three dimensional structural studies of α-n-acetylgalactosaminidase (α-naga) in α-naga deficiency...
TRANSCRIPT

Journal of Dermatological Science (2005) 37, 15—20
www.intl.elsevierhealth.com/journals/jods
Three dimensional structural studies ofa-N-acetylgalactosaminidase (a-NAGA) in a-NAGAdeficiency (Kanzaki disease): different genemutations cause peculiar structural changesin a-NAGAs resulting in different substratespecificities and clinical phenotypes
Takuro Kanekuraa,*, Hitoshi Sakurabab, Fumiko Matsuzawac,Seiichi Aikawac, Hirofumi Doic, Yoshio Hirabayashid,Noriko Yoshiia, Tomoko Fukushigea, Tamotsu Kanzakia
aDepartment of Dermatology, Kagoshima University Graduate School of Medical and Dental Sciences,8-35-1 Sakuragaoka, Kagoshima 890-8520, JapanbDepartment of Clinical Genetics, The Tokyo Metropolitan Institute of Medical Science, Tokyo MetropolitanOrganization for Medical Research, 3-18-22 Honkomagome, Bunkyo-ku, Tokyo 113-8613, JapancCelestar Lexico-Sciences, Inc., MTG D-17, 1-3 Nakase, Mihama-ku, Chiba 261-8501, JapandLaboratory for Cell Glycobiology, Frontier Research Program, The Institute for Physical andChemical Research, 2-1 Hirosawa, Wako 341-0198, Japan
Received 24 June 2004; received in revised form 17 September 2004; accepted 22 September 2004
KEYWORDSa-N-acetylgalac-
tosaminidasedeficiency;
Kanzaki disease;Homology modeling
Summary
Background: Kanzaki disease (OMIM#104170) is attributable to a deficiency in a-N-acetylgalactosaminidase (a-NAGA; E.C.3.2.1.49), which hydrolyzes GalNAca1-O-Ser/Thr. Missense mutations, R329W or R329Q were identified in two Japanese Kanzakipatients. Although they are on the same codon, the clinical manifestation was moresevere in R329W because an amino acid substitution led to protein instability resultingin structural change, which is greater in R329W than in R329Q.Objective: To examine whether the different clinical phenotypes are attributable tothe two mutations.Methods: Plasma a-NAGA activity and urinary excreted glycopeptides were mea-sured and three-dimensional models of human a-NAGA and its complexes withGalNAca1-O-Ser and GalNAca1-O-Thr were constructed by homology modeling.
* Corresponding author. Tel.: +81 99 275 5388; fax: +81 99 275 1134.E-mail address: [email protected] (T. Kanekura).
0923-1811/$30.00 # 2004 Japanese Society for Investigative Dermatology. Published by Elsevier Ireland Ltd. All rights reserved.doi:10.1016/j.jdermsci.2004.09.005

16 T. Kanekura et al.
Results: Residual enzyme activity was significantly higher in the R329Q- than theR329W mutant (0.022 � 0.005 versus 0.005 � 0.001 nmol/h/ml: p < 0.05); the urin-ary ratios of GalNAca1-O-Ser:GalNAca1-O-Thr were 2:10 and 8:10, respectively.GalNAca1-O-Ser/Thr fit tightly in a narrow space of the active site pocket of a-NAGA. GalNAca1-O-Thr requires a larger space to associate with a-NAGA because ofthe side chain (CH3–) of the threonine residue.Conclusion: Our findings suggest that the association of a-NAGAwith its substrates isstrongly affected by the amino acid substitution at R329 and that the association withGalNAca1-O-Thr is more highly susceptible to structural changes. The residual mutantenzyme in R329W could not associate with GalNAca1-O-Thr and GalNAca1-O-Ser.However, the residual mutant enzyme in R329Q catalyzed GalNAca1-O-Ser to someextent. Therefore, the urinary ratio of GalNAca1-O-Ser:GalNAca1-O-Thr was lowerand the clinical phenotype was milder in the R329Q mutation. Structural analysisrevealed biochemical and phenotypic differences in these Kanzaki patients with theR329Q and R329W mutation.# 2004 Japanese Society for Investigative Dermatology. Published by Elsevier IrelandLtd. All rights reserved.
1. Introduction
Kanzaki disease is an inherited autosomal recessivedisorder caused by a deficiency in a-N-acetylgalac-tosaminidase (a-NAGA; E.C.3.2.1.49), which hydro-lyzes GalNAca1-O-Ser/Thr [1]. The enzymaticdefect results in the lysosomal accumulation ofGalNAca1-O-Ser/Thr [2] and the increased urinaryexcretion of O-linked sialylglycopeptides that con-tain a-N-acetylgalactosaminyl moieties [3—5]. Thedisorder was first described in 1989 by Kanzaki et al.[1] in a 46-year-old Japanese woman who had dif-fuse angiokeratoma, mild intellectual impairment,and vacuolization of various kinds of cells. Theinfantile form of the disease was reported indepen-dently as Schindler disease [6,7]. Using genomic DNAisolated from the lymphoblasts of the first Japanesepatient with Kasnzaki disease, the molecular anom-aly was identified as a single C-to-T transition atnucleotide 985 that predicted an arginine-to-tryp-tophan substitution in amino acid residue 329(designated R329W) of the a-NAGA polypeptides[8]. More recently, a second Japanese woman witha-NAGA deficiency was reported; she had a G-to-Amutation at nucleotide 986 resulting in an arginine-to-glutamine substitution in residue 329 (designatedR329Q) [5]. Although the mutation was on the samecodon in both patients, they manifested differentclinical phenotypes. Angiokeratoma corporis diffu-sum, a dermatological sign of Kanzaki disease, wasmilder in the patient with the R329Q mutationcompared to the patient with R329W [5,9]. Whilethe patient with R329W manifested somewhat slowpsychomotor activity and an IQ of 70, the patientwith R329Q had an IQ of 112 [5,9].
These observations raise the question of how theR329W and R329Q mutations produce the moleculardefects in a-NAGA and whether the different clin-
ical phenotypes are attributable to the differentmutations. In a previous study [10], we constructeda three-dimensional model of human a-NAGA anddeduced the structural changes caused by R329Wand R329Q in patients with Kanzaki disease. Humana-NAGA consists of two domains. Domain I (aminoacid residues 18—311) has an (a/b)8-barrel struc-ture with the active site (D115 and D217) anddomain II (residues 312—404) has a b-sandwich foldconsisting of eight anti-parallel b-strands. R329 islocated on the 10th b-strand in domain II, facingdomain I, and forms hydrogen bonds with the neigh-boring amino acid residues T274, T306, and I312.Both R329W and R329Q cause disruption of thehydrogen bonds, which results in instability of theenzyme protein. Furthermore, R329W introduces abulkier side chain leading to steric clashes, andcauses more drastic changes than R329Q.
In this study, we carefully determined the a-NAGA activities in these two patients and analyzedurinary excreted glycopeptides. Furthermore, weconstructed three-dimensional structural modelsof human a-NAGA and its complexes with Gal-NAca1-O-Ser and GalNAca1-O-Thr by homology-modeling. We attempted to answer the above ques-tions from a structural perspective.
2. Materials and methods
2.1. Patients
Patient one was a 59-year-old woman with theR329W mutation. She presented with severe angio-keratoma, her IQ was 70 [1,4,8,9]. Patient two was a47-year-old woman with the R329Q mutation. Thedegree of her angiokeratoma was moderate and herIQ was 112 [5].

Three dimensional structural studies of a-N-acetylgalactosaminidase 17
2.2. Assay of plasma a-NAGA activity
Plasma a-NAGA activity was determined fluorome-trically with 4-methylumbelliferyl-a-N-acetylgalac-tosaminide (Seikagaku Corp., Tokyo, Japan) aspreviously described [5].
2.3. Analysis of urinary excretedglycopeptides
Urinary excreted glycopeptides were purified andmeasured by high-performance liquid chromatogra-phy. The chemical structure of these substances wasdetermined by amino acid analysis, carbohydrateanalysis, sequential enzymatic hydrolysis, and pro-ton nuclear-resonance spectroscopy, as describedpreviously [1].
2.4. Structural modeling of complexes ofhuman a-NAGA with GalNAca1-O-Ser andGalNAca1-O-Thr
Structural models of these complexes were based oncrystallographic data on chicken a-NAGA and theGalNAc complex (PDB code; 1KTC) [11]. For model-ing we used the molecular modeling softwareSYBYL/COMPOSER and BIOPOLIMER (TRIPOS, Moun-tain View, CA, USA). We employed the chicken a-NAGA backbone structure to model human a-NAGA.After generating and adjusting each side chain, weperformed the energy minimization procedure tooptimize conformations and side chain rotamersas described previously [12]. The GalNAc residuewas replaced with GalNAca1-O-Thr or GalNAca1-O-Ser to prepare the structural models of thecomplexes.
3. Results
3.1. Plasma a-NAGA activity
Plasma a-NAGA activity was measured carefully andrepeatedly. Our assay showed that both patientsmanifested slight residual enzyme activity; it waslower in the patient with the R329W than the
Table 1 Profile of the patients
Case Age/sex Mutation Angiokeratoma
1 59/F R329W +++2 47/F R329Q ++
* a-NAGA activity in plasma (nmol/h/ml). The difference betwe** Ratio of amino acid (AA) residues in urinary glyco-AA (Ser/Thr
patient with the R329Q mutation (mean � S.E. offive separate determinations: 0.005 � 0.001 nmol/h/ml versus 0.022 � 0.005 nmol/h/ml). The differ-ence was statistically significant at p < 0.05. Theactivity in normal subjects ranges from 0.94 to4.19 nmol/h/ml.
3.2. Measurement of urinary excretedamino acid O-glycosides
The ratio of GalNAca1-O-Ser to GalNAca1-O-Thr was8:10 and 2:10 in the patient with the R329W and theR329Q mutation, respectively. The results of plasmaa-NAGA activity and the ratio of urinary excretedamino acid O-glycosides are summarized with clin-ical profiles in Table 1.
3.3. Three-dimensional structural model ofthe complex of human a-NAGA withGalNAca1-O-Ser and GalNAca1-O-Thr
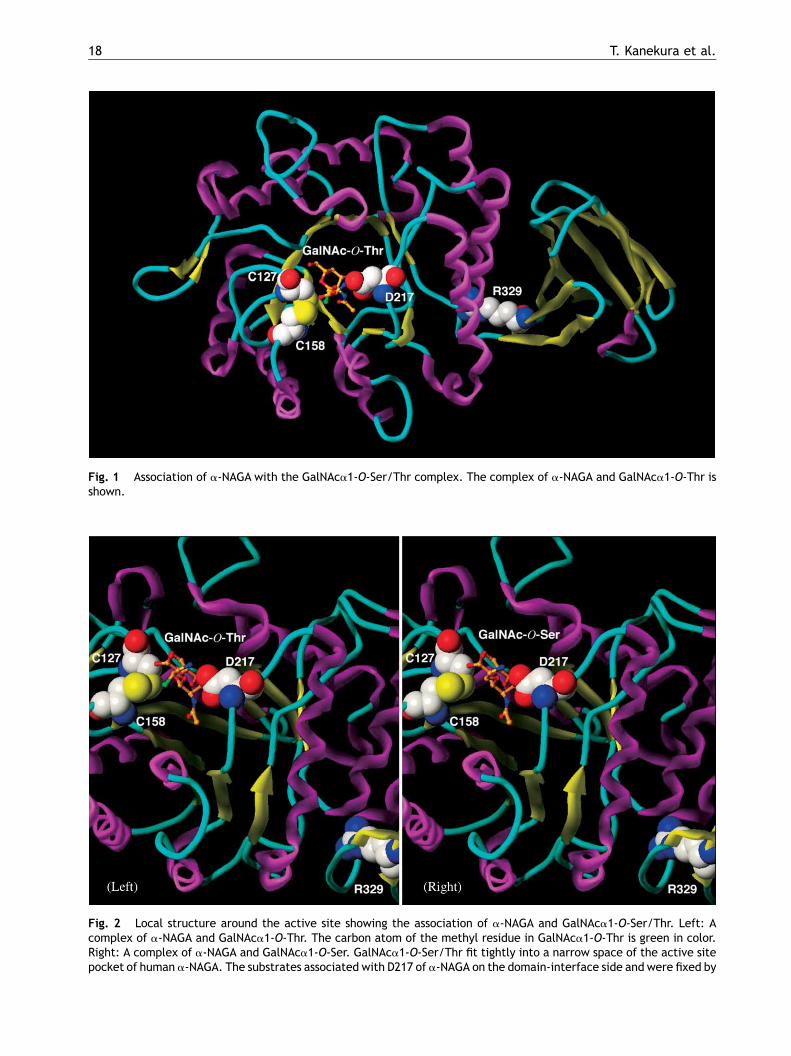
We constructedmodels of complexes of human wild-type a-NAGA with substrates and examined thebinding affinity between a-NAGA and GalNAca1-O-Ser and GalNAca1-O-Thr. GalNAca1-O-Ser/Thr wasfixed by D217 on the domain interface side in thebarrel structure and by C127 and C158, which form adisulfide bond, on the opposite side. GalNAca1-O-Ser/Thr fit tightly into a narrow space; the methylresidue in GalNAca1-O-Thr was adjacent to C158(Fig. 1). Compared to GalNAca1-O-Ser (Fig. 2,right), GalNAca1-O-Thr required extra space forthe methyl residue to bind to a-NAGA (Fig. 2,left).
4. Discussion
We examined the effect of amino acid substitutionsR329W and R329Q on the association with a-NAGAand its substrates from a structural perspective.According to our model, GalNAca1-O-Ser/Thr fittightly into a narrow space of the active site pocketof human a-NAGA. The substrates associated withD217 of a-NAGA on the domain-interface side andwere fixed by C127 and C158, which form a disulfide
I.Q. a-NAGA activity* Ser/Thr ratio**
70 0.005 � 0.001 8/10112 0.022 � 0.005 2/10
en case 1 and 2 is statistically significant at p < 0.05.).
18 T. Kanekura et al.
Fig. 1 Association of a-NAGA with the GalNAca1-O-Ser/Thr complex. The complex of a-NAGA and GalNAca1-O-Thr isshown.
Fig. 2 Local structure around the active site showing the association of a-NAGA and GalNAca1-O-Ser/Thr. Left: Acomplex of a-NAGA and GalNAca1-O-Thr. The carbon atom of the methyl residue in GalNAca1-O-Thr is green in color.Right: A complex of a-NAGA and GalNAca1-O-Ser. GalNAca1-O-Ser/Thr fit tightly into a narrow space of the active sitepocket of human a-NAGA. The substrates associated with D217 of a-NAGA on the domain-interface side and were fixed by

Three dimensional structural studies of a-N-acetylgalactosaminidase 19
bond, on the opposite side. GalNAca1-O-Thrrequires a large space to bind a-NAGA because ofa methyl residue; GalNAca1-O-Ser, on the otherhand, has a hydrogen atom instead of the methylresidue and requires less space for binding. Conse-quently, the association of a-NAGA with GalNAca1-O-Thr may be more susceptible to structuralchanges of a-NAGA than is the association of a-NAGAwith GalNAca1-O-Ser. We previously demonstratedthat an amino acid substitution at R329 leads tostructural changes and results in protein instability[10]. It is also possible that the R329mutants are lessstable thermodynamically or are not folded prop-erly. Immunoblot analysis showed that the matureform of a-NAGAwas decreased in cells from patientswith lysosomal a-N-acetylgalactosaminidase defi-ciency [4]. Our results were compatible with thestructural prediction and the instability of theexpressed protein appears to be the primary factorin causing the disorder.
Sensitive and careful assay detected a smallamount of residual a-NAGA activity in both patients.These residual enzyme activities may play a role inthe manifestation of the biochemical and clinicalfeatures in the patients with the different types ofmutation. Compared with R329Q, besides the loss ofhydrogen bonds, the R329W mutation introduces abulkier side chain resulting in steric clashes andcauses more drastic changes than R329Q [10]. Thestructural changes in the R329W mutant have agreater effect on the association with a-NAGA andits substrates than those of the R329Q mutant.Based on the results of urinary glycopeptide analy-sis, we posit that the residual mutant enzyme in theR329W mutation cannot hydrolyze both GalNAca1-O-Thr and GalNAca1-O-Ser. On the other hand, theresidual mutant enzyme in the R329Q mutation cancatalyze GalNAca1-O-Ser to some extent although itis unable to cleave GalNAca1-O-Thr, which is sus-ceptible to structural changes in the associationwith a-NAGA. The accumulation of GalNAca1-O-Ser in addition to GalNAca1-O-Thr in the R329Wmutant may result in the more severe phenotype.Clinically, the patient with R329W mutation mani-fested more prominent angiokeratomas and lowerIQ compared to the patient with R329Q (Table 1)[5,9].
In conclusion, we demonstrated that R329W andR329Q substitutions lead to structural changes in a-NAGA. This, in turn, resulted in the instability of theexpressed enzyme protein and the disturbance of
C127 and C158, which form a disulfide bond, on the opposite sibecause of amethyl residue, which is located adjacent to C158methyl residue and requires less space for binding (right). An aand results in protein instability. The R329W mutation cause
binding between mutant a-NAGA and GalNAc-O-Ser/Thr. The different gene mutations producedpeculiar conformational abnormalities in theenzyme and resulted in the expression of distinctclinical phenotypes.
Acknowledgements
This work was partly supported by grants from theTokyo Metropolitan Government, Japan Society forthe Promotion of Science, and the Ministry ofHealth, Labor and Welfare of Japan.
References
[1] Kanzaki T, Yokota M, Mizuno N, Matsumoto Y, Hirabayashi Y.Novel lysosomal glycoamino acid storage disease with angio-keratoma corporis diffusum. Lancet 1989;i:875—7.
[2] Kanda A, Tsuyama S, Murata F, Kodama K, Hirabayashi Y,Kanzaki T. Immunoelectron microscopic analysis of lysoso-mal deposits in a-N-acetylgalactosaminidase deficiency withangiokeratoma corporis diffusum. J Dermatol Sci2002;29:42—8.
[3] Hirabayashi Y, Matsumoto Y, Matsumoto M, Toida T, Iida N,Matsubara T, et al. Isolation and characterization of majorurinary amino acid O-glycosides and a dipeptide O-glycosidefrom a new lysosomal storage disorder (Kanzaki disease).Excessive excretion of serine- and threonine-linked glycan inthe patient urine. J Biol Chem 1990;265:1693—701.
[4] Kanzaki T, Wang AM, Desnick RJ. Lysosomal a-N-acetylga-lactosaminidase deficiency, the enzymatic defect in angio-keratoma corporis diffusum with glycopeptiduria. J ClinInvest 1991;88:707—11.
[5] Kodama K, Kobayashi H, Abe R, Ohkawara A, Yoshii N,Yotsumoto S, et al. A new case of a-N-acetylgalactosamini-dase deficiency with angiokeratoma corporis diffusum, withMeniere’s syndrome and without mental retardation. Br JDermatol 2001;144:363—8.
[6] Schindler D, Bishop DF, Wolfe DE, Wang AM, Egge H, LemieuxRU, et al. Neuroaxonal dystrophy due to lysosomal a-N-acetylgalactosaminidase deficiency. N Engl J Med1989;320:1735—40.
[7] Desnick RJ, Wang AM. Schindler disease: an inherited neu-roaxonal dystrophy due to a-N-acetylgalactosaminidasedeficiency. J Inherit Metab Dis 1990;13:549—59.
[8] Wang AM, Kanzaki T, Desnick RJ. The molecular lesion in thea-N-acetylgalactosaminidase gene that causes angiokera-toma corporis diffusum with glycopeptiduria. J Clin Invest1994;94:839—45.
[9] Kanzaki T, Yokota M, Irie F, Hirabayashi Y, Wang AM, DesnickRJ. Angiokeratoma corporis diffusum with glycopeptiduriadue to deficient lysosomal a-N-acetylgalactosaminidaseactivity. Clinical, morphologic, and biochemical studies.Arch Dermatol 1993;129:460—5.
de. GalNAca1-O-Thr requires a large space to bind a-NAGA(left). GalNAca1-O-Ser has a hydrogen atom instead of themino acid substitution at R329 leads to structural changess more drastic conformational changes than R329Q [8].

20 T. Kanekura et al.
[10] Sakuraba H, Matsuzawa F, Aikawa S, Doi H, Kotani M, NakadaH, et al. Structural and immunocytochemical studies of a-N-acetylgalactosaminidase deficiency (Schindler/Kanzaki dis-ease). J Hum Genet 2004;49:1—8.
[11] Garman SC, Hannick L, Zhu A, Garboczi DN. The 1.9 Astructure of a-N-acetylgalactosaminidase: molecular basis
of glycosidase deficiency diseases. Structure 2002;10:425—34.
[12] Sakuraba H, Matsuzawa F, Aikawa S, Doi H, Kotani M,Lin H, et al. Molecular and structural studies of theGM2 gangliosidosis 0 variant. J Hum Genet 2002;47:176—83.