vol 21 6 2017 - institut de l'information scientifique et
TRANSCRIPT

Volume 1 - Number 1 May - September 1997
Volume 21 - Number 6 June 2017

The PDF version of the Atlas of Genetics and Cytogenetics in Oncology and Haematology is a reissue of the original articles published in collaboration with
the Institute for Scientific and Technical Information (INstitut de l’Information Scientifique et Technique - INIST) of the French National Center for Scientific
Research (CNRS) on its electronic publishing platform I-Revues.
Online and PDF versions of the Atlas of Genetics and Cytogenetics in Oncology and Haematology are hosted by INIST-CNRS.
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
Scope
The Atlas of Genetics and Cytogenetics in Oncology and Haematology is a peer reviewed on-line journal in open access,
devoted to genes, cytogenetics, and clinical entities in cancer, and cancer-prone diseases.
It is made for and by: clinicians and researchers in cytogenetics, molecular biology, oncology, haematology, and pathology.
One main scope of the Atlas is to conjugate the scientific information provided by cytogenetics/molecular genetics to the
clinical setting (diagnostics, prognostics and therapeutic design), another is to provide an encyclopedic knowledge in cancer
genetics. The Atlas deals with cancer research and genomics. It is at the crossroads of research, virtual medical university
(university and post-university e-learning), and telemedicine. It contributes to "meta-medicine", this mediation, using
information technology, between the increasing amount of knowledge and the individual, having to use the information.
Towards a personalized medicine of cancer.
It presents structured review articles ("cards") on:
1- Genes,
2- Leukemias,
3- Solid tumors,
4- Cancer-prone diseases, and also
5- "Deep insights": more traditional review articles on the above subjects and on surrounding topics.
It also present
6- Case reports in hematology and
7- Educational items in the various related topics for students in Medicine and in Sciences.
The Atlas of Genetics and Cytogenetics in Oncology and Haematology does not publish research articles.
See also: http://documents.irevues.inist.fr/bitstream/handle/2042/56067/Scope.pdf
Editorial correspondance
Jean-Loup Huret, MD, PhD,
Genetics, Department of Medical Information,
University Hospital
F-86021 Poitiers, France
phone +33 5 49 44 45 46
Editor, Editorial Board and Publisher See:http://documents.irevues.inist.fr/bitstream/handle/2042/48485/Editor-editorial-board-and-publisher.pdf
The Atlas of Genetics and Cytogenetics in Oncology and Haematology is published 12 times a year by ARMGHM, a non
profit organisation, and by the INstitute for Scientific and Technical Information of
the French National Center for Scientific Research (INIST-CNRS) since 2008.
The Atlas is hosted by INIST-CNRS (http://www.inist.fr)
Staff: Vanessa Le Berre
Philippe Dessen is the Database Directorof the on-line version (Gustave Roussy Institute – Villejuif – France).
Publisher Contact: INIST-CNRS
Mailing Address: Catherine Morel, 2,Allée du Parc de Brabois, CS 10130, 54519 Vandoeuvre-lès-Nancy France.
Email Address:[email protected]
Articles of the ATLAS are free in PDF format, and metadata are available on the web in Dublin Core XML format and freely
harvestable.A Digital object identifier (DOI®), recorded at the International Agency CrossRefhttp://www.crossref.org/ is
assigned to each article.
http://AtlasGeneticsOncology.org
© ATLAS - ISSN 1768-3262

Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6)
Atlas of Genetics and Cytogenetics
in Oncology and Haematology
INIST-CNRS
OPEN ACCESS JOURNAL
Editor-in-Chief Jean-Loup Huret (Poitiers, France) Lymphomas Section Editor Antonino Carbone (Aviano, Italy)
Myeloid Malignancies Section Editor Robert S. Ohgami (Stanford, California)
Bone Tumors Section Editor Judith Bovee (Leiden, Netherlands)
Head and Neck Tumors Section Editor Cécile Badoual (Paris, France)
Urinary Tumors Section Editor Paola Dal Cin (Boston, Massachusetts)
Pediatric Tumors Section Editor Frederic G. Barr (Bethesda, Maryland)
Cancer Prone Diseases Section Editor Gaia Roversi (Milano, Italy)
Cell Cycle Section Editor João Agostinho Machado-Neto (São Paulo, Brazil)
DNA Repair Section Editor Godefridus Peters (Amsterdam, Netherlands)
Hormones and Growth factors Section Editor Gajanan V. Sherbet (Newcastle upon Tyne, UK)
Mitosis Section Editor Patrizia Lavia (Rome, Italy)
WNT pathway Section Editor Alessandro Beghini (Milano, Italy)
B-cell activation Section Editors Anette Gjörloff Wingren and Barnabas Nyesiga (Malmö,
Sweden)
Oxidative stress Section Editor Thierry Soussi (Stockholm, Sweden/Paris, France)
Board Members
Sreeparna
Banerjee Department of Biological Sciences, Middle East Technical University, Ankara, Turkey; [email protected]
Alessandro
Beghini Department of Health Sciences, University of Milan, Italy; [email protected]
Judith Bovée 2300 RC Leiden, The Netherlands; [email protected]
Antonio Cuneo Dipartimento di ScienzeMediche, Sezione di Ematologia e Reumatologia Via Aldo Moro 8, 44124 - Ferrara, Italy;
Paola Dal Cin Department of Pathology, Brigham, Women's Hospital, 75 Francis Street, Boston, MA 02115, USA; [email protected]
François
Desangles
IRBA, Departement Effets Biologiques des Rayonnements, Laboratoire de Dosimetrie Biologique des Irradiations, Dewoitine C212,
91223 Bretigny-sur-Orge, France; [email protected]
Enric Domingo Molecular and Population Genetics Laboratory, Wellcome Trust Centre for Human Genetics, Roosevelt Dr. Oxford, OX37BN, UK
Ayse Elif Erson-
Bensan Department of Biological Sciences, Middle East Technical University, Ankara, Turkey; [email protected]
Ad Geurts van
Kessel
Department of Human Genetics, Radboud University Medical Center, Radboud Institute for Molecular Life Sciences, 6500 HB
Nijmegen, The Netherlands; [email protected]
Oskar A. Haas Department of Pediatrics and Adolescent Medicine, St. Anna Children's Hospital, Medical University Vienna, Children's Cancer
Research Institute Vienna, Vienna, Austria. [email protected]
Anne Hagemeijer Center for Human Genetics, University Hospital Leuven and KU Leuven, Leuven, Belgium; [email protected]
Nyla Heerema Department of Pathology, The Ohio State University, 129 Hamilton Hall, 1645 Neil Ave, Columbus, OH 43210, USA;
Sakari Knuutila Hartmann Institute and HUSLab, University of Helsinki, Department of Pathology, Helsinki, Finland; [email protected]
Lidia Larizza Lab Centro di Ricerche e TecnologieBiomedicheIRCCS-IstitutoAuxologico Italiano Milano, Italy; l.larizza@auxologico
Roderick Mc Leod Department of Human, Animal Cell Lines, Leibniz-Institute DSMZ-German Collection of Microorganisms, Cell Cultures,
Braunschweig, Germany; [email protected]
Cristina Mecucci Hematology University of Perugia, University Hospital S.Mariadella Misericordia, Perugia, Italy; [email protected]
Fredrik Mertens Department of Clinical Genetics, University and Regional Laboratories, Lund University, SE-221 85 Lund, Sweden;
Konstantin Miller Institute of Human Genetics, Hannover Medical School, 30623 Hannover, Germany; [email protected]
Felix Mitelman Department of Clinical Genetics, University and Regional Laboratories, Lund University, SE-221 85 Lund, Sweden;
Hossain Mossafa Laboratoire CERBA, 95066 Cergy-Pontoise cedex 9, France; [email protected]
Stefan Nagel Department of Human, Animal Cell Lines, Leibniz-Institute DSMZ-German Collection of Microorganisms, Cell Cultures,
Braunschweig, Germany; [email protected]
Florence
Pedeutour
Laboratory of Solid Tumors Genetics, Nice University Hospital, CNRSUMR 7284/INSERMU1081, France;
Susana Raimondi Department of Pathology, St. Jude Children's Research Hospital, 262 Danny Thomas Place, Mail Stop 250, Memphis, Tennessee
38105-3678, USA; [email protected]
Clelia Tiziana
Storlazzi Department of Biology, University of Bari, Bari, Italy; [email protected]
Sabine Strehl CCRI, Children's Cancer Research Institute, St. Anna Kinderkrebsforschunge.V., Vienna, Austria; [email protected]
Nancy Uhrhammer Laboratoire Diagnostic Génétique et Moléculaire, Centre Jean Perrin, Clermont-Ferrand, France; [email protected]
Dan L. Van Dyke Mayo Clinic Cytogenetics Laboratory, 200 First St SW, Rochester MN 55905, USA; [email protected]
Roberta Vanni Universita di Cagliari, Dipartimento di ScienzeBiomediche(DiSB), CittadellaUniversitaria, 09042 Monserrato (CA) - Italy;
Franck Viguié Service d'Histologie-Embryologie-Cytogénétique, Unité de Cytogénétique Onco-Hématologique, Hôpital Universitaire Necker-Enfants
Malades, 75015 Paris, France; [email protected]

Atlas of Genetics and Cytogenetics
in Oncology and Haematology
INIST-CNRS
OPEN ACCESS JOURNAL
Volume 21, Number 6, June 2017
Table of contents
Gene Section
GSKIP (GSK3-beta interaction protein) 197 Christine Bellanné-Chantelot, Isabelle Plo
KNL1 (cancer susceptibility candidate 5) 200 Masato Takimoto, Jean-Loup Huret
ATG2B (Autophagy-related 2B) 205 Christine Bellanné-Chantelot, Isabelle Plo
Leukaemia Section
Primary mediastinal B-cell lymphoma (PMBL) 208 Luis Miguel Juárez Salcedo, Samir Dalia
t(2;11)(p21;q23) without KMT2A (MLL) rearrangement 210 Ana L. Ruano, Shashirekha Shetty
t(1;12)(p36;p13) ETV6/PRDM16 215 Francois P. Duhoux, Hélène A. Poirel
t(1;1)(p36;p36) PRDM16/SKI 218 Jean-Loup Huret
t(1;3)(p36;q21) PSMD2/PRDM16 ??? 220 Jean-Loup Huret
t(1;7)(p36;p12) IKZF1/PRDM16 223 Jean-Loup Huret
Case Report Section
A pediatric case of acute lymphoblastic leukemia with t(2;9)(q12;q34) (RANBP2/ABL1 fusion) 225 Marc De Braekeleer, Nadia Guéganic, Alexandra Schifferli, Joëlle Tchinda

Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 197
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
GSKIP (GSK3-beta interaction protein) Christine Bellanné-Chantelot, Isabelle Plo
Département de Génétique, Hpitaux Universitaires Pitié-Salpétrière-Charles Foix, Paris (CBC);
INSERM UMR1170, Institut Gustave Roussy, Villejuif, (CBC, IP), France. christine.bellanne-
[email protected]; [email protected]
Published in Atlas Database: October 2016
Online updated version : http://AtlasGeneticsOncology.org/Genes/GSKIPID64074ch14q32.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68250/10-2016-GSKIPID64074ch14q32.pdf DOI: 10.4267/2042/68250
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract GSK3beta interaction protein (GSKIP) is a negative
regulator of GSK3B (GSK3 beta) which is a highly
conserved serine-threonine kinase involved in many
cellular processes including glycogen metabolism,
proliferation, differentiation, and development.
GSKIP directly interacts with GSK3B through its C-
terminal conserved GSK3B -binding domain (GID)
and negatively regulates GSK3B in the Wnt/ beta -
catenin signaling pathway. The overexpression of
GSKIP may result in the activation of the Wnt
pathway involved in hematopoietic stem cell
homeostasis and normal megakaryopoiesis and in
the development of leukemia stem cells in acute
myeloid leukemia (AML). In a mouse model,
GSK3B allelic deletion results in a myelodysplastic
syndrome that, when combined with GSK3A
deletion, leads to AML
The germline duplication of ATG2B and GSKIP,
both located in 14q32.2, predisposes to the
development of familial myeloproliferative
neoplasms with autosomal dominant inheritance, in
particular essential thrombocythemia progressing to
leukemia. Overexpression of ATG2B and GSKIP
enhances megakaryocyte progenitor differentiation
by increasing progenitor sensitivity to
thrombopoietin. Both genes cooperate with somatic
JAK2, MPL and CALR mutations and their
overexpression provides a growth advantage to
hematopoietic cells carrying these driver mutations
that may explain the familial aggregation and the
progression of essential thrombocythemia to
myelofibrosis and leukemia.
Keywords
GSKIP; Myeloproliferative neoplasms (MPN);
essential thrombocythemia; myelofibrosis;
leukemia; predisposition; ATG2B/GSKIP;
chromosome 14; CNV; autophagy; Wnt/beta-
catenin pathway
Identity
Other names: C14orf129
HGNC (Hugo): GSKIP
Location : 14q32.2
Location (base pair)
GSKIP starts at 96.831.073 and ends at 96.853.629
bp (according to hg19-Feb_2009)
Local order
centromere to telomere.
Note
cooperates with ATG2B, also located in 14q32.2 and
included in the 700 kb duplication
NC_000014.9:g.96.163.103_96.857.129dup (on
Assembly GRCh37)
DNA/RNA
Description The GSKIP gene consists of 2 non-coding exons and
2 exons, spanning a coding region of 3433 bp.
Transcription There are four transcripts that differ by their 5'UTR
and encode the same protein. The longest transcript
(NM_001271904) of the GSKIP gene has a total

GSKIP (GSK3-beta interaction protein) Bellanné-Chantelot C, Plo I
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 198
total length of 1050 nucleotides.
Pseudogene
Not yet identified.
Protein
Description
The protein encoded by the GSKIP gene is the
GSK3-beta interaction protein of 139 amino acids,
with a calculated molecular mass of 15.648 kDa.
Expression
Expression of GSKIP has been detected in various
normal human tissues (bone marrow, whole blood,
thymus, brain, heart, muscle, colon, kidney, liver,
lung, pancreas, thyroid, salivary and adrenal glands,
skin, ovary, uterus, placenta, prostate and testis). The
gene is overexpressed in bone, colon and rectum.
In hematopoietic cells, GSKIP is expressed in
CD34+ purified hematopoietic progenitors and
CD36+ erythroblasts or CD41+ megakaryocytes
derived from CD34+ progenitors cultured in vitro
(Saliba et al, 2016)
Localisation
GSKIP is localized in the cytoplasm and nucleus.
Function
GSKIP belongs to the family of A-kinase anchoring
proteins (AKAPs) that bind serine/threonine kinase
(PKA). These AKAPs proteins interact with the
regulatory domain of PKA and facilitate their
phosphorylation. GSKIP directly interacts with
GSK3B through its C-terminal conserved GSK3B-
binding domain (GID; amino acid 115-139) and
negatively regulates GSK3B in the Wnt/beta-catenin
signaling pathway (Chou et al, 2006). The
overexpression of GSKIP may mimic activation of
the Wnt pathway involved in hematopoietic stem
cell homeostasis and normal megakaryopoiesis (Li et
al, 2008) and in the development of leukemia stem
cells in AML (Wang. et al, 2010).
It has recently been shown in a mouse model that
Gsk3b allelic deletion results in a myelodysplastic
syndrome that, when combined with GSK3A
deletion, leads to AML (Guezguez et al, 2016).
Mutations
Germinal
A germline 14q32.2 head-to-tail duplication of 700
kb has been associated with familial myeloid
malignancies (Saliba et al , 2015). The germline
duplication includes the genes TCL1A, GSKIP,
ATG2B, BDKRB1, BDKRB2 and the first exon of
AK7. The overexpression of ATG2B and GSKIP
that are expressed in myeloid cells, enhances
hematopoietic progenitor differentiation,
particularly of megacaryocytes. The development of
myeloid malignancies required the cooperation of
both genes with the myeloproliferative neoplasms
(MPN) driver JAK2 Val617Phe mutation, MPL or
CALR mutations. The mechanism of cooperation
between ATG2B and GSKIP with MPN driver
mutations remains unknown.
The germline duplication with the same distal and
proximal breakpoints has only been identified in
MPN families originated from West Indies
(Martinique) suggesting a founder effect.
Implicated in
Familial myeloproliferative neoplasms (MPN)
Disease
Familial MPN originated from West-indies
(Martinique) and in particular, essential
thrombocythemia progressing to myelofibrosis
and/or acute myeloid leukemia and primary
myelofibrosis may be linked to ATG2B/GSKIP
germline duplication. The predisposition is highly
penetrant (80%) and is characterized by an earlier
age of MPN onset in comparison to sporadic cases
(41 years versus > 60 years). The spectrum of
acquired driver mutations (JAK2 Val617Phe, MPL and
CALR mutations) is similar to the spectrum of
mutations in sporadic MPN cases.
Prognosis
The percentage of transformation is close to 50% in
these familial MPN cases and is related to the
detection of mutations affecting epigenetic regulator
genes such as TET2 IDH1 or IDH2.
Acute myeloid leukemia (AML)
Disease
AML originated from West-indies (Martinique) may
be linked to ATG2B/GSKIP germline duplication.
Prognosis
The prognosis of the disease is also linked to the
detection of acquired mutations in TET2, IDH1 or in
IDH2. No TP53 mutation was found, contrary to
what was observed in AML evolving from MPN,
suggesting a different pathway for leukemic
transformation.
References Chou HY, Howng SL, Cheng TS, Hsiao YL, Lieu AS, Loh JK, Hwang SL, Lin CC, Hsu CM, Wang C, Lee CI, Lu PJ, Chou CK, Huang CY, Hong YR. GSKIP is homologous to the Axin GSK3beta interaction domain and functions as a negative regulator of GSK3beta. Biochemistry. 2006 Sep 26;45(38):11379-89
Guezguez B, Almakadi M, Benoit YD, Shapovalova Z, Rahmig S, Fiebig-Comyn A, Casado FL, Tanasijevic B, Bresolin S, Masetti R, Doble BW, Bhatia M. GSK3 Deficiencies in Hematopoietic Stem Cells Initiate Pre-neoplastic State that Is Predictive of Clinical Outcomes of

GSKIP (GSK3-beta interaction protein) Bellanné-Chantelot C, Plo I
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 199
Human Acute Leukemia. Cancer Cell. 2016 Jan 11;29(1):61-74
Li D, August S, Woulfe DS. GSK3beta is a negative regulator of platelet function and thrombosis. Blood. 2008 Apr 1;111(7):3522-30
Saliba J, Saint-Martin C, Di Stefano A, Lenglet G, Marty C, Keren B, Pasquier F, Valle VD, Secardin L, Leroy G, Mahfoudhi E, Grosjean S, Droin N, Diop M, Dessen P, Charrier S, Palazzo A, Merlevede J, Meniane JC, Delaunay-Darivon C, Fuseau P, Isnard F, Casadevall N, Solary E, Debili N, Bernard OA, Raslova H, Najman A, Vainchenker W, Bellanné-Chantelot C, Plo I. Germline
duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet. 2015 Oct;47(10):1131-40
Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010 Mar 26;327(5973):1650-3
This article should be referenced as such:
Bellanné-Chantelot C, Plo I. GSKIP (GSK3-beta interaction protein). Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):197-199.

Gene Section Review
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 200
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
KNL1 (cancer susceptibility candidate 5) Masato Takimoto, Jean-Loup Huret
Institute for Genetic Medicine, Hokkaido University, Sapporo, Hokkaido, Japan.
[email protected] (MT); Genetics, Dept Medical Information, University of Poitiers, CHU
Poitiers Hospital, F-86021 Poitiers, France. [email protected] (JLH)
Published in Atlas Database: October 2016
Online updated version : http://AtlasGeneticsOncology.org/Genes/AF15q14ID318.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68251/10-2016-AF15q14ID318.pdf DOI: 10.4267/2042/68251
This article is an update of : Takimoto M. CASC5 (cancer susceptibility candidate 5). Atlas Genet Cytogenet Oncol Haematol 2013;17(1) Takimoto M. CASC5 (Cancer Sensitibity Candidate 5). Atlas Genet Cytogenet Oncol Haematol 2007;11(1) Huret JL, Charrin C. AF15q14 (ALL1 fused gene from 15q14). Atlas Genet Cytogenet Oncol Haematol 2000;4(2)
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract
Review on KNL1, with data on DNA, on the protein
encoded, and where the gene is implicated.
Keywords
KNL1
Identity
Other names: CT29, CASC5, AF15Q14, D40,
PPP1R5, hKNL-1, hSpc105, AF15q14, KIAA1570
HGNC (Hugo): KNL1
Location: 15q15.1
DNA/RNA
Note
Whole genomic size is about 70 kbp, but consists of
27 exons.
Transcription
KNL1 mRNA expression is dominant in normal
human testis and slight expression are observed in
other organs, such as placenta. Analysis on cancer
cell lines, such as HeLa, gave single band with 8.5
kb. There is another alternative splicing site at the 5'
side of this gene that generates a short exon with 78
bp in cDNA. There are potential other alternative
splicing at cancer cell lines.
Analysis on testis mRNA shows two bands with size
of approximately 6 and 8,5 kb which are probably
derived from the two isoforms.
Protein
Description
Encodes 1833 amino acids and 2342 amino acids.
The KLN1 protein contains: conserved motifs,
which are the following: a (S/G)ILK motif (aa 25-
28), a RRVSF motif (aa 57-61), and, for BUB3
recognition, MELT repeats (aa 140-161, 308-329,
474-494, 562-582, 750-769, 859-882, 902-924, 940-
962, 1025-1044, 1073-1096, 1114-1136, 1152-
1174). The Bubs recognition KI motifs
KI(D/N)XXXF(L/I)XXLK, are KIDTTSFLANLK
(aa 202-213) for BUB1, and KIDFNDFIKRLK (aa
238-249) for BUB1B: (BUBR1); a nuclear
localization signal (aa 1789-1803); a coiled coil
region (aa 1942-2133) and the ZWINT (Zwint-1)
binding region (aa 1834 or 19811 for a smaller
region -2108); and RWD repeats (aa 2109- 2353)
With the NSL1 (hMis14)-binding region (aa 2109-
2316), according to Kiyomitsu et al., 2011, VEGA
checking, and SwissProt.

KNL1 (cancer susceptibility candidate 5) Takimoto M, Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 201
Figure 1. Domain structure of KNL1 protein. Brown arrows indicate protein interactions, and blue arrows and line with T-shape indicate phosphorylation and dephosphorylation, respectively. MT :microtubules. KT : kinetochore.
Expression
KNL1 protein expressions with molecular weight of
approximately 250 kDa and 300 kDa are observed in
human testicular germ cells and cancer cell lines.
Localisation
In germ cell of testis, significant high expressions of
KNL1 protein are observed in nucleus of
spermatocytes and slightly in spermatogonia. It is
noteworthy that round spermatids express
significantly high KNL1 protein in their pre-
acrosome. As KNL1 protein has no hydrophobic
signal peptide in its amino terminus, it probably
localizes outer surface of pre-acrosome membrane of
spermatids inside of the cells..
In mitosis KNL1 protein is localized in kinetochore
in a human cancer cell line.
Function
KNL1 is a large of kinetochore protein, constituting
KMN (KNL1/ MIS12 complex/ NDC80 complex)
network. KMN network is the central hub of outer
kinetochore, not only connecting mitotic
chromosomes and spindles but also coordinating
microtubule-binding, chromosome congression and
spindle assembly checkpoint (SAC) signaling. There
are two microtubule binding activity in KMN
network, one in NDC80 and the other in KNL1.
KNL1 protein binds directly microtubule through its
far N-terminal region, consisting of 68 amino acids,
in vitro, and indirectly through the interactions with
MIS12 and NDC80 complexes at its C-terminus.
KNL1 depletion affects metaphase chromosome
congression. In C. elegans,
The depletion preclude metaphase chromosomal
congression, while in yeast, Drosophila and human
cells, the depletions show partial alignment
phenotypes, in which some chromosomes congress
to the equator but many chromosomes remain
stranded near the spindle poles. In the N-terminus of
KNL1 protein, AURKB (Aurora kinase B)
phosphorylates SILK and RVSF motifs, and then
disrupts the interaction between KNL1 and protein
phosphatase I (PP1), which also binds the motifs.
KNL1 augments Aurora B kinase activity that
phosphorylates outer kinetochore protein, such as
NDC80, resulting in reduction its microtubule-
binding activity. Protein phosphatase 2A ( PP2A) is
recruited by BUB1B (BubR1), one of SAC protein.
Both PP1 and PPA2 are suggested to counteract
Aurora B kinase activity. Especially, PP1 was shown
to stabilize microtubule attachments to kinetochores
probably through KNL1, while Aurora B kinase
destabilizes microtubule-binding by NDC80

KNL1 (cancer susceptibility candidate 5) Takimoto M, Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 202
phosphorylation as described. The destabilized
binding of KNL1 to microtubules is presumably
important for correcting and eliminating erroneous
kinetochore-microtubule attachment during SAC.
It is suggested that the bindings with microtubule
and with PP1 also play roles in SAC silencing.
Although their binding sites are in close proximity,
one of their bindings do not affect the other, and they
contribute independently to the silencing of SAC.
Two KI motifs, KI1 and KI2, localized in the N-
terminus, bind SAC protein, BUB1 and BUB1B,
respectively, through tetratricopeptide repeats
(TPRs) of the proteins, resulting into folding the
motifs into short alpha-helices. Although BUB1
fragment with mutation in KI-binding domain was
not able to bind to KNL1 in vitro, BUB1 and BUB1B
mutants with KI-binding sites were able to attach to
kinetochore. Mutations in the BUB3-binding domain
(BUB3-BD) in BUB1 and BUB1B prevent
kinetochore localization of the SAC protein. BUB1
fragment consisting only of N-terminus with TPRs
does not localize to kinetochore and longer
fragments that accommodate BUB3-BD did. Those
results suggest that BUB3-BD of BUB1 and
BUB1B, rather than TPRs, is critical for their
recruitment to kinetochore and that the interaction
between TPRs of Bub proteins and KI-motif of
KNL1 might play a subsidiary role in the localization
of BUB1 and BUB1B to kinetochore. In response to
SAC signal, the first step of this response is
phosphorylation of MELT motifs of KNL1, located
in the N-terminal and central region, by Mps1
kinase, and then the phosphorylated MELT motifs
bind BUB3/BUB1complex, mediating SAC
signaling. KNL1- BUB3-BUB1(KBB) complex
binds MXD1 (MAD1)/ MAD2L1 (MAD2) complex,
and then, together with MAD1 phosphorylation by
Msp1, the binding leads to CDC20/MAD2
formation, an essential part of Mitotic Checkpoint
Complex (MCC) that inhibits Anaphase Promoting
Complex/Cyclosome (APC/C).
Recently, it suggested that there are two pathways
for recruiting MAD1-MAD2 that results in SAC
activation. One is the pathway through KBB, as
described above, the other is KNTC1 (ROD)-RW10-
ZWILCH, (RZZ) complex, which interacts with
KNL1 through ZWINT (Zwint-1 protein). The
former is required for SAC activation when
kinetochores are misaligned but is not essential when
kinetochores are unattached from microtubules. The
latter binds SPDL1 (Spindly protein) and MAD1-
MAD2, and causes the anaphase-onset delay in
response to unattached kinetochore independently of
the former.
It was suggested that the binding of KNL1 with
microtubules and with PP1 contribute to silencing of
SAC, in which motor protein dynein, moving along
on microtubules, is suggested to work to strip
MAD1-MAD2. The C-terminal region of KNL1
interacts with MIS12 and Zwint-1 protein, through
RWD motif and coiled-coiled region, respectively.
The binding with the former plays role in connecting
inner kinetochore with KNL1 and the latter mediates
the interaction of KNL1 with RZZ complex which
works in SAC regulation as described above.
Implicated in
Leukemia
A small subset of leukemia with a t(11;15)(q23;q14)
has been described for long and has often be referred
as: t(11;15)(q23;q15) MLL/AF15q14. KMT2A,
(previous symbol: mixed leukemia gene (MLL)) is
translocated with KNL1 (previous symbol CASC5,
originally described as AF15q14), which makes
research of published cases often arduous.
t(11;15)(q23;q15) and/or KMT2A/KNL1
t(11;15)(q23;q14-15) Data on 16 cases with a t(11;15)(q23;q14-15),
according to (Yang et al., 2014) are the following:
there was 2 of myelodysplastic syndrome (MDS)
cases, 10 acute myeloid leukemia (AML) cases (2
M1, 4 M2, 3 M4, and 1 NOS), and 4 acute
lymphoblastic leukemia (ALL) cases. Mean age of
the patients was 20.6 years (range 1-54); there were
11 males and 5 females. Abnormalities of
chromosome 3 were seen in 10 out of 16 cases. Out
of 8 patients for whom clinical data were available,
only 3 are in complete remission, whereas 5 patients
died with a mean survival period of 10.4 months.
t(11;15)(q23;q15) and KMT2A/KNL1 Of 7 cases with a t(11;15)(q23;q15) and
KMT2A/KNL1 hybrid gene and fusion protein
((Chinwalla et al., 2003; Kuefer et al., 2003; Meyer
et al., 2006; Yang et al., 2014). Diagnosis was:
therapy related MDS (t-MDS) in 2 cases, AML in 4
cases (1 M2, 2 AML-M4, 1 AML-NOS), and 1 de
novo T-ALL. Sex ratio was 5M:1F; There was 3
children and 4 adult patients. Of three cases with data
on survival, patients died at: 8 mths, 8 mths, and 22
mths. Of four cases with documented karyotypes: the
karyotype was a complex karyotype with markers in
two cases, abnormalities of chromosome 3 were seen
in three cases, +21 in two cases. KMT2A (MLL)
exon 8, 9, or 10 were fused to exon 10, 11 or 12 of
KNL1, the fusion protein contains the 1362 or 1418
first aa from MLL with aa 1796, 1818 or 1819 from
KNL1 (according to authors and/or VEGA).
A t(3;15;p14;q15) KNL1/ ADAMTS9-AS2 is
mentioned, without further data in the ChiTARS
database (Gorohovski et al., 2016) as a chimeric EST
(dbEST Id:12413828; accession BQ375909).
Lung cancer
In one study on primary lung cancer, KNL1 mRNA
expression was observed in more than 40% of the
cases, which is the highest among all the different
types of cancers examined. The study also revealed

KNL1 (cancer susceptibility candidate 5) Takimoto M, Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 203
that clinicopathological findings correlates with
KNL1 expression. KNL1 mRNA expression is more
frequent in the tumors with low differentiation than
the ones with moderate and high differentiation.
Further, the tumors derived from smoker express
higher incidence of KNL1 mRNA than the ones from
non-smoker.
Spermatogenesis
KNL1 mRNA was highly expressed in normal testis.
As KNL1 protein expressions were observed in
spermatogonia and spermatocytes in seminiferous
tube of human testes, this protein may also play a
role in cell division as a kinetochore protein in
meiotic cells. It is noteworthy that KNL1 protein is
also significantly expressed in pre-acrosome of
spermatids, especially from its early stage,
suggesting that KNL1 might be playing important
role in the formation of acrosome, an essential
organelle for fertilization. KNL1 expressions in
testes of the patients with infertility were
significantly lower than normal ones.
Microcephaly
Disease
Autosomal recessive primary microcephaly
(MCPH) is a very rare neuro-developmental disorder
with brain size reduction, no structural malformation
of the brain at birth, mild-to moderate mental
retardation and absence of other neurological or
somatic disease.
There are 12 genetic loci responsible for MCPH, and
it was suggested that one of the responsible genetic
locus, MCPH4, resides on chromosome 15.
Subsequently, the study on the patients with MCPH4
of consanguineous families in Morocco revealed that
a homozygous missense mutation was observed in
exon 18 of KNL1 gene.
This point mutation caused skipping this exon in
splicing reaction in the mRNA maturation of KNL1,
suggesting that the affected nucleotide is a part of
Exonic splicing enhancers.
The mutation resulted in frame-shift and truncation
of KNL1 protein.
As RWD repeats at near its carboxy terminus were
deleted by this mutation, the truncated protein is no
longer able to bind to Mis12, leading to the defected
recruitment of KNL1 protein to kinetochore. One of
the patients in the Morroco families has
cryptorchidism in addition to microcephaly. The
results of studies on MCH4 is a direct demonstration
that KNL1 is essential to cell division in vivo.
References Blencowe BJ. Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci. 2000 Mar;25(3):106-10
Bollen M. Kinetochore signalling: the KIss that MELTs Knl1. Curr Biol. 2014 Jan 20;24(2):R68-70
Caldas GV, DeLuca JG. KNL1: bringing order to the kinetochore. Chromosoma. 2014 Jun;123(3):169-81
Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006 Dec 1;127(5):983-97
Chinwalla V, Chien A, Odero M, Neilly MB, Zeleznik-Le NJ, Rowley JD. A t(11;15) fuses MLL to two different genes, AF15q14 and a novel gene MPFYVE on chromosome 15. Oncogene. 2003 Mar 6;22(9):1400-10
Espeut J, Cheerambathur DK, Krenning L, Oegema K, Desai A. Microtubule binding by KNL-1 contributes to spindle checkpoint silencing at the kinetochore. J Cell Biol. 2012 Feb 20;196(4):469-82
Faesen AC, Musacchio A. The (phospho) needle in the (MELT) Haystack. Mol Cell. 2015 Mar 5;57(5):765-6
Genin A, Desir J, Lambert N, Biervliet M, Van Der Aa N, Pierquin G, Killian A, Tosi M, Urbina M, Lefort A, Libert F, Pirson I, Abramowicz M. Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum Mol Genet. 2012 Dec 15;21(24):5306-17
Gorohovski A, Tagore S, Palande V, Malka A, Raviv-Shay D, Frenkel-Morgenstern M. ChiTaRS-3.1-the enhanced chimeric transcripts and RNA-seq database matched with protein-protein interactions. Nucleic Acids Res. 2017 Jan 4;45(D1):D790-D795
Hayette S, Tigaud I, Vanier A, Martel S, Corbo L, Charrin C, Beillard E, Deleage G, Magaud JP, Rimokh R. AF15q14, a novel partner gene fused to the MLL gene in an acute myeloid leukaemia with a t(11;15)(q23;q14). Oncogene. 2000 Sep 7;19(38):4446-50
Jamieson CR, Govaerts C, Abramowicz MJ. Primary autosomal recessive microcephaly: homozygosity mapping of MCPH4 to chromosome 15. Am J Hum Genet. 1999 Nov;65(5):1465-9
Kerres A, Vietmeier-Decker C, Ortiz J, Karig I, Beuter C, Hegemann J, Lechner J, Fleig U. The fission yeast kinetochore component Spc7 associates with the EB1 family member Mal3 and is required for kinetochore-spindle association. Mol Biol Cell. 2004 Dec;15(12):5255-67
Kiyomitsu T, Murakami H, Yanagida M. Protein interaction domain mapping of human kinetochore protein Blinkin reveals a consensus motif for binding of spindle assembly checkpoint proteins Bub1 and BubR1. Mol Cell Biol. 2011 Mar;31(5):998-1011
Kops GJ, Saurin AT, Meraldi P. Finding the middle ground: how kinetochores power chromosome congression. Cell Mol Life Sci. 2010 Jul;67(13):2145-61
Krenn V, Wehenkel A, Li X, Santaguida S, Musacchio A. Structural analysis reveals features of the spindle checkpoint kinase Bub1-kinetochore subunit Knl1 interaction. J Cell Biol. 2012 Feb 20;196(4):451-67
Kuefer MU, Chinwalla V, Zeleznik-Le NJ, Behm FG, Naeve CW, Rakestraw KM, Mukatira ST, Raimondi SC, Morris
SW. Characterization of the MLL partner gene AF15q14 involved in t(11;15)(q23;q14). Oncogene. 2003 Mar 6;22(9):1418-24
Liu D, Vleugel M, Backer CB, Hori T, Fukagawa T, Cheeseman IM, Lampson MA. Regulated targeting of protein phosphatase 1 to the outer kinetochore by KNL1 opposes Aurora B kinase. J Cell Biol. 2010 Mar 22;188(6):809-20

KNL1 (cancer susceptibility candidate 5) Takimoto M, Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 204
Meyer C, Schneider B, Jakob S, Strehl S, Attarbaschi A, Schnittger S, Schoch C, Jansen MW, van Dongen JJ, den Boer ML, Pieters R, Ennas MG, Angelucci E, Koehl U, Greil J, Griesinger F, Zur Stadt U, Eckert C, Szczepański T, Niggli FK, Schäfer BW, Kempski H, Brady HJ, Zuna J, Trka J, Nigro LL, Biondi A, Delabesse E, Macintyre E, Stanulla M, Schrappe M, Haas OA, Burmeister T, Dingermann T, Klingebiel T, Marschalek R. The MLL recombinome of acute leukemias. Leukemia. 2006 May;20(5):777-84
Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol. 2004 Nov;6(11):1135-41
Przewloka MR, Zhang W, Costa P, Archambault V, D'Avino PP, Lilley KS, Laue ED, McAinsh AD, Glover DM. Molecular analysis of core kinetochore composition and assembly in Drosophila melanogaster. PLoS One. 2007 May 30;2(5):e478
Sasao T, Itoh N, Takano H, Watanabe S, Wei G, Tsukamoto T, Kuzumaki N, Takimoto M. The protein encoded by cancer/testis gene D40/AF15q14 is localized in spermatocytes, acrosomes of spermatids and ejaculated spermatozoa. Reproduction. 2004 Dec;128(6):709-16
Sasao T, Takimoto M, Itoh N, Maeda T, Tanaka T, Masumori N, Tsukamoto T. Testis cancer gene D40 expression and its relationship with clinicopathological features in infertile men. Int J Urol. 2011 Feb;18(2):175-9
Silió V, McAinsh AD, Millar JB. KNL1-Bubs and RZZ
Provide Two Separable Pathways for Checkpoint Activation at Human Kinetochores. Dev Cell. 2015 Dec 7;35(5):600-13
Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005 Aug;5(8):615-25
Takimoto M, Wei G, Dosaka-Akita H, Mao P, Kondo S, Sakuragi N, Chiba I, Miura T, Itoh N, Sasao T, Koya RC, Tsukamoto T, Fujimoto S, Katoh H, Kuzumaki N. Frequent expression of new cancer/testis gene D40/AF15q14 in lung cancers of smokers. Br J Cancer. 2002 Jun 5;86(11):1757-62
Taylor SS, Hussein D, Wang Y, Elderkin S, Morrow CJ. Kinetochore localisation and phosphorylation of the mitotic checkpoint components Bub1 and BubR1 are differentially regulated by spindle events in human cells. J Cell Sci. 2001 Dec;114(Pt 24):4385-95
Wei G, Takimoto M, Yoshida I, Mao PZ, Koya RC, Miura T, Kuzumaki N. Chromosomal assignment of a novel human gene D40. Nucleic Acids Symp Ser. 1999;(42):71-2
Welburn JP, Vleugel M, Liu D, Yates JR 3rd, Lampson MA, Fukagawa T, Cheeseman IM. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010 May 14;38(3):383-92
Yang JJ, Park TS, Lee ST, Seo JY, Oh SH, Cho EH, Strehl S, Mühlegger N, Dworzak MN, Zuna J, Pospisilova D, Meyer C, Marschalek R, Kim HJ, Kim SH. Molecular characterization and clinical impact of t(11;15)(q23;q14-15) MLL-CASC5 rearrangement. Haematologica. 2014 Jan;99(1):e11-3
This article should be referenced as such:
Takimoto M, Huret JL. KNL1 (cancer susceptibility candidate 5). Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):200-204.

Gene Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 205
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
ATG2B (Autophagy-related 2B) Christine Bellanné-Chantelot, Isabelle Plo
Département de Génétique, Hôpitaux Universitaires Pitié-Salpétrière-Charles Foix, Paris (CBC);
INSERM UMR1170, Institut Gustave Roussy, Villejuif, (CBC, IP), France. christine.bellanne-
[email protected]; [email protected]
Published in Atlas Database: October 2016
Online updated version : http://AtlasGeneticsOncology.org/Genes/ATG2BID55326ch14q32.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68252/10-2016-ATG2BID55326ch14q32.pdf DOI: 10.4267/2042/68252
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract Autophagy is a cellular process involved in the
sequestration of cytosolic components and their
degradation by lysosomes. Autophagy has been
involved in physiological responses to stress or
aging and in the development of many human
diseases including solid and haematological cancers.
In humans, 16 autophagy-related genes are known.
The ATG2B protein is involved in the late steps of
the autophagy process i.e. the formation of
autophagosomes that fuse with lysosomes before
degradation. Loss-of -function (frameshift) acquired
mutations of ATG2B have been identified in gastric
and colorectal carcinomas with high microsatellite
instability. Both pharmacologic and genetic
evidence indicate that autophagy plays pleiotropic
functions in hematopoietic cell homeostasis and
leukemogeneis. Autophagy could exert two opposite
roles (cell death and survival) depending on the
nature of the hematopoietic malignancy.
The germline duplication of ATG2B and GSKIP,
both located in 14q32.2, predisposes to the
development of familial myeloproliferative
neoplasms with autosomal dominant inheritance, in
particular essential thrombocythemia progressing to
leukemia. Overexpression of ATG2B and GSKIP
enhances megakaryocyte progenitor differentiation
by increasing progenitor sensitivity to
thrombopoietin. Both genes cooperate with somatic
JAK2, MPL and CALR mutations and their
overexpression provides a growth advantage to
hematopoietic cells carrying these driver mutations
that may explain the familial aggregation and the
progression of essential thrombocythemia to
myelofibrosis and leukemia.
Keywords
ATG2B; Myeloproliferative neoplasms (MPN);
essential thrombocythemia; myelofibrosis;
leukemia; predisposition; ATG2B/GSKIP;
chromosome 14; CNV; autophagy; Wnt/beta-
catenin pathway
Identity
Other names: C14orf103
HGNC (Hugo): ATG2B
Location : 14q32.2
Location (base pair)
ATG2B starts at 96.745.539 and ends at 96.829.738
bp (on Assembly GRCh37)
Local order
telomere to centromere.
Note
cooperates with GSKIP, also located in 14q32.2 and
included in the 700 kb duplication
NC_000014.9:g.96.163.103_96.857.129dup (on
Assembly GRCh37)
DNA/RNA
Description
The ATG2B gene consists of 42 exons spanning a
region of 82.08 kb.

ATG2B (Autophagy-related 2B) Bellanné-Chantelot C, Plo I
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 206
Transcription
A single mRNA transcript (NM_018036.6) of the
ATG2B gene, with a total length of 6234
nucleotides, has been annotated.
Pseudogene
Not yet identified.
Protein
Description
The protein encoded by the ATG2B gene is the
autophagy-related protein 2 homolog B of 2078
amino acids, with a calculated molecular mass of
232.8 kDa
Expression
Expression of ATG2B has been detected in various
normal human tissues (bone marrow, whole blood,
thymus, brain, heart, muscle, colon, kidney, liver,
lung, pancreas, thyroid, salivary and adrenal glands,
skin, ovary, uterus, placenta, prostate and testis).
In hematopoietic cells, ATG2B is expressed in
CD34+ purified hematopoietic progenitors and
CD36+ erythroblasts or CD41+ megakaryocytes
derived from CD34+ progenitors cultured in vitro
(Saliba et al, 2016)
Localisation
ATG2B is mainly localized in the nucleus.
Function
Autophagy is an intracellular degradation system by
which cytoplasmic materials are enclosed by the
autophagosomes and transferred to lysosomes before
degradation. The autophagy process has been
extensively studied in yeast; 35 autophagy-related
genes (ATG) have been identified, of which 16 are
currently known in humans . This cellular process is
a highly conserved among species. In humans, two
ATG2 proteins, ATG2A and ATG2B, have
redundant functions and are required for
autophagosome formation (Velikkakath AK et al ,
2012).
Homology 44.5% of human ATG2B residues are identical to
those of human ATG2A.
Mutations
Germinal
A germline 14q32.2 head-to-tail duplication of 700
kb has been associated with familial myeloid
malignancies (Saliba et al , 2015). The germline
duplication includes the genes TCL1A, GSKIP,
ATG2B, BDKRB1, BDKRB2 and the first exon of
AK7. The overexpression of ATG2B and GSKIP
that are expressed in myeloid cells, enhances
hematopoietic progenitor differentiation,
particularly of megacaryocytes. The development of
myeloid malignancies required the cooperation of
both genes with the myeloproliferative neoplasms
(MPN) driver JAK2 Val617Phe mutation, MPL or
CALR mutations. The mechanism of cooperation
between ATG2B and GSKIP with MPN driver
mutations remains unknown.
The germline duplication with the same distal and
proximal breakpoints has only been identified in
MPN families originated from West Indies
(Martinique) suggesting a founder effect.
Somatic
A loss-of-function somatic mutation (c.3120delA,
p.Lys1040fs) in gastric carcinomas (15.6%) and in
colorectal carcinomas (11.6%) (Klionsky DJ, 2009).
Implicated in
Familial myeloproliferative neoplasms (MPN)
Disease
Familial MPN, in particular, essential
thrombocythemia progressing to myelofibrosis
and/or acute myeloid leukemia and primary
myelofibrosis, with autosomal dominant inheritance
and originated from West-indies (Martinique) may
be linked to ATG2B/GSKIP germline duplication.
The predisposition is highly penetrant (80%) and is
characterized by an earlier age of MPN onset in
comparison to sporadic cases (41 years versus > 60
years). The spectrum of acquired driver mutations
(JAK2 Val617Phe, MPL and CALR mutations) is
similar to the spectrum of mutations in sporadic
MPN cases.
Prognosis
The percentage of transformation is close to 50% in
these familial MPN cases and is related to the
detection of mutations affecting epigenetic regulator
genes such as TET2 IDH1 or IDH2.
Acute myeloid leukemia (AML)
Disease
AML originated from West-indies (Martinique) may
be linked to ATG2B/GSKIP germline duplication.
Prognosis
The prognosis of the disease is also linked to the
detection of acquired mutations in TET2, IDH1 or in
IDH2.
No TP53 mutation was found, contrary to what was
observed in AML evolving from MPN, suggesting a
different pathway for leukemic transformation.
Gastric carcinoma
Loss-of-functions somatic mutations in ATG genes
(ATG2B, ATG5, ATG9B and ATG12) are identified

ATG2B (Autophagy-related 2B) Bellanné-Chantelot C, Plo I
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 207
in 28% of gastric carcinomas with high
microsatellite instability. These mutations may
contribute to cancer development by deregulating
the autophagy process (Kang et al, 2009).
Colorectal cancer
Loss-of-functions somatic mutations in ATG genes
(ATG2B, ATG5, ATG9B and ATG12) are identified
in 28% of colorectal carcinomas with high
microsatellite instability. These mutations may
contribute to cancer development by deregulating
the autophagy process (Kang et al, 2009).
References Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, Ahn CH, Yoo NJ, Lee SH. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite
instability. J Pathol. 2009 Apr;217(5):702-6
Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007 Nov;8(11):931-7
Saliba J, Saint-Martin C, Di Stefano A, Lenglet G, Marty C, Keren B, Pasquier F, Valle VD, Secardin L, Leroy G, Mahfoudhi E, Grosjean S, Droin N, Diop M, Dessen P, Charrier S, Palazzo A, Merlevede J, Meniane JC, Delaunay-Darivon C, Fuseau P, Isnard F, Casadevall N, Solary E, Debili N, Bernard OA, Raslova H, Najman A, Vainchenker W, Bellanné-Chantelot C, Plo I. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet. 2015 Oct;47(10):1131-40
Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012 Mar;23(5):896-909
This article should be referenced as such:
Bellanné-Chantelot C, Plo I. ATG2B (Autophagy-related 2B). Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):205-207.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 208
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
Primary mediastinal B-cell lymphoma (PMBL) Luis Miguel Juárez Salcedo, Samir Dalia
Principe de Asturias University Hospital, Madrid, Spain (LMJS); Oncology and Hematology, Mercy
Clinic Joplin, Joplin, MO, USA (SD); [email protected].
Published in Atlas Database: September 2016
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/PrimMediastiBLymphomID1477.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68253/09-2016-PrimMediastiBLymphomID1477.pdf DOI: 10.4267/2042/68253
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract Review on primary mediastinal B-cell lymphoma,
with data on clinics, and the genes involved.
Keywords
primary mediastinal B-cell lymphoma; diffuse large
B-cell lymphoma
Identity
Other names
Thymic large B-cell lymphoma
Primary mediastinal (thymic) large B-cell
lymphoma
Mediastinal Large B-cell lymphoma
Clinics and pathology
Disease
PMBL is a subtype of diffuse large B-cell lymphoma
(DLBCL) that arises in the thymus. It accounts for 2-
4% on non-Hodgkin lymphoma and 10% of DLBCL.
It is epidemiology, clinically and biologically
distinct from the other subtypes of DLBCL. Similar
to nodular sclerosis Hodgkin lymphoma (NSHL)
arising in the mediastinum, it is likely derived from
thymic B cells (Dunleavy et al., 2015)
Phenotype/cell stem origin An origin from medullary thymic B cells has been
proposed for this disease.
PMBL has a B-cell phenotype and express CD20 and
pan B-cell markers such as CD79a, CD 45, CD 19
and CD22, but tumor cells do not express
surface immunoglobulin, therefore, monoclonality
cannot be established by Κ and λ staining. B-cell
transcription factors including PAX-5, OCT2 and
BOB1 are typically strongly expressed. CD23
expression is present in almost 66% of cases; CD30
is expressed in 78% whereas CD 15 is usually
negative, although one third of patients are positive.
High expression of BCL2 and PD1 has been
described (Bledsoe et al., 2016).
CD21 and class I and/or II histocompatibility
molecules have been claimed to be absent. Bcl-2
protein seems to be generally expressed, while
fragmentary data are available concerning the
occurrence of some molecules, such as CD10,
MUM1/IRF4, PAX5/BSAP (B-cell Specific
Activating Protein), Bcl-6.
Epidemiology
Typically presents in adolescents and young adults
with a median age of 35 years and a female
predominance with a male.female ratio of 1:2.
(Gaulard et al., 2008).
Clinics
PMBL is an aggressive disease manifested by a
localized, bulky mediastinal mass, often with pleural
and pericardial effusions.
Symptoms at diagnosis are caused by the mediastinal
mass, and complications such as superior vena cava
syndrome are common at presentation.
Regional lymph nodes may be involved, but spread
to distant nodal sites is uncommon. Less frequent,
the disease involves extranodal sites, including the
lung, kidneys, gastrointestinal organs or brain.
Pathology
Morphologically, the thymic B-cells are medium to
large cells having round or lobulated nuclei and
abundant cytoplasm. In most cases,

Primary mediastinal B-cell lymphoma (PMBL) Juárez Salcedo LM, Dalia S
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 209
compartmentalizing sclerosis is observed, and
sometimes tumor cells can resemble Hodgkin/Reed
Sternberg cells.
The nodal architecture is typically diffuse, with
occasional cases showing focal nodularity, and
necrosis is sometimes seen.
Treatment
In making decisions about the initial treatment one
must consider the long-term complications of
mediastinal radiation in this population of patients
who are predominantly young women.
R-CHOP followed by radiation has been effective in
low-risk patients.
In high-risk disease and high rate of primary
refractory disease DA- EPOCH-R without radiation
is the best treatment option, followed or not of
autologous stem cell transplantation.
Prognosis
Although the international prognosis index (IPI) is
useful in DLBCL, its use in PMBL could be limited
by the young age distribution and its typical
mediastinal presentation.
Lactate dehydrogenase level, male sex, performance
status and advanced-stage disease may be useful
predictors of survival.
Genetics
Note
Among the most common genetic alterations in
PMBL are abnormalities on chromosome 9p and 2p.
The 9p region encodes JAK2, which then activates
the STAT6 through phosphorylation. This STAT 6
phosphorylated can transcriptionally repress BCL6.
Also in 9p region, CD274 and PDCD1LG2
(programmed death ligands (PDLs) 1 and 2
respectively) are rearranged at a frequency of 20%.
Gains or amplifications of REL may be seen at 2p.
One third of cases may have gains in chromosome
X.
New two recurrent mutations have been identified;
one of these is the recurrent somatic coding-
sequence mutation in the PTPN1 gene (also found in
Hodgkin lymphoma cases) and the recurrent point
mutation in the XPO1 (exportin 1 gene or CRM1),
which results in the Glu571Lys missense
substitution, in refractory/relapsed PMBL (Jardin et
al., 2016).
The XPO1 mediate the translocation of numerous
RNAs and cellular regulatory proteins, including
tumor suppressor proteins (TP53, BRCA1, NPM1,
APC and FOXO).
References Bishop PC, Wilson WH, Pearson D, Janik J, Jaffe ES, Elwood PC. CNS involvement in primary mediastinal large B-cell lymphoma. J Clin Oncol. 1999 Aug;17(8):2479-85
Bledsoe JR, Redd RA, Hasserjian RP, Soumerai JD, Nishino HT, Boyer DF, Ferry JA, Zukerberg LR, Harris NL, Abramson JS, Sohani AR. The immunophenotypic spectrum of primary mediastinal large B-cell lymphoma reveals prognostic biomarkers associated with outcome. Am J Hematol. 2016 Oct;91(10):E436-41
Dunleavy K, Wilson WH. Primary mediastinal B-cell lymphoma and mediastinal gray zone lymphoma: do they require a unique therapeutic approach? Blood. 2015 Jan 1;125(1):33-9
Gaulard P, Harris NL, Pileri SA, et al. Primarymediastinal (thymic) large B-cell lymphoma.. WHO Classification of Tumours of Hae-matopoietic and Lymphoid Tissues In:Swerdlow SH, Campo E, Harris NL, Jaffe ES,Pileri SA, Stein H, Thiele J, Vardiman JW, editors. Lyon: IARCPress; 2008. pp 250-251.2.
Jardin F, Pujals A, Pelletier L, Bohers E, Camus V, Mareschal S, Dubois S, Sola B, Ochmann M, Lemonnier F, Viailly PJ, Bertrand P, Maingonnat C, Traverse-Glehen A, Gaulard P, Damotte D, Delarue R, Haioun C, Argueta C, Landesman Y, Salles G, Jais JP, Figeac M, Copie-Bergman C, Molina TJ, Picquenot JM, Cornic M, Fest T, Milpied N, Lemasle E, Stamatoullas A, Moeller P, Dyer MJ, Sundstrom C, Bastard C, Tilly H, Leroy K. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma Am J Hematol 2016 Sep;91(9):923-30
This article should be referenced as such:
Juárez Salcedo LM, Dalia S. Primary mediastinal B-cell lymphoma (PMBL). Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):208-209.

Leukaemia Section Review
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 210
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
t(2;11)(p21;q23) without KMT2A (MLL) rearrangement Ana L. Ruano, Shashirekha Shetty
Robert J. Tomisch Pathology, Laboratory Medicine Institute, Cleveland Clinic, Cleveland, Ohio
(ALR, SS), and University Hospitals, Case Western Reserve University, Cleveland, Ohio (SS),
USA; [email protected] and [email protected]
Published in Atlas Database: August 2016
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0211p21q23ID1333.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68254/08-2016-t0211p21q23ID1333.pdf DOI: 10.4267/2042/68254
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract Forty five cases carrying the t(2;11)(p21;q23) have
been reported in the literature, mostly in
myelodysplastic syndromes (MDS) and acute
myeloid leukemia (AML). Some of these cases
involve rearrangements of the the MLL gene (also
known as KMT2A), on 11q23, which confers a more
aggressive behavior in myeloid neoplasms. Several
individual case reports, as well as series such as 19
cases reported by Bousquet et al., 2008 and 7 cases
by Dvorak et al., 2014, describe myeloid neoplasms
carrying the t(2;11)(p21;q23)
without an MLL gene rearrangement, with possible
prognostic implications. The authors of this paper
describe two additional cases from their institution.
Keywords
Myelodysplastic syndrome, acute myeloid leukemia,
t(2;11).
Identity
Other names
Acute myeloid leukemia and myelodysplastic
syndrome with t(2;11)(p21;q23) without MLL gene
rearrangement.
Representative images of karyotype showing rearrangement between chromosomes 2 and 11, and FISH images showing lack of MLL gene rearrangement.

t(2;11)(p21;q23) without KMT2A (MLL) rearrangement Ruano AL, Shetty S
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 211
Clinics and pathology
Disease
Phenotype/cell stem origin
Myeloid blasts.
Thirty two case are available: 2 chronic
myeloproliferative cases, 18 myelodysplastic
syndromes (MDS), and 12 cases of acute myeloid
leukemia (AML). Diagnoses were: chronic
myelogenous leukemia harbouring the
t(9;22)(q34;q11) (CML, 1 case), polycythemia vera
(PV, 1 case), myelodysplastic/myeloproliferative
overlap syndrome (MDS/MPN, 1 case), refractory
anemia (RA, 1 case), refractory anemia with excess
blasts (RAEB, RAEB-I, RAEB-II, 5 cases),
refractory cytopenia with multilineage dysplasia
(RCMD, 2 cases), RCMD with ring sideroblasts
(RCMD-RS, 1 case), unclassifiable MDS (MDS-U,
1 case), low grade MDS NOS (7 cases); AML NOS
(3 cases), AML-M0 (1 case), AML-M1 (1 case),
AML-M2 (3 cases), AML-M4 (2 cases) and AML-
M5 (2 cases). Ten of the twelve AML cases were
classified either as having multilineage dysplasia or
as arising from MDS (chronic myelomonocytic
leukemia , refractory anemia). In two cases,
dysplasia could not be assessed (Harrison et al.,
1998; Gozzetti et al., 2003; Royer-Pekora et al.,
2003; Bousquet et al., 2008; Dvorak et al., 2014;
McCormick et al., 2014; Ruano and Shetty,
unpublished observation)
The authors of this paper describe two additional
cases, above included (Ruano and Shetty,
unpublished observation): one in a patient diagnosed
with myelodysplastic/myeloproliferative
(MDS/MPN) overlap syndrome best classified as
atypical chronic myeloid leukemia (CML),
BCR/ABL negative; another patient presented with
acute monoblastic leukemia, later classified as AML
with myelodysplasia-related changes after obtaining
knowledge on the cytogenetic findings.
Epidemiology
Two cases consisted of a 44 year old female and a 54
year old male (Ruano and Shetty, unpublished
observation).
Altogether, there were 26 male and 6 female
patients. Median age at diagnosis was 56-57 years
(range 37-74).
Clinics
The authors describe a case of a 54 year old male that
initially presented with ankle swelling and left upper
quadrant discomfort noted to have marked
leukocytosis (WBC=237,000/µL) and macrocytic
anemia (Hb=10.1 g/dL ; MCV=101.9 fl) associated
with massive splenomegaly.
This patient was diagnosed with atypical CML,
BCR/ABL negative. The second patient is a 44 year
old female with no significant medical history that
presented with upper and lower extremity pain and
found to be pancytopenic with 2% circulating blasts.
She was eventually diagnosed with acute myeloid
leukemia with myelodysplasia-related changes.
Pathology
Five of the seven cases reported by Dvorak et al.,
2014 showed marked megakaryocytic dysplasia.
The case described by McCormick et al., 2014
showed a hypercellular bone marrow (95%) with
increased megakaryoctyes ranging from small
hypolobated forms to large and normally lobated.
Bone marrow aspirate showing intermediate size blasts with fine nuclear chromatin and moderate basophilic cytoplasm with occasional small azurophiic granules and vacuoles. The blasts were diffusely positive for alpha-naphthyl butyrate esterase and
negative for myeloperoxidase cytochemistries (insert).

t(2;11)(p21;q23) without KMT2A (MLL) rearrangement Ruano AL, Shetty S
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 212
Hypercellular bone marrow (>95%) showing sheets of immature mononuclear cells with fine chromatin and prominent nucleoli, consistent with blasts.
From the cases reported by the authors of this paper,
the bone marrow aspirate and biopsy from the 54
year old patient with atypical CML, BCR/ABL
negative showed granulocytic hyperplasia with left
shift with occasional dyserythropoiesis and
dysgranulopoiesis. There was more prominent
dysmegakaryopoiesis consisting of small,
hypolobated forms with occasional
micromegakaryocytes. Blasts represented 3% of
bone marrow elements. One year later, the patient
presented with acute myeloid leukemia with 40%
circulating blasts expressing CD5, CD7, CD13,
CD33, CD34, CD117, and HLA-DR, with dim
expression of CD56. Two follow-up bone marrows
showed persistent involvement by AML.
The bone marrow aspirate and biopsy from the 44
year old female with AML showed 89% blasts
expressing CD13 (dim), CD33, CD38, CD45 (dim),
CD56, CD64, CD65 (bright), HLA-DR, and CD117
(on a subset). performed on the bone marrow aspirate
shows 69% blasts which are positive for CD4, CD13
(dim), CD33, CD38, CD45 (dim), CD56, CD64,
CD65 (bright), HLA-DR, and CD117 (subset).
Cytochemical stains showed that the blasts were
negative for myeloperoxidase and positive for alpha-
naphthyl butyrate esterase. See Figure 2 and 3.
Treatment The patient reported by Gozzeti et al., 2003 was
treated with cytarabine for 7 days, achieving a partial
remission. His disease progressed afterwards for
what he was treated with various
chemotherapeutic regimens but died 1 year after
diagnosis. The patient reported by Royer-Pokora et
al., 2003 was treated with imatinib achieving
complete response that lasted for 22 months, when
the case report was written.
The seven patients reported by Dvorak et al., 2014
were treated either with symptomatic or
cytoreductive therapy. One patient with deletion 5q
was treated with lenalidomide but had only a partial
response to treatment. Two patients underwent
peripheral stem cell transplantation, after which one
had stable disease but the other transformed to AML.
The median overall survival for this small cohort was
72 months and at the time of publication, 2 patients
were alive and 5 had died.
The patient reported by McCormick et al., 2014
remained clinically stable 70 months after initial
diagnosis, requiring only periodic phlebotomies.
The 54 year old patient described by this page's
authors was initially treated with hydroxyurea after
being diagnosed with atypical CML. He transformed
to AML 14 months later and was treated with 7+3,
ara-C and idarubicin. Due to persistent disease he
was started on a new cycle of chemotherapy which
was complicated by fever and altered mental status
eventually leading to patient's death approximately 1
month later. The 44 year old patient diagnosed with
AML with myelodysplasia related changes was
treated with 7+3 regimen, with cytarabine and
daunorubicin and had 2 negative bone marrows at 14
days and 1 month after treatment was started.
She remained clinically stable but was lost to follow-
up 1 year after diagnosis.

t(2;11)(p21;q23) without KMT2A (MLL) rearrangement Ruano AL, Shetty S
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 213
Prognosis
Dvorak et al., 2014 determined that the median
survival of MDS patients harbouring the
t(2;11)(p21;q23) was significantly greater than that
of MDS patients with complex karyotype or trisomy
8 as sole abnormality (72 vs 7.5 and 57 months,
respectively, p=0.0007). Other studies have placed
the t(2;11)(p21;q23) in the intermediate risk
category. However, the MLL gene status was not
evaluated. Studies in larger groups of patients, also
possibly including patients with AML, are needed in
order to determine if the t(2;11)(p21;q23) without
MLL rearrangement truly conveys a better
prognosis.
Cytogenetics
Cytogenetics morphological
The case described by Harrison et al., 1998 showed
a 46XX, t(2;11)(p21;q23), del(5q)(q13q33)
[24]/46XX[1] karyotype and was negative for MLL
gene rearrangements by Southern Blot. The case
described by Gozzetti et al., 2003 had a 46XX,
t(2;11)(p21;q23)[14] karyotype and was negative for
an MLL rearrangement by FISH. The CML case
reported by Royer-Pekora et al., 2003 initially had
both, a t(9;22)(q34;q11) and a t(2;11)(p21;q23) on
all analysed cells. After the patient achieved major
molecular and cytogenetic response to imatinib, a
new clone containing only the t(2;11) emerged. This
translocation was not present on skin fibroblasts
from which the authors conclude that it was not
present constitutionally.
Among the 19 cases reported by Bousquet et al.,
2008, the t(2;11)(p21;q23) was the sole cytogenetic
abnormality in 5 cases, and was associated with
other abnormalities in the other 14 cases including
deletion 5q (8 cases) and chromosome 7
abnormalities (4 cases). All cases were negative for
MLL gene rearrangements by FISH, and an
alternative breakpoint located downstream from
MLL was identified by PCR-based molecular
techniques, without a definitively identified gene.
They also describe overexpression of microRNA
(MiR) -125b in their patient series.
All seven cases reported by Dvorak et al., 2014
showed the t(2;11)(p21;q23). In 2 cases, this was the
only cytogenetic abnormality, in 4 cases a deletion
of 5q was also present in the main clone, and in 1
patient the deletion 5q was present in a subclone.
They were all negative for MLL gene
rearrangements by FISH.
Cytogenetic analysis from the case described by
McCormick et al., 2014 showed four lines: one
showing a 46XY normal male karyotype, two
showing the t(2;11)(p21;q23-24), one of which also
showed del(5)(q15q31), and a fourth one showing
only the del5q. There was no MLL gene
rearrangement detected by FISH and allele specific
polymerase chain reaction (PCR) was negative for a
JAK2V617F mutation. However, a mutation in
JAK2 exon 12 was identified.
From the cases described by the authors of this page,
the karyotype of the 54 year old male with atypical
CML was reported as 46,XY,t(2;11)(p21;q23). The
karyotype of the 44 year old female with AML was
reported as
47,XX,t(2;11)(p21;q23),+8[16]/47,idem,+i(8)(q10)[
4]. Both cases were negative for MLL gene
rearrangement by FISH (see Figure 1).
Genes involved and proteins
Note
The gene on 11q23 involved in these translocations
has not been yet identified. Bousquet et al., 2008
determined through FISH that the breakpoint in their
cases appeared to be located downstream from the
MLL region. By PCR, they assessed expression of
several genes/sequence tags known to be located in
this region but did not find altered expression of any
of them. They did find, however, that their series of
patients showed overexpression of miR -125b (6- to
90-fold) when compared to other patients with MDS
or AML without the translocation. McCormick et al.,
2014 also found a 200-fold overexpression of miR-
125b in their case.
miR-125b originates from chromosomes 11q24
(MIR125B1 ) and 21q21 (MIR125B2 ) It is a
regulator of normal hematopoiesis and exerts its
oncogenic effect through several mechanisms
including arrest of myeloid/monocytic
differentiation, promoting stem cell renewal and
targeting intermediaries in the TP53 pathway.
Besides AML/MDS with t(2;11)(p21;q23), miR-
125b has also found to play a role in pediatric
acutepromyelocytic leukemia , acute
megakaryoblastic leukemia of trisomy 21, and
precursor B-cell acute lymphoblastic leukemia (B-
ALL) (McCormick et al., 2014).
References Bousquet M, Quelen C, Rosati R, Mansat-De Mas V, La Starza R, Bastard C, Lippert E, Talmant P, Lafage-Pochitaloff M, Leroux D, Gervais C, Viguié F, Lai JL, Terre C, Beverlo B, Sambani C, Hagemeijer A, Marynen P, Delsol G, Dastugue N, Mecucci C, Brousset P. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J Exp Med. 2008 Oct 27;205(11):2499-506
Dvorak P, Lysak D, Vokurka S, Michalova K, Sarova I, Jonasova A, Hruba M, Rykovska A, Subrt I. The translocation t(2;11)(p21;q23) without MLL gene rearrangement--a possible marker of good prognosis in myelodysplastic syndrome patients. Hematol Oncol. 2014 Jun;32(2):82-6

t(2;11)(p21;q23) without KMT2A (MLL) rearrangement Ruano AL, Shetty S
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 214
Fleischman EW, Reshmi S, Frenkel MA, Konovalova WI, Guleva GP, Kulagina OE, Konstantinova LN, Tupitsyn NN, Rowley JD. MLL is involved in a t(2;11)(p21;q23) in a patient with acute myeloblastic leukemia. Genes Chromosomes Cancer. 1999 Feb;24(2):151-5
Gozzetti A, Tozzuoli D, Crupi R, Raspadori D, Fabbri A, Lauria F. A case of acute myelogenous leukemia: myelodysplastic syndrome with t(2;11)(p21;q23) without MLL rearrangement. Cancer Genet Cytogenet. 2003 Jul 15;144(2):177-8
Harrison CJ, Cuneo A, Clark R, Johansson B, Lafage-Pochitaloff M, Mugneret F, Moorman AV, Secker-Walker LM. Ten novel 11q23 chromosomal partner sites. European 11q23 Workshop participants. Leukemia. 1998 May;12(5):811-22
McCormick SR, Higgins RR, Grutkoski PS, Bousquet M, Quelen C, Bartholomaus LM, Brousset P. Myeloid
neoplasm with translocation t(2;11)(p21;q23-24), elevated microRNA 125b-1, and JAK2 exon 12 mutation. Br J Haematol. 2015 Apr;169(2):290-3
Royer-Pokora B, Hildebrandt B, Redmann A, Herold C, Kronenwett R, Haas R, Drechsler M, Wieland C. Simultaneous occurrence of a t(9;22) (Ph) with a t(2;11) in a patient with CML and emergence of a new clone with the t(2;11) alone after imatinib mesylate treatment. Leukemia. 2003 Apr;17(4):807-10
This article should be referenced as such:
Ruano AL, Shetty S. t(2;11)(p21;q23) without KMT2A (MLL) rearrangement. Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):210-214.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 215
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
t(1;12)(p36;p13) ETV6/PRDM16 Francois P. Duhoux, Hélène A. Poirel
Cliniques universitaires Saint-Luc, Université catholique de Louvain / helene.antoine-
Published in Atlas Database: August 2016
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0112p36p13ID1697.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68255/08-2016-t0112p36p13ID1697.pdf DOI: 10.4267/2042/68255
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract Review on t(1;12)(p36;p13) translocations, with data
on clinics, and the genes involved.
Keywords
chromosome 1; chromosome 12; t(1;12)(p36;p13);
PRDM16; ETV6
Clinics and pathology
Disease
Acute myeloid leukaemia (AML).
Phenotype/cell stem origin
AML M4 according to immunophenotyping vs M1
according to cytology.
Epidemiology
Found in one case of AML in a 46 years old female
Duhoux et al., 2012).
Cytology
Leukemic infiltrate of myeloid origin (blasts I and
II); high proportion of myeloid blast cells and
restricted percentage of monocytes.
Prognosis
The patient died 12 months after diagnosis.
Cytogenetics
Cytogenetics morphological
The t(1;12)(p36;p13.2) was the sole anomaly.
Genes involved and proteins
PRDM16 (PR domain containing 16)
Location
1p36.32
DNA/RNA
11 splice variants
Protein
1276 amino acids and smaller proteins. Contains a
N-term PR domain; 7 Zinc fingers, a proline-rich
domain, and 3 Zinc fingers in the C-term. Binds
DNA. Transcription activator; PRDM16 has an
intrinsic histone methyltransferase activity.
PRDM16 forms a transcriptional complex with
CEBPB. PRDM16 plays a downstream regulatory
role in mediating TGFB signaling (Bjork et al.,
2010). PRDM16 induces brown fat determination
and differentiation. PRDM16 is expressed
selectively in the earliest stem and progenitor
hematopoietic cells, and is required for the
maintenance of the hematopoietic stem cell pool
during development.
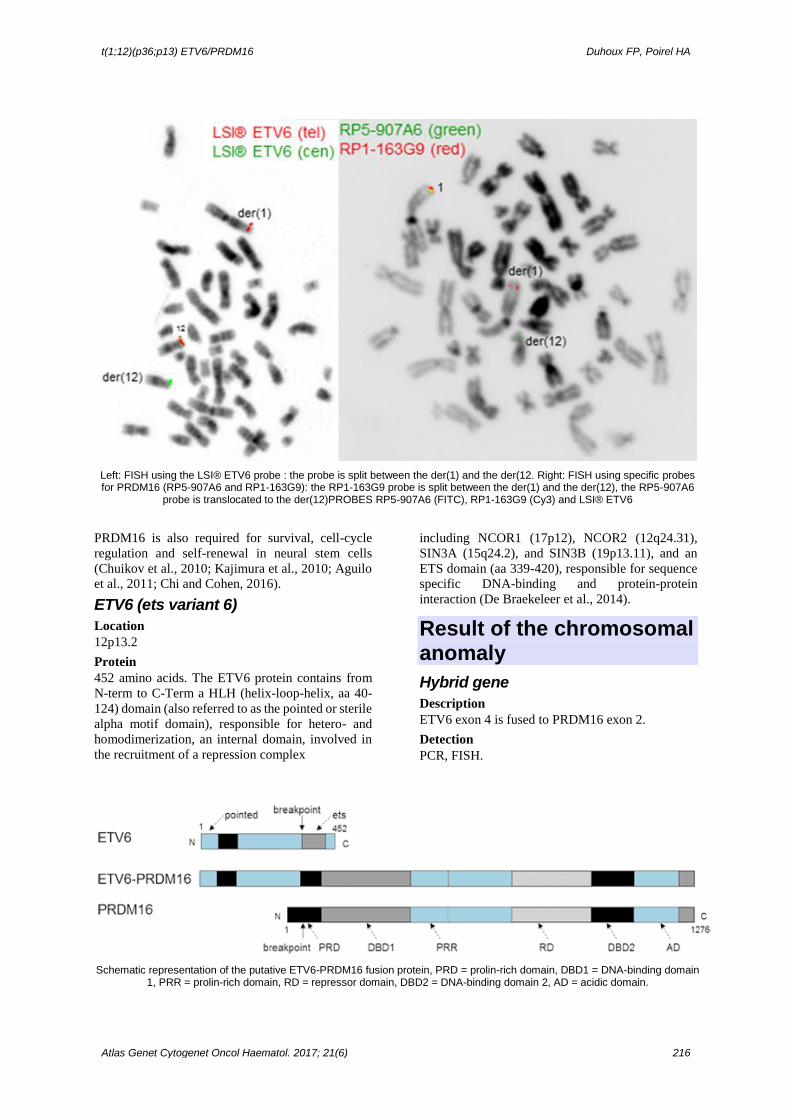
t(1;12)(p36;p13) ETV6/PRDM16 Duhoux FP, Poirel HA
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 216
Left: FISH using the LSI® ETV6 probe : the probe is split between the der(1) and the der(12. Right: FISH using specific probes for PRDM16 (RP5-907A6 and RP1-163G9): the RP1-163G9 probe is split between the der(1) and the der(12), the RP5-907A6
probe is translocated to the der(12)PROBES RP5-907A6 (FITC), RP1-163G9 (Cy3) and LSI® ETV6
PRDM16 is also required for survival, cell-cycle
regulation and self-renewal in neural stem cells
(Chuikov et al., 2010; Kajimura et al., 2010; Aguilo
et al., 2011; Chi and Cohen, 2016).
ETV6 (ets variant 6)
Location
12p13.2
Protein
452 amino acids. The ETV6 protein contains from
N-term to C-Term a HLH (helix-loop-helix, aa 40-
124) domain (also referred to as the pointed or sterile
alpha motif domain), responsible for hetero- and
homodimerization, an internal domain, involved in
the recruitment of a repression complex
including NCOR1 (17p12), NCOR2 (12q24.31),
SIN3A (15q24.2), and SIN3B (19p13.11), and an
ETS domain (aa 339-420), responsible for sequence
specific DNA-binding and protein-protein
interaction (De Braekeleer et al., 2014).
Result of the chromosomal anomaly
Hybrid gene
Description
ETV6 exon 4 is fused to PRDM16 exon 2.
Detection
PCR, FISH.
Schematic representation of the putative ETV6-PRDM16 fusion protein, PRD = prolin-rich domain, DBD1 = DNA-binding domain 1, PRR = prolin-rich domain, RD = repressor domain, DBD2 = DNA-binding domain 2, AD = acidic domain.

t(1;12)(p36;p13) ETV6/PRDM16 Duhoux FP, Poirel HA
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 217
References Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, Snoeck HW. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011 May 12;117(19):5057-66
Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. 2010 Mar 1;19(5):774-89
Chi J, Cohen P. The Multifaceted Roles of PRDM16: Adipose Biology and Beyond. Trends Endocrinol Metab. 2016 Jan;27(1):11-23
Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010 Oct;12(10):999-1006
De Braekeleer E, Douet-Guilbert N, De Braekeleer M.. ETV6 (ets variant 6). Atlas Genet Cytogenet Oncol Haematol. 2014; 18(12):886-899.
Duhoux FP, Ameye G, Montano-Almendras CP, Bahloula
K, Mozziconacci MJ, Laibe S, Wlodarska I, Michaux L, Talmant P, Richebourg S, Lippert E, Speleman F, Herens C, Struski S, Raynaud S, Auger N, Nadal N, Rack K, Mugneret F, Tigaud I, Lafage M, Taviaux S, Roche-Lestienne C, Latinne D, Libouton JM, Demoulin JB, Poirel HA; Groupe Francophone de Cytogénétique Hématologique (GFCH); Belgian Cytogenetic Group for Haematology and Oncology (BCG-HO).. PRDM16 (1p36) translocations define a distinct entity of myeloid
malignancies with poor prognosis but may also occur in lymphoid malignancies. Br J Haematol. 2012 Jan;156(1):76-88. doi: 10.1111/j.1365-2141.2011.08918.x. Epub 2011 Nov 3.
Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM.. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009 Aug 27;460(7259):1154-8. Epub 2009 Jul 29.
This article should be referenced as such:
Duhoux FP, Poirel HA. t(1;12)(p36;p13) ETV6/PRDM16. Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):215-217.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 218
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
t(1;1)(p36;p36) PRDM16/SKI Jean-Loup Huret
Medical Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021
Poitiers, France. [email protected]
Published in Atlas Database: September 2016
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0101p36p36ID1642.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68256/09-2016-t0101p36p36ID1642.pdf DOI: 10.4267/2042/68256
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract Review on t(1;1)(p36;p36) PRDM16/SKI
translocations, with data on clinics, and the genes
involved.
Keywords
chromosome 1; t(1;1)(p36;p36), SKI; PRDM16
Clinics and pathology
Disease
Acute myeloid leukemia (AML)
Phenotype/cell stem origin
Only one case to date (Masetti et al 2014).
Clinics
An 8-year-old child was diagnosed with acute
monoblastic leukemia with differentiation (AML-
M5b).
Prognosis
5 years after the allograft, the patient was alive and
disease-free.
Cytogenetics
Additional anomalies
The patient also had a t(7;21)(p22;q22) RUNX1 /
USP42, and a del(5)(q14q34)
Genes involved and proteins
PRDM16 (PR domain containing 16)
Location
1p36.32
DNA/RNA
11 splice variants
Protein
1276 amino acids and smaller proteins. Contains a
N-term PR domain; 7 Zinc fingers, a proline-rich
domain, and 3 Zinc fingers in the C-term.
Binds DNA.
Transcription activator; PRDM16 has an intrinsic
histone methyltransferase activity. PRDM16 forms a
transcriptional complex with CEBPB.
PRDM16 plays a downstream regulatory role in
mediating TGFB signaling (Bjork et al., 2010).
PRDM16 induces brown fat determination and
differentiation.
PRDM16 is expressed selectively in the earliest stem
and progenitor hematopoietic cells, and is required
for the maintenance of the hematopoietic stem cell
pool during development. PRDM16 is also required
for survival, cell-cycle regulation and self-renewal in
neural stem cells (Chuikov et al., 2010; Kajimura et
al., 2010; Aguilo et al., 2011; Chi and Cohen, 2016).
SKI (SKI proto-oncogene)
Location

t(1;1)(p36;p36) PRDM16/SKI Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 219
1p36.33
Protein
Negative regulator of the TGFB)/Smad signaling.
SKI and SKIL (SnoN) are transcriptional co-
repressors which interact with Smads, and repress
transcription induced by TGFB.
TGFB signaling suppresses tumor cell proliferation
at early stages of tumorigenesis but promotes
epithelial-to-mesenchymal transition (EMT), tumor
invasion, and metastasis at late malignant stages.
SKI interacts with the Hippo pathway.
SKI inhibites the transcription activity of TAZ as
well as its ability to promote transformation and
EMT (Rashidian et al., 2015).
High expression of SKI results in activation of
hematopoietic stem cells transcriptional programs
and an HSC autonomous repopulating advantage
that favors myeloid development through hepatocyte
growth factor (HGF) signaling and independently of
its ability to inhibit TGFB signaling. SKI is highly
expressed in chronic myeloid leukemia (CML) and
acute myeloid leukemia (AML) (Singbrant et al.,
2014). SKI has a critical role in the development of
neuronal and muscle cells or tissues. SKI mutations
have been identified in Shprintzen-Goldberg
syndrome (SGS) (Schepers et al., 2015).
Result of the chromosomal anomaly
Hybrid gene
Description
5' PRDM16 ? 3' SKI; breakpoint: AAG CTA GCC
AAA A - GT CTC CTC TGA
Fusion protein
Description
Fusion of PRDM16 exon 1 (MRSKARARKLAK) to
SKI exon 2 (VSSEPPASIRPK.)
References
Masetti R, Togni M, Astolfi A, Pigazzi M, Indio V, Rivalta B, Manara E, Rutella S, Basso G, Pession A, Locatelli F. Whole transcriptome sequencing of a paediatric case of de novo acute myeloid leukaemia with del(5q) reveals RUNX1-USP42 and PRDM16-SKI fusion transcripts. Br J Haematol. 2014 Aug;166(3):449-52
Rashidian J, Le Scolan E, Ji X, Zhu Q, Mulvihill MM, Nomura D, Luo K. Ski regulates Hippo and TAZ signaling to suppress breast cancer progression. Sci Signal. 2015 Feb 10;8(363):ra14
Schepers D, Doyle AJ, Oswald G, Sparks E, Myers L, Willems PJ, Mansour S, Simpson MA, Frysira H, Maat-Kievit A, Van Minkelen R, Hoogeboom JM, Mortier GR, Titheradge H, Brueton L, Starr L, Stark Z, Ockeloen C, Lourenco CM, Blair E, Hobson E, Hurst J, Maystadt I, Destrée A, Girisha KM, Miller M, Dietz HC, Loeys B, Van Laer L. The SMAD-binding domain of SKI: a hotspot for de novo mutations causing Shprintzen-Goldberg syndrome. Eur J Hum Genet. 2015 Feb;23(2):224-8
Singbrant S, Wall M, Moody J, Karlsson G, Chalk AM, Liddicoat B, Russell MR, Walkley CR, Karlsson S. The SKI proto-oncogene enhances the in vivo repopulation of hematopoietic stem cells and causes myeloproliferative disease. Haematologica. 2014 Apr;99(4):647-55
Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, Snoeck HW. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011 May 12;117(19):5057-66
Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. 2010 Mar 1;19(5):774-89
Chi J, Cohen P. The Multifaceted Roles of PRDM16: Adipose Biology and Beyond. Trends Endocrinol Metab. 2016 Jan;27(1):11-23
Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010 Oct;12(10):999-1006
Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009 Aug 27;460(7259):1154-8
This article should be referenced as such:
Huret JL. t(1;1)(p36;p36) PRDM16/SKI. Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):218-219.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 220
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
t(1;3)(p36;q21) PSMD2/PRDM16 ??? Jean-Loup Huret
Medical Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021
Poitiers, France. [email protected]
Published in Atlas Database: September 2016
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0103ID1002.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68257/09-2016-t0103ID1002.pdf DOI: 10.4267/2042/68257
This article is an update of : Hess JL. t(1;3)(p36;q21). Atlas Genet Cytogenet Oncol Haematol 2002;6(3) Cornillet-Lefebvre P, Daliphard S, Struski S. t(1;3)(p36;q21). Atlas Genet Cytogenet Oncol Haematol 2001;5(1) Huret JL. t(1;3)(p36;q21). Atlas Genet Cytogenet Oncol Haematol 1997;1(1)
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Identity
This entity probably does not exist: 1- PSMD2 sits in 3q27, while the breakpoint is in
3q21;
2- PSMD2, a protein of the proteasome is well
known by its alias, RPN1, while the true RPN1, a
protein involved in N-glycosylation, sitting in 3q21,
is better known by its full name: Ribophorin I.
The translocation is therefore likely to be
t(1;3)(p36;q21) RPN1/PRDM16
Clinics and pathology Disease
Myeloid lineage (MDS, AML, therapy related AML,
CML, MPD); features similar to those of the
3q21q26 syndrome including normal or elevated
platelet count at diagnosis, megakaryocytic
hyperplasia and dysplasia. Very rarely in lymphoid
lineage
t(1;3)(p36;q21) G-banding (left) - Courtesy Diane H. Norback, Eric B. Johnson, and Sara Morrison-Delap, UW Cytogenetic Services; R-banding (right) Courtesy Pascale Cornillet-Lefebvre and Stéphanie Struski (above) and Christiane Charrin (below)

t(1;3)(p36;q21) PSMD2/PRDM16 ??? Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 221
Phenotype/cell stem origin
of 39 cases, there were: 22 myelodysplastic
syndromes (MDS) (17/22 transformed into
refractory acute myeloid leukemia (AML) of -M1 or
-M4 type), 8 de novo AML, 3 therapy-related MDS,
2 polycythemia vera, 1 essential thrombocythemia, 1
chronic myelogenous leukemia (CML), 1 multiple
myeloma, 1 waldenstrom's macroglobulinemia
Epidemiology
patients are aged: 30-80 yrs
Clinics
Roughly 50% of patients present with MDS, another
10% with therapy associated MDS, 25% with de
novo AML, and the remainder with a range of other
myeloproliferative disorders. The majority of MDS
patients transform into AML with a short
preleukemic phase.
Blood data: frequent thrombocytosis or normal
platelet count
Cytology
frequently characterized by
dysmegakaryocytopoiesis
Pathology
The pathology is typical of MDS, often with a
prominent monocytic component. Trilineage
dysplasia. Acute leukemias that evolve usually show
the morphology of M4 AML.
Treatment
Patients are treated with conventional chemotherapy
for AML.
Prognosis
Very poor so far: from 16 cases, median survival was
6 mths in AML, 20 mths in MDS
Cytogenetics
Note
Other rearrangements showing similar clinical
features include inv(3)(q21q26), t(3;3)(q21;q26),
t(3;5)(q21;q31), t(3;8)(q21;q24), and
t(3;21)(q26;q22). The breakpoints in 3q21 cluster in
a 50 kb region centromeric to the breakpoint in
inv(3)(q21;q26) and the ribophorin gene (RPN1).
The breakpoints at 1p36 are clustered in a 90 kb
region at 1p36.3.
Additional anomalies
del (5q) in 5 of 20 cases (1/4)
Genes involved and proteins
Mechanisms of Oncogenesis : The available data
suggest that transcription of MEL1 (MDS1/EVI1 -
like gene) is activated as a result of translocation
bringing the gene just 3' to RPN1 gene at 3q21.
MEL1 is a 1257 amino acid protein that is
homologous (63% similar in amino acid sequence)
to EVI. The mechanism of activation of MEL1 is
similar to EVI1 that is activated by juxtaposition 3'
to RPN1 in the t(3;3)(q21;q26) and 5' to RPN1 in the
inv(3)(q2126). It appears that MEL1 is normally
expressed in uterus and kidney and not in normal
hematopoietic cells or in leukemias that lack the
t(1;3)(p36;q31 The MEL1 protein contains 2 DNA
binding domains (7 C2H2 zinc finger repeats at the
amino terminus and 3 zinc finger repeats at the
carboxyl terminus). The amino terminal domain of
MEL1 contains a PRD domain, a motif also found in
the same location in the MDS1/EV1 protein but not
in MDS1). This is of interest because this domain is
also found in RIZ, PRDI-BF1, and egl-43 and is
homologous to the SET (Suvar3-9, Enhancer of
zeste, Trithorax) domain that present in MLL.
Inclusion of this domain in EVI1 appears to convert
EVI1 from a transcriptional repressor to an activator.
Therefore MEL1 may be a transcriptional activator.
The target genes of MEL1 have not been identified.
References Bitter MA, Neilly ME, Le Beau MM, Pearson MG, Rowley JD. Rearrangements of chromosome 3 involving bands 3q21 and 3q26 are associated with normal or elevated platelet counts in acute nonlymphocytic leukemia. Blood. 1985 Dec;66(6):1362-70
Bloomfield CD, Garson OM, Volin L, Knuutila S, de la Chapelle A. t(1;3)(p36;q21) in acute nonlymphocytic leukemia: a new cytogenetic-clinicopathologic association. Blood. 1985 Dec;66(6):1409-13
Grigg AP, Gascoyne RD, Phillips GL, Horsman DE. Clinical, haematological and cytogenetic features in 24 patients with structural rearrangements of the Q arm of chromosome 3. Br J Haematol. 1993 Jan;83(1):158-65
Huang S, Shao G, Liu L. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J Biol Chem. 1998 Jun 26;273(26):15933-9
Marsden KA, Pearse AM, Collins GG, Ford DS, Heard S, Kimber RI. Acute leukemia with t(1;3)(p36;q21), evolution to t(1;3)(p36;q21), t(14;17)(q32;q21), and loss of red cell A and Le(b) antigens. Cancer Genet Cytogenet. 1992 Nov;64(1):80-5
Mochizuki N, Shimizu S, Nagasawa T, Tanaka H, Taniwaki M, Yokota J, Morishita K. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood. 2000 Nov 1;96(9):3209-14

t(1;3)(p36;q21) PSMD2/PRDM16 ??? Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 222
Moir DJ, Jones PA, Pearson J, Duncan JR, Cook P, Buckle VJ. A new translocation, t(1;3) (p36;q21), in myelodysplastic disorders. Blood. 1984 Aug;64(2):553-5
Secker-Walker LM, Mehta A, Bain B. Abnormalities of 3q21 and 3q26 in myeloid malignancy: a United Kingdom Cancer Cytogenetic Group study. Br J Haematol. 1995 Oct;91(2):490-501
Shimizu S, Suzukawa K, Kodera T, Nagasawa T, Abe T,
Taniwaki M, Yagasaki F, Tanaka H, Fujisawa S, Johansson B, Ahlgren T, Yokota J, Morishita K. Identification of breakpoint cluster regions at 1p36.3 and 3q21 in
hematologic malignancies with t(1;3)(p36;q21). Genes Chromosomes Cancer. 2000 Mar;27(3):229-38
Welborn JL, Lewis JP, Jenks H, Walling P. Diagnostic and prognostic significance of t(1;3)(p36;q21) in the disorders of hematopoiesis. Cancer Genet Cytogenet. 1987 Oct;28(2):277-85
This article should be referenced as such:
Huret JL. t(1;3)(p36;q21) PSMD2/PRDM16 ???. Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):220-222.

Leukaemia Section Short Communication
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 223
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
t(1;7)(p36;p12) IKZF1/PRDM16 Jean-Loup Huret
Medical Genetics, Dept Medical Information, University of Poitiers, CHU Poitiers Hospital, F-86021
Poitiers, France. [email protected]
Published in Atlas Database: August 2016
Online updated version : http://AtlasGeneticsOncology.org/Anomalies/t0107p36p12ID1656.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68258/08-2016-t0107p36p12ID1656.pdf DOI: 10.4267/2042/68258
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Abstract Review on t(1;7)(p36;p12) translocations, with data
on clinics, and the genes involved.
Keywords
chromosome 1; chromosome7; t(1;7)(p36;p12);
PRDM16; IKZF1
Clinics and pathology
Disease
A t(1;7)(p36;p12) was found in a case of
myelodysplastic syndrome (MDS) (Duhoux et al.,
2012).
Clinics
A 66-year-old male patient who died 49 months after
diagnosis.
Cytogenetics
Cytogenetics morphological
A +8 was present.
Genes involved and proteins
PRDM16 (PR domain containing 16)
Location
1p36.32
DNA/RNA
11 splice variants
Protein
1276 amino acids and smaller proteins. Contains a
N-term PR domain; 7 Zinc fingers, a proline-rich
domain, and 3 Zinc fingers in the C-term. Binds
DNA. Transcription activator; PRDM16 has an
intrinsic histone methyltransferase activity.
PRDM16 forms a transcriptional complex with
CEBPB. PRDM16 plays a downstream regulatory
role in mediating TGFB signaling (Bjork et al.,
2010). PRDM16 induces brown fat determination
and differentiation. PRDM16 is expressed
selectively in the earliest stem and progenitor
hematopoietic cells, and is required for the
maintenance of the hematopoietic stem cell pool
during development. PRDM16 is also required for
survival, cell-cycle regulation and self-renewal in
neural stem cells (Chuikov et al., 2010; Kajimura et
al., 2010; Aguilo et al., 2011; Chi and Cohen, 2016).
IKZF1 (Ikaros family zinc finger 1)
Location
7p12.2
DNA/RNA
Numerous splice variants
Protein
519 amino acids. Contains 6 Zn fingers (act as DNA-
binding domain, and dimerization domain).
Transcription regulator. IKZF1 is involved in
chromatin remodeling complexes, such as
nucleosome-remodeling and histone deacetylation
(NuRD), and can both enhance and repress gene
transcription. IKZF1 plays a key role in
hematopoietic stem cell maintenance, B- and T-
lymphopoiesis, erythropoiesis and the fetal-to-adult
hemoglobin switch.
ETV6 and IKZF1 are components of a network of
heptad transcription factors (ERG, FLI1, GATA2,
LMO2, LYL1, RUNX1, and TAL1 (SCL). This
heptad acts in combination to regulate genes in

t(1;7)(p36;p12) IKZF1/PRDM16 Huret JL
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 224
hematopoietic stem and progenitor cells) that
regulate the expression of a number of hematopoietic
genes and whose high expression in acute myeloid
leukemia is associated with poor prognosis
(Unnikrishnan et al., 2016). IKZF1 deletions are
associated with unfavorable prognosis in childhood
B-cell precursor acute lymphoblastic leukemia
(ALL) (Boer et al., 2016), and is associated with a
higher relapse risk and worse survival in adults with
common B-cell ALL (Yao et al., 2016). IKZF1
mutations were found in cases of common variable
immunodeficiency syndrome with progressive B
lymphopenia and an increased risk of acute
lymphoblastic leukemia (Kuehn et al., 2016).
Result of the chromosomal anomaly
Hybrid gene
Description
5' IKZF1 - 3' PRDM16
Transcript
IKZF1 exon 3 joined to PRDM16 exon 3.
References Boer JM, van der Veer A, Rizopoulos D, Fiocco M, Sonneveld E, de Groot-Kruseman HA, Kuiper RP, Hoogerbrugge P, Horstmann M, Zaliova M, Palmi C, Trka J, Fronkova E, Emerenciano M, do Socorro Pombo-de-Oliveira M, Mlynarski W, Szczepanski T, Nebral K, Attarbaschi A, Venn N, Sutton R, Schwab CJ, Enshaei A, Vora A, Stanulla M, Schrappe M, Cazzaniga G, Conter V, Zimmermann M, Moorman AV, Pieters R, den Boer ML. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. 2016 Jan;30(1):32-8
Unnikrishnan A, Guan YF, Huang Y, Beck D, Thoms JA, Peirs S, Knezevic K, Ma S, de Walle IV, de Jong I, Ali Z, Zhong L, Raftery MJ, Taghon T, Larsson J, MacKenzie KL, Van Vlierberghe P, Wong JW, Pimanda JE. A quantitative proteomics approach identifies ETV6 and IKZF1 as new regulators of an ERG-driven transcriptional network.
Nucleic Acids Res. 2016 Dec 15;44(22):10644-10661
Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, Snoeck HW. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011 May 12;117(19):5057-66
Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. 2010 Mar 1;19(5):774-89
Chi J, Cohen P. The Multifaceted Roles of PRDM16: Adipose Biology and Beyond. Trends Endocrinol Metab. 2016 Jan;27(1):11-23
Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010 Oct;12(10):999-1006
Duhoux FP, Ameye G, Montano-Almendras CP, Bahloula K, Mozziconacci MJ, Laibe S, Wlodarska I, Michaux L, Talmant P, Richebourg S, Lippert E, Speleman F, Herens C, Struski S, Raynaud S, Auger N, Nadal N, Rack K, Mugneret F, Tigaud I, Lafage M, Taviaux S, Roche-Lestienne C, Latinne D, Libouton JM, Demoulin JB, Poirel HA. PRDM16 (1p36) translocations define a distinct entity of myeloid malignancies with poor prognosis but may also occur in lymphoid malignancies. Br J Haematol. 2012 Jan;156(1):76-88
Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, Spiegelman BM. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature. 2009 Aug 27;460(7259):1154-8
Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, Maffucci P, Pierce KR, Abbott JK, Voelkerding KV, South ST, Augustine NH, Bush JS, Dolen WK, Wray BB, Itan Y, Cobat A, Sorte HS, Ganesan S, Prader S, Martins TB, Lawrence MG, Orange JS, Calvo KR, Niemela JE, Casanova JL, Fleisher TA, Hill HR, Kumánovics A, Conley ME, Rosenzweig SD. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med. 2016 Mar 17;374(11):1032-1043
Yao QM, Liu KY, Gale RP, Jiang B, Liu YR, Jiang Q, Jiang H, Zhang XH, Zhang MJ, Chen SS, Huang XJ, Xu LP, Ruan GR. Prognostic impact of IKZF1 deletion in adults with common B-cell acute lymphoblastic leukemia. BMC Cancer. 2016 Apr 11;16:269
This article should be referenced as such:
Huret JL. t(1;7)(p36;p12) IKZF1/PRDM16. Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):223-224.

Case Report Section
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 225
Atlas of Genetics and Cytogenetics in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
A pediatric case of acute lymphoblastic leukemia with t(2;9)(q12;q34) (RANBP2/ABL1 fusion) Marc De Braekeleer, Nadia Guéganic, Alexandra Schifferli, Joëlle Tchinda
Cytogenetics Laboratory, Faculty of Medicine, University of Brest, France
[email protected] (MdeB, NG)) Department of Hematology/Oncology, University
Children's Hospital Basel, Switzerland (AS) University Children's Hospital Zurich, Switzerland
(JT).
Published in Atlas Database: December 2015
Online updated version : http://AtlasGeneticsOncology.org/Reports/t0209q12q34BraekeleerID100084.html
Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68259/12-2015-t0209q12q34BraekeleerID100084.pdf DOI: 10.4267/2042/68259
This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2017 Atlas of Genetics and Cytogenetics in Oncology and Haematology
Clinics
Age and sex 21 months old female patient.
Previous history
no preleukemia
no previous malignancy
no inborn condition of note
Organomegaly
No hepatomegaly , no splenomegaly , no enlarged
lymph nodes , no central nervous system
involvement (t)
Blood
WBC : 77.5 (N: 6-17.5)X 109/l
HB : 29 (N: 105-135)g/dl
Platelets : 69 (N: 150-450)X 109/l
Blasts : 76%
Bone marrow : Hypercellular marrow, with 93.7%
blasts (small to middle-sized cells with large nucleus
and minimal cytoplasm).%
Cyto-Pathology Classification
Phenotype
Pre-B acute lymphoblastic leukemia.
Immunophenotype
cTDT, cCD79a, cIgM, CD19 and CD20 positive.
Rearranged Ig Tcr not performed.
Diagnosis Pre-B acute lymphoblastic leukemia.
Survival
Date of diagnosis 01-2014
Treatment
Protocol AIEOP-BFM ALL 2009 high risk.
Complete remission : Treatment related death : no
Relapse : no
Status A
Last follow up 12-2015
Survival 23 +months
Karyotype Sample bone marrow.
Banding G banding.
Results
46,XX,t(2;9)(q12-14;q34),add(5)(p14)[5]/46,sl,-
7,+mar[2]/46,XX[3]
Other molecular cytogenetics technics
fluorescence in situ hybridization(FISH) analysis
using ETV6-RUNX1, 5'MLL-3'MLL, CEP4,
CEP10, CEP17, 5'IGH-3'IGH, 3'TCF3-5'TCF3,
BCR-ABL1.

A pediatric case of acute lymphoblastic leukemia with t(2;9)(q12;q34) (RANBP2/ABL1 fusion)
De Braekeleer M, et al.
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 226
FISH experiments with BAC clones located in bands
2q12.1 to 2q14.2.
Other molecular cytogenetics results
All negative, except for BCR-ABL1 with 3 ABL1
signals and 2 BCR signals.
A split signal on der(2) and der(9) was found with
RP11-622D1, RP11-347H10, RP11-259O12, RP11-
348G16 and RP11-953L12. These BAC clones
overlap the RANBP2 gene and allow refinement of
the breakpoint to a 25kb region covering the 5' end
and the first three exons of RANBP2.
Other Molecular Studies
Technics: MLPA.
Results: Negative.
GTG banding showing chromosomes 2 and 9 and the derivatives der(2) and der(9).
FISH with BACs RP11-953L12 (spectrum green, located in 2q12 and containing RANBP2) and RP11-83J21 (spectrum orange, located in 9q34 and containing the 3' part of ABL1) and CEP9 (in aqua) showing one fusion signal on der(2).
No fusion is detected on der(9) because RP11-83J21 does not cover the 5' part of ABL1.
Comments We present here a unique case of pediatric acute
lymphoblastic leukemia. This fusion gene was
identified in another case by RNA-sequencing
(Roberts et al., 2012).

A pediatric case of acute lymphoblastic leukemia with t(2;9)(q12;q34) (RANBP2/ABL1 fusion)
De Braekeleer M, et al.
Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6) 227
References Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, Becksfort J, Finney RP, Teachey DT, Maude SL, Tse K, Moore R, Jones S, Mungall K, Birol I, Edmonson MN, Hu Y, Buetow KE, Chen IM, Carroll WL, Wei L, Ma J, Kleppe M, Levine RL, Garcia-Manero G, Larsen E, Shah NP, Devidas M, Reaman G, Smith M, Paugh SW, Evans WE, Grupp SA, Jeha S, Pui
CH, Gerhard DS, Downing JR, Willman CL, Loh M, Hunger SP, Marra MA, Mullighan CG. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012 Aug 14;22(2):153-66
This article should be referenced as such:
De Braekeleer M, Guéganic N, Schifferli A, Tchinda J. A pediatric case of acute lymphoblastic leukemia with t(2;9)(q12;q34) (RANBP2/ABL1 fusion). Atlas Genet Cytogenet Oncol Haematol. 2017; 21(6):225-227.

Atlas of Genetics and Cytogenetics
in Oncology and Haematology
INIST-CNRS OPEN ACCESS JOURNAL
Instructions to Authors
Policies- Instructions to Authors
See http://documents.irevues.inist.fr/bitstream/handle/2042/48486/Instructions-to-authors.pdf
Manuscripts submitted to the Atlas must be submitted solely to the Atlas.
The Atlas publishes "cards", "deep insights", "case reports", and "educational items".
Cards are structured review articles. Detailed instructions for these structured reviews can be found at:
http://AtlasGeneticsOncology.org/Forms/Gene_Form.html for reviews on genes,
http://AtlasGeneticsOncology.org/Forms/Leukaemia_Form.html for reviews on leukaemias,
http://AtlasGeneticsOncology.org/Forms/SolidTumour_Form.html for reviews on solid tumours,
http://AtlasGeneticsOncology.org/Forms/CancerProne_Form.html for reviews on cancer-prone diseases.
According to the length of the paper, cards are divided, into "reviews" (texts exceeding 2000 words), "mini reviews" (between),
and "short communications" (texts below 400 words).
Deep Insights are written as traditional papers, made of paragraphs with headings, at the author's convenience.
Case Reports in haematological malignancies are dedicated to recurrent –but rare- chromosomes abnormalities in
leukaemias/lymphomas; see http://atlasgeneticsoncology.org//BackpageAuthors.html#CASE_REPORTS .
It is mandatory to use the specific "Submissionformfor Case reports":
see http://AtlasGeneticsOncology.org/Reports/Case_Report_Submission.html.
Educational Items must be didactic, give full information and be accompanied with iconography.
Research articlesThe Atlas of Genetics and Cytogenetics in Oncology and Haematology does not publish research articles.
Authorship All authors should qualify for authorship according to the ICMJE criteria.
Editorial Ethics see http://documents.irevues.inist.fr/bitstream/handle/2042/56068/Policies-editorial-ethics.pdf for: Peer Review Process /
Commissioned papers vs Unsolicited papers /Responsibility for the reviewers / Editorial responsibilities / Conflict of interest-
Competing interests/ Privacy and Confidentiality - Iconography / Protection of Human Subjects and Animals in Research /
Duplicate Publication/ Plagiarism / Retracting a publication
Subscription The Atlas is FREE!
Costs/Page Charge There is NO page charge.
PubMed Central Once the paper online, authors are encouraged to send their manuscript to PubMed Central
http://www.ncbi.nlm.nih.gov/pmc/ with reference to the original paper in the Atlas in
http://documents.irevues.inist.fr/handle/2042/15655
Corporate patronage, sponsorship and advertising
Enquiries should be addressed to [email protected].
Rules, Copyright Notice and Disclaimer
http://documents.irevues.inist.fr/bitstream/handle/2042/48487/Copyright-sponsorship.pdf
Property As "cards" are to evolve with further improvements and updates from various contributors, the property of the cards
belongs to the editor, and modifications will be made without authorization from the previous contributor (who may,
nonetheless, be asked for refereeing); contributors are listed in an edit history manner. Authors keep the rights to use further
the content of their papers published in the Atlas, provided that the source is cited.
Copyright The information in the Atlas of Genetics and Cytogenetics in Oncology and Haematology is issued for general
distribution. All rights are reserved. The information presented is protected under international conventions and under national
laws on copyright and neighbouring rights. Commercial use is totally forbidden. Information extracted from the Atlas may be
reviewed, reproduced or translated for research or private study but not for sale or for use in conjunction with commercial
purposes. Any use of information from the Atlas should be accompanied by an acknowledgment of the Atlas as the source,
citing the uniform resource locator (URL) of the article and/or the article reference, according to the Vancouver convention.
Reference to any specific commercial products, process, or service by trade name, trademark, manufacturer, or otherwise, does
not necessarily constitute or imply its endorsement, recommendation, or favouring. The views and opinions of contributors
and authors expressed herein do not necessarily state or reflect those of the Atlas editorial staff or of the web site holder, and
shall not be used for advertising or product endorsement purposes. The Atlas does not make any warranty, express or implied,
including the warranties of merchantability and fitness for a particular purpose, or assumes any legal liability or responsibility
for the accuracy, completeness, or usefulness of any information, and shall not be liable whatsoever for any damages incurred
as a result of its use. In particular, information presented in the Atlas is only for research purpose, and shall not be used for
diagnosis or treatment purposes. No responsibility is assumed for any injury and/or damage to persons or property for any use
or operation of any methods products, instructions or ideas contained in the material herein.
See also "Uniform Requirements for Manuscripts Submitted to Biomedical Journals: Writing and Editing for Biomedical
Publication - Updated October 2004": http://www.icmje.org.
http://AtlasGeneticsOncology.org© ATLAS - ISSN 1768-3262

Atlas of Genetics and Cytogenetics
in Oncology and Haematology
INIST-CNRS
OPEN ACCESS JOURNAL