dipartimento di biotecnologie cellulari ed ematologia...
TRANSCRIPT

1
Dipartimento di Biotecnologie Cellulari ed Ematologia
Sezione di Ematologia
DOTTORATO DI RICERCA IN SCIENZE EMATOLOGICHE XXVI CICLO
Coordinatore Prof. Robin Foà
TESI DI DOTTORATO
La Malattia Minima Residua nelle Sindromi Linfoproliferative Acute e Croniche.
Relatore Dottoranda Prof.ssa Anna Guarini Dott.ssa Irene Della Starza
Anno Accademico 2012-2013

2
INDICE
Introduzione................................................................................................pag.5
I°Progetto: Whole-genome amplification (WGA) per la valutazione di target molecolari ed il monitoraggio della malattia minima residua nella leucemia acuta linfoide.
La Leucemia Acuta Linfoide....................................................................................pag.10
La ricombinazione genica.......................................................................................pag.20
I riarrangiamenti genici del B Cell Receptor............................................................pag.25
I riarrangiamenti genici del T Cell Receptor............................................................pag.31
Obiettivo dello studio............................................................................................pag.38
Materiali e Metodi..................................................................................................pag.39
Pazienti e campioni studiati.....................................................................................pag.39
Estrazione del DNA.................................................................................................pag.39
Whole Genome Amplification..................................................................................pag.39
Screening dei riarrangiamenti genici Ig/TCR..........................................................pag.40
Sequenziamento e analisi del gene........................................................................pag.42
Disegno dei primers ed analisi RQ-PCR.................................................................pag.42
Risultati..................................................................................................................pag.44
Riarrangiamenti genici Ig/TCR identificati al momento della diagnosi: DNA genomico
vs DNA amplificato..................................................................................................pag.44
Confronto della quantificazione della malattia e risultati di MMR tra DNA genomico
e amplificato............................................................................................................pag.46
Conclusioni...........................................................................................................pag.48
Bibliografia............................................................................................................pag.51

3
II°Progetto: Confronto di due strategie di RQ-PCR per la valutazione della malattia minima residua nelle malattie linfoproliferative: correlazione tra lo stato mutazionale dei geni delle immunoglobuline e performance di RQ-PCR.
La Leucemia Linfatica Cronica…............................................................................pag.63
Stato mutazionale dei geni delle catene pesanti delle immunoglobuline.............pag.71
Obiettivo dello studio ….......................................................................................pag.75 Materiali e Metodi..................................................................................................pag.76
Pazienti e campioni studiati....................................................................................pag.76
Estrazione del DNA e analisi IgH............................................................................pag.76
Sequenziamento e analisi del gene........................................................................pag.77
Disegno di primer e sonde per l'analisi RQ-PCR...................................................pag.78
Risultati..................................................................................................................pag.80
Riarrangiamenti del gene IGH e analisi di mutazione.............................................pag.80
Confronto tra diverse strategie di disegno di set di primers/probe per la valutazione
RQ-PCR..................................................................................................................pag.82
Correlazione tra il carico mutazionale IGH e le performance di RQ-PCR..............pag.84
Conclusioni…........................................................................................................pag.86 Bibliografia............................................................................................................pag.89

4
III°Progetto: La malattia minima residua condiziona la prognosi dei pazienti affetti da linfoma follicolare: risultati del trial FIL FOLL05.
Il Linfoma Follicolare.............................................................................................pag. 97
Il gene BCL-2 nel linfoma follicolare.....................................................................pag.104
Obiettivo dello studio.........................................................................................pag.108 Materiali e Metodi...............................................................................................pag.109
Pazienti e campioni studiati................................................................................. pag.109
Screening del riarrangiamento BCL2/IGH@........................................................pag.109
Sequenziamento e analisi del gene.....................................................................pag.110
Analisi RQ-PCR....................................................................................................pag.111
Risultati...............................................................................................................pag.112
Analisi qualitativa e burden tumorale dei pazienti all'arruolamento....................pag.112
Valutazione della MMR ai diversi time points.......................................................pag.115
Conclusioni….....................................................................................................pag.118 Bibliografia.........................................................................................................pag.121
Pubblicazioni......................................................................................................pag.131
Abstracts.............................................................................................................pag.133

5
Introduzione
La Malattia Minima Residua (MMR)
La genetica delle malattie neoplastiche rappresenta uno dei campi che ha visto il
maggiore sviluppo negli ultimi anni, offrendo grandi speranze sul fronte di una completa
comprensione della genesi di queste patologie ed aprendo nuove possibilità
diagnostiche e terapeutiche. Ogni neoplasia origina necessariamente da una o più
alterazioni del DNA e dalla trasformazione del corredo genetico della cellula colpita.
Comprendere la natura e le cause di queste trasformazioni permette di identificare il
tipo di patologia cui ci troviamo di fronte e di studiare la possibilità di intervenire
direttamente sull'origine della malattia, con trattamenti specifici.
Le maggiori conoscenze cliniche e biologiche in campo oncoematologico hanno
permesso lo sviluppo di nuovi farmaci e di strategie terapeutiche potenzialmente
sempre più efficaci, tuttavia ancora un significativo numero di pazienti ricade a causa
della persistenza di cellule neoplastiche residue, indicate con il termine di “malattia
minima residua” (MMR).
L' uso dello studio della malattia minima residua come marker di risposta molecolare al
trattamento, può migliorare la valutazione della risposta clinica, guidare la selezione
delle strategie terapeutiche e, possibilmente, indicare l'esito clinico a lungo termine
(Cazzaniga G et al, Haematologica 2005) in un certo numero di patologie ematologiche.
Lo studio della MMR viene oggi eseguito nell’ambito delle leucemie acute linfoblastiche
(LAL) e nelle malattie linfoproliferative croniche, sulla base di protocolli che ne regolano
il monitoraggio.
Nelle LAL la MMR è fondamentale per guidare e modificare la gestione terapeutica dei
pazienti (Bruggemann M et al, Blood 2006). La riduzione del carico tumorale durante e
dopo il trattamento di induzione fornisce informazioni cruciali sulla risposta alla terapia e
rischio di recidiva. La negatività della MMR ai diversi tempi del trattamento è risultata
significativamente associata ad una bassa incidenza di recidive (3-15% a tre anni), ed
al contrario un incremento di 5-10 volte degli eventi (39-86% a tre anni) si è osservato
nei casi di MMR positiva (Cazzaniga G et al, Clin. Exp. Heamatol. 2003). Questo
permette di identificare pazienti "a basso rischio" e "ad alto rischio", che possono trarre
profitto dalla riduzione o dalla intensificazione della terapia, rispettivamente (Vidriales

6
MB et al, Blood 2003; Flohor T et al, LeuKemia 2008; Bassan R et al, Blood 2009).
Il dato della MMR nella LAL del bambino e dell'adulto è risultato essere un fattore
prognostico indipendente da altri parametri clinici e biologici caratterizzanti la malattia
all’esordio. L’analisi della malattia minima a tempi più distanti dalla diagnosi è risultata
ancora più significativa nell’identificare i pazienti a rischio di recidiva, fornendo le basi
per un suo utilizzo nei protocolli clinici.
L'ampia introduzione di nuove strategie terapeutiche efficaci ha permesso che un
numero crescente di pazienti con neoplasie ematologiche potesse essere indagato per
la valutazione della MMR.
Nella leucemia linfatica cronica (LLC) diversi studi hanno dimostrato che i criteri di
risposta morfologici non sono sufficientemente sensibili per predire il risultato e che i
pazienti che abbiamo conseguito una eradicazione rilevabile della MMR, presentano
una sopravvivenza prolungata (Moreno C et al, Blood 2006; Böttcher S et al, J Clin
Oncol 2012).
Nonostante questo, il ruolo clinico della MMR nella LLC resta da chiarire.
Nel Linfoma follicolare (LF) il monitoraggio della MMR è risultato essere un fattore
predittivo ben consolidato dell'andamento clinico della malattia post-trapianto (Ladetto
M et al, Blood 2008), al contrario il suo ruolo dopo terapia convenzionale è ancora in
discussione, sebbene molti studi sono stati in grado di dimostrarne il valore prognostico
(Corradini P et al, J Clin Oncol 2004; Rown JR et al, Biol Blood Marrow Transplant
2007; Procházka V et al, J Clin Oncol, 2011; Ladetto M et al, Blood 2013).
La biologia variabile dei tumori maligni delle cellule B influenza non solo
l'interpretazione dei dati di MMR per le decisioni cliniche, ma ha anche implicazioni per
gli aspetti tecnici in campo diagnostico.
Solo l’impiego di tecniche dotate di adeguata sensibilità, specificità nel riconoscimento
delle cellule patologiche, di stabilità dei marcatori identificati e di riproducibilità, può
permettere di distinguere tra loro i pazienti in remissione sulla base dei diversi livelli di
MMR permettendo così, una più precisa definizione di “stato di remissione”.
Attualmente, la tipizzazione immunofenotipica tramite citofluorimetria insieme allo studio
molecolare effettuato utilizzando la Polymerase Chain Reaction (PCR) sono le due
tecniche che possiedono i requisiti necessari allo studio della MMR dimostrando una
sensibilità di almeno 10-4 (ovvero capacità di rilevare una cellula neoplastica ogni 104
cellule normali), alta specificità, riproducibilità, espressione quantitativa del risultato ed
applicabilità.

7
L’introduzione nella diagnostica molecolare della PCR quantitativa (RQ-PCR) ha
ampliato le conoscenze sul significato della MMR, grazie alla possibilità di individuare e
quantizzare sequenze specifiche di DNA e DNA complementare (cDNA), attraverso
l'utilizzo di sonde fluoromarcate, la cui frammentazione durante la fase di allungamento
della reazione di amplificazione, permette emissione di fluorescenza che viene captata
e quantizzata, portando ad un'elevata riproducibilità e specificità dei risultati.
Le cellule leucemiche possono essere distinte dalle normali cellule ematopoietiche sulla
base di specifici patterns di espressione antigenica evidenziabili tramite analisi
immunofenotipica, analizzando la presenza di aberrazioni cromosomiche che risultano
in trascritti di un gene di fusione o nell’espressione aberrante di trascritti, e ancora
valutando il riarrangiamento dei geni delle IGH e del TCR nelle regioni giunzionali
paziente-specifiche, considerate markers tumorali specifici, simili ad impronte digitali
che differiscono in lunghezza e composizione per clone linfocitario e conseguentemente
per ogni paziente affetto da neoplasia linfoide.
L'analisi molecolare quantitativa della MMR (RQ-PCR) attraverso l'utilizzo di sonde
allele-specifiche (ASO), è in grado di raggiungere limiti di sensibilità riproducibili di 1x10-
5, tuttavia l'applicabilità del metodo è limitata ai pazienti con riarrangiamenti genici
IG/TCR aventi regioni giunzionali adatte a raggiungere una sensibilità sufficiente.
Questo tipo di valutazione della MMR presenta però un certo numero di limitazioni, tra
cui il fallimento nell'identificare il marcatore molecolare nei casi somaticamente
ipermutati, dove il basso livello di infiltrazione tumorale non consente un approccio
quantitativo adeguato.
Inoltre, l'evoluzione di diversi subcloni leucemici durante il trattamento, nonché la
presenza di oligoclonalità alla diagnosi che non viene detectata molecolarmente dagli
ASO, può anche essere un motivo per potenziali risultati falsi negativi.
Pertanto, si è resa necessaria l'introduzione di tecniche molecolari alternative, che negli
ultimi tempi si stanno sempre più diffondendo in campo oncoematologico..
Il metodo di sequenziamento di seconda generazione, denominato “next-generation
sequencing (NGS)” permette l'identificazione delle cellule B o T clonogeniche con alta
sensibilità e specificità ed è risultato adatto per il rilevamento di MMR, come
recentemente dimostrato in pazienti con LLC (Logan AC et al, Leukemia 2013).
La valutazione della MMR mediante NGS può non solo superare alcuni svantaggi dei
metodi basati sulla PCR, come la necessità di primers specifici del paziente, ma esso
ha il potenziale per raggiungere un livello di sensibilità superiore (fino a 1x10-6) con un

8
migliore range di quantificabilità della malattia.
Inoltre, l'approccio NGS permette di analizzare la diversità genetica e l'eterogeneità
clonogenica che possono contribuire alla nostra attuale comprensione della biologia
della malattia e della cinetica di ricaduta (Gawad C et al, Blood 2012).
Nonostante questi vantaggi, va osservato che entrambi i metodi (NGS e ASO-RQ-PCR)
richiedono un campione contenente un'infiltrazione tumorale significativa per
l'identificazione del clone alla diagnosi ed entrambi risentono dell'evoluzione clonale.
Pertanto è ragionevole ipotizzare che l'uso di un approccio NGS darà un valore
aggiunto alle attuali tecniche di valutazione della MMR consentendo l'identificazione di
un marcatore molecolare nella stragrande maggioranza delle neoplasie linfoidi.
In conclusione, grazie all'integrazione di tecniche multidisciplinari ci si sta sempre più
avvicinando ad un sistema in grado di tradurre rapidamente le informazioni necessarie
per una diagnosi efficace ed una più precisa valutazione della risposta al trattamento,
con il fine di elaborare terapie mirate adatte al singolo paziente e di predire la ricaduta
prima della manifestazione clinica.

9
I° Progetto: Whole-genome amplification (WGA) per la valutazione di target molecolari ed il monitoraggio della malattia minima residua nella leucemia acuta linfoide.

10
La Leucemia Acuta Linfoide La leucemia acuta linfoide rappresenta un gruppo clinicamente e biologicamente
eterogeneo di malattie che originano dai precursori linfoidi, caratterizzate da morfologia
indifferenziata. La trasformazione leucemica genera una progenie di blasti linfoidi
leucemici che hanno subito un blocco maturativo in una fase precoce del processo di
differenziazione. Le basi fisiopatologiche dei sintomi e segni delle LAL consistono in
una soppressione della normale emopoiesi, nell'infiltrazione e colonizzazione degli
organi linfoidi e non e nella liberazione di linfochine e mediatori dell'infiammazione sia
delle cellule leucemiche che delle cellule normali, con conseguente anemia
(emoglobina ridotta nel 75% dei casi), aumento del numero dei leucociti (da 10.000 ad
oltre 100.000/mm3 nel 66% dei casi), e piastrinopenia (numero delle piastrine ridotto
nell’ 80% dei casi).
La caratteristica principale delle LAL è la presenza in circolo di cellule blastiche
leucemiche (linfoblasti) in percentuale variabile da meno del 10% ad oltre il 90%.
L’accumulo extramidollare di linfoblasti può risultare in diversi siti e specialmente a
livello di linfonodi, fegato, sistema nervoso centrale, testicoli ed ossa (Hoelzer D et al,
Hematology 2002).
La LAL è la più frequente sindrome neoplastica nei bambini, con una prevalenza
nell’età di 3-4 anni, mentre rappresenta il 20% delle leucemie dell’adulto, con una
prognosi migliore nei bambini rispetto agli adulti.
L'eterogeneità delle LAL ha reso necessaria la messa a punto di criteri classificativi che
permettessero di identificare gruppi di pazienti con caratteristiche e prognosi differenti.
I marcatori clinici che definiscono la prognosi dei pazienti affetti da LAL sono
rappresentati dall'età, dal numero di globuli bianchi, dalla risposta al trattamento, dalla
presenza di marcatori citogenetici e molecolari, dalla farmaco resistenza e dalla
presenza di MMR dopo terapia.
La diagnosi e la conseguente classificazione della malattia è resa possibile mediante la
valutazione di una combinazione di fattori morfologici, citochimici, immunologici,
citogenetici e molecolari.
L’esame morfologico degli strisci di sangue venoso periferico e del midollo osseo, in
relazione a criteri definiti dal sistema di classificazione FAB (French-American-British)
consente di riconoscere 3 sottotipi distinti di LAL noti come L1, L2, L3. La forma
predominante è la L2 con un’incidenza pari al 65-70%, rispetto alle forme L1 e L3 la cui

11
incidenza è rispettivamente del 25-30% e del 5-10%. La forma L1 è prevalente nei
bambini, mentre la forma L2 nell’adulto (Guglielmi C et al, Leukemia 1997).
La forma L3 interessa le cellule B mature ed è per questo che viene considerata non
una leucemia linfoblastica in senso stretto, ma la leucemizzazione di un particolare tipo
di linfoma non-Hodgkin detto linfoma di Burkitt.
Nell’ambito dell’analisi citochimica per la valutazione della LAL viene usato un pannello
costituito da diverse reazioni. Tutte le LAL sono negative alla mieloperossidasi (MPO),
tranne una piccola percentuale di casi (3-5%) che risultano essere MPO+ ed in cui è
stata riscontrata un’alta incidenza di ricadute. Il 95% di LAL L1 e L2 sono positive alla
“deossinucleotidil transferasi terminale” (TdT) e la sua presenza è utile nella diagnosi
differenziale con i casi di linfocitosi reattiva. Più dell’80% di blasti in casi di LAL T
mostrano una forte positività alla fosfatasi acida e all’α-naftil-acetato esterasi (ANAE).
L’analisi immunofenotipica permette non solo di confermare la diagnosi di LAL, ma
anche di classificarne le diverse forme e di monitorarne la malattia minima residua
durante e dopo la terapia (Campana D et al, Blood 1995).
I blasti cellulari dei pazienti affetti da LAL sono caratterizzati dall’espressione di antigeni
di superficie ed intrascitoplasmatici che corrispondono a marcatori di maturazione
fisiologica dei progenitori cellulari linfoidi, per cui le LAL vengono distinte
fenotipicamente in B o T, in base all’appartenenza alla linea cellulare ed al loro stadio di
maturazione.
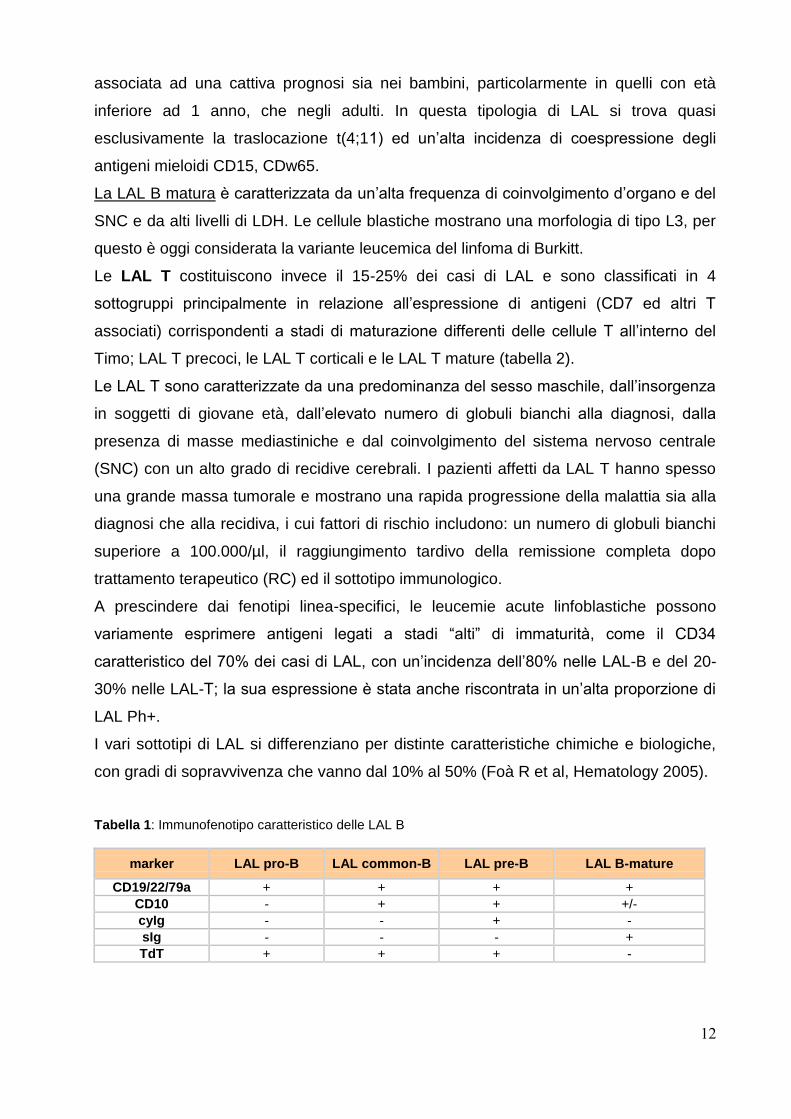
Le LAL B rappresentano circa l’80-85% dei casi di LAL e possono essere classificate in
base all’espressione di antigeni caratteristici della linea B (CD79a intracitoplasmatico e
CD19 di superficie) del CD10 e delle immunoglobuline citoplasmatiche (cyIg) e di
membrana (sIg) in 4 sottogruppi, LAL pro-B, LAL B common, LAL pre-B e LAL B mature
(tabella 1).
La LAL B common è il più frequente sottotipo di LAL B dell’adulto (60%), la
sopravvivenza è del 30-35% e può essere paragonata alla LAL pre-B, poiché non
esistono differenze rilevanti. Rispetto ai bambini, gli adulti mostrano una più bassa
incidenza di fattori prognostici favorevoli (iperdiploidia, presenza del trascritto TEL/AML-
1) ed una predominanza di fattori sfavorevoli (BCR-ABL+). Probabilmente negli adulti vi
è una più alta resistenza alla terapia ed una sfavorevole farmacocinetica dei
chemioterapici.
La LAL pro-B, nota come LAL CALLA negativa o LAL pre pre B (CD10-, CD19+, cyIgM-
, sIgM-, CD24+/-) è riscontrata nell’11% degli adulti e nel 25% dei bambini, ed è
12
associata ad una cattiva prognosi sia nei bambini, particolarmente in quelli con età
inferiore ad 1 anno, che negli adulti. In questa tipologia di LAL si trova quasi
esclusivamente la traslocazione t(4;11) ed un’alta incidenza di coespressione degli
antigeni mieloidi CD15, CDw65.
La LAL B matura è caratterizzata da un’alta frequenza di coinvolgimento d’organo e del
SNC e da alti livelli di LDH. Le cellule blastiche mostrano una morfologia di tipo L3, per
questo è oggi considerata la variante leucemica del linfoma di Burkitt.
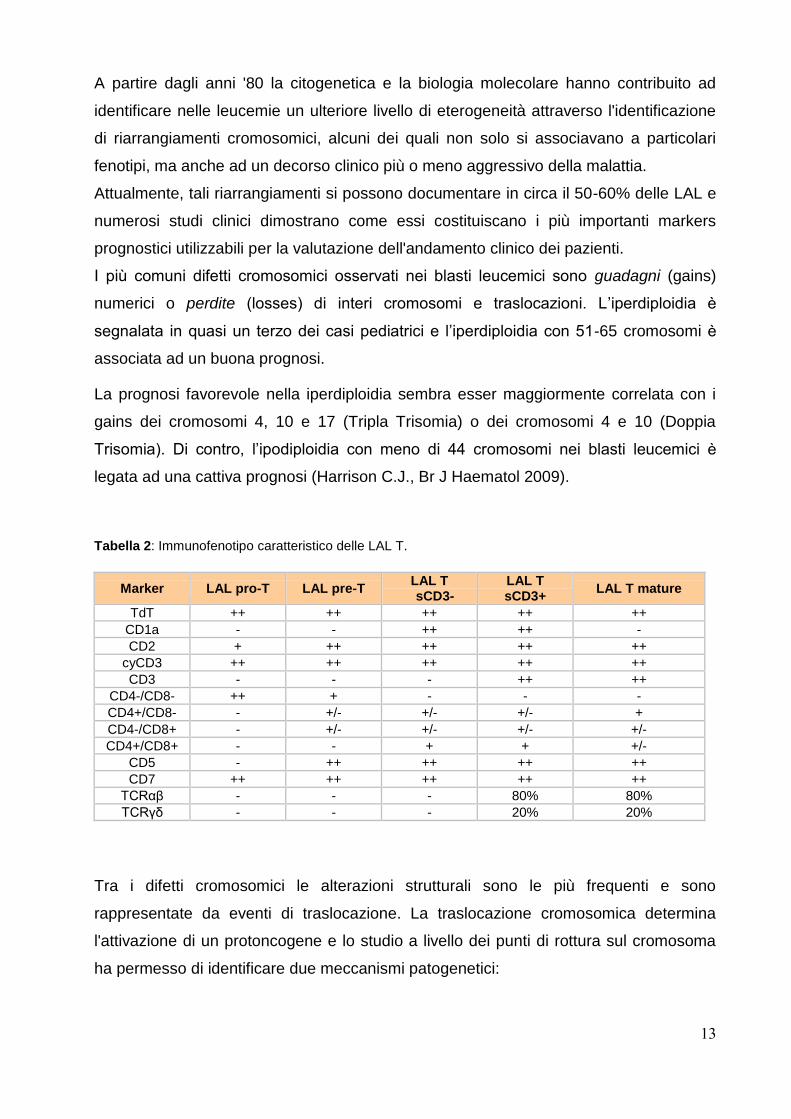
Le LAL T costituiscono invece il 15-25% dei casi di LAL e sono classificati in 4
sottogruppi principalmente in relazione all’espressione di antigeni (CD7 ed altri T
associati) corrispondenti a stadi di maturazione differenti delle cellule T all’interno del
Timo; LAL T precoci, le LAL T corticali e le LAL T mature (tabella 2).
Le LAL T sono caratterizzate da una predominanza del sesso maschile, dall’insorgenza
in soggetti di giovane età, dall’elevato numero di globuli bianchi alla diagnosi, dalla
presenza di masse mediastiniche e dal coinvolgimento del sistema nervoso centrale
(SNC) con un alto grado di recidive cerebrali. I pazienti affetti da LAL T hanno spesso
una grande massa tumorale e mostrano una rapida progressione della malattia sia alla
diagnosi che alla recidiva, i cui fattori di rischio includono: un numero di globuli bianchi
superiore a 100.000/µl, il raggiungimento tardivo della remissione completa dopo
trattamento terapeutico (RC) ed il sottotipo immunologico.
A prescindere dai fenotipi linea-specifici, le leucemie acute linfoblastiche possono
variamente esprimere antigeni legati a stadi “alti” di immaturità, come il CD34
caratteristico del 70% dei casi di LAL, con un’incidenza dell’80% nelle LAL-B e del 20-
30% nelle LAL-T; la sua espressione è stata anche riscontrata in un’alta proporzione di
LAL Ph+.
I vari sottotipi di LAL si differenziano per distinte caratteristiche chimiche e biologiche,
con gradi di sopravvivenza che vanno dal 10% al 50% (Foà R et al, Hematology 2005).
Tabella 1: Immunofenotipo caratteristico delle LAL B
marker LAL pro-B LAL common-B LAL pre-B LAL B-mature
CD19/22/79a + + + +
CD10 - + + +/-
cyIg - - + -
sIg - - - +
TdT + + + -
13
A partire dagli anni '80 la citogenetica e la biologia molecolare hanno contribuito ad
identificare nelle leucemie un ulteriore livello di eterogeneità attraverso l'identificazione
di riarrangiamenti cromosomici, alcuni dei quali non solo si associavano a particolari
fenotipi, ma anche ad un decorso clinico più o meno aggressivo della malattia.
Attualmente, tali riarrangiamenti si possono documentare in circa il 50-60% delle LAL e
numerosi studi clinici dimostrano come essi costituiscano i più importanti markers
prognostici utilizzabili per la valutazione dell'andamento clinico dei pazienti.
I più comuni difetti cromosomici osservati nei blasti leucemici sono guadagni (gains)
numerici o perdite (losses) di interi cromosomi e traslocazioni. L’iperdiploidia è
segnalata in quasi un terzo dei casi pediatrici e l’iperdiploidia con 51-65 cromosomi è
associata ad un buona prognosi.
La prognosi favorevole nella iperdiploidia sembra esser maggiormente correlata con i
gains dei cromosomi 4, 10 e 17 (Tripla Trisomia) o dei cromosomi 4 e 10 (Doppia
Trisomia). Di contro, l’ipodiploidia con meno di 44 cromosomi nei blasti leucemici è
legata ad una cattiva prognosi (Harrison C.J., Br J Haematol 2009).
Tabella 2: Immunofenotipo caratteristico delle LAL T.
Marker LAL pro-T LAL pre-T LAL T
sCD3- LAL T sCD3+
LAL T mature
TdT ++ ++ ++ ++ ++
CD1a - - ++ ++ -
CD2 + ++ ++ ++ ++
cyCD3 ++ ++ ++ ++ ++
CD3 - - - ++ ++
CD4-/CD8- ++ + - - -
CD4+/CD8- - +/- +/- +/- +
CD4-/CD8+ - +/- +/- +/- +/-
CD4+/CD8+ - - + + +/-
CD5 - ++ ++ ++ ++
CD7 ++ ++ ++ ++ ++
TCRαβ - - - 80% 80%
TCRγδ - - - 20% 20%
Tra i difetti cromosomici le alterazioni strutturali sono le più frequenti e sono
rappresentate da eventi di traslocazione. La traslocazione cromosomica determina
l'attivazione di un protoncogene e lo studio a livello dei punti di rottura sul cromosoma
ha permesso di identificare due meccanismi patogenetici:

14
1. in seguito alla traslocazione il protoncogene è sottoposto al controllo di nuovi
enhancer che aumentano la velocità di trascrizione dei geni da esso regolati;
2. la traslocazione cromosomica può determinare la formazione di un mRNA di fusione
che porta alla sintesi di una proteina chimerica dotata di nuove proprietà.
La maggior parte di tali alterazioni ha un profondo effetto sui meccanismi di controllo dei
processi di proliferazione, differenziamento, maturazione e sopravvivenza dei
progenitori emopoietici midollari. Circa il 90% delle LAL in età pediatrica rivela anomalie
cromosomiche clonali, il 50% di queste sono rappresentate da traslocazioni (Harrison
CJ et al, Rev Clin Exp Hematol 2002).
Le principali anomalie cromosomiche delle LAL sono: BCR/ABL t(9;22)(q34;q11),
ALL1/AF4 t(4;11)(q21;q23), E2A/PBX1 t(1;19)(q23;p13), TEL/AML1 t(12;21)(p13;q22),
c-MYC/IgH t(8;14)(q24;q32), SIL/TAL1 t(1;14)(p32;q11) (tabella 3).
Tutte le leucemie con anomalie del cariotipo hanno una prognosi peggiore, in
particolare quelle con la t(4;11) e con la t(9;22).
L’incidenza delle traslocazioni è diversa tra adulti e bambini. La t(9;22)(q34;q11) è la
traslocazione più comune nelle LAL B common e nelle LAL pre-B, con un’incidenza del
25-30% nelle leucemie acute linfoidi dell’adulto e del 3-5% in quelle pediatriche. La
conseguenza molecolare di questa traslocazione è la formazione di un gene ibrido
BCR/ABL codificante per una proteina oncogenica di fusione con attività tirosin-chinasica
costitutiva, in grado di attivare molteplici vie di trasduzione del segnale che influenzano
la crescita, la sopravvivenza e le proprietà adesive dei progenitori linfoidi. Il gene c-abl,
presente sul cromosoma 9, contiene 11 esoni e codifica per una proteina di 145 Kd
(p145) appartenente alla famiglia delle tirosin-chinasi, enzimi in grado di catalizzare il
trasferimento di un gruppo fosfato dall’adenosin trifosfato (ATP) al residuo di serina o
tirosina di proteine substrato. Il gene BCR, presente sul cromosoma 22, codifica per una
proteina di 160 Kd (p160) associata ad attività serin-treonin-chinasica. L’acquisizione
della capacità trasformante da parte di c-abl può dipendere dal fatto che la sequenza del
I° esone BCR aumenta l’attivazione della tirosin-chinasi quando viene fusa al II° esone di
ABL (Cimino G et al, Haematologica 2006).
Il punto di rottura sul cromosoma 22 avviene all’interno di un’area detta zona bcr
(breakpoint cluster region) comprendente 4 esoni chiamati b1, b2, b3 e b4, mentre il
punto di rottura sul cromosoma 9 avviene in un’area molto vasta che si trova al 5’ del II°
esone di ABL. La maggior parte dei punti di rottura del bcr cadono in una zona detta
“major breakpoint cluster region” (M-bcr) ed interessa gli esoni b2 e b3 con produzione

15
di una proteina di 210 Kd (p210), presente nel 50% dei casi di LAL Ph+. Nella restante
metà dei casi, la rottura sul cromosoma 22 non cade all’interno della M-bcr, ma cade
più vicino al 5’ del gene BCR, in una regione localizzata nel I° introne e definita “minor
breakpoint cluster region” (m-bcr). Questo porta alla formazione di un gene ibrido più
corto che codifica per una proteina di 190 Kd (p190). Entrambe le proteine (p210 e
p190) sono dotate di attività autocatalitica, poiché si autofosforilano in tirosina, questo è
l’evento critico per il controllo della proliferazione, della progressione attraverso il ciclo
cellulare e dei processi di differenziazione cellulare. Alcuni studi attribuiscono alla p190
una maggiore attività trasformante dovuta ad una diversa configurazione spaziale tra le
regioni biochimicamente rilevanti di BCR e ABL.
Negli ultimi anni inoltre è stato individuato un nuovo punto di rottura che coinvolge gli
esoni 19 e 20 (originariamente denominati c3 e c4) del gene BCR. Tale punto è
localizzato distalmente al M-bcr ed è denominato µ-BCR. Il gene chimerico c3a2
derivante dalla giustapposizione dei geni e19 a2 BCRJABL, codifica per un proteina di
230 Kd (p230) che è stata riscontrata in alcuni pazienti affetti da Leucemia Mieloide
Cronica Neutrofila, una rara malattia mieloproliferativa caratterizzata dalla presenza di
un elevato numero di granulociti maturi.
Il trascritto TEL/AML1 t(12;21)(p13;q22), è presente in meno dell’1% dei pazienti adulti
e nel 20-30% dei casi pediatrici. La traslocazione determina la formazione di un
prodotto chimerico che coinvolge il gene AML1, codificante per una proteina che si lega
a specifiche sequenze del DNA regolando l’espressione di altri geni quali il gene della
mieloperossidasi, il gene del GM-CSF ed i geni della linea T, ed il gene TEL che
codifica per un fattore di trascrizione (Hübuer J et al, Leukemia 2004).
Il trascritto ALL1/AF4 t(4;11)(q21;q21;q23), è presente nel 5-7% sia degli adulti che dei
pediatrici, esclusi i bambini con età inferiore ad 1 anno, dove è riscontrato nel 60% dei
casi.
Il gene ALL1, localizzato nella regione 11q23 del cromosoma 11, è costituito da 36
esoni distribuiti su un segmento di 100 kb e codifica per una proteina con una massa
molecolare di 431 KDa. E’ un gene promiscuo che può fondersi con più di 30 partners
differenti e le sue mutazioni non sono associate con una linea leucemica definita. Nelle
traslocazioni dell’ 11q23 i punti di rottura sono localizzati in una regione di 8.5 Kb detta
“breakpoint cluster region (bcr)”, compresa tra gli esoni 5 ed 11 (Cimino G et al, Blood
2000). Tali traslocazioni portano alla formazione di prodotti costituiti dalla porzione N-
terminale di ALL1 e da sequenze codificanti poste sui rispettivi geni “partners”. Nelle

16
t(4;11) e t(11;19) il gene ALL1 si riarrangia con il gene AF4 ed il gene ENL, posti
rispettivamente sui cromosomi 4 e 19 e si ipotizza che il prodotto chimerico abbia la
funzione di fattore di trascrizione. Altri geni sono coinvolti in traslocazioni con ALL1:
AF1, AF6, AF10, AF9, AF17, AFX ELL.
La traslocazione t(1;19)(q23;p13), genera un fattore di trascrizione derivato dalla
fusione del gene E2A, localizzato nel punto di rottura sul cromosoma 19, codificante
fattori che favoriscono il legame delle Ig, e PBX1, un gene homeobox posto sul braccio
lungo del cromosoma 1 (Foà R et al, Br J Haematol 2003). Tale traslocazione si associa
spesso ad età pediatrica, è presente nel 25% delle LAL pre-B e nel 1% delle LAL early
pre-B; le forme associate con mutazioni di N-Ras ed inattivazione del gene TP53
presentano una prognosi più sfavorevole rispetto ai casi in cui non si osservano queste
alterazioni.
Una osservazione chiave è che più dei due terzi di casi pediatrici con LAL B presentano
alterazioni genetiche che modificano il normale processo di maturazione linfoide
(Mullighan C. Best Pract Res Clin Haematol 2011).
Tabella 3: Principali traslocazioni presenti nelle LAL.
Traslocazione Gene coinvolto Patologia
t(9;22)(q34;q11) BCR/ABL LAL common-B e pre-B
t(4;11)(q21;q23) ALL1/AF4 LAL pre-B
t(8;14)(q24;q32) MYC/IgH LAL B
t(12;21)(p13;q22) TEL/AML1 LAL B
t(1;19)(q23;p13) E2A/PBX1 LAL pre-B
t(10;14)(q24;q11) HOX11/TRD LAL T
t(1;14)(p32;q11) SIL/TAL1 LAL T
Lo sviluppo dei linfociti B dai precursori staminali midollari è regolato da numerosi fattori
di trascrizione che inducono il commitment del lineage linfocitario, la repressione dei
lineage alternativi, la maturazione linfocitaria. I geni targets sono PAX5 (paired box 5),
IKZF1 (gene codificante per il fattore di trascrizione linfoide IKAROS), EBF1 (early-B
factor 1) e LEF1 (lymphoid enhancer factor 1). Queste alterazioni genetiche si
presentano generalmente in eterozigosi ed includono delezioni focali o ampie,
mutazioni e traslocazioni. Quella più comune colpisce il gene PAX5 (circa il 30% dei
casi) anche sottoforma di traslocazioni con altri geni come TEL, senza però incidere
sulla prognosi. Le alterazioni di IKZF1 sono meno comuni, si presentano più

17
frequentemente come delezioni che mutazioni e sono associate a sottogruppi di
bambini con LAL ad alto rischio di ricaduta (Mullighan C. Best Pract Res Clin Haematol
2011). Queste alterazioni possono direttamente influenzare il trascrittoma della cellula
leucemica oppure possono essere associate ad alterazioni genetiche aggiuntive che
inducono l’attivazione di chinasi.
Nel 50% dei casi pediatrici, sono stati riscontrati dei riarrangiamenti che alterano
l’espressione del gene CRLF2 (cytokine receptor like factor 2), il quale è localizzato
nella regione pseudoautosomica 1 (PAR1) di Xp/Yp. Le alterazioni genetiche di questa
banda includono l’inserimento (juxtaposition) nel locus IgH (14q32), generando il
trascritto IgH@-CRLF2 o la delezione focale di PAR1 che sovrappone gli elementi
regolatori del gene del recettore purinergico P2RY8 a CRLF2, generando il trascritto
chimerico P2RY8-CRLF2. I mediatori a valle (downstream) del segnale di CRLF2 non
sono ancora ben caratterizzati ma probabilmente coinvolgono i geni della famiglia delle
Janus Kinase; infatti recentemente, numerosi studi, hanno dimostrato come fino al 50%
dei casi con CRLF2 riarrangiato presentano concomitanti mutazioni attivanti i geni JAK1
o JAK2 (Mullighan C. Best Pract Res Clin Haematol, 2011; Hertzberg L et al, Blood
2010) Queste forme sono potenzialmente sensibili ai farmaci inibitori di JAK,
attualmente in studi preclinici.
Nei pazienti affetti da LAL-T, le traslocazioni più ricorrenti interessano prevalentemente
le regioni regolatorie dei geni codificanti per il T Cell Receptor, che mappano sul
cromosoma 14 ed in un 30% di casi sono riscontrate alterazioni del gene TAL-1. In un
numero ristretto di casi il locus tal-1, posto sul braccio corto del cromosoma 1, (1p32) è
coinvolto in una traslocazione definita tal-t, con un locus TCR [t(1;14) o t(1;7)], mentre
nella maggior parte dei casi è presente una delezione (tal-d) che determina la rottura
della zona 5’ di tal-1 e giustappone quest’ultimo ad un altro gene, chiamato SIL,
localizzato sempre sul cromosoma 1p, ma in posizione più centromerica rispetto a tal-1
(Nirmala K et al, Leukemia Res 2002). In entrambi i casi tal-1 è sottoposto al controllo di
un nuovo promotore rappresentato da TCR o SIL. I pazienti che presentano tal-d o tal-t
non differiscono significativamente dagli altri casi di LAL T in cui non sono evidenti
alterazioni citogenetiche a livello del cromosoma 1p32.
Soltanto poche aberrazioni cromosomiche e molecolari mostrano una chiara
correlazione con la prognosi di LAL negli adulti. Queste includono la traslocazione
t(4;11) e la traslocazione t(9;22). Un’altra aberrazione con impatto prognostico

18
sfavorevole sembra essere l’ipoploidia, mentre la traslocazione t(10;14), il 12p ed
un’alta iperploidia sembrano rappresentare fattori prognostici favorevoli.
Le cellule leucemiche possono essere distinte dalla controparte normale inoltre,
attraverso l'analisi del riarrangiamento dei geni che codificano per le immunoglobuline o
il T Cell Receptor (Ig/TCR) (Vitale A et al, Cur Opinion in Oncol 2006).
Durante l’ontogenesi dei B e dei T linfociti, i geni Ig e TCR vengono assemblati
mediante un processo di riarrangiamento somatico. I segmenti genici separati
codificanti le regioni V, D, J vengono riuniti per formare un unico esone codificante la
regione variabile. Lo studio di questi riarrangiamenti è divenuto il metodo più sensibile
per valutare la clonalità di un’espansione linfoide. Il riarrangiamento dei geni che
codificano per la catena pesante delle Ig è stato riscontrato nel 90-95% di LAL B,
mentre il riarrangiamento dei geni che codificano per il TCR è stato riscontrato nel 95%
di LAL T e nel 50-70% di LAL B (Foroni L et al, Best Pract Res Clin Haematol 2002).
Poiché tali ricombinazioni sono di origine clonale, l’analisi della configurazione genica
delle Ig e del TCR può essere usata per valutare la persistenza di cloni maligni i cui
riarrangiamenti sono stati determinati al momento della diagnosi.
La resistenza farmacologica può essere un importante fattore nel fallimento della
terapia in caso di LAL. Nelle LAL del bambino è stata riscontrata una maggiore
resistenza in soggetti iperdiploidi con età superiore ai 10 anni, rispetto a pazienti più
giovani, mentre nelle LAL dell’adulto, soprattutto nei casi BCR/ABL+, è stata riscontrata
una resistenza ai farmaci cortisonici (prednisone) associata con un basso grado di
remissione completa (66%vs 84%)(Foà R et al, Rew Cli Exp Hemat 2002).
La presenza della MMR dopo terapia è un importante fattore prognostico che permette
di stimare il rischio di recidiva nei singoli pazienti. Nonostante i significativi progressi
ottenuti nella terapia delle LAL, soprattutto pediatriche, il 30% dei bambini e più della
metà degli adulti presenta recidiva della malattia. Nella maggior parte dei casi alla
diagnosi viene riscontrata la presenza di cloni multipli aventi alterazioni genetiche
distinte, le quali possono influenzare in maniera decisiva la risposta al trattamento e
quindi il rischio di recidiva. E' dimostrato che le due fasi della malattia (diagnosi e
recidiva) condividono origini comuni clonali ancestrali, ma mostrano differenze nella
natura delle alterazioni genetiche. In meno del 10% dei casi pediatrici alla recidiva viene
identificato un clone completamente differente da ciò che è stato visto alla diagnosi
(Mullighan C. Best Pract Res Clin Haematol 2011).
La recidiva è quindi espressione della persistenza di un clone leucemico “resistente”

19
alla terapia convenzionale e di conseguenza il monitoraggio della MMR può contribuire
alla comprensione della storia biologica della malattia stessa (Cazzaniga G et al, Rew
Clin Exp Hemat 2003). Non è ancora noto per quanto tempo le cellule leucemiche
persistano durante la fase di remissione della malattia. I criteri convenzionali per
stabilire la remissione in pazienti affetti da LAL sono basati sull’esame morfologico di
campioni di sangue midollare ed i pazienti vengono considerati in completa remissione
quando gli aspirati midollari contengono meno del 5% di blasti. I pazienti adulti con LAL
che non vanno in remissione entro 4-5 settimane dall’inizio della terapia hanno una
cattiva prognosi. Dato che la scomparsa dei blasti è un importante fattore predittivo di
sopravvivenza, la risposta alla terapia è oggi valutata precocemente, entro 2 settimane
per gli adulti ed entro 7 giorni per i bambini (Foà R et al, Rew Cli Exp Hemat 2002).
L’approccio molecolare allo studio della MMR in pazienti affetti da LAL ha assunto
un’importanza rilevante, data anche la crescente rapidità ed il continuo affinamento
delle tecniche di analisi. Tale strategia consente di dimostrare una remissione
molecolare precoce in risposta alla terapia, fondamentale per valutare la prognosi e la
sopravvivenza dei pazienti, potendoli stratificare in funzione del rischio relativo di
ricaduta.

20
La ricombinazione genica
I geni delle immunoglobuline sono presenti in configurazione germinale in tutte le cellule
di un organismo, ma solo i linfociti B esprimono questi geni in forma funzionalmente
riarrangiata capace, durante lo sviluppo, di dare origine a proteine funzionali. In un
linfocita B in via di maturazione, la prima ricombinazione si verifica nel locus della
catena pesante (heavy chain, H) μ e porta al congiungimento di uno dei segmenti D con
uno dei segmenti JH, accompagnato dall’eliminazione del tratto di DNA interposto.
Successivamente alla ricombinazione DJH, uno dei segmenti VH in posizione 5’ rispetto
al complesso DJH, riarrangia dando origine ad un’unità codificante VHDJH. In questa
fase, tutti i segmenti in posizione 5’ rispetto al segmento D riarrangiato vengono
eliminati, come tutti i segmenti VH a valle (Jung D et al, Annu Rev Immunol 2006).
I geni della regione C (regione costante) rimangono separati dal complesso VH DJH da
un introne: in questo modo il gene riarrangiato a partire dall‘estremità 5’ sarà costituito
da un esone leader, una sequenza promotrice, un introne, un segmento VH DJH, un
altro introne e diversi segmenti C. Terminato il riarrangiamento l’RNA polimerasi si lega
al promotore e inizia a trascrivere il gene: il trascritto di RNA primario viene sottoposto
ad un processo di poliadenilazione differenziale, splicing delle sequenze introniche e la
formazione di RNA messaggero, il quale esce dal nucleo, si lega ai ribosomi e viene
tradotto in proteina. Nella catena pesante inizialmente vengono trascritti sia il segmento
genico Cμ che il segmento Cδ, la processazione seguente porta la formazione di un
messaggero contenente il trascritto Cμ o Cδ. La proteina prodotta dal gene riarrangiato
su uno dei due cromosomi inibisce irreversibilmente il riarrangiamento nell’altro
cromosoma, secondo il meccanismo dell’esclusione allelica (Daly J et al, EMBOJ 2007).
Questo meccanismo assicura che i linfociti B funzionali non contengano mai più di
un’unità VH-D-JH e una unità VL-JL. I prodotti proteici di un riarrangiamento funzionale
danno un segnale di feedback negativo che previene il riarrangiamento sul secondo
allele. L’espressione della forma transmembrana, ma non della forma secreta della
catena µ porta al silenziamento dell’altro allele della catena pesante ed innesca il
riarrangiamento dei geni della catena leggera k. La ricombinazione VH DJH, nel locus
della catena pesante delle immunoglobuline si verifica solo nei precursori B linfocitari e
rappresenta una tappa critica nell’espressione delle Ig, poiché solo il gene V
riarrangiato verrà successivamente trascritto. Quando la cellula ha completato il
riarrangiamento della catena pesante è classificata come cellula pre-B. La maturazione

21
da cellula pre-B a linfocita maturo richiede il riarrangiamento produttivo della catena
leggera k o λ, con meccanismi sostanzialmente analoghi a quelli della catena pesante.
L’espressione della catena k sul BCR dà un segnale di silenziamento ed esclusione
allelica del secondo allele della catena leggera (Langerak AW. Crit Rev Immunol 2006).
Se il riarrangiamento k non è produttivo su entrambi gli alleli inizia il riarrangiamento dei
geni della catena λ. Se anche questo non è produttivo, il linfocita B interrompe la
maturazione e muore per apoptosi.
Dato che dal riarrangiamento dei geni della regione variabile vengono prodotti due
diversi RNA messaggero per le catene pesanti, l’ulteriore maturazione del linfocita B
porta alla coespressione in membrana di IgD e IgM dotate della stessa specificità
antigenica, che caratterizza i linfociti B maturi. Gli stadi successivi della maturazione del
linfocita B sono sotto il controllo dell’antigene e nel corso di tale processo maturativo
una cellula B può iniziare ad esprimere sulla membrana e secernere IgG, IgA, IgE al
posto delle IgM e IgD (figura 1).
Figura 1: Eventi di ricombinazione e trascrizionali per la produzione di una catena pesante completa IgH. Fonte: Jayanta Chaudhuri & Frederick W. Alt. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nature Reviews Immunology July 2004;4, 541-552.
Per ogni singola cellula B ognuna di queste classi di immunoglobuline ha una diversa
regione costante nelle catene pesanti, ma presenta le stesse regioni variabili, quelle

22
cioè formate nella cellula precursore e che hanno costituito il sito combinatorio
dell’antigene. Poiché ogni catena pesante conferisce ad un anticorpo una differente
funzione effettrice, la stessa regione variabile può essere coinvolta in differenti tipi di
reazioni immunitarie, ma sempre specifiche per quel determinato antigene. Questo
fenomeno, detto cambio di classe (switch isotipico), dipende dal coinvolgimento di brevi
sequenze di DNA, regioni di switch, localizzate in posizione 5’ rispetto ad ogni
segmento CH, per cui il DNA viene riarrangiato avvicinando il gene codificante una
determinata regione costante al segmento riarrangiato VHDJH.
Il clivaggio della ricombinazione V(D)J viene iniziato da due ricombinasi denominate
rispettivamente RAG1 e RAG2, enzimi in grado di riconoscere sequenze segnale di
ricombinazione specifiche (RSS), localizzate a valle dei segmenti V, ad entrambi i lati
dei segmenti D ed a monte dei segmenti J (figura 2) (Sen R et al, Curr Opin Immunol
2006).
Figura 2: Schema rappresentativo delle ricombinasi RAG1 e RAG2. Fonte: David G. Schatz & Yanhong Ji. Recombination centres and the orchestration of V(D)J recombination. Nature Reviews Immunology April 2011;11, 251-263.
Ogni RSS contiene un eptamero palindromico conservato e un nonamero conservato
ricco in A-T, separati da una sequenza spaziatrice di 12 o 23 paia di basi che
corrispondono rispettivamente ad uno o a due giri dell’elica del DNA, per questa ragione
vengono chiamate sequenze segnale di ricombinazione a un giro oppure sequenze
segnale a due giri. Le sequenze segnale con spaziatori a un giro possono unirsi
solamente con sequenze segnale con spaziatori a due giri. Nel DNA della catena

23
pesante le sequenze segnale dei segmenti VH e JH possiedono spaziatori a due giri,
mentre le RSS fiancheggianti i segmenti DH hanno spaziatori a un giro; nel DNA della
catena leggera k la sequenza segnale dei segmenti V ha uno spaziatore a un giro,
mentre quella dei segmenti J è a due giri, nel DNA della catena λ invece accade il
contrario, le RSS di V hanno uno spaziatore a due giri, mentre quelle di J sono a un
giro. Questa regola assicura il riarrangiamento VDJ o VJ nell’ordine corretto, evitando la
giunzione tra segmenti dello stesso tipo.
Successivamente al riconoscimento si ha l’avvicinamento delle sequenze RSS con le
sequenze codificanti e la formazione di un’ansa di DNA intercalato che viene tagliata a
livello giunzionale da RAG1 e RAG2, con la formazione di sequenze nucleotidiche
palindromiche (nucleotidi P) (Jackson KJ et al, BMC Immunol 2004). La giunzione tra
le sequenze codificanti è spesso imprecisa, nonostante l’esatto riconoscimento delle
sequenze segnale, questa flessibilità porta alla formazione di numerosi riarrangiamenti
non produttivi, ma anche di numerose combinazioni produttive che codificano per
diversi amminoacidi a livello di ciascuna giunzione, contribuendo alla diversità
anticorpale.
Può inoltre avvenire l’aggiunta di nucleotidi, detti nucleotidi N, alle estremità libere
delle sequenze codificanti VDJ da parte dell’enzima desossiribonucleotidiltransferasi
(TdT).
L’unione delle sequenze codificanti e delle RSS viene catalizzata da enzimi del normale
processo di riparazione del DNA. Dopo una ricombinazione delle catene leggere e
successiva espressione insieme con le catene pesanti in superficie per formare il BCR, i
geni RAG-1 e RAG-2 sono nuovamente down-modulati. Possono comunque essere
riattivati quando il recettore è autoreattivo in risposta ad autoantigeni. Questo porta ad
una nuova espressione delle proteine RAG e ad una ricombinazione sulla catena
leggera k per eliminare la self-reattività (recupero del recettore). In questo modo le
cellule esprimeranno una IgM di membrana modificata con una catena leggera diversa
e non avranno autoreattività.
I meccanismi con cui il DNA in configurazione germinale del TCR nei linfociti T viene
riarrangiato per formare geni funzionali sono simili a quelli coinvolti nel riarrangiamento
dei geni delle Ig: sono state identificate le sequenze segnale di riconoscimento,
separate da sequenze spaziatrici che fiancheggiano ciascun segmento V, D e J simili a
quelle dei linfociti B.
Tutti i riarrangiamenti dei geni del TCR seguono la regola di giunzione osservata per i

24
geni delle immunoglobuline: le cellule pre-T esprimono i geni attivanti la ricombinazione
(RAG-1/2) che riconoscono le sequenze segnale eptameriche e nonameriche
catalizzando le giunzioni V-D-J con gli stessi meccanismi osservati nei geni delle
immunoglobuline.
Sebbene i linfociti B e T utilizzino meccanismi simili per i riarrangiamenti genici delle
regioni variabili, i geni delle Ig non vengono normalmente riarrangiati nei linfociti T e i
geni del TCR non vengono riarrangiati nei linfociti B. Probabilmente il sistema delle
ricombinasi è regolato in modo che, in ogni tipo cellulare, si verifichi solo il
riarrangiamento del DNA che codifica il recettore corretto.
Il riarrangiamneto VDJ avviene nel caso della catena pesante delle immunoglobuline
(IgH), del TCR β e del TCR δ, il riarrangiamento diretto dei segmenti V-J avviene per la
catena leggera delle immunoglobuline (IgK e IgL), per il TCR α e per il TCR γ. Le
diverse combinazioni VDJ rappresentano il repertorio genico di ricombinazione che è
stimato essere 2x106 molecole per le immunoglobuline, 3x106 molecole per il TCRαβ e
5x103 per il TCRγδ. La grande diversità anticorpale è data dalla giunzione combinatoria
VDJ, dalla flessibilità giunzionale, dall’inserzione random di nucleotidi P ed N dando
origine a regioni altamente diversificate che contribuiscono ad un repertorio totale delle
molecole delle Ig e del TCR superiore a 1012 (Van Dongen JJM et al, Leukemia 2003).
I linfociti B inoltre estendono il loro repertorio immunoglobulinico sul riconoscimento
dell’antigene nei centri germinativi attraverso l’ipermutazione somatica, un processo che
porta alla maturazione dell’affinità degli anticorpi prodotti in risposta ad un antigene
proteico (Inlay MA et al, J Immunol 2006). Questo processo avviene nei centri
germinativi dei follicoli linfoidi ed è il risultato dell’ipermutazione somatica dei geni delle
Ig nelle cellule B in divisione, seguita dalla selezione dei linfociti B ad alta affinità da
parte dell’antigene presentato dalle cellule dendritiche follicolari. Anche se il processo di
ipermutazione produce mutazioni nell’intera regione variabile, la maggior parte delle
mutazioni cade nei CDR e questo riflette il ruolo dell’antigene nella selezione dei linfociti
B con recettori ad affinità più elevata nel corso della maturazione.

25
I riarrangiamenti del B Cell Receptor
La catena pesante H
Il locus della catena pesante delle immunoglobuline (IgH) è localizzato sul cromosoma
14q32.3, in un’area ricoprente approssimativamente 1250 kb.
Sono stati identificati complessivamente 46-52 segmenti VH che possono essere
raggruppati secondo la loro omologia in 6-7 sottogruppi VH. In aggiunta, sono stati
descritti 30 segmenti genici VH non funzionali, ai quali vanno inseriti 27 DH funzionali e
6 JH (figura 3).
I segmenti genici VH più frequentemente utilizzati dalle cellule B normali e patologiche
appartengono alla famiglia VH3 (30-50%), VH4 (20-30%) e VH1 (10-20%), ricoprendo
così il 75-95% di tutto il repertorio VH usato (Camacho FI et al, Blood 2003).
Nelle LAL pre-B sono usati in maniera relativamente frequente anche i segmenti del
VH6.
I segmenti genici VH sono composti da 3 regioni cerniera (Framework region, FR) e 2
regioni determinanti la complementarietà (Complementary determin region, CDRs).
Gli FRs sono caratterizzati dalla loro similarità tra le varie famiglie VH, mentre i CDRs
sono molto diversi all’interno della stessa famiglia VH; i CDRs rappresentano le
sequenze target preferite per le ipermutazioni somatiche nel corso della reazione del
centro germinale, processo che ne aumenta la variabilità.
Gli FRs sono meno toccati da tali mutazioni, possono essere ritrovate sostituzioni
nucleotidiche, specialmente nei linfociti B, solo sotto un forte evento mutazionale.
Basandosi sull’omologia di sequenza i 27 segmenti DH possono essere raggruppati in 7
famiglie, tutte le famiglie comprendono almeno 4 membri, tranne la VII che consiste di
un solo segmento posto a monte della regione JH (DH7-27).
La ricombinazione tra i segmenti DH e JH risulterà nella formazione di giunzioni
incomplete DH-JH, che possono essere facilmente messe in evidenza in cellule pre-B
CD10+/CD19+ derivate dal midollo e quindi in un subset (20-25%) di LAL pre-B che
mostrano un genotipo immaturo (Van Dongen JJM et al, Leukemia 2003).
E’ stata messa in evidenza una maggior frequenza di espressione dei segmenti DH2,
DH3, DH7-27 comprendendo rispettivamente il 36%, il 33% ed il 19% di tutti i segmenti
identificati.
Tuttavia possono essere riscontrati riarrangiamenti incompleti DH-JH anche nelle
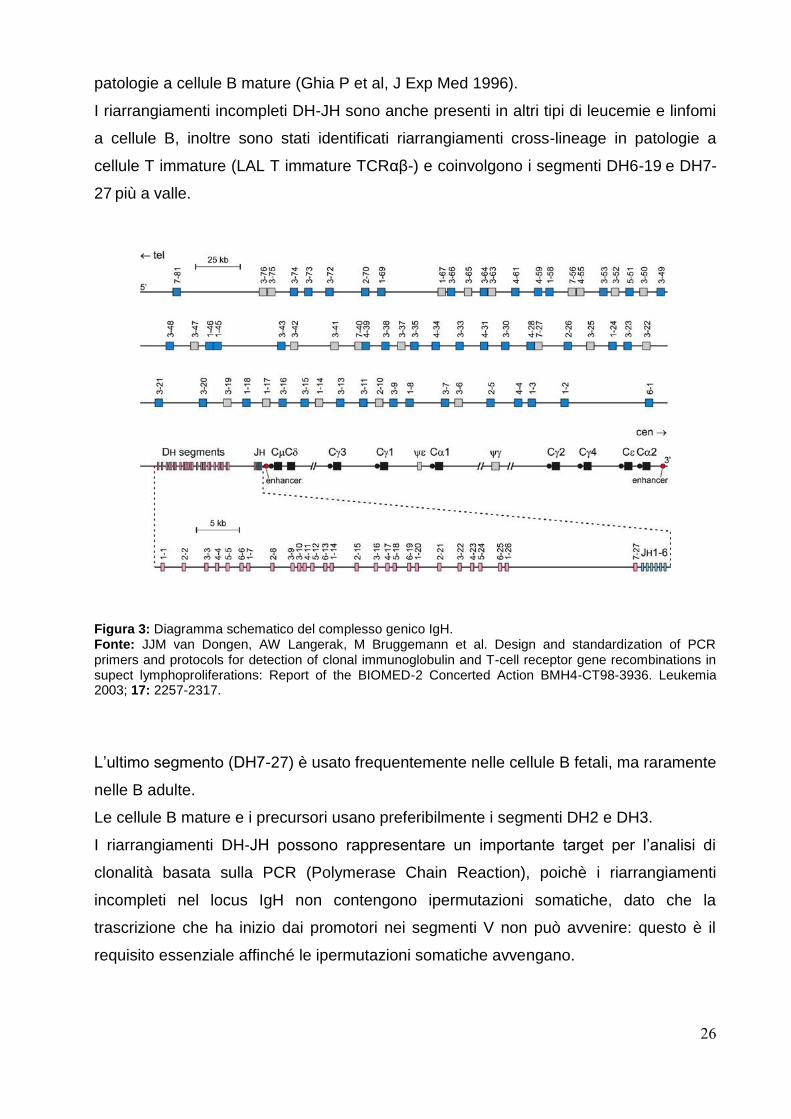
26
patologie a cellule B mature (Ghia P et al, J Exp Med 1996).
I riarrangiamenti incompleti DH-JH sono anche presenti in altri tipi di leucemie e linfomi
a cellule B, inoltre sono stati identificati riarrangiamenti cross-lineage in patologie a
cellule T immature (LAL T immature TCRαβ-) e coinvolgono i segmenti DH6-19 e DH7-
27 più a valle.
Figura 3: Diagramma schematico del complesso genico IgH. Fonte: JJM van Dongen, AW Langerak, M Bruggemann et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in supect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257-2317.
L’ultimo segmento (DH7-27) è usato frequentemente nelle cellule B fetali, ma raramente
nelle B adulte.
Le cellule B mature e i precursori usano preferibilmente i segmenti DH2 e DH3.
I riarrangiamenti DH-JH possono rappresentare un importante target per l’analisi di
clonalità basata sulla PCR (Polymerase Chain Reaction), poichè i riarrangiamenti
incompleti nel locus IgH non contengono ipermutazioni somatiche, dato che la
trascrizione che ha inizio dai promotori nei segmenti V non può avvenire: questo è il
requisito essenziale affinché le ipermutazioni somatiche avvengano.

27
La catena leggera K
Il locus della catena leggera Igk posizionato sul cromosoma 2p11.2, contiene molti
segmenti genici distinti Vk, raggruppati in 7 famiglie Vk, oltre a 5 segmenti genici Jk a
monte della regione Ck.
Le Vk1, Vk2 e Vk3 sono famiglie multi membri includenti sia i segmenti genici funzionali
che gli pseudogeni, mentre la altre famiglie (Vk4, Vk5, Vk7) contengono un singolo
segmento o pochi segmenti (Vk6). Tutti i segmenti genici Vk sono suddivisi in due
grandi cluster, uno posto immediatamente a monte e nello stesso verso dei segmenti
Jk, l’altro posto in maniera più distanziata ed in senso inverso, rispettivamente definiti
prossimale e distale.
L’ultimo cluster costituisce i riarrangiamenti così detti invertiti, che sono richiesti per
formare le giunzioni Vk-Jk coinvolgenti i geni Vk del cluster distale.
In aggiunta ai segmenti Vk e Jk, ci sono altri elementi nel Igk locus che possono essere
coinvolti nella ricombinazione.
Il Kde, approssimativamente 24 Kb a valle della regione Jk-Ck, può riarrangiare ai
segmenti Vk (Vk-Kde), ma anche ad un introne nella regione Jk-Ck (IRSS-Kde)(figura
4).
Entrambi i tipi di riarrangiamento portano all’inattivazione dell’allele Igk, attraverso la
delezione dell’esone Ck (IRSS-Kde) oppure dell’intera area Jk-Ck (Vk-Kde)(Beishuizen
A et al, Leukemia 1994).
Dato che la ricombinazione Igk inizia nelle cellule pre B del midollo osseo, i
riarrangiamenti Igk possono anche essere evidenziati nelle LAL pre-B (40-60% dei casi)
e coinvolgono il Kde per il 35-50%.
Nelle LAL pre-B dei bambini, la ricombinazione Vk-Kde predomina sull’IRSS-Kde,
mentre nelle LAL degli adulti le delezioni riguardano esclusivamente Vk-Kde (Van der
Velden VHJ et al, Leukemia 2002).
Nelle leucemie croniche a cellule B, i riarrangiamenti Igk sono più frequenti, essendo
evidenziabili in tutti i casi Igk+ e λ+.
Per definizione i riarrangiamenti funzionali Vk-Jk sono riscontrati su almeno un allele
nelle leucemie B-cell k+, il secondo allele non codificante è in configurazione germinale
(circa 50% dei casi), contiene un riarrangiamento Vk-Jk fuori marker di lettura (20%)
oppure è inattivato dal riarrangiamento del Kde (circa 30% dei casi).
I riarrangiamenti Kde (virtualmente) si trovano in tutte le leucemie B-cell λ+ (85% degli
alleli), con una predominanza di ricombinazione dell’IRSS-Kde su Vk-Kde.

28
Questo implica che tutte le leucemie λ+ contengono un riarrangiamento Kde, mentre
riarrangiamenti Vk-Jk potenzialmente funzionali, sono rari.
Diversi studi hanno mostrato che l’uso del segmento genico Vk è identico tra diverse
popolazioni di cellule B normali e patologiche e questo ampiamente riflette il numero di
segmenti genici disponibili in ogni famiglia.
In entrambi i riarrangiamenti Vk-Jk o Vk-Kde, i segmenti genici delle prime quattro
famiglie predominano.
Figura 4: Diagramma schematico del complesso genico IgK. Fonte: JJM van Dongen, AW Langerak, M Bruggemann et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in supect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257-2317.
L’uso del gene Vk2 sembra essere più elevato nelle LAL pre B rispetto alle
linfoproliferazioni B più mature o anche alle cellule B normali.
Il cluster distale ed invertito Vk è raramente usato nei riarrangiamenti Vk-Jk, mentre i
segmenti pseudogenici Vk non sono mai coinvolti, anche nei casi λ+.
Poco si sa sull’uso del segmento genico Jk, è stato messo in evidenza che i segmenti
Jk più usati sono Jk1, Jk2 e Jk4 (van der Burg M et al, Blood 2001).
I riarrangiamenti Vk-Jk possono essere importanti target di PCR per quei tipi di
proliferazioni B in cui le ipermutazioni somatiche possono ostacolare l’amplificazione del

29
target VH-JH.
Le ricombinazioni coinvolgenti il Kde sono però probabilmente più rilevanti, dato che la
delezione delle sequenze coinvolte nell’introne risulta nella rimozione dell’attivatore
delle Igk, che è ritenuto essere essenziale affinché il processo di ipermutazione
somatica avvenga.
La catena leggera λ
I riarrangiamenti genici IGλ sono presenti nel 5-10% dei tumori a cellule B IgK+ ed in
tutte le malignità Igλ+. Il locus IGλ è posizionato sul cromosoma 22q11.2, contiene 73-
74 geni Vλ, tra cui 30-33 sono funzionali (Figura 5). Sulla base dell' omologia di
sequenza, i geni Vλ possono essere raggruppati in 11 famiglie (10 contenenti segmenti
genici funzionali Vλ) e tre clan. I membri della stessa famiglia tendono ad essere
raggruppati sul cromosoma.
I geni Jλ e Cλ sono organizzati in tandem con un segmento Jλ che precede un gene Cλ.
Ci sono sette segmenti genici J-Cλ, di cui J-Cλ1, J-Cλ2, J-Cλ3, e J-Cλ7 sono funzionali
e codificano i quattro isotipi IGλ. Esiste tuttavia un variazione polimorfica del numero di
segmenti genici J-Cλ, poiché alcuni individui possono trasportare fino a 11 di loro su un
allele, a causa di un'amplificazione della regione Cλ2-Cλ3 (van der Burg M et al, J
Immunol 2002).
Figura 5: Diagramma schematico del complesso genico Igλ. Fonte: JJM van Dongen, AW Langerak, M Bruggemann et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in supect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257-2317.

30
Diversi studi hanno dimostrato che il repertorio genico IGλ di cellule B normali e maligne
è condizionato (Tu m̈kaya T et al. Leukemia 2001).
Oltre il 90 % dei geni Vλ utilizzati dalle cellule B normali appartengono alle famiglie Vλ1,
Vλ2 e Vλ3, comprendenti il 60% dei geni funzionali. Inoltre, tre geni (2-14, 1-40 e 2-8)
rappresentano circa la metà del repertorio espresso. Mentre le cellule B normali usano i
segmenti genici J-Cλ1, J-Cλ2, e J-Cλ3 in proporzioni più o meno equivalenti, le cellule B
neoplastiche tendono ad utilizzare prevalentemente i segmenti genici J-Cλ2 e J–Cλ3.
Sia nelle cellule B normali che maligne, il segmento J-Cλ7 è usato molto raramente
(1%).
Contrariamente a quanto avviene nel topo, nei riarrangiamenti IGλ umani vi è una certa
diversità giunzionale a causa dell' attività esonucleasica e dell'aggiunta di nucleotidi N
nei riarrangiamenti.(Farner NL et al, J Immunol 1999; Ignatovich O et al, J Mol Biol
1999).
Questa diversità giunzionale è tuttavia molto meno estesa di quella del locus IGH, ed un
numero di riarrangiamenti derivano direttamente dall'accoppiamento dei segmenti
genici Vλ e Jλ della linea germinale.
Il locus IGλ potrebbe rappresentare una complementare alternativa al locus IGH per gli
studi di clonalità sulle cellule B.

31
I riarrangiamenti genici del T Cell Receptor (TCR)
La catena γ
I riarrangiamenti del TCRγ sono stati usati a lungo per la valutazione molecolare della
clonalità linfoide e rappresentano il prototipo di un repertorio ristretto di target
molecolare.
Il TCRγ è un target preferenziale per l’analisi di clonalità, poiché è riarrangiato ad uno
stadio precoce dello sviluppo linfoide T, probabilmente appena dopo il TCRδ, in
entrambi i precursori TCRαβ e TCRγδ. La catena gamma è riarrangiata in più del 90%
delle LAL T, nelle leucemie LGL (Large Granular Lymphocyte) e nelle T-PLL (Leucemie
Prolinfocitiche a cellule T), nel 50-75% di T-NHL (Linfomi non Hodking a cellule T)
periferici e nelle micosi fungoide, ma non nelle proliferazioni NK, è anche riarrangiata
nella maggior parte di LAL-B, ma molto meno nei B-NHL (Linfomi non Hodking a cellule
B)(Szczepanski T et al, Leukemia 1998; Szczepanski T et al, Leukemia 1999).
Il TCRγ contiene un limitato numero di segmenti Vγ e Jγ.
Il locus umano del TCRγ è situato sul cromosoma 7p14 e contiene 14 segmenti Vγ, per
10 di questi è stato dimostrato il riarrangiamento.
Il repertorio Vγ espresso include solo sei geni Vγ (Vγ2, Vγ3, Vγ4, Vγ5, Vγ8 e Vγ9), ma il
riarrangiamento avviene anche con i segmenti Vγ7, Vγ10 e Vγ11.
Il riarrangiamento di VγB (anche conosciuto come Vγ12) è così eccezionale che
raramente è usato nella diagnostica molecolare.
I segmenti Vγ riarrangianti possono essere suddivisi in quelli appartenenti alla famiglia
VγI (Vγ2, Vγ3, Vγ4, Vγ5, Vγ7 e Vγ8, con un omologia >90% ed ancora più elevata tra
Vγ2 e Vγ4 e tra Vγ3 e Vγ5) e nei singoli membri Vγ9, Vγ10 e Vγ111.
Il locus del TCRγ contiene 5 segmenti Jγ: Jγ1.1 (JP1), Jγ1.2 (JP), Jγ1.3 (Jγ1), Jγ2.1
(JP2) e Jγ2.3 (Jγ2) di cui Jγ1.3 e Jγ2.3 sono altamente omologhi, come Jγ1.1 e Jγ2.1
(figura 6).
Il locus TCRγ non contiene segmenti D e dimostra addizioni di nucleotidi relativamente
1 -Il segmento Vγ4 è circa 40bp più lungo degli altri membri della VγI ed i riarrangiamenti Vγ4 sono
relativamente comuni in entrambe le cellule linfoidi fisiologiche e patologiche.
-E’ stata descritta una delezione interstiziale di circa 170 bp all’estremità 3’ del segmento Vγ2 in diversi casi
di LAL T e questa rappresenta approssimativamente il 5% dei riarrangiamenti.

32
limitate (Griesinger F et al, J Clin Imvest 1989).
La lunghezza giunzionale V-J del TCRγ varia da 20-30 bp, paragonata
approssimativamente a 60 bp per le IgH e il TCRδ completo.
La capacità di distinguere i riarrangiamenti clonali da quelli policlonali dipende dalla
complessità del repertorio policlonale.
Mentre lo stretto repertorio in configurazione germinale del TCRγ facilita l’amplificazione
molecolare, la limitata diversità giunzionale dei riarrangiamenti complica la distinzione
tra i prodotti di PCR clonali e policlonali (Kode J et al, Leuk Lymphoma 2004).
In generale, le popolazioni clonali minori che usano i riarrangiamenti più frequenti come
VγI-Jγ1.3/2.3 sono a rischio di essere persi tra il repertorio policlonale, mentre le
combinazioni rare possono essere messe in evidenza con una sensibilità maggiore.
Tuttavia è possibile che linfociti T policlonali occasionali dimostranti rari riarrangiamenti
Vγ-Jγ possono essere scambiati per un riarrangiamento clonale, a causa dell’assenza
di un background policlonale per quel tipo di riarrangiamento.
Un‘ulteriore possibile sorgente di popolazioni clonali minori risultano dalla presenza di
linfociti T esprimenti TCRγδ+ che dimostrano riarrangiamenti canonici TCRγ, ma non
dimostrano l’addizione di nucleotidi N.
Figura 6: Diagramma schematico del complesso genico TCRγ sulla banda cromosomica 7p14. Fonte: JJM van Dongen, AW Langerak, M Bruggemann et all. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in supect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003 17, 2257-2317.
Il riarrangiamento TCRγ canonico umano più comunemente riconosciuto coinvolge i
segmenti Vγ9-Jγ1.2 ed avviene nell’1% di linfociti T del sangue.

33
Risulta quindi estremamente importante analizzare i prodotti molecolari del TCRγ
usando tecniche elettroforetiche ad alta risoluzione o usando criteri di separazione degli
stessi prodotti che vanno al di là del peso molecolare, per ridurre il rischio di risultati
falsi positivi.
E’ anche importante conoscere il profilo dei riarrangiamenti canonici e le situazioni in cui
loro più comunemente avvengono; i Vγ9-Jγ1.2 si trovano principalmente nel sangue
periferico ed aumentano la frequenza con l’età, poiché vi è l’accumulo di linfociti T TCR
γδ+.
Diversamente dal TCRδ il gamma non è deleto nelle cellule αβ+, dal momento che
questo tipo di riarrangiamento avviene in entrambi i precursori αβ e γδ, la sua
identificazione non può essere usata per la determinazione del tipo di linea cellulare.
La catena α/δ
I geni della catena α/δ del TCR si trovano in un unico complesso locus genico
localizzato sul cromosoma 14. I geni della catena δ sono assemblati nei timociti
CD4CD8 doppi negativi, mentre quelli della catena α nei timociti CD4CD8 doppi positivi,
questo probabilmente dovuto in parte all'attività specifica dei rispettivi enhancer (Eδ e
Eα) durante le fasi di maturazione della cellula T (Krangel M. S.et al, Immunol Rev
1998).
Il repertorio genico della catena α contiene 80 segmenti genici V, 61 segmenti genici J
ed 1 segmento genico C, mentre i segmenti genici D sono assenti. Tra i segmenti genici
V e J della catena α è localizzato il locus della catena δ, più precisamente sulla banda
14q11.2, costituito da un numero limitato di segmenti; 8 elementi V, 4 elementi J, 3
elementi D. Almeno 5 degli 8 segmenti genici V possono riarrangiare con I segmenti Jα
ed alcuni segmenti Vα possono in rari casi riarrangiare con la catena δ. II
riarrangiamento di Vα con i segmenti genici di Jα causa la delezione dell’intero locus
intermedio del TCRδ (figura 7).
Il TCRδ V101S1 (Vδ1), il TCRδ V102S1 (Vδ2) ed il TCRAD V17S1 (Vδ3) sono usati
esclusivamente nei riarrangiamenti del TCRδ, mentre il TCRαδ V6S1 (Vδ4), il TCRαδ
V21S1 (Vδ5) ed il TCRαδ V17S1 (Vδ6) possono essere usati sia nella catena delta che
nella catena alpha del TCR
Il TCRαδ V28S1 (Vδ7) ed il TCRαδ V14S1 (Vδ8) sono usati raramente nei
riarrangiamenti del δ (Verschuren MC et al, Immunology 1998).

34
Il repertorio in configurazione germinale delle cellule T γδ+ è piccolo paragonato a
quello di cellule T αβ+ e l’intero repertorio di ricombinazione è più limitato dato il
riarrangiamento preferenziale nel sangue periferico e nel timocita di cellule Tγδ+.
Alla nascita il repertorio di cellule T γδ+ presenti nel sangue di cordone ombelicale è
ampio, con nessuna restrizione apparente o preferenziale di particolari combinazioni
Vγ/Vδ.
Durante l’infanzia tale repertorio nel sangue periferico è formato così che negli adulti
predominano cellule portanti il riarrangiamento Vγ9/Vδ2.
I repertori Vδ1 e Vδ2 diventano ristretti con l’età portando all’insorgenza di cellule Vδ1+
e Vδ2+ oligoclonali nell’intestino.
Le cellule T γδ+ sono distribuite attraverso i tessuti linfoidi umani, con una maggiore
espressione di particolari segmenti Vδ in alcune localizzazioni anatomiche umane.
Molte cellule T γδ+ intraepiteliali presenti nell’intestino tenue e nel colon esprimono
Vδ1. Similarmente Vδ1 è espresso anche da cellule spleniche normali, mentre cellule T
γδ+ della pelle esprimono Vδ2.
Tuttavia il piccolo numero di segmenti genici VDJ adatti per la ricombinazione limita la
potenziale diversità di ricombinazione, il CDR3 o la diversità giunzionale è estesa data
l’aggiunta di regioni N, regioni P e delezioni casuali mediate dalle ricombinasi.
Il locus del TCRδ è il primo di tutti i loci del TCR a riarrangiare durante l’ontogenesi
della cellula T.
Il primo evento è un riarrangiamento Dδ2-Dδ3, seguito da un Vδ2-(Dδ1-Dδ2)-Dδ3 per
ottenere alla fine un riarrangiamento Vδ-Dδ-Jδ.
Riarrangiamenti immaturi (Vδ2-Dδ3 o Dδ2-Dδ3) si trovano nel 70% di LAL pre B,
mentre c’è una predominanza di riarrangiamenti maturi comprendenti sia la forma
incompleta Dδ2-Jδ1 che completa Vδ1, Vδ2, Vδ3-Jδ1 riscontrati nelle LAL T(Schneider
M et al, Br J Haematol 1997).
Le LAL T γδ+ formano un gruppo di LAL relativamente piccolo (10-15% di LAL T) e
costituiscono un 2% di tutte le LAL.
I riarrangiamenti Vδ1-Jδ1 predominano nelle LAL T γδ+; il Vδ1 non è mai stato trovato
riarrangiare con un altro segmento Jδ che non sia Jδ1.
Le catene Vδ1-Jδ1-Cδ1 sono quasi sempre legate alle famiglie VγI e VγII ricombinate a
Jγ2.3-Cγ2. L’uso di questo gene correla con l’origine timica immatura di queste cellule
leucemiche.
Molti linfomi a cellule T esprimono TCRαβ, mentre una minoranza esprime TCRγδ e

35
comprende diverse entità distinte.
I linfomi periferici che esprimono TCRγδ comprendono l’8-13% di tutti i linfomi T
periferici e sono stati documentati riarrangianti Vδ1-Jδ oltre ad altri riarrangiamenti di Vδ
a Jδ1.
Figura 7: Diagramma schematico del complesso genico del TCRα/δ. Fonte: JJM van Dongen, AW Langerak, M Bruggemann et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in supect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257-2317.
Il linfoma epatoslpenico a cellule T γδ+ è derivato da cellule T TCRγδ+ spleniche che
normalmente esprimono Vδ1.
Si tratta di un’entità non comune che esibisce caratteristiche clinico-patologiche distinte.
Il linfoma cutaneo a cellule T TCRγδ+ esprime Vδ2 e sembra perciò rappresentare
un’espansione clonale di cellule T γδ+ che normalmente risiedono nella pelle.
Altre proliferazioni clonali γδ includono proliferazioni LGL γδ+ CD3+ che comprendono
circa il 5% di tutte le LGL CD3+ e spesso mostrano riarrangiamenti Vδ1-Jδ1.
La catena β
I riarrangiamenti genici del TCRβ si verificano non solo in quasi tutti i tumori maligni
delle cellule T mature, ma anche in circa l'80% delle LAL T CD3- ed il 95 % delle LAL T
CD3+(Langerak AW et al, Leukemia 1999).
I riarrangiamenti del TCRβ non sono ristretti alle malignità di linea T, poichè circa un
terzo delle LAL pre-B portano riarrangiamenti del TCRβ (Szczepanski T et al, Leukemia
1999).
La loro frequenza è molto più bassa nelle proliferazioni a cellule B mature (0-7%) (van
Dongen JJM et al, Clin Chim Acta 1991).

36
Il locus umano del TCRβ è localizzato sul braccio lungo del cromosoma 7, a banda
7q34 e si estende su una regione di 685 kb. In contrasto con i loci del TCRγ e del
TCRδ, il cluster genico della regione V è molto più complesso (Figura 8). Contiene circa
65 elementi genici Vβ suddivisi in 30 sottogruppi. Le più grandi famiglie, Vβ5, Vβ6, Vβ8,
e Vβ13 raggiungono una dimensione di sette, nove, cinque e otto membri,
rispettivamente. 12 famiglie Vβ contengono solo un singolo membro (Arden B et al,
Immunogenetics 1995).
Di tutti gli elementi genici Vβ, 39-47 sono qualificati come funzionali ed appartengono a
23 famiglie. 10-16 elementi genici sono classificati come pseudogeni. Inoltre, un gruppo
di sei geni orfani Vβ non funzionali sono stati riportati localizzati sul braccio corto del
cromosoma 9 (9p21)(Charmley P et al, Immunogenetics 1993).
Essi non vengono rilevati in trascritti. Tutti i geni Vβ, tranne uno, si trovano a monte di
due cluster Dβ-Jβ-Cβ. La Figura 8 mostra come entrambi i segmenti genici Cβ (Cβ1 e
Cβ2) sono preceduti da un gene Dβ (Dβ1 e Dβ2) ed un cluster Jβ, che comprende sei
(Jβ1.1-Jβ1.6) e sette (Jβ2.1-Jβ2.7) segmenti Jβ funzionali. I loci della regione Jβ sono
classificati in due famiglie secondo la loro localizzazione genomica, e non vi è omologia
di sequenza (Wei S et al, Immunogenetics 1994).
A causa del grande repertorio codificato dalla linea germinale, la diversità combinatoria
dei riarrangiamenti genici del TCRβ è ampia rispetto ai riarrangiamenti del TCRγ e del
TCRδ. Il repertorio principale delle molecole TCRβ è ulteriormente prorogato dall'
aggiunta di una media di 3,6 e 4,6 nucleotidi alle giunzioni Vβ-Dβ e Dβ-Jβ
rispettivamente e dalla delezione di una media di 3.6 (Vβ), 3.8 (5'Dβ), 3.7 (3'Dβ), e 4.1
(Jβ) nucleotidi (Rowen L et al, Science 1996).
Durante la maturazione delle cellule T, il riarrangiamento del TCRβ avviene in due fasi
consecutive: Dβ-Jβ e Vβ-Dβ-Jβ, con un intervallo di 1-2 giorni tra questi due processi.
Il segmento genico Dβ1 può aderire sia a Jβ1 che ai segmenti genici Jβ2, mentre il
segmento genico Dβ2 unisce generalmente solo segmenti genici Jβ2 per la sua
posizione nel locus genico (Langerak AW et al, Leukemia 1999).
Tuttavia, a causa della presenza di due cluster consecutivi Dβ-Jβ, è anche possibile
che due riarrangiamenti siano rilevabili su un unico allele: un riarrangiamento
incompleto Dβ2- Jβ2 ed un riarrangiamento completo o incompleto nella regione Dβ1–
Jβ1.
Nei riarrangiamenti del TCRβ è riscontrato l'uso di una distribuzione non casuale dei
segmenti genici. Negli individui sani, alcune famiglie Vβ predominano nel repertorio

37
delle cellule T periferiche (ad esempio Vβ1-Vβ5), mentre altri sono solo raramente
utilizzati (ad esempio Vβ11, Vβ16, Vβ18 e Vβ23).
Figura 8: Diagramma schematico del complesso genico TCRβ. Fonte: JJM van Dongen, AW Langerak, M Bruggemann et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in supect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003; 17: 2257-2317.
I valori medi del repertorio Vβ sembrano essere stabili durante l'invecchiamento, anche
se vi è l'aumento della deviazione standard nei soggetti anziani (Van den Beemd MWM
et al, Cytometry 2000).
La rappresentazione dei segmenti Jβ è tutt'altro che uniforme. La famiglia Jβ2 è usata
più frequentemente rispetto alla famiglia Jβ1 (72 vs 28%). In particolare, la percentuale
di Jβ2.1 è superiore alle altre (24 %), seguita da Jβ2.2 (11%) e Jβ2.3 e Jβ2.5 (10%
ciascuno) (Jores R et al, J Immunol 1993).
I riarrangiamenti del TCRβ differiscono tra le categorie di tumori maligni delle cellule T.
Riarrangiamenti completi Vβ-Jβ1 e incompleti Dβ-Jβ2 sono stati riscontrati più
frequentemente in casi di LAL T TCRαβ+, rispetto ai casi di LAL T CD3- e LAL T
TCRγδ+. Più in generale, nelle LAL T, la regione del TCRβ Dβ-Jβ1 è relativamente
frequentemente coinvolta nei riarrangiamenti, in contrasto con i riarrangiamenti nelle
LAL pre B che coinvolgono esclusivamente la regione Dβ-Jβ2 del TCRβ (Szczepanski T
et al, Leukemia 1999).

38
Obiettivo dello studio
L'analisi dell'intero genoma ha recentemente ampliato la possibilità di una precisa
caratterizzazione genomica delle malattie. Un pre-requisito per un tale approccio è la
disponibilità di una quantità sufficiente di DNA germinale e di DNA delle cellule tumorali
e la loro buona qualità, poiché la mancanza di quantità e qualità appropriate di DNA
porta ad una restrizione del tipo e del numero di saggi genetici che possono essere
eseguiti. Un approccio affidabile per aumentare la quantità di DNA è la sua
amplificazione utilizzando un metodo di amplificazione dell'intero genoma (WGA).
Il risultato di questa procedura è l’ottenimento di grandi quantità di DNA di partenza di
alta qualità, utile in tutti quei casi in cui è richiesta la disponibilità indefinita di materiale
per una qualsiasi analisi molecolare (es. genotipizzazione, Real time PCR, sequencing
ecc).
L' uso della malattia minima residua come marker di risposta molecolare al trattamento,
può migliorare la valutazione della risposta clinica, guidare la selezione delle strategie
terapeutiche e, possibilmente, indicare l'esito clinico a lungo termine (Cazzaniga G et al,
Haematologica 2005) in un certo numero di malattie ematologiche.
Nel presente studio, è stato applicato l'utilizzo del metodo WGA, con lo scopo di
confrontare i risultati ottenuti con il DNA genomico ed il DNA amplificato al momento
della diagnosi e durante il follow-up clinico dei pazienti, e valutare sia l'applicabilità che
l'affidabilità del DNA amplificato in questo tipo di analisi.
A questo scopo, sono stati studiati 20 casi di LAL dell'adulto sia mediante PCR che RQ
-PCR su DNA genomico e DNA amplificato utilizzando i riarrangiamenti genici Ig/TCR
come marcatori di MMR.

39
Materiali e Metodi
Pazienti e campioni studiati
Venti pazienti adulti affetti da LAL, di età compresa tra 18-34, pervenuti presso il nostro
centro, sono stati inseriti nello studio. Sono stati analizzati i relativi campioni di sangue
midollare sia alla diagnosi che durante il follow-up. Tutti i pazienti sono stati arruolati
nel protocollo GIMEMA 1308 ed hanno dato il loro consenso informato secondo le
linee guida istituzionali. La diagnosi è stata stabilita attraverso criteri morfologici,
citochimici e immunologici, secondo le classificazioni “French-American-British” (FAB)
e “World Health Organization”” (WHO). Aspirati di midollo osseo alla diagnosi e durante
il follow-up sono stati raccolti in provette contenenti citrato di sodio e mantenuti a
temperatura ambiente fino al successivo processamento.
I campioni di follow-up sono stati correlati alla fase IA dell'induzione (giorno +33),
secondo il protocollo AIEOP pediatrico LAL 2000.
Estrazione del DNA
Per l'analisi molecolare, le cellule del midollo osseo sono state separate mediante Ficoll
su gradiente di densità ed il DNA è stato isolato usando il kit di purificazione del DNA
Wizard Genomic DNA Purification Kit (Promega Corp., Madison, Wisconsis, USA). La
determinazione della purezza e della concentrazione del DNA estratto è stata valutata
mediante lo spettrofotometro Epperndorf BIOPhotometer (Eppendorf AG, Hamburg,
Germany), ed i campioni che presentavano un rapporto di A260/A280 compreso tra 1.8
e 1.9, sono stati diluiti con acqua deionizzata al fine di ottenere una concentrazione
standard di DNA di 100µg/mL. La qualità del DNA è stata valutata a seguito di una corsa
elettroforetica su gel d’agarosio dove un DNA genomico di buona qualità presentava un
sola banda ad alto peso molecolare.
Whole Genome Amplification (WGA)
L'amplificazione dell'intero genoma (WGA) è stata effettuata utilizzando il Qiagen
REPLI-g Mini Kit (QIAGEN GmbH, Hilden, Germania), secondo il protocollo del
produttore. Il modello consisteva di 40 ng di DNA genomico umano estratto da un pellet
di cellule mononucleate. La miscela di reazione è stata preparata utilizzando due

40
differenti tamponi ed una master mix contenente, rispettivamente, i random esameri e la
Phi29 DNA polimerasi, per ottenere un volume finale di 30 microlitri. I campioni sono
stati quindi incubati a 30°C per 16 h, dopo inattivazione termica a 65°C per 10 minuti.
Questo metodo si basa su un sistema di amplificazione multiplo di spostamento, in cui
vi è l'associazione di esameri casuali per la singola molecola del filamento bersaglio.
Esso consiste in una amplificazione isotermica, poiché la reazione avviene ad una
temperatura di 30°C con una DNA polimerasi (Phi29) ad alta processività. Nel momento
in cui la Phi 29 comincia l'elongazione, i filamenti di DNA a monte vengono spostati e
possono quindi servire da stampo per nuovi eventi di annealing dei primers. Questo
darà origine ad un network di DNA a struttura ramificata, generando un'abbondanza di
copie della molecola di DNA originale.
Screening dei riarrangiamenti genici Ig/TCR
Il DNA genomico ed il DNA amplificato ottenuto dai campioni della diagnosi sono stati
amplificati mediante reazione di amplificazione Polymerase Chain Reaction (PCR)
utilizzando il termociclatore GeneAmp® PCR System 9700 (Applied Biosystems, Foster
City, CA, USA). La PCR è stata eseguita utilizzando il set di primers del BIOMED-1 per
Ig kappa/Kde, (Vk-Kde, introne-Kde), per i riarrangiamenti completi ed incompleti della
catena delta del TCR (TCRD; Vd-(Dd)-Jd1, DD2-Jd1, Vd2-DD3, DD2-DD3) e della
catena gamma (TCRG; Vg-Jg1.3/2.3, Vg-Jg1.1/2.1)(Van Dongen JJM et al, Leukemia
1999).
I riarrangiamenti completi ed incompleti della catena pesante delle immunoglobuline
(VH- (DH)-JH, DH-JH) sono stati identificati utilizzando cinque primers per le famiglie
VH e sette primers per le famiglie DH in combinazione con un JH consensus (Van
Dongen JJM et al, Leukemia 2003). Per I riarrangiamenti completi ed incompleti della
catena beta del TCR (TCRB; Dβ-Jβ e Vβ-Dβ-Jβ), sono stati utilizzati i rispettivi set di
primer per la multiplex-PCR del BIOMED-2 (Van Dongen JJM et al, Leukemia 2003).
Le condizioni di amplificazione di ciascun riarrangiamento sono riportate nelle tabelle 4
e 5.
I prodotti di amplificazione ottenuti sono stati ulteriormente esaminati mediante analisi
degli omo/eterodimeri al fine di discriminare le amplificazioni derivate da popolazioni di
cellule linfoidi monoclonali o policlonali (Langerak AW et al, Leukemia 1997; Borowitz
MJ et al, Blood 2008). I prodotti di PCR biclonali o biallelici sono stati separati mediante

41
escissione degli ampliconi dal gel di poliacrilammide o per clonazione del DNA
attraverso il pMOS Blunt end Cloning KIT (Amersham Biosciences Europe GMBH. MI,
Italy).
Tabella 4: Schema di amplificazione dei riarrangiamenti IgH (A) e IgK (B).
Tabella 5: Schema di amplificazione dei riarrangiamenti del TCRγ/δ (C), del TCRα (D) e del TCRβ (E).
(A) Denaturazione
Annealing Estensione N° Cicli
2’ 94°C
30’’ 94°C 45’’ 60°C 1’ 72°C 35
7’ 72°C
(B) Denaturazione Annealing Estensione N° Cicli
3’ 92°C
45’’ 92°C 1', 30’’ 60°C 2’ 72°C 40
10’ 72°C
(C) Denaturazione Annealing Estensione N° Cicli
1',30” 94°C 1' 60°C 1',30” 72°C 1
30’’ 94°C 30’’ 60°C 1',30” 72°C 35
6’ 72°C
(D) Denaturazione Annealing Estensione N° Cicli
7’ 95°C
45’’ 95°C 45’’ 60°C 1',30” 72°C 35
10’ 72°C
(E) Denaturazione Annealing Estensione N° Cicli
7’ 95°C
30’’ 94°C 45’’ 60°C 1',30” 72°C 35
10’ 72°C

42
Sequenziamento e analisi del gene
I prodotti di PCR sono stati sequenziati direttamente secondo metodo Sanger con
marcatura fluorescente utilizzando il kit di reazione BigDye Terminator v3.1 Cycle
Sequencing Kit (Applied Biosystems, Foster City, CA, USA) e il termociclatore
GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA, USA). Durante la
sintesi del filamento di DNA i didesossinucleotidiltrifosfati (ddNTP) marcati con quattro
differenti fluorocromi vengono, mediante l’enzima DNA polimerasi, inseriti al filamento
stampo impedendo l’aggiunta di ulteriori nucleotidi. Al termine della reazione di
sequenza la miscela di singoli frammenti di DNA ottenuta è stata purificata utilizzando il
kit DyeEx 2.0 Spin Kit (QIAGEN, Valencia, CA, USA) che permette di eliminare i ddNTP
fluorescenti in eccesso non incorporati attraverso il principio cromatografico della gel-
filtrazione su colonnine contenenti resina. I frammenti di DNA purificati sono stati
sottoposti ad elettroforesi capillare mediante il sequenziatore ABI PRISM® 3100-Avant
Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). L’acquisizione dei dati
relativi ai campioni è stata eseguita utilizzando il software 3100-Avant Data Collection
Software e le sequenze sono state analizzate mediante il programma ABI PRISM® DNA
Sequencing Analysis Software (Versione 3.7), e visualizzate come elettroferogrammi.
L'interpretazione dei risultati è stata eseguita allineando le sequenze nucleotidiche
ottenute alla directory IMGT(international ImMunoGeneTics information system) per
l'identificazione di riarrangiamenti IGHV, IGKV, TCRA, TCRB, TCRD, TCRG.
Disegno dei primers e analisi RQ-PCR
Le regioni giunzionali paziente-specifiche dei prodotti di PCR risultati clonali sono stati
identificati come potenziali target per la valutazione della MMR quantitativa (Langerak
AW et al, Leukemia 1997). Gli Oligonucleotidi relativi a tali regioni (ASO primer) sono
stati disegnati utilizzando il programma Primer Express ABI PRISM Primer Design
Software(Applied Biosystems, Foster City, CA).
Gli ASO primers sono stati testati per specificità e sensibilità con l'obiettivo di
selezionare per ogni paziente uno o due oligonucleotidi con una sensibilità di almeno
10-4 e un range quantitativo di 10-4 per il primo primer e di 5x10-4 per l'eventuale
secondo primer identificato (Van der Velden VHJ et al, Leukemia 2007).
Le curve standard per le valutazioni preliminari degli oligo costruiti e le successive
analisi quantitative, sono state preparate a partire da diluizioni seriali del campione della

43
diagnosi, nel DNA estratto da un pellet di cellule mononucleate proveniente da un pool
di sangue venoso periferico (PBL) di 5 donatori sani e portato alla concentrazione di
100 μg/ml.
I saggi sono stati allestiti per raggiungere una gamma di sensibilità e quantificabilità
≥10-4, con uno slope della curva compreso tra 3.1 e 3.9 ed un coefficiente di
correlazione ≥0,98, secondo le linee guida della ESG-MRD–ALL (Van der Velden VHJ
et al, Leukemia 2007).
Le reazioni sono state preparate utilizzando i reagenti del kit TaqMan PCR Core
Reagent (PE Applied Biosystems, Foster City CA, USA) e processate nell' ABIPrism
7300-7500 alle seguenti condizioni: un'incubazione iniziale di 2 minuti a 50°C e 10
minuti di denaturazione a 95°C, seguite da 50 cicli di denaturazione a 95°C per 15
secondi ed annealing a 58-63°C per 1 minuto. Sono state rispettate le linee guida per
l'analisi di RQ-PCR della MMR per tutti i riarrangiamenti ed i pazienti studiati, ESG-
MRD-ALL (European Study Group on MRD Detection for ALL)(Van der Velden VHJ et
al, Leukemia 2007).

44
Risultati Riarrangiamenti genici Ig/TCR identificati al momento della diagnosi: DNA genomico vs
DNA amplificato
Dei 20 casi di LAL esaminati, 16 hanno mostrato un fenotipo a cellule B e 4 un fenotipo
a cellule T. Tutti i campioni sono stati valutati per i ririarrangiamenti genici Ig/TCR.
Lo screening è stato eseguito a cieco sia su DNA genomico che su DNA amplificato e
successivamente sono stati confrontati i risultati. Su DNA genomico in 18/20 (90%)
pazienti è stato identificato almeno un riarrangiamento clonale, mentre in 2/20 (10%)
casi non è stato identificato alcun marcatore. In 10/18 (55,5%) pazienti è stato rilevato
un doppio marker, che ha permesso di caratterizzare e seguire con maggiore precisione
la malattia. Sono stati trovati e sequenziati 87 riarrangiamenti, con un valore medio di
4.8 riarrangiamenti/caso ed un massimo di 8 riarrangiamenti genici in un caso. Sono
state utilizzate solo 28/87(32,2%) ricombinazioni, quelle che nelle prove preliminari per
la valutazione della sonda allele-specifica hanno dato la più alta sensibilità e specificità.
Riarrangiamenti IGH (IGHVDJ e IGHDJ) sono stati rilevati in 16/87 casi (18,4%) e 9/16
(56,2%) sono stati utilizzati per l'analisi RQ-PCR. IGK-KDE è stato il secondo
riarrangiamento più comune identificatio, 21/87 (24,1%), e 2/21 riarrangiamenti (9,5%)
sono stati utilizzati per l'analisi RQ-PCR. I riarrangiamenti del TCRγ/δ sono stati
identificati in 42/87 casi (48,3%) e 12/42 (28,6%) sono risultati un utile marker per
l'analisi RQ-PCR. Infine, 8/87 (9,2%) riarrangiamenti del TCRα/β sono stati identificati e
5/8 (62,5%) sono stati utilizzati per la valutazione della MMR. Lo screening effettuato
sui prodotti WGA non ha dato differenze sia nel tipo di marker identificato che nelle
caratteristiche del riarrangiamento; le ricombinazioni identificate avevano le stesse
delezioni, inserzioni e regioni N del corrispondente DNA genomico (Tabella 6).
Il numero di nucleotidi N incorporati è un fattore importante per ottenere una sensibile
analisi di RQ-PCR; per i riarrangiamenti IGH, IGK-KDE, TCRγ/δ e TCRα/β, il numero
medio di nucleotidi N è stato 19, 5, 12 e 14 rispettivamente, identico nei target
individuati sia su DNA genomico che amplificato.
Nei test preliminari, sensibilità e specificità di 10-4/10-5 per l'analisi quantitativa con
almeno un marcatore, sono state ottenute nel 96,4% (27/28) degli oligonucleotidi
specifici disegnati. In un solo caso (3,6%), il primer identificato aveva una bassa
sensibilità (5x10-4).
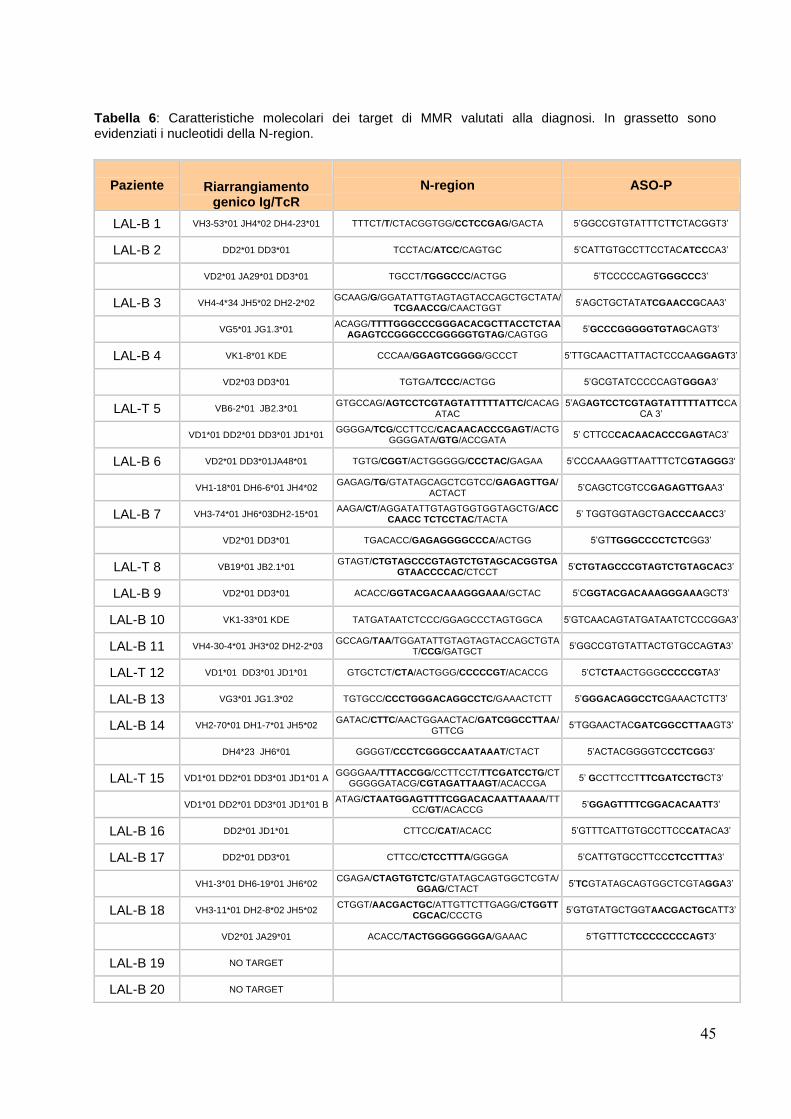
45
Tabella 6: Caratteristiche molecolari dei target di MMR valutati alla diagnosi. In grassetto sono evidenziati i nucleotidi della N-region.
Paziente
Riarrangiamento genico Ig/TcR
N-region ASO-P
LAL-B 1 VH3-53*01 JH4*02 DH4-23*01 TTTCT/T/CTACGGTGG/CCTCCGAG/GACTA 5’GGCCGTGTATTTCTTCTACGGT3’
LAL-B 2 DD2*01 DD3*01 TCCTAC/ATCC/CAGTGC 5’CATTGTGCCTTCCTACATCCCA3’
VD2*01 JA29*01 DD3*01 TGCCT/TGGGCCC/ACTGG 5’TCCCCCAGTGGGCCC3’
LAL-B 3 VH4-4*34 JH5*02 DH2-2*02 GCAAG/G/GGATATTGTAGTAGTACCAGCTGCTATA/
TCGAACCG/CAACTGGT 5’AGCTGCTATATCGAACCGCAA3’
VG5*01 JG1.3*01 ACAGG/TTTTGGGCCCGGGACACGCTTACCTCTAA
AGAGTCCGGGCCCGGGGGTGTAG/CAGTGG 5’GCCCGGGGGTGTAGCAGT3’
LAL-B 4 VK1-8*01 KDE CCCAA/GGAGTCGGGG/GCCCT 5’TTGCAACTTATTACTCCCAAGGAGT3’
VD2*03 DD3*01 TGTGA/TCCC/ACTGG 5’GCGTATCCCCCAGTGGGA3’
LAL-T 5 VB6-2*01 JB2.3*01 GTGCCAG/AGTCCTCGTAGTATTTTTATTC/CACAG
ATAC 5’AGAGTCCTCGTAGTATTTTTATTCCA
CA 3’
VD1*01 DD2*01 DD3*01 JD1*01 GGGGA/TCG/CCTTCC/CACAACACCCGAGT/ACTG
GGGGATA/GTG/ACCGATA 5’ CTTCCCACAACACCCGAGTAC3’
LAL-B 6 VD2*01 DD3*01JA48*01 TGTG/CGGT/ACTGGGGG/CCCTAC/GAGAA 5’CCCAAAGGTTAATTTCTCGTAGGG3'
VH1-18*01 DH6-6*01 JH4*02 GAGAG/TG/GTATAGCAGCTCGTCC/GAGAGTTGA/
ACTACT 5’CAGCTCGTCCGAGAGTTGAA3’
LAL-B 7 VH3-74*01 JH6*03DH2-15*01 AAGA/CT/AGGATATTGTAGTGGTGGTAGCTG/ACC
CAACC TCTCCTAC/TACTA 5’ TGGTGGTAGCTGACCCAACC3’
VD2*01 DD3*01 TGACACC/GAGAGGGGCCCA/ACTGG 5’GTTGGGCCCCTCTCGG3’
LAL-T 8 VB19*01 JB2.1*01 GTAGT/CTGTAGCCCGTAGTCTGTAGCACGGTGA
GTAACCCCAC/CTCCT 5’CTGTAGCCCGTAGTCTGTAGCAC3’
LAL-B 9 VD2*01 DD3*01 ACACC/GGTACGACAAAGGGAAA/GCTAC 5’CGGTACGACAAAGGGAAAGCT3’
LAL-B 10 VK1-33*01 KDE TATGATAATCTCCC/GGAGCCCTAGTGGCA 5’GTCAACAGTATGATAATCTCCCGGA3’
LAL-B 11 VH4-30-4*01 JH3*02 DH2-2*03 GCCAG/TAA/TGGATATTGTAGTAGTACCAGCTGTA
T/CCG/GATGCT 5’GGCCGTGTATTACTGTGCCAGTA3’
LAL-T 12 VD1*01 DD3*01 JD1*01 GTGCTCT/CTA/ACTGGG/CCCCCGT/ACACCG 5’CTCTAACTGGGCCCCCGTA3’
LAL-B 13 VG3*01 JG1.3*02 TGTGCC/CCCTGGGACAGGCCTC/GAAACTCTT 5’GGGACAGGCCTCGAAACTCTT3’
LAL-B 14 VH2-70*01 DH1-7*01 JH5*02 GATAC/CTTC/AACTGGAACTAC/GATCGGCCTTAA/
GTTCG 5’TGGAACTACGATCGGCCTTAAGT3’
DH4*23 JH6*01 GGGGT/CCCTCGGGCCAATAAAT/CTACT 5’ACTACGGGGTCCCTCGG3’
LAL-T 15 VD1*01 DD2*01 DD3*01 JD1*01 A GGGGAA/TTTACCGG/CCTTCCT/TTCGATCCTG/CT
GGGGGATACG/CGTAGATTAAGT/ACACCGA 5’ GCCTTCCTTTCGATCCTGCT3’
VD1*01 DD2*01 DD3*01 JD1*01 B ATAG/CTAATGGAGTTTTCGGACACAATTAAAA/TT
CC/GT/ACACCG 5’GGAGTTTTCGGACACAATT3’
LAL-B 16 DD2*01 JD1*01 CTTCC/CAT/ACACC 5’GTTTCATTGTGCCTTCCCATACA3’
LAL-B 17 DD2*01 DD3*01 CTTCC/CTCCTTTA/GGGGA 5’CATTGTGCCTTCCCTCCTTTA3’
VH1-3*01 DH6-19*01 JH6*02 CGAGA/CTAGTGTCTC/GTATAGCAGTGGCTCGTA/
GGAG/CTACT 5’TCGTATAGCAGTGGCTCGTAGGA3’
LAL-B 18 VH3-11*01 DH2-8*02 JH5*02 CTGGT/AACGACTGC/ATTGTTCTTGAGG/CTGGTT
CGCAC/CCCTG 5’GTGTATGCTGGTAACGACTGCATT3’
VD2*01 JA29*01 ACACC/TACTGGGGGGGGA/GAAAC 5’TGTTTCTCCCCCCCCAGT3’
LAL-B 19 NO TARGET
LAL-B 20 NO TARGET
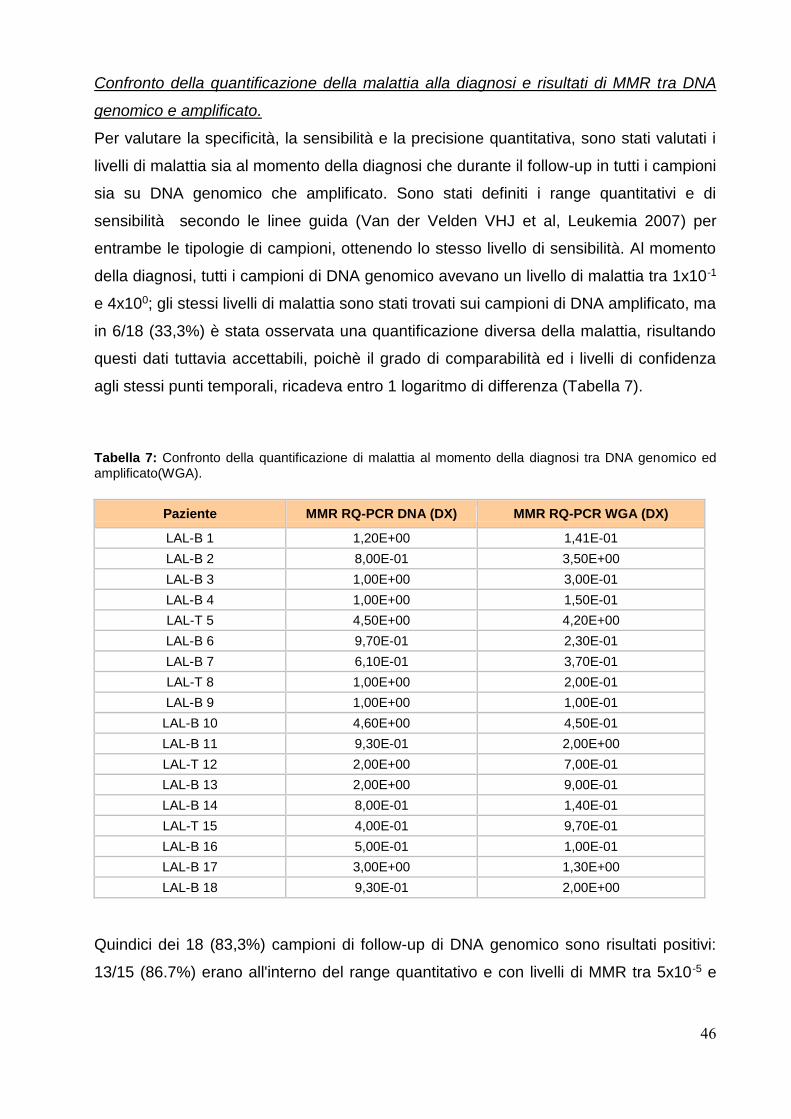
46
Confronto della quantificazione della malattia alla diagnosi e risultati di MMR tra DNA
genomico e amplificato.
Per valutare la specificità, la sensibilità e la precisione quantitativa, sono stati valutati i
livelli di malattia sia al momento della diagnosi che durante il follow-up in tutti i campioni
sia su DNA genomico che amplificato. Sono stati definiti i range quantitativi e di
sensibilità secondo le linee guida (Van der Velden VHJ et al, Leukemia 2007) per
entrambe le tipologie di campioni, ottenendo lo stesso livello di sensibilità. Al momento
della diagnosi, tutti i campioni di DNA genomico avevano un livello di malattia tra 1x10-1
e 4x100; gli stessi livelli di malattia sono stati trovati sui campioni di DNA amplificato, ma
in 6/18 (33,3%) è stata osservata una quantificazione diversa della malattia, risultando
questi dati tuttavia accettabili, poichè il grado di comparabilità ed i livelli di confidenza
agli stessi punti temporali, ricadeva entro 1 logaritmo di differenza (Tabella 7).
Tabella 7: Confronto della quantificazione di malattia al momento della diagnosi tra DNA genomico ed amplificato(WGA).
Paziente MMR RQ-PCR DNA (DX) MMR RQ-PCR WGA (DX)
LAL-B 1 1,20E+00 1,41E-01
LAL-B 2 8,00E-01 3,50E+00
LAL-B 3 1,00E+00 3,00E-01
LAL-B 4 1,00E+00 1,50E-01
LAL-T 5 4,50E+00 4,20E+00
LAL-B 6 9,70E-01 2,30E-01
LAL-B 7 6,10E-01 3,70E-01
LAL-T 8 1,00E+00 2,00E-01
LAL-B 9 1,00E+00 1,00E-01
LAL-B 10 4,60E+00 4,50E-01
LAL-B 11 9,30E-01 2,00E+00
LAL-T 12 2,00E+00 7,00E-01
LAL-B 13 2,00E+00 9,00E-01
LAL-B 14 8,00E-01 1,40E-01
LAL-T 15 4,00E-01 9,70E-01
LAL-B 16 5,00E-01 1,00E-01
LAL-B 17 3,00E+00 1,30E+00
LAL-B 18 9,30E-01 2,00E+00
Quindici dei 18 (83,3%) campioni di follow-up di DNA genomico sono risultati positivi:
13/15 (86.7%) erano all'interno del range quantitativo e con livelli di MMR tra 5x10-5 e
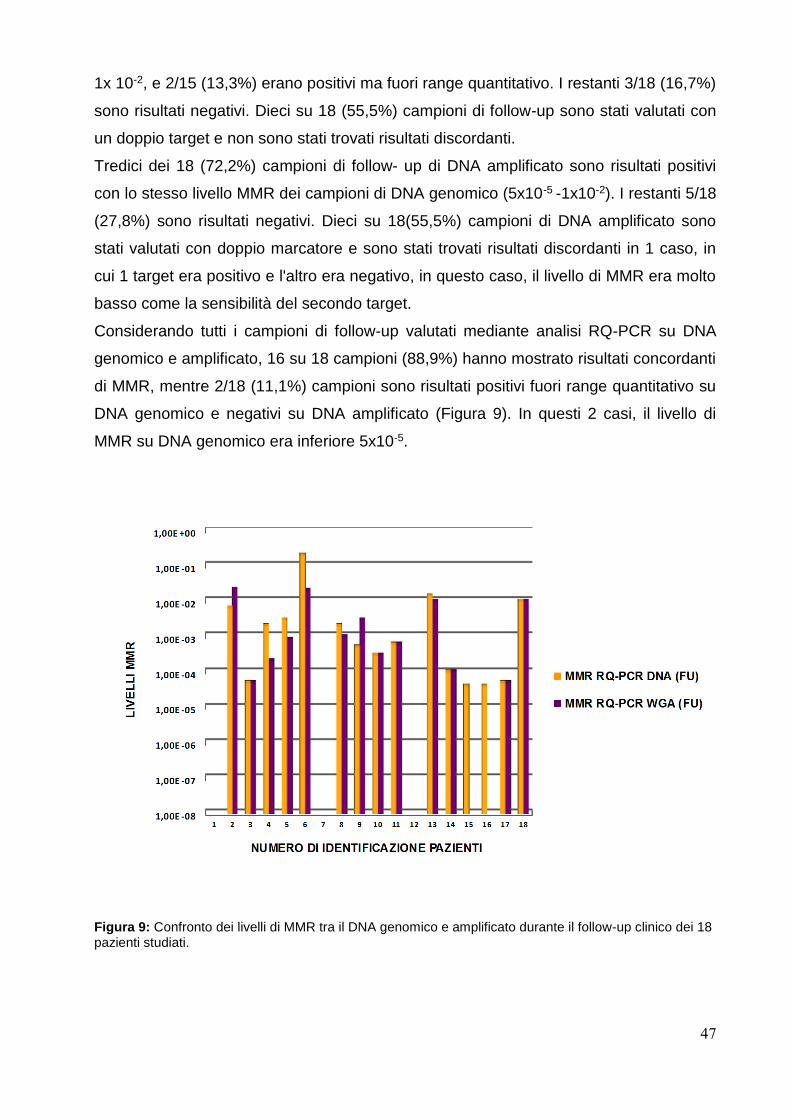
47
1x 10-2, e 2/15 (13,3%) erano positivi ma fuori range quantitativo. I restanti 3/18 (16,7%)
sono risultati negativi. Dieci su 18 (55,5%) campioni di follow-up sono stati valutati con
un doppio target e non sono stati trovati risultati discordanti.
Tredici dei 18 (72,2%) campioni di follow- up di DNA amplificato sono risultati positivi
con lo stesso livello MMR dei campioni di DNA genomico (5x10-5 -1x10-2). I restanti 5/18
(27,8%) sono risultati negativi. Dieci su 18(55,5%) campioni di DNA amplificato sono
stati valutati con doppio marcatore e sono stati trovati risultati discordanti in 1 caso, in
cui 1 target era positivo e l'altro era negativo, in questo caso, il livello di MMR era molto
basso come la sensibilità del secondo target.
Considerando tutti i campioni di follow-up valutati mediante analisi RQ-PCR su DNA
genomico e amplificato, 16 su 18 campioni (88,9%) hanno mostrato risultati concordanti
di MMR, mentre 2/18 (11,1%) campioni sono risultati positivi fuori range quantitativo su
DNA genomico e negativi su DNA amplificato (Figura 9). In questi 2 casi, il livello di
MMR su DNA genomico era inferiore 5x10-5.
Figura 9: Confronto dei livelli di MMR tra il DNA genomico e amplificato durante il follow-up clinico dei 18 pazienti studiati.

48
Conclusioni Lo studio della MMR è oggi eseguito nel contesto di diversi tumori come la LAL e le
malattie linfoproliferative, per la cui gestione i protocolli clinici sono basati sul
monitoraggio biologico della malattia.
Nelle LAL la MMR è fondamentale per guidare e modificare la gestione terapeutica dei
pazienti (Bruggemann M et al, Blood 2006). La riduzione del carico tumorale durante e
dopo il trattamento di induzione fornisce informazioni cruciali sulla risposta alla terapia e
rischio di recidiva. Questo permette di identificare pazienti "a basso rischio" e "ad alto
rischio", che possono trarre profitto dalla riduzione o dalla intensificazione della terapia,
rispettivamente (Vidriales MB et al, Blood 2003; Flohor T et al, Leukemia 2008; Bassan
R et al, Blood 2009).
L’approccio molecolare allo studio della MMR in pazienti affetti da LAL ha assunto
un’importanza rilevante, data anche la crescente rapidità ed il continuo affinamento
delle tecniche di analisi. Tale strategia consente di dimostrare una remissione
molecolare precoce in risposta alla terapia. Tuttavia un pre-requisito fondamentale è la
disponibilità di una quantità sufficiente di DNA germinale e di DNA delle cellule tumorali.
La mancanza di quantità e qualità appropriate di DNA porta ad una restrizione del tipo e
del numero di saggi genetici che possono essere eseguiti. L' amplificazione dell'intero
genoma (WGA) può rappresentare un metodo affidabile per superare questo problema.
Diversi lavori hanno riportato l'uso del WGA per l'analisi di mutazione dei tumori umani
(Hughes S et al, Cyt Gen Res 2004; Hughes S et al, Pro Biop Mol Biol 2005). Inoltre, il
WGA può essere utilizzato in altri campi, come la diagnosi genetica preimpianto (Jiao Z
et al, Pren Diagn 2003; Ao A et al, J Ass Rep Gen 1998; Kristjansson K et al, Nature
Gen 1994; Handyside AH et al, Mol Hum Rep 2004) e la diagnosi prenatale (Martel-
Petit V et al, Pren Diagn 2001; Sekizawa A et al, Obst Gynecol 1996; Lovisa L et al,
Rev Art Hum Mut 2006; Niap HT et al, J Obstet Gynaecol Res 2010; Nathan RT et al,
Mol Hum Rep 2011).
La valutazione dell'applicabilità della tecnologia WGA nel contesto dell'analisi della
MMR ha un importante valore clinico, pertanto in questo studio ne è stato applicato
l'uso all'analisi molecolare quantitativa in campioni di sangue midollare provenienti da
pazienti adulti affetti da LAL, valutati al momento della diagnosi e durante il follow-up
clinico, con lo scopo di confrontare i risultati ottenuti con quelli derivati da DNA
genomico originale, e stimarne applicabilità ed affidabilità in questo tipo di analisi. Lo

49
screening molecolare alla diagnosi non ha dato differenze nel tipo di target evidenziato;
i riarrangiamenti derivati da DNA genomico ed amplificato avevano le stesse delezioni,
inserzioni e regioni N. E' stata osservata una piccola discrepanza nella quantificazione
della malattia in 6 casi, ma il livello di differenza tra DNA genomico ed amplificato era
entro 1 logaritmo e quindi non significativo. Questi risultati concordanti hanno
dimostrato la robustezza del WGA, in questo punto di valutazione, poichè ha permesso
l'amplificazione e l'analisi di tutti i campioni diagnostici, riproducendo una copia fedele ai
campioni di DNA genomico, senza l'aggiunta di ulteriori mutazioni che avrebbero reso
l'analisi inaccettabile. Risultati simili sono stati ottenuti recentemente in una piccola
coorte di 10 pazienti affetti da leucemia mielomonocitica cronica in cui questa tecnica di
amplificazione ha permesso la genotipizzazione di 25 geni associati alla leucemia
nonostante le basse quantità di DNA disponibile (Rinke J et al,Clin Chem 2013).
Nella valutazione della MMR al follow up, 2 casi positivi analizzati da DNA genomico
sono risultati negativi con il WGA. In questi due casi, i livelli di MMR sul DNA genomico
erano al di sotto 5x10-5, va sottolineato che tale valutazione di MMR, diversa tra DNA
genomico e WGA, non si traduce in una diversa classificazione nel gruppo di rischio per
il paziente.
Se il livello di MMR è molto basso, l'amplificazione del DNA di una quota minima di
malattia non avviene con il metodo WGA. Anche se è stato dimostrato che
l'amplificazione con il WGA produce quantità accettabili di substrato amplificato
indipendentemente dalle quote di DNA genomico di partenza (Dean FB et al, Pro Nat
Aca Scien USA 2002), alcuni studi hanno riportato una correlazione positiva tra quantità
crescenti di DNA genomico e rendimenti più elevati di DNA amplificato ottenuti dopo
WGA. Inoltre, tracce di contaminanti o enzimi nei campioni di DNA genomico possono
competere all'interno della reazione di amplificazione del WGA e creare delle
interferenze, in particolare quando le concentrazioni di DNA stampo sono basse
(Bergen AW et al, BMC Biotech 2005).
Studi preliminari hanno anche dimostrato che i rendimenti del WGA dipendono dalla
qualità del DNA stampo usato nella reazione e che uno stampo di DNA povero può
diminuire la precisione dell'analisi molecolare [Bergen AW et al, BMC Biotech 2005;
Sun G et al, Legal Medicine (Tokyo)2005].
Il WGA rappresenta l'unico modo per aumentare significativamente la quantità di DNA
che può essere derivato da campioni clinici poveri (Hughes S et al, Pro Biop Mol Biol
2005). Inoltre, la possibilità di amplificare i campioni anche quando il livello di MMR è

50
molto basso potrebbe essere utile per l'identificazione dei cloni resistenti e per l'analisi
di mutazioni genetiche che inducono farmaco-resistenza.
In un recente studio (Rosenquinst R et al, Leukemia 2013) è stato riportato che il WGA
come fonte per l'analisi mutazionale sottostima la frequenza di alcune mutazioni (FLT3
e NPM1) in un'ampia coorte di pazienti con leucemia mieloide acuta in età pediatrica,
suggerendo una certa prudenza nell'utilizzare il WGA, in particolare per l'analisi di
determinate mutazioni. E' evidente che questo metodo deve essere considerato uno
strumento per amplificare il materiale senza sostituire il DNA genomico.
Per concludere, il WGA fornisce una fonte di DNA che permette:1) l'identificazione di
target fedeli per la valutazione della MMR in tutti i pazienti; 2) una quantificazione della
malattia sensibile e precisa al momento della diagnosi; 3) una quantificazione della
MMR paragonabile al DNA genomico per valori compresi tra 4x100 e 5x10-5.
Il metodo WGA apre la strada per ampliare notevolmente le possibilità di analisi in tutti
quei casi in cui la quantità di DNA è un fattore limitante.

51
Bibliografia
1. Ao, A., Wells, D., Handyside, A.H., Winston, R.M. & Delhanty, J.D.
Preimplantation genetic diagnosis of inherited cancer: familial adenomatous
polyposis coli. Journal of Assisted Reproduction and Genetics 1998; 15: 140-144.
2. Arden, B., Clark, S.P., Kabelitz, D., Mak, T.W. Human T-cell receptor variable
gene segment families. Immunogenetics 1995; 42: 455–500
3. Bassan, R., Spinelli., O, Oldani, E., Intermesoli, T., Tosi, M., Peruta, B., Rossi,
G., Borlenghi, E., Pogliani, E.M., Terruzzi, E., Fabris, P., Cassibba, V.,
Lambertenghi-Deliliers, G., Cortelezzi, A., Bosi, A., Gianfaldoni, G., Ciceri, F.,
Bernardi, M., Gallamini, A., Mattei, D., Di Bona, E., Romani, C., Scattolin, A.M.,
Barbui, T. & Rambaldi, A. Improved risk classification for risk-specific therapy based
on the molecular study of minimal residual disease (MRD) in adult acute
lymphoblastic leukemia (ALL). Blood 2009; 113: 4153-4162.
4. Beishuizen A, de Bruijn MA, Pongers-Willemse MJ, Verhoeven MA, van Wering
ER, Hählen K, Breit TM, de Bruin-Versteeg S, Hooijkaas H, van Dongen JJ.
Heterogeneity in junctional regions of immunoglobulin Kappa deleting element
rearrangements in B cell leukemias; a new molecular target for detection of minimal
residual disease. Leukemia 1997; 11: 2200-2207.
5. Bergen, A.W., Qi, Y., Haque, K.A., Welch, R.A. & Chanock, S.J. Effects of DNA
mass multiple displacement whole genome amplification and genotyping
performance. BMC Biotechnology 2005; 5, 24.
6. Blanco, L., Bernad, A., Lázaro, J.M., Martín, G., Garmendia, C. & Salas, M.
Highly efficient DNA synthesis by the phage phi 29 DNA polymerase. Symmetrical
mode of DNA replication. Journal of Biological Chemistry 1989; 264: 8935-8940.
7. Borowitz, M.J., Devidas, M., Hunger, S.P., Bowman, W.P., Carroll, A.J., Carroll,
W.L., Linda, S., Martin, P.L., Pullen, D.J., Viswanatha, D., Willman, C.L., Winick, N.
& Camitta, B.M.; Children's Oncology Group. Clinical significance of minimal
residual disease in childhood acute lymphoblastic leukemia and its relationship to
other prognostic factors: a Children's Oncology Group study. Blood 2008; 111:
5477-5485.
8. Brüggemann, M., Raff, T., Flohr, T., Gökbuget, N., Nakao, M., Droese, J.,

52
Lüschen, S., Pott, C., Ritgen, M., Scheuring, U., Horst, H.A., Thiel, E., Hoelzer, D.,
Bartram, C.R. & Kneba M; German Multicenter Study Group for Adult Acute
Lymphoblastic Leukemia. Clinical significance of minimal residual disease
quantification in adult patients with standard-risk acute lymphoblastic leukemia.
Blood 2006;107: 1116-23.
9. Brüggemann, M., Schrauder, A., Raff, T., Pfeifer, H., Dworzak, M., Ottmann,
O.G., Asnafi, V., Baruchel, A., Bassan, R., Benoit, Y., Biondi, A., Cavé, H.,
Dombret, H., Fielding, A.K., Foà, R., Gökbuget, N., Goldstone, A.H., Goulden, N.,
Henze, G., Hoelzer, D., Janka-Schaub, G.E., Macintyre, E.A., Pieters, R.,
Rambaldi, A., Ribera, J.M., Schmiegelow, K., Spinelli, O., Stary, J., von
Stackelberg, A., Kneba, M., Schrappe, M. & van Dongen, J.J.; European Working
Group for Adult Acute Lymphoblastic Leukemia (EWALL); International Berlin-
Frankfurt-Münster Study Group (I-BFM-SG). Standardized MRD quantification in
European ALL trials: proceedings of the Second International Symposium on MRD
assessment in Kiel, Germany, 18-20 September 2008. Leukemia 2008; 24: 521-
535.
10. Camacho FI, Algara P, Rodríguez A, Ruíz-Ballesteros E, Mollejo M, Martínez N,
Martínez-Climent JA, González M, Mateo M, Caleo A, Sánchez-Beato M,
Menárguez J, García-Conde J, Solé F, Campo E, Piris MA. Molecular heterogeneity
in MCL defined by the use of specific VH genes and the frequency of somatic
mutations. Blood 2003; 101: 4042–4046.
11. Campana, D., Pui, C.H. Detection of minimal residual disease in acute
leukaemia: methodologic advances and clinical significance. Blood 1995; 85:1416-
1434.
12. Cazzaniga, G., Gaipa, G., Rossi, V., Biondi, A. Minimal residual disease as a
surrogate marker for risk assignment to all patients. Reviews in Clinical and
Experimental Hematology. 2003; 7(3):292-323.
13. Cazzaniga, G., & Biondi, A. Molecular monitoring of childhood acute
lymphoblastic leukemia using antigen receptor gene rearrangements and
quantitative polymerase chain reaction technology. Haematologica 2005; 90: 382-
390.
14. Charmley, P., Wei, S., Concannon, P. Polymorphisms in the TCRB- V2 gene
segments localize the Tcrb orphon genes to human chromosome 9p21.

53
Immunogenetics 1993; 38: 283–286.
15. Cimino, G., Elia, L., Rapanotti, M.C., Sprovieri, T., Mancini, M., Cuneo, A.,
Mecucci, C., Fioritoni, G., Carotenuto, M., Morra, E., Liso, V., Annino, L., Saglio, G.,
De Rossi, G., Foà, R., Mandelli, F. A prospective study of residual-disease
monitoring of the ALL1/AF4 transcript in patients with t(4;11) acute lymphoblastic
leukemia. Blood. 2000 Jan 1;95(1):96-101.
16. Cimino, G., Pane, F., Elia, L., Finolezzi, E., Fazi, P., Annino, L., Meloni, G.,
Mancini, M., Tedeschi, A., Di Raimondo, F., Specchia, G., Fioritoni, G., Leoni, P.,
Cuneo, A., Mecucci, C., Saglio, G., Mandelli, F., Foà, R.; GIMEMA Leukemia
Working Party.The role of BCR/ABL isoforms in the presentation and outcome of
patients with Philadelphia-positive acute lymphoblastic leukemia: a seven-year
update of the GIMEMA 0496 trial. Haematologica. 2006; 91(3):377-80.
17. Daly, J., Licence, S., Nanou, A., Morgan, G., Mårtensson, I.L. Transcription of
productive and nonproductive VDJ-recombined alleles after IgH allelic exclusion.
The EMBO Journal. 2007; 26(19):4273-82.
18. David, G., Schatz & Yanhong, Ji. Recombination centres and the orchestration of
V(D)J recombination. Nature Reviews Immunology April 2011;11: 251-263.
19. Dean, F.B., Hosono, S., Fang, L., Wu, X., Faruqi, A.F., Bray-Ward, P., Sun, Z.,
Zong, Q., Du, Y., Du, J., Driscoll, M., Song, W., Kingsmore, S.F., Egholm, M. &
Lasken RS. Comprehensive human genome amplification using multiple
displacement amplification. Proceedings of the National Academy of Sciences USA
2002, 99: 5261–5266.
20. Esteban, J.A., Salas, M. & Blanco L. (1993) Fidelity of phi 29 DNA polymerase.
Comparison between protein-primed initiation and DNA polymerization. Journal of
Biological Chemistry 2002; 268: 2719-2722.
21. Farner, N.L., Dorner, T., Lipsky, P.E. Molecular mechanisms and selection
influence the generation of the human V lambda J lambda repertoire. The Journal
of Immunology 1999; 162: 2137–2145.
22. Foà, R. and Vitale, A. Towards an integrated classification of adult acute
lymphoblastic leukaemia. Reviews in Clinical and Experimental Hematology 2002:
6(2):181-199.
23. Foà R, Vitale A, Mancini M, Cuneo A, Mecucci C, Elia L, Lombardo R, Saglio G,
Torelli G, Annino L, Specchia G, Damasio E, Recchia A, Di Raimondo F, Morra E,
Volpe E, Tafuri A, Fazi P, Hunger SP, Mandelli F. E2A-PBX1 fusion in adult acute

54
lymphoblastic leukaemia: biological and clinical features. British Journal of
Haematology. 2003 Feb; 120(3):484-7.
24. Foà, R., Vitale, A., Chiaretti, S and Guarini, A. A broad and integrated diagnostic
work-up for a modern management of Acute Lymphoblastic Leukaemia (ALL).
Hematology 2005; 10 Suppl 1: 55-62.
25. Foroni, L. and Hoffbrand, A.V. Molecular analysis of minimal residual disease in
adult acute lymphoblastic leukaemia. Best Practice & Research Clinical
Haematology 2002; 15: 71-90.
26. Flohr, T., Schrauder, A., Cazzaniga, G., Panzer-Grümayer, R., van der Velden,
V., Fischer, S., Stanulla, M., Basso, G., Niggli, F.K., Schäfer, B.W., Sutton, R.,
Koehler, R., Zimmermann, M., Valsecchi, M.G., Gadner, H., Masera, G., Schrappe,
M., van Dongen, J.J., Biondi, A. & Bartram, C.R.; International BFM Study Group (I-
BFM-SG). Minimal residual disease-directed risk stratification using real-time
quantitative PCR analysis of immunoglobulin and T-cell receptor gene
rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for
childhood acute lymphoblastic leukemia. Leukemia 2008; 22: 771-782.
27. Gawad C, Pepin F, Carlton VE, Klinger M, Logan AC, Miklos DB, Faham M,
Dahl G, Lacayo N. Massive evolution of the immunoglobulin heavy chain locus in
children with B precursor acute lymphoblastic leukemia. Blood 2012; 120: 4407–
4417.
28. Ghia, P., ten Boekel, E., Sanz, E., de la Hera, A., Rolink, A., Melchers,
F.Ordering of human bone marrow B lymphocyte precursors by single-cell
polymerase chain reaction analyses of the rearrange-ment status of the
immunoglobulin H and L chain gene loci. The Journal of Experimental Medicine
1996; 184: 2217–2229.
29. Gökbuget, N., Kneba, M., Raff, T., Trautmann, H., Bartram, C.R., Arnold, R.,
Fietkau, R., Freund, M., Ganser, A., Ludwig, W.D., Maschmeyer, G., Rieder, H.,
Schwartz, S., Serve, H., Thiel, E., Brüggemann, M. & Hoelzer, D. Adult patients
with acute lymphoblastic leukemia and molecular failure display a poorprognosis
and are candidates for stem cell transplantation and targeted therapies. German
Multicenter Study Group for Adult Acute Lymphoblastic Leukemia. Blood 2012, 120:
1868-76.
30. Griesinger F, Greenberg JM, Kersey JH. T cell receptor gamma and delta

55
rearrangments in hematologic malignancies. Relantionship to lymphoid
differentiation. The Journal of Clinical Investigation. 1989:506-516.
31. Guglielmi C, Cordone I, Boecklin F, Masi S, Valentini T, Vegna ML, Ferrari A,
Testi AM, Foa R. Immunophenotype of adult and childhood acute lymphoblastic
leukemia: changes at first relapse and clinico-prognostic implications. Leukemia.
1997 Sep;11(9):1501-7.
32. Handyside, A.H., Robinson, M.D., Simpson, R.J., Omar, M.B., Shaw, M.A.,
Grudzinskas, J.G. & Rutherford A. Isothermal whole genome amplification from
single and small numbers of cells: a new era for preimplantation genetic diagnosis
of inherited disease. Molecular Human Reproduction 2004; 10: 767–772.
33. Harrison, C.J., Foroni, L. Cytogenetics and molecular genetics of acute
lymphoblasitc leukaemia. Reviews in Clinical and Experimental Hematology 2002;
6:91-113.
34. Harrison, C.J. Cytogenetics of pediatric and adolescent acute lymphoblastic
leukemia. British Journal of Haematology 2009, 144:147-56.
35. Hertzberg L, Vendramini E, Ganmore I, Cazzaniga G, Schmitz M, Chalker J,
Shiloh R, Iacobucci I, Shochat C, Zeligson S, Cario G, Stanulla M, Strehl S, Russell
LJ, Harrison CJ, Bornhauser B, Yoda A, Rechavi G, Bercovich D, Borkhardt A,
Kempski H, te Kronnie G, Bourquin JP, Domany E, Izraeli S. Down syndrome acute
lymphoblastic leukemia: highly heterogeneous disease in which aberrant
expression of CRLF2 is associated with mutated JAK2: a report of I- BFM Study
Group. Blood 2010,115:1006-17.
36. Hoelzer D, Gökbuget N, Ottmann O, Pui CH, Relling MV, Appelbaum FR, van
Dongen JJ, Szczepański T. Acute lymphoblastic leukaemia. Hematology 2002;162-
192.
37. Hübner, S., Cazzaniga, G., Flohr, T., van der Velden, V.H., Konrad, M.,
Pötschger, U., Basso, G., Schrappe, M., van Dongen, J.J., Bartram, C.R., Biondi,
A., Panzer-Grümayer, E.R. High incidence and unique features of antigen receptor
gene rearrangements in TEL-AML1-positive leukemias. Leukemia 2004
Jan;18(1):84-91.
38. Hughes, S., Arneson, N., Done, S. & Squire, J. The use of whole genome
amplification in the study of human disease. Progress in Biophysics and Molecular
Biology 2005; 88: 173-189.

56
39. Hughes, S., Lim, G., Beheshti, B., Bayani, J., Marrano, P., Huang, A. & Squire,
J.A. Use of whole genome amplification and comparative genomic hybridisation to
detect chromosomal copy number alterations in cell line material and tumour tissue.
Cytogenetic and Genome Research 2004; 105: 18-24.
40. Ignatovich, O., Tomlinson, I.M., Popov, A.V., Bruggemann, M., Winter, G.
Dominance of intrinsic genetic factors in shaping the human immunoglobulin
Vlambda repertoire. Journal of Molecular Biology 1999; 294: 457– 465.
41. Inlay, M.A., Gao, H.H., Odegard, V.H., Lin, T., Schatz, D.G., Xu, Y. Roles of the
Ig kappa light chain intronic and 3' enhancers in Igk somatic hypermutation. The
Journal of Immunology 2006 Jul 15;177(2):1146-51.
42. Jackson, K.J., Gaeta, B., Sewell, W., Collins, A.M. Exonuclease activity and P
nucleotide addition in the generation of the expressed immunoglobulin repertoire.
BMC Immunology 2004 Sep 2;5:19.
43. Jayanta Chaudhuri & Frederick, W. Alt. Class-switch recombination: interplay of
transcription, DNA deamination and DNA repair. Nature Reviews Immunology July
2004;4, 541-552.
44. Jiao, Z., Zhou, C., Li, J., Shu, Y., Liang, X., Zhang, M. & Zhuang, G. Birth of
healthy children after preimplantation diagnosis of betathalassemia by whole-
genome amplification. Prenatal Diagnosis 2003; 23: 646–651.
45. Jores, R., Meo, T. Few V gene segments dominate the T cell receptor beta-chain
repertoire of the human thymus. The Journal of Immunology 1993; 151: 6110–
6122.
46. Jung D., Giallourakis C., Mostoslavsky R., Alt F.W. Mechanism and Control of
V(D)J Recombination at the Immunoglobulin Heavy Chain Locus. Annual Review of
Immunology. 2006; 24: 542-70.
47. Kode J, Dudhal N, Banavali S, Advani S Chiplunkar S. Clonal T-cell receptor
gamma and delta gene rearrangments in T-cell acute lymphoblastic leukemia at
diagnosis: predictor of prognosis and response to chemotherapy. LeuKemia
lymphoma 2004; 45; 125-133.
48. Krangel, M. S., C. Hernandez-Munain, P. Lauzurica, M. McMurry, J. L. Roberts,
and X. P. Zhong. Developmental regulation of V(D)J recombination at the TCR α/δ
locus. Immunological Reviews1998; 165: 131–147.
49. Kristjansson, K., Chong, S.S., Van den Veyver, I.B., Subramanian, S., Snabes,
M.C. & Hughes, M.R. Preimplantation single cell analyses of dystrophin gene

57
deletions using whole genome amplification. Nature Genetics 1994; 6: 19–23.
50. Langerak, A.W., Szczepański, T., van der Burg, M., Wolvers-Tettero, I.L. & van
Dongen, J.J. Heteroduplex PCR analysis of rearranged T cell receptor genes for
clonality assessment in suspect T cell proliferations. Leukemia 1997; 11: 2192-
2199.
51. Langerak A.W., Wolvers-Tettero I.L.M., van Dongen J.J.M. Detection of T cell
receptor beta (TCRB) gene rearrangement patterns in T cell malignancies by
Southern blot analysis. Leukemia 1999; 13: 965–974.
52. Langerak A.W., van Dongen J.J.M. Recombination in the human IGK locus.
Critical Reviews in Immunology. 2006; 26(1):23-42.
53. Lovisa, L. & Syva¨nen, A.C. Multiple Displacement Amplification To Create a
Long-Lasting Source of DNA for Genetic Studies. Review Article Human Mutation
2006; 27: 603-614.
54. Martel-Petit, V., Petit, C., Marchand, M., Fleurentin, A., Fontaine, B., Miton, A.,
Lemarie, P., Philippe, C. & Jonveaux, P. Use of the Kleihauer test to detect fetal
erythroblasts in the maternal circulation. Prenatal Diagnosis 2001; 21: 106-111.
55. Mullighan C. Genomic Profiling of B-progenitor acute lymphoblastic leukemia.
Best Practise & Research Clinical Haematology 2011; 24:489-503.
56. Nirmala K1, Rajalekshmy KR, Raman SG, Shanta V, Rajkumar T. PCR-
heteroduplex analysis of TCRγδ and TAL-1 deletions in T-acute lymphoblastic
leukemias: implications in the detection of minimal residual disease. Leukemia
Research. 2002; 26:335-343.
57. Nathan R. Treff, Jing Su1, Xin Tao, Lesley E. Northrop, and Richard T. Scott Jr.
Single-cell whole-genome amplification technique impacts the accuracy of SNP
microarray-based genotyping and copy number analyses. Molecular Human
Reproduction 2011;17: 335–343.
58. Niap H. Tan, Rodger Palmer and Rubin Wang. Evaluation of the efficacy of
constitutional array-based comparative genomic hybridization in the diagnosis of
aneuploidy using genomic and amplified DNA. The Journal of Obstetric and
Gynaecology Research 2010; 36:19–26.
59. Paez, J.G., Lin, M., Beroukhim, R., Lee, J.C., Zhao, X., Richter, D.J., Gabriel, S.,
Herman, P., Sasaki, H., Altshuler, D., Li, C., Meyerson, M. & Sellers, W.R. Genome
coverage and sequence fidelity of phi29 polymerase-based multiple strand
displacement whole genome amplification. Nucleic Acids Research 2004; 32, 71.

58
60. Pongers-Willemse, M.J., Seriu, T., Stolz, F., d’Aniello, E., Gameiro, P., Pisa, P.,
Gonzalez, M., Bartram, C.R., Panzer-Grümayer, E.R., Biondi, A., San Miguel, J.F.
& van Dongen, J.J.M. Primers and protocols for standardized detection of minimal
residual disease in acute lymphoblastic leukemia using immunoglobulin and T cell
receptor gene rearrangements and TAL1 delitions as PCR target: report of the
BIOMED-1 Concerted Action: investigation of minimal residual disease in acute
leukemia. Leukemia 1999; 13, 110-118.
61. Rinke, J., Schafer, V., Schimdt, M., Ziermann, J., Kohlmann, A., Hochhaus, A. &
Ernsta, T. Genotyping of 25 leukemia-associated genes in a single work flow by
next-generation sequencing technology with low amounts of input template DNA.
Clinical Chemistry 2013; 59(8): 1238-1250.
62. Rosequinst, R., Ehrencrona, H., Hasle, H., Palle, J., & Kanduri, M. Whole-
genome-amplified DNA as a source for mutational analysis understimates the
frequency of mutations in pediatric acute myeloid leukemia. Leukemia 2013; 27:
510-512.
63. Rowen, L., Koop B.F., Hood, L. The complete 685-kilobase DNA sequence of the
human beta T cell receptor locus. Science 1996; 272: 1755–1762.
64. Schneider, M., Panzer, S., Stolz, F., Fischer, S., Gadner, H., Panzer- Grumayer,
E.R. Crosslineage TCR delta rearrangements occur shortly after the DJ joinings of
the IgH genes in childhood precursor B ALL and display age-specific
characteristics. British Journal of Haematology 1997; 99: 115–121.
65. Sekizawa, A., Watanabe, A., Kimura, T., Saito, H., Yanaihara, T. & Sato, T.
Prenatal diagnosis of the fetal RhD blood type using a single fetal nucleated
erythrocyte from maternal blood. Obstetrics & Gynecology 1996; 87, 501-505.
66. Sen R., Oltz E. Genetic and epigenetic regulation of IgH gene assembly. Current
Opinion in Immunology. 2006 Jun;18(3):237-42.
67. Sermon, K., Lissens, W., Joris, H., Van Steirteghem, A. & Liebaers, I. Adaptation
of the primer extension preamplification (PEP) reaction for preimplantation
diagnosis: single blastomere analysis using short PEP protocols. Molecular Human
Reproduction 1996; 2: 209–212.
68. Sun G, Kaushal R, Pal P, Wolujewicz M, Smelser D, Cheng H, Lu M,
Chakraborty R, Jin L, Deka R. Whole-genome amplification: relative efficiencies of
the current methods. Legal Medicine (Tokyo) 2005; 7: 279-286.

59
69. Szczepański, T., van der Velden, V.H., Raff, T., Jacobs, D.C., van Wering, E.R.,
Brüggemann, M., Kneba, M. & van Dongen, J.J. Comparative analysis of T-cell
receptor gene rearrangements at diagnosis and relapse of T-cell acute
lymphoblastic leukemia (T-ALL) shows high stability of clonal markers for
monitoring of minimal residual disease and reveals the occurrence of second T-
ALL. Leukemia 2003; 17: 2149-2156.
70. Szczepański T, Langerak AW, Wolvers-Tettero IL, Ossenkoppele GJ, Verhoef G,
Stul M, Petersen EJ, de Bruijn MA, van't Veer MB, van Dongen JJ. Immunoglobulin
and T cell receptor gene rearrangement patterns in acute lymphoblastic leukemia
are less mature in adults than in children: implications for selection of PCR targets
for detection of minimal residual disease. Leukemia 1998; 12: 1081–1088.
71. Szczepański T, Beishuizen A, Pongers-Willemse MJ, Hählen K, Van Wering ER,
Wijkhuijs AJ, Tibbe GJ, De Bruijn MA, Van Dongen JJ. Cross-lineage T-cell
receptor gene rearrangements occur in more than ninety percent of childhood
precursor-B-acute lymphoblastic leukemias: alterna- tive PCR targets for detection
of minimal residual disease. Leukemia 1999; 13: 196–205.
72. Szczepański, T., Willemse, M.J., Brinkhof, B., van Wering, E.R., van der Burg, M.
& van Dongen, J.J. Comparative analysis of Ig and TCR gene rearrangements at
diagnosis and at relapse of childhood precursor-B-ALL provides improved
strategies for selection of stable PCR targets for monitoring of minimal residual
disease. Blood 2002; 99: 2315-2323.
73. Tan, N.H., Palmer, R. & Wang, R. Evaluation of the efficacy of constitutional
array-based comparative genomic hybridization in the diagnosis of aneuploidy
using genomic and amplified DNA. Journal of Obstetrics and Gynaecology
Research 2010; 36: 19–26.
74. Treff, N.R., Su, J., Tao, X., Northrop, L.E., & Scott Jr., R.T. Single-cell whole-
genome amplification technique impacts the accuracy of SNP microarray-based
genotyping and copy number analyses. Molecular Human Reproduction 2011; 17:
335–343.
75. Tümkaya T1, van der Burg M, Garcia Sanz R, Gonzalez Diaz M, Langerak AW,
San Miguel JF, van Dongen JJ. Immunoglobulin lambda isotype gene
rearrangements in B-cell malignancies. Leukemia 2001; 15: 121–127.
76. van den Beemd R1, Boor PP, van Lochem EG, Hop WC, Langerak AW,

60
Wolvers-Tettero IL, Hooijkaas H, van Dongen JJ. Flow ctometric analysis of the Vb
repertoire in healthy controls. Cytometry 2000; 40: 336–345.
77. van der Burg M, Barendregt BH, van Gastel-Mol EJ, Tumkaya T, Langerak AW,
van Dongen JJ. Unraveling of the polymorphic C lambda 2–C lambda 3
amplification and the Ke+Oz-polymorph- ism in the human Ig lambda locus. The
Journal of Immunology 2002; 169: 271– 276.
78. van der Burg M, Tumkaya T, Boerma M, de Bruin-Versteeg S, Langerak AW, van
Dongen JJM. Ordered recombination of immunoglobulin light chain genes occurs at
the IGK locus but seems less strict at the IGL locus. Blood 2001; 97: 1001–1008.
79. Van der Velden VHJ, Willemse MJ, van der Schoot CE, van Wering ER, van
Dongen JJM. Immunoglobulin kappa deleting element rearrangements in precursor-
B acute lymphoblastic leukemia are stable targets for detection of minimal residual
disease by real-time quantitative PCR. Leukemia 2002; 16: 928– 936.
80. van der Velden, V.H., Cazzaniga, G., Schrauder, A., Hancock, J., Bader, P.,
Panzer-Grumayer, E.R., Flohr, T., Sutton, R., Cave, H., Madsen, H.O., Cayuela,
J.M., Trka, J., Eckert, C., Foroni, L., Zur Stadt, U., Beldjord, K., Raff, T., van der
Schoot, C.E. & van Dongen, J.J.; European Study Group on MRD detection in ALL
(ESG-MRD-ALL). Analysis of minimal residual disease by Ig/TCR gene
rearrangements: guidelines for interpretation of real-time quantitative PCR data.
Leukemia 2007; 21: 604-611.
81. van der Velden, V.H., Hochhaus, A., Cazzaniga, G., Szczepanski, T., Gabert, J.
& van Dongen, J.J. Detection of minimal residual disease in hematologic
malignancies by real-time quantitative PCR: principles, approaches, and laboratory
aspects. Leukemia 2003; 17: 1013-1034.
82. van der Velden, V.H., Wijkhuijs, J.M. & van Dongen, J.J. Non-specific
amplification of patient-specific Ig/TCR gene rearrangements depends on the time
point during therapy: implications for minimal residual disease monitoring.
Leukemia 2008; 22: 641-644.
83. van Dongen JJM, Wolvers-Tettero ILM. Analysis of immunoglobu- lin and T cell
receptor genes. Part II: possibilities and limitations in the diagnosis and
management of lymphoproliferative diseases and related disorders. Clin Chim Acta
1991; 198: 93–174.
84. van Dongen JJM, Wolvers-Tettero ILM. Analysis of immunoglo- bulin and T cell

61
receptor genes. Part I: basic and technical aspects. Clin Chim Acta 1991; 198: 1–
91.
85. JJM van Dongen, AW Langerak, M Bru ̈ggemann, PAS Evans, M Hummel, FL
Lavender,E Delabesse, F Davi,E Schuuring, R Garc'ıa-Sanz, JHJM van Krieken, J
Droese, D Gonza ́lez, C Bastard, HE White, M Spaargaren, M Gonza ́lez, A
Parreira, JL Smith, GJ Morgan, M Kneba and EA Macintyre. Design and
standardization of PCR primers and protocols for detection of clonal
immunoglobulin and T-cell receptor gene recombinations in supect
lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936.
Leukemia 2003 17, 2257-2317.
86. Verschuren MC, Wolvers-Tettero IL, Breit TM, van Dongen JJ. T- cell receptor V
delta–J alpha rearrangements in human thymo- cytes: the role of V delta–J alpha
rearrangements in T-cell receptor-delta gene deletion. Immunology 1998; 93: 208–
212.
87. Vidriales, M.B., Pérez, J.J., López-Berges, M.C., Gutiérrez, N., Ciudad, J., Lucio,
P., Vazquez, L., García-Sanz, R., del Cañizo, M.C., Fernández-Calvo, J., Ramos,
F., Rodríguez, M.J., Calmuntia, M.J., Porwith, A., Orfao, A. & San-Miguel, J.F.
Minimal residual disease in adolescent (older than 14 years) and adult acute
lymphoblastic leukemias: early immunophenotypic evaluation has high clinical
value. Blood 2003; 101: 4695-4700.
88. Vitale A, Guarini A, Chiaretti S and Foà R. The changing scene of adult acute
lymphoblastic leukaemia. Current Opinion in Oncology 2006; 18:652-659.
89. Wei S, Charmley P, Robinson MA, Concannon P. The extent of the human
germline T-cell receptor V beta gene segment repertoire. Immunogenetics 1994;
40: 27–36.

62
II° Progetto: Confronto di due strategie di RQ-PCR per la valutazione della malattia minima residua nelle malattie linfoproliferative: correlazione tra lo stato mutazionale dei geni delle immunoglobuline e performance di RQ-PCR.

63
La Leucemia Linfatica Cronica La Leucemia Linfatica cronica (LLC) è una neoplasia ematologica caratterizzata da
un’espansione clonale di una popolazione linfocitaria CD19+/CD5+ (figura 1)(Chiorazzi
N. et al, Hematology 2006).
Negli ultimi anni, grazie alla identificazione delle diverse caratteristiche biologiche
ascrivibili alla cellula leucemica della LLC, è notevolmente cambiata la concezione di
tale patologia. Per lungo tempo si pensava infatti che le cellule di LLC derivassero da
linfociti B immaturi, immunologicamente incompetenti e con un basso potenziale
proliferativo.
Oggi è stato invece ampiamente dimostrato in diversi studi, che le cellule di LLC non
sono in uno stato di quiescenza (Messmer B.T. et al, J Clin Invest 2005) bensì
presentano un determinato turn-over ed una anormalità dei geni che soprassiedono alla
funzione della apoptosi. La nuova concezione della LLC pertanto, vede tale patologia
come una malattia da accumulo non più statica ma dinamica (Chiorazzi N. et al, Annu
Rev Immunol 2003), caratterizzata dalla presenza di cellule mature, antigenicamente
esperte e immunologicamente competenti.
Il 95% dei pazienti presenta una proliferazione clonale che coinvolge i linfociti di tipo B
(LLC-B), mentre solo una piccola percentuale di pazienti ha una proliferazione clonale
di linfociti di tipo T (LLC-T). Anche dopo il riconoscimento dei numerosi disordini
linfoproliferativi cronici che coinvolgono i linfociti B maturi, la LLC rimane di gran lunga
la patologia più diffusa.
E’ ,infatti, la forma di leucemia più comune nella popolazione adulta dei paesi
occidentali, dove rappresenta circa il 25-30% di tutte le leucemie. La LLC è più
frequente negli uomini rispetto alle donne con un rapporto di circa 2:1 e il tasso di
incidenza annua è di circa 2-6 nuovi casi per 100,000 abitanti ogni anno ed aumenta
con l’età (Rozman C. et al, N Engl J Med 1995).
Pur essendo l’età media alla diagnosi di 65 anni, recentemente è stato riportato un
aumento nell’incidenza tra i giovani in quanto un terzo di nuovi casi sono diagnosticati
nella popolazione al di sotto dei 55 anni di età (De Lima M. et al, Semin Oncol 1998).
I decessi non correlati alla LLC e lo sviluppo di seconde neoplasie sono predominanti
nei pazienti anziani mentre gli effetti diretti della leucemia prevalgono nei pazienti
giovani.
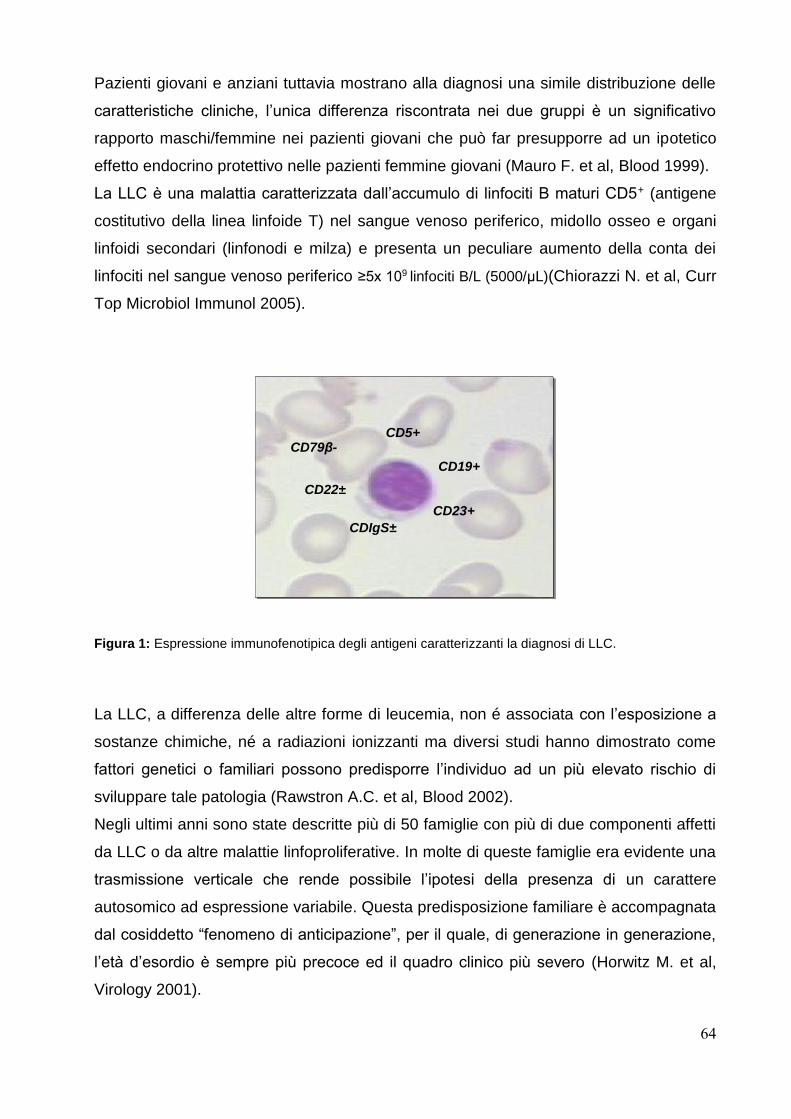
64
Pazienti giovani e anziani tuttavia mostrano alla diagnosi una simile distribuzione delle
caratteristiche cliniche, l’unica differenza riscontrata nei due gruppi è un significativo
rapporto maschi/femmine nei pazienti giovani che può far presupporre ad un ipotetico
effetto endocrino protettivo nelle pazienti femmine giovani (Mauro F. et al, Blood 1999).
La LLC è una malattia caratterizzata dall’accumulo di linfociti B maturi CD5+ (antigene
costitutivo della linea linfoide T) nel sangue venoso periferico, midollo osseo e organi
linfoidi secondari (linfonodi e milza) e presenta un peculiare aumento della conta dei
linfociti nel sangue venoso periferico ≥5x 109 linfociti B/L (5000/μL)(Chiorazzi N. et al, Curr
Top Microbiol Immunol 2005).
Figura 1: Espressione immunofenotipica degli antigeni caratterizzanti la diagnosi di LLC.
La LLC, a differenza delle altre forme di leucemia, non é associata con l’esposizione a
sostanze chimiche, né a radiazioni ionizzanti ma diversi studi hanno dimostrato come
fattori genetici o familiari possono predisporre l’individuo ad un più elevato rischio di
sviluppare tale patologia (Rawstron A.C. et al, Blood 2002).
Negli ultimi anni sono state descritte più di 50 famiglie con più di due componenti affetti
da LLC o da altre malattie linfoproliferative. In molte di queste famiglie era evidente una
trasmissione verticale che rende possibile l’ipotesi della presenza di un carattere
autosomico ad espressione variabile. Questa predisposizione familiare è accompagnata
dal cosiddetto “fenomeno di anticipazione”, per il quale, di generazione in generazione,
l’età d’esordio è sempre più precoce ed il quadro clinico più severo (Horwitz M. et al,
Virology 2001).
CD23+
CD22±
CD79β-
CDIgS±
CD19+
CD5+

65
Il rischio globale di sviluppare questa malattia è 7-9 volte maggiore tra familiari di primo
grado di pazienti affetti da LLC, rispetto al resto della popolazione.
Dal punto di vista clinico e biologico la LLC con predisposizione familiare è molto simile
ai casi sporadici, anche se è stata riscontrata un’alta proporzione di femmine rispetto ai
pazienti maschi con storia familiare di LLC. Dal momento che la LLC è più
frequentemente diagnosticata nei maschi è possibile presupporre che le femmine
possano avere un numero maggiore di geni o geni più penetranti che predispongono di
più all’insorgenza della LLC (Mauro F.R. et al, Haematologica 2006).
La LLC è spesso asintomatica, nel 25-30% dei casi non sono riscontrabili né sintomi
clinici né segni obiettivi di malattia; in tali casi la diagnosi di LLC avviene in seguito ad
accertamenti casuali di laboratorio.
Le più comuni caratteristiche cliniche includono linfoadenopatia (87%), splenomegalia
(54%), ed epatomegalia (14%). Circa il 10% dei pazienti presentano un' anemia
emolitica autoimmune con test di Coombs positivo. Il 10% dei pazienti possono avere
ipogammaglobulinemia, ed un altro 15% può avere ipergammaglobulinemia o
gammopatia monoclonale. Molti pazienti asintomatici sono identificati sulla base della
sola linfocitosi rilevata con l’emocromo completo.
Con il progredire della malattia possono comparire altri sintomi, che non sono
caratteristici della leucemia linfatica cronica ma sono comuni ad altre patologie
linfoproliferative, e sono conseguenti all’invasione del midollo osseo da parte di linfociti
neoplastici: la stanchezza, associata a pallore cutaneo e palpitazioni sono una
conseguenza dell’anemia, mentre le manifestazioni emorragiche sono secondarie alla
riduzione delle piastrine (Catovsky D. et al, Ann Hematol 1991).
Inoltre l’accumulo dei linfociti patologici ostacola la normale produzione da parte del
midollo osseo di linfociti e granulociti neutrofili, in questo modo si crea uno stato di
immunodeficienza che predispone l’individuo malato all’insorgenza di infezioni.
Infine in un 5% di pazienti la malattia si può manifestare associata a fenomeni
autoimmuni, cioè produzione di anticorpi contro antigeni propri, in particolare antigeni di
globuli rossi e piastrine, dando origine a patologie concomitanti quali l’anemia emolitica
autoimmune, la piastrinopenia autoimmune o più raramente l’associazione di entrambi
(sindrome di Fisher-Evans).
L’infiltrazione del midollo osseo può avvenire secondo quattro configurazioni: nodulare,
diffuso, interstiziale e misto. Il pattern nodulare suggerisce uno stadio precoce della
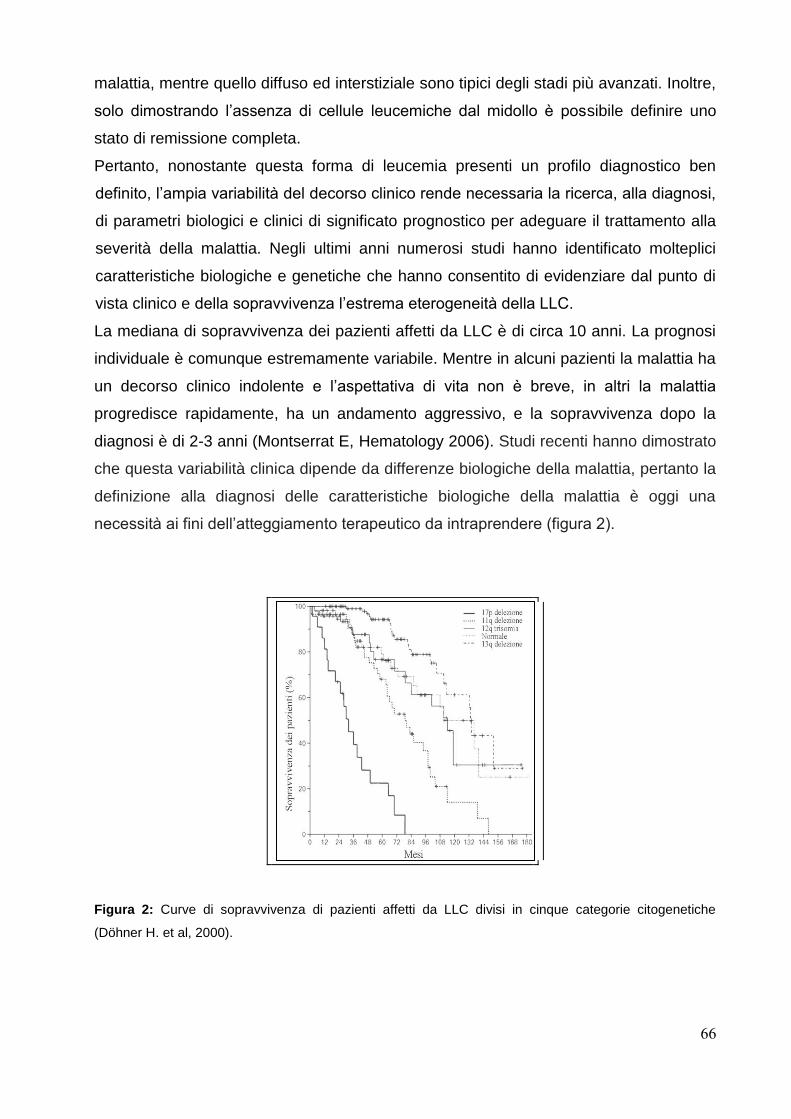
66
malattia, mentre quello diffuso ed interstiziale sono tipici degli stadi più avanzati. Inoltre,
solo dimostrando l’assenza di cellule leucemiche dal midollo è possibile definire uno
stato di remissione completa.
Pertanto, nonostante questa forma di leucemia presenti un profilo diagnostico ben
definito, l’ampia variabilità del decorso clinico rende necessaria la ricerca, alla diagnosi,
di parametri biologici e clinici di significato prognostico per adeguare il trattamento alla
severità della malattia. Negli ultimi anni numerosi studi hanno identificato molteplici
caratteristiche biologiche e genetiche che hanno consentito di evidenziare dal punto di
vista clinico e della sopravvivenza l’estrema eterogeneità della LLC.
La mediana di sopravvivenza dei pazienti affetti da LLC è di circa 10 anni. La prognosi
individuale è comunque estremamente variabile. Mentre in alcuni pazienti la malattia ha
un decorso clinico indolente e l’aspettativa di vita non è breve, in altri la malattia
progredisce rapidamente, ha un andamento aggressivo, e la sopravvivenza dopo la
diagnosi è di 2-3 anni (Montserrat E, Hematology 2006). Studi recenti hanno dimostrato
che questa variabilità clinica dipende da differenze biologiche della malattia, pertanto la
definizione alla diagnosi delle caratteristiche biologiche della malattia è oggi una
necessità ai fini dell’atteggiamento terapeutico da intraprendere (figura 2).
Figura 2: Curve di sopravvivenza di pazienti affetti da LLC divisi in cinque categorie citogenetiche
(Döhner H. et al, 2000).

67
Lo stato non mutato della regione variabile della catena pesante delle immunoglobuline
(IgHV), la positività della cellula leucemica per l’antigene di attivazione CD38 e per la
molecola di trasduzione del segnale ZAP-70, le alterazioni citogenetiche quali la
delezione dei cromosomi 17p e 11q, la trisomia del cromosoma 12 e le mutazioni a
carico del gene TP53 e del gene ATM, identificano una malattia clinicamente più
aggressiva che richiede specifici trattamenti terapeutici. L’individuazione di tale
distintivo profilo biologico pertanto permette di identificare precocemente alla diagnosi
due gruppi di pazienti con prognosi differente, uno a prognosi sfavorevole per i quali
devono essere indicati trattamenti più aggressivi, e pazienti a prognosi favorevole o con
malattia stabile che possono avere un decorso clinico benigno senza necessitare di
alcun trattamento terapeutico. Parametri quali profilo morfologico e immunofenotipico di
LLC tipica, assenza dell’espressione dell’antigene CD38, IgHV mutate, assenza di
mutazioni del gene TP53, assenza di delezione del 17p e del11q e la presenza della
delezione del 13q sono stati associati a stabilità della malattia (Guarini A. et al, Blood
2003).
Un profilo biologico ben definito è stato anche identificato in un sottogruppo di pazienti
affetti da LLC in fase di regressione spontanea della malattia (Del Giudice I. et al, Blood
2009).
I numerosi sforzi volti a cercare dei criteri per definire la prognosi della malattia si
incrociano con la ricerca dei meccanismi patogenetici che ne stanno alla base.
La disregolazione del processo di morte cellulare programmata (apoptosi) è ormai
largamente riconosciuto come uno dei meccanismi principali nella patogenesi di
numerosi tumori; nella LLC essa assume un ruolo particolarmente importante. Le cellule
di LLC, infatti, non possiedono un’elevata capacità replicativa e sono per lo più bloccate
nelle fasi G0/G1 del ciclo cellulare ed il loro accumulo pertanto è legato alla perdita della
capacità di andare incontro ad apoptosi. E’ importante quindi capire se la resistenza
all’apoptosi sia legata a fattori intrinseci alla cellula o dipenda da messaggi esterni che
giungono dal microambiente in cui vive (Ghia P. et al., Adv. Cancer Res 2000; Collins
R.J. et al., Br. J. Haematol 1989). Per quanto riguarda i fattori intrinseci, molto
importante è l’equilibrio tra fattori pro- e anti-apoptotici. Tra questi, i principali regolatori
dell’apoptosi sono delle proteine appartenenti alla famiglia Bcl-2 (B-cell lymphoma-2)
che giocano un ruolo cruciale in questo meccanismo inibendo (Bcl-2, Bcl-xL, Bcl-w, Bfl-
1 e Mcl-1) o promuovendo (Bax, Bak, Bcl-xS, Bid, Bik e Hrk) l’apoptosi.
L’eterodimerizzazione tra membri pro- e anti-apoptotici di questa famiglia ed i livelli

68
relativi di entrambi i tipi di proteine, può determinare la predisposizione a rispondere ad
un determinato stimolo apoptotico (Packham G. et al, Immunology 2005).
Altri fattori intrinseci critici per il controllo dell’apoptosi sono rappresentati dalle proteine
p53 e ATM, la cui mancata o carente espressione dovuta sia alla presenza di delezione
delle regioni cromosomiche in cui mappano i geni che le codificano, sia a mutazioni
presenti nella regione genica, può alterare il fisiologico processo apoptotico.
La sopravvivenza dei linfociti B leucemici non è dovuta solo alla loro capacità di
resistere all’apoptosi, mediante i meccanismi precedentemente discussi, ma risulta
influenzata anche dall’ambiente circostante e dalle cellule che lo compongono (cellule
stromali e cellule “nurse-like”) (Caligaris-Cappio F. et al, Br. J. Haematol 2003). È stato
infatti dimostrato che le cellule B neoplastiche non sopravvivono in coltura e vanno
rapidamente incontro a morte. Linfociti T e diversi tipi di cellule aderenti chiamate
“cellule stromali” sono i principali elementi del microambiente che conferiscono
condizioni favorevoli per la crescita e la sopravvivenza delle cellule B leucemiche. Studi
in vivo indicano che alcune citochine prodotte dai linfociti T (IL4, INFα, INFγ) inibiscono
la risposta apoptotica delle cellule B neoplastiche, mediante up-regolazione di Bcl-2. Le
chemochine (CXCL e CCL) inoltre, presenti nel microambiente, e i loro recettori (CXCR
e CCR) rappresentano un gruppo di molecole che giocano un ruolo essenziale nella
circolazione dei linfociti e nell’attrazione degli stessi verso il sito di infiammazione.
L’infiltrazione dei linfociti B leucemici nel midollo osseo e negli organi linfatici è regolata
dalle interazioni delle chemochine con i loro recettori espressi dalle cellule B di LLC.
Dati recenti indicano che le cellule B neoplastiche esprimono specifici recettori per
chemochine e rispondono in maniera selettiva ad alcune di esse raggiungendo, così,
sedi precise (Trentin L. et al, Blood 1993; Trentin L. et al, Blood 2004)
Nonostante sia stata effettuata un’estesa caratterizzazione molecolare delle cellule di
LLC, si continuano a studiare altri meccanismi che possano svolgere un ruolo in tale
patologia. A tale scopo molti lavori hanno focalizzato la loro attenzione sui micro-RNA
(mirRNA), molecole che costituiscono l' 1-3% del genoma dei vertebrati e regolano
centinaia di geni differenti. I mirRNA sono una classe di piccoli RNA non codificanti che
modulano l’espressione genica a livello post-trascrizionale di diversi geni coinvolti nella
proliferazione cellulare, nel differenziamento, nell’apoptosi e svolgono funzioni
regolatorie nell'organogenesi. Alcuni di questi miR sono presenti in regioni genomiche
coinvolte in traslocazioni e delezioni presenti in leucemie e linfomi e questo dato ha
suggerito un possibile ruolo dei miR nella linfomagenesi. In particolare è stato riportato

69
un possibile coinvolgimento nell’oncogenesi della LLC del miR-15a e miR-16-1 (Fulci V.
et al, Blood 2007) presenti fisiologicamente sulla regione cromosomica 13q14 che
invece risulta deleta nella maggior parte dei casi di LLC. L'assenza o la down
regolazione dei miR-15 e miR-16 provoca la sovraespressione della oncoproteina Bcl2
e la conseguente deregolazione del processo apoptotico delle cellule leucemiche.
Per molti anni il trattamento iniziale della LLC è stata la somministrazione, in modo
continuo o ciclico, di agenti chemioterapici. Ma negli ultimi due decenni il trattamento
terapeutico dei pazienti con LLC è radicalmente cambiato. L’introduzione degli analoghi
delle purine come la Fludarabina (Keating MJ. et al, Hematology 1991), e degli anticorpi
monoclonali come le molecole chimeriche Campath-1H (anti-CD52) e Rituximab (anti-
CD20) (Lundin J. et al, Blood 2002; Pedersen I.M. et al, Blood 2002) nel trattamento di
questa forma di leucemia hanno mostrato un importante aumento sia della percentuale
di risposte complete, inclusa quella molecolare, sia di sopravvivenza libera da
progressione. Recentemente alcuni studi hanno anche mostrato come la
chemioimmunoterapia oggi può essere considerata il miglior trattamento per molti
pazienti con LLC (Abrisqueta P. et al, Blood 2009). Ma nonostante ciò molti pazienti
comunque progrediscono o mostrano complicazioni dovuti al trattamento terapeutico o
resistenza alla terapia iniziale, mostrando così una prognosi infausta. Pertanto la
sperimentazione clinica oggi ha come obiettivo l’utilizzo di nuovi agenti terapeutici utili in
questa fascia di pazienti con LLC. Gli anticorpi monoclonali come Ofatumumab
(Humax-CD20) e GA-101 (anti-CD20, RO 5072759) hanno permesso di ottenere un
miglioramento della risposta al trattamento (Montserrat E. et al, Hematology Ed 2010), e
l’uso di agenti immunomodulatori come la Lenalidomide, che agiscono sul
microambiente essenziale per preservare le cellule B neoplastiche dalla apoptosi,
hanno già mostrato avere una attività terapeutica nei pazienti con LLC. A questi
trattamenti terapeutici si sono aggiunti nuovi farmaci ad attività anti-tirosinchinasica
(ibrutinib, etc) capaci di inibire il pathway del BCR che svolge un ruolo molto importante
nella progressione della malattia e che dai primi dati sembrano produrre risultati molto
significativi nel controllo della malattia.(Byrd JC, N Engl J Med. 2013)
Diversi studi attualmente sono in fase di sperimentazione al fine di determinare il ruolo
di questi nuovi agenti terapeutici nella LLC non solo come terapia di prima linea ma
anche come terapia di mantenimento.
L’importanza di un corretto inquadramento nosologico riveste rilevanti implicazioni
clinico-prognostiche dal momento che ad ogni disordine linfoproliferativo si può

70
associare un andamento clinico diverso, un approccio terapeutico differenziato e una
prognosi differente.
Attraverso l’organizzazione di Consensus Conference, principalmente dall’“International
Working Group on CLL”, sono emerse indicazioni per la standardizzazione della
diagnosi, della stadiazione e dei criteri di valutazione della risposta al trattamento che
sono adottati a livello internazionale dai principali Istituti di Ematologia e Oncologia (Br J
Haematol, 2004).

71
Stato mutazionale dei geni delle catene pesanti
delle immunoglobuline
Come linfociti B maturi, le cellule di LLC esprimono il recettore delle cellule B (BCR)
sulla loro superficie, costituito da immunoglobuline (IgM e IgD) in associazione a
molecole non polimorfe Igα ed Igβ che mediante le sequenze ITAM (immunoreceptor
tyrosine-based activation motif) presenti nella loro coda citoplasmatica sono in grado di
mediare la funzione di trasduzione del segnale.
Durante la maturazione, i linfociti B proliferanti all’interno del centro germinativo
linfonodale, vanno incontro al processo dell’ipermutazione somatica a carico dei geni
riarrangiati delle catene pesanti e leggere delle immunoglobuline (Ig). Tali mutazioni si
concentrano nelle regioni variabili (V), principalmente in corrispondenza delle regioni
che determinano la complementarietà (CDR), e la presenza di mutazioni correla con un
aumento della affinità degli anticorpi rivolti verso l’antigene che ha dato origine alla
risposta.
Un importante progresso nella caratterizzazione della LLC, non solo a livello biologico
ma anche prognostico, è l’analisi molecolare delle sequenze nucleotidiche dei geni che
codificano per la regione variabile delle catene pesanti delle Ig. Circa il 50% di pazienti
affetti da LLC presentano mutazioni somatiche nella regione variabile delle catene
pesanti delle Ig riarrangiate.
Nel 1999, due studi pubblicati simultaneamente da Damle R.N. et al. e da Hamblin T.J.
et al. definirono l’importanza prognostica dello stato mutazionale dei geni IgVH nella
LLC.
In seguito la rilevanza prognostica dello stato mutazionale IgVH è stata confermata da
numerose pubblicazioni e, ad oggi, è considerato il miglior marcatore capace di predire
la progressione della malattia nei pazienti affetti da LLC.
Più in particolare sono stati identificati due gruppi di pazienti caratterizzati da una
configurazione mutata e non mutata dei geni IgHV. E’ stato ampiamente dimostrato che
questi due gruppi di pazienti hanno un andamento clinico significativamente diverso.
Mentre il gruppo con IgHV non mutate dimostra una più rapida progressione di malattia
e una prognosi sfavorevole (Keating M.J. et al, Hematology 2003), i pazienti con
ipermutazioni delle IgHV hanno un andamento clinico più indolente ed una prognosi
significativamente migliore, suggerendo che il differente stadio di maturazione delle

72
cellule, valutato secondo lo stato mutazionale dei geni IgHV può definire distinti
meccanismi patogenetici della LLC.
La mediana di sopravvivenza dei pazienti appartenenti al gruppo dei non mutati è di
circa 9-10 anni, mentre la sopravvivenza mediana del gruppo dei mutati è di circa 24
anni (Damle R.N. et al, Blood 1999; Hamblin. TJ et al, Blood 1999). Pazienti con IgVH
non mutate presentano caratteristiche sfavorevoli: morfologia atipica delle cellule del
sangue venoso periferico, caratteristiche citogenetiche quali trisomia del 12 e delezione
dell’11q e del 17p, evoluzione clonale, progressione della malattia e infine resistenza
alla terapia rispetto ai casi con geni IgVH mutati (Ghia P. et al, Crit Rev Oncol Hematol
2007).
La definizione di stato mutazionale mutato o non mutato è basato su un cut-off
arbitrariamente definito: le sequenze sono considerate non mutate se differiscono dalla
sequenza del gene in configurazione germinale in percentuale <2%.
Lo studio dello stato mutazionale è effettuato analizzando le sequenze IgVH ottenute dal
sequenziamento diretto di frammenti amplificati utilizzando 6/7 primers disegnati per
riconoscere i geni membri delle famiglie VH.
Il sequenziamento automatico delle catene pesanti e leggere delle immunoglobuline
oltre ad essere un importante strumento a livello prognostico, permette l’identificazione
dei diversi geni V(D)J riarrangiati utilizzati dalle cellule di LLC. Così i progressi
nell’identificazione dei geni Ig, il loro riarrangiamento, e le loro modificazioni a seguito
dell’attivazione e differenziazione delle cellule B ha fornito uno strumento utile per
studiare il grado di competenza, l’esperienza antigenica e lo stadio di maturazione delle
cellule B.
I recettori delle cellule B di LLC di vari pazienti sono spesso strutturalmente omologhi,
suggerendo che gli antigeni legati al recettore sono simili e rilevanti nella patogenesi
della LLC.
Le cellule di LLC che presentano uno stato non mutato dei geni che codificano per le
catene pesanti delle immunoglobuline utilizzano principalmente i geni appartenenti alla
famiglia VH1 all’interno della quale quello più frequentemente utilizzato è il gene VH1-69.
Le cellule di LLC che presentano invece uno stato mutato dei geni che codificano per le
catene pesanti delle immunoglobuline utilizzano principalmente i geni appartenenti alle
famiglie VH3 e VH4. All’interno di queste famiglie i geni più frequentemente utilizzati
sono i geni VH4-34, VH3-07 e VH3-23 la cui frequenza relativa può cambiare a seconda
delle caratteristiche cliniche delle varie coorti di pazienti studiate e diversa

73
localizzazione geografica. Anche l’utilizzo delle specifiche famiglie geniche VL e dei geni
appartenenti alle diverse famiglie appare non casuale come per il repertorio VH. Queste
caratteristiche possono indicare che i precursori delle cellule B ricevono diversi stimoli
da distinti tipi di antigeni prima della trasformazione leucemica e/o che i precursori sono
trasformati in cellule leucemiche in diversi stadi di maturazione (Chiorazzi N. et al, Annu
Rev Immunol 2003). La natura di questi antigeni è per ora sconosciuta, sebbene siano
possibili alcune speculazioni: è possibile che virus latenti o batteri commensali attivino
particolari cloni delle cellule B attraverso il BCR. La LLC potrebbe risultare, direttamente
o indirettamente, da specifiche infezioni ripetute. In alternativa è possibile che siano
antigeni o autoantigeni a provocare l’espansione clonale. Le cellule di LLC presentano
frequentemente recettori polireattivi, che legano antigeni multipli inclusi gli autoantigeni.
Questo meccanismo è possibile nei casi non mutati e in alcuni casi di LLC mutati con
geni IgVH codificanti per recettori polireattivi. Inoltre l’associazione di simili segmenti
V(D)J, con un unico residuo giunzionale, specialmente nella giunzione VL-JL, è stata
vista in anticorpi diretti contro polisaccaridi (Casadevall A. et al, J Exp Med 1991) o
contro determinati apteni chimici (Milner E.C. et al, J Immunol 1982; Wysocki L.J.et al, J
Exp Med 1987) così come la presenza di un’arginina nella giunzione VL-JL è
caratteristica dell’anticorpo umano diretto contro l’antigene capsulare polisaccaridico
dell’Haemophilus Influenzae di tipo B (Insel R.A. et al, Int Rev Immunol 1992).
Il sequenziamento automatico delle immunoglobuline permette, inoltre, di studiare la
composizione, la lunghezza e le implicazioni del CDR3 tra i diversi cloni delle cellule B.
La lunghezza del CDR3, infatti, varia a seconda della famiglia VH utilizzata nel
riarrangiamento del clone di LLC: (VH4>VH1>VH3) e del gene appartenete a tale
famiglia. La lunghezza media del CDR3 dei casi che esprimono il gene VH3-07 è molto
corta, mentre la lunghezza media dei casi che esprimono il gene VH1-69 è molto più
lunga (Chiorazzi N. et al, Curr Top Microbiol Immunol.2005).
Un'altra rilevante caratteristica riscontrata sulle cellule di LLC è la presenza di particolari
combinazioni dei geni VH/VL: il gene VH1-69 si associa più frequentemente con la
catena leggera codificata dal gene Vκ3-20, i geni VH4-34 e VH4-39, si trovano in
associazione con le catene leggere codificate dai geni Vκ2-30 e Vκ1-39 rispettivamente
(Stamatopoulos K. et al, Blood 2005) e il gene VH3-21 si ritrova invece in associazione
con le catene leggere codificate dal gene Vλ3-21 o Vλ2-14 (Tobin G. et al, Blood 2003).
Tutte queste particolarità riscontrate nella struttura dei diversi BCR sono il possibile
risultato di una selezione da parte di diversi epitopi antigenici.

74
Nonostante non sia ancora chiaro, molti studi supportano l’idea che la stimolazione
antigenica sia un prerequisito essenziale per l’evoluzione delle cellule di LLC anche nei
casi che non esibiscono mutazioni sui geni IgVH. Inoltre recenti studi sui segnali di
competenza hanno rilevato che i casi non mutati di LLC tendono ad esprimere una
maggiore quantità di BCR e rispondono meglio alla stimolazione se comparati ai casi
mutati di LLC. Questi risultati portano all’idea che i casi non mutati mantengano la loro
abilità di rispondere alla stimolazione del BCR, mentre i mutati divengono simili a cellule
anergiche. Sono stati proposti numerosi modelli per spiegare la derivazione delle cellule
mutate e non mutate. In particolare un importante studio di Chiorazzi N. e Ferrarini M.
ha proposto due modelli. Il primo ipotizza che le cellule mutate possano derivare da
cellule B stimolate da antigeni T-dipendenti che dirigono le cellule attraverso la classica
reazione che avviene nel centro germinativo, mentre le cellule non mutate potrebbero
derivare da cellule B presenti nella zona marginale e che siano guidate da un processo
T-indipendente che non richiede la presenza delle cellule T o di mutazioni somatiche. Il
secondo modello suggerisce la derivazione di entrambe i casi mutati e non mutati da
cellule B della zona marginale che rispondono ad una stimolazione T indipendente che
possono o meno sviluppare mutazioni somatiche. Oppure che le cellule possano
derivare da entrambe i modelli (Chiorazzi N. et al, Annu Rev Immunol 2003). Infine, è
possibile che la stimolazione antigenica possa promuovere un'evoluzione intraclonale
che porta ad un accumulo di mutazioni deleterie al DNA e quindi ad un più aggressivo
decorso clinico.
In conclusione gli studi sullo stato mutazionale delle catene pesanti delle Ig, sui
riarrangiamenti VHDJH, sulle combinazioni VH/VL e sulle caratteristiche funzionali del
BCR hanno fornito considerevoli informazioni riguardo le caratteristiche delle cellule B
leucemiche e sulla possibile patogenesi di questa patologia, ma ulteriori studi sono
necessari per confermare le varie ipotesi sui meccanismi di trasformazione leucemica.

75
Obiettivo dello Studio
Nell’ambito della LLC è stato messo in evidenza come pazienti aventi una MMR
negativa abbiano una sopravvivenza libera da progressione più lunga ed una migliore
sopravvivenza globale. Pertanto la MMR viene identifica come un marcatore
prognostico in diversi protocolli terapeutici.
La valutazione molecolare di RQ-PCR della MMR richiede l'identificazione al momento
della diagnosi di bersagli molecolari specifici del paziente, come i riarrangiamenti genici
della catena pesante delle immunoglobuline (IGH), sui quali sono costruiti primer e
sonde, utilizzati per il monitoraggio della malattia durante il follow- up. La presenza di
mutazioni somatiche nella regione IGH durante la maturazione delle cellule B nel centro
germinativo, presenti in circa la metà dei pazienti con LLC e nel 20% dei pazienti con
linfoma mantellare (LCM), potrebbe influenzare l'analisi di MMR. Pertanto, diverse
strategie per la progettazione di primer e sonde sono state sviluppate per ottimizzare
l'analisi molecolare, in base alle caratteristiche del gene IGH.
L'obiettivo di questo studio è stato quello di confrontare due strategie di costruzione
primers/sonda su una serie di LLC e LCM con un alto carico mutazionale IGH per
identificare la tecnica di elezione nel monitoraggio della MMR in queste neoplasie.

76
Materiali e Metodi Pazienti e campioni studiati
Questo studio è stato eseguito su due serie di campioni. La prima serie è stata fornita
dalla Divisione di Ematologia dell'Università di Torino e consisteva di 14 campioni di
pazienti affetti da leucemia linfatica cronica (n=7) e linfoma mantellare (n=7), senza
criteri di selezione a parte la disponibilità di materiale. La seconda serie è stata fornita
dalla Divisione di Ematologia dell'Università' “Sapienza” di Roma e consisteva di 11
campioni di pazienti affetti da leucemia linfatica cronica, selezionati sulla base di una
percentuale elevata di mutazioni (>5%) della regione variabile della catena pesante
delle immunoglobuline (IGHV).
La diagnosi di LLC è stata effettuata sulla base della presenza di più di 5.000 linfociti/μL
patologici presenti nel sangue periferico e di criteri morfologici e immunofenotipici
(CD5+/CD20+, CD23+, bassa intensità di espressione delle sIg e CD22, CD10-).
La diagnosi di linfoma mantellare, così come quella di tutti i linfomi, è istologica ed è
stata effettuata sull’esame morfologico ed immunoistochimico del tessuto linfonodale
patologico.
I linfociti neoplastici del linfoma mantellare esprimono la positività ai marcatori di linea
B (CD19, CD20, CD79a) e per il CD5, antigene normalmente espresso sulla
popolazione linfocitaria T e presente anche sulle cellule di LLC. La diagnosi
differenziale con tale patologia è possibile grazie all’uso del CD23 e del CD20
(positivi nella LLC e negativi nel linfoma mantellare).
Estrazione del DNA e analisi IGH
L’analisi eseguita è stata effettuata sulle cellule mononucleate del sangue periferico
(PBMC: periferal blood mononucleated cell) dei pazienti affetti da LLC/LCM isolate
mediante la separazione su gradiente di densità Ficoll. A partire da un pellet di cellule
contenenti circa 5-10 x106 PBMC è stato estratto il DNA genomico mediante Wizard
Genomic DNA Purification Kit (Promega Corp., Madison, Wisconsis, USA).
La determinazione della purezza e della concentrazione del DNA estratto è stata
valutata mediante lo spettrofotometro Epperndorf BIOPhotometer (Eppendorf AG,
Hamburg, Germany), ed i campioni che presentavano un rapporto di A260/A280
compreso tra 1.8 e 1.9, sono stati diluiti con acqua deionizzata al fine di ottenere una

77
concentrazione standard di DNA di 100µg/mL. La qualità del DNA è stata valutata a
seguito di una corsa elettroforetica su gel d’agarosio dove un DNA genomico di buona
qualità presentava una sola banda ad alto peso molecolare.
Un totale di 300 ng di DNA genomico sono stati amplificati mediante reazione di
amplificazione Polymerase Chain Reaction (PCR) utilizzando il termociclatore
GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA, USA).
L’amplificazione è stata ottenuta mediante un’unica PCR utilizzando i primers di senso
relativi alla regione Leader e FR1 della regione variabile ed un primer antisenso
consensus relativo alla regione FR4 della regione di giunzione delle immunoglobuline
(Ghia P et al, Leukemia 2007).
Le condizioni di PCR per l’amplificazione del DNA genomico sono state le seguenti:
viene eseguita una denaturazione iniziale a 95°C per 5’, successivamente si effettuano
denaturazione, annealing ed estensione per 35 cicli rispettivamente a 95°C per 30’’,
60°C per 30’’ e 72°C per 30”. Alla fine dei cicli si esegue un’estensione a 72°C per 10’.
La separazione e l’identificazione degli amplificati di PCR è stata eseguita mediante
corsa elettroforetica su gel di agarosio al 2% in TBE 1X, contenente 5 µl/100ml di
bromuro d’etidio (SIGMA-ALDRICH, St. Louis; MO, USA) come agente intercalante del
DNA.
Sequenziamento e analisi del gene
I prodotti di PCR sono stati sequenziati direttamente secondo metodo Sanger con
marcatura fluorescente utilizzando il kit di reazione BigDye Terminator v3.1 Cycle
Sequencing Kit (Applied Biosystems, Foster City, CA, USA) e il termociclatore
GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA, USA). Durante la
sintesi del filamento di DNA i didesossinucleotiditrifosfati (ddNTP) marcati con quattro
differenti fluorocromi vengono, mediante l’enzima DNA polimerasi, inseriti al filamento
stampo impedendo l’aggiunta di ulteriori nucleotidi. Al termine della reazione di
sequenza la miscela di singoli frammenti di DNA ottenuta è stata purificata utilizzando il
kit DyeEx 2.0 Spin Kit (QIAGEN, Valencia, CA, USA) che permette di eliminare i ddNTP
fluorescenti in eccesso non incorporati attraverso il principio cromatografico della gel-
filtrazione su colonnine contenenti resina. I frammenti di DNA purificati sono stati
sottoposti ad elettroforesi capillare mediante il sequenziatore ABI PRISM® 3100-Avant
Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). L’acquisizione dei dati

78
relativi ai campioni è stata eseguita utilizzando il software 3100-Avant Data Collection
Software e le sequenze sono state analizzate mediante il programma ABI PRISM® DNA
Sequencing Analysis Software (Versione 3.7), e visualizzate come elettroferogrammi.
L'interpretazione dei risultati è stata eseguita allineando le sequenze nucleotidiche
ottenute alla directory IMGT (international ImMunoGeneTics information system) per
l'identificazione di riarrangiamenti IGH ed il calcolo del carico mutazionale.
Disegno di primer e sonde per l'analisi RQ -PCR
I Primer e le sonde sono state progettate per tutti i campioni secondo due diverse
strategie, e precisamente il metodo A ed il metodo B, adottati dai due diversi centri
ematologici di provenienza, Torino e Roma.
La differenza principale tra i due metodi è rappresentata dal posizionamento dei primers
e delle sonde sulle regioni VDJ. In particolare, nel metodo A i primers di senso e
antisenso sono progettati sulla specifica sequenza del paziente, mentre nel metodo B
solo l'oligonucleotide di senso è sequenza-specifico, essendo la sonda ed il primer
antisenso in configurazione germinale.
Per il metodo A (figura 3), il primer di senso è stato costruito sulla seconda regione
determinante la complementarietà (CDR2) o FR3, mentre il primer antisenso è sempre
costruito sulla regione altamente ipervariabile CDR3 (Ladetto M et al, Biol Blood Marrow
Transplant 2000).Le sonde di senso per l' analisi di RQ- PCR sono state costruite sulla
regione FR3 (Ladetto M et al, Biol Blood Marrow Transplant 2000).
Per il metodo B (figura 4), l'analisi della RQ-PCR è stata effettuata con solo un primer
paziente-specifico, in combinazione con la sonda fluorescente ed il primer reverse,
disegnati sulla regione germinale di ogni riarrangiamento (Cazzaniga G et al,
Haematologica; 2005; Donovan JW et al, Blood 2000). L' innesco di senso è
posizionato sulla regione CDR3 ed il primer antisenso insieme con la sonda sulla
regione FR4, dove è inclusa la porzione del JH. Le curve standard per le valutazioni
preliminari degli oligo costruiti sono state preparate a partire da diluizioni seriali del
campione della diagnosi, nel DNA estratto da un pellet di cellule mononucleate
proveniente da un pool di sangue venoso periferico (PBL) di 5 donatori sani e portato
alla concentrazione di 100 μg/ml.
I saggi sono stati allestiti per raggiungere una gamma di sensibilità e quantificabilità
≥10-4, con uno slope della curva compreso tra 3.1 e 3.9 ed un coefficiente di

79
correlazione ≥0,98, secondo le linee guida della ESG-MRD–ALL (van der Velden VHJ
et al, Leukemia 2007).
Le reazioni sono state preparate utilizzando i reagenti del kit TaqMan PCR Core
Reagent (PE Applied Biosystems, Foster City CA, USA) e processate nell' ABIPrism
7900 (metodo A) e nell' ABIPrism 7300-7500 (metodo B) alle seguenti condizioni:
un'incubazione iniziale di 2 minuti a 50°C e 10 minuti di denaturazione a 95°C, seguite
da 42 cicli (metodo A) e 50 cicli (metodo B) di denaturazione a 95°C per 15 secondi ed
annealing a 60-62°C per 1 minuto e a 58-63°C, rispettivamente, per il metodo A e B.
Per entrambi i metodi sono state rispettate le linee guida per l'analisi di RQ-PCR per i
riarrangiamenti IGH, ESG-MRD-ALL (European Study Group on MRD Detection for
ALL) al fine di stabilire i range quantitativi e di sensibilità per tutti i pazienti (van der
Velden VHJ et al, Leukemia 2007).
Figura 3: metodo A; il primer di senso è stato costruito sulla seconda regione determinante la complementarietà (CDR2), mentre il primer antisenso è sempre costruito sulla regione altamente ipervariabile CDR3 (Ladetto M et al, Biol Blood Marrow Transplant 2000).Le sonde di senso per l' analisi di RQ- PCR sono state costruite sulla regione FR3 (Ladetto M et al, Biol Blood Marrow Transplant 2000).
Figura 4: metodo B; l' innesco di senso paziente-specifico è posizionato sulla regione CDR3 ed il primer antisenso insieme con la sonda, in configurazione germinale, sulla regione FR4, dove è inclusa la porzione del JH (Cazzaniga G et al, Haematologica; 2005; Donovan JW et al, Blood 2000).
L FR1 FR2 FR4 FR3 CDR1 CDR2 CDR3
primer 5’ patient specific
primer 3’ patient specific
Consensus/ specific probe derived from FR3
Q R R Q
FR1 FR2 FR4 FR3 CDR1 CDR2 CDR3 L
primer 5’ patient
specific
primer 3’
consensu
Consensus
probe
Q R R Q
JH

80
Risultati Riarrangiamenti del gene IGH e analisi di mutazione
Su 25 campioni di pazienti affetti da LLC (18) e LCM (7) analizzati al momento della
diagnosi, sono state trovate venticinque ricombinazioni: 12 appartenenti alla famiglia
genica IGHV3, 6 a IGHV4, 5 a IGHV1, 1 a IGHV2 e 1 a IGHV6.
I geni più frequentemente osservati sono stati: IGHV3-30 (3/12), IGHV3-7 (3/12), IGHV4
-34 (3/6), IGHV1-46 (2/5) e IGHV1-8 (2/5).
Tra i riarrangiamenti genici IGHD, le famiglie più comuni identificate sono state: IGHD3
(11/25), seguita da IGHD2 (4/25) e IGHD6 (4/25). Il segmento IGHD2-15 era il gene più
frequentemente utilizzato nella famiglia IGHD2 (75%).
Per i riarrangiamenti genici IGHJ, è stato riscontrato un utilizzo frequente del IGHJ4
(14/25) e IGHJ6 (5/25) (Tabella 1 e Tabella 1 supplementare).
In base all' analisi dello stato mutazionale, 22/25 (88%) campioni (17 LLC, 5 LCM)
avevano mutazioni >2% e di conseguenza sono stati classificati come mutati, mentre i
restanti 3 (12%, 1 LLC, 2 LCM) hanno mostrato mutazioni <2% e sono stati classificati
come non mutati. Venti dei 25 (80%) campioni (15 LLC e 5 LCM) hanno mostrato un
carico mutazionale >5%.
I geni della regione del IGHJ hanno mostrato una percentuale di mutazioni da 0 a
29,4%, con un valore mediano del 10,42%. 23/25 (92%) campioni presentavano
mutazioni >2% (16 LLC e 7 LCM) e 18/25 (72%) hanno mostrato una carico mutazionale
>5% (13 LLC e 5 LCM)(Tabella 2).
Tabella 1: Frequenza in % delle famiglie IGH osservata nei 25 campioni studiati.
Famiglia IGH
IGHV1 IGHV2 IGHV3 IGHV4 IGHV6 IGHD1 IGHD2 IGHD3 IGHD4 IGHD6 IGHJ3 IGHJ4 IGHJ5 IGHJ6
% 20 (5/25)
4 (1/25)
48 (12/25)
24 (6/25)
4 (1/25)
12 (3/25)
16 (4/25)
44 (11/25)
12 (3/25)
16 (4/25)
8 (2/25)
56 (14/25)
12 (3/25)
20 (5/25)
Tabella 2: % di mutazione sulle regioni V e J della catena pesante delle Ig nei 25 campioni studiati.
Patologia Campioni % Mut. IGHV>2%
% Mut. IGHV<2%
% Mut. IGHV>5%
% Mut. IGHJ>2%
% Mut. IGHJ<2%
% Mut. IGHJ>5%
LLC LCM Toale
18 7 25
94 (17/18) 71 (5/7)
88 (22/25)
5 (1/18) 27 (2/7)
12 (3/25)
83 (15/18) 71 (5/7)
80 (20/25)
89 (16/18) 100 (7/7) 92 (23/25)
11 (2/18) 0 (0/18) 8 (2/25)
72 (13/18) 71 (5/7)
72 (18/25)
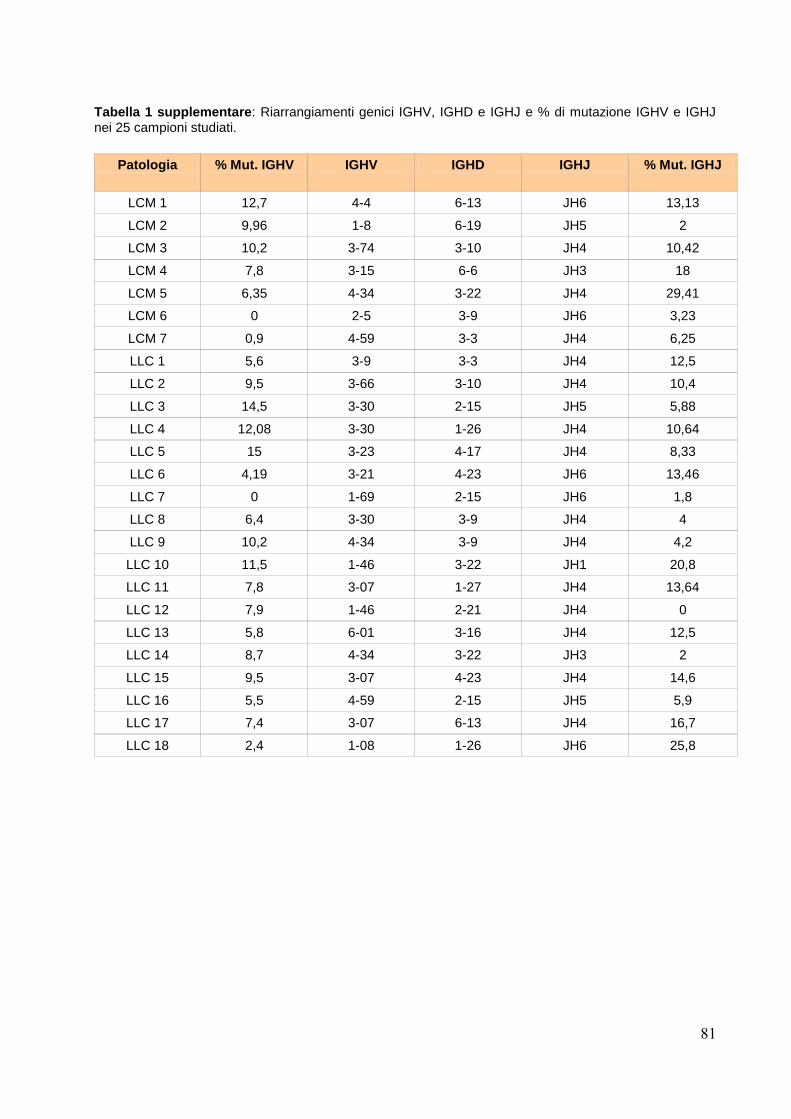
81
Tabella 1 supplementare: Riarrangiamenti genici IGHV, IGHD e IGHJ e % di mutazione IGHV e IGHJ nei 25 campioni studiati.
Patologia
% Mut. IGHV
IGHV IGHD IGHJ % Mut. IGHJ
LCM 1 12,7 4-4 6-13 JH6 13,13
LCM 2 9,96 1-8 6-19 JH5 2
LCM 3 10,2 3-74 3-10 JH4 10,42
LCM 4 7,8 3-15 6-6 JH3 18
LCM 5 6,35 4-34 3-22 JH4 29,41
LCM 6 0 2-5 3-9 JH6 3,23
LCM 7 0,9 4-59 3-3 JH4 6,25
LLC 1 5,6 3-9 3-3 JH4 12,5
LLC 2 9,5 3-66 3-10 JH4 10,4
LLC 3 14,5 3-30 2-15 JH5 5,88
LLC 4 12,08 3-30 1-26 JH4 10,64
LLC 5 15 3-23 4-17 JH4 8,33
LLC 6 4,19 3-21 4-23 JH6 13,46
LLC 7 0 1-69 2-15 JH6 1,8
LLC 8 6,4 3-30 3-9 JH4 4
LLC 9 10,2 4-34 3-9 JH4 4,2
LLC 10 11,5 1-46 3-22 JH1 20,8
LLC 11 7,8 3-07 1-27 JH4 13,64
LLC 12 7,9 1-46 2-21 JH4 0
LLC 13 5,8 6-01 3-16 JH4 12,5
LLC 14 8,7 4-34 3-22 JH3 2
LLC 15 9,5 3-07 4-23 JH4 14,6
LLC 16 5,5 4-59 2-15 JH5 5,9
LLC 17 7,4 3-07 6-13 JH4 16,7
LLC 18 2,4 1-08 1-26 JH6 25,8

82
Confronto tra diverse strategie di disegno di set di primers/probe per la valutazione RQ -
PCR
24/25 (96%) campioni sono risultati valutabili in RQ-PCR con almeno un metodo e 5/25
(20 %) campioni con entrambi i metodi.
Ventitre dei 25 (92 %) campioni (17 LLC e 6 LCM) sono risultati valutabili utilizzando il
metodo A, mentre 6/25 (24 %)(1 LLC e 5 LCM) sono risultati valutabili con il metodo B
(Tabella 3).
In particolare, 1 (4 %) campione (caso LLC9) non è stato valutabile per entrambi i
metodi ed un altro caso (caso LCM1) non è stato valutabile con il metodo A, ma è
risultato valutabile con il metodo B. Al contrario, il metodo A è risultato “vincente” in 18
dei 19 casi che non sono stati valutati con il metodo B.
Un confronto della fattibilità dei due approcci è mostrato nelle tabelle supplementari 2 e
3.
Tabella 3: Grado di successo della RQ-PCR secondo il metodo A ed il metodo B.
Patologia Pazienti Pz valutati con il
metodo B
Pz valutati con il
metodo A
Pz valutati con
entrambi i metodi
Pz valutati con almeno 1 metodo
Pz non valutabili
LLC LCM
Totale
18 7 25
1 5 6
17 6 23
1 4 5
17 7 24
1 0 1

83
Tabella 2 supplementare: Determinazione della curva standard. Valutazione della sensibilità e del range
quantitativo secondo il metodo A.
Patologia sensibilità Range Quantitativo
Slope Coefficiente Correlazione
LCM 1 NV NV
LCM 2 10-4 10-4 3,2 0,99
LCM 3 10-5 10-4 3,8 1
LCM 4 10-5 10-4 3,6 1
LCM 5 10-5 10-4 3,7 1
LCM 6 10-5 10-5 3,2 1
LCM 7 10-4 10-4 3,5 1
LLC 1 10-5 10-4 3,6 1
LLC 2 10-4 10-4 3,4 1
LLC 3 10-4 10-4 3,9 1
LLC 4 10-5 10-4 3,3 1
LLC 5 10-4 10-4 3,7 0,99
LLC 6 10-4 10-4 3,6 1
LLC 7 10-5 5x10-5 3,3 1
LLC 8 10-5 10-5 3,5 0,99
LLC 9 NV NV
LLC 10 10-4 10-4 3,6 0,99
LLC 11 10-4 10-4 3,1 1
LLC 12 10-5 5x10-5 3,4 0,99
LLC 13 10-4 10-4 3,2 0,99
LLC 14 10-5 10-4 3,5 1
LLC 15 10-5 10-4 3,5 1
LLC 16 10-5 10-4 3,8 1
LLC 17 10-4 10-4 3,5 0,99
LLC 18 10-4 10-4 3,4 1
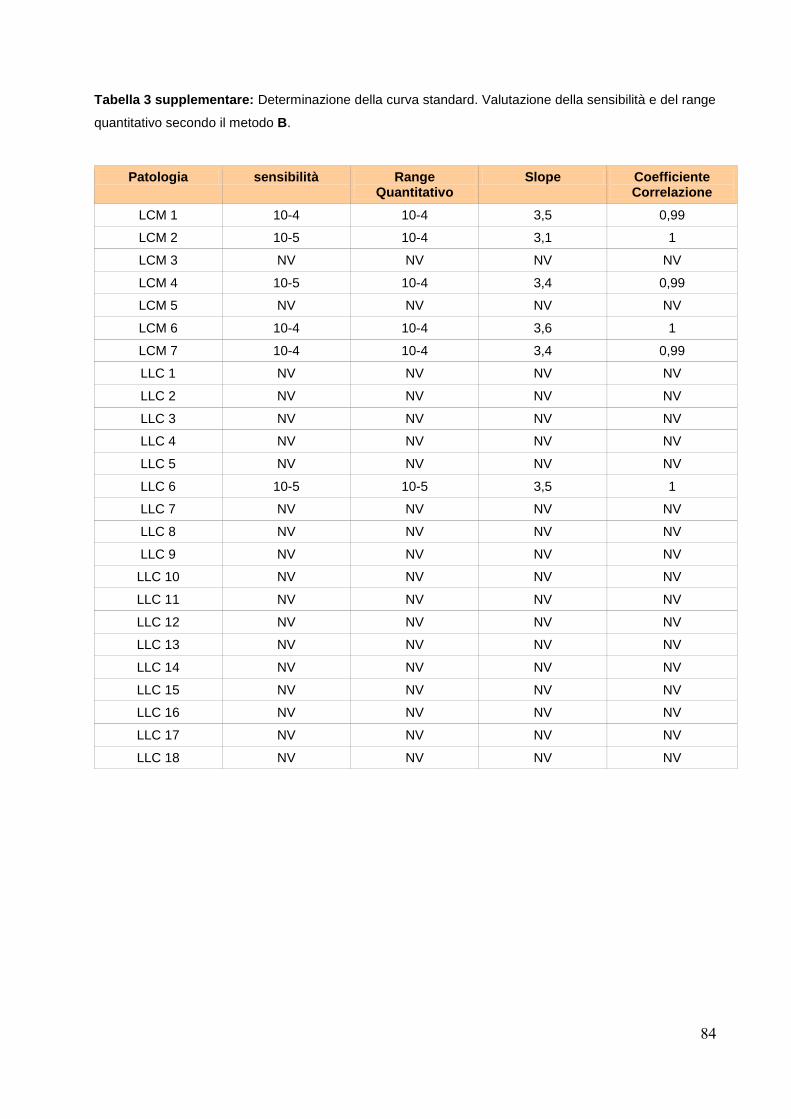
84
Tabella 3 supplementare: Determinazione della curva standard. Valutazione della sensibilità e del range
quantitativo secondo il metodo B.
Patologia sensibilità Range Quantitativo
Slope Coefficiente Correlazione
LCM 1 10-4 10-4 3,5 0,99
LCM 2 10-5 10-4 3,1 1
LCM 3 NV NV NV NV
LCM 4 10-5 10-4 3,4 0,99
LCM 5 NV NV NV NV
LCM 6 10-4 10-4 3,6 1
LCM 7 10-4 10-4 3,4 0,99
LLC 1 NV NV NV NV
LLC 2 NV NV NV NV
LLC 3 NV NV NV NV
LLC 4 NV NV NV NV
LLC 5 NV NV NV NV
LLC 6 10-5 10-5 3,5 1
LLC 7 NV NV NV NV
LLC 8 NV NV NV NV
LLC 9 NV NV NV NV
LLC 10 NV NV NV NV
LLC 11 NV NV NV NV
LLC 12 NV NV NV NV
LLC 13 NV NV NV NV
LLC 14 NV NV NV NV
LLC 15 NV NV NV NV
LLC 16 NV NV NV NV
LLC 17 NV NV NV NV
LLC 18 NV NV NV NV

85
Correlazione tra il carico mutazionale IGH e le performance di RQ-PCR
Per spiegare il diverso andamento dei due metodi, sono stati correlati i risultati di RQ-
PCR con lo stato ed il carico mutazionale delle regioni VH e JH.
Il metodo A è stato eseguibile indipendentemente dal carico mutazionale di VH e JH.
Infatti, solo 2/22 (9%) casi mutati IGHV e 2/21 (9,5 %) casi mutati IGJH sono risultati
non valutabili.
Al contrario, lo scarso rendimento del metodo B è risultato evidente per la presenza di
casi mutati IGHV/J: 18/22 (81,8 %), casi IGHV mutati e 17/21 (80,9 %) casi IGJH mutati
non erano valutabili. Vale la pena notare che in una serie di CLL con IGHV/J in
configurazione germinale, il metodo B ha avuto successo in 21/23 casi (91 %).
Tuttavia, non è stata trovata una correlazione specifica tra il carico di mutazioni nella
regione VH e JH e le prestazioni del metodo B. Infatti, i 6 casi valutabili con il metodo B
mostravano una configurazione IGHV germinale in 1 caso ed una vasta gamma di
mutazioni IGHV (0,9-12,7 %) negli altri 5 casi. Allo stesso modo, il carico mutazionale
del JH è stato del 2%, 3,23%, 6,25 %, 13.46 %, 16.13 % e 18 %, rispettivamente.

86
Conclusioni Nelle malattie linfoproliferative croniche, l'uso della MMR è stato finora un comune end
point nella valutazione della risposta in molti studi clinici, permettendo lo studio della
cinetica di deplezione delle cellule tumorali e la previsione di recidiva, con la possibilità
di rappresentare ben presto uno strumento di guida terapeutica (Rawstron AC et al,
Leukemia 2013).
Solo l’impiego di tecniche dotate di adeguata sensibilità, specificità nel riconoscimento
delle cellule patologiche, di stabilità dei marcatori utilizzati e di riproducibilità, può
permettere di distinguere tra loro i pazienti in remissione sulla base dei diversi livelli di
MMR permettendo così, una più precisa definizione di “stato di remissione”.
Nell’ambito della LLC è stato messo in evidenza come pazienti aventi una MMR
negativa abbiano una sopravvivenza libera da progressione più lunga e una migliore
sopravvivenza globale. Pertanto la MMR viene identificata come un marcatore
prognostico in una malattia considerata non ancora eradicabile, in diversi protocolli
terapeutici.
La valutazione molecolare di RQ-PCR della MMR richiede l'identificazione al momento
della diagnosi di bersagli molecolari specifici del paziente, come i riarrangiamenti genici
della catena pesante delle immunoglobuline (IGH), sui quali sono costruiti primers e
sonde, utilizzati per il monitoraggio della malattia durante il follow- up. La presenza di
mutazioni somatiche nella regione IGH durante la maturazione delle cellule B nel centro
germinativo, presenti in circa la metà dei pazienti con LLC e nel 20% dei pazienti con
linfoma mantellare (MCL), potrebbe influenzare l'analisi di MMR. Pertanto, diverse
strategie per la progettazione di primers e sonde sono state sviluppate per ottimizzare
l'analisi molecolare, in base alle caratteristiche del gene IGH.
Lo scopo di questo studio è stato quello di confrontare due diversi approcci nella
progettazione di primers/sonda specifici per eseguire le analisi quantitative e valutarne
l'applicabilità, specificità e sensibilità nel contesto delle malattie linfoproliferative a
cellule B caratterizzate da un carico mutazionale variabile delle regioni IGHV e IGHJ
della catena pesante delle immunoglobuline.
La differenza principale tra il metodo A ed il metodo B risiedeva sia nel diverso
posizionamento dei primers e delle sonde su sequenze IGH sia nella specificità del
sistema.
Nel metodo A, entrambi i primers senso e antisenso erano specifici per la sequenza

87
(come potrebbe essere anche la sonda in casi difficili). Nel metodo B, solo
l'oligonucleotide senso era paziente-specifico, mentre le sonde ed il primer antisenso
erano in configurazione germinale. Per quanto riguarda il posizionamento del set
primers/sonda sulla sequenza, nel metodo A i primers senso e antisenso erano
posizionati sulla regione del CDR2 e CDR3 rispettivamente, mentre la sonda era posta
sulla regione FR3; nel metodo B il primer senso era posizionato sulla regione CDR3 ed
il primer antisenso insieme con la sonda sulla regione FR4, dove è inclusa la porzione
del JH. Pertanto, i due metodi avevano una singola regione in comune, la CDR3, dove il
metodo A pone l'oligonucleotide antisenso ed il metodo B quello di senso. Il metodo B
deriva dalle linee guida del Gruppo europeo di studio sulla valutazione della MMR per le
LAL, una neoplasia che non è influenzata da mutazioni di sequenza, e permette
valutabilità di analisi in >90% dei casi.
Il confronto tra le due strategie di progettazione ha mostrato che il 92% dei campioni
sono stati valutati utilizzando il metodo A, mentre solo il 24% sono stati esaminati con
metodo B, dimostrando che il metodo A rappresenta una strategia più potente per
l'analisi RQ-PCR per le malattie linfoproliferative a cellule B, quali LLC o LCM, dove le
mutazioni a carico delle regioni IGHV e IGHJ si verificano in un notevole numero di casi.
Le mutazioni sono più frequenti nelle regioni CDR1 e CDR2, che nel FR3 e meno
frequenti nelle regioni FR1 e FR2. Su queste basi, il metodo A è risultato efficace
indipendentemente dallo stato mutazionale IGHV/J, grazie alla capacità di eludere le
zone colpite dalle mutazioni ed evitando un annealing abortivo o improprio del primer
sul CDR. Al contrario, la prestazione del metodo B è stata influenzata dallo stato
mutazionale IGHV/J, poiché l'annealing del primer antisenso e della sonda è
certamente condizionato dal loro posizionamento sulla regione JH.
Inoltre, la lunghezza del CDR3 varia a seconda dell'età, della fase di maturazione delle
cellule B e lo stato ipermutato del gene VH. Infatti, la lunghezza della regione CDR3
aumenta durante la vita fetale fino alla nascita e non continua nella vita adulta nei topi e
nell'uomo (Xue W et al, Human Immunology 1997). Questo è dovuto alla riduzione della
lunghezza delle catene pesanti come cellula B matura (Rosner K et al, Immunology
2001). Inoltre, anticorpi mutati hanno regioni CDR3 brevi rispetto anticorpi non mutati.
Queste caratteristiche sono riflesse dalle differenze nel profilo VDJ tra LLC (Duke VM et
al, Haematologica 2003), in quanto la LLC mutata ha un CDR3 più breve della LLC non
mutata.
Per il metodo A, un breve CDR3, purché riconosciuto all'interno di un riarrangiamento

88
VDJ, è sufficiente per un corretto annealing del primer antisenso. Al contrario, per il
metodo B, un breve CDR3 influenza la funzionalità d'innesco del primer di senso e
quindi il successo delle analisi RQ-PCR.
Per concludere possiamo dire che: i) la presenza di mutazioni somatiche in malattie
linfoproliferative a cellule B è un marcatore fenotipico che influenza la sua analisi; ii) le
diverse strategie di posizionamento del primer/sonda influenzano fortemente il
successo o il fallimento di RQ-PCR; iii) nessun carico mutazionale specifico è stato
identificato come responsabile del successo o del fallimento delle performance.
E' quindi necessario riformulare le strategie molecolari per valutare la MMR secondo la
biologia e le caratteristiche del recettore delle cellule B della malattia in esame, in un
momento in cui il monitoraggio MMR sta diventando sempre più frequentemente
utilizzato per la gestione dei pazienti con malattie linfoproliferative a cellule B.

89
Bibliografia
1. Abrisqueta P1, Pereira A, Rozman C, Aymerich M, Giné E, Moreno C, Muntañola
A, Rozman M, Villamor N, Hodgson K, Campo E, Bosch F, Montserrat E.
Improving survival in patients with chronic lymphocytic leukemia (1980-2008): the
Hospital Clinic of Barcelona experience. Blood. 2009, 114:2044-50.
2. Adderson EE, Shackelford PG, Insel RA, Quinn A, Wilson PM, Carroll
WL.Immunoglobulin light chain variable region gene sequences for human
antibodies to Haemophilus influenzae type b capsular polysaccharide are
dominated by a limited number of V kappa and V lambda segments and VJ
combinations. The Journal of Clinical Investigation 1992 Mar; 89(3):729-38.
3. Bottcher S., Ritgen M, Fischer K, Stilgenbauer, S., Busch, R.M., Fingerle-
Rowson, G., Fink, A.M., Bühler, A., Zenz, T., Wenger, M.K., Mendila, M.,
Wendtner, C.M., Eichhorst, B.F., Döhner, H., Hallek, M.J. & Kneba, M.. Minimal
residual disease quantification is an independent predictor of progression-free
and overall survival in chronic lymphocytic leukemia: a multivariate analysis from
the randomized GCLLSG CLL8 trial. Journal Clinical Oncology 2012; 30: 980-
988.
4. Brochet X, Lefranc MP, Giudicelli V. IMGT/V-QUEST: the highly customized and
integrated system for IG and TR standardized V-J and V-D-J sequence analysis.
Nucleic Acids Research 2008; 36: W503–W508.
5. Brüggemann M, Gökbuget N, Kneba M. Acute lymphoblastic leukemia:
monitoring minimal residual disease as a therapeutic principle. Seminars in
Oncology 2012; 39: 47-57.
6. Caligaris-Cappio F.: Role of the microenvironment in chronic lymphocytic
leuckamia. British Journal of Haematology 2003, 123:380-383.
7. Casadevall A, Scharff MD. The mouse antibody response to infection with
Cryptococcus neoformans: VH and VL usage in polysaccharide binding
antibodies. The Journal of Experimental Medicine 1991 Jul 1;174(1):151-60.

90
8. Catovsky D, Matutes E, Buccheri V, Shetty V, Hanslip J, Yoshida N, Morilla R. A
classification of acute leukemia for thhe 1990s. Annals of Hematology 1991 Feb;
62(1):16-21.
9. Cazzaniga G, Biondi A. Molecular monitoring of childhood acute lymphoblastic
leukemia using antigen receptor gene rearrangements and quantitative
polymerase chain reaction technology. Haematologica 2005; 90: 382-390.
10. Chiorazzi N., Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned
from studies of the B cell antigen receptor. Annual Review of Immunology 2003,
21:841-94.
11. Chiorazzi N, Allen SL, Ferrarini M.Clinical and laboratory parameters that define
clinically relevant B-CLL subgroups. Current Topics in Microbiology and
Immunology 2005; 294:109-33. Review.
12. Chiorazzi N, Ferrarini M.Evolving view of the in-vivo kinetics of chronic
lymphocytic leukemia B cells. Hematology 2006: 273-8, 512. Review.
13. Coad JE, Matutes E, Catovsky D.Splenectomy in lymphoproliferative disorders: a
report on 70 cases and review of the literature. Leukemia Lymphoma 1993 Jul;
10(4-5):245-64. Review.
14. Collins R.J., Verschuer L.A., Harmon B.V., Prentice R.L., Pope J.H. and Kerr J.F.
Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia
cells following their culture in vitro. British Journal of Haematology 1989; 71:343-
350.
15. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A,
Budman D, Dittmar K, Kolitz J, Lichtman SM, Schulman P, Vinciguerra VP, Rai
KR, Ferrarini M, Chiorazzi N. Ig VH gene mutation status and CD38 expression
as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999;
94:1840–7.
16. De Lima M, O'Brien S, Lerner S, Keating MJ. Chronic lymphocytic leukemia in
the young patient. Seminars in Oncology 1998 Feb; 25(1):107-16. Review.
17. Del Giudice I, Chiaretti S, Tavolaro S, De Propris MS, Maggio R, Mancini F,
Peragine N, Santangelo S, Marinelli M, Mauro FR, Guarini A, Foà R. Spontaneus

91
clinical remission in chronic lymphocytic leucemia: clinical and biologic features
of 9 cases. Blood 2009; 114: 638-646.
18. Donovan JW, Ladetto M, Zou G, Neuberg D, Poor C, Bowers D, Gribben JG.
Immunoglobulin heavy-chain consensus probes for real-time PCR quantification
of residual disease in acute lymphoblastic leukemia. Blood 2000; 95: 2650-2658.
19. Duke VM1, Gandini D, Sherrington PD, Lin K, Heelan B, Amlot P, Mehta AB,
Hoffbrand AV, Foroni L. V(H) gene usage differs in germline and mutated B-cell
chronic lymphocytic leukemia. Haematologica 2003; 88: 1259-1271.
20. Fenk R, Haas R, Kronenwett R. Molecular Monitoring of Minimal Residual
Disease in Patients with Multiple Myeloma. Hematology 2004; 9: 17-33.
21. Ferrero S, Drandi D, Mantoan B, Ghione P, Omedè P, Ladetto M. Minimal
residual disease detection in lymphoma and multiple myeloma: impact on
therapeutic paradigms. Hematological Oncology 2011; 29: 167-176.
22. Fulci V, Chiaretti S, Goldoni M, Azzalin G, Carucci N, Tavolaro S, Castellano L,
Magrelli A, Citarella F, Messina M, Maggio R, Peragine N, Santangelo S, Mauro
FR, Landgraf P, Tuschl T, Weir DB, Chien M, Russo JJ, Ju J, Sheridan R,
Sander C, Zavolan M, Guarini A, Foà R, Macino G. Quantitative technologies
establish a novel microRNA profile of chronic lymphocytic leukemia. Blood 2007;
109: 4944-51.
23. Ghia P. and Caligaris-Cappio F. The indispensable role of microenvironment in
the natural history of low-grade B-cell neoplasms. Advances in Cancer Research
2000; 79:157- 173.
24. Ghia P, Ferreri AM, Caligaris-Cappio F. Chronic lymphocytic leukemia. Critical
Reviews in Oncology/Hematology 2007; 64:234-46.
25. Ghia P, Stamatopoulos K, Belessi C, Moreno C, Stilgenbauer S, Stevenson F,
Davi F, Rosenquist R; European Research Initiative on CLL. ERIC
recommendations on IGHV gene mutational status analysis in chronic
lymphocytic leukemia. Leukemia 2007; 21:1–3.
26. Guarini A, Gaidano G, Mauro FR, Capello D, Mancini F, De Propris MS, Mancini
M, Orsini E, Gentile M, Breccia M, Cuneo A, Castoldi G, Foa R. Chronic

92
lymphocytic leukemia patients with highly stable and indolent disease show
distinctive phenotypic and genotypic features. Blood 2003; 102:1035–1041.
27. Hamblin T.J., Davis Z., Gardiner A., Oscier D.G., Stevenson F.K. Unmutated
IgV(H) genes are associated with a more aggressive form of chronic lymphocytic
leukemia. Blood 1999; 94:1848–54.
28. Horwitz MS.Adenovirus immunoregulatory genes and their cellular targets.
Virology. 2001 Jan 5; 279(1):1-8. Review.
29. Insel RA, Adderson EE, Carroll WL. The repertoire of human antibody to the
Haemophilus influenzae type b capsular polysaccharide. International Reviews of
Immunology 1992; 9(1):25-43.
30. Kalina T, Flores-Montero J, van der Velden VH, Martin-Ayuso M, Böttcher S,
Ritgen M, Almeida J, Lhermitte L, Asnafi V, Mendonça A, de Tute R, Cullen M,
Sedek L, Vidriales MB, Pérez JJ, te Marvelde JG, Mejstrikova E, Hrusak O,
Szczepański T, van Dongen JJ, Orfao A; EuroFlow Consortium (EU-FP6, LSHB-
CT-2006-018708). EuroFlow standardization of flow cytometer instrument
settings and immunophenotyping protocols. Leukemia 2012; 26: 1986–2010.
31. Keating MJ, Chiorazzi N, Messmer B, Damle RN, Allen SL, Rai KR, Ferrarini M,
Kipps TJ. Biology and treatment of Chrinic Lymphocytic Leucemia. Hematology
2003, 2003:153-175.
32. Ladetto M, Donovan JW, Harig S, Trojan A, Poor C, Schlossnan R, Anderson
KC, Gribben JG. Real Time Polymerase Chain Reaction of Immunoglobulin
rearrangements for quantitative evaluation of minimal residual disease in multiple
myeloma. Biology of Blood and Marrow Transplantation 2000; 6: 241-253.
33. Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene
F, Wu Y, Gemrot E, Brochet X, Lane J, Regnier L, Ehrenmann F, Lefranc G,
Duroux P. IMGT, the international ImMunoGeneTics information system. Nucleic
Acids Research 2009; 37: D1006-D1012.
34. Logan AC, Zhang B, Narasimhan B, Carlton V, Zheng J, Moorhead M, Krampf
MR, Jones CD, Waqar AN, Faham M, Zehnder JL, Miklos DB. Minimal residual
disease quantification using consensus primers and high-throughput IGH
sequencing predicts post-transplant relapse in chronic lymphocytic leukemia.
Leukemia 2013; 27: 1659–1665.

93
35. Lundin J, Kimby E, Björkholm M, Broliden PA, Celsing F, Hjalmar V, Möllgård L,
Rebello P, Hale G, Waldmann H, Mellstedt H, Osterborg A. Phase II trials
subcutaneous anti-CD52 monoclonal antibody alentuzumab (Campath-1H as
first-line treatment for patients with B-cell chronic lymphocytic leukaemia (B-CLL).
Blood 2002; 100: 768-773.
36. Mauro FR, Foa R, Giannarelli D, Cordone I, Crescenzi S, Pescarmona E, Sala R,
Cerretti R, Mandelli F. Clinical characteristics and outcome of young chronic
lymphocytic leukemia patients: a single institution study of 204 cases. Blood
1999; 15:448-54.
37. Mauro FR, Giammartini E, Gentile M, Sperduti I, Valle V, Pizzuti A, Guarini A,
Giannarelli D, Foà R. Clinical features and outcome of familial chronic
lymphocytic leukemia. Haematologica. 2006; 91:1117-20.
38. Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, Murphy EJ,
Koduru P, Ferrarini M, Zupo S, Cutrona G, Damle RN, Wasil T, Rai KR,
Hellerstein MK, Chiorazzi N. In vivo measurements document the dynamic
cellular kinetics of chronic lymphocytic leukemia B cells. The Journal of Clinical
Investigation 2005 Mar; 115(3):755-64.
39. Milner EC, Capra JD. VH families in the antibody response to p-
azophenylarsonate: correlation between serology and amino acid sequence. The
Journal of Immunology.1982 Jul; 129(1):193-9.
40. Montserrat E. New prognostic markers in CLL. Hematology 2006;279-84.
41. Montserrat E., Moreno C. New monoclonal antibodies and immunomodulatory
agents for chronic lymphocytic leukemia therapy. Hematology 2010; 4:93-96.
42. Moreno, C., Villamor, N., Colomer, D., Esteve, J., Giné, E., Muntañola, A.,
Campo, E., Bosch, F. & Montserrat, E. Clinical significance of minimal residual
disease, as assessed by different techniques, after stem cell transplantation for
chronic lymphocytic leukemia. Blood 2006; 107: 4563-4569.
43. Packham G, Stevenson FK. Bodyguards and assassins: Bcl-2 family proteins
and apoptosis control in chronic lymphocytic leukaemia. Immunology. 2005 Apr;
114(4):441-9. Review.
44. Pedersen I.M., Buhl A.M., Klausen P., Geisler CH., Jurlander J. The Chimerical
anti-CD20 antibody rituximab induces apoptosis in B-cell chronic lymphocytic
leukaemia through a p38 mitogen activated-protein-kinase dependent
mechanism. Blood 2002; 99: 1314-1319.

94
45. Pott C. Minimal residual disease detection in mantle cell lymphoma: technical
aspects and clinical relevance. Seminars in Hematology 2011; 48: 172-184.
46. Rosner K, Winter DB, Tarone RE, Skovgaard GL, Bohr VA, Gearhart PJ. Third
complementarity-determining region of mutated VH immunoglobulin genes
contains shorter V, D, J, P, and N components than non-mutated genes.
Immunology 2001; 103: 179-187.
47. Rawstron AC, Yuille MR, Fuller J, Cullen M, Kennedy B, Richards SJ, Jack AS,
Matutes E, Catovsky D, Hillmen P, Houlston RS. Inherited predisposition to CLL
is detectable as subclinical monoclonal B-lymphocyte expansion. Blood 2002 Oct
1; 100(7):2289-90.
48. Rawstron AC, Böttcher S, Letestu R, Villamor N, Fazi C, Kartsios H, de Tute RM,
Shingles J, Ritgen M, Moreno C, Lin K, Pettitt AR, Kneba M, Montserrat E,
Cymbalista F, Hallek M, Hillmen P, Ghia P; European Research Initiative in CLL.
Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC)
update on the international harmonised approach for flow cytometric residual
disease monitoring in CLL. Leukemia 2013; 27: 142-149.
49. Rozman C, Montserrat E. Chronic lymphocytic leukemia. The New England
Journal of Medicine 1995 Oct 19; 333(16):1052-7. Review. No abstract available.
Erratum in: The New England Journal of Medicine 1995 Nov 30; 333(22):1515.
50. Stamatopoulos K, Belessi C, Hadzidimitriou A, Smilevska T, Kalagiakou E, Hatzi
K, Stavroyianni N, Athanasiadou A, Tsompanakou A, Papadaki T, Kokkini G,
Paterakis G, Saloum R, Laoutaris N, Anagnostopoulos A, Fassas
A.Immunoglobulin light chain repertoire in chronic lymphocytic leukemia. Blood
2005 Nov 15; 106(10):3575-83. Epub 2005 Aug 2.
51. Trentin L, Cabrelle A, Facco M, Carollo D, Miorin M, Tosoni A, Pizzo P, Binotto
G, Nicolardi L, Zambello R, Adami F, Agostini C, Semenzato G. Homeostatic
chemokines drive migration of malignant B cells in patients with Non-Hodgkin’s
Lymphomas. Blood 2004; 104:502-508.
52. Trentin L, Zambello R, Agostini C, Siviero F, Adami F, Marcolongo R, Raimondi
R, Chisesi T, Pizzolo G, Semenzato G. Expression and functional role of tumor
necrosis factor receptors on leukemc cells from patients with type B chronic
lymphoproliferative disorders. Blood 1993; 81:752-75.
53. Tobin G, Thunberg U, Johnson A, Eriksson I, Söderberg O, Karlsson K, Merup
M, Juliusson G, Vilpo J, Enblad G, Sundström C, Roos G, Rosenquist R.Chronic

95
lymphocytic leukemias utilizing the VH3-21 gene display highly restricted
Vlambda2-14 gene use and homologous CDR3s: implicating recognition of a
common antigen epitope. Blood. 2003 Jun 15; 101(12):4952-7. Epub 2003 Feb
13.
54. Van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, Van
Dongen JJ. Detection of minimal residual disease in hematologic malignancies
by real-time quantitative PCR: principles, approaches, and laboratory aspects.
Leukemia 2003; 17: 1013-1034.
55. van der Velden VH, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-
Grumayer ER, Flohr T, Sutton R, Cave H, Madsen HO, Cayuela JM, Trka J,
Eckert C, Foroni L, Zur Stadt U, Beldjord K, Raff T, van der Schoot CE, van
Dongen JJ; European Study Group on MRD detection in ALL (ESG-MRD-ALL).
Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines
for interpretation of real-time quantitative PCR data. Leukemia 2007; 21: 604–
611.
56. Wysocki LJ, Gridley T, Huang S, Grandea AG 3rd, Gefter ML. Single germline
VH and V kappa genes encode predominating antibody variable regions elicited
in strain A mice by immunization with p-azophenylarsonate. The Journal of
Experimental Medicine 1987 Jul 1;166(1):1-11.
57. Xue W, Luo S, Adler WH, Schulze DH, Berman JE. Immunoglobulin Heavy
Chain Junctional Diversity in Young and Aged Humans. Human Immunology
1997; 57: 80-92.

96
III° Progetto: La malattia minima residua condiziona la prognosi dei pazienti affetti da linfoma follicolare: risultati del trial FIL FOLL05.

97
Il Linfoma Follicolare
Il linfoma follicolare (LF) è un linfoma a basso grado che origina dalle cellule B
centrofollicolari dei centri germinativi dei linfonodi, rappresenta il 20-25% di tutti i linfomi
non Hodgkin ed è il più frequente linfoma a cellule B dei paesi occidentali mentre è raro
nei paesi asiatici.
La neoplasia colpisce in egual modo entrambi i sessi ed insorge mediamente intorno ai
50/60 anni. Meno del 10% dei pazienti ha età inferiore ai 30 anni.
La sopravvivenza dei pazienti è molto variabile, con una media di 8-10 anni e tende ad
aumentare, grazie all'introduzione dell'immunoterapia.
La diagnosi di malattia si basa sull'esame istologico di biopsie escissionali di linfonodi o
di altro campione chirurgico.
La diagnosi istologica si rifà ai criteri classificativi stabiliti dalla “World Health
Organization” (WHO) (Jaffe ES, Hematology 2009). I LF sono caratterizzati da un
pattern di crescita follicolare oppure follicolare e diffuso.
Sulla base del numero medio di centroblasti presenti nella lesione neoplastica, si
distinguono secondo la classificazione WHO i LF di grado 1, con una predominanza di
piccole cellule (0-5 centroblasti per campo); quelli di grado 2, di tipo “misto” (6-15) e
quelli di grado 3, LF “a grandi cellule” (più di 15).
I LF di grado 3 si suddividono in 3a e 3b. I 3a esprimono ancora una componente
centrocitaria, i 3b mostrano invece un “tappeto di centroblasti” e sono maggiormente
assimilabili al linfoma non Hodgkin diffuso a grandi cellule.
Il grado istologico del tumore, è un utile indicatore del decorso clinico della malattia.
In questo senso, l'ultima versione della classificazione WHO, approvata nel 2008, ha
raggruppato insieme i LF di grado 1, 2 e 3a essendo simili dal punto di vista
prognostico; diversificandoli nettamente dai LF di grado 3b che mostrano una prognosi
decisamente peggiore a causa della loro aggressività. In particolare tali neoplasie
mostrano spesso cariotipi complessi e mutazioni di geni come TP53.
Da un punto di vista immunofenotipico, le cellule tumorali dei LF esprimono
immunoglobuline di superficie (soprattutto IgM e IgD), le catene leggere delle Ig kappa
o lambda ed antigeni pan B quali CD19, CD20, CD22, CD79a e b. Inoltre risultano
negative per CD5 e CD43 ed in circa il 60% dei casi positive per il CD10. La positività
per il CD10 si è dimostrata utile per distinguere il LF dai linfomi della zona marginale; la
negatività per CD5 e CD43 per distinguerlo dalla leucemia linfatica cronica.

98
Il marcatore distintivo del LF è la traslocazione dei cromosomi 14 e 18
[t(14;18)(q32;q21)]. Questa alterazione genetica è presente nel 70-95% dei casi di
malattia (Yunis JJ et al, N Engl J Med 1987; Cleary ML et al, Cell 1986) e causa la
giustapposizione del gene anti-apoptotico BCL-2 (18q21) alla regione JH del gene della
catena pesante delle immunoglobuline (Ig) (14q32). Questo si traduce nell'aumentata
espressione del gene e nell'accumulo della proteina bcl-2 nelle cellule tumorali. Si
produce infatti una proteina di fusione bcl-2/IGH funzionale.
L'alterazione citogenetica nasce negli stadi iniziali di sviluppo della cellule B, durante il
meccanismo di riarrangiamento delle Ig (Kuppers R et al, N Engl J Med 1999) ed è
generalmente presente già al momento dell'esordio.
Altre traslocazioni possono presentarsi nei casi di LF e queste sono le t(2;18)(p11;q21)
e la t(18;22)(q21;q11), che vedono rispettivamente la traslocazione di BCL-2 accanto ai
geni delle catene leggere kappa e lambda delle Ig (IGΚ) e (IGL).
Nell'ultimo decennio la tecnica del “gene expression profiling” ha permesso
l'identificazione di set di geni con un valore prognostico indipendente dai convenzionali
parametri clinici e patologici (Dave SS et al, N Engl J Med 2004; Glas AM et al, J Clin
Oncol 2007).
In particolare sono stati definiti due profili di espressione genica distinti, chiamati
“immune-response 1” e “immune-response 2” associati rispettivamente ad una
prolungata e ad una ridotta sopravvivenza.
I geni associati a buona prognosi sono espressi da cellule T, quelli associati a cattiva
prognosi sono espressi preferenzialmente da macrofagi e cellule dendritiche.
Da qui la recente ipotesi per cui i LF si svilupperebbero grazie alle alterazioni genetiche
presenti nelle cellule tumorali e alle modificazioni di quella rete di interazioni
immunologiche che connette le cellule maligne alle altre cellule del microambiente
tumorale (de Jong D, J Clin Oncol 2005)(figura 1).
In particolare, la presenza nella massa tumorale di cellule T regolatorie (Treg) è
associata ad una sopravvivenza prolungata (Carreras J et al, Blood 2006).
Le cellule T helper follicolari (TFH) sono altrettanto importanti nel LF (King C et al, Annu
Rev Immunol 2008) in quanto associate ad una maggiore sopravvivenza dei pazienti.
Al contrario i macrofagi promuoverebbero la crescita tumorale e questo probabilmente
grazie alla produzione di citochine e chemochine.
Le cellule dendritiche follicolari, infine, favorirebbero la neoplasia prevenendo l'apoptosi.
La risposta immunitaria, comunque, differisce da individuo ad individuo e non solo per

99
la differente esposizione ai vari antigeni, ma anche a causa di varianti genetiche
ereditarie come nel caso dei polimorfismi di singoli nucleotidi.
Figura 1. Un modello di doppio pathway per il linfoma follicolare. Come primo evento, l'apoptosi-resistenza in assenza di una stimolazione antigenica viene data dalla traslocazione t(14;18) o da eventi biologici genomici equivalenti. Successivamente, alterazioni precoci secondarie producono una intrinseca dicotomia prognostica. Nel pathway a cattiva prognosi, le alterazioni genomiche guidano il processo verso uno stato immunologico attivato ed un instabile corredo genetico. Le cellule follicolari dendritiche attivate (FDC) e le cellule T attivate giocano un ruolo dominante. In alternativa, nel pathway a buona prognosi le alterazioni genomiche provocano un'ulteriore stabilizzazione delle cellule tumorali in un contesto immunologico inattivo delle FDCs e dei linfociti T. Le alterazioni genomiche si accumulano ad un ritmo lento con relativa resistenza alla trasformazione e buona prognosi.
Fonte: de Jong D: “Molecular pathogenesis of follicular lymphoma: a cross talk of genetic and immunologic factors”. The Journal of Clinical Oncology 2005, 23:6358–6363.
La stadiazione viene definita applicando il sistema di Ann Arbor, basato sulla
valutazione del numero dei siti coinvolti e sulla presenza di malattia al disopra ed al di
sotto del diaframma. Il sistema definisce quattro stadi:
•Stadio I: coinvolgimento di una sola stazione linfonodale (chiamato stadio IE se c'è
un'unica localizzazione extranodale);
•Stadio II: coinvolgimento di due o più stazioni linfonodali dallo stesso lato del
diaframma (stadio IIE se c’è una limitata localizzazione extranodale per contiguità);
•Stadio III: coinvolgimento di linfonodi da ambedue i lati del diaframma;
•Stadio IV: localizzazione extranodale estesa (midollo osseo, fegato, polmone).
Ogni stadio viene definito A oppure B in base all’assenza o presenza di sintomi
sistemici.

100
Nell'80% dei casi i pazienti si collocano, al momento della diagnosi, negli stadi III e IV,
ma ciò non è necessariamente associato ad una prognosi severa. Solo un 15-20% dei
pazienti si trova agli stadi iniziali di malattia (I e II).
Una volta diagnosticata la malattia i pazienti vengono inclusi in una delle tre categorie di
rischio definite dal “Follicular Lymphoma International Prognostic Index” (FLIPI) o dal
“Follicular Lymphoma International Prognostic Index-2” (FLIPI-2), che sono associate
ad una diversa attesa di vita (Solal-Céligny P et al, Blood 2004).
Nel FLIPI i cinque fattori clinici che determinano la stratificazione dei pazienti nelle varie
categorie di rischio sono: l'età (superiore ai 60 anni), i livelli di emoglobina (inferiori a
120g/l) il numero delle aree linfonodali interessate (superiori a 4), lo stadio di Ann Arbor
(avanzato) ed i livelli di LDH (superiori alla norma).
Nel FLIPI-2 sono: l'età, i livelli di emoglobina (inferiori a 120g/l), la grandezza delle aree
linfonodali interessate, il coinvolgimento del midollo osseo, ed i livelli di β2-
microglobulina.
In base al punteggio, che va da 0 a 5, i pazienti vengono definiti a basso (0-1),
moderato (2) o alto (3-5) rischio. Diversi studi hanno confermato la validità della
valutazione prognostica FLIPI nel predire correttamente la sopravvivenza globale e la
sopravvivenza in assenza di progressione di malattia (Tan D et al, Hematol Oncol Clin,
2008).
Da un punto di vista clinico a volte le condizioni dei pazienti affetti da LF sono buone: si
osservano linfoadenopatie generalizzate asintomatiche con interessamento nodale e
splenico. Nel 50% dei casi di LF di grado 1 si osserva anche un coinvolgimento
midollare, coinvolgimento che è presente anche nel 30% dei casi di grado 2 e 3
(Armitage JO et al., J Clin Oncol 1998).
I sintomi sistemici sono rari e solamente in un 10% dei casi si osserva leucemizzazione.
Solitamente il decorso clinico è indolente per alcuni anni ma poi, frequentemente, si
osserva la trasformazione della malattia in una forma istologica più aggressiva,
caratterizzata da una crescita più rapida ed una minore risposta ai chemioterapici
(Gallagher CJ et al., J Clin Oncol 1986). La trasformazione verso un tipo istologico più
aggressivo è un evento terminale comune (Bastion Y et al., J Clin Oncol 1997) che si
verifica nel 20-60% dei pazienti (Yuen AR et al., J Clin Oncol 1995; Montoto S et al., J
Clin Oncol 2007). Questa percentuale raggiunge il 70% nei casi esaminati all'autopsia
(Garvin D et al, Cancer 1983).
In particolare linfomi diffusi a grandi cellule (DLCL) oppure linfomi di Burkitt che

101
originano da LF di grado 1 o 2, così come DLCL che nascono da LF di grado 3 sono
considerati linfomi trasformati; al contrario la progressione di un LF di grado 1/2 in un LF
di grado 3 non è sempre considerata come una trasformazione (Bernstein SH et al,
Hematology 2009).
La patogenesi di questo processo rimane ampiamente sconosciuta. Le attuali
conoscenze della biologia della trasformazione suggeriscono il coinvolgimento di fattori
genetici, epigenetici e del microambiente, in particolare le mutazioni di TP53 (Sander
CA et al., Blood 1993), l'inattivazione genetica e/o epigenetica del gene CDKN2A/p16
(Pinyol M et al., Blood 1998), le traslocazioni coinvolgeneti il protoncogene BCL6
(Akasaka T et al., Blood 2003), le alterazioni che coinvolgono il cromosoma 1p36
(Martinez-Climent JA et al., Blood 2003), e cambiamenti nell'espressione di MYC
(Lossos IS et al., Proc. Natl. Acad. Sci. 2002). Sono stati evidenziati cambiamenti
acquisiti nel numero di copie di cromosomi (Fitzgibbon J et al., Leukemia 2007). Inoltre,
è stata riscontrata un'associazione tra progressione a DLCL ed un'aberrante
ipermutazione somatica (Rossi D et al, Haematologica 2006), un meccanismo di
instabilità genetica derivante dal funzionamento anomalo dell' ipermutazione somatica
fisiologica (Pasqualucci L et al., Nature 2001).
Il profilo genomico del LF trasformato mostra in parte delle similitudini con quello del
DLCL de novo ed in parte presenta combinazioni uniche di geni alterati con implicazioni
diagnostiche e terapeutiche (Pasqualucci L et al, Cell Rep 2014).
La strategia terapeutica per la cura del LF dipende da numerosi fattori, primo tra tutti lo
stadio di malattia.
Una minoranza dei pazienti è affetta da LF localizzato, cioè in stadio I e II. Per questi la
terapia standard è la radioterapia con irradiazione dei campi coinvolti (IFRT)
(Guadagnolo BA et al, InterJ Rad Oncol 2006; Peterson PM et al, J Clin Oncol 2004).
Il possibile beneficio derivante dall’aggiunta di anticorpi monoclonali anti-CD20 alla
radioterapia in questi pazienti è in corso di valutazione.
Circa l’80% dei pazienti è affetto da una malattia in stadio III e IV e la maggior parte di
questi è clinicamente asintomatico (Tan D et al, Hematol/Oncol Clin North America
2008).
Per questi pazienti si attua la strategia del “watching and waiting”: non si interviene con
un trattamento finché non insorgono i primi sintomi sistemici. Alcuni dati indicano tale
approccio come il migliore in termini di sopravvivenza generale e tasso di
trasformazione istologica. Questa strategia è supportata anche dall'evidenza clinica di

102
casi che mostrano regressione spontanea di malattia (Friedberg JW et al., J Clin Oncol
2008).
Al contrario, quando il paziente deve essere trattato, si procede solitamente con un
approccio immuno-chemioterapico.
Un esempio di strategia immuno-chemioterapica è il trattamento standard di prima linea
R-CHOP che combina la polichemioterapia CHOP [Cyclophosphamide,
Hydroxydaunorubicin (doxorubicin), Oncovin (vincristine),Prednisone], all'utilizzo
dell'anticorpo monoclonale anti-CD20 denominato Rituximab. Questa terapia si è
dimostrata superiore alla sola chemioterapia, sia come percentuali di risposta sia come
tassi di sopravvivenza libera da malattia (Hiddemann W et al, Blood 2005; Vitolo U et al,
Critical Rev Oncol/Hematol 2008)
Il Rituximab si è rivelato molto efficace anche in monoterapia e nella terapia di
mantenimento portata avanti dopo il trattamento immuno-chemioterapico.
Lo Zevalin, invece, è un farmaco radio immunoconiugato costituito da un anticorpo
monoclonale anti-CD20 coniugato con l'isotopo radioattivo β emittente Ittrio 90. La
sensibilità dei linfomi all'irradiazione e la specificità degli anticorpi anti-CD20 spiegano il
razionale dell'impiego nonché il successo di questi farmaci, la cui collocazione nella
strategia terapeutica del LF è in corso di definizione.
La chemioterapia ablativa e la radioterapia seguite da un autotrapianto di cellule
staminali si sono dimostrate efficaci in pazienti con malattia recidivata e/o in fase
avanzata.
Il trapianto di midollo allogenico è una strategia destinata a pazienti giovani che
risultano essere refrattari a molteplici linee terapeutiche, ma comporta rischi maggiori
rispetto al trapianto autologo di cellule staminali a causa della maggiore tossicità.
Nuovi approcci terapeutici sono oggi in fase avanzata di studio. Tra questi vi è la
realizzazione di vaccini paziente specifici, disegnati per provocare risposte umorali o
cellulari contro le immunoglobuline clonali di superficie, ed i nuovi inibitori orali della
fosfatidil-inositolo chinasi delta (PI3Kδ) o Idelalisib, della Bruton tirosin chinasi (BTK) o
Ibrutinib.
Il monitoraggio molecolare della malattia minima residua (MMR) è uno strumento
altrettanto fondamentale per valutare l'efficacia della strategia terapeutica nei pazienti
con LF.
A volte pazienti in apparente regressione clinica di malattia nascondono una certa
quantità di cellule malate identificabili grazie alla ricerca di alterazioni cellulari tipiche

103
della malattia.
Nel LF l'analisi della MMR viene eseguita attraverso la valutazione della
presenza/assenza del trascritto ibrido BCL-2/IGH e della determinazione della sua
quantità nelle cellule del sangue periferico e del sangue midollare.

104
Il gene BCL-2 nel linfoma follicolare
I membri della famiglia BCL-2 (B cell CLL/Lymphoma-2) regolano il pathway
mitocondriale della morte cellulare programmata (apoptosi) grazie ad una serie di
complesse interazioni che influenzano l'integrità della membrana mitocondriale esterna
(Green DR et al, Cancer Cell 2002).
La famiglia BCL-2 è composta da proteine antiapoptotiche e proapoptotiche che hanno
in comune quattro domini di omologia chiamati BH1, BH2, BH3 e BH4 (Kvansakul M et
al, Cell Death Differ 2008).
Le proteine anti-apoptotiche sono proteine integrali di membrana presenti sulla
membrana mitocondriale esterna, ma possono essere presenti anche nel citoplasma o
nella membrana del reticolo endoplasmatico. Fanno parte di questo gruppo le proteine
a1, bcl-2, bcl-XL, bcl-w e mcl-1; le molecole pro-apoptotiche sono bax e bak(figura 2).
Figura 2: La grande famiglia della proteina bcl-2. Questa famiglia comprende proteine pro-sopravvivenza (evidenziate nelle caselle verdi) e proteine pro-apoptotiche (evidenziate in rosso). Oltre a questa suddivisione funzionale della famiglia vi è una suddivisione strutturale che divide le proteine in proteine multidominio e proteine BH3-only. In a) sono evidenziati i membri della famiglia che condividono quattro domini BCL-2 di omologia, denominati BH (proteine multidominio). Le proteine bcl-2-antagonist/killer (bak) e bcl-2-associati proteina X (bax) sono essenziali per l'apoptosi attivata attraverso il pathway mitocondriale. In b) sono evidenziati i membri che mostrano solo il dominio BH3, le proteine BH3-only. Fonte: Lessene G, Czabotar PE, Colman PM. “BCL-2 family antagonists for cancer therapy” Nat Rev Drug Discov, 2008 Dec;7(12):989-1000.
Altre proteine che svolgono un ruolo altrettanto importante nella regolazione

105
dell’apoptosi sono le proteine “BH3 only”, chiamate così perché mancano dei domini
BH1, BH2 e BH4.
Nei mammiferi sono note 8 proteine di questo tipo, le più importanti sono bad, bix e bim.
L’interazione tra proteine pro-apoptotiche e anti-apoptotiche determina il destino della
cellula. L’induzione dell’apoptosi è un processo complesso che nei diversi tessuti viene
controllato da differenti membri della famiglia BCL-2 e generato in risposta a diversi
stimoli.
La proteina bcl-2, ad esempio, serve per la sopravvivenza di linfociti B e T maturi (Veis
DJ et al, Cell 1993).
La conformazione germinale del gene BCL-2 è composta da tre esoni con un grande
introne di 225Kb presente tra l'esone 2 e l'esone 3.
La proteina bcl-2 wild tipe ha un peso molecolare di 26kDa e svolge una funzione anti-
apoptotica. Tale proteina tende ad eterodimerizzare con bax a formare un complesso
ad alto peso molecolare bcl-2/bax.
Se nelle cellule bax è presente in eccesso allora si formano degli omodimeri di bax che
accelerano il processo di morte cellulare; al contrario, se è bcl-2 ad essere presente in
eccesso, gli eterodimeri bcl-2/bax sono la specie prevalente e la morte cellulare viene
bloccata.
Le traslocazioni che coinvolgono il gene BCL-2 sono comuni nelle neoplasie linfoidi
umane.
Questi riarrangiamenti avvengono sempre nei primi stadi dell'ontogenesi della cellula B
e permettono alla cellula di maturare sino allo stadio di cellula IgM+ e IgD+.
La traslocazione più comune è la t(14;18)(q32;q21) che causa la giustapposizione della
regione 18q21 all'IGH(figura 3).
Occasionalmente la regione 18q21 è traslocata sull'IGΚ [t(2;18) (p11;q21)] o sull'IGL
[t(18;22)(q21;q11)].
Le rotture sul 18q21 sono presenti in quasi tutti i casi di linfoma follicolare e nel 30% dei
DLCL.
In generale ogni traslocazione porta ad una deregolazione dell'espressione del gene
BCL-2 e conseguentemente alla presenza costitutiva di alti livelli della proteina.
Il riarrangiamento t(14;18)(q32;q21) permette l'unione del gene BCL-2 alla regione 3'
non trascritta del segmento J dell'IGH. Tale traslocazione non altera la sequenza
codificante di BCL-2 che rimane intatta. Il cDNA (DNA complementare) risultante è
pertanto rappresentato dalla fusione dei tre esoni di BCL-2 (al lato 3' del trascritto) con
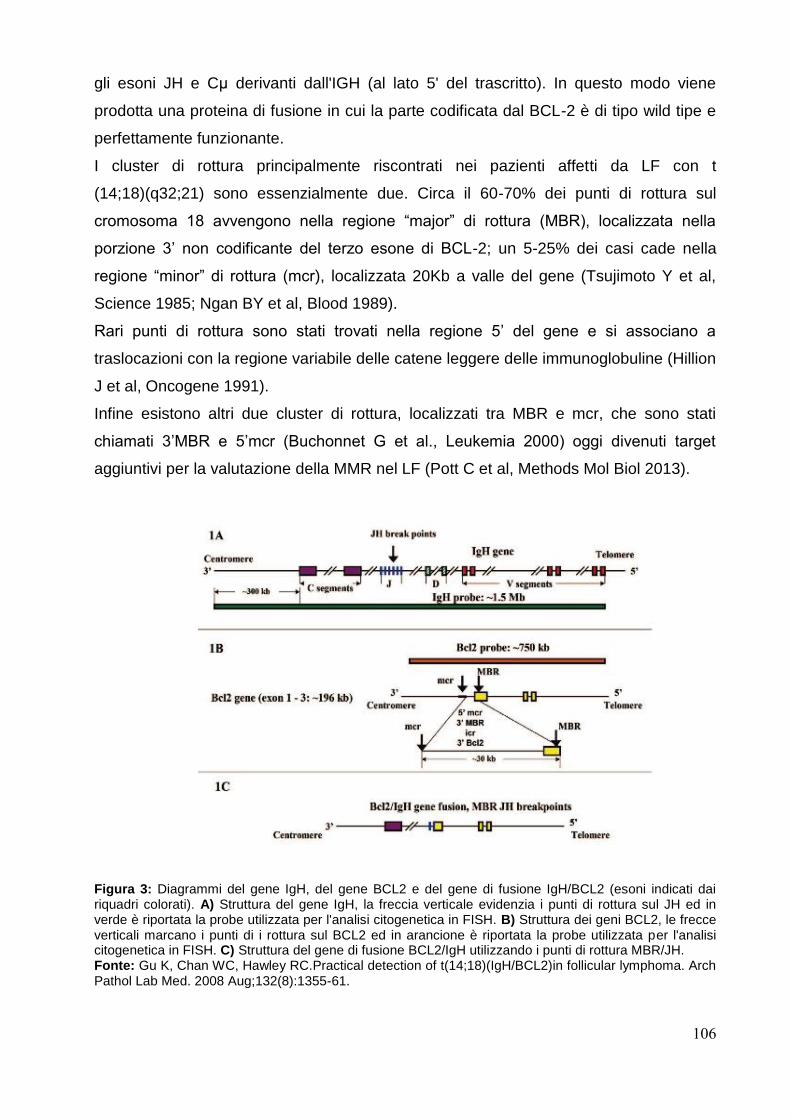
106
gli esoni JH e Cμ derivanti dall'IGH (al lato 5' del trascritto). In questo modo viene
prodotta una proteina di fusione in cui la parte codificata dal BCL-2 è di tipo wild tipe e
perfettamente funzionante.
I cluster di rottura principalmente riscontrati nei pazienti affetti da LF con t
(14;18)(q32;21) sono essenzialmente due. Circa il 60-70% dei punti di rottura sul
cromosoma 18 avvengono nella regione “major” di rottura (MBR), localizzata nella
porzione 3’ non codificante del terzo esone di BCL-2; un 5-25% dei casi cade nella
regione “minor” di rottura (mcr), localizzata 20Kb a valle del gene (Tsujimoto Y et al,
Science 1985; Ngan BY et al, Blood 1989).
Rari punti di rottura sono stati trovati nella regione 5’ del gene e si associano a
traslocazioni con la regione variabile delle catene leggere delle immunoglobuline (Hillion
J et al, Oncogene 1991).
Infine esistono altri due cluster di rottura, localizzati tra MBR e mcr, che sono stati
chiamati 3’MBR e 5’mcr (Buchonnet G et al., Leukemia 2000) oggi divenuti target
aggiuntivi per la valutazione della MMR nel LF (Pott C et al, Methods Mol Biol 2013).
Figura 3: Diagrammi del gene IgH, del gene BCL2 e del gene di fusione IgH/BCL2 (esoni indicati dai riquadri colorati). A) Struttura del gene IgH, la freccia verticale evidenzia i punti di rottura sul JH ed in verde è riportata la probe utilizzata per l'analisi citogenetica in FISH. B) Struttura dei geni BCL2, le frecce verticali marcano i punti di i rottura sul BCL2 ed in arancione è riportata la probe utilizzata per l'analisi citogenetica in FISH. C) Struttura del gene di fusione BCL2/IgH utilizzando i punti di rottura MBR/JH. Fonte: Gu K, Chan WC, Hawley RC.Practical detection of t(14;18)(IgH/BCL2)in follicular lymphoma. Arch Pathol Lab Med. 2008 Aug;132(8):1355-61.

107
Numerosi studi hanno messo in evidenza che, qualunque sia il cluster di rottura, JH6 è
il segmento “joining” più frequentemente coinvolto nelle traslocazioni (Jager U et al.,
Blood 2000).
Esistono opinioni discordanti riguardo una eventuale correlazione tra la localizzazione
del punto di rottura sul cromosoma 18 e le caratteristiche della malattia o la
sopravvivenza dei pazienti (Lopez-Guillermo A et al, Blood, 1999; Buchonnet G et al.,
Leukemia, 2002).
Una serie di esperimenti ha inoltre dimostrato che la proteina bcl-2 coopera con c-Myc
nella trasformazione di precursori di cellule B (Vaux et al, Nature 1998) ma, in assenza
di un oncogene addizionale, BCL-2 non promuove la proliferazione cellulare.
Quindi la t(14;18) non è sufficiente per lo sviluppo del LF (Bende RJ et al., Leukemia
2007); per poter crescere in vitro, le cellule LF hanno bisogno di segnali stimolatori che
vengono forniti da cellule immunoregolatorie quali i linfociti T e le cellule dendritiche
follicolari.
Su queste osservazioni si basa l'ipotesi che vede il LF come una malattia progressiva
che parte con l'acquisizione della traslocazione durante le prime fasi di sviluppo delle
cellule B nel midollo osseo (Martinez A et al, Cur Ematol Malign Rep 2008).
Il vantaggio di sopravvivenza, che hanno le cellule BCL-2 positive nei CG, faciliterebbe
l'insorgenza di eventi genetici addizionali, che a loro volta contribuirebbero alla
modulazione del microambiente CG. Per un certo periodo di tempo le cellule
neoplastiche riceverebbero i segnali di crescita e di sopravvivenza dalle cellule
circostanti, e così si svilupperebbe una malattia incurabile ma indolente.
Successivamente, particolari alterazioni geniche secondarie permetterebbero alle
cellule di sopravvivere al di fuori dei CG, definendo una malattia molto aggressiva.
La traslocazione t(14;18)(q32;q21), però, è stata ritrovata anche in individui sani
indipendentemente dal sesso, razza ed età, con punti di rottura simili a quelli dei
pazienti con LF e, ad oggi, il significato biologico di questo evento non è noto (Biagi JJ
et al., Blood 2002; Schmitt C et al, Leukemia Res 2006).
In particolare questa evidenza solleva interrogativi riguardo l’autentico significato di una
positività riscontrata in un’analisi molecolare, poichè ad oggi non esiste nessun
collegamento tra la positività, negli individui sani, per la presenza di questa
traslocazione, ed il successivo sviluppo di LF.

108
Obiettivo dello studio
Il marcatore molecolare distintivo del LF è la traslocazione dei cromosomi 14 e 18
[t(14;18)(q32;q21)], con conseguente giustapposizione del gene anti-apoptotico BCL-2
(18q21) alla regione JH del gene della catena pesante delle Ig (14q32).
La MMR viene solitamente valutata con tecniche di biologia molecolare, che vanno ad
analizzare la presenza, l'assenza e la quantità del trascritto ibrido BCL-2/IGH@
rilevabile in cellule del sangue periferico e del sangue midollare.
Nel LF il monitoraggio della MMR è risultato essere un fattore predittivo ben consolidato
dell'andamento clinico della malattia post-trapianto (Ladetto M et al,Blood 2008).
Studi che dimostrano il valore prognostico della analisi di MMR qualitativa dopo terapia
convenzionale sono sempre più numerosi (Corradini P et al, J Clin Oncol 2004;Rown
JR et al, Biol Blood Marrow Transplant 2007; Procházka V et al, J Clin Oncol, 2011;
Ladetto M et al, Blood 2013).
Al contrario, pochi lavori affrontano l'analisi quantitativa della MMR nel LF (Rambaldi A
et al, Blood 2005; Goff L et al, J Clin Oncol 2009).
Lo scopo di questo studio è stato quello di valutare il valore prognostico della MMR
quantitativa nell'ambito del protocollo FOLL05 (NCT00774826), i cui risultati terapeutici
sono stati recentemente pubblicati (Federico M. et al, J Clin Oncol. 2013).

109
Materiali e Metodi
Pazienti e campioni studiati
Lo studio prospettico, randomizzato, multicentrico di fase III FOLL05 (NCT00774826) è
stato condotto su un totale di 504 pazienti, di età compresa tra 18 e 75 anni, con una
diagnosi istologica di LF di grado I, II e IIIa secondo la classificazione WHO e di stadio
II- IV secondo il sistema Ann Arbor.
Il protocollo terapeutico comprendeva tre bracci di trattamento: 8 dosi di rituximab (R)
per 8 cicli, in combinazione con 8 cicli di CVP(Cyclophosphamide, Vincristine,
Prednisone)(braccio di controllo), o con 6 cicli di CHOP(Cyclophosphamide, Vincristine,
Doxorubicin, Prednisone) o FM (Fludarabine, Mitoxantrone).
L’analisi molecolare per la ricerca del riarrangiamento del BCL2/IGH@ è stata eseguita
per tutti i punti previsti dal protocollo, su DNA estratto da un pellet di cellule
mononucleate di sangue midollare mediante Wizard Genomic DNA Purification Kit
(Promega Corp, Madison, Wisconsis, USA).
Screening del riarrangiamento BCL-2/IGH@
L'analisi qualitativa del riarrangiamento del BCL2/IGH@ è stata eseguita
prospetticamente al momento della diagnosi, a sei settimane dopo la fine del
trattamento, e ogni sei mesi durante il secondo e terzo anno di follow-up. Tutte le analisi
qualitative sono state centralizzate ed eseguite presso il laboratorio di Ematologia di
Pisa.
I 4 laboratori (Roma, Torino, Bologna, Pisa) della FIL-MRD-NETWORK (rete della
Fondazione Italiana Linfomi, fondata nel 2009), dopo standardizzazione inter-
laboratori delle metodiche, hanno eseguito retrospettivamente l’analisi molecolare
quantitativa per il riarrangiamento BCL2/IGH@ sui campioni centralizzati dal
laboratorio di Pisa alla diagnosi ed al momento della risposta (sei settimane dalla fine
del trattamento), allo scopo di valutare se la determinazione della quantità della
malattia in questi due time-points aggiunga informazioni rispetto la MMR valutata
qualitativamente (presenza/assenza del trascritto).
In primo luogo, i campioni sono stati riamplificati e sequenziati per confermare il dato
di partenza.

110
Il DNA è stato amplificato mediante reazione di amplificazione Polymerase Chain
Reaction (PCR) utilizzando il termociclatore GeneAmp® PCR System 9700 (Applied
Biosystems, Foster City, CA, USA).
L’amplificazione è stata ottenuta mediante nested PCR utilizzando il set di primers
relativo alla regione di MBR, di mcr ed alla regione di giunzione delle immunoglobuline
(Gribben JG et al. Blood 1991).
Le condizioni di PCR per l’amplificazione della regione di MBR sono state le seguenti:
-I round: denaturazione, annealing ed estensione per 27 cicli rispettivamente a 94°C per
1’, 55°C per 1’ e 72°C per 1. Alla fine dei cicli si esegue un’estensione a 72°C per 10’.
-II round: denaturazione, annealing ed estensione per 30 cicli rispettivamente a 94°C
per 1’, 58°C per 1’ e 72°C per 1. Alla fine dei cicli si esegue un’estensione a 72°C per
10’.
Le condizioni di PCR per l’amplificazione della regione di mcr sono state le seguenti:
-I round: 1 ciclo con denaturazione a 94°C per 3', annealing a 58°C per 1', estensione a
72°C per 1', seguito da denaturazione, annealing ed estensione per 30 cicli
rispettivamente a 94°C per 1’, 58°C per 1’ e 72°C per 1'. Alla fine dei cicli si esegue
un’estensione a 72°C per 10’.
-II round: 1 ciclo con denaturazione a 94°C per 3', annealing a 60°C per 1', estensione a
72°C per 1', seguito da denaturazione, annealing ed estensione per 30 cicli
rispettivamente a 94°C per 45”, 60°C per 30” e 72°C per 30”. Alla fine dei cicli si esegue
un’estensione a 72°C per 10’.
La separazione e l’identificazione degli amplificati di PCR è stata eseguita mediante
corsa elettroforetica su gel di agarosio al 2% in TBE 1X, contenente 5 µl/100ml di
bromuro d’etidio (SIGMA-ALDRICH, St. Louis; MO, USA) come agente intercalante del
DNA.
Sequenziamento e analisi del gene
I prodotti di PCR sono stati sequenziati direttamente secondo metodo Sanger con
marcatura fluorescente utilizzando il kit di reazione BigDye Terminator v3.1 Cycle
Sequencing Kit (Applied Biosystems, Foster City, CA, USA) e il termociclatore
GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, CA, USA). Durante la
sintesi del filamento di DNA i didesossinucleotiditrifosfati (ddNTP) marcati con quattro
differenti fluorocromi vengono, mediante l’enzima DNA polimerasi, inseriti nel filamento

111
stampo impedendo l’aggiunta di ulteriori nucleotidi. Al termine della reazione di
sequenza la miscela di singoli frammenti di DNA ottenuta è stata purificata utilizzando il
kit DyeEx 2.0 Spin Kit (QIAGEN, Valencia, CA, USA) che permette di eliminare i ddNTP
fluorescenti in eccesso non incorporati attraverso il principio cromatografico della gel-
filtrazione su colonnine contenenti resina. I frammenti di DNA purificati sono stati
sottoposti ad elettroforesi capillare mediante il sequenziatore ABI PRISM® 3100-Avant
Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). L’acquisizione dei dati
relativi ai campioni è stata eseguita utilizzando il software 3100-Avant Data Collection
Software e le sequenze sono state analizzate mediante il programma ABI PRISM® DNA
Sequencing Analysis Software (Versione 3.7), e visualizzate come elettroferogrammi.
L'interpretazione dei risultati è stata eseguita allineando le sequenze nucleotidiche
ottenute alla directory IMGT (international ImMunoGeneTics information system) per
l'identificazione del riarrangiamento genico BCL2/IGH@.
Analisi RQ-PCR
L'analisi è stata eseguita utilizzando i rispettivi primers e la probe per la RQ-PCR
(Ladetto M et al, Exp Hematol. 2001).
Le curve standard per l'analisi quantitativa, sono state preparate a partire da diluizioni
seriali della linea cellulare DOHH-2, nel DNA estratto da un pellet di cellule
mononucleate proveniente da un pool di sangue venoso periferico (PBL) di donatori
sani e portato alla concentrazione di 100 μg/ml. I saggi sono stati allestiti per
raggiungere una gamma di sensibilità e quantificabilità ≥10-4, con uno slope della curva
compreso tra 3.1 e 3.9 ed un coefficiente di correlazione ≥0,98, secondo le linee guida
della ESG-MRD–ALL (van der Velden VHJ et al, Leukemia 2007).
Le reazioni sono state preparate utilizzando i reagenti del kit TaqMan PCR Core
Reagent (PE Applied Biosystems, Foster City CA, USA) e processate nell' ABIPrism
7300-7500 alle seguenti condizioni: un'incubazione iniziale di 2 minuti a 50°C e 10
minuti di denaturazione a 95°C, seguite da 42 cicli di denaturazione a 95°C per 15
secondi ed annealing a 60°C per 1 minuto. Sono state rispettate le linee guida per
l'analisi di RQ-PCR della MMR per tutti i campioni studiati, ESG-MRD-ALL (European
Study Group on MRD Detection for ALL)(van der Velden VHJ et al, Leukemia 2007).

112
Risultati
I risultati dell'analisi di MMR qualitativa e quantitativa di questo studio sono stati
recentemente sottomessi per pubblicazione in un lavoro dal titolo “Minimal residual
disease after conventional treatment significantly impacts on progression-free survival
of patients with follicular lymphoma: the FIL FOLL05 trial”.
(Galimberti S et al, Clinical Cancer Research).
Analisi qualitativa e burden tumorale molecolare dei pazienti all'arruolamento
Alla diagnosi, 415/504 pazienti eleggibili per il trattamento sono stati valutati per
BCL2/IGH@ mediante PCR qualitativa, 220 dei quali (53%) sono risultati positivi per il
marcatore molecolare.
Lo stato molecolare alla diagnosi non correlava significativamente con il coinvolgimento
istologico midollare: 43% dei casi senza coinvolgimento midollare all'analisi
microscopica, mostrava un marcatore molecolare, mentre il 50% dei casi con
infiltrazione midollare è risultato BCL2/IGH@ negativo.
Il rilevamento del gene di fusione BCL2/IGH@ prima della terapia ha condizionato in
modo significativo la qualità della risposta: il tasso di remissione completa (CR-
complete remission) era 77,9% per i casi PCR-negativi contro il 68,2% per i casi PCR-
positivi (p= 0,027).
Tuttavia, la presenza del marcatore molecolare prima del trattamento non ha avuto un
impatto significativo sulla sopravvivenza libera da progressione (PFS: progression free
survival) a 3-anni: 69% per i casi MMR-positivi contro il 61% per i casi MMR-negativi;
p=0,085).
Il burden tumorale è stato valutato mediante PCR quantitativa in 105 casi BCL2/IGH@
positivi, il valore mediano è stato di 3 x 10-3 copie (range:0,000018-6). Il numero di
copie BCL2/IGH@ non correlava con lo stadio, la performance, l'età >65 anni o il sesso,
ma era significativamente più alto nei pazienti con alto score prognostico FLIPI e
punteggio FLIPI2.
Tra i pazienti con elevato burden tumorale, il livello di risposta complessivo (OR-overall
survival) era significativamente inferiore nei casi con bassa massa tumorale (38,9% vs
76,6%, p=0.006). Quando è stata eseguita un'analisi statistica (curva ROC) al fine di

113
trovare il valore di BCL2/IGH@ che meglio prediva la ricaduta, 1 x 10-4 copie è risultato
essere il valore più predittivo. Infatti, il 22% dei casi aventi un numero di copie <1 x 10-4
è recidivato contro il 78% dei pazienti con un valore di copie >1 x 10-4 (p=0.033).
Questo risultato ha mostrato un chiaro vantaggio anche in termini di PFS a 3 anni (80%
contro il 59% per i casi con elevato carico tumorale; p=0,015)(figura 4).
Quando questo valore, al di sopra del quale si parla di elevato burden tumorale
molecolare all’arruolamento, è stato incluso nell'analisi multivariata insieme con le
variabili prognosticamente rilevanti già identificate nello studio clinico pubblicato (qualità
della risposta, tipo di trattamento e il punteggio FLIPI) (Federico M. et al, J Clin Oncol.
2013), il tumor burden molecolare elevato ha mostrato un impatto prognostico
indipendente e negativo sulla sopravvivenza libera da progressione (HR = 1.38, 95%
CI:1.1-14.2, p = 0,03) (tabella 1).
Figura 4: la PFS è significativamente più lunga nei pazienti con livelli di BCL2/IGH@<1x10-4, prima del
trattamento (linea tratteggiata).p=0.015
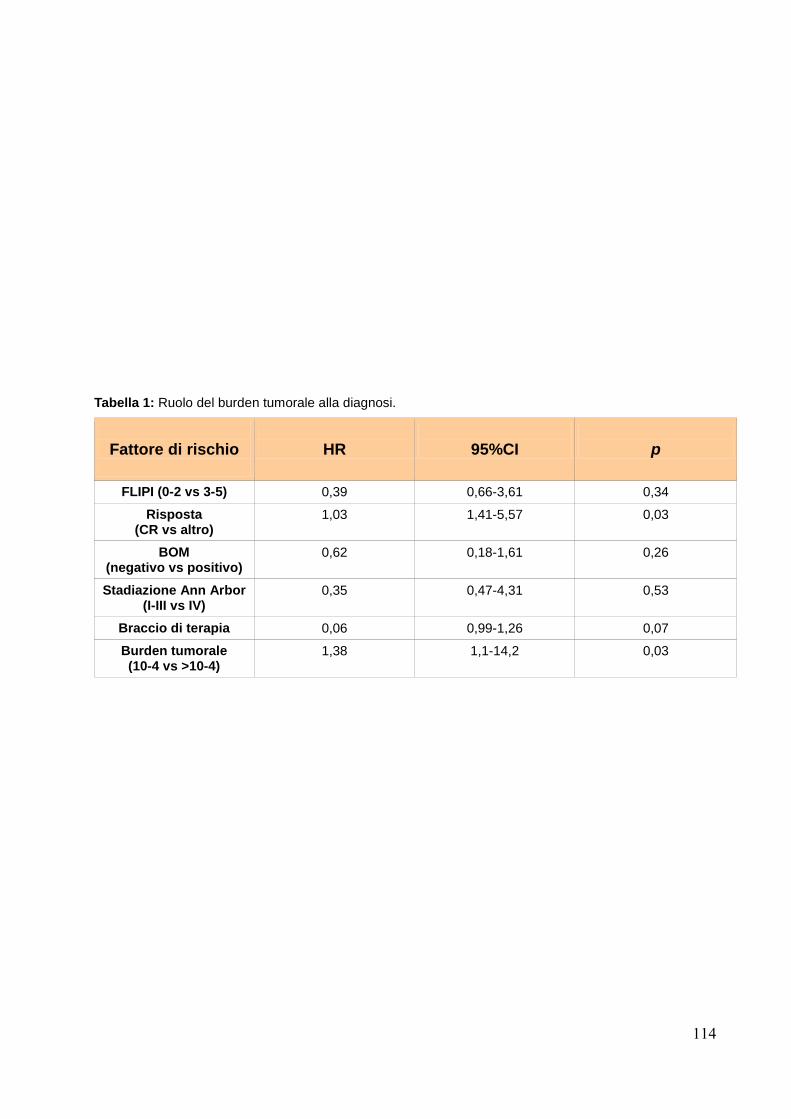
114
Tabella 1: Ruolo del burden tumorale alla diagnosi.
Fattore di rischio
HR
95%CI
p
FLIPI (0-2 vs 3-5) 0,39 0,66-3,61 0,34
Risposta (CR vs altro)
1,03 1,41-5,57 0,03
BOM (negativo vs positivo)
0,62 0,18-1,61 0,26
Stadiazione Ann Arbor (I-III vs IV)
0,35 0,47-4,31 0,53
Braccio di terapia 0,06 0,99-1,26 0,07
Burden tumorale (10-4 vs >10-4)
1,38 1,1-14,2 0,03

115
Valutazione della MMR ai diversi time points
Al primo punto temporale di osservazione molecolare (6 settimane dopo la fine della
terapia) 3 pazienti sono usciti dal protocollo e 63 campioni non sono stati inviati al
laboratorio molecolare referente, così 154 dei 220 casi precedentemente PCR-positivi
sono stati rivalutati mediante PCR qualitativa: 109 (70.8%) hanno raggiunto la negatività
della PCR.
A + 6 settimane, la conversione alla negatività della MMR, non è risultata correlabile
significativamente con le caratteristiche cliniche dei pazienti, la qualità della risposta
clinica o il braccio di terapia.
La negatività della MMR al termine del trattamento è stata correlata con una minore
probabilità di recidiva, ma senza significatività statistica (33% contro 41%, p=0,363).
Analogamente, la PFS a 3 anni è stata più lunga per i pazienti che hanno raggiunto una
negatività molecolare rispetto a chi ha mantenuto il marcatore molecolare (68,4% contro
54,4%), ma questa differenza non ha raggiunto la significatività statistica (p=0,143).
La valutazione molecolare a 12 mesi ha evidenziato 63 pazienti MMR-negativi e 24
MMR- positivi; dopo 24 mesi, 46 casi sono stati MMR negativi e 19 MMR-positivi.
La PFS è risultata significativamente condizionata dallo stato molecolare a 12 e 24
mesi, con un valore del 66% a 3 anni per i casi PCR negativi contro un 41% per i casi
PCR positivi a 12 mesi (p=0.015)(figura 5) ed un valore dell'84% contro 50% per i casi
PCR negativi e positivi, rispettivamente, a 24 mesi (p=0.014).
La negatività della MMR a 12 e 24 mesi dalla fine del trattamento ha comportato una
migliore PFS sia nei pazienti in CR che in quelli in remissione parziale (PR-partial
remission)(3 anni PSF=72% per i casi CR/PCR- vs 32% per i casi CR/PCR+, ed il 62%
per i casi PR/PCR- vs 25% per i casi PR/ PCR+; p=0.001)(figura 6).
Quando la negatività molecolare a 12 mesi nei pazienti con o senza remissione
completa è stata inserita in un'analisi multivariata, la negatività della MMR ha
mantenuto il suo impatto favorevole sulla sopravvivenza libera da progressione (HR=
1.5, 95% CI:1.1-14.2-0,19, p=0.03), indipendentemente dalla risposta clinica (Tabella
2).
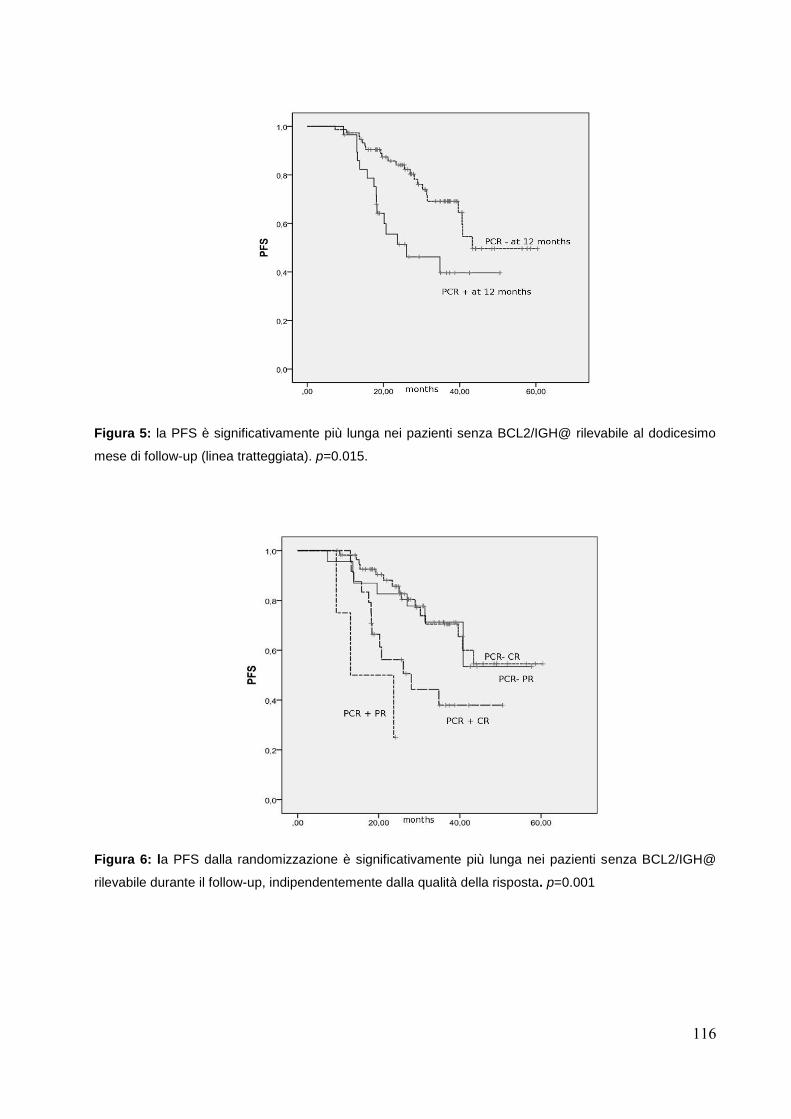
116
Figura 5: la PFS è significativamente più lunga nei pazienti senza BCL2/IGH@ rilevabile al dodicesimo
mese di follow-up (linea tratteggiata). p=0.015.
Figura 6: la PFS dalla randomizzazione è significativamente più lunga nei pazienti senza BCL2/IGH@
rilevabile durante il follow-up, indipendentemente dalla qualità della risposta. p=0.001
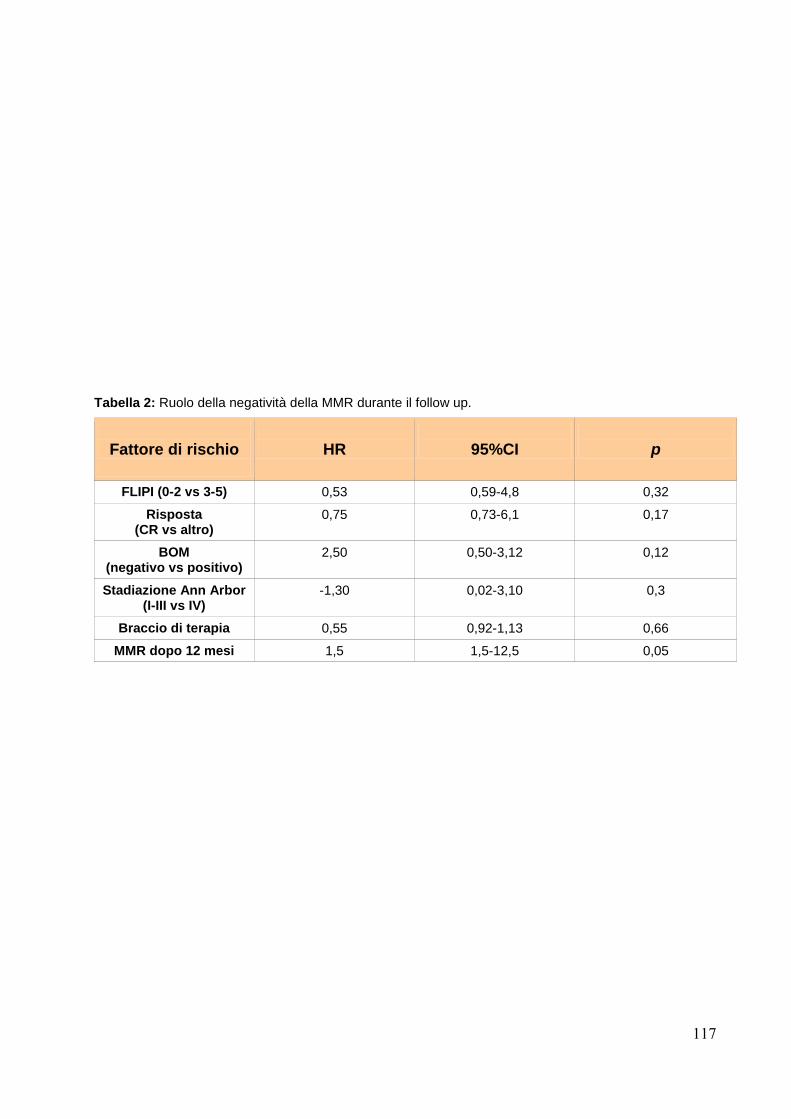
117
Tabella 2: Ruolo della negatività della MMR durante il follow up.
Fattore di rischio
HR
95%CI
p
FLIPI (0-2 vs 3-5) 0,53 0,59-4,8 0,32
Risposta (CR vs altro)
0,75 0,73-6,1 0,17
BOM (negativo vs positivo)
2,50 0,50-3,12 0,12
Stadiazione Ann Arbor (I-III vs IV)
-1,30 0,02-3,10 0,3
Braccio di terapia 0,55 0,92-1,13 0,66
MMR dopo 12 mesi 1,5 1,5-12,5 0,05

118
Conclusioni
La traslocazione t(14;18) (q32;q21) è la caratteristica genetica del LF.
Il rilevamento di tale traslocazione può facilitare la diagnosi di linfoma follicolare e può
essere utilizzata per monitorare la risposta alla terapia ed il livello di malattia residua.
Sono passati più di 20 anni da quando sono state fatte le prime osservazioni sul valore
della negatività molecolare nell'outcome dei pazienti con LF (Gribben JG et al, N Engl J
Med. 1991). Da allora una serie di studi ha confermato il valore critico del
raggiungimento della remissione molecolare in contesti disparati; in trial singoli e
multicentrici, con trattamenti convenzionali ed intensificati, trattamenti privi di Rituximab
e con Rituximab. Inoltre, la remissione molecolare ha confermato un elevato valore
prognostico anche in studi con un prolungato follow-up (Corradini P et al, J Clin Oncol
2004; Rown JR et al, Biol Blood Marrow Transplant 2007). Questo suggerisce che in
pazienti con persistente negatività molecolare, le cellule tumorali (se presenti) sono
chiaramente sotto controllo stabile e possibilmente permanente (Ladetto M et al, Blood
2008; Procházka V et al, J Clin Oncol, 2011; Ladetto M et al, Blood 2013).
Il ruolo della MMR dopo terapia convenzionale nel LF è comunque ancora non
codificato nella pratica clinica, e rimane ancora oggetto di studio nell’ambito di trial
clinici sperimentali.
Negli ultimi anni sono stati sviluppati diversi metodi di analisi quantitativa (Dolken L et
al, Biotechniques 1998; Sanchez-Vega B et al, Mod Pathol 2002).
Questi metodi si sono rivelati un valido strumento per una valutazione accurata delle
cellule BCL2/IgH@ positive nel sangue midollare o nel sangue periferico e per un
migliore monitoraggio molecolare della MMR in diversi protocolli terapeutici (Summers
KE et al, Br J Haematol. 2002).
Una quantificazione precisa delle cellule BCL2/IgH@ positive alla diagnosi, può aiutare
a definire la probabilità di risposta alla chemioterapia convenzionale con o senza
l'aggiunta di Rituximab, inoltre, il monitoraggio molecolare della MMR permette
l'identificazione precoce dei pazienti con un rischio notevolmente più elevato di recidiva
della malattia (Rambaldi A et a, Blood 2005; Goff L et al, J Clin Oncol 2009).
L’obiettivo di questo progetto è di definire il valore della MMR quantitativa nell’ambito
del trattamento immunochemioterapico di prima linea del LF, il protocollo FOLL05, che
prevedeva come end-point secondario la valutazione della MMR qualitativa prima del

119
trattamento, post-terapia, e durante il follow-up.
Le analisi qualitative e quantitative sono state condotte secondo le linee guida europee
per il rilevamento della MMR nel linfoma (van der Velden VHJ et al, Leukemia 2007).
La sensibilità raggiunta dalle nostre valutazioni è stata di 1x10-5, paragonabile a quanto
riportato da altri autori (van Oers MH et al, J Clin Oncol. 2010; Rambaldi A et al, Blood.
2005).
Sono stati considerati solo campioni di sangue midollare provenienti da pazienti
BCL2/IGH@ positivi al momento dell'arruolamento.
Questo potrebbe essere rilevante, perché sono state riportate, differenze superiori a 1
logaritmo in favore di campioni di sangue midollare rispetto a campioni di sangue
periferico, (Leonard BM et al, Blood 1998), inoltre i valori di MMR da sangue periferico
sono risultati meno predittivi (Gribben JG et al, Blood 1994).
Il riarrangiamento BCL2/IGH@ è stato rilevato nel 53 % dei pazienti e la sua specificità
è stata confermata nei casi persistentemente PCR-positivi dopo terapia, mediante
sequenziamento alla diagnosi e dopo trattamento, in modo da rendere improbabile la
presenza di un falso target molecolare durante il follow-up.
Nel dibattito ancora aperto sul ruolo prognostico della MMR quantitativa nel LF, i nostri
risultati mostrano come la massa tumorale alla diagnosi significativamente condizioni la
qualità della risposta e della PFS, in accordo ed a supporto di quanto osservato
precedentemente (Rambaldi A et a, Blood 2005; Goff L et al, J Clin Oncol 2009).
La scomparsa del marcatore molecolare dopo terapia positivamente condiziona la
risposta dei pazienti con LF, con una significatività statistica del follow-up a lungo
termine.
Infatti, la PFS a 3 anni è stata più lunga per i pazienti che hanno raggiunto negatività
molecolare solo dopo 2 mesi dalla fine della terapia (68,4 % contro 54,4 %), e la
significatività statistica è aumentata quando tale negatività viene mantenuta dopo
ulteriori 6 o 12 mesi. Il minor impatto della negatività molecolare alla fine del trattamento
potrebbe essere giustificato da una troppo breve intervallo di tempo trascorso tra
l'ultimo ciclo di Rituximab e la determinazione della MMR.
Questo dato potrebbe essere probabilmente spiegato con la farmacocinetica del
Rituximab, dato che tracce di anticorpi circolanti potrebbero essere trovate fino a 6 mesi
dopo l'ultima somministrazione (Berinstein Nl et al, Ann Oncol 1998). Ciò è
ulteriormente confermato dall'osservazione che un miglior valore predittivo si osserva
se la valutazione della MMR post-terapia viene eseguita dopo 60-90 giorni (Ladetto M

120
et al, Blood 2012).
Infine, i nostri dati sostengono il ruolo della negatività della MMR non solo nei pazienti
che raggiungono una remissione completa (CR), ma anche in quelli con remissione
parziale (PR): la PFS a 3 anni è stata del 62 % per i casi in PR PCR-negativi contro il
32 % per i pazienti in CR ma ancora PCR-positivi dopo 12 mesi di follow-up.
Questi dati sono in linea con quelli già riportati nel linfoma mantellare, dove il valore
della MMR è risultato altamente predittivo per la durata della risposta,
indipendentemente dalla risposta clinica (risposta a 2 anni: 94% per i pazienti in
CR/PCR- ed il 100% per i pazienti in PR/PCR-, rispetto al 71% per i pazienti in
CR/PCR+ ed il 51% per i pazienti in PR/PCR +)(Pott C et al, Blood 2010).
Due principali risultati sono quindi emersi: 1) il burden tumorale al momento della
diagnosi condiziona in modo significativo la qualità della risposta e la PFS, e 2)
l'ottenimento della negatività molecolare qualitativa a 12 mesi dalla fine del trattamento
correla con un risultato migliore (3 anni PFS 66% vs 41, p = 0.015).
In conclusione, i risultati di questo ampio studio supportano il valore dell'analisi
qualitativa e quantitativa della MMR sia alla diagnosi (burden tumorale molecolare), che
al momento della valutazione della risposta clinica (ridefinizione della risposta clinica
completa e parziale), che nel follow-up nei pazienti con LF trattati con i tradizionali
schemi chemio-immunoterapici.

121
Bibliografia
1. Armitage JO & Weisenburger DD: “New approach to classifying non-Hodgkin’s
lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s
Lymphoma Classification Project”. The Journal of Clinical Oncology 1998;
16:2780-2795.
2. Akasaka, T., Lossos, I.S., and Levy, R. BCL6 gene translocation in follicular
lymphoma: a harbinger of eventual transformation to diffuse aggres- sive
lymphoma. Blood 2003;102: 1443–1448.
3. Bastion Y, Sebban C, Berger F, Felman P, Salles G, Dumontet C, Bryon PA,
Coiffier B: “Incidence, predictive factors, and outcome of lymphoma
transformation in follicular lymphoma patients”. The Journal of Clinical Oncology
1997;15:1587–1594.
4. Bende RJ, Smit LA, van Noesel CJ: “Molecular pathways in follicular lymphoma”.
Leukemia 2007, 21:18–29.
5. Berinstein NL, Grillo-López AJ, White CA, Bence-Bruckler I, Maloney D,
Czuczman M, Green D, Rosenberg J, McLaughlin P & Shen D. Association of
serum Rituximab (IDEC-C2B8) concentration and anti-tumor response in the
treatment of recurrent low-grade or follicular non-Hodgkin's lymphoma. Annals of
Oncology. 1998; 9(9):995-1001.
6. Bernstein SH & Burack WR: “The incidence, natural history, biology, and
treatment of transformed lymphomas”. Hematology 2009; 542-551.
7. Biagi JJ & Seymour JF: “Insights into the molecular pathogenesis of follicular
lymphoma arising from analysis of geographic variation”. Blood 2002; 99:4265-
4275.
8. Brown JR, Feng Y, Gribben JG, Neubeg D, Fisher DC, Mauch P, Nadler LM,
Freedman AS. Long-term survival after autologous bone marrow transplan-
tation for follicular lymphoma in first remission. Biology of Blood and Marrow
Transplantation. 2007; 13:1057- 1065.
9. Buchonnet G, Lenain P, Ruminy P, Lepretre S, Stamatoullas A, Parmentier F,
Jardin F, Duval C, Tilly H, Bastard C: “Characterisation of BCL2-JH

122
rearrangements in follicular lymphoma: PCR detection of 3’ BCL2 breakpoints
and evidence of a new cluster”. Leukemia 2000; 14:1563-1569.
10. Buchonnet G, Jardin F, Jean N, Bertrand P, Parmentier F, Tison S, Lepretre S,
Contentin N, Lenain P, Stamatoullas-Bastard A, Tilly H, Bastard C: “Distribution
of BCL2 breakpoints in follicular lymphoma and correlation with clinical features:
specific subtypes or same disease?”. Leukemia 2002; 16:1852–1856.
11. Carreras J, Lopez-Guillermo A, Fox BC, Colomo L, Martinez A, Roncador G,
Montserrat E, Campo E, Banham AH: “High numbers of tumor-infiltrating
FOXP3-positive regulatory T cells are associated with improved overall survival
in follicular lymphoma”. Blood 2006; 108: 2957–2964.
12. Cleary ML, Smith SD, Sklar J: “Cloning and structural analysis of cDNAs for bcl-2
and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18)
translocation”. Cell 1986; 47:19–28.
13. Corradini P, Ladetto M, Zallio F, Astolfi M, Rizzo E, Sametti S, Cuttica A, Rosato
R, Farina L, Boccadoro M, Benedetti F, Pileri A, Tarella C. Long-term follow-up
of indolent lymphoma patients treated with high-dose sequential chemotherapy
and au- tografting: evidence that durable molecular and clinical remission
frequently can be attained only in follicular subtypes. The Journal of Clinical
Oncology 2004; 22:1460- 1468.
14. Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, Fisher RI,
Braziel RM, Rimsza LM, Grogan TM, Miller TP, LeBlanc M, Greiner TC,
Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S,
Holte H, Delabie J, Connors JM, Lansdorp PM, Ouyang Q, Lister TA, Davies AJ,
Norton AJ, Muller-Hermelink HK, Ott G, Campo E, Montserrat E, Wilson WH,
Jaffe ES, Simon R, Yang L, Powell J, Zhao H, Goldschmidt N, Chiorazzi M,
Staudt LM: “Prediction of survival in follicular lymphoma based on molecular
features of tumor-infiltrating immune cells”. The New England Journal of
Medicine 2004; 351:2159–2169.
15. de Jong D: “Molecular pathogenesis of follicular lymphoma: a cross talk of
genetic and immunologic factors”. The Journal of Clinical Oncology 2005;
23:6358–6363.
16. Dolken L, Schuler F, Dolken G. Quantitative de- tection of t(14;18)-positive cells

123
by real-time quantitative PCR using fluorogenic probes. Bio-techniques. 1998;
25:1058-1064.
17. Federico M, Luminari S, Dondi A, Tucci A, Vitolo U, Rigacci L, Di Raimondo F,
Carella AM, Pulsoni A, Merli F, Arcaini L, Angrilli F, Stelitano C, Gaidano G,
Dell'olio M, Marcheselli L, Franco V, Galimberti S, Sacchi S, Brugiatelli M. R-
CVP versus R-CHOP versus R-FM for the initial treatment of patients with
advanced-stage follicular lymphoma: results of the FOLL05 trial conducted by
the Fondazione Italiana Linfomi.The Journal of Clinical Oncology 2013;
31(12):1506-13.
18. Fitzgibbon J, Iqbal S, Davies A, O'shea D, Carlotti E, Chaplin T, Matthews J,
Raghavan M, Norton A, Lister TA, Young BD: “Genome-wide detection of
recurring sites of uniparental disomy in follicular and transformed follicular
lymphoma”. Leukemia 2007; 21:1514-1520.
19. Friedberg JW, Cohen P, Chen L, Robinson KS, Forero-Torres A, La Casce AS,
Fayad LE, Bessudo A, Camacho ES, Williams ME, van der Jagt RH, Oliver JW,
Cheson BD: “Bendamustine in patients with rituximab-refractory indolent and
transformed non-Hodgkin’s lymphoma: results from a phase II multicenter,
single-agent study”. The Journal of Clinical Oncology 2008; 26:204-210.
20. Gallagher CJ, Gregory WM, Jones AE, Stansfeld AG, Richards MA, Dhaliwal
HS, Malpas JS, Lister TA: “Follicular lymphoma: prognostic factors for response
and survival”. The Journal of Clinical Oncology 1986; 4:1470–1480.
21. Garvin AJ, Simon RM, Osborne CK, Merrill J, Young RC, Berard CW: “An
autopsy study of histologic progression in non-Hodgkin’s lymphomas. 192 cases
from the National Cancer Institute”. Cancer 1983; 52:393–398.
22. Glas AM, Knoops L, Delahaye L, Kersten MJ, Kibbelaar RE, Wessels LA, van
Laar R, van Krieken JH, Baars JW, Raemaekers J, Kluin PM, van't Veer LJ, de
Jong D: “Gene-expression and immunohistochemical study of specific T-cell
subsets and accessory cell types in the transformation and prognosis of follicular
lymphoma”. The Journal of Clinical Oncology 2007; 25:390–398.
23. Goff L, Summers K, Iqbal S, Kuhlmann J, Kunz M, Louton T, Hagenbeek A,
Morschhauser F, Putz B, Lister A, Rohatiner A. Quantitative PCR analysis for
Bcl-2/IgH in a phase III study of Yttrium-90 Ibritumomab Tiuxetan as

124
consolidation of first remission in patients with follicular lymphoma. The Journal
of Clinical Oncology 2009; 27(36):6094-100.
24. Green DR & Evan G: “A matter of life and death”. Cancer Cell 2002; 1:19–30.
25. Gribben JG, Freedman AS, Woo SD, Blake K, Shu RS, Freeman G, Longtine
JA, Pinkus GS, Nadler LM. All advanced stage non Hodgkin's lymphoma with a
polymerase chain reaction amplifiable breakpoint of bcl-2 have residual cells
containing the bcl-2 rearrangment at evaluation and after treatment. Blood 1991;
78: 3275-80.
26. Gribben JG, Freedman AS, Neuberg D, Roy DC, Blake K, Woo SD, Grossbard
ML, Rabinowe SN, Coral F, Freeman G, Ritz J, Nadler LM. Immunologic purging
of marrow assessed by PCR before autologous bone marrow transplantation for
B-cell lymphoma. The New England Journal of Medicine 1991; 325: 1525-1533.
27. Gribben JG, Neuberg D, Freedman AS, Gimmi Cd, Pesek KW, Barber M,
Saporito L, Woo SD, Coral F, Spector N. Detection by polymerase chain reaction
of residual cells with the bcl-2 translocation is associated with increased risk of
relapse after autologous bone marrow transplantation for B-cell lymphoma.
Blood. 1993; 81(12): 3449-57.
28. Gribben JG, Neuberg D, Barber M, Moore J, Pesek KW, Freedamn AS, Nadler
LM. Detection of residual lymphoma cells by polymerase chain reaction in
peripheral blood is significantly less predictive for relapse than detection in bone
marrow. Blood. 1994; 83(12): 3800-7.
29. Gu K, Chan WC, Hawley RC.Practical detection of t(14;18)(IgH/BCL2) in
follicular lymphoma. Archives of Pathology & Laboratory Medicine. 2008 Aug;
132(8):1355-61.
30. Guadagnolo BA, Li S, Neuberg D, Ng A, Hua L, Silver B, Stevenson MA, Mauch
P: “Long-term outcome and mortality trends in early-stage, grade 1-2 follicular
lymphoma treated with radiation therapy”. International Journal of Radiation
Oncology 2006; 64: 928–934.
31. Hiddemann W, Kneba M, Dreyling M, Schmitz N, Lengfelder E, Schmits R,
Reiser M, Metzner B, Harder H, Hegewisch-Becker S, Fischer T, Kropff M, Reis
HE, Freund M, Wörmann B, Fuchs R, Planker M, Schimke J, Eimermacher H,
Trümper L, Aldaoud A, Parwaresch R, Unterhalt M: “Frontline therapy with
rituximab added to the combination of cyclophosphamide, doxorubicin,

125
vincristine, and prednisone (CHOP) significantly improves the outcome for
patients with advanced-stage follicular lymphoma compared with therapy with
CHOP alone: results of a prospective randomized study of the German Low-
Grade Lymphoma Study Group”. Blood 2005; 106: 3725–3732.
32. Hillion J, Mecucci C, Aventin A, Leroux D, Wlodarska I, Van Den Berghe H,
Larsen CJ: “A variant translocation t(2;18) in follicular lymphoma involves the 5’
end of bcl-2 and Ig kappa light chain gene”. Oncogene 1991; 6: 169–172.
33. Jaffe ES: “The 2008 WHO classification of lymphomas: implications for clinical
practice and translational research”. Hematology 2009; 523-531.
34. Jager U, Bocskor S, Le T, Mitterbauer G, Bolz I, Chott A, Kneba M, Mannhalter
C, Nadel B: “Follicular lymphomas’ BCL-2/IgH junctions contain templated
nucleotide insertions: novel insights into the mechanism of t(14;18)
translocation”. Blood 2000; 95: 3520-3529.
35. King C, Tangye SG, Mackay CR: “T follicular helper (TFH) cells in normal and
dysregulated immune responses”. Annual Review of Immunology 2008; 26: 741–
766.
36. Kvansakul M, Yang H, Fairlie WD, Czabotar PE, Fischer SF, Perugini MA,
Huang DC, Colman PM: “Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like
domain-swapped dimer that binds a highly selective subset of BH3-containing
death ligands”. Cell Death and Differentiation 2008; 15: 1564–1571.
37. Kuppers R, Klein U, Hansmann ML, Rajewsky K: “Cellular origin of human B-cell
lymphomas”. The New England Journal of Medicine 1999; 341: 1520–1529.
38. Ladetto M, Sametti S, Donovan JW, Ferrero D, Astolfi M, Mitterer M, Ricca I,
Drandi D, Corradini P, Coser P, Pileri A, Gribben JG, Tarella C. A validated real-
time quantitative PCR approach shows a correlation between tumor burden and
successful ex vivo purging in follicular lymphoma patients. Experimental
Hematology 2001; 29: 183-193.
39. Ladetto M, Drandi D, Compagno M,Astolfi M, Volpato F, Voena C, Novarino A,
Pollio B, Addeo A, Ricca I, Falco P, Cavallo F, Vallet S, Corradini P, Pileri A,
Tamponi G, Palumbo A, Bertetto O, Boccadoro M, Tarella C. PCR-detectable
nonneoplastic Bcl-2/IgH rearrangements are common in normal subjects and
cancer patients at diagnosis but rare in subjects treated with chemotherapy.The

126
Journal of Clinical Oncology 2003; 21(7): 1398-403.
40. Ladetto M, De Marco F, Benedetti F, Vitolo U, Patti C, Rambaldi A, Pulsoni A,
Musso M, Liberati AM, Olivieri A, Gallamini A, Pogliani E, Rota Scalabrini D,
Callea V, Di Raimondo F, Pavone V, Tucci A, Cortelazzo S, Levis A, Boccadoro
M, Majolino I, Pileri A, Gianni AM, Passera R, Corradini P, Tarella C; Gruppo
Italiano Trapianto di Midollo Osseo (GITMO); Intergruppo Italiano Linfomi (IIL).
Prospective, multicenter randomized GITMO/IIL trial comparing intensive (R-
HDS) versus conventional (CHOP-R) chemoimmunotherapy in high-risk follicular
lymphoma at diagnosis: the superior disease control of R-HDS does not
translate into an overall survival advantage. Blood 2008; 111(8): 4004-13
41. Ladetto M, Lobetti-Bodoni C, Mantoan B, et al. PCR-Based Minimal Residual
Disease (MRD) detection is a Strong Independent Outcome Predictor also in
Rituximab-Intensive Non-ASCT-Based Programs: Results From the ML17638
Multicenter Randomised Phase III Trial for Elderly Follicular Lymphoma (FL)
Patients of the Fondazione Italiana Linfomi (FIL) Blood (ASH Annual Meeting
Abstracts), 2012; 120: 787.
42. Léonard BM, Hétu F, Busque L,Gyger M, Bélanger R, Perreault C, Roy DC.
Lymphoma cell burden in progenitor cell grafts measured by competitive
polymerase chain reaction: less than one log difference between bone marrow
and peripheral blood sources. Blood. 1998; 91(1): 331-9.
43. Lessene G, Czabotar PE, Colman PM. “BCL-2 family antagonists for cancer
therapy” Nature Reviews Drug Discovery, 2008 Dec; 7(12): 989-1000.
44. Lopez-Guillermo A, Cabanillas F, McDonnell TI, McLaughlin P, Smith T, Pugh
W, Hagemeister F, Rodriguez MA, Romaguera JE, Younes A, Sarris AH, Preti
HA, Lee MS: “Correlation of bcl-2 rearrangement with clinical characteristics and
outcome in indolent follicular lymphoma”. Blood 1999; 93:3081–3087.
45. Lossos, I.S., Alizadeh, A.A., Diehn, M., Warnke, R., Thorstenson, Y., Oefner,
P.J., Brown, P.O., Botstein, D., and Levy, R. Transformation of follicular
lymphoma to diffuse large-cell lymphoma: alternative patterns with increased or
decreased expression of c-myc and its regulated genes. Proceedings of the
National Academy of Science of USA 2002; 99: 8886–8891.
46. Martinez A, Carreras J, Campo E: “The Follicular Lymphoma Microenvironment:
From Tumor Cell to Host Immunity”. Current Hematologic Malignancy Reports

127
2008; 3: 179–186.
47. Martinez-Climent, J.A., Alizadeh, A.A., Segraves, R., Blesa, D., Rubio-
Moscardo, F., Albertson, D.G., Garcia-Conde, J., Dyer, M.J., Levy, R., Pinkel, D.,
and Lossos, I.S. Transformation of follicular lymphoma to diffuse large cell
lymphoma is associated with a heterogeneous set of DNA copy num- ber and
gene expression alterations. Blood 2003; 101: 3109–3117.
48. Montoto S, Davies AJ, Matthews J, Calaminici M, Norton AJ, Amess J,
Vinnicombe S, Waters R, Rohatiner AZ, Lister TA: “Risk and clinical implications
of transformation of follicular lymphoma to diffuse large B-cell lymphoma”. The
Journal of Clinical Oncology 2007; 25: 2426–2433.
49. Ngan BY, Nourse J, Cleary ML: “Detection of chromosomal translocation
t(14;18) within the minor cluster region of bcl-2 by poly- merase chain reaction
and direct genomic sequencing of the enzymatically amplified DNA in follicular
lymphomas”. Blood 1989; 73: 1759–1762.
50. Pasqualucci, L., Neumeister, P., Goossens, T., Nanjangud, G., Chaganti, R. S.,
Kuppers, R., and Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in
B-cell diffuse large- cell lymphomas. Nature 2001; 412: 341-346.
51. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB,
Ouillette P, Trifonov V, Rossi D, Tabbò F, Ponzoni M, Chadburn A, Murty VV,
Bhagat G, Gaidano G, Inghirami G, Malek SN, Rabadan R, Dalla-Favera R.
Genetics of follicular lymphoma transformation. Cell Rep. 2014 Jan 16; 6(1):130-
40.
52. Peterson PM, Gaspodarowicz M, Tsang R: “Long term outcome in stage I and II
follicular lymphoma following treatment with involved field radiation therapy
alone”. The Journal of Clinical Oncology 2004; 22: 653-667.
53. Pinyol, M., Cobo, F., Bea, S., Jares, P., Nayach, I., Fernandez, P.L., Montserrat,
E., Cardesa, A., and Campo, E. p16(INK4a) gene inactivation by deletions,
mutations, and hypermethylation is associated with transformed and aggressive
variants of non-Hodgkin’s lymphomas. Blood 1998; 91: 2977–2984.
54. Pott C, Hoster E, Delfau-Larue MH, Beldjord K, Böttcher S, Asnafi V, Plonquet A,
Siebert R, Callet-Bauchu E, Andersen N, van Dongen JJ, Klapper W, Berger F,
Ribrag V, van Hoof AL, Trneny M, Walewski J, Dreger P, Unterhalt M,

128
Hiddemann W, Kneba M, Kluin-Nelemans HC, Hermine O, Macintyre E, Dreyling
M. Molecular remission is an independent predictor of clinical outcome in
patients with mantle cell lymphoma after combined immunochemotherapy: a
European MCL intergroup study. Blood. 2010; 115(16): 3215-23.
55. Pott C, Brüggemann M, Ritgen M, van der Velden VH, van Dongen JJ, Kneba
M.MRD detection in B-cell non-Hodgkin lymphomas using Ig gene
rearrangmentsand chromosomal translocations as targets for real-time
quantitative PCR. Methods Mol Biol 2013; 971:175-200.
56. Procházka V, Papajík T."Molecular remission in follicular lymphoma: is the era of
residual disease monitoring over? The Journal of Clinical Oncology 2011;
29(11):e318; author reply e319.
57. Rambaldi A, Carlotti E, Oldani E, Della Starza I, Baccarani M, Cortelazzo S,
Lauria F, Arcaini L, Morra E, Pulsoni A, Rigacci L, Rupolo M, Zaja F, Zinzani PL,
Barbui T, Foa R. Quantitative PCR of bone marrow BCL2/IGH@+ cells at
diagnosis predicts treatment response and long-term outcome in follicular non-
Hodgkin lymphoma. Blood 2005; 105(9): 3428-33.
58. Rossi D, Berra E, Cerri M, Deambrogi C, Barbieri C, Franceschetti S, Lunghi M,
Conconi A, Paulli M, Matolcsy A, Pasqualucci L, Capello D, Gaidano G. Aberrant
somatic hypermutation in transformation of follicular lymphoma and chronic
lymphocytic leukemia to diffuse large B-cell lymphoma. Haematologica 2006; 91:
1405–1409.
59. Sanchez-Vega B, Vega F, Hai S, Medeiros LJ, Luthra R. Real-time
t(14;18)(q32;q21) PCR assay combined with high-resolution capillary electro-
phoresis: a novel and rapid approach that allows accurate quantitation and size
determination of bcl-2/JH fusion sequences. Mod Pathol. 2002; 15: 448-453.
60. Sander CA, Yano T, Clark HM, Harris C, Longo DL, Jaffe ES, Raffeld M:”p53
mutation is associated with progression in follicular lymphomas”. Blood 1993; 82:
1994-2004.
61. Schmitt C, Balogh B, Grundt A, Buchholtz C, Leo A, Benner A, Hensel M, Ho
AD, Leo E: “The bcl-2/IgH rearrangement in a population of 204 healthy
individuals: Occurrence, age and gender distribution, breakpoints, and detection
method validity”. Leukemia Research 2006, 30:745–750.

129
62. Solal-Céligny P, Roy P, Colombat P, White J, Armitage JO, Arranz-Saez R, Au
WY, Bellei M, Brice P, Caballero D, Coiffier B, Conde-Garcia E, Doyen C,
Federico M, Fisher RI, Garcia-Conde JF, Guglielmi C, Hagenbeek A, Haïoun C,
LeBlanc M, Lister AT, Lopez-Guillermo A, McLaughlin P, Milpied N, Morel P,
Mounier N, Proctor SJ, Rohatiner A, Smith P, Soubeyran P, Tilly H, Vitolo U,
Zinzani PL, Zucca E, Montserrat E:”Follicular lymphoma international prognostic
index”. Blood 2004; 104: 1258–1265.
63. Summers KE, Davies AJ, Matthews J, et al. The relative role of peripheral blood
and bone marrow for monitoring molecular evidence of disease in follicular
lymphoma by quantitative real-time poly- merase chain reaction. British Journal
of Haematology. 2002; 118: 563-566.
64. Swerdlow SH: “Pediatric follicular lymphomas, marginal zone lymphomas, and
marginal zone hyperplasia”. American Journal of Clinical Pathology 2004; 122:
98-109.
65. Tan D & Horning SJ: “Follicular Lymphoma: Clinical Features and Treatment”.
Hematology/Oncology Clinics of North America 2008; 22: 863–882.
66. Tsujimoto Y, Cossman J, Jaffe E, Croce CM: “Involvement of the bcl-2 gene in
human follicular lymphoma”. Science 1985; 228: 1440–1443.
67. van der Velden VH, Cazzaniga G, Schrauder A, Hancock J, Bader P, Panzer-
Grumayer ER, Flohr T, Sutton R, Cave H, Madsen HO, Cayuela JM, Trka J,
Eckert C, Foroni L, Zur Stadt U, Beldjord K, Raff T, van der Schoot CE, van
Dongen JJ; European Study Group on MRD detection in ALL (ESG-MRD-ALL).
Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines
for interpretation of real-time quantitative PCR data. Leukemia. 2007; 21(4): 604-
11.
68. van Dongen JJ, Langerak AW, Brüggemann M, Evans PA, Hummel M, Lavender
FL, Delabesse E, Davi F, Schuuring E, García-Sanz R, van Krieken JH, Droese
J, González D, Bastard C, White HE, Spaargaren M, González M, Parreira A,
Smith JL, Morgan GJ, Kneba M, Macintyre EA. Design and standardization of
PCR primers and protocolos for detection of clonal immunoglobulin and T-cell
receptor gene recombinations in suspect lymphoproliferationc: report of the

130
BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003; 17(12): 2257-
317
69. van Oers MH, Tönnissen E, Van Glabbeke M, et al. BCL-2/IgH polymerase chain
reaction status at the end of induction treatment is not predictive for progression-
free survival in relapsed/resistant follicular lymphoma: results of a prospective
randomized EORTC 20981 phase III intergroup study. The Journal of Clinical
Oncology 2010; 28(13): 2246-52.
70. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ: “Bcl-2-deficient mice
demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and
hypopigmented hair”. Cell 1993; 75: 229–240.
71. Vitolo U, Ferreri AJ, Montoto S: “Follicular lymphomas”. Critical Reviews in
Oncology/Hematology 2008; 66: 248–261.
72. Yuen AR, Kamel OW, Halpern J, Horning SJ: “Long-term survival after histologic
transformation of low-grade follicular lymphoma”. The Journal of Clinical
Oncology 1995; 13:1726–1733.
73. Yunis JJ, Frizzera G, Oken MM, McKenna J, Theologides A, Arnesen M:
“Multiple recurrent genomic defects in follicular lymphoma. A possible model for
cancer”. The New England Journal of Medicine 1987; 316: 79–84.

131
Pubblicazioni 1.Immunocompetent cell functions in Ph+ acute lymphoblastic leukemia patients
on prolonged Imatinib maintenance treatment.
Roberta Maggio, Nadia Peragine , Maria Stefania De Propris, Antonella Vitale Loredana
Elia, Elisabetta Calabrese , Irene Della Starza , Stefania Intoppa, Maria Laura Milani ,
Anna Guarini , Robin Foa.` Cancer Immunol Immunother, 2011; 60:599-607.
2.Comparison of two RQ-PCR strategies for minimal residual disease evaluation
in lymphoproliferative disorders: Correlation between immunoglobulin gene
mutation load and RQ-PCR performance.
Irene Della Starza, Marzia Cavalli, Ilaria Del Giudice, Daniela Barbero, Barbara
Mantoan , Elisa Genuardi , Claudia Mannu, Anna Gazzola, Elena Ciabatti , Anna
Guarini, Robin Foà , Sara Galimberti , Pierpaolo Piccaluga , Gianluca Gaidano, Marco
Ladetto, Luigia Monitillo on behalf of the FIL-MRD network. Hematol Oncol. 2013 Nov
19. [Epub ahead of print]
3.Whole-genome amplification for the detection of molecular targets and minimal
residual disease monitoring in acute lymphoid leukemia.
Irene Della Starza, Lucia Anna De Novi, Vittorio Nunes, Ilaria Del Giudice, Caterina
Ilari, Marilisa Marinelli, Alina Delia Negulici, Antonella Vitale,Sabina Chiaretti, Robin
Foà, Anna Guarini. Br J Haematol. 2014 Jan 22. [Epub ahead of print]
4.Minimal residual disease monitoring in chronic lymphocytic leukemia patients:
a comparative analysis of flow cytometry and ASO IgH RQ-PCR approaches
Raponi S, Della Starza I, De Propris MS, Del Giudice I, Mauro FR, Marinelli M, Di Maio
V, Foà R, Guarini A. Br J Hematol (in press).
5.Chlorambucil plus Rituximab with or without Maintenance Rituximab as First-
Line Treatment for Elderly Chronic Lymphocytic Leukemia Patients.
Robin Foà, Ilaria Del Giudice, Antonio Cuneo, Giovanni Del Poeta, Stefania Ciolli,
Francesco Di Raimondo, Francesco Lauria, Emanuele Cencini, Gian Matteo Rigolin,
Agostino Cortelezzi, Francesco Nobile, Vincenzo Callea, Maura Brugiatelli, Massimo

132
Massaia, Stefano Molica, Livio Trentin, Rita Rizzi, Giorgina Specchia, Francesca Di
Serio, Lorella Orsucci, Achille Ambrosetti, Marco Montillo, Pier Luigi Zinzani, Felicetto
Ferrara, Fortunato Morabito, Maria Angela Mura, Silvia Soriani, Nadia Peragine,
Simona Tavolaro, Silvia Bonina, Marilisa Marinelli, Maria Stefania De Propris, Irene
Della Starza, Alfonso Piciocchi, Alessandra Alietti, Eva J. Runggaldier, Enrica Gamba,
Francesca Romana Mauro, Sabina Chiaretti, and Anna Guarini. Am J Hematol. 2014
Jan 11. [Epub ahead of print]
6."Fludarabine plus alemtuzumab (FluCam) front-line treatment in young patients
with chronic lymphocytic leukemia (CLL) and an adverse biologic profile.".Mauro
FR, Molica S, Laurenti L, Cortelezzi A, Carella AM, Zaja F, Chiarenza A, Angrilli F,
Nobile F, Marasca R, Musolino C, Brugiatelli M, Piciocchi A, Vignetti M, Fazi P, Gentile
G, De Propris MS, Starza ID, Marinelli M, Chiaretti S, Del Giudice I, Nanni M, Albano F,
Cuneo A, Guarini A, Foà R; GIMEMA (Gruppo Italiano Malattie EMatologiche
dell’Adulto) Working Party for chronic lymphoproliferative disorders.
Leuk Res. 2014 Feb;38(2):198-203. Epub 2013 Nov 19.
7.Minimal Residual Disease conditions the outcome of Patients Affected by
Follicular Lymphoma: results from the FIL FOLL05 trial.
Sara Galimberti, Stefano Luminari, Elena Ciabatti, Francesca Guerrini, Alessandra
Dondi, Luigi Marcheselli, Marco Ladetto, Pier Paolo Piccaluga, Anna Gazzola, Claudia
Mannu, Luigia Montillo, Barbara Mantoan, Ilaria Del Giudice, Irene Della Starza,
Marzia Cavalli, Susanna Grassi, Luca Arcaini, Alessandra Tucci, Giuseppe Alberto
Palumbo, Luigi Rigacci, Alessandro Pulsoni, Umberto Vitolo, Daniele Vallisa, Giovanni
Bertoldero, Gianluca Gaidano, Mario Petrini, Massimo Federico. Submitted Clin
Cancer Res.
8.DNA extraction methods significantly influence the amount and purity of
lymphoma genomic material. Advices from the Fondazione Italiana Linfomi-MRD
Network.
Mannu Claudia, Gazzola Anna, Ciabatti Elena, Cavalli Marzia, Della Starza Irene,
Genuardi Elisa, Mantoan Barbara, Monitillo Luigia, del Giudice Ilaria, Ladetto Marco,
Gaidano Gianluca, Galimberti Sara, Piccaluga PierPaolo. Submitted Leuk Lymphoma.

133
Abstracts
1.HHV8-Related and HHV8-Unrelated primary effusions lymphomas: similarities
and differences.
V.Ascoli, G.Marangi, I.Cozzi, V.Giannelli, M.Merli, F.Petrachi, C.Lorusso, G Della
Grotta, C. Danese, I. Della Starza, R.Guarini. (Poster I Congresso SIAPEC Società di
Anatomia Patologica) 2012.
2.Comparison of two RQ-PCR strategies for minimal residual disease evaluation
in lymphoproliferative disorders: Correlation between immunoglobulin gene
mutation load and RQ-PCR performance.
Irene Della Starza, Marzia Cavalli, Ilaria Del Giudice, Daniela Barbero, Barbara
Mantoan, Elisa Genuardi, Claudia Mannu, Anna Gazzola, Elena Ciabatti, Anna Guarini,
Robin Foà , Sara Galimberti, Pierpaolo Piccaluga, Gianluca Gaidano, Marco Ladetto,
Luigia Monitillo on behalf of the FIL-MRD network (EHA European Hematology
Association) 2013.
3.Immunoglobulin gene rearrangments in Asian patients with Chronic
Lymphocytic Leukemia.
Marilisa Marinelli, Ilaria Del Giudice, Caterina Ilari, Irene Della Starza, Silvia Bonina,
Yok-Lam Kwong, Thomas Chan, Kit-Fai Wong, Anna Guarini, Eric Tse, Robin Foà (EHA
European Hematology Association) 2013.
4.Prognostic role of BCL-2 molecular monitoring in patients with
early stage follicular lymphoma.
Alessandro Pulsoni, Irene Della Starza, Giorgia Annechini, Federico De Angelis,
Gianna Maria D’Elia, Pasqualina D’Urso, Sara Panfilio, Marzia Cavalli, Lavinia Grapulin
and Robin Foà. Poster 12th International Conference On Malignant Lymphoma (ICML
2013).

134