2008 cir. res
TRANSCRIPT

Sympathetic Activation Causes Focal Adhesion SignalingAlteration in Early Compensated Volume Overload
Attributable to Isolated Mitral Regurgitation in the DogAbdelkarim Sabri, Khadija Rafiq, Rachid Seqqat, Mikhail A. Kolpakov, Ray Dillon, Louis J. Dell’italia
Abstract—We reported that left ventricular (LV) dilatation after 4 weeks of isolated mitral regurgitation (MR) in the dogsis marked by extracellular matrix loss and an increase in adrenergic drive. Given that extracellular matrix proteins andtheir receptor integrins influence �-adrenergic receptor (�-AR) responses in vitro, we tested whether �1-AR activationmodulates focal adhesion (FA) signaling and LV remodeling in these same dogs with isolated MR. Normal dogs werecompared with dogs with MR of a 4-week duration and with MR dogs treated with �1-AR blockade (�1-RB)(extended-release metoprolol succinate, 100 mg QD) that was started 24 hours after MR induction. In MR LVs, adecrease in collagen accumulation compared with normal dogs was associated with a decrease in FA kinase tyrosinephosphorylation, along with FA kinase interaction with adapter and cytoskeletal proteins, p130Cas and paxillin,respectively, as determined by immunoprecipitation assays. There was increased phosphorylation of stress relatedmolecules p38 mitogen-activated protein kinase (MAPK) and Hsp27 and survival signaling kinases extracellularsignal-regulated kinase 1/2 and AKT, with no evidence of cardiomyocyte apoptosis. �1-RB attenuated FA signaling lossand prevented p38 MAPK, Hsp27, and AKT phosphorylation induced by MR and significantly increased LV epicardialcollagen content. However, �1-RB did not improve LV endocardial collagen loss or LV dilatation induced by MR.Isolated myocytes from normal and MR dog hearts treated with �1- or �2-AR agonists demonstrated no difference inFA kinase, p38 MAPK, Hsp27, or AKT phosphorylation. These results showed that chronic stimulation of �1-AR duringearly compensated MR impairs FA signaling that may affect myocyte/fibroblast–extracellular matrix scaffoldingnecessary for LV remodeling. (Circ Res. 2008;102:1127-1136.)
Key Words: �-adrenergic receptors � focal adhesion � volume overload � mitral regurgitation � apoptosis
Myocardial remodeling in response to pressure or vol-ume overload (VO) is an adaptation of myocardial
structure to accommodate chronic changes in myocardialdemand. This remodeling is thought ultimately to drive amaladaptive process of ventricular dilatation and pump dys-function that contributes to the pathogenesis of heart failure.In contrast to pressure overload, the VO of isolated mitralregurgitation (MR) results in left ventricular (LV) dilatation,side-to-side slippage of cells, and degradation of extracellularmatrix (ECM) proteins.1,2 In addition to these structuralchanges, there is development of cardiac hypertrophy andsubsequent LV dysfunction in MR that are improved by�-adrenergic receptor blockade (�-RB)3 but not by angioten-sin-converting enzyme inhibition or angiotensin type 1 recep-tor blockade,4,5 suggesting that the adrenergic system is morecentral to the pathophysiology of VO of isolated MR.Moreover, studies in humans and dogs have demonstrated anincrease in adrenergic drive even in a mild compensated state
of isolated MR,6,7 most likely attributable to the earlyachievement of preload reserve.
�-Adrenergic receptors (�-ARs) belong to the large familyof G protein–coupled receptors that is involved in positiveinotropic, chronotropic, and lusitropic responses throughactivation of G protein.8 In the failing hearts, several defectsin �-AR signaling have been detected, including receptordownregulation and uncoupling from the stimulatory G pro-tein,9 increased level of inhibitory G protein subunits,10
decreased adenylate cyclase activity, and increased �-ARkinase-1 expression and activity.11,12 �-AR signaling mayalso be affected by ECM proteins. Herein, plating myocyteson laminin substrate as opposed to glass selectively reduced�1-AR and enhanced �2-AR regulation of ICa.13 In addition,activation of integrin signaling blocks adult cardiomyocyteapoptosis induced by �1-AR activation.14
The ability of integrins as ECM receptors to regulatecytoskeletal architecture has been well characterized in vitro
Original received July 31, 2006; resubmission received September 11, 2007; revised resubmission received March 11, 2008; accepted March 12, 2008.From the Cardiovascular Research Center (A.S., K.R., R.S., M.A.K.), Department of Anatomy & Cell Biology, Temple University, Philadelphia, Pa;
Auburn University of Veterinary Medicine (R.D.), Ala; and Departments of Medicine and Physiology and Biophysics (L.J.D.), University of Alabama,Birmingham.
Correspondence to Abdelkarim Sabri, PhD, Cardiovascular Research Center, Temple University, MRB 801, 3420 N Broad St, Philadelphia, PA 19140.E-mail [email protected]
© 2008 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.107.163642
1127 by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

and in vivo.15,16 In addition, integrin signaling has beenimplicated in G protein–coupled receptor–, hormone-, andgrowth factor–induced alterations in gene transcription incardiac myocytes.15,16 Integrins are a family of heterodimerictransmembrane receptors (composed of � and � subunits)containing extracellular ligand binding domains that showbinding specificity for ECM components and a short cyto-plasmic domain that serves to couple integrins with the actincytoskeleton.16 Binding of a matrix protein to an integrinheterodimer typically results in the activation of the nonre-ceptor tyrosine kinase, focal adhesion kinase (FAK). Acti-vated FAK, in turn, recruits the nonreceptor tyrosine kinasec-Src, the multifunctional adapter molecule Grb2, p130Cas,paxillin, and other signaling intermediates.15,16 Integrin �1and FAK knockout result in defective heart development andearly embryonic lethality.17,18 Interestingly, in mice withmyocyte-restricted FAK inactivation, an eccentric cardiachypertrophy develops with age and in response to pressureoverload stimuli, suggesting that downregulation of focaladhesion (FA) signaling is associated with the developmentof eccentric cardiac hypertrophy.19 In a similar study, persis-tent challenge of mice with myocyte-restricted FAK inacti-vation leads to enhanced cardiac fibrosis and cardiac dysfunc-tion in comparison with challenged genetic controls.20
Pressure overload–induced cardiac hypertrophy is accom-panied by enhanced expression of ECM proteins, integrins,and enhanced activation of FA signaling that correlates withadvancement of hypertrophy.21–23 However, little is knownabout integrin signaling during VO-induced cardiac hypertro-phy that is associated with a decrease in ECM accumulationas during isolated MR in the dog.1 We reported an increase incatecholamine release into the LV interstitial fluid space after4 weeks of MR.7 We extended this work using samples fromthese same dogs and now show for the first time thatincreased adrenergic drive impairs FA signaling early in thecourse of LV remodeling in the VO of isolated MR.
Materials and MethodsMitral valve regurgitation was induced in conditioned mongrel dogsof either sex (19 to 26 kg) by chordal rupture using a fluoroscopicguided catheterization method previously described in our laborato-ry.1,7 Dogs were randomly assigned to 1 of 5 groups: (1) unoperated
controls (n�6); (2) 2 and 4 weeks of MR (2W-MR and 4W-MR;n�6); and (3) 2 and 4 weeks of MR treated with �1-AR blocker(extended-release metoprolol succinate, 100 mg PO, once daily;n�6) starting 24 hours after MR induction. In these same animals,we have previously reported the results of M-mode echocardiogra-phy, which was performed in the conscious state at baseline and atthe time of euthanasia.1 Animals were maintained at a deep plane ofgeneral anesthesia using isoflurane and were mechanically venti-lated. At the end of the in vivo experiments, the heart was arrestedwith intracardiac injection of KCl and quickly extirpated and placedin phosphate-buffered ice slush, and the coronaries were flushed withoxygenated Krebs solution. A portion of the LV (midmyocardium)was cut and snap-frozen in liquid nitrogen for subsequent biochem-ical studies. This study was approved by the Animal ServicesCommittees at the University of Alabama at Birmingham andAuburn University.
An expanded Materials and Methods section is available in theonline data supplement at http://circres.ahajournals.org.
ResultsEffects of �1-RB on LV Remodeling and FunctionFollowing MRMean arterial pressure was significantly decreased comparedwith controls in both 4W-MR and 4W-MR��1-RB dogs(Table). Heart rate, LV, end-diastolic pressure, and meansystemic vascular resistance did not differ from controls in4W-MR and 4W-MR��1-RB dogs. However, cardiac outputwas significantly decreased in 4W-MR��1-RB dogs com-pared with controls. There was a trend toward increases in LVmass in 4W-MR and 4W-MR��1-RB dogs compared withcontrols, but this did not achieve statistical significance. Wehave previously shown in these same 4W-MR dogs a markedincrease in catecholamine release into the LV interstitial fluidspace that was significantly attenuated by �1-RB.7 The restingheart rate was measured under general anesthesia approxi-mately 24 hours after the last dose of metoprolol and, thus,may not reflect the effect of �1-RB on heart rate in theconscious state. However, in these same dogs, heart rateresponse to stellate stimulation was decreased in 4W-MR��1-RB compared with 4W-MR dogs,7 suggestive of anegative chronotropic effect of �1-RB.
LV end-diastolic dimension increased similarly in both4W-MR and 4W-MR��1-RB compared with baseline (Ta-ble). In addition, cardiac function assessed by LV fractional
Table. LV Hemodynamics and Echocardiography in Control, 4W-MR, and 4W-MR��1-RB Dogs
Control (n�6) 4W-MR (n�6) 4W-MR��1-RB (n�6)
Heart rate, bpm 94�6 86�6 83�5
Mean arterial pressure, mm Hg 84�3 65�4* 62�3*
LV end diastolic pressure, mm Hg 4�1 8�1 6�2
Systemic vascular resistance, dyne � sec � cm�5 2007�219 1606�240 2342�176
Cardiac output, L/min 3.6�0.4 3.8�0.6 2.1�0.1*
LV mass, g/kg 4.4�0.2 5.0�0.3 5.0�0.2
LV end-diastolic dimension, % change 9.2�2.9† 17.7�4.7†
LV end-systolic dimension, % change �7.4�5.8 0.1�3.6
LV end-diastolic wall thickness, % change �12.0�8.5 �10.2�5.8
Wall thickness/end-diastolic dimension, % change �18.6�9.3† �20.9�6.5†
Fractional shortening, % change 32.7�11.3† 34.3�13.9†
Values presented as means�SEM. *P�0.05 vs control, †P�0.05, % change vs baseline study.
1128 Circulation Research May 9, 2008
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

shortening was significantly increased from baseline in the4W-MR group, because LV end-systolic dimension remainedunchanged (Table). However, this increase was not affectedby �1-RB. In a similar fashion, response of LV dP/dt tostellate stimulation was similar in 4W-MR and 4W-MR��1-RB dogs, whereas stellate-stimulated heart ratechange was attenuated in 4W-MR��1-RB compared withcontrol and 4W-MR dogs.7 There was a significant decreasein LV end-diastolic wall thickness decreased significantly in4W-MR group compared with baseline, along with a signif-icant increase in LV end-diastolic dimension, indicatingeccentric LV remodeling. However, these changes were notaffected by �1-RB treatment.
�1-RB Reduces Interstitial Collagen DegradationInduced by MRQuantitative evaluation of myocardial interstitial collagenrevealed a significant decrease in volume percentage ofcollagen for untreated 4W-MR hearts compared with controlsat both the endocardium and epicardium levels (Figure 1).Moreover, the decrease in collagen accumulation was evidentat the endocardium at 2 weeks after MR (Figure I in theonline data supplement) and was sustained for 4 months afterMR (data not shown). �1-RB prevented collagen loss by�68% in the epicardium induced by 4 weeks of MR but hasno detectable effect on interstitial endocardium collagen.
�1-RB Prevents FAK Signaling DownregulationInduced by MRSeveral studies have shown that pressure overload–inducedcardiac hypertrophy is associated with an increase in integrinsignaling.22,23 However, these models of cardiac hypertrophywere associated with an increase rather than a decrease inECM deposition. Because maximal cardiac hypertrophy re-
quires ECM, we examined whether components of theintegrin-signaling cascade were altered after 4W-MR. Weexamined tyrosine phosphorylation of FAK and correlatedthese with ECM changes. In control dogs, there was basalFAK tyrosine phosphorylation as determined by blotting withanti-phosphotyrosine antibodies (Figure 2A). MR inductionfor 4 weeks led to a significant decrease in tyrosine phos-phorylation of FAK without a decrease in FAK expression,because the same blot reblotted with anti-FAK antibodies
*P<.05 vs. control#P<.05 vs. 4W-MR
0
1
2
3
Control
LV endocardium
LV epicardium
4W-MR
#
*
Vol
ume
% c
olla
gen
4W-MR+β1-RB
**
Figure 1. Volume percentage of collagen of LV endocardiumand epicardium in control, 4W-MR, and 4W-MR��1-RB dogs.*P�0.05 vs control; #P�0.05, epicardium in 4W-MR vs4W-MR��1-RB dogs.
A
B
Y397 FAK
Y861 FAK
Y576 FAK
FAK
Ctrl 4W-MR4W-MR+ β1-RB
Y925 FAK
Ctrl 4W-MR4W-MR+ β1-RB
IP:FAKP-Tyr
FAK
0.0
0.4
0.8
1.2
FAK
pho
spho
ryla
tion
(fold
incr
ease
ove
r con
trol)
*
#
Ctrl 4W-MR 4W-MR+ β1-RB
Figure 2. �1-RB prevents FAK tyrosine downregulationinduced by MR. A, LV extracts from control, 4W-MR, and4W-MR��1-RB dogs were immunoprecipitated (IP) with anti-FAK antibodies and immunoblotted with anti-phosphotyrosineantibodies. Top, Representative autoradiogram (with eachlane from a single gel exposed for the same duration). Bot-tom, Fold induction (n�6 each group). *P�0.05, 4W-MR vscontrol; #P�0.05, 4W-MR��1-RB vs 4W-MR. B, Representa-tive immunoblots showing accumulation of phospho-FAKTyr397, -576/577, -861, and -925 in LV extracts from control,4W-MR, and 4W-MR��1-RB. Blots were stripped and blottedwith anti-FAK antibodies.
Sabri et al Focal Adhesion Signaling in Early Volume Overload 1129
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

showed equal FAK expression levels between control andMR dogs (Figure 2A). A similar decrease in FAK tyrosinephosphorylation was observed at 2 weeks after MR (supple-mental Figure II). The lack of quantitative changes in totalFAK expression between control and MR hearts was alsoassociated with a lack of detectable changes in FAK distri-bution, as evaluated by double immunostaining using anti-FAK polyclonal antibodies (supplemental Figure III). FAKimmunolabeling was localized throughout the myocardium ofcontrol hearts, including the cardiomyocytes that also stainedpositively for sarcomeric �-actin, and some staining of theinterstitial space. There was also FAK staining of endothelialcells and the medial layer of blood vessels (data not shown).In 4W-MR hearts, there was no qualitative change in FAKimmunolabeling compared with normal hearts. Thus, FAKexpression and distribution were not significantly affected by4W-MR. Treatment with �1-RB significantly reduced thedecrease in FAK tyrosine phosphorylation induced by MR at4 weeks but did not reach statistical significance in 2W-MRdogs compared with controls (Figure 2A and supplementalFigure II). Because FAK can be tyrosine-phosphorylated on anumber of tyrosine residues, including Tyr397, -578/577,-861, and -925 in response to various stimuli, we nextmapped the phosphorylation site on FAK using series ofwell-characterized phospho-specific antibodies.24 FAK-Tyr397, -578/577, -861, and -925 phosphorylation decreasedsignificantly in 4W-MR dogs compared with controls andtreatment with �1-RB improved the decrease in FAK-Tyr397,-578/577, -861 tyrosine phosphorylation sites without anydetectable differences between these different sites. However,�1-RB was without significant effect on FAK-Tyr925 phos-phorylation, suggesting a different mechanism of regulationof this FAK tyrosine phosphorylation site (Figure 2B).
Consistent with a reduction in FAK tyrosine phosphorylation,there was a decreased association between FAK and p130Cas, andFAK and paxillin, 2 important components of FA complex thatassociate with and are phosphorylated by FAK,15,16 in 4W-MRdogs compared with controls (Figure 3A). Treatment with�1-RB prevented both FAK–paxillin and FAK-p130Cas dissoci-ation. To investigate whether p130Cas and paxillin are alsoaffected by 4W-MR, analysis of p130Cas and paxillin tyrosinephosphorylation was performed. p130Cas and paxillin tyrosinephosphorylation was also decreased in 4W-MR dogs and corre-lated with those of FAK (Figure 3B and 3C). Treatment with�1-RB prevented the decrease in paxillin tyrosine phosphoryla-tion but did not reach statistical significance for p130Cas phos-phorylation. These data indicate that the loss of collagen follow-ing 4W-MR is associated with a decrease in FAKphosphorylation and its association with downstream signalingmolecules. However, this downregulation seems to be selectivefor FAK and FAK associated molecules as tyrosine phosphor-ylation of Pyk2, a FA-related kinase with strong homology toFAK, was not significantly affected in 2W-MR (supplementalFigure IV) and 4W-MR (Figure 3D) hearts.
�1-RB Prevents Alteration of FAK DownstreamSignaling Induced by MRPutative signaling pathways downstream from FAK/p130Cas/paxillin could involve activation of the extracellular signal-
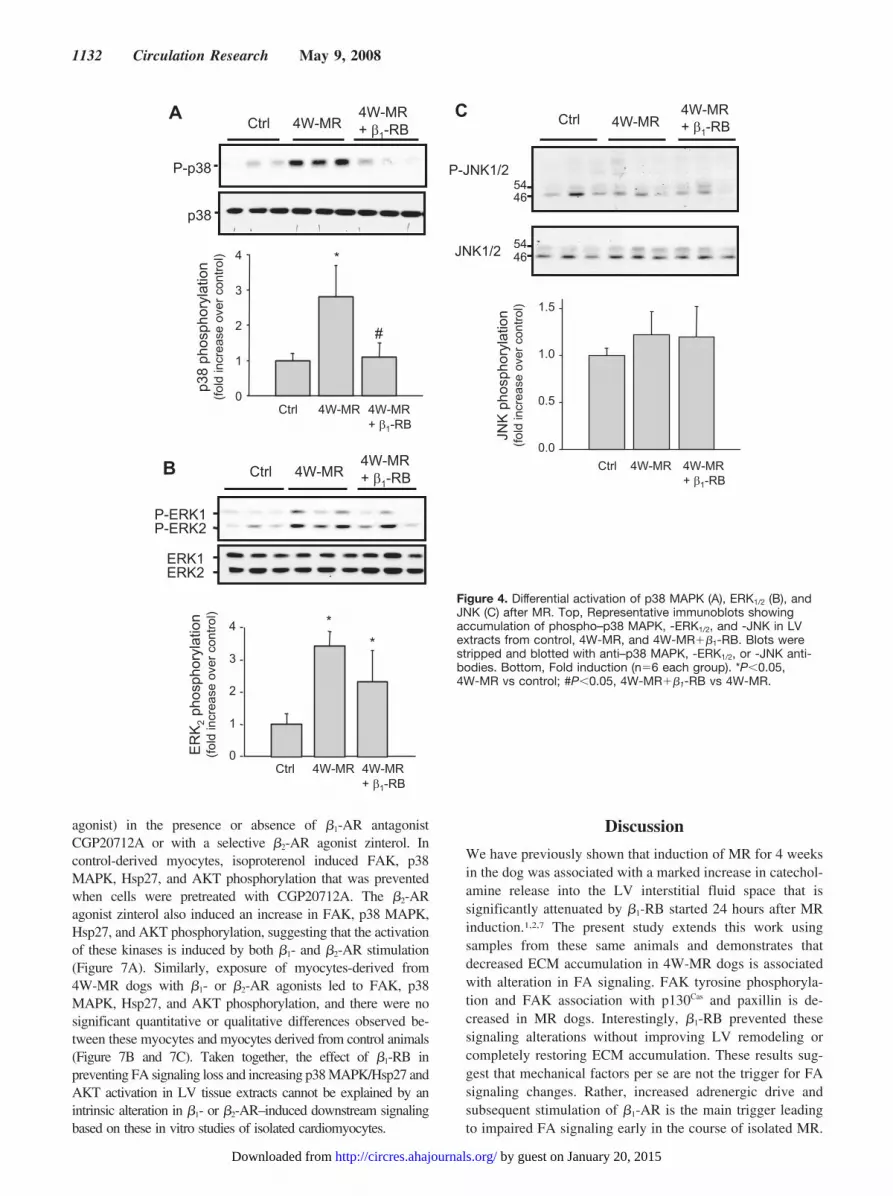
regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK),and p38 mitogen-activated protein kinase (MAPK) pathways,each of which has been implicated in hypertrophic signaltransduction.25–27 Therefore, we examined whether the loss ofFA signaling observed in MR dogs was associated with changesin p38 MAPK, ERK1/2, and JNK phosphorylation by immuno-blot analysis. Control animals presented basal p38 MAPK andERK1/2 phosphorylation and induction of MR for 4 weeks led toan increase in p38 MAPK and ERK1/2 phosphorylation (Figure4A and 4B). However, 4 weeks of MR had no detectable effecton JNK phosphorylation state (Figure 4C). A similar increase inp38 MAPK and ERK1/2 phosphorylation was also observed inthe 2W-MR group (supplemental Figure V and data not shown).Interestingly, �1-RB completely abolished p38 MAPK phos-phorylation induced after 4W-MR, whereas activation of ERK1/2
was not significantly affected at 2 and 4 weeks after MR (Figure4A and 4B and supplemental Figure V). This suggests that theVO of isolated MR differentially regulates the activation of these3 MAP-kinases with p38 MAPK and ERK1/2 being activated inresponse to MR and that p38 MAPK is dependent on �1-ARstimulation.
MR-Induced FA Signaling Alteration Is NotAssociated With Cell Death by ApoptosisBoth loss of ECM and/or loss of FAK signaling have beenshown to lead to cell death termed anoikis.28–30 To examinewhether apoptosis occurs after ECM loss and subsequent FAalteration during induction of MR, we measured caspase-3activity and DNA fragmentation by ELISA and TUNEL assay incontrol, 4W-MR, and 4W-MR��1-RB groups (Figure 5). Wedid not detect any difference among control, 4W-MR, and4W-MR��1-RB in either caspase-3 activity (normal, 249�13relative fluorescence unit (RFU)/min per milligram protein;4W-MR, 242�27 RFU/min per milligram protein; 4W-MR��1-RB, 198�26 RFU/min per milligram protein), DNAfragmentation (normal, 5.2�0.9 optical density (OD)410 toOD500/mg protein; 4W-MR, 5.8�1.4 OD410 to OD500/mg protein;4W-MR��1-RB, 4.00�0.9 OD410 to OD500/mg protein), or thepercentage of TUNEL-positive cardiomyocytes (normal,0.026�0.011%; 4W-MR, 0.021�0.002%; 4W-MR��1-RB,0.03�0.015%). These data showed that MR at this earlycompensatory hypertrophy stage is not associated with myocyteapoptosis despite a loss of ECM and FA signaling.
MR-Induced AKT and HSP27 Phosphorylation IsPrevented by �1-RBThe absence of apoptotic markers in MR hearts despite thedecrease in FA signaling led us to hypothesize that othercompensatory signaling molecules may be activated to counter-act the effect of ECM loss. One of the molecules that has beenshown to play a role in survival and protection of myocytesagainst apoptosis is the phosphatidylinositol 3-kinase/AKT path-way.31 As FAK has been identified as the major site for bindingof phosphatidylinositol 3-kinase, the inositol lipid products ofwhich are key mediators of Akt activation,32 we next examinedwhether induction of MR is associated with an increase in AKTphosphorylation and whether �1-RB prevents this activation.Immunoblotting with anti–phospho-AKT at the Ser473 residue,which has been shown to be required for maximal activation of
1130 Circulation Research May 9, 2008
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

AKT,33 showed an increase in AKT phosphorylation following4W-MR (Figure 6A). The amount of total Akt expression wasnot different between the 2 groups of animals. The increase inAKT phosphorylation was abrogated in 4W-MR��1-RB dogs,suggesting that �1-AR stimulation mediates AKT activation.Interestingly, these data also showed that FAK and AKTphosphorylation are differentially regulated by �1-ARs in theMR model, with AKT phosphorylation being independent fromFAK activation.
Another molecule that has been shown to be downstreamof p38 MAPK and to confer protection and antiapoptoticproperties for the heart is heat shock protein (Hsp)27.34 MRinduction for 4 weeks increased Hsp27 phosphorylation, asassessed by immunoblot (Figure 6B). As for p38 MAPKactivation, �1-RB abolished Hsp27 activation induced by
4W-MR (Figure 6B). These data, together, suggest thatstimulation of �1-AR is involved in the activation of antiapo-ptotic pathways, AKT and Hsp27, that may compensate forthe loss of ECM and FA signaling in early MR.
�-AR Signaling Is Not Altered inMR-Isolated CardiomyocytesBecause �1-RB reduced FA signaling loss and prevented p38MAPK, Hsp27, and AKT activation, we thought that alterationin �-AR signaling may occur in cardiomyocytes at this earlystage of MR. To test this hypothesis, myocytes were isolatedfrom LV of control and 4W-MR dogs and plated on laminin-coated dishes for 6 hours in 5% FBS DMEM. Myocytes werethen switched to serum-free medium for 1 hour before theirtreatment with isoproterenol (a nonspecific �1- and �2-AR
A
B
P-Tyr
IP: p130Cas
IP: p130Cas
p130
Cas
phos
phor
ylat
ion
(fold
incr
ease
ove
r con
trol)
*
0.0
0.4
0.8
1.2
Ctrl 4W-MR4W-MR+ β1-RB
Ctrl 4W-MR 4W-MR+ β1-RB
Ctrl 4W-MR4W-MR+ β1-RBIP:FAK
P-Tyr
p130Cas
Paxillin
FAK
C
D
Ctrl 4W-MR4W-MR+ β1-RB
IP: Paxillin
P-Tyr
paxillin
Pax
illin
pho
spho
ryla
tion
(fold
incr
ease
ove
r con
trol)
0.0
0.4
0.8
1.2
*
#
Ctrl 4W-MR 4W-MR+ β1-RB
Ctrl 4W-MR4W-MR+ β1-RB
p-Pyk2
Pyk2
0.0
0.4
0.8
1.2
1.6
Pyk
2 ph
osph
oryl
atio
n(fo
ld in
crea
se o
ver c
ontro
l)
Ctrl 4W-MR 4W-MR+ β1-RB
Figure 3. Alteration of FAK downstream signaling induced by MR is prevented by �1-RB. A, FAK immunoprecipitates from LVlysates of control, 4W-MR, and 4W-MR��1-RB dogs were resolved on SDS-PAGE and blotted with anti-phosphotyrosine,p130Cas, paxillin, or FAK antibodies. B and C, p130Cas or paxillin immunoprecipitates from control, 4W-MR, or 4W-MR��1-RB LVextracts were resolved on SDS-PAGE and blotted with anti-phosphotyrosine, anti-p130Cas (B), or anti-paxillin (C) antibodies. Top,Representative autoradiograms (with each lane from a single gel exposed for the same duration). Bottom, Fold induction (n�6each group). *P�0.05, 4W-MR vs control; #P�0.05, 4W-MR��1-RB vs 4W-MR. D, Representative immunoblot showing accumu-lation of phospho-Pyk2 Tyr-402 in LV extracts from control, 4W-MR, and 4W-MR��1-RB. Blot was stripped and blotted with anti-Pyk2 antibodies.
Sabri et al Focal Adhesion Signaling in Early Volume Overload 1131
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from
agonist) in the presence or absence of �1-AR antagonistCGP20712A or with a selective �2-AR agonist zinterol. Incontrol-derived myocytes, isoproterenol induced FAK, p38MAPK, Hsp27, and AKT phosphorylation that was preventedwhen cells were pretreated with CGP20712A. The �2-ARagonist zinterol also induced an increase in FAK, p38 MAPK,Hsp27, and AKT phosphorylation, suggesting that the activationof these kinases is induced by both �1- and �2-AR stimulation(Figure 7A). Similarly, exposure of myocytes-derived from4W-MR dogs with �1- or �2-AR agonists led to FAK, p38MAPK, Hsp27, and AKT phosphorylation, and there were nosignificant quantitative or qualitative differences observed be-tween these myocytes and myocytes derived from control animals(Figure 7B and 7C). Taken together, the effect of �1-RB inpreventing FA signaling loss and increasing p38 MAPK/Hsp27 andAKT activation in LV tissue extracts cannot be explained by anintrinsic alteration in �1- or �2-AR–induced downstream signalingbased on these in vitro studies of isolated cardiomyocytes.
Discussion
We have previously shown that induction of MR for 4 weeksin the dog was associated with a marked increase in catechol-amine release into the LV interstitial fluid space that issignificantly attenuated by �1-RB started 24 hours after MRinduction.1,2,7 The present study extends this work usingsamples from these same animals and demonstrates thatdecreased ECM accumulation in 4W-MR dogs is associatedwith alteration in FA signaling. FAK tyrosine phosphoryla-tion and FAK association with p130Cas and paxillin is de-creased in MR dogs. Interestingly, �1-RB prevented thesesignaling alterations without improving LV remodeling orcompletely restoring ECM accumulation. These results sug-gest that mechanical factors per se are not the trigger for FAsignaling changes. Rather, increased adrenergic drive andsubsequent stimulation of �1-AR is the main trigger leadingto impaired FA signaling early in the course of isolated MR.
A
B
P-ERK1P-ERK2
ERK1ERK2
Ctrl 4W-MR4W-MR+ β1-RB
ER
K2
phos
phor
ylat
ion
(fold
incr
ease
ove
r con
trol)
0
1
2
3
4 *
Ctrl 4W-MR 4W-MR+ β1-RB
*
P-p38
p38
Ctrl 4W-MR4W-MR+ β1-RB
p38
phos
phor
ylat
ion
(fold
incr
ease
ove
r con
trol) *
#
0
1
2
3
4
Ctrl 4W-MR 4W-MR+ β1-RB
C Ctrl 4W-MR4W-MR+ β1-RB
P-JNK1/2
4654
4654JNK1/2
0.0
0.5
1.0
1.5
JNK
pho
spho
ryla
tion
(fold
incr
ease
ove
r con
trol)
Ctrl 4W-MR 4W-MR+ β1-RB
Figure 4. Differential activation of p38 MAPK (A), ERK1/2 (B), andJNK (C) after MR. Top, Representative immunoblots showingaccumulation of phospho–p38 MAPK, -ERK1/2, and -JNK in LVextracts from control, 4W-MR, and 4W-MR��1-RB. Blots werestripped and blotted with anti–p38 MAPK, -ERK1/2, or -JNK anti-bodies. Bottom, Fold induction (n�6 each group). *P�0.05,4W-MR vs control; #P�0.05, 4W-MR��1-RB vs 4W-MR.
1132 Circulation Research May 9, 2008
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

In this early stage of isolated MR, loss of FAK tyrosinephosphorylation was associated with the loss of FAK interactionwith p130Cas and paxillin, which are important docking sites forother signaling molecules that play an additional role in survivalsignaling.15,16 This is consistent with FAK involvement in thetyrosine phosphorylation of p130Cas and paxillin and indicatesthat MR induces downregulation of FA signaling, as well as
destruction of the FA complex. This is in stark contrast to theactivation of FA signaling reported in the early compensatedstages of experimentally induced pressure overload in vivo or inisolated cardiomyocytes subjected to pulsatile mechanical
A
B
pAKT
AKT
AK
T ph
osph
oryl
atio
n(fo
ld in
crea
se o
ver c
ontro
l) *
#
0
1
2
3
4
Ctrl 4W-MR 4W-MR+ β1-RB
Ctrl 4W-MR 4W-MR+ β1-RB
P-Hsp27
Hsp27
Ctrl 4W-MR 4W-MR+ β1-RB
Hsp
27 p
hosp
hory
latio
n(fo
ld in
crea
se o
ver c
ontro
l)
*
#
0
1
2
3
4
Ctrl 4W-MR 4W-MR+ β1-RB
Figure 6. MR-induced AKT (A) and Hsp27 (B) phosphorylation isprevented by �1-RB. Top, Representative immunoblots showingaccumulation of phospho-AKT and -Hsp27 in LV extracts fromcontrol, 4W-MR, and 4W-MR��1-RB. Blots were stripped andreblotted with anti-AKT or -Hsp27 antibodies. Bottom, Foldinduction (n�6 each group). *P�0.05, 4W-MR vs control;#P�0.05, 4W-MR��1-RB vs 4W-MR.
A
B
Cas
pase
3 a
ctiv
ity
(RFU
/min
/mg)
0
100
200
300
Ctrl 4W-MR 4W-MR+ β1-RB
DN
A F
ragm
enta
tion
OD
410-
OD
500/
mg
prot
ein
0
2
4
6
8
Ctrl 4W-MR 4W-MR+ β1-RB
C
% o
f TU
NE
L po
sitiv
e nu
clei
0
0.025
0.050
Ctrl 4W-MR 4W-MR+ β1-RB
Figure 5. Induction of MR is not associated with myocardial cellapoptosis. LV homogenates from the control, 4W-MR, and4W-MR��1-RB groups were assessed for apoptosis. A,Caspase-3 activity was measured using fluorogenic substrate.Results are expressed as RFU per minute per milligram of pro-tein. B, DNA fragmentation as measured by anti-histone anti-body ELISA. Results are expressed as relative OD410 to OD500
per milligram of protein. Values are means�SE. C, Morphometryof TUNEL staining of LV sections from control, 4W-MR, and4W-MR��1-RB.
Sabri et al Focal Adhesion Signaling in Early Volume Overload 1133
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

C
0.0
0.5
1.0
1.5
2.0
Hsp
27 p
hosp
hory
latio
n(fo
ld in
crea
se o
ver c
ontro
l)
Ctrl Iso Zint Iso+CGP
0.0
1.0
2.0
3.0
Ctrl Iso Zint Iso+CGP
AKT
phos
phor
ylat
ion
(fold
incr
ease
ove
r con
trol)
0.0
0.5
1.0
1.5
2.0
p38
phos
phor
ylat
ion
(fold
incr
ease
ove
r con
trol)
Ctrl Iso Zint Iso+CGP
Control4W-MR
0.0
0.5
1.0
1.5
2.0
Ctrl Iso Zint Iso+CGP
FAK
phos
phor
ylat
ion
(fold
incr
ease
ove
r con
trol)
**
* **
*
***
Ctrl Iso Zint Iso+CGP
P-AKT
P-p38
P-Hsp27
P-AKT
P-p38
P-Hsp27
AKT
p38
Hsp27 Hsp27
p38
AKT
Y397-FAK
FAK
Y397-FAK
FAK
Ctrl Iso Zint Iso+CGP
A- Control derived Myocytes B- 4W-MR derived Myocytes
Figure 7. Preservation of �-AR signaling in MR-isolated cardiomyocytes. Cardiac myocytes were isolated from control (A) or 4W-MR(B) LV as described in Materials and Methods. A and B, Representative immunoblots showing accumulation of phospho-FAK, –p38MAPK, -Hsp27, and -AKT after treatment with 10 �mol/L isoproterenol (Iso) or 1 �mol/L zinterol (Zint) for 5 minutes compared withcontrol (Ctrl): 1 �mol/L CGP20712A was added 30 minutes before isoproterenol stimulation. C, Summary graphs represent quantitativedata from 4 independent experiments from cells derived from 4 different animals. *P�0.05 vs control.
1134 Circulation Research May 9, 2008
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

stretch.22,23,35 Interestingly, the decrease in FAK and paxillintyrosine phosphorylation was not associated with their cleavageor caspase-3 activation in 2W-MR (supplemental Figure II anddata not shown) and 4W-MR (Figures 2A and 5A) LVs,suggesting that dephosphorylation and disruption of the FA com-plex occurs before caspase-3 activation and FAK/paxillin degrada-tion. In the failing and dilated hearts in which myocyte apoptosiswas identified, FA protein (FAK and paxillin) cleavage has beendetected and may involve an increase in caspase-3 activity.36
The role of the constitutive phosphorylation of FA proteins innormal cellular function is not fully understood but may beimportant for maintaining cell survival and FA integrity in theresting state.16,37 In mice with a selective inactivation of FAK incardiomyocytes, an eccentric cardiac hypertrophy develops withage and even in the face of angiotensin II infusion or inductionof pressure overload with transaortic constriction.19 In anotherstudy, persistent challenge of similar transgenic mice withtransaortic constriction leads to enhanced cardiac fibrosis andcardiac dysfunction in comparison with challenged geneticcontrols.20 Despite these cardiac structural changes, both studiesfailed to detect an increase in myocyte apoptosis. This is incontrast to findings in myocyte-restricted deletion of the �1
integrin in adult mouse hearts in which dilated cardiomyopathyand concomitant heart failure were observed.38 These findings inmice, in addition to our present study, showed that impaired FAsignaling is associated with the development of eccentric cardiachypertrophy. Recent studies in cardiomyocytes in vitro have dem-onstrated that disruption of FAK signaling prevented hypertrophicresponses induced by G protein–coupled receptors and promotedcardiomyocyte apoptosis by anoikis.29,30,39
Protection against apoptosis in this early adaptive phase ofisolated MR could be explained by an increase in survivalsignaling pathways ERK1/2, AKT, and Hsp27 activation thatcounter proapoptotic events resulting from the loss of FAsignaling. In addition, there was no increase in JNK phosphor-ylation, which has been shown to mediate �1-AR–inducedcardiomyocyte apoptosis.40 Surprisingly, �1-RB prevented bothAKT and Hsp27 stimulation, implicating �1-AR stimulation inthe activation of these survival signaling pathways that areindependent of FAK activation. Nevertheless, there is emergingevidence that prolonged activation of some survival signalingpathways may have deleterious effects on cardiomyocyte sur-vival. Herein, induction of an activated Akt1 gene in the mouseheart induced adaptive cardiac hypertrophy in the acute phaseand dilated cardiomyopathy in the chronic phase, suggesting thatAkt and Akt-dependent signaling pathways are involved in bothphysiological and pathological cardiac growth.41 These dataemphasize that cardiac apoptosis control is multifactorial andthat the balance between pro- and antiapoptotic pathways overtime may dictate the transition form heart hypertrophy to failure.
In this study, we showed that �1-RB prevented FA signal-ing loss and p38 MAPK, Hsp27, and AKT activation inducedby 4W-MR. However, we could not detect a significantdifference in �1- or �2-AR expression by immunoblot ofplasma membrane proteins prepared from control- or MR-derived cardiomyocytes (data not shown). A defect in �-ARdownstream signaling is also unlikely because myocytesderived from control and MR hearts showed similar �1- and�2-AR responses in FAK, AKT, p38 MAPK, and Hsp27
phosphorylation. Although these data indicate an intact �-ARsignaling mechanism of cardiomyocytes in vitro, it is note-worthy that cardiomyocytes were isolated from the whole LVand were stimulated with submaximal concentrations ofagonists, which may not allow detecting differences in kinasephosphorylation between control- and MR-derived cardio-myocytes. Furthermore, cardiomyocytes were grown on lami-nin substrate, which may not reproduce the marked loss ofECM in MR hearts in vivo. This is especially importantbecause plating myocytes on laminin substrate, as opposed toglass, selectively reduced �1-AR and enhanced �2-AR regu-lation of ICa.13 Thus, our short-term in vitro studies inMR-derived cardiomyocytes plated on laminin may negatethe loss of ECM in MR hearts with increased adrenergic drivein vivo.
The selective downregulation of FA signaling in thecompensated phase of MR was associated by a small amountof LV dilatation and increased LV fractional shortening. Thisoccurs in spite of the renin–angiotensin system and adrener-gic drive activation and is in stark contrast to the activation ofFA signaling in experimentally induced pressure overload.22,23
In these dogs, �1-RB decreased catecholamine release into theinterstitial fluid in response to electric and angiotensin II stim-ulation7 and restored FA signaling without affecting LV end-di-astolic dimension to wall thickness ratio and wall stress indexes.Our data suggest that prolonged and excessive adrenergic driveis a potential mediator of FA signaling alteration early in theadaptive phase of VO. Consistent with these findings, prolongedstimulation of �-ARs has been shown to promote disruption of�1-integrin signaling in cultured cardiomyocytes and stimulationof �1-integrin signaling was efficient to protect cardiomyocytesagainst �-AR–induced apoptosis.14 However, �1-RB did notprevent a marked loss of endocardial collagen, which couldexplain the failure to improve short-term diastolic remodelingand function. Long-term therapy of VO with �1-AR doesimprove isolated cardiomyocyte function but has no effect oninterstitial collagen loss and LV dilatation and remodeling(preliminary data and elsewhere3). The primary loss of myocyte/fibroblast–ECM scaffolding and their interaction offers a newtarget, in addition to �1-AR, to attenuate excessive adrenergicdrive in the VO of MR.
Sources of FundingThis work was supported by NIH grant HL76799 (to A.S.), NIHSpecialized Centers of Clinically Oriented Research in CardiacDysfunction grant P50HL077100 (to L.J.D.), American Heart Asso-ciation Grant 0430301N (to A.S.), Department of Veteran Affairs (toL.J.D.), and, in part, by a grant from AstraZeneca (to L.J.D.).
DisclosuresNone.
References1. Tallaj J, Wei C-C, Hankes GH, Holland M, Rynders P, Dillon AR, Ardell JL,
Armour JA, Lucchesi PA, Dell’Italia LJ. {beta}1-adrenergic receptorblockade attenuates angiotensin II-mediated catecholamine release into thecardiac interstitium in mitral regurgitation. Circulation. 2003;108:225–230.
2. Stewart J, James A. Wei C-C, Brower GL, Rynders PE, Hankes GH,Dillon AR, Lucchesi PA, Janicki JS, Dell’Italia LJ. Cardiac mast cell- andchymase-mediated matrix metalloproteinase activity and left ventricularremodeling in mitral regurgitation in the dog. J Mol Cell Cardiol. 2003;35:311–319.
Sabri et al Focal Adhesion Signaling in Early Volume Overload 1135
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

3. Tsutsui H, Spinale FG, Nagatsu M, Schmid PG, Ishihara K, DeFreyte G,Cooper IVG, Carabello BA. Effects of chronic �-adrenergic blockade onthe left ventricular and cardiocyte abnormalities of chronic canine mitralregurgitation. J Clin Invest. 1994;93:2639–2648.
4. Dell’italia LJ, Balcells E, Meng QC, Su X, Schultz D, Bishop SP, MachidaN, Straeter-Knowlen IM, Hankes GH, Dillon R, Cartee RE, Oparil S.Volume-overload cardiac hypertrophy is unaffected by ACE inhibitor treatmentin dogs. Am J Physiol Heart Circ Physiol. 1997;273:H961–H970.
5. Perry GJ, Wei C-C, Hankes GH, Dillon SR, Rynders P, Mukherjee R,Spinale FG, Dell’Italia LJ. Angiotensin II receptor blockade does notimprove left ventricular function and remodeling in subacute mitral re-gurgitation in the dog. J Am Coll Cardiol. 2002;39:1374–1379.
6. Mehta RH, Supiano MA, Oral H, Grossman PM, Montgomery DS, SmithMJ, Starling MR. Compared with control subjects, the systemic sympa-thetic nervous system is activated in patients with mitral regurgitation. AmHeart J. 2003;145:1078–1085.
7. Hankes GH, Ardell JL, Tallaj J, Wei CC, Aban I, Holland M, Rynders P,Dillon R, Cardinal R, Hoover DB, Armour JA, Husain A, Dell’Italia LJ.beta1-Adrenoceptor blockade mitigates excessive norepinephrine releaseinto cardiac interstitium in mitral regurgitation in dog. Am J Physiol HeartCirc Physiol. 2006;291:H147–H151.
8. Steinberg SF. The molecular basis for distinct {beta}-adrenergic receptorsubtype actions in cardiomyocytes. Circ Res. 1999;85:1101–1111.
9. Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, RasmussenR, Zera P, Menlove R, Shah P, Jamieson S, Stinson E. Beta1- andbeta2-adrenergic-receptor subpopulations in nonfailing and failing humanventricular myocardium: coupling of both receptor subtypes to musclecontraction and selective beta1-receptor down-regulation in heart failure.Circ Res. 1986;59:297–309.
10. Feldman AM, Cates AE, Veazey WB, Hershberger RE, Bristow MR,Baughman KL, Baumgartner WA, Van Dop C. Increase of the40,000-mol wt pertussis toxin substrate (G protein) in the failing humanheart. J Clin Invest. 1988;82:189–197.
11. Roth DA, Urasawa K, Helmer GA, Hammond HK. Down-regulation ofcardiac guanosine 5�-triphosphate-binding proteins in right atrium andleft ventricle in pacing-induced congestive heart failure. J Clin Invest.1993;91:939–949.
12. Ungerer M, Kessebohm K, Kronsbein K, Lohse MJ, Richardt G. Acti-vation of �-adrenergic receptor kinase during myocardial ischemia. CircRes. 1996;79:455–460.
13. Wang YG, Samarel AM, Lipsius SL. Laminin binding to beta1-integrinsselectively alters beta1- and beta2-adrenoceptor signalling in cat atrialmyocytes. J Physiol (Lond). 2000;527:3–9.
14. Communal C, Singh M, Menon B, Xie Z, Colucci WS, Singh K. beta1integrins expression in adult rat ventricular myocytes and its role in theregulation of beta-adrenergic receptor-stimulated apoptosis. J CellBiochem. 2003;89:381–388.
15. Ross RS. The extracellular connections: the role of integrins in myo-cardial remodeling. J Card Fail. 2002;8:S326–S331.
16. Schaller MD. Biochemical signals and biological responses elicited by thefocal adhesion kinase. Biochim Biophys Acta. 2001;1540:1–21.
17. Bloch W, Forsberg E, Lentini S, Brakebusch C, Martin K, Krell HW,Weidle UH, Addicks K, Fassler R. beta1 integrin is essential for teratomagrowth and angiogenesis. J Cell Biol. 1997;139:265–278.
18. Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, NomuraS, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility andenhanced focal adhesion contact formation in cells from FAK-deficientmice. Nature. 1995;377:539–544.
19. Peng X, Kraus MS, Wei H, Shen TL, Pariaut R, Alcaraz A, Ji G, ChengL, Yang Q, Kotlikoff MI, Chen J, Chien K, Gu H, Guan JL. Inactivationof focal adhesion kinase in cardiomyocytes promotes eccentric cardiachypertrophy and fibrosis in mice. J Clin Invest. 2006;116:217–227.
20. DiMichele LA, Doherty JT, Rojas M, Beggs HE, Reichardt LF, Mack CP,Taylor JM. Myocyte-restricted focal adhesion kinase deletion attenuatespressure overload-induced hypertrophy. Circ Res. 2006;99:636–645.
21. Terracio L, Rubin K, Gullberg D, Balog E, Carver W, Jyring R, Borg T.Expression of collagen binding integrins during cardiac development andhypertrophy. Circ Res. 1991;68:734–744.
22. Franchini KG, Torsoni AS, Soares PH, Saad MJ. Early Activation of themulticomponent signaling complex associated with focal adhesion kinaseinduced by pressure overload in the rat heart. Circ Res. 2000;87:558–565.
23. Bayer AL, Heidkamp MC, Patel N, Porter MJ, Engman SJ, Samarel AM.PYK2 expression and phosphorylation increases in pressure overload-induced left ventricular hypertrophy. Am J Physiol Heart Circ Physiol.2002;283:H695–H706.
24. Ruest PJ, Roy S, Shi E, Mernaugh RL, Hanks SK. Phosphospecificantibodies reveal focal adhesion kinase activation loop phosphorylation innascent and mature focal adhesions and requirement for the autophos-phorylation site. Cell Growth Differ. 2000;11:41–48.
25. Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, KlevitskyR, Hewett TE, Jones SP, Lefer DJ, Peng C-F, Kitsis RN, Molkentin JD.The MEK1-ERK1/2 signaling pathway promotes compensated cardiachypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350.
26. Ramirez MT, Sah VP, Zhao X-L, Hunter JJ, Chien KR, Brown JH. TheMEKK-JNK pathway is stimulated by alpha1-adrenergic receptor andRas activation and is associated with in-vitro and in-vivo cardiac hyper-trophy. J Biol Chem. 1997;272:14057–14061.
27. Wang Y, Huang S, Sah VP, Ross Jr. J, Brown JH, Han J, Chien KR.Cardiac muscle cell hypertrophy and apoptosis induced by distinctmembers of the p38 mitogen-activated protein kinase family. J BiolChem. 1998;273:2161–2168.
28. Frisch S, Francis H. Disruption of epithelial cell-matrix interactionsinduces apoptosis. J Cell Biol. 1994;124:619–626.
29. Heidkamp MC, Bayer AL, Kalina JA, Eble DM, Samarel AM.GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal ratventricular myocytes. Circ Res. 2002;90:1282–1289.
30. Rafiq K, Kolpakov MA, Abdelfettah M, Streblow DN, Hassid A, Dell’Italia LJ,Sabri A. Role of protein-tyrosine phosphatase SHP2 in focal adhesion kinasedown-regulation during neutrophil cathepsin G-induced cardiomyocytesAnoikis. J Biol Chem. 2006;281:19781–19792.
31. O’Neill BT, Abel ED. Akt1 in the cardiovascular system: friend or foe?J Clin Invest. 2005;115:2059–2064.
32. Chan TO, Rittenhouse SE, Tsichlis PN. AKT/PKB and other D3phosphoinisitide-regulated kinases: kinase activation by phosphoinosi-tide-dependent phosphorylation. Annu Rev Biochem. 1999;68:965–1014.
33. Bayascas JR, Alessi DR. Regulation of Akt/PKB Ser473 phosphorylation.Mol Cell. 2005;18:143–145.
34. Hollander JM, Martin JL, Belke DD, Scott BT, Swanson E, Krish-namoorthy V, Dillmann WH. Overexpression of wild-type heat shockprotein 27 and a nonphosphorylatable heat shock protein 27 mutantprotects against ischemia/reperfusion injury in a transgenic mouse model.Circulation. 2004;110:3544–3552.
35. Torsoni AS, Constancio SS, Nadruz W Jr, Hanks SK, Franchini KG.Focal adhesion kinase is activated and mediates the early hypertrophicresponse to stretch in cardiac myocytes. Circ Res. 2003;93:140–147.
36. Melendez J, Welch S, Schaefer E, Moravec CS, Avraham S, Avraham H,Sussman MA. Activation of pyk2/related focal adhesion tyrosine kinaseand focal adhesion kinase in cardiac remodeling. J Biol Chem. 2002;277:45203–45210.
37. Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88:1112–1119.
38. Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, OmuraM, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, RossRS. Cardiac myocyte-specific excision of the {beta}1 integrin generesults in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464.
39. Taylor JM, Rovin JD, Parsons JT. A role for focal adhesion kinase inphenylephrine-induced hypertrophy of rat ventricular cardiomyocytes.J Biol Chem. 2000;275:19250–19257.
40. Remondino A, Kwon SH, Communal C, Pimentel DR, Sawyer DB, SinghK, Colucci WS. {beta}-adrenergic receptor-stimulated apoptosis incardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. CircRes. 2003;92:136–138.
41. Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS,Walsh K. Disruption of coordinated cardiac hypertrophy and angio-genesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118.
1136 Circulation Research May 9, 2008
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from

CIRCRESAHA/2007/163642/R2
1
SUPPLEMENTARY INFORMATION
MATERIALS & METHODS
Materials: Monoclonal antibodies to FAK, paxillin and p130Cas used for Western blot
analysis were obtained from BD Biosciences, and 4G10 monoclonal antibodies for
phosphotyrosine and phospho-FAK (Tyr397) were from Upstate Biotechnology.
Polyclonal antibodies for total FAK (used for immunoprecipitation), p38-MAPK, ERK1/2,
AKT, and HSP27 were from Santa Cruz Biotechnology. Polyclonal antibodies for
phospho-FAK (Tyr576), -FAK (Tyr925), p38-MAPK (Thr180/Tyr182), -ERK1/2
(Thr202/Tyr204), -AKT (Ser473), and -Hsp27 (Ser82) were from Cell Signaling.
Polyclonal antibodies for phospho-FAK (Tyr861) and sarcomeric α-actin were from
Sigma. All other chemicals were from standard suppliers.
Cardiac myocytes isolation and culture. Myocyte preparation was performed as
described previously with some modification.1 After hearts were removed, a portion of
the left ventricle was excised and a branch of the left anterior descending coronary artery
was cannulated. The tissue initially was perfused with Tyrode solution; pH 7.4 at 37°C.
After 10-15 min, the perfusion was switched to a Ca2+-free solution. At ~3-5 min,
collagenase (0.4 mg/ml; type II, Worthington Biochemical) and BSA (0.5 mg/ml; Sigma)
were added and perfusion was continued for another 10-12 min. Digested tissue was
sliced away from the subepicardial area, placed in 10 ml of enzyme solution, and swirled.
The supernatant was collected, and 10 ml of fresh Ca2+-free solution with 0.4 mg/ml
collagenase and 0.5 mg/ml BSA was added to the slurry and gently bubbled in a water
bath maintained at 37°C. Supernatant was collected for six subsequent washes. After

CIRCRESAHA/2007/163642/R2
2
5 min of settling, the final pellet was washed in 10 ml of incubation buffer containing (in
mM) 118 NaCl, 4.8 KCl, 1.2 MgSO4, 1.2 KH2PO4, 0.68 glutamine, 11 glucose,
20 NaHCO3, 5 HEPES, 5 pyruvate, 10 taurine, and 0.5 CaCl2 plus 2% BSA. After another
30 min of settling, the pellet was washed a second time with incubation buffer now
containing 1 mM CaCl2 and was allowed to equilibrate at room temperature for 60 min.
Myocytes were plated at a density of ~0.5 × 105 cells/cm2 on plastic culture dishes coated
with laminin (10 µg/ml; BD Bioscience). The culture medium consisted of Eagle's
minimum essential medium (GIBCO) with the following additions: nonessential amino
acids (GIBCO), vitamins (2X; GIBCO); 10 µg/ml insulin (Sigma), 10 µg/ml transferrin
(GIBCO); 5% fetal bovine serum, and 100 U/ml penicillin-100 µg/ml streptomycin. Cells
were incubated at 37°C in 5% CO2 in a humidified environment for 6 hrs then medium
was switched to serum free DMEM/F12 1hr before treatments.
Immunoprecipitation and immunoblot analysis. Extraction of proteins from LV or
cultured cells was performed as previously described.2,3 Lysates were clarified by
centrifugation at 12,000 rpm, and the supernatants (1 mg of protein/ml) were subjected to
immunoprecipitation with corresponding antibodies. After overnight incubation at 4°C,
protein A- or G-agarose beads were added and left for an additional 3 hrs.
Immunocomplexes were then subjected to SDS-polyacrylamide gel electrophoresis
(PAGE) followed by western blot analysis according to methods published previously or
to the manufacturer's instructions.2,3 Each panel in each figure represents results from a
single gel exposed for a uniform duration, with bands detected by enhanced
chemiluminescence and quantified by laser scanning densitometry.

CIRCRESAHA/2007/163642/R2
3
Caspase-3 Assay. Caspase 3 activity was measured with CaspACE assay system
(Promega, Madison, WI). In brief, heart lysates were prepared by dounce
homogenization in lysis buffer provided with the kit. The lysates were centrifuged at
15,000g for 20 minutes at 4 oC, and the supernatants containing 100 µg protein were used
for caspase-3 assay. Caspase-3 activity was examined by measurement of the rate of
cleavage of fluorogenic conjugated substrate MCA-Val-Asp-Gln-Met-Asp-Gly-Trp-Lys-
(DNP)-NH2. The specificity of the assay was confirmed by addition of the specific
caspase-3 inhibitor Z-DQMD-FMK in the reaction mixture at a concentration of 50 µM
during the incubation.
Apoptotic cell death ELISA. Cell death detection ELISA kit (Roche Applied Science,
Indianapolis, IN) was used to quantitatively determine the apoptotic DNA fragmentation
by measuring the cytosolic histone-associated mono- and oligo-nucleosomes fragments
associated with apoptotic cell death.
Evaluation of apoptosis in tissue sections. TUNEL labeling using fluorescein-labeled
dUTP was performed on 5-µm sections from LV fixed by 10% formalin per the
manufacturer’s protocol (In Situ Cell Death Detection kit; Roche Applied Science,
Indianapolis, IN). Sections were then counterstained with DAPI for 15 minutes to
visualize nuclei. Digital photographs were taken under fluorescence microscopy at x200
magnification. Twenty-five to thirty random high-power fields from each heart sample
were chosen and blindly quantified.
Immunohistochemistry. Frozen Sections from MR and control LVs were fixed in 4%
paraformaldehyde/PBS 30 min at 4 °C. After washes in PBS, sections were preincubated
with 2% BSA in PBS for 30 min at room temperature, followed by overnight incubation

CIRCRESAHA/2007/163642/R2
4
with the primary anti-FAK polyclonal antibody. After washes in PBS, sections were then
incubated with monoclonal anti-α-actin (sarcomeric) antibodies followed by incubations
with Alexa-conjugated anti-mouse (-rabbit) Ig antibodies. After 2 washes in PBS, cells
were incubated with DAPI for 30 min at room temperature, mounted, and then examined
using an epifluorescence microscope.
Data Analysis. All data are presented as mean±SEM. One-way repeated-measures
ANOVA. A value of P<0.05 was considered significant
SUPPLEMENTARY RESULTS
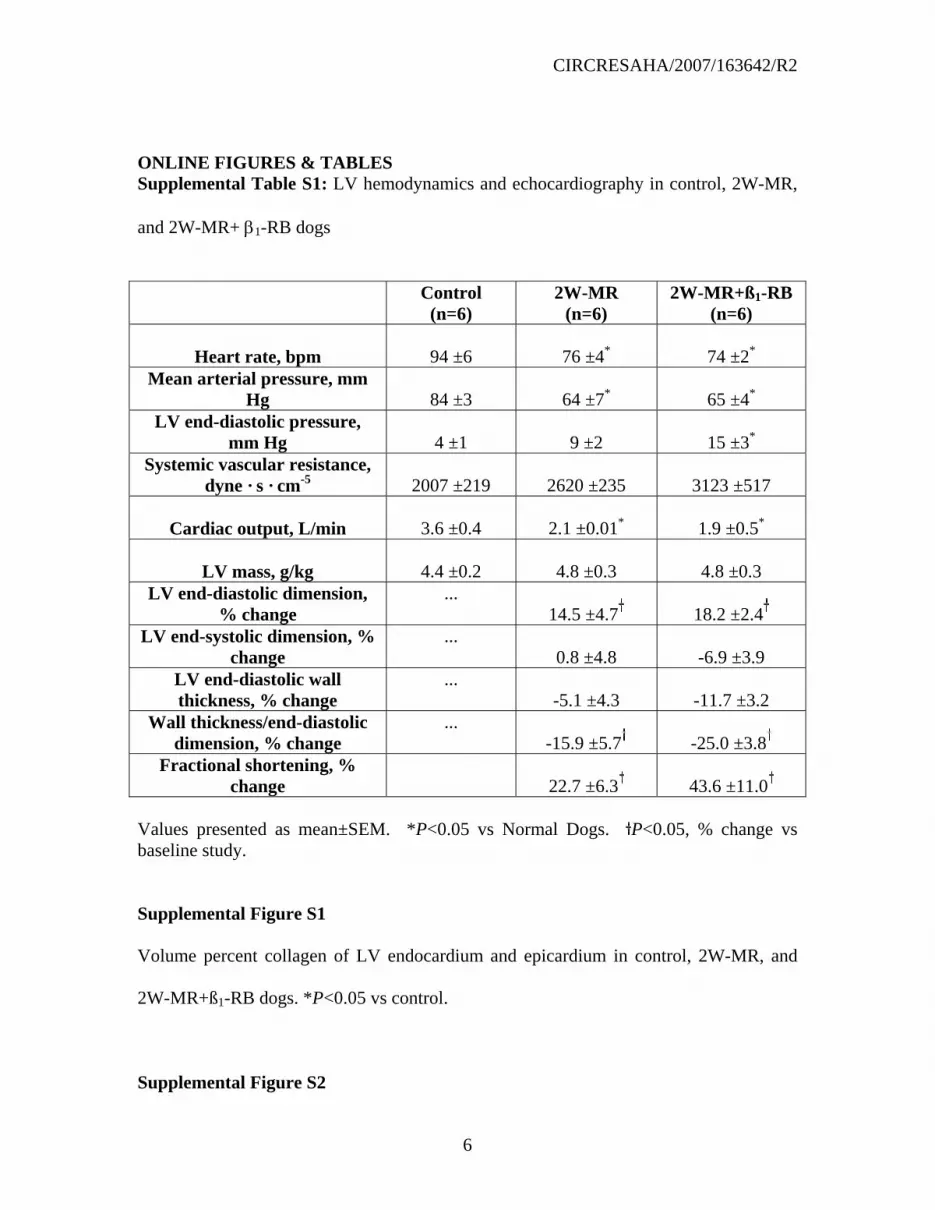
LV remodeling and cardiac function in 2W-MR and 2W-MR+β1-RB animals.
Hemodynamics.
After 2 weeks of MR, mean arterial pressure, heart rate, and cardiac output were
significantly decreased compared with controls, whereas LV end-diastolic pressure and
mean systemic vascular resistance did not significantly differ from controls in the
untreated 2W-MR dogs. There was a trend toward increases in LV mass in 2W-MR and
2W-MR+β1-RB dogs but this did not achieve statistical significance. β1-RB had no
significant effect on cardiac output, heart rate, LV end-diastolic pressure, and mean
systemic vascular resistance compared to untreated 2W-MR (Supplemental Table S1).
However, there was a significant increase in LV end-diastolic pressure in 2W-MR+β1-RB
dogs compared to normal animals.
Echo LV remodeling and function

CIRCRESAHA/2007/163642/R2
5
Compared with baseline both 2W-MR and 2W-MR+β1-RB groups had a similar increase
in LV end-diastolic dimension compared to baseline (Supplemental Table S1). In
addition, cardiac function assessed by LV fractional shortening was significantly
increased from baseline in 2W-MR group, as LV end-systolic dimension remained
unchanged (Supplemental Table S1). However, this increase was not affected in 2W-
MR+β1-RB group. Eccentric LV remodeling, as measured by LV wall thickness/end-
diastolic dimension was decreased similarly in 2W-MR and 2W-MR+β1-RB group.
REFERENCES
1. Pacioretty LM, Gilmour RF, Jr. Restoration of transient outward current by
norepinephrine in cultured canine cardiac myocytes. Am J Physiol Heart Circ
Physiol. 1998;275:H1599-1605.
2. Rafiq K, Kolpakov MA, Abdelfettah M, Streblow DN, Hassid A, Dell'Italia LJ,
Sabri A. Role of Protein-tyrosine phosphatase SHP2 in focal adhesion kinase
down-regulation during neutrophil cathepsin G-induced cardiomyocytes anoikis.
J Biol Chem. 2006;281:19781-19792.
3. Sabri A, Alcott SG, Elouardighi H, Pak E, Derian C, Andrade-Gordon P, Kinnally
K, Steinberg SF. Neutrophil cathepsin G promotes detachment-induced
cardiomyocyte apoptosis via a protease-activated receptor-independent
mechanism. J Biol Chem. 2003;278:23944-54.
CIRCRESAHA/2007/163642/R2
6
ONLINE FIGURES & TABLES Supplemental Table S1: LV hemodynamics and echocardiography in control, 2W-MR,
and 2W-MR+ β1-RB dogs
Control
(n=6) 2W-MR
(n=6) 2W-MR+ß1-RB
(n=6)
Heart rate, bpm
94 ±6
76 ±4*
74 ±2* Mean arterial pressure, mm
Hg
84 ±3
64 ±7*
65 ±4* LV end-diastolic pressure,
mm Hg
4 ±1
9 ±2
15 ±3* Systemic vascular resistance,
dyne · s · cm-5
2007 ±219
2620 ±235
3123 ±517
Cardiac output, L/min
3.6 ±0.4
2.1 ±0.01*
1.9 ±0.5*
LV mass, g/kg
4.4 ±0.2
4.8 ±0.3
4.8 ±0.3 LV end-diastolic dimension,
% change ...
14.5 ±4.7
18.2 ±2.4 LV end-systolic dimension, %
change ...
0.8 ±4.8
-6.9 ±3.9 LV end-diastolic wall thickness, % change
... -5.1 ±4.3
-11.7 ±3.2
Wall thickness/end-diastolic dimension, % change
... -15.9 ±5.7
-25.0 ±3.8
Fractional shortening, % change
22.7 ±6.3
43.6 ±11.0
Values presented as mean±SEM. *P<0.05 vs Normal Dogs. P<0.05, % change vs baseline study.
Supplemental Figure S1
Volume percent collagen of LV endocardium and epicardium in control, 2W-MR, and
2W-MR+ß1-RB dogs. *P<0.05 vs control.
Supplemental Figure S2

CIRCRESAHA/2007/163642/R2
7
Left ventricular extracts from control, 2W-MR, and 2W-MR+β1-RB dogs were
immunoprecipitated (IP) with anti-FAK antibodies and immunoblotted with anti-
phosphotyrosine antibodies. Top, representative autoradiogram (with each lane from a
single gel exposed for the same duration). Bottom, fold induction, n=6 each group,
*P<0.05 2W-MR vs. control.
Supplemental Figure S3
LV sections from control (A, C, E) and MR (B, D, F) dogs double stained with anti-FAK
(green) and sarcomeric α-actin (red) antibodies. Nuclei were visualized with DAPI
staining (Blue). FAK labeling was localized around cardiomyocytes with some staining
of the interstitial space. No qualitative change in FAK distribution was observed between
control and MR LVs. Bar represents 50 µm.
Supplemental Figure S4
Top, representative immunoblot showing accumulation of phospho-Pyk2 Tyr-402 in LV
extracts from control, 2W-MR, and 2W-MR+β1-RB. Blot was stripped and blotted with
anti-Pyk2 antibodies. Bottom, fold induction, n=6 each group.
Supplemental Figure S5
Top, representative immunoblots showing accumulation of phospho-ERK1/2 in LV
extracts from control, 2W-MR, and 2W-MR+β1-RB. Blots were stripped and blotted with
anti-ERK1/2 antibodies. Bottom, fold induction, n=6 each group, *P<0.05 2W-MR vs.
control.

Vol
ume
% C
olla
gen
0
1
2
3
4
Control 2 W-MR 2W-MR+ β1-AR
**
LV endocardiumLV epicardium
Supplemental Figure S1

P-Tyr
FAK
IP: FAKCtrl 2W-MR
2W-MR+ β1-RB
Supplemental Figure S2
FAK
pho
spho
ryla
tion
(fold
incr
ease
ove
r con
trol)
Ctrl 2W-MR 2W-MR+ β1-RB
0.2
0.4
0.6
0.8
1.0
1.2
**

Sham 4W-MR
FAK
α-Actin
FAK/α-Actin
Supplemental Figure S3
0.0
0.4
0.8
1.2
Pyk
2 ph
osph
oryl
atio
n(fo
ld in
crea
se o
ver c
ontro
l)p-Pyk2
Pyk2
Ctrl 2W-MR2W-MR+ β1-RB
Ctrl 2W-MR 2W-MR+ β1-RB
Supplemental Figure S4

Supplemental Figure S5
ER
K2
phos
phor
ylat
ion
(fold
incr
ease
ove
r con
trol)
Ctrl 2W-MR 2W-MR+ β1-RB
0
1
2
3
Ctrl 2W-MR 2W-MR+ β1-RB
p-ERK1p-ERK2
ERK1ERK2
*
*

Dell'italiaAbdelkarim Sabri, Khadija Rafiq, Rachid Seqqat, Mikhail A. Kolpakov, Ray Dillon and Louis J.Compensated Volume Overload Attributable to Isolated Mitral Regurgitation in the Dog
Sympathetic Activation Causes Focal Adhesion Signaling Alteration in Early
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2008 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.107.1636422008;102:1127-1136; originally published online March 20, 2008;Circ Res.
http://circres.ahajournals.org/content/102/9/1127World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org/content/suppl/2008/03/20/CIRCRESAHA.107.163642.DC1.htmlData Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on January 20, 2015http://circres.ahajournals.org/Downloaded from