universitÀ di roma “sapienza” - padis.uniroma1.itpadis.uniroma1.it/bitstream/10805/2109/1/tesi...
TRANSCRIPT

UNIVERSITÀ DI ROMA “SAPIENZA”
Scuola di Dottorato in Scienze Mediche Sperimentali e Cliniche
Dottorato di Ricerca in “Medicina Molecolare”
XXV ciclo
TESI
Regolazione trascrizionale del microRNA onco-soppressore
let-7c nella leucemia mieloide acuta
Silvia Careccia
Tutor: Prof. Massimo Levrero
Relatore esterno: Dott.ssa Maria Giulia Rizzo
Coordinatore: Prof. Alberto Gulino

2

3
INDICE
Aspetti generali…………………………………………………………………………..…….. 5
Introduzione…………………………………………………………………………………….…. 7
Capitolo 1. La Leucemia Mieloide Acuta……………………………………………. 7
1.1 Le leucemie………………………………………………………………………… 7
1.2 La leucemia mieloide acuta: caratteristiche generali………………… 8
1.3 La classificazione Franco-Americana-Britannica………………………. 9
1.4 Patogenesi molecolare della leucemia mieloide acuta……………….. 11
1.5 Anomalie citogenetiche nella leucemia mieloide acuta……………… 13
1.6 La leucemia promielocitica acuta……………………………………………. 14
1.7 Altre aberrazioni cromosomiche comuni nelle LAM………………….. 16
1.8 Mutazioni geniche nelle LAM…………………………………………………… 18
Capitolo 2. L’Epigenetica…………………………………………………………………… 22
2.1 L’organizzazione della cromatina nella cellula eucariota……………. 22
2.2 Epigenetica e trascrizione………………………………………………………. 23
2.2.1 La metilazione del DNA………………………………………………… 23
2.2.2 Le modificazioni post-traduzionali degli istoni…………………… 25
Capitolo 3. I microRNA………………………………………………………………….…… 31
3.1 I micro-RNA: biogenesi e meccanismi d’azione………………………….. 31
3.2 I miRNA intronici…………………………………………………………………… 34
3.3 miRNA e meccanismi epigenetici……………………………………………. 35
3.3.1 miRNA e metilazione del DNA…………………………………………….. 35
3.3.2 miRNA e modificazioni istoniche………………………………………… 36
3.4 MicroRNA e tumori………………………………………………………………… 37
3.4.1 MicroRNA e leucemie…………………….…………..…………………… 37
3.4.2 MicroRNA e tumori solidi………………………………………………… 39
3.5 La famiglia di microRNA let-7…………………………………………………. 41
3.5.1 Aspetti generali sulla famiglia let……………………………………… 41

4
3.5.2 Let-7 e tumori…………………………………………………………………. 44
Obiettivi della ricerca…………………………………………………………………….. 48
Materiali e Metodi…………………………………………………………………………. 49
• Linee cellulari e condizioni di coltura……………………………………………. 49
• Preparazione di RNA e cDNA………………………………………………………. 50
• Stem-loop RT-PCR………………………………………………………………………. 50
• Real Time PCR (qRT-PCR)…………………………………………………………… 51
• Analisi bioinformatica…………………………………………………………………. 52
• 5’ Rapid Amplification of cDNA Ends (5’ RACE)……………………………… 52
• Costrutti luc e saggi luciferasici……………………………………………………. 53
• Trasfezioni transienti…………………………………………………………………… 54
• Immunoprecipitazione della cromatina (ChIP)……………………………….. 55
• Analisi statistica…………………………………………………………………………… 59
Risultati…………………………………………………………………………………………….. 60
Identificazione di un nuovo TSS per il miRNA let-7c…………………………………... 60
Analisi dell’attività trascrizionale del promotore intronico del let-7c in cellule di leucemia promielocitica acuta…………………………………………………….. 61
Modificazioni epigenetiche del promotore del gene ospite del let-7c LINC00478 dopo trattamento con ATRA……………………………………………………. 62
Modificazioni epigenetiche del promotore intronico del let-7c dopo trattamento con ATRA……………………………………………………………………………… 64
Attività trascrizionale del promotore intronico del let-7c in tumori solidi………………………………………………………………………………………………………… 67
Correlazione dell’espressione del gene ospite LINC00478 e del let-7c in diversi tipi di tumore………………………………………………………………………………… 67
Discussione e conclusioni………………………………………...……………………… 70
Bibliografia…………………………………………………………………………………………… 75

5
ASPETTI GENERALI
La leucemia mieloide acuta (LAM) rappresenta un gruppo eterogeneo di disordini
ematopoietici, caratterizzati da distinte lesioni genetiche, in cui si ha un blocco del
processo differenziativo in uno stadio specifico della maturazione ematopoietica. Il
corretto differenziamento dipende da geni che codificano per fattori di trascrizione, la
cui mancata regolazione potrebbe, quindi, essere importante per lo sviluppo delle
neoplasie. E’ stato comunque dimostrato che i fattori di trascrizione non sono i soli
regolatori chiave dell’espressione genica. Meccanismi epigenetici come metilazione
del DNA, modificazioni post-traduzionali degli istoni, rimodellamento dei nucleosomi
ed espressione di piccoli RNA regolatori, contribuiscono alla regolazione
dell’espressione genica e alla determinazione della specificità cellulare e tissutale. La
de-regolazione di questi meccanismi coopera con le alterazioni genetiche
all’insorgenza e alla progressione tumorale. Recentemente, è stata identificata una
nuova rete di circuiti regolatori che operano a livello post-trascrizionale: i microRNA
(miRNA). I miRNA sono RNA non codificanti di dimensioni microscopiche (19-25
nucleotidi), regolatori e modulatori dell’espressione genica, coinvolti in importanti
processi cellulari come l’apoptosi, la proliferazione ed il differenziamento cellulare.
Alcuni miRNA mostrano espressione ubiquitaria, mentre altri sono limitati a certi
stadi dello sviluppo o a certi tessuti e tipi cellulari. In particolare, numerosi dati
dimostrano che i miRNA sono espressi differentemente in cellule ematopoietiche in
vivo e suggeriscono che possano giocare un ruolo importante nel differenziamento
ematopoietico. Il gruppo in cui ho svolto la mia attività di ricerca ha precedentemente
dimostrato che in blasti di leucemia acuta promielocitica (LAP), un sottotipo di LAM,
un ristretto gruppo di miRNA è differenzialmente espresso ed è modulato dal
trattamento differenziante con acido retinoico tutto-trans (ATRA). Inoltre, il confronto
dell’espressione di questi miRNA in promielociti normali e in blasti di LAP alla
diagnosi, ha rivelato in questi ultimi una ridotta espressione del miRNA let-7c. Il let-
7c appartiene alla famiglia di miRNA let-7, che svolge ruoli importanti in varie attività
cellulari, e diversi membri di questa famiglia sono specificamente repressi in vari tipi
di tumori. Il let-7c è un miRNA intronico localizzato all’interno di un introne del gene
non-codificante proteine LINC00478 e nostri precedenti dati indicano che la sua
trascrizione, in cellule di LAP, sembra essere sotto il controllo del promotore del gene

6
ospite. Sulla base di quanto sopradetto, lo scopo di questa tesi è stato quello di
individuare i possibili meccanismi molecolari alla base della regolazione
trascrizionale del miRNA let-7c in cellule mieloidi. I nostri dati mostrano che, in
cellule di LAP, l’incremento dell’espressione del let-7c indotta dall’ATRA potrebbe
essere mediata da meccanismi epigenetici. In maniera interessante, abbiamo anche
identificato, per il let-7c, oltre al promotore del gene ospite LINC00478, un nuovo sito
di inizio di trascrizione. Questo nuovo promotore ha una propria attività
trascrizionale. Tuttavia, trattamenti con ATRA, che inducono sul promotore del gene
ospite LINC00478/let-7c uno stato di maggiore attivazione trascrizionale, data da una
conformazione più aperta della cromatina, non portano a modulazioni significative
negli stessi marcatori epigenetici sul promotore intronico. Inoltre, abbiamo dati che
suggeriscono che il promotore intronico abbia un ruolo nel guidare la trascrizione del
let-7c in contesti tumorali diversi da quello leucemico in cui la trascrizione del let-7c
sembra essere principalmente a carico del promotore del gene ospite.
Questi risultati potrebbero contribuire ad una migliore comprensione del complesso
programma trascrizionale coinvolto nella transizione dal normale differenziamento
mieloide a quello patologico e potrebbero permettere l’identificazione di nuovi
bersagli molecolari, e quindi di nuove terapie, per le leucemie mieloidi acute.

7
INTRODUZIONE
Capitolo 1. La leucemia mieloide acuta
1.1 Le leucemie
Per leucemie si intende un gruppo eterogeneo di malattie neoplastiche, che
prevedono un qualsiasi processo di alterazione proliferativa a carattere progressivo
ed irreversibile, delle cellule emopoietiche del midollo osseo. Le leucemie hanno
origine dalla trasformazione maligna di una cellula progenitrice staminale
eamatopoietica, con alterazione della proliferazione e del differenziamento della
stessa e delle cellule derivanti mieloidi e linfoidi (globuli bianchi) e/o eritroidi
(globuli rossi) e/o megacariociti (piastrine).
Nelle leucemie, i blasti presentano un vantaggio proliferativo nei confronti del tessuto
normale; in questo modo vengono prodotte più cellule di quante ne muoiano e
queste, accumulandosi nel midollo osseo, determinano un’alterazione della
proliferazione e del differenziamento delle normali cellule ematopoietiche.
L’entità del difetto maturativo dei precursori leucemici e la rapidità di insorgenza
della malattia nei pazienti consente di separare nettamente le forme acute,
caratterizzate da elementi cellulari poco differenziati, dalle leucemie croniche che
presentano elementi ben definiti, e un decorso più lento e stabile nel tempo. Tale
distinzione è di notevole importanza non solo sul piano biologico, ma anche e
soprattutto sul piano clinico, per le implicazioni di ordine prognostico e terapeutico
che da essa derivano.
Dal punto di vista qualitativo la proliferazione neoplastica può interessare i
precursori della linea cellulare linfopoietica (leucemia linfoide), oppure quelli della
linea mielopoietica (leucemia non linfoide o mieloide). Le leucemie quindi, in base al
tipo cellulare coinvolto ed alle caratteristiche cliniche, possono essere suddivise in
leucemie mieloidi acute (LAM) e croniche (LCM), e linfoidi acute (LAL) e croniche
(LCL).

8
1.2 La leucemia mieloide acuta: caratteristiche generali
Per il presente lavoro, particolare importanza rivestono le leucemie mieloidi acute
(LAM), che rappresentano il 25% circa delle leucemie diagnosticate negli adulti. Le
LAM sono forme rapidamente progressive, che coinvolgono cellule immature le quali
proliferano in maniera afinalistica ed incontrollata, e sono incapaci di maturare in
maniera ordinata e di adempiere alle proprie funzioni. La LAM si può definire un
disordine ematopoietico clonale, causato da alterazioni genetiche a carico delle
cellule staminali ematopoietiche. A causa delle mutazioni genetiche accumulate, i
blasti leucemici possono anche inibire il differenziamento dei precursori mieloidi
normali del midollo, a causa di specifiche chemochine prodotte dai blasti tumorali
(Youn et al, 2000). Sul piano biologico questa proliferazione aberrante, originatasi a
livello del midollo osseo, si diffonde poi agli altri organi con potenzialità
emolinfopoietica (milza, linfonodi, fegato), invadendo successivamente tutti i tessuti
dell’organismo. In questo modo, in assenza di trattamenti la LAM determina
emorragie, infiltrazioni in altri organi e infezioni fatali entro un anno dalla diagnosi
(Estey e Döhner, 2006; Lowenberg et al, 1999).
Negli ultimi 30 anni, il trattamento delle LAM si è basato su combinazioni di
chemioterapici quali le antracicline, principalmente daunorubicina, idarubicina e
citarabina (Tallman et al, 2005; Ganetsky et al, 2012). Il trattamento si divide in due
fasi: una prima fase di induzione, che ha come obbiettivo la remissione della malattia
(con una possibile post-induzione), e una seconda fase di terapia post-remissione.
Generalmente, il trattamento delle LAM include almeno un ciclo di chemioterapia
intensiva di induzione, seguita da un addizionale ciclo di intensivo di terapia di
consolidamento, a cui fa seguito una terapia di mantenimento.
Il tasso di remissione e la sopravvivenza alle LAM varia grandemente in base all’età
del paziente e alla citogenetica dei blasti leucemici, ma rimane comunque il più basso
di tutte le leucemie (Estey e Döhner, 2006); alcuni studi mostrano infatti come il tasso
di sopravvivenza a 5 anni sia del 55% per pazienti con una citogenetica “favorevole”,
il 24% per pazienti a rischio intermedio, e solo il 5% per pazienti con citogenetica
“sfavorevole” (Byrd et al, 2002).

9
1.3 La classificazione Franco-Americana-Britannica
Nelle LAM, l’accumulo dei blasti leucemici si origina principalmente a causa
dell’incapacità dei precursori mieloidi di maturare completamente, perciò i sistemi di
classificazione tradizionali hanno utilizzato la morfologia cellulare per definire
diversi sottotipi di LAM. Questi sottotipi sono definiti sulla base dello stadio in cui si
verifica il blocco del differenziamento normale. Le notevoli divergenze nella
nomenclatura classificativa adoperata fino ad oggi, sono state superate dagli studi
effettuati da un gruppo cooperativo che ha elaborato un sistema di classificazione
ormai universalmente adottato: la classificazione Franco–Americana–Britannica
(FAB, Bennett et al, 1985).
In base a questa convenzione, i principali criteri su cui si deve basare una corretta
classificazione delle leucemie acute sono rappresentati da:
- morfologia delle cellule leucemiche del midollo osseo e del sangue periferico
dopo colorazione panottica (May–Grünwald, Wright, Romanovsky);
- comportamento di alcune reazioni citochimiche capaci di mettere in evidenza
attività enzimatiche e/o prodotti del metabolismo intracellulare con aspetti
peculiari per determinate linee cellulari.
Per quanto riguarda le LAM, il gruppo cooperatore FAB ha identificato almeno i
seguenti sottotipi:
M0 = (leucemia mieloblastica acuta altamente indifferenziata) Vi è predominanza di
blasti di aspetto completamente indifferenziato. L’identificazione può avvenire solo
per mezzo di anticorpi monoclonali specifici.
M1 = (leucemia mieloblastica acuta non differenziata) Vi è una prevalenza di blasti
indifferenziati; il nucleo è rotondeggiante od ovale con uno o più nucleoli evidenti e
cromatina finemente distribuita; citoplasma con scarsi granuli azzurrofili e rarissimi
corpi di Auer. La quota mieloide maturante è inferiore al 10% e quella monocitaria al
20%. Le indagini citochimiche dimostrano la positività della mieloperossidasi (MPO)
nel 3% dei blasti; anche la naftolo AS-D acetato esterasi (NASD-AE) risulta positiva,
ma in maniera debole e senza inibizione da parte del NaF.
M2 = (leucemia mieloblastica acuta differenziata) Vi è predominanza di blasti con
evidenti segni di differenziamento granulocitario; il nucleo è con più nucleoli e
cromatina finemente distribuita; il citoplasma è ricco di granulazioni azzurrofile e

10
corpi di Auer. La quota mieloide maturante è superiore al 10% e spesso presenta
anomalie morfologiche. Intensa risulta la positività alla MPO e alla NASD-AE non
inibita dal NaF.
M3 = (leucemia promielocitica acuta) E’ caratterizzata dalla predominanza di blasti di
tipo promielocitico con nucleo irregolare, talora reniforme o bilobato; il citoplasma è
ricchissimo di granulazioni azzurrofile associate a numerosi corpi di Auer. Le indagini
citochimiche dimostrano la massima positività alla MPO, mentre risulta positiva e non
inibita dal NaF la NASD-AE.
M4 = (leucemia mielomonocitica acuta) Vi è presenza nel sangue periferico e nel
midollo osseo di blasti ad orientamento mieloide e monocitario, questi ultimi in
proporzione variabile, ma sempre superiore al 20%. La positività alla MPO è intensa
negli elementi della linea mieloide, mentre risulta praticamente negativa nei
precursori monocitari. I blasti monocitici presentano, però, una spiccata positività per
l’alfa naftilacetato esterasi (ANAE) e per la NASD-AE con inibizione, nel caso di questa
ultima, da parte del NaF.
M5 = (leucemia monocitica acuta) Vi è prevalenza di elementi della linea monocitaria.
Si distinguono: una varietà A (M5A) con prevalenza di elementi più indifferenziati di
tipo monoblastico caratterizzati da grossa taglia, nucleo rotondo od ovale con nucleoli
evidenti, ampio citoplasma basofilo in cui si osservano fini granulazioni azzurrofile;
una varietà B (M5B) in cui le cellule presentano aspetti più differenziati, di tipo
promonocitico–monocitico. Per l’identificazione di tale varietà risultano
indispensabili le indagini citochimiche che dimostrano un’intensa positività della
NASD-AE, fortemente inibita dal NaF, e dell’ANAE.
M6 = (eritroleucemia) E’ una varietà molto rara, caratterizzata dalla proliferazione di
elementi leucemici appartenenti alle linee eritroblastiche e granuloblastiche. La
percentuale di cellule eritroidi midollari deve essere superiore al 50%; inoltre i blasti
mieloidi devono rappresentare almeno il 30% della popolazione non eritroblastica
midollare. Le indagini citochimiche possono mettere in evidenza in una percentuale
variabile di eritroblasti una spiccata positività alla colorazione Schiff-acido periodico
(PAS).
M7 = (leucemia megacarioblastica) E’ una varietà abbastanza rara, caratterizzata da
blasti che possono presentare un aspetto indifferenziato o talora di tipo linfoide. Le
indagini citochimiche possono evidenziare una PAS positività ed una positività focale
della ANAE senza tuttavia aspetti di specificità.

11
1.4 Patogenesi molecolare della leucemia mieloide acuta
Oltre alle differenze morfologiche e citochimiche manifestate dai blasti leucemici, la
LAM è una patologia estremamente eterogenea sotto il profilo genetico. E’ ormai
generalmente accettato che la patogenesi molecolare delle leucemie sia, almeno nella
maggior parte dei casi, un processo multi-stadio, nel quale l’accumulo di distinte
lesioni genetiche porta alla crescita clonale di precursori immaturi ed all’insorgenza
della malattia. L’attenzione della ricerca contro il cancro è stata tradizionalmente
rivolta allo studio di oncogeni e onco-soppressori che regolano la proliferazione e la
morte cellulare. Sebbene l’alterazione di questi processi può essere un evento-chiave
anche nella patogenesi della LAM, è la distruzione del normale differenziamento
cellulare ad avere rilevanza cruciale nell’insorgenza delle leucemie. I difetti dei blasti
leucemici possono includere anche un’evasione dalla morte cellulare programmata,
instabilità genomica e la disseminazione multi-organo delle cellule leucemiche.
Per quanto riguarda la distruzione dei normali processi di proliferazione e morte
cellulare, uno degli eventi patogenetici più importanti nelle LAM è l’alterazione dei
meccanismi di trasduzione del segnale. La crescita clonale di precursori ematopoietici
immaturi è dovuta ad una loro aumentata proliferazione e resistenza all’apoptosi, alla
quale certamente contribuisce l’attivazione costitutiva e/o aberrante delle vie del
segnale controllate dai recettori di fattori di crescita. Sebbene la proliferazione sia
regolata nei precursori ematopoietici normali dai fattori di crescita e dai segnali di
adesione, nelle cellule leucemiche può essere avviata in modo autonomo. In effetti,
l’attivazione aberrante di molecole implicate nella trasduzione del segnale è stata
riscontrata circa nel 50% delle LAM primarie (Steffen et al, 2005). Questa
proliferazione aberrante è spesso il risultato di mutazioni geniche che influenzano le
vie del segnale, e colpiscono i recettori tirosina-chinasici di membrana Flt3 e Kit,
oppure le proteine che ricevono il segnale a valle da questi recettori, come N-Ras e K-
Ras (Kottaridis et al, 2001; Döhner, 2007). Queste lesioni possono anche alterare la
sopravvivenza cellulare, principalmente stimolando la via del segnale della fosfatidil-
inositolo trifosfato chinasi, e comportando un’evasione dei blasti dai programmi
apoptotici. La caratterizzazione delle anomalie delle vie del segnale ha concentrato
l’attenzione sulla proliferazione cellulare come potenziale bersaglio terapeutico nelle
LAM. Tuttavia, l’ostacolo principale a questa strategia è rappresentato dai molti modi
attraverso i quali nelle LAM il segnale delle chinasi può essere attivato.

12
Il blocco del differenziamento è un’altra tipica anomalia dei blasti leucemici, e le LAM
rappresentano un eccellente modello per studiare il legame tra la regolazione del
differenziamento e la progressione tumorale. Per attivare i profili di espressione
genica necessari per il differenziamento dei vari stipiti ematopoietici, è necessario un
corretto bilancio di fattori di trascrizione generali e linea-specifici. Frequentemente,
nelle LAM le lesioni genetiche colpiscono, direttamente o indirettamente, questi
fattori di trascrizione, causando un difetto nel differenziamento ematopoietico.
Fondamentalmente, due tipi di lesioni genetiche contraddistinguono questa leucemia
e contribuiscono al fenotipo dei blasti leucemici: riarrangiamenti cromosomici e
mutazioni geniche. I riarrangiamenti cromosomici possono distruggere il normale
differenziamento a vari livelli. Fattori di trascrizione importanti possono essere
direttamente implicati nella traslocazione, oppure i riarrangiamenti coinvolgono un
coattivatore che risulta espresso in maniera aberrante. Le mutazioni puntiformi
possono colpire i fattori di trascrizione ematopoietici quali PU.1 e C/EBPα, e possono
condurre così al blocco del normale differenziamento mieloide (Martens 2011;
Mròzec et al, 2007). Può essere importante considerare che molti pazienti di LAM che
hanno mutazioni in PU.1 e C/EBPα non hanno traslocazioni cromosomiche. E’
possibile quindi che le proteine di fusione tipicamente associate a queste leucemie
distruggano le funzioni di PU.1 e C/EBPα. In effetti, è stato dimostrato che la proteina
di fusione AML1/ETO, prodotta dalla traslocazione t(8;21) (Vedi anche Capitolo 1.5
Anomalie citogenetiche nella leucemia mieloide acuta), distrugge la funzione di
C/EBPα (Westendorf et al, 1998).
Il contributo dei diversi eventi mutazionali che portano al cancro può variare nelle
LAM, e le caratteristiche dei blasti leucemici possono differire da un caso ad un altro
sulla base delle lesioni geniche che si sono prodotte. Perciò possono esistere casi in
cui l’accumulo dei blasti è principalmente determinato da un’inappropriata
proliferazione in assenza di normali fattori di crescita, e da un auto-rinnovamento
incontrollato delle cellule staminali, piuttosto che da un blocco del differenziamento,
e altri in cui il principale difetto dei blasti è l’incapacità di raggiungere un
differenziamento terminale.
In passato la LAM era considerata una neoplasia “monolitica”, che prevedeva un
trattamento chemioterapico pressoché unico per tutti i pazienti. Oggi, la grande
diversità genetica che lo studio delle LAM ha svelato lascia spazio alla prospettiva di
terapie personalizzate.

13
1.5 Anomalie citogenetiche nella leucemia mieloide acuta
L’eterogeneità genetica delle LAM richiama il problema di un’appropriata
classificazione sia per la comprensione dei meccanismi che intervengono nella
patogenesi della malattia, che per il valore prognostico che ne può derivare e la
definizione di strategie terapeutiche mirate. Sebbene il valore diagnostico del dato
morfologico e cito-chimico sia rimasto tuttora inalterato, appare sempre più
importante affiancare alla classificazione FAB anche una classificazione citogenetica,
basata sull’identificazione di riarrangiamenti non casuali del cariotipo. Le aberrazioni
cromosomiche sono una caratteristica frequentemente riscontrata nelle leucemie.
Tali riarrangiamenti possono essere evidenziati, oltre che con la citogenetica
tradizionale, anche con l’ibridazione in situ fluorescente (FISH), che impiega sonde
molecolari specifiche per i geni coinvolti nell’anomalia, o con la RT-PCR, che utilizza
sequenze nucleotidiche complementari a quelle localizzate alle estremità 3’ e 5’ del
gene di fusione. Un’altra possibilità consiste nella dimostrazione della proteina
chimerica, utilizzando un anticorpo monoclonale specifico. Le anomalie del cariotipo
presenti nella LAM comprendono traslocazioni reciproche e non, inversioni, trisomie,
monosomie e delezioni. Traslocazioni reciproche e inversioni peri-/paracentriche
sono presenti nel 40% circa dei pazienti affetti da LAM. Un ampio numero di studi ha
chiaramente dimostrato un ruolo centrale di questi riarrangiamenti genici nella
leuchemiogenesi. Tali studi hanno evidenziato che: geni di fusione specifici correlano
strettamente con specifici fenotipi tumorali; le terapie di successo sono associate ad
un decremento o alla eradicazione della chimera associata alla malattia; i costrutti dei
geni di fusione sono in grado di generare in modelli animali disordini simili a quelli
osservati nelle leucemie umane; il silenziamento dei trascritti di fusione in vitro
contrasta la leuchemiogenesi e la proliferazione cellulare, e può indurre il
differenziamento (Mitelman et al, 2007). L’importanza delle anomalie citogenetiche è
sottolineata dal fatto che, ad oggi, la principale stratificazione del rischio dei pazienti
di LAM è definita su criteri citogenetici, dividendo i pazienti in tre grandi gruppi
prognostici: pazienti con una prognosi favorevole, che portano specifiche aberrazioni
cromosomiche quali t(8;21), t(15;17) e inv16, a prognosi intermedia (cariotipo
normale), e a prognosi sfavorevole (anomalie del cariotipo complesse).
Molti di questi riarrangiamenti coinvolgono loci genici codificanti attivatori
trascrizionali, i quali generano una proteina di fusione che mantiene la capacità di

14
legarsi al DNA, ma la cui attività trascrizionale è aberrante. L’alterata regolazione dei
geni bersaglio necessari per un normale sviluppo mieloide conduce infine alla
trasformazione leucemica.
1.6 La leucemia promielocitica acuta
Di particolare interesse per questo lavoro di tesi è la leucemia promielocitica acuta
(LAP). La traslocazione t(15;17) è tipicamente riscontrata in più del 95% dei casi di
leucemia promielocitica acuta ,che rappresenta circa il 10% di tutte le LAM ed ha
caratteristiche morfologiche corrispondenti ai sottotipi FAB M3 (80%) o vM3 (20%,
variante ipogranulare). La t(15;17) genera l’oncogene di fusione PML/RARα, che ha
un ruolo centrale nella leuchemiogenesi dei blasti LAP (Melnich e Lich, 1999).
Il gene PML codifica per una proteina che regola la senescenza e l’apoptosi: è ormai
dimostrato che PML sopprime la crescita cellulare se overespresso e, ad oggi, è
considerato un oncosoppressore sia nelle leucemie che nei tumori solidi (Gurrieri et
al, 2004). PML è caratterizzato da una particolare distribuzione all’interno di
determinate regioni nucleari chiamate PODs (PML Oncogenic Domains) o NB
(Nuclear Bodies, corpi nucleari), dove ha un ruolo centrale come “impalcatura” per
l’assemblaggio e il disassemblaggio delle proteine che compongono i NB. Nella
t(15;17) i NB risultano disgregati e PML assume una localizzazione micro-diffusa
(Melnich e Lich, 1999; Carracedo et al, 2011).
Il gene RARα, invece, codifica per un membro della famiglia dei recettori nucleari per
i retinoidi (RARs). In assenza di acido retinoico tutto-trans (ATRA), RARα si lega ai
siti RARE (Retinoic Acid Responsive Elements) dei geni bersaglio come etero-dimero
insieme ad un’altra proteina ad essa correlata, il recettore dei retinoidi X (RXR).
L’etero-dimero normale RARα/RXR recluta complessi di co-repressione e reprime
così la trascrizione dei geni bersaglio. Un cambiamento conformazionale, causato dal
legame con ATRA a concentrazioni fisiologiche, promuove la dissociazione dei co-
repressori e stimola il reclutamento di co-attivatori. L’onco-proteina PML/RARα
agisce invece come un repressore costitutivo che è insensibile alle concentrazioni
fisiologiche di ATRA (Licht, 2006). Numerosi studi hanno dimostrato che il solo
PML/RARα transgenico può indurre LAP in modelli murini (Brown et al, 1997;
Grisolano et al, 1997). PML/RARα compete con RARα, formando omodimeri e
sequestrando RXR. In questo modo PML/RARα lega gli elementi RARE sui geni

15
bersaglio e media la repressione trascrizionale tramite il reclutamento di DNA metil-
trasferasi (DNMT) e/o la de-acetilazione degli istoni per mezzo di complessi N-
CoR/Sin3A/HDACs (Di Croce et al, 2002; Insinga et al, 2005; Martens et al, 2010). Di
notevole importanza è che blasti di LAP PML/RARα-positivi sono indotti a
differenziare dal trattamento con dosi farmacologiche di ATRA (da 10-7 a 10-6 M): la
LAP è per questo un modello particolarmente interessante nello studio del cancro
umano, essendo la prima neoplasia maligna dell’uomo ad essere efficacemente
trattata con un induttore del differenziamento cellulare. Dosi farmacologiche di acido
retinoico sono necessarie per innescare la degradazione di PML/RARα e il
riassemblaggio dei corpi nucleari (Yoshida et al, 1996; Nervi et al, 1998). E’
generalmente accettato che questo trattamento sia anche in grado di indurre un
cambiamento conformazionale di PML/RARα; in entrambi i casi, l’ATRA induce la
dissociazione dei complessi co-repressori e il reclutamento di proteine con attività di
acetilasi istonica (HAT) sui geni bersaglio, aprendo così le strutture cromatiniche e
consentendo il ripristino della trascrizione genica (Grignani et al, 1998; Martens et al,
2010).
L’introduzione di ATRA come terapia principale di trattamento della LAP ha
trasformato questo tipo di leucemia dallo stato di neoplasia quasi sempre ad esito
fatale, a malattia altamente curabile, con un tasso di remissione clinica (CR) intorno al
90%. L’ATRA di per sé non è curativo, poiché le remissioni ATRA-indotte sono solo
transitorie, ma è utile come trattamento prima e in combinazione alle terapie
citotossiche intensive, principalmente a base di antracicline, necessarie ad ottenere
remissioni prolungate, con una sopravvivenza globale e libera da malattia intorno al
70-80% (Hu, 2011).
E’ interessante notare che il gene di fusione reciproco RARα/PML è anch’esso
espresso nei blasti di LAP e codifica per una proteina di fusione. Sebbene le sue
proprietà funzionali rimangano poco conosciute, sistemi animali transgenici
suggeriscono che possa avere un ruolo nel promuovere il fenotipo leucemico (He et
al, 2000; Zimonjic et al, 2000).
Esistono infine rari casi di LAP che non hanno la t(15;17), i quali sono sempre
caratterizzati da riarrangiamenti che coinvolgono la ricombinazione di RARα con
partner localizzati su altri siti cromosomici. La t(11;17)(q23;q12) genera una fusione
tra RARα e il gene PLZF (Promyelocytic Leukemia Zinc Finger), il quale codifica per un
membro della famiglia dei fattori di trascrizione POZ/zinc-finger (Chen et al, 1993).

16
La t(5;17) fonde il gene della nucleofosmina (NPM) a RARα (Redner et al, 1996). Altre
rare traslocazioni associate alle LAP sono la t(11;17)(q13;q12) che fonde la proteina
dell’apparato mitotico nucleare (NuMa) a RARα (Wells et al, 1997), e la t(17;17) nella
quale RARα è fuso al trasduttore di segnale e attivatore trascrizionale STAT5b
(Arnould et al, 1999). Alcune varianti di LAP, generalmente le LAP PLZF/RARα e
STAT5b positive, si sono dimostrate parzialmente o totalmente resistenti alla terapia
differenziante con ATRA (Licth et al, 1995; Cheng et al, 1999). Altre varianti, come le
LAP NPM1/RARα e NuMA/ RARα positive, sono invece responsive all’ATRA.
1.7 Altre aberrazioni cromosomiche comuni delle LAM
Il più frequente riarrangiamento cromosomico nelle LAM umane è la traslocazione
bilanciata tra i cromosomi 8 e 21, t(8;21), riscontrata in circa 10% di tutte le LAM.
Questa traslocazione produce il gene chimerico e la proteina di fusione costitutita da
una porzione del fattore di trascrizione AML1 fusa alla proteina co-repressoria ETO.
La risultante proteina di fusione AML1/ETO funziona come repressore trascrizionale
dei geni regolati da AML1, reclutando sui promotori bersaglio complessi di co-
repressione trascrizionali come SMRT, N-Cor e Sin3A e proteine con attività di istone
deacetilasi (HDACs) (Nervi et al, 2008).
Un altro riarrangiamento relativamente frequente è l’inversione cromosomica
inv(16), che viene riscontrata approssimativamente nell’8% dei casi di LAM, e causa
la fusione di una porzione ammino-terminale della proteina core binding factor β
(CBFβ) con un tratto carbossi-terminale della catena pesante della miosina del
muscolo liscio (MYH11). Normalmente, CBFβ interagisce col fattore ti trascrizione
AML1, modulandone l’attività regolatoria sui promotori dei geni bersaglio. In
contrasto, la proteina di fusione CBFβ-MYH11 continua a legarsi ad AML1,
conferendole però un’abberrante attività repressoria (Lutterbach et al, 1999).
Il gene MLL (Mixed Lineage Leukemia) è implicato almeno nel 10% delle leucemie
acute, sia LAL che LAM, e nei pazienti LAM le traslocazioni MLL sono generalmente
associate ad una prognosi sfavorevole (Eguchi et al, 2005). In condizioni normali,
MLL è un enzima con attività istone metil-trasferasi che funziona come un regolatore
positivo globale della trascrizione genica. Nelle leucemie acute, le traslocazioni
cromosomiche possono fondere MLL ad uno di oltre 50 geni partner, producendo una
proteina di fusione MLL che funziona come un potente oncogene (Krivtsov e

17
Armstrong, 2007). Sebbene i meccanismi d’azione molecolari e i bersagli genomici
specifici delle proteine di fusione MLL nelle LAM non siano stati ad oggi chiaramente
definiti, si ritiene che il dominio ammino-terminale MLL serva per reclutare
l’oncoproteina su loci genici specifici, mentre il partner di fusione funzioni come
un’unità effettrice, sostenendo una trans-attivazione aberrante del gene bersaglio
(Yokohama et al, 2010).
Esistono infine numerose altre traslocazioni bilanciate meno comuni, che
colletivamente costituiscono il 6% circa dei casi di LAM. Tra queste, sono importanti
le traslocazioni che coinvolgono i geni Hox, codificanti per fattori trascrizionali con un
ruolo importante nello sviluppo ematopoietico (Argiropoulos e Humphries, 2007). La
traslocazione meglio caratterizzata è la t(7;11), che genera il prodotto di fusione
NUP98/HOXA9. NUP98 è una nucleoporina facente parte del complesso del poro
nucleare, che può trovarsi fusa anche con altri membri dei geni Hox nelle leucemie,
come nella traslocazione NUP98/HOXD13 (Gough et al, 2011). HOXA9 è un membro
dei geni Hox espresso nei progenitori ematopoietici precoci, la cui espressione scende
durante il differenziamento ematopoietico, fino ad essere non rilevabile nelle cellule
terminalmente differenziate (Argiropoulos e Humphries, 2007). L’oncoproteina di
fusione NUP98/HOXA9 può promuovere la leuchemiogenesi conferendo un vantaggio
proliferativo e una inibizione del differenziamento nei blasti attraverso l’iper-
espressione dei geni bersaglio di HOXA9 (Yassin et al, 2009).
Le principali aberrazioni cromosomiche nelle LAM, e le onco-proteine che ne
risultano, sono riassunte in Tabella 1. In totale, approssimativamente il 35% delle
LAM è caratterizzato da traslocazioni cromosomiche ben definite. Circa la metà dei
pazienti LAM manifesta invece un cariotipo normale, mentre la rimanente parte ha
anomalie cariotipiche complesse. Tuttavia, negli ultimi anni numerose altre
anormalità genetiche sono state identificate nei blasti di LAM, che sfuggono
all’identificazione tramite analisi citogenetiche. Queste lesioni comprendono
mutazioni geniche oppure un’espressione aberrante di specifici geni, che hanno
svelato l’esistenza di un’enorme eterogeneità all’interno dei sotto-gruppi di LAM
definiti a livello citogenetico.
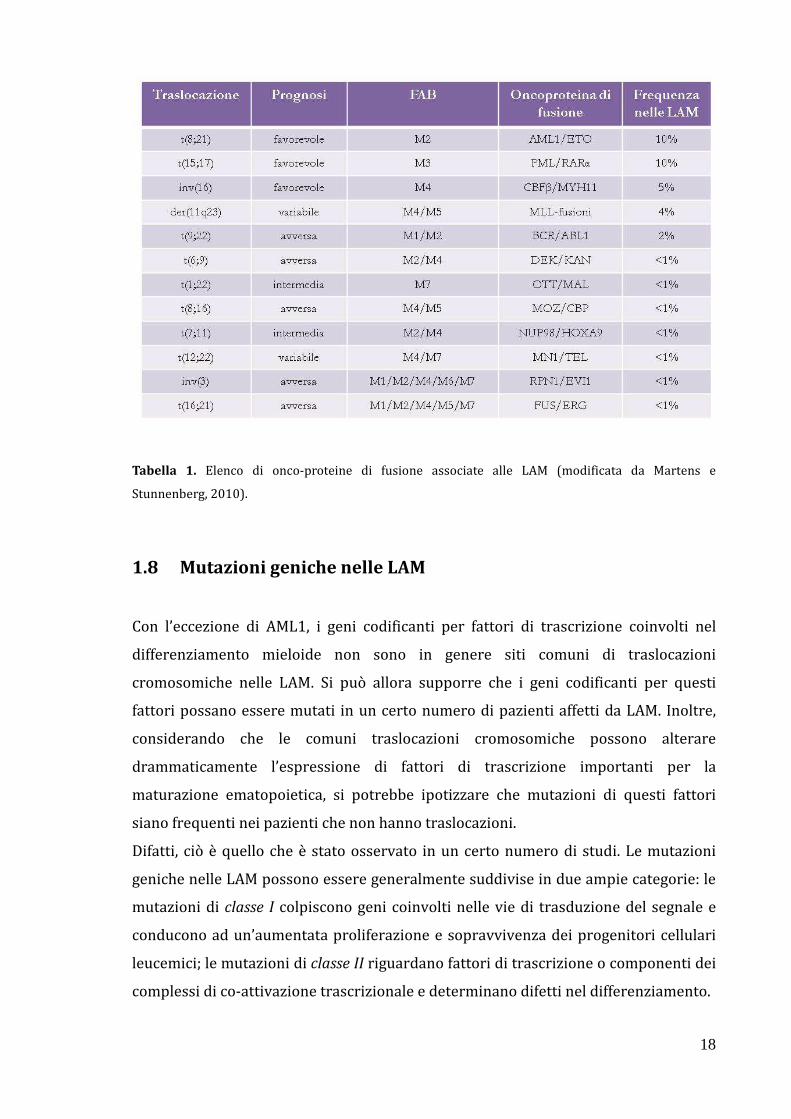
18
Tabella 1. Elenco di onco-proteine di fusione associate alle LAM (modificata da Martens e
Stunnenberg, 2010).
1.8 Mutazioni geniche nelle LAM
Con l’eccezione di AML1, i geni codificanti per fattori di trascrizione coinvolti nel
differenziamento mieloide non sono in genere siti comuni di traslocazioni
cromosomiche nelle LAM. Si può allora supporre che i geni codificanti per questi
fattori possano essere mutati in un certo numero di pazienti affetti da LAM. Inoltre,
considerando che le comuni traslocazioni cromosomiche possono alterare
drammaticamente l’espressione di fattori di trascrizione importanti per la
maturazione ematopoietica, si potrebbe ipotizzare che mutazioni di questi fattori
siano frequenti nei pazienti che non hanno traslocazioni.
Difatti, ciò è quello che è stato osservato in un certo numero di studi. Le mutazioni
geniche nelle LAM possono essere generalmente suddivise in due ampie categorie: le
mutazioni di classe I colpiscono geni coinvolti nelle vie di trasduzione del segnale e
conducono ad un’aumentata proliferazione e sopravvivenza dei progenitori cellulari
leucemici; le mutazioni di classe II riguardano fattori di trascrizione o componenti dei
complessi di co-attivazione trascrizionale e determinano difetti nel differenziamento.

19
Nell’ambito della classe I, di notevole rilevanza sono le mutazioni che colpiscono il
gene FLT3. Il recettore tirosina-chinasi FLT3 e il suo ligando sono importanti per la
proliferazione e il differenziamento dei progenitori ematopoietici più immaturi. Nei
blasti di pazienti LAM, FLT3 è iper-espresso nel 60-92% dei casi (Drexler, 1996;
Rosnet et al, 1996). Le mutazioni somatiche in FLT3 comportano un’attivazione
costitutiva del recettore, e sono state identificate all’interno di due domini funzionali
del recettore, il dominio giusta-membrana (Juxtamembrane; JM) e il dominio tirosina-
chinasico (TKD; Nakao et al, 1996; Yamamoto et al, 2001). Il dominio JM, che ha un
ruolo cruciale nell’auto-inibizione dell’attività chinasica del recettore, è
frequentemente distrutto da duplicazioni interne in tandem (Internal Tandem
Duplications; ITD), presenti nel 28-34% dei pazienti a cariotipo normale, mentre le
mutazioni puntiformi in questo dominio sono rare (Kottaridis et al, 2001). Nelle LAM,
il loop di attivazione nel lobo carbossi-terminale del TKD può subire mutazioni
puntiformi, piccole delezioni o inserzioni (Mead et al, 2007). Sotto il profilo clinico, le
mutazioni di FLT3 sono rilevanti sia per il loro impatto prognostico, sia perché FLT3
costitutivamente attivo rappresenta un interessante bersaglio per la terapia
molecolare delle LAM. Numerosi studi hanno infatti chiaramente evidenziato come
pazienti di LAM a cariotipo normale con mutazioni FLT3-ITD abbiano un decorso
significativamente peggiore rispetto a pazienti che mancano di FLT3-ITD (Kottaridis
et al, 2001). Anche nella LAP, dove queste mutazioni sono relativamente frequenti
rispetto agli altri sottotipi FAB, la presenza di FLT3-ITD è associata ad un rischio
maggiore di fallimento delle terapie (Kainz et al, 2002).
Nei pazienti di LAM, mutazioni geniche sono state riscontrate anche nei geni RAS, con
una incidenza maggiore per N-RAS, sporadicamente in K-RAS e solo molto raramente
in H-RAS (Döhner, 2007). Ad oggi, sebbene non sia stato attribuito un chiaro
significato prognostico alla presenza di queste mutazioni nelle LAM, mutazioni di RAS
possono rappresentare un altro obbiettivo di terapie molecolari.
Le mutazioni nel gene della nucleofosmina 1 (NPM1) sono relativamente frequenti
nei blasti di pazienti LAM, e rappresentano la più comune alterazione genetica
riscontrata nei pazienti a cariotipo normale (Falini et al, 2005). NPM1 è
un’abbondante fosfo-proteina che, in condizioni fisiologiche, è localizzata nei nucleoli
e fa la spola tra nucleo e citoplasma. NPM1 ha un ruolo in numerosi processi biologici,
quali la biogenesi dei ribosomi, la risposta allo stress, la stabilità genomica, la
trascrizione genica e la regolazione dell’attività di importanti onco-soppressori come

20
p53 e ARF (Grisendi et al, 2006). Queste attività sono legate sia al controllo della
proliferazione e sopravvivenza cellulare, che al differenziamento. Per questo motivo,
le mutazioni di NPM1 nelle LAM possono considerarsi al confine tra classe I e classe
II. Queste mutazioni generalmente cadono nell’esone 12 del gene, e comportano un
accumulo anomalo della proteina nel citoplasma (Falini et al, 2005). Il 40% dei
pazienti con mutazioni in NPM1 sono anche portatori di FLT3-ITD. Alcuni studi hanno
evidenziato come mutazioni in NPM1 possano rappresentare marcatori di prognosi
favorevole, ma solo in assenza di FLT3-ITD (Schnittger et al, 2005; Thiede et al,
2006).
Nell’ambito della classe II, di rilievo sono le mutazioni che colpiscono il fattore
trascrizionale C/EBPα, una proteina chiave nei processi di differenziamento mieloide.
Sono stati identificati due tipi di mutazioni nel gene di C/EBPα nelle LAM: 1)
mutazioni non-senso, che colpiscono la porzione N-terminale della proteina,
causando la formazione di una isoforma tronca a funzione di dominante negativo; 2)
mutazioni nel domino C-terminale a cerniera di leucina, che causano un’inefficace
capacità di legare il DNA o di formare omo-dimeri. Alcuni studi evidenziano che le
mutazioni in C/EBPα nelle LAM sono associate ad esito favorevole della malattia
(Estey e Doher, 2006; Mròzec et al, 2007).
Le mutazioni parziali in tandem del gene MLL (Partial tandem Duplications, PTD)
sono state le prime mutazioni riportate avere un ruolo prognostico nelle LAM a
cariotipo normale, e sono associate ad una durata più breve di remissione clinica. A
differenza delle proteine chimeriche di fusione MLL, MLL-PTD conserva tutti i domini
funzionali della proteina. La presenza di MLL-PTD è associata al silenziamento
dell’allele MLL normale, probabilmente attraverso meccanismi epigenetici (Estey e
Doher, 2006; Mròzec et al, 2007).
Le mutazioni del gene Wilms’ Tumor 1 (WT1) nelle LAM furono identificate per la
prima volta nel 1998 (King-Underwood e Pritchard-Jones, 1998). WT1 controlla sia la
quiescenza dei progenitori ematopoietici che il differenziamento delle cellule mielo-
monocitiche; perciò le mutazioni che distruggono le sue funzioni normali possono
alterare sia la capacità proliferativa che indurre un blocco del differenziamento nei
blasti leucemici.
Esistono infine altre mutazioni geniche, come le mutazioni dell’ubiquitina ligasi CBL,
la cui prevalenza e rilevanza clinica non sono state ancora chiaramente determinate
(Caligiuri et al, 2007). Inoltre, un certo numero di geni può avere un’espressione

21
deregolata nei blasti di LAM, senza necessariamente essere mutato o oggetto di
traslocazioni. I geni BAALC, ERG, MN1 e EVI1, risultano di frequente iper-espressi, e la
loro espressione deregolata può avere un significato prognostico (Mròzec et al, 2007).

22
Capitolo 2. L’epigenetica
2.1 L’organizzazione della cromatina nella cellula eucariota Nelle cellule eucariotiche il DNA si presenta come una complessa struttura
nucleoproteica che prende il nome di “cromatina”. L’unità fondamentale della
cromatina viene definita nucleosoma. Il livello organizzativo minimo della cromatina
è il “core” nucleosomale in cui 146 paia di basi di DNA si avvolgono intorno ad un
complesso di 8 proteine chiamate “istoni”. Questo ottamero è costituito dagli istoni
H2A, H2B, H3, H4 che presenti in duplice copia formano un tetramero (H3)2(H4)2 e
due eterodimeri H2A-H2B (Kelley, 1973; Kornberg et al, 1974; Roark et al, 1974;
Luger et al,1997). Gli istoni sono piccole proteine basiche, costituite da un dominio
globulare e da un segmento di 20-35 residui all’N-terminale ricco di aminoacidi
basici, la coda istonica, che protrude dal nucleosoma (Peterson and Laniel, 2004). Il
dominio globulare del “core” istonico è composto da 3 α-eliche coinvolte nei contatti
istone-istone ed istone-DNA (Arents et al, 1991). A livello molecolare le interazioni
del DNA con le proteine istoniche sono dovute a legami idrogeno che si instaurano tra
i gruppi fosfato del DNA e i residui amminici degli istoni, e ad interazioni
elettrostatiche con la catena basica laterale. Infatti, in tutti gli istoni sono presenti
molti residui aminoacidici carichi positivamente che interagiscono facilmente con
l’impalcatura fosfodiesterica del DNA che ha carica negativa. Gli istoni H3 ed H4 sono
sorprendentemente ben conservati attraverso l’evoluzione suggerendo quindi una
loro cruciale funzione. Gli altri istoni sono meno conservati e sono state trovate
varianti con diverse proprietà. Tra un nucleosoma e l’altro c’è un tratto di DNA
“linker” (DNA d’unione) che, negli eucarioti superiori, contiene un ulteriore istone,
H1, chiamato istone "linker", che non sembra importante per la formazione della
particella nucleosomale, ma pare coinvolto nella formazione di strutture di ordine
superiore. Si pensa che l'istone H1 stabilizzi le strutture cromatiniche mediante la
neutralizzazione delle cariche elettrostatiche del DNA di unione, attraverso il suo
dominio carbossi-terminale molto ricco di residui amminoacidici carichi
positivamente (Wolffe, 1997).

23
2.2 Epigenetica e trascrizione
L’epigenetica è la scienza che studia i cambiamenti ereditari nell’espressione genica
che avvengono senza apportare variazioni nella sequenza del DNA. I meccanismi
epigenetici sono essenziali per il normale sviluppo e per il mantenimento di un
modello di espressione genica tessuto-specifica nei mammiferi. Alterazioni dei
processi epigenetici possono portare ad alterate funzioni geniche e allo sviluppo di
malattie come il cancro, sindromi pediatriche, nonché fattori che contribuiscono a
malattie autoimmuni e all'invecchiamento (Rodenhiser and Mann, 2006).
L’organizzazione del DNA nelle fibre di cromatina costituisce un ostacolo al legame
con le proteine coinvolte nella trascrizione, per cui queste strutture devono essere
dinamiche ed in grado di compiere transizioni finemente regolate della loro
conformazione; tali cambiamenti della struttura cromatinica, facilitando o impedendo
l’accesso dei fattori trascrizionali al DNA nucleosomale, rappresentano quindi un
importante meccanismo di regolazione. La regolazione della trascrizione a livello
epigenetico coinvolge sia modificazioni post-traduzionali degli istoni, ad opera di
complessi che modificano covalentemente alcuni loro specifici residui aminoacidici,
che la metilazione del DNA. Questi meccanismi molecolari collaborano in modo
coordinato agli eventi di apertura e chiusura della cromatina stessa.
2.2.1 La metilazione del DNA
Nelle cellule eucariote la metilazione del DNA, che interessa la base citosina
nell’ambito del dinucleotide citosina – guanina (“CpG”), influenza la trascrizione del
gene interessato (Razin and Cedar, 1991). Dal punto di vista biochimico, questa
modificazione consiste nella sostituzione, operata dall’enzima DNA metiltrasferasi
(DNMT), dell’atomo di ossigeno legato al carbonio in posizione 5 della base citosina
con un gruppo metile, con formazione di 5-metilcitosina. Circa l’80% dei dinucleotidi
CpG presenti nel genoma dei mammiferi sono metilati (Cheung and Lau, 2005),
mentre il restante 20% risulta non metilato e prevalentemente situato, con elevata
densità (“CpG Island”), nelle regioni promotrici di geni costitutivi o inducibili.
Il fatto che ogni potenziale sito di metilazione nel genoma sia simmetrico implica
l’ereditabilità di tale modificazione: infatti, quando viene metilata la base citosina su

24
uno dei due filamenti di DNA, viene metilata anche la stessa base sul filamento
complementare. Dopo la duplicazione dell’acido nucleico, DNMT specifiche ricreano il
profilo di metilazione del filamento di DNA parentale in quello neo-sintetizzato.
Questa modificazione epigenetica è implicata in molti processi, quali la regolazione
trascrizionale e la modulazione della struttura della cromatina. Inoltre, essa esercita
un effetto stabilizzante che promuove e garantisce l'integrità del genoma, oltre ad una
corretta espressione temporale e spaziale dei geni durante lo sviluppo (Song et al,
2005).
La metilazione del DNA può reprimere la trascrizione (Figura 1) mediante
meccanismi diretti (tra cui l’inibizione di fattori della trascrizione che legano il DNA)
o indiretti, dovuti essenzialmente agli effetti di proteine che si legano alla base
citosina metilata nell’ambito del dinucleotide CG: ad esempio, le proteine MeCP2 e
MBDs si legano a CpG metilate e reprimono la trascrizione grazie all’interazione con
l’enzima istone deacetilasi (HDAC); a sua volta, quest’ultimo deacetila specifici residui
aminoacidici localizzati all’estremità amino-terminale delle proteine istoniche,
portando ad un rimodellamento della cromatina che risulta meno accessibile agli
enzimi della trascrizione, con conseguente repressione dell’espressione genica
(Jaenisch and Bird, 2003).

Figura 1: La metilazione come meccanismo epigenetico. La metilazione del DNA può portare al
silenziamento trascrizionale; ciò è dovuto al cambiamento di conformazione della cromatina, che passa
da uno stato più “aperto”, attivo dal punto di vista trascrizionale
2.2.2 Le modificazioni post
Le modificazioni post-traduzionali degli istoni includono l’acetilazione di lisine (K), la
metilazione di lisine ed arginine (R), la fosforilazione di serine (S) e treonine (T),
l’ubiquitinazione e la sumolazione di
queste modificazioni sono state riscontrate nelle code istoniche N
l’eccezione dell’ubiquitinazione che si verifica nelle code C
e H2B (Peterson and Laniel, 2004). Una
modificazioni post-traduzionali degli istoni è riportata in
La metilazione come meccanismo epigenetico. La metilazione del DNA può portare al
silenziamento trascrizionale; ciò è dovuto al cambiamento di conformazione della cromatina, che passa
da uno stato più “aperto”, attivo dal punto di vista trascrizionale, ad uno più “chiuso”.
cazioni post-traduzionali degli istoni
traduzionali degli istoni includono l’acetilazione di lisine (K), la
metilazione di lisine ed arginine (R), la fosforilazione di serine (S) e treonine (T),
l’ubiquitinazione e la sumolazione di K, e l’ADP-ribosilazione. La maggior parte di
queste modificazioni sono state riscontrate nelle code istoniche N
l’eccezione dell’ubiquitinazione che si verifica nelle code C-terminali degli istoni H2A
e H2B (Peterson and Laniel, 2004). Una rappresentazione schematica delle principali
traduzionali degli istoni è riportata in Figura 2.
25
La metilazione come meccanismo epigenetico. La metilazione del DNA può portare al
silenziamento trascrizionale; ciò è dovuto al cambiamento di conformazione della cromatina, che passa
, ad uno più “chiuso”.
traduzionali degli istoni includono l’acetilazione di lisine (K), la
metilazione di lisine ed arginine (R), la fosforilazione di serine (S) e treonine (T),
ribosilazione. La maggior parte di
queste modificazioni sono state riscontrate nelle code istoniche N-terminali, con
terminali degli istoni H2A
rappresentazione schematica delle principali
Figura 2.

Figura 2: Rappresentazione delle principali modificazioni post
istoniche ( da Spivakov and Fisher, 2007)
Acetilazione: L’acetilazione dell’istone H2A in lisina 5 (K5), dell’istone H2B in lisina
12, 15 e 20 (K12, K15, K20), dell’istone
dell’istone H4 in lisina 5 e 8 (K5, K8) sono modificazioni che promuovono
l’attivazione trascrizionale. L’acetilazione delle code amino
consiste nel trasferimento della porzione acetile
residui conservati di lisina. Questo tipo di modificazione neutralizza le cariche
positive delle code istoniche indebolendo la loro interazione con i gruppi fosfato del
DNA carichi negativamente. Ciò determina un rilas
nucleosomale che permette l’accesso dei fattori trascrizionali promuovendo alti livelli
di trascrizione genica. Così, un’aumentata acetilazione istonica è associata
all’attivazione della trascrizione genica, mentre una sua diminuzio
una generale inibizione della trascrizione.
L’acetilazione è promossa da enzimi ad attività istone acetiltransferasica (HAT), a
localizzazione nucleare o citoplasmatica. Le HAT che catalizzano reazioni a livello
nucleare sono principalme
acetiltransferasi localizzate nel citoplasma invece regolano l’acetilazione degli istoni
Rappresentazione delle principali modificazioni post-traduzionali a carico delle code
istoniche ( da Spivakov and Fisher, 2007)
: L’acetilazione dell’istone H2A in lisina 5 (K5), dell’istone H2B in lisina
12, 15 e 20 (K12, K15, K20), dell’istone H3 in lisina 4, 9,14 e 27 (K4, K9, K14, K27) e
dell’istone H4 in lisina 5 e 8 (K5, K8) sono modificazioni che promuovono
l’attivazione trascrizionale. L’acetilazione delle code amino-terminali degli istoni
consiste nel trasferimento della porzione acetile dell’acetil-CoA sul gruppo ε
residui conservati di lisina. Questo tipo di modificazione neutralizza le cariche
positive delle code istoniche indebolendo la loro interazione con i gruppi fosfato del
DNA carichi negativamente. Ciò determina un rilassamento della struttura
nucleosomale che permette l’accesso dei fattori trascrizionali promuovendo alti livelli
di trascrizione genica. Così, un’aumentata acetilazione istonica è associata
all’attivazione della trascrizione genica, mentre una sua diminuzio
una generale inibizione della trascrizione.
L’acetilazione è promossa da enzimi ad attività istone acetiltransferasica (HAT), a
localizzazione nucleare o citoplasmatica. Le HAT che catalizzano reazioni a livello
nucleare sono principalmente coinvolte nella regolazione trascrizionale. Le
acetiltransferasi localizzate nel citoplasma invece regolano l’acetilazione degli istoni
26
traduzionali a carico delle code
: L’acetilazione dell’istone H2A in lisina 5 (K5), dell’istone H2B in lisina
H3 in lisina 4, 9,14 e 27 (K4, K9, K14, K27) e
dell’istone H4 in lisina 5 e 8 (K5, K8) sono modificazioni che promuovono
terminali degli istoni
CoA sul gruppo ε-NH3+ dei
residui conservati di lisina. Questo tipo di modificazione neutralizza le cariche
positive delle code istoniche indebolendo la loro interazione con i gruppi fosfato del
samento della struttura
nucleosomale che permette l’accesso dei fattori trascrizionali promuovendo alti livelli
di trascrizione genica. Così, un’aumentata acetilazione istonica è associata
all’attivazione della trascrizione genica, mentre una sua diminuzione è associata con
L’acetilazione è promossa da enzimi ad attività istone acetiltransferasica (HAT), a
localizzazione nucleare o citoplasmatica. Le HAT che catalizzano reazioni a livello
nte coinvolte nella regolazione trascrizionale. Le
acetiltransferasi localizzate nel citoplasma invece regolano l’acetilazione degli istoni

27
neo-sintetizzati, controllando la loro traslocazione nucleare. Oggi si contano sei
famiglie di proteine che presentano attività istone acetiltransferasica (Roth et al,
2001; Ehrenhofer-Murray, 2004). Fra queste, la famiglia di GCN5 e quella di p300
sono quelle meglio caratterizzate. I membri della famiglia GCN5 sono tutti co-
attivatori trascrizionali. Fra di essi troviamo la p300/Cbp Associated Protein (PCAF),
del quale saggi in vitro e in vivo hanno dimostrato la sua interazione con i membri
della famiglia di p300 (Schiltz et al, 1999). La famiglia di p300 include p300 e la
proteina di legame CREB (Creb Binding Protein, CBP), due proteine nucleari che
coordinano e regolano l’apparato trascrizionale della cellula. Fin dalle prime ricerche
è stato chiaro il ruolo di entrambe queste proteine quali co-attivatori della
trascrizione, a cui è seguita l’identificazione della loro attività acetiltransferasica
(Giordano and Avantaggiati, 1999). p300 e CBP sono due proteine altamente
omologhe, che contengono molti motivi strutturali comuni e che si sovrappongono da
un punto di vista funzionale.
L’acetilazione degli istoni è un processo reversibile, infatti sono stati isolati enzimi ad
attività istone deacetilasica (HDAC) (Figura 3). Le HDAC rimuovono i gruppi acetili
dai residui N-terminali di lisina compattando la cromatina con conseguente inibizione
trascrizionale; le HDAC sono suddivise in quattro diverse classi, in base all’omologia
di sequenza e all’organizzazione del dominio (Dokmanovic, 2007).
Gli enzimi appartenenti alla classe I sono le HDAC1, 2, 3, e 8; quelli di classe II sono le
HDAC 4, 5, 6, 7, 9 e 10 (de Ruijter et al, 2003). Un terzo gruppo di HDAC, la famiglia
Sirtuin, il cui membro principale è la proteina Sir1 strutturalmente non correlata alle
altre due famiglie, richiede il NAD+ come cofattore nella reazione di deacetilazione. Le
Sirtuin sono conservate dal punto di vista evoluzionistico negli eucarioti, nei
procarioti e negli archea, dove svolgono diverse funzioni nello sviluppo, nella
formazione dell’eterocromatina e nell’apoptosi (Buck et al, 2004). L’unica HDAC
appartenente alla classe IV ad oggi è l’HDAC11, di cui si conosce ben poco oltre alla
sua capacità di interagire con l’HDAC6 (Verdin et al, 2003).

28
Figura 3: L’acetilazione delle code istoniche è correlata all’attivazione trascrizionale; è un processo
reversibile catalizzato dalle istone acetitasi (HAT) e dalle istone deacetilasi (HDAC). In seguito
all’azione delle HAT la cromatina assume una conformazione prona alla trascrizione; viceversa,
l’azione delle HDAC rende la cromatina non accessibile al macchinario di trascrizione.
Metilazione: mentre l’acetilazione è generalmente associata con l’attivazione
trascrizionale, la metilazione degli istoni è legata sia ad attivazione che ad inibizione
trascrizionale. La diversa regolazione sulla trascrizione a carico di questo tipo di
modificazione dipende dal residuo aminoacidico istonico coinvolto e dal numero di
gruppi metilici associati a ciasuna lisina (mono-, di-, trimetilazione). In particolare, la
metilazione delle lisine è sia pro-attivatoria che inibitoria. Per esempio, la
trimetilazione dell’istone H3 in lisina 9 (K9), lisina 27 (K27) e lisina 79 (K79), e la
trimetilazione dell’istone H4 in lisina 20 (K20) è associata alla formazione
dell’eterocromatina e quindi a repressione genica, mentre la di- e trimetilazione
dell’istone H3 nelle lisine 4 e 36 (K4, K36) è legata all’attivazione trascrizionale
(Martin and Zhang, 2005; Barski et al, 2007). La metilazione delle arginine, come
quella dell’istone H3 in arginina 17 (R17) e dell’istone H4 in arginina 3 (R3), è invece
associata solo all’attivazione trascrizionale (Zhang and Reinberg, 2001; Peterson and
Laniel, 2004). Inoltre, come già descritto in precedenza, alcune di queste lisine
possono anche essere acetilate (ad esempio la K4, K9 e K27 dell’istone H3). Questa
alternanza tra acetilazione e metilazione ha spesso come risultato un cambiamento

29
nello stato di attivazione genica. La metilazione è promossa dagli enzimi istone
metiltransferasi (HMT) (Ehrenhofer-Murray, 2004). Un gruppo lisinico ε-NH3+ può
essere mono-, di- o trimetilato. Grazie alla struttura cristallografica delle HMT e allo
studio del loro sito catalitico è stato possibile capire come un enzima possa essere in
grado di catalizzare una mono-, ma non una di- o trimetilazione. Nelle monometilasi
come Set7/9, il gruppo ε-NH3+ è legato così fortemente nel sito attivo che non c’è
ulteriore spazio per un gruppo metilico ingombrante (Xiao et al, 2003; Ehrenhofer-
Murray, 2004). Perciò l’enzima può catalizzare solo la mono-metilazione. Invece le di-
e trimetilasi mostrano una maggiore flessibilità nel loro sito attivo. A differenza
dell’acetilazione, che risulta nella neutralizzazione delle cariche positive nel gruppo ε-
NH3, la metilazione lascia le cariche intatte. Mentre l’acetilazione degli istoni viene
regolata in maniera dinamica dall’azione delle HAT ed HDAC, la metilazione degli
istoni sembra essere chimicamente più stabile. Attualmente, comunque, sono stati
isolati diversi enzimi istone demetilasi (HDM), che sono in grado di demetilare l’H3 in
K4, in K27 e in K36.
Fosforilazione: è associata ad attivazione trascrizionale, alla condensazione dei
cromosomi durante la mitosi, e al riparo da danno al DNA (Sauve et al, 1999). E’ stato
dimostrato che la fosforilazione dell’istone H3 sulla serina 10 (S10) in collaborazione
con altre modificazioni (per esempio, l’acetilazione in K14) induce l’inizio della
trascrizione (Lo et al, 2001), mentre da sola o in combinazione con altre
fosforilazioni, recluta fattori responsabili della condensazione e della segregazione
cromosomica (Wei et al, 1999). Una delle risposte cellulari precoci dopo l’induzione
della rottura di entrambi i filamenti della doppia elica di DNA (Double Strand Break) è
la fosforilazione della Serina 139 (S139) dell’estremo C-terminale dell’istone H2AX,
una variante minore dell’istone H2A, in corrispondenza del sito di danno (Rogakou et
al, 1998). Studi di colocalizzazione proteica hanno mostrato che la fosforilazione
dell’istone H2AX è necessaria per il reclutamento di proteine coinvolte nella
riparazione del danno e nel blocco del ciclo cellulare (Karlsson and Stenerlow, 2004).
poli-ADP-ribosilazione: è una reazione catalizzata dall’enzima nucleare
poli(ADPribosio) polimerasi (PARP). E’ stato visto che l’ADP-ribosilazione dell’istone
H1 promuove la decondensazione della fibra cromatinica, mentre la modificazione
dell’istone H2B porta ad una parziale dissociazione del DNA dal core istonico. La sua
azione, quindi, determina un riarrangiamento nucleosomale e trasforma la cromatina

30
in una struttura trascrizionalmente attiva (de Murcia et al, 1988; Kraus and Lis,
2003).
Ubiquitinazione: è una modificazione post-traduzionale grazie alla quale una catena
(poliubiquitinazione), una singola molecola (monoubiquitinazione) o piu’ di una
singola molecola (multiubiquitinazione) di ubiquitina viene legata a proteine
bersaglio. L’ubiquitinazione delle proteine ha una funzione precisa: indica che queste
molecole possono essere degradate dal proteosoma. Gli istoni H2A, H2B, H3, H1 e la
variante H2A.Z possono essere monoubiquitinati (Osley et al, 2006). Il ruolo della
monoubiquitinazione è ancora poco noto, dal momento che l’aggiunta di una singola
molecola di ubiquitina non è sufficiente ad indirizzare le proteine verso la
degradazione proteosomale. Alcune evidenze sperimentali suggeriscono che
l’ubiquitinazione degli istoni possa avere un ruolo nel controllo della trascrizione
attraverso la regolazione della metilazione di alcuni residui specifici.
Sumolazione: consiste nel legame covalente della proteina SUMO (Small Ubiquitin-
related MOdifier) a residui di lisina delle proteine bersaglio. Questa modificazione
coinvolge diverse proteine, regolandone in generale la localizzazione e l’attività. Nel
caso degli istoni, il legame della proteina SUMO si oppone all’acetilazione, attribuendo
alla sumolazione un ruolo di inibizione della trascrizione (Iniguez-Lluhi, 2006). Per
esempio, la sumolazione dell’istone H4 media il silenziamento genico attraverso il
reclutamento di istone deacetilasi e della proteina eterocromatinica 1 (HP1) (Shiio
and Eisenman, 2003).

31
Capitolo 3. I microRNA
3.1 I microRNA: biogenesi e meccanismi d’azione
L'identificazione negli anni '90 dei microRNA (miRNA) ha aperto una nuova era nella
comprensione dei processi regolatori post-trascrizionali dell’espressione genica. I
miRNA sono una ampia classe di piccoli RNA non codificanti formati da 19-25
nucleotidi che regolano negativamente l’espressione genica a livello post-
trascrizionale, inducendo la degradazione di specifici RNA messaggeri (mRNA), o
impedendone la traduzione in proteina. E’ stato inoltre dimostrato che in lievito, nelle
piante, e negli animali i miRNA sono in grado di indurre silenziamento trascrizionale
mediante modificazioni del DNA e/o della cromatina (Noma et al, 2004). I miRNA
riconoscono specificamente gli mRNA bersaglio mediante un appaiamento
complementare delle basi. Tali RNA non sono perfettamente complementari tra loro
(vi sono uno o più disappaiamenti) e, nella maggior parte dei casi, i miRNA provocano
un blocco nella traduzione, senza causare la degradazione del mRNA bersaglio
(Bartel, 2009).
Le prime evidenze dell’esistenza e del ruolo dei miRNA nella regolazione traduzionale
furono osservate nei nematodi. In Caenorhabditis elegans (C.elegans) furono
identificati piccoli RNA in grado di ridurre la sintesi proteica senza modificare i livelli
di accumulo di specifici mRNA. Il primo gene codificante per uno di questi piccoli RNA
ad essere descritto, lin-4, è un regolatore essenziale della divisione cellulare allo
stadio larvale. Esso da origine ad un corto RNA di 22 nucleotidi di lunghezza,
perfettamente complementare ad un tratto della regione 3’ non tradotta (3’UTR)
dell’mRNA codificante per la proteina LIN-14, precedentemente dimostrata, tramite
approccio genetico, essere controllata da lin-4 (Lee et al., 1993). Attualmente sono
stati individuati, mediante tecniche di clonazione tradizionale in associazione ad
ausili bioinformatici, 2042 miRNA maturi (1600 precursori) nell’uomo ed è stato
stimato che circa il 30% dei geni umani sono regolati da miRNA (Bartel, 2004).
Pertanto, attualmente i miRNA costituiscono la più grande classe di geni regolatori. I
miRNA attivi nella regolazione dei loro mRNA bersaglio sono definiti miRNA “maturi”.
I geni di miRNA sono dispersi in tutti i cromosomi umani eccetto che nel cromosoma
Y.

32
La biogenesi dei miRNA ha inizio con la trascrizione di una breve sequenza genomica
che dà origine ad un “microRNA primario” (pri-miRNA). Esso viene trascritto nel
nucleo della cellula dalla RNA polimerasi II (RNAPol II) oppure, meno
frequentemente, dalla RNA polimerasi III (RNAPol III; Borchert et al, 2006). I pri-
miRNA, così come avviene per gli altri mRNA di classe II, subiscono modificazioni
post-trascrizionali quali l’aggiunta del cappuccio di 7-metil-guanosina all’estremità 5’
e della coda di poli(A) all’estremità 3’ (Lee et al, 2002). La loro lunghezza può variare
da qualche centinaia fino ad alcune migliaia di basi e possono contenere uno o più
precursori dei miRNA (pre-miRNA) in forma policistronica. I pri-miRNA si ripiegano
fino ad assumere una tipica struttura secondaria caratterizzata da uno o più diversi
ripiegamenti a forcina (“stem-loop”), ognuno dei quali darà in seguito origine ad un
miRNA diverso. L’evento di maturazione successivo avviene ad opera di un complesso
multi-proteico (Microprocessor) di cui fanno parte l’RNasi di tipo III Drosha e la
proteina di legame all’RNA a doppio filamento DiGeorge syndrome critical region 8
gene DGCR8 (Landthaler et al, 2004). I pri-miRNA vengono riconosciuti da Drosha a
livello dei ripiegamenti a forcina e tagliati alla base di tali strutture. Come risultato
del taglio si ha la formazione dell’intermedio successivo, il pre-miRNA, della
lunghezza di 60-70 nucleotidi e caratterizzato da una peculiare estremità di 2
nucleotidi a singolo filamento alle terminazioni 3’ (Lee et al, 2003). Questa estremità
asimmetrica al 3’ è legata dalla proteina Esportina-5, che permette la traslocazione
nel citoplasma del pre-miRNA (Lund et al, 2004; Zeng, 2006). In Drosophila e nei
mammiferi esistono anche vie non canoniche di processamento dei miRNA, che
portano alla formazione di un pre-miRNA attraverso meccanismi indipendenti da
Drosha. In genere, questi meccanismi riguardano miRNA localizzati in brevi introni
(miRtrons), trasportati nel citoplasma direttamente dopo lo splicing (Berezikov et al,
2007). Il pre-miRNA, traslocato nel citoplasma, viene riconosciuto a livello della
protrusione al 3’ da un complesso multienzimatico con attività endonucleasica,
chiamato Dicer, che completa la maturazione dei miRNA tagliandolo nella forma
finale a doppio filamento di 22 nucleotidi con 2 nucleotidi sporgenti all’estremità 3’
(Meister e Tuschl, 2004). A questo punto i miRNA vengono incorporati nel complesso
ribonucleoproteico dei miRNA (miRNP), noto anche come complesso silenziatore
indotto da RNA (RISC). Quando il miRNA viene caricato sul RISC uno dei due filamenti
viene allontanato e degradato. Il filamento che viene a far parte del RISC è in genere
quello con l’estremità 5’ con energia libera minore (Schwarz et al, 2003). La

33
regolazione della produzione dei miRNA può avvenire durante tutte le fasi della loro
biogenesi.
Fin dalla loro scoperta, l’interesse per il ruolo rappresentato dai miRNA nella
regolazione genica è cresciuto esponenzialmente. Numerosi dati sperimentali hanno
dimostrato che i miRNA agiscono in numerosi processi cellulari, quali proliferazione,
apoptosi, differenziamento, trasduzione del segnale ed in processi fisiologici come il
metabolismo, la embriogenesi e l’organogenesi (Kloosterman and Plasterk, 2006;
Alvarez-Garcia and Miska, 2005).
I miRNA possono avere profili di espressione specifici per stadi di sviluppo, tessuti e
patologie; ciascun tessuto può così essere caratterizzato e da uno specifico e distinto
profilo di espressione di un gruppo di miRNA (Rana, 2007).
Il controllo dell’espressione genica avviene a livello post-trascrizionale, inducendo la
degradazione di specifici mRNA oppure impedendo la traduzione della proteina. Il
meccanismo d’azione che porta al silenziamento genico dipende dal grado di
complementarità tra il miRNA e il suo mRNA bersaglio. Il grado di complementarietà,
che si limita ad una regione di 6-8 nucleotidi all’estremità 5’ del miRNA detta “seed”, è
una caratteristica fondamentale per il destino dell’mRNA dopo il reclutamento del
RISC. Una ridotta omologia di sequenza porta generalmente alla repressione
traduzionale senza la degradazione dell’mRNA bersaglio, mentre se l’appaiamento
alla regione 3’UTR degli mRNA bersaglio è perfetto, come nei miRNA delle piante o in
alcuni miRNA animali, si verifica il taglio del messaggero, catalizzato dal complesso
RISC reclutato dal miRNA stesso, che porta infine alla degradazione dell’mRNA
bersaglio (Ambros, 2004).
Il meccanismo tramite cui RISC regola la traduzione è tutt’ora oggetto di attivo
dibattito; a tal riguardo sono stati proposti tre modelli:
• la repressione dell’inizio della traduzione (Pillai et al, 2005);
• la repressione della fase di allungamento della traduzione (Maroney et al,
2006; Nottrott et al, 2006);
• la destabilizzazione del trascritto attraverso un accorciamento della coda di
poli(A) (Wu et al, 2006).
Comunque il processo, qualora le informazioni di sequenza non siano sufficienti ad
indirizzare il meccanismo regolatorio, può essere influenzato anche da altri fattori,
quali la composizione proteica del RISC a cui è associato il miRNA, il pattern proteico

34
a cui il miRNA è correlato o il promotore che guida la trascrizione del gene bersaglio
(Filipowicz et al, 2008).
3.2 I miRNA intronici
I geni dei miRNA sono presenti nel genoma come unità independenti (miRNA
intergenici) o localizzati all’interno di introni di altri geni, definiti “ospite”, nello
stesso orientamento o nell’orientamento opposto (miRNA intronici; Rodriguez et al,
2004). Dati recenti riportano che circa il 50% dei geni per i miRNA si trova in regioni
intergeniche, mentre il restante 50% risiede in sequenze introniche di specifici geni
(Griffiths-Jones, 2007; Saini et al, 2008).
Mentre i miRNA che risiedono nell’introne di un gene nell’ orientamento antisenso
(opposto a quello della trascrizione) sono, per definizione, trascritti
indipendentemente dal gene ospite, fino a poco tempo fa si credeva che i miRNA
intronici orientati nello stesso senso del gene che li conteneva, fossero prodotti da un
trascritto comune a quello del gene ospite, ovvero dipendessero dal promotore del
gene ospite per la loro trascrizione (Bartel, 2004).
Se ciò fosse vero, l’espressione di questi miRNA potrebbe essere semplicemente
dedotta dal profilo di espressione del loro gene ospite. In effetti, in esperimenti di
microarray nell’uomo, è stata trovata una buona correlazione tra i livelli
d’espressione dei miRNA intronici e dei loro geni ospite (Baskerville and Bartel,
2005).
Sebbene diversi dati sperimentali esistano a supporto di questo modello di biogenesi
di “trascrizione comune” dei miRNA intronici con i loro geni ospite, un crescente
numero di evidenze mostra che molti miRNA intronici, con orientamento “senso”,
sono trascritti come unità trascrizionali indipendenti. Aboobaker e colleghi hanno
trovato che il pattern di ibridazione in situ di miR-7 in Drosophila è diverso da quello
del suo gene ospite bancal: mentre bancal è espresso ubiquitariamente, miR-7 ha un
modello molto particolare di espressione spazio-temporale, suggerendo differenze
nella regolazione in cis di questo miRNA e del suo gene ospite (Aboobaker et al,
2005). Nell’uomo, le nuove strategie “high throughput” per la caratterizzazione di
promotori, come la ChIP-seq per valutare le modificazioni istoniche e la presenza di
RNA polimerasi II, in combinazione o meno con l’analisi del posizionamento dei
nucleo somi, suggeriscono che circa un terzo dei miRNA intronici possiede un

35
promotore proprio indipendente da quello del gene ospite (Ozsolak et al, 2008;
Corcoran et al, 2009; Wang et al, 2009).
Tuttavia, non è ancora chiaro se la trascrizione dei miRNA intronici indipendente da
quella dei geni ospite sia un eccezione o piùttosto la regola.
3.3 miRNA e meccanismi epigenetici
Cambiamenti epigenetici, come la metilazione del DNA e la modificazione degli istoni,
giocano un ruolo molto importante nel rimodellamento della cromatina e nella
regolazione dell’espressione di geni codificanti per proteine nelle neoplasie umane
(Esteller, 2007). Tali meccanismi interessano anche la regolazione trascrizionale dei
miRNA e sono considerati meccanismi chiave nella modulazione della loro
espressione (Lujambio et al., 2008. Weber et al, 2007).
3.3. 1 miRNA e metilazione del DNA
Così come avviene per i geni codificanti proteine, anomali profili di metilazione di
isole CpG, situate all’interno o nelle vicinanze di un gene codificante per un miRNA,
possono determinare un’alterata espressione di alcuni miRNA, generando alterazioni
patogeniche, inclusa la tumorigenesi (Jones and Baylin, 2002).
Circa il 50% dei geni dei miRNA sono associati a isole CpG. Alcuni di questi miRNA
sono stati trovati essere metilati in alcuni tipi di tumore come il promotore del miR-
124a, ipermetilato in linee cellulari di cancro al colon ma non nel tessuto sano
(Lujambio et al, 2007). Nei tumori mammari è stato dimostrato che l’ipermetilazione
del promotore del miR-31 e del suo gene ospite, il lncRNA LOC554202, è uno dei
principali meccanismi che ne determinano il suo silenziamento (Augoff et al, 2012).
La ridotta espressione del mir-34b/c, componente cruciale nella via del segnale
dell’oncosoppressore p53 (Corney et al, 2007), correla con l’ipermetilazione dell’
isola CpG presente sul suo promotore nella maggior parte delle neoplasie al colon-
retto (Toyota et al, 2008; Lujambio et al, 2008). L’ipermetilazione delle isole CpG del
miR-34b/c, del miR-148 e del miR-9 risultano inoltre essere associate allo sviluppo di
metastasi in numerose neoplasie umane (Lujambio et al, 2008). Grady e colleghi
hanno trovato che il trattamento combinato di 5-AzaC, sostanza che demetila il DNA,
con l’inibitore delle HDAC tricostatina A (TSA) poteva ripristinare i livelli

36
d’espressione del miR-342 e del suo gene ospite EVL, (Grady et al, 2008). Per quanto
riguarda le leucemie, un esempio eccellente è il miR-223, un miRNA espresso
esclusivamente nelle cellule mieloidi. Fazi e colleghi hanno dimostrato che in blasti
AML con traslocazione t(8;21) che genera l’oncoproteina di fusione AML1/ETO, il
miR-223 è ipo-espresso. Ciò è dovuto al legame di AML1/ETO che, reclutando sul suo
promotore un complesso epigenetico che comprende le DNMT e le HDAC, ne causa il
silenziamento (Fazi et al, 2007).
3.3.2 miRNA e modificazioni istoniche
Le deacetilasi istoniche (HDAC) e le proteine del gruppo polycomb (PcG) sono gli
enzimi regolatori delle modificazioni istoniche più studiati. La perdita di acetilazione
nelle code istoniche, mediata dalle HDAC, risulta nella repressione genica. Le HDAC
sono state spesso trovate over-espresse in vari tipi di cancro (Halkidou et al, 2004;
Song et al, 2005), così sono diventate uno dei maggiori bersagli per le terapie
epigenetiche.
In un lavoro di Scott e colleghi, è stato dimostrato che trattando una linea di tumore al
seno con un inibitore delle HDAC, i livelli d’espressione di 27 miRNA venivano
rapidamente alterati (Scott et al, 2006). La metilazione del DNA e le modificazioni
istoniche lavorano spesso insieme nel regolare l’espressione dei miRNA. Ad esempio,
l’espressione del miRNA onco-soppressore miR-127 può essere altamente
incrementata dalla demetilazione del promotore e dall’ inibizione delle HDAC in
cellule tumorali umane (Saito et al, 2006).
E’ interessante notare che potrebbe esistere una regolazione a feedback; infatti,
alcuni studi hanno mostrato che un ristretto gruppo di miRNA può a sua volta
regolare l’espressione di enzimi chiave del macchinario epigenetico. Per esempio,
HDAC1 è stato dimostrato essere direttamente down-regolato dal miR-449a (Noonan
et al, 2009). I membri della famiglia del miR-29 (a/b/c) sono stati riportati reprimere
l’espressione degli enzimi DNMT3a e 3b appaiandosi al 3’UTR del loro trascritto
(Fabbri et al, 2007).
Questo ruolo nella regolazione a “feedback” indica una complessa rete tra microRNA
e macchinario epigenetico che aumenta la complessità delle conseguenze dell’alterata
regolazione epigenetica dei miRNA, attribuendogli un ruolo anche come controllori
della struttura cromatinica.
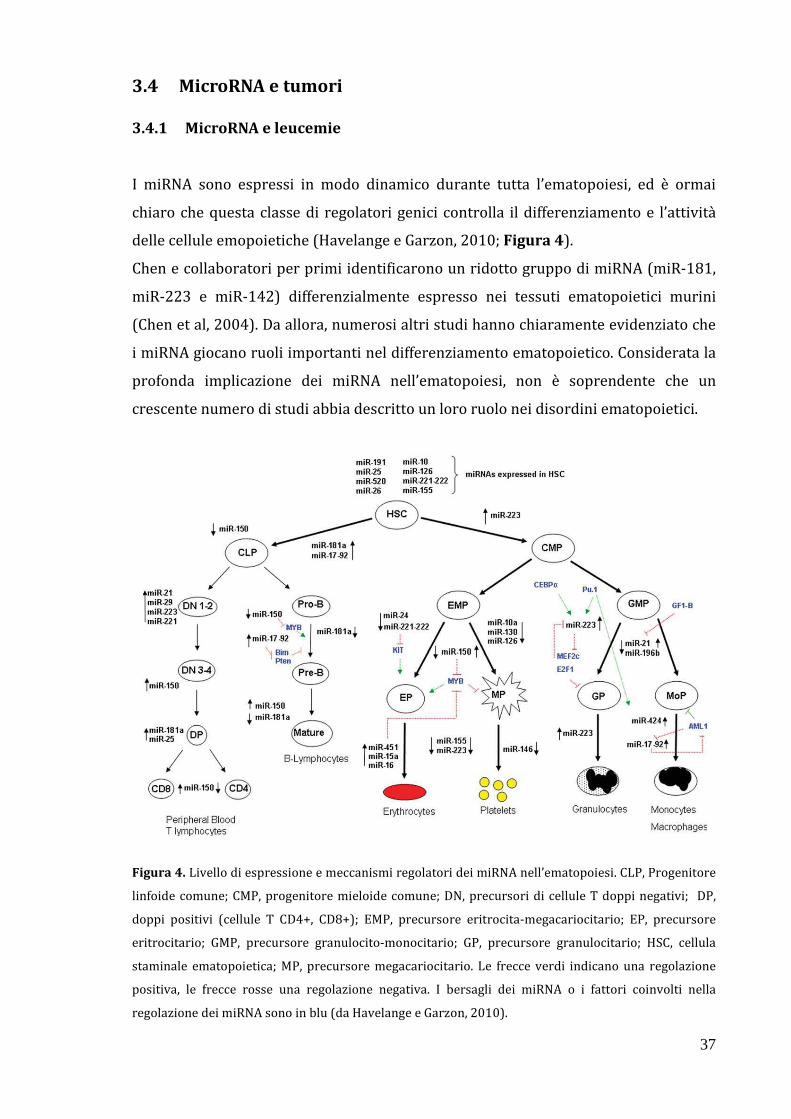
37
3.4 MicroRNA e tumori
3.4.1 MicroRNA e leucemie
I miRNA sono espressi in modo dinamico durante tutta l’ematopoiesi, ed è ormai
chiaro che questa classe di regolatori genici controlla il differenziamento e l’attività
delle cellule emopoietiche (Havelange e Garzon, 2010; Figura 4).
Chen e collaboratori per primi identificarono un ridotto gruppo di miRNA (miR-181,
miR-223 e miR-142) differenzialmente espresso nei tessuti ematopoietici murini
(Chen et al, 2004). Da allora, numerosi altri studi hanno chiaramente evidenziato che
i miRNA giocano ruoli importanti nel differenziamento ematopoietico. Considerata la
profonda implicazione dei miRNA nell’ematopoiesi, non è soprendente che un
crescente numero di studi abbia descritto un loro ruolo nei disordini ematopoietici.
Figura 4. Livello di espressione e meccanismi regolatori dei miRNA nell’ematopoiesi. CLP, Progenitore
linfoide comune; CMP, progenitore mieloide comune; DN, precursori di cellule T doppi negativi; DP,
doppi positivi (cellule T CD4+, CD8+); EMP, precursore eritrocita-megacariocitario; EP, precursore
eritrocitario; GMP, precursore granulocito-monocitario; GP, precursore granulocitario; HSC, cellula
staminale ematopoietica; MP, precursore megacariocitario. Le frecce verdi indicano una regolazione
positiva, le frecce rosse una regolazione negativa. I bersagli dei miRNA o i fattori coinvolti nella
regolazione dei miRNA sono in blu (da Havelange e Garzon, 2010).

38
Nelle leucemie, e nei tumori in generale, i miRNA possono funzionare da oncogeni
(onco-miR) attraverso vari meccanismi, e principalmente tramite la repressione di
proteine oncosoppressorie, oppure avere un ruolo di oncosoppressori, bersagliando
mRNA oncogenici. Le cause della deregolazione dell’espressione di specifici miRNA
nello sviluppo tumorale non sono ancora del tutto note. La maggior parte dei geni che
trascrivono per i miRNA risiede in particolari regioni genomiche conosciute come siti
fragili, che nelle cellule cancerose sono spesso soggette ad alterazioni (Calin et al,
2004). Il ruolo dei miRNA nelle leucemie fu per la prima volta evidenziato dal gruppo
di Calin nel 2002, il quale mostrò che pazienti affetti da una comune forma di
leucemia, la leucemia linfocitica cronica delle cellule B (LCL), presentavano
frequentemente delezioni o silenziamenti del cluster intronico di miRNA che
includeva miR-15a e miR-16-1 (Calin et al, 2002).
Il locus (13q14) dove mappano questi due miRNA risulta essere deleto in più del 65%
dei casi di LCL, come nel 16-40% dei mielomi multipli e nel 60% dei carcinomi
prostatici. In un altro studio, è stato dimostrato che il miR-15a e il miR-16-1 regolano
in maniera negativa il gene anti-apoptotico Bcl2, la cui iper-espressione è stata
riscontrata in molti tipi di tumori, inclusi le leucemie ed i linfomi (Cimmino et al,
2005). Il miR-223 potrebbe avere funzioni oncosoppressorie, almeno in alcuni
sottotipi di LAM: è stato dimostrato che AML1/ETO reprime l’espressione del miR-
223 tramite silenziamento epigenetico, e l’espressione ectopica di questo miR in
cellule di LAM è in grado di promuovere il differenziamento granulocitario (Fazi et al,
2007). Al contrario, il miR-155 rappresenta un classico esempio di onco-miR nelle
leucemie: l’espressione aberrante di questo miRNA è stata riscontrata in specifici
sottogruppi di LAM (Garzon et al, 2008a; Garzon et al, 2008b), e l’espressione forzata
del miR-155 in cellule staminali ematopoietiche (HSC) murine causa mielo-
proliferazione, splenomegalia e cambiamenti displastici (O’Connel et al, 2008). La
grande variabilità nel decorso ed esito della malattia nei pazienti di LAM, anche
all’interno di sottogruppi definiti a livello morfologico o citogenetico, ha suggerito la
possibilità che l’espressione di gruppi specifici di miRNA possa essere associata a
particolari sotto-tipi di leucemia e avere un significato prognostico. Ad esempio, Isken
e collaboratori hanno analizzato 50 casi di pazienti LAM e 12 campioni di controllo
per l’espressione di 154 miRNA umani. Questo studio mostra una iper-espressione
dei miR-34a, miR-221 e miR-222 nelle cellule di LAM rispetto a precursori
CD34+(Isken et al, 2008). Specifici profili di espressione di miRNA possono

39
consentire di discriminare le LAM dalle LAL, come è stato mostrato da uno studio nel
quale sono stati identificati 27 miRNA espressi differenzialmente tra LAL e LAM (Mi
et al, 2007). Tra questi miRNA, i miR-128a, -128b, - 223, e let-7b potevano distinguere
le LAL dalle LAM con un’accuratezza diagnostica del 97-99%. Garzon e collaboratori
hanno analizzato l’espressione di miRNA in cellule di LAP nel differenziamento
granulocitario indotto da ATRA (Garzon et al, 2007). In questo studio, il trattamento
con ATRA nella linea cellulare di LAP NB4 induceva una diminuita espressione del
miR-181b e un aumento di espressione dei miR-15a, -15b, -16, -107, -223, -342, let-
7a, let-7c e let-7d, suggerendo per questi ultimi una possibile funzione di
oncosoppressori (Garzon et al, 2007). Il nostro gruppo di ricerca ha recentemente
confermato ed esteso questi risultati in vivo in blasti primari di LAP (Careccia et al,
2009). In questo lavoro, campioni derivati da pazienti LAP, trattati con successo con
terapie a base di chemioterapici e ATRA, mostravano una diminuita espressione del
miR-181b, e un’aumentata espressione dei miR-15b, -16, -107, -223, -342 e let-7c.
Inoltre, è stato identificato uno specifico profilo di espressione di un ristretto gruppo
di miRNA che distingueva i blasti LAP dai promielociti normali. In particolare,
l’espressione del miR-342 e del let-7c risultavano significativamente ridotte nei blasti
leucemici rispetto a promielociti normali differenziati in vitro da progenitori CD34+.
Abbiamo dimostrato che, in cellule di LAP, i promotori di questi due miRNA erano
bersaglio dell’oncoproteina PML/RARα e, in risposta al trattamento con ATRA,
l’aumentata espressione del miR-342 e del let-7c era associata al distacco di
PML/RARα dai loro promotori (Careccia et al, 2009). Questi risultati suggerivano che
specifici profili di espressione di miRNA potevano distinguere le cellule di LAP dai
promielociti normali, e che la repressione trascrizionale dei miR-342 e let-7c ad
opera dell’oncoproteina PML/RARα poteva rappresentare un evento importante nella
leuchemiogenesi delle LAP.
3.4.2 MicroRNA e tumori solidi
E’ stato stimato che circa il 30% dei geni umani è regolato dai miRNA (Bartel, 2004).
Analisi di espressione hanno dimostrato che molti miRNA sono finemente regolati
durante lo sviluppo ed in modo tessuto-specifico (He and Hannon, 2004; Miska,
2005). Al contrario, un crescente numero di lavori mostra che un profilo di
espressione aberrante dei miRNA è associato con molte malattie umane,

40
specialmente con il cancro (Anindo and Yaqinuddin, 2012). Infatti, i miRNA possono
funzionare sia come oncogeni che come oncosoppressori e il ruolo ad essi associato
dipende dalla funzione di inibire l’espressione di geni soppressori tumorali o di
oncogeni. Esistono tuttavia anche dei casi in cui uno stesso miRNA funzioni sia come
oncogene che come oncosoppressore, ruolo che dipende dal contesto cellulare
specifico in cui si trova il miRNA: un esempio è il miR-29a. Il suo ruolo di soppressore
tumorale è stato dimostrato in tumori solidi come neuroblastoma, sarcoma, tumore
gastrico e tumore al cervello (Xu et al, 2009; Cui et al., 2011). In contrasto, il miR-29a
risulta avere un ruolo da promotore dell’oncogenesi nelle leucemie mieloidi acute
(AML) (Han et al, 2010) e nelle leucemie linfocitiche croniche del tipo B-cellule (B-
CLL) (Santanam et al, 2010).
Alcuni esempi di miRNA promotori dell’oncogenesi sono:
• il cluster miRNA 17-92, che regola la proliferazione, l’apoptosi e lo sviluppo
cellulare (Pospisil et al, 2011) e che risulta sovraespresso in diversi tipi di
tumori (Dixon-McIver et al, 2008; Volinia et al, 2006).
• Il miRNA-21 che risulta over-espresso in differenti tipi di tumore solido con la
funzione di inibire l’espressione di differenti geni soppressori tumorali con
conseguente aumento della proliferazione e/o diminuzione dall’apoptosi
(Huang et al, 2013).
• Il miRNA-155 che risulta altamente espresso in differenti tipi di tumori. Viene
processato a partire da un esone di un RNA non codificante, BIC (B Cell
Integration Cluster), che originariamente è stato descritto come il sito di
integrazione di DNA virale nei linfomi indotti da virus (Clurman and Hayward,
1989). Nel cancro alla mammella, la sua elevata espressione è associata ad un
aumento della sopravvivenza cellulare e della resistenza ai chemioterapici
(Kong et al, 2010). Nel tumore al colon, il miR-155 targhetta molti componenti
del sistema di riparo del DNA causando un elevato tasso di mutazioni al DNA e
l’instabilità di micro satelliti (Valeri et al, 2010).
• Il miRNA-30d regola la proliferazione, l’apoptosi, la senescenza, e la
migrazione nelle cellule tumorali. Il locus cromosomico che ospita il miR-30d è
stato trovato amplificato in più del 30% di molti tipi di tumore solido umano
ed è stato dimostrato che la sua inibizione blocca la crescita tumorale in vivo
(Li et al, 2012).

41
Esempi di miRNA che al contrario svolgono un ruolo di soppressori tumorali sono:
• I miRNA della famiglia let-7, la cui espressione risulta ridotta in differenti tipi
di tumori e di cui si conosce ampiamente in letteratura l’importante ruolo di
soppressori tumorali, ad esempio nel cancro al polmone. È stato visto infatti
che l’espressione endogena di questo miRNA riduce in vivo la crescita del
tumore in modelli di tumore al polmone (Kumar et al, 2008; Roush and Slack,
2008).
• Il miRNA-34, regolato trascrizionalmente da p53, è frequentemente represso
nel carcinoma al pancreas ed è implicato nella regolazione del ciclo cellulare,
dell’apoptosi e del riparo del DNA (Chang et al, 2007).
• I miR-143 e miR-145, i cui livelli di espressione risultano ridotti in differenti
tipi di tumori e il cui ripristino, nel tumore alla prostata e nel tumore colon-
rettale, riduce significativamente la crescita del tumore sia in vitro che in vivo
(Clape et al, 2009; Chen et al, 2009).
3.5 La famiglia di micro-RNA let-7
3.5.1 Aspetti generali sulla famiglia let-7
Particolarmente rilevante per l’argomento di questo lavoro di tesi, è il microRNA let-
7c, appartenente alla famiglia di miRNA let-7. Il gene lethal (let-7) fu inizialmente
scoperto come un gene essenziale per lo sviluppo in C.elegans, e fu uno dei primi
miRNA identificati (Reinhart et al, 2000). La famiglia di miRNA let-7, spesso presente
in copie multiple nel genoma, è molto conservata nelle specie animali (Tabella 2).
Per distinguere tra le diverse isoforme, una lettera posta dopo let-7 distingue i diversi
componenti della famiglia, che differiscono leggermente nella sequenza del miRNA
maturo. Un numero posto alla fine del nome può indicare che la stessa sequenza è
presente in copie multiple nel genoma. Ad esempio, nell’uomo esistono 10 sequenze
mature di let-7, e 13 loci genici. Tre precursori diversi codificano per lo stesso let-7a
maturo (let-7a-1, let-7a-2, let-7a-3), e precursori di due loci genici diversi producono
il let-7f (let-7f-1 e let-7f-2).

42
Tabella 2. Organizzazione schematica dei membri di let-7 nelle diverse specie. I numeri nella tabella
rappresentano il numero di copie annotate in mirBase (http://microrna.sanger.ac.uk/sequences/). Il
Cluster 1 è costituito dal miR-100, 99, let-7, e miR-125; il Cluster 2 è rappresentato dal let-7a, d e let-7f.
Il Cluster 3 è composto dal let-7a-3 e let-7b, mentre il Cluster 4 è composto dal let-7f e miR-98 (da
Roush e Slack, 2008).
In C.elegans, il let-7 è un gene con funzioni eterocroniche (geni le cui mutazioni
causano trasformazioni fatali allo sviluppo temporale della cellula). Durante gli stadi
larvali dello sviluppo di C.elegans, le cellule dell’ipodermide, conosciute come cellule
di giunzione (“seam cells”), si dividono in modo analogo alle cellule staminali, con una
cellula figlia che differenzia e l’altra che mantiene le proprietà auto-rinnovanti e
continua il programma proliferativo. Alla transizione tra stadio larvale L4 e adulto, la
cellule figlia auto-rinnovante cessa di proliferare e differenzia. Cellule di giunzione
mutanti con perdita di funzione per let-7, falliscono nell’uscire dal ciclo cellulare a
questo stadio, e subiscono divisioni cellulari extra (Reinhart et al, 2000). La
maggioranza degli animali con perdita di funzione per let-7 muore a causa di questi
difetti nello sviluppo, e il mutante nullo è stato perciò definito lethal (let). Il ruolo
cruciale ricoperto da let-7 nello sviluppo di C.elegans è suggerito anche dalla sua
espressione strettamente regolata nel tempo, che comincia ad essere rilevata allo
stadio L3, e raggiunge il suo picco massimo durante lo stadio L4, in accordo con il suo
ruolo nel promuovere l’uscita dal ciclo cellulare e il differenziamento terminale delle
cellule di giunzione alla fine dello stadio larvale L4 (Esquela-Kersher et al, 2005).
Sebbene molte caratteristiche dei miRNA let-7 sembrino altamente conservate nelle
specie, esistono anche differenze cospicue passando da C.elegans agli organismi più

43
complessi. Ad esempio, il numero dei membri della famiglia è maggiore nei vertebrati,
e la sola sequenza matura del let-7a è identica tra tutte le specie animali, dal C.elegans
all’uomo. Gli altri membri hanno identica “seed”, ma differiscono in varia misura nei
nucleotidi rimanenti (Roush e Slack, 2008). Anche i meccanismi regolatori
dell’espressione temporale di let-7 possono differire tra invertebrati e organismi
superiori. Alcuni studi hanno mostrato che negli eucarioti inferiori, come in C.elegans
e Drosophila, la regolazione avviene a livello della trascrizione del pri-let-7 (Sempere
et al, 2002; Johnson et al, 2003), mentre nei mammiferi intervengono meccanismi più
complessi, e la regolazione può avvenire sia livello trascrizionale che post-
trascrizionale (Newman et al, 2008; Viswanathan et al, 2008).
Ad ogni modo, la comparsa di let-7 maturo negli stadi avanzati dell’embriogenesi è
una caratteristica conservata in molti organismi, come nel tessuto cerebrale di topo e
Danio rerio (Lagos-Quintana et al, 2002; Wulczyn et al, 2007). In contrasto con la loro
espressione nei tessuti differenziati, forme mature di let-7 sono assenti in cellule
staminali embrionali umane e murine, e un tema comune sembra un loro aumento di
espressione in seguito al differenziamento (Lagos-Quintana et al, 2002; Sempere et al,
2004; Mineno et al, 2006; Wulczyn et al, 2007), rispecchiando l’espressione di let-7
nelle cellule di giunzione differenzianti di C.elegans. In effetti, una delle funzioni
principali del let-7 sembra quella di promuovere il differenziamento cellulare:
l’espressione del let-7a, let-7c e let-7e maturi è aumentata durante lo sviluppo
cerebrale di topo (Wulczyn et al, 2007), e in un altro studio il let-7 è stato trovato
espresso in progenitori cellulari di tessuto mammario indotti a differenziare (Ibarra
et al, 2007). Un’altra funzione della famiglia let-7 è il controllo del ciclo cellulare: la
perdita di let-7 in C.elegans e Drosophila conduce ad una iper-proliferazione cellulare
(Reinhart et al, 2000; Caygill e Johnston, 2008) e, allo stesso modo, il blocco
d’espressione di let-7 nella linea di carcinoma polmonare umana A549 induce
un’aumentata proliferazione, mentre la sua iper-espressione blocca la progressione
del ciclo cellulare (Johnson et al, 2007). Alcuni studi suggeriscono che bassi livelli di
let-7 potrebbero essere usati come una caratteristica diagnostica di certe popolazioni
di cellule staminali: ad esempio, una bassa espressione di let-7c è stata usata per
arrichire le cellule staminali in una linea di epitelio mammario murino Comma-Dβ,
mentre un’iper-espressione di let-7c riduceva la sotto-popolazione delle cellule auto-
rinnovanti (Ibarra et al, 2007).

44
Questi dati suggeriscono che la regolazione del differenziamento e della
proliferazione cellulare sono funzioni conservate della famiglia let-7 durante
l’evoluzione delle specie. Tuttavia, sebbene i miRNA della famiglia let-7 abbiano
caratteristici “pattern” di espressione temporali durante lo sviluppo, un ruolo diretto
del let-7 nello sviluppo dei vertebrati non è stato ad oggi chiaramente dimostrato. Il
ritardo nell’identificazione di queste funzioni è probabilmente dovuto alle difficoltà
associate al silenziamento dei molteplici membri della famiglia nello stesso animale, e
alla possibile ridondanza dei membri di una famiglia così numerosa (Roush e Slack,
2008).
3.5.2 Let-7 e tumori
Le proprietà di de-differenziamento e di aumento della proliferazione appena
descritte, sono anche caratteristiche tipiche delle cellule tumorali. In effetti, la ridotta
espressione di vari membri della famiglia let-7 è stata associata a molti tumori umani
(Büssing et al, 2008) ed alle cellule staminali tumorali (Yu et al, 2007), suggerendo un
ruolo di questi miRNA come oncosoppressori nei tumori umani. In particolare,
l’espressione dei miRNA let-7 è stata trovata ridotta in linee cellulari e tessuti primari
di carcinoma polmonare (Takamizawa et al, 2004; Johnson et al, 2007) e, in pazienti
affetti da carcinoma polmonare a cellule non piccole, associata a cattiva prognosi
(Takamizawa et al, 2004). Questi dati sono in accordo con l’osservazione che diversi
membri della famiglia let-7 mappano su siti cromosomici frequentemente deleti nei
tumori polmonari (Calin et al, 2004). Potenziali meccanismi molecolari di una
funzione oncosoppressoria dei miRNA let-7 emergono da diversi studi, nei quali è
stato riportato che let-7 regola numerosi oncogeni. E’ stato dimostrato che K-RAS e N-
RAS sono bersagli molecolari di let-7, suggerendo che questi miRNA possano
funzionare da oncosoppressori nei carcinomi polmonari in vivo riducendo
l’espressione di RAS (Johnson et al, 2005). Un altro bersaglio molecolare di let-7 è
l’oncogene MYC, sebbene la rilevanza in vivo di questa interazione debba essere
ancora completamente chiarita (Leucci et al, 2008). Il cluster altamente conservato
del let-7a, let-7d e let-7f può essere coinvolto nei linfomi a cellule B, poiché sembra
esser regolato direttamente dall’oncogene v-myc (v-myc myelocytomatosis viral
oncogene homolog). E’ stato dimostrato che let-7a reprime l’espressione di MYC nel
linfoma di Burkitt, suggerendo l’esistenza di un circuito a “feedback” negativo

45
(Sampson et al, 2007; Chang et al, 2008). Numerosi gruppi hanno riportato che un
altro importante oncogene, HMGA2, è regolato negativamente da membri della
famiglia let-7 (Lee e Dutta, 2007; Mayr et al, 2007). Nei tumori le mutazioni di HMGA2
frequentemente causano delezioni del 3’ UTR, portando all’eliminazione dei siti di
legame per let-7, e alla iper-espressione della proteina HMGA2 che promuove il
cancro (Mayr et al, 2007). Numerosi lavori hanno identificato altri bersagli dei miRNA
let-7 che supportano la potenziale rilevanza di questa famiglia di miRNA nel controllo
del ciclo cellulare in cellule tumorali: CDC25a e CDK6, due regolatori fondamentali
nella progressione del ciclo cellulare, si sono rivelati bersagli diretti di let-7 (Johnson
et al, 2007), così come la ciclina D1 (Schultz et al, 2008).
Studi recenti riportano che due proteine in grado di legare l’RNA, Lin28 e Lin28B,
sono in grado di bloccare il corretto processamento e maturazione dei precursori
della famiglia let-7, suggerendo che l’aberrante aumento di espressione di Lin28 e
Lin28B, frequentemente riscontrata nei tumori umani, possa promuovere la
tumorigenesi tramite la repressione di let-7 (Viswanathan et al, 2009). La proteina
Lin28 lega il trascritto primario e il precursore a forcina del microRNA (pri-let-7 e
pre-let-7) inibendo il taglio da parte di Drosha. Lin28 è in grado anche di promuovere
l’uridilazione terminale del pre-let-7, mediante una uridil-transferasi 3’terminale,
determinando la degradazione del trascritto. Questo indica che Lin28 inibisce la
processazione attuata da Dicer (Viswanathan et al, 2008; Viswanathan and Daley,
2010). Inoltre, il ripristino dell’espressione di let-7 in cellule di leucemia mieloide
cronica K-562 mediante inibizione dell’espressione di Lin28, diminuisce la capacità
proliferativa e quella di formare colonie in soft agar, e stimola modificazioni
morfologiche indicative di un’induzione del differenziamento (Newman et al, 2008;
Viswanathan et al, 2008; Viswanathan et al, 2009).
Nonostante la ridotta espressione dei membri della famiglia let-7 sia stata
generalmente associata ad un aumento della capacità trasformante in vitro in vari tipi
tumorali, alcuni dati contrastanti in vari tipi di cancro suggeriscono che la funzione
della famiglia let-7 non sia ancora completamente chiarita, e che singoli membri della
famiglia let-7 possano avere ruoli distinti. E’ possibile infatti che i 13 loci genici di let-
7 nell’uomo non esistano semplicemente per avere diverse funzioni nei vari tessuti,
ma potrebbero avere differenti funzioni nella stessa cellula. Alcuni lavori riportano
che in certi tumori alcune isoforme di let-7 sono iper-espresse mentre altre sono
perse. Ad esempio, in uno studio sul mesotelioma maligno, è stato trovato iper-

46
espresso il let-7b, mentre l’espressione del let-7g era fortemente ridotta (Guled et al,
2009).
Per quanto riguarda il let-7c, particolarmente importante per il lavoro di questa tesi,
è un miRNA intronico localizzato sul cromosoma 21 in un introne del gene non-
codificante proteine LINC00478, all’interno del quale si trovano anche il miR-99a and
miR-125b-2, ed è espresso ubiquitariamente in tessuti murini ed umani (Ro et al,
2007). Let-7c presenta funzioni oncosoppressive che correlano con il suo
coinvolgimento nella carcinogenesi (Shell et al, 2007). E’ stato individuato un gruppo
di LOG (let-7–regulated oncofetal genes) sovra-espressi in molti carcinomi umani la
cui sovra-espressione correlava con la perdita di espressione di let-7. L’RNA di questi
geni era ipo-regolato da una sovra-espressione esogena del let-7(Boyerinas et al,
2008). Questa attività di oncosoppressore è stata riportata anche in modelli di
tumore della prostata e del polmone. In particolare, nei carcinomi polmonari la
modulazione dell’espressione del let-7c è strettamente correlata a quella
dell’oncogene RAS, la cui attivazione è un evento cruciale per l’insorgenza della
neoplasia (Johnson et al, 2005; Ozen et al, 2008). Il let-7c è stato mostrato essere
anche direttamente implicato nell’epatocarcinoma, essendo la sua espressione
diminuita durante la progressione tumorale (Pineau et al, 2010).
Nei tumori ematopoietici, il let-7c è regolato negativamente in pazienti di LAM con
t(8;21), inv(16) e con Linfoma di Burkitt (Jongen-Lavrencic et al, 2008; Leucci et al,
2008). Jongen-Lavrencic e colleghi, hanno valutato l’espressione di 260 miRNA in 215
casi LAM di nuova diagnosi, appartenenti a diversi gruppi citogenetici. In questo
studio hanno mostrato che una ridotta espressione di let-7b e let-7c consentiva di
discriminare le LAM con t(8;21) e inv(16) dagli altri gruppi definiti a livello
molecolare, suggerendo un potenziale ruolo onco-soppressivo di let-7b e let-7c in
questi sottotipi di leucemia (Jongen-Lavrencic et al, 2008). Inoltre, nostri dati recenti
mostrano che l’espressione ectopica del let-7c è sufficiente a indurre il
differenziamento mieloide in linee cellulari e blasti primari derivati da pazienti di
AML di nuova diagnosi (Pelosi et al, 2012).
In conclusione, la funzione della famiglia di miRNA let-7 come oncosoppressore in
molti tipi di cancro, incluse le leucemie, rende estremamente importante sia la
comprensione di come i vari membri let-7 vengano regolati, che l’ulteriore
definizione di quali siano i loro bersagli nella tumorigenesi e i meccanismi molecolari

47
nei quali sono implicati. E’ possibile infatti che la dettagliata comprensione di questi
meccanismi possa condurre allo sviluppo di nuove terapie contro il cancro.

48
Obiettivi della ricerca
Dati recenti suggeriscono che una significativa porzione del genoma umano è regolata
da miRNA. Numerosi studi hanno, inoltre, dimostrato che l’espressione dei miRNA è
spesso deregolata nelle malattie umane, incluse quelle che riguardano il tessuto
ematopoietico. Comunque, sebbene l’importanza biologica dei miRNA sta diventando
sempre più chiara, la regolazione della loro espressione non è ancora pienamente
conosciuta. Nel laboratorio in cui è stata svolta questa tesi, è stata osservata un’espressione
coordinata del let-7c con il suo gene ospite LINC00478, che è un bersaglio del
repressore trascrizionale PML/RARα . Inoltre, in seguito a trattamento con ATRA, che
porta alla rimozione di PML/RARα, il let-7c mostrava una coordinata espressione con
il suo gene ospite suggerendo, quindi, che l’espressione del miRNA fosse regolata
esclusivamente dal promotore del suo gene ospite (Careccia et al., 2009). Il let-7c è un
miRNA intronico, e fino a poco tempo fa era opinione diffusa che tali miRNA, a
differenza di quelli intergenici che presentano elementi regolatori propri, fossero
semplicemente trascritti dal promotore del gene ospite con un meccanismo mediato
dalla RNApol II, e maturati in seguito, tramite il canonico macchinario di “splicing”
dell’RNA. Tuttavia, recentemente è stato dimostrato come la regolazione dei miRNA
intronici sia molto più complessa (vedi Discussione), ammettendo l’esistenza anche di
una loro regolazione indipendente dal gene ospite.
Per quanto sopradetto, lo scopo di questa ricerca è quello di valutare l’esistenza di un
promotore per il let-7c in grado di regolarlo indipendentemente dal suo gene ospite e
di identificare i possibili meccanismi di regolazione trascrizionale coinvolti nella
ridotta espressione del let-7c in blasti di LAP rispetto a promielociti normali.

49
Materiali e metodi Linee cellulari e condizioni di coltura Le linee cellulari leucemiche NB4, HEL, HL-60, KG-1, K-562, SKNO-1, THP-1, U937 e
UF-1 sono state cresciute in terreno di coltura RPMI + Glutamax (Gibco, Life
Technologies) contenente il 10% di siero bovino fetale (FBS) o 20% per le BV-173,
GDM-1, KG-1a, KASUMI-1 e PL-21 inattivato per 1 ora a 56 °C (Hyclone, Perbio, Logan,
Utah), e 1% di penicillina-streptomicina (Cambrex Bio-Science, Verviers, Belgio). Le
OCI-AML3 sono state coltivate in alpha-MEM (Life Technologies Invitrogen, Carlsbad,
CA, USA) con l’aggiunta del 20% di siero bovino fetale (FBS, Hyclone, Perbio, Logan,
Utah) e 1% di penicillina-streptomicina (Cambrex Bio-Science, Verviers, Belgio). Le
cellule, che crescono in sospensione, sono state piastrate ad una concentrazione
compresa tra 0,1-1 x 106 cellule/mL.
Le linee cellulari di adenocarcinoma polmonare H1299 e A549, di adenocarcinoma di
mammella MCF-7 e di carcinoma ovarico A2780 sono state cresciute in RPMI (Life
Technologies Invitrogen, Carlsbad, CA, USA) con il 10% di FBS (Cambrex Bio-Science,
Verviers, Belgium) e 1% di penicillina-streptomicina (Cambrex Bio-Science, Verviers,
Belgio). Le linee di adenocarcinoma di mammella SKBR3 e MDA-231, di
neuroblastoma SH-SY5Y, di epatocarcinoma Hep-G2 e di carcinoma colorettale HCT-
116 sono state cresciute in Dulbecco’s modified Eagles medium (DMEM; Life
Technologies Invitrogen, Carlsbad, CA, USA) contenente il 10% di FBS o 15% FBS per
le SH-SY5Y, (Cambrex Bio-Science, Verviers, Belgium) e 1% di penicillina-
streptomicina (Cambrex Bio-Science, Verviers, Belgio). La linea di adenocarcinoma di
prostata LNCaP è stata cresciuta in RPMI con 10% FBS (Cambrex Bio-Science,
Verviers, Belgium), 1% di penicillina-streptomicina (Cambrex Bio-Science, Verviers,
Belgio), e con l’aggiunta di glucosio allo 0,35%. Tutte le linee sono state coltivate alla
temperatura di 37 °C, in atmosfera con 5% di CO2 e umidità al 90%.
I trattamenti con acido retinoico tutto-trans (ATRA; Sigma, St Louis, MO) sono stati
fatti alla concentrazione di 1 µM per 24h nei saggi luciferasici e 72h per l’analisi di
ChIP.

50
Preparazione di RNA e cDNA
L’estrazione dell’RNA totale dalle cellule analizzate è stata effettuata con il metodo
che impiega il reagente Trizol®, seguendo il protocollo fornito dalla ditta (Reagent,
GibcoBRL).
Per la preparazione di cDNA, è stato usato 1 μg di RNA totale per campione, in una
miscela di reazione contenente i seguenti componenti:
• RT-buffer 1X [Tris-SO4 pH 9,1; (NH4)2SO4 90 mM];
• 200 nM dNTP;
• 150 ng di esameri casuali (sequenze casuali di nucleotidi in grado di appaiarsi a
qualsiasi sequenza di RNA);
• 2,5 μM ditiotreitolo (DTT);
• 2 unità RNasi Out;
• 10 unità retrotrascrittasi del virus della leucemia murina (M-MLV, 10 u/μl, Roche
Diagnostics).
La miscela di reazione è stata portata ad un volume finale di 30 μl con acqua RNasi
free ed i campioni incubati per 1 h a 37°C.
Stem-loop RT-PCR
L’espressione del let-c maturo è stata misurata tramite un metodo che prevede una
retrotrascrizione che fa uso di primers “stem-loop”, seguita dall’analisi mediante
Real-Time PCR (stem loop RT-PCR; Chen et al, 2005). Per ogni campione, sono stati
retro-trascritti 50 ng di RNA totale in una mix di reazione così composta:
• dNTP mix (100 mM) 0,15 µl
• Multiscribe™ RT (50U/µl) 1,00 µl
• 10 X RT Buffer 1,5 µl
• Rnase inhibitor (20 U/µl) 0,19 µl
alla quale sono stati aggiunti 3 µL di primer microRNA-specifico per hsa-let-7c,
oppure per lo “small-non coding” RNA U19 (RNU19), usato come normalizzatore

51
interno (0,25 mM; Applied Biosystems), e acqua RNasi-free fino al volume finale di 15
µL. La mix di reazione è stata incubata 5 minuti in ghiaccio, poi 30’ a 16°C, 30’ a 42°C,
e 5’ a 85°C.
Real Time PCR
Le reazioni di Real-Time PCR (qRT-PCR), sono state effettuate utilizzando un
termociclatore della ditta Applied Biosystem modello 7500 Real Time PCR System
SDS v1,2.
L’espressione del let-7c è stata misurata mediante primers e sonda specifici,
sintetizzati dalla ditta Applied Biosystem. Le reazioni di amplificazione per ciascun
campione sono state effettuate in triplicato in un volume finale di 20 μl contenente: 1
μl del prodotto di retro-trascrizione specifico per il miRNA, 10μl di TaqMan®
Universal PCR Master Mix 2X (Applied Biosystem), 1 μl di una mix di sonda
TaqMan® 200 nM, primer senso e primer antisenso 20x (Applied Biosystem) e acqua
DNasi ed RNasi-free. Il ciclo di amplificazione usato è quello standard, che prevede
una prima fase di 2 minuti a 50°C, una seconda fase di 10 minuti a 95°C seguita da
una successione di 40 cicli a 95°C per 15 secondi e 60°C per 1 minuto ognuno. I dati
ottenuti sono stati analizzati con il metodo comparativo dei ΔΔCt o delle fold change.
Per l’espressione di let-7c, la normalizzazione è stata fatta con sonde e primers
specifici per lo “small-non coding” RNU19 (Applied Biosystem).
L’espressione di LINC00478 è stata misurata con il metodo Sybr Green®; le reazioni
di amplificazione, per ciascun campione, sono state effettuate in triplicato in un
volume finale di 20 μl contenente: 1 μl del prodotto di retro-trascrizione, 10 μl di Sybr
Green® Master Mix 2x, primers specifici 300 nM (FW -
CCATGGTGATGTGAAGATGAGAA; RV - CCTGAAGATCTGGTTTGCAGAA) e acqua fino al
volume di 20 μl. Il ciclo di amplificazione usato è quello standard, incluso lo stadio
finale di dissociazione. I dati ottenuti sono stati analizzati con il metodo comparativo
dei ΔΔCt. La normalizzazione è stata fatta con primers specifici per il gene GAPDH
(FW - TCCCTGAGCTGAACGGGAAG; RV - GGAGGAGTGGGTGTCGCTGT).

52
Analisi bioinformatica
Al fine di identificare un putativo nuovo inizio della trascrizione, una regione di 10kb
a monte del pre-let-7c è stata sottomessa a vari motori di ricerca in grado di
riconoscere elementi regolatori caratteristici dei promotori. In particolare sono stati
usati:
Promoter Scan v.1.7 (http://www-bimas.cit.nih.gov/molbio/proscan/);
BDGP (http://www.fruitfly.org/seq_tools/promoter.html);
FPROM(http://www.softberry.ru/berry.phtmlgroup=programs&subgroup=promoter
&topic=fprom).
È stata inoltre eseguita, con il software MatInspector Professional di Genomatix
(http://www.genomatix.de), una ricerca di siti di legame per specifici fattori di
trascrizione nella regione al 5’ del putativo TSS identificato in silico.
Per identificare la presenza di isole CpG è stato usato il software CpG Island
searcher(http://cpgislands.usc.edu/).
5‘ rapid amplification of cDNA ends (5‘ RACE)
Questa procedura, il cui protocollo è fornito dal kit commerciale della Invitrogen,
permette la sintesi e l’amplificazione della regione 5’ terminale del cDNA mediante
PCR.
La sintesi del primo filamento di cDNA è stata effettuata partendo da 5μg di RNA
estratto dalla linea cellulare LNCaP con Trizol (Invitrogen) trattato con DNasi I,
utilizzando primers specifici (GSP1) [GSP1: 5’-ACGTAAAATGTCTTCTGAC-3’]. La
reazione è catalizzata da una trascrittasi inversa fornita dal kit (SuperScript TM II RT)
in presenza di PCR buffer, MgCl2, dNTPs e DTT. Dopo la sintesi è stato ottenuto un
doppio filamento ibrido formato da mRNA-cDNA. L’emifilamento di mRNA è stato
degradato attraverso l’utilizzo di una RNAsi mix. A questo punto il cDNA a singolo
filamento è stato purificato mediante l’utilizzo di una colonna S.N.A.P., fornita dal kit. I
componenti del buffer, i dNTPs non utilizzati, gli enzimi ed i primer rimasti in
soluzione sono stati così rimossi.

53
E’ stata quindi aggiunta all’estremità 3’ del cDNA, attraverso l’utilizzo della TdT
(Terminal deoxynucleotidil Transferase) e in presenza di dCTP, una coda
omopolimerica di poli(C) che permette di creare un sito di attacco per un primer di
ancoraggio (anchor primer) che sarà utilizzato nella successiva reazione di
amplificazione.
Il cDNA poliC è stato successivamente amplificato tramite una reazione di PCR
usando un primer senso GSP-2 [GSP2: 5’-AGGGTTTGGACCAGGATCT-3’] che appaia a
monte del primer GSP-1 ed un primer antisenso AAP (Abridged Anchor Primer),
fornito nel kit, complementare alla coda di poliC.
Il prodotto di PCR è stato controllato mediante elettroforesi su gel d’agarosio e la
dimensione della banda amplificata è stata valutata dal confronto con il marker 1Kb
DNA ladder plus (Invitrogen). Successivamente, è stata eseguita una nested PCR con
un primer senso GSP3 che appaia a monte del primer GSP2 [GSP3: 5’
CAAAATGCTATACACAGTGCCG-3’] ed un primer antisenso AUAP (Abridged Universal
Amplification Primer) che appaia con l’AAP.
Il prodotto di PCR è stato quindi clonato nel TA cloning vector pCR2.1 (Invitrogen) e
sequenziato.
Costrutti luc e saggi luciferasici
Il frammento di 1.7 Kb del promotore intronico del let-7c, contenente i siti RARE non
canonici, la TATA box e la sequenza CAAT, è stato amplificato tramite PCR
convenzionale utilizzando DNA genomico umano usando i seguenti primers:
FW: 5’-AAAAGAGCTCGCAGTTTGTGTCCCATTGAA-3’
RV: 5’-AAAACTCGAGGAGAATTGAAGCCTGCCTTG-3’.
Il frammento di 2 Kb del promotore del gene ospite è stato amplificato allo stesso
modo con i seguenti primers:
FW: 5’-AAAAAGCTAGCGACAGCCACAGT-3’
RV: 5’-AAAACTCGAGAACCAGCCAGTG-3’.
I prodotti amplificati sono stati separati mediante elettroforesi su gel d’agarosio “low
melting” (SeaPlaque GTG). Le banda corrispondenti ai frammenti d’interesse sono

54
state isolate e purificate utilizzando il QIAquick Gel Extraction Kit (Qiagen) e clonate
nel vettore pGL3 Basic (Promega) a monte del gene della luciferasi.
Per i saggi di attività luciferasica, la linea cellulare NB4 è stata trasfettata mediante
elettroporazione con Nucleofector 4D system (Lonza, Walkersville, MD,USA) con 0,4
µg dei costrutti luc contenenti il frammento di promotore intronico del let-7c (pGL3-
let-7c-intronic prom), il frammento di promotore del gene ospite (pGL3-LINC00478
prom), il vettore pGL3-Basic vuoto (Ctrl) e 10 ng di Renilla luciferasi pRL-TK. Il
vettore pRL-TK, che fornisce un’espressione costitutiva della luciferasi Renilla, è stato
co-trasfettato come controllo interno per correggere le differenze sia nell’efficienza di
trasfezione che di raccolta delle cellule. Dopo 16 ore dall’elettroporazione è stato fatto
un cambio di terreno e le cellule sono state trattate con ATRA 1µM. 24 ore dopo il
trattamento sono state raccolte.
Le linee cellulari H1299 e LNCaP sono state trasfettate transientemente con
Lipofectamine® 2000 (Invitrogen) in dish da 60 mm, usando 2 µg dei vettori pGL3-
let-7c-intronic prom, pGL3-LINC00478 prom, il vettore pGL3 Basic vuoto e 10 ng di
Renilla luciferasi pRL-TK. Le cellule sono state raccolte 24 ore dalla trasfezione.
L’attività luciferasica è stata misurata e normalizzata con l’attività della Renilla,
usando il kit Dual Luciferase assay (Promega). L’attività luciferasica normalizzata del
controllo (pGL3 Basic) è stata posta uguale a 1. Per ogni esperimento, i punti sono
stati fatti in triplicato.
Trasfezioni transienti
La linea cellulare NB4 è stata trasfettata mediante elettroporazione con Nucleofector
4D system (Lonza, Walkersville, MD,USA). Per ogni campione, 2-4 x 106
cellule/cuvetta sono state centrifugate a 200 g a temperatura ambiente (RT) per 10’. I
pellet sono stati risospesi in 100 µL di buffer di elettroporazione SF + Supplement 1
(Lonza) cui sono stati aggiunti 0,4 µg dei costrutti luc, 10 ng di Renilla luciferasi pRL-
TK, usata come normalizzatore interno, e trasferiti in cuvetta. Il programma di
elettroporazione usato è stato il EH-100. Dopo l’elettroporazione, le cellule sono state
incubate per 10’ a RT, poi piastrate alla densità di 1 milione cellule/mL in terreno
senza antibiotici. Dopo 16 ore è stato fatto un cambio di terreno e le cellule sono state
piastrate in terreno completo alla densità di 0,1-0,2 milioni/mL. L’efficienza di

55
trasfezione è stata verificata elettroporando le cellule in cuvetta alle medesime
condizioni con 0,4 µg di plasmide pmaxGFP® (Lonza), e misurando la percentuale di
cellule GFP-positive a 24 ore dalla trasfezione tramite conta al microscopio. Nei vari
esperimenti, l’efficienza di trasfezione è stata del 59% ±5%.
Le linee cellulari H1299 e LNCaP sono state trasfettate transientemente con
Lipofectamine® 2000 (Invitrogen) in dish da 60 mm secondo il protocollo fornito
dalla ditta.
Immunoprecipitazione della cromatina (ChIP) L’immunoprecipitazione della cromatina (ChIP) è una tecnica che permette
l’individuazione, in sistemi cellulari, dei legami esistenti tra le proteine e specifiche
regioni del DNA. La tecnica si basa sul fatto che è possibile convertire in legami
covalenti le deboli interazioni tra i fattori trascrizionali ed il DNA. L'utilizzo della ChIP
prevede che le cellule siano inizialmente fissate con formaldeide all’1% che permette
di convertire in legami covalenti le deboli interazioni tra i fattori trascrizionali ed il
DNA genomico (crosslink del DNA). Il DNA legato covalentemente alle proteine viene
sottoposto a “sonicatura” e cioè ridotto, con l’utilizzo di ultrasuoni, a frammenti di
dimensioni comprese fra 500 e 2000 paia di basi. Dopo “sonicatura” il materiale
ottenuto è utilizzato in esperimenti di immunoprecipitazione con anticorpi specifici
per la proteina di interesse. La cromatina immunoprecipitata è poi sottoposta ad una
reazione di “reverse crosslinking” per scindere il legame tra il fattore proteico
selezionato nell’immunoprecipitazione e la sua sequenza bersaglio. Una successiva
reazione di PCR con sequenze di innesco specifiche sul DNA immunoprecipitato
consente poi di identificare le sequenze di DNA connesse con le proteine di interesse
(Gurtner et al, 2003).
a) “Crosslinking” con formaldeide
La formaldeide è stata aggiunta direttamente al terreno di coltura delle cellule ad una
concentrazione finale dell’1% per 10 minuti alla temperatura di 22°C. Per evitare che
la formazione di troppi legami mascheri i siti antigenici delle proteine stesse, la
reazione è stata bloccata aggiungendo glicina 0,125 M per 5 minuti a 22°C. Quindi le
cellule sono state raccolte, lavate con PBS 1X freddo, e centrifugate per ottenere i
pellet cellulari.
b) Preparazione dei nuclei

56
Il pellet cellulare è stato risospeso in 5 volumi di tampone di lisi (PIPES 5 mM pH 8;
KCl 85 mM; NP40 0,5%), e lasciato in ghiaccio per 30 minuti. I nuclei sono stati quindi
separati meccanicamente con l’apposito pestello “Dounce” (Wheaton Instruments) e
recuperati centrifugando per 10 minuti a 2000 rpm a 4°C. Il pellet nucleare è stato
risospeso in 500-1000 μl di tampone di “sonicatura” (SDS 0,1%; EDTA 10 mM; Tris-
HCl 50 mM pH 8).
c) Frammentazione della cromatina
La cromatina contenuta nei nuclei isolati è stata frammentata con il sonicatore
(Bandelin Sonopuls HD 2070) in ghiaccio per 1 minuto alla massima potenza. I
parametri di “sonicatura” (tempo e potenza) variano da campione a campione. Le
dimensioni dei frammenti di DNA ottenuti sono stati controllati caricando 5 μl della
soluzione di cromatina su un gel d’agarosio allo 0,8%. Quindi i detriti cellulari sono
stati rimossi centrifugando a 14000 rpm per 10 minuti a 4°C. Il supernatante è stato
successivamnte diluito 10 volte nel tampone di diluizione (SDS 0,01%; TritonX-100
1,1%; EDTA 1,2 mM; Tris-HCl 16,7 mM pH 8,1; NaCl 16,7 mM).
d)“Pre-clearing” della cromatina
La cromatina “sonicata” è stata incubata per 1 ora in agitazione continua a 4 °C con 20
μl di proteina G coniugata alle biglie di agarosio (Pierce) per eliminare la frazione di
DNA con sequenze altamente ripetute che possono legarsi aspecificamente alle resine
usate nell’immunoprecipitazione. Successivamente i campioni sono stati sottoposti a
breve centrifugazione a 14000 rpm e il sovranatante recuperato.
e) Immunoprecipitazione della cromatina
Per la successiva immunoprecipitazione il sovranatante è stato diviso in aliquote
uguali da 60 μg di DNA ed immunoprecipitato, a 4 °C in agitazione per una notte, con i
seguenti anticorpi specifici: anti-PML (PG-M3 and H-238), anti-RXR (∆N 197), anti-
p300 (N-15) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); anti-HDAC1 (3284)
(Sigma-Aldrich, St Louis, MO, USA) anti-H3K9me3 (ab8898) (Abcam, Cambridge, UK);
anti-H3K4me2 (07-030), anti-H3K4me3 (07-473), anti-H3K9ac (07-352), anti-
H3K14ac (07-353), anti-H3K27ac (07-360) (Millipore, Billerica, MA, USA). In un
campione (il No Ab) non è stato aggiunto nessun anticorpo.
f) Raccolta degli immunocomplessi
Per la raccolta degli immunocomplessi sono stati aggiunti 30 μl di proteina G saturata
(vedi avanti Saturazione della proteina G) agli immunocomplessi e incubati per 2 ore
a 4°C. In questo modo gli immunocomplessi che si sono formati tra gli anticorpi e le

57
loro proteine bersaglio, eventualmente legate al DNA, si legano attraverso il loro
frammento cristallizzabile (FC) alla proteina G e possono in tal modo essere raccolti
centrifugando a 14000 rpm. Dopo centrifugazione, il supernatante che conterrà la
cromatina non immunoprecipitata è stato rimosso, e gli immunocomplessi legati alle
biglie sono stati sottoposti a 10 lavaggi. E’ stato recuperato solamente il supernatante
dei campioni No Ab, che viene utilizzato come “input”, che sarà trattato dal “reverse
crosslinking” in poi. L’“input”, amplificato attraverso la PCR, fornisce infatti sia una
misura della quantità di cromatina che era contenuta nei diversi campioni al
momento dell’immunoprecipitazione, sia informazioni sulla specificità
dell’amplificazione di una sequenza bersaglio ottenuta dalla PCR.
g) Saturazione della proteina G
Contemporaneamente all’immunoprecipitazione della cromatina (punto e), alle
stesse condizioni di tempo e temperatura, la quantità di proteina G necessaria per la
raccolta degli immunocomplessi (30 μl per campione) è stata saturata, incubandola
con 1 mg/ml di DNA di salmon-sperma “sonicato” e 1 mg/ml di albumina sierica
bovina (BSA), in un volume finale di 60 μl a campione raggiunto aggiungendo
tampone di immunoprecipitazione (SDS 0,1 %, EDTA 10mM, Tris HCl 50 mM pH 8,
desossicolato 0,5%, LiCl 150 mM). Lo scopo di questo passaggio è quello di
neutralizzare i siti della proteina G e dell’agarosio coniugato cui si può legare in
maniera aspecifica il DNA da immunoprecipitare o la proteina anticorpale dando
risultati falsamente positivi.
h) Lavaggi degli immunocomplessi ed eluizione
Gli immunocomplessi sono stati lavati 4 volte con un apposito tampone di lavaggio a
bassa salinità (SDS 0,1 %,Triton X-100 1%, EDTA 2 mM, Tris-HCl 20 mM pH 8.1, NaCl
150mM), 4 volte con un tampone di lavaggio ad alta salinità (SDS 0,1%, Triton X-100
1%, EDTA 2 mM, Tris-HCl 20 mM pH 8.1, NaCl 500 mM) e 2 volte con TE 1X (Tris-HCl
10 mM, EDTA 1 mM pH 8.0). Ciascun lavaggio è stato fatto in agitazione continua a
4°C per 5 minuti seguito da centrifugazione alla velocità di 3000 rpm per 3 minuti a
4°C. Gli immunocomplessi sono stati poi eluiti dalla resina con 200 μl di tampone di
eluizione (SDS 1%; NaHCO3 0,1 M) per 2 volte a temperatura ambiente in agitazione
continua per 10 minuti e centrifugati brevemente a 14000 rpm per recuperare il
supernatante.
i)“Reverse crosslinking”

58
Infine, aggiungendo NaCl 200 mM per 5 ore a 65°C all’eluato, abbiamo ottenuto il
distacco dell’anticorpo e della proteina bersaglio dal DNA (“reverse crosslinking”). I
residui proteici sono stati successivamente eliminati con un trattamento con
proteinasi K (Roche Diagnostics, Mannheim, Germania), aggiungendo 10 μl di EDTA
0,5 M, 20 μl di Tris-HCl 1 M pH 6.8, 2 μl di proteinasi K 10 mg/ml, per 2 ore a 45°C.
l) Estrazione del DNA
La purificazione del DNA è stata ottenuta aggiungendo ad ogni campione 1 volume di
fenolo:cloroformio:alcol isoamilico 25:24:1, centrifugando a temperatura ambiente
per 10 minuti a 14000 rpm e recuperando la fase superiore contenente gli acidi
nucleici. Il DNA è stato quindi precipitato a -20°C per tutta la notte aggiungendo
nell’ordine: 1/10 di NaAc 3 M pH 7, 1 μl di glicogeno (che permetterà di visualizzare
più facilmente il pellet di DNA) e 2,5 volumi di etanolo al 100%. Dopo centrifugazione,
il DNA è stato lavato in etanolo al 70% e risospeso in 30 μl di acqua bidistillata
autoclavata (gli input in 100 μl, poiché molto concentrati).
m) Quantificazione dell’immunoprecipitato mediante PCR
Il risultato della ChIP è stato analizzato eseguendo una Real Time-PCR con il metodo
Sybr Green® sul DNA ottenuto in ogni immunoprecipitazione specifica con primers
complementari ai promotori dei geni esaminati. L’amplificazione del DNA avviene
solo se la proteina, bersaglio dell’anticorpo utilizzato, era legata alla sequenza di DNA
amplificata al momento dell’immunoprecipitazione.
Le sequenze dei primers utilizzati sono le seguenti:
C21 RARE FW: 5’ –CAGCTTCGCGGTAATT– 3’
C21 RARE RV: 5’ –TCCTCCTAGACCCTGACAGCAG– 3’
ER4 FW: 5’ –CCAATTCCTTGAAACCAGTAACTAAAC– 3’
ER4 RV: 5’ –GCCATGAATGTAGTATTACGAGCCTAT– 3’
DR3 FW: 5’ –CCTGATAAGACAATGAAACCAAAACA– 3’
DR3 RV: 5’ –CAAATGTGAAATAAGAGGCAGCAT– 3’
#1 FW: 5’ –GGTTCTTTTTAGTGCCGCTCAGT– 3’
#1 RV: 5’ –CACAATTTCCTTAAGCAGAATTTAAAGC– 3’
#2 FW: 5’ – CTTGTACCTCCATGCTGCCTC– 3’
#2 RV: 5’– AAGCCAGTGAAAGTTGGTGTACC– 3’
#3 FW: 5’– CGCTCAGTATGTTTGGTTTCTGTAC– 3’
#3 RV: 5’- AGCCAAACAGGAAAGCAAGC– 3’

59
Analisi statistica
Per ogni esperimento, i risultati sono espressi come media aritmetica di 3
esperimenti indipendenti, ± l’errore standard della media (Standard Error of the
Mean, S.E.M.). Le analisi statistiche sono state condotte usando il test t di Student.
L’analisi di correlazione è stat eseguita usando il coefficiente di correlazione di
Pearson.

60
RISULTATI
Identificazione di un nuovo TSS per il miRNA let-7c
Allo scopo di verificare l’esistenza di un nuovo promotore in grado di regolare il let-7c
indipendentemente dal suo gene ospite, abbiamo eseguito un’analisi in silico della
regione genomica che si estende per 10 kb a monte della sequenza del pre-miRNA
usando gli algoritmi: ProScan v.1.7, BDGP and F/PROM. Questi strumenti
bioinformatici hanno predetto un singolo putativo TSS, non ancora descritto,
localizzato circa 700 bp a monte del pre-let-7c, all’interno del sesto introne del gene
ospite del let-7c, LINC00478 (Figura 5a). L’analisi bioinformatica ha inoltre rivelato
che questa regione presenta elementi regolatori tipici di promotori associati alla RNA
Polimerasi II e siti di legame per fattori di trascrizione altamente conservati, come
TATA box e CAAT box. Per validare sperimentalmente questo putativo TSS, abbiamo
utilizzato la tecnica della “5’ Rapid Amplification of cDNA Ends” (RACE).
A causa della natura labile dei pri-miRNA dovuta al loro rapido processamento, non
siamo stati in grado di ottenere un prodotto di 5’ RACE dalla linea cellulare di LAP
NB4, nella quale l’espressione del let-7c è molto bassa. Per questo motivo, abbiamo
analizzato i livelli di espressione del let-7c in una serie di linee cellulari sia di tumore
solido che leucemiche. I nostri dati mostrano che fra le lineee cellulari analizzate le
LNCaP, un carcinoma della prostata, presentano i maggiori livelli di espressione di let-
7c (Figura 5b). Mediante analisi di 5’ RACE eseguita in questa linea cellulare,
abbiamo ottenuto un singolo prodotto di amplificazione il cui sequenziamento ha
permesso di localizzare esattamente il TSS: 668bp a monte del pre-let-7c, molto
vicino al TSS identificato in silico (Figura 5c).

Figura 5. Identificazione di un nuovo TSS intronico del let
a) Rappresentazione schematica del
putativo TSS (freccia azzurra)
Le distanze indicate sono relative
primers GSP-1 e GSP-2 usati nella 5’ RACE
indicate. Le barre di errore indicano la S.E.M. (n=3);
amplificazione del cDNA ottenuto
prodotto di amplificazione del c
dei primers, linea 3: controllo
Analisi dell’attività trascrizionale del promotore intronico del let
in cellule di leucemia promielo
Per valutare se il nuovo promotore del let
intrinseca, abbiamo clonato, da DNA genomico umano di placenta, un frammento di
1700bp a monte del nuovo TSS intronico del let
vettore “reporter” con attività
abbiamo clonato in un vettore luciferasico anche un frammento di 2000bp a monte
del 5’UTR del gene ospite
Abbiamo quindi eseguito saggi di tr
trasfettate transientemente con il costrutto
pGL3-LINC00478 prom
6a, l’attività trascrizionale
Identificazione di un nuovo TSS intronico del let-7c.
Rappresentazione schematica della regione a monte del pre-let-7c. In figura sono mostrati il
(freccia azzurra) identificato, la TATA box (bottone verde) e la CAAT box
Le distanze indicate sono relative al pre-let-7c. Le frecce (nera e gialla) indicano le posizioni dei
2 usati nella 5’ RACE; b) Livello di espressione (2^-ΔCt)
Le barre di errore indicano la S.E.M. (n=3); c) 5’-RACE su LNCaP
ottenuto usando i primer GSP-2 e Abridged Anchor Primer
prodotto di amplificazione del cDNA senza la coda di poli- (-dC) usato come controllo per la specificità
controllo negativo senza templato, linea M: marker 1kb.
Analisi dell’attività trascrizionale del promotore intronico del let
in cellule di leucemia promielocitica acuta
Per valutare se il nuovo promotore del let-7c possedesse un’attività trascrizionale
intrinseca, abbiamo clonato, da DNA genomico umano di placenta, un frammento di
1700bp a monte del nuovo TSS intronico del let-7c (vedi Materiali e Metodi
vettore “reporter” con attività luciferasica (pGL3-let-7c-intronic prom
abbiamo clonato in un vettore luciferasico anche un frammento di 2000bp a monte
del 5’UTR del gene ospite (pGL3-LINC00478 prom).
Abbiamo quindi eseguito saggi di transattivazione in cellule umane di LAP NB4
trasfettate transientemente con il costrutto pGL3-let-7c-intronic prom
LINC00478 prom e con il vettore reporter vuoto (Ctrl). Come mostrato in Figura
trascrizionale basale del vettore pGL3-let-7c-intronic prom
61
. In figura sono mostrati il
e la CAAT box (ottagono blu).
gialla) indicano le posizioni dei
ΔCt) del let-7c nelle linee
LNCaP. Linea 1: prodotto di
Abridged Anchor Primer (AAP), linea 2:
dC) usato come controllo per la specificità
Analisi dell’attività trascrizionale del promotore intronico del let-7c
7c possedesse un’attività trascrizionale
intrinseca, abbiamo clonato, da DNA genomico umano di placenta, un frammento di
Materiali e Metodi) in un
intronic prom). Analogamente,
abbiamo clonato in un vettore luciferasico anche un frammento di 2000bp a monte
ansattivazione in cellule umane di LAP NB4
intronic prom, il costrutto
Come mostrato in Figura
intronic prom è maggiore

rispetto al controllo rappresentato dal vettore vuoto.
LINC00478 prom ha un’attività luciferasica
intronico (Figura 6a). Dopo trattamento con ATR
del gene ospite è incrementata. Questi dati suggeriscono che l’aumento dei livelli
d’espressione del pri-
principalmente dovuto all’attività del promotore del gene
Figura 6. Attività trascrizionale del promotore intronico del let
ospite LINC00478 in linee cellulari leucemiche.
a) Saggi di attività luciferasica sulla
luciferasici contenenti il promotore intronico del let
gene ospite LINC00478 (pGL3
trattate e dopo 24 ore di trattamento con ATR
trasfezione con l’attività del vettore
(n=3); b) Analisi dell’espressione
in cellule NB4 non trattate e dopo
per l’espressione di GAPDH ed espressi come
errore indicano la S.E.M. (n=3)
Modificazioni epigenetiche del promotore del gene ospite del let
LINC00478 dopo trattamento con ATRA
Per studiare il contributo trascrizionale dei due promotori del let
gene ospite, in cellule di LAP, abbiamo studiato le modificazi
regioni in cellule NB4 prima e dopo trattamento con ATRA.
rispetto al controllo rappresentato dal vettore vuoto. Tuttavia, il
un’attività luciferasica ~2 volte più elevata rispetto al promotore
). Dopo trattamento con ATRA soltanto l’attività del promotore
del gene ospite è incrementata. Questi dati suggeriscono che l’aumento dei livelli
-let-7c osservati in cellule NB4 dopo ATRA (
principalmente dovuto all’attività del promotore del gene ospite.
Attività trascrizionale del promotore intronico del let-7c e del promotore del gene
in linee cellulari leucemiche.
Saggi di attività luciferasica sulla linea cellulare NB4 trasfettata transientemente con
luciferasici contenenti il promotore intronico del let-7c (pGL3-let-7c intronic prom
pGL3-LINC00478 prom). L’attività luciferasica è stata
trattate e dopo 24 ore di trattamento con ATRA 1µM. I valori sono stati normalizzati per l’efficienza di
trasfezione con l’attività del vettore co-trasfettato renilla pRLTK. Le barre di errore indicano la S.E.M.
Analisi dell’espressione, misurata tramite qRT-PCR, di pri-let-7c e del gene os
non trattate e dopo 24 ore di trattamento con ATRA 1µM. I valori sono stati normalizzati
per l’espressione di GAPDH ed espressi come fold change rispetto al campione non trattato
errore indicano la S.E.M. (n=3).
Modificazioni epigenetiche del promotore del gene ospite del let
LINC00478 dopo trattamento con ATRA
Per studiare il contributo trascrizionale dei due promotori del let
gene ospite, in cellule di LAP, abbiamo studiato le modificazioni epigenetiche su tali
regioni in cellule NB4 prima e dopo trattamento con ATRA. L’analisi con il software
62
Tuttavia, il vettore pGL3-
~2 volte più elevata rispetto al promotore
A soltanto l’attività del promotore
del gene ospite è incrementata. Questi dati suggeriscono che l’aumento dei livelli
7c osservati in cellule NB4 dopo ATRA (Figura 6b) sia
e del promotore del gene
transientemente con i costrutti
7c intronic prom) ed il promotore del
sica è stata misurata in cellule non
I valori sono stati normalizzati per l’efficienza di
Le barre di errore indicano la S.E.M.
e del gene ospite LINC00478
I valori sono stati normalizzati
campione non trattato. Le barre di
Modificazioni epigenetiche del promotore del gene ospite del let-7c
Per studiare il contributo trascrizionale dei due promotori del let-7c, intronico e del
oni epigenetiche su tali
L’analisi con il software

63
CpG Island Searcher (vedi Materiali e Metodi) ha rivelato che sia sul promotore del
gene ospite che sul promotore intronico del let-7c non sono presenti isole CpG. Data
l’assenza di isole CpG su entrambi i promotori abbiamo escluso un coinvolgimento
delle DNMT, quindi della metilazione del DNA, per la regolazione trascrizionale del
let-7c.
Precedentemente, avevamo dimostrato che PML/RARα legava il sito RARE presente
sul promotore del gene ospite del let-7c in maniera ATRA-sensibile, e che il suo
rilascio era accompagnato da un incremento dell’acetilazione dell’istone H3 (Careccia
et al, 2009). In questo lavoro di tesi noi confermiamo questi dati e mostriamo una co-
localizzazione di RXR con PML, il cui legame, così come per PML, è ridotto dopo
trattamento con ATRA (Figura 7b). Inoltre, osserviamo la rimozione del legame
dell’istone deacetilasi 1 (HDAC1) dal promotore del gene ospite LINC00478, ed un
incremento dell’istone acetil trasferasi p300. In accordo con questi risultati, l’analisi
di altri marcatori epigenetici della conformazione della cromatina mostra un notevole
incremento dell’istone H3 acetilato in lisina 9, lisina 14 e lisina 27 (H3K9ac, H3K14ac
e H3K27ac), ed una diminuzione del tri-metilato in lisina 9 (H3K9me3). In cellule non
trattate, inoltre, ridotti livelli di marcatori di promotori attivi, come il di- e tri-
metilato in lisina 4 (H3K4me2, H3K4me3), confermano, a livello basale, uno stato di
repressione trascrizionale di questo promotore. Al contrario trattamenti con ATRA
determinano un aumento di H3K4me2, ed una riduzione di H3K4me3 (Figura 7b).
Questi dati indicano, quindi, che dopo trattamento con ATRA il promotore del gene ospite
LINC00478 presenta uno stato della cromatina trascrizionalmente più permissivo.
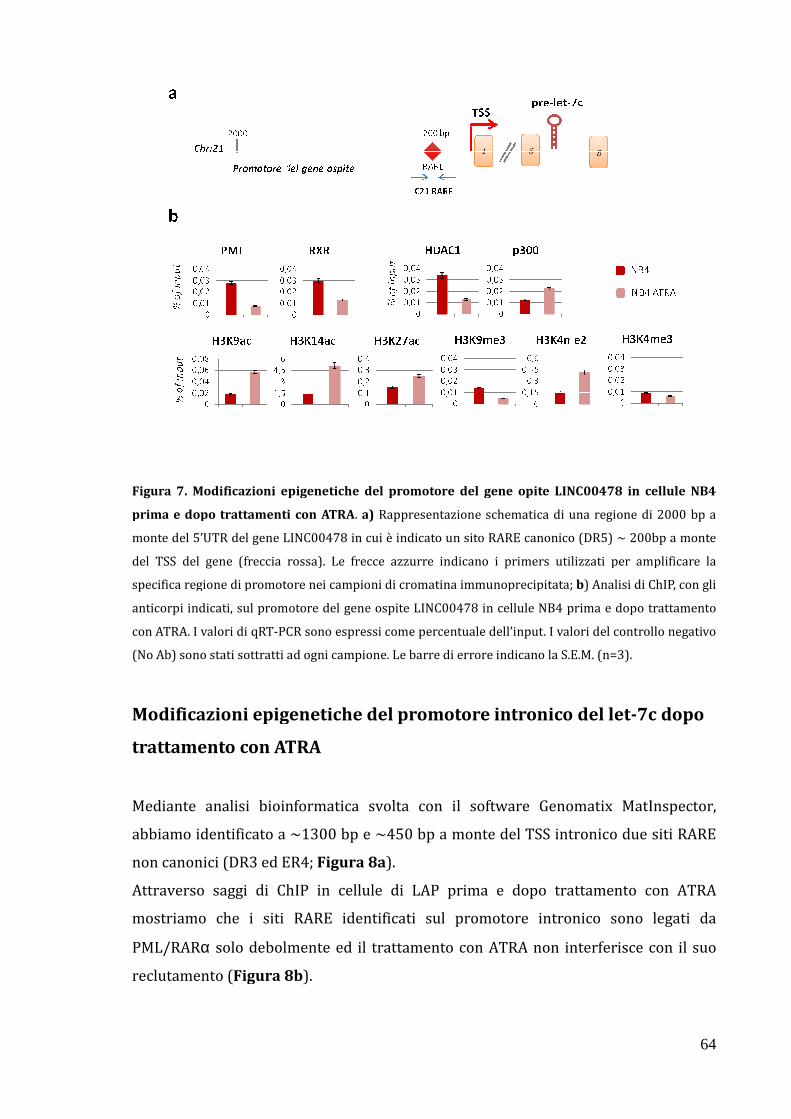
Figura 7. Modificazioni epigenetiche
prima e dopo trattamenti con ATRA
monte del 5’UTR del gene LINC00478
del TSS del gene (freccia rossa)
specifica regione di promotore
anticorpi indicati, sul promoto
con ATRA. I valori di qRT-PCR
(No Ab) sono stati sottratti
Modificazioni epigenetiche del promotore intronico del let
trattamento con ATRA
Mediante analisi bioinformatica svolta con il software Genomatix
abbiamo identificato a ~1300 bp e ~450 bp a monte del TSS intronico due siti RARE
non canonici (DR3 ed ER4;
Attraverso saggi di ChIP in cellule di LAP prima e dopo trattamento con ATRA
mostriamo che i siti RARE identificati sul prom
PML/RARα solo debolmente ed il trattamento con ATRA
reclutamento (Figura 8b
Modificazioni epigenetiche del promotore del gene opite LINC00478 in cellule NB4
prima e dopo trattamenti con ATRA. a) Rappresentazione schematica di una
LINC00478 in cui è indicato un sito RARE canonico
(freccia rossa). Le frecce azzurre indicano i primers utilizzati per amplificare
regione di promotore nei campioni di cromatina immunoprecipitata;
promotore del gene ospite LINC00478 in cellule NB4 prima e dopo trattamento
PCR sono espressi come percentuale dell’input. I valori del
ad ogni campione. Le barre di errore indicano la S.E.M. (n=3)
Modificazioni epigenetiche del promotore intronico del let
trattamento con ATRA
analisi bioinformatica svolta con il software Genomatix
abbiamo identificato a ~1300 bp e ~450 bp a monte del TSS intronico due siti RARE
non canonici (DR3 ed ER4; Figura 8a).
Attraverso saggi di ChIP in cellule di LAP prima e dopo trattamento con ATRA
mostriamo che i siti RARE identificati sul promotore intronico sono legati da
solo debolmente ed il trattamento con ATRA non interferisce con
Figura 8b).
64
l promotore del gene opite LINC00478 in cellule NB4
di una regione di 2000 bp a
in cui è indicato un sito RARE canonico (DR5) ~ 200bp a monte
ers utilizzati per amplificare la
romatina immunoprecipitata; b) Analisi di ChIP, con gli
prima e dopo trattamento
valori del controllo negativo
Le barre di errore indicano la S.E.M. (n=3).
Modificazioni epigenetiche del promotore intronico del let-7c dopo
analisi bioinformatica svolta con il software Genomatix MatInspector,
abbiamo identificato a ~1300 bp e ~450 bp a monte del TSS intronico due siti RARE
Attraverso saggi di ChIP in cellule di LAP prima e dopo trattamento con ATRA
otore intronico sono legati da
non interferisce con il suo

Inoltre, dopo ATRA non si hanno modulazioni di H3K9ac, H3K14ac, H3K9me3,
H3K4me2 e H3K4me3
Figura 8. Modificazioni epigenetiche del promotore intronico del let
dopo trattamenti con ATRA. a)
cui sono indicati il nuovo TSS identificato attraverso
canonici (DR3 and ER4) presenti a monte del TSS
amplificate mediante qRT-PCR
promotore intronico del let
cromatina è stata immunoprecipitata con gli anticorpi indicati
percentuale dell’input. I valori del
barre di errore indicano la S.E.M. (n=3)
Per verificare se altre putative sequenze regolatorie di let
(Oszolak et al, 2008; Corcoran et al, 2009) potessero rispondere epigeneticamente
all’ATRA, abbiamo analizzato queste regioni per il legame di PML/RAR
significative modificazioni istoniche.
I nostri dati mostrano uno scenario epigenetico molto simile a quello del nostro
promotore intronico. Infatti, PML/RAR
trattamenti con ATRA non determinano modulazioni di H3K14ac e H3K4me3
9b-d).
Inoltre, dopo ATRA non si hanno modulazioni di H3K9ac, H3K14ac, H3K9me3,
(Figura 8b).
Modificazioni epigenetiche del promotore intronico del let-7c
dopo trattamenti con ATRA. a) Rappresentazione schematica della regione a monte del
cui sono indicati il nuovo TSS identificato attraverso 5’RACE (freccia nera)
presenti a monte del TSS. Le frecce azzurre indicano la posizione delle regioni
PCR nei campioni di cromatina immunoprecipitata
let-7c in cellule NB4 prima e dopo trattamento con ATRA 1 µM per 72h. La
cromatina è stata immunoprecipitata con gli anticorpi indicati. I valori di qRT
valori del controllo negativo (No Ab) sono stati sottratt
barre di errore indicano la S.E.M. (n=3).
Per verificare se altre putative sequenze regolatorie di let-7c, indicate da altri gruppi
(Oszolak et al, 2008; Corcoran et al, 2009) potessero rispondere epigeneticamente
alizzato queste regioni per il legame di PML/RAR
significative modificazioni istoniche.
I nostri dati mostrano uno scenario epigenetico molto simile a quello del nostro
promotore intronico. Infatti, PML/RARα e HDAC1 non legano queste regioni, e
trattamenti con ATRA non determinano modulazioni di H3K14ac e H3K4me3
65
Inoltre, dopo ATRA non si hanno modulazioni di H3K9ac, H3K14ac, H3K9me3,
7c in cellule NB4 prima e
a della regione a monte del pre-let-7c in
era)e i due siti RARE non
e indicano la posizione delle regioni
nei campioni di cromatina immunoprecipitata; b) Analisi di ChIP sul
in cellule NB4 prima e dopo trattamento con ATRA 1 µM per 72h. La
I valori di qRT-PCR sono espressi come
sottratti ad ogni campione. Le
7c, indicate da altri gruppi
(Oszolak et al, 2008; Corcoran et al, 2009) potessero rispondere epigeneticamente
alizzato queste regioni per il legame di PML/RARα ed alcune
I nostri dati mostrano uno scenario epigenetico molto simile a quello del nostro
e HDAC1 non legano queste regioni, e
trattamenti con ATRA non determinano modulazioni di H3K14ac e H3K4me3 (Figura
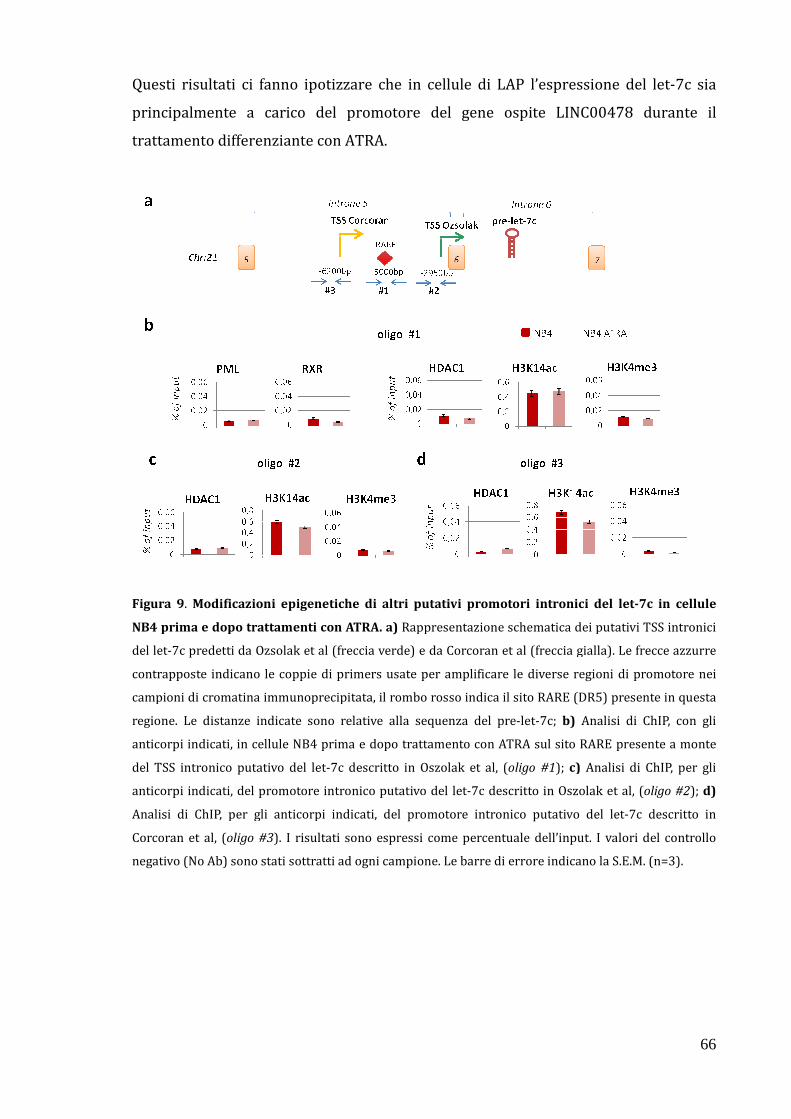
Questi risultati ci fanno ipotizzare che in cellule di LAP l’espressione del let
principalmente a carico del promotore del gene ospite LINC00478 durante il
trattamento differenziante con ATRA.
Figura 9. Modificazioni epigenetiche di altri putativ
NB4 prima e dopo trattamenti con ATRA
del let-7c predetti da Ozsol
contrapposte indicano le coppie di primers usate per
campioni di cromatina immunoprecipitata
regione. Le distanze indicate sono relative alla sequenza del pre
anticorpi indicati, in cellule NB4 prima e dopo trattamento con ATRA
del TSS intronico putativo
anticorpi indicati, del promotore intronico
Analisi di ChIP, per gli anticorpi indicati,
Corcoran et al, (oligo #3). I risultati sono espressi come
negativo (No Ab) sono stati
Questi risultati ci fanno ipotizzare che in cellule di LAP l’espressione del let
principalmente a carico del promotore del gene ospite LINC00478 durante il
ento differenziante con ATRA.
Modificazioni epigenetiche di altri putativi promotori intronic
NB4 prima e dopo trattamenti con ATRA. a) Rappresentazione schematica dei putativi TSS
Ozsolak et al (freccia verde) e da Corcoran et al (freccia gialla
contrapposte indicano le coppie di primers usate per amplificare le diverse
campioni di cromatina immunoprecipitata, il rombo rosso indica il sito RARE
regione. Le distanze indicate sono relative alla sequenza del pre-let-7c; b)
in cellule NB4 prima e dopo trattamento con ATRA sul sito RARE presente a monte
del let-7c descritto in Oszolak et al, (oligo #1); c)
del promotore intronico putativo del let-7c descritto in Oszolak et al,
per gli anticorpi indicati, del promotore intronico putativ
). I risultati sono espressi come percentuale dell’input.
sottratti ad ogni campione. Le barre di errore indicano la S.E.M. (n=3)
66
Questi risultati ci fanno ipotizzare che in cellule di LAP l’espressione del let-7c sia
principalmente a carico del promotore del gene ospite LINC00478 durante il
intronici del let-7c in cellule
Rappresentazione schematica dei putativi TSS intronici
freccia gialla). Le frecce azzurre
diverse regioni di promotore nei
(DR5) presente in questa
b) Analisi di ChIP, con gli
sul sito RARE presente a monte
c) Analisi di ChIP, per gli
in Oszolak et al, (oligo #2); d)
utativo del let-7c descritto in
percentuale dell’input. I valori del controllo
Le barre di errore indicano la S.E.M. (n=3).

Attività trascrizionale del promotore intronico del let
solidi
Per valutare il potenziale coinvolgimento del promotore del gene ospite e/o di quello
intronico nel guidare
abbiamo trasfettato i costrutti luciferasici con la regione del promotore intronico o
del gene ospite in una linea cellulare di adenocarcinoma di prostata (LNCaP) o in una
di adenocarcinoma polmonare (H1299). I saggi di trans attivazione mostrano che
nelle linee cellulari di t
(Figura 10). Inoltre, è da notare che, differentemente dalle cellule leucemiche,
l’attività del promotore intronico è più elevata di quella del gene ospite. Ciò
suggerisce un potenziale ruolo della r
trascrizione del let-7c preferenzialmente in questi contesti cellulari.
Figura 10. Attività trascrizionale dei promotori intronico e d el gene ospite in linee cellulari
solido. Saggi di attività luci
di prostata LNCaP trasfettate transientemente con
intronico del let-7c ed il promotore del gene ospite LINC00478
24h dopo la trasfezione. I valori sono stati normalizzati per l’efficienza di trasfezione
vettore co-trasfettato renilla
Correlazione dell’espressione del gene osp
in diversi tipi di tumore
Per approfondire il contributo relativo dei due promotori in diversi istotipi tumorali,
abbiamo valutato la correlazione tra l’espressione del gene ospite LINC00478 e del
ascrizionale del promotore intronico del let
Per valutare il potenziale coinvolgimento del promotore del gene ospite e/o di quello
l’espressione del let-7c in contesti tumorali non leucemici,
costrutti luciferasici con la regione del promotore intronico o
del gene ospite in una linea cellulare di adenocarcinoma di prostata (LNCaP) o in una
di adenocarcinoma polmonare (H1299). I saggi di trans attivazione mostrano che
nelle linee cellulari di tumore solido analizzate entrambi i promotori sono attivi
). Inoltre, è da notare che, differentemente dalle cellule leucemiche,
l’attività del promotore intronico è più elevata di quella del gene ospite. Ciò
suggerisce un potenziale ruolo della regione regolatoria intronica nel guidare la
7c preferenzialmente in questi contesti cellulari.
Attività trascrizionale dei promotori intronico e d el gene ospite in linee cellulari
Saggi di attività luciferasica sulle linee di adenocarcinoma polmonare H1299
trasfettate transientemente con i costrutti luciferasici contenenti il promotore
7c ed il promotore del gene ospite LINC00478. L’attività lucifera
I valori sono stati normalizzati per l’efficienza di trasfezione
renilla pRLTK. Le barre di errore indicano la S.E.M. (n=3)
Correlazione dell’espressione del gene ospite LINC00478 e del let
in diversi tipi di tumore
er approfondire il contributo relativo dei due promotori in diversi istotipi tumorali,
abbiamo valutato la correlazione tra l’espressione del gene ospite LINC00478 e del
67
ascrizionale del promotore intronico del let-7c in tumori
Per valutare il potenziale coinvolgimento del promotore del gene ospite e/o di quello
7c in contesti tumorali non leucemici,
costrutti luciferasici con la regione del promotore intronico o
del gene ospite in una linea cellulare di adenocarcinoma di prostata (LNCaP) o in una
di adenocarcinoma polmonare (H1299). I saggi di trans attivazione mostrano che
umore solido analizzate entrambi i promotori sono attivi
). Inoltre, è da notare che, differentemente dalle cellule leucemiche,
l’attività del promotore intronico è più elevata di quella del gene ospite. Ciò
egione regolatoria intronica nel guidare la
7c preferenzialmente in questi contesti cellulari.
Attività trascrizionale dei promotori intronico e d el gene ospite in linee cellulari di tumore
H1299 e adenocarcinoma
i costrutti luciferasici contenenti il promotore
luciferasica è stata misurata
I valori sono stati normalizzati per l’efficienza di trasfezione con l’attività del
Le barre di errore indicano la S.E.M. (n=3).
ite LINC00478 e del let-7c
er approfondire il contributo relativo dei due promotori in diversi istotipi tumorali,
abbiamo valutato la correlazione tra l’espressione del gene ospite LINC00478 e del
68
let-7c in linee cellulari di LAM di vari sottotipi FAB (Tabella 3 )ed in linee cellulari di
diversi tipi di tumore solido (Tabella 4).
Linea cellulare Sottotipo FAB KG1a M0
KG1 M1
HL60 M2
Kasumi-1 M2
SKNO-1 M2
NB4 M3
UF-1 M3
PL-21 M3
GDM-1 M4
OCI-AML3 M4
THP-1 M5
U937 pseudo M5
HEL M6
K562 CML
BV173 CML
Tabella 3. Linee cellulari AML usate e relativo sottotipo FAB
Linea cellulare Istotipo tumorale LNCaP Adenocarcinoma di prostata
DU-145 Carcinoma di prostata
PC-3 Carcinoma di prostata
SK-BR-3 Carcinoma mammario
MCF-7 Adenocarcinoma mammario
MDA-MB-231 Carcinoma mammario
A-549 Carcinoma polmonare
H1299 Carcinoma polmonare a cellule non piccole SH-SY5Y Neuroblastoma
A2780 Carcinoma ovarico
Hep-G2 Epatocarcinoma
HCT-116 Carcinoma colorettale
Tabella 4. Linee cellulari di tumore solido usate e relativo istotipo tumorale.
I nostri dati mostrano una significativa correlazione tra espressione del let-7c maturo
e quella del suo gene ospite in un contesto cellulare leucemico (Figura 11a). Al
contrario, nelle varie linee derivanti da tumori solidi non si osserva tale correlazione
(Figura 11b).
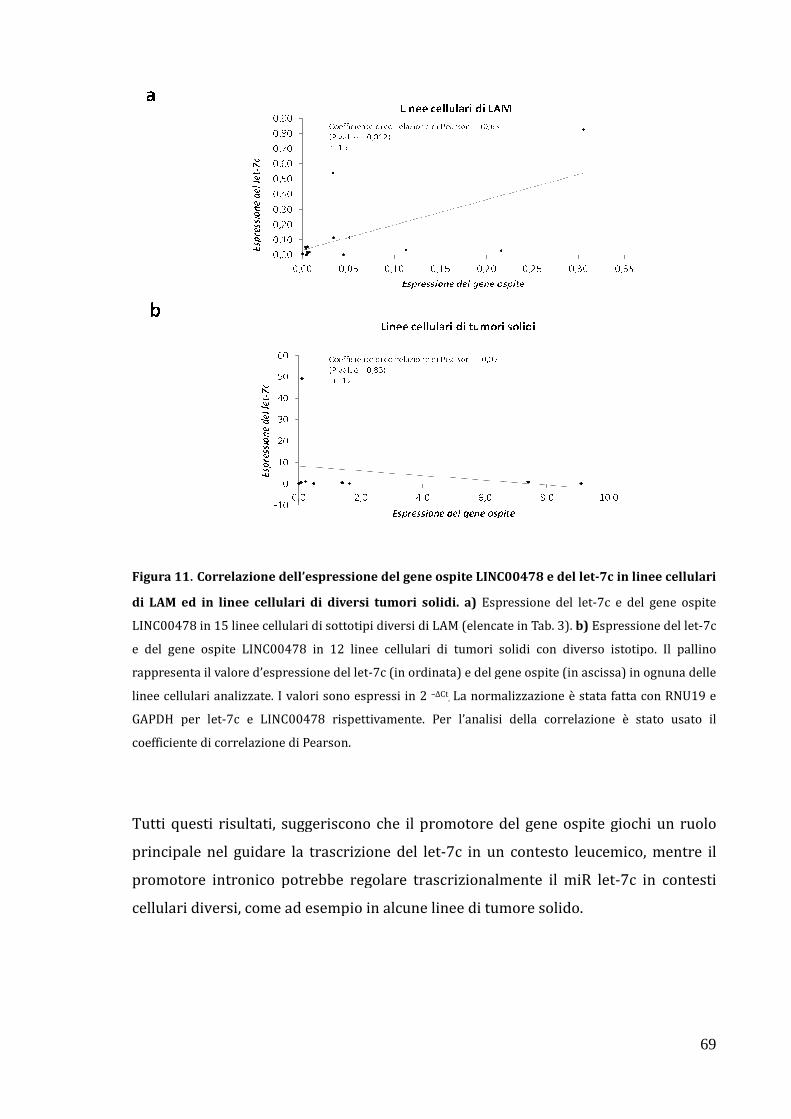
Figura 11. Correlazione dell’espressione del gene ospite LINC00478 e del let
di LAM ed in linee cellulari di diversi tumori solidi.
LINC00478 in 15 linee cellulari di
e del gene ospite LINC00478
rappresenta il valore d’espressione del let
linee cellulari analizzate. I valori
GAPDH per let-7c e LINC00478
coefficiente di correlazione di Pearson.
Tutti questi risultati, suggeriscono che il promotore del gene ospite giochi un ruolo
principale nel guidare la trascrizione del let
promotore intronico potrebbe regolare trascrizionalmente il miR let
cellulari diversi, come ad esempio in alcune linee di tumore solido.
Correlazione dell’espressione del gene ospite LINC00478 e del let
di LAM ed in linee cellulari di diversi tumori solidi. a) Espressione del
linee cellulari di sottotipi diversi di LAM (elencate in Tab. 3).
LINC00478 in 12 linee cellulari di tumori solidi con diverso istotipo
d’espressione del let-7c (in ordinata) e del gene ospite (in ascissa)
I valori sono espressi in 2 –ΔCt. La normalizzazione è stata fatta con
LINC00478 rispettivamente. Per l’analisi della correlazione è stato usato il
coefficiente di correlazione di Pearson.
Tutti questi risultati, suggeriscono che il promotore del gene ospite giochi un ruolo
principale nel guidare la trascrizione del let-7c in un contesto leucemico, mentre il
ore intronico potrebbe regolare trascrizionalmente il miR let
cellulari diversi, come ad esempio in alcune linee di tumore solido.
69
Correlazione dell’espressione del gene ospite LINC00478 e del let-7c in linee cellulari
l let-7c e del gene ospite
LAM (elencate in Tab. 3). b) Espressione del let-7c
diverso istotipo. Il pallino
gene ospite (in ascissa) in ognuna delle
La normalizzazione è stata fatta con RNU19 e
lla correlazione è stato usato il
Tutti questi risultati, suggeriscono che il promotore del gene ospite giochi un ruolo
7c in un contesto leucemico, mentre il
ore intronico potrebbe regolare trascrizionalmente il miR let-7c in contesti
cellulari diversi, come ad esempio in alcune linee di tumore solido.

70
Discussione e Conclusioni
Recenti evidenze suggeriscono che una significativa porzione del genoma umano è
regolata dai miRNA. Inoltre, alcuni studi hanno dimostrato che l’espressione dei
miRNA è spesso deregolata nelle malattie umane, incluse quelle che riguardano il
sistema ematopoietico (Ambros, 2004; Bartel, 2004; Alvarez-Garcia and Miska, 2005).
Comunque, sebbene l’importanza biologica dei miRNA ormai sia chiara, i meccanismi
della loro regolazione necessitano di ulteriori studi. Quindi, l’identificazione di
elementi genetici e meccanismi epigenetici responsabili della regolazione
trascrizionale dei miRNA potrebbe aiutarci a capire le vie del segnale alla base della
biologia dei miRNA.
Nel laboratorio in cui si è svolto questo lavoro di tesi, è stato dimostrato che il miRNA
let-7c è ipoespresso in pazienti di LAP di nuova diagnosi rispetto a promielociti
normali derivanti da un sistema di differenziamento granulocitario in vitro (Careccia
et al, 2009), e che l’espressione ectopica dilet-7c è sufficiente a indurre il
differenziamento mieloide di linee cellulari e blasti primari derivati da pazienti di
LAM di nuova diagnosi (Pelosi et al, 2012). Questi dati sottolineano, quindi,
l’importanza di studiare i meccanismi molecolari coinvolti nella regolazione
dell’espressione di let-7c in cellule leucemiche.
Il let-7c, membro della famiglia let-7, è un miRNA intronico localizzato sul cromosoma
21 in un introne del gene LINC00478, all’interno del quale si trovano anche il miR-99a
and miR-125b-2. La famiglia di miRNA let-7 presenta una conservazione elevata in
tutta la filogenesi animale, da C. Elegans all’uomo. Ciò suggerisce che i membri di
questa famiglia di miRNA siano essenziali regolatori dell’espressione genica in vari
organismi (Hertel et al, 2012).
Fino a poco tempo fa, era generalmente accettato che i miRNA intronici fossero
trascritti come conseguenza della trascrizione del loro gene-ospite (Rodriguez et al,
2004; Kim and Kim, 2007; Liang et al, 2007; Saini et al, 2007; Gromak, 2012). In
accordo con questo modello, recentemente abbiamo osservato in cellule leucemiche
una coordinata regolazione dell’espressione di let-7c con il suo gene ospite,
suggerendo che la sua trascrizione potesse essere controllata dallo stesso promotore.
In maniera interessante, inoltre, abbiamo osservato un reclutamento di PML/RARα su
siti RARE nel promotore del gene ospite, ed un suo rilascio dopo trattamento con

71
ATRA, in parallelo ad un aumentata espressione di let-7c (Careccia et al, 2009). Ad
oggi, tuttavia, sta emergendo il concetto che il processamento dal trascritto primario
del gene ospite del miRNA non è l’unica via per la generazione di miRNA intronici
(Baskerville and Bartel, 2005).
Un’ipotesi interessante suggerisce che quando un miRNA intronico “nasce” utilizzi
inizialmente il TSS del suo gene ospite e che possa acquisire un promotore proprio,
indipendente, più tardi durante l’evoluzione (Ozsolak et al, 2008). Secondo tale
ipotesi, i miRNA intronici evolutivamente più “vecchi” (come, ad esempio, il let-7c)
avrebbero perciò una maggiore probabilità di avere promotori propri indipendenti
rispetto a miRNA relativamente più recenti.
Infatti, con lo sviluppo di sempre più nuove strategie “high throughput” per la
caratterizzazione di promotori, come la ChIP-seq per valutare le modificazioni
istoniche, in combinazione o meno con l’analisi del posizionamento dei nucleosomi
(Ozsolak et al, 2008; Wang et al, 2009, Monteys et al, 2010), sono stati identificati
numerosi miRNA intronici che potrebbero essere espressi da promotori indipendenti
dal gene ospite. Per quanto riguarda il let-7c, alcuni studi indicano diversi putativi siti
intronici di inizio di trascrizione (Oszolak et al, 2008; Corcoran et al, 2009; Wang et al,
2009; Monteys et al, 2010). In particolare, Ozsolak e collaboratori hanno identificato
un putativo nuovo promotore prossimale del let-7c, combinando la mappatura dei
nucleosomi con i rimodellamenti della cromatina in alcune linee cellulari tumorali. In
questo lavoro, è stato identificato un putativo TSS intronico per il let-7c, situato nel
quinto introne del gene ospite LINC00478. Altri autori, invece, suggeriscono che il let-
7c potrebbe essere regolato, mediante un meccanismo RNA polimerasi III-mediato, da
un proprio promotore intronico il cui TSS è localizzato nel sesto introne del gene
ospite LINC00478 (Monteys et al, 2010).
Da quanto sopra riportato, si evince che il reale promotore del let-7c deve essere
ancora ben identificato e caratterizzato. Per cercare, quindi, un possibile nuovo
promotore intronico per il let-7c, abbiamo effettuato un’analisi in silico su una regione
genomica immediatamente “a monte” del let-7c, identificando un nuovo putativo TSS
localizzato a circa 700 bp dal pre-let-7c. Questa regione è stata quindi validata
sperimentalmente mediante 5’ RACE e, clonata a monte del gene della luciferasi, ha
mostrato avere un’attività trascrizionale. Questi dati suggeriscono quindi che per la
trascrizione del let-7c bisogna considerare almeno due promotori distinti: il

72
promotore distale del gene ospite LINC00478, ed il promotore prossimale intronico
specifico del miRNA.
In questo studio di tesi abbiamo anche confermato il legame di PML/RARα sul
promotore del gene ospite del let-7c e mostrato una co-localizzazione di RXR con
PML. Questi risultati sono in accordo con recenti ritrovamenti indicanti che RXR è
presente associato a PML/RARα in complessi funzionali (Martens et al, 2010).
Abbiamo quindi verificato se PML/RARα fosse in grado di legare il promotore
intronico del let-7c dove, mediante analisi bioinformatica, abbiamo trovato due siti
RARE non canonici (DR3 ed ER4). Sebbene i siti DR5, DR2 e DR1 siano i più comuni
siti di legame per RXR/RARα in condizioni fisiologiche, ci sono evidenze che
suggeriscono che PML-RARα-RXR abbia una più estesa capacità di legare il DNA e che
sia in grado di legare anche ripetizioni dirette o evertite con spaziatura variabile
(Martens et al, 2010). I nostri dati tuttavia mostrano che sul promotore intronico da
noi identificato PML-RARα è reclutato solo debolmente, ed il trattamento con ATRA
non interferisce con il suo reclutamento.
Abbiamo inoltre analizzato alcuni dei principali enzimi implicati nelle modificazioni
epigenetiche e l’acetilazione e metilazione degli istoni su entrambi i promotori del let-
7c in cellule di LAP NB4. I nostri dati mostrano che dopo trattamenti con ATRA, solo il
promotore del gene ospite presenta una conformazione più aperta della cromatina ed
un aumento dei marcatori epigenetici che correlano con uno stato di maggiore
attivazione trascrizionale. Sorprendentemente, dopo trattamento con ATRA
osserviamo sul promotore del gene ospite una diminuzione di H3K4me3, noto
marcatore di promotori trascrizionalmente attivi. Ciò sembrerebbe in conflitto con le
altre modificazioni epigenetiche analizzate. Comunque, deve essere considerato che il
promotore del gene ospite del let-7c non ha isole CpG, e che il profilo delle
modificazioni epigenetiche di tali promotori non è esattamente uguale a quello dei
promotori con isole CpG. Ad esempio, segnali di H3K4me3 più deboli sono stati
descritti essere associati a promotori senza isole CpG (Wang et al, 2009). In ogni caso,
nel nostro sistema sperimentale questo tipo di modificazione istonica non sembra
avere un ruolo rilevante nei cambiamenti di conformazione della cromatina sul
promotore del LINC00478.
Al contrario, il promotore intronico dopo trattamenti con ATRA non mostra
modulazione alcuna negli stessi marcatori epigenetici. Questi dati sono in linea con i

73
dati di luciferasi sui promotori di let-7c (vedi Analisi dell’attività trascrizionale del
promotore intronico del let-7c in cellule di leucemia promielocitica acuta), in cui solo
l’attività del promotore del gene ospite è incrementata da trattamenti con ATRA.
Questi risultati indicano che il promotore del gene ospite abbia un ruolo
predominante nella regolazione del let-7c durante il differenziamento indotto da
ATRA in cellule di LAP, mentre il promotore intronico potrebbe avere un ruolo nel
guidare la trascrizione del let-7c in contesti tumorali diversi da quello leucemico.
Infatti, i saggi di luciferasi effettuati indicano che l’attività del promotore intronico è
più elevata di quella del gene ospite nelle linee cellulari di tumore solido LNCaP e
H1299, suggerendo che questo promotore possa avere un ruolo importante nel
guidare la trascrizione del let-7c in contesti cellulari diversi dalle leucemie.A
rafforzare questa ipotesi, abbiamo dati preliminari che mostrano una significativa
correlazione tra l’espressione del gene ospite LINC00478 e del let-7c in linee cellulari
di LAM di vari sottotipi FAB. Al contrario, nelle varie linee derivanti da tumori solidi
non si osserva tale correlazione. Poiché l’espressione dei microRNA può essere
regolata a livello post-trascrizionale, ed in particolare per la famiglia let-7 sono ben
noti i meccanismi di regolazione post-trascrizionale (vedi anche Capitolo 3.5.2 Let-7 e
tumori), per validare questi nostri risultati preliminari, sarà nostro interesse
analizzare l’espressione del pri-miR nelle varie linee leucemiche e di tumore solido
mettendola in relazione con l’espressione del miRNA maturo e del gene ospite.
La nozione secondo cui promotori intronici siano in grado di guidare l’espressione di
specifici miRNA è una scoperta relativamente recente, che si propone di ampliare le
nostre (poche) conoscenze sulla regolazione trascrizionale dei miRNA intronici. Per i
miRNA caratterizzati da un doppio promotore, quello del gene ospite e quello
intronico, sorge un numero sempre maggiore di domande a proposito dei contributi
forniti da ogni promotore ai livelli di espressione dei miRNA, ed è ancora ben lontano
dall’essere determinato quale o quali siano i meccanismi responsabili del potenziale
uso alternativo di questi promotori nella regolazione dei miRNA.
Questi dati sottolineano quindi una complessa regolazione dell’espressione di let-7c
che potrebbe avere un ruolo importante nella deregolazione e patogenesi dei tumori.
Inoltre, l’uso alternativo di questi due promotori potrebbe rappresentare un nuovo
meccanismo regolatorio dell’espressione del let-7c in differenti tessuti o nei vari
sottotipi di AML.

74
Infine, il contributo fornito dalle due tipologie di promotori (gene ospite ed intronico)
nel controllo dell’espressione del let-7c potrebbe essere dipendente dal contesto
cellulare ,e ulteriori e dettagliati studi andranno svolti per verificare questa ipotesi.
L’analisi dei meccanismi di regolazione epigenetica del promotore del gene ospite,
che sembra essere responsabile della trascrizione del let-7c in cellule leucemiche,
apre nuove possibilità nell’ambito di una terapia basata sull’impiego di farmaci in
grado di modificare la cromatina. Questi dati potrebbero, quindi, contribuire ad una
migliore comprensione dei complessi meccanismi molecolari coinvolti nella
transizione dal normale differenziamento mieloide a quello patologico e potrebbero
permettere l’identificazione di nuovi bersagli e quindi di nuove terapie per le
leucemie mieloidi acute. Infine, se sarà verificata l’ipotesi del ruolo del promotore
intronico nel guidare la trascrizione del let-7c in relazione al contesto cellulare, sarà
interessante analizzare i possibili meccanismi che sono alla base della sua regolazione
in tali contesti e studiare così le cause della sua de-regolazione nei vari tipi di tumore.

75
BIBLIOGRAFIA
Aboobaker AA, Tomancak P, Patel N, Rubin GM, Lai EC: Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. Proc Natl Acad Sci USA 2005, 102:18017-18022.
Alvarez-Garcia I, Miska EA (2005). MicroRNA functions in animal development and human disease. Development 132: 4653-4662.
Ambros V (2004). The functions of animal microRNAs. Nature 431: 350-355.
Anindo MI, Yaqinuddin A (2012). Insights into the potential use of microRNAs as biomarker in cancer Int J Surg. 10(9):443-9.
Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN (1991). The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc Natl Acad Sci U S A 88(22):10148-52
Argiropoulos B, Humphries RK (2007) Hox genes in hematopoiesis and leukemogenesis. Oncogene 26: 6766-6776.
Arnould C, Philippe C, Bourdon V, Gr goire MJ, Berger R, Jonveaux P (1999). The signal transducer and activator of transcription STAT5b gene is a new partner of retinoic acid receptor alpha in acute promyelocytic-like leukaemia. Hum Mol Genet 8: 1741-1749.
Augoff K, McCue B, Plow EF, Sossey-Alaoui K (2012). miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triplenegative breast cancer. Mol.
Cancer 11: 5
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z et al (2007). High-resolution profiling of histone methylations in the human genome. Cell 129(4):823-37
Bartel DP (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281-297.

76
Bartel DP (2009). MicroRNAs: target recognition and regulatory functions. Cell 136: 215-233.
Baskerville S, Bartel DP (2005). Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11: 241-247.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR et al (1985). Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med 103: 620-625.
Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC (2007). Mammalian mirtron genes. Mol Cell 28: 328-336.
Borchert GM, Lanier W, Davidson BL (2006). RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 13: 1097-1101.
Boyerinas B, Park SM, Shomron N, Hedegaard MM, Vinther J, Andersen JS et al (2008). Identification of let-7-regulated oncofetal genes. Cancer Res 68: 2587-2591.
Brown D, Kogan S, Lagasse E, Weissman I, Alcalay M, Pelicci PG et al (1997). A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci U S A 94: 2551-2556.
Buck SW, Gallo CM, Smith JS (2004). Diversity in the Sir2 family of protein deacetylases. J
Leukoc Biol. 75(6):939-50.
Bussing I, Slack FJ, Grosshans H (2008). let-7 microRNAs in development, stem cells and cancer. Trends Mol Med 14: 400-409.
Byrd JC, Mrozek K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC et al (2002). Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood 100: 4325-4336.
Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ et al (2007). Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood 110: 1022-1024.

77
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E et al (2002). Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 99: 15524-15529.
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S et al (2004). Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 101: 2999-3004.
Careccia S, Mainardi S, Pelosi A, Gurtner A, Diverio D, Riccioni R et al (2009). A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene 28: 4034-4040.
Carracedo A, Ito K, Pandolfi PP (2011). The nuclear bodies inside out: PML conquers the cytoplasm. Curr Opin Cell Biol 23: 360-366.
Caygill EE, Johnston LA (2008). Temporal regulation of metamorphic processes in Drosophila by the let-7 and miR-125 heterochronic microRNAs. Curr Biol 18: 943-950.
Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM et al (2008). Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 40: 43-50.
Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, et al. (2007) Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26(5):745-52
Clape C, Fritz V, Henriquet C, Apparailly F, Fernandez PL, Iborra F et al (2009). miR-143 interferes with ERK5 signaling, and abrogates prostate cancer progression in mice. PLoS ONE 26;4(10):e7542.
Chen X, Guo X, Zhang H, Xiang Y, Chen J, Yin Y et al (2009). Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 28(10):1385-92
Chen Z, Brand NJ, Chen A, Chen SJ, Tong JH, Wang ZY et al (1993). Fusion between a novel Kruppel-like zinc finger gene and the retinoic acid receptor-alpha locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. EMBO J 12: 1161-1167.

78
Chen CZ, Li L, Lodish HF, Bartel DP (2004). MicroRNAs modulate hematopoietic lineage differentiation. Science 303: 83-86.
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT et al (2005). Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33: e179.
Cheng GX, Zhu XH, Men XQ, Wang L, Huang QH, Jin XL et al (1999). Distinct leukemia phenotypes in transgenic mice and different corepressor interactions generated by promyelocytic leukemia variant fusion genes PLZF-RARalpha and NPM-RARalpha. Proc Natl
Acad Sci U S A 96: 6318-6323.
Cheung P and Lau P (2005). Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol. 19: 563-573
Corcoran DL, Pandit KV, Gordon B, Bhattacharjee A, Kaminski N, Benos PV (2009). Features of mammalian microRNA promoters emerge from polymerase II chromatin immunoprecipitation data. PLoS One 4: e5279
Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY (2007). MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res. 67: 8433–8438.
Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M et al (2005). miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102: 13944-13949.
Clurman BE, Hayward WS (1989). Multiple protooncogene activations in avian leukosis virus induced lymphomas: evidence for stage-specific events. Mol Cell Biol 9(6): 2657–2664
Cui Y, Su WY, Xing J, Wang YC, Wang P, Chen XY, et al (2011).. MiR-29a Inhibits Cell Proliferation and Induces Cell Cycle Arrest through the Downregulation of p42.3 in Human Gastric Cancer. PLoS One 6(10): e25872
de Murcia G, Huletsky A, Poirier GG (1988). Modulation of chromatin structure by poly(ADP-ribosylation). Biochem Cell Biol 66(6):626-35.

79
de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 370 (Pt 3):737-49.
Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M et al (2002). Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295: 1079-1082.
Dixon-McIver A, East P, Mein CA, Cazier JB, Molloy G, Chaplin T et al (2008). Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS
One 3(5): e2141
Dokmanovic M, Clarke C, Marks PA (2007). Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 5(10):981-9
Döhner H (2007). Implication of the molecular characterization of acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 412-419.
Drexler HG (1996). Expression of FLT3 receptor and response to FLT3 ligand by leukemic cells. Leukemia 10: 588-599.
Eguchi M, Eguchi-Ishimae M, Greaves M (2005). Molecular pathogenesis of MLL-associated leukemias. Int J Hematol 82: 9-20.
Ehrenhofer-Murray AE (2004). Chromatin dynamics at DNA replication, transcription and repair. Eur J Biochem. 271(12):2335-2349. Review
Esquela-Kerscher A, Johnson SM, Bai L, Saito K, Partridge J, Reinert KL et al (2005). Post-embryonic expression of C. elegans microRNAs belonging to the lin-4 and let-7 families in the hypodermis and the reproductive system. Dev Dyn 234: 868-877.
Esteller M (2007). Cancer epigenomics: DNA methylomes and histone-modification maps. Nat
Rev Genet. 8(4):286-98.
Estey E, Dohner H (2006). Acute myeloid leukaemia. Lancet 368: 1894-1907.

80
Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al (2007). MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 104(40):15805-10
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L et al (2005). Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 352: 254-266.
Fazi F, Racanicchi S, Zardo G, Starnes LM, Mancini M, Travaglini L et al (2007). Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer
Cell 12: 457-466.
Filipowicz W, Bhattacharyya SN, Sonenberg N (2008). Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102-114.
Ganetsky A (2012). The role of decitabine for the treatment of acute myeloid leukemia. Ann
Pharmacother 46: 1511-1517.
Garzon R, Pichiorri F, Palumbo T, Visentini M, Aqeilan R, Cimmino A et al (2007). MicroRNA gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene 26: 4148-4157.
Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C et al (2008a). Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci U S A 105: 3945-3950.
Garzon R, Volinia S, Liu CG, Fernandez-Cymering C, Palumbo T, Pichiorri F et al (2008b). MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 111: 3183-3189.
Giordano A, Avantaggiati ML (1999). p300 and CBP: partners for life and death. J Cell Physiol. 181(2):218-30.
Gough SM, Slape CI, Aplan PD (2011). NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood 118: 6247-6257.

81
Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD et al (2008). Epigenetic silencing of the intronic microRNA hsamiR-342 and its host gene EVL in colorectal cancer. Oncogene 27: 3880–3888.
Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M et al (1998). Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature 391: 815-818.
Griffiths-Jones S (2007). Annotating noncoding RNA genes. Annu Rev Genomics Hum Genet 8: 279-298.
Grisendi S, Mecucci C, Falini B, Pandolfi PP (2006). Nucleophosmin and cancer. Nat Rev Cancer 6: 493-505.
Grisolano JL, Wesselschmidt RL, Pelicci PG, Ley TJ (1997). Altered myeloid development and acute leukemia in transgenic mice expressing PML-RAR alpha under control of cathepsin G regulatory sequences. Blood 89: 376-387.
Gromak N (2012). Intronic microRNAs: a crossroad in gene regulation. Biochem Soc Trans 40: 759-761.
Guled M, Lahti L, Lindholm PM, Salmenkivi K, Bagwan I, Nicholson AG et al (2009). CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma -A miRNA microarray analysis. Genes Chromosomes Cancer 48: 615-623.
Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ et al (2004). Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst 96: 269-279.
Halkidou,K. Gaughan L, Cook S, Leung HY, Neal DE, Robson CN (2004). Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 59: 177–189.
Han YC, Park CY, Bhagat G, Zhang J, Wang Y, Fan JB et al. (2010). microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J Exp Med 207(3): 475–489

82
Havelange V, Garzon R (2010). MicroRNAs: emerging key regulators of hematopoiesis. Am J
Hematol 85: 935-942.
He LZ, Bhaumik M, Tribioli C, Rego EM, Ivins S, Zelent A et al (2000). Two critical hits for promyelocytic leukemia. Mol Cell 6: 1131-1141.
He L, Hannon GJ (2004). MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev
Genet. 5(7):522-31.
Hertel J, Bartschat S, Wintsche A, Otto C (2012). Evolution of the let-7 microRNA family. RNA
Biol. 9(3):231-41.
Hu J (2011). Arsenic in the treatment of newly diagnosed acute promyelocytic leukemia: current status and future research direction. Front Med 5: 45-52.
Huang Y, Yang YB, Zhang XH, Yu XL, Wang ZB, Cheng XC (2013). MicroRNA-21 gene and cancer. Med Oncol 30(1):376.
Ibarra I, Erlich Y, Muthuswamy SK, Sachidanandam R, Hannon GJ (2007). A role for microRNAs in maintenance of mouse mammary epithelial progenitor cells. Genes Dev 21: 3238-3243.
Iniguez-Lluhi JA (2006). For a healthy histone code, a little SUMO in the tail keeps the acetyl away. ACS Chem Biol. 1(4):204-6
Insinga A, Pelicci PG, Inucci S (2005). Leukemia-associated fusion proteins. Multiple mechanisms of action to drive cell transformation. Cell Cycle 4: 67-69.
Isken F, Steffen B, Merk S, Dugas M, Markus B, Tidow N et al (2008). Identification of acute myeloid leukaemia associated microRNA expression patterns. Br J Haematol 140: 153-161.
Jaenisch R and Bird A (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33 Suppl.: 245-254
Johnson SM, Lin SY, Slack FJ (2003). The time of appearance of the C. elegans let-7 microRNA

83
is transcriptionally controlled utilizing a temporal regulatory element in its promoter. Dev
Biol 259: 364-379.
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A et al (2005). RAS is regulated by the let-7 microRNA family. Cell 120: 635-647.
Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D et al (2007). The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res 67: 7713-7722.
Jones PA and Baylin SB (2002). The fundamental role of epigenetic events in cancer. Nat Rev
Genet. 3(6):415-28.
Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ, Lowenberg B (2008). MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood 111: 5078-5085.
Kainz B, Heintel D, Marculescu R, Schwarzinger I, Sperr W, Le T et al (2002). Variable prognostic value of FLT3 internal tandem duplications in patients with de novo AML and a normal karyotype, t(15;17), t(8;21) or inv(16). Hematol J 3: 283-289.
Karlsson KH, Stenerlow B (2004). Focus formation of DNA repair proteins in normal and repair-deficient cells irradiated with high-LET ions. Radiat Res. 161(5):517-27.
Kelley RI (1973). Isolation of a histone IIb1-IIb2 complex. Biochem Biophys Res Commun., 54(4):1588-94.
Kim YK, Kim VN (2007). Processing of intronic microRNAs. Embo J 26: 775-783.
King-Underwood L, Pritchard-Jones K (1998). Wilms' tumor (WT1) gene mutations occur mainly in acute myeloid leukemia and may confer drug resistance. Blood 91: 2961-2968.
Kloosterman WP, Plasterk RH (2006). The diverse functions of microRNAs in animal development and disease. Dev Cell 11: 441-450.
Kong W, He L, Coppola M, et al. MicroRNA-155regulates cell survival, growth and

84
chemosensitivity by targeting FOXO3a in breast cancer. JBiol Chem, 2010
Kornberg RD, Thomas JO (1974). Chromatin structure; oligomers of the histones. Science 84(139):865-8
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA et al (2001). The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 98: 1752-1759.
Kraus WL, Lis JT (2003). PARP goes transcription. Cell 113(6):677-83.
Krivtsov AV, Armstrong SA (2007). MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7: 823-833.
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002). Identification of tissue-specific microRNAs from mouse. Curr Biol 12: 735-739.
Landthaler M, Yalcin A, Tuschl T (2004). The human DiGeorge syndrome critical region gene 8 and Its D. melanogaster homolog are required for miRNA biogenesis. Curr Biol 14: 2162-2167.
Lee Y, Jeon K, Lee JT, Kim S, Kim VN (2002). MicroRNA maturation: stepwise processing and subcellular localization. EMBO J 21: 4663-4670.
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J et al (2003). The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415-419.
Lee YS, Dutta A (2007). The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev 21: 1025-1030.
Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P, Bellan C et al (2008). MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol 216: 440-450.

85
Liang Y, Ridzon D, Wong L, Chen C (2007). Characterization of microRNA expression profiles in normal human tissues. BMC Genomics 8: 166.
Li N, Kaur S, Greshock J, Lassus H, Zhong X, Wang Y et al (2012). A combined array-based comparative genomic hybridization and functional library screening approach identifies mir-30d as an oncomir in cancer. Cancer Res 72(1):154-64.
Licht JD, Chomienne C, Goy A, Chen A, Scott AA, Head DR et al (1995). Clinical and molecular characterization of a rare syndrome of acute promyelocytic leukemia associated with translocation (11;17). Blood 85: 1083-1094.
Licht JD (2006). Reconstructing a disease: What essential features of the retinoic acid receptor fusion oncoproteins generate acute promyelocytic leukemia? Cancer Cell 9: 73-74.
Lo WS, Duggan L, Emre NC, Belotserkovskya R, Lane WS, Shiekhattar R, Berger SL (2001). Snf1--a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 293(5532):1142-6.
Lowenberg B, Downing JR, Burnett A (1999). Acute myeloid leukemia. N Engl J Med 341: 1051-1062.
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389(6648):251-60.
Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F et al (2007). Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 67: 1424–1429.
Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D et al (2008). A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 105: 13556-13561.
Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U (2004). Nuclear export of microRNA precursors. Science 303: 95-98.

86
Lutterbach B, Hou Y, Durst KL, Hiebert SW (1999). The inv(16) encodes an acute myeloid leukemia 1 transcriptional corepressor. Proc Natl Acad Sci U S A 96: 12822-12827.
Maroney PA, Yu Y, Fisher J, Nilsen TW (2006). Evidence that microRNAs are associated with translating messenger RNAs in human cells. Nat Struct Mol Biol 13: 1102-1107.
Martens JH, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, Ferrara F et al (2010). PML-RARalpha/RXR Alters the Epigenetic Landscape in Acute Promyelocytic Leukemia. Cancer Cell 17: 173-185.
Martens JH (2011). Acute myeloid leukemia: a central role for the ETS factor ERG. Int J
Biochem Cell Biol 43: 1413-1416.
Martin C, Zhang Y (2005). The diverse functions of histone lysine methylation. Nat Rev Mol
Cell Biol 6(11):838-49.
Mayr C, Hemann MT, Bartel DP (2007). Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 315: 1576-1579.
Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE (2007). FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood 110: 1262-1270.
Meister G, Tuschl T (2004). Mechanisms of gene silencing by double-stranded RNA. Nature 431: 343-349.
Melnick A, Licht JD (1999). Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93: 3167-3215.
Mi S, Lu J, Sun M, Li Z, Zhang H, Neilly MB et al (2007). MicroRNA expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc Natl
Acad Sci U S A 104: 19971-19976.
Mineno J, Okamoto S, Ando T, Sato M, Chono H, Izu H et al (2006). The expression profile of microRNAs in mouse embryos. Nucleic Acids Res 34: 1765-1771.

87
Mitelman F, Johansson B, Mertens F (2007). The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7: 233-245.
Monteys AM, Spengler RM, Wan J, Tecedor L, Lennox KA, Xing Y et al (2010). Structure and activity of putative intronic miRNA promoters. RNA 16: 495-505
Mrozek K, Dohner H, Bloomfield CD (2007). Influence of new molecular prognostic markers in patients with karyotypically normal acute myeloid leukemia: recent advances. Curr Opin
Hematol 14: 106-114.
Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K et al (1996). Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 10: 1911-1918.
Nervi C, Ferrara FF, Fanelli M, Rippo MR, Tomassini B, Ferrucci PF et al (1998). Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARalpha fusion protein. Blood 92: 2244-2251.
Nervi C, Fazi F, Grignani F (2008). Oncoproteins, heterochromatin silencing and microRNAs: a new link for leukemogenesis. Epigenetics 3: 1-4.
Newman MA, Thomson JM, Hammond SM (2008). Lin-28 interaction with the Let-7 precursor loop mediates regulated microRNA processing. RNA 14: 1539-1549.
Noma K, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S et al (2004). RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet 36: 1174-1180.
Noonan EJ, Place RF, Pookot D, Basak S, Whitson JM, Hirata H, et al (2009). miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 28(14):1714-24.
Nottrott S, Simard MJ, Richter JD (2006). Human let-7a miRNA blocks protein production on actively translating polyribosomes. Nat Struct Mol Biol 13: 1108-1114.
O'Connell RM, Rao DS, Chaudhuri AA, Boldin MP, Taganov KD, Nicoll J et al (2008). Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J Exp Med 205: 585-594.

88
Osley MA, Fleming AB, Kao CF (2006). Histone ubiquitylation and the regulation of transcription. Results Probl Cell Differ 41:47-75.
Ozen M, Creighton CJ, Ozdemir M, Ittmann M (2008). Widespread deregulation of microRNA expression in human prostate cancer. Oncogene 27: 1788-1793.
Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG et al (2008). Chromatin structure analyses identify miRNA promoters. Genes Dev 22: 3172-3183.
Pelosi A, Careccia S, Lulli V, Romania P, Marziali G, Testa U et al (2012). miRNA let-7c promotes granulocytic differentiation in acute myeloid leukemia. Oncogene, Sep 10. doi: 10.1038/onc.2012.398. [Epub ahead of print]
Peterson CL and Laniel MA (2004). Histones and histone modifications. Curr. Biol. 14(14): R546-51.
Pillai RS, Bhattacharyya SN, Artus CG, Zoller T, Cougot N, Basyuk E et al (2005). Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 309: 1573-1576.
Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B et al (2010). miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A 107: 264-269.
Pospisil V, Vargova K, Kokavec J, Rybarova J, Savvulidi F, Jonasova A et al (2011). Epigenetic silencing of the oncogenic miR-17-92 cluster during PU.1-directed macrophage differentiation. Embo J 30(21): 4450–4464
Rana TM (2007). Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol 8: 23-36.
Razin, A. and Cedar, H. (1991). DNA methylation and gene expression. Microbiol. Rev. 55: 451-58.
Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ (1996). The t(5;17) variant of acute promyelocytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood 87: 882-886.

89
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE et al (2000). The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901-906.
Ro S, Park C, Young D, Sanders KM, Yan W (2007). Tissue-dependent paired expression of miRNAs. Nucleic Acids Res 35: 5944-5953.
Roark DE, Geoghegan TE, Keller GH (1974). A two-subunit histone complex from calf thymus. Biochem Biophys Res Commun. 59(2):542-7.
Rodenhiser D, Mann M (2006). Epigenetics and human disease: translating basic biology into clinical applications CMAJ 174(3):341-8.
Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A (2004): Identification of mammalian microRNA host genes and transcription units. Genome Res 14:1902-1910.
Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273(10):5858-68.
Rosnet O, Buhring HJ, Marchetto S, Rappold I, Lavagna C, Sainty D et al (1996). Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 10: 238-248.
Roth SY, Denu JM, Allis CD (2001). Histone acetyltransferases. Annu. Rev. Biochem. 70: 81–120
Roush S, Slack FJ (2008). The let-7 family of microRNAs. Trends Cell Biol 18: 505-516.
Saini HK, Griffiths-Jones S, Enright AJ (2007). Genomic analysis of human microRNA transcripts. Proc Natl Acad Sci U. S. A. 104: 17719-17724.
Saini HK, Enright AJ, Griffiths-Jones S (2008). Annotation of mammalian primary microRNAs. BMC Genomics 9: 564.

90
Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P et al (2007). MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res 67: 9762-9770.
Santanam U, Zanesi N, Efanov A, Costinean S, Palamarchuk A, Hagan JP et al (2010). Chronic lymphocytic leukemia modeled in mouse by targeted miR-29 expression. Proc Natl Acad Sci U
S A 107(27): 12210–12215.
Sauve DM, Anderson HJ, Ray JM, James WM, Roberge M (1999). Phosphorylation-induced rearrangement of the histone H3 NH2-terminal domain during mitotic chromosome condensation. J Cell Biol 145(2):225-35.
Schiltz RL, Mizzen CA, Vassilev A, Cook RG, Allis CD, Nakatani Y (1999). Overlapping but distinct patterns of histone acetylation by the human coactivators p300 and PCAF within nucleosomal substrates. J Biol Chem. 274(3):1189-92.
Schnittger S, Schoch C, Kern W, Mecucci C, Tschulik C, Martelli MF et al (2005). Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 106: 3733-3739.
Schultz J, Lorenz P, Gross G, Ibrahim S, Kunz M (2008). MicroRNA let-7b targets important cell cycle molecules in malignant melanoma cells and interferes with anchorage-independent growth. Cell Res 18: 549-557.
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD (2003). Asymmetry in the assembly of the RNAi enzyme complex. Cell 115: 199-208.
Scott GK, Mattie MD, Berger CE, Benz SC, Benz CC (2006).Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 66(3):1277-81.
Sempere LF, Dubrovsky EB, Dubrovskaya VA, Berger EM, Ambros V (2002). The expression of the let-7 small regulatory RNA is controlled by ecdysone during metamorphosis in Drosophila melanogaster. Dev Biol 244: 170-179.
Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V (2004). Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5: R13.

91
Shell S, Park SM, Radjabi AR, Schickel R, Kistner EO, Jewell DA et al (2007). Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci U S A 104: 11400-11405.
Shiio Y, Eisenman RN (2003), Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA 100(23):13225-30.
Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, et al (2005). Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS 113: 264–268.
Song F, Smith JF, Kimura MT, Morrow AD, Matsuyama T, Nagase H et al (2005). Association of tissue-specific differentially methylated regions (TDMs) with differential gene expression. Proc. Natl. Acad. Sci. U.S. A. 102: 3336-3341
Steffen B, Muller-Tidow C, Schwable J, Berdel WE, Serve H (2005). The molecular pathogenesis of acute myeloid leukemia. Crit Rev Oncol Hematol 56: 195-221.
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H et al (2004). Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 64: 3753-3756.
Tallman MS, Gilliland DG, Rowe JM (2005). Drug therapy for acute myeloid leukemia. Blood 106: 1154-1163.
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M et al (2006). Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 107: 4011-4020.
Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y et al (2008). Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res. 68: 4123–4132.
Valeri N, Gasparini P, Fabbri M, Braconi C, Veronese A, Lovat F et al (2010). Modulation of mismatch repair and genomic stability by miR-155. Proc Natl Acad Sci U S A 107(15): 6982–6987

92
Verdin E, Dequiedt F, Kasler HG (2003). Class II histone deacetylases: versatile regulators.
Trends Genet. 19(5):286-93
Viswanathan SR, Daley GQ, Gregory RI (2008). Selective blockade of microRNA processing by Lin28. Science 320: 97-100.
Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S et al (2009). Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 41: 843-848.
Viswanathan SR, Daley GQ (2010). Lin28: A microRNA regulator with a macro role. Cell 140: 445-449.
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F et al (2006). A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U
S A 103(7): 2257–2261
Wang X, Xuan Z, Zhao X, Li Y, Zhang MQ (2009). High-resolution human core-promoter prediction with CoreBoost_HM. Genome Res 19: 266-275.
Weber B, Stresemann C, Brueckner B, Lyko F (2007). Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 6(9):1001-5.
Wei Y, Yu L, Bowen J, Gorovsky MA, Allis C, (1999). Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 97(1):99-109.
Wells RA, Catzavelos C, Kamel-Reid S (1997). Fusion of retinoic acid receptor alpha to NuMA, the nuclear mitotic apparatus protein, by a variant translocation in acute promyelocytic leukaemia. Nat Genet 17: 109-113.
Westendorf JJ, Yamamoto CM, Lenny N, Downing JR, Selsted ME, Hiebert SW (1998). The t(8;21) fusion product, AML-1-ETO, associates with C/EBP-alpha, inhibits C/EBP-alpha-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol 18: 322-333.
Wolffe AP (1997). Histone H1. Int J Biochem Cell Biol. 29(12):1463-6.

93
Wu L, Fan J, Belasco JG (2006). MicroRNAs direct rapid deadenylation of mRNA. Proc Natl
Acad Sci U S A 103: 4034-4039.
Wulczyn FG, Smirnova L, Rybak A, Brandt C, Kwidzinski E, Ninnemann O et al (2007). Post-transcriptional regulation of the let-7 microRNA during neural cell specification. FASEB J 21: 415-426.
Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, et al (2003) Structure and catalytic mechanism of the human histone methyltransferase. Nature 421(6923):652-6.
Xu H, Cheung IY, Guo HF, Cheung NK (2009). miR-29 modulates expression of immunoinhibitory molecule B7-H3: potential implications for immune based therapy of human solid tumors. Cancer Res 69(15): 6275–6281
Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S et al (2001). Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 97: 2434-2439.
Yassin ER, Sarma NJ, Abdul-Nabi AM, Dombrowski J, Han Y, Takeda A et al (2009). Dissection of the transformation of primary human hematopoietic cells by the oncogene NUP98-HOXA9. PLoS One 4: e6719.
Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML (2010). A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 17: 198-212.
Yoshida H, Kitamura K, Tanaka K, Omura S, Miyazaki T, Hachiya T et al (1996). Accelerated degradation of PML-retinoic acid receptor alpha (PML-RARA) oncoprotein by all-trans-retinoic acid in acute promyelocytic leukemia: possible role of the proteasome pathway. Cancer Res 56: 2945-2948.
Youn BS, Mantel C, Broxmeyer HE (2000). Chemokines, chemokine receptors and hematopoiesis. Immunol Rev 177: 150-174.
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S et al (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917-1920.

94
Zeng Y (2006). Principles of micro-RNA production and maturation. Oncogene 25: 6156-6162.
Zhang Y, Reinberg D (2001). Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15(18):2343-60. Review
Zimonjic DB, Pollock JL, Westervelt P, Popescu NC, Ley TJ (2000). Acquired, nonrandom chromosomal abnormalities associated with the development of acute promyelocytic leukemia in transgenic mice. Proc Natl Acad Sci U S A 97: 13306-13311.