ace activity is modulated by the enzyme α-galactosidase a
TRANSCRIPT

ORIGINAL ARTICLE
ACE activity is modulated by the enzyme α-galactosidase A
Elice Carneiro Batista & Luiz Roberto Carvalho & Dulce Elena Casarini &Adriana Karaoglanovic Carmona & Edson Lucas dos Santos & Elton Dias da Silva &
Robson Augusto dos Santos & Clovis Ryuichi Nakaie & Maria Verônica Munoz Rojas &
Suzana Macedo de Oliveira & Michael Bader & Vânia D’Almeida &
Ana Maria Martins & Kely de Picoly Souza & João Bosco Pesquero
Received: 14 July 2010 /Revised: 7 September 2010 /Accepted: 13 September 2010 /Published online: 13 October 2010# Springer-Verlag 2010
Abstract Fabry disease is a multisystem X-linked disorderresulting from α-galactosidase A (α-GalA) gene mutationsleading to the accumulation of globotriaosylceramidemainly in endothelium compromising heart, kidney, andbrain. In Fabry patients, progressive renal failure isfrequently treated with angiotensin I-converting enzyme(ACE) inhibitors. We were interested in the possibleinteractions between ACE inhibitors therapy and the onlycausative therapy for Fabry disease, the enzyme replace-ment therapy (ERT) using recombinant human α-GalA(rhα-GalA). Our results suggest that ACE activity wassignificantly inhibited in plasma of Fabry patients and theblood pressure level decreased just after ERT (at the end of
the rhα-GalA infusion). Interestingly, 2 weeks later, ACEactivity was significantly upregulated and the plasma levelsof angiotensin II increased in the patients treated with rhα-GalA following the elevations of ACE activity. The sameinhibitory effect on ACE activity was also observed in ratsafter rhα-GalA infusion. Furthermore, ACE activity inCHO cells transfected with the human ACE was inhibiteddose and time-dependently by rhα-GalA. In vitro, theincubation of plasma from healthy volunteers with rhα-GalA significantly reduced ACE activity. Finally, rhα-GalAalso inhibited ACE activity and released galactose residuesfrom purified rabbit lung ACE dose-dependently. Insummary, our results suggest that rhα-GalA interacts with
E. C. Batista :A. K. Carmona : E. D. da Silva :C. R. Nakaie :S. M. de Oliveira : J. B. PesqueroDepartment of Biophysics, Universidade Federal de São Paulo,São Paulo, Brazil
L. R. CarvalhoNephrology Department, Hospital São Vicente de Paulo,Passo Fundo, Brazil
D. E. CasariniDepartment of Medicine, Division of Nephrology,Universidade Federal de Sao Paulo,São Paulo, Brazil
E. L. dos Santos :K. de Picoly SouzaSchool of Environmental and Biological Science,Federal University of Grande Dourados,Dourados, MS, Brazil
R. A. dos SantosDepartment of Physiology and Biophysics,Universidade Federal de Minas Gerais,Belo Horizonte, Minas Gerais, Brazil
M. V. M. RojasPersonalized Genetic Health, Genzyme do Brasil,São Paulo, SP, Brazil
M. BaderMax-Delbrück-Center for Molecular Medicine (MDC),Robert-Rössle Str. 10,13125 Berlin, Germany
V. D’AlmeidaDepartment of Biosciences,Universidade Federal de Sao Paulo,São Paulo, Brazil
A. M. MartinsDepartment of Pediatrics, Universidade Federal de Sao Paulo,São Paulo, Brazil
J. B. Pesquero (*)Department of Biophysics, Escola Paulista de Medicina,Universidade Federal de Sao Paulo,04023-062 São Paulo, SP, Brazile-mail: [email protected]
J Mol Med (2011) 89:65–74DOI 10.1007/s00109-010-0686-2

ACE and inhibits its activity, possibly by removing thegalactose residues from the enzyme. This modulation mighthave profound impact on the clinical outcome of Fabrypatients treated with rhα-GalA.
Keywords ACE . Angiotensin . Biochemistry . Biology .
Blood pressure . Angiotensin I-converting enzymeα-galactosidase A . Fabry disease . ACE inhibitors
Introduction
Fabry disease is an X-linked lysosomal storage disordercaused by a deficient activity of the enzyme α-galactosidaseA (α-GalA) [1]. The α-GalA gene is located at Xq22[2, 3]. Decreased activity of α-GalA leads to incompletemetabolism and ongoing accumulation of glycosphingo-lipids, mainly globotriaosylceramide (Gb-3 or GL3) invarious cell types and tissues [1, 4–6] as vascular endothe-lium, renal cells, cardiac myocytes, and neural cells [2, 7–9].Fabry disease is progressive with potential vital organinvolvement and life-threatening complications. Renal mani-festations occur relatively early in the course of disease and,in many patients, progress to renal failure [10–13].
Enzyme replacement therapy (ERT) for Fabry diseasehas been available since 2001. Studies with agalsidase αand β have shown that ERT may significantly slow the rateof renal dysfunction [14–16]. ERT for Fabry disease is achronic treatment, requiring constant infusions of therecombinant human enzyme [9] and, despite its high cost,currently it is the only specific treatment that can improvethe quality of life of Fabry patients and change the naturalhistory of the disease [9, 17–19]. Furthermore, to amelioraterenal damage, Fabry patients are often treated with blockersof the renin-angiotensin system (RAS), such as antagonistsof the angiotensin II (AngII) receptor, AT1, and inhibitorsof the angiotensin I-converting enzyme (ACE). ACE, apivotal component of the RAS, is expressed in the plasmamembrane of vascular endothelial cells (in particularly inthe lung), epithelial cells of renal proximal tubules,gastrointestinal tract, heart and in various regions of thebrain [19–22], the main tissues affected in Fabry disease.
In this context, this study was aimed at evaluating apossible interaction between recombinant human α-GalA(rhα-GalA) and ACE by: (1) measuring the effect of rhα-GalA infusion in Fabry patients on plasma ACE activity;(2) evaluating the effect of rhα-GalA in vitro on ACEactivity in the plasma of healthy control subjects, in Chinesehamster ovary (CHO) cells expressing human ACE and inpurified rabbit lung ECA; (3) evaluating the in vivo effectof rhα-GalA infusion on ACE activity and blood pressureof rats.
Material and methods
Volunteers
Thirteen Fabry patients (39±4 years old, four women andnine men) under ERTwith the rhα-GalA (Fabrazyme®) weredivided in Fabry—before ERT and Fabry—after ERT. TheFabry group not under ERT (Fabry—without ERT) wascomposed of other 17 patients (27±4 years old, 15 womenand two men) and the control group was composed of 21healthy subjects (30±2 years old, 13 women and eight men).
Blood collection
Venous blood samples from Fabry patients under standardERT (Fabrazyme® 1 mg/kg/every other week) before andafter infusion, and from Fabry patients that never receivedERT were obtained from the forearm at rest. The patientsare treated at CREIM (Centro de Referência em ErrosInatos do Metabolismo—São Paulo) and Hospital SãoVicente de Paulo (Passo Fundo). Plasma was collected intoS-Monovette 5.0 ml tube containing EDTA (Sarstedt®) anda cocktail of inhibitors EDTA free (Roche®) for theanalysis of angiotensins. The samples used for the analysisof plasma ACE activity were collected into S-Monovette5.0 mL tube containing citrate solution (Sarstedt®). Bloodwas collected from Fabry patients under ERT before and atthe end of the 4-h infusion with rhα-GalA. All individualstaking part in this study gave informed written consent, theprotocol was in accordance with legal requirements and theDeclaration of Helsinki, and it was approved by the EthicsCommittee of the Universidade Federal de São Paulo(Protocol nr. 2020/07).
Plasma ACE activity
Plasma ACE activity on Abz-FRK(Dnp)P-OH was deter-mined under the optimal experimental conditions (buffer,pH, chloride, and zinc ions) previously established forrecombinant wild-type ACE [23]. Enzymatic activity wascontinuously monitored with a Hitachi F-2000 fluorimeterby measuring the fluorescence (λex=320 nm and λem=420 nm) for 5–10 min. Before starting the reaction by theaddition of the substrate, the plasma was preincubated for5 min in the cuvette at 37°C in the assay buffer. For ACEactivity determination, 5 μl of plasma was incubated with10 μM Abz-FRK(Dnp)P-OH at 37°C in 0.1 M Tris-HCl,pH 7.0, containing 50 mM NaCl and 10 μM ZnCl2, in afinal volume of 1.0 ml. The slope was converted into nmol ofsubstrate hydrolyzed per minute based on a calibration curveobtained by complete hydrolysis of the peptide as reportedpreviously [23]. ACE activity is reported as μM/min/μl ofserum. The measurements were performed in duplicate. The
66 J Mol Med (2011) 89:65–74

amount of AngI and AngII in plasma was evaluated asdescribed elsewhere [24].
ACE activity in CHO-ACE cells
Chinese hamster ovary cells transfected with the humansomatic ACE gene has been used as a model of high ACEactivity [25]. Cells were cultured in high glucose DMEMand plated in 12-well plates (Corning) at 0.75×105 cells perwell (90% confluence on the day of the experiment). Thecells were washed with HBSS buffer (140 mM NaCl, 5 mMKCl, 0.1 mM CaCl2, 0.63 mM MgSO4, 1 mM Na2HPO4,and 6.1 mM glucose) containing 10 μM ZnCl2 at pH 7.4and incubated in the same buffer with 10 μM Abz-FRK(Dnp)P-OH. Aliquots (1 ml) of culture supernatant werecollected at different times (0–6 min), the reaction wasstopped at 0°C and then transferred to a 1-cm-pathlengthcuvette. The final fluorescence relative to each incubationtime (λex=320 nm and λem=420 nm) was measured in aHitachi F-2000 fluorimeter. The arbitrary fluorescence unitswere converted into micromoles of substrate hydrolyzedbased on a calibration curve obtained using a standardsolution of complete hydrolyzed substrate [23]. The activitieswere inhibited by 2.5 μM of the specific ACE inhibitorlisinopril used as control in the assays. The protein contentof each well was determined by the method of Lowry et al.[26] using bovine serum albumin as standard. Measure-ments were performed in triplicate and ACE activity valueswere reported as μmol of substrate hydrolyzed per minute ina volume of 1.0 ml per μg protein (μM/min μg of protein).
Galactose measurement
Purified rabbit lung ACE (Sigma®) was incubated with 1,10, and 100 μg of rhα-GalA and the amount of galactosereleased was quantified by using a galactose assay kit(BioVision) according to the manufacturer’s recommenda-tion (measurements were performed in triplicate).
ACE activity in the rat ileum preparation
Wistar rats (300–350 g, n=5) were treated with rhα-GalA(Fabrazyme® 1 mg/kg body weight injected intravenously).Twenty four hours later, the animals were euthanized andthe ileum removed. The organ was suspended in a 5 mlchamber containing Tyrode solution kept at 37°C andbubbled with a 95% O2 and 5% CO2 mixture. Thecomposition of the Tyrode solution was (mM): NaCl 137,KCl 2.7, CaCl2 1.36, MgCl2 0.49, NaH2PO4 0.36,NaHCO3, and C6H12O6. The organs were equilibrated for60 min before the beginning of the experiments. After30 min (equilibration period), AngI or AngII (10−7 M finalconcentration in the chamber) was administered. The organ
bath was washed before and after administration of thepeptides. Measurements were performed in duplicate andthe index of conversion of AngI to AngII (ACE activity)was expressed by the ratio of the Ang I/Ang II effects.
Direct blood pressure measurements
Wistar rats (300–350 g, n=6) were instrumented for directblood pressure measurements. The animals were anesthe-tized with ketamine/xylasine (30 mg/kg and 10 mg/kg,respectively) for cannulation of the femoral vein and artery.Thus, AngI or AngII (10−8 M) were administered intra-arterially and the effect on blood pressure was measuredand quantified. Recombinant α-GalA (Fabrazyme®) wasadministered intravenously in the animals and 4 h later thesame procedure was performed with the administration ofAngI and AngII (10−8 M) as described above. The finalresult (AngI conversion in AngII or ACE activity) wasdetermined by the ratio between the effect of the AngInormalized by effect of the Ang II.
Blood pressure measurements by telemetry
Wistar rats (300–320 g, n=6) were instrumented fortelemetry blood pressure measurements. The pressuresensor catheter (TA11PA-C40 blood pressure device—DataSciences International, St. Paul, MN, USA) was implantedvia the right femoral artery into the abdominal aorta and thetransmitter was placed in a subcutaneous pocket along theright flank. All rats were allowed to recover from thesurgery for 7 days before baseline blood pressure valueswere recorded for 24 days. The rats received normal chow(0.25% NaCl) and water. The data from the TA11PA-C40device were transmitted via radio frequency signals to areceiver below the home cage and thereafter collected usingthe Dataquest ART system, version 2.1 (Data SciencesInternational), which allowed us to detect, collect, andanalyze signals from several animals simultaneously. Themean arterial pressure was recorded using the DATAQUESTsoftware (ART 2.1). Data from blood pressure were monitoredfor 2 h daily for 24 consecutive days. The chronic evaluationof blood pressure was used to analyze the effect of rhα-GalAadministered intravenously in the animals. rhα-GalA wasadministered every 6 days (200 μL of rhα-GalA solution,Fabrazyme® 1 μg/g body weight, n=5). Control animalsreceived only saline at the same time. Blood pressure wasalso evaluated after acute administration of the ACE inhibitorlisinopril intraperitonially (10 mg/kg body weight).
Statistical analysis
The data were subjected to two-way analyses of variance(one-way ANOVA), followed by Tukey’s post-test. Data were
J Mol Med (2011) 89:65–74 67

expressed as means±standard error of means. Differenceswere considered significant at p<0.05. Analysis wasperformed using GraphPad.Prism™.
Results
rhα-GalA alters, in vivo, ACE activity and angiotensin Iand II concentration in the plasma of Fabry patients
Blood samples were collected from control subjects, fromFabry patients naïve to rhα-GalA infusion (without ERT)and from Fabry patients under standard treatment (1 mg/kgrhα-GalA every other week—Fabrazyme®) just before andafter the completion of one ERT session. Results show that,independently of age, gender, and gene mutation (Table 1),ACE activity in Fabry patients without ERT was notdifferent from the one in the control subjects (Fig. 1a).However, ACE activity was significantly higher in theplasma of Fabry patients receiving ERT just before thebeginning of the infusion 2 weeks after the last ERT whencompared to the values observed in control and patientswithout ERT treatment. Surprisingly, a drastic reduction inACE activity was observed at the end of the infusion ofrhα-GalA (Fig. 1a). When blood pressure levels weremeasured in these patients under either condition, a markeddecrease was observed after rhα-GalA infusion (Fig. 1d).The concentration of AngI and AngII was evaluated in theplasma of all groups (Fig. 1b–c). In agreement to the higherACE activity found in the plasma of the patients beforerhα-GalA infusion, the concentration of AngII was alsosignificantly increased in this condition. After infusion,AngII concentration significantly decreased to valuessimilar to the ones of patients without treatment (Fig. 1c).No significant alteration was observed in the levels of AngIin any of these conditions (Fig. 1b).
rhα-GalA inhibits plasma ACE activity in vitro
Plasma samples of healthy control subjects were incubateddirectly with rhα-GalA at different time intervals. Similarlyto the in vivo measurements, it was possible to observe adirect inhibitory effect of the recombinant rhα-GalA onACE activity (Fig. 2a). In order to check the inhibitoryeffect of rhα-GalA on ACE activity in a different system,CHO-ACE expressing cells were used. Results show thatAsCE activity in CHO-ACE cells incubated with rhα-GalAdecreased proportionally to the concentration of therecombinant enzyme and to time (Fig. 2b–c, respectively).As a control, the cells were also incubated with mannitol(osmolar diuretic substance present in the composition ofrhα-GalA- Fabrazyme®) and with sodium phosphate, bothin the same concentration present in the rhα-GalA solution.
In both cases, no change was observed on ACE activity(data not shown).
Animal assays
In order to better characterize the inhibitory effect of rhα-GalA on ACE activity, we evaluated the inhibition of AngIto AngII conversion by ACE in a rat ileum preparation.Therefore, the contractile effect of AngI administered to theisolated ileum preparation 24 h after treatment of theanimals with rhα-GalA was measured. The results showthat AngI conversion was partially blocked by the treat-ment, again evidencing the inhibitory effect of rhα-GalAon ACE activity (Fig. 3a). On the contrary, the effect ofAngII was not changed by rhα-GalA treatment (data notshown). In addition, the vasoconstrictor effect of AngI andAngII injected intravenously in vivo in awake rats wasevaluated 4 h after rhα-GalA administration. At this timepoint, there was no alteration promoted by rhα-GalAadministration in the basal blood pressure value in the rats.However, the results show a reduction in the constrictresponse of AngI after rhα-GalA treatment (Fig. 3b and c).No changes were observed in the vasoconstriction effect ofAngII (Fig. 3b).
rhα-GalA alters blood pressure in rats
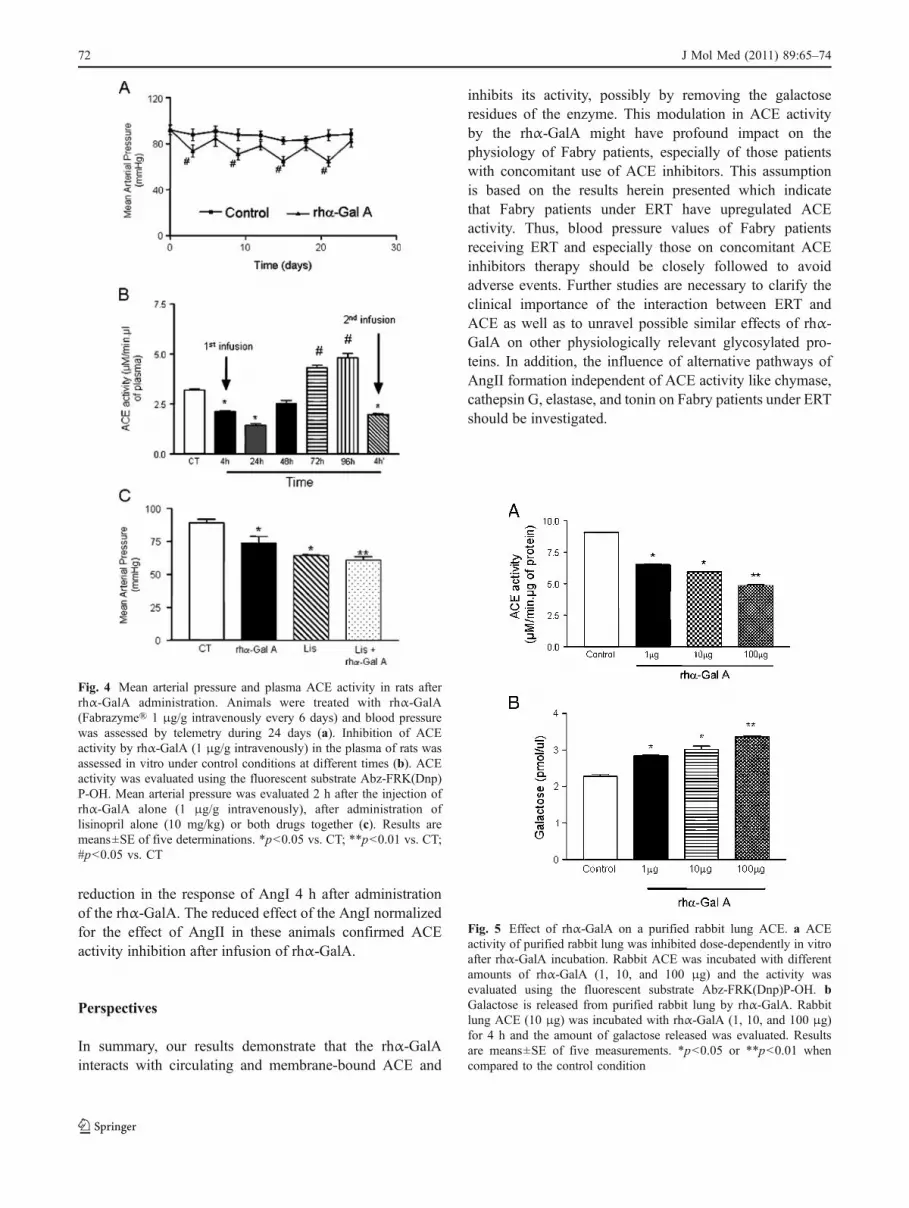
Telemetry blood pressure measurements show that afterrhα-GalA infusion in rats the blood pressure levels andACE activity were significantly decreased (Fig. 4a–b). Inthe period of 6 days after rhα-GalA infusion, bloodpressure values returned to the baseline. The second rhα-GalA administration 1 week later also inhibited ACEactivity significantly (Fig. 4b). When the long-term effectof rhα-GalA on ACE activity was evaluated, a similarprofile was observed as in humans, with a significantdecrease in ACE activity 4 and 24 h after administration ofrhα-GalA and an increase in ACE activity before the nexttreatment (Fig. 4b). No alteration in ACE activity wasobserved in control animals receiving saline (data notshown). A correlation between ACE activity and bloodpressure could be visualized. The concomitant administra-tion of rhα-GalA and lisinopril, an ACE inhibitor, did notpotentiate the hypotensive effect in the animals (Fig. 4c).
rhα-GalA inhibits the activity and releases galactose fromthe purified rabbit lung ACE In addition to the in vitroinhibitory effect of rhα-GalA on plasma ACE, we couldshow that the recombinant enzyme was also able to inhibitdose dependently the activity of purified rabbit lung ACE(Fig. 5a). Furthermore, during the 4-h incubation, rhα-GalAcould also release residues of galactose from ACE in adose-dependent manner (Fig. 5b).
68 J Mol Med (2011) 89:65–74

Discussion
In Fabry disease, the accumulation of Gb3 leads topremature death due to progressive organ failure includingend-stage renal disease [5, 27]. Baseline proteinuria and
glomerular filtration rate are important factors that affectthe rate of loss of kidney function. Fabry patients withbaseline proteinuria >1 g should control urine proteinexcretion with the use of ACE inhibitors and/or AT1receptor blockers. These substances are effective in providing
Table 1 Gender, age, α-GalA mutation, activity, and ACE activity in the Fabry patients and control subjects
Patients Controls
N Gender Age(years)
Mutation ACE activity(μM/min.μL)
α-GalA activity(μmol/L.h)
Time underERT (months)
N Gender Age(years)
ACE activity(μM/min.μL)
1 M 40 Sub.exon1 (TGG-TAG) 8.40 ND 24 1 F 31 5.18
2 M 50 Sub.exon1 (TGG-TAG) 5.14 0.03 24 2 M 24 5.62
3 F 43 Sub.exon1 (TGG-TAG) 10.11 1.16 16 3 M 26 6.98
4 F 45 Sub.exon1 (TGG-TAG) 8.63 0.23 26 4 F 26 5.38
5 M 63 Del.exon7 (CGC^TCTTAt ACC ATC)
9.03 0.40 8 5 F 25 5.53
6 M 45 Del.exon7 (CGC^TCTTAt ACC ATC)
10.60 0.45 8 6 F 23 7.35
7 M 31 Del.exon7 (CGC^TCTTAt ACC ATC)
9.25 0.40 8 7 F 28 5.46
8 F 20 Sub.exon7 (TGG-TAG) 8.09 2.00 19 8 F 26 5.97
9 M 28 Del.exon6 (CAG^GATAaggacgtaattgccatcaatcAG GAC)
9.76 0.20 4 9 F 21 5.79
10 M 32 Sub.exon7 (CGA-CAA) 11.65 ND 4 10 F 25 4.83
11 M 50 Sub.exon7 (CGA-CAA) 8.62 0.20 25 11 M 28 5.25
12 F 43 Sub.exon7 (CGA-CAA) 8.32 1.00 6 12 M 37 6.41
13 M 15 Sub.exon7 (CGA-CAA) 7.75 ND 16 13 F 29 6.17
14 F 19 Sub.exon1 (TGG-TAG) 9.61 6.41 -ERT 14 F 29 5.20
15 F 17 Sub.exon1 (TGG-TAG) 3.33 6.42 -ERT 15 M 40 6.43
16 F 23 Sub.exon1 (TGG-TAG) 4.75 1.50 -ERT 16 M 29 5.74
17 F 25 Sub.exon1 (TGG-TAG) 3.37 ND -ERT 17 M 40 7.54
18 F 32 Sub.exon1 (TGG-TAG) 4.30 1.67 -ERT 18 M 29 5.94
19 F 12 Sub.exon1 (TGG-TAG) 1.56 3.18 -ERT 19 F 24 6.11
20 F 20 Del.exon7 (CGC^TCTTAt ACC ATC)
4.51 5.31 -ERT 20 F 47 5.44
21 F 15 Del.exon7 (CGC^TCTTAt ACC ATC)
3.14 1.30 -ERT 21 F 27 5.73
22 M 36 Del.exon7 (CGC^TCTTAt ACC ATC)
4.14 0.61 -ERT
23 F 62 Del.exon7 (CGC^TCTTAt ACC ATC)
8.57 6.16 -ERT
24 M 15 Del.exon7 (CGC^TCTTAt ACC ATC)
6.10 1.44 -ERT
25 F 51 Del.exon7 (CGC^TCTTAt ACC ATC)
3.24 3.82 -ERT
26 F 24 Del.exon7 (CGC^TCTTAt ACC ATC)
5.23 3.75 -ERT
27 F 31 Del.exon7 (CGC^TCTTAt ACC ATC)
4.38 4.61 -ERT
28 F 15 Del.exon1 (CAT^CTGGGc TGC)
4.28 1.38 -ERT
29 F 48 Del.exon1 (CAT^CTGGGc TGC)
11.29 2.57 -ERT
30 F 17 Sub.exon1 (TGG-TAG) 3.74 2.28 -ERT
ND not determined
-ERT naive patients without ERT
J Mol Med (2011) 89:65–74 69

renoprotection and controlling blood pressure due to theirwell-known actions on glomerular hemodynamics and theirinteractions with cytokines [28–32], although their use maybe challenging because patients with Fabry disease oftenhave relatively low blood pressure [33]. In addition, patientsunder ACE inhibitor therapy can be more sensitive to thehypotensive effect of rhα-GalA [34]. Interestingly, ERTalone has been shown to improve renal function as one of itsbeneficial effects which include the reduction in Gb3deposits in the kidneys, heart, and skin, and decrease ofGb3 urinary excretion [14, 15, 35, 36]. Additionally,Schiffmann et al. [36] reported that ERT performed everyweek rather than every 2 weeks can improve the decline inrenal function in patients with a severe decrease in GFR. Themechanistic basis and the impact of these findings were notcarefully evaluated yet, but a link to the RAS was intriguing.
ACE is expressed in the plasma membrane of differentcells, many of them affected in Fabry disease by theaccumulation of Gb3 [19–22]. ACE is involved in thegeneration of AngII, a potent vasoconstrictor, from AngI,an inactive precursor. Thus, based on the assumption thatthe RAS and its components could interfere with the effectsof ERT in Fabry disease, we sought to analyze the activityand concentration of components of this system in theplasma of Fabry patients. The first parameter we evaluatedwas the activity of ACE and we could not find anydifference in the plasma activity of this enzyme in the
patients when compared to healthy subjects. However, atthe end of the rhα-GalA infusion (4 h after the beginning),ACE activity drastically decreased to much lower values,suggesting an inhibitory role of rhα-GalA upon ACE. Thus,the beneficial effects of rhα-GalA therapy could be linked toits capacity of inhibiting ACE activity and consequently theamount of AngII produced in Fabry patients. The observedupregulation of ACE activity 2 weeks after ERT may be dueto a compensatory increase in ACE expression.
In order to further explore these intriguing findings andto prove this hypothesis, we performed in vivo and in vitroexperiments and we present herein strong evidences for aninteraction between the recombinant enzyme rhα-galactosidase A and ACE. Our data point to an inhibitoryaction of the recombinant enzyme on ACE activity, whichis probably mediated by the release of the galactose residuesfrom the ACE molecule. This assumption is based on the factthat the main activity of rhα-GalA is to catalyze the hydrolysisof α-galactosides. All forms of ACE are heavily glycosylatedand differences in glycosylation affect its enzymatic activity[37–41]. The degree of ACE glycosylation is important forthe catalytic properties of the enzyme [37]. In addition,glycosylation plays an important role in the release of ACEfrom the membrane, possibly by affecting the folding of theprotein and its recognition by other enzymes involved in itstransport and cleavage [42]. Kost et al. [43] demonstratedthat asialo-ACE or agalacto-ACE was able to form a dimer,
Fig. 1 ACE activity, serum AngI, and AngII concentration and meanarterial pressure in Fabry patients. ACE activity (a), serum angiotensinI (b) and II (c) of control subjects, Fabry patients ERT naïve, andFabry patients under ERT before and after the infusion. ACE activitywas evaluated using the fluorescent substrate Abz-FRK(Dnp)P-OH.Mean arterial pressure of Fabry patients before and after rhα-GalA
infusion (d). *p<0.001 Fabry patients before infusion vs. Fabry ERT-naïve and -control subjects; **p<0.001 Fabry patients after infusionvs. Fabry ERT and control subjects; ***p<0.001 Fabry patients beforeinfusion vs. Fabry patients after infusion; #p<0.05 Fabry patientsbefore infusion vs. Fabry patients after infusion. Values are expressedas means±SE of 16 determinations
70 J Mol Med (2011) 89:65–74

whereas deglycosylated ACE and sequentially desialylatedand degalactosylated ACE failed to dimerize. In thebiomembrane system studied by the authors, the somaticACE forms dimers via carbohydrate-mediated interactions,providing evidence for the existence of a carbohydrate-recognizing domain on the ACE molecule. The same kind ofresult has been shown by Gralnick et al. [44] for vonWillebrand factor (vWF). These authors have shown that thepenultimate galactose in the vWF molecule was responsiblefor multimer formation and activity. On the other hand,studies from Conroy et al. [45], Baudin et al. [37], and Orthet al. [46] have shown that deglycosylation of maturesomatic ACE does not alter ACE enzymatic activity.
Decreased ACE activity implies reduction in the con-version of AngI to AngII, a potent vasopressor [47, 48]. Wewere able to demonstrate this inhibitory effect of rhα-GalAin in vivo and in vitro models. For instance, rhα-GalAtreatment in Fabry patients reduced plasma ACE activity; invitro incubation of plasma from healthy volunteers withrhα-GalA inhibited ACE activity; incubation of CHO-ACEcells with rhα-GalA decreased ACE activity; treatment ofrats with rhα-GalA decreased ACE activity, as measured bythe conversion of AngI to AngII and finally, incubation ofrhα-GalA with purified ACE partially inhibited ACEactivity. Direct blood pressure measurements in rats showed
Fig. 3 Inhibitory effect of rhα-GalA on ACE activity in vivo in rats.rhα-GalA (Fabrazyme®) was injected intraperitonially in rats and itsinhibitory effect on ACE activity was evaluated by the contractileresponse of AngI and AngII in an ileum preparation. AngI and Ang II(10−7 M) were administrated to the rat ileum preparation before(control, CT) and after treatment of the animals 24 h earlier with 1 μg/grhα-GalA (a). Direct hypertensive effect of AngI and AngII injectedintravenously on the blood pressure in awaken rats (b) and ratio ofblood pressure effect for AngI/AngII after intravenous administrationof 10−8 M AngI or AngII (c). Results are means±SE of six preparations.*p<0.05 when compared to control values
Fig. 2 Direct in vitro inhibition of ACE activity by rhα-GalA in theplasma of healthy control volunteers and in CHO-ACE cells. Plasmafrom healthy volunteers was incubated with 1 μg of rhα-GalA for 1,2, and 4 h (a). Results are means±SE of ten determinations. Dose-response curve for the inhibitory effect of α-GalA on ACE activity inCHO-ACE cells (b). Cells were incubated for 1 h at 37°C withdifferent amounts of rhα-GalA. ACE activity in CHO-ACE cellsincubated at 37°C with 1 μg of the rhα-GalA for 2 and 4 h (c). ACEactivity was evaluated using the fluorescent substrate Abz-FRK(Dnp)P-OH. Results are means±SE of ten determinations in a or fourdeterminations b and c. *p<0.05 vs. control subjects (CT); **p<0.01vs. CT; #p<0.001 vs. CT
J Mol Med (2011) 89:65–74 71
reduction in the response of AngI 4 h after administrationof the rhα-GalA. The reduced effect of the AngI normalizedfor the effect of AngII in these animals confirmed ACEactivity inhibition after infusion of rhα-GalA.
Perspectives
In summary, our results demonstrate that the rhα-GalAinteracts with circulating and membrane-bound ACE and
inhibits its activity, possibly by removing the galactoseresidues of the enzyme. This modulation in ACE activityby the rhα-GalA might have profound impact on thephysiology of Fabry patients, especially of those patientswith concomitant use of ACE inhibitors. This assumptionis based on the results herein presented which indicatethat Fabry patients under ERT have upregulated ACEactivity. Thus, blood pressure values of Fabry patientsreceiving ERT and especially those on concomitant ACEinhibitors therapy should be closely followed to avoidadverse events. Further studies are necessary to clarify theclinical importance of the interaction between ERT andACE as well as to unravel possible similar effects of rhα-GalA on other physiologically relevant glycosylated pro-teins. In addition, the influence of alternative pathways ofAngII formation independent of ACE activity like chymase,cathepsin G, elastase, and tonin on Fabry patients under ERTshould be investigated.
Fig. 5 Effect of rhα-GalA on a purified rabbit lung ACE. a ACEactivity of purified rabbit lung was inhibited dose-dependently in vitroafter rhα-GalA incubation. Rabbit ACE was incubated with differentamounts of rhα-GalA (1, 10, and 100 μg) and the activity wasevaluated using the fluorescent substrate Abz-FRK(Dnp)P-OH. bGalactose is released from purified rabbit lung by rhα-GalA. Rabbitlung ACE (10 μg) was incubated with rhα-GalA (1, 10, and 100 μg)for 4 h and the amount of galactose released was evaluated. Resultsare means±SE of five measurements. *p<0.05 or **p<0.01 whencompared to the control condition
Fig. 4 Mean arterial pressure and plasma ACE activity in rats afterrhα-GalA administration. Animals were treated with rhα-GalA(Fabrazyme® 1 μg/g intravenously every 6 days) and blood pressurewas assessed by telemetry during 24 days (a). Inhibition of ACEactivity by rhα-GalA (1 μg/g intravenously) in the plasma of rats wasassessed in vitro under control conditions at different times (b). ACEactivity was evaluated using the fluorescent substrate Abz-FRK(Dnp)P-OH. Mean arterial pressure was evaluated 2 h after the injection ofrhα-GalA alone (1 μg/g intravenously), after administration oflisinopril alone (10 mg/kg) or both drugs together (c). Results aremeans±SE of five determinations. *p<0.05 vs. CT; **p<0.01 vs. CT;#p<0.05 vs. CT
72 J Mol Med (2011) 89:65–74

Sources of funding This work was supported by Fundação deAmparo a Pesquisa do Estado de São Paulo (FAPESP, 2008/06676-8),Conselho Nacional de Desenvolvimento Científico e Tecnológico(CNPq), Coordenação de Aperfeiçoamento de Pessoal de NívelSuperior (Probral CAPES/DAAD, 239/06), Deutsche AkademischeAustauschdienst (DAAD/PROBRAL) and Deutsche Forschungsge-meinschaft (BA 1374/16-1). Elice Carneiro Batista was supported by adoctoral fellowship from CAPES PROEX (Nr.33009015001).
Disclosures María Verónica Muñoz Rojas works as medical manager,Genzyme do Brasil. Fabrazyme is manufactured by Genzyme for Fabrydisease patient treatment.
References
1. Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL,Lastes L (1967) Enzymatic defect in Fabry’s disease Ceramide-trihexosidase deficiency. N Engl J Med 276(21):1163–1167
2. BloomD, Speijer D, Linthosrt GE, Donker-KoopmanWG, StrijlandA, Aerts JMFG (2003) Recombinant enzyme therapy for Fabrydisease: absence of editing of humanα-galactosidase AmRNA. AmJ Hum Genet 72:23–31
3. Clarke TR (2007) Narrative review: Fabry disease. Ann InternMed 146:425–433
4. Brady RO, Schiffmann R (2000) Clinical features of and recentadvances in therapy for Fabry disease. Jama 284:2771–2775
5. Desnick RJ, Ioannou YA, Eng CM (2001) α-Galactosidase Adeficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS,Valle D (eds) The metabolic and molecular basis of inheriteddisease, 8th edn. McGraw-Hill, New York, pp 3733–3774
6. Matsuzawa F, Aikawa SI, Doi H, Okumiya T, Sakuraba H (2005)Fabry disease: correlation between structural changes in α-galactosidase, and clinical and biochemical phenotypes. HumGenet 117:317–338
7. MacDermot KD, Holmes A, Miners AH (2001) Anderson–Fabrydisease: clinical manifestations and impact of disease in a cohortof 98 hemizygous males. J Med Genet 38:750–760
8. Altarescu GM, Goldfarb LG, Park KY, Kaneski C, Jeffries N,Litvalk S, Nagle JW, Schiffmann R (2001) Identification of fifteennovel mutations and genotype–phenotype relationship in Fabrydisease. Clin Genet 60(46):51
9. Desnick RS, Brady R, Barrenger J, Collins AJ, Germain DP,Goldman M, Grabowsiki G, Packman S, Wilcose WR (2003)Fabry disease, an under-recognized multisystemic disorder: expertrecommendations for diagnosis, management, and enzyme replace-ment therapy. Ann Intern Med 138:338–346
10. Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM,Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin HA III etal (2002) Natural history of Fabry renal disease: influence of alfa-galactosidase A activity and genetic mutations on clinical course.Medicine 81:122–138
11. MacDermot KD, Holmes A, Miners AH (2001) Natural history ofFabry disease in affected males and obligate carrier females. JInherit Metab Dis 24(2):13–14
12. Vedder AC, Linthorst GE, van Breemen MJ, Groener JEM,Bemelman FJ, Strijland A, Mannens MMAM, Aerts JMFG,Hollak CEM (2007) The Dutch Fabry cohort: diversity of clinicalmanifestations and Gb3 levels. J Inherit Metab Dis 30:68–78
13. Aerts JM, Groener JE, Kuiper S, Donker-Koopman WE, StrijlandA, Ottenhoff R, Van Roomen C, Mirzaian M, Wijburg FA,Linthorst GE et al (2008) Elevated globotriaosylsphingosine is ahallmark of Fabry disease. PNAS 105(8):2812–2817
14. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S,Caplan L, Linthorst GE, Desnick RJ (2001) Safety and efficacy ofrecombinant human alfa-galactosidase A replacement therapy inFabry’s disease. N Engl J Med 345:9–16
15. Schiffmann R, Kopp JB, Austin HA III, Sabnis S, Moore DF,Weibel T, Balow JE, Brady RO (2001) Enzyme replacementtherapy in Fabry disease: a randomized controlled trial. JAMA285:2743–2749
16. MacDermot KD, Holmes A, Miners AH (2001) Anderson–Fabrydisease: clinical manifestations and impact of disease in a cohortof 60 obligate carrier females. J Med Genet 38:769–775
17. Vedder AC, Linthorst GE, Houge G, Groener JEM, Ormel EE,Bouma BJ, Aerts JMFG, Hirth A, Hollak CEM (2007) Treatmentof Fabry disease: outcome of a comparative trial with agalsidasealfa or beta at a dose of 0.2 mg/kg. PLoS ONE 2(7):e598.doi:10.1371/journal.pone.0000598
18. Eng CM, Germain DP, DP BM, Warnock DG, Wanner C, HopkinRJ, Bultas J, Lee P, Sims K et al (2006) Fabry disease: guidelinesfor the evaluation and management of multiorgan systeminvolvement. Genet Med 8(9):539–548
19. Griedling KK, Murphy TJ, Alexander RW (1993) Molecularbiology of the renin-angiotensin system. Circulation 87:1816–1828
20. Cushman DW, Cheung HS (1971) Concentration of angiotensinconverting enzyme in tissues of rat. BBA 250:261–265
21. Mendelsohn FAO, Allen AM, Chai SY, McKinley MJ, OldfieldBJ, Paxinos G (1990) The brain angiotensin system: insights frommapping its components. Trends Endocrinol Metab 1:189–197
22. Carluccio M, Soccio M, De Caterina R (2001) Aspects of genepolymorphisms in cardiovascular disease: the renin-angiotensinsystem. Eur J Clin Invest 31:476–488
23. Araujo MC, Melo RL, Cesari MH, Juliano MA, Juliano L,Carmona AK (2000) Peptidase specificity characterization of C-and N-terminal catalytic sites of angiotensin I-converting enzyme.Biochemistry 39:8519–8525
24. Ronchi FA, Andrade MC, Carmona AK, Krieger JE, Casarini DE(2005) N-domain angiotensin-converting enzyme isoform expres-sion in tissues of Wistar and spontaneously hypertensive rats. JHypertens 23(10):1793–1794
25. Sabatini RA, Bersanetti PA, Farias SL, Juliano L, Juliano MA,Casarini DE, Carmona AK, Paiva ACM, Pesquero JB (2007)Determination of angiotensin I-converting enzyme activity in cellsin culture using fluorescence resonance energy transfer peptides.Anal Biochem 363:255–262
26. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Proteinmeasurement with the Folin phenol reagent. J Biol Chem 1:265–275
27. Hoffmann B (2009) Fabry disease: recent advances in pathology,diagnosis, treatment and monitoring. Orphanet J Rare Dis 4:21
28. Wright JT, Bakris G, Wright JT, Bakris G, Greene T, Agodoa LY,Appel LJ, Charleston J, Cheek D, Douglas-Baltimore JG (2002)African American study of kidney disease and hypertension studygroup: effect of blood pressure lowering and antihypertensivedrug class on progression of hypertensive kidney disease: resultsfrom the AASK Trial. JAMA 288:2421–2431
29. Remuzzi G, Ruggenenti P, Perico N (2002) Chronic renaldiseases: renoprotective benefits of renin-angiotensin systeminhibition. Ann Intern Med 136:604
30. Rule AD, Larson TS, Bergstralh EJ, Slezak JM, Jacobsen SJ,Cosio FG (2004) Using serum creatinine to estimate glomerularfiltration rate: accuracy in good health and in chronic kidneydisease. Ann Intern Med 12:929–937
31. Ruggenenti P, Perna A, Gherardi G, Benini R, Remuzzi G (2000)Chronic proteinuric nephropathies: outcomes and response totreatment in a prospective cohort of 352 patients with differentpatterns of renal injury. Am J Kidney Dis 35:1155–1165
J Mol Med (2011) 89:65–74 73

32. Sarafidis PA, Khosla N, Bakris GL (2007) Antihypertensivetherapy in the presence of proteinuria. Am J Kidney Dis 49:12–26
33. Warnock DG (2009) Optimizing treatment to reach therapeuticgoals in Fabry disease. Clinical Therapeutics 31, Supplement A
34. Tahir H, Jackson LL, Warnock DG (2007) Antiproteinuric therapyand Fabry nephropathy: sustained reduction of proteinuria inpatients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol 18:2609–2617
35. Sakuraba H, Murata-Ohsawa M, Kawashima I, Tajima Y, KotaniM, Ohshima T, Chiba Y, Takashiba M, Jigami Y, Fukushige T(2006) Comparison of the effects of agalsidase alfa and agalsidasebeta on cultured human Fabry fibroblasts and Fabry mice. J HumGenet 51:180–188
36. Schiffmann R, Askari H, Timmons M, Robinson C, Benko W,Brady RO, Ries M (2007) Weekly enzyme replacement therapymay slow decline of renal function in patients with Fabry diseasewho are on long-term biweekly dosing. J Am Soc Nephrol18:1576–1583
37. Baudin B, Alves N, Pilon A, Beneteau-Burnat B, Giboudeau J(1997) Structural and biological roles of glycosylation inpulmonary angiotensin I-converting enzyme. Glycobiology 7:565–570
38. Ramchandran R, Kasturi S, Douglas JG, Sen I (1996)Metalloprotease-mediated cleavage secretion of pulmonary ACEby vascular endothelial and kidney epithelial cells. Am J Physiol271:H744–H751
39. Sadhukhan R, Santhamma KR, Reddy P, Peschon JJ, Black RA,Sen I (1999) Unaltered cleavage and secretion of angiotensin-converting enzyme in tumor necrosis factor-alphaconvertingenzyme-deficient mice. J Biol Chem 274:10511–10516
40. Soubrier F, Alhenc-Gelas F, Hubert C, Allegrini J, John M,Tregear G, Corvol P (1988) Two putative active centers in humanangiotensin I converting enzyme revealed by molecular cloning.Proc Natl Acad Sci 85:9386–9390
41. Ripka JE, Ryan JW, Valido FA, Chung AYK, Peterson CM, UrryRL (1993) N-glycosylation of forms of angiotensin convertingenzyme from four mammalian species. Biochem Biophys ResCommun 196:503–508
42. Yu XC, Sturrock ED, Wu Zh, Biemann K, Ehlers MRW, RiordanJF (1997) Identification of N-linked glycosylation sites in humantestis angiotensin-converting enzyme and expression of an activedeglycosylated form. J Biol Chem 272:3511–3519
43. Kost OA, Bovin NV, Chemodanova EE, Nasonov VV, Orth TA(2000) New feature of angiotensin-converting enzyme: carbohydrate-recognizing domain. J Mol Recognit 13:360–369
44. Gralnick HR, Kramer WS, McKeown LP, Garfinkel L, Pinot A,Williams SB, Krutzsch H (1996) Platelet adhesion at high shearrates: the roles of von Willebrand factor/GPIb and the beta 1integrin alpha 2 beta 1. Thromb Res 81(1):113–119
45. Conroy JM, Hartley JM, Soffer RL (1978) Canine pulmonaryangiotensin-converting enzyme: Physicochemical, catalytic andimmunological properties. Biochim Biophys Acta 524:403–412
46. Orth T, Voronov S, Binevski P, Saenger W, Kost OA (1998)Glycosylation of bovine pulmonary angiotensin-converting enzymemodulates its catalytic properties. FEBS Lett 431:255–258
47. Corvol P, Williams TA, Soubrier F (1995) Dipeptidyl dipeptidase:angiotensin-converting enzyme. Meth Enzymol 248:283–305
48. Ehlers MRW, Riordan JF (1989) Angiotensin-converting enzyme:new concepts concerning its biological role. Biochemistry28:5318–5322
74 J Mol Med (2011) 89:65–74