 1185
rs17237827 0.025 A>G 1274
rs17231450 0.050 C>A 1318 miR-150∗
rs17231457 0.050 A>C 1536 miR-571
ACE None
ACE2 None
AGT
rs61762526 0.007 G>T 33
rs5042 0.008 C>T 40 miR-486-3p
rs4753 0.021 G>C 158 miR-337-5p
rs61751079 0.002 G>A 159 miR-639, -539
rs5043 0.021 T>C 167
rs61751080 0.002 T>A 168
rs11684 N.D. C>T 184 miR-103, -107
rs1803103 N.D. G>T 187 miR-483-3p
rs15022 N.D. G>T 188 miR-483-3p
rs1803104 N.D. A>G 316
rs1803106 N.D. A>C 338 miR-129-5p miR-335∗
rs61751081 0.002 C>T 350
rs5044 0.029 T>G 423 miR-1283, -606
rs61762525 0.004 C>T 473 miR-548l, -559
rs7079 0.312 C>A 556 miR-218-1∗, -584
rs55720804 0.035 C>- 573
International Journal of Hypertension 9
Table 2: Continued.
RAS Component dbSNP# Hetero dbSNP bp from Stop Codon Loss of Function miRSNP Gain of Function miRSNP
rs61751082 0.023 C>- 575
REN rs11799601 0.500 C>A 48 miR-326, -330-5p
r11571124 0.041 C>T 145 miR-138 miR-150∗
ATP6AP2 (PRR) rs5963816 0.296 A>T 266 miR-664
rs6609080 0.186 A>G 358 miR-1179
rs9062 N.D. G>T 654 miR-508-5p miR-410
rs10536 0.334 A>G 761 miR-802 miR-140-3p, -497∗
rs1060063 0.340 T>C 809
MAS1 (MasR) None
LNPEP (AT4R)
rs17087239 0.004 C>T 61 miR-992, -15b
rs39602 0.488 C>G 217
rs75912980 0.180 T>A 364 miR-1225-5p, -9
rs1057808 0.014 T>C 408 miR-922, -15b
rs3756618 0.028 A>T 501 miR-22∗, -26b∗ miR-223
rs35838718 N.D. A>- 561
rs79818663 0.105 G>T 594
rs77639920 N.D. C>T 614 miR-664∗
rs62377081 N.D. G>T 695 miR-302a∗, -1264 miR-548l∗
Source: http://www.patrocles.org/.
Table 3: Physiological ramifications of RAS miRSNPs.
RAS Component Loss of FunctionmiRSNP
Physiological ResultGain of
FunctionmiRSNP
Physiological Result
AGTR1 (AT1R) AT1R↑HypertensionCardiac/vascularremodelingAtherosclerosis
AT1R↓AntihypertensiveAntiremodelingAnti-atherosclerosis
ACE ACE↑ ACE↓AGT AGT↑ AGT↓REN REN↑ REN↓ATP6AP2 (PRR) PRR↑ PRR↓AGTR2 (AT2R) AT2R↑
AntihypertensiveAntiremodelingAntiatherosclerosis
AT2R↓ HypertensionCardiac/vascularremodelingAtherosclerosis
ACE2 ACE2↑ ACE2↓MAS1 (MasR) MasR↑ MasR↓LNPEP (AT4R) AT4R↑ AT4R↓
11. Conclusion
miRNAs have been increasingly implicated in the control ofvarious biological processes, including cell differentiation,cell proliferation, development and apoptosis, and manypathological processes such as cancer, Alzheimer’s disease,and cardiovascular disease [34–36]. Importantly, severalstudies which have now demonstrated that polymorphismsat the miRNA target site in the 3′-UTRs of transcribedmRNAs can have detrimental effects given that miRSNPscan lead to the modulation of gene expression [79, 83, 100–106]. In support of this hypothesis, a recent study suggestedthat differences in SNP allele frequency among ethnic groupsaccount for differences in gene expression [129]. Therefore,we speculate that miRSNPs can modulate RAS phenotypicgene expression diversities, at least in part, through alterationof miRNA target binding capability, ultimately leading to
differences in the susceptibility to complex genetic disorders,such as cardiovascular disease.
References
[1] L. Kruglyak and D. A. Nickerson, “Variation is the spice oflife,” Nature Genetics, vol. 27, no. 3, pp. 234–236, 2001.
[2] S. T. Sherry, M. Ward, and K. Sirotkin, “dbSNP - database forsingle nucleotide polymorphisms and other classes of minorgenetic variation,” Genome Research, vol. 9, no. 8, pp. 677–679, 1999.
[3] J. C. Venter, M. D. Adams, E. W. Myers et al., “The sequenceof the human genome,” Science, vol. 291, no. 5507, pp. 1304–1351, 2001.
[4] H. Nuckel, U. H. Frey, M. Bau et al., “Association of a novelregulatory polymorphism (−938C>A) in the BCL2 genepromoter with disease progression and survival in chronic

10 International Journal of Hypertension
lymphocytic leukemia,” Blood, vol. 109, no. 1, pp. 290–297,2007.
[5] M. Krawczak, J. Reiss, and D. N. Cooper, “The mutationalspectrum of single base-pair substitutions in mRNA splicejunctions of human genes: causes and consequences,” HumanGenetics, vol. 90, no. 1-2, pp. 41–54, 1992.
[6] D. P. Bartel, “MicroRNAs: target Recognition and RegulatoryFunctions,” Cell, vol. 136, no. 2, pp. 215–233, 2009.
[7] M. De Gasparo, K. J. Catt, T. Inagami, J. W. Wright, and TH.Unger, “International union of pharmacology. XXIII. Theangiotensin II receptors,” Pharmacological Reviews, vol. 52,no. 3, pp. 415–472, 2000.
[8] W. G. Thomas and F. A. O. Mendelsohn, “Angiotensinreceptors: form and function and distribution,” InternationalJournal of Biochemistry and Cell Biology, vol. 35, no. 6, pp.774–779, 2003.
[9] L. Hunyady and K. J. Catt, “Pleiotropic AT1 receptorsignaling pathways mediating physiological and pathogenicactions of angiotensin II,” Molecular Endocrinology, vol. 20,no. 5, pp. 953–970, 2006.
[10] P. K. Mehta and K. K. Griendling, “Angiotensin II cellsignaling: physiological and pathological effects in the car-diovascular system,” American Journal of Physiology, vol. 292,no. 1, pp. C82–C97, 2007.
[11] T. S. Elton and M. M. Martin, “Angiotensin II type 1 recep-tor gene regulation: transcriptional and posttranscriptionalmechanisms,” Hypertension, vol. 49, no. 5, pp. 953–961, 2007.
[12] C. Oro, H. Qian, and W. G. Thomas, “Type 1 angiotensinreceptor pharmacology: signaling beyond G proteins,” Phar-macology and Therapeutics, vol. 113, no. 1, pp. 210–226, 2007.
[13] E. R. Porrello, L. M. Delbridge, and W. G. Thomas, “Theangiotensin II type 2 (AT2) receptor: an enigmatic seventransmembrane receptor,” Frontiers in Bioscience, vol. 14, pp.958–972, 2009.
[14] E. S. Jones, A. Vinh, C. A. McCarthy, T. A. Gaspari,and R. E. Widdop, “AT2 receptors: functional relevance incardiovascular disease,” Pharmacology and Therapeutics, vol.120, no. 3, pp. 292–316, 2008.
[15] M. A. Crackower, R. Sarao, G. Y. Oudit et al., “Angiotensin-converting enzyme 2 is an essential regulator of heartfunction,” Nature, vol. 417, no. 6891, pp. 822–828, 2002.
[16] M. C. Chappell, J. Gregory Modralt, D. I. Diz, and C. M.Ferrario, “Novel aspects of the renal renin-angiotensin sys-tem: angiotensin-(1-7),ACE2 and blood pressure regulation,”Contributions to Nephrology, vol. 143, pp. 77–89, 2004.
[17] M. K. Raizada and A. J. Ferreira, “ACE2: a new target for car-diovascular disease therapeutics,” Journal of CardiovascularPharmacology, vol. 50, no. 2, pp. 112–119, 2007.
[18] M. Iwai and M. Horiuchi, “Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axisvs. ACE2-angiotensin-(1-7)-Mas receptor axis,” HypertensionResearch, vol. 32, no. 7, pp. 533–536, 2009.
[19] A. H. J. Danser, “Local renin-angiotensin systems,” Molecularand Cellular Biochemistry, vol. 157, no. 1-2, pp. 211–216,1996.
[20] M. C. Petrie, N. Padmanabhan, J. E. McDonald, C. Hillier,J. M. C. Connell, and J. J. V. McMurray, “Angiotensin con-verting enzyme (ACE) and non-ACE dependent angiotensinII generation in resistance arteries from patients with heartfailure and coronary heart disease,” Journal of the AmericanCollege of Cardiology, vol. 37, no. 4, pp. 1056–1061, 2001.
[21] P. B. M. W. M. Timmermans, P. C. Wong, A. T. Chiu, and W.F. Herblin, “Nonpeptide angiotensin II receptor antagonists,”
Trends in Pharmacological Sciences, vol. 12, no. 2, pp. 55–62,1991.
[22] P. B. M. W. M. Timmermans, P. C. Wong, A. T. Chiu etal., “Angiotensin II receptors and angiotensin II receptorantagonists,” Pharmacological Reviews, vol. 45, no. 2, pp. 205–251, 1993.
[23] S. Y. Chai, R. Fernando, G. Peck et al., “The angiotensinIV/AT4 receptor,” Cellular and Molecular Life Sciences, vol. 61,no. 21, pp. 2728–2737, 2004.
[24] P. M. L. Vanderheyden, “From angiotensin IV binding site toAT4 receptor,” Molecular and Cellular Endocrinology, vol. 302,no. 2, pp. 159–166, 2009.
[25] A. H. J. Danser, “(Pro)renin receptors: are they biologicallyrelevant?” Current Opinion in Nephrology and Hypertension,vol. 18, no. 1, pp. 74–78, 2009.
[26] G. Nguyen and D. N. Muller, “The biology of the (pro)reninreceptor,” Journal of the American Society of Nephrology, vol.21, no. 1, pp. 18–23, 2010.
[27] A. H. J. Danser, “The increase in renin during renininhibition: does it result in harmful effects by the (pro)reninreceptor,” Hypertension Research, vol. 33, no. 1, pp. 4–10,2010.
[28] N. Bushati and S. M. Cohen, “MicroRNA functions,” AnnualReview of Cell and Developmental Biology, vol. 23, pp. 175–205, 2007.
[29] J. Brennecke, A. Stark, R. B. Russell, and S. M. Cohen,“Principles of microRNA-target recognition,” PLoS Biology,vol. 3, no. 3, article e85, 2005.
[30] M. Kiriakidou, G. S. Tan, S. Lamprinaki, M. De Planell-Saguer, P. T. Nelson, and Z. Mourelatos, “An mRNA m7Gcap binding-like motif within human Ago2 represses trans-lation,” Cell, vol. 129, no. 6, pp. 1141–1151, 2007.
[31] D. T. Humphreys, B. J. Westman, D. I. K. Martin, andT. Preiss, “MicroRNAs control translation initiation byinhibiting eukaryotic initiation factor 4E/cap and poly(A) tailfunction,” Proceedings of the National Academy of Sciences ofthe United States of America, vol. 102, no. 47, pp. 16961–16966, 2005.
[32] S. Bagga, J. Bracht, S. Hunter et al., “Regulation by let-7 andlin-4 miRNAs results in target mRNA degradation,” Cell, vol.122, no. 4, pp. 553–563, 2005.
[33] R. C. Friedman, K. K.-H. Farh, C. B. Burge, and D. P.Bartel, “Most mammalian mRNAs are conserved targets ofmicroRNAs,” Genome Research, vol. 19, no. 1, pp. 92–105,2009.
[34] T. Dalmay, “MicroRNAs and cancer,” Journal of InternalMedicine, vol. 263, no. 4, pp. 366–375, 2008.
[35] W. J. Lukiw, “Micro-RNA speciation in fetal, adult andAlzheimer’s disease hippocampus,” NeuroReport, vol. 18, no.3, pp. 297–300, 2007.
[36] E. van Rooij, L. B. Sutherland, N. Liu et al., “A signaturepattern of stress-responsive microRNAs that can evokecardiac hypertrophy and heart failure,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 103, no. 48, pp. 18255–18260, 2006.
[37] E. Berezikov, V. Guryev, J. van de Belt, E. Wienholds, R. H.A. Plasterk, and E. Cuppen, “Phylogenetic shadowing andcomputational identification of human microRNA genes,”Cell, vol. 120, no. 1, pp. 21–24, 2005.
[38] I. Rigoutsos, T. Huynh, K. Miranda, A. Tsirigos, A. McHardy,and D. Platt, “Short blocks from the noncoding parts of thehuman genome have instances within nearly all known genesand relate to biological processes,” Proceedings of the National

International Journal of Hypertension 11
Academy of Sciences of the United States of America, vol. 103,no. 17, pp. 6605–6610, 2006.
[39] A. Krek, D. Grun, M. N. Poy et al., “CombinatorialmicroRNA target predictions,” Nature Genetics, vol. 37, no.5, pp. 495–500, 2005.
[40] L. S. Hon and Z. Zhang, “The roles of binding site arrange-ment and combinatorial targeting in microRNA repression ofgene expression,” Genome Biology, vol. 8, no. 8, article R166,2007.
[41] R. F. Place, L.-C. Li, D. Pookot, E. J. Noonan, and R.Dahiya, “MicroRNA-373 induces expression of genes withcomplementary promoter sequences,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 105, no. 5, pp. 1608–1613, 2008.
[42] D. H. Kim, P. Sætrom, O. Snøve Jr., and J. J. Rossi,“MicroRNA-directed transcriptional gene silencing in mam-malian cells,” Proceedings of the National Academy of Sciencesof the United States of America, vol. 105, no. 42, pp. 16230–16235, 2008.
[43] Y. Lee, M. Kim, J. Han et al., “MicroRNA genes aretranscribed by RNA polymerase II,” EMBO Journal, vol. 23,no. 20, pp. 4051–4060, 2004.
[44] X. Cai, C. H. Hagedorn, and B. R. Cullen, “Human microR-NAs are processed from capped, polyadenylated transcriptsthat can also function as mRNAs,” RNA, vol. 10, no. 12, pp.1957–1966, 2004.
[45] W. Filipowicz, S. N. Bhattacharyya, and N. Sonenberg,“Mechanisms of post-transcriptional regulation by microR-NAs: are the answers in sight?” Nature Reviews Genetics, vol.9, no. 2, pp. 102–114, 2008.
[46] Y. Altuvia, P. Landgraf, G. Lithwick et al., “Clustering andconservation patterns of human microRNAs,” Nucleic AcidsResearch, vol. 33, no. 8, pp. 2697–2706, 2005.
[47] A. Rodriguez, S. Griffiths-Jones, J. L. Ashurst, and A. Bradley,“Identification of mammalian microRNA host genes andtranscription units,” Genome Research, vol. 14, no. 10, pp.1902–1910, 2004.
[48] Y.-K. Kim and V. N. Kim, “Processing of intronic microR-NAs,” EMBO Journal, vol. 26, no. 3, pp. 775–783, 2007.
[49] G. Song and L. Wang, “MiR-433 and miR-127 arise fromindependent overlapping primary transcripts encoded by themiR-433-127 locus,” PLoS ONE, vol. 3, no. 10, Article IDe3574, 2008.
[50] Y. Lee, C. Ahn, J. Han et al., “The nuclear RNase III Droshainitiates microRNA processing,” Nature, vol. 425, no. 6956,pp. 415–419, 2003.
[51] T. Du and P. D. Zamore, “MicroPrimer: the biogenesis andfunction of microRNA,” Development, vol. 132, no. 21, pp.4645–4652, 2005.
[52] D. S. Schwarz, G. Hutvagner, T. Du, Z. Xu, N. Aronin, and P.D. Zamore, “Asymmetry in the assembly of the RNAi enzymecomplex,” Cell, vol. 115, no. 2, pp. 199–208, 2003.
[53] A. Khvorova, A. Reynolds, and S. D. Jayasena, “FunctionalsiRNAs and miRNAs exhibit strand bias,” Cell, vol. 115, no.2, pp. 209–216, 2003.
[54] L. Peters and G. Meister, “Argonaute Proteins: mediators ofRNA Silencing,” Molecular Cell, vol. 26, no. 5, pp. 611–623,2007.
[55] N. H. Tolia and L. Joshua-Tor, “Slicer and the argonautes,”Nature Chemical Biology, vol. 3, no. 1, pp. 36–43, 2007.
[56] T. M. Rana, “Illuminating the silence: understanding thestructure and function of small RNAs,” Nature ReviewsMolecular Cell Biology, vol. 8, no. 1, pp. 23–36, 2007.
[57] J. G. Doench and P. A. Sharp, “Specificity of microRNAtarget selection in translational repression,” Genes and Devel-opment, vol. 18, no. 5, pp. 504–511, 2004.
[58] B. P. Lewis, C. B. Burge, and D. P. Bartel, “Conservedseed pairing, often flanked by adenosines, indicates thatthousands of human genes are microRNA targets,” Cell, vol.120, no. 1, pp. 15–20, 2005.
[59] A. Grimson, K. K.-H. Farh, W. K. Johnston, P. Garrett-Engele, L. P. Lim, and D. P. Bartel, “MicroRNA targetingspecificity in mammals: determinants beyond seed pairing,”Molecular Cell, vol. 27, no. 1, pp. 91–105, 2007.
[60] C. B. Nielsen, N. Shomron, R. Sandberg, E. Hornstein, J.Kitzman, and C. B. Burge, “Determinants of targeting byendogenous and exogenous microRNAs and siRNAs,” RNA,vol. 13, no. 11, pp. 1894–1910, 2007.
[61] D. P. Bartel, “MicroRNAs: target recognition and regulatoryfunctions,” Cell, vol. 136, no. 2, pp. 215–233, 2009.
[62] L. Wu, J. Fan, and J. G. Belasco, “MicroRNAs directrapid deadenylation of mRNA,” Proceedings of the NationalAcademy of Sciences of the United States of America, vol. 103,no. 11, pp. 4034–4039, 2006.
[63] I. Behm-Ansmant, J. Rehwinkel, T. Doerks, A. Stark, P.Bork, and E. Izaurralde, “mRNA degradation by miRNAsand GW182 requires both CCR4:NOT deadenylase andDCP1:DCP2 decapping complexes,” Genes and Development,vol. 20, no. 14, pp. 1885–1898, 2006.
[64] A. Eulalio, J. Rehwinkel, M. Stricker et al., “Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing,” Genes and Development, vol. 21, no.20, pp. 2558–2570, 2007.
[65] A. J. Giraldez, Y. Mishima, J. Rihel et al., “Zebrafish MiR-430promotes deadenylation and clearance of maternal mRNAs,”Science, vol. 312, no. 5770, pp. 75–79, 2006.
[66] D. T. Humphreys, B. J. Westman, D. I. K. Martin, andT. Preiss, “MicroRNAs control translation initiation byinhibiting eukaryotic initiation factor 4E/cap and poly(A) tailfunction,” Proceedings of the National Academy of Sciences ofthe United States of America, vol. 102, no. 47, pp. 16961–16966, 2005.
[67] R. S. Pillai, S. N. Bhattacharyya, C. G. Artus et al., “Molec-ular biology: inhibition of translational initiation by let-7microRNA in human cells,” Science, vol. 309, no. 5740, pp.1573–1576, 2005.
[68] R. Thermann and M. W. Hentze, “Drosophila miR2induces pseudo-polysomes and inhibits translation initia-tion,” Nature, vol. 447, no. 7146, pp. 875–878, 2007.
[69] G. Mathonnet, M. R. Fabian, Y. V. Svitkin et al., “MicroRNAinhibition of translation initiation in vitro by targeting thecap-binding complex eIF4F,” Science, vol. 317, no. 5845, pp.1764–1767, 2007.
[70] M. Wakiyama, K. Takimoto, O. Ohara, and S. Yokoyama,“Let-7 microRNA-mediated mRNA deadenylation and trans-lational repression in a mammalian cell-free system,” Genes &Development, vol. 21, no. 15, pp. 1857–1862, 2007.
[71] T. P. Chendrimada, K. J. Finn, X. Ji et al., “MicroRNAsilencing through RISC recruitment of eIF6,” Nature, vol.447, no. 7146, pp. 823–828, 2007.
[72] P. Alexiou, M. Maragkakis, G. L. Papadopoulos, M. Reczko,and A. G. Hatzigeorgiou, “Lost in translation: an assessmentand perspective for computational microrna target identifi-cation,” Bioinformatics, vol. 25, no. 23, pp. 3049–3055, 2009.
[73] M. Maragkakis, P. Alexiou, G. L. Papadopoulos et al., “Accu-rate microRNA target prediction correlates with protein

12 International Journal of Hypertension
repression levels,” BMC Bioinformatics, vol. 10, article 1471,p. 295, 2009.
[74] R. C. Friedman, K. K.-H. Farh, C. B. Burge, and D. P.Bartel, “Most mammalian mRNAs are conserved targets ofmicroRNAs,” Genome Research, vol. 19, no. 1, pp. 92–105,2009.
[75] S. Lall, D. Grun, A. Krek et al., “A genome-wide map ofconserved microRNA targets in C. elegans,” Current Biology,vol. 16, no. 5, pp. 460–471, 2006.
[76] D. Gaidatzis, E. van Nimwegen, J. Hausser, and M. Zavolan,“Inference of miRNA targets using evolutionary conservationand pathway analysis,” BMC Bioinformatics, vol. 8, no. 1, p.69, 2007.
[77] D. E. Kuhn, M. M. Martin, D. S. Feldman, A. V. Terry Jr., G. J.Nuovo, and T. S. Elton, “Experimental validation of miRNAtargets,” Methods, vol. 44, no. 1, pp. 47–54, 2008.
[78] M. M. Martin, E. J. Lee, J. A. Buckenberger, T. D.Schmittgen, and T. S. Elton, “MicroRNA-155 regulateshuman angiotensin II type 1 receptor expression in fibrob-last,” The Journal of Biological Chemistry, vol. 281, no. 27, pp.18277–18284, 2006.
[79] M. M. Martin, J. A. Buckenberger, J. Jiang et al., “The humanangiotensin II type 1 receptor +1166 A/C polymorphismattenuates microRNA-155 binding,” The Journal of BiologicalChemistry, vol. 282, no. 33, pp. 24262–24269, 2007.
[80] S. E. Sansom, G. J. Nuovo, M. M. Martin, et al., “MiR-802regulates human angiotensin II type 1 receptor expressionin intestinal epithelial C2BBe1 cells ,” American Journal ofPhysiology Gastrointestinal and Liver Physiology. In press.
[81] D. E. Kuhn, G. J. Nuovo, M. M. Martin et al., “Humanchromosome 21-derived miRNAs are overexpressed in downsyndrome brains and hearts,” Biochemical and BiophysicalResearch Communications, vol. 370, no. 3, pp. 473–477, 2008.
[82] D. E. Kuhn, G. J. Nuovo, A. V. Terry Jr. et al., “Chromosome21-derived microRNAs provide an etiological basis for aber-rant protein expression in human down syndrome brains,”The Journal of Biological Chemistry, vol. 285, no. 2, pp. 1529–1543, 2010.
[83] P. Sethupathy, C. Borel, M. Gagnebin et al., “HumanmicroRNA-155 on chromosome 21 differentially interactswith its polymorphic target in the AGTR1 3′ untrans-lated region: a mechanism for functional single-nucleotidepolymorphisms related to phenotypes,” American Journal ofHuman Genetics, vol. 81, no. 2, pp. 405–413, 2007.
[84] J. C. Murdoch, J. C. Rodger, and S. S. Rao, “Down’ssyndrome: an atheroma free model?” British Medical Journal,vol. 2, no. 6081, pp. 226–228, 1977.
[85] S. Yla-Herttuala, J. Luoma, T. Nikkari, and T. Kivimaki,“Down’s syndrome and atherosclerosis,” Atherosclerosis, vol.76, no. 2-3, pp. 269–272, 1989.
[86] D. Kapell, B. Nightingale, A. Rodriguez, J. H. Lee, W. B.Zigman, and N. Schupf, “Prevalence of chronic medicalconditions in adults with mental retardation: comparisonwith the general population,” Mental Retardation, vol. 36, no.4, pp. 269–279, 1998.
[87] C. C. Draheim, J. A. McCubbin, and D. P. Williams, “Dif-ferences in cardiovascular disease risk between nondiabeticadults with mental retardation with and without Downsyndrome,” American Journal on Mental Retardation, vol.107, no. 3, pp. 201–234, 2002.
[88] A. K. Greene, S. Kim, G. F. Rogers, S. J. Fishman, B. R. Olsen,and J. B. Mulliken, “Risk of vascular anomalies with Downsyndrome,” Pediatrics, vol. 121, no. 1, pp. e135–e140, 2008.
[89] P. A. Baird and A. D. Sadovnick, “Causes of death to age 30 inDown syndrome,” American Journal of Human Genetics, vol.43, no. 3, pp. 239–248, 1988.
[90] L. Brattstrom, E. Englund, and A. Brun, “Does Downsyndrome support homocysteine theory of arteriosclerosis?”The Lancet, vol. 1, no. 8529, pp. 391–392, 1987.
[91] S. M. Pueschel, W. Y. Craig, and J. E. Haddow, “Lipids andlipoproteins in persons with Down’s syndrome,” Journal ofIntellectual Disability Research, vol. 36, no. 4, pp. 365–369,1992.
[92] F. Licastro, A. Marocchi, S. Penco et al., “Does Down’s syn-drome support the homocysteine theory of atherogenesis?.Experience in elderly subjects with trisomy 21,” Archives ofGerontology and Geriatrics, vol. 43, no. 3, pp. 381–387, 2006.
[93] G. Goi, C. Baquero-Herrera, F. Licastro, G. Dogliotti, and M.M. Corsi, “Advanced oxidation protein products (AOPP) andhigh-sensitive C-reactive protein (hs-CRP) in an “atheroma-free model”: Down’s syndrome,” International Journal ofCardiology, vol. 113, no. 3, pp. 427–429, 2006.
[94] T. Boettger, N. Beetz, S. Kostin et al., “Acquisition of thecontractile phenotype by murine arterial smooth muscle cellsdepends on the Mir143/145 gene cluster,” Journal of ClinicalInvestigation, vol. 119, no. 9, pp. 2634–2647, 2009.
[95] K. R. Cordes, N. T. Sheehy, M. P. White et al., “MiR-145and miR-143 regulate smooth muscle cell fate and plasticity,”Nature, vol. 460, no. 7256, pp. 705–710, 2009.
[96] A. Daugherty, M. W. Manning, and L. A. Cassis, “AngiotensinII promotes atherosclerotic lesions and aneurysms inapolipoprotein E-deficient mice,” Journal of Clinical Investi-gation, vol. 105, no. 11, pp. 1605–1612, 2000.
[97] L. Bao, M. Zhou, L. Wu et al., “PolymiRTS Database: linkingpolymorphisms in microRNA target sites with complextraits,” Nucleic Acids Research, vol. 35, no. 1, pp. D51–D54,2007.
[98] M. Hariharan, V. Scaria, and S. K. Brahmachari, “dbSMR:a novel resource of genome-wide SNPs affecting microRNAmediated regulation,” BMC Bioinformatics, vol. 10, article108, 2009.
[99] S. Hiard, C. Charlier, W. Coppieters, M. Georges, and D.Baurain, “Patrocles: a database of polymorphic miRNA-mediated gene regulation in vertebrates,” Nucleic AcidsResearch, vol. 38, supplement 1, pp. D640–D651, 2009.
[100] J. F. Abelson, K. Y. Kwan, B. J. O’Roak et al., “Medicine:sequence variants in SLITRK1 are associated with Tourette’ssyndrome,” Science, vol. 310, no. 5746, pp. 317–320, 2005.
[101] H. He, K. Jazdzewski, W. Li et al., “The role of microRNAgenes in papillary thyroid carcinoma,” Proceedings of theNational Academy of Sciences of the United States of America,vol. 102, no. 52, pp. 19075–19080, 2005.
[102] A. Clop, F. Marcq, H. Takeda et al., “A mutation creating apotential illegitimate microRNA target site in the myostatingene affects muscularity in sheep,” Nature Genetics, vol. 38,no. 7, pp. 813–818, 2006.
[103] S. Zuchner, G. Wang, K.-N. Tran-Viet et al., “Mutationsin the novel mitochondrial protein REEP1 cause hereditaryspastic paraplegia type 31,” American Journal of HumanGenetics, vol. 79, no. 2, pp. 365–369, 2006.
[104] P. J. Mishra, R. Humeniuk, P. J. Mishra, G. S. A.Longo-Sorbello, D. Banerjee, and J. R. Bertino, “A miR-24 microRNA binding-site polymorphism in dihydrofolatereductase gene leads to methotrexate resistance,” Proceedingsof the National Academy of Sciences of the United States ofAmerica, vol. 104, no. 33, pp. 13513–13518, 2007.

International Journal of Hypertension 13
[105] B. D. Adams, H. Furneaux, and B. A. White, “The micro-ribonucleic acid (miRNA) miR-206 targets the humanestrogen receptor-α (ERα) and represses ERαmessenger RNAand protein expression in breast cancer cell lines,” MolecularEndocrinology, vol. 21, no. 5, pp. 1132–1147, 2007.
[106] M. S. Nicoloso, H. Sun, R. Spizzo et al., “Single-nucleotidepolymorphisms inside microRNA target sites influencetumor susceptibility,” Cancer Research, vol. 70, no. 7, pp.2789–2798, 2010.
[107] B. Baudin, “Polymorphism in angiotensin II receptor genesand hypertension,” Experimental Physiology, vol. 90, no. 3,pp. 277–282, 2005.
[108] A. Bonnardeaux, E. Davies, X. Jeunemaitre et al.,“Angiotensin II type 1 receptor gene polymorphisms inhuman essential hypertension,” Hypertension, vol. 24, no. 1,pp. 63–69, 1994.
[109] K. Kainulainen, M. Perola, J. Terwilliger et al., “Evidence forinvolvement of the type 1 angiotensin II receptor locus inessential hypertension,” Hypertension, vol. 33, no. 3, pp. 844–849, 1999.
[110] W. Y. S. Wang, R. Y. L. Zee, and B. J. Morris, “Associationof angiotensin II type 1 receptor gene polymorphism withessential hypertension,” Clinical Genetics, vol. 51, no. 1, pp.31–34, 1997.
[111] G. Kobashi, A. Hata, K. Ohta et al., “A1166C variant ofangiotensin II type 1 receptor gene is associated with severehypertension in pregnancy independently of T235 variant ofangiotensinogen gene,” Journal of Human Genetics, vol. 49,no. 4, pp. 182–186, 2004.
[112] J.-G. Wang and J. A. Staessen, “Genetic polymorphisms inthe renin-angiotensin system: relevance for susceptibility tocardiovascular disease,” European Journal of Pharmacology,vol. 410, no. 2-3, pp. 289–302, 2000.
[113] Z. Jiang, W. Zhao, F. Yu, and G. Xu, “Association ofangiotensin II type 1 receptor gene polymorphism withessential hypertension,” Chinese Medical Journal, vol. 114, no.12, pp. 1249–1251, 2001.
[114] K. Ono, T. Mannami, S. Baba, N. Yasui, T. Ogihara, andN. Iwai, “Lack of association between angiotensin II type 1receptor gene polymorphism and hypertension in Japanese,”Hypertension Research, vol. 26, no. 2, pp. 131–134, 2003.
[115] P. Palatini, G. Ceolotto, F. Dorigatti et al., “Angiotensin IItype 1 receptor gene polymorphism predicts development ofhypertension and metabolic syndrome,” American Journal ofHypertension, vol. 22, no. 2, pp. 208–214, 2009.
[116] A. P. R. M. Osterop, M. J. M. Kofflard, L. A. Sandkuijl et al.,“AT1 receptor A/C1166 polymorphism contributes to cardiachypertrophy in subjects with hypertrophic cardiomyopathy,”Hypertension, vol. 32, no. 5, pp. 825–830, 1998.
[117] O. A. Makeeva, K. V. Puzyrev, E. N. Pavlukova et al., “ACE andAGTR1 genes polymorphisms in left ventricular hypertrophypathogenesis in humans,” Molekulyarnaya Biologiya, vol. 38,no. 6, pp. 990–996, 2004.
[118] T. D. J. Smilde, M. W. Zuurman, H. L. Hillege et al., “Renalfunction dependent association of AGTR1 polymorphism(A1166C) and electrocardiographic left-ventricular hyper-trophy,” American Journal of Hypertension, vol. 20, no. 10, pp.1097–1103, 2007.
[119] M. Lajemi, C. Labat, S. Gautier et al., “Angiotensin II type1 receptor-153A/G and 1166A/C gene polymorphisms andincrease in aortic stiffness with age in hypertensive subjects,”Journal of Hypertension, vol. 19, no. 3, pp. 407–413, 2001.
[120] J. Dıez, C. Laviades, J. Orbe et al., “The A1166C polymor-phism of the AT1 receptor gene is associated with collagentype I synthesis and myocardial stiffness in hypertensives,”Journal of Hypertension, vol. 21, no. 11, pp. 2085–2092, 2003.
[121] A. Benetos, S. Gautier, S. Ricard et al., “Influence ofangiotensin-converting enzyme and angiotensin II type 1receptor gene polymorphisms on aortic stiffness in nor-motensive and hypertensive patients,” Circulation, vol. 94, no.4, pp. 698–703, 1996.
[122] L. Tiret, H. Blanc, J.-B. Ruidavets et al., “Gene poly-morphisms of the renin-angiotensin system in relation tohypertension and parental history of myocardial infarctionand stroke: the PEGASE study,” Journal of Hypertension, vol.16, no. 1, pp. 37–44, 1998.
[123] C.-K. Wu, C.-T. Tsai, J.-J. Hwang et al., “Renin-angiotensinsystem gene polymorphisms and diastolic heart failure,”European Journal of Clinical Investigation, vol. 38, no. 11, pp.789–797, 2008.
[124] J. Lin, F. B. Hu, L. Qi, and G. C. Curhan, “Genetic polymor-phisms of angiotensin-2 type 1 receptor and angiotensinogenand risk of renal dysfunction and coronary heart disease intype 2 diabetes mellitus,” BMC Nephrology, vol. 10, no. 1,article 9, 2009.
[125] O. Amir, R. E. Amir, H. Paz, E. Attias, M. Sagiv, andB. S. Lewis, “Relation between AT1R gene polymorphismand long-term outcome in patients with heart failure,”Cardiology, vol. 112, no. 2, pp. 151–157, 2009.
[126] G. T. Jones, A. R. Thompson, F. M. van Bockxmeer et al.,“Angiotensin II type 1 receptor 1166C polymorphism is asso-ciated with abdominal aortic aneurysm in three independentcohorts,” Arteriosclerosis, Thrombosis, and Vascular Biology,vol. 28, no. 4, pp. 764–770, 2008.
[127] P. McColgan, G. E. Peck, R. M. Greenhalgh, and P. Sharma,“The genetics of abdominal aortic aneurysms: a comprehen-sive meta-analysis involving eight candidate genes in over16,700 patients,” International Surgery, vol. 94, no. 4, pp. 350–358, 2009.
[128] V. A. Cameron, T. J. Mocatta, A. P. Pilbrow et al.,“Angiotensin type-1 receptor A1166C gene polymorphismcorrelates with oxidative stress levels in human heart failure,”Hypertension, vol. 47, no. 6, pp. 1155–1161, 2006.
[129] R. S. Spielman, L. A. Bastone, J. T. Burdick, M. Morley,W. J. Ewens, and V. G. Cheung, “Common genetic variantsaccount for differences in gene expression among ethnicgroups,” Nature Genetics, vol. 39, no. 2, pp. 226–231, 2007.
Submit your manuscripts athttp://www.hindawi.com
Stem CellsInternational
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
MEDIATORSINFLAMMATION
of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Behavioural Neurology
EndocrinologyInternational Journal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Disease Markers
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
BioMed Research International
OncologyJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Oxidative Medicine and Cellular Longevity
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
PPAR Research
The Scientific World JournalHindawi Publishing Corporation http://www.hindawi.com Volume 2014
Immunology ResearchHindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Journal of
ObesityJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Computational and Mathematical Methods in Medicine
OphthalmologyJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Diabetes ResearchJournal of
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Research and TreatmentAIDS
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Gastroenterology Research and Practice
Hindawi Publishing Corporationhttp://www.hindawi.com Volume 2014
Parkinson’s Disease
Evidence-Based Complementary and Alternative Medicine
Volume 2014Hindawi Publishing Corporationhttp://www.hindawi.com