cd44-positive cancer stem cells expressing cellular prion...
TRANSCRIPT

Tumor and Stem Cell Biology
CD44-Positive Cancer Stem Cells Expressing Cellular PrionProtein Contribute to Metastatic Capacity in ColorectalCancer
Lei Du1, Guanhua Rao3, Hongyi Wang2, Baowei Li1, Weili Tian3, Jiantao Cui2, Leya He4, Brian Laffin5,Xiuyun Tian2, Chunyi Hao2, Hongmin Liu1, Xin Sun1, Yushan Zhu3, Dean G. Tang5, Maryam Mehrpour1,6,Youyong Lu2, and Quan Chen1,3
AbstractCancer stem cells are implicated in tumor progression, metastasis, and recurrence, although the exact
mechanisms remain poorly understood. Here, we show that the expression of cellular prion protein (PrPc,PRNP) is positively correlated with an increased risk of metastasis in colorectal cancer. PrPc defines asubpopulation of CD44-positive cancer stem cells that contributes to metastatic capacity. PrPcþCD44þ
colorectal cancer stem cells displayed high liver metastatic capability, unlike PrPc�CD44þ stem cells, thatwas inhibited by RNAi-mediated attenuation of PrPc. Notably, administration of PrPc monoclonal antibodiessignificantly inhibited tumorigenicity and metastasis of colorectal cancer stem cells in mouse models oforthotopic metastasis. PrPc promoted epithelial to mesenchymal transition (EMT) via the ERK2 (MAPK1)pathway, thereby conferring high metastatic capacity. Our findings reveal the function of PrPc in regulatingEMT in cancer stem cells, and they identify PrPc as candidate therapeutic target in metastatic colorectalcancer. Cancer Res; 73(8); 2682–94. �2013 AACR.
IntroductionCancer is a major cause of mortality, and most human
cancer-related deaths are related to metastasis (1, 2). Thedifficulty in diagnosing and therapeutically eliminatingmetastasis is related to our limited understanding of themolecules specifically expressed on the surface of metastaticcancer cells. Accumulating evidence suggests that a subsetof cancer cells, operationally termed cancer stem cells (CSC)or tumor-initiating cells, possesses enhanced ability toregenerate tumors and survive anticancer therapies (3).CSCs were first identified in the hematopoietic system (4),
and have now been reported in most human solid tumors onthe basis of their functional properties (5–7). ColorectalCSCs (CCSC) have been distinguished using CD44 or CD133either alone (8–10), or combined with other markers, such asEpCAM, CD166, CD29, CD24, LGR5 and aldehyde dehydro-genase 1 (ALDH1; refs. 11–13). Previous studies by ourselvesand other authors have shown that as few as 100 CD44þ
cancer cells isolated from patients were able to develop intoa heterogeneous tumor, and that spheroids derived from asingle CD44þ cancer cell could recapitulate the heteroge-neous hierarchy of tumor cells (8, 14). These studies indicatethat CCSCs may trigger the formation of colorectal cancer(CRC), and that CD44 is a functionally important surfacebiomarker of CCSCs. Importantly, an antibody specific toCD44 selectively eradicated leukemic stem cells in vivo byaltering the fate of leukemic stem cells (15). Monoclonalantibodies targeting surface antigens specifically expressedin CSCs offer a potentially powerful strategy to eliminateCSCs with minimal toxicity. Besides CD44, monoclonalantibodies targeting CD123 and CD47 have been reportedto eradicate leukemia stem cells (16).
Recent evidence suggests that CSCs may be endowedwith enhanced disseminating capacity enabling them toinvade neighboring tissues or metastasize to distant organs(17–19). Although CD44 and some other adhesion mole-cules, such as E-cadherin, could act as migration suppres-sors by restricting cancer cells to their primary sites (20),these same molecules with certain modifications (e.g.,splice variants of CD44) or in combination with otheradhesion molecules, such as CD26, may result in metastaticphenotypes (21–24).
Authors' Affiliations: 1State Key Laboratory of Biomembrane and Mem-brane Biotechnology, Institute of Zoology, Chinese Academy of Sciences;2Laboratory ofMolecular Oncology, Key Laboratory of Carcinogenesis andTranslational Research (Ministry of Education), Peking University CancerHospital & Institute, Beijing; 3Tianjin Key Laboratory of Protein Sciences,College of Life Sciences, Nankai University, Tianjin; 4Cancer ResearchCenter, Tongji Hospital, Tongji Medical College, Huazhong University ofScience and Technology, Wuhan, China, 5Department of Molecular Car-cinogenesis, University of Texas M.D. Anderson Cancer Center, SciencePark, Smithville, Texas; and 6INSERM U984, Facult�e de Pharmacie, Uni-versit�e Paris-Sud, Chatenay-Malabry, France
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
L. Du, G. Rao, H. Wang, and B. Li contributed equally to this work.
Corresponding Author: Quan Chen, Institute of Zoology, CAS, 1 BeichenWest Road, Chaoyang District, Beijing 100101 China. Phone: 86-10-6480-7321; Fax: 86-10-6480-7321; E-mail: [email protected] and Youyong Lu,Peking, University Cancer Hospital & Institute. Phone: 86-10-8819-6765;E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-12-3759
�2013 American Association for Cancer Research.
CancerResearch
Cancer Res; 73(8) April 15, 20132682
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

Cellular prion protein (PrPc) is a highly conserved glyco-protein present in all vertebrates and has the same proteinsequence as the scrapie prion protein (PrPsc), a pathogenicfactor in scrapie in sheep, bovine spongiform encephalop-athy in cattle, and kuru in human beings (25, 26). In additionto the central involvement of PrPc in prion diseases, severallines of evidence have intriguingly implicated PrPc in humantumors including glioblastoma, breast, prostate, gastric andcolorectal cancers (CRC; 27). PrPc interacts homophilicallywith itself or heterotypically with components of the extra-cellular matrix (ECM) such as laminin and vitronectin, tomediate cell adhesion (28). PrPc expression is required forthe self-renewal of CD34þ hematopoietic stem cells (29, 30).However, despite tantalizing evidence linking PrPc tohuman cancers, its physiologic function and exact role incancer development remain elusive. Here, we report thathigh levels of PrPc expression (in PrPcþ cells) identify afunctionally distinct subpopulation of CD44þ CCSCs, whichcontributes to CRC initiation and metastasis. Our findingsthus reveal hitherto-unknown roles of PrPc in CCSCs andepithelial–mesenchymal transition (EMT), and suggest thatPrPc could provide a potential therapeutic target in meta-static CRCs.
Materials and MethodsCells, animals, and samplesFresh colon samples from patients with CRC and paired
normal subjects were collected from the tumor bank ofBeijing Cancer Hospital (Beijing, China), as approved by theResearch Ethics Board at the Beijing Institute for CancerResearch. A tissue microarray (TMA) was constructed asdescribed previously (31, 32). The NCI-60 human tumor cellline panel was kindly provided by Dr. Jacque Grassi (NationalCancer Institute, Bethesda, MD). SW480 and MDA-MB-231cell lines were purchased from American Type CultureCollection, authenticated using polyphasic (genotypic andphenotypic) testing every 6 months, and cultured accordingto the manufacturer's instructions. Human primary CRCcells were isolated from patients and cultured in withserum-free medium. All CCSCs were subcultured for fewerthan 20 passages. Four-week-old female nude mice (BALB/c-nu/nu) and 5-week-old female Nonobese diabetic/severecombined immunodeficient (NOD/SCID) mice were pur-chased from Chinese Academy of Medical Sciences (Beijing,China), and all experiments were conducted under standardconditions in accordance with the National and Institutionalregulations.
Tumorigenesis and metastasis in orthotopic xenograftmodelsNOD/SCID mice were anesthetized with pentobarbital and
the cecumswere exteriorized by laparotomy. Various numbersof CD44þPrPcþ, CD44þPrPc�, CD44�PrPcþ, and CD44�PrPc�
subpopulations freshly sorted from patients, or 50,000 DsRed-labeled PrPc overexpressing or knockdown CCSCs wereinjected into the cecal wall. All the animals were monitoredfor up to 80 to 100 days or until tumor-associated deathoccurred. Primary tumors and metastasis were evaluated by
a whole-body fluorescence imaging system (In-Vivo FX PRO,Carestream).
ResultsThe PrPc expression level is correlated with clinical andpathologic features and outcomes in CRC patients
On the basis of our previous observations that CD44 is arobust marker for CCSCs (14), we hypothesized that additionalfactors may exist that are coexpressed with CD44 in thepromotion of migration and metastasis of CSCs. We thereforecompared gene expression in colorectal cancer, liver metas-tases, and normal tissues by cDNA array conducted in our lab.In addition, we conducted a CD44 coexpression analysis fromOncomine datasets (Ki_Colon, Koinuma_Colon, and Smith_Colorectal_2 datasets; refs., 33–35). Interestingly, we foundthat several genes were in fact coexpressed with CD44 andupregulated in metastases; one of these genes was PRNP (datanot shown), which encodes PrPc.
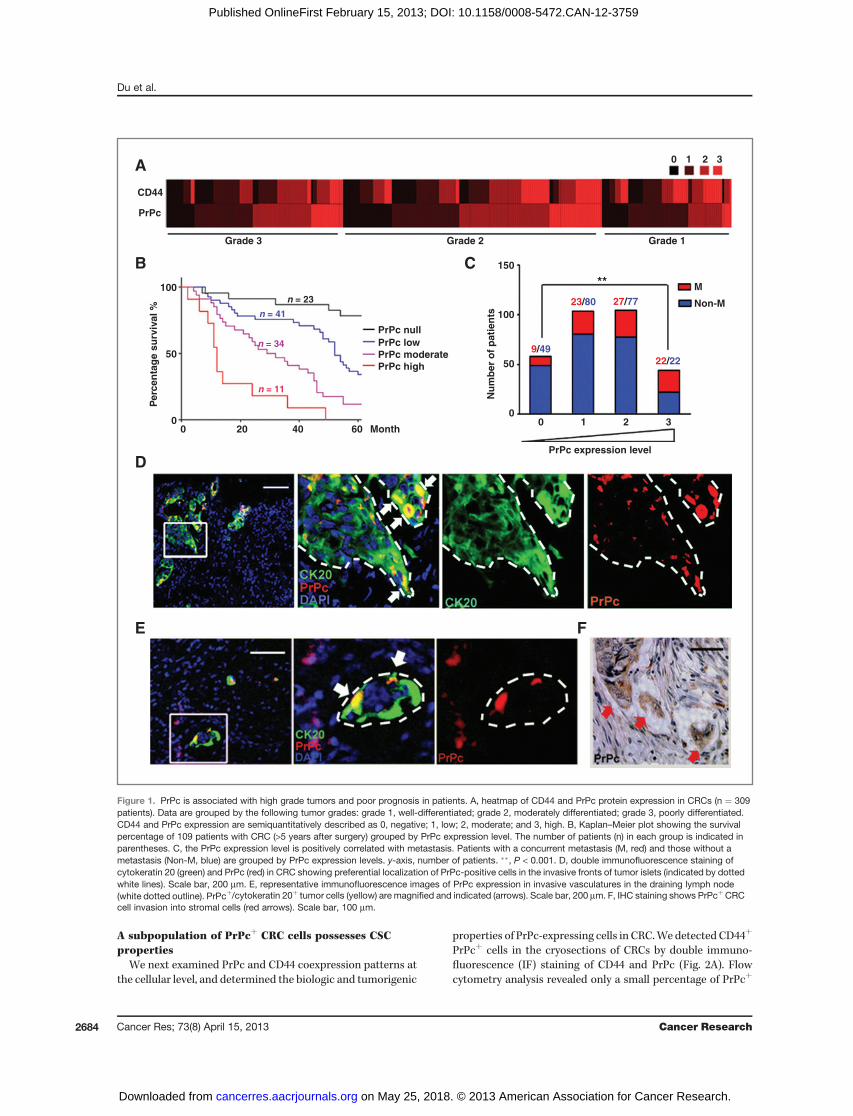
To find out whether PrPc and CD44 are associated with theclinical and pathologic features, we carried out immunohis-tochemical (IHC) staining in a TMA containing 309 colorectaltumor specimens. CD44 and PrPc were found to be coex-pressed in most of the tumors, especially in those that werepoorly differentiated (correlation coefficient ¼ 0.73; Fig. 1A).The PrPc expression level was significantly higher in poorlydifferentiated than in moderately and well-differentiated sam-ples (P < 0.001; Fig. 1A and Supplementary Fig. S1A), whereasthe level of CD44 was not associated with clinical stage (P ¼0.143; Fig. 1A). This observation was supported by examiningmultiple Oncomine cDNA microarray datasets, which revealedthat the PRNP mRNA expression levels were significantlyelevated in high-grade and advanced-stage CRCs (Supplemen-tary Fig. S1B). Importantly, the patients with CRC with thehighest PrPc expression were also found to have the poorestprognosis, with a median survival of only 12 months (Fig. 1B),which was significantly (P < 0.0001) shorter than that ofpatients whose primary tumor was PrPc-negative (mediansurvival >5 years).
CRC-related death is usually attributable to recurrenceand distant metastasis. Out of 309 specimens, there were 228primary CRCs without metastasis versus 81 cases withconcurrent distant metastasis. Strikingly, we found that PrPcwas highly expressed in the primary tumors of the patientswith liver or lung metastasis (P ¼ 0.0007; Fig. 1C). OncominecDNA microarray data analysis also showed a close corre-lation of the PrPc level with disease recurrence and earlypatient death (Supplementary Fig. S1C). We also observedthat PrPc expression was most prominent at the interfaceand invasion fronts of tumor nodules (identified by cytoker-atin 20 staining; Fig. 1D), which may constitute a reservoir ofmobile CSCs (36). In the lymph nodes that had been invadedby CRC cells, PrPcþ cells were also observed in vascularinvasions (Fig. 1E and Supplementary Fig. S1D). In addition,high levels of PrPc were present in CRC cells and could beseen invading the tumor stroma (Fig. 1F). Collectively, thesedata indicate that high PrPc expression is associated withadvanced clinical stages, increased metastatic risk, andreduced patient survival.
PrPcþ Metastatic Cancer Stem Cells
www.aacrjournals.org Cancer Res; 73(8) April 15, 2013 2683
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759
A subpopulation of PrPcþ CRC cells possesses CSCproperties
We next examined PrPc and CD44 coexpression patterns atthe cellular level, and determined the biologic and tumorigenic
properties of PrPc-expressing cells in CRC.We detected CD44þ
PrPcþ cells in the cryosections of CRCs by double immuno-fluorescence (IF) staining of CD44 and PrPc (Fig. 2A). Flowcytometry analysis revealed only a small percentage of PrPcþ
A
CD44
PrPc
PrPc null
9/49
23/80 27/77
22/22
PrPc lowPrPc moderatePrPc high
n = 23
n = 41
n = 34
n = 11
PrPc expression level
Nu
mb
er o
f p
atie
nts
Per
cen
tag
e su
rviv
al %
150
100
50
0
100
50
0
Grade 3 Grade 2 Grade 1
M
Non-M
**
0 1 2 3
00 20 40 60 Month
1 2 3
B
D
E F
C
Figure 1. PrPc is associated with high grade tumors and poor prognosis in patients. A, heatmap of CD44 and PrPc protein expression in CRCs (n ¼ 309patients). Data are grouped by the following tumor grades: grade 1, well-differentiated; grade 2, moderately differentiated; grade 3, poorly differentiated.CD44 and PrPc expression are semiquantitatively described as 0, negative; 1, low; 2, moderate; and 3, high. B, Kaplan–Meier plot showing the survivalpercentage of 109 patients with CRC (>5 years after surgery) grouped by PrPc expression level. The number of patients (n) in each group is indicated inparentheses. C, the PrPc expression level is positively correlated with metastasis. Patients with a concurrent metastasis (M, red) and those without ametastasis (Non-M, blue) are grouped by PrPc expression levels. y-axis, number of patients. ��, P < 0.001. D, double immunofluorescence staining ofcytokeratin 20 (green) and PrPc (red) in CRC showing preferential localization of PrPc-positive cells in the invasive fronts of tumor islets (indicated by dottedwhite lines). Scale bar, 200 mm. E, representative immunofluorescence images of PrPc expression in invasive vasculatures in the draining lymph node(white dotted outline). PrPcþ/cytokeratin 20þ tumor cells (yellow) are magnified and indicated (arrows). Scale bar, 200 mm. F, IHC staining shows PrPcþCRCcell invasion into stromal cells (red arrows). Scale bar, 100 mm.
Du et al.
Cancer Res; 73(8) April 15, 2013 Cancer Research2684
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

Figure 2. TheCD44þPrPcþ subpopulation ofCRCcells possessesCSCproperties. A, double immunofluorescence staining ofCD44 (green) andPrPc (red) in arepresentative CRC cryosection. The section was counterstained with 40,6-diamidino-2-phenylindole (DAPI; blue). Scale bar, 100 mm. B, a representative4-color flow cytometric analysis of a CRC. Cells were gated byCD45 negativity and EpCAMpositivity to remove leukocytes and stromal cells (left andmiddle)before the indicated subpopulations were analyzed and sorted out on the basis of PrPc andCD44 expression (right). C, Venn diagram illustrating the degree ofoverlap between the CD44 and PrPc populations. The percentage of each subpopulation is shown with the median values (M) and ranges indicated. D,clonogenic efficiency of indicated subpopulations freshly isolated from CRC (day 42). Single colonies are magnified to show the detailed morphology.Colonieswith diameter�0.5mmwere counted for statistical analysis. ��,P < 0.001; ���,P < 0.0001. E, representative H&E images of regenerated tumors fromCD44þPrPcþ and CD44þPrPc� subpopulations in NOD/SCID mice (day 90). Note that the cancer cells from CD44þPrPcþ subpopulation have infiltrated intotheunderlyingmuscle tissue. Scale bar, 100mm.F, growth curve of xenograft tumor initiated from1�106CD44RNAi (black), PrPcRNAi (blue), CD44andPrPcRNAi (bright red) CRC cells in nude mice (n ¼ 6). Parental (brown) and scramble (pink) shRNA cells were used as controls. �, P < 0.01; ���, P < 0.0001.
PrPcþ Metastatic Cancer Stem Cells
www.aacrjournals.org Cancer Res; 73(8) April 15, 2013 2685
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759
cells in the CD45�EpCAMþCD44þ population in patient tumors(Fig. 2B), and the abundance of CD44þPrPcþ cells ranged from0.02% to 10.83% (median 0.28%; Fig. 2C and SupplementaryTable S1). Interestingly, the subpopulation of CD44þPrPcþ cellsin the 6 patients with concomitant liver metastasis was signif-icantly higher than that in the 25 primary CRC patients withoutmetastasis (3.19% vs. 0.79%, mean, P ¼ 0.03; SupplementaryTable S1). Furthermore, PrPc and CD44 were closely associatedin the NCI-60 human tumor cell line panel (correlation coeffi-cient ¼ 0.68; Supplementary Fig. S2A–S2C).
We next asked whether PrPc is associated with CD44 incontributing to CSC properties. The abilities to form anchor-age-independent spheres/spheroids in vitro and to regenerateserially transplantable xenograft tumors in immunodeficientmice are 2 of the hallmarks of CSCs (37). We therefore usedfluorescence-activated cell sorting (FACS) to purify 4 sub-populations: CD44þPrPcþ, CD44þPrPc�, CD44�PrPcþ, andCD44�PrPc� from patient tumors (Fig. 2B). The purity of eachsubpopulation was more than 97%, as assessed by post-sortFACS and fluorescence microscopic analyses (data not shown).As expected, CD44þPrPcþ CRC cells displayed significantlygreater ability to form colonies compared with CD44þPrPc� orCD44�PrPcþ cells (P¼ 0.02 andP¼ 0.007, respectively),whereasCD44�PrPc� cancer cells failed to generate colonies (Fig. 2D).
To evaluate their tumorigenic potential, the CD44þPrPcþ,CD44þPrPc�, CD44�PrPcþ, and CD44�PrPc� subpopulationsisolated from 9 patients (i.e., cases 22–30 and SupplementaryTable S1) were subcutaneously injected intoNOD/SCID mice.As predicted by their clonogenicity data, xenograft tumors
were generated from the CD44þPrPcþ, CD44þPrPc�, andCD44�PrPcþ cells, but not from the CD44�PrPc� cells in 6 of9 cases (Supplementary Table S2). In limited-dilution assays,the CD44þPrPcþ cells displayed greater tumor-initiatingcapacity than the other subpopulations (P < 0.001) (Supple-mentary Table S2). We frequently observed that tumor cellshad invaded the underlying muscle tissue in tumors regener-ated from CD44þPrPcþ (but not in those regenerated fromCD44þPrPc�) cells (Fig. 2E). To further test their self-renewalcapabilities in vivo, 100 CD44þPrPcþ cancer cells purified froma first-generation xenograft tumor were transplanted intoNOD/SCID mice. Our findings indicated that 8 of 9 transplan-tations grew secondary xenograft tumors that recapitulatedthe phenotype of the original tumor, and even higher tumor-igenic capacities were detected in the third transplantation(Supplementary Fig. S2D and data not shown).
Similar tumor-initiating capacities were also observed innude mice. We isolated the 4 subpopulations prepurified fromCD45� tumor cells, and injected them subcutaneously intonude mice (cases 1–11, Supplementary Table S1). Tumordevelopment was observed in 8 cases (Supplementary Fig.S2E). Again, the CD44þPrPcþ CRC cells exhibited the greatesttumor-initiating capacity (P < 0.05; Supplementary Table S2).As shown in Supplementary Figure S2E, the reconstitutedtumors were histologically heterogeneous. PrPc and CD44coexpression was detected in only a minor subset of cellsin regenerated tumors (Supplementary Fig. S2F–S2G), alsosuggesting that the CD44þPrPcþ cells were capable of self-renewal.
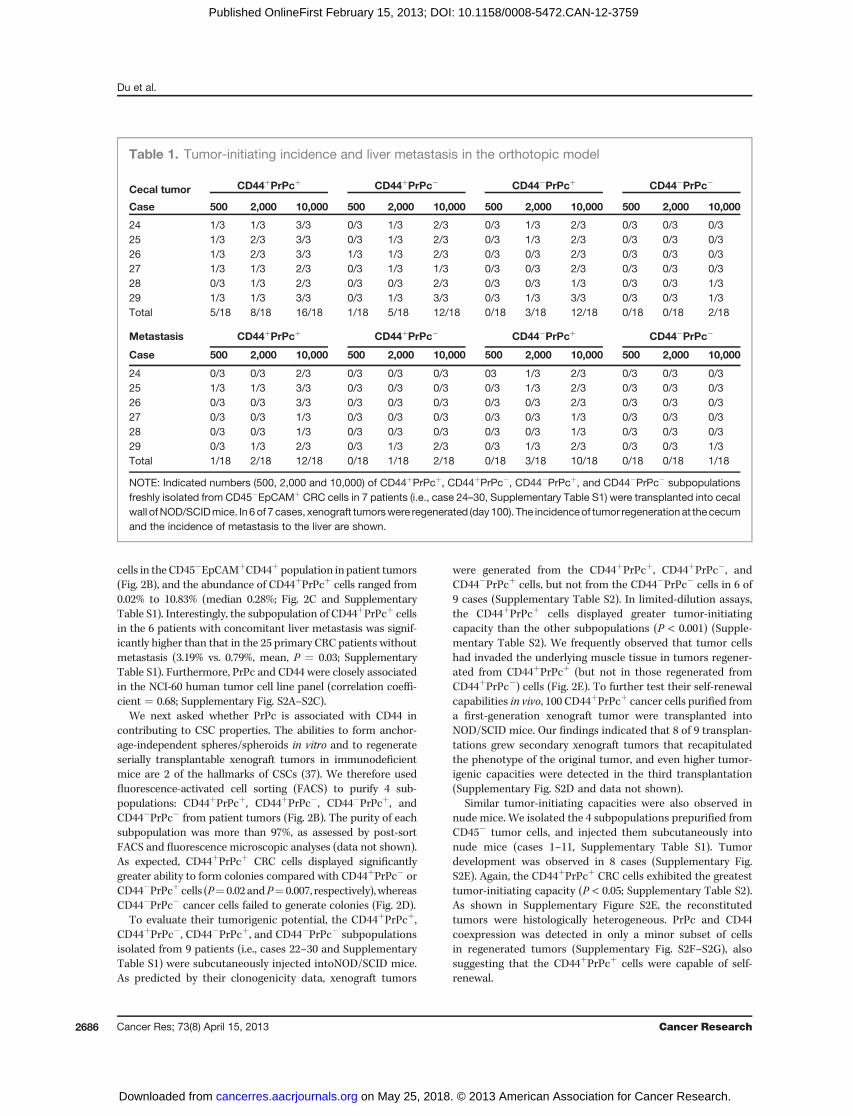
Table 1. Tumor-initiating incidence and liver metastasis in the orthotopic model
Cecal tumor CD44þPrPcþ CD44þPrPc� CD44�PrPcþ CD44�PrPc�
Case 500 2,000 10,000 500 2,000 10,000 500 2,000 10,000 500 2,000 10,000
24 1/3 1/3 3/3 0/3 1/3 2/3 0/3 1/3 2/3 0/3 0/3 0/325 1/3 2/3 3/3 0/3 1/3 2/3 0/3 1/3 2/3 0/3 0/3 0/326 1/3 2/3 3/3 1/3 1/3 2/3 0/3 0/3 2/3 0/3 0/3 0/327 1/3 1/3 2/3 0/3 1/3 1/3 0/3 0/3 2/3 0/3 0/3 0/328 0/3 1/3 2/3 0/3 0/3 2/3 0/3 0/3 1/3 0/3 0/3 1/329 1/3 1/3 3/3 0/3 1/3 3/3 0/3 1/3 3/3 0/3 0/3 1/3Total 5/18 8/18 16/18 1/18 5/18 12/18 0/18 3/18 12/18 0/18 0/18 2/18
Metastasis CD44þPrPcþ CD44þPrPc� CD44�PrPcþ CD44�PrPc�
Case 500 2,000 10,000 500 2,000 10,000 500 2,000 10,000 500 2,000 10,000
24 0/3 0/3 2/3 0/3 0/3 0/3 03 1/3 2/3 0/3 0/3 0/325 1/3 1/3 3/3 0/3 0/3 0/3 0/3 1/3 2/3 0/3 0/3 0/326 0/3 0/3 3/3 0/3 0/3 0/3 0/3 0/3 2/3 0/3 0/3 0/327 0/3 0/3 1/3 0/3 0/3 0/3 0/3 0/3 1/3 0/3 0/3 0/328 0/3 0/3 1/3 0/3 0/3 0/3 0/3 0/3 1/3 0/3 0/3 0/329 0/3 1/3 2/3 0/3 1/3 2/3 0/3 1/3 2/3 0/3 0/3 1/3Total 1/18 2/18 12/18 0/18 1/18 2/18 0/18 3/18 10/18 0/18 0/18 1/18
NOTE: Indicated numbers (500, 2,000 and 10,000) of CD44þPrPcþ, CD44þPrPc�, CD44�PrPcþ, and CD44�PrPc� subpopulationsfreshly isolated from CD45�EpCAMþ CRC cells in 7 patients (i.e., case 24–30, Supplementary Table S1) were transplanted into cecalwall ofNOD/SCIDmice. In 6of 7 cases, xenograft tumorswere regenerated (day100). The incidenceof tumor regenerationat the cecumand the incidence of metastasis to the liver are shown.
Du et al.
Cancer Res; 73(8) April 15, 2013 Cancer Research2686
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

Figure 3. PrPc confers greater metastatic capacity. A, 10,000 freshly sorted CD44þPrPcþ cells from CRCs were transplanted into the cecal wall of NOD/SCIDmice. The regenerated cecal tumor (black arrow) and the liver metastatic nodules (white arrow) are shown. B, metastatic nodules in the liver formed afterinjecting 2,000 of the cells into the cecal wall (day 100). Metastatic cancer cells are distinguished from normal mouse liver tissue by having a highernucleus/cytoplasm ratio. Scale bar, 300 mm. C, metastatic efficiency. The number of cecal tumors with liver metastasis (white bar) vs. cecal tumors withoutmetastasis (black bar) determined 100 days after CRC cell injections. D, metastatic areas were determined by Image-Pro Plus software based on H&Estaining. The metastatic area was normalized to CD44þPrPcþ cells. ��, P < 0.001. E, PrPcþ cells exhibit a higher metastatic index than PrPc� cells.Metastatic lesions were determined under a microscope on the basis of H&E staining, and the metastatic index was defined as the number of metastaticnodules per primary tumor volume. ���, P < 0.0001. F, cell migration assays with the indicated subpopulations freshly isolated from CRCs. Each samplewas analyzed in triplicate, and the experiment was carried out using 7 patient tumors. ���, P < 0.0001. G, PrPc and CD44 expression in migrated vs.nonmigrated CRC cells determined by flow cytometry analysis. This experiment was conducted in triplicate using 6 patient tumors.
PrPcþ Metastatic Cancer Stem Cells
www.aacrjournals.org Cancer Res; 73(8) April 15, 2013 2687
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759
Du et al.
Cancer Res; 73(8) April 15, 2013 Cancer Research2688
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759
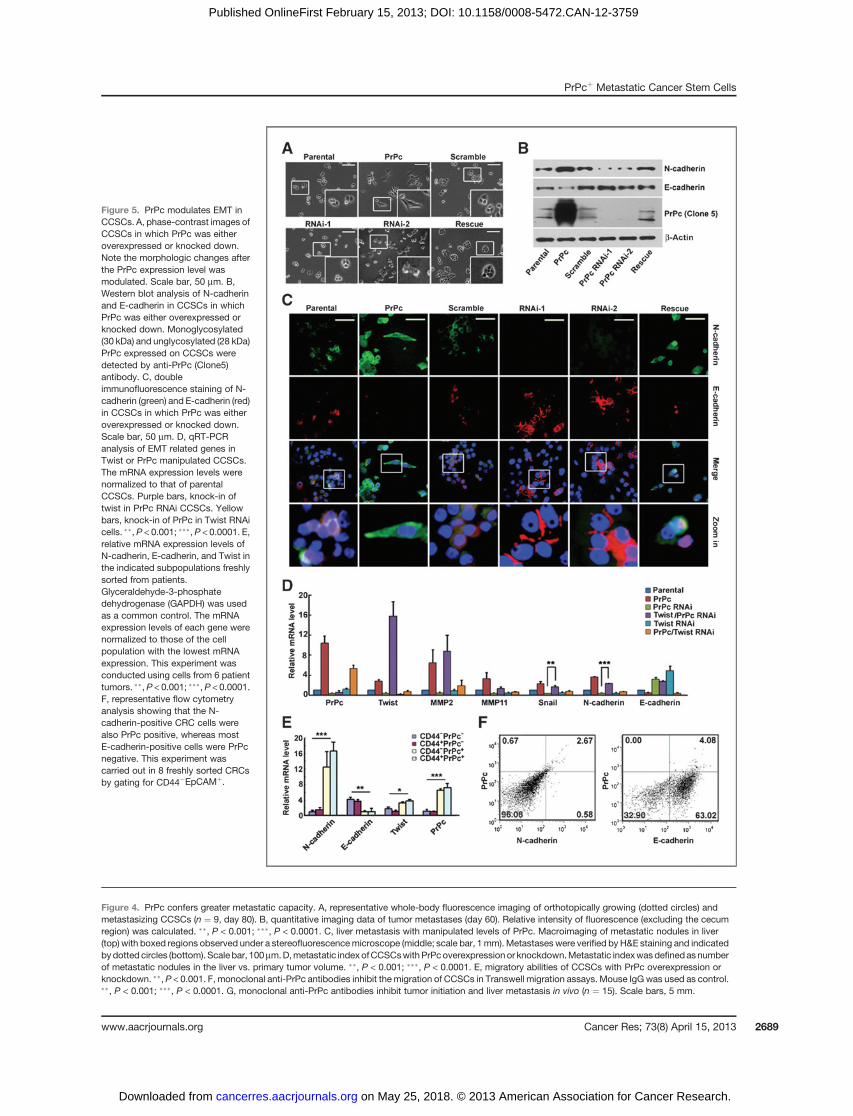
Figure 4. PrPc confers greater metastatic capacity. A, representative whole-body fluorescence imaging of orthotopically growing (dotted circles) andmetastasizing CCSCs (n ¼ 9, day 80). B, quantitative imaging data of tumor metastases (day 60). Relative intensity of fluorescence (excluding the cecumregion) was calculated. ��, P < 0.001; ���, P < 0.0001. C, liver metastasis with manipulated levels of PrPc. Macroimaging of metastatic nodules in liver(top) with boxed regions observed under a stereofluorescencemicroscope (middle; scale bar, 1mm). Metastaseswere verified byH&E staining and indicatedbydotted circles (bottom). Scale bar, 100mm.D,metastatic indexofCCSCswithPrPcoverexpression or knockdown.Metastatic indexwasdefinedasnumberof metastatic nodules in the liver vs. primary tumor volume. ��, P < 0.001; ���, P < 0.0001. E, migratory abilities of CCSCs with PrPc overexpression orknockdown. ��, P < 0.001. F, monoclonal anti-PrPc antibodies inhibit themigration of CCSCs in Transwell migration assays. Mouse IgG was used as control.��, P < 0.001; ���, P < 0.0001. G, monoclonal anti-PrPc antibodies inhibit tumor initiation and liver metastasis in vivo (n ¼ 15). Scale bars, 5 mm.
Figure 5. PrPc modulates EMT inCCSCs. A, phase-contrast images ofCCSCs in which PrPc was eitheroverexpressed or knocked down.Note the morphologic changes afterthe PrPc expression level wasmodulated. Scale bar, 50 mm. B,Western blot analysis of N-cadherinand E-cadherin in CCSCs in whichPrPc was either overexpressed orknocked down. Monoglycosylated(30 kDa) and unglycosylated (28 kDa)PrPc expressed on CCSCs weredetected by anti-PrPc (Clone5)antibody. C, doubleimmunofluorescence staining of N-cadherin (green) and E-cadherin (red)in CCSCs in which PrPc was eitheroverexpressed or knocked down.Scale bar, 50 mm. D, qRT-PCRanalysis of EMT related genes inTwist or PrPc manipulated CCSCs.The mRNA expression levels werenormalized to that of parentalCCSCs. Purple bars, knock-in oftwist in PrPc RNAi CCSCs. Yellowbars, knock-in of PrPc in Twist RNAicells. ��, P < 0.001; ���,P < 0.0001. E,relative mRNA expression levels ofN-cadherin, E-cadherin, and Twist inthe indicated subpopulations freshlysorted from patients.Glyceraldehyde-3-phosphatedehydrogenase (GAPDH) was usedas a common control. The mRNAexpression levels of each gene werenormalized to those of the cellpopulation with the lowest mRNAexpression. This experiment wasconducted using cells from 6 patienttumors. ��,P < 0.001; ���,P < 0.0001.F, representative flow cytometryanalysis showing that the N-cadherin-positive CRC cells werealso PrPc positive, whereas mostE-cadherin-positive cells were PrPcnegative. This experiment wascarried out in 8 freshly sorted CRCsby gating for CD44�EpCAMþ.
PrPcþ Metastatic Cancer Stem Cells
www.aacrjournals.org Cancer Res; 73(8) April 15, 2013 2689
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

Du et al.
Cancer Res; 73(8) April 15, 2013 Cancer Research2690
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

As in leukemic stem cells (15), knocking down CD44 in CRCcells significantly inhibited tumor regeneration (14). To inves-tigate the role of PrPc in tumor initiation, we isolated primaryCRC cells from patients and cultured them in ultra-low attach-ment dishes without serum. Remarkably, PrPc knockdown inthe CRC cells also retarded tumor growth in nude mice,although the inhibitory effect was slightly less apparent thanwith CD44 knockdown (Fig. 2F). However, combined knock-down of both CD44 and PrPc completely abolished tumordevelopment (Fig. 2F). Taken together, these data suggest thatthe CD44þPrPcþ subpopulation of CRC cells does possess CSCproperties.
PrPcþ CSC is more metastaticBecause PrPc is highly expressed in metastatic tissues, we
attempted to find out whether it contributes to metastasisin CRC. To recapitulate human cancers with greater fidelity,we transplanted 500, 2,000 and 10,000 cells from each of theCD44þPrPcþ, CD44þPrPc�, CD44�PrPcþ and CD44�PrPc�
subpopulations isolated from 7 patients (i.e., case 24–30,Supplementary Table S1), into the cecal wall of NOD/SCIDmice (38). As shown in Table 1, 6 of the 7 patient-derivedCRC cells produced cecal tumors, which was consistentwith the results obtained from subcutaneously-implantedtumor cells (Supplementary Table S2). The CD44þPrPcþ
cells clearly formed tumors with the greatest efficiency(P < 0.01; Table 1 and Supplementary Table S2). Remarkably,macroscopic and microscopic analysis revealed that livermetastases were almost exclusively observed in the miceimplanted with PrPcþ cells (i.e., CD44þPrPcþ and CD44�
PrPcþ; Fig. 3A–C; Table 1). Furthermore, semiquantitativemeasurement of the metastatic area and the metastatic indexconfirmed that the tumors derived from PrPcþ CRC cells werehighly metastatic (Fig. 3D–E).The spleen transplantation model is another commonly
used approach to test the proliferative andmetastatic potentialof CSCs (39, 40). CD44þPrPcþ, CD44þPrPc�, CD44�PrPcþ, andCD44�PrPc� subpopulations were sorted from the tumors of10 patients (i.e., cases 12–21, Supplementary Table S1), and 500cells from each subpopulation were then transplanted into thespleen of nude mice. Similar to the results obtained using theorthotopic model, liver metastasis occurred within 70 days in28 of 30 cases of CD44þPrPcþ, 3 of 30 cases of CD44þPrPc�, 15of 30 cases of CD44�PrPcþ, and 2 of 30 cases of CD44�PrPc�
cells (Supplementary Fig. S3A–S3B). These results suggest thatPrPcþ is intimately associated with the CRC liver metastasis.Transwell analysis showed that CD44þPrPcþ cells displayedabout 16-fold greater migratory ability than CD44þPrPc� cells(Fig. 3F).Most of themigrating cells (84.25� 3.14%)were foundto be PrPc-positive, versus only 1.25� 0.22% of the nonmigrant
cells (Fig. 3G). Collectively, these observations imply that PrPcis crucial for CCSCmigration in vitro and for metastasis in vivo.
PrPc is functionally important for cell migration andcancer metastasis
Because CSC is generally rare and it is difficult to label andtrace its growth in animals, we generated a cell line of CD44þ
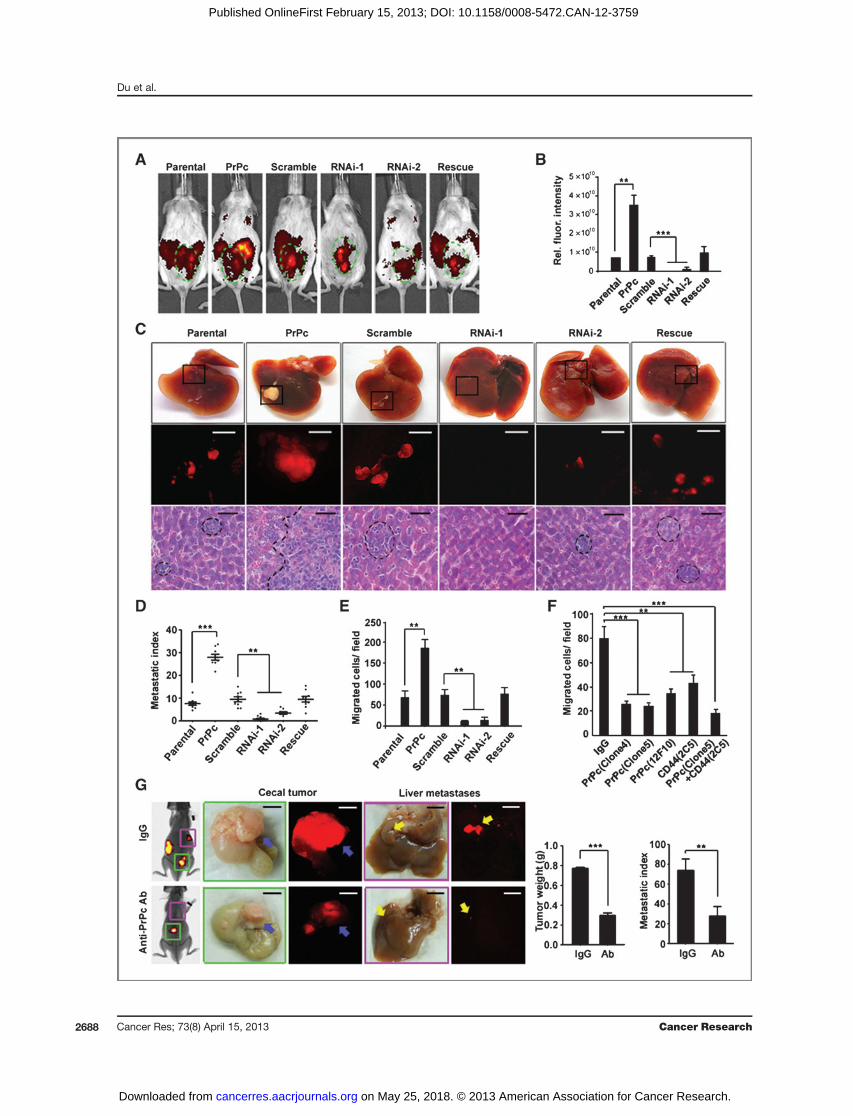
CCSCs isolated from a CRC patient. These cells have all thecharacteristics of CSCs including constitutive expression ofCD44, and the capacity to form spheres and generate xenografttumors (14). The CCSCs were grown in ultra-low attachmentdishes without serum, and were subcultured once a week forfewer than 20 passages. To investigate the unique role of PrPcin metastasis, we used this cell line prelabeled with DsRed andinwhich PrPc had either been knocked downby shRNAs orwasoverexpressed via the pBABE-PURO retrovirus system. Asshown in Fig. 4A–C, PrPc knockdown strongly inhibited tumorgrowth and liver metastasis as assessed by whole-body fluo-rescence imaging and hematoxylin and eosin (H&E) staining,suggesting that PrPc play a central role in CSC metastasis. Incontrast, both tumor growth and liver metastasis were pro-moted by PrPc overexpression. Moreover, the level of PrPcexpression was correlated with the metastatic index (Fig. 4D).Similar results were observed using the spleen metastasismodel (Supplementary Fig. S4A–S4C). In addition, Transwellmigration assays revealed that the levels of PrPc correlatedwith themigratory capacity of CCSCs (Fig. 4E). It is noteworthythat the PrPc levels did not alter the cell-cycle profiles (Sup-plementary Fig. S4D). Using CRC cells with CSC properties, wewere able to show that PrPc is functionally required for themigration of CCSCs and contributes to CRC metastasis.
PrPc antibodies inhibit migration and metastasisSurface markers unique to CSCs would provide ideal ther-
apeutic targets on these cells, and indeed monoclonal anti-bodies have shown therapeutic efficacy in both preclinicaltumormodels andpatientswith cancer (41).Wewere thereforeinterested in addressing the question of whether PrPc could beused as a therapeutic target, in a manner similar to CD44 (15).We generated several monoclonal antibodies against PrPc, outof which we selected the IgG1 antibody. The specificity of theantibodies was verified by ELISA, Western blotting (WB), andIHC staining (Supplementary Fig. S4E–S4G). The migration ofCD44þ CCSCs in vitro was strongly inhibited after they hadbeen treatedwith 1mg/mL anti-PrPc antibodies. The inhibitoryeffect obtained was greater than that associated with the anti-CD44 antibody (�1.7 fold). A combination of 1 mg/mL anti-CD44 and 1 mg/mL anti-PrPc antibodies was able to inhibitmigration by up to approximately 80% (Fig. 4F); whereas PrPc
Figure 6. PrPc-regulated EMT is ERK2 dependent. A, Western blot analysis of the proteins indicated in CCSCs in which PrPc was either overexpressed orknocked down. B, parental CCSCs and those overexpressing PrPcwere treated with the ERK2 inhibitor U0126 for 24 hours, and awhole-cell lysate was usedfor Western Blot analysis of the molecules indicated. C, CCSCs were treated with 5 mg/mL anti-PrPc antibody (Ab), 2 mg/ml laminin (LN), and a combinationof Ab and LN for 24 hours, after which cells were harvested for Western Blot analysis. D, phase-contrast images of CCSCs maintained under the conditionsindicated. Note that the anti-PrPc antibodies rescued the morphologic changes in the presence of LN. Scale bar, 100 mm. E, Double immunofluorescencestaining of N-cadherin (green) and E-cadherin (red) in CCSCs in the presence of 10 mmol/L U0126, 2 mg/mL LN, and indicated dose of anti-PrPcantibodies (mg/mL). Scale bar, 20 mm. F, migration of CCSCs in the presence of U0126, 2 mg/mL LN, and indicated dose of anti-PrPc antibodies (mg/mL).
PrPcþ Metastatic Cancer Stem Cells
www.aacrjournals.org Cancer Res; 73(8) April 15, 2013 2691
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

antibodies did not show any effect on cell migration in PrPcknockdown cells (data not shown).
It was essential to find out whether PrPc monoclonal anti-bodies can prevent CRC metastases in vivo. NOD/SCID micebearing orthotopic implantations of CCSCs were administered200 mg antibodies intraperitoneally on a weekly basis. ELISAanalysis revealed that the concentration of anti-PrPc antibodyin the serum of tumor-bearing animals was maintained at 0.87to 1 mg/mL (data not shown). The orthotopic and metastatictumors were monitored by whole-body fluorescence imagingevery week and after 8 to 10weeks themice were sacrificed andunderwent pathology examinations. The presence of anti-PrPcantibodies dramatically decreased tumorigenicity and livermetastases (Fig. 4G), whereas their bodyweight did notdecrease (not shown). CCSCs precoated with anti-PrPc anti-bodies before orthotopic implantation produced a similarinhibitory effect (Supplementary Fig. S4H).
Tomake sure that the observedmetastasis-inhibiting effectsof anti-PrPc antibodies were not due to inhibition of primarytumor growth, we preinoculated the CCSCs orthotopically intothe cecal wall for 3 weeks. Mice with equivalent tumors werethen grouped and injected with 100 or 200 mg antibodiesweekly. The results clearly showed that the anti-PrPc antibodyinhibited tumor growth and metastasis at the lower dosage(100 mg/week) and eliminated tumor growth at higher dosage(200 mg/week; Supplementary Fig. S4I).
PrPc promotes EMT in CCSCsWe consistently observed that the PrPc-overexpressing
CCSCs exhibited an elongated mesenchymal-like morphology,whereas the PrPc knockdown cells were flatter (Fig. 5A; Sup-plementaryFig. S4G).Also, PrPcþCRCcells generatedbranchingcolonies with loose cell–cell interaction (see Fig. 2D), charac-teristic of the EMT phenotype. This prompted us to investigatewhether PrPc promotes EMT, a known mechanism of CSCmigration and invasion. Cells undergoing EMT retain stemcell functionality and form invasive front colonies (42). We firstused quantitative reverse transcription-PCR (qRT-PCR; primersshown in Supplementary Table S3) to measure several EMT-related genes by manipulating PrPc levels. In PrPc overexpres-sing cells, mesenchymal genes such as N-cadherin and Twistwere significantlyupregulated,whereasE-cadherin, anepithelialmarker, was downregulated (Supplementary Fig. S5A). In addi-tion, there were increased levels of matrix metalloproteinase 2(MMP2) andMMP11 (Supplementary Fig. S5A), whichmay havecontributed to the metastasis observed in PrPc overexpressingcells. In contrast, knockdown of PrPc resulted in a reversedexpression pattern (Supplementary Fig. S5A). Western blotanalysis and double immunofluorescence staining further con-firmed the correlation of PrPc with the expression of EMT-related molecules (Fig. 5B and C; Supplementary Fig. S5B).
Twist is a key transcription factor regulating mesenchymalcell fate, differentiation, andmorphogenesis (43). Because PrPcexpression had been found to influence the expression of Twist(Supplementary Fig. S5A), we reasoned that PrPc might reg-ulate EMT via Twist. To test this, Twist was ectopicallyexpressed in CCSCs in which PrPc was stably knocked downby shRNA. We observed that constitutively overexpressed
Twist was indeed able to induce EMT in PrPc-knocked downCCSCs, leading to increased levels of Snail and N-cadherin(Fig. 5D). However, overexpression of PrPc in the Twist RNAicells was unable to restore EMT, probably because Snail andN-cadherin expression was lower than in the parental cells(Fig. 5D). Overall, we conclude that PrPc regulates the EMTphenotype by modulating Twist.
Next, we used qRT-PCR to measure the mRNA levels ofTwist, N-cadherin, and E-cadherin in CD44þPrPcþ, CD44þ
PrPc�, CD44�PrPcþ, and CD44�PrPc� subpopulations of CRCcells freshly purified from 6 patients. The results showed thatboth Twist and N-cadherin were expressed at significantlyhigher levels in the PrPc-positive (i.e. CD44þPrPcþ and CD44�
PrPcþ) CRC cells than in the PrPc� subpopulations (Fig. 5E).Furthermore, approximately 82% of N-cadherinþ cells werealso PrPcþ, whereas more than 93% of E-cadherinþ cells werePrPc� in CD45�EpCAMþ CRC cells as analyzed by flow cyto-metry (Fig. 5F). These findings suggest that PrPc is positivelycorrelated with the mesenchymal properties of cells from CRCpatients.
Evidence that PrPc-induced EMT is ERK2 dependentEMT is regulated by the TGF-b and ERK signaling pathways
(44). To understand the molecular mechanisms of PrPc-induced EMT, we first assessed the activation of these 2pathways (45). Knocking down PrPc seemed to result in areduction of both Phospho-smad2 and ERK1/2; however, over-expression of PrPc activated ERK1/2 but not smad2, suggestingthat ERK signaling may act downstream of PrPc (Fig. 6A). Insupport of this, we found that U0126, an inhibitor of ERKsignaling, suppressed the expression of several EMT-associat-ed molecules (Fig. 6B) as well as EMT itself, especially whenPrPc was overexpressed.
Laminin is a PrPc ligand (46), which, we reasoned, couldactivate events occurring downstream of PrPc, such as EMT.Laminin did enhance both ERK2phosphorylation and the EMTphenotype (Fig. 6C and D). Interestingly, PrPc antibodyblocked the laminin-induced ERK activation, EMT proteinexpression and EMTmorphology in a dose-dependentmanner,(Fig. 6C and D), probably via interference with the interactionbetween laminin and its receptor PrPc. Immunofluorescencestaining confirmed that the anti-PrPc antibody attenuated thelaminin-induced N-cadherin expression (Fig. 6E). In addition,Transwell migration assays revealed that U0126 blocked thePrPc overexpression-inducedmigration, and that the anti-PrPcantibody also attenuated laminin-enhanced migration (Fig.6F). Our findings thus suggest that ERK2 signaling pathway isinvolved in PrPc-induced EMT.
DiscussionIt has been shown that CSCs contribute to both tumor
initiation and cancer metastasis in many tumor systems.However, in most cases, the underlying mechanisms are notwell understood. In this study, we have identified a distinctPrPcþ subpopulation in CD44þ CCSCs that displays enhancedcapacity to regenerate tumors. These results emphasize thefunctionalities of PrPc as an "enrichment" factor for CCSC. Ourdata show that PrPc is a novel CCSCbiomarker, which, by itself,
Du et al.
Cancer Res; 73(8) April 15, 2013 Cancer Research2692
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

functionally contributes to both CRC tumorigenicity andmetastasis. Interestingly, the PrPcþ CRC cells tend to localizeat the invasion fronts in patient tumors, are more mesenchy-mal, and behave like other cancer cells undergoing EMT,properties that have probably given these cells a greatercapacity to disseminate and metastasize. These observationsindicate PrPc's role in regulating CCSC likely occurringthrough its ability to promote EMT and the exact mechanismwarrants future investigations. In terms of the mechanisminvolved, our work implies that PrPc may regulate EMTthrough ERK2- and Twist-dependent signaling pathways.Importantly, the interaction of PrPc with its ligand, laminin,directly activates ERK2 signaling and promotes EMT. There-fore, conceptually, interference with PrPc expression/func-tions, the PrPc-laminin interaction or its downstream signalingevents,may be a promising avenue in developing new therapiestargeting PrPcþ CRC cell-mediated metastasis.Previous work has shown that PrP1, a PrPc ortholog in the
zebra fish, regulates the stability of E-cadherin mediated celladhesion (47), suggesting that PrPc expression may endow thecell with properties of EMT, which is a developmentallyconserved regulatory mechanism. In addition, PrPc-null(PRNP0/0) cells display less migratory activity than PRNPþ/þ
cells (48). The more aggressive phenotype of PRNPþ/þ cells isconsistent with our observations that PrPc contributes toCCSC migration. Recently, CD26þ CRC cells have also beenfound to possess CSC properties, with greatly increased met-astatic capacity, and, intriguingly, like PrPc, CD26 also func-tions as an adhesion molecule that binds to ECM (24). Thesefindings, together with our present study, strongly suggest thatthe enhanced tumorigenic and metastatic capacities of CCSCsare probably determined by the CSC-intrinsic expression offunctional molecules such as PrPc and CD26, which in turnmediate the interactions of CSCswith theirmicroenvironment.Future work will attempt to elucidate the potential relation-ship between PrPcþ and CD26þ CCSCs.Previous studies have shown that the expression of PrPc is
positively correlated with invasiveness and drug resistance inboth gastric and breast cancers (27, 49). In addition, Liao andcolleagues have reported that endoglin and PrPc are enrichedin mammosphere-initiating cells (50). Although PrPc has beenreported to be highly expressed in some CRCs (51), as far as weare aware this is the first report linking PrPc to CCSCs, and ourdata help to explain how PrPc is associated with cancermetastasis. The level of PrPc was inversely correlated withpatient survival, suggesting a potential prognostic value inCRC. It has recently emerged that monoclonal antibodies
targeting surface molecules expressed in CSCs show powerfultherapeutic efficacy by eliminating CSCs and leading totumor resolution (16). Our findings therefore have significantclinical implications, as PrPcþ CSCs may provide importantnew targets for therapy in metastatic CRC. Indeed, PrPcmonoclonal antibodies display strong inhibitory effects onCCSC tumorigenesis and metastasis in orthotopic xenograftmodels, suggesting possible therapeutic applications in theclinic. Overall, our findings throw new light on the mechan-isms underlying CRC cell metastasis and are of clinicalimportance for the design of therapeutic strategies for treat-ing CRC.
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: L. Du, B. Laffin, M. Mehrpour, Q. ChenDevelopment of methodology: L. Du, G. Rao, B. Li, Y. Zhu, D.G. TangAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.):G. Rao, H.Wang, B. Li, W. Tian, J.T. Cui, L. He, X. Tian,C. Hao, H. Liu, Y. Zhu, D.G. Tang, Y. LuAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): L. Du, G. Rao, H. Wang, B. Li, B. Laffin, D.G. Tang,M. MehrpourWriting, review, and/or revision of the manuscript: L. Du, G. Rao, B. Li,M. Mehrpour, Y. Lu, Q. ChenAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): G. Rao, B. Li, X. Sun, D.G. TangStudy supervision: Y. Lu, Q. Chen
AcknowledgmentsThe authors would like to thank Drs. G. Ge and K.-J. Wu for providing Twist
plasmids, Dr. S. L. Holbeck for providing CMA slides, Dr. J. Grassi for providingantibody against PrPc (SAF69), Mrs. X. Zhang, T. Zhao, J. Wang, and L. Dou forFACS assistance, Mrs. Y. Zhou and X. Shi for whole-body fluorescence imagingassays, Dr. W. Zhang and Dr. C. Han for biostatistical analysis, Dr. A. Lu, Dr. W.Xue, and Dr. L. Zhou for pathologic determination, and Dr. A. Zhou for hissuggestions and revision of our manuscript.
Grant SupportThis work was supported by initiative of stem cell and regenerative medicine
from Chinese Academy of Sciences (XDA01040409), the 973 project from theMinistry of Science and Technology (MOST) of China (2009CB512800 and2010CB912204), and a grant from the National Natural Science Foundation ofChina (NSFC; 81130045 to Q. Chen and 31000614 to L. Du), from MOST2010CB529403 (X. Tian), MOST 2010CB912204 (Y. Zhu), and from InstitutNational de la Sant�e et de la Recherche M�edicale, Partenariat Hubert Curien- Programme Francais de Coop�eration avec la Chine, Fondation Franco-Chinoisepour la Science et ses Applications (M. Mehrpour).
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received September 26, 2012; revised January 31, 2013; accepted February 2,2013; published OnlineFirst February 15, 2013.
References1. Sporn MB. The war on cancer. Lancet 1996;347:1377–81.2. Jemal A, Thun MJ, Ward EE, Henley SJ, Cokkinides VE, Murray TE.
Mortality from leading causes by education and race in the UnitedStates, 2001. Am J Prev Med 2008;34:1–8.
3. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cellbiology to cancer. Nat Rev Cancer 2003;3:895–902.
4. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as ahierarchy that originates from a primitive hematopoietic cell. Nat Med1997;3:730–7.
5. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF.Prospective identification of tumorigenic breast cancer cells. ProcNatlAcad Sci U S A 2003;100:3983–8.
6. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al.Identification of a cancer stem cell in human brain tumors. Cancer Res2003;63:5821–8.
7. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S,et al. Identification of bronchioalveolar stem cells in normal lung andlung cancer. Cell 2005;121:823–35.
PrPcþ Metastatic Cancer Stem Cells
www.aacrjournals.org Cancer Res; 73(8) April 15, 2013 2693
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

8. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, et al. Phenotypiccharacterization of humancolorectal cancer stemcells. ProcNatl AcadSci U S A 2007;104:10158–63.
9. Ricci-Vitiani L, Lombardi DG, Pilozzi E, BiffoniM, TodaroM, PeschleC.Identification and expansion of human colon-cancer-initiating cells.Nature 2007;445:111–5.
10. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cellcapable of initiating tumour growth in immunodeficient mice. Nature2007;445:106–10.
11. Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, Appelman H,et al. Aldehyde dehydrogenase 1 is a marker for normal andmalignant human colonic stem cells (SC) and tracks SC over-population during colon tumorigenesis. Cancer Res 2009;69:3382–9.
12. Takeda K, Kinoshita I, Shimizu Y, Matsuno Y, Shichinohe T, Dosaka-Akita H. Expression of LGR5, an Intestinal Stem Cell Marker, DuringEach Stage of Colorectal Tumorigenesis. Anticancer Res 2011;31:263–70.
13. Vermeulen L, Todaro M, de Sousa MF, Sprick MR, Kemper K, PerezAM, et al. 14. Single-cell cloning of colon cancer stem cells reveals amulti-lineage differentiation capacity. Proc Natl Acad Sci U S A2008;105:13427–32.
14. Du L,Wang H, He L, Zhang J, Ni B, Wang X, et al. CD44 is of functionalimportance for colorectal cancer stem cells. Clin Cancer Res2008;14:6751–60.
15. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44eradicates human acute myeloid leukemic stem cells. Nat Med2006;12:1167–74.
16. Majeti R. Monoclonal antibody therapy directed against human acutemyeloid leukemia stem cells. Oncogene 2011;30:1009–19.
17. Dalerba P, Clarke MF. Cancer stem cells and tumor metastasis: firststeps into uncharted territory. Cell Stem Cell 2007;1:241–2.
18. WichaMS.Cancer stemcells andmetastasis: lethal seeds.ClinCancerRes 2006;12:5606–7.
19. Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, et al.Distinct populations of cancer stem cells determine tumor growth andmetastatic activity in human pancreatic cancer. Cell Stem Cell2007;1:313–23.
20. Gao AC, LouW, Dong JT, Isaacs JT. CD44 is a metastasis suppressorgene for prostatic cancer located on human chromosome 11p13.Cancer Res 1997;57:846–9.
21. Bankfalvi A, Krassort M, Buchwalow IB, Vegh A, Felszeghy E, Piffko J.Gains and losses of adhesion molecules (CD44, E-cadherin, and beta-catenin) during oral carcinogenesis and tumour progression. J Pathol2002;198:343–51.
22. Jothy S. CD44 and its partners in metastasis. Clin Exp Metastasis2003;20:195–201.
23. Martin TA, Harrison G, Mansel RE, Jiang WG. The role of the CD44/ezrin complex in cancer metastasis. Crit Rev Oncol Hematol2003;46:165–86.
24. Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK, et al.A subpopulation of CD26þ cancer stem cells with metastaticcapacity in human colorectal cancer. Cell Stem Cell 2010;6:603–15.
25. Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M,et al. Mice devoid of PrP are resistant to scrapie. Cell 1993;73:1339–47.
26. Klein MA, Aguzzi A. The neuroimmune interface in prion diseases.News Physiol Sci 2000;15:250–5.
27. Mehrpour M, Codogno P. Prion protein: from physiology to cancerbiology. Cancer Lett 2010;290:1–23.
28. Aguzzi A, Polymenidou M. Mammalian prion biology: one century ofevolving concepts. Cell 2004;116:313–27.
29. Dodelet VC, Cashman NR. Prion protein expression in human leuko-cyte differentiation. Blood 1998;91:1556–61.
30. Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein isexpressed on long-term repopulating hematopoietic stem cells andis important for their self-renewal. Proc Natl Acad Sci U S A2006;103:2184–9.
31. Tang Z, Zhao M, Ji J, Yang G, Hu F, He J, et al. Overexpression ofgastrin and c-met protein involved in human gastric carcinomas andintestinal metaplasia. Oncol Rep 2004;11:333–9.
32. Wang HY, Zhang JY, Cui JT, Tan XH, Li WM, Gu J, et al. Expressionstatus of S100A14 and S100A4 correlates with metastatic potentialand clinical outcome in colorectal cancer after surgery. Oncol Rep2010;23:45–52.
33. Ki DH, Jeung HC, Park CH, Kang SH, Lee GY, Lee WS, et al. Wholegenome analysis for liver metastasis gene signatures in colorectalcancer. Int J Cancer 2007;121:2005–12
34. Koinuma K, Yamashita Y, Liu W, Hatanaka H, Kurashina K, Wada T,et al. Epigenetic silencing of AXIN2 in colorectal carcinoma withmicrosatellite instability. Oncogene 2006;25:139–46.
35. Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, et al.Experimentally derived metastasis gene expression profile predictsrecurrence and death in patients with colon cancer. Gastroenterology2010;138:958–68
36. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A,et al. The EMT-activator ZEB1 promotes tumorigenicity by repres-sing stemness-inhibiting microRNAs. Nat Cell Biol 2009;11:1487–95.
37. Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr HA,Delaloye JF, et al. Interactions between cancer stem cells and theirniche govern metastatic colonization. Nature 2012;481:85–9.
38. Fu X, Besterman JM, Monosove A, Hoffman RM. Models of humanmetastatic colon cancer in nude mice orthotopically constructed byusing histologically intact patient specimens. Proc Natl Acad Sci U SA1991;88:9345–9.
39. Brand MI, Casillas S, Dietz DW, Milsom JW, Vladisavljevic A. Devel-opment of a reliable colorectal cancer liver metastasis model. J SurgRes 1996;63:425–32.
40. Giavazzi R, Jessup JM, Campbell DE, Walker SM, Fidler IJ. Experi-mental nude mouse model of human colorectal cancer liver metasta-ses. J Natl Cancer Inst 1986;77:1303–8.
41. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S,et al. Anti-CD47 antibody synergizes with rituximab to promotephagocytosis and eradicate non-Hodgkin lymphoma. Cell2010;142:699–713.
42. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion:migrating cancer stem cells - an integrated concept of malignanttumour progression. Nat Rev Cancer 2005;5:744–9.
43. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C,et al. Twist, a master regulator of morphogenesis, plays an essentialrole in tumor metastasis. Cell 2004;117:927–39.
44. Zavadil J, B€ottinger EP. TGF-b and epithelial-to-mesenchymal transi-tions. Oncogene 2005;24:5764–74.
45. Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 2010;38:114–27.
46. Rieger R, Edenhofer F, Lasmezas CI, Weiss S. The human 37-kDalaminin receptor precursor interactswith the prion protein in eukaryoticcells. Nat Med 1997;3:1383–8.
47. Malaga-Trillo E, Sempou E. PrPs: Proteins with a purpose: Lessonsfrom the zebrafish. Prion 2009;3:129–33.
48. Muras AG, Hajj GN, Ribeiro KB, Nomizo R, Nonogaki S, Chammas R,et al. Prion protein ablation increases cellular aggregation and embo-lization contributing to mechanisms of metastasis. Int J Cancer2009;125:1523–31.
49. Pan Y, Zhao L, Liang J, Liu J, Shi Y, Liu N, et al. Cellular prion proteinpromotes invasion and metastasis of gastric cancer. FASEB J2006;20:1886–8.
50. Liao MJ, Zhang CC, Zhou B, Zimonjic DB, Mani SA, Kaba M, et al.Enrichment of a population ofmammary gland cells that formmammo-spheres and have in vivo repopulating activity. Cancer Res2007;67:8131–8.
51. Antonacopoulou AG, Palli M, Marousi S, Dimitrakopoulos FI, Kyria-kopoulou U, Tsamandas AC, et al. Prion protein expression and theM129V polymorphism of the PRNP gene in patients with colorectalcancer. Mol Carcinog 2010;49:693–9.
Du et al.
Cancer Res; 73(8) April 15, 2013 Cancer Research2694
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759

2013;73:2682-2694. Published OnlineFirst February 15, 2013.Cancer Res Lei Du, Guanhua Rao, Hongyi Wang, et al. Contribute to Metastatic Capacity in Colorectal CancerCD44-Positive Cancer Stem Cells Expressing Cellular Prion Protein
Updated version
10.1158/0008-5472.CAN-12-3759doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2013/02/14/0008-5472.CAN-12-3759.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/73/8/2682.full#ref-list-1
This article cites 51 articles, 14 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/73/8/2682.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/73/8/2682To request permission to re-use all or part of this article, use this link
on May 25, 2018. © 2013 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 15, 2013; DOI: 10.1158/0008-5472.CAN-12-3759