synthetic studies towards the synthesis …d-scholarship.pitt.edu/16753/1/buck-thesis.pdf32 1.3...
TRANSCRIPT

SYNTHETIC STUDIES TOWARDS THE SYNTHESIS OF ANATOXIN-A(S)
by
Eric E. Buck
B.S., University of Minnesota, 2006
Submitted to the Graduate Faculty of
The Kenneth P. Dietrich School of Arts and Sciences
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy
University of Pittsburgh
2012

ii
UNIVERSITY OF PITTSBURGH
The Kenneth P. Dietrich School of Arts and Sciences
This thesis was presented
by
Eric E. Buck
It was defended on
November 29th
, 2012
and approved by
Scott Nelson, Professor, Department of Chemistry
Kazunori Koide, Associate Professor, Department of Chemistry
Billy Day, Professor, Department of Pharmaceutical Science
Thesis Director: Peter Wipf, Distinguished University Professor, Department of Chemistry

iii
Copyright © by Eric E. Buck
2012

iv
Three routes toward the total synthesis of the neurotoxin anatoxin-a(s) have been explored. They
focus on the assembly of the backbone utilizing both Pd(0)- and Pd(II)-mediated processes. Our
current route leads to a late-stage precursor of anatoxin-a(s), hydroxy guanidine 134, in 18 steps
and in 8.0% overall yield utilizing an amidoalkylation step in the key cyclization.
SYNTHETIC STUDIES TOWARDS THE SYNTHESIS OF ANATOXIN-A(S)
Eric E. Buck, PhD
University of Pittsburgh, 2012

v
TABLE OF CONTENTS
1.0 SYNTHETIC STUDIES TOWARDS THE SYNTHESIS OF ANATOXIN-A(S) ......... 1
1.1 INTRODUCTION ................................................................................................................ 1
1.1.1 Cyanobacteria ................................................................................................... 1
1.1.2 Strategies for the formation of anatoxin-a(s)’s guanidine backbone:
Inspiration from related heterocycles. ....................................................................... 8
1.2 RESULTS AND DISCUSSION ......................................................................................... 20
1.2.1 Route 1: Palladium(0)-catalyzed diaminations ........................................... 21
1.2.2 Route 2: Palladium(II)-catalyzed amidocarbonylations ............................ 24
1.2.3 Route 3: Palladium(II)-catalyzed amidoalkylations ................................... 32
1.3 CONCLUSION ................................................................................................................... 50
2.0 EXPERIMENTAL ............................................................................................................. 51
3.0 REFERENCES ................................................................................................................... 90
APPENDIX A .............................................................................................................................. 94

vi
LIST OF TABLES
Table 1. Diamination of butadiene derivatives. ............................................................................ 18
Table 2. Conditions for the oxidation of silyl enol ether 72. ........................................................ 23
Table 3. Amidocarbonylation conditions. ..................................................................................... 26
Table 4. Conditions for attempted Curtius rearrangement of carboxylic acid 87. ........................ 30
Table 5. Conditions for attempted Hofmann rearrangement of amide 97. ................................... 32
Table 6. Screening conditions for key amidoalkylation of guanidine 99. .................................... 35
Table 7. Pd(II)-mediated cyclization of MOM-derivative 121. .................................................... 42
Table 8: Exploration of different bases to optimize phosphorylation........................................... 46
Table 9: Solvent screen under the phosphorylation conditions. ................................................... 47
Table 10: Exploring different phosphorylation conditions. .......................................................... 48

vii
LIST OF FIGURES
Figure 1. Structure of the cytotoxin aplysiatoxin (1). ..................................................................... 2
Figure 2. The structure of microcystin LR (2). ............................................................................... 3
Figure 3. The structure of nodularin (3). ......................................................................................... 3
Figure 4. Structure of cylindrospermopsin (4). ............................................................................... 4
Figure 5. Selected structures of neurotoxins produced by cyanobacteria. ...................................... 4
Figure 6: Structure identification of cyclization compounds and comparable urea compounds 81
and 82. ........................................................................................................................................... 27

viii
LIST OF SCHEMES
Scheme 1. Proposed mechanism of action of anatoxin-a(s). .......................................................... 6
Scheme 2. Biosynthesis of anatoxin-a(s) (8). ................................................................................. 7
Scheme 3: Synthesis of guanidine 15. ............................................................................................ 8
Scheme 4: Possible synthetic strategies for the synthesis of anatoxin-a(s). ................................... 9
Scheme 5: Two possible pathways for nucleopalladation. ............................................................. 9
Scheme 6. Synthesis of (+)-preussin. ............................................................................................ 10
Scheme 7. The synthesis of urea derivatives using the Pd2(dba)3/Xantphos catalyst system....... 11
Scheme 8: Intramolecular amination of alkenes through aminopalladation. ................................ 12
Scheme 9. Synthesis of (±)-alchorneine. ...................................................................................... 13
Scheme 10. Stereoselective formation of cyclic carbamates. ....................................................... 13
Scheme 11. Asymmetric synthesis of (-)-anatoxin-a. ................................................................... 14
Scheme 12. Synthesis of (+)-prosopinine. .................................................................................... 15
Scheme 13. Asymmetric intramolecular aminopalladation. ......................................................... 16
Scheme 14. Overman’s 2nd
generation catalysts. .......................................................................... 16
Scheme 15. Diamination of terminal olefins and proposed pathway. .......................................... 17
Scheme 16: Synthesis of bicyclic guanidine. ................................................................................ 18
Scheme 17. Shi's proposed mechanism for the diamination of olefins. ....................................... 19
Scheme 18. 1st Generation retrosynthetic analysis of anatoxin-a(s) (8). ...................................... 21

ix
Scheme 19. Attempted synthesis of silyl enol ether 68. ............................................................... 22
Scheme 20. Synthesis of TES silyl enol ether 72. ........................................................................ 23
Scheme 21. Ma's work on DMPU derivatives. ............................................................................. 24
Scheme 22. 2nd
Generation retrosynthetic analysis of anatoxin-a(s) (8). ..................................... 25
Scheme 23. Synthesis of allylic guanidine 78. ............................................................................. 25
Scheme 24. Proposed mechanism of amidocarbonylation. .......................................................... 28
Scheme 25. Synthesis of carboxylic acid 87 and proposed mechanism of ester isomerization. .. 29
Scheme 26. Radical decarboxylation of thiohydroxamate esters 94 and 95. ................................ 31
Scheme 27. Formation of amide 97. ............................................................................................. 31
Scheme 28. 3rd
Generation retrosynthetic analysis of anatoxin-a(s) (8). ...................................... 33
Scheme 29. Synthesis of thiourea 101. ......................................................................................... 33
Scheme 30. Synthesis of guanidine 99. ........................................................................................ 34
Scheme 31. Proposed catalytic cycle of amidoalkylation. ............................................................ 37
Scheme 32. Synthesis of alkyl azide 113. ..................................................................................... 38
Scheme 33. Synthesis of methyl carbamate 116. .......................................................................... 39
Scheme 34. Synthesis of N-hydroxy guanidine 9. ........................................................................ 40
Scheme 35. Improved synthesis of 99 and synthesis of MOM derivative 121............................. 41
Scheme 36: Trans-aminopalladation (AP) vs. cis-aminopalladation. .......................................... 43
Scheme 37. Synthesis of hydroxy guanidine 134. ........................................................................ 44
Scheme 38. A model system synthesis of 134 to explore phosphorylation conditions. ............... 45
Scheme 39: Attempted phosphorylation of 134............................................................................ 49

x
ACKNOWLEDGMENTS
I would like to first express my gratitude towards Dr. Wipf for providing me the opportunity to
work in his lab over the past several years. He has provided me with an environment that has
helped me develop into a better chemist, scientist, and person. I would be remiss if I did not
mention the wonderful molecule of the month competitions. The discussions and the freedom to
express my creativity during these events are some of my favorite memories of grad school. I
will never miss a β-leaving group no matter where my studies take me.
I would like to thank Dr. Nelson, Dr. Koide, and Dr. Day for being on my committee. I
have always enjoyed discussing not only my own chemistry during our various meetings but also
for their willingness to provide me with excellent feedback. I would like to give a special thanks
to Dr. Day for all the assistance he gave me on the BST3 LC-MS. That machine was invaluable
to my research. I am also very grateful to Dr. Floreancig. Even under the shortened timeline he
was always willing to discuss the chemistry and to provide extremely useful feedback. Thank
you.
I would like to take the time to acknowledge my fellow graduate students in the Wipf
group. I would like to thank Chenbo and Chad and later Nolan for providing some of the best
discussions as we worked on MOM. I could not thank Adam enough. Your role as a mentor and
friend cannot be adequately expressed in words. Last but not least I would like to thank Chris

xi
and Nate. For being there from day one. Kara you have my gratitude for willing to wait until
noon before badgering me.
Lastly, I need to thank my family for always being there. Thanks to my nephew who calls
me his friend Uncle Eric. You helped me more than you know little buddy.

xii
ABBREVIATIONS
Ac acetyl
AIBN 2,2’-azo bisisobutyronitrile
atm atmosphere
ATR Attenuated Total Reflectance
Boc tert-butoxycarbonyl
Bn benzyl
Bz benzoyl
Cbz benzyloxycarbonyl
DABCO 1,4-diazabicyclo[2.2.2]octane
dba dibenzylideneacetone
DCC dicyclohexylcarbodiimide
DCE 1,2-dichloroethane
DCM dichloromethane
DIAD diisopropyl azodicarboxylate
DIC diisopropyl carbodiimide
DIPA diisopropylamine
DIPEA diisopropylethylamine
DMAP N,N-4-dimethylaminopyridine

xiii
DMF N,N-dimethylformamide
DMPU N,N-dimethyl propylene urea
DMSO dimethylsulfoxide
DPPA diphenylphosphoryl azide
dr diastereomeric ratio
DSS 4,4-dimethyl-4-silapentane-1-sulfonic acid
EDA ethylenediamine
EDCI 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
ee enantiomeric excess
FDA Food and Drug Administration
Fmoc 9-fluorenylmethoxycarbonyl
HMPA hexamethylphosphoramide
IBBO (+)-2,2’-isopropylidenebis[(4R)-4-benzyl-2-
oxazoline)]
LD50 lethal dose required to kill 50% of the population
LDA lithium diisopropylamine
m-CPBA meta-chloroperbenzoic acid
MOM methoxymethyl
Ms methanesulfonyl
MVK methyl vinyl ketone
ND not determined
NMM N-methylmorpholine
NMP N-methylproline

xiv
NR no reaction
Ph phenyl
phthNH phthalimide
phthNK potassium phthalimide
PIDA phenyliodonium diacetate
PIFA phenyliodonium bis(trifluoroacetate)
PNS peripheral nervous system
Pyr pyridine
TBAB tetra-n-butylammonium bromide
TBAF tetra-n-butylammonium fluoride
TBDPS tert-butyldiphenylsilyl
TEA triethylamine
TES triethylsilyl
Tf trifluoromethanesulfonyl
TFA trifluoroacetic acid
TFAA trifluoroacetic anhydride
THF tetrahydrofuran
THP 2-tetrahydropyranyl
TMS trimethylsilyl
Ts p-toluenesulfonyl

1
1.0 SYNTHETIC STUDIES TOWARDS THE SYNTHESIS OF ANATOXIN-A(S)
1.1 INTRODUCTION
1.1.1 Cyanobacteria
Cyanobacteria, or blue-green algae, are prokaryotic microorganisms predominately found in
ponds, lakes, rivers and oceanic waters. Fossil records date these photosynthetic microorganisms
as far back as 2.15 billion years ago, and their ability to produce oxygen was a large factor in
turning earth’s early atmosphere into an oxygen-rich system.1 A rapid growth of cyanobacteria
can occur when large amounts of sunlight and food rich in nitrogen and phosphorus are present.
This rapid growth is referred to as an algal bacterial bloom and is often detrimental to
surrounding plant, animal, and human life.2,3
One example of a harmful algal bloom occurred in
1993 when a sudden increase in the salinity of several freshwater bodies of water in Florida, due
to a tidal surge, caused a dramatic decrease in local plant life.4 In the past 20 years,
cyanobacteria have emerged as interesting sources of biologically active secondary metabolites
that are commonly referred to as cyanotoxins. These cyanotoxins are classified into two groups
according to their modes of toxicity: cytotoxins that are assayed using cultured mammalian cells,
and highly lethal biotoxins that are assayed using small animals.3

2
While the cytotoxins are typically less deadly than the biotoxins, they are known for their
antialgal, antimycotic, and antibacterial properties. Cytotoxins also exhibit modest antitumor
activity.3 The presence of blue-green algae of the genus Lyngbya majuscula, which produces the
protein kinase C activator aplysiatoxin (1, Figure 1), on the surface of the red alga G.
coronopifolia was eventually determined to be the cause of food poisoning and dermatitis in
Hawaii in 1994 (Figure 1).4,5
Figure 1. Structure of the cytotoxin aplysiatoxin (1).
Biotoxins are highly lethal metabolites that are further divided into hepatotoxins and
neurotoxins. Hepatotoxins, or molecules that target the liver, include three common types:
microcystins, nodularins, and cylindrospermopsins. Microcystins are identified by their
macrocyclic heptapeptide backbone. The presence of five non-proteinogenic amino acids is
characteristic to microcystins, while the two proteinogenic amino acids are varied throughout the
microcystin family. Microcystin LR (microcystin leucine arginine) (2, Figure 2) is the most
lethal and abundant hepatotoxin with an LD50 of 25-125 g/Kg (mice). This natural product was
responsible for 50 fatal human poisonings at a hemodialysis center in Brazil.6 Its mechanism of
action consists of blocking protein phosphatases, leading to hemorrhaging of the liver.3
Another family of hepatotoxins, the nodularins, resembles the microcystins structurally
and has a characteristic pentapeptide macrocycle. These secondary metabolites are inhibitors of

3
serine/threonine protein phosphatase 1 and 2A, and this inhibition will lead to hemorrhaging.6
Nodularin (2, Figure 3), the parent compound in this family, possesses an LD50 of 50 – 70 g/Kg
(mice).7
Figure 2. The structure of microcystin LR (2).
Figure 3. The structure of nodularin (3).
Cylindrospermopsin (4, Figure 4), isolated from C. raciborskii, is a unique member of the
hepatotoxin family due to its lack of a macrocyclic peptide backbone. In 1979, it was
responsible for the hospitalization of 140 children on Palm Island, Queensland, Australia, and it
can also be found in Europe and America. Cylindrospermopsin exhibits an LD50 of 2.1 mg/Kg
(mice) and its mechanism of action is to block protein synthesis, leading to kidney and liver
failure. Recently, there is new evidence suggesting that protein synthesis inhibition is not the
only mechanism responsible for its toxicity.8,9

4
Figure 4. Structure of cylindrospermopsin (4).
In addition to producing cytotoxins, cyanobacteria produce several neurotoxins, in
particular anatoxin-a (5) and homoanatoxin-a (6), the saxitoxins (7), and the organophosphate
anatoxin-a(s) (8, Figure 5). Anatoxin-a, produced by several strains of Anabaena, is a neurotoxic
alkaloid with an LD50 of 200 – 250 g/Kg (mice). It binds to nicotinic receptors, leading to
respiratory failure within a few hours.2,6
As a result, 5 is commonly known as “very fast death
factor”. To date, there are several reported total syntheses of anatoxin-a and its structural analog
homoanatoxin-a, with the most recent being Stockman’s approach published in 2008.10
Homoanatoxin-a, isolated from Oscillatoria formosa, has a similar mechanism of action as
anatoxin-a.2
Figure 5. Selected structures of neurotoxins produced by cyanobacteria.

5
The saxitoxins, also known as paralytic shellfish poisons, block voltage gated sodium
channels, leading to neuromuscular paralysis and death by respiratory failure. With an LD50 of
10 g/Kg in mice, saxitoxin (7) is the most lethal of the saxitoxins and has been linked to a
number of sheep deaths in Australia.2,6
Anatoxin-a(s) (8) is an organophosphate isolated from Anabeana flos-aque in North
America, A. lemmermannii in Denmark, and Anabeana crassa in South America. In North
America, it has been linked to the deaths of dogs, birds, and swine; in Denmark it has been
associated with the death of wild birds.2,11,12
Anatoxin-a(s) was also detected in the Faxinal
Reservior (the main reservoir for the Caxias do Dul, Brazil), which commonly experiences
Anabeana crassa blooms. To date there are no reports of neurotoxicity caused by anatoxin-a(s)
in Brazil.13
It is a strong anticholinesterase (LD50 of 20 - 40 g/Kg in mice) that acts only in the
peripheral nervous system (PNS) and has a similar mechanism of action as the insecticide
paraoxon and the chemical weapon sarin.14-16
It is proposed that anatoxin-a(s) irreversibly
phosphorylates a serine residue in the active site of acetylcholinesterase. As shown in Scheme 1,
the hydroxyl functionality of an acetylcholinesterase serine residue adding into the phosphate
moiety of anatoxin-a(s), which will then lead to the release of hydroxy guanidine 9. One could
envision activation of the guanidine functionality could occur prior to serine hydroxyl addition to
help promote phosphorylation. It is this irreversible phosphorylation which leads to the build up
of acetylcholine in the synaptic cleft.17
This accumulation of acetylcholine leads to
neuromuscular paralysis and eventually death by respiratory failure. Symptoms of anatoxin-a(s)
poisoning include muscle weakness, respiratory distress, rhinorrhea, and eventually convulsions
followed by death.2

6
Scheme 1. Proposed mechanism of action of anatoxin-a(s).
In 1992, Moore proposed a biosynthesis of anatoxin-a(s) (Scheme 2). An arginine
molecule undergoes a stereoselective oxidation at the -position (10) followed by a nucleophilic
displacement by the guanidine functionality to form cyclic guanidine 11. Activation of the
hydroxy group to promote displacement probably occurs prior to nucleophilic displacement;
however, this exact mechanism is unknown at this time. Another methylene oxidation at the -
position (12) precedes a possible retro-aldol mechanism that releases a unit of glycine (13) and
aldehyde 14. Feeding experiments with two deuterium atoms incorporated at the C-3 position
and their subsequent replacement, gives some support to the proposed oxidation and retro-aldol
mechanism. However, by this proposed mechanism there should be a deuterium atom attached
to the aldehyde. The mechanism by which the dimethylamine portion is added to intermediate
14 to give 15 remains unknown. The proposed mechanism by Moore, involving a reductive
amination and dimethylation pathway, does not explain the loss of the remaining deuterium atom
that was observed during the feeding experiments. With the backbone framework in place,
tertiary amine 15 undergoes another selective oxidation (9) followed by phosphorylation,
probably by an enzymatic-mediated pathway, of the resulting hydroxyguanidine to provide
anatoxin-a(s) (8).18-21
To date, there is no information provided on the specific enzymes
involved in the biosynthesis of anatoxin-a(s).

7
Scheme 2. Biosynthesis of anatoxin-a(s) (8).
To determine the absolute stereochemistry of anatoxin-a(s), Moore prepared guanidine 15
(and its enantiomer) from L-asparagine (or D-asparagine, respectively) (Scheme 3). First,
carboxylic acid 16 was synthesized in two-steps from N-benzyloxycarbonyl-L-asparagine.22
Treatment of 16 with N-hydroxysuccinimide in the presence of DCC formed the N-
hydroxysuccinimide ester, which upon exposure to dimethyl amine gave amide 17. Next,
removal of the Boc group with TFA followed by Cbz deprotection with Pd/C and H2 gave the
corresponding diamine. Reduction of the amide with borane gave 18. Formation of the
guanidine 19 was accomplished by the treatment of 18 with S,S-dimethyl-N-
tosyliminodithiocarbonimidate in refluxing ethanol, which after removal of the toluenesulfonyl

8
group with HBr, gave guanidine 15. The circular dichroism (CD) spectrum of 15 matched the
CD spectrum of 15 that was generated from a natural source of anatoxin-a(s).
Scheme 3: Synthesis of guanidine 15.
1.1.2 Strategies for the formation of anatoxin-a(s)’s guanidine backbone: Inspiration
from related heterocycles.
The formation of nitrogen containing five-membered rings and related heterocycles can
be accomplished through metal-mediated methods, which provide an opportunity to control the
regioselectivity of the carbon-nitrogen bond forming event as well as the potential for reagent
controlled stereoselectivity.23
The challenge to synthesizing anatoxin-a(s) comes from the N-O
bond to be attached to the lone stereocenter present in the molecule. Two distinct strategies were
envisioned: formation of the carbon-nitrogen bond through a pre-tethered olefin or through a
diamination sequence (Scheme 4). For developing a synthetic strategy towards anatoxin-a(s), we
turned to the use of aminopalladation methods that have been developed for the construction of
guanidines, pyrrolidines, and ureas. A second strategy for the construction of 1,2-diamino
containing compounds is the diamination of olefins.

9
Scheme 4: Possible synthetic strategies for the synthesis of anatoxin-a(s).
One could envision two possible pathways for the amination of unactivated olefins
through palladium catalysis. The first pathway consists of cis-nucleopalladiation, or the
formation of a palladium-nitrogen complex (24), prior to the cyclization event (Scheme 5). The
second pathway involves the precoordination of palladium to the olefin (26), thereby activating it
towards nucleophilic addition and is generally referred to as trans-nucleopalladation. This
possibility of two different mechanistic pathways makes the development of an enantioselective
process difficult to achieve. Also, when applied to the construction of guanidines through a
similar transformation, the regioselectivity of the catalyst between the two available nitrogens
will play a key role in the outcome of the cyclization.
Scheme 5: Two possible pathways for nucleopalladation.

10
One strategy for the construction of nitrogen-containing heterocycles is through
palladium-catalyzed intramolecular ring closure of olefin containing aliphatic amines or related
nitrogen functionality. The Wolfe group in 2006 used this strategy for the synthesis of the natural
product (+)-preussin (Scheme 6).24
Treatment of amino-olefin 28 with phenylbromide in the
presence of the Pd(OAc)2/DPEphos catalyst system generated pyrrolidine 29 in 62% yield. A
likely mechanism involves oxidative insertion into the aryl bromide bond by Pd(0) followed by
formation of the aminopalladium complex. This complex then proceeds through a cis-
nucleopalladation pathway to give 29, after reductive coupling. This cyclization added the
flexibility to install various aryl groups at a late stage in order to probe the biological activity of
different (+)-preussin analogs. Pyrrolidine 29 was subjected to a one-pot reduction and
deprotection to afford (+)-preussin in 95% yield (96% ee).
Scheme 6. Synthesis of (+)-preussin.
The Wolfe group expanded on their pyrrolidine formation by optimizing the previous
reaction conditions for use in the construction of cyclic ureas.25
They treated arylbromide 30
with Pd2(dba)3/Xantphos, which undergoes oxidative insertion of the arylbromide bond (Scheme
7). The resulting palladium complex undergoes amidopalladation of allylic urea 31, followed by
nitrogen alkylation, and the formation of alkyl-palladium complex 32. This complex then
undergoes a reductive elimination to generate cyclic urea 33 in 88% yield and a 12:1 dr. The use
of base is necessary to promote amidopalladation. In 2012, they reported an enantioselective

11
variant of this methodology using the diastereomeric phosphoramidite ligand, (S)-Siphos-PE to
give 35 in 81% yield (92% ee).26
These examples by the Wolfe group all report a mechanism
that proceeds through cis-nucleopalladation mechanism and the formation of that complex is
facilitated by the presence of base.
Scheme 7. The synthesis of urea derivatives using the Pd2(dba)3/Xantphos catalyst system.
In 2011, the Stahl group reported their mechanistic studies of Wacker-type intramolecular
amination of alkenes (Scheme 8).27
Treatment of tosylamide 36 with Pd(OAc)2 in the presence
of pyridine produce pyrrolidine 38 in 87% yield. During the course of their mechanistic studies,
they observed that the rate of the reaction did not depend on the concentration of O2 but did show
a dependence on the concentrations of palladium catalyst and substrate. This observation
provided evidence toward a cis-aminopalladation pathway by which the mechanism would
proceed through likely intermediate 37. The Stahl group followed this work with a report in
2012 on an enantioselective variation of this reaction. Using the chiral pyridine oxazoline ligand

12
(S)-41 and Pd(TFA)2 they were able to form pyrrolidine 40 in 93% yield and 98% ee.
Scheme 8: Intramolecular amination of alkenes through aminopalladation.
Interestingly, they reported a low yield and almost a complete loss of enantioselectivity when
using Pd(OAc)2. They attributed this observation to the acetate ligands promoting a change in
the mechanistic pathway from a cis-nucleopalladation sequence to a trans-nucleopalladation
pathway. This is one of the few examples indicating the influence of the anionic ligands on the
mechanistic pathway of this type of cyclization.
A variation of the nucleopalladation of olefins is through the activation of the olefin by
the presence of a leaving group (typically carbonates) in the -position of the olefin. The Büchi
group reported in 1989 the use of this strategy in the synthesis of (±)-alchorneine and (±)-
isoalchorneine (Scheme 9).28
First, methoxy guanidine 42 was subjected to a Pd(0)-mediated
annulation that could proceed through either a -allyl Pd(II) intermediate or a cis-
amidopalladation complex to generate methoxy cyclic guanidine 43 in 81% yield. Methoxy

13
cyclic guanidine 43 was treated with two equivalents of PdCl2(CH3CN)2 in CH2Cl2 at 40 C to
give (±)-alchorneine in 46% yield.
Scheme 9. Synthesis of (±)-alchorneine.
In 1998, an enatioselective version of this strategy was applied by Trost as a method for
the desymmetrization of meso-alcohols. Treatment of a solution of allylic diol 44 and TsNCO in
THF with Pd2(dba)3/L-45 led to oxazolidin-2-one 48 in 84% yield and 99% ee (Scheme 10).29
A
significant enhancement of enantiomeric enrichment was observed upon the addition of an
external base, which has been shown to facilitate formation of the amidopalladium complex.
Scheme 10. Stereoselective formation of cyclic carbamates.

14
The reaction likely proceeds through dicarbamate 46, followed by formation of a cis-
amidopalladation complex (47). The regioselectivity of the cis-amidopalladation complex is
controlled through the use of chiral ligand L-45. The carbamate then cyclizes and, after
elimination of the carbamate, results in the stereoselective formation of oxazolidinone 48.
Another possibility would be the formation of a -allyl intermediate as the first-step, followed by
the formation a cis-amidopalladation complex. This complex would then undergo cyclization to
give 48.
In 1999, the Trost group applied this strategy to the asymmetric synthesis of the
cyanotoxin (–)-anatoxin-a by utilizing a similar -allyl Pd(II) complex to promote cyclization
(Scheme 11).30
Their end game strategy for the synthesis of (–)-anatoxin-a involved treatment of
allylic carbonate 49 with (dba)3Pd2CHCl3/L-51 to give [2.4.1] bicyclic amine 50 in 90% yield
and 88% ee. Pyrrolidine derivative 50 was then transformed into (-)-anatoxin-a.
Scheme 11. Asymmetric synthesis of (-)-anatoxin-a.
The Hirai group reported in 1997 the synthesis of (+)-prosopinine and (+)-palustrine
utilizing a key Pd(II)-mediated cyclization of urethanes.31
Urethane 52 was treated with
PdCl2(CH3CN)2 in THF to give fused piperdine 54 in 77% yield as a single diastereomer
(Scheme 12). The authors propose Pd(II) coordinates to the olefin (53), thereby activating it
towards nucleophilic addition. The oxygen of the methoxymethylether assists in the

15
organization of the transition state through the chelation of Pd(II). This is an example of a trans-
aminopalladation pathway, although it should be noted the authors do not provide any
experimental evidence for this pathway. Although the Hirai group used a similar strategy to
promote cyclization as the Trost group, the ability of these cyclizations to proceed through
different mechanistic pathways increases the difficulty of developing enantioselective conditions
for this cyclization. This difficulty will be compounded in the synthesis of guanidines due to the
presence of a second nitrogen that can participate in coordination with the metal catalyst.
Scheme 12. Synthesis of (+)-prosopinine.
In 2002, the Overman group developed a set of chiral Pd(II) catalysts to induce
asymmetric annulation of allylic ureas in the synthesis of oxazolines.32
They treated allylic urea
55 with 5 mol% of FOP-OTFA in 1:1 CH2Cl2/CH3NO2 to give urea 56 in 96% yield and 90% ee
(Scheme 13). FOP-OTFA is produced in situ by treating the corresponding Pd-I complex with
AgOTFA. Although a variety of silver salts were investigated, the best result was achieved with
silver trifluoroacetate. In 2005, a 2nd
generation catalyst was developed that replaced the
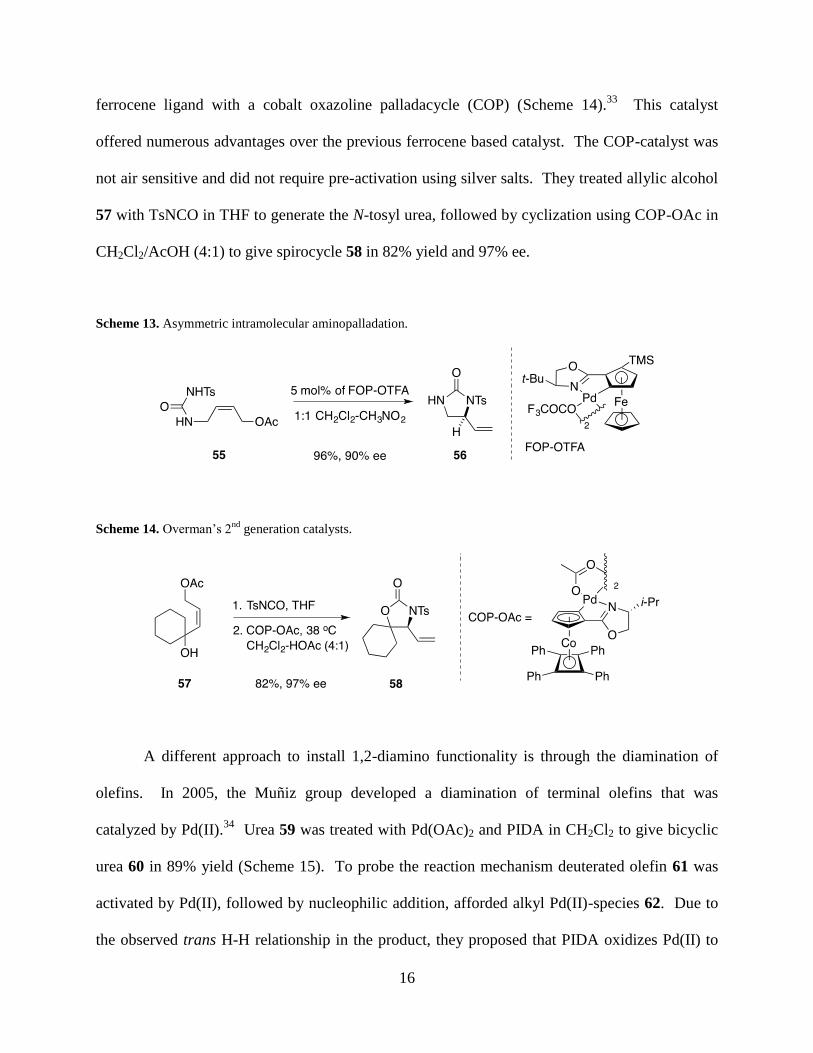
16
ferrocene ligand with a cobalt oxazoline palladacycle (COP) (Scheme 14).33
This catalyst
offered numerous advantages over the previous ferrocene based catalyst. The COP-catalyst was
not air sensitive and did not require pre-activation using silver salts. They treated allylic alcohol
57 with TsNCO in THF to generate the N-tosyl urea, followed by cyclization using COP-OAc in
CH2Cl2/AcOH (4:1) to give spirocycle 58 in 82% yield and 97% ee.
Scheme 13. Asymmetric intramolecular aminopalladation.
Scheme 14. Overman’s 2nd
generation catalysts.
A different approach to install 1,2-diamino functionality is through the diamination of
olefins. In 2005, the Muñiz group developed a diamination of terminal olefins that was
catalyzed by Pd(II).34
Urea 59 was treated with Pd(OAc)2 and PIDA in CH2Cl2 to give bicyclic
urea 60 in 89% yield (Scheme 15). To probe the reaction mechanism deuterated olefin 61 was
activated by Pd(II), followed by nucleophilic addition, afforded alkyl Pd(II)-species 62. Due to
the observed trans H-H relationship in the product, they proposed that PIDA oxidizes Pd(II) to

17
Pd(IV), and subsequent nucleophilic addition of the remaining secondary nitrogen with
concomitant loss of Pd(II) gives diamination product 63 as a single diastereomer.
Scheme 15. Diamination of terminal olefins and proposed pathway.
Muñiz followed their work on the cyclization of ureas by applying a similar strategy to
the synthesis of bicyclic guanidines in 2008.35
In their communication, they treated guanidine 64
with Pd(OAc)2 in the presence of Copper bromide to give bicyclic guanidine 65 in 99% yield
(Scheme 16). They did note that under these reaction conditions it was possible to isolate
substantial quantities of the related urea compound. Switching to copper chloride eventually
solved that issue. Also, the authors specifically only examined examples in which the two
nitrogens that had potential to cyclization first had the same substitution to avoid issues with
regioselectivity. There are few examples in the literature that address this issue of
regioselectivity in guanidine synthesis.

18
Scheme 16: Synthesis of bicyclic guanidine.
In 2007, the Shi group published a Pd(0)-catalyzed diamination of diene and trienes.36
The various dienes combined with di-tert-butyldiaziridinone (66) were treated with 10 mol% of
Pd(PPh3)4 in benzene at reflux to provide cyclic ureas in moderate to excellent yields (Table 1).
Both electron-rich and conjugated dienes were well tolerated and electron-deficient dienes gave
Table 1. Diamination of butadiene derivatives.

19
the desired cyclic ureas, albeit in a modest yield compared to electron rich dienes. When
conjugated trienes were used, the reaction took place at the central olefin selectively. One
noteworthy example involves the use of 2-trimethysilyloxybutadiene (67) to give the only
example of a monosubstituted cyclic urea in excellent yield (68, Table 1). The authors
proposethat the reaction first involves Pd(0) undergoing an oxidative insertion into the N-N bond
of di-tert-butyldiaziridinone followed by coordination of the diene to Pd(II) (Scheme 17). Next,
stepwise carbon-nitrogen bond formation generates the -allyl Pd(II) intermediate followed by
reductive elimination to regenerate Pd(0) and provide the product. Once formed these urea
compounds can be cleaved to reveal the diamino compounds that can be reclosed to form
guanidines.
Scheme 17. Shi's proposed mechanism for the diamination of olefins.

20
We wanted to explore two distinct pathways for the construction of anatoxin-a(s)’s
guanidine core. One pathway is through a nucleopalladation pathway that can cyclize onto
activated or unactived olefins. The challenges associated with the development of an
enantioselctive method for this cyclization include controlling a cis- or trans-nucleopalladation
pathway. Also, for this strategy to be successful for the synthesis of anatoxin-a(s), a
regioselective cyclization must be developed that can differentiate two distinct nitrogens. These
same challenges are also present in proceeding through a diamination strategy.
1.2 RESULTS AND DISCUSSION
Anatoxin-a(s) (8) is a structurally interesting organophosphate that contains a pentacyclic
guanidine moiety with a N-O bond attached to the only stereocenter. This functionality provides
a unique environment to expand on current metal-catalyzed carbon-nitrogen bond forming
methodology. Current methods focus on the formation of pyrrolidines and cyclic ureas through
various metal-catalyzed processes. However, there is a general lack of methodology for the
formation of cyclic guanidine rings with the most notable being the seminal work by Büchi28
and
the work of the Muñiz group on the Pd(II)-catalyzed formation of bicyclic guanidines,37
on
metal-catalyzed amidocarbonylations and amidoalkylations involving acyclic guanidine
precursors. The methodology developed by the Muñiz group relies on identical guanidine
nitrogens and does not address the regiochemistry in the cyclization. The ability to differentiate
between two electronically different nitrogens still remains a synthetic challenge. Also the
challenge of proceeding through a cis- or trans-nucleopalladation is a concern toward
establishing an enantioselective ring cyclization. Shi’s recently developed methodology for the

21
diamination of butadiene derivatives to give pentacyclic ureas would provide a facile route to
assemble the anatoxin-a(s) backbone.36
1.2.1 Route 1: Palladium(0)-catalyzed diaminations
The initial synthetic efforts towards the total synthesis of the cyanotoxin anatoxin-a(s) (8)
began by utilizing the diamination work developed by Shi (Table 1, vide supra). A
retrosynthetic analysis of anatoxin-a(s) resulted in silyl enol ether 68. Silyl enol ether 68 is
synthesized from di-tert-butyl-diaziridinone 66 and 2-trimethylsilyloxybutadiene 67 using Pd(0)
catalysis (Scheme 18).
Scheme 18. 1st Generation retrosynthetic analysis of anatoxin-a(s) (8).
To apply Shi’s diamination protocol to a synthesis of anatoxin-a(s), methyl vinyl ketone
was treated with TMSCl and triethylamine to give 2-trimethylsilyloxybutadiene 67 in 48% yield
(Scheme 19). Di-tert-butyl-diaziridinone 66 was synthesized from di-tert-butylurea according to
the procedure reported by Shi.38
2-Trimethylsilyloxybutadiene 67 and di-tert-butyldiaziridinone
66 were subjected to catalytic Pd(0) in benzene at 65 °C to give silyl enol ether 68. However,

22
Scheme 19. Attempted synthesis of silyl enol ether 68.
attempts at isolation of silyl enol ether 68 only resulted in desilylated ketone 70. To counteract
the instability of silyl enol ether 68, it was carried forward without purification. When subjected
to standard ozonolysis conditions, only decomposition of silyl enol ether 68 was observed.
Surmising that the difficulty with the subsequent ozonolysis was due to the inherent
instability of the TMS silyl enol ether, the more stable triethyl silyl enol ether was selected.
Methyl vinyl ketone in THF was subjected to a solution of LDA/ HMPA at -78 °C followed by
silylation of the enolate with TESCl and warming to 0 °C to provide 2-triethylsilyloxybutadiene
71 in 85% yield (Scheme 20). Next, a solution of 2-triethylsilyloxybutadiene 71 and di-tert-
butyldiaziridinone 66 in toluene or THF at 65 °C was treated with catalytic Pd(PPh3)4 to provide
TES enol ether 72 in 40-67% yield. With TES enol ether 72 in hand, conditions towards the
installation of the required nitrogen functionality were explored (Table 2). Ozone was bubbled
through a solution of TES enol ether 72 in CH2Cl2/CH3OH at -78 C but after treatment with

23
Scheme 20. Synthesis of TES silyl enol ether 72.
Me2S only decomposition of the starting material was observed. Treatment of 72 with aqueous
OsO4 in the presence or absence of NaIO4 in THF did not result in any dihydroxylated product or
carboxylic acid 69. Treatment of 72 with buffered m-CPBA in CH2Cl2 followed by the addition
of 1 M HCl in CH3OH gave -hydroxy ketone 73 in 5% yield (Entry 4, Table 2).39
In light of
the difficulties pertaining to the oxidation of silyl enol ether 72 and the modest yield of the
diamination reaction, an alternative route was chosen in which the backbone of anatoxin-a(s)
would be assembled through a key Pd(II) catalyzed amidocarbonylation reaction.
Table 2. Conditions for the oxidation of silyl enol ether 72.

24
1.2.2 Route 2: Palladium(II)-catalyzed amidocarbonylations
Previous work in the Wipf group involving amidocarbonylation reactions catalyzed by
Pd(II) was pursued by Gil Ma (Scheme 21).40
Working towards the development of new cyclic
urea derivatives as alternatives to DMPU, 4-bromo-1-butene was treated with neat methylamine
to give N-methyl-3-butenylamine. The resulting homoallyic amine was treated with phosgene to
give the corresponding chloroamidate, which was trapped with methylamine in the presence of
diisopropylethylamine to provide urea 74 in 66% yield. Urea 74 was subjected to PdCl2 (10 mol
%) and CuCl2 (3 equiv) in CH3OH under 1 atm of CO to generate alkyl-Pd(II) species 75,
followed by CO insertion, and trapping of the acyl Pd(II) intermediate with CH3OH to afford
methyl ester 76 in 89% yield. By revising our previous retrosynthetic scheme to utilize a similar
amidocarbonylation, one can envision that anatoxin-a(s) can be synthesized from methyl ester 77
(Scheme 22). Methyl ester 77 is the result of a Pd(II)-catalyzed amidocarbonylation of allylic
guanidine 78.
Scheme 21. Ma's work on DMPU derivatives.40

25
Scheme 22. 2nd
Generation retrosynthetic analysis of anatoxin-a(s) (8).
The synthesis of allylic guanidine 78 commenced with the treatment of allylamine with
CbzNCS41
to give thiourea 79 in 99% yield (Scheme 23). The electron-withdrawing nature of
the Cbz protecting group facilitated the formation of the guanidine functionality by increasing
the electrophilic nature of the thiourea. Treatment of thiourea 79 and H2NOBn•HCl with EDCI
gave allylic guanidine 78 in 76% yield. The use of H2NOBn•HCl serves three purposes: 1) The
strong electron donating nature of the amine facilitates guanidine formation, 2) to differentiate
the electronics of the two nitrogens for the key amidocarbonylation step, and 3) to install the
required N-O bond that is present in the natural product.
Scheme 23. Synthesis of allylic guanidine 78.
With access to guanidine 78, conditions for the key amidocarbonylation transformation
catalyzed by Pd(II) were investigated. Two requirements for this reaction were considered: the
regiochemistry had to be controlled and ligands for the development of an asymmetric variant

26
needed to be screened. Allyl guanidine 78 was treated with 10 mol% of Pd(OAc)2 and CuCl2 (3
equiv) in CH3OH under 1 atm of CO to give a 2.2:1 mixture of methyl esters 77 and 80 in a
combined 76% yield (Entry 1, Table 3). Other Pd(II) complexes gave complex mixtures of
Table 3. Amidocarbonylation conditions.
products that were not isolated. The addition of chiral ligands or other additives to affect the
isomeric ratio only succeeded in increasing the ratio of undesired regioisomer 77 without any
significant improvement in ee. Compounds 77 and 80 were assigned based on the chemical shift
of the C2-H of the Boc-derivatives 77-Boc and 80-Boc. By comparing the chemical shift of the
C2-H of 77-Boc ( 3.92-3.82) and C2-H of 80-Boc ( 4.49) and of comparable urea compounds
8142
and 82.43
Although the guanidine and urea compounds share groups with similar electronics

27
attached to the nitrogen adjacent to the stereocenter, the electronics between the guanidine
compounds and the ureas are different. This comparison was used as a preliminary structure
assignment while the synthesis was carried forward. Ideally a crystal structure would have been
obtained to unambiguously assign the structures but no crystal of these urea compounds or
derivatives were obtained.
Figure 6: Structure identification of cyclization compounds and comparable urea compounds 81 and 82.
The amidocarbonylation is thought to proceed through a Wacker-type cyclization
pathway outlined below (Scheme 24).44
First, Pd(II) may coordinate to olefin 83, thereby
activating it towards nucleophilic addition, or, alternatively, Pd(II) forms N-Pd(II)amido species
84 with concomitant loss of HX. Next, amidoalkylation forms alkyl-Pd(II) species 85, followed
by CO insertion to give acyl-Pd(II) species 86. Subsequent reductive elimination forms methyl
ester 77 and Pd(0), which is reoxidized by CuCl2 to Pd(II). Due to the prevalence of the
undesired methyl ester, by addition of the weaker nucleophile, it is thought that the reaction
proceeds through amido-Pd(II) species 83 and not by simple Lewis activation of the olefin 84.

28
Scheme 24. Proposed mechanism of amidocarbonylation.
Despite the preliminary results of the amidocarbonylation giving the undesired
regioisomer, it was decided to proceed with the hydrolysis of the methyl esters; however, when a
solution of methyl esters 77 and 80 were subjected to KOTMS in THF, only carboxylic acid 87
was observed in 55 – 69% yield (Scheme 25). Initially, KOTMS deprotonates to the methyl
ester of regioisomer 80, followed by ring opening to form the ,-unsaturated ester 89. The ring
opening of ester 80 is facilitated by the electron withdrawing nature of the Cbz-protecting group.
At this juncture, the negative charge is delocalized between N1 (89) and N2 (90). N2 then adds
back into the --unsaturated ester to give the desired methyl ester 77. Calculations show that
77 is the thermodynamically more stable compound by 5.3 Kcal/mol.* The isomerization is
followed by hydrolysis.
* Calculations preformed using MMFF conformer distribution followed by PM3 equilibration of geometry using Spartan 10

29
Scheme 25. Synthesis of carboxylic acid 87 and proposed mechanism of ester isomerization.
With a route to carboxylic acid 87, conditions to install the required nitrogen
functionality by a Curtius rearrangement were explored. 45
Exposing of carboxylic acid 87 to
DPPA and triethylamine in tert-BuOH at reflux resulted in decomposition of starting material
(Entry 1, Table 4). In a more stepwise approach towards formation of the acyl azide, carboxylic
acid 87 was treated with oxalyl chloride followed by the addition of sodium azide. To the
resulting solution was added tert-BuOH and the reaction mixture was heated at reflux; however,
only decomposition of starting material was observed (Entry 2, Table 4). In 2005, the Lebel
group reported a mild one-pot procedure for the synthesis of Boc-protected amines by treating
carboxylic acids with sodium azide, Boc2O, and TBAB in the presence of Zn(OTf)2 via a Curtius
rearrangement.46
Unfortunately, when carboxylic acid 87 was exposed to these conditions, no
reaction of the starting material was observed (Entry 3, Table 4). Due to the unforeseen
difficulty in accomplishing the Curtius rearrangement, an alternative method for the installation
of the nitrogen functionality on the side chain was examined.

30
Table 4. Conditions for attempted Curtius rearrangement of carboxylic acid 87.
In 2008, the Renaud group reported the synthesis of alkyl azides using the radical
decarboxylation of thiohydroxamate esters.47
These conditions were applied to the coupling of
N-hydroxydithiocarbamates 92 and 93 with carboxylic acid 87 in the presence of DCC and
DMAP. The corresponding thiohydroxamate esters 94 and 95 were produced in 76% and 56%
yield, respectively (Scheme 26). Unfortunately, exposing thiohydroxamate esters 94 and 95 to
AIBN in benzene at reflux in the presence of phenyl sulfonylazide slowly decomposed the
thiohydroxamate esters and failed to give desired alkyl azide 96. A possible explanation is that
upon the generation of the primary radical, a 1,2-H migration occurs to produce a more stable
tertiary radical or a ring-opening event occurs to produce an N-centered radical that subsequently
decomposes.

31
Scheme 26. Radical decarboxylation of thiohydroxamate esters 94 and 95.
After the radical decarboxylation failed to provide alkyl azide 96, a Hofmann
rearrangement was explored for the installment of the required nitrogen functionality.48,49
A
mixture of methyl esters 77 and 80 in CH3OH at -78 C was treated with liquid NH3 to give
amide 97 in 64% yield (Scheme 27). In the presence of 5 equiv of Pb(OAc)4 and triethylamine
in tert-BuOH/ DMF at 100 C none of desired rearranged Boc-protected amine 97 was observed
(Table 5). Switching Pb(OAc)4 to PIFA only resulted in decomposition of starting material.
Scheme 27. Formation of amide 97.

32
Examination of the less active PIDA led to similar results (Entry 3, Table 5). Due to the
troubling preliminary results of the key amidocarbonylation cyclization and the difficulty of
installing the required nitrogen functionality, an alternative route that utilized a Pd(II)-mediated
amidoalkylation was pursued.
Table 5. Conditions for attempted Hofmann rearrangement of amide 97.
1.2.3 Route 3: Palladium(II)-catalyzed amidoalkylations
Our previous retrosynthetic analysis was revised toward an approach to anatoxin-a(s) from olefin
98. Olefin 98 can be prepared by a Pd(II)-catalyzed amidoalkylation of allylic guanidine 99
(Scheme 28). Mono protection of commercially available 2-butenediol using TBDPSCl in the

33
Scheme 28. 3rd
Generation retrosynthetic analysis of anatoxin-a(s) (8).
presence of imidazole gave (Z)-4-(tert-butyldiphenylsilyloxy)but-2-en-1-ol in 94% yield
(Scheme 29).50
Next, bromide displacement of the free alcohol with Br2, PPh3 and imidazole in
THF afforded the allylic bromide in 91% yield.51
The allylic bromide was displaced with
PhthNK in DMF at 80 C to provide phthalamide 100 in 75% yield. Phthalimide 100 was
carried through a two-step procedure of phthalimide removal with hydrazine in EtOH at reflux
and trapping the resulting allylic amine with CbzNCS41
in CH2Cl2 to yield thiourea 101 in 96%
yield over the two steps. Thiourea 101 was coupled with H2NOBnHCl in the presence of
diisopropylethylamine and EDCI to afforded guanidine 102 in 71% yield (Scheme 30). The
guanidine was subjected to a two-step procedure to install the benzyl moiety required for the key
Scheme 29. Synthesis of thiourea 101.

34
amidoalkylation step. The guanidine was treated with TBAF in THF followed by the addition of
BzCl to the crude alcohol in the presence of pyridine and DMAP to provide guanidine 99 in 22%
yield over the two steps. It is unclear why the isolated yield over the two steps was low, but
perhaps exposing 102 to unbuffered TBAF led to cleavage of the Cbz group. Unfortunately no
side-products were isolated that would provide insight on this issue. Due to the poor yield for
Scheme 30. Synthesis of guanidine 99.
guanidine 99, the synthetic sequence was changed. First, thiourea 101 was deprotected with
TBAF, followed by benzoylation of the resulting allylic alcohol to give thiourea 103, and
guanidine formation with H2NOBnHCl in the presence of diisopropylethylamine and EDCI to
afford guanidine 99 in 38% yield over three steps.
With guanidine 99 in hand, suitable conditions to effect the key amidoalkylation
catalyzed by Pd(II) were explored. Initially, treatment of guanidine 99 with 20 mol% of
PdCl2(CH3CN)2 in THF (0.05 M) followed by Boc protection of the free amine gave olefin 104

35
in 59% yield over the two steps (Entry 2, Table 6). The subsequent Boc protection was due to
the unforeseen polarity of the unprotected compound. Although compound 98 behaves normally
Table 6. Screening conditions for key amidoalkylation of guanidine 99.

36
by TLC analysis, purification of 98 by chromatography on SiO2 required a substantially more
polar mobile phase than was required for TLC analysis. Increasing the concentration of the
initial Pd(II)-mediated cyclization reaction resulted in an increased 79% yield of olefin 104. The
use of a Pd(0) catalyst resulted in a poor 17% yield of olefin 105 (Entry 1, Table 6). Using the
palladacycle catalyst developed by Overman (Scheme 14),33
a 1:1 ratio of 104 and 105 in a 70%
combined yield with olefin 104 in 48% ee was obtained. The use of Josiphos ligand A
completely shut down the catalytic cycle (Entry 6, Table 6).52
Subjecting guanidine 99 to
PdCl2(CH3CN)2/pyBox B catalyst system in 1,2-dichloroethane at 85 C gave the desired olefin
in 67% yield and 2% ee (Entry 8, Table 6). Calculations show that isomer 104 is
thermodynamically more stable than 105 by 1.91 Kcal/mol.†
In 2002, the Overman group proposed a mechanism for a similar transformation
involving the cyclization of carbamate derivatives.32
The Pd(II) initially coordinates to olefin 99,
directed by chelation from the benzoyl carbonyl oxygen, and activating the olefin towards
intramolecular nucleophilic attack (106). An intramolecular nucleophilic addition of the more
electron rich nitrogen of the guanidine moiety and release of HCl forms alkyl-Pd(II) intermediate
107. Finally, elimination of olefin 98 regenerates Pd(II) that can re-enter the catalytic cycle
(Scheme 31).
Facilitated by a route to racemic olefin 104, the completion of the synthesis of anatoxin-
a(s) was pursued. A solution of olefin 104 in CH3OH/CH2Cl2 buffered with 10 equiv of pyridine
was cooled to -78 C (Scheme 32). Ozone was bubbled through the solution followed by
treatment with NaBH4 to afford alcohol 108 in 61% yield. In the absence of pyridine only
† Calculations preformed using MMFF conformer distribution followed by PM3 equilibration of geometry using Spartan 10

37
Scheme 31. Proposed catalytic cycle of amidoalkylation.
decomposition of starting material was observed. A recent report from Dussault hypothesizes
that pyridine could potentially add to the carbonyl oxide that is formed after the break down of
the initial ozonide to generate intermediate 109.53
The zwitterionic adduct 109 then adds to a
second equivalent of carbonyl oxide (110). Then, decomposition of 111 would produce
2equivalents of the aldehyde (112) and release an equivalent of O2 and pyridine. The authors
observed a general trend of increased yields when pyridine was added to these reactions.
Alcohol 108 was mesylated with MsCl and triethylamine in CH2Cl2 followed by treatment of the
crude mesylate with sodium azide in DMF at 65 C to give alkyl azide 113 in 77% yield over the
two steps.

38
Scheme 32. Synthesis of alkyl azide 113.
With a synthesis of azide 113 realized, conditions for reduction and subsequent
dimethylation were investigated (Scheme 33). Initially, several conditions were explored to
reduce the azide to the amine. Either Pd/C and H2 or standard Staudinger conditions (not shown)
gave the corresponding amine quantitatively. Surprisingly, the subsequent dimethylation of the
free amine proved difficult. Several conditions were surveyed to achieve the dimethylation.
Mostly variations of the Eschweiler-Clarke conditions utilizing formaldehyde and NaCNBH3
were tried. However, only the primary amine was reisolated. A possible explanation for this
difficulty is the ability of the guanidine nitrogens or the O-benzyl oxygen to form hydrogen
bonds to the free amine and preventing nucleophilic addition into formaldehyde. A Merck
Molecular Force Field (MMFF) lowest energy conformation calculation of the primary amine
showed the presence of hydrogen bonding with the N-OBn oxygen. Accordingly, azide 113 was
reduced by Pd/C and H2 with in situ trapping of the resulting primary amine with di-tert-
butyldicarbonate to give tert-butylcarbamate 115 in 62% yield. Methylation of tert-

39
butylcarbamate 115 was achieved in 79% yield by treating a solution of tert-butylcarbamate 115
and 20 equiv of iodomethane in DMF at -20 C with NaH and warming to room temperature to
give methylcarbamate 116.
Scheme 33. Synthesis of methyl carbamate 116.
Methyl carbamate 116 was subjected to a solution of CH2Cl2/TFA (4:1) to remove both
Boc groups followed by a selective methylation of the secondary amine in the presence of the
exposed guanidine nitrogen under Eschweiler-Clarke conditions to afford dimethylamine
117.54,55
Treatment of 117 with Pd/C and H2 at 60 psi gave an inseparable mixture of N-hydroxyl
guanidine 9 and guanidine 118 in 35% and 56% yield, respectively, over the three steps. The 1H-
NMR spectra of 9 and 118 matched the values for those compounds that were reported in the
isolation paper (Scheme 34).17
The original structures were assigned based on enriched 50% 13
C
and 90% 15
N, which was used to confirm the guanidine core of anatoxin-a(s). The structural
assignment was supported by FAB MS-MS and high-resolution data. The location of the
hydroxyl group was determined based on similar chemical shifts observed in neosaxitoxin and

40
saxitoxin. Also, Moore and coworkers prepared synthetic 15 (the single enantiomer of 118,
Scheme 3), which matched the spectral data of 15 that had been obtained from degradation of
anatoxin-s(s).
Scheme 34. Synthesis of N-hydroxy guanidine 9.
Since the major product under the only conditions that succeeded in removing the benzyl
group was 118, we decided to exchange the benzyl group with a more labile functionality. To
that extent, starting with thiourea 101, the synthetic route would be optimized and the benzyl
group would be replaced with a functionality that would be more amendable to a late-stage
cleavage.
Due to the low yields in the formation of the cyclization precursors using TBAF for silyl
deprotection, we switched to the milder HF•pyr. Benzoylation of the alcohol produced thiourea
103 in 82% yield over the two steps (Scheme 35). Following our previously optimized
conditions for guanidine formation, treatment of thiourea 103 with benzyl hydroxyl amine in the
presence of diisopropylethylamine gave benzyl derivative 99 in 74% yield. Initially several silyl
groups (TES, TBS, TBDPS) and THP were tried in place of the benzyl moiety but after

41
cyclization, the substrate was too labile to be viable. To install the methoxy methyl
functionality, we first treated commercially available hydroxy phthalimide 119 with freshly
prepared chloromethyl methyl ether to give methoxy methyl protected hydroxy phthalimide 120.
Treatment of 120 with hydrazine gave MOM-protected N-hydroxy amine, which when added to
a solution of 103 in the presence of EDCI and diisopropylethylamine, gave guanidine 121 in
81% yield.
Scheme 35. Improved synthesis of 99 and synthesis of MOM derivative 121.
Next, subjecting guanidine derivative 121 to our previously optimized cyclization
conditions with PdCl2(CH3CN)2 gave a single spot by TLC. After Boc-protection of the exposed
guanidine nitrogen, olefin 122 was isolated in 95% yield as the sole regioisomer (assigned based
on the chemical shift of the C2-H) (Table 7). During the cyclization studies with benzylated
guanidine derivative 99, treatment of 99 with Overman’s chiral palladium(II) catalyst, COP-
OAc. Unfortunately, when 99 was subjected to those conditions, a mixture of regioisomers was

42
observed. After SFC separation of the regioisomers, a 3:1 e.r. was observed by chiral SFC. For
MOM derivative 121 we switched to the (S)-(+)-COP-Cl catalyst and observed only a single
regioisomer of the cyclization products (122 or 123) was observed. Analysis by chiral SFC
showed that isomer 122 was produced in a 3:1 e.r. This effect of the anionic ligand
Table 7. Pd(II)-mediated cyclization of MOM-derivative 121.
influencing the outcome of the reaction is similar to what was reported by Stahl for the
cyclization of aliphatic amines to generate pyrrolidine compounds (Scheme 36).56
This effect
was attributated to a change in the mechanistic pathway from a trans-aminopalladation pathway
to a cis-aminopalladation pathway (Scheme 5). In their reported cyclization with Pd(TFA)2, a
90% yield (96% ee) and a 9:1 ratio of trans- vs. cis-aminopalladation (AP) products was
observed; however, when Pd(OAc)2 was used as a catalayst a decrease in yield,
enantioselectivity (48%, 20% ee), and a reversal of the ratio of trans- vs. cis-AP products (1:9)

43
was observed. Interestingly, separating the trans products (125 and 126) still showed that these
compounds were produced in a high enanteomeric ratio. They concluded that the poor
enantioselectivity they observed was for the cis-aminopalladation pathway, while high
enantioselectivities were observed for the trans-aminopalladation pathway.
Scheme 36: Trans-aminopalladation (AP) vs. cis-aminopalladation.
NHTs
D
Ph
PdIIX2 / LNTs
NMe
N
O
Ph
L
Pd(TFA)2: 93%, 98% ee (9:1 trans/cis)
Pd(OAc)2: 48%, 20% ee (1:9 trans*/cis)
trans* (9:1 e.r.)
3Å M. S., toluene
H Ph
+
NTs
D Ph
NTs
D Ph
NTs
H Ph
+ +
trans-AP products cis-AP products
125 126 127 128124
1 atm O2
With a route established for the production of 122 and conditions for a moderately
enantioselective cyclization developed, completion of a synthesis of anatoxin-a(s) was pursued.
To achieve that we bubbled ozone through a solution of 122 that had been buffered with 10
equiv of pyridine, as we had determined to be required in the previous route (Scheme 37). After
treatment of the ozonide intermediate with NaBH4 alcohol 129 was isolated in 78% yield. Next,
mesylation of the alcohol with methane sulfonyl chloride, followed by nucleophilic displacement
of the mesylate with NaN3 gave alkyl azide 130 in 87% yield over the two steps. When azide
130 was reduced under the previous conditions for azide reduction (Pd/C, H2) lower and
irreproducible yields were observed. However, by switching to Lindlar’s catalyst wesaw an
improved and reproducible 80% yield of carbamate 131. Presumably, the lower yield was due to

44
competitive cleavage of the Cbz group, which was not observed when utilizing the less reactive
Lindlar’s catalyst. With an efficient route to carbamate 131 established, a two-step procedure to
install the dimethyl amine functionality was applied. Methylation of the carbamate with CH3I
and NaH gave, after exposure to TFA, methyl amine 132 in 73% yield over the two steps.
Scheme 37. Synthesis of hydroxy guanidine 134.
Exposing methyl amine 132 to reductive amination conditions produced dimethyl amine
133 in 66% yield. Cleavage of the methoxy methyl functionality proceeded smoothly to give
hydroxy guanidine 134 in 74% yield. With the successful cleavage of the methoxy methyl group
we next explored conditions for the successful phosphorylation of 134.
Before exploring phosphorylation conditions, a model system of 134 was developed as
phosphorylation of 134 would not be trivial. Towards that end commercially available ethylene
thiourea (135) was treated with HI in CH3OH to give alkylated thioimidate 136 in 93% yield
(Scheme 38). Installation of the Cbz group proceeded smoothly by treating 136 with CbzCl and

45
triethylamine to give 137 in 99% yield. As was worked out during the synthesis of 134, the Cbz
group is installed first to facilitate the addition of the electron rich nitrogen functionality. To
finish the synthesis of the guanidine core, 119 was exposed to hydrazine and the resulting amine
was added to a solution of 137, which after treatment with AgNO3 gave guanidine 138 in 74%
yield. Finally, removal of the MOM group was achieved using TMSBr to give our desired
model system 139 in 99% yield.
Scheme 38. A model system synthesis of 134 to explore phosphorylation conditions.
With a facile synthesis of the model system achieved, hydroxyl guanidine 139 was
subjected to conditions to install the biologically important phosphate moiety. The most
common conditions for the installation of phosphate groups typically involve the use of
chlorophosphates as the phosphorylating agent. Initially, 139 was treated with dimethyl
chlorophosphate in CH3CN with triethylamine and 140 was isolated in a 31% yield (Entry 1,
Table 8). Efforts to improve this yield were initially focused on utilizing different bases. First,
deprotonation of 139 with NaH at 0 oC, followed the addition of dimethyl chlorophosphate and
warming to rt did not yield 140. Switching to NaHMDS we were able to isolate 140 in a similar
33% yield (Entry 3, Table 8) but no product was detected when KHMDS was used. A decrease

46
in the isolated yield of 140 was observed when DBU was used as the base (Entry 5, Table 8) and
the use of diisopropylethylamine did not produce phosphorylated guanidine 142. Interestingly,
the use of diphenyl chlorophosphate was observed to give a slightly higher yield under the initial
conditions, which may be evidence that the low yield was due to the stability of dimethyl
chlorophosphate (Entry 7, Table 8).
Table 8: Exploration of different bases to optimize phosphorylation.
With no observable improvement to the phosphorylation when utilizing different bases,
solvent effects on the phosphorylation of 139 were explored. It was known at this time that
hydroxyguanidine compound 134 was not soluble in CH3CN, which added another potential
hurdle to the success of this reaction. Using our initial phosphorylation conditions, while
increasing the equiv of dimethyl chlorophosphate from two to five equivalents, we observed a

47
37% yield of 140 (Entry 1, Table 9). Unfortunately, using these same conditions with different
solvents (DMF, NMP, PhCN) resulted in no observable product formation.
Table 9: Solvent screen under the phosphorylation conditions.
With a small variation of yield observed dependent on the equivalents of the
phosphorylation agent used and no improvement of the reaction utilizing different solvents,
different phosphorylating reagents were explored. First, a coupling strategy was explored by
treating 139 with dimethyl phosphate in the presence of diisopropyl carbodiimide (DIC) (Entry
1, Table 10). Under these conditions no observable product was detected and the starting
material had disappeared by LC-MS analysis of the reaction mixture (Entry 1, Table 10). Next, a
more reactive phosphorylating agent than a chlorophosphate was used. To this end, a solution of

48
Table 10: Exploring different phosphorylation conditions.
dimethyl phosphate and triethylamine was treated with TFAA, followed by the addition of
methyl imidazole, which generated the highly active dimethyl methyl-imidazole phosphate. The
addition of this phosphorylating agent to 139 produced the desired dimethyl phosphate 140 in an
improved 49% yield (Entry 2, Table 10). Attempts at using phenyl dichlorophosphate followed
by the addition of methanol (Entry 3, Table 10) and the addition dibenzyl
diethylphosphoramidite to 139 followed m-CPBA did not yield did not yield any observable
product formation (Entry 4, Table 10).
Hydroxy guanidine 134 was subjected to the two phosphorylation conditions that had
been successful on the model system (139) (Scheme 39). Unfortunately, subjecting 134 to
dimethyl chlorophosphate in the presence of triethylamine did not lead to successful
phosphorylation. Exposure of 134 to the more active dimethyl methylimidazole phosphate did
not result in any detectable (by TLC, 1H-NMR, or LC-MS) amount of 135.

49
Scheme 39: Attempted phosphorylation of 134.
Unable to achieve phosphorylation of 134 under our previously worked out conditions,
we proceeded to try other methods of phosphorylation. Subjecting 134 to dimethyl
chlorophosphonate in a variety of solvents and different bases did not produce any observable
trace of 134. Exposure of 134 to either dibenzyl N,N-diethyl phosphoramidite or dimethyl N,N-
diethyl phosphoramidite in the presence of triethylamine resulted in the intermediate mass
potentially being detected by direct inject mass spectroscopic analysis of the reaction solution
([M + H]+). No
1H-NMR data of this intermediate could be obtained. The addition of m-CPBA
to the solution resulted in what appeared to be oxidation of the intermediate to the mass expected
for 140/141 as detected by LC-MS analysis of the reaction solution. Unfortunately, this mass
was never detected after work-up of the reaction mixture. Without spectroscopic proof of the
intermediate or the resulting product, it is uncertain what occured under these conditions. Also,
attempts at using a coupling strategy involving DCC and dimethyl phosphate resulted in loss of
the starting material with no detectable product formation. In 2004, the Jones group published a
procedure for the phosphorylation of alcohols utilizing N-phosphoryl oxazolidinones as transfer
reagents.57,58
Under their conditions, they are able to achieve phosphorylation of primary,
secondary, tertiary, and aromatic alcohols. Unfortunately, under their reported conditions we
saw no detectable phosphorylation of 134. The use of a harder base to promote anion formation
also did not yield 140.

50
1.3 CONCLUSION
Anatoxin-a(s) (8) is a unique organophosphate and one of the strongest PNS-anticholinesterase
agents. The backbone framework was assebled using an array of Pd(0) and Pd(II) metal-
mediated reactions. The Initial effort towards the total synthesis of anatoxin-a(s) centered around
assembling the heterocyclic scaffold using Shi’s diamination procedure of 2-trimethylsilyloxy-
butadiene (67) to give silyl enol ether 68 (Scheme 19, vide supra); however, difficulties in
functionalizing silyl enol ether 68 led to different methods to construct the framework of
anatoxin-a(s) being explored. A key amidocarbonylation reaction catalyzed by Pd(II) was
pursued but preliminary results showed that the undesired regioisomer was the preferred product.
Also, as in the 1st generation route, installation of the dimethylamine side-chain present in the
natural product proved difficult. A 3rd
generation route involved a key amidoalkylation of
guanidine 121, which led to advanced intermediate 134 in 18 steps and 8.0% overall yield. In
this route, a new methodology for the regioselective cyclization of unsymmetrical guanidines
was developed. Using the catalyst system developed by Overman, (R)-(–)-COP-Cl, we were
able to achieve a 3:1 enantiomeric ratio for the key palladium-mediated cyclization sequence.
Although a variety of conditions were attempted to phosphorylate 134, none of the conditions
tried were successful in generating phosphate 140.

51
2.0 EXPERIMENTAL
General. All glassware was dried in an oven at 140 C for 2 h or flame dried prior to use and all
moisture-sensitive reactions were performed under a dry N2 atmosphere unless otherwise stated.
THF and Et2O were distilled from sodium/benzophenone ketyl; triethylamine, pyridine, and
diisopropylethylamine were distilled from CaH2 and stored over KOH; DMF was through
activated neutral alumina and dried for 4 days over 3 Å molecular sieves; CH2Cl2 and toluene
were purified by passage through an activated alumina filtration system; 1,2-dichloroethane was
distilled over CaH2; CbzNCS,41
di-tert-butylurea,38
66,38
67,47
71,47
(Z)-4-(tert-
Butyldiphenylsilyloxy)but-2-en-1-ol,50
(Z)-(4-Bromobut-2-enyloxy)(tert-butyl)diphenylsilane,51
were prepared according to literature procedures.
Reactions were monitored by thin-layer chromatography using pre-coated silica gel 60
F254 plates (EMD, 250 µm thickness) and visualization was accomplished with a 254 nm UV
light and by staining with a KMnO4 solution (1.5 g of KMnO4 and 1.5 g of K2CO3 in 100 mL of
a 0.1% NaOH solution) or Vaughn’s reagent (4.8 g of (NH4)6Mo7O24•4 H2O and 0.2 g of
Ce(SO4)2 in 100 mL of a 3.5 N H2SO4 solution). Flash chromatography was performed using
SiO2 (Silicycle, Silia-P Flash Silica Gel, 40-63 µm) or Florisil (purchased from Fischer
chemical and used as received, 100 - 200 mesh).

52
1H and
13C NMR spectra were obtained on a Bruker advance 300 MHz in CDCl3 unless
otherwise noted. Chemical shifts (δ) were reported in parts per million with the residual solvent
peak used as an internal standard δ 1H /
13C (Solvent); 7.26 /77.23 (CDCl3) and are tabulated as
follows: chemical shift, multiplicity (s = singlet, bs = broad singlet, d = doublet, dd = doublet of
doublets, ddd = doublet of doublet of doublets, dddd = doublet of doublet of doublet of doublets,
dt = doublet of triplets, t = triplet, td = triplet of doublets, bt = broad triplet, tt = triplet of triplets,
at = apparent triplet, q = quartet, m = multiplet), number of protons, and coupling constant(s).
Infrared spectra were determined as neat solids or oils on a Smiths Detection IdentifyIR FT-IR
spectrometer. Mass spectra were obtained on a Micromass Autospec double focusing
instrument. Melting points were determined using a Laboratory Devices Mel Temp II in open
capillary tubes and are uncorrected. Enantiomeric ratios were determined using a Mettler Toledo
AG – Berger SFCTM
Analytix.
1,3-Di-tert-butyl-4-(1-(trimethylsilyloxy)vinyl)imidazolidin-2-one (68)36
and 4-acetyl-1,3-di-
tert-butylimidazolidin-2-one (70). A solution of 66 (0.500 g, 2.94 mmol) in toluene (8.80 mL)
was treated with Pd(PPh3)4 (0.339 g, 0.294 mmol). To the mixture was added 67 (1.67 g, 5.87
mmol) and the resulting solution was heated to 65 ˚C for 1 h. The reaction solution was cooled

53
to rt and filtered through SiO2 (9:1 hexanes/EtOAc) to give 68 (174 mg, 19 %): 1H NMR (300
MHz, benzene-d6) δ 4.33 (d, 1 H, J = 1.5 Hz), 4.07 (d, 1 H, J = 1.5 Hz ) 3.71 (dd, 1 H, J = 3.0,
9.0 Hz), 3.10 (at, 1 H, J = 8.4, 9.0 Hz), 3.00 (dd, 1 H, J = 3.0, 8.4 Hz), 1.51 (s, 9 H), 1.34 (s, 9
H), 0.15 (s, 9 H); and 70: IR (ATR) 1718, 1679 cm-1
; 1H NMR (300 MHz, benzene-d6) δ 3.72
(dd, 1 H, J = 4.8, 10.2 Hz), 2.85 (dd, 1 H, J = 9.0, 9.9 Hz ) 2.54 (dd, 1 H, J = 4.5, 9.0 Hz), 1.83
(s, 3 H), 1.28 (s, 9 H), 1.20 (s, 9 H); 13
C NMR (75 MHz, benzene-d6) δ 208.3, 160.6, 61.6, 54.5,
53.4, 43.7, 28.5, 27.5, 24.5; HRMS (ES+) m/z calcd for C13H25N2O2 ([M + H]
+) 241.1916, found
241.1915.
1,3-Di-tert-butyl-4-(1-(triethylsilyloxy)vinyl)imidazolidin-2-one (72). A solution of 66 (50.0
mg, 0.294 mmol) and Pd(PPh3)4 (33.9 mg, 0.0294 mmol) in THF (0.100 mL) was treated with 71
(108 mg, 0.587 mmol) and heated to 65 ˚C for 1 h. The reaction solution was cooled to room rt
and filtered through Florisil (9:1 hexanes/EtOAc) to give 72 (48.1 mg, 46%): IR (ATR) 1690
cm-1
; 1H NMR (300 MHz, benzene-d6) δ 4.26 (d, 1 H, J = 1.8 Hz), 4.06 (d, 1 H, J = 1.5 Hz ),
3.74 (dd, 1 H, J = 3.3, 9.3 Hz), 3.11 (dd, 1 H, J = 8.4, 9.3 Hz), 3.02 (dd, 1 H, J = 3.0, 8.1 Hz),
1.51 (s, 9 H), 1.35 (s, 9 H), 0.98 (s, 9 H), 0.65 (m, 6 H); 13
C NMR (75 MHz, CDCl3) δ 160.9,

54
160.3, 88.3, 55.7, 54.0, 52.9, 47.2, 28.7, 27.6, 6.9, 5.0; HRMS (ES+) m/z calcd for
C19H38N2O3SiNa ([M + Na]+) 377.2600, found 377.2590.
1,3-Di-tert-butyl-4-(2-hydroxyacetyl)imidazolidin-2-one (73). A solution of 72 (109 mg,
0.307 mmol) in CH2Cl2 (1.5 mL) and 5% aq. NaHCO3 (1.5 mL) was cooled to 0 ˚C, treated with
m-CPBA (151 mg, 0.613 mmol), stirred at rt overnight. The reaction was diluted with sat.
NaHCO3 and extracted with CH2Cl2. The combined organic extracts were washed with sat.
Na2SO3, sat. NaHCO3, brine, dried (Na2SO4), and concentrated. The crude residue was
dissolved in CH3OH (1.0 mL) and treated with 1.5 M HCl (1.0 mL). The reaction mixture was
stirred overnight and extracted with CH2Cl2 (3 x 20 mL). The combined organic layers were
washed with water, dried (Na2SO4), concentrated, and purified by chromatography on SiO2 (1:1
hexanes/EtOAc) to give 73 (3.9 mg, 5%): IR (ATR) 3396, 1702 cm-1
; 1H NMR (300 MHz,
benzene-d6) δ 4.35 (dd, 1 H, J = 5.1, 19.8 Hz), 4.13 (dd, 1 H, J = 4.8, 19.8 Hz ) 3.72 (dd, 1 H, J =
4.2, 9.9 Hz), 2.86 (t, 1 H, J = 5.1 Hz), 2.74 (t, 1 H, J = 9.6 Hz), 2.44 (dd, 1 H, J = 4.2 9.3 Hz),
1.18 (s, 9 H), 1.13 (s, 9 H); HRMS (EI+) m/z calcd for C13H24N2O3 ([M]
+.) 256.1787, found
256.1779.

55
1-Benzyloxycarbonyl-3-allylthiourea (79). To a solution of CbzNCS41
(0.831 g, 4.30 mmol) in
CH2Cl2 (164 mL) at 0 C was added allylamine (0.330 mL, 4.30 mmol). The solution was
warmed to rt, stirred for 1.5 h, and quenched with 1 M HCl (10.0 mL). The organic layer was
washed with water (10 mL) and brine (10 mL), dried (Na2SO4), concentrated, and purified by
chromatography on SiO2 (4:1, hexanes/EtOAc) to give 79 (1.07 g, 99%) as a white solid: Mp
61.3-63.5 C; IR (ATR) 3252, 3164, 1715, 1534 cm
-1.
1H NMR (300 MHz, CDCl3) 9.71 (s, 1
H), 8.11 (s, 1 H), 7.43–7.33 (m, 5 H), 5.92 (dddd, 1 H, J = 5.7, 5.7, 10.5, 17.1 Hz), 5.32–5.21 (m,
2 H), 5.18 (s, 2 H), 4.30 (tt, 2 H, J = 1.2, 5.7 Hz); 13
C NMR (75 MHz, CDCl3) 179.3, 152.7,
134.7, 132.1, 128.9, 128.8, 128.3, 117.7, 68.2, 48.0; HRMS (EI+) m/z calcd for C12H14N2O2S
([M]+.
) 250.0776, found 250.0786.

56
1- Benzyloxycarbonyl-2-benzyloxy-3-allylguanidine (78). To a solution of 79 (1.50 g, 6.87
mmol), O-benzylhydroxylamine (1.25 mL, 10.3 mmol), and diisopropylethylamine (1.26 mL,
7.56 mmol) in CH2Cl2 (68.7 mL) at 0 C was added EDCI (2.63 g, 13.7 mmol, 2 equiv). The
reaction mixture was stirred for 1 h, warmed to rt, stirred for 14 h, and quenched with 1 M HCl
(5 mL). The organic solution was washed with water (10 mL) and brine (10 mL), dried
(Na2SO4), concentrated, and purified by chromatography on SiO2 (2:1, hexanes/EtOAc to 1:1
hexanes/EtOAc) to give 78 (1.78 g, 76%) as a clear oil: IR (ATR) 3386, 1726, 1640 cm-1
; 1H
NMR (300 MHz, CDCl3) 7.96 (s, 1 H), 7.48–7.29 (m, 10 H), 6.41 (bs, 1 H), 5.99 (dddd, 1 H, J
= 1.2, 5.7, 11.4, 15.9 Hz), 5.20 (m, 2 H), 5.15 (s, 2 H) 4.90 (s, 2 H), 3.79-3.75 (m, 2 H); 13
C
NMR (75 MHz, CDCl3) 153.0, 148.1, 137.7, 135.2, 134.4, 128.9, 128.8, 128.5, 128.4, 128.1,
116.1, 76.0, 67.8, 43.4; HRMS (EI+) m/z calcd for C19H21N3O3
([M]
+.) 339.1583, found
339.1581.

57
Methyl 3,4-(1-benzyloxy-2-benzyloxycarbonyl)guanidinobutanoate (77) and methyl 3,4-(1-
benzyloxycarbonyl-2-benzyloxy)guanidinobutanoate (80). A 50-mL, 3-necked, round-
bottomed flask fitted with rubber septum and a three-way hose adapter (with stopcock, equipped
with a vacuum inlet, and a balloon filled with CO) was charged with Pd(OAc)2 (56.1 mg, 0.250
mmol), CuCl2 (1.02 g, 7.49 mmol), and degassed CH3OH (4.0 mL). The flask was evacuated
and back-filled with CO (3x), cooled to 0 C, and stirred for 0.5 h. To this mixture was added a
solution of 78 (848 mg, 2.50 mmol) in degassed CH3OH (4.32 mL). The reaction mixture was
stirred at 0 C for 18 h, warmed to rt, stirred for 4 h, and concentrated. The residue was
dissolved in EtOAc, and filtered through celite. The brownish filtrate was washed with sat.
NaHCO3 (1 x 5 mL), ethylenediamine (1 x 1 mL), and sat. NaHCO3 (1 x 5 mL). The washing
procedure was repeated until the organic layer remained yellow. The organic solution was dried
(Na2SO4), concentrated, and purified by chromatography on florisil (1:1, hexanes/EtOAc to
100% EtOAc) to give 77 and 80 (750 mg, 76%) in a isomeric ratio of 2.2:1 as a yellow oil that
was not further separated: 1H NMR (300 MHz, CDCl3) 7.43–7.26 (m, 20 H), 5.26 (ad, 2 H, J =
1.5 Hz, minor isomer 77), 5.16 (s, 2 H, major isomer 77), 5.07 (d, 1 H, J = 11.4 Hz, major isomer
77), 5.00 (d, 2 H, J = 11.4 Hz, major isomer 77), 4.98 (s, 2 H, minor isomer 80).

58
Methyl 3,4-(1-benzyloxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidinobutanoate
(77-Boc) and methyl 3,4-(1-benzyloxycarbonyl-2-benzyloxy-3-tert-butoxycarbonyl)
guanidinobutanoate (80-Boc). A 50-mL, 3-necked, round-bottomed flask fitted with rubber
septum and a three-way hose adapter (with stopcock, equipped with a vacuum inlet, and a
balloon filled with CO) was charged with Pd(OAc)2 (166 mg, 0.737 mmol), CuCl2 (3.00 g, 22.1
mmol), and degassed CH3OH (12.0 mL). The flask was evacuated and back-filled with CO (3x),
cooled to 0 C, and stirred for 0.5 h. To this mixture was added a solution of 78 (2.50 g, 7.37
mmol) in degassed CH3OH (12.6 mL) and the resulting solution was stirred at 0 C for 18 h,
warmed to rt, stirred for 4 h, and concentrated. The residue was dissolved in EtOAc, filtered
through celite, and the brownish filtrate was washed with sat. NaHCO3 (10 mL),
ethylenediamine (1 mL), and sat. NaHCO3 (10 mL). The washing procedure was repeated until
the organic layer remained yellow. The organic solution was dried (Na2SO4) and concentrated
under reduced pressure. The crude mixture of 77 and 80 was dissolved in THF (73.7 mL) and
treated with Boc2O (1.93 g, 8.84 mmol, 1.2 equiv). To this solution was added triethylamine
(2.30 mL, 1.66 mmol) followed by DMAP (182 mg, 1.47 mmol, 0.2 equiv) and the resulting
mixture was stirred for 24 h, treated with sat. NH4Cl (20 mL), and extracted with EtOAc (3 x 20
mL). The combined organic extracts were dried (Na2SO4), concentrated, and purified by
chromatography on SiO2 (2:1, hexanes/EtOAc) to give 77-Boc (1.40 g, 38%) as a white solid:

59
Mp 123.3–124.5 C;
IR (ATR) 1752, 1724, 1687, 1631 cm-1
; 1H NMR (300 MHz, CDCl3)
7.39–7.25 (m, 10 H), 5.02 (s, 2 H), 4.95 (d, 1 H, J = 10.5 Hz), 4.81 (d, 1 H, J = 10.5 Hz), 3.92–
3.82 (m, 2 H), 3.62 (s, 3 H), 3.47–3.42 (m, 1 H), 2.77 (dd, 1 H, J = 3.9, 16.5 Hz), 2.32 (dd, 1 H, J
= 8.4, 16.5 Hz), 1.48 (s, 9 H); 13
C NMR (75 MHz, CDCl3) 170.8, 160.0, 149.7, 136.4, 135.4,
129.5, 129.0, 128.7, 128.6, 128.4, 128.0, 83.9, 67.9, 52.2, 28.2; HRMS (EI+) m/z calcd for
C26H31N3O7 ([M]+) 497.2162, found 497.2160: and 80-Boc (0.834 g, 23%) as a yellow oil: IR
(ATR) 1733, 1724, 1709, 1653 cm-1
; 1H NMR (300 MHz, CDCl3) 7.48–7.25 (m, 10 H), 5.30
(d, 1 H, J = 12.6 Hz), 5.23 (d, 1 H, J = 12.3 Hz), 5.05 (s, 2 H), 4.49 (ddd, 1 H, J = 3.3, 6.9, 12.9
Hz), 3.78–3.76 (m, 1 H), 3.68 (s, 3), 3.67 (dd, 1 H, J = 3.9, 6.9 Hz), 2.87 (dd, 1 H, J = 3.3, 16.2
Hz), 2.46 (dd, 1 H, J = 9.9, 16.2 Hz); 13
C NMR (75 MHz, CDCl3) 170.6, 151.6, 151.0, 141.9,
137.4, 135.6, 129.9, 128.9, 128.7, 128.5, 128.3, 128.2, 128.0, 82.9, 68.3, 52.7, 52.2, 50.0, 37.2,
28.0; HRMS (ES+) m/z calcd for C26H31N3O7Na ([M + Na]
+) 520.2060, found 520.2038.
3,4-(1-Benzyloxy-2-benzyloxycarbonyl)guanidinobutanoic acid (87). To a solution of 77 and
70 (0.350 g, 0.881 mmol) in Et2O/THF (35.2 mL, 10:1 v/v) was added KOTMS (0.314 g, 2.20
mmol) and the resulting mixture was stirred for 3 h. After the addition of water (10 mL) the
biphasic solution was vigorously stirred for 5 min, the aqueous phase was separated, acidified
(pH = 2) with 1 M HCl, and extracted with EtOAc (5 x 10 mL). The combined organic extracts

60
were dried (Na2SO4) and concentrated to give of 87 (233 mg, 69%) as a powder: Mp 150.1–
151.9 C; IR (ATR) 3340, 1702, 1638, 1610 cm-1
. 1H NMR (300 MHz, CDCl3) 7.43–7.26 (m,
10 H), 5.16 (s, 2 H), 5.05 (d, 1 H, J = 11.4 Hz), 4.98 (d, 1 H, J = 11.1 Hz), 3.86–3.77 (m, 1 H),
3.67 (dd, 1 H, J = 7.8, 9.6 Hz), 3.17 (dd, 1 H, J = 7.2, 9.6 Hz), 2.63 (dd, 1 H, J = 3.9, 16.8 Hz),
2.19 (dd, 1 H, J = 9.3, 16.8 Hz); 13
C NMR (75 MHz, CDCl3) 173.2, 166.7, 164.3, 137.0,
136.0, 130.0, 129.0, 128.7, 128.6, 128.3, 128.1, 78.8, 67.5, 57.3, 45.3, 34.5; HRMS (EI+) m/z
calcd for C20H21N3O5 ([M]+.
) 383.1481, found 383.1479.
Methyl-3,4-(1-benzyloxy-2-benzyloxycarbonyl)guanidinebutanoyloxy(methyl)
carbamodithioate (94). A solution of 87 (0.250 g, 0.652 mmol) in CH2Cl2 (10.0 mL) at 0 C
was treated with DCC (161 mg, 0.717 mmol) and DMAP (8.05 mg, 0.0652 mmol). After 0.5 h
the flask was protected from light and a solution of 9247
(89.5 mg, 0.652 mmol) in CH2Cl2 (3.0
mL) was added slowly. The mixture was warmed to rt, stirred for 24 h, filtered through celite,
concentrated, and purified by chromatography on SiO2 (1:1, hexanes/EtOAc) to give of 94 (248
mg, 76%) as a foam: IR (ATR) 3319, 1621, 1569, 1536 cm-1
; 1H NMR (300 MHz, CDCl3)
8.03 (bs, 1 H), 7.46–7.29 (m, 10 H), 5.17 (s, 2 H), 5.02 (s, 2 H), 3.95–3.86 (m, 1 H), 3.72 (dd, 1
H, J = 8.0, 9.9 Hz), 3.65 (s, 3 H), 3.27 (dd, 1 H, J = 6.3, 9.9 Hz), 2.76 (dd, 1 H, J = 4.5, 16.8 Hz),

61
2.53 (s, 3 H), 2.33 (dd, 1 H, J = 9.0, 16.8 Hz); 13
C NMR (75 MHz, CDCl3) 196.9, 167.4,
166.5, 164.5, 137.0, 136.0, 130.2, 129.2, 128.8, 128.6, 128.4, 128.1, 78.8, 67.5, 56.9, 45.0, 42.8,
32.4, 19.0; MS (ES+) m/z 525 ([M + Na]
+, 70), 503 (100), 247 (80), 212 (85).
Methyl-3,4-(1-benzyloxy-2-benzyloxycarbonyl)guanidinobutanoyloxy(phenyl)
carbamodithioate (95). A solution of 87 (0.250 g, 0.652 mmol) in CH2Cl2 (10.0 mL) at 0 C
was treated with DCC (161 mg, 0.717 mmol) and DMAP (8.05 mg, 0.0652 mmol). After 0.5 h
the flask was protected from light and a solution of 9347
(0.130 g, 0.652 mmol) in CH2Cl2 (3.0
mL) was added slowly. The mixture was warmed to rt, stirred for 24 h, filtered through celite,
concentrated, and purified by chromatography on SiO2 (1:1, hexanes/EtOAc) to give 95 (207 mg,
56%) as a yellow oil: IR (ATR) 3360, 1653, 1603, 1577 cm-1
; 1H NMR (300 MHz, CDCl3)
7.99 (bs, 1 H), 7.48–7.26 (m, 15 H), 5.17 (s, 2 H), 5.03 (d, 1 H, J = 11.4 Hz), 4.98 (d, 1 H, J =
11.4 Hz), 3.89–3.80 (m, 1 H), 3.69 (dd, 1 H, J = 7.8, 9.9 Hz), 3.28 (dd, 1 H, J = 6.6, 9.9 Hz),
2.78 (dd, 1 H, J = 3.9, 16.8 Hz), 2.58 (s, 3 H), 2.32 (dd, 1 H, J = 9.9 Hz, 16.5 Hz); 13
C NMR (75
MHz, CDCl3) 197.6, 167.3, 166.6, 164.5, 137.0, 136.0, 130.0, 129.8, 129.0, 128.7, 128.6,
128.5, 128.4, 128.0, 78.7, 77.4, 67.5, 57.1, 45.0, 32.3, 19.8; HRMS (ES+) m/z calcd for
C28H29N4O5S2 ([M + H]+) 565.1579, found 565.1559.

62
3,4-(1-Benzyloxy-2-benzyloxycarbonyl)guanidinobutanamide (97). A sealed tube was
charged with a solution of 77 and 80 (0.250 g, 0.629 mmol, 1:1) in CH3OH (2.1 mL), cooled to -
78 C, and treated with liquid ammonia (2.1 mL). The tube was sealed, slowly warmed to rt
over 1 h, stirred 13 h, and concentrated to give 97 (153 mg, 64%) as a white solid: Mp 141.8–
143.5 C; IR (ATR) 3362, 3178, 1668, 1649, 1579 cm-1
; 1H NMR (300 MHz, CDCl3) 7.93
(bs, 1 H), 7.44–7.26 (m, 10 H), 5.65 (bs, 1 H), 5.23 (bs, 1 H), 5.18 (dd, 1 H, J = 12.6 Hz), 5.14
(dd, 1 H, J = 12.6 Hz), 5.02 (s, 2 H), 3.90–3.81 (m, 1 H), 3.65 (dd, 1 H, J = 7.8, 9.9 Hz), 3.23
(dd, 1 H, J = 6.0, 9.9 Hz), 2.41 (dd, 1 H, J = 4.2, 15.6 Hz), 2.10 (dd, 1 H, J = 9.0, 15.3 Hz); 13
C
NMR (75 MHz, CDCl3) 171.8, 166.3, 164.3, 137.1, 135.9, 130.0, 129.0, 128.7, 128.6, 128.4,
128.1, 78.5, 67.4, 57.3, 45.0, 35.1; HRMS (ES+) m/z calcd for C20H22N4O4Na ([M + Na]
+)
405.1539, found 405.1525.

63
(Z)-2-(4-(tert-Butyldiphenylsilyloxy)but-2-enyl)phthalimide (100). To a solution of (Z)-(4-
bromobut-2-enyloxy)(tert-butyl)diphenylsilane (13.4 g, 34.5 mmol) in DMF (98.5 mL) was
added potassium phthalimide (9.78 g, 51.7 mmol). The resulting mixture was heated to 80 C
for 25.5 h, cooled to rt, concentrated, and diluted with CH2Cl2 (100 mL). The crude solution was
filtered through SiO2, concentrated, and purified by chromatography on SiO2 (5:1,
hexanes/EtOAc) to give 100 (11.8 g, 75%) as a pale yellow oil: IR (ATR) 3068, 1709 cm-1
; 1H
NMR (300 MHz, CDCl3) 7.84–7.69 (m, 8 H), 7.48–7.37 (m, 6 H), 5.85-5.76 (m, 1 H), 5.54–
5.45 (m, 1 H), 4.49 (td, 2 H, J = 0.6, 6.0 Hz), 4.20 (td, 2 H, J = 0.6, 6.9 Hz), 1.09 (2, 9 H); 13
C
NMR (75 MHz, CDCl3) 168.0, 135.8, 134.1, 133.7, 132.3, 129.9, 127.9, 124.2, 123.4, 60.4,
35.2, 27.0, 19.3; HRMS (EI+) m/z calcd for C27H26NO3Si ([M – 15]
+) 440.1682, found
440.1694.

64
(Z)-4-(3-(Benzyloxycarbonyl)thiourea)but-2-enyl-tert-butyldiphenylsilyloxane (101). To a
solution of 100 (11.3 g, 24.9 mmol) in EtOH (258 mL) was added hydrazine monohydrate (2.54
g, 49.8 mmol). The mixture was heated at reflux for 4 h, cooled to 0 C, filtered, and
concentrated. The crude residue was added at 0 C to a solution of CbzNCS41
(7.21 g, 37.3
mmol) in CH2Cl2 (258 mL). The solution was stirred for 15 h while slowly warming to rt,
washed with 1 M HCl (25 mL), water (25 mL), and brine (25 mL). The organic phase was dried
(Na2SO4) and concentrated. The crude material was purified by chromatography on SiO2 (9:1,
hexanes/EtOAc) to give 101 (12.3 g, 96%) as a yellow oil: IR (ATR) 3248, 3287, 1715, 1526
cm-1
; 1H NMR (300 MHz, CDCl3) 9.54 (bs, 1 H), 8.25 (s, 1 H), 7.72–7.67 (m, 4 H), 7.45–7.37
(m, 11 H), 5.86–5.78 (m, 1 H), 5.56-5.48 (m, 1 H), 5.18 (s, 2 H), 4.32 (dd, 2 H, J = 0.9, 6 Hz),
4.14 (t, 2 H, J = 5.4 Hz), 1.08 (s, 9 H); 13
C NMR (75 MHz, CDCl3) 179.0, 152.6, 135.7, 134.7,
133.6, 133.4, 129.9, 129.2, 128.9, 128.5, 127.9, 124.4, 77.2, 68.3, 60.4, 43.1, 27.0, 19.3; HRMS
(ES+) m/z calcd for C29H34N2O3SiSNa ([M + Na]
+) 541.1957, found 541.1957.

65
(Z)-4-(2-(Benzyloxy)-3-(benzyloxycarbonyl)guanidino)but-2-enyl-tert-butyldiphenylsilylane
(102). To a solution of 101 (1.00 g, 1.99 mmol) and O-benzylhydroxylamine hydrochloride
(0.397 g, 2.39 mmol) in CH2Cl2 (13.3 mL) was added diisopropylethylamine (0.697 mL, 4.18
mmol). The resulting mixture was cooled to 0 C, treated with EDCI (6.41 g, 33.4 mmol, 1.5
equiv), stirred for 1 h, warmed to rt, and stirred for 41 h. The solution was washed with 1 M HCl
(5 mL), water (5 mL), and brine (5 mL). The organic phase was dried (Na2SO4), concentrated,
and purified by chromatography on SiO2 (2:1, hexanes/EtOAc) to give 102 (0.84 g, 71%) as a
yellow oil: IR (ATR) 3390, 1728, 1646 cm-1
; 1H NMR (300 MHz, CDCl3) 7.92 (s, 1 H),
7.76–7.74 (m, 4 H), 7.47–7.32 (m, 16 H), 6.23 (bt, 1 H, J = 5.4 Hz), 5.81–5.75 (m, 1 H), 5.56–
5.50 (m, 1 H), 5.15 (s, 2 H), 4.85 (s, 2 H), 4.37–4.34 (m, 2 H), 3.62 (t, 2 H, J = 5.7 Hz), 1.12 (s, 9
H); 13
C NMR (75 MHz, CDCl3) 152.9, 147.9, 137.7, 135.8, 135.3, 133.8, 132.0, 129.9, 129.4,
128.9 (2), 128.6, 128.5, 128.1, 127.9, 127.8, 126.8, 76.1, 67.8, 60.5, 38.4, 27.0, 19.3; HRMS
(EI+) m/z calcd for C37H41N3O4 ([M]
+.) 591.3097, found 591.3080.

66
(Z)-4-(3-(Benzyloxycarbonyl)thioureido)but-2-enyl benzoate (103). A solution of 102 (8.19
g, 16.3 mmol) in THF (54.3 mL) was treated with TBAF (32.6 mL, 32.6 mmol, 1 M in THF) and
stirred for 24 h. The solution was diluted with water (40 mL), extracted with EtOAc (3 x 40 mL),
dried (Na2SO4), and concentrated. The crude oil was dissolved in pyridine (163 mL) and cooled
to 0 °C. The resulting solution was treated with DMAP (1.01 g, 8.16 mmol) and BzCl (0.508 mL,
4.38 mmol) was added over 5 min via syringe. The solution was warmed to rt, stirred for 12.5 h,
treated with sat. NH4Cl (50 mL), and stirred for 1 h. The mixture was diluted with H2O (25 mL)
and extracted with EtOAc (3 x 50 mL). The combined organic extracts were washed with 1 M
HCl (3 x 25 mL), dried (Na2SO4), concentrated, and purified by chromatography on SiO2 (4:1,
hexanes/EtOAc) to give 103 as a white solid: Mp 75.2–76.8 °C; IR (ATR) 3287, 1713, 1521
cm-1
; 1H NMR (300 MHz, CDCl3) 9.73 (bs, 1 H), 8.15 (s, 1 H), 8.07–8.04 (m, 2 H), 7.57 (mt,
1 H, J = 7.5 Hz), 7.47–7.36 (m, 7 H), 5.93–7.75 (m, 2 H), 5.17 (s, 2 H), 4.95 (d, 2 H, J = 6.6 Hz),
4.47 (t, 2 H, J = 6.0 Hz); 13
C NMR (75 MHz, CDCl3) 179.4, 166.6, 152.6, 134.7, 133.3, 129.9,
129.1, 129.0, 128.7, 128.6, 128.5, 127.9, 68.4, 60.6, 42.9; HRMS (ES+) m/z calcd for
C20H20N2O4SNa ([M + Na]+) 407.1041, found 407.1021.

67
(Z)-4-(2-(Benzyloxy)-3-(benzyloxycarbonyl)guanidino)but-2-enyl benzoate (99). To a
solution of 103 and O-benzylhydroxylamine hydrochloride (3.15 g, 19.6 mmol, 1.2 equiv) in
CH2Cl2 (81.5 mL) was added diisopropylamine (5.98 mL, 35.8 mmol). The resulting mixture
was cooled to 0 °C, treated with EDCI (6.25 g, 32.6 mmol), stirred for 1 h, warmed to rt, and
stirred for 12 h. The solution was washed with 1 M HCl (80 mL), water (80 mL), and brine (80
mL). The organic phase was dried (Na2SO4), concentrated, and purified by chromatography on
SiO2 (2:1 hexanes/EtOAc) to give 99 (4.93 g, 50%) over two steps as a yellow oil: IR (ATR)
3388, 1726, 1646, 1452 cm-1
; 1H NMR (300 MHz, CDCl3) 8.08–8.03 (m, 2 H), 7.90 (bs, 1 H),
7.56 (tt, 1 H, J = 1.2, 6.6 Hz), 7.46–7.24 (m, 12 H), 6.37 (bt, 1 H, J = 5.4 Hz), 5.84–5.72 (m, 2
H), 5.12 (s, 2 H), 4.93 (d, 2 H, J = 5.4 Hz), 4.87 (s, 2 H), 3.87 (t, 2 H, J = 5.7 Hz); 13
C NMR (75
MHz, CDCl3) 153.0, 147.8, 137.7, 135.2, 131.0, 129.9, 128.9 (2), 128.6, 128.5 (2), 128.1,
126.4, 76.11, 67.9, 60.9, 38.3; HRMS (EI+) m/z calcd for C27H27N3O5 ([M]
+.) 473.1951, found
473.1951.

68
3,4-(1-Benzyloxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidinobut-1-ene (104). A
solution of 99 (25.0 mg, 0.0528 mmol) in THF (0.264 mL) was treated with PdCl2(CH3CN)2
(2.77 mg, 0.0106 mmol, 0.2 equiv) and stirred overnight. To the solution was added Boc2O
(14.3 mg, 0.0634 mmol), DMAP (2.61 mg, 0.0211 mmol), and triethylamine (8.24 µL, 0.0581
mmol). The reaction mixture was stirred for 6 h, quenched with sat. NH4Cl (2 mL), extracted
with EtOAc (3 x 2 mL), the combined organic extracts were dried (Na2SO4), and concentrated.
The crude mixture was purified by chromatography on SiO2 (1:1, hexanes/EtOAc) to give 104
(20.0 mg, 84%) as a yellow oil: IR (ATR) 1761, 1692 cm-1
; 1H NMR (300 MHz, CDCl3)
7.38–7.18 (m, 10 H), 5.79 (ddd, 1 H, J = 8.7, 9.9, 17.1 Hz), 5.39 (d, 1 H, J = 17.1 Hz), 5.33 (d, 1
H, J = 10.5 Hz), 5.01 (d, 1 H, J = 9.9 Hz), 5.00 (d, 1 H, J = 12.3 Hz), 4.85 (d, 1 H, J = 11.7 Hz),
4.85 (d, 1 H, J = 10.5 Hz), 4.02 (aq, 1 H, J = 8.4 Hz), 3.83 (dd, 1 H, J = 8.1, 10.2 Hz), 3.39 (dd, 1
H, J = 8.7, 10.5 Hz), 1.49 (s, 9 H). 13
C NMR (75 MHz, CDCl3) 160.3, 149.7, 149.4, 136.3,
135.4, 133.7, 129.4, 128.8, 128.6, 128.5, 128.5, 128.0, 122.2, 83.7, 77.9, 67.9, 63.7, 46.9, 28.2;
HRMS (EI+) m/z calcd for C25H29N3O5 ([M]
+.) 451.2107, found 451.2106.
General procedure for chiral ligand screen in the amidoalkylation of compound 104. A
solution of PdCl2(CH3CN)2 (0.2 equiv) and ligand (0.4 equiv) in THF was stirred for 40 min. To
this mixture was added a solution of 99 in THF and the resulting solution was stirred overnight.
After the addition of THF (to a final concentration of 0.2 M), Boc2O (1.2 equiv), DMAP (0.4

69
equiv), and triethylamine (1.1 equiv), the mixture was stirred for 6–24 h, quenched with sat.
NH4Cl, and extracted with EtOAc (3x). The combined organic extracts were dried (Na2SO4),
concentrated, and purified by chromatography on SiO2 (1:1, hexanes/EtOAc).
2,3-(1-Benzyloxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidinopropanol (108).
Ozone was bubbled through a solution of 104 (2.62 g, 5.80 mmol) and pyridine (4.69 mL, 58.0
mmol) in CH3OH/CH2Cl2 (69.6 mL/ 46.4 mL, 0.05 M) at -78 C for 15 min. Argon was bubbled
through the solution for 5 min, NaBH4 (329 mg, 8.70 mmol) was added, the mixture was stirred
for 1 h, and slowly warmed to rt overnight. The solution was quenched with water (25 mL),
extracted with EtOAc (4 x 25 mL), the combined organic extracts were dried (Na2SO4),
concentrated, and purified by chromatography on SiO2 (1:1 hexanes/EtOAc to 100% EtOAc) to
give 108 (1.51 g, 57%) as a white foam: IR (ATR) 3440, 1752, 1690 cm-1
; 1H NMR (300 MHz,
CDCl3) 7.44–7.27 (m, 10 H), 5.09 (s, 2 H), 4.95 (s, 2 H), 3.73–3.59 (m, 2 H), 3.57 (dd, 1 H, J =
3.3, 12.3 Hz), 3.51–3.44 (m, 1 H), 3.40 (dd, 1 H, J = 2.4, 12.3 Hz), 1.98 (bs, 1 H), 1.47 (s, 9 H);
13C NMR (75 MHz, CDCl3) 160.0, 151.2, 149.7, 136.3, 135.7, 129.7, 129.0, 128.7, 128.5,
128.3, 127.9, 83.6, 77.5, 67.9, 59.7, 59.4, 44.2, 28.0; HRMS (EI+) m/z calcd for C24H29N3O6
([M]+.
) 455.2056, found 455.2074.

70
2,3-(1-Benzyloxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidin-1-azido-propane
(113). To a solution of 108 (0.500 g, 1.08 mmol), triethylamine (0.467 mL, 3.29 mmol), and
DMAP (27.1 mg, 0.220 mmol) in CH2Cl2 (5.40 mL) at 0 C was added MsCl (0.174 mL, 2.20
mmol). The reaction mixture was stirred for 4.5 h at 0 C, warmed to rt, and stirred for 1.5 h.
The solution was diluted with CH2Cl2 (10 mL), and washed with sat. Na2CO3 (10 mL), brine (10
mL), dried (Na2SO4), and concentrated. The crude residue was dissolved in DMF (5.50 mL),
treated with NaN3 (79.3 mg, 1.23 mmol), and placed in a preheated 65 C oil bath for 6 h. The
solution was cooled to rt, diluted with EtOAc, washed with water, dried (Na2SO4), and
concentrated. The crude material was purified by chromatography on SiO2 (1:1 hexanes/EtOAc)
to give 113 (405 mg, 77%) as a clear oil: IR (ATR) 2102, 1756, 1690 cm-1
; 1H NMR (300
MHz, CDCl3) 7.43–7.26 (m, 10 H), 5.11 (d, 1 H, J = 12.3 Hz), 5.06 (d, 1 H, J = 12.3 Hz), 4.96
(d, 1 H, J = 11.1 Hz), 4.76 (d, 1 H, J = 11.1 Hz), 3.70 (dd, 1 H, J = 8.1, 10.2 Hz), 3.51 (dd, 1 H, J
= 5.4, 10.2 Hz), 3.47–3.40 (m, 1 H), 3.32 (d, 2 H, J = 4.5 Hz), 1.49 (s, 9 H); 13
C NMR (75 MHz,
CDCl3) 159.9, 149.7, 149.6, 136.5, 135.7, 129.6, 129.1, 128.8, 128.6, 128.4, 128.0, 84.0, 78.2,
67.9, 57.3, 50.2, 45.0, 28.1; HRMS (EI+) m/z calcd for C24H28N6O5 ([M]
+) 480.2121, found
480.2132.

71
2,3-(1-Benzyloxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidino-1-(N-tert-
butoxycarbonyl)propylamine (115). To a solution of 113 (0.183 mg, 0.381 mmol), Boc2O
(103 mg, 0.457 mmol), and Pd/C (40.5 mg, 0.0381 mmol) in EtOAc (3.81 mL) was added
triethylamine (0.0703 mL, 0.495 mmol). The reaction vessel was evacuated and back-filled with
H2 (3x), stirred for 2 h, and the hydrogen balloon was removed. The reaction mixture was stirred
for an additional 3 h and filtered through celite. The organic solution was washed with water (3
x 5 mL), dried (Na2SO4), concentrated, and purified by chromatography on SiO2 (1:1,
hexanes/EtOAc) to give 113 (0.110 g, 52%): IR (ATR) 3364, 1757, 1703 cm-1
; 1H NMR (300
MHz, CDCl3) 7.45–7.21 (m, 10 H), 5.12 (d, 1 H, J = 12.3 Hz), 5.07 (d, 1 H, J = 12.3 Hz), 4.97
(d, 1 H, J = 11.4 Hz), 4.93 (d, 1 H, J = 11.1 Hz), 4.10 (bs, 1 H), 3.68 (dd, 1 H, J = 8.1, 9.9 Hz),
3.54–3.49 (m, 1 H), 3.46–3.41 (m, 1 H), 3.37 (m, 1 H), 3.00–2.94 (m, 1 H), 1.46 (s, 9 H), 1.41 (s,
9 H); 13
C NMR (75 MHz, CDCl3) 159.9, 156.2, 149.8, 136.5, 135.8, 130.1, 129.2, 128.8,
128.6, 128.4, 128.0, 83.8, 79.9, 68.0, 58.3, 44.7, 39.2, 28.4, 28.1; HRMS (ES+) m/z calcd for
C24H38N4O7Na ([M + Na]+) 577.2638, found 577.2604.

72
2,3-(1-Benzyloxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidino-1-(N-tert-
butoxycarbonyl, N-methyl)propylamine (116). A solution of 115 (50.0 mg, 0.090 mmol) and
CH3I (0.112 mL, 1.80 mmol) in DMF (1.80 mL) at -20 ºC was treated with NaH (3.42 mg, 0.135
mmol). After 30 min, the solution was warmed to 0 ºC, stirred for 30 min, then warmed to rt.
After 30 min, the reaction was quenched with water, extracted with EtOAc, and the combined
organic extracts were dried (Na2SO4) and concentrated. The crude residue was purified by
chromatography on SiO2 (1:1 hexanes/EtOAc) to give 116 (40.4 mg, 79%): IR (ATR) 1759,
1688 cm-1
; 1H NMR (300 MHz, CDCl3, 50 ºC) 7.41–7.26 (m, 10 H), 5.11 (d, 1 H, J = 12.3
Hz), 5.06 (d, 1 H, J = 12.3 Hz), 5.00 (d, 1 H, J = 11.1 Hz), 4.81 (d, 1 H, J = 10.8 Hz), 3.66 (d, 2
H, J = 6.6 Hz), 3.59–3.55 (m, 1 H), 3.35 (d, 2 H, J = 6.3 Hz), 2.75 (s, 3 H), 1.47 (s, 9 H), 1.44 (s,
9 H); 13
C NMR (75 MHz, CDCl3) 159.9, 149.9, 149.6, 136.9, 136.0, 129.7, 129.0, 128.8,
128.6, 128.5, 128.0, 83.8, 78.1, 67.9, 57.7, 48.7, 45.9, 28.6, 28.3; HRMS (ES+) m/z calcd for
C30H40N4O7Na ([M + Na]+) 591.2795, found 591.2757.

73
2,3-(1-Benzyloxy-2-benzyloxycarbonyl)guanidinodimethylpropylamine (117),
5-((Dimethylamino)methyl)-2-iminioimidazolidin-1-olate (9), and 1-(2-iminoimidazolidin-
4-yl)-N,N-dimethylmethanamine (118).17
A solution of 116 (0.100 g, 0.176 mmol) in THF
(1.41 mL) was treated with TFA (0.352 mL) and stirred for 1 h. The reaction mixture was
quenched with NaHCO3 (3 mL), extracted with EtOAc (5 x 3 mL), dried (Na2SO4), and
concentrated. The crude residue was dissolved in distilled EtOAc (1.06 mL), treated with
paraformaldehyde (6.67 mg, 0.211 mmol) and NaBH3CN (24.4 mg, 0.369 mmol). The resulting
mixture was stirred overnight, quenched with NaHCO3 (3 mL), extracted with EtOAc (5 x 3
mL), dried (Na2SO4), and concentrated to give 117 as a white solid: IR (ATR) 3371, 1655, 1610
cm-1
; 1H NMR (300 MHz, CDCl3) 8.07 (bs, 1 H), 7.43 (s, 5 H), 7.37–7.27 (m, 5 H), 5.18 (s, 2
H), 5.10 (d, 1 H, J = 12.0 Hz), 5.05 (d, 1 H, J = 12.0 Hz), 3.92 (t, 1 H, J = 9.0 Hz), 3.61 (q, 1 H,
J = 9.4 Hz), 3.37 (t, 1 H, J = 9.6 Hz), 2.61 (dd, 1 H, J = 9.0, 13.8 Hz), 2.43 (s, 3 H), 2.40 (s, 3 H),
2.17 (ad, 1 H, J = 14.4 Hz); HRMS (ES+) m/z calcd for C21H26N4O3Na ([M + Na]
+) 405.1903,
found 405.1867. A solution of 117 in MeOH (20 mL) was passed through an H-cube, fitted with
a Pd/C cartridge at 60 psi, and at a flow rate of 1 mL/min. The solution was concentrated and the
residue was purified by HPLC chromatography (flow rate: 0.7 mL/min, column 3 micron C18
100 x 4.6 mm, mobile phase 95:5 water (0.1% TFA): MeOH, wavelength 254 nm) to give an
inseparable mixture of 9 (9.71 mg, 35%): 1H NMR (600 MHz, D2O (DSS)) 4.51–4.46 (m, 1

74
H), 3.94 (dd, 1 H, J = 13.8, 15.0 Hz), 3.75 (dd, 1 H, J = 7.2, 14.4 Hz), 3.52–3.46 (m, 2 H), 3.00
(s, 6 H); and 118 (13.9 mg, 56%): 1H NMR (600 MHz, D2O (DSS)) 4.58–4.53 (m, 1 H), 3.98
(dd, 1 H, J = 9.6, 9.6 Hz), 3.52–3.46 (m, 2 H), 3.38 (dd, 1 H, J = 4.8, 13.8 Hz), 2.97 (s, 6 H).
(Z)-4-(3-((Benzyloxy)carbonyl)thioureido)but-2-en-1-yl benzoate (103). Seven 50 mL plastic
tubes were each charged with 101 (6 x 1.00 g, 1.93 mmol and 1 x 0.88 g, 1.67 mmol) in THF (6
x 19.3 mL and 1 x 17.2 mL). Each of these solutions were treated with HF•pyr (6 x 1.45 mL,
9.64 mmol and 1 x 1.25 mL, 8.43 mmol) and stirred for 5 h. The reactions were combined into a
2-L Erlenmeyer flask, quenched with sat NaHCO3 (0.500 L), and extracted with EtOAc (3 x
0.100 L). The organic layer was dried (Na2SO4), concentrated, and the crude yellow solid was
dissolved in freshly distilled pyridine (133 mL), treated with DMAP (654 mg, 5.30 mmol), and
cooled to 0 ºC. To the resulting solution was added BzCl (1.85 mL, 15.9 mmol) over 5 min via
syringe. The solution was warmed to rt and stirred overnight. To the solution was added sat.
NH4Cl (100 mL), the reaction was stirred for 1 h, and extracted with EtOAc (3 x 100 mL). The
combined organic extracts were washed with water (3 x 100 mL), dried (Na2SO4), concentrated,
and purified by chromatography on SiO2 (4:1 hexanes/EtOAc) to give 4.19 g (82%) of 103 as a
white solid: IR (ATR) 3289, 1709, 1515 cm-1
; 1H NMR (300 MHz, Acetone – d6) 9.96 (s, 1

75
H), 9.85 (s, 1 H), 8.06-8.03 (m, 2 H), 7.64 (tt, 1 H, J = 1.5, 6.6 Hz), 7.54-7.49 (m, 2 H), 7.45-
7.35 (m, 5 H), 5.93-5.82 (m, 2 H), 5.21 (s, 2 H), 5.02 (d, 2 H, J = 5.4 Hz), 4.53 (t, 2 H, J = 5.4
Hz); 13
C NMR (75 MHz, Acetone – d6) 180.8, 166.6, 154.1, 136.5, 133.9, 131.2, 130.2, 129.4
(2), 129.2, 128.9, 127.8, 68.2, 61.3, 43.0; HRMS (ES+) m/z calcd for C20H20N2O4SNa ([M +
Na]+) 407.1041, found 407.1021.
(Z)-4-(2-(Benzyloxy)-3-(benzyloxycarbonyl)guanidino)but-2-enyl benzoate (99). A solution
of 103 (1.00 g, 2.60 mmol) and O-benzylhydroxylamine hydrochloride (648 mg, 3.38 mmol, 1.3
equiv) in CH2Cl2 (13.0 mL) was treated with diisopropylethylamine (1.43 mL) and EDCI (648
mg, 3.38 mmol). The reaction was stirred overnight, quenched with water (15.0 mL), extracted
with CH2Cl2 (3 x 15.0 mL), dried (Na2SO4), and concentrated. The crude material was purified
by chromatography on SiO2 (2:1 hexanes/EtOAc) to give 1.23 g (74%) of 99 as a yellow oil. 1H
spectra for 99 matched with previously recorded data; 1H NMR (300 MHz, CDCl3) 8.06–8.04
(m, 2 H), 7.90 (bs, 1 H), 7.54 (tt, 1 H, J = 1.2, 7.2 Hz), 7.43–7.28 (m, 12 H), 6.37 (bt, 1 H, J =
5.2 Hz), 5.82–5.72 (m, 2 H), 5.09 (s, 2 H), 4.92 (d, 2 H, J = 5.6 Hz), 4.86 (s, 2 H), 3.85 (t, 2 H, J
= 5.6 Hz);

76
N-(Methoxymethoxy)phthalimide (120).59
A solution of N-hydroxyphthalimide (75.0 g, 446
mmol) and diisoproylethylamine (81.9 mL, 491 mmol) in DMF (686 mL) was treated with a
freshly prepared solution of MOMCl60
in toluene (195 mL, 535 mmol, 2.75 M). The solution
was stirred for 5 h, diluted with CH2Cl2 (500 mL), and washed with sat. NaHCO3 (5 x 100 mL).
The organic phase was dried (Na2SO4) and concentrated to give 120 (91.4 g, 99.0 %) as a white
solid. The spectra matched those previously reported: 1H NMR (400 MHz, CDCl3) 7.87 (s, 4
H), 5.09 (s, 2 H), 3.58 (s, 3 H): 13
C NMR (100 MHz, CDCl3) 163.9, 134.7, 129.3, 123.7,
101.6, 58.0; HRMS (ES+) m/z calcd for C10H10NO4 ([M + H]
+) 208.0604, found 208.0603.
(Z)-4-(2-(Methoxymethoxy)-3-(benzyloxycarbonyl)guanidino)but-2-enyl benzoate (121). A
solution of 120 (943 mg, 4.55 mmol) in CH2Cl2 (4.55 mL) and CH3OH (0.592 mL) was treated
with hydrazine monohydrate (223 L, 4.55 mmol). After 5.5 h the resulting precipitate was
filtered and the filtrate was washed with 5 N aq. ammonia (3 x 5 mL). The aqueous layer was

77
extracted with CH2Cl2 (3 x 5 mL), dried (Na2SO4), and concentrated. The resulting crude oil
was dissolved in CH2Cl2 (3.50 mL), treated with a solution of 103 (0.500 g, 1.30 mmol),
diisopropylethylamine (0.543 mL, 3.25 mmol), and EDCI (534 mg, 2.73 mmol) in CH2Cl2 (3.00
mL). The solution was stirred overnight, quenched with water (10 mL), extracted with EtOAc (3
x 10 mL), dried (Na2SO4), and purified by chromatography on SiO2 (2:1 hexanes/EtOAc) to give
121 (337 mg, 61%) as a pale yellow oil: IR (ATR) 3385, 1715, 1646 cm-1
; 1H NMR (300 MHz,
Acetone – d6) 8.49 (s, 1 H), 8.04 (dd, 2 H, J = 1.2, 8.4 Hz), 7.65–7.60 (m, 1 H), 7.52–7.49 (m,
2 H), 7.43–7.33 (m, 5 H), 6.59 (bt, 1 H, J = 5.2 Hz), 5.86–5.76 (m, 2 H), 5.17 (s, 2 H), 5.00 (d, 2
H, J = 5.6 Hz), 4.87 (s, 2 H), 3.92 (t, 2 H, J = 6.0 Hz), 3.36 (s, 3 H); 13
C NMR (100 MHz,
Acetone – d6) 166.5, 153.8, 148.6, 136.7, 133.8, 132.1, 131.2, 130.1, 129.3 (2), 129.1, 129.0,
126.6, 98.7, 68.0, 56.6, 38.6; HRMS (ES+) m/z calcd for C22H25N3O5Na ([M + Na]
+) 450.1641,
found 450.1656.
3,4-(1-Methoxymethoxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)-guanidinobut-1-ene
(122). A solution of 121 (325 mg, 0.760 mmol) in THF (3.80 mL) was treated with
PdCl2(CH3CN)2 (19.7 mg, 0.0760 mmol) and stirred overnight. To the solution was added
Boc2O (205 mg, 0.912 mmol), DMAP (37.5 mg, 0.304 mmol), and triethylamine (0.162 mL,
1.14 mmol). The reaction was stirred for 4 h, quenched with sat. NaHCO3 (5 mL), extracted with
EtOAc (3 x 5 mL), dried (Na2SO4), and purified by chromatography on SiO2 (2:1

78
hexanes/EtOAc) to give 122 (308 mg, 95.0%) as a yellow oil: IR (ATR) 1761, 1696 cm-1
; 1H
NMR (300 MHz, Acetone – d6) 7.44–7.30 (m, 5 H), 5.93 (ddd, 1 H, J = 8.4, 10.2, 17.1 Hz),
5.44 (d, 4 H, J = 17.1 Hz), 5.32 (d, 1 H, J = 10.5 Hz), 5.13 (d, 1 H, J = 12.3 Hz), 5.07 (d, 1 H, J =
12.6 Hz), 4.73 (d, 1 H, J = 7.5 Hz), 4.61 (d, 1 H, J = 7.5 Hz), 4.19 (q, 1 H, J = 8.1 Hz), 3.94 (dd,
1 H, J = 8.4, 10.2 Hz), 3.49 (dd, 1 H, J = 8.1, 10.2 Hz), 3.30 (s, 3 H), 1.48 (s, 9 H); 13
C NMR
(100 MHz, Acetone – d6) 159.8, 150.3, 150.2, 137.9, 135.4, 129.0, 128.9, 128.5, 120.5, 101.7,
83.3, 67.5, 63.7, 56.7, 47.9, 28.0; HRMS (ES+) m/z calcd for C20H27N3O6Na ([M + Na]
+)
428.1798, found 428.1802. Retention time of enantiomers was determined by chiral SFC
analysis (chiralpak-IC: 250 × 4.5 mm; 2.0 mL/min; 20% methanol; 220 nm detection; Rt1 6.52
min, Rt2 8.39 min).
3,4-(1-Methoxymethoxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)-guanidinobut-1-ene
(S-122). A solution of 121 (0.500 g, 0.702 mmol) in CH2Cl2 (1.94 mL) was treated with (S)-(+)-
COP-Cl (42.8 mg, 0.0292 mmol) and stirred 3 h. To the solution was added THF (3.91 mL),
Boc2O (0.309 mg, 1.40 mmol), DMAP (57.8 mg, 0.468 mmol), and triethylamine (0.249 mL,
1.75 mmol). The reaction was stirred overnight, quenched with sat. NaHCO3 (6 mL), extracted
with EtOAc (3 x 6 mL), dried (Na2SO4), and concentrated. The crude material was purified by
chromatography on SiO2 (2:1 hexanes/EtOAc) to give 359 mg (76%) of S-122 as a yellow oil:
72.5:27.5 er was determined by chiral SFC analysis (chiralpak-IC: 250 × 4.5 mm; 2.0 mL/min;

79
20% methanol; 220 nm detection; Rt1 6.49 min, Rt2 8.45 min); [α]20
D +14.7 (c 1.00, CHCl3); IR
(ATR) 1761, 1694 cm-1
; 1H NMR (400 MHz, Acetone – d6) 7.43–7.29 (m, 5 H), 5.94 (ddd, 1
H, J = 8.4, 10.4, 16.8 Hz), 5.45 (d, 1 H, J = 17.2 Hz), 5.33 (d, 1 H, J = 10.8 Hz), 5.11 (d, 1 H, J =
12.4 Hz), 5.06 (d, 1 H, J = 12.8 Hz), 4.73 (d, 1 H, J = 7.2 Hz), 4.61 (d, 1 H, J = 7.6 Hz), 4.22 (q,
1 H, J = 8.0 Hz), 3.96 (dd, 1 H, J = 8.0, 10.0 Hz), 3.51 (dd, 1 H, J = 8.0, 10.4 Hz), 3.31 (s, 3 H),
1.49 (s, 9 H); 13
C NMR (100 MHz, Acetone – d6) 159.9, 150.5, 150.4, 138.1, 135.7, 129.1,
129.0, 128.6, 120.5, 101.9, 83.5, 67.7, 63.9, 56.8, 48.0, 28.1; HRMS (ES+) m/z calcd for
C20H27N3O6Na ([M + Na]+) 428.1798, found 428.1762.
3,4-(1-Methoxymethoxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)-guanidinobut-1-ene
(R-122). A solution of 121 (0.500 g, 1.17 mmol) in CH2Cl2 (1.94 mL) was treated with (R)-(-)-
COP-Cl (42.8 mg, 0.0292 mmol) and stirred 3 h. To the solution was added THF (3.91 mL),
Boc2O (0.309 mg, 1.40 mmol), DMAP (57.7 mg, 0.468 mmol), and triethylamine (0.166 mL,
1.17 mmol). The reaction was stirred overnight, quenched with sat. NaHCO3 (6 mL), extracted
with EtOAc (3 x 6 mL), dried (Na2SO4), and concentrated. The crude material was purified by
chromatography on SiO2 (2:1 hexanes/EtOAc) to give 351 mg (74%) of R-122 as a yellow oil:
27.0:73.0 er was determined by chiral SFC analysis (chiralpak-IC: 250 × 4.5 mm; 2.0 mL/min;
20% methanol; 220 nm detection; Rt1 6.49 min, Rt2 8.38 min); [α]20
D -15.1 (c 1.00, CHCl3)
(46% ee); IR (ATR) 1761, 1694 cm-1
; 1H NMR (400 MHz, Acetone – d6) 7.43–7.23 (m, 5 H),

80
5.94 (ddd, 1 H, J = 8.0, 10.0, 16.8 Hz), 5.45 (d, 1 H, J = 17.0 Hz), 5.33 (d, 1 H, J = 9.8 Hz), 5.11
(d, 1 H, J = 12.4 Hz), 5.06 (d, 1 H, J = 12.8 Hz), 4.73 (d, 1 H, J = 7.6 Hz), 4.61 (d, 1 H, J = 7.6
Hz), 4.23 (q, 1 H, J = 8.0 Hz), 3.96 (dd, 1 H, J = 8.4, 10.4 Hz), 3.51 (dd, 1 H, J = 8.0, 10.4 Hz),
3.31 (s, 3 H), 1.49 (s, 9 H); 13
C NMR (100 MHz, Acetone – d6) 159.9, 150.5, 150.4, 138.1,
135.7, 129.1, 129.0, 128.6, 120.5, 101.9, 83.5, 67.7, 63.9, 56.8, 48.0, 28.1; HRMS (ES+) m/z
calcd for C20H27N3O6Na ([M + Na]+) 428.1798, found 428.1768.
2,3-(1-Methoxymethoxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidinopropanol
(129). Ozone was bubbled for 5 min through a solution of 122 (1.00 g, 2.47 mmol) in
CH2Cl2/CH3OH 9.87 mL/ 14.8 mL) and pyridine (1.99 mL, 24.7 mmol) at -78 oC. Argon was
bubbled through the solution for 5 min, NaBH4 (0.111 g, 2.96 mmol) was added, the solution
was warmed to 0 oC, and stirred for 4 h. The reaction was quenched with water (25 mL),
extracted with CH2Cl2 (3 x 30 mL), dried (Na2SO4), and purified by chromatography on SiO2
(1:1 hexanes/EtOAc to 100% EtOAc) to give 129 (0.570 g, 1.39 mmol) as a yellow oil: IR
(ATR) 1758, 1696 cm-1
; 1H NMR (300 MHz, Acetone – d6) 7.44–7.30 (m, 5 H), 5.11 (d, 1 H,
J = 12.6 Hz), 5.06 (d, 1 H, J = 12.6 Hz), 4.78 (d, 1 H, J = 7.5 Hz), 4.70 (d, 1 H, J = 7.8 Hz),
3.94–3.73 (m, 4 H), 3.42 (s, 3 H), 1.49 (s, 9 H); 13
C NMR (100 MHz, Acetone – d6) 160.0,
151.3, 150.4, 137.9, 129.0, 128.8, 128.5, 102.1, 83.3, 67.6, 61.0, 60.1, 57.1, 45.5, 28.1; HRMS
(ES+) m/z calcd for C19H27N3O7Na ([M + Na]
+) 432.1747, found 432.1740.

81
2,3-(1-Methoxymethoxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidin-1-azido-
propane (130). A solution of 129 (0.250 g, 0.611 mmol) and triethylamine (0.260 mL, 1.83
mmol) in CH2Cl2 (3.05 mL) at 0 oC was treated with MsCl (0.0351 mL, 0.444 mmol) followed
by DMAP (9.13 mg, 0.0740 mmol). After 1 h the reaction was quenched with NaHCO3 (3 mL),
extracted with CH2Cl2 (3 x 3 mL), dried (Na2SO4), and concentrated. The residue was dissolved
in DMF (3.05 mL), treated with 3 Å molecular sieves, triethylamine (0.260 mL, 1.83 mmol), and
NaN3 (60.1 mg, 0.916 mmol). The reaction was placed in a preheated 50 oC oil bath, stirred
overnight, diluted with EtOAc (3 mL), washed with water (3 x 3 mL), dried (Na2SO4), and
purified by chromatography on SiO2 (1:1 hexanes/EtOAc) to give 130 (0.230 g, 86.7%) as a
clear oil: IR (ATR) 2104, 1756, 1689 cm-1
; 1H NMR (400 MHz, Acetone – d6) 7.44–7.42 (m,
2 H), 7.39–7.29 (m, 3 H), 5.12 (d, 1 H, J = 12.8 Hz), 5.08 (d, 1 H, J = 12.8 Hz), 4.77 (d, 1 H, J =
7.6 Hz), 4.66 (d, 1 H, J = 7.6 Hz), 3.97–3.90 (m, 3 H), 3.69 (q, 1 H, J = 10.4 Hz), 3.65–3.62 (m,
1 H), 3.43 (s, 3 H), 1.49 (s, 9 H); 13
C NMR (100 MHz, Acetone – d6) 159.9, 150.7, 150.3,
138.2, 129.1, 129.0, 128.6, 102.5, 83.5, 67.6, 59.3, 57.0, 50.6, 45.7, 28.1; HRMS (ES+) m/z
calcd for C19H26N6O6Na ([M + Na]+) 457.1812, found 457.1803.

82
2,3-(1-Methoxymethoxy-2-benzyloxycarbonyl-3-tert-butoxycarbonyl)guanidin-1-(N-tert-
butoxycarbonyl)propylamine (131). A round-bottomed flask charged with 130 (0.500 g, 1.15
mmol), Lindlar's catalyst (245 mg, 0.115 mmol), Boc2O (381 mg, 1.73 mmol), and EtOAc (11.5
mL) was evacuated and backfilled with H2 (3x). After 24 h the H2 atmosphere was removed, the
vessel was placed under nitrogen atmosphere, and stirred overnight. The reaction solution was
filtered through celite and purified by chromatography on SiO2 (2:1 hexanes/EtOAc) to give 468
mg (80.0%) of 131 as an oil: IR (ATR) 3377, 1760, 1700 cm-1
; 1H NMR (600 MHz, Acetone –
d6) 7.43 (d, 2 H, J = 7.2 Hz), 7.37 (t, 3 H, J = 7.2 Hz), 7.31 (m, 1 H), 5.11 (d, 1 H, J = 12.0
Hz), 5.07 (d, 1 H, J = 12.6 Hz), 4.82 (d, 1 H, J = 7.8 Hz), 4.65 (d, 1 H, J = 7.8 Hz), 3.89 (dd, 1 H,
J = 9.0, 9.6 Hz), 3.83–3.79 (m, 1 H), 3.69 (ddd, 1 H, J = 2.4, 7.8, 14.4 Hz), 3.65 (dd, 1 H, J =
7.2, 9.6 Hz), 3.45 (s, 3 H), 3.25 (tt, 1 H, J = 5.4, 5.4, 14.4 Hz), 1.48 (s, 9 H), 1.41 (s, 9 H); 13
C
NMR (100 MHz, Acetone – d6) 159.9, 157.1, 150.9, 150.5, 138.1, 129.1, 129.0, 128.6, 102.3,
83.4, 79.3, 67.6, 60.1, 57.2, 46.0, 40.0, 28.5, 28.1; HRMS (ES+) m/z calcd for C24H36N4O8Na
([M + Na]+) 531.2431, found 531.2418.

83
2,3-(1-Methoxymethoxy-2-benzyloxycarbonyl)guanidin-1-(N-methyl)propylamine (132). A
solution of 131 (0.500 g, 0.983 mmol) and CH3I (1.24 mL, 19.7 mmol) in DMF (98.3 mL) at 0
oC was treated with NaH (24.8 mg, 0.983 mmol) and stirred for 1 h. The reaction was warmed to
rt, stirred for 3 h, quenched with sat. NaHCO3 (10 mL), extracted with EtOAc (3 x 10 mL), dried
(Na2SO4), and concentrated. The crude material was purified by chromatography on SiO2 (1:1
hexanes/EtOAc), dissolved in CH2Cl2 (7.86 mL), treated with TFA (1.97 mL) and stirred for 1 h.
The solution was quenched with NaHCO3 (25 mL), extracted with EtOAc (3 x 25 mL), dried
(Na2SO4), and purified by chromatography on SiO2 (95:5 CH2Cl2/MeOH with 1% Et3N) to give
0.230 g (73%) of 132 as a white solid: IR (ATR) 3485, 3372, 1650, 1599 cm-1
; 1H NMR (400
MHz, CD3CN) 7.37–7.27 (m, 5 H), 5.05 (s, 2 H), 4.98 (d, 1 H, J = 7.6 Hz), 4.80 (d, 1 H, J =
7.6 Hz), 3.79–3.72 (m, 1 H), 3.57 (dd, 1 H, J = 8.8, 9.6 Hz), 3.48 (s, 3 H), 3.44 (dd, 1 H, J = 9.2,
9.6 Hz), 2.84 (dd, 1 H, J = 5.6, 12.4 Hz), 2.79 (dd, 1 H, J = 3.6, 12.8 Hz), 2.38 (s, 3 H); 13
C
NMR (100 MHz, CD3CN) 168.1, 164.7, 138.7, 129.4, 128.8, 128.7, 102.0, 67.3, 62.0, 57.6,
51.2, 44.2, 36.8; HRMS (ES+) m/z calcd for C15H23N4O4 ([M + H]
+) 323.1719, found 323.1714.

84
2,3-(1-Methoxymethoxy-2-benzyloxycarbonyl)guanidin-1-(N-dimethyl)propylamine (133).
A solution of 132 in freshly distilled EtOAc (14.0 mL) was treated with paraformaldehyde (44.1
mg, 1.40 mmol). After 30 min NaCNBH3 (0.111 mg, 1.68 mmol) was added and the reaction
was stirred overnight. The reaction was quenched with water (15 mL), extracted with EtOAc (3
x 15 mL), dried (Na2SO4) and purified by chromatography on SiO2 (97:3 CH2Cl2/MeOH with
1% Et3N) to give 310 mg (66.1%) of 133 as an oil: IR (ATR) 3371, 1650, 1601 cm-1
; 1H NMR
(300 MHz, Acetone – d6) 8.23 (s, 1 H), 7.40–7.28 (m, 5 H), 5.08 (s, 2 H), 4.98 (d, 1 H, J = 7.5
Hz), 4.88 (d, 1 H, J = 7.5 Hz), 3.86–3.71 (m, 2 H), 3.52 (s, 3 H), 3.37 (dd, 1 H, J = 8.4, 8.7 Hz),
2.73 (dd, 1 H, J = 3.9, 12.3 Hz), 2.52 (dd, 1 H, J = 8.7, 12.3 Hz), 2.24 (s, 6 H); 13
C NMR (100
MHz, Acetone – d6) 168.1, 164.6, 138.7, 129.1, 128.6, 128.4, 101.7, 66.9, 60.9, 60.6, 57.1,
46.4, 45.8; HRMS (ES+) m/z calcd for C16H24N4O4 ([M + Na]
+) 359.1695, found 359.1717.

85
2,3-(1-Hydroxy-2-benzyloxycarbonyl)guanidin-1-(N-dimethyl)propylamine (134). A
solution of 133 in CH3OH (0.297 mL, 0.1 M) was treated with a solution of HCl in 1,4-dioxane
(0.0743 mL, 0.149 mmol, 4M solution in 1,4-dioxane), placed in a preheated 50 oC oil bath, and
stirred for 48 h. The solution was concentrated and purified by chromatography on C18 (100%
water) to give 6.4 mg (74%) of 134 as a white solid. Diagnostic peaks for 134: IR (ATR) 3431-
2891, 1745, 1646 cm-1
; 1H NMR (600 MHz, D2O) 7.35–7.31 (m, 5 H), 5.21 (s, 2 H), 4.54 (m,
1 H), 4.03 (dd, 1 H, J = 6.4, 7.2 Hz), 3.69 (dd, 1 H, J = 4.8, 9.6 Hz), 3.60–3.57 (m, 1 H), 3.40
(dd, 1 H, J = 3.2, 9.2 Hz), 2.24 (s, 6 H); 13
C NMR (125 MHz, MeOD) 160.1, 153.1, 135.9,
129.9, 129.8, 129.7, 70.5, 59.8, 58.7; HRMS (ES+) m/z calcd for C14H21N4O3 ([M + H]
+)
293.1614, found 293.1620.
2-(Methylthio)-4,5-dihydro-1H-imidazole (136).61
A solution of ethylenethiourea (1.00 g, 9.79
mmol), CH3OH (1.19 mL), and HI (2.66 mL, 14.7 mmol, 47% wt solution) was placed in a
preheated 75 oC oil bath and stirred overnight. The solution was cooled to rt, treated with 40%

86
NaOH solution until a pH of 11 was reached, and extracted with Et2O (3 x 5 mL). The organic
solution was dried (MgSO4), and concentrated to give 874 mg (77%) of 136 as a solid. The
spectra matched those previously reported: 1H NMR (300 MHz, CDCl3) 5.18 (s, 1 H), 3.42 (s,
4 H), 2.23 (s, 3 H).
2-(Methylthio)-3-benzyloxycarbonyl-4,5-dihydroimidazole (137). A solution of 136 (0.200 g,
1.72 mmol) and triethylamine (0.367 mL, 2.58 mmol) in THF (4.30 mL) was treated with CbzCl
(0.294 mL, 2.07 mmol) and stirred overnight. The reaction mixture was quenched with sat.
NH4Cl (5 mL), extracted with EtOAc, dried (MgSO4), and purified by chromatography on SiO2
(100% CH2Cl2 to 90:10 CH2Cl2/MeOH) to give 427 mg (99%) of 137 as a white solid: Mp 64.7-
66.2 oC; IR (ATR) 1709, 1579 cm
-1;
1H NMR (300 MHz, CDCl3) 7.36–7.32 (m, 5 H), 5.18 (s,
2 H), 3.86 (s, 3 H); 13
C NMR (100 MHz, CDCl3) 158.9, 151.2, 135.1, 128.2, 128.0, 127.8,
67.3, 53.4, 47.0, 14.6; HRMS (ES+) m/z calcd for C12H15N2O2S ([M + H]
+) 251.0854, found
251.0846.

87
2-(Methylthio)-3-benzyloxycarbonyl-4,5-dihydroimidazole (138). A solution of 120 (1.14 g,
5.49 mmol) in CH3CN (11.0 mL) was treated with hydrazine monohydrate (0.523 mL, 5.49
mmol). After 2 h the precipitate was filtered, the filtrate was treated with 137 (275 mg, 5.49
mmol), and Et3N (0.199 mL, 0.137 mmol). The solution was cooled 0 oC, treated with AgNO3
(236 mg, 1.37 mmol), and stirred overnight. The solution was filtered through celite,
concentrated, and purified by chromatography on SiO2 (95% CH2Cl2 5% MeOH) to give 225 mg
(74%) of 138 as a white solid: 1H NMR (300 MHz, CDCl3) 7.43–7.40 (m, 2 H), 7.35–7.28 (m,
3 H), 5.21 (s, 2 H), 4.99 (s, 2 H), 3.87 (t, 2 H, J = 7.2 Hz), 3.43–3.38 (m, 5 H); 13
C NMR (75
MHz, CDCl3) 152.6, 150.7, 135.5, 128.1, 127.8, 127.7, 98.4, 67.4, 56.7, 45.1, 39.8.
2-(N-Hydroxy)-3-benzyloxycarbonyl-4,5-dihydroguanidine (139). A solution of 138 (10.0
mg, 0.0358 mmol) in CH2Cl2 (0.179 mL) at 0 oC was treated with TMSBr (0.0241 mL, 0.179
mmol). After 4 h the solution was quenched with water (1 mL), extracted with CH2Cl2 (3 x 1
mL), and the aqueous layer was concentrated to give 3.42 mg (99%) of 139 as a white solid: 1H

88
NMR (300 MHz, CD3OD) 7.43–7.36 (m, 5 H), 5.31 (s, 2 H), 4.17 (dd, 2 H, J = 8.1, 10.5 Hz),
3.77 (dd, 2 H, J = 6.9, 9.3 Hz).
2-(N-Dimethylphosphate)-3-benzyloxycarbonyl-4,5-dihydroguanidine (140). A solution of
139 (50.0 mg, 0.213 mmol) in CH3CN (1.07 mL) was treated with dimethyl chlorophosphate
(0.0477 mL, 0.425 mmol) and Et3N (0.0302 mL, 2.12 mmol). After 3 days, the reaction was
purified by column chromatography on SiO2 (9:1 ethyl acetate/acetone to 1:1 ethyl
acetate/acetone) to give 22.5 mg (31%) of 140 as a clear oil: IR (ATR) 3336, 1743, 1702 cm-1
;
1H NMR (400 MHz, CD3CN) 7.46–7.33 (m, 5 H), 5.93 (s, 1 H), 5.18 (s, 2 H), 3.90 (dd, 2 H, J
= 7.6, 88 Hz), 3.74 (s, 3 H), 3.71 (s, 3 H), 3.44–3.40 (m, 2 H); 31
P NMR (162 MHz, CD3CN)
1.37; HRMS (ES+) m/z calcd for C13H19N3O6P ([M + H]
+) 344.1011, found 344.0994.
A solution of dimethyl phosphate (0.0756 mL, 0.777 mmol) and triethylamine (0.166 mL, 1.17
mmol) in CH3CN (3.12 mL) at 0 oC was treated with TFAA (0.119 mL, 0.855 mmol). The
solution was warmed to rt, stirred for 10 min, at rt, cooled to 0 oC, treated with a solution of 1-
methylimidazole (70.9 mg, 0.855 mmol) in CH3CN (0.775 mL), and stirred overnight. A portion
of the above solution (3.32 mL, 0.662 mmol, 0.2 M) was added to a solution of 139 (130 mg,
0.553 mmol) in CH3CN (0.360 mL) at 0 oC. The resulting solution was treated with
triethylamine (0.0932 mL, 0.663 mmol), stirred overnight, and purified by column

89
chromatography on SiO2 (9:1 ethyl acetate/acetone to 1:1 ethyl acetate/acetone) to give 93.6 mg
(49.3 %) of 140 as an oil: 1H NMR (400 MHz, CD3CN) 7.48–7.36 (m, 5 H), 6.12 (s, 1 H),
5.21 (s, 2 H), 3.94 (t, 2 H, J = 7.6 Hz), 3.79 (s, 3 H), 3.77 (s, 3 H), 3.46 (t, 2 H, J = 7.6 Hz); 31
P
NMR (162 MHz, CD3CN) 1.71.
2-(N-Diphenylphosphate)-3-benzyloxycarbonyl-4,5-dihydroguanidine (141). A solution of
139 (50.0 mg, 0.213 mmol) in CH3CN (1.06 mL) was treated with diphenyl chlorophosphate
(0.223 mL, 1.06 mmol) and triethylamine (0.0302 mL, 0.213 mmol). After 3 h the resulting
solution was purified by column chromatography on SiO2 (7:3 EtOAc / hexanes) to give 39.6 mg
(40%) of 141 as a yellow oil: 1H NMR (400 MHz, CD3CN) 7.47–7.18 (m, 15 H), 6.02 (s, 1
H), 5.22 (s, 2 H), 3.91 (dd, 2 H, J = 7.6, 8.8 Hz), 3.40 (dd, 2 H, J = 7.6, 8.8 Hz); 31
P NMR (162
MHz, CD3CN) -10.4.

90
3.0 REFERENCES
(1) Rasmussen, B.; Fletcher, I. R.; Brocks, J. J.; Kilburn, M. R. Nature 2008, 455, 1101-
1104.
(2) Aráoz, R.; Molgó, J.; Tandeau de Marsac, N. Toxicon 2010, 56, 813-828.
(3) Jaiswal, P.; Singh, P. K.; Prasanna, R. Can. J. Microbiol. 2008, 54, 701-717.
(4) Harr, K. E.; Szabo, N. J.; Cichra, M.; Phlips, E. J. Toxicon 2008, 52, 385-388.
(5) Nagai, H.; Yasumoto, T.; Hokama, Y. Toxicon 1996, 37, 753-761.
(6) Briand, J.-F.; Jacquet, S.; Bernard, C.; Humbert, J.-F. Vet. Res. 2003, 34, 361-377.
(7) Gulledge, B. M.; Aggen, J. B.; Eng, H.; Sweimeh, K.; Chamberlin, A. R. Bioorg. Med.
Chem. Lett. 2003, 13, 2907-2911.
(8) Moore, R. E.; Ohtani, I.; Runnegar, M. T. J. Am. Chem. Soc. 1992, 114, 7941-7942.
(9) Runnegar, M. T.; Xie, C.; Snider, B. B.; Wallace, G. A.; Weinreb, S. M.; Kuhlenkamp, J.
Toxicol. Sci. 2002, 67, 81-87.
(10) Roe, S. J.; Stockman, R. A. Chem. Commum. 2008, 3432-3434.
(11) Henriksen, P.; Carmichael, W. W.; An, J.; Moestrup, Ø. Toxicon 1997, 35, 901-913.
(12) Onodera, H.; Oshima, Y.; Henriksen, P.; Yasumoto, T. Toxicon 1997, 35, 1645-1648.
(13) Becker, V.; Ihara, P.; Yunes, J. S.; Huszar, V. L. M. J. Appl. Phycol. 2010, 22, 235-241.

91
(14) van Apeldoorn, M. E.; van Egmond, H. P.; Speijers, G. J. A.; Bakker, G. J. I. Mol. Nutr.
Food Res. 2007, 51, 7-60.
(15) Tuovinen, K.; Kaliste-Korhonen, E.; Raushel, F. M.; Hänninen, O. Toxicology 1999, 134,
169-178.
(16) Petrikovics, I.; Papahadjopoulos, D.; Hong, K.; Cheng, T. C.; Baskin, S. I.; Jiang, J.;
Jaszberenyi, J. C.; Logue, B. A.; Szilasi, M.; McGuinn, W. D.; Way, J. L. Toxicol. Sci.
2004, 77, 258-262.
(17) Matsunaga, S.; Moore, R. E.; Niemezura, W. P. J. Am. Chem. Soc. 1989, 111, 8021-8023.
(18) Moore, B. S.; Ohtani, I.; Koning, C. B. d.; Moore, R. E. Tetrahedron Lett. 1992, 33,
6595-6598.
(19) Hemscheidt, T.; Burgoyne, D. L.; Moore, R. E. J. Chem. Soc., Chem. Commun. 1995,
205-206.
(20) Shimizu, Y. Annu. Rev. Microbiol. 1996, 50, 431-465.
(21) Moore, B. S. Nat. Prod. Rep. 1999, 16, 653-674.
(22) Waki, M.; Kitajima, Y.; Izumiya, N. Synthesis 1981, 266-268.
(23) Minatti, A.; Muñiz, K. Chem. Soc. Rev. 2007, 36, 1142-1152.
(24) Bertrand, M. B.; Wolfe, J. P. Org. Lett. 2006, 8, 2353-2356.
(25) Wolfe, J. P. Synlett 2008, 19, 2913-2937.
(26) Hopkins, B. A.; Wolfe, J. P. Angew. Chem. Int. Ed. 2012, ASAP.
(27) Ye, X.; Liu, G.; Popp, B. V.; Stahl, S. S. J. Org. Chem. 2011, 76, 1031-1044.
(28) Büchi, G.; Rodriguez, A. D.; Yakushijin, K. J. Org. Chem. 1989, 54, 4494-4496.
(29) Trost, B. M.; Patterson, D. E. J. Org. Chem. 1998, 63, 1339-1341.
(30) Trost, B. M.; Oslob, J. D. J. Am. Chem. Soc. 1999, 121, 3057-3064.

92
(31) Hirai, Y.; Watanabe, J.; Nozaki, T.; Yokoyama, H.; Yamaguchi, S. J. Org. Chem. 1997,
62, 776-777.
(32) Overman, L. E.; Remarchuck, T. P. J. Am. Chem. Soc. 2002, 124, 12-13.
(33) Kirsch, S. F.; Overman, L. E. J. Org. Chem. 2005, 70, 2859-2861.
(34) Streuff, J.; Hövelmann, C. H.; Nieger, M.; Muñiz, K. J. Am. Chem. Soc. 2005, 127,
14586-14587.
(35) Hövelmann, C. H.; Streuff, J.; Brelot, L.; Muñiz, K. Chem. Commum. 2008, 2334-2336.
(36) Du, H.; Zhao, B.; Shi, Y. J. Am. Chem. Soc. 2007, 129, 762-763.
(37) Hövelmann, C. H.; Streuff, J.; Brelot, L.; Muñiz, K. Chem. Commun. 2008, 2334-2336.
(38) Du, H.; Zhao, B.; Shi, Y. Org. Syn. 2009, 86, 315.
(39) Chavan, S. P.; Thakkar, M.; Jogdand, G. F.; Kalkote, U. R. J. Org. Chem. 2006, 71,
8986-8988.
(40) Ma, G., Design and Synthesis of Organic Molecules with New Physical and Biological
Properties. University of Pittsburgh, Pittsburgh, 2005.
(41) Lanman, B. A.; Overman, L. E.; Paulini, R.; White, N. S. J. Am. Chem. Soc. 2007, 129,
12896-12900.
(42) Khuong-Huu, F.; Le Forestier, J.-P.; Goutarel, R. Tetrahedron 1972, 28, 5207-5220.
(43) MacNevin, C. J.; Moore, R. L.; Liotta, D. C. J. Org. Chem. 2008, 73, 1264-1269.
(44) McDonald, R. I.; Liu, G.; Stahl, S. S. Chem. Rev. 2011, 111, 2981-3019.
(45) Shioiri, T.; Ninomiya, K.; Yamada, S.-i. J. Am. Chem. Soc. 1972, 94, 6203-6205.
(46) Lebel, H.; Leogane, O. Org. Lett. 2005, 7, 4107-4110.
(47) Nyfeler, E.; Renaud, P. Org. Lett. 2008, 10, 985-988.

93
(48) Loudon, G. M.; Radhakrishna, A. S.; Almound, M. R.; Blodgett, J. K.; Boutin, R. H. J.
Org. Chem. 1984, 49, 4272-4276.
(49) Simons, S. S., Jr J. Org. Chem. 1973, 38, 414-416.
(50) Trost, B. M.; Wasner, J.; Meyer, A. J. Am. Chem. Soc. 2007, 129, 14556-14557.
(51) Lemieux, R. M.; Devine, P. N.; Mechelke, M. F.; Meyers, A. I. J. Org. Chem. 1999, 64,
3585-3591.
(52) Blaser, H.-U.; Brieden, W.; Pugin, B.; Spindler, F.; Studer, M.; Togni, A. Top. Catal.
2002, 19, 3-16.
(53) Willand-Charnley, R.; Fisher, T. J.; Johnson, B. M.; Dussault, P. H. Org. Lett. 2012, 14,
2242-2245.
(54) Eschweiler, W. Ber. 1905, 38, 880-887.
(55) Clarke, H. T.; Gillespie, H. B.; Weisshaus, S. Z. J. Am. Chem. Soc. 1933, 55, 4571-4587.
(56) Weinstein, A. B.; Stahl, S. S. Angew. Chem. Int. Ed. 2012, 51, ASAP.
(57) Jones, S.; Smanmoo, C. Tetrahedron Lett. 2004, 45, 1585-1588.
(58) Jones, S.; Smanmoo, C. Org. Lett. 2005, 7, 3271-3274.
(59) Canham, S. M.; France, D. J.; Overman, L. E. J. Am. Chem. Soc. 2010, 132, 7876-7877.
(60) Berliner, M.; Belecki, K. Org. Syn. 2007, 84, 102-107.
(61) Denk, M. K.; Ye, X. Tetrahedron Lett. 2005, 46, 7597-7599.

94
APPENDIX A
SELECTED 1H AND
13C SPECTRA

95

96

97

98

99

100

101

102

103

104

105

106

107

108

109

110

111

112

113

114

115

116

117

118

119

120

121

122