wee1 is a biological target of the mir-17-92 cluster in leukemia
TRANSCRIPT

Loyola University ChicagoLoyola eCommons
Master's Theses Theses and Dissertations
2011
WEE1 Is a Biological Target of the miR-17-92Cluster in LeukemiaSonia Susan OlikaraLoyola University Chicago
This Thesis is brought to you for free and open access by the Theses and Dissertations at Loyola eCommons. It has been accepted for inclusion inMaster's Theses by an authorized administrator of Loyola eCommons. For more information, please contact [email protected].
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 License.Copyright © 2011 Sonia Susan Olikara
Recommended CitationOlikara, Sonia Susan, "WEE1 Is a Biological Target of the miR-17-92 Cluster in Leukemia" (2011). Master's Theses. Paper 570.http://ecommons.luc.edu/luc_theses/570

LOYOLA UNIVERSITY CHICAGO
WEE1 IS A BIOLOGICAL TARGET OF
THE MIR-17-92 CLUSTER IN LEUKEMIA
A THESIS SUBMITTED TO
THE FACULTY OF THE GRADUATE SCHOOL
IN CANDIDACY FOR THE DEGREE OF
MASTER OF SCIENCE
PROGRAM IN MOLECULAR BIOLOGY
BY
SONIA OLIKARA
CHICAGO, ILLINOIS
DECEMBER 2011

Copyright by Sonia Olikara, 2011 All rights reserved.

iii
ACKNOWLEDGMENTS
First and foremost, I would like to thank my mentor, Nancy Zeleznik-Le, Ph.D.,
for helping me improve my scientific and critical thinking skills. I am grateful for the
guidance and encouragement she provided during my time in the lab. Her constant
optimism kept me going even when I felt discouraged. I have learned a great deal from
her that I will take with me in my future career.
I would also like to acknowledge my committee members, Manuel Diaz, M.D.
and Charles Hemenway, M.D., Ph.D., for being a source of many useful suggestions and
interesting ideas. In addition, I am greatly appreciative of past and current lab members
including Nick, Laurie, Noah, Alyson, Yousaf, Shubin, and Adam for welcoming me to
the lab and helping me learn new techniques.
Thank you to the Molecular Biology Program at Loyola University Chicago for
giving me the opportunity to earn my Master’s degree. I value the knowledge I have
acquired through my coursework and the many opportunities the program has provided to
develop my scientific communication skills. I am also thankful for the members of the
Hematologic Malignancy group for their helpful advice.
Finally, I would like to thank my family. I am deeply appreciative of my parents
and brother for motivating me to succeed and instilling in me the value of education. I
am also exceptionally grateful for my fiancé, Blake, for his unwavering support and
encouragement during my Master’s degree.

To my family

v
TABLE OF CONTENTS
ACKNOWLEDGMENTS iii LIST OF TABLES vi LIST OF FIGURES vii ABSTRACT viii CHAPTER 1: LITERATURE REVIEW 1 Mixed Lineage Leukemia 1 MicroRNAs 4 miR-17-92 Cluster 6 WEE1 9 CHAPTER 2: METHODS 13 MicroRNA Target Gene Prediction 13 Cloning of Wild-Type Luciferase Reporter Construct 13 Cloning of Mutant Luciferase Reporter Constructs 15 Luciferase Reporter Assays 17 Cell Culture 18 RNA Isolation and cDNA Synthesis 19 TaqMan MicroRNA Assays 21 Nuclear and Cytoplasmic Protein Extraction 21 Western Blot Analysis 22 CHAPTER 3: RESULTS 25 Aim: Determine whether WEE1 is a bona fide target of the miRNAs within the miR-17-92 cluster in MLL fusion leukemias 25 Determination of whether miR-17-92 Specifically Targets the 3’ Untranslated Region of WEE1 26 Determination of miR-17-92 Expression Levels in MLL Fusion Leukemia Cell Lines Compared to Non-MLL Fusion Leukemia Cell Lines 32 Determination of WEE1 Expression Levels in MLL Fusion Leukemia Cell Lines Compared to Non-MLL Fusion Leukemia Cell Lines 33 Determination of the Relationship between miR-17-92 Expression and WEE1 Expression 37 CHAPTER 4: DISCUSSION 39 REFERENCES 49 VITA 56

vi
LIST OF TABLES
Table Page
1. Primers used to amplify the 3’ UTR of WEE1 from human genomic DNA 13
2. Primers used to add flanking restriction sites to the 3’ UTR of WEE1 14
3. Primers used to mutate the putative miR-17, miR-20a, and miR-18a target site in the 3’ UTR of WEE1 16
4. Primers used to mutate the putative miR-19a and miR-19b target site in the 3’ UTR of WEE1 17

vii
LIST OF FIGURES
Figure Page
1. Mixed Lineage Leukemia 2
2. Maturation of MicroRNAs and Mechanisms for Downregulating Target Gene Expression 5
3. The miR-17-92 Cluster 8
4. Role of WEE1 in Cell Cycle Progression 11 5. Predicted miR-17-92 Binding Sites in the 3’ UTR of WEE1 27
6. Luciferase Reporter and Mutagenesis Assays 29
7. Quantification of Endogenous miR-17 and miR-19a Levels 34
8. Endogenous Nuclear and Cytoplasmic WEE1 Protein Expression 36
9. Negative Correlation between WEE1 Expression and miR-17 or miR-19a
Expression 38
10. Model of the Regulation of WEE1 by the miR-17-92 Cluster 46

viii
ABSTRACT
MicroRNAs (miRNAs, miRs) are 18-24 nucleotide single-stranded RNAs that
bind to complementary sites in the 3’ untranslated region (UTR) of their mRNA targets.
They act together with the RNA-induced silencing complex (RISC) to post-
transcriptionally regulate target gene expression by either repressing translation or
directly cleaving mRNA targets. The polycistronic miR-17-92 cluster, which encodes
miR-17, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92a, is overexpressed in
several solid tumors and hematopoietic malignancies. Recently, it was shown that miR-
17-92 is overexpressed in leukemias arising from chromosomal translocations involving
the Mixed Lineage Leukemia (MLL) gene. Since MLL fusion leukemias are categorized
as being particularly aggressive, discovering targets of the miR-17-92 cluster may shed
light on potential novel therapeutics.
In the present study, we used target gene prediction algorithms to predict potential
targets of miRNAs within the miR-17-92 cluster. WEE1, a kinase that inhibits cell cycle
progression, was identified as a possible target of five of the six miRNAs of the cluster.
We hypothesized that high miR-17-92 expression in MLL fusion leukemias causes
downregulation of WEE1, which promotes cell cycle progression and contributes to
leukemogenesis.
Through luciferase reporter assays and mutagenesis studies, we found that miR-
17, miR-20a, and miR-18a specifically target nucleotides 465 to 487 of the 3’ UTR of

ix
WEE1, while miR-19a and miR-19b exert control on WEE1 by targeting nucleotides
1069 to 1091. Surprisingly, we saw no significant increase in endogenous miR-17 or
miR-19a expression as detected by TaqMan MicroRNA Assays in the established MLL
fusion leukemia cell lines we tested (MV-4-11, RS4;11, THP-1, MonoMac6) as
compared to the non-MLL fusion leukemia cell lines (K-562, HL-60, U-937). We
believe that miR-17-92 expression levels may be dependent on factors in addition to the
presence of a MLL fusion protein, especially in cell lines. Notably, while we did not
detect a direct relationship between MLL rearrangement status and miR-17-92
expression, we observed a negative correlation between endogenous miR-17 or miR-19a
expression and endogenous WEE1 protein expression as detected by Western blot
analysis in the same panel of cell lines. The results of this project suggest that WEE1 is a
valid biological target of the miR-17-92 cluster in leukemia.

1
CHAPTER 1
LITERATURE REVIEW
Mixed Lineage Leukemia
The Mixed Lineage Leukemia (MLL) gene was first identified for its involvement
in chromosomal translocations at chromosome 11, band q23 that cause acute lymphoid
leukemias, acute myeloid leukemias, or mixed lineage leukemias, also known as
myeloid-lymphoid leukemias1. Chromosomal rearrangements between nonhomologous
chromosomes create an in-frame fusion between MLL and one of more than 60 different
fusion partners2 (Figure 1A). A chimeric protein is produced in which the amino-
terminus of MLL is fused with the carboxyl-terminus of the fusion partner3.
MLL fusion partner genes encode either nuclear proteins or cytoplasmic proteins,
with different functions. Over 80% of MLL fusion leukemias possess a MLL fusion
partner gene that encodes a nuclear protein such as AF4, AF9, ENL, ELL, AF10, and
AF5q314. Many of these nuclear fusion partners are normally present in a transcriptional
elongation complex, while others are transcription factors. Many of the cytoplasmic
fusion partners have the ability to dimerize or multimerize. Certain fusion partners are
more prevalent in certain types of leukemia. For example, MLL-AF4 in more common in
acute lymphoid leukemia5, while MLL-AF9 is more common in acute myeloid leukemia.
Leukemias arising from chromosomal translocations of the MLL gene account for
5 to 10% of all human acute leukemias6. MLL rearrangements are the most common

2
Figure 1. Mixed Lineage Leukemia
!"#$%&'()*+&,*'
-.*%/%#%/)0'1*)�%2)3%&'
!"#$%&'()*+&,*'
!""#$%&'()#
415.%%6#'7,8*,##$%&'9%/)$&' (:9'!$&;,*#' 423<)3%&'9%/)$&'
=>1'9%/)$&'
*'+,-./01#!""#?*,)68%$&+'
-0"#+,*'7,;$%&'!"
#"
(A) Wild-type MLL and the MLL fusion protein after undergoing chromosomal translocation. (B) Event-free survival for patients with wild-type MLL and various MLL rearrangements. Figure was adapted from Pieters et al. 200710.
$%&'()*+,"!""#
!"""-,.--./0,1,/)2"
3%1,"4-51"6%.0/52%2"78,.-29"

3
chromosome abnormalities found in infant leukemias and in patients previously treated
with DNA topoisomerase II inhibitors7, 8. Compared to other leukemias, MLL fusion
leukemias are particularly aggressive and have poor prognosis9, 10. Event-free survival of
leukemia patients bearing MLL translocations including t(9;11), t(4;11), and t(11;19) is
significantly worse than for leukemia patients with wild-type MLL10 (Figure 1B).
Wild-type MLL is a member of the Trithorax group of proteins. MLL and other
Trithorax proteins are involved in maintaining active gene expression. These proteins
antagonize the effect of Polycomb proteins, which are responsible for negatively
regulating gene expression. Mll homozygous null mice have defects in embryonic
hematopoiesis and are embryonic lethal, suggesting a crucial role for MLL in
development11.
MLL is a very large protein that contains numerous functional domains3 (Figure
1A). Some domains are retained in the fusion protein including the AT-hooks and the
repression domain. The AT-hooks bind AT-rich DNA12. The repression domain
contains a CXXC domain, which binds non-methylated CpG DNA13. Other domains
such as the four plant homeodomains (PHD), transcriptional activation domain, and
histone methyltransferase domain (SET) are not retained in the fusion protein that causes
leukemia. The individual MLL plant homeodomains, each of which fold into a structure
that coordinates two zinc ions, have differing functions. Some are required for
homodimerization and another for binding to cyclophilin Cyp3314. The activation
domain interacts with the CREB-binding protein (CBP), which recruits transcriptional

4
activators and possesses intrinsic histone acetyltransferase activity15. The SET domain is
responsible for trimethylation of lysine 4 on histone H316, 17.
MicroRNAs
MicroRNAs (miRNAs, miRs) are approximately 22 nucleotide noncoding RNAs
that function as antisense regulators of messenger RNAs (mRNAs)18. Victor Ambros
first discovered this class of small RNAs in C. elegans in 1993. The lin-4 miRNA was
shown to regulate the transition from the first larval stage to the second by
downregulating lin-14 mRNA19. In 2000, C. elegans let-7 miRNA was shown to regulate
the transition from the late larval stage to adult cell fates20. Today over 500 miRNA
genes have been discovered in the human genome alone with notably high degrees of
sequence conservation across species21.
Many miRNAs display distinct temporal and spatial expression patterns, while
others are constitutively transcribed18. RNA Polymerase II transcribes miRNA genes22,
which are located either within intergenic regions or within the introns of pre-mRNAs18
(Figure 2A). The transcribed pri-miRNA forms a stem loop structure. Drosha cleaves
the ends of the stem to produce 60-70 nucleotide pre-miRNAs23, 24. Ran-GTP and
Exportin5 facilitate active transport of the miRNA precursor from the nucleus to the
cytoplasm25, 26. Dicer then cleaves off the loop to reveal a miRNA:miRNA duplex27.
Helicase unwinds the duplex and the miRNA strand whose 5’ end is more tightly paired
is degraded28, 29. The other miRNA strand, which represents the mature miRNA, is
loaded on the RNA-induced silencing complex (RISC), where it scans for perfect or near

5
Figure 2. Maturation of MicroRNAs and Mechanisms for Downregulating Target Gene Expression
Target mRNAs from loci unrelated to miRNA gene
!"
#" $"
(A) Maturation of miRNAs. miRNA genes are transcribed by RNA polymerase II and then cleaved by Drosha. The resulting pre-miRNA is transported into the cytoplasm, cleaved by Dicer, unwound by Helicase, and loaded onto the RISC complex. Figure was adapted from Bartel 200422. (B) and (C) Mechanisms utilized by miRNAs together with the RISC complex to downregulate target gene expression. Figures were adapted from Bartel 200422. (B) Extensive complementarity in the coding region or 3’ UTR specifies mRNA cleavage. (C) Short complementary segments in the 3’ UTR specifies translational repression.
%&'"(("
)*+!"
),*+!"),*+!-"
)*+!"

6
perfect complementary mRNA sequences30. The seed region, which encompasses
nucleotides 2-7 of the miRNA, determines targeting of the miRNA to target mRNA31.
miRNAs utilize different mechanisms to downregulate gene expression of their
targets. When there is a high degree of complementarity to the mRNA sequence, the
target mRNA is cleaved within the miRNA-binding site, which results in decreased
mRNA and protein levels24 (Figure 2B). However, when there is a lower degree of
complementarity to the mRNA sequence, translation is repressed, which results in
decreased protein levels alone24 (Figure 2C). Translation is thought to be repressed by
the slowing or stalling of ribosomes or by the degradation of newly synthesized
polypeptide.
miRNAs are often abnormally expressed in many cancers including leukemias.
For example, it was shown that MLL fusion proteins induce overexpression of miR-196b,
which contributes to leukemogenesis32. Determining which miRNAs are upregulated and
downregulated in different subtypes of leukemia serves as a valuable diagnostic marker
and may be useful in developing novel therapeutics33. Techniques for replacing
underexpressed miRNAs or inhibiting overexpressed miRNAs both in vitro and in vivo
are currently being developed.
miR-17-92 Cluster
Although many miRNAs are present alone in the genome, some are located in
clusters containing multiple miRNAs. miRNA clusters are expressed together as a long
precursor and are then processed into individual miRNAs34. The miR-17-92 cluster is
located within an 800 base-pair region of chromosome 13 within the third intron of a

7
primary transcript called C13orf2535 (Figure 3A). The only known function of this
transcript is to produce these miRNAs. The miR-17-92 cluster encodes six miRNAs
(miR-17, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92a)36. The miRNAs of the
miR-17-92 cluster share a great deal of sequence homology, suggesting that they might
be able to act collectively on a common target. Gene duplications have produced miR-
106a-363 and miR-106b-25, two paralogs of the miR-17-92 cluster36. miRNAs from
these three clusters are placed into families based on conserved seed sequences31 (Figure
3B).
The miR-17-92 cluster is crucial for normal development of the heart, lungs, and
immune system. Knockout of this cluster has been shown to result in smaller embryos
and immediate postnatal death due to ventricular septal defects in the heart and severely
hypoplastic lungs37. It has been demonstrated that the miR-17-92 cluster is highly
expressed in embryonic lung tissue, but decreases over time38. Additionally, this cluster
is involved in promoting progression from pro-B to pre-B cells37.
The miR-17-92 cluster acts as an oncogene39 and is overexpressed in
hematopoietic malignancies and solid tumors including those originating from the breast,
colon, lung, pancreas, prostate, and stomach40. Several target genes with important roles
in cell cycle progression, apoptosis, and angiogenesis have been identified and
experimentally validated for this cluster. The E2F family of transcription factors, which
when expressed at high levels can induce apoptosis, is one such target41. The miR-17-92
cluster suppresses apoptosis by downregulating E2F1, E2F2, and E2F3. The pro-
apoptotic gene Bim has also been shown to be a direct target of the miR-17-92 cluster42.

8
Figure 3. The miR-17-92 Cluster
!""#$%#
&#'$#
!"
#"
(A) The miR-17-92 cluster is located within an intron on chromosome 13. Figure was adapted from Mendell 200836. (B) miRNAs are aligned to show nucleotide sequence conservation. Nucleotides that are conserved within a miRNA family are highlighted in blue. The seed sequences (nucleotides 2-7) of each miRNA are in bold print.
miR-17 CAAAGUGCUUACAGUGCAGGUAG miR-20a UAAAGUGCUUAUAGUGCAGGUAG miR-20b CAAAGUGCUCAUAGUGCAGGUAG miR-106a AAAAGUGCUUACAGUGCAGGUAG miR-106b UAAAGUGCUGACAGUGCAGAU miR-93 CAAAGUGCUCUUCGUGCAGGUAG miR-18a UAAGGUGCAUCUAGUGCAGAUAG miR-18b UAAGGUGCAUCUAGUGCAGUUAG miR-19a UGUGCAAAUCUAUGCAAAACUGA miR-19b UGUGCAAAUCCAUGCAAAACUGA miR-25 CAUUGCACUUGUCUCGGUCUGA miR-92a UAUUGCACUUGUCCCGGCCUGU miR-363 AAUUGCACGGUAUCCAUCUGUA

9
Another target that has been validated is the cyclin-dependent kinase inhibitor p21, which
is a negative regulator of the G1/S checkpoint43. The miR-17-92 cluster is also known to
downregulate expression of the tumor suppressor Pten44. Finally, the anti-angiogenic
proteins Tsp1 and CTGF are both negatively regulated by the miR-17-92 cluster45. High
levels of the miR-17-92 cluster have been shown to increase the number of leukemia
stem cells, block differentiation, and enhance proliferation, while low levels of the miR-
17-92 cluster increase differentiation and decrease self-renewal46.
Each subtype of acute myeloid leukemia and acute lymphoid leukemia has a
unique miRNA expression profile. The Chen group used bead-based miRNA expression
profiling assays and TaqMan qPCR assays to show that the individual miRNAs of the
miR-17-92 cluster are specifically upregulated in MLL rearranged leukemias, but not in
the other subtypes that they tested47, 48. Furthermore, this cluster is overexpressed in
mouse leukemia cells with MLL-ELL and MLL-ENL fusions, although the expression is
not quite as high as in the corresponding human leukemias49.
WEE1
Wee1 was first discovered in the fission yeast S. pombe as a mitotic inhibitor that
controls the G2/M transition50. Fission yeast deficient in Wee1 prematurely enter mitosis
and divide at a small size. The kinase was named “Wee” in reference to the small size of
fission yeast lacking Wee1. The catalytic domain of the human gene was cloned in
199151, and the full-length gene was identified in 199552. Wee1 has also been found in S.
cerevisiae, Xenopus, Drosophila, and C. elegans.

10
Human WEE1 is a protein kinase that adds an inhibitory phosphate on Tyr15 of
Cyclin dependent kinase 1 (Cdk1) during interphase53. WEE1 holds Cdk1 in an inactive
state until the G2/M transition of the cell cycle. The function of WEE1 is antagonized by
Cdc25 phosphatase, which removes the inhibitory phosphate at the onset of the M
phase54.
As shown in Figure 4, the pathway from inactive Cdk1 to fully active Cdk1
requires several molecular players. During interphase, Cdk1 associates with Cyclin B
and is phosphorylated by Cdk-activating kinase (CAK) on Thr161. Simultaneously,
WEE1 phosphorylates Cdk1 on Tyr15, while the WEE kinase family member, MYT1,
phosphorylates Cdk1 on Thr14 and Tyr15. These inhibitory phosphorylations on Thr14
and Tyr15 dominantly inhibit Cdk1 until the onset of mitosis, at which point Cdc25
dephosphorylates Thr14 and Tyr15. Cdk1 becomes active and triggers the rapid switch
into mitosis54. Upon entry into mitosis, WEE1 is inactivated by phosphorylation on
Ser53 and Ser123 by Plk1 and Cdk155. WEE1 is subsequently ubiquitinated and
degraded55. WEE1 levels remain low throughout M phase and G1 phase.
The WEE family of kinases consists of WEE1, WEE2, and MYT1. While WEE1
and WEE2 solely phosphorylate Cdk1 on Tyr1553, MYT1 is a dual-specificity kinase that
phosphorylates Cdk1 on both Thr14 and Tyr1556. WEE1 and WEE2 are soluble proteins
that localize to the nucleus56. Conversely, MYT1 is membrane-associated and localizes
to the cytoplasmic endoplasmic reticulum and Golgi apparatus56. Importantly, in
Xenopus, it was found that Wee1 is 10-fold more active than Wee257.

11
!"#$%&'()*+%
Figure 4. Role of WEE1 in Cell Cycle Regulation
,-.$%
%%%%%%%%%#$/$%%%%%%#$0%%%%%%"$1%
,-.$%
%%%%%%%%%#$/$%%%%%%#$0%%%%%%"$1%
,-.$%
%%%%%%%%%#$/$%%%%%%#$0%%%%%%"$1%
2% 2% 2% 2%
344$%&'()*+%
,567'(%8%
,567'(%8% ,567'(%8%,9&% ,-6:1%
;(<+=>?)*+% !'<@*'*%
During interphase, Cdk1 associates with Cyclin B. Cdk-activating kinase (CAK) then adds an activating phosphate to Cdk1 on Thr161. WEE1 adds an inhibitory phosphate to Cdk1 on Tyr15, while MYT1 adds two inhibitory phosphates to Cdk1 on Thr14 and Tyr15. At the onset of mitosis, Cdc25 phosphatase removes the inhibitory phosphates, thereby fully activating Cdk1 and promoting mitosis.%

12
The kinase domain of WEE1 is highly conserved across WEE family members.
All WEE family members possess five conserved amino acid residues within their kinase
domain that are unique to this family of kinases58. The noncatalytic regions of these
proteins are important for regulation, protein-protein interactions, and subcellular
localization.
miR-195 was recently shown to target two sites in the 3’ UTR of WEE1 in human
embryonic stem cells59. Since WEE1 levels control the rate of human embryonic stem
cell division, downregulation of WEE1 by miR-195 promotes the cell cycle and increases
cell proliferation59. This study was the first to show a miRNA regulating WEE1.
Nevertheless, it is still important to discover novel miRNAs that target WEE1. It is
conceivable that in different types of cells, other unique miRNAs target WEE1 for
downregulation. For this reason, we set out to validate WEE1 as a target of the miR-17-
92 cluster.

13
CHAPTER 2
METHODS
MicroRNA Target Gene Prediction
Prediction algorithms including TargetScan (www.targetscan.org), MicroCosm
Targets (www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5), PicTar (pictar.mdc-
berlin.de), and miBridge (sitemaker.umich.edu/mibridge/target_predictions) were used to
predict potential biological targets of the miR-17-92 cluster.
Cloning of Wild-Type Luciferase Reporter Construct
In order to amplify the 3’ UTR of WEE1 (NM_003390.3), the following PCR was
set up: 40.1 uL dH2O, 5 uL 10x Cloned Pfu buffer, 0.4 uL 25 mM dNTPs, 1 uL 1ug/uL
human genomic DNA, 1.25 uL 10 uM Forward Primer (Table 1), 1.25 uL 10 uM Reverse
Primer (Table 1), 1 uL Cloned Pfu DNA Polymerase (Stratagene, Catalog #600159-81).
The PCRs were run in the PCR Express Thermal Cycler (Hybaid) according to the
following protocol: one cycle of 95°C for 45 seconds, 40 cycles of 95°C for 45 seconds,
62.6°C for 45 seconds, 72°C for 3 minutes, one cycle of 72°C for 10 minutes, and hold at
4°C. The band at 939 bp was cut out and gel extracted using the QIAEX II Gel
Extraction Kit (Qiagen, Catalog #20051).
Table 1. Primers used to amplify the 3’ UTR of WEE1 from human genomic DNA Amplicon Primers (5’ to 3’) WEE1 3’ UTR (nucleotides 2418 - 3356 of NM_003390.3)
Forward: ATGTTACACCAGCCTTTCCAGGGT Reverse: AGACAATTAAGGTAAGCTCAGAGTGA

14
In order to add restriction sites to the ends of the above PCR product to facilitate
cloning into the reporter vector, the following PCR was set up: 40.1 uL dH2O, 5 uL 10x
Cloned Pfu buffer, 0.4 uL 25 mM dNTPs, 1 uL of the above PCR product, 1.25 uL 10
uM Forward Primer (Table 2), 1.25 uL 10 uM Reverse Primer (Table 2), 1 uL Cloned Pfu
DNA Polymerase. The PCRs were run according to the following protocol: one cycle of
95°C for 45 seconds, 40 cycles of 95°C for 45 seconds, 65.2°C for 45 seconds, 72°C for
3 minutes, one cycle of 72°C for 10 minutes, and hold at 4°C. The band at 969 bp was
cut out and gel extracted.
Table 2. Primers used to add flanking restriction sites to the 3’ UTR of WEE1 Amplicon Primers (5’ to 3’) WEE1 3’ UTR (nucleotides 2418SpeI - 3356HindIII)
Forward: TCTCTCTCTACTAGTATGTTACACCAGCCTTTCCAGGGT Reverse: TCTCTCTCTAAGCTTAGACAATTAAGGTAAGCTCAGAGTGA
The 3’ UTR of WEE1 that was now flanked by SpeI and HindIII restriction sites
was digested with SpeI and HindIII at 37°C for 2 hours along with the pMIR-REPORT
Luciferase vector (Applied Biosystems, Catalog #AM5795). The gel extracted digests
were then ligated using T4 DNA Ligase (New England Biolabs, Catalog #M0202L) at
16°C overnight to create a construct with the 3’ UTR of WEE1 immediately following
the luciferase gene. The ligation was electroporated into homemade DH10!
Electrocompetent Cells using 1 uL ligation reaction and 20 uL cells using the following
electroporator settings: 4 k" resistance, 330 µF capacitance. The transformants were
miniprepped using the GeneJET Plasmid Miniprep Kit (Fermentas, Catalog #K0503).
The clone was confirmed by sequencing (ACGT, Inc.).

15
Cloning of Mutant Luciferase Reporter Constructs
In order to mutate the putative miR-17-92 binding sites in the 3’ UTR of WEE1,
three mutant luciferase reporter constructs were generated. For one of the constructs, the
putative miR-17, miR-20a, and miR-18a target site was mutated. In another of the
constructs, the putative miR-19a and miR-19b target site was mutated. The third mutant
construct had all the putative miR-17-92 target sites mutated.
To produce the construct with the putative miR-17, miR-20a, and miR-18a target
site mutated (referred to as miR-17 Mut), a modified site-directed mutagenesis protocol
with non-overlapping primers was used to set up the following PCR using an error proof
polymerase: 32.5 uL dH2O, 10 uL 5x Phusion HF Buffer, 1 uL 10 mM dNTPs, 2.5 uL 10
uM Forward Primer (Table 3), 2.5 uL 10 uM Reverse Primer (Table 3), 1 uL 10 ng/uL
template (wild-type luciferase reporter construct), 0.5 uL Phusion DNA Polymerase
(Finnzymes, Catalog #F-530S). The PCRs were run according to the following Touch
Down PCR protocol: one cycle of 98°C for 30 seconds, 20 cycles of 98°C for 10 seconds,
touch down from 60°C to 50°C in 0.5°C increments for 30 seconds, 72°C for 8 minutes,
25 cycles of 98°C for 10 seconds, 50°C for 30 seconds, 72°C for 8 minutes, one cycle of
72°C for 10 minutes, and hold at 4°C. The PCR product was DpnI treated at 37°C for 1
hour to digest the parental DNA template. It was then electroporated into DH10!
Electrocompetent Cells using 1 uL PCR product and 20 uL cells. The transformants were
miniprepped, and the plasmid DNA was sequenced. Since stray mutations can be
introduced during site-directed mutagenesis, the mutated region was ligated into the

16
pMIR-REPORT Luciferase vector using the SpeI and HindIII restriction sites used
previously.
Table 3. Primers used to mutate the putative miR-17, miR-20a, and miR-18a target site in the 3’ UTR of WEE1 Amplicon Primers (5’ to 3’) Luciferase WEE1 3’ UTR miR-17 Mut pMIR-REPORT
Forward: GACTTGTATATCCCACTGGGAGACAGGGGTAGGCATTGCA TGAACCATGGGATG Reverse: GCCAATCAATGTTAATAAAACACAAGTCAAAGACAATGTA CCACATGTTTTAGACC
To generate the construct with the putative miR-19a and miR-19b target site
mutated (referred to as miR-19 Mut) and the construct with all the putative miR-17-92
target sites mutated (referred to as miR-17,19 Mut), the following PCRs were set up: 39.5
uL dH2O, 5 uL 10x Cloned Pfu buffer, 1 uL 10 mM dNTPs, 1 uL 10 ng/uL template
(wild-type luciferase reporter construct or miR-17 Mut), 1.25 uL 10 uM Forward Primer
(Table 4), 1.25 uL 10 uM Reverse Primer (Table 4), 1 uL Cloned Pfu DNA Polymerase.
The PCRs were run according to the following protocol: one cycle of 95°C for 45
seconds, 40 cycles of 95°C for 45 seconds, gradient from 55°C to 70°C for 45 seconds,
72°C for 2 minutes, one cycle of 72°C for 10 minutes, and hold at 4°C. The PCR
products from all three annealing temperatures were pooled together and PCR purified.
Each of the inserts and the pMIR-REPORT Luciferase vector were digested with SpeI
and HindIII at 37°C for 2 hours. The gel extracted digests were then ligated overnight at
16°C using a 3:1 molar ratio of insert to vector. The ligations were electroporated into
homemade DH10! Electrocompetent Cells using 2 uL ligation reaction and 20 uL cells.
The transformants were miniprepped and all clones were confirmed by sequencing.

17
Table 4. Primers used to mutate the putative miR-19a and miR-19b target site in the 3’ UTR of WEE1 Amplicon Primers (5’ to 3’) Luciferase WEE1 3’ UTR miR-19 Mut pMIR-REPORT
Forward: TCTCTCTCTACTAGTATGTTACACCAGCCTTTCCAGGGT Reverse: CCTTTATTAAGCTTAGACAATTAAGGTAAGCTCAGAGTGAC TTTTAATATGCCAATCAATGTTAATAAAACACAAGTCAAAGACAAT GTACCACATGTTTTAGACC
Luciferase WEE1 3’ UTR miR-17,19 Mut pMIR-REPORT
Forward: TCTCTCTCTACTAGTATGTTACACCAGCCTTTCCAGGGT Reverse: CCTTTATTAAGCTTAGACAATTAAGGTAAGCTCAGAGTGAC TTTTAATATGCCAATCAATGTTAATAAAACACAAGTCAAAGACAAT GTACCACATGTTTTAGACC
Luciferase Reporter Assays
The wild-type and mutant luciferase reporter constructs as well as miR-17
MSCV-PIG (Jianjun Chen, University of Chicago), miR-17-19b MSCV-PIG (J. Chen),
miR-17-92 MSCV-PIG (J. Chen), MSCV-PIG (J. Chen), and pRL-TK (Promega, Catalog
#e2241) were midiprepped using the Plasmid Midi Kit (Qiagen, Catalog #12145).
60,000 HEK293T cells were plated into each of 16 wells of a 24-well plate (TPP,
Catalog #92024). Each well was transfected 24 hours later using 6 ng control vector
(pRL-TK), 120 ng Luciferase WEE1 3’ UTR (Wild-Type, miR-17 Mut, miR-19 Mut, or
miR-17,19 Mut), and 600 ng miRNA (miR-17, miR-17-19b, miR-17-92, or Empty
Vector). The CalPhos Mammalian Transfection Kit (Clontech, Catalog #631312) was
used for the transfections. The medium was removed from each well the following day
and replaced with fresh complete medium (DMEM (HyClone), 10% FBS, and 1%
Pen/Strep).
The reporter assay was performed using the Dual-Luciferase Reporter Assay
System (Promega, Catalog #E1910). 42 hours after transfection, the medium was
removed from each well of the 24-well plate and the cells were gently washed with 200
uL sterile 1x PBS. 100 uL 1x Passive Lysis Buffer (1600 uL dH2O, 400 uL 5x Passive

18
Lysis Buffer) was added to each well and the plate was rocked on a rocking platform at
room temperature for 15 minutes. Since the experiment was performed in triplicate, three
20 uL aliquots of each of the 16 cell lysates were transferred to a white round-bottom 96-
well plate (Costar). The plate was centrifuged at 1500 rpm for 5 minutes at room
temperature to bring the cell lysates to the bottom of the wells.
Using the Microplate Luminometer (Veritas), both injectors were flushed three
times with dH2O, three times with 70% Ethanol, three times with dH2O, and then three
times with air. The first injector was primed with Luciferase Assay Reagent II
(lyophilized Luciferase Assay Substrate in Luciferase Assay Buffer II) and the second
injector was primed with Stop & Glo Reagent (48 uL 50x Stop & Glo Substrate, 2352 uL
Stop & Glo Buffer). The assay was run using the luminometer, which dispensed 100 uL
Luciferase Assay Reagent II into each well, measured firefly luciferase, dispensed 100 uL
Stop & Glo Reagent into each well, and measured Renilla luciferase. After the run, both
injectors were flushed again. The data was analyzed by determining the relative
luciferase (firefly luciferase: Renilla luciferase) and normalizing to the wild-type
luciferase reporter. The experiment was performed in triplicate and repeated three times.
Cell Culture
Four MLL fusion leukemia cell lines (MV-4-11, RS4;11, THP-1, MonoMac6),
three non-MLL fusion leukemia cell lines (K-562, HL-60, U-937), and one non-leukemia
cell line (HEK293T) were thawed from liquid nitrogen. MV-4-11, K-562, and HL-60
cells were cultured in IMDM (Gibco), 10% FBS, and 1% Pen/Strep. RS4;11, THP-1,
MonoMac6, and U-937 cells were cultured in RPMI-1640 (HyClone), 10% FBS, 1%

19
Pen/Strep, and 0.05 mM 2-mercaptoethanol. HEK293T cells were cultured in DMEM
(HyClone), 10% FBS, and 1% Pen/Strep. All cells were cultured in a 37°C incubator
with 5% carbon dioxide. Each cell line was expanded from a 10 cm2 flask to two 75 cm2
flasks. The cell pellets were resuspended in 3 mL sterile 1x PBS and then counted using
the Bright Line Counting Chamber (Hausser Scientific Company).
For TaqMan MicroRNA Assays, 2,500,000 cells were aliquoted per microfuge
tube for each cell line. The cell pellets were resuspended in 500 uL TRI Reagent (Sigma,
Catalog #T9424) by repeated pipetting. The resuspended cell lysates were stored at -
80°C until RNA was isolated.
For Western blotting, 1,000,000 cells were aliquoted per microfuge tube for each
cell line. Cell pellets were incubated on ice for immediate use in nuclear and cytoplasmic
protein extraction.
RNA Isolation and cDNA Synthesis
RNA was isolated from MV-4-11, RS4;11, THP-1, MonoMac6, K-562, HL-60,
U-937, and HEK293T cells according to the manufacturer’s protocol (Sigma). To
summarize, cells that had been resuspended in TRI Reagent and stored at -80°C were
briefly thawed in a room temperature water bath. The samples were then allowed to
stand at room temperature for 5 minutes. 100 uL of chloroform was added to each
sample and the tubes were shaken vigorously for 15 seconds. The samples were allowed
to stand at room temperature for 10 minutes. The tubes were centrifuged at 12,000 x g
for 15 minutes at 4°C. The colorless aqueous phase (upper layer) containing the RNA
was transferred to a fresh microfuge tube by pipetting. 250 uL of isopropanol was added

20
and mixed. The samples were allowed to stand at room temperature for 10 minutes. The
tubes were centrifuged at 12,000 x g for 10 minutes at 4°C. 500 uL of 75% ethanol was
added to the RNA pellet and vortexed. The tubes were centrifuged at 7,500 x g for 5
minutes at 4°C. The supernatant was discarded and then the RNA pellet was air-dried in
the hood for 10 minutes. The RNA pellet was reuspended in 20 uL of RNase-free water.
The tube was incubated in a 55°C water bath for 10 minutes with periodic mixing. The
isolated RNA was stored at -80°C.
cDNA was reverse transcribed from total isolated RNA using the TaqMan
MicroRNA Reverse Transcription Kit (Applied Biosystems, Catalog #4366596) and
primers specific to each miRNA from the TaqMan MicroRNA Assay Kit (Applied
Biosystems). 10 ug of RNA was used for each reaction. For each 15 uL reverse
transcription reaction, the following components were combined on ice: 0.15 uL 100 mM
dNTPs (with dTTP), 1 uL MultiScribe Reverse Transcriptase (50 U/uL), 1.5 uL 10x
Reverse Transcription Buffer, 0.19 uL RNase Inhibitor (20 U/uL), 4.16 uL Nuclease-free
water, 5 uL total RNA diluted to a concentration of 2 ng/uL (isolated from either MV-4-
11, RS4;11, THP-1, MonoMac6, K-562, HL-60, U-937, or HEK293T cells), and 3 uL RT
primer (specific to either U6 snRNA (constitutively expressed control), miR-17, or miR-
19a). The reactions were incubated on ice for 5 minutes and subsequently loaded in the
PCR Express Thermal Cycler (Hybaid). The following program was run: 16°C for 30
minutes, 42°C for 30 minutes, 85°C for 5 minutes, and hold at 4°C. The cDNA was
stored at -20°C until the Real-Time PCR step.

21
TaqMan MicroRNA Assays
During the PCR amplification step, AmpliTaq Gold DNA polymerase was used to
amplify target cDNA using sequence-specific primers. For each 20 uL reaction, the
following components were combined on ice: 10 uL TaqMan 2x Universal PCR Master
Mix, No AmpErase UNG (Applied Biosystems, Catalog #4324018), 7.67 uL Nuclease-
free water, 1 uL 20x TaqMan MicroRNA Assay mix (specific to either U6 snRNA, miR-
17, or miR-19a), and 1.33 uL of the cDNA from the reverse transcription step. Since
reactions were performed in triplicate, 20 uL of complete PCR master mix was dispensed
into each of three wells of a clear 96-well PCR plate. The plate was sealed with an
optical adhesive cover and centrifuged at 1500 rpm for 5 minutes. Real-time PCR was
performed using the ABI 7300 Real-Time PCR System (Applied Biosystems) using the
following program: one cycle of 95°C for 10 minutes followed by 40 cycles of 95°C for
15 seconds and 60°C for 60 seconds. The data was analyzed with ABI Prism 7300
software. miR-17 and miR-19a expression levels were determined relative to U6 snRNA
levels using the 2-##Ct method60. Relative expression was arbitrarily normalized to MV-4-
11 expression levels. The assay was performed in triplicate and repeated two to five
times.
Nuclear and Cytoplasmic Protein Extraction
All steps for nuclear and cytoplasmic protein extraction were performed on ice.
1,000,000 cell aliquots from MV-4-11, RS4;11, THP-1, MonoMac6, K-562, HL-60, U-
937, and HEK293T cells that were pelleted according to the protocol above were
resuspended in 400 uL cold Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM

22
EDTA, 1 mM DTT, 1:100 Protease inhibitor cocktail (Sigma, Catalog #P8340)) by gentle
pipetting. The cells were allowed to swell on ice for 15 minutes. 25 uL of 10% Nonidet
NP-40 (Calbiochem, Catalog #492015) was added and then the tubes were vigorously
vortexed for 10 seconds. The tubes were centrifuged at 14,000 rpm for 15 minutes at
4°C. The supernatants containing the cytoplasmic extracts were snap frozen in 30 uL
aliquots and stored at -80°C until further analysis. The nuclear pellet was resuspended in
50 uL ice-cold Buffer C (20 mM HEPES pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM DTT,
1:100 Protease inhibitor cocktail (Sigma, Catalog #P8340)). The tubes were vigorously
rocked for 15 minutes on a rotator (Glas-Col) at 4°C and then centrifuged at 14,000 rpm
for 5 minutes at 4°C. The supernatants containing the nuclear extracts were snap frozen
in 30 uL aliquots and stored at -80°C until further analysis.
Western Blot Analysis
SDS-polyacrylamide (10%) gels (10% Bottom-Resolving; 4% Top-Stacking) with
ten wells each were poured. The protein samples were prepared by combining 30 uL
cytoplasmic or nuclear extracts with 10 uL Sample Loading Buffer (2.5 mL 1 M Tris pH
6.8, 1 g SDS, 0.05 g Bromophenol Blue, 5 mL Glycerol, 2.5 mL H2O, 500 mM DTT).
The samples were vortexed, boiled at 95°C for 5 minutes, briefly centrifuged, and then
loaded in the wells. 10 uL of Precision Plus Protein Kaleidoscope Standard (Bio-Rad,
Catalog #161-0375) was loaded as a marker. The gels were electrophoresed at 150 V, 0.5
A for 65 minutes using 1x Running Buffer (5x Running Buffer: 15.1 g Tris base, 94 g
Glycine, 50 mL 10% SDS, final volume brought up to 1 L using dH2O).

23
Immobilon Transfer Membranes (Millipore, Catalog #IPVH00010) were pre-
incubated with methanol for 5 minutes and then washed twice with dH2O. The
membranes and gels were then equilibrated in 1x CAPS Buffer (100 mL 10x CAPS
Buffer (22.13 g CAPS, final volume brought up to 1 L using dH2O, pH 11.0), 100 mL
Methanol, 800 mL dH2O) for 25 minutes. Gels were transferred to membranes at 70 V,
0.5 A for 2 hours using 1x CAPS Buffer.
The membranes were each blocked in 5% blocking solution (TBS-T (75 mL 2 M
NaCl, 20 mL 1 M Tris pH 7.5, 904 mL dH2O, 1 mL Tween-20), 5% w/v BSA) for 1 hour
at room temperature on the Belly Dancer (Stovall Life Science). The membranes were
then incubated in primary antibody mixture (TBS-T, 5% w/v BSA, WEE1 rabbit
polyclonal antibody (Cell Signaling, Catalog #4936, 1:1000 dilution)) with gentle
shaking on the Orbitron Rotator II (Boekel Scientific) at 4°C overnight. The membranes
were washed three times for 10 minutes each with 15 mL TBS-T. The membranes were
then incubated in secondary antibody mixture (TBS-T, 5% w/v BSA, ECL Anti-Rabbit
IgG-HRP Antibody (GE Healthcare, Catalog #NA934V, 1:3000 dilution)) for 2 hours at
room temperature on the Belly Dancer. The membranes were washed five times for 15
minutes each with 15 mL TBS-T.
Using the SuperSignal West Pico Chemiluminescent Substrate Kit (Thermo
Scientific, Catalog #34078), the Stable Peroxide Solution and the Luminol Enhancer
Solution were mixed at a 1:1 ratio to prepare the substrate working solution. The
membranes were incubated with substrate working solution for 5 minutes at room
temperature on the Belly Dancer. The excess reagent was drained and the membranes

24
were wrapped in clear plastic wrap. The membranes were developed using the FujiFilm
LAS-3000 Luminescent Image Analyzer with Image Reader LAS-3000 software.
In order to normalize for loading, the membranes were washed two times for 5
minutes each with 15 mL TBS-T and then stripped by performing two 10 minute
incubations with 15 mL Mild Stripping Buffer (15 g Glycine, 1 g SDS, 10 mL Tween-20,
final volume brought up to 1 L using dH2O, pH 2.2). The membranes were subsequently
washed two times for 10 minutes each with 15 mL PBS followed by two washes for 5
minutes each with 15 mL TBS-T. The membranes were blocked with 5% blocking
solution as described above and then incubated with the appropriate primary and
secondary antibody mixtures. !-Actin mouse monoclonal antibody (Sigma, Catalog
#A5441, 1:5000 dilution) was used as the loading control primary antibody followed by
ECL Anti-Mouse IgG-HRP Antibody (GE Healthcare, Catalog #NA931V, 1:3000
dilution). Band intensity from Western blot images was quantified using MultiGauge
V3.0 software (FujiFilm) by subtracting background from band intensity. WEE1
expression was determined relative to !-Actin expression and was arbitrarily normalized
to MV-4-11 expression levels. The experiment was repeated twice.

25
CHAPTER 3
RESULTS
Aim: Determine whether WEE1 is a bona fide target of the miRNAs within the miR-17-92 cluster in MLL fusion leukemias
It was recently shown that overexpression of the miR-17-92 cluster is a signature
of MLL fusion leukemias48. Because leukemias caused by rearrangement of the MLL
gene are particularly aggressive and have poor prognosis, we predict that identification of
mRNA transcripts targeted for downregulation by this miRNA cluster will shed light on
novel molecular targets for this disease.
Bioinformatic miRNA target gene prediction algorithms were used to predict
potential targets of miR-17, miR-18a, miR-19a, miR-20a, miR-19b, and miR-92a.
TargetScan predicts targets by searching for sites within human untranslated regions that
match the seed region of miRNAs. MicroCosm Targets functions by identifying high
complementarity between miRNAs and 3’ UTRs while favoring complementarity in the
seed region. PicTar predicts targets of human miRNAs based on conservation of the
target sequence across species. miBridge searches for both 5’ UTR and 3’ UTR target
sites in the same mRNA.
For each miRNA of the cluster, we compiled a list of the thirty target genes with
the highest total context score from TargetScan, the twenty target genes with the lowest
P-value from MicroCosm Targets, the twenty target genes with the highest PicTar score
from PicTar, and all predicted target genes from miBridge. The data was synthesized to

26
assemble a list of gene targets that appeared in more than one prediction program. Both
MicroCosm Targets and miBridge predicted WEE1 to be a high probability target of
miR-17 and miR-20a. WEE1 also was identified as a likely target of miR-18a, miR-19a,
and miR-19b in one prediction program each.
The putative miR-17-92 target sites within the 3’ UTR of WEE1 are shown in
Figure 5A. Since miR-17, miR-20a, and miR-18a are highly homologous, it is not
surprising that they are all predicted to target the same region in the 3’ UTR of WEE1.
Similarly, miR-19a and miR-19b share a common target site. The predicted alignment
between WEE1 mRNA and each of the individual miRNAs of the cluster shows that there
is a high degree of sequence complementarity within each paring especially in the seed
region (Figure 5B). We hypothesized that the cluster exerts combinatorial control on this
target, thereby amplifying the effects of downregulation. The goal of this aim is to
elucidate whether WEE1 is a valid target of miR-17-92 in MLL fusion leukemias.
Determination of whether miR-17-92 Specifically Targets the 3’ Untranslated Region of WEE1
A study published earlier this year (after my project had been initiated)
demonstrated that relative luciferase activity decreased by 21% when a luciferase reporter
construct containing the 3’ UTR of WEE1 was co-transfected with miR-17 compared to
when a luciferase reporter construct without the 3’ UTR was co-transfected with miR-
1761. Similarly, the authors found a 15% reduction in luciferase activity with miR-20a61.
While these results suggest that WEE1 is a target of miR-17 and miR-20a, they fail to
identify the specific region of binding within the 3’ UTR and also neglect to investigate
whether the rest of the miR-17-92 cluster exerts a regulatory effect on WEE1.

27
Figure 5. Predicted miR-17-92 Binding Sites in the 3’ UTR of WEE1
!"#$%&'(!"#$)*+'(!"#$%,+(-+./01(2"10(
!"#$%3+'(!"#$%34(-+./01(2"10(
!"
#"
(A) Sequence of WEE1. The protein coding sequence is indicated in blue and the 3’ UTR is the region from the stop codon to the poly-A tail. The putative miR-17-92 target sites are highlighted in yellow. (B) Predicted alignment between WEE1 mRNA and miR-17, miR-20a, miR-18a, miR-19a, and miR-19b. The microRNA seed sequence is indicated in pink.

28
To establish whether WEE1 is a biological target of the entire miR-17-92 cluster
and to determine the specific regions being targeted, we performed a luciferase reporter
assay along with mutagenesis studies. First, the 3’ UTR of WEE1 with the putative miR-
17-92 binding sites intact was amplified from human genomic DNA and cloned into the
pMIR-REPORT Luciferase vector immediately following the luciferase gene (Figure
6A). We also generated three mutant versions of the reporter construct that disrupt
binding between the miRNA and the mRNA by specifically mutating the six nucleotides
where the seed region of the miRNA is predicted to bind. Each guanine was converted to
a thymine and vice versa. Each cytosine was converted to an adenine and vice versa.
One mutant possesses mutations in the putative miR-17, miR-20a, and miR-18a target
site while another mutant contains mutations in the putative miR-19a and miR-19b target
site (Figure 6A). The third mutant has all the putative miR-17-92 target sites mutated
(Figure 6A).
We co-expressed the wild-type or one of three mutant reporter constructs with
miR-17, miR-17-19b, miR-17-92, or empty vector in HEK293T cells. Renilla luciferase,
which was under the control of the HSV TK promoter, was also co-expressed for each
condition as a control. 42 hours after transfection, the cells were lysed and firefly and
Renilla luciferase activity were measured. Firefly luciferase activity was normalized
relative to Renilla luciferase activity to control for differences in transfection efficiency.
Results are presented from one representative experiment performed in triplicate. Three
independent experiments were conducted.

29
Figure 6. Luciferase Reporter and Mutagenesis Assays
!"#$%&'()&*
+,-*.'/0/1&'*
2334*56*789* !"#$%&$'()*+,-.*)/"*./)"0*+1*)
!"#$%&'()&*
+,-*.'/0/1&'*
2334*56*789*!"#$%&2)(3+2)%4+)
*+,-.*)/"*./)!5*+*.6)
!"#$%&'()&*
+,-*.'/0/1&'*
2334*56*789*!"#$%'+2)%'7)
*+,-.*)/"*./)!5*+*.6)
!"#$%&'()&*
+,-*.'/0/1&'*
2334*56*789*899)*+,-.*)/"*./)
!5*+*.6)
!"
#"
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1.8
$%&'()*+,"
-%.(/01"2341"
/54"67)"
-%.(/841"
/89"67)"
!&&"67)"
(A) Schematic of wild-type and mutant luciferase reporter constructs. (B)-(E) Relative luciferase activity measured 42 hours after the co-transfection of one of the reporter constructs with miR-17 (B), miR-17-19b (C), miR-17-92 (D), or empty vector (E) along with Renilla luciferase in HEK293T cells. The data is presented as relative firefly luciferase: Renilla luciferase normalized to the wild-type construct, with error bars showing standard deviation. * p<0.05; ** p<0.01; *** p<0.001.
:"
;"
<"
$%&'()*+,"
$%&'()*+,"
$%&'()*+,"
-%.(/01"2341"
/54"67)"
-%.(/01"2341"
/54"67)"
-%.(/01"2341"
/54"67)"
-%.(/841"
/89"67)"
-%.(/841"
/89"67)"
-%.(/841"
/89"67)"
!&&"67)"
!&&"67)"
!&&"67)"
-%.(/0" -%.(/0(/89"
-%.(/0(82" <-+)*"=,>)?@"
AAA"
AAA"
AAA"
AAA"
AAA"
AAA"
A"
A"A"
AA"AAA"
.,&4BC,"D7>%E,@4F,"!>BC%)*"
.,&4BC,"D7>%E,@4F,"!>BC%)*"
.,&4BC,"D7>%E,@4F,"!>BC%)*"
.,&4BC,"D7>%E,@4F,"!>BC%)*"

30
Since the alignment score between WEE1 and the individual miRNAs of the miR-
17-92 cluster is high, we hypothesized that WEE1 is a physiological target of miR-17-92.
If WEE1 is in fact a target, when the wild-type reporter construct is co-expressed with
any of the miRNAs of the cluster, the miRNA will bind to the 3’ UTR and repress
translation of luciferase, resulting in low luminescence. On the other hand, co-expression
of a reporter plasmid containing mutated binding sites with one of the miRNAs of the
cluster will inhibit binding and result in relatively higher luminescence.
As seen in Figure 6B, when the putative miR-17 binding site is mutated in the
reporter construct (miR-17, miR-20a, and miR-18a target site mutated or all target sites
mutated), relative luciferase activity increases upon overexpression of miR-17 as
compared to when the putative binding site is intact (wild-type or miR-19a and miR-19b
target site mutated). There is a statistically significant increase in luciferase activity
(p<0.001) in both cases. This indicates that WEE1 is a valid target of miR-17. Because
luciferase activity does not increase when the putative miR-19a and miR-19b site is
mutated, we can deduce that miR-17 specifically targets the predicted miR-17 target site
(Figure 6B).
The miR-17-19b construct expresses all of the miRNAs present in the miR-17-92
cluster, except for miR-92a. All five of the miRNAs within miR-17-19b are predicted to
target WEE1. When the miR-17, miR-20a, and miR-18a target site is mutated in the
reporter construct, overexpression of miR-17, miR-20a, and miR-18a leads to an increase
in luciferase activity (Figure 6C). This suggests that WEE1 is a target of at least one of
these miRNAs. Notably, the change in luciferase activity is quite similar to the change in

31
luciferase activity for miR-17 alone, indicating that perhaps miR-20a and miR-18a are
less important than miR-17 in regulating WEE1. When the miR-19a and miR-19b target
site is mutated in the reporter construct, overexpression of miR-19a and miR-19b causes
a small, but statistically significant increase in luciferase activity (Figure 6C). This
implies that WEE1 is a target of miR-19a, miR-19b, or both. Finally, when all of the
predicted target sites are mutated, co-expression of all five miRNAs increases luciferase
activity by a larger margin, indicating that possibly all five miRNAs collectively regulate
WEE1 expression (Figure 6C).
As shown in Figure 6D, co-expression of the entire miR-17-92 cluster with any of
the mutant reporter constructs results in a statistically significant increase in luciferase
activity. Since the reporter construct with all of the predicted target sites mutated had the
greatest increase in luciferase activity relative to the wild-type construct, this provides
further evidence that the entire miR-17-92 cluster is acting jointly to downregulate WEE1
expression.
Surprisingly, there were small, but statistically significant increases in luciferase
activity in the presence of the empty vector (Figure 6E). This observation led us to
predict that high endogenous miR-17-92 levels in HEK293T cells could be exerting an
effect on this assay. It was later confirmed by TaqMan MicroRNA Assays that
HEK293T cells have relatively high endogenous levels of miR-17 and miR-19a (Figure
7A, B).
From the luciferase reporter and mutagenesis assays, we can conclude that miR-
17 specifically targets nucleotides 465 to 487 of the 3’ UTR of WEE1, which leads to

32
downregulation of WEE1. miR-20a and miR-18a also conceivably regulate WEE1 by
targeting the same region in the 3’ UTR, albeit to a lesser extent than miR-17. Finally,
miR-19a and miR-19b exert control on WEE1 by targeting nucleotides 1069 to 1091 of
the 3’ UTR, although this regulation is likely less influential than that of miR-17.
Determination of miR-17-92 Expression Levels in MLL Fusion Leukemia Cell Lines Compared to Non-MLL Fusion Leukemia Cell Lines
Previous studies have shown that the miR-17-92 cluster is particularly
upregulated in samples from patients with acute leukemias bearing MLL rearrangements
compared to patient samples with other common translocations47, 48. ChIP analysis
showed that MLL fusion proteins upregulate expression of the miR-17-92 cluster by
directly binding to the locus promoter region48. From these prior studies, we
hypothesized that overexpression of miR-17-92 would also be a signature of MLL fusion
leukemia cell lines.
To quantify endogenous miR-17-92 expression in MLL fusion leukemia cell lines
and non-MLL fusion leukemia cell lines, we isolated RNA, synthesized cDNA using a
looped reverse transcription primer specific to each miRNA, and performed TaqMan
MicroRNA Assays using a variety of cell lines. The MLL fusion leukemia cell lines
included those with MLL-AF4 fusions (MV-4-11 and RS4;11) and MLL-AF9 fusions
(THP-1 and MonoMac6). These cell lines represent the most common MLL
translocations. The non-MLL fusion leukemia cell lines chosen included one with a
BCR-ABL gene fusion (K-562), one with amplified c-Myc (HL-60), and a histiocytic
lymphoma (U-937). We also included a non-leukemia cell line (HEK293T) as a control.

33
We selected two representative miRNAs, miR-17 and miR-19a, to quantify. miR-
17 was chosen because miR-17, miR-20a, and miR-18a have highly homologous
sequences and are predicted to target the same site in the 3’ UTR of WEE1. Similarly,
since miR-19a and miR-19b differ by only one nucleotide and share the same target site,
miR-19a was selected as the second representative miRNA. Ubiquitously expressed U6
snRNA served as the internal control. The assay was performed in triplicate and repeated
two to five times.
Based on results from the bead-based miRNA expression profiling assay and the
TaqMan qPCR assay47, 48, we expected the MLL fusion leukemia cell lines to have high
levels of miR-17 and miR-19a and the non-MLL fusion leukemia cell lines and the non-
leukemia control cell line to have low expression of these two miRNAs.
Surprisingly, our results showed no significant increase in miR-17 (Figure 7A) or
miR-19a (Figure 7B) expression in the MLL fusion leukemia cell lines compared to the
non-MLL fusion leukemia cell lines and the non-leukemia control. The cell lines with
the highest expression of these two miRNAs were HL-60 and U-937 cells, both of which
are non-MLL fusion leukemia cells.
From the TaqMan MicroRNA Assays, we can conclude that miR-17-92
expression levels in leukemia cell lines are dependent on factors in addition to the
presence of a MLL translocation.
Determination of WEE1 Expression Levels in MLL Fusion Leukemia Cell Lines Compared to Non-MLL Fusion Leukemia Cell Lines
Western blot analysis was conducted to examine endogenous WEE1 protein
levels in MLL fusion leukemia cell lines and non-MLL fusion leukemia cell lines. To

34
Figure 7. Quantification of Endogenous miR-17 and miR-19a Levels
0 0.5
1 1.5
2 2.5
3 3.5
!"#$%&"'()*+",,-./
'.0'1
-!234'
5./
.5$67'
8(9:;<='
>2;<4'
8?27@'
92A7
:'
=8B23'
!CDE33'
5F2D233'
G'
0 0.5
1 1.5
2 2.5
3 3.5
!"#$%&"'()*+",,-./
'.0'1
-!23;$'
5F2D233'
!CDE33'
=8B23'
5./
.5$67'
92A7
:'
8?27@'
>2;<4'
8(9:;<='
H'
(A) Endogenous expression levels of miR-17 (A) and miR-19a (B) in four MLL fusion leukemia cell lines (MV-4-11, RS4;11, THP-1, MonoMac6), three non-MLL fusion leukemia cell lines (K-562, HL-60, U-937), and one non-leukemia cell line (HEK293T). RNA was isolated from each cell line, cDNA was synthesized, and expression levels of miR-17 and miR-19a were quantified with TaqMan MicroRNA Assays. Shown are average expression levels relative to U6 snRNA and normalized to MV-4-11. Error bars indicate standard deviation.

35
enrich for WEE1, which localizes to the nucleus, nuclear and cytoplasmic proteins were
extracted from the same panel of cell lines used for quantification of miR-17 and miR-
19a levels (MV-4-11, RS4;11, THP-1, MonoMac6, K-562, HL-60, U-937, and
HEK293T). Nuclear and cytoplasmic extracts were electrophoresed on SDS-
polyacrylamide gels, transferred to PVDF membranes, and probed for WEE1 expression
using an antibody that is reactive with human WEE1. The membranes were then stripped
and re-probed for !-Actin as a loading control. The nuclear WEE1 and !-Actin bands
were quantified using MultiGauge V3.0 software. Two independent experiments were
performed and the results from one representative experiment are presented.
We originally hypothesized that the MLL fusion leukemia cell lines, which we
expected to have high miR-17-92 levels, would have low WEE1 expression if WEE1
were indeed a target of miR-17-92. Conversely, we predicted that the non-MLL fusion
leukemia cell lines, which we expected to have low miR-17-92 levels, would have high
WEE1 expression.
Western blot analysis revealed that, as expected, WEE1 is present in the nucleus62
(Figure 8A), but not in the cytoplasm (Figure 8C). WEE1 expression from nuclear
extracts was quantified relative to !-Actin expression (Figure 8B). Notably, there was no
connection between endogenous WEE1 expression and MLL rearrangement status. For
example, the cell lines with the highest relative expression of WEE1, MonoMac6 and
THP-1, were in fact both MLL fusion leukemia cell lines. Strikingly, the cell lines with
the lowest relative expression of WEE1 were the same cell lines with the highest relative
expression of miR-17 and miR-19a, namely HL-60 and U-937.

36
Figure 8. Endogenous Nuclear and Cytoplasmic WEE1 Protein Expression
0
0.5
1
1.5
2
2.5
3
!"#$#%%&
'($)%%&
*+,#%&
!-.
-!/01&
2#31
4&
+5#16&
7#89:&
+;2489*&
'<=/>?<&;@AB<CCD-.
&-E&F
;;%&
G&
!"#$#%%&
'($)%%&
*+,#%&
!-.
-!/01&
2#31
4&
+5#16&
7#89:&
+;2489*&
H&
F;;%&
F;;%&
I#H0>.&
J&F;;%&
I#H0>.&
(A) Western blot analysis of endogenous WEE1 from nuclear extracts of the indicated cell lines. A shorter exposure (upper) and longer exposure (lower) of the WEE1 blot are shown. !-Actin served as a loading control. (B) Quantification of WEE1 expression levels from nuclear extracts relative to !-Actin levels. Relative expression was normalized to MV-4-11. Band intensity was quantified using MultiGauge V3.0 software. (C) Western blot analysis of endogenous WEE1 from cytoplasmic extracts of the same cell lines used in (A) and (B). !-Actin was used as a loading control.

37
From the Western blot analysis, we can conclude that in the cell lines we tested,
endogenous WEE1 expression is dependent on factors in addition to the presence of a
MLL fusion protein.
Determination of the Relationship between miR-17-92 Expression and WEE1 Expression
We initially hypothesized that high endogenous expression of miR-17-92 in MLL
fusion leukemia cell lines would correspond with low endogenous expression of WEE1
in the same cell lines. While we did not see a direct relationship between MLL
rearrangement status and miR-17-92 expression, we were interested in discerning
whether a relationship existed between miR-17-92 expression and WEE1 expression.
Relative expression of miR-17 that was determined by TaqMan MicroRNA
Assays was plotted against relative expression of WEE1 that was determined by
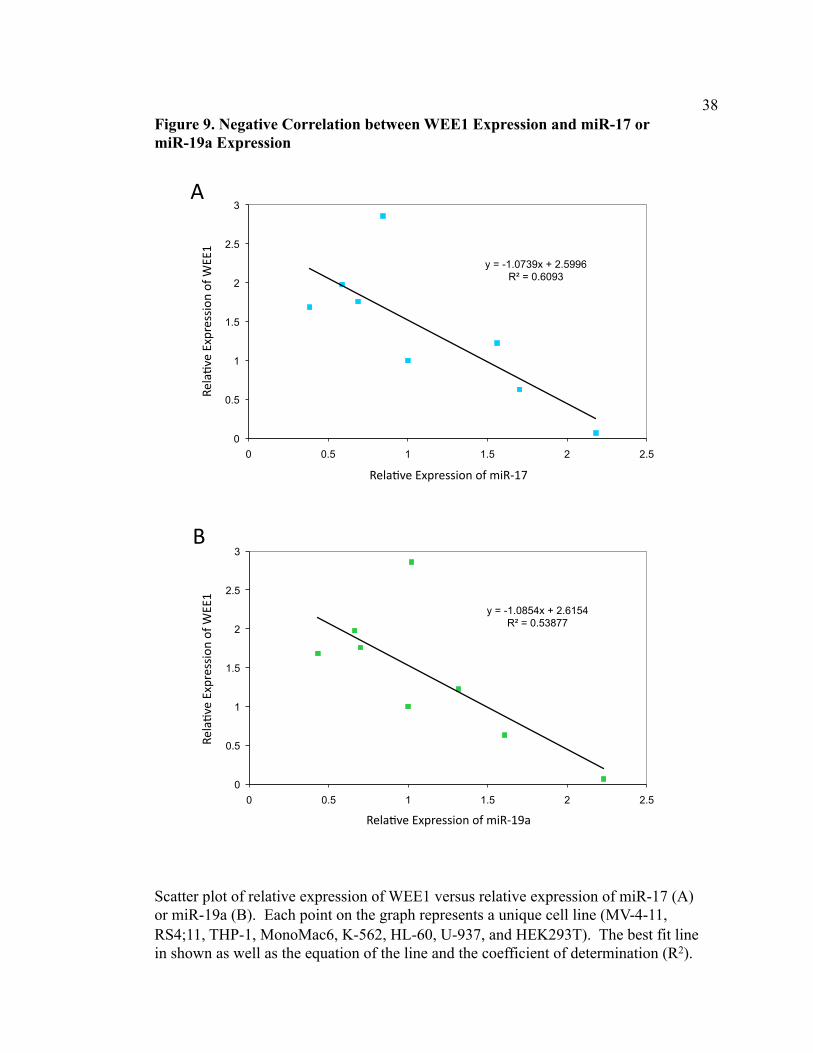
quantification of Western blot analysis. We observed a negative correlation between the
two variables with a coefficient of determination (R2) of 0.6093 (Figure 9A). In general,
as miR-17 levels increased, there was a corresponding decrease in WEE1 expression
levels. Similarly, we saw a negative correlation between relative expression of miR-19a
and relative expression of WEE1 with a coefficient of determination (R2) of 0.5388
(Figure 9B). This result strengthens our conclusion that WEE1 is a valid target of the
miR-17-92 cluster.
38
Figure 9. Negative Correlation between WEE1 Expression and miR-17 or miR-19a Expression
y = -1.0739x + 2.5996 R! = 0.6093
0
0.5
1
1.5
2
2.5
3
0 0.5 1 1.5 2 2.5
!"#$%&"'()*+",,-./
'.0'1
((2'
!"#$%&"'()*+",,-./'.0'3-!425'
6'
Scatter plot of relative expression of WEE1 versus relative expression of miR-17 (A) or miR-19a (B). Each point on the graph represents a unique cell line (MV-4-11, RS4;11, THP-1, MonoMac6, K-562, HL-60, U-937, and HEK293T). The best fit line in shown as well as the equation of the line and the coefficient of determination (R2).
!"#$%&"'()*+",,-./
'.0'1
((2'
!"#$%&"'()*+",,-./'.0'3-!427$'
8'
y = -1.0854x + 2.6154 R! = 0.53877
0
0.5
1
1.5
2
2.5
3
0 0.5 1 1.5 2 2.5

39
CHAPTER 4
DISCUSSION
Identifying targets of the miR-17-92 cluster is important for expanding our
understanding of the complex molecular pathways that regulate gene expression and are
dysregulated in leukemia and cancer in general. Previous studies have experimentally
validated a range of gene targets of the miR-17-92 cluster in various systems. The
individual miRNAs of this cluster have been shown to promote cell proliferation by
downregulating p2143 and Pten44, suppress apoptosis by downregulating E2Fs41 and
Bim42, and induce angiogenesis in solid tumors by downregulating Tsp1 and CTGF45.
Since combined haploinsufficiency of Bim and Pten only partially mimics the oncogenic
effects of miR-17-92 overexpression44, it is likely that additional targets contribute to
these effects. As such, we were interested in seeking novel targets of the miR-17-92
cluster. In this study, we set out to ascertain which gene transcripts were being targeted
for downregulation by the miR-17-92 cluster in MLL fusion leukemias.
We used bioinformatic miRNA target gene prediction algorithms to predict
additional targets of miR-17-92. WEE1, a protein kinase that adds an inhibitory
phosphate to Cdk1, was identified as a possible target of miR-17, miR-20a, miR-18a,
miR-19a, and miR-19b. According to MicroCosm Targets, among all the miRNAs
predicted to target WEE1, miR-17 had the second highest score, miR-20a had the ninth
highest score, and miR-18a had the thirtieth highest score. It is conceivable, therefore,

40
that multiple miRNAs from the miR-17-92 cluster collectively regulate WEE1
expression.
Our objective for this thesis was to experimentally validate the computationally
predicted target WEE1. We hypothesized that overexpression of the miR-17-92 cluster
in MLL fusion leukemias leads to downregulation of WEE1, which promotes cell cycle
progression and eventually results in leukemia.
Luciferase reporter assays are considered the “gold standard” in validating
miRNA gene targets44, 63, 64. We performed luciferase reporter assays in which we co-
transfected a wild-type or mutant luciferase reporter construct with various combinations
of miRNAs from the miR-17-92 cluster. We observed statistically significant increases
in luciferase activity when a putative miRNA binding site was mutated in the reporter
construct compared to when the site was intact.
From our experiments, it appeared as though miR-17 is the most crucial regulator
of WEE1 expression. The change in luciferase activity for miR-17, miR-20a, and miR-
18a together was quite similar to the change in luciferase activity for miR-17 alone,
indicating that perhaps miR-20a and miR-18a are less important. Furthermore, while the
increase in luciferase activity for miR-19a and miR-19b was statistically significant, it
was not as sizeable as for miR-17. To better decipher the individual contribution of each
miRNA of the miR-17-92 cluster to WEE1 regulation, it would be interesting to follow
up these studies by expressing each miRNA individually in this assay.
Unexpectedly, we observed small increases in luciferase activity in the presence
of the empty vector. This suggests that endogenous miR-17-92 expression in HEK293T

41
cells might be slightly affecting the assay. It would be interesting to repeat this
experiment in a cell line with low endogenous levels of miR-17 and miR-19a, for
example, in K-562 cells. These cells would have to be nucleofected with the appropriate
constructs because they cannot be transfected easily.
Overall, from the luciferase reporter assays, we were able to experimentally
confirm our hypothesis that WEE1 is a bona fide target of the miR-17-92 cluster.
Importantly, through mutagenesis studies, we found the specific region in the 3’ UTR of
WEE1 that miR-17, miR-20a, and miR-18a target (nucleotides 465 to 487) and the
specific region in the 3’ UTR of WEE1 that miR-19a and miR-19b target (nucleotides
1069 to 1091).
TaqMan MicroRNA Assays are a useful technique for quantifying endogenous
levels of miRNAs in tissues ranging from cell lines to patient samples65. A panel of cell
lines was selected for this study including MLL fusion leukemia cell lines (MV-4-11,
RS4;11, THP-1, MonoMac6), non-MLL fusion leukemia cell lines (K-562, HL-60, U-
937), and a control non-leukemia cell line (HEK293T). We isolated RNA from each of
the cell lines, prepared cDNA, and then assayed for endogenous miR-17 and miR-19a
expression using TaqMan MicroRNA Assays.
Surprisingly, HL-60 and U-937, both of which are non-MLL fusion leukemia cell
lines, displayed the highest expression of these two miRNAs. This is not entirely
unexpected, though, because HL-60 cells have amplification of the c-Myc gene, which
has been shown to directly transactivate transcription of the miR-17-92 cluster41.

42
Likewise, U-937 cells are derived from lymphoma cells, and many lymphomas possess
amplification of the human locus encoding the miR-17-92 cluster35.
The difference between the relative miR-17-92 expression levels we observed and
those that the Chen group observed47, 48 may be attributed to inherent differences in
expression profiles between cell lines and patient samples. The method of creating new
cell lines requires immortalizing primary human cells, a process that can cause changes in
gene expression66. Interestingly, TaqMan qPCR data from the Chen group showed that
patient samples better followed the expected trend of high miR-17-92 levels in MLL
fusion leukemias and low miR-17-92 levels in non-MLL fusion leukemias than cell
lines47, 48. In fact, they found that miR-17 and miR-20a expression was high in all the
leukemia cell lines they tested regardless of MLL rearrangement status47, 48. While the
Chen group tested ME1, KASUMI-1, and NB4 as their non-MLL fusion leukemia cell
lines, we tested K-562, HL-60, and U-937 cells. We did, however, have overlap in the
MLL fusion leukemia cell lines tested. Common cell lines included MV-4-11, THP-1,
and MonoMac6. The Chen group observed high expression of miR-17 and miR-20a in
all three of these cell lines47, 48. We also observed fairly high expression of miR-17 and
miR-19a in these cell lines, but unlike the Chen group, we did not mean-center our
expression data, which makes a direct comparison between the two sets of data difficult.
To further validate WEE1 as a target of the miR-17-92 cluster, we conducted
Western blot analysis to assess the relative endogenous expression of WEE1 across a
panel of MLL fusion leukemia cell lines (MV-4-11, RS4;11, THP-1, MonoMac6), non-
MLL fusion leukemia cell lines (K-562, HL-60, U-937), and a control non-leukemia cell

43
line (HEK293T). We hypothesized that cell lines with high expression levels of miR-17-
92 would express lower levels of WEE1.
We chose to look at WEE1 protein expression using Western blot analysis rather
than WEE1 mRNA expression using qRT-PCR because miRNA gene targeting always
downregulates protein expression, but only sometimes downregulates mRNA expression.
It allowed us to explore whether either of the mechanisms for downregulation, mRNA
cleavage or translational repression, occurred. mRNA cleavage takes place when the
mRNA has extensive complementarity to the miRNA24, 67. Translational repression
occurs when there is insufficient complementarity for mRNA cleavage, but adequate
complementarity at multiple sites within the 3’ UTR for translational repression24, 67.
Translational repression is also the more common mechanism used by metazoan
miRNAs. Since the individual miRNAs of the miR-17-92 cluster are metazoan miRNAs
and because there is not perfect complementarity between the miRNAs of the miR-17-92
cluster and the 3’ UTR of WEE1, we hypothesized that WEE1 mRNA levels remained
constant upon miRNA targeting, but WEE1 protein levels decreased.
To enrich for WEE1, which localizes to the nucleus62, we prepared nuclear and
cytoplasmic protein extracts. As predicted, we detected WEE1 in the nuclear extracts,
but not in the cytoplamic extracts. Remarkably, the two cell lines with the highest
endogenous expression of miR-17 and miR-19a, HL-60 and U-937, displayed the lowest
endogenous expression of WEE1.
Interestingly, WEE1 electrophoresed slower than expected. While the predicted
molecular weight for WEE1 is 72 kDa, product literature for the WEE1 antibody

44
indicates that the apparent molecular weight on SDS-polyacrylamide gels is typically 95
to 100 kDa. In our Western blot analysis, WEE1 was detected at approximately 150 kDa,
which raises the possibility of WEE1 dimerization or the presence of post-translational
modifications.
We were interested to see if a relationship existed between endogenous miR-17 or
miR-19a expression and endogenous WEE1 expression. Plotting the relative expression
of miR-17 versus the relative expression of WEE1 revealed a negative correlation
between the two variables. A similar negative correlation was observed for the
relationship between the relative expression of miR-19a and the relative expression of
WEE1. That is, as the expression of one of the two miRNAs increased, the expression of
WEE1 decreased. We concluded that the regulation was likely occurring at physiological
levels because we observed this trend while studying endogenous expression rather than
by overexpression.
While the data indicates that there is a correlation between miR-17-92 levels and
WEE1 expression levels, it does not necessarily imply causation between the variables.
However, causation was established through the luciferase reporter assays, which showed
that luciferase activity increased upon co-expression of the miR-17-92 cluster with a
reporter plasmid containing mutations in all of the putative miR-17-92 binding sites.
Nevertheless, observing a negative correlation between miR-17-92 and WEE1 in the cell
lines we tested provides further evidence in support of WEE1 being a valid target of the
miR-17-92 cluster.

45
According to the model shown in Figure 10A, when expression of the miR-17-92
cluster is low, WEE1 is translated normally. As such, WEE1 functions to prevent cells
from entering mitosis until they are ready. Conversely, when expression of the miR-17-
92 cluster is high, miR-17, miR-20a, or miR-18a bind to nucleotides 465 to 487 and miR-
19a or miR-19b bind to nucleotides 1069 to 1091 of the 3’ UTR of WEE1 (Figure 10B).
It is conceivable that different molecules of WEE1 mRNA have different combinations of
miRNAs from the miR-17-92 cluster bound at these target sites. Together with the RISC
complex, the miR-17-92 cluster represses translation of WEE1. WEE1 is a critical
regulator of the G2/M transition, one of the restriction checkpoints. At each checkpoint,
cell cycle progression stalls if flaws are detected. Because WEE1 inhibits cell cycle
progression, it is an anti-proliferative protein. Consequently, downregulation of WEE1
by the miR-17-92 cluster would have pro-proliferative functional effects.
In summary, we have established WEE1 as a novel target of the entire miR-17-92
cluster except for miR-92a. Moreover, we have determined the exact location within the
3’ UTR of WEE1 that miR-17, miR-20a, miR-18a, miR-19a, and miR-19b target. While
we did not observe a relationship between miR-17-92 expression and MLL rearrangement
status in the cell lines we tested, we demonstrated a specific inverse relationship between
endogenous miR-17-92 expression and endogenous WEE1 expression in these cell lines.
The results from this project present several new possibilities for future
investigations. It remains to be determined the exact contribution of each miRNA from
the miR-17-92 cluster to WEE1 regulation. This can be established by studying each
miRNA individually in a luciferase reporter assay. To determine whether miR-17-92 is

46
Figure 10. Model of the Regulation of WEE1 by the miR-17-92 Cluster
!"#$%%&#
'()*# '()*#
!"#$%%&#
+,'-&./#012/#&32# +,'-&42/#&45#
(A) In the absence of the miR-17-92 cluster, WEE1 is translated as normal and WEE1 protein levels remain high. (B) In the presence of high expression of the miR-17-92 cluster in some types of leukemia, miR-17, miR-20a, or miR-18a bind to their target site in the 3’ UTR of WEE1 and miR-19a or miR-19b bind to their target site. Together with the RISC complex, the miR-17-92 cluster inhibits translation of WEE1, which corresponds to a decrease in WEE1 protein levels. Since WEE1 inhibits cell cycle progression, downregulation of WEE1 promotes cell cycle progression and contributes to leukemogenesis.
!#
6#

47
regulating WEE1 by mRNA cleavage or by translational repression, it would be useful to
investigate WEE1 mRNA levels by qRT-PCR. If mRNA levels are unchanged by
changes in miR-17-92 expression, one can deduce that downregulation is occurring via
translational repression. If, however, WEE1 mRNA levels co-vary with WEE1 protein
levels, this would suggest that mRNA cleavage is occurring. Additionally, it would be
interesting to investigate the expression of WEE1 in samples from patients with different
subtypes of leukemia to determine whether a negative correlation exists between miR-17-
92 and WEE1 in patient samples. Another possible avenue for future research would be
to explore whether WEE1 is a target of the miR-17-92 cluster in other types of cancers
including solid tumors. A positive finding would suggest that there are broader
implications for this regulatory relationship. It also remains to be resolved whether
abnormal miRNA expression causes development of the cancer or whether the cancer
causes abnormal miRNA expression.
The conclusions from this research suggest that the miR-17-92 cluster and WEE1
are potential therapeutic targets for leukemia. Researchers are already working on
developing efficient delivery mechanisms to administer antisense oligonucleotides to
inhibit miRNAs such as the oncogenic miR-17-92 cluster68. One of the challenges of this
approach is that all of the miRNAs of the cluster would have to be inhibited
simultaneously due to the functional redundancy of the cluster. Furthermore, developing
a mechanism to reintroduce WEE1 into leukemia patients with downregulated WEE1
could prove beneficial in inhibiting uncontrolled cell proliferation. It is my hope that

48
targeted cancer therapies such as these will continue to make their way from the bench to
the bedside.

49
REFERENCES 1. Ziemin-van der Poel S, McCabe NR, Gill HJ, Espinosa R,3rd, Patel Y, Harden A,
Rubinelli P, Smith SD, LeBeau MM, Rowley JD. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci U S A 1991 Dec 1;88(23):10735-9.
2. Meyer C, Kowarz E, Hofmann J, Renneville A, Zuna J, Trka J, Ben Abdelali R, Macintyre E, De Braekeleer E, De Braekeleer M, Delabesse E, de Oliveira MP, Cave H, Clappier E, van Dongen JJ, Balgobind BV, van den Heuvel-Eibrink MM, Beverloo HB, Panzer-Grumayer R, Teigler-Schlegel A, Harbott J, Kjeldsen E, Schnittger S, Koehl U, Gruhn B, Heidenreich O, Chan LC, Yip SF, Krzywinski M, Eckert C, Moricke A, Schrappe M, Alonso CN, Schafer BW, Krauter J, Lee DA, Zur Stadt U, Te Kronnie G, Sutton R, Izraeli S, Trakhtenbrot L, Lo Nigro L, Tsaur G, Fechina L, Szczepanski T, Strehl S, Ilencikova D, Molkentin M, Burmeister T, Dingermann T, Klingebiel T, Marschalek R. New insights to the MLL recombinome of acute leukemias. Leukemia 2009 Aug;23(8):1490-9.
3. Popovic R, Zeleznik-Le NJ. MLL: How complex does it get? J Cell Biochem 2005 May 15;95(2):234-42.
4. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 2007 Nov;7(11):823-33.
5. Pui CH, Frankel LS, Carroll AJ, Raimondi SC, Shuster JJ, Head DR, Crist WM, Land VJ, Pullen DJ, Steuber CP. Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia with the t(4;11)(q21;q23): A collaborative study of 40 cases. Blood 1991 Feb 1;77(3):440-7.
6. Daser A, Rabbitts TH. Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev 2004 May 1;18(9):965-74.
7. Tkachuk DC, Kohler S, Cleary ML. Involvement of a homolog of drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992 Nov 13;71(4):691-700.
8. Thirman MJ, Gill HJ, Burnett RC, Mbangkollo D, McCabe NR, Kobayashi H, Ziemin-van der Poel S, Kaneko Y, Morgan R, Sandberg AA. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. N Engl J Med 1993 Sep 23;329(13):909-14.

50
9. Mann G, Attarbaschi A, Schrappe M, De Lorenzo P, Peters C, Hann I, De Rossi G, Felice M, Lausen B, Leblanc T, Szczepanski T, Ferster A, Janka-Schaub G, Rubnitz J, Silverman LB, Stary J, Campbell M, Li CK, Suppiah R, Biondi A, Vora A, Valsecchi MG, Pieters R, Interfant-99 Study Group. Improved outcome with hematopoietic stem cell transplantation in a poor prognostic subgroup of infants with mixed-lineage-leukemia (MLL)-rearranged acute lymphoblastic leukemia: Results from the interfant-99 study. Blood 2010 Oct 14;116(15):2644-50.
10. Pieters R, Schrappe M, De Lorenzo P, Hann I, De Rossi G, Felice M, Hovi L, LeBlanc T, Szczepanski T, Ferster A, Janka G, Rubnitz J, Silverman L, Stary J, Campbell M, Li CK, Mann G, Suppiah R, Biondi A, Vora A, Valsecchi MG. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (interfant-99): An observational study and a multicentre randomised trial. Lancet 2007 Jul 21;370(9583):240-50.
11. Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci U S A 1998 Sep 1;95(18):10632-6.
12. Zeleznik-Le NJ, Harden AM, Rowley JD. 11q23 translocations split the "AT-hook" cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc Natl Acad Sci U S A 1994 Oct 25;91(22):10610-4.
13. Birke M, Schreiner S, Garcia-Cuellar MP, Mahr K, Titgemeyer F, Slany RK. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res 2002 Feb 15;30(4):958-65.
14. Fair K, Anderson M, Bulanova E, Mi H, Tropschug M, Diaz MO. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol Cell Biol 2001 May;21(10):3589-97.
15. Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol 2001 Apr;21(7):2249-58.
16. Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to hox gene promoters. Mol Cell 2002 Nov;10(5):1107-17.
17. Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 2002 Nov;10(5):1119-28.

51
18. Ambros V. microRNAs: Tiny regulators with great potential. Cell 2001 Dec 28;107(7):823-6.
19. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993 Dec 3;75(5):843-54.
20. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in caenorhabditis elegans. Nature 2000 Feb 24;403(6772):901-6.
21. Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA 2003 Feb;9(2):175-9.
22. Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004 Jan 23;116(2):281-97.
23. Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J 2002 Sep 2;21(17):4663-70.
24. Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci U S A 2003 Aug 19;100(17):9779-84.
25. Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 2003 Dec 15;17(24):3011-6.
26. Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science 2004 Jan 2;303(5654):95-8.
27. Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III drosha initiates microRNA processing. Nature 2003 Sep 25;425(6956):415-9.
28. Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003 Oct 17;115(2):209-16.
29. Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD. Asymmetry in the assembly of the RNAi enzyme complex. Cell 2003 Oct 17;115(2):199-208.
30. Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in drosophila cells. Nature 2000 Mar 16;404(6775):293-6.

52
31. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005 Jan 14;120(1):15-20.
32. Popovic R, Riesbeck LE, Velu CS, Chaubey A, Zhang J, Achille NJ, Erfurth FE, Eaton K, Lu J, Grimes HL, Chen J, Rowley JD, Zeleznik-Le NJ. Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood 2009 Apr 2;113(14):3314-22.
33. Mian YA, Zeleznik-Le NJ. MicroRNAs in leukemias: Emerging diagnostic tools and therapeutic targets. Curr Drug Targets 2010 Jul;11(7):801-11.
34. Lau NC, Lim LP, Weinstein EG, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in caenorhabditis elegans. Science 2001 Oct 26;294(5543):858-62.
35. Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M. Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res 2004 May 1;64(9):3087-95.
36. Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell 2008 Apr 18;133(2):217-22.
37. Ventura A, Young AG, Winslow MM, Lintault L, Meissner A, Erkeland SJ, Newman J, Bronson RT, Crowley D, Stone JR, Jaenisch R, Sharp PA, Jacks T. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008 Mar 7;132(5):875-86.
38. Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol 2007 Oct 15;310(2):442-53.
39. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature 2005 Jun 9;435(7043):828-33.
40. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 2006 Feb 14;103(7):2257-61.

53
41. O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-myc-regulated microRNAs modulate E2F1 expression. Nature 2005 Jun 9;435(7043):839-43.
42. Koralov SB, Muljo SA, Galler GR, Krek A, Chakraborty T, Kanellopoulou C, Jensen K, Cobb BS, Merkenschlager M, Rajewsky N, Rajewsky K. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell 2008 Mar 7;132(5):860-74.
43. Ivanovska I, Ball AS, Diaz RL, Magnus JF, Kibukawa M, Schelter JM, Kobayashi SV, Lim L, Burchard J, Jackson AL, Linsley PS, Cleary MA. MicroRNAs in the miR-106b family regulate p21/CDKN1A and promote cell cycle progression. Mol Cell Biol 2008 Apr;28(7):2167-74.
44. Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell 2003 Dec 26;115(7):787-98.
45. Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a myc-activated microRNA cluster. Nat Genet 2006 Sep;38(9):1060-5.
46. Wong P, Iwasaki M, Somervaille TC, Ficara F, Carico C, Arnold C, Chen CZ, Cleary ML. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer Res 2010 May 1;70(9):3833-42.
47. Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, Chen P, Wang Y, Yan M, Qian Z, Neilly MB, Jin J, Zhang Y, Bohlander SK, Zhang DE, Larson RA, Le Beau MM, Thirman MJ, Golub TR, Rowley JD, Chen J. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc Natl Acad Sci U S A 2008 Oct 7;105(40):15535-40.
48. Mi S, Li Z, Chen P, He C, Cao D, Elkahloun A, Lu J, Pelloso LA, Wunderlich M, Huang H, Luo RT, Sun M, He M, Neilly MB, Zeleznik-Le NJ, Thirman MJ, Mulloy JC, Liu PP, Rowley JD, Chen J. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc Natl Acad Sci U S A 2010 Feb 23;107(8):3710-5.
49. Li Z, Luo RT, Mi S, Sun M, Chen P, Bao J, Neilly MB, Jayathilaka N, Johnson DS, Wang L, Lavau C, Zhang Y, Tseng C, Zhang X, Wang J, Yu J, Yang H, Wang SM, Rowley JD, Chen J, Thirman MJ. Consistent deregulation of gene expression between human and murine MLL rearrangement leukemias. Cancer Res 2009 Feb 1;69(3):1109-16.

54
50. Russell P, Nurse P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987 May 22;49(4):559-67.
51. Igarashi M, Nagata A, Jinno S, Suto K, Okayama H. Wee1(+)-like gene in human cells. Nature 1991 Sep 5;353(6339):80-3.
52. Parker LL, Sylvestre PJ, Byrnes MJ,3rd, Liu F, Piwnica-Worms H. Identification of a 95-kDa WEE1-like tyrosine kinase in HeLa cells. Proc Natl Acad Sci U S A 1995 Oct 10;92(21):9638-42.
53. McGowan CH, Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J 1993 Jan;12(1):75-85.
54. O'Farrell PH. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol 2001 Dec;11(12):512-9.
55. Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J 1995 May 1;14(9):1878-91.
56. Kornbluth S, Sebastian B, Hunter T, Newport J. Membrane localization of the kinase which phosphorylates p34cdc2 on threonine 14. Mol Biol Cell 1994 Mar;5(3):273-82.
57. Leise W,3rd, Mueller PR. Multiple Cdk1 inhibitory kinases regulate the cell cycle during development. Dev Biol 2002 Sep 1;249(1):156-73.
58. Mueller PR, Coleman TR, Dunphy WG. Cell cycle regulation of a xenopus Wee1-like kinase. Mol Biol Cell 1995 Jan;6(1):119-34.
59. Qi J, Yu JY, Shcherbata HR, Mathieu J, Wang AJ, Seal S, Zhou W, Stadler BM, Bourgin D, Wang L, Nelson A, Ware C, Raymond C, Lim LP, Magnus J, Ivanovska I, Diaz R, Ball A, Cleary MA, Ruohola-Baker H. microRNAs regulate human embryonic stem cell division. Cell Cycle 2009 Nov 15;8(22):3729-41.
60. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 2001 Dec;25(4):402-8.
61. Trompeter HI, Abbad H, Iwaniuk KM, Hafner M, Renwick N, Tuschl T, Schira J, Muller HW, Wernet P. MicroRNAs MiR-17, MiR-20a, and MiR-106b act in concert to modulate E2F activity on cell cycle arrest during neuronal lineage differentiation of USSC. PLoS One 2011 Jan 20;6(1):e16138.

55
62. McGowan CH, Russell P. Cell cycle regulation of human WEE1. EMBO J 1995 May 15;14(10):2166-75.
63. Clancy JL, Nousch M, Humphreys DT, Westman BJ, Beilharz TH, Preiss T. Methods to analyze microRNA-mediated control of mRNA translation. Methods Enzymol 2007;431:83-111.
64. Stark A, Brennecke J, Russell RB, Cohen SM. Identification of drosophila MicroRNA targets. PLoS Biol 2003 Dec;1(3):E60.
65. Benes V, Castoldi M. Expression profiling of microRNA using real-time quantitative PCR, how to use it and what is available. Methods 2010 Apr;50(4):244-9.
66. Drexler HG, Macleod RA. History of leukemia-lymphoma cell lines. Hum Cell 2010 Aug;23(3):75-82.
67. Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002 Sep 20;297(5589):2056-60.
68. Scherr M, Venturini L, Battmer K, Schaller-Schoenitz M, Schaefer D, Dallmann I, Ganser A, Eder M. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res 2007;35(22):e149.

56
VITA
The author, Sonia Olikara, was born in Columbus, Indiana on March 28, 1983 to
Cherian and Marina Olikara. She received a Bachelor of Arts degree with a double major
in Biochemistry and Spanish from DePauw University in Greencastle, Indiana in May
2004. As an undergraduate student, Sonia was awarded a Science Research Fellowship,
where she investigated the molecular phylogenies of rotifers through gene sequencing
and began working towards determining the structure of long-chain acyl-CoA
dehydrogenase and gamma-glutamylcysteine synthetase.
From July 2005 to July 2009, Sonia worked as a Research Technologist at the
University of Chicago in the laboratory of Steve Goldstein, M.D., Ph.D. During that
time, she studied how sumoylation regulates potassium ion channels at the plasma
membrane.
In August 2009, Sonia entered the Molecular Biology Program at Loyola
University Chicago in Maywood, Illinois. She later joined the laboratory of Nancy
Zeleznik-Le, Ph.D., where she examined the role of the miR-17-92 cluster in MLL fusion
leukemias.
After completing her Master of Science degree, Sonia will join the Integrated
Graduate Program in the Life Sciences as a Ph.D. student at Northwestern University.
She plans to study cancer genetics and epigenetics during her doctoral degree.

THESIS APPROVAL SHEET
The thesis submitted by Sonia Olikara has been read and approved by the following committee: Nancy Zeleznik-Le, Ph.D., Director Professor of Medicine Oncology Institute Loyola University Chicago Manuel Diaz, M.D. Professor of Medicine and Molecular Biology Program Director Oncology Institute Loyola University Chicago Charles Hemenway, M.D., Ph.D. Ronald McDonald House Charities Endowed Professor in Pediatric Hematology/Oncology Oncology Institute Loyola University Chicago The final copies have been examined by the director of the thesis and the signature which appears below verifies the fact that any necessary changes have been incorporated and that the thesis is now given final approval by the committee with reference to content and form. The thesis is therefore accepted in partial fulfillment of the requirements for the degree of Master of Science. ________________ ___________________________________________ Date Director’s Signature
