the crystal structure of tgf-e33 and comparison to tgf-p2
TRANSCRIPT

Protein Science (19%), 5:1261-1271. Cambridge University Press. Printed in the USA. Copyright 0 1996 The Protein Society
The crystal structure of TGF-E33 and comparison to TGF-P2: Implications for receptor binding
~~~ ~~~ ~~ ~~
PEER R.E. MITTL, JOHN P. PRIESTLE, DAVID A. COX, GARY MCMASTER, NICO CERLETTI, AND MARKUS G. GRUTTER Ciba-Geigy Ltd., Pharmaceutical Research, CH-4002 Basle, Switzerland
(RECEIVED February 7, 1996; ACCEPTED April 11, 1996)
Abstract
Transforming growth factors /? belong to a group of cytokines that control cellular proliferation and differentia- tion. Five isoforms are known that share approximately 75% sequence identity, but exert different biological ac- tivities. The structure of TGF-/?3 was solved by X-ray crystallography and refined to a final R-factor of 17.5% at 2.0 A resolution. Comp~ison with the structure of TGF-02 (Schlunegger MP, Grutter MG, 1992, Nature 358430- 434; Daopin S, Piez KA, Ogawa Y, Davies DR, 1992, Science 252369-373) reveals a virtually identical central core. Differences exist in the conformations of the N-terminal a-helix and in the P-sheet loops. In TGF-03, the N-terminal cu-helix has moved = 1 A away from the central core. This movement can be correlated with the muta- tion of Leu 17 to Val and Ala 47. to Pro in TGF-63. The /?-sheet loops rotate as a rigid body 9" around an axis that runs approximately parallel to the dimer axis. If these differences are recognized by the TGF-0 receptors, they might account for the individual cellular responses. A molecule of the precipitating agent dioxane is bound in a crystal contact, forming a hydrogen bond with Trp 32. This dioxane may occupy a carbohydrate-binding site, because dioxane possesses some structural similarity with a carbohydrate. The dioxane is in contact with two tryp- tophans, which are often involved in carbohydrate recognition.
Keywords: crystal contact; dioxane; proteoglycan; receptor recognition; TGF-/?
Transforming growth factors f i are a group of multifunctional cytokines that play an important role in the development and repair of tissue (MassaguC, 1990; Roberts & Sporn, 1990). To date, five isoforms have been described (TGF-/?I-TGF-/?S) that are closely related to one another structurally and functionally. TGF-04 is thought to be the chicken homologue of TGF-/?I (At- tisano et al., 1994). The TGF-/? isoforms possess similar biolog- ical activities and belong to a superfamily of cytokines that also include the Mullerian inhibitory substance, inhibindactivins, bone morphogenic proteins, and D r o s ~ ~ ~ ~ l ~ decapentaplegic gene product (Burt, 1992).
TGF-03, like the other isoforms, is clearly an important endog- enous mediator of growth, maintenance, and repair processes in developing embryos, neonates and adults. The various in vitro and in vivo pharmacological actions of exogenous TGF-/?3 allow the prediction of possible clinical applications in a number of differ- ent indication areas, including wound healing (for acute and
Reprint requests to: Markus 0. Grutter, Ciba-Geigy Ltd., Pharma- ceuticai Research, CH-4002 Basle, Switzerland; e-rnail: [email protected].
Abbreviutions: E-factor, temperature-factor; Dxn, dioxan; hGH, hu- man growth hormone; hGHbp, human growth hormone binding pro- tein; TGF-p, transforming growth factor p; RMSD, RMS deviation; CHO, Chinese hamster ovary.
chronic skin wounds, gastrointestinal ulcers) and chemoprotec- tion (Cox, 1995).
The TGF-6 isoforms signal through two different membrane- bound receptors that are known to be Ser/Thr-kinases (Lin et al., 1992). The type I and type I1 receptors consist of a short cysteine-rich extracellular domain with two potential N-glycosyla- tion sites, a single membrane-spanning region, and an intracel- lular kinase domain. The binding of TGF-/? to the extracellular domain of receptor-I1 induces the assembling of the ternary TGF-/?:receptor-1:receptor-I1 complex. Whether the complex signals as a heterodimer or heterotetramer is presently unclear (Yamashita et al., 1994). In the ternary complex, the kinase do- main of receptor-I1 phosphorylates receptor-I, which in turn propagates the signal transduction (Wrana et al., 1994). The type 111 receptor, which is a membrane-anchored proteoglycan with- out any apparent signaling motif (Ldpez-Casillas et al., 1991; Wang et al., 1991), eliminates the biological differences of TGF-P isoforms (Lopez-Casillas et ai., 1993).
Lin et al. (1995) showed that the soluble extracellular domain of receptor 11 binds TGF-P1 and TGF-fi3 with apparent disso- ciation constants of 200 pM and 500 pM, respectively, but the recognition of TGF-02 had to be supported by the presence of receptor-111. Different receptor-I1 isoforms, which might be gen- erated by alternative splicing, contain an insertion/deletion in
1261

1262
the extracellular domain and are able to select between TGF-01 and TGF-fl2 (Suzuki et al., 1994). These data suggest that dif- ferences in binding affinities of the different isoforms are due to subtle differences in their three-dimensional structures.
The structure of human TGF-02 has been analyzed by X-ray crystallography (Daopin et al., 1992, 1993; Schlunegger & Grut- ter, 1992, 1993). The secondary structural elements of bovine TGF-01 expressed in CHO cells have been assigned by NMR (Archer et al., 1993). We report the structure of TGF-03 from two different crystal forms at 3.3 A and 2.0 A resolution (Kinemage 1).
Results and discussion
Chain con formation
TGF-03 forms a dimer of two identical subunits. Each subunit consists of a single 112-residue polypeptide chain that has to be considered as a single domain. The core of the subunit consists of three disulfide bridges forming a knot-like structure (Schlun- egger & Grutter, 1992). Together with nerve growth factor, platelet-derived growth factor (Murray-Rust et al., 1993), and chorionic gonadotropin (Lapthorn et al., 1994), TGF-03 belongs to the class of cysteine-knot proteins. The disulfide bridges Cys 44-Cys 109 and Cys 48-Cys 11 1 form a ring wide enough for the disulfide bridge Cys 15-Cys 78 to pass through (Fig. 1; see Kinemage 1). This disulfide knot replaces the hydrophobic core commonly found in globular proteins and forms a rigid structural entity. As already found in TGF-02 (Daopin et al., 1992; Schlunegger & Grutter, 1993) and TGF-61 (Archer et al., 1993), TGF-03 also possesses three a-helices and four 0-sheet strands. The 0-sheet consists of two separated antiparallel &sheets, which can be subdivided into nine pieces (Fig. 2). These @-sheets run approximately antiparallel to each other, but lack the hydrogen bonding pattern for a four-stranded @-sheet. The three a-helices are inserted at the N-terminus (HI) and into the loops between the &sheet strands a/b (H,) and b/c (H3). The /3-sheet strands c/d form a type 11' hairpin loop (Sibanda et al., 1989), with an arginine at the second position. Pro 85 causes strand d to wrap around strand c, such that strand d forms hydrogen bonds with strand c from both sides. The over- all chain-fold topology has similarity with a hand (Fig. 3). The palm represents the disulfide core and the fingers the four &sheet strands. Helices HI and H, are mimicked by the thumb and the
P.R.E. Mitt1 et al.
wrist, respectively. An additional disulfide bridge (Cys 7-Cys 16) enables a tight packing of helix HI onto the disulfide core.
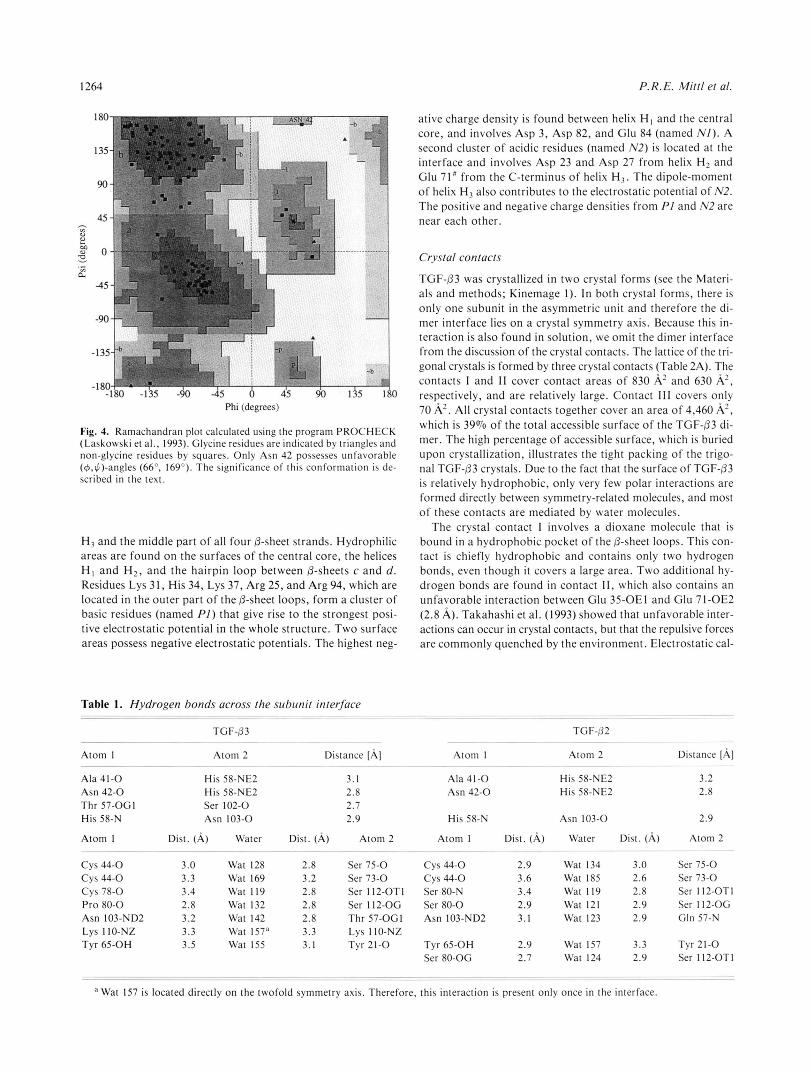
A Ramachandran plot shows that most of the non-glycine res- idues cluster in the a-helical and @-sheet regions (Fig. 4). Only Asn 42 possesses unfavorable dihedral main-chain angles (4 = 66", $ = 169"), but well-defined density. Its main-chain atoms are involved in hydrogen bonds that form a short piece of four- stranded antiparallel 0-sheet (Ala 41-0. . .Val 106-N, Asn 42- N. . .Leu 20-0, Phe43-N. . .Arg 18-0) and it also participates in the formation of the dimer interface (Table 1). In TGF-02, this residue adopts a similar conformation. It is likely that this unfavorable main-chain conformation is crucial for the forma- tion of the interface, and therefore conserved within TGF-02 and TGF-3. A cis-peptide bond occurs before Pro 36, which shows the same conformation in TGF-/32 and TGF-PI and is conserved in the sequence of all TGF-@ isoforms known so far (Fig. 2). The cis-conformation is stabilized by a hydrogen bond between Glu 35-0 and Leu 89-N.
Most of the side-chain torsion angles cluster around the stag- gered conformations, but the side chain of Cys 16, which connects helix H I with the central core, adopts an eclipsed conformation (xl = 242"). A more relaxed staggered conformation cannot be obtained due to the packing of helix H, onto the disulfide core. During the refinement, alternative side-chain conformations for His 34, Ser 59, and Ser 112 emerged. Ser 59 and Ser 112 are lo- cated inside the protein in close proximity to each other. The ability of the Oy-atoms to adopt different conformations, al- though these residues are not exposed to the bulk solvent, indi- cates a relatively loose packing in the interior of the protein. The two alternate Ser 59 side-chain conformations are stabilized by hydrogen bonds to Cys 11 1 (Ser 59-OG. . .Cys 11 1-0) or t o Thr 56 (Ser 59-OG.. .Thr 56-N, Ser 59-OG. . .Thr 56-OGI). The side chain of Ser 112 interacts with Asp 55 in both conforma- tions (Ser 112-OG. . .Asp 55-OD2). His 34 is located at the sur- face of the protein and participates in a cluster of positively charged residues. The two conformations of His 34 reflect the normal flexibility of surface residues.
Dimer formation
TGF-03 is composed of two identical subunits that are related by crystal symmetry. The whole dimer can be generated by ap- plying the symmetry operator (x - y , - y , 113 - t) and the unit
Fig. 1. Stereo plot of the TGF-03 disulfide knot. The disulfide bridges between Cys 44 and Cys 109 and between Cys 48 and Cys 1 1 1 form a ring that is wide enough for the disul- fide bridge between Cys 15 and Cys 78 to pass through. The 2F0 - F, map was contoured at 1.5 a.

TGF-03 crystal structure 1263
Protein I source 1 1 10 2 0 3 0 40 50 TGF-O 1 I human I ALDTNYCFSST--EKNCCVRQLYIDFRKDLGWKWIHEPKGYH~FCLGPCPY TGF-b2 ALDAAYCFRNV--QDNCCLRPLYIDFKRDLGWKWIHEPKGYNANFCAGACPY h u m TGF-P3
GVGQEYCFGNN--GPNCCVKPLYINFRKDLGWKWIHEPKGYEANYCLGNCPY x./aevis TGF-P5
DLDTDYCFGPGTDEKNCCVRPLYIDFRKDLQWKWIHEPKGYMANFCMGPCPY chicken TGF-P4
ALDTNYCFRNL--EENCCVRPLYIDFRQDLGWKWVHEPKGYYANFCSGPCPY human
Secondary structure *********** ***cccccccc##################cccccccccc Motif HHHHHH aaaa a a a H H H H H bbb bbb bbb
Protein 51 60 70 80 90 100 110
TGF-P 1 LWSSDTQHSRVLSLYNTINPEASASPCCVSQDLEPLTILYYIGKTPKIEQLSNMI~SCKCS TGF-P2
IWSLDTQYSKVLALYNQHNPGASAAPCCVPQALEPLPIVYWGRKPKVEQLSNMIVRSCKCS
IWSMDTQYSKVLSLYNQNNPGASISPCCVPDVLEPLPIIYWGRTAKVEQLSNMWRSCNCS TGF-P5
IWSADTQYTKVLALYNQHNPGASRAPCCVPQTLDPLPIIYWGRNVRVEQLSNMWRACKCS TGF-P4
LRSADTTHSTVLGLYNTLNPEASASPCCVPQDLEPLTILYWGRTPKVEQLSNMWKSCKCS TGF-P3
Secondary structure cccccccccccccccccccccccccccccccccc##################cccccccccc Motif
HHHHHHHHHHHH cccc cccccccccc dddddddddddd dddd
Fig. 2. Multiple sequence alignment of TGF-/3 isoforms. Rows 2-5, sequences of TGF-PI-TGF-fl5; row 6 , secondary struc- ture assignment of TGF-63 calculated using the program PROCHECK ( H , a-helix; a-d, &strands) (Laskowski et al., 1993); row 7, assignment of the central core (C), N-terminal a-helix (*), and &sheet loops (#).
cell shift (l/2/0). The interface is formed by interactions be- tween residues from helix H, and the central &sheet from the second subunit. The two subunits are separated by a cavity that is filled with water molecules. This cavity is bridged by a disul- fide between Cys 77 and Cys 77” (# refers to residues from the second subunit). The mutation of Cys 77 to Ser in TGF-01 (Amatayakul-Chantler et al., 1994) and thecorresponding Cys 80 in the closely related activin A (Husken-Hindi et al., 1994) ab- rogates dimer formation, indicating that these disulfide bridges are important for maintaining the quaternary structures and thereby the biological activities of these cytokines. Although the interface covers an area of 1,250 A 2 , which is 11% of the ac- cessible surface of the whole molecule, the interactions between the subunits are relatively weak and the disulfide bridge donates a large portion of binding energy. Only eight direct hydrogen bonds are distributed over the contact area (Table I ) . The clos-
est distances are found between the segments 41-42, 57-58, and 102-103, which also possess low B-factors. Most of the inter- actions are formed between main-chain atoms. Only the side chains of Thr 57 and His 58 are involved in hydrogen bonds be- tween the subunits that are not bridged by water molecules. The interface is populated by 29 water molecules; 23 of them are lo- cated in the cavity between the subunits and 6 water molecules bind in the cleft between helix H3 and the &sheet loops on the protein surface. In Table I , the 13 water molecules that partici- pate in hydrogen bonds between both subunits are listed.
Charged residues and hydrophobic surfaces
The surface of TGF-63 is mainly hydrophobic (Fig. 5 ) , which might explain the low solubility of this protein in concentrated buffer solutions. Hydrophobic areas cover the surfaces of helix
Fig. 3. Ribbon representation of the TGF-fl3 dimer (drawn using MOLSCRIPT, Kraulis 119911). The two individual subunits are colored light and dark grey. The disulfide bridges are depicted as ball-and- stick models.
1264 P. R. E. Mitt1 et al.
Phi (degrees)
Fig. 4. Ramachandran plot calculated using the program PROCHECK (Laskowski et al., 1993). Glycine residues are indicated by triangles and non-glycine residues by squares. Only Asn 42 possesses unfavorable (@,$)-angles (66". 169"). The significance of this conformation is de- scribed in the text.
H3 and the middle part of all four &sheet strands. Hydrophilic areas are found on the surfaces of the central core, the helices HI and H2, and the hairpin loop between &sheets c and d. Residues Lys 31, His 34, Lys 37, Arg 25, and Arg 94, which are located in the outer part of the 0-sheet loops, form a cluster of basic residues (named P I ) that give rise to the strongest posi- tive electrostatic potential in the whole structure. Two surface areas possess negative electrostatic potentials. The highest neg-
Table 1. Hydrogen bonds across the subunit interface -~ ~ ~~ ~
~~ - ~ _ _ _ ~ ~ ~~~
~~
TCF-63 . " ..~
Atom 1 Atom 2 Distance [A]
Ala 41 -0 His 58-NE2 3.1 Asn 42-0 His 58-NE2 2.8 Thr 57-OGI Ser 102-0 2.7 His 58-N Asn 103-0 2.9
_ _ _ ~ ~
~ ~" -~___ ~ ~~
Atom I
CYS 44-0 CYS 44-0 CYS 78-0 Pro 80-0 Asn 103-ND2
Tyr 65-OH LYS I IO-NZ
Dist. (A)
3.0 3.3 3.4 2.8 3.2 3.3 3.5
~ ~ ~ ~ ~ _ _ _
Water
Wat 128 Wat 169 Wat 119 Wat 132 Wat 142 Wat 157" Wat 155
~ ~~~~
Dist. (A)
2.8 3.2 2.8 2.8 2.8 3.3 3.1
~~ ~~
Atom 2 ~ ~ ~~
Ser 75-0 Ser 73-0 Ser 1 12-OT 1 Ser 112-OG Thr 57-OGI
Tyr 21-0 LYS 1 IO-NZ
ative charge density is found between helix H I and the central core, and involves Asp 3, Asp 82, and Glu 84 (named N I ) . A second cluster of acidic residues (named N2) is located at the interface and involves Asp 23 and Asp 27 from helix Hz and Glu 71' from the C-terminus of helix H,. The dipole-moment of helix H, also contributes to the electrostatic potential of N2. The positive and negative charge densities from PI and N2 are near each other.
Crystal contacts
TGF-03 was crystallized in two crystal forms (see the Materi- als and methods; Kinemage 1). In both crystal forms, there is only one subunit in the asymmetric unit and therefore the di- mer interface lies on a crystal symmetry axis. Because this in- teraction is also found in solution, we omit the dimer interface from the discussion of the crystal contacts. The lattice of the tri- gonal crystals is formed by three crystal contacts (Table 2A). The contacts I and I1 cover contact areas of 830 A2 and 630 A', respectively, and are relatively large. Contact 111 covers only 70 A'. All crystal contacts together cover an area of 4,460 A', which is 39% of the total accessible surface of the TGF-03 di- mer. The high percentage of accessible surface, which is buried upon crystallization, illustrates the tight packing of the trigo- nal TGF-63 crystals. Due to the fact that the surface of TGF-03 is relatively hydrophobic, only very few polar interactions are formed directly between symmetry-related molecules, and most of these contacts are mediated by water molecules.
The crystal.contact I involves a dioxane molecule that is bound in a hydrophobic pocket of the &sheet loops. This con- tact is chiefly hydrophobic and contains only two hydrogen bonds, even though it covers a large area. Two additional hy- drogen bonds are found in contact 11, which also contains an unfavorable interaction between Glu 35-OE1 and Glu 71-OE2 (2.8 A). Takahashi et al. (1993) showed that unfavorable inter- actions can occur in crystal contacts, but that the repulsive forces are commonly quenched by the environment. Electrostatic cal-
His 58-N Asn 103-0
Atom I Dist. ( A ) Water Dist. (A) ~~ ~- . ~ ~~
CYS 44-0 2.9 Wat 134 3.0 c y s 44-0 3.6 Wat 185 2.6 Ser 80-N 3.4 Wat 119 2.8 Ser 80-0 2.9 Wat 121 2.9 Asn 103-ND2 3. I Wat 123 2.9
Tyr 65-OH 2.9 Wat 157 3.3 Ser 80-OG 2.7 Wat 124 2.9
~~~~
.~ - ~~~ " ~
Wat 157 is located directly on the twofold symmetry axis. Therefore, this interaction is present only once in the interface. ~ ~~
~~ ~~
~~
Distance [A] 3.2 2.8
2.9
Atom 2
Ser 75-0 Ser 73-0 Ser 1 12-OT I Ser 112-OG Gln 57-N
Tyr 21-0 Ser 1 12-OTI
~ ~~~~

TGF-133 crystal structure 1265
P1’
N2’ N2
culations show that this is the case in the trigonal TGF-03 crys- with Lys 37 that belongs to PI. Glu 71 from the symmetry-related tals as well. The environment of Glu 35 possesses a positive molecule participates in cluster N2 and exerts a negative poten- electrostatic potential, due to the basic residues of cluster PI tial. A similar situation is found in contact 111, where Arg 94 nearby. Glu 35 also participates in an intramolecular salt bridge and Lys 107 come very close (Arg 94-NH2. . .Lys 107-NZ,
I1
111
Table 2. Crystal contacts of TGF-/33 (tri) and of TGF-03 (hex)
Buried area Contact No. of Distance Contact [A2] partners Residues involved waters Polar interactions [AI
A. TGF-f13 (tri)” I b 830 a:al a: 25-29, 31-32 11 Gln 26-0.. .Lys 31-NZ 2.8
b 1 ~ 2 b: 50-51, 64, 67-68, 70 Lys 31-NZ. ..Gin 26-0 2.8 c:b, c: 29, 32, 90, 92, 97,
Dxn
d:e4 d: 65, 71-72 f:g, e: 35, 89, 87 e:d6 f: 11-13, 20, 26-27 g:f7 g: 85, 94-100
h:is h: 94 i:h9 i: 107
630
70
B. TGF-fl3 (hex)‘ I 5 80 a: 69, 71-72
b: 92-95 C: 11-13, 18-20, 27,43 d: 35, 94, 96, 98, 100
Glu 12-0.. .Thr 95-OG1 Glu 35-OE1.. .Glu 71-OE2
3.3 2.8
2
Asp 27-ODl . . .Arg 94-NH1 Asn 69-0DI. 3 -Arg 94-NE Glu 71-OE1.. .Val 92-0
Ala 72-0. . -Thr 95-OG1 Glu 71-OEl...Arg 94-N
3.3 2.9 2.6 2.7 3.1
a Given in the column “Contact partners” are the fragments that interact with each other. The operator that relates the two fragments is specified as the symmetry operator plus unit cell translations. 1, ( -x , y - x , 2/3 - z ) + (O/O/O); 2, (-y, x - y , 2/3 + z ) + (1/2/-1); 3, (y -x , -x , 1/3 + z) + (-l/l/O); 4, ( X , y, Z) + (l/l/O); 5 , ( X -y, -y, 1/3 - Z) + (l/l/O), 6, ( X , y, Z) + (-l/-l/O); 7, ( X -y . -y, 113 - Z) + (O/l/O); 8, ( y - X , -X ,
1/3+z) + (-I/O/O); 9, (y , x, -z) + (-2/0/0). This contact appears only once per monomer because it encompasses the twofold symmetry axis. Given in the column “Contact partners” are the fragments that interact with each other. The operator that relates the two fragments is speci-
fied as the symmetry operator plus unit cell translations. 1, ( y , x, 1/3 - z ) + (O/-l/l); 2, ( X - y , x , 116 + z ) + (O/-l/O); 3, ( y , X , 113 - z ) + (l/O/l); 4, ( y , y - X , 5/6 + Z) + (l/l/-1).

1266 P.R.E. Mitt1 et al.
3.6 A). Lys 107 is located close to the negative electrostatic po- tential of cluster N1 and participates in a salt bridge with Glu 84, whereas Arg 94 belongs to cluster PI and is certainly positively charged.
A different situation is found in the hexagonal TGF-03 crystals. These crystals are very loosely packed (V, = 5.08 A3 Da"). The lattice is built up by a single crystal contact that is mainly hy- drophilic and involves one salt bridge and several hydrogen bonds (Table 2B). The residues that participate in the forma- tion of this contact are similar to those in contact I1 of the trig- onal crystals. The observation that the same sets of residues are involved in crystal contacts in different lattices can be explained by the high charge density of these areas. Hydrophobic inter- actions do not seem to be as important for the formation of these crystals as they are for the trigonal crystals. These obser- vations indicate that the trigonal TGF-83 crystals are rather sta- bilized by the attraction of complementary charged surfaces and hydrophobic interactions than by the effects of individual resi- dues. In the trigonal crystal form, dioxane might be as important for keeping symmetry-related molecules together by hydrophobic interactions as, for example, Ca2+ ions are for the formation of ferritin crystals (Lawson et al., 1991) or phosphate ions are for bovine pancreatic trypsin inhibitor crystals (Wlodawer, 1984).
Comparison with TGF-P2
The structure of TGF-P2 was solved independently by Daopin et ai. (1992) and Schlunegger and Griitter (1992). Because both structures are more or less identical (Daopin et al., 1994), even though the proteins were folded by different routes, we focus on the comparison with the structure of human TGF-02 puri- fied from Escherichia coli and folded in vitro (Brookhaven Pro- tein Data Bank accession code 1TFG). Human TGF-fi2 and TGF-03 share 79% sequence identity. The superposition of the whole dimers reveal an RMSD of 1.27 A for all 224 Ca-atoms. Given the high sequence identity, a much lower RMSD would be expected on the basis of a statistical approach (Chothia & Lesk, 1987), indicating large structural rearrangements or crys- tallization effects. The omission of the outer parts of the @-sheet loops and helix H l yield a central protein core (residues 15-22,
41-84, 103-1 12, and the identical residues from the second sub- unit) that superimposes with an RMSD of 0.36 A. Helix H1 (residues 1-14) and the P-sheets loops that also include helix H2 (residues 23-40 and 85-102) superimpose as rigid bodies with RMSD of 1.03 and 0.67 A, respectively.
The superposition of the central cores reveal that the &sheet loops of TGF-P3 are rotated 9" away from the second subunit relative to TGF-/32 (Fig. 6). The hinge axis runs through the Ca-atom of residue 41, approximately parallel to the twofold dimer axis. In TGF-03, helix H I has moved = l A away from the central core. Due to the fact that these parts have high B-factors, the differences could also be caused by crystal forces. In the medium-resolution TGF-03 structure, the orientation of helix H1 is identical to that in the high-resolution structure, and the P-sheet loops show only small deviations (Fig. 7) despite their different crystal packings. Both TGF-P3 crystal packings are dif- ferent from the crystal packing of TGF-P2 crystals. We there- fore conclude that the 0-sheet loops form a flexible structural entity that adopt different orientations in TGF-f12 and TGF-03.
The movement of helix H I can be correlated with the substi- tution of individual residues. In TGF-62, the large side chain of Leu 17 is accommodated easily in the cleft between helix H l and the central core (Fig. 8). Due to the branching at the CP- atom, the mutation Val 17 +Leu (all mutations are given in the order .+ residueTGFg2) places Val 17-032 in van der Waals contact with the main chain of helix H1 (Val 17-CG2. . Leu 2-0, 3.2 A). The mutation Pro 47 .+ Ala ruptures the H-bond Ala 47-N- .Asp 13-0 (2.8 A), which is present between helix H1 and the central core in TGF-P2. Due to the absence of the amide proton in TGF-P3, Glu 13-0 has to move to van der Waals distance away from Pro 47-N (3.4 A). These movements are transferred to the conformation of helix H I . The orientation of this a-helix is also affected by the mutation of Arg 52 .+ Trp and Glu 12 --t Gln. These mutations influence important inter- actions between helix HI and the central core. In TGF-P2, Arg 52 forms a salt bridge with the side chain of Glu 12, which itself is hydrogen bonded to Tyr 6 (Tyr 6-OH. - .Glu 12-OE1). A further hydrogen bond is formed between Arg 52 and Cys 15 (Arg 52-NE. . .Cys 15-0), which is also present in TGF-fl2 (Trp 52-NE1 -Cys 15-0). The sequence comparison of TGF-
Fig. 6. Superposition of TGF-03 and TGF-82, based on the residues from the central core (residues 15-22, 41-84, 103-112, 15#-22#, 41#-84#, 103#-112#) and viewed parallel to the twofold dimer axis. For clarity, only the central core for TGF-03 is shown. Different conformations are found for the N-terminal a-helix (residues 1-14) and the 8-sheets loops (residues 23-40 and 85-102), which are shown in red for TGF-03 and in green for TGF-02.

TGF-63 crystal structure
93'
1267
93 93
Fig. 7. Stereo plot of the TGF-63 Cor-backbones from the hexagonal (thin lines) and the trigonal (bold lines) space groups. For the superposition, only residues 15-22,41-84, 103-1 12, 15#-22#, 41#-84', and 103#-112" were taken into account. The major differences occur around residue 68, due to the weak electron density of this region in the TGF-63 (hex) structure.
61, TGF-62, and TGF-63 reveals that Val 17 and Pro 47 are different only in TGF-02, whereas Trp 52 is different only in TGF-63 (Fig. 2).
The dimer interfaces of TGF-62 and TGF-63 are virtually identical. His 58 is a central residue for the dimer formation, because it participates in three direct hydrogen bonds with the neighboring subunit. This residue is also present in TGF-62, where it forms similar interactions (Table I ) . In TGF-P1 and TGF-65, it is replaced by a tyrosine, suggesting that the dimer interface of these cytokines look different. In TGF-63, a hydro- gen bond is also present between Thr 57-OGl and Ser 102-0, which is disrupted by the replacement of Thr 57 with Gln in TGF-62. The replacement of Pro 80 against Ser in TGF-02 ex- erts a rearrangement of water molecules that populate the cav- ity between the subunits. In TGF-62, Wat 124 bridges the interface by forming hydrogen bonds with Ser 80-OG and the C-terminus from the second subunit. Due to the Pro 80 + Ser mutation, this water molecule is absent in TGF-63. The remain-
ing interface water molecules are conserved in TGF-62 and TGF-63.
Dioxane binding
A dioxane molecule is bound in a hydrophobic pocket near the outer part of the 6-sheet loops (Kinemage 1). The binding site is formed by several hydrophobic side chains, among them Trp 30 and Trp 32. Dioxane forms a short hydrogen bond with Trp 32 (Trp 32-NEl. . .Dxn-Ol, 2.7 A). Dxn-C5 is in contact with the side chain of Trp 30 (Trp 30-CZ3. . .Dxn-CS, 3.4 A). The con- tact area with the molecule that donates the hydrogen bond is 210 A 2 , which is 75% of the total accessible dioxane surface. Compared with the medium-resolution TGF-63 structure, the side chain of Trp 32 rotates 150" around xz in order to provide space for the binding of dioxane (Fig. 9).
A well-defined binding site like this for a non-natural ligand is quite surprising. Dioxane is contacted by two tryptophans,
Fig. 8. Superposition of helix H I from TGF-03 (bold lines) and TGF-02 (thin lines). For simplicity, the main chain is shown for residues 1-17 and 46-52, but only the side chains for residues 12, 17.47, and 52 are indicated. The superposition is identical to that in Figures 6 and 7. Mutations are given in the order residueTGF.p3 - residueTGFp*. Hydrogen bonds are indicated by broken lines and general distances by dotted lines.

1268 P.R.E. Mitt1 et al.
Fig. 9. Binding site of the dioxane that is buried in a crystal contact. The main chains are indicated by tubes in light and dark grey for the two independent subunits. Residues that contact the dioxane in the TGF-03 (tri) structure are shown as ball-and- stick models (drawn using MOLSCRIPT, Kraulis [1991]). In the TGF-P3 (hex) structure, the side chain of Trp 32 has rotated 150” around x2, which is shown in thin lines.
which are often involved in the recognition of carbohydrates (Vyas, 1991) and possesses some structural similarity with a py- ranose. In order to test whether a carbohydrate would fit into this binding site, we positioned various carbohydrate molecules in this site using the dioxane six-membered ring as a template. The model shown in Figure 10 and Kinemage 1 demonstrates that the dioxane-binding site is sufficiently large to accommo- date a carbohydrate molecule without structural rearrangements. A carbohydrate recognition site might be crucial for the biolog- ical activities of TGF-/3 because receptor-I, receptor-11, and re- ceptor-I11 are all highly glycosylated.
Potential receptor recognition sites
TGF-03 binds to a number of different glycoproteins. Among these are the membrane-bound type I, type 11, and type 111 re-
ceptors and the soluble proteoglycans decorin, biglycan, fibro- modulin, and lumican. Structural or physicochemical differences must be important for their biological functions, because TGF- p2 and TGF-P3 induce biological effects with different poten- cies (Graycar et al., 1989). Knowing the structure of TGF-/32, Schlunegger and Griitter (1992) proposed residues I-13,25-35, 67-73, and 91-96 to be involved in receptor binding. The super- position of the two TGF-b structures reveals that three of these segments indeed possess different conformations and are there- fore good candidates for receptor recognition. Only residues 67-73 show the identical conformation in both structures. If ei- ther receptor-I or receptor-I1 would recognize the &sheet loops (residues 22-40 and 85-102) or helix HI (residues 1-14), the dif- ferent conformations of these fragments might alter the assem- bling of the ternary complex, depending on the bound TGF-/3 isoform. Under the assumption that helix H, would be involved
Fig. 10. The carbohydrate was modeled based on the structure shown in Figure 9. Potential hydrogen bonds are indicated as broken lines. Because the hydrogen bond between Trp 32 and the ligand should also be present in the carbohydrate:TGF-P3 complex, two possible orientations for the C5 atom remain. Due to the fact that Dxn-C5 is a t van der Waals distance to Trp 30, no substituent larger than a hydrogen can be accommodated at this site. A suitable model can therefore only be obtained with a pentose, where the 1’-hydroxyl group points toward Tyr 90. In this model, the 2’- and 3’-hydroxyl groups are in equatorial configurations and form favorable interactions with the side chains of Glu 99 and Tyr 90. An equatorial configuration is also required for the 1’-hydroxyl group, otherwise it would collide with the protein. Only the configuration of the 4‘-hydroxyl is not fixed. The carbohydrates that satisfy these requirements best are P-D-xylo-pyranoside and or-L-arabino-pyranoside, depending on the configuration of the 4’-hydroxyl group.

TGF-03 crystal structure 1269
in this recognition, Val 17, Pro 47, and Arg 52 (Leu 17, Ala 47, and Trp 52 in TGF-02) are good candidates for the determina- tion of the biological specificity.
Structural (De Vos et al., 1992; Clackson & Wells, 1995) and mutational studies (Cunningham & Wells, 1993) of the hGH: hGHbp-complex have shown that the distribution of hydrophilic and hydrophobic areas are important for receptor recognition, which might also be the case for the TGF-0:receptor complex. The positive electrostatic potentials around cluster PI and the negative potentials around NI are common features of TGF-02 and TGF-03, but the negative potential around N2 is present only in TGF-03. The absence of N2 in TGF-02 might be caused by the replacement of Gln 26 -+ Arg. In TGF-02, the electrostatic potential of N2 is counterbalanced by Arg 26. Residues 69-73 from the other subunit contribute to the electrostatic potential around N2. Qian et al. (1994) showed that the deletion of these residues had a profound effect on the biological activities of TGF-0 1, indicating that this area might be involved in the rec- ognition. It is possible that Asp 23, Gln 26, and Asp 27 are in- volved in the recognition as well, because they possess different electrostatic potentials in TGF-02 and TGF-03, and are located close to residues 69-73.
Materials and methods
Crystallization and data collection
Biologically active human TGF-03 was prepared by in vitro fold- ing of monomeric protein obtained from E. coli inclusion bod- ies and purified as described by Cerletti (1995). Initial crystals were obtained using the empirical crystallization matrix derived by Jancarik and Kim (1991) with the vapor diffusion method. A mixture of 2 pL protein (3-6 mg/mL in 10 mM acetic acid) and 2 pL reservoir solution was equilibrated at room tempera- ture (20-22 "C) against a buffer consisting of 30% (w/v) poly- ethylene glycol 4000, 200 mM magnesium chloride, and 100 mM Tris/HCL, pH 8.4. Crystals grew within 2-4 days as hexagonal bipyramids reaching a maximum size of 0.4 x 0.4 x 0.3 mm3. These crystals were fragile and difficult t o handle. The crystals belonged to space group P6,22 with unit-cell dimensions of a = b = 77.8 A, c = 143.2 A, a = = 90°, y = 120°, and con- tained one subunit per asymmetric unit with V, = 5.08 A 3 Da" (Matthews, 1968). Due to their high solvent content, these crys- tals diffracted only to 3.3-A resolution. A second crystal form
Table 3. Characteristics of crystals and data
Crystal form
Space group Unit cell
vm Resolution limit Completeness
Rmerge
Unique reflections Multiplicity
was discovered using dioxane as the precipitating agent. The res- ervoir solution consisted of 15% (v/v) dioxane and 100 mM so- dium acetate, pH 5.0. To reproduce large X-ray quality crystals, seeding was indispensable. The seed crystals had typical sizes of 0.03 x 0.03 X 0.1 mm3 and were washed for 15 min in 10 mM acetic acid before they were pipetted into the crystallization drop. The crystals grew as hexagonal rods up to sizes of 0.15 x 0.15 x 0.5 mm3. The space group was determined to be P3,21 (a = b = 49.3 A, c = 78.9 A, a = 0 = 90°, y = 120") with one subunit per asymmetric unit. They were densely packed (V, = 2.25 A 3 Da") and stable for at least one day in 10 mM acetic acid. In 100 mM acetic acid, they dissolved within 1 h.
Although these crystals are relatively small, they diffract up to 2.0-A resolution. Diffraction data were collected on a FAST area detector (Enraf-Noninus) using CuK, radiation generated by a rotating anode generator (model FR571) and evaluated using the program MADNES (Pflugrath & Messerschmidt, 1987). Each data frame covered a rotation angle of 0.1 O and was exposed for 100 min. Results from the data collection are given in Table 3.
Structure solution and refinement
One subunit of the TGF-(32 structure (Schlunegger & Griitter, 1993) served as a starting model for solving the structure of TGF-03 by molecular replacement, using the data from the hex- agonal crystals. The cross-rotation function was calculated using the program ALMN (CCP4, 1994) with data between 15 and 3.3 A resolution and yielded a single solution (4.2 a) for the ro- tational angles a = O.O", 0 = 54.8", and y= 2.8". After refine- ment of this solution, the translation function was calculated using the program TFFC (CCP4, 1994) with data from 15 to 3.3 A resolution. The maximum was found for a fractional shift of 0.75, 0.99, and 0.40. Subsequently, this partially refined structure was used to solve the structure in the trigonal crystal form at high resolution. Two cross-rotation functions were cal- culated using the medium-resolution TGF-03 or the TGF-02 structures as search molecules. The function calculated with TGF-03 (hex) (data from 8 to 3.5 A resolution, Patterson ra- dius 6-30 A) yielded a clear 6 . 5 ~ solution for the rotational an- gles a = 92.9", /3 = 142.4", y = 2.8", that coincided with the solution found using the TGF-02 structure at a = 261 .O", 0 = 164.1", y = 261.0" (6.2 u peak, data from 8 to 3.5 A resolution). Fifteen cycles of Patterson correlation refinement (X-PLOR;
Hexagonal crystals "______ P6122 u = b = 77.8 A , c = 143.2 A a = p = 90", y = 120" 5.08 A' Da"
99.9% (03-3.30 A) 100.0% (3.36-3.30 A ) 13.2% 4,232 9.7
3.3 A
Trigonal crystals
P3221 a = b = 49.3 A, c = 78.9 A
2.25 A 3 Da"
76.0% (10.0-2.0 A) 37.4% (2.1-2.0 A ) 7.0% 6,323 3.7
a = B = 90", y = 120"
2.0 A

1270 P.R.E. Mitt1 et al.
Briinger, 1990) with data between 15 and 3.5 A slightly increased the correlation coefficient from 0.124 to 0.126 for the roughly oriented TGF-P3 (hex) search model. The three-dimensional translation function was calculated for the two enantiomorphic space groups P3121 and H221, using data from 20-6 A resolu- tion. With P3,21, a 8 . 9 ~ maximum was found for a fractional shift of 0.08,0.76,0.14. This maximum was resolution indepen- dent and higher than the maximum obtained for P3,21 (5.9 a).
The approximately positioned search model had an R-factor of 45.9% in the resolution range 15-3 A, which was reduced to 41.4% by 25 cycles of rigid-body refinement, treating the en- tire model as one group. Refinement was continued by simulated annealing using X-PLOR (Briinger et al., 1987) on a Silicon Graphics workstation. At the beginning, the B-factors were set uniformly to 20 A2. After each manual intervention (program 0) (Jones et al., 1991), the structure was subsequently improved by 40 cycles of restrained least-squares refinement and 0.25 ps of molecular dynamics kept at 300 K . After additional 80 cycles of least-squares refinement, 20 cycles of individual B-factor re- finement were performed. When the R-factor dropped to just below 30%, an area of strong positive electron density near the side chain of Trp 32 emerged, which was interpreted as a dioxane molecule. During refinement, the dioxane conformation was restrained using values obtained by Koritsanszky et al. (1991). During the last refinement cycles, two alternate side-chain con- formations for His 34, Ser 59, and Ser 112 became visible and were refined as such. The refinement converged at an R-factor of 17.5% (7.0-2.0 A resolution), with good geometry. The RMSD from standard geometry for bond lengths and angles were 0.007 A and 1.41", respectively. The final structure in- cluded 890 protein atoms, 78 water molecules, and 6 dioxane atoms. Details on the refined structures of TGF-03 are given in Table 4.
Quality of the structures
The average coordinate errors of the final TGF-P3 models ac- cording to a Luzzati plot (Luzzati, 1952) were estimated to be 0.23 A for the high-resolution structure and 0.35 A for the medium-resolution structure. In the high-resolution structure, all atoms except the side chains of Arg 9 and Arg 94 are well de- fined in the electron density. Because these residues are located at the protein surface, they are likely to be very mobile. The av- erage B-factor for the whole structure including water molecules is 29.6 A*. Most of the residues from the central core possess
Table 4. Structure characteristics
Structure TGF-03 (hex) TGF-03 (tri)
Resolution 10.0-3.3 A 7.0-2.0 A R-factor 22.8% 17.5% Stereo chemistry RMSband = 0.016 A RMSbond = 0.007 A
No. of waters None 78 Average B-values All a toms 40.1 A 2 Main-chain, 24.2 A'
RMS,,,,, 1.92" RMS,,,,, = 1.41"
Side-chain, 31.8 A' Water, 42.2 A 2
Dioxane, 43.5 A' "___
main-chain B-factors below 20 A 2 (residues 15-22,36-39,43-45, 57-66, 76-82, 84-90, 104-112). Values above 30 A 2 are ob- served only for helix H, (residues 1-12), the &sheet loops (res- idues 25-27 and 92-96), and the C-terminus of helix H, (residues 70-73).
Map calculation
Accessible surfaces were calculated by the method of Lee and Richards (1971) with a probe radius of 1.4 A. The electrostatic potentials and hydrophobicity maps were calculated using the program MAPROP from the 0 program package (Kleywegt & Jones, 1994). For calculating the electrostatic potentials, the minimum and maximum distance cut-off values were set to 1 A and 12 A , respectively, and a power of 1.5 was used. For the hy- drophobicity maps, the default values were kept.
References
Amatayakul-Chantler S, Qian SW, Gakenheimer K, Bottinger EP, Roberts AB, Sporn MB. 1994. [Ser77] transforming growth factor-81. J Bioi Chem 269:27687-27691.
Archer SJ, Bax A, Roberts AB, Sporn MB, Ogawa Y, Piez KA, Weather- bee JA, Tsang MLS, Lucas R, Zheng BL, Wenker J, Torchia DA. 1993. Transforming growth factor-61: Secondary structure as determined by heteronuclear magnetic resonance spectroscopy. Biochemlstry32:1164- 1171.
Attisano L, Wrana JL, Lopez-Casillas F, Massague J . 1994. TGF-8 recep- tors and action. Biochim Biophys Acfa 1222:71-80.
Brunger AT. 1990. Extension of molecular replacement: A new search strategy based on Patterson correlation refinement. Acta Crystallogr A 46:46-57.
Brunger AT, Kuriyan J, Karplus M. 1987. Crystallographic R-factor refine- ment by molecular replacement. Science 235:458-460.
Burt DW. 1992. Evolutionary grouping of the transforming growth factor- 6 superfamily. Biochem Biophys Res Commun 184:590-595.
CCP4. 1994. Collaborative Computational Project, No. 4. The CCP4 suite: Programs for protein crystallography. Acfa Crystallogr D 50:760-763.
Cerletti N. 1995. Novel process for the production of biologically active di- meric protein. PCTAppl. No. E P 95/02718.
Chothia C, Lesk AM. 1987. Theevolution of protein structures. ColdSpring Hurbor Symp Quanr Bioi 52:399-405.
Clackson T, Wells JA . 1995. A hot spot of binding energy in a hormone- receptor interface. Science 267:383-386.
Cox DA. 1995. Transforming growth factor-beta 3. Cell BIOI Int 19:357-371, Cunningham BC, Wells JA . 1993. Comparison of a structural and a func-
tional epitope. J Mol Bioi 234554-563. Daopin S, Davies D, Schlunegger MP, Grutter MG. 1994. Comparison of
two crystal structures of TGF-82: The accuracy of refined protein struc- tures. Acta Crystallogr D 50~85-92.
Daopin S. Li M, Davies DR. 1993. Crystal structure of TGF-02 refined at 1.8 A resolution. Proteins Struct Funct Genet 17~176-192.
Daopin S, Piez KA, Ogawa Y, Davies DR. 1992. Crystal structure of trans-
257:369-373. forming growth factor-82: An unusual fold for the superfamily. Science
De Vos AM, Ultsch M, Kossiakoff AA. 1992. Human growth hormone and extracellular domain of its receptor: Crystal structure of the complex. Science 255~306-312.
Craycar JL, Miller DA, Arrick BA, Lyons RM, Moses HL, Derynck R. 1989. Human transforming growth factor-&: Recombinant expression, puri- fication, and biological activities in comparison with transforming growth factor-pl and -B2. Mol Endocrinol3:1977-1986.
Husken-Hindi P, Tsuchida K , Park M, Corrigan AZ, Vaughan JM, Vale WW, Fischer WH. 1994. Monomeric activin A retains high receptor binding affinity but exhibits low biological activity. J Biol Chem 269:19380- 19384.
Jancarik J, Kim SH. 1991. Sparse matrix sampling: A screening method for crystallization of proteins. J Appl Crystallogr 24:409-41 I .
Jones TA, Zou JY, Cowan SW, Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crysfallogr A 47:110-119.
Kleywegt GJ, Jones TA. 1994. Detection, delineation, measurement and dis-

TGF-03 crystal structure
play of cavities in macromolecular structures. Acta Crystallogr D 50: 178-185.
Koritsanszky T, Strumpel MK, Buschmann J, Luger P, Hansen NK, Pichon- Pesme V. 1991. A study of the anomeric effect on an electronic scale:
J A m Chem SOC 113:9148-9154. The electron density of 1 ,Cdioxane and trans-2,5-dichloro-l ,4-dioxane.
Kraulis P. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24:946-950.
Lapthorn AJ, Harris DC, Littlejohn A, Lustbader JW, Canfield RE, Ma- chin KJ, Morgan FJ, Isaacs NW. 1994. Crystal structure of human cho-
Laskowski RA, MacArthur MW, Moss DS, Thornton JM. 1993. rionic gonadotropin. Nature 369:455-461.
PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr 26:282-291.
Lawson DW, Artymiuk PJ, Yewdall SJ, Smith JMA, Livinston JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, Thomas CD, Shaw WV, Harrison PM. 1991. Solving the structure of human H ferritin by genet- ically engineering intermolecular crystal contacts. Nature 349:541-544.
Lee B, Richards FM. 1971. The interpretation of protein structures: Esti- mation of static accessibility. J Mol Biol55:379-384.
Lin HY, Moustakas A, Knaus P, Wells RG, Henis YI, Lodish HF. 1995. The soluble exoplasmic domain of type I1 transforming growth factor (TGF)-P receptor. JBiol Chem 270~2747-2754.
Lin HY, Wang YF, Ng-Eaton E, Weinberg R, Lodish H. 1992. Expression cloning of the TGF-0 type I 1 receptor, a functional transmembrane ser- ine/threonine kinase. Cell 68:775-785.
Lopez-Casillas F, Cheifetz S, Doody J, Andres JL, Lane WS, Massague J. 1991. Structure and expression of the membrane proteoglycan betagly- can, a component of the TGF-P receptor system. Cell 67:785-795.
Lopez-Casillas F, Wrana JL, Massague J. 1993. Betaglycan presents ligand to the TGF-P signalling receptor. Cell 73:1435-1444.
Luzzati PV. 1952. Traitment statistique des erreurs dans la determination des structures cristallines. Acta Crystallogr 5:802-810.
Massague J. 1990. The transforming growth factor-0 family. Annu Rev Cell Biol 6:597-641.
Matthews BW. 1968. X-ray structure of proteins. J Mol Biol33:461-467. Murray-Rust J, McDonald NQ, Blundell TL, Hosang M, Oefner C, Win-
kler F, Bradshaw RA. 1993. Topological similarities in TGF-P2, PDGF-
1271
BB and NGF define a superfamily of polypeptide growth factors. Struc- ture 1:153-159.
Pflugrath JW, Messerschmidt A. 1987. Crystal orientation and X-ray pat- tern prediction of area-detector diffractometer systems in macromolec- ular crystallography. Acta Crystdlogr 20:306-315.
Qian SW, Burmester JK, Sun PD, Huang A, Ohlsen DL, Suardet L, Flanders KC, Davies D, Robets AB, Sporn B. 1994. Characterization of mutated transforming growth factor-Os which possess unique biological proper- ties. Biochemistry 33:12298-12304.
Roberts AB, Sporn MB. 1990. The transforming growth factor-Os. In: Sporn MB, Roberts AB, eds. Peptide growth factors and their receptors I . Ber-
Schlunegger MP, Griitter MG. 1992. An unusual feature revealed by the crys- lin: Springer-Verlag. pp 419-472.
tal structure at 2.2 A resolution of human transforming growth factor- 02. Nature 358:430-434.
Schlunegger MP, Griitter MG. 1993. Refined crystal structure of human transforming growth factor-02 at 1.95 resolution. J Mol Biol231: 445-458.
Sibanda BL, Blundell TL, Thornton JM. 1989. Conformation of @-hairpins in protein structures. J Mol Biol206:759-717.
Suzuki A, Shioda N, Maeda T, Tada M, Ueno N. 1994. Cloning of an mouse TGF-P type I1 receptor gene. FEBS Let 355:19-22.
Takahashi T, Endo S, Nagayama K. 1993. Stabilization of protein crystals by electrostatic interactions as revealed by a numerical approach. JMol Biol234:421-432.
Vyas NK. 1991. Atomic features of protein-carbohydrate interactions. Curr Opin Struct Biol 1:132-740.
Wang XF, Lin HY, Ng-Eaton E, Downward J, Lodish HF, Weinberg RA.
ceptor. Cell 67:797-805. 1991. Expression cloning and characterization of the TGF-0 type I11 re-
Wlodawer A. 1984. Structure of bovine pancreatic trypsin inhibitor: Results of joint neutron and X-ray refinement of crystal form 11. JMol Biol180:
Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. 1994. Mechanism of activation of the TGF-0 receptor. Nature 370:341-347.
Yamashita H, ten Dijke P, Franzen P, Miyazono K, Heldin CH. 1994. For- mation of hetero-oligomeric complexes of type I and type 11 receptors for transforming growth factor-0. J Biol Chem 269:20172-20178.
301-329.