2010 missiou circ res
TRANSCRIPT

TRAF5 Deficiency Accelerates Atherogenesis in Mice byIncreasing Inflammatory Cell Recruitment and Foam
Cell FormationAnna Missiou,* Philipp Rudolf,* Peter Stachon, Dennis Wolf, Nerea Varo, Peter Aichele,
Christian Colberg, Natalie Hoppe, Sandra Ernst, Christian Munkel, Carina Walter, Benjamin Sommer,Ingo Hilgendorf, Hiroyasu Nakano, Christoph Bode, Andreas Zirlik
Rationale: Tumor necrosis factor receptor–associated factors (TRAFs) are cytoplasmic adaptor proteins for theTNF/interleukin-1/Toll-like receptor superfamily. Ligands of this family comprise multiple important cytokinessuch as TNF�, CD40L, and interleukin-1� that promote chronic inflammatory diseases such as atherosclerosis.We recently reported overexpression of TRAF5 in murine and human atheromata and that TRAF5 promotesinflammatory functions of cultured endothelial cells and macrophages.
Objective: This study tested the hypothesis that TRAF5 modulates atherogenesis in vivo.Methods and Results: Surprisingly, TRAF5�/�/LDLR�/� mice consuming a high-cholesterol diet for 18 weeks
developed significantly larger atherosclerotic lesions than did TRAF5�/�/LDLR�/� controls. Plaques ofTRAF5-deficient animals contained more lipids and macrophages, whereas smooth muscle cells and collagenremained unchanged. Deficiency of TRAF5 in endothelial cells or in leukocytes enhanced adhesion ofinflammatory cells to the endothelium in dynamic adhesion assays in vitro and in murine vessels imaged byintravital microscopy in vivo. TRAF5 deficiency also increased expression of adhesion molecules and chemokinesand potentiated macrophage lipid uptake and foam cell formation. These findings coincided with increasedactivation of JNK and appeared to be independent of TRAF2. Finally, patients with stable or acute coronaryheart disease had significantly lower amounts of TRAF5 mRNA in blood compared with healthy controls.
Conclusions: Unexpectedly, TRAF5 deficiency accelerates atherogenesis in mice, an effect likely mediated byincreased inflammatory cell recruitment to the vessel wall and enhanced foam cell formation. (Circ Res. 2010;107:757-766.)
Key Words: TRAF5 � inflammation � atherosclerosis � monocyte recruitment � foam cells
Atherosclerosis is one of the leading causes of deathworldwide. The concept that inflammatory and immu-
nologic mechanisms drive atherogenesis emerged from bothextensive laboratory studies and from clinical observationsdemonstrating associations between biomarkers of inflamma-tion and cardiovascular events.1,2 These findings have spurredthe quest for antiinflammatory or immunomodulatory treat-ments to fight atherosclerosis and its sequelae. Selectivemodulation of key signaling pathways may afford a fruitfulstrategy to achieve that goal.
Tumor necrosis factor (TNF) receptor–associated factors(TRAFs) represent important signal transducers for the TNF/interleukin (IL)-1/Toll-like receptor (TLR) superfamily, sev-
eral members of which potently promote atherogenesis. Weand others found expression of TRAFs in cells typicallyresident in the atherosclerotic plaques.3–6 However, only afew reports have investigated TRAF-dependent functions inthe context of vascular disease: TRAF3 and TRAF2 havebeen implicated in transducing shear stress,7,8 and Luo et alrecently demonstrated that activation of TNFR2 mediatesischemia-induced atherogenesis by inducing TRAF2-dependent survival pathways.9 Endothelial cells (ECs) andsmooth muscle cells from unstable plaque regions of humancarotid arteries overexpress TRAF4.4 TRAF6 promotes neo-intima formation on balloon injury in the carotid artery ofmice as well as development of in-stent stenosis in rab-
Original received February 22, 2010; revision received July 12, 2010; accepted July 14, 2010. In June 2010, the average time from submission to firstdecision for all original research papers submitted to Circulation Research was 14.5 days.
From the Department of Cardiology (A.M., P.R., P.S., D.W., C.C., N.H., S.E., C.M., C.W., B.S., I.H., C.B., A.Z.); Spemann Graduate School ofBiology and Medicine (A.M., A.Z.); Faculty of Biology (A.M.); and Institute for Medical Microbiology and Hygiene, Department of Immunology (P.A.),University of Freiburg, Germany; Department of Clinical Chemistry (N.V.), University of Navarra, Pamplona, Spain; and Department of Immunology(H.N.), Juntendo University, School of Medicine, Tokyo, Japan.
*Both authors contributed equally to this work.Correspondence to Andreas Zirlik, MD, Department of Cardiology, University of Freiburg, Breisacherstrasse 33, 79106 Freiburg, Germany. E-mail
[email protected]© 2010 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.110.219295
757

bits.10,11 In accord, Donners et al observed reduced neointimaformation and leukocyte infiltration on carotid artery ligationin mice lacking the binding site for TRAF6 on CD40 onleukocytes.12 The same group recently showed that thesemice crossed with Apolipoprotein E-deficient mice developsignificantly smaller atherosclerotic lesions on a high-choles-terol diet (HCD).13 Finally, we recently reported attenuationof atherogenesis in mice deficient for TRAF1.14 Thesefindings suggest that TRAFs participate in vascular disease,but the function of TRAF5 in this context remains unsettled.
Two independent groups identified TRAF5 as a 64-kDaprotein interacting with LT�R (lymphotoxin � receptor) andCD40.15 Further studies implicated TRAF5 with the activa-tion of CD30, CD27, LMP-1 (latent membrane protein 1),HVEM (herpes virus entry mediator), Ox40, 4-1BB, double-stranded RNA-dependent protein kinase PKR, and NIK(nuclear factor �B kinase).16,17 TRAF5 contains a RINGfinger followed by 5 Zn2� finger motifs at its N terminus,crucial for downstream signaling and the conserved TRAFdomain at its C terminus, needed for self-association andrecognition of cognate receptors. Both TRAF2 and TRAF5participate in nuclear factor (NF)�B and c-Jun N-terminalkinase (JNK) signaling on engagement of TNFR1, CD30, andCD40.18 Increasing evidence indicates that impairment ofJNK and NF�B activation resulting from TRAF5 deficiencycan be compensated for by TRAF2 and vice versa.19 How-ever, a nonredundant role for TRAF5 has been recentlypostulated in LMP-1–mediated JNK activation,20 as well asglucocorticoid tumor necrosis factor receptor (GITR)-
induced NF�B, extracellular signal-regulated kinase(ERK)1/2, and p38 stimulation.21 TRAF5-deficient micedisplayed impaired CD40-stimulated B-cell proliferation andmild defects in affinity maturation of IgG antibodies.22 Ourgroup recently showed overexpression of TRAF5 in humanand mouse atheromata and dependency of CD40L and TNF�-induced IL-6 expression in ECs and macrophages on TRAF5in line with a proinflammatory role.3 Therefore, this studytested the hypothesis that TRAF5 deficiency attenuates mu-rine atherogenesis in vivo.
MethodsAn expanded Methods section is available in the Online DataSupplement at http://circres.ahajournals.org.
In Vivo StudyTRAF5-deficient mice were back-crossed 5 times to C57/BL6 (TheJackson Laboratory) and crossed with LDLR�/� mice (The JacksonLaboratory). Eight-week-old male littermates of TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� consumed HCD for 18 weeks(Ssniff based on Research Diets D12108). Fetal liver cell (FLC)transplantations for evaluation of TRAF2 function in atherosclerosiswere performed similarly as previously described.23 At the end of thestudy, mice were harvested and histologically analyzed as describedpreviously.14,24,25
Cell Isolation and CultureMurine ECs, thioglycollate-elicited peritoneal macrophages, andbone marrow–derived macrophages (BMMs) were isolated andcultured as described previously.3 Murine leukocytes for flowcytometric analysis were isolated from blood obtained by retroorbitalbleeding. For generation of murine splenocytes, mice were eutha-nized and their spleens harvested and crushed.
Western BlottingMurine ECs and BMMs were stimulated with TNF� (20 ng/mL) andIL-1� (10 ng/mL) for 24 hours, lysed, separated by SDS-PAGEunder reducing conditions, and blotted to poly(vinylidene difluoride)membranes as described previously.3 Western blots were analyzeddensitometrically using ImageJ (NIH).
Enzyme-Linked Immunoabsorbent AssayMouse keratinocyte-derived chemokine (KC) and monocyte che-moattractant protein (MCP)-1 were analyzed in the supernatants ofcell cultures using commercially available ELISA Kits (R&D)according to the instructions of the manufacturer.
Flow CytometryCells were washed in 0.1% BSA and preincubated with mouseFc-Block (�CD16/32, ebioscience) before antibodies were added.Intracellular staining for FoxP3 was performed using the mouseT-cell regulatory staining kit (ebioscience). Samples were analyzedwith a fluorescence-activated cell sorter (FACS Calibur, BectonDickinson). The mean fluorescence indices (MFI) were quantifiedusing the FlowJo software.
Cytometric Bead AssayFor analysis of inflammatory markers and chemokines, the cytomet-ric bead assay for murine inflammation and chemokines (BD) wasused. For detection of p-JNK, p-ERK, and p-p38 the murinesignaling cytometric bead assay was used.
Dynamic Adhesion AssaysDynamic adhesion assays in the flow chamber were performed asdescribed previously.25 Adherent and rolling leukocytes were filmedunder the microscope and counted by blinded investigators.
Non-standard Abbreviations and Acronyms
ACS acute coronary syndrome
BMM bone marrow–derived macrophage
CHD coronary heart disease
EC endothelial cell
ERK extracellular signal-regulated kinase
FACS fluorescence-activated cell sorting
FLC fetal liver cell
HCD high-cholesterol diet
ICAM intercellular adhesion molecule
IL interleukin
JNK c-Jun N-terminal kinase
KC keratinocyte-derived chemokine
LMP latent membrane protein
MCP monocyte chemoattractant protein
NF�B nuclear factor �B
Th1/Th2 T helper cell 1/2
TLR Toll-like receptor
TNF tumor necrosis factor
TNFR tumor necrosis factor receptor
TRAF tumor necrosis factor receptor-associated factor
Treg regulatory T cell
VCAM vascular adhesion molecule
VLA very late antigen
758 Circulation Research September 17, 2010

Cell-Spreading AssayActin polymerization was evaluated in murine peritoneal macro-phages as previously described.14
Intravital MicroscopyMice were anesthetized with ketamine and xylazin, intraperitoneallyinjected with 200 ng of murine TNF�, and the cremaster muscle wasprepared as described previously.26 Venules with diameters between30 and 50 �m were observed and recorded through a high sensitivitycamera system (AxioCam MRm, Carl Zeiss), and adhering androlling leukocytes were quantified by blinded investigators.
Lipid-Uptake AssayBMMs were isolated as described above. After differentiation, cellswere starved for 2 hours and stimulated with 10 �g/mL Alexa488-labeled acetylated LDL in RPMI for 1 hour. Analysis was performedby fluorescence-activated cell sorting (FACS).
Quantitative RT-PCR of Murine SamplesTotal RNA was isolated from organs according to a modified TRIzolprotocol and transcribed to cDNA, and targets were analyzed byquantitative RT-PCR as detailed in the Online Data Supplement.
Immunologic TestingT-cell proliferation and activation were quantified by FACS usingantibodies against KLRG-1, CD62L, and CD44 (ebioscience) inresponse to �CD2/CD28 stimulation. Intracellular expression ofIL-2, TNF�, and interferon-� was quantified by FACS (BDPharmingen).
Data AnalysisData of at least 3 experiments were pooled and presented asmeans�SEM. Statistical analysis used the Student’s 2-tailed t testfor paired or unpaired values (where appropriate). A probabilityvalue of �0.05 was considered statistically significant.
Clinical StudyA total of 325 patients undergoing coronary angiography wereincluded in the Tumor Necrosis Factor in Cardiovascular Risk Study(TRAFICS), registered as UKF000748, and approved by the localInstitutional Review Board of the University of Freiburg. Patientswere divided into 3 groups: no coronary heart disease (No CHD),stable coronary heart disease (CHD), and acute coronary syndrome(ACS). TRAF5 and GAPDH transcripts were quantified by real-timeRT PCR and analyzed as detailed in the Online Data Supplement.
ResultsTRAF5 Deficiency Accelerates Atherosclerosisin MiceTo test the hypothesis that TRAF5 modulates murine athero-sclerosis in vivo, TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice consumed HCD for 18 weeks. All miceappeared healthy, reproduced according mendelian ratios,and had no obvious abnormalities and a normal life span.Weights and leukocyte counts did not differ at the end of thestudy. Cholesterol and triglyceride levels did not differ atbaseline between both groups but were slightly higher inTRAF5�/�LDLR�/� animals after HCD (N�10 per group,P�0.05; Online Table I).
Surprisingly, TRAF5-deficient animals developed signifi-cantly larger atherosclerotic plaques in aortic roots(0.113�0.01 mm2 versus 0.163�0.02 mm2, N�12 per group,P�0.02, Figure 1A) and arches (0.051�0.01 mm2 versus0.133�0.02 mm2, N�8 per group, P�0.01, Figure 1B)compared with control animals on consumption of HCD.
Similar results were obtained in en face analysis of abdominalaortas (N�16, Figure 1C). Analysis of composition revealedmost prominently an increase in macrophage content in aorticTRAF5�/�/LDLR�/� plaques (P�0.005). Lipid contenttended to increase, whereas collagen, smooth muscle cells,and T-cell numbers remained unchanged (Figure 2A through2D). These features resemble those associated with unstableplaques in humans.27
TRAF5 Deficiency Promotes Rolling and Adhesionof Inflammatory CellsDuring early atherogenesis leukocytes roll and adhere toactivated endothelium before migrating into the intima.TRAF5 deficiency on either ECs or leukocytes enhancedadhesion by up to 198�10% compared with wild-typecontrols in the flow chamber (N�3 each, P�0.05, Figure3A). We also observed significantly increased rolling ofleukocytes in the same model (N�3 each, P�0.05, Figure3A). Accordingly, in vivo adhesion of leukocytes to theendothelium was significantly enhanced in TRAF5-deficientmice on intraperitoneal injection of 200 ng TNF� (N�3 pergroup, P�0.05; Figure 3B) as assessed by intravital micros-copy. Rolling also tended to increase in this model (N�3 pergroup, P�0.13; Figure 3B).
Interactions of adhesion molecules on ECs with integrinson immune cells represent the main underlying mechanism ofadhesion. TRAF5-deficient ECs expressed significantly moreintercellular adhesion molecule (ICAM)-1 compared withrespective controls (N�9, P�0.05, Figure 4A). Alike,TRAF5-deficient aortic tissue expressed significantly moreVCAM-1 and by tendency more ICAM-1 transcripts, asassessed by quantitative RT-PCR (N�3, Figure 4B). TRAF-deficient monocytes also expressed higher amounts ofCD49d, a component of VLA-4, the ligand for VCAM-1,involved in both rolling and firm adhesion (N�3, P�0.004,Figure 4C).
TRAF5 Deficiency Enhances Migration and theExpression of ChemokinesTo test whether TRAF5 also modulates in vivo migration, weinduced sterile peritonitis by instillation of 4% thioglycollatein TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice.After 72 hours, significantly more inflammatory cells accu-mulated in the peritoneal cavity of TRAF5�/�/LDLR�/�
mice (N�3, P�0.0007, Figure 5A). Chemokines such as KCand MCP-1 attract leukocytes and additionally cause rapidactivation of �1- and �2-integrins, which pave the way forfirm adhesion. Therefore, we analyzed chemokine expressionin serum and aortic lysates from TRAF5�/�/LDLR�/� andTRAF5�/�/LDLR�/� mice challenged with TNF�, demon-strating increased MCP-1 expression in mice deficient forTRAF5 (Figure 5B and 5C). Similarly, ECs and BMMs,produced significantly more MCP-1 and KC (N�8 each,P�0.05, Online Figure I, A and B). In accord, increasedMCP-1 and KC expression was detected in aortic plaquesfrom TRAF5-deficient mice on HCD (Figure 5D and 5E).These data revealed an additional mechanism for enhancedadhesion and augmented atherogenesis in these mice.
Missiou et al TRAF5 and Atherosclerosis 759

TRAF5 Deficiency Increases MacrophageLDL UptakeOnce resident in the intima, monocytes differentiate intomacrophages, imbibe modified lipids, and become foam cells.TRAF5-deficient BMMs internalized significantly more fluo-
rescently labeled acetylated LDL than control cells as as-sessed by fluorescence microscopy and FACS (Figure 6A).Mechanistically, we found higher levels of the scavengerreceptor CD36 on TRAF5-deficient macrophages comparedwith controls (N�4, P�0.001, Figure 6B). In contrast, the
Figure 1. TRAF5 deficiency acceler-ates murine atherosclerosis. A and B,TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice consumed HCD for 18weeks and intimal lesion size in the aor-tic root (A) and arch (B), as well as lipiddeposition (oil red O) in the abdominalaortas (C) were quantified. Left, Pooleddata�SEM (N�12, 8, and 16 per group,respectively). Right, Images of represen-tative sections stained for lipid deposi-tion (oil red O).
Figure 2. TRAF5-deficient plaquescontain more macrophages and lipids.A through D, Sections of the aorticarches and roots of mice treated asdescribed above were subjected to anal-ysis of macrophage-, smooth musclecell–, lipid-, and collagen-specific stain-ing. Mac-3–, �-actin–, oil red O–, andpicrosirius red–positive staining in rela-tion to total wall area is presented asmeans�SEM.
760 Circulation Research September 17, 2010

expression of TLR4, also known to mediate modified LDLuptake, was similar between TRAF5-deficient and competentmacrophages28 (Figure 6C), and the expression of scavengerreceptor A even decreased (N�5, P�0.003, Figure 6D). Inblood of TRAF5-deficient animals, ABCA1, another lipidtransporter, was significantly increased on monocytes (N�5,P�0.025, Figure 6E).
TRAF5 Deficiency Enhances Actin PolymerizationDifferences in adhesion and lipid uptake may also rely onvariations of actin cytoskeleton formation.29 Peritoneal mac-rophages from TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice were allowed to adhere to serum-coated glasscoverslips and stained with phalloidin. Both TRAF5-deficient
and -competent cells spread over time. Interestingly, TRAF5-deficient leukocytes formed significantly more raffles, sug-gesting that TRAF5 deficiency promotes actin polymeriza-tion (Online Figure II).
TRAF5 Deficiency Does Not Alter Levels ofInflammatory Cytokines but Reduces TRegulatory CellsBecause differences in inflammatory gene expression and/orT-cell and macrophage subtypes may greatly influenceatherogenesis, we also characterized these features. Withinflammatory mediators being below the detection limit inmice on HCD, we challenged mice with intraperitonealinjection of TNF�. Under these conditions, TRAF5�/�/
Figure 3. TRAF5 deficiency promotesinflammatory cell recruitment to theendothelium. A, Adhesion and rolling ofPMA-activated thioglycollate-elicitedperitoneal leukocytes obtained fromTRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice were quantified on TNF�-activated ECs isolated from TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� miceunder flow conditions (0.5 dyn/cm2, N�3each). Adherent and rolling leukocyteswere counted under the microscope.Pooled data represent means�SEM. B,Venules (30 to 50 �m) of the cremastermuscle were evaluated for adhesion androlling of leukocytes in TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice 4hours after IP administration of 200 ng ofTNF� (N�3). Leukocyte numbers werecounted manually and are presented atleft. Representative images are dis-played at right. Data represent themeans�SEM.
Figure 4. TRAF5 deficiency enhancesadhesion molecule expression in ECsand macrophages. A, TRAF5-deficientand -competent murine ECs were stimu-lated with or without 20 ng/mL TNF�and 10 ng/mL IL-1�. Cell lysates wereanalyzed for ICAM-1 protein expressionby Western blotting. Pooled densitomet-ric data adjusted for GAPDH expressionare given as means�SEM (top), andrepresentative blots are shown (bottom)(N�9). B, TRAF5�/�/LDLR�/� andTRAF5�/�/LDLR�/� mice consumedHCD for 8 weeks, total RNA wasextracted from aortas, and ICAM-1 andVCAM-1 transcripts were quantified byRT-PCR. Data are presented as meanratios of target/GAPDH�SEM (N�3). C,Murine monocytes were obtained fromTRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice, injected with 200 ng ofTNF� or vehicle IP 4 hours before isola-tion and analyzed for CD49d expressionby FACS. Pooled data as means�SEMare shown (n�3).
Missiou et al TRAF5 and Atherosclerosis 761

LDLR�/� mice expressed similar amounts of TNF�, IL-6,interferon-�, IL-10, and IL-12p70 in serum and arterial tissue(Online Figure III, A and B). We further analyzed leukocyteand monocyte subsets in the spleen and blood of these mice(Online Figure III, C and D). We found no difference innumbers of B cells (CD20), CD4� and CD8� T cells,monocytes, Ly6chigh (Gr1high) and Ly6clow (Gr1low) mono-cytes, and neutrophils. In contrast, the spleens of TRAF5�/�/LDLR�/� mice challenged with TNF� had fewer regulatoryT cell (Treg) cells. Because differences in T cell function mayalso modulate the atherogenic response, we tested T cellproliferation and activation. Interestingly, both CD4� andCD8� T cells from TRAF5�/�/LDLR�/� animals prolifer-ated more than those of respective controls animals. Thiscoincided with increased T cell activation (CD44high,CD62Llow) and proliferation markers (KLRG-1) in TRAF5-deficient CD8� T cells at baseline and following mildstimulation with �CD3/CD28 (Online Figure IV, A and B).
However, on stimulation with �CD3/CD28 for 3 days, thesecells did not produce different amounts of TNF�, IL-2, andinterferon-�, suggesting no relevant bias in T helper cell 1/2(Th1/Th2) cytokine expression in these animals (OnlineFigure IVC).
TRAF5 Deficiency Enhances JNK ActivationWe observed increased JNK activation following 10 minutesof TNF� stimulation in TRAF5-deficient ECs compared withcontrols, whereas ERK and p38 phosphorylation remainedunchanged (Online Figure V, A and B). Similar findings forp-JNK were obtained in BMMs (Online Figure V, C) and asa trend also in aortic lysates (Online Figure V, D). In contrast,splenocytes (Online Figure V, E) containing predominantly Band T cells and resident peritoneal lymphocytes (OnlineFigure V, F) showed decreased JNK phosphorylation whenTRAF5-deficient, suggesting a cell type–specific TRAF5-dependent regulation of JNK activation.
Figure 5. TRAF5 deficiency promotesmigration and chemokine expression.A, Sterile peritonitis was induced by instil-lation of 4% thioglycollate in TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice.After 72 hours, cells were counted underthe microscope (N�3). Data representmeans�SEM. B and C, Serum (B) andarterial tissue (C) of TRAF5�/�/LDLR�/�
and TRAF5�/�/LDLR�/� mice 4 hoursafter IP administration of 200 ng of TNF�were assayed for MCP-1 expression byELISA and cytometric bead array, respec-tively (N�5 each). Data representmeans�SEM. D, Sections of the aorticroots from TRAF5�/�/LDLR�/� andTRAF5�/�/LDLR�/� mice consuming HCDfor 18 weeks were stained for MCP-1 andquantified by image analysis software.Data represent means�SEM. E, TRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� miceconsumed HCD for 8 weeks, total RNA wasextracted from aortas, and KC transcriptswere quantified by RT-PCR. Data are pre-sented as mean ratios of KC/GAPDH�SEM(N�3).
Figure 6. TRAF5 deficiency promoteslipid uptake. A, BMMs obtained fromTRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice were incubated with fluo-rescent-labeled acetylated LDL (acLDL).Lipid uptake was quantified by FACS.Data represent means�SEM (N�4). Bthrough D, Murine TRAF5-deficient and-competent BMMs were stimulated withTNF� 20 ng/mL or vehicle IP and ana-lyzed for CD36 (B), TLR4 (C), and scav-enger receptor A (SR-A) (D) expressionby FACS and Western blotting. Datarepresent means�SEM (N�4). E, Inblood from TRAF5�/�/LDLR�/� andTRAF5�/�/LDLR�/� mice, CD115-positive cells were tested for ABCA1expression by FACS. Data representmean fluorescent intensity�SEM (N�4).
762 Circulation Research September 17, 2010
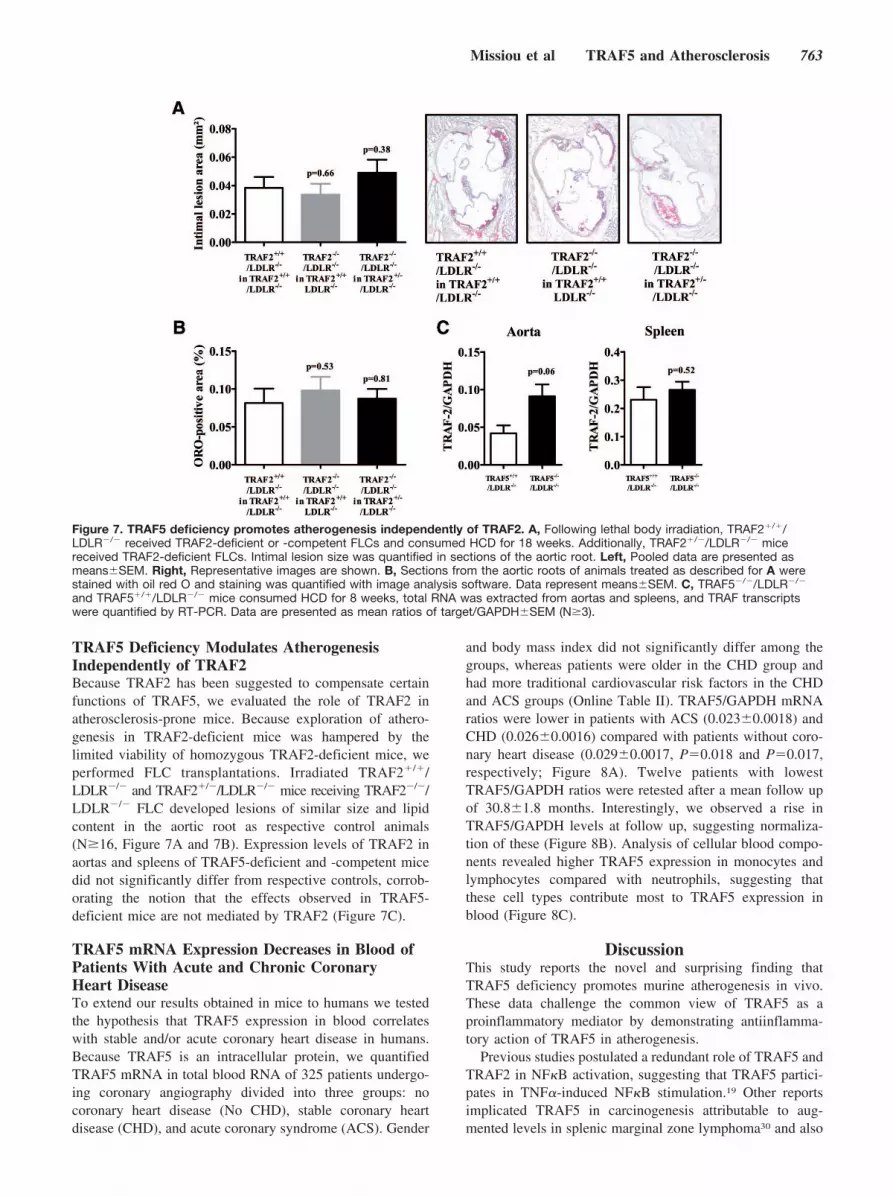
TRAF5 Deficiency Modulates AtherogenesisIndependently of TRAF2Because TRAF2 has been suggested to compensate certainfunctions of TRAF5, we evaluated the role of TRAF2 inatherosclerosis-prone mice. Because exploration of athero-genesis in TRAF2-deficient mice was hampered by thelimited viability of homozygous TRAF2-deficient mice, weperformed FLC transplantations. Irradiated TRAF2�/�/LDLR�/� and TRAF2�/�/LDLR�/� mice receiving TRAF2�/�/LDLR�/� FLC developed lesions of similar size and lipidcontent in the aortic root as respective control animals(N�16, Figure 7A and 7B). Expression levels of TRAF2 inaortas and spleens of TRAF5-deficient and -competent micedid not significantly differ from respective controls, corrob-orating the notion that the effects observed in TRAF5-deficient mice are not mediated by TRAF2 (Figure 7C).
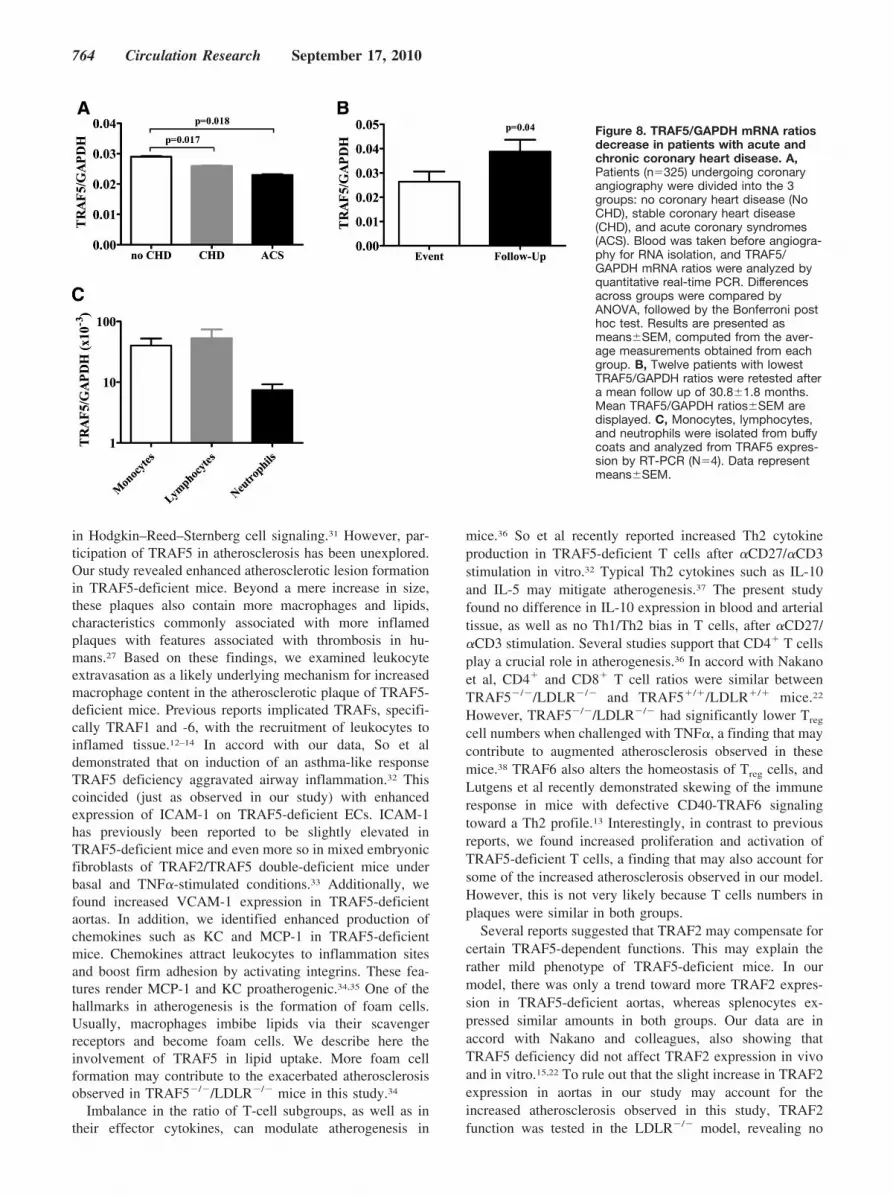
TRAF5 mRNA Expression Decreases in Blood ofPatients With Acute and Chronic CoronaryHeart DiseaseTo extend our results obtained in mice to humans we testedthe hypothesis that TRAF5 expression in blood correlateswith stable and/or acute coronary heart disease in humans.Because TRAF5 is an intracellular protein, we quantifiedTRAF5 mRNA in total blood RNA of 325 patients undergo-ing coronary angiography divided into three groups: nocoronary heart disease (No CHD), stable coronary heartdisease (CHD), and acute coronary syndrome (ACS). Gender
and body mass index did not significantly differ among thegroups, whereas patients were older in the CHD group andhad more traditional cardiovascular risk factors in the CHDand ACS groups (Online Table II). TRAF5/GAPDH mRNAratios were lower in patients with ACS (0.023�0.0018) andCHD (0.026�0.0016) compared with patients without coro-nary heart disease (0.029�0.0017, P�0.018 and P�0.017,respectively; Figure 8A). Twelve patients with lowestTRAF5/GAPDH ratios were retested after a mean follow upof 30.8�1.8 months. Interestingly, we observed a rise inTRAF5/GAPDH levels at follow up, suggesting normaliza-tion of these (Figure 8B). Analysis of cellular blood compo-nents revealed higher TRAF5 expression in monocytes andlymphocytes compared with neutrophils, suggesting thatthese cell types contribute most to TRAF5 expression inblood (Figure 8C).
DiscussionThis study reports the novel and surprising finding thatTRAF5 deficiency promotes murine atherogenesis in vivo.These data challenge the common view of TRAF5 as aproinflammatory mediator by demonstrating antiinflamma-tory action of TRAF5 in atherogenesis.
Previous studies postulated a redundant role of TRAF5 andTRAF2 in NF�B activation, suggesting that TRAF5 partici-pates in TNF�-induced NF�B stimulation.19 Other reportsimplicated TRAF5 in carcinogenesis attributable to aug-mented levels in splenic marginal zone lymphoma30 and also
Figure 7. TRAF5 deficiency promotes atherogenesis independently of TRAF2. A, Following lethal body irradiation, TRAF2�/�/LDLR�/� received TRAF2-deficient or -competent FLCs and consumed HCD for 18 weeks. Additionally, TRAF2�/�/LDLR�/� micereceived TRAF2-deficient FLCs. Intimal lesion size was quantified in sections of the aortic root. Left, Pooled data are presented asmeans�SEM. Right, Representative images are shown. B, Sections from the aortic roots of animals treated as described for A werestained with oil red O and staining was quantified with image analysis software. Data represent means�SEM. C, TRAF5�/�/LDLR�/�
and TRAF5�/�/LDLR�/� mice consumed HCD for 8 weeks, total RNA was extracted from aortas and spleens, and TRAF transcriptswere quantified by RT-PCR. Data are presented as mean ratios of target/GAPDH�SEM (N�3).
Missiou et al TRAF5 and Atherosclerosis 763
in Hodgkin–Reed–Sternberg cell signaling.31 However, par-ticipation of TRAF5 in atherosclerosis has been unexplored.Our study revealed enhanced atherosclerotic lesion formationin TRAF5-deficient mice. Beyond a mere increase in size,these plaques also contain more macrophages and lipids,characteristics commonly associated with more inflamedplaques with features associated with thrombosis in hu-mans.27 Based on these findings, we examined leukocyteextravasation as a likely underlying mechanism for increasedmacrophage content in the atherosclerotic plaque of TRAF5-deficient mice. Previous reports implicated TRAFs, specifi-cally TRAF1 and -6, with the recruitment of leukocytes toinflamed tissue.12–14 In accord with our data, So et aldemonstrated that on induction of an asthma-like responseTRAF5 deficiency aggravated airway inflammation.32 Thiscoincided (just as observed in our study) with enhancedexpression of ICAM-1 on TRAF5-deficient ECs. ICAM-1has previously been reported to be slightly elevated inTRAF5-deficient mice and even more so in mixed embryonicfibroblasts of TRAF2/TRAF5 double-deficient mice underbasal and TNF�-stimulated conditions.33 Additionally, wefound increased VCAM-1 expression in TRAF5-deficientaortas. In addition, we identified enhanced production ofchemokines such as KC and MCP-1 in TRAF5-deficientmice. Chemokines attract leukocytes to inflammation sitesand boost firm adhesion by activating integrins. These fea-tures render MCP-1 and KC proatherogenic.34,35 One of thehallmarks in atherogenesis is the formation of foam cells.Usually, macrophages imbibe lipids via their scavengerreceptors and become foam cells. We describe here theinvolvement of TRAF5 in lipid uptake. More foam cellformation may contribute to the exacerbated atherosclerosisobserved in TRAF5�/�/LDLR�/� mice in this study.34
Imbalance in the ratio of T-cell subgroups, as well as intheir effector cytokines, can modulate atherogenesis in
mice.36 So et al recently reported increased Th2 cytokineproduction in TRAF5-deficient T cells after �CD27/�CD3stimulation in vitro.32 Typical Th2 cytokines such as IL-10and IL-5 may mitigate atherogenesis.37 The present studyfound no difference in IL-10 expression in blood and arterialtissue, as well as no Th1/Th2 bias in T cells, after �CD27/�CD3 stimulation. Several studies support that CD4� T cellsplay a crucial role in atherogenesis.36 In accord with Nakanoet al, CD4� and CD8� T cell ratios were similar betweenTRAF5�/�/LDLR�/� and TRAF5�/�/LDLR�/� mice.22
However, TRAF5�/�/LDLR�/� had significantly lower Treg
cell numbers when challenged with TNF�, a finding that maycontribute to augmented atherosclerosis observed in thesemice.38 TRAF6 also alters the homeostasis of Treg cells, andLutgens et al recently demonstrated skewing of the immuneresponse in mice with defective CD40-TRAF6 signalingtoward a Th2 profile.13 Interestingly, in contrast to previousreports, we found increased proliferation and activation ofTRAF5-deficient T cells, a finding that may also account forsome of the increased atherosclerosis observed in our model.However, this is not very likely because T cells numbers inplaques were similar in both groups.
Several reports suggested that TRAF2 may compensate forcertain TRAF5-dependent functions. This may explain therather mild phenotype of TRAF5-deficient mice. In ourmodel, there was only a trend toward more TRAF2 expres-sion in TRAF5-deficient aortas, whereas splenocytes ex-pressed similar amounts in both groups. Our data are inaccord with Nakano and colleagues, also showing thatTRAF5 deficiency did not affect TRAF2 expression in vivoand in vitro.15,22 To rule out that the slight increase in TRAF2expression in aortas in our study may account for theincreased atherosclerosis observed in this study, TRAF2function was tested in the LDLR�/� model, revealing no
Figure 8. TRAF5/GAPDH mRNA ratiosdecrease in patients with acute andchronic coronary heart disease. A,Patients (n�325) undergoing coronaryangiography were divided into the 3groups: no coronary heart disease (NoCHD), stable coronary heart disease(CHD), and acute coronary syndromes(ACS). Blood was taken before angiogra-phy for RNA isolation, and TRAF5/GAPDH mRNA ratios were analyzed byquantitative real-time PCR. Differencesacross groups were compared byANOVA, followed by the Bonferroni posthoc test. Results are presented asmeans�SEM, computed from the aver-age measurements obtained from eachgroup. B, Twelve patients with lowestTRAF5/GAPDH ratios were retested aftera mean follow up of 30.8�1.8 months.Mean TRAF5/GAPDH ratios�SEM aredisplayed. C, Monocytes, lymphocytes,and neutrophils were isolated from buffycoats and analyzed from TRAF5 expres-sion by RT-PCR (N�4). Data representmeans�SEM.
764 Circulation Research September 17, 2010

modulation of atherogenesis in vivo. Mechanistically, oursignaling data suggest that enhanced JNK activation inTRAF5-deficient ECs and macrophages may mediate someof the effects observed. Macrophage JNK2 deletion waspreviously reported to suffice to reduce atherogenesis andfoam cell formation in mice.39 In contrast to our data, Nakanoet al22 reported similar activation of JNK in TRAF5-deficientand -competent fibroblasts, whereas Kraus et al20 founddecreased activation in B cells following CD40 stimulation.Interestingly, in our study, JNK was more activated in ECs,macrophages, and, by trend, also in aortas of TRAF5-deficient animals, whereas it tended to be less activated insplenocytes and resident peritoneal cells, the latter beingpredominant reservoirs for lymphocytes, suggesting a celltype–specific TRAF5-dependent regulation of JNK activa-tion. It may very well be that increased JNK activation inTRAF5-deficient macrophages and ECs mediates some of theproatherogenic functions observed in this study, whereas adecreased JNK activation in lymphocytes mediates some ofthe immunologic effects of TRAF5 previously observed.
To test the hypothesis that TRAF5 may also associate withclinical disease, we performed a pilot clinical study in 325patients undergoing coronary angiography. Intriguingly,TRAF5/GAPDH mRNA ratios significantly decreased in theblood of patients with chronic and acute coronary heartdisease. These preliminary data support the notion thatTRAF5 represents a protective marker in atherosclerosis.
In summary, the present study identifies TRAF5 as anantiinflammatory modulator in murine atherosclerosis.TRAF5 deficiency accelerates atherogenesis, most likely bypromoting increasing leukocyte recruitment to the vessel walland enhancing foam cell formation. Our findings shed newlight on the functional role of TRAFs in chronic inflamma-tory diseases such as atherosclerosis and may ultimately giverise to the development of novel therapeutic options inatherosclerosis.
AcknowledgmentsWe thank Dr Peter Libby (Brigham and Women’s Hospital, Boston,Mass) for financial support of the generation of TRAF5�/�/LDLR�/� mice, expert advice on experimental design, and criticalreview of the manuscript; Dr Michael Reth (Max-Planck-Institute inFreiburg) for comentoring the PhD thesis of A.M.; Dr Uwe Schon-beck (Pfizer) for important and fruitful discussions on the topic; DrJens Stein (University of Bern, Switzerland) for excellent trainingand advice on intravital microscopy; and Dr Marie Follo for expertadvice on qPCR.
Sources of FundingThis work was supported by research grants from the GermanResearch Foundation to A.Z. (DFG ZI743/3-1 and 3-2). A.M. wasfunded by the Excellence Initiative of the German Research Foun-dation (DFG GSC-4, Spemann Graduate School).
DisclosuresNone.
References1. Hansson GK, Libby P. The immune response in atherosclerosis: a
double-edged sword. Nat Rev Immunol. 2006;6:508–519.2. Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874.
3. Zirlik A, Bavendiek U, Libby P, MacFarlane L, Gerdes N, Jagielska J,Ernst S, Aikawa M, Nakano H, Tsitsikov E, Schonbeck U. TRAF-1, -2,-3, -5, and -6 are induced in atherosclerotic plaques and differentiallymediate proinflammatory functions of CD40L in endothelial cells. Arte-rioscler Thromb Vasc Biol. 2007;27:1101–1107.
4. Slevin M, Elasbali AB, Miguel Turu M, Krupinski J, Badimon L, GaffneyJ. Identification of differential protein expression associated with devel-opment of unstable human carotid plaques. Am J Pathol. 2006;168:1004–1021.
5. Mukundan L, Bishop GA, Head KZ, Zhang L, Wahl LM, Suttles J. TNFreceptor-associated factor 6 is an essential mediator of CD40-activatedproinflammatory pathways in monocytes and macrophages. J Immunol.2005;174:1081–1090.
6. Han S, Yoon K, Lee K, Kim K, Jang H, Lee NK, Hwang K, Young LeeS. TNF-related weak inducer of apoptosis receptor, a TNF receptorsuperfamily member, activates NF-kappa B through TNF receptor-associated factors. Biochem Biophys Res Commun. 2003;305:789–796.
7. Urbich C, Mallat Z, Tedgui A, Clauss M, Zeiher AM, Dimmeler S.Upregulation of TRAF-3 by shear stress blocks CD40-mediated endothe-lial activation. J Clin Invest. 2001;108:1451–1458.
8. Yamawaki H, Lehoux S, Berk BC. Chronic physiological shear stressinhibits tumor necrosis factor-induced proinflammatory responses inrabbit aorta perfused ex vivo. Circulation. 2003;108:1619–1625.
9. Luo D, Luo Y, He Y, Zhang H, Zhang R, Li X, Dobrucki WL, SinusasAJ, Sessa WC, Min W. Differential functions of tumor necrosis factorreceptor 1 and 2 signaling in ischemia-mediated arteriogenesis and angio-genesis. Am J Pathol. 2006;169:1886–1898.
10. Miyahara T, Koyama H, Miyata T, Shigematsu H, Inoue J, Takato T,Nagawa H. Inflammatory signaling pathway containing TRAF6 con-tributes to neointimal formation via diverse mechanisms. Cardiovasc Res.2004;64:154–164.
11. Miyahara T, Koyama H, Miyata T, Shigematsu H, Inoue J, Takato T,Nagawa H. Inflammatory responses involving tumor necrosis factorreceptor-associated factor 6 contribute to in-stent lesion formation in astent implantation model of rabbit carotid artery. J Vasc Surg. 2006;43:592–600.
12. Donners MM, Beckers L, Lievens D, Munnix I, Heemskerk J, Janssen BJ,Wijnands E, Cleutjens J, Zernecke A, Weber C, Ahonen CL, Benbow U,Newby AC, Noelle RJ, Daemen MJ, Lutgens E. The CD40-TRAF6 axisis the key regulator of the CD40/CD40L system in neointima formationand arterial remodeling. Blood. 2008;111:4596–4604.
13. Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A,Seijkens T, Engel D, Cleutjens J, Keller AM, Naik SH, Boon L, OufellaHA, Mallat Z, Ahonen CL, Noelle RJ, de Winther MP, Daemen MJ,Biessen EA, Weber C. Deficient CD40-TRAF6 signaling in leukocytesprevents atherosclerosis by skewing the immune response toward anantiinflammatory profile. J Exp Med. 2010;207:391–404.
14. Missiou A, Kostlin N, Varo N, Rudolf P, Aichele P, Ernst S, Munkel C,Walter C, Stachon P, Sommer B, Pfeifer D, Zirlik K, MacFarlane L, WolfD, Tsitsikov E, Bode C, Libby P, Zirlik A. Tumor necrosis factorreceptor-associated factor 1 (TRAF1) deficiency attenuates atherosclero-sis in mice by impairing monocyte recruitment to the vessel wall.Circulation. 2010;121:2033–2044.
15. Nakano H, Oshima H, Chung W, Williams-Abbott L, Ware CF, Yagita H,Okumura K. TRAF5, an activator of NF-kappaB and putative signaltransducer for the lymphotoxin-beta receptor. J Biol Chem. 1996;271:14661–14664.
16. Ha H, Han D, Choi Y. TRAF-mediated TNFR-family signaling. CurrProtoc Immunol. 2009;Chapter 11:Unit11 19D.
17. Xie P, Kraus ZJ, Stunz LL, Bishop GA. Roles of TRAF molecules in Blymphocyte function. Cytokine Growth Factor Rev. 2008;19:199–207.
18. Aizawa S, Nakano H, Ishida T, Horie R, Nagai M, Ito K, Yagita H,Okumura K, Inoue J, Watanabe T. Tumor necrosis factor receptor-associated factor (TRAF) 5 and TRAF2 are involved in CD30-mediatedNFkappaB activation. J Biol Chem. 1997;272:2042–2045.
19. Tada K, Okazaki T, Sakon S, Kobarai T, Kurosawa K, Yamaoka S,Hashimoto H, Mak TW, Yagita H, Okumura K, Yeh WC, Nakano H.Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-inducedNF-kappa B activation and protection from cell death. J Biol Chem.2001;276:36530–36534.
20. Kraus ZJ, Nakano H, Bishop GA. TRAF5 is a critical mediator of in vitrosignals and in vivo functions of LMP1, the viral oncogenic mimic ofCD40. Proc Natl Acad Sci U S A. 2009;106:17140–17145.
21. Esparza EM, Lindsten T, Stockhausen JM, Arch RH. Tumor necrosisfactor receptor (TNFR)-associated factor 5 is a critical intermediate of
Missiou et al TRAF5 and Atherosclerosis 765

costimulatory signaling pathways triggered by glucocorticoid-inducedTNFR in T cells. J Biol Chem. 2006;281:8559–8564.
22. Nakano H, Sakon S, Koseki H, Takemori T, Tada K, Matsumoto M,Munechika E, Sakai T, Shirasawa T, Akiba H, Kobata T, Santee SM,Ware CF, Rennert PD, Taniguchi M, Yagita H, Okumura K. Targeteddisruption of Traf5 gene causes defects in CD40- and CD27-mediatedlymphocyte activation. Proc Natl Acad Sci U S A. 1999;96:9803–9808.
23. Zheng Y, Ouaaz F, Bruzzo P, Singh V, Gerondakis S, Beg AA. NF-kappaB RelA (p65) is essential for TNF-alpha-induced fas expression butdispensable for both TCR-induced expression and activation-induced celldeath. J Immunol. 2001;166:4949–4957.
24. Bavendiek U, Zirlik A, LaClair S, MacFarlane L, Libby P, Schonbeck U.Atherogenesis in mice does not require CD40 ligand from bone marrow-derived cells. Arterioscler Thromb Vasc Biol. 2005;25:1244–1249.
25. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U,Ahrens I, Ernst S, Bassler N, Missiou A, Patko Z, Aikawa M, SchonbeckU, Bode C, Libby P, Peter K. CD40 ligand mediates inflammationindependently of CD40 by interaction with Mac-1. Circulation. 2007;115:1571–1580.
26. Baez S. An open cremaster muscle preparation for the study of bloodvessels by in vivo microscopy. Microvasc Res. 1973;5:384–394.
27. Sukhova GK, Schonbeck U, Rabkin E, Schoen FJ, Poole AR, BillinghurstRC, Libby P. Evidence for increased collagenolysis by interstitial colla-genases-1 and -3 in vulnerable human atheromatous plaques. Circulation.1999;99:2503–2509.
28. Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, WitztumJL, Bae YS, Miller YI. Lipoprotein accumulation in macrophages viatoll-like receptor-4-dependent fluid phase uptake. Circ Res. 2009;104:1355–1363.
29. Sakr SW, Eddy RJ, Barth H, Wang F, Greenberg S, Maxfield FR, TabasI. The uptake and degradation of matrix-bound lipoproteins by macro-phages require an intact actin Cytoskeleton, Rho family GTPases, andmyosin ATPase activity. J Biol Chem. 2001;276:37649–37658.
30. Ruiz-Ballesteros E, Mollejo M, Rodriguez A, Camacho FI, Algara P,Martinez N, Pollan M, Sanchez-Aguilera A, Menarguez J, Campo E,Martinez P, Mateo M, Piris MA. Splenic marginal zone lymphoma:
proposal of new diagnostic and prognostic markers identified after tissueand cDNA microarray analysis. Blood. 2005;106:1831–1838.
31. Horie R, Watanabe T, Ito K, Morisita Y, Watanabe M, Ishida T,Higashihara M, Kadin M. Cytoplasmic aggregation of TRAF2 andTRAF5 proteins in the Hodgkin-Reed-Sternberg cells. Am J Pathol.2002;160:1647–1654.
32. So T, Salek-Ardakani S, Nakano H, Ware CF, Croft M. TNF receptor-associated factor 5 limits the induction of Th2 immune responses.J Immunol. 2004;172:4292–4297.
33. Zhang L, Blackwell K, Thomas GS, Sun S, Yeh WC, Habelhah H.TRAF2 suppresses basal IKK activity in resting cells and TNFalpha canactivate IKK in TRAF2 and TRAF5 double knockout cells. J Mol Biol.2009;389:495–510.
34. Boisvert WA, Santiago R, Curtiss LK, Terkeltaub RA. A leukocytehomologue of the IL-8 receptor CXCR-2 mediates the accumulation ofmacrophages in atherosclerotic lesions of LDL receptor-deficient mice.J Clin Invest. 1998;101:353–363.
35. Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, Rollins BJ.Absence of monocyte chemoattractant protein-1 reduces atherosclerosisin low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–281.
36. Galkina E, Ley K. Immune and inflammatory mechanisms of atheroscle-rosis. Annu Rev Immunol. 2009;27:165–197.
37. Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF,Soubrier F, Esposito B, Duez H, Fievet C, Staels B, Duverger N,Scherman D, Tedgui A. Protective role of interleukin-10 in atheroscle-rosis. Circ Res. 1999;85:e17–e24.
38. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, ZollJ, Merval R, Esposito B, Cohen JL, Fisson S, Flavell RA, Hansson GK,Klatzmann D, Tedgui A, Mallat Z. Natural regulatory T cells control thedevelopment of atherosclerosis in mice. Nat Med. 2006;12:178–180.
39. Ricci R, Sumara G, Sumara I, Rozenberg I, Kurrer M, Akhmedov A,Hersberger M, Eriksson U, Eberli FR, Becher B, Boren J, Chen M,Cybulsky MI, Moore KJ, Freeman MW, Wagner EF, Matter CM, LuscherTF. Requirement of JNK2 for scavenger receptor A-mediated foam cellformation in atherogenesis. Science. 2004;306:1558–1561.
Novelty and Significance
What Is Known?● Tumor necrosis factor receptor-associated factors (TRAFs) mediate
signaling for a variety of cytokines previously implicated in chronicinflammatory diseases such as atherosclerosis.
● TRAF5 is overexpressed in atherosclerotic lesions and mediates proinflam-matory gene expression in cells commonly found in the atheromata.
● TRAF5 promotes NF�B and JNK signaling in lymphocytes, suggestingthat it is a proinflammatory mediator in these cell types.
What New Information Does This Article Contribute?● TRAF5 deficiency accelerates atherogenesis in mice, suggesting that
TRAF5 signaling may be antiinflammatory and atheroprotective inthis context in vivo.
● Mechanistically, deficiency of TRAF5 enhances adhesion of inflam-matory cells to the endothelium and promotes lipid uptake.
● Accordingly, patients with stable or acute coronary heart diseaseexpress significantly lower amounts of TRAF5 mRNA in bloodcompared with healthy controls.
It is well accepted that atherosclerosis is an inflammatorydisease. Several members of the TNF/IL-1/Toll-like receptor
superfamily have been implicated in atherogenesis. Unfortu-nately, unselective inhibition of cytokines such as CD40L,although clinically effective, is associated with undesired sideeffects, limiting their therapeutic potential. Modulation of centralsignaling molecules such as TRAFs that bundle downstreamsignaling in a function-specific manner may potentially over-come some of these limitations. The present study demonstratesthat TRAF5-deficient mice develop greater and more inflamedatherosclerotic lesions, identifying TRAF5 signaling as potentialantiinflammatory and antiatherosclerotic event. Our data chal-lenge previous reports that suggest a proinflammatory role ofTRAF5 in lymphocytes; this emphasizes the importance ofevaluating such molecules in vivo. In line with our findings inmice, patients with stable or acute coronary heart disease havelower levels of TRAF5 mRNA in their blood compared withhealthy controls. These data suggest enhancement of TRAF5signaling as a potential new therapeutic strategy for atheroscle-rosis and other chronic inflammatory diseases. Further studiescould focus on ways to specifically and selectively modulate thissignaling pathway.
766 Circulation Research September 17, 2010