a new incorporation mechanism for trivalent actinides into bioapatite: a trlfs and exafs study
TRANSCRIPT

A New Incorporation Mechanism for Trivalent Actinides intoBioapatite: A TRLFS and EXAFS StudyKiel Holliday,*,†,‡ Stephanie Handley-Sidhu,§ Kathy Dardenne,† Joanna Renshaw,§ Lynne Macaskie,⊥
Clemens Walther,† and Thorsten Stumpf*,†
†Institute for Nuclear Waste Disposal, Karlsruhe Institute for Technology, 1 Hermann-von Helmholtz-platz,Eggenstein-Leopoldshafen 76344, Germany‡Lawrence Livermore National Lab, 7000 East Avenue, Livermore, California 94551, United States§School of Geography, Earth and Environmental Sciences, ⊥School of Biosciences, University of Birmingham, Edgbaston,Birmingham, B15 2TT United Kingdom
*S Supporting Information
ABSTRACT: One of the most toxic byproducts of nuclear power andweapons production is the transuranics, which have a high radiotoxicityand long biological half-life due to their tendency to accumulate in theskeletal system. This accumulation is inhomogeneous and has beenassociated with the chemical properties and structure of the bonematerial rather than its location or function. This suggests a chemicaldriving force to incorporation and requires an atomic scale mechanisticunderstanding of the incorporation process. Here we propose a newincorporation mechanism for trivalent actinides and lanthanides intosynthetic and biologically produced hydroxyapatite. Time-resolvedlaser fluorescence spectroscopy and extended X-ray absorption finestructure have been used to demonstrate that trivalent actinides andlanthanides incorporate into the amorphous grain boundaries ofapatite. This incorporation site can be used to explain patterns in uptake and distribution of radionuclides in the mammalianskeletal system.
■ INTRODUCTIONOne of the most toxic byproducts of nuclear power andweapons production is the transuranics due to their longbiological half-life and high radiotoxicity. For instance, Pu, Am,and Cm have been identified as more carcinogenic than Ra atsimilar activities due to the inhomogeneous nature of theirdistribution in the skeletal system.1−3 As much as 70% of theseradionuclides are deposited into the bone material leading tolong biological half-lives.4,5 This inhomogeneous distributionhas been shown to be dependent on the chemical propertiesand structure of the bone and not its location or function.2,4
This would suggest that incorporation and retention ofradionuclides within biological hard tissue, consisting mostlyof hydroxyapatite, is dependent on the chemical incorporationmechanism independent of biological function. The chemicalform and solubility of the transuranics such as Pu, Am, and Cmhas been shown to have considerable influence on uptake in theskeletal system.6,7 This has led to the investigation of differenttreatments, such as DTPA, to limit the retention of theseradionuclides.8 These treatments while limiting uptake in theliver have little effect on the uptake by the skeletal system,despite evidence that long-term deposition into hard tissue cantake days.8,9
There is, therefore, a need to develop an atomic scalemechanistic understanding of the incorporation process at thesolid−solution interface so that treatment can better addressthe process of actinide deposition in biological hard tissue. Byusing apatite, the main component of bone material, one canstudy a well-defined single component approximating thebehavior of biological hard tissue. Previous work has beenperformed with lanthanide homologues of the trivalentactinides on postincorporation heat treated samples of apatiteby X-ray diffraction (XRD),10−13 extended X-ray absorptionfine structure (EXAFS),14 and time-resolved laser fluorescencespectroscopy (TRLFS).15−17 These studies on heated samplesare irrelevant to the incorporation at room temperature since ithas been shown that the speciation is different before heattreatment.18,19 Two key studies have focused on the speciationof Eu3+ (an analogue for trivalent actinide ions) in apatite atroom temperature by TRLFS. Both studies concluded that Eu3+
was incorporated into the C3 symmetry Ca(I) site ofapatite.18,19 In the first study, all reported emission spectrahad more peaks than would be permitted by C3 symmetry,
Received: January 2, 2012Revised: February 3, 2012Published: February 7, 2012
Article
pubs.acs.org/Langmuir
© 2012 American Chemical Society 3845 dx.doi.org/10.1021/la300014a | Langmuir 2012, 28, 3845−3851

which was explained as emission lines arising from impurities inthe naturally occurring apatite samples used for the study.18
The second study eliminated this possibility by using syntheticapatite and produced similar emission spectra.19 It was alsoconcluded to be Eu3+ substituting on the Ca(I) site because ofbeing trapped by the coprecipitation process. Although siteselective excitation was used in this later study, the emissionspectra had a poor resolution at room temperature, and it is notpossible to clearly resolve the splitting to make a definitivestatement on the symmetry of the Eu3+. Only one study wasperformed under similar conditions with actinides, namely Cm,where it was concluded that Cm3+ incorporates into the apatitestructure unlike other phosphate mineral phases such asTh4P6O23, ZrP2O7, and Zr2O(PO4)2 in which only surfacesorption was evident.20 No speculation was made on the site ofincorporation in the Cm study. This study is aimed at resolvingconflicting information on the site of incorporation forlanthanides in synthetic apatite and tests the applicability ofthe proposed mechanism to actinides and biologically producedapatite.
■ METHODSHydroxyapatite Synthesis. Hydroxyapatite was synthesized by
coprecipitation of calcium and phosphate at basic pH. All chemicalswere reagent grade and purchased from Alpha Aesar. Concentratedsolutions of calcium chloride and sodium hydrogen phosphate weremade by dissolving in 18 MΩ water. These solutions were then mixedin a 5:3 calcium to phosphate ratio and brought to pH 11 with sodiumhydroxide. The solution was stirred for 24 h before the solid wascollected via filtration. This solid was dried and subsequently heated to700 °C to improve crystallinity.Bioapatite Synthesis. Serratia sp. NCIMB 40259 was used with
kind permission of Isis Innovation, Oxford, UK.21 Biofilm-loaded foamcubes (prewashed in isotonic saline; ∼4.5 mg dry cell mass/cube) wereplaced in flasks containing 50 mL of TAPSO/NaOH buffer (50 mM;pH 9.2) with 5 mM glycerol 2-phosphate (G2P) and 1 mM CaCl2.The volume lost to evaporation was replaced daily with the samesolutions over the 8 day synthesis, after which the material wasdislodged by a gentle tapping and squeezing action. The remainingsolid was washed with acetone, air-dried, and characterized byscanning electron microscope and X-ray diffraction.22
X-ray Diffraction (XRD). Samples were ground to a fine powderand spread in a thin layer over a low-background sample holder (singlecrystal silicon wafer) with the aid of methanol. The powder XRDpatterns were collected on a Bruker D8 Advance diffractometer, usinga Cu anode (wavelength Kα1 at 0.1540598 nm). Patterns were takenwith use of an acceleration of 40 mV and current of 40 mA over arange from 10 to 120° 2θ with a step size of 0.01° 2θ and 4 s per step.Phases were identified with Bruker-AXS EVA software.Batch Studies. Batch studies were performed simply by placing the
solution containing the doping ion in contact with hydroxyapatite,while controlling ionic strength, solid to liquid ratio, pH, and dopingion concentration. The solution containing the doping ion was 2.0mM sodium perchlorate to maintain ionic strength and was adjusted topH of 6 with sodium hydroxide and perchloric acid as needed beforebeing added to hydroxyapatite at a solid to liquid ratio of 2.0 g/L. TheCm concentration was 0.08 μM (taken from stock solution) and theEu concentration was 1.0 μM. Samples were occasionally stirred overthe contact time of 21 days. The isotopic composition of the long-livedcurium solution (20.0 μM) is 97.3% Cm-248, 2.6% Cm-246, 0.04%Cm-245, 0.02% Cm-247, and 0.009% Cm-244 in 1.0 M HClO4.Time-Resolved Laser Fluorescence Spectroscopy (TRLFS).
TRLFS of Eu3+ is a versatile structural probe in both crystalline andamorphous solids. The nondegenerate (5D0 → 7F0) transition givesrise to a single emission line for each species allowing for determiningthe number of nonequivalent sites. This transition can then beselectively excited to obtain an emission spectrum from each unique
Eu3+ environment. The splitting of subsequent transitions in theemission spectrum (5D0 →
7F1−6) is determined by the local symmetryof the Eu3+ and follows the selection rules determined by Judd-Ofelttheory. The fluorescence lifetime can also be related to the number ofwaters in the immediate coordination sphere by Horrocks’ equation23
allowing for the distinction between surface sorbed and incorporatedspecies. Further detail of Eu3+ TRLFS can be found in severalthorough reviews.24−26
TRLFS of Eu3+ is rather weak because of the multiple transitionsand not suited for trace level investigations. By using Cm3+ thedetection limit is lowered to 107 atoms mm2− and expands the study toactinides, which are relevant to radiotoxicity issues. The 6D7/2 →
8S7/2transition of Cm3+ is sensitive to spectral shifts due to the ligand fieldand the 8S7/2 ground state can be split depending on ligand strengthand symmetry. Once again the fluorescence lifetime can be related tothe number of water molecules by a simple equation. In this case theKimura equation.27 Further information on Cm3+ TRLFS can befound in the reviews by Edelstein28 or Geipel.29
TRLFS was performed by using a pulsed (20 Hz) XeCl-eximer laser(Lambda Physics, EMG, 308 nm) pumped dye laser (LambdaScanmate). The following dyes were used: QUI for UV excitation,Rhodamine 6G for direct excitation of Eu, and Rhodamine B for directexcitation of Cm. Indirect excitation of Eu and Cm was performed at394.0 and 396.6 nm, respectively. The range used for the directexcitation of Eu was 575−582 nm, and the direct excitation of Cm wasperformed at 600−612 nm. The laser wavelength was monitored witha Toptica WS7 wavemeter (>10−5 nm accuracy). Fluorescencemeasurements were detected by an optical multichannel analyzerthat consists of a polychrometer with 300/600/1200 lines/mmgratings (Jobin Yvon) and an intensified, gated photodiode array(Spectroscopy Instruments). Maximum resolution at 300 and 1200lines/mm was measured to be 0.9 and 0.2 nm, respectively. Thedetection system was calibrated with a neon lamp (Pen Ray 6032).The samples were cooled to <20 K by a helium refrigerated cryostat(CTI-cryogenics) to improve resolution. For the discrimination ofRayleigh and Raman scattering the minimum gate delay between laserpulse and camera gating was set to 1.0 μs. The gate width of thecamera was fixed at 10 ms to ensure the collection of the entirefluorescence signal. Fluorescence lifetime measurements were madewith a delay time step between 15 and 200 μs and a total of 60 stepswere taken for each lifetime measurement.
Extended X-ray Absorption Fine Structure (EXAFS). Euro-pium L3-edge and L2-edge spectra were recorded at the INE beamlineat ANKA (Germany), using a double-crystal monochromator (DCM)equipped with a pair of Si(111) crystals. The DCM is operated infixed-exit mode. The incident intensity is held constant by means of apiezo-driven feedback system. The parallel alignment of the crystalfaces is detuned to ∼70% of the maximum beam intensity. Thesamples powders were mounted in Kapton tape. Measurements weremade in fluorescence mode, using a five-element Ge detector with asample orientation of 45° to the incident beam. The energy calibrationwas performed with a Fe metal foil (K edge at 7112 eV). EXAFS dataanalysis was performed with standard procedures in Athena andArtemis30 interfaces of the iFEFFIT software.31 The phase andamplitude data used for data analysis are calculated for a 15 atomcluster derived from the undoped apatite structure. Single pathscattering files for phase and amplitude are used too for comparison.The k-range used for the fit is 2.669−8.715 Å−1 and fits are performedin the R-space on the k2-weighted data. The overall scaling factor S0
2
was held constant at 0.85 after its determination on a standard sample.Distances are determined with an error of 0.02 Å and coordinationnumbers with an error of 20%.
■ RESULTS AND DISCUSSION
TRLFS of Eu3+-Doped Hydroxyapatite. This study usessite-selective TRLFS of Eu doped at 100 ppm in synthetichydroxyapatite to eliminate extraneous peaks from multiplesites or impurities. Measurements were performed at deeptemperatures (<20 K) for improved resolution. Despite
Langmuir Article
dx.doi.org/10.1021/la300014a | Langmuir 2012, 28, 3845−38513846
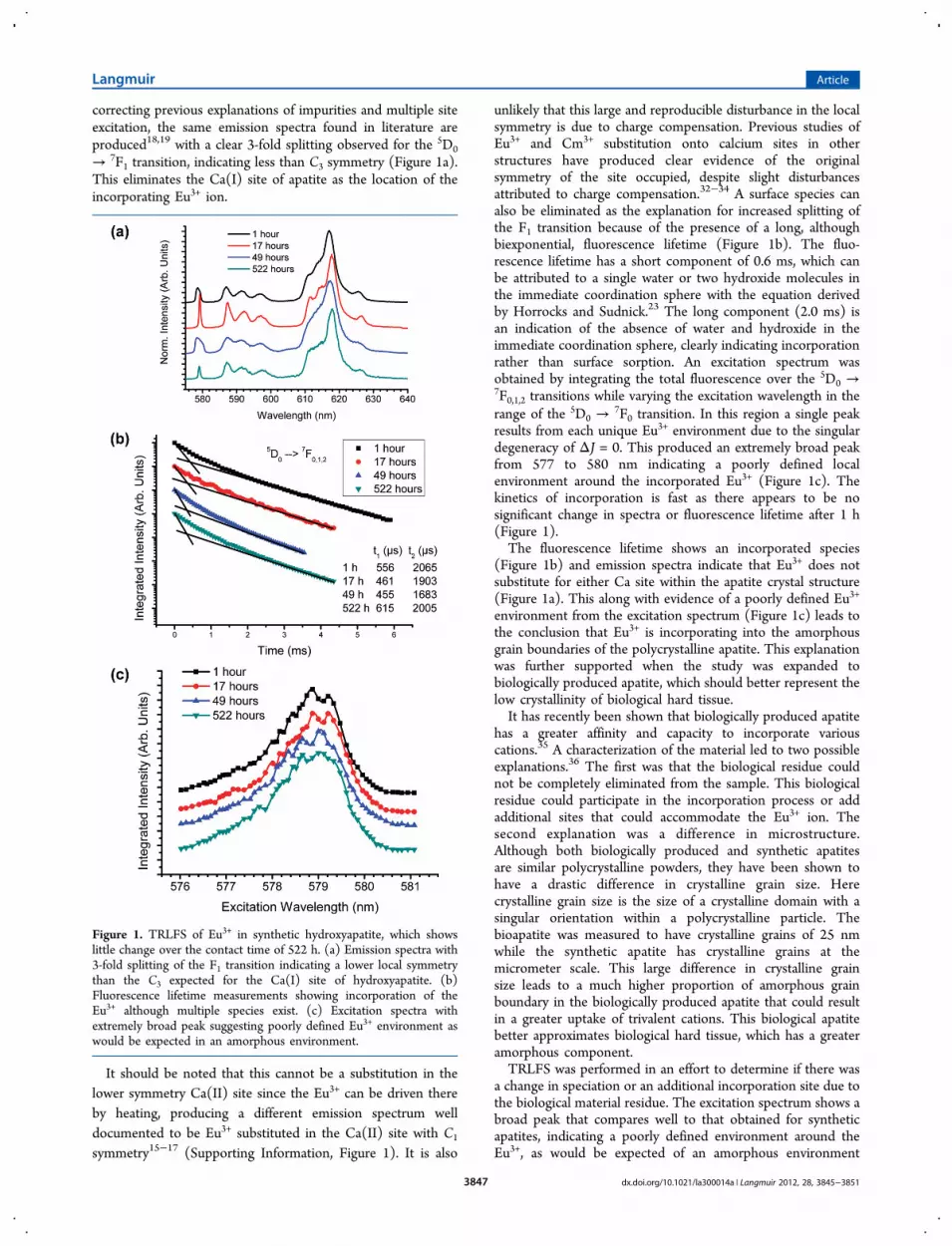
correcting previous explanations of impurities and multiple siteexcitation, the same emission spectra found in literature areproduced18,19 with a clear 3-fold splitting observed for the 5D0
→ 7F1 transition, indicating less than C3 symmetry (Figure 1a).This eliminates the Ca(I) site of apatite as the location of theincorporating Eu3+ ion.
It should be noted that this cannot be a substitution in thelower symmetry Ca(II) site since the Eu3+ can be driven thereby heating, producing a different emission spectrum welldocumented to be Eu3+ substituted in the Ca(II) site with C1
symmetry15−17 (Supporting Information, Figure 1). It is also
unlikely that this large and reproducible disturbance in the localsymmetry is due to charge compensation. Previous studies ofEu3+ and Cm3+ substitution onto calcium sites in otherstructures have produced clear evidence of the originalsymmetry of the site occupied, despite slight disturbancesattributed to charge compensation.32−34 A surface species canalso be eliminated as the explanation for increased splitting ofthe F1 transition because of the presence of a long, althoughbiexponential, fluorescence lifetime (Figure 1b). The fluo-rescence lifetime has a short component of 0.6 ms, which canbe attributed to a single water or two hydroxide molecules inthe immediate coordination sphere with the equation derivedby Horrocks and Sudnick.23 The long component (2.0 ms) isan indication of the absence of water and hydroxide in theimmediate coordination sphere, clearly indicating incorporationrather than surface sorption. An excitation spectrum wasobtained by integrating the total fluorescence over the 5D0 →7F0,1,2 transitions while varying the excitation wavelength in therange of the 5D0 →
7F0 transition. In this region a single peakresults from each unique Eu3+ environment due to the singulardegeneracy of ΔJ = 0. This produced an extremely broad peakfrom 577 to 580 nm indicating a poorly defined localenvironment around the incorporated Eu3+ (Figure 1c). Thekinetics of incorporation is fast as there appears to be nosignificant change in spectra or fluorescence lifetime after 1 h(Figure 1).The fluorescence lifetime shows an incorporated species
(Figure 1b) and emission spectra indicate that Eu3+ does notsubstitute for either Ca site within the apatite crystal structure(Figure 1a). This along with evidence of a poorly defined Eu3+
environment from the excitation spectrum (Figure 1c) leads tothe conclusion that Eu3+ is incorporating into the amorphousgrain boundaries of the polycrystalline apatite. This explanationwas further supported when the study was expanded tobiologically produced apatite, which should better represent thelow crystallinity of biological hard tissue.It has recently been shown that biologically produced apatite
has a greater affinity and capacity to incorporate variouscations.35 A characterization of the material led to two possibleexplanations.36 The first was that the biological residue couldnot be completely eliminated from the sample. This biologicalresidue could participate in the incorporation process or addadditional sites that could accommodate the Eu3+ ion. Thesecond explanation was a difference in microstructure.Although both biologically produced and synthetic apatitesare similar polycrystalline powders, they have been shown tohave a drastic difference in crystalline grain size. Herecrystalline grain size is the size of a crystalline domain with asingular orientation within a polycrystalline particle. Thebioapatite was measured to have crystalline grains of 25 nmwhile the synthetic apatite has crystalline grains at themicrometer scale. This large difference in crystalline grainsize leads to a much higher proportion of amorphous grainboundary in the biologically produced apatite that could resultin a greater uptake of trivalent cations. This biological apatitebetter approximates biological hard tissue, which has a greateramorphous component.TRLFS was performed in an effort to determine if there was
a change in speciation or an additional incorporation site due tothe biological material residue. The excitation spectrum shows abroad peak that compares well to that obtained for syntheticapatites, indicating a poorly defined environment around theEu3+, as would be expected of an amorphous environment
Figure 1. TRLFS of Eu3+ in synthetic hydroxyapatite, which showslittle change over the contact time of 522 h. (a) Emission spectra with3-fold splitting of the F1 transition indicating a lower local symmetrythan the C3 expected for the Ca(I) site of hydroxyapatite. (b)Fluorescence lifetime measurements showing incorporation of theEu3+ although multiple species exist. (c) Excitation spectra withextremely broad peak suggesting poorly defined Eu3+ environment aswould be expected in an amorphous environment.
Langmuir Article
dx.doi.org/10.1021/la300014a | Langmuir 2012, 28, 3845−38513847

(Figure 2a). The fluorescence lifetime was also similar for bothbiologically produced and synthetic apatite (Figure 2b). The
most compelling evidence was that the fluorescence emissionspectra are identical (Figure 2c). This illustrates that the samespeciation exists in both samples, which eliminates biologicalresidue as a possible explanation for the increase in uptake inthe case of trivalent actinides and lanthanides. The mostprobable explanation is, therefore, the increased amorphousgrain boundary proportion in biologically produced apatiteresults in an increase in the uptake of cations.Structural Determination with EXAFS. Extended X-ray
absorption fine structure (EXAFS) was performed as acomplementary probe of the local environment and structure
of the incorporated Eu3+. It also is able to provide uniqueinformation not available by TRLFS such as bond distance andcoordination number. The results for synthetic and biologicallyproduced apatite were similar with the exception that traceamounts of Fe present in biologically produced apatite onlyallowed for measurements up to k = 6.1 Å−1 (SupportingInformation, Table 1). Because of this, only results for synthetichydroxyapatite are presented and compared to expected valuesfor that of the Ca(I) site in hydroxyapatite (ICSD #22059) inTable 1.
The distance from the incorporated Eu3+ to nearest neighboroxygens was fit with a single shell, because a two-shell approachdid not significantly improve the fit or lower the Debye−Wallerfactor (Figure 3). For comparison, distances in the referencedCa(I) site of hydroxyapatite are given as a weighted average,although it is recognized that the nearest neighbor oxygenatoms occur at two distinct distances. The first shell oxygenatoms were pulled closer to the Eu3+ atom than the Ca(I) sitecrystal structure by 0.16 Å. It could be argued that this was adistortion due to the incorporation of Eu3+ as a solid solutionalthough the effect is larger than expected for the difference inradii (0.04 Å) when compared to other solid solutions.37 Thiseffect is consistent with an amorphous material that would havemore flexibility in bond distances. The coordination numberhas also dropped from 9 in the solid solution to 7.9. This wasalso measured in EXAFS studies of Eu phosphate glass,38
although it should be noted that this is not significantlydifferent given the large uncertainty in coordination number inEXAFS measurements. Nevertheless, a drop of the coordina-tion number is fully consistent with a diminution of the bondlength. The next shell was fit with phosphate and shows nosignificant difference from the hydroxyapatite, although nonecould be expected given previous studies of amorphousphosphates.38
Definitive differences arise in the Eu−Ca shell as comparedto the Ca(I)−Ca distance. Here any effect due to theisomorphic substitution of Eu3+ for Ca would have dissipated.The Eu3+ data show an increase in coordination number whencompared to the Ca(I) site; however, the most strikingdifference was an increase in the Eu−Ca interaction distance by0.65 Å longer than the Ca(I)−Ca distance. It should be notedthat these results are also inconsistent with substitution on theCa(II) site as was already established for samples after heattreatment.18 This can only be explained by a complete breakwith the crystal structure of hydroxyapatite and eliminates the
Figure 2. TRLFS of Eu3+ in biologically produced apatite as comparedto synthetic apatite. (a) Excitation spectra with broad peak comparableto previous analysis of synthetic apatite suggesting amorphousenvironment. (b) Lifetime measurements comparable to syntheticapatite confirming incorporation. (c) Emission spectra from bioapatiteas compared to the synthetic hydroxyapatite indicating the sameincorporated Eu3+ environment suggesting the same incorporationmechanism and excluding biological residue as an incorporation site.
Table 1. EXAFS Fitted Parameters Compared to That of theCa(I) Sitea
bond R (Å) N σ2 (Å2) Eo (eV)
Eu−O 2.39 7.9 0.004 4.6Ca(I)−O 2.55 9.0Eu−P 3.21 4.2 0.0002 10.7Ca(I)−P 3.21 3.0Eu−Ca 4.11 3.7 0.0004 11.8Ca(I)−Ca 3.44 2.0
aTypical error associated with coordination number is 20% whilereported bond distances are ±0.02 Å. The k-range of fitting was2.669−8.715 Å−1. The R-range of fitting was 1.341−4.262 Å. Thegoodness of fit can be expressed as the difference of the fit andexperimental data as a percent referred to as the residual, which was1.2% for hydroxyapatite.
Langmuir Article
dx.doi.org/10.1021/la300014a | Langmuir 2012, 28, 3845−38513848

possibility of Eu3+ incorporating through isomorphic sub-stitution within apatite at room temperature. The EXAFS datathereby confirmed what was suggested by TRLFS. Eu3+ isincorporating into the amorphous grain boundaries of syntheticand biologically produced hydroxyapatite.Actinide Containing Material: Cm3+ Studies. TRLFS
was performed on Cm3+ containing hydroxyapatite andbiologically produced hydroxyapatite in the same way that itwas done for Eu3+ apatites. The emission spectra after UVexcitation shows an extremely broad (fwhm =10 nm) peakcentered at 607 nm for synthetic and bioapatite, establishingthe same local environment for trivalent actinides in both formsof apatite (Figure 4a). The large peak width suggests a range oflocal environments that would also be expected for Cm3+
incorporated into the amorphous grain boundaries as wasestablished for Eu3+ by TRLFS and EXAFS. The maximumemission wavelength was slightly higher in energy than the 609nm that was previously reported.20 This is likely due to thepublished spectrum being collected at a longer delay time,which has the effect of shifting the peak to lower energy. Thiseffect occurs because the range of species present exhibitshorter lifetimes at higher energies. The peak reported here at607 nm still shows a stronger bathochromic shift than otherexamples of crystalline Cm3+-doped phosphates, such as the 8-fold coordinated LuPO4
39 and 9-fold coordinated LaPO4.40
The excitation spectrum produced the same extremely broadpeak as UV excitation with a large shoulder at higher energies(Figure 4b). This shoulder was shown to be due to “hot band”excitation of the higher excited states and the large increase in
intensity reinforces the assumption that a large range of Cm3+
environments exists. The fluorescence lifetimes in both sampleswere similar and fit to a biexponential lifetime (Figure 4c). Theshort-lived component of 235 μs can be attributed to twowaters by the Kimura equation.27 The longer lived lifetime of850 μs shows a complete loss of the hydration sphere indicatingincorporation into the bulk. These results compare well to thatobtained with Eu3+.Lastly, direct excitation was used to probe the region of Cm3+
fluorescence. It can be seen through fluorescence line
Figure 3. EXAFS data with fit of synthetic hydroxyapatite. (a) FTmagnitude (thick solid line), imaginary part (thin solid line), and fitresults (open triangles and circles). FT is performed in the range1.341−4.262 Å. (b) k2-weighted Eu L3 EXAFS of the sample (solidline) and fit result (open circles). Data were fit in the range 2.669−8.715 Å−1. The residual of the fit was 1.2%.
Figure 4. TRLFS of Cm3+ in synthetic apatite and bioapatite. (a)Emission spectra with broad peak due to poorly defined Cm3+
environment centered at 607 nm. (b) Fluorescence lifetime measure-ment indicating incorporation and multiple environments. (c)Excitation spectrum of biologically produced hydroxyapatite indicatingthe range of species as would be expected from an amorphousenvironment with large shoulder due to hot band excitation.
Langmuir Article
dx.doi.org/10.1021/la300014a | Langmuir 2012, 28, 3845−38513849

narrowing that the broad emission spectrum shown by UVexcitation is in no way limited to the resolution of theinstrument (Figure 5). This demonstrated that the broad peak
emission spectrum was due to a continuum of closely relatedsites. Such spectra would be expected from a sample with apoorly defined Cm3+ environment that has no long-range order.This is presented as further evidence that trivalent actinide andlanthanide incorporate into the amorphous grain boundaries ofhydroxyapatite.
■ CONCLUSIONS AND IMPLICATIONS
We propose a new mechanism for trivalent actinide andlanthanide uptake into apatites: incorporation into theamorphous grain boundaries of the polycrystalline apatite andno incorporation onto a calcium site within the crystal structureas previously proposed. This has been demonstrated in thecurrent study by TRLFS and EXAFS of synthetic andbiologically produced hydroxyapatite doped with Eu3+ andCm3+. This is in direct conflict with previous assumptions thatincorporation took place as an isomorphic substitution on theCa(I) site creating a solid solution. This new conclusion notonly explains the current data presented in this paper, but alsopreviously unexplained phenomena presented in the TRLFS ofother Eu:apatite studies performed by incorporating ions atroom temperature.18,19
This proposed mechanism for the incorporation of trivalentactinides helps to understand the incorporation process anddistribution of transuranics throughout the skeletal system oforganisms exposed to transuranic elements. It has been shownthat Pu, Am, and Cm have an inhomogeneous distribution inbiological hard tissue leading to high local concentrations andconsequently a higher radiotoxicity.1−4 This finding can now berevisited with the insight that these areas of high localconcentration are most likely areas of low crystallinity. Inareas of high crystallinity it would be assumed that surfacesorption would be the main mode of retention. Because of theslow growth of bones, it has been shown that incorporation cantake days in areas where surface sorption is dominant.9 Thisnew insight would allow one to distinguish situations where atreatment to desorb radionuclides might be successful fromthose where incorporation has most likely already taken place.Understanding the processes that govern the sorption andincorporation of radionuclides in biological hard tissue is
envisioned as the first step in designing treatment to decreasethe amount of radionuclides retained over long time periods byorganisms exposed to trivalent actinides.
■ ASSOCIATED CONTENT*S Supporting InformationA table giving the EXAFS fitted parameter of synthetic apatite(OH-Ap) and biologically produced apatite (Bio-Ap) and afigure showing the TRLFS emission spectrum of sintered Eu3+-doped hydroxyapatite. This material is available free of chargevia the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] (K.H.) or [email protected](T.S.).NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSWe would like to thank Sebastian Buchner for technicalassistance with TRLFS measurements and Claire Mennan forassistance with preparation of Serratia sp. This work wascofinanced by the Helmholtz Gemeinschaft DeutscherForschungszentren (HGF) by supporting the Helmholtz-Hochschul-Nachwuchsgruppe “Aufklarung geochemischer Re-aktionsmechanismen an der Wasser/Mineralphasen Grenzfl-ache” and the EPSRC (EP/G063699/1). We thank the ANKAsynchrotron source for providing the beamtime. This work wasperformed under the auspices of the U.S. Department ofEnergy by Lawrence Livermore National Laboratory underContract DE-AC52-07NA27344 and by the Department ofHomeland Security, Domestic Nuclear Detection Office underContract HSHQDC-07-C-00034.
■ REFERENCES(1) Priest, N. D.; Jackson, S. Plutonium in bone: a high resolutionautoradiographic study using plutonium-241. Int. J. Radiat. Biol. 1977,32, 325−350.(2) Polig, E.; Bruenger, F. W.; Lloyd, R. D.; Miller, S. C.Microdistribution of 239Pu in the beagle skeleton. Health Phys. 1998,75, 251−258.(3) Wood, M. D.; Beresford, N. A.; Semenov, D. V.; Yankovich, T.L.; Copplestone, D. Radionuclide transfer to reptiles. Radiat. Environ.Biophys. 2010, 49, 509−530.(4) McInroy, J. F.; Boyd, H. A.; Eutsler, B. C.; Romero, D. Part IV:preparation and analysis of the tissues and bones. Health Phys. 1985,49, 587−621.(5) Thomas, R. G.; Healy, J. W.; McInroy, J. F. Plutoniumpartitioning among internal organs. Health Phys. 1984, 46, 839−844.(6) Fouillit, M.; et al. Comparative tissue uptake and cellulardeposition of three different plutonium chemical forms in rats. Int. J.Radiat. Biol. 2004, 80, 683−689.(7) Guilmette, R. A.; Kanapilly, G. M.; Lundgren, D. L.; Eidson, A. F.Biokinetics of inhaled 244Cm oxide in the rat: effect of heat treatmentat 1150 °C. Health Phys. 1984, 46, 845−858.(8) Guilmette, R. A.; Muggenburg, B. A. Decorporation therapy forinhaled plutonium nitrate using repeatedly and continuouslyadministered DTPA. Int. J. Radiat. Biol. 1993, 63, 395−403.(9) Talbot, R. J.; Newton, D.; Warner, A. J. Metabolism of injectedplutonium in two healthy men. Health Phys. 1993, 65, 41−46.(10) Fleet, M. E.; Pan, Y. Site preference of Nd in fluorapatite[Ca10(PO4)6F2]. J. Solid State Chem. 1994, 112, 78−81.(11) Fleet, M. E.; Pan, Y. Site preference of rare earth elements influorapatite. Am. Mineral. 1995, 80, 329−335.
Figure 5. Fluorescence line narrowing in TRLFS of Cm3+ containingbioapatite from direct excitation illustrating the presence of acontinuum of related environments as would be expected by theincorporation into amorphous grain boundaries.
Langmuir Article
dx.doi.org/10.1021/la300014a | Langmuir 2012, 28, 3845−38513850

(12) Fleet, M. E.; Pan, Y. Rare earth elements in apatite: uptake fromH2O-bearing phosphate-fluoride melts and the role of volatilecomponents. Geochim. Cosmochim. Acta 1997, 61, 4745−4760.(13) Fleet, M. E.; Liu, X.; Pan, Y. Rare-earth elements in chlorapatite[Ca10(PO4)6Cl2]: Uptake, site preference, and degradation ofmonoclinic structure. Am. Mineral. 2000, 85, 1437−1446.(14) Martin, P.; Carlot, G.; Chevarier, A.; Den-Auwer, C.; Panczer,G. Mechanisms involved in thermal diffusion of rare earth elements inapatite. J. Nucl. Mater. 1999, 275, 268−276.(15) Ryan, F. M.; Warren, R. W.; Hopkins, R. H.; Murphy, J.Selective site laser excitation and ESR studies of Nd3+ ions inCa5(PO4)3F. J. Electrochem. Soc. 1978, 125, 1493−1498.(16) Ternane, R.; Trabelsi-Ayedi, M.; Kbir-Ariguib, N.; Piriou, B.Luminescent properties of Eu3+ in calcium hyroxyapatite. J. Lumin.1999, 81, 165−170.(17) Graeve, O. A.; Kanakala, R.; Madadi, A.; Williams, B. C.; Glass,K. C. Luminescence variations in hydroxyapatites doped with Eu2+ andEu3+ ions. Biomaterials 2010, 31, 4259−4267.(18) Gaft, M.; et al. Eu3+ luminescence in high-symmetry sites ofnatural apatite. J. Lumin. 1997, 72−74, 572−574.(19) Karbowiak, M.; Hubert, S. Site-selective emission spectra ofEu3+:Ca5(PO4)3F. J. Alloys Compd. 2000, 302, 87−93.(20) Cavellec, R.; Lucas, C.; Simoni, E.; Hubert, S.; Edelstein, N.Structural characterization of sorption complexes of Cm(III) at thephosphate minerals-solution interface using laser spectrofluorimetry.Radiochim. Acta 1998, 82, 221−225.(21) Finlay, J. A.; et al. Phosphate release and heavy metalaccumulation by biofilm-immobilized and chemically-coupled cells ofa Citrobacter sp. pre-grown in continuous culture. Biotechnol. Bioeng.1999, 63, 87−97.(22) Yong, P.; Macaskie, L. E.; Sammons, R. L.; Marquis, P. M.Synthesis of nanophase hydroxyapatite by a Serratia sp from waste-water containing inorganic phosphate. Biotechnol. Lett. 2004, 26,1723−1730.(23) Horrocks, W. D. W.; Sudnick, D. R. Lanthanide ion probes ofstructure in biology. Laser-induced luminescence decay constantsprovide a direct measure of the number of metal-coordinated watermolecules. J. Am. Chem. Soc. 1979, 101, 334−340.(24) Choppin, G. R.; Peterman, D. R. Applications of lanthanideluminescence spectroscopy to solution studies of coordinationchemistry. Coord. Chem. Rev. 1998, 174, 283.(25) Hemmila, I.; Laitala, V. Progress in Lanthanides as LuminescentProbes. J. Fluoresc. 2005, 15, 529.(26) Bunzli, J-C.G.; Piguet, C. Taking advantage of luminescentlanthanide ions. Chem. Soc. Rev. 2005, 34, 1048.(27) Kimura, T.; Choppin, G. R. Luminescence study ondetermination of the hydration number of Cm(III). J. Alloys Compd.1994, 213/214, 313−317.(28) Edelstein, N. M.; Klenze, R.; Fanghanel, T.; Hubert, S. Opticalproperties of Cm(III) in crystals and solutions and their application toCm(III) speciation. Coord. Chem. Rev. 2006, 250, 948.(29) Geipel, G. Some aspects of actinide speciation by laser-inducedspectroscopy. Coord. Chem. Rev. 2006, 250, 844.(30) Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS:data analysis for X-ray absorption spectroscopy using IFEFFIT. J.Synchrotron Rad. 2005, 12, 537.(31) Newville, M. IFEFFIT: interactive XAFS analysis and FEFFfitting. J. Synchrotron Rad. 2001, 8, 322.(32) Schmidt, M.; Stumpf, T.; Fernandes, M. M.; Walther, C.;Fanghanel, T. Charge compensation in solid solutions. Angew. Chem.,Int. Ed. 2008, 47, 5846−5850.(33) Fernandes, M. M.; et al. Site-selective time-resolved laserfluorescence spectroscopy of Eu3+ in calcite. J. Colloid Interface Sci.2008, 321, 323−331.(34) Stumpf, T.; Fanghanel, T. A time-resolved laser fluorescencespectroscopy (TRLFS) study of the interaction of trivalent actinides(Cm(III)) with calcite. J. Colloid Interface Sci. 2002, 249, 119−122.(35) Handley-Sidhu, S.; Renshaw, J. C.; Yong, P.; Kerley, R.;Macaskie, L. E. Nano-crystalline hydroxyapatite bio-mineral for the
treatment of strontium from aqueous solutions. Biotechnol. Lett. 2011,33, 79−87.(36) Handley-Sidhu, S.; et al. Uptake of Sr2+ and Co2+ into biogenichydroxyapatite: implications for biomineral ion exchange synthesis.Environ. Sci. Technol. 2011, 45, 6986−6990.(37) Holliday, K.; Hartmann, T.; Poineau, F.; Kennedy, J. R.;Czerwinski, K. Synthesis and characterization of zirconia-magnesiainert matrix fuel: uranium homolog studies. J. Nucl. Mater. 2009, 393,224−229.(38) Mountjoy, G.; et al. A rare earth L3-edge EXAFS and L1-edgeXANES study of Ce, Nd and Eu phosphate glasses and crystals in thecomposition range from metaphosphate to ultraphosphate. J. Non-Cryst. Solids 2001, 279, 20−27.(39) Murdoch, K. M.; Edelstein, N. M.; Boatner, L. A.; Abraham, M.M. Excited state absorption and fluorescence line narrowing studies ofCm3+ in LuPO4. J. Chem. Phys. 1996, 105, 2539−2547.(40) Holliday, K. S., et al. Site-selective time resolved laserfluorescence spectroscopy of Eu and Cm doped LaPO4. Radiochim.Acta DOI: 10.1524/ract.2012.1900.
Langmuir Article
dx.doi.org/10.1021/la300014a | Langmuir 2012, 28, 3845−38513851