structural studies of lumazine synthases – thermostability
TRANSCRIPT

Center for Structural Biochemistry Department of Biosciences at Novum
Karolinska Institutet, S-141 57 Huddinge, Sweden
Structural Studies of Lumazine Synthases – Thermostability,
Catalytic Mechanism and Molecular Assembly
Xiaofeng Zhang
Stockholm 2005

Cover Illustration: Electron density of the active site of lumazine synthase from the hyperthermophilic bacterium Aquifex aeolicus.
All previously published papers were reproduced with permission from the publisher.
Published and printed by Karolinska University Press Box 200, SE-171 77 Stockholm, Sweden © Xiaofeng Zhang, 2005ISBN 91-7140-605-0



ABSTRACTRiboflavin, also known as vitamin B2, is biosynthesized in plants, bacteria,
archaea and fungi. The primary biological function of riboflavin is related to its existence as a component of the two coenzymes, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), which play an important role for electron transfer in energy metabolism.
This project is mainly focused on structural studies of lumazine synthase (LS) from the hyperthermophilic bacterium Aquifex aeolicus (LSAQ). The enzyme is involved in the penultimate step of biosynthesis of riboflavin. The aim of this study is to gain insights into the structural basis of thermostability, catalytic mechanism as well as the molecular assembly of the enzyme. Methods used for these studies include X-ray crystallography, electron microscopy (EM), small angle X-ray scattering (SAXS) and differential scanning calorimetry (DSC).
Lumazine synthase from the hyperthermophile A. aeolicus displays dramatic stability against high temperature. The calorimetric melting profile indicates an apparent melting temperature (Tm) of 120 C. The factors that determine the thermostability of A. aeolicus LS were revealed by structural comparisons (Paper I, 2001).
In the second last step of riboflavin biosynthesis, lumazine synthase catalyzes the formation of 6-7-dimethyl-8-ribityllumazine, which is subsequently converted to riboflavin. In light of the structural studies of the enzyme in complexes with inhibitors (four complex structures were studied in this work), which were designed to mimic the substrates, reaction intermediate and the product at different stages of the reaction, a structural model of the catalytic process, which illustrates binding of substrates, enantiomer specificity, proton abstraction/donation, inorganic phosphate elimination, formation of the Schiff base and cyclisation, was proposed (Paper II, 2003).
Lumazine synthase assumes at least four assembly forms, namely the virus-like icosahedral capsid with a diameter of about 160 Å, the pentameric form, the stacking pentamers and larger capsids with a diameter of about 300 Å (metamorphosis of the enzyme is reviewed in Appendix A). The pH and/or buffer dependence of the assembly states of LS from B. subtilis, A. aeolicus and a designed mutant LS from A. aeolicus(structure determined by cryo-EM in manuscript IV) were studied using small angle x-ray scattering (SAXS) and cryo-EM. The results indicate that multiple assembly states are a general feature of lumazine synthases. Furthermore, the catalytic function of the enzyme is closely correlated with the assembly state (Manuscript, III).
Sequence alignment revealed that an insertion of 1–4 residues after Gly138 is unique for the pentameric lumazine synthases. Structural comparisons and modeling studies suggested that this insertion may inhibit the formation of icosahedral capsids. The structure of lumazine synthase from A. aeolicus with a four-residue’s insertion (IDEA) is studied by cryo-EM. It is shown that the mutant forms large capsids with a diameter of 292 Å. The analysis of the subunit interactions indicated that the assembly of the mutant does not follow the theory of “quasi-equivalence”, because the contact surfaces are non-equivalent. Compared to that of the wild type enzyme, the pentamer of the mutant is widened. The expanded pentameric structure provides a model for an alternative conformation of the LS pentamer as it could also be formed during the catalytic reaction in the T=1 capsid (Manuscript IV)

List of Publications I. Zhang, X., Meining, W., Fischer, M., Bacher, A. & Ladenstein, R. (2001). X-ray
structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile Aquifex aeolicus at 1.6 Å resolution: determinants of thermostability revealed from structural comparisons. J. Mol. Biol. 306: 1099-1114
II. Zhang, X., Meining, W., Cushman, M., Haase, I., Fischer, M., Bacher, A. & Ladenstein, R. (2003). A structure-based model of the reaction catalyzed by lumazine synthase from Aquifex aeolicus. J. Mol. Biol. (328): 167-182
III. Zhang, X., Konarev, P., Svergun, D. I., Xing, L., Cheng, H., Haase, I., Fischer, M., Bacher, A. Ladenstein, R. & Meining, W. (2005) Multiple assembly states of lumazine synthase - a model relating catalytic function and molecular assembly. Manuscript
IV. Nilsson, J., Xing, L., Zhang, X., Bergman, L., Haase, I., Fischer, M., Bacher, A. Meining, W., Ladenstein, R. & Cheng, H., A 180 subunit complex of a lumazine synthase mutant violates quasi-equivalence in capsid assembly. Manuscript

CONTENTS1 Introduction .................................................................................................. 1
1.1 Structure of Proteins........................................................................... 1 1.2 X-ray Scattering ................................................................................. 1
1.2.1 Solution Scattering (Small angle X-ray Scattering) .................. 2 1.2.2 Crystal Scattering....................................................................... 4 1.2.3 The Electron Density and the Phase Problem ........................... 8
1.3 Protein X-ray Crystallography........................................................... 8 1.3.1 Protein Crystallization and Data Collection .............................. 8 1.3.2 Symmetry and Symmetry Operations ..................................... 11 1.3.3 Phasing Methods...................................................................... 12 1.3.4 Patterson Function and Patterson Map .................................... 13 1.3.5 Rotation, Translation and Molecular Replacement................. 14 1.3.6 Density Modification ............................................................... 15 1.3.7 Model Building and Crystallographic Refinement ................. 16
1.4 Hyperthermophiles and Proteins from Hyperthermophiles............. 18 1.4.1 Hyperthermophiles................................................................... 18 1.4.2 Aquifex aeolicus ...................................................................... 19 1.4.3 Protein Stability and Folding ................................................... 19 1.4.4 Dominant Forces of Protein Stability ...................................... 21
1.5 Riboflavin and Enzymes Involved in Riboflavin Biosynthesis....... 23 1.5.1 Riboflavin................................................................................. 23 1.5.2 Enzymes involved in Riboflavin Biosynthesis........................ 24 1.5.3 Crystal Structures of Lumazine Synthase................................ 25 1.5.4 Catalytic Mechanism of Lumazine Synthase .......................... 28 1.5.5 Molecular Assembly of Lumazine Synthase........................... 31
2 Crystal Structure of Lumazine Synthase from Aquifex aeolicus – Determinants of Thermostability (Paper I) ................................................................................. 35
2.1 Results and Discussion..................................................................... 35 2.2 Conclusions ...................................................................................... 39
3 Structures of Aquifex aeolicus Lumazine Synthase in Complex with Inhibitors – A Model of the Reaction Process (Paper II) ........................................................ 40
3.1 Results and Discussion..................................................................... 40 3.2 Conclusions ...................................................................................... 45
4 Multiple Assembly states of Lumazine Synthases – A Model Relating Catalytic Function and Molecular Assembly (Paper III)................................................. 46
4.1 Results and Discussion..................................................................... 46 4.2 Conclusions ...................................................................................... 51
5 A 180 subunit complex of a lumazine synthase mutant violates quasi-equivalence in capsid assembly (Paper IV).............................................................................. 52
5.1 Results and Discussion..................................................................... 52 5.2 Conclusions ...................................................................................... 55
6 Acknowledgements .................................................................................... 57 7 References .................................................................................................. 59

LIST OF ABBREVIATIONS
3D Three-dimensional ASA Solvent accessible surface area AU Asymmetric unit B-factor Temperature factor ( 2 28B x , where 2x is the mean
square displacement of the atom from its equilibrium position) Da Dalton DM Density modification DSC Differential scanning calorimetry EM Electron microscopy Fc Calculated structure factorFo Observed structure factorFAD Flavin-adenine dinucleotide FMN Riboflavin mononucleotide LS Lumazine synthase LSAQ Lumazine synthase from Aquifex aeolicusLSAQ-IDEA Mutant of lumazine synthase from Aquifex aeolicus with the IDEA
insertionLSBS Lumazine synthase from Bacillus subtilisMAD Multiple-wavelength anomalous dispersion MIR Multiple isomorphous replacement ML Maximum likelihood MR Molecular replacement NMR Nuclear magnetic resonance spectroscopy NCS Non-crystallographic symmetries ORF Open-reading frame RDL 6,7-dioxo-5H-8-ribitylaminolumazine RNOP 6-ribitylamino-5-nitroso-2,4(1H,3H)-pyrimidine-dione RNO2P 6-ribitylamino-5-nitro-2,4(1H,3H)-pyrimidine-dione RPL 3-(7-hydroxy-8-ribityllumazine-6-yl)propionic acid RPP 5-(6-D-ribitylamino-2,4(1H,3H)pyrimidine-dione-5-yl)pentyl-1-
phosphonic acid RS Riboflavin synthase SAD Single-wavelength anomalous dispersion SAXS Small angle X-ray scattering SIR Single isomorphous replacement T Triangulation numberTm Melting temperature WT Wild type

1
1 INTRODUCTION 1.1 STRUCTURE OF PROTEINS
Proteins are essential for all organisms. A special class of proteins, namely the enzymes, is involved in catalyzing almost all biochemical reactions in living cells. The function and activity of proteins depend on their three-dimensional (3D) structures. Therefore the knowledge of protein structures is a key for the understanding of many fundamental biological problems. After the discovery of the double helix structure of the DNA molecule and the determination of the first 3D protein structure of myoglobin by X-ray crystallography in the middle of last century1-3, solving 3D structures of biological macromolecules has become one of the most demanding tasks in the field of biosciences. Thanks to the development and application of many new techniques such as gene sequencing and cloning, protein expression, purification and high-throughput crystallization, multi-dimensional nuclear magnetic resonance spectroscopy (NMR), cryogenic technique, the increasing accessibility of synchrotron X-ray beams as well as the development of computer programs, the determination of protein structures is becoming a standard technique. To date, more than 30,000 protein structures have been determined and all these structures together provide us with quite a comprehensive source of information for insights into many essential biological processes of the living systems.
Electron microscopy (EM), NMR spectroscopy and X-ray crystallography are the most important methods for protein structure determination. Using techniques such as 2D crystallization, sample freezing and image reconstruction, EM is now widely applied to study, at low resolution (usually at nm level), the structure of biological macromolecules including viral proteins, membrane proteins and complexes, which can virtually have a molecular weight up to millions of Daltons.4-6
NMR is a versatile technique used not only to determine protein structures in solution but also to study enzyme kinetics, ligand binding and molecular dynamics. However, the application of NMR for high-resolution structural analysis is limited to proteins with a molecular weight of less than 25–30 kDa.7-9 X-ray diffraction is the first and, until now, the most powerful technique to solve even very large structures of biological macromolecules. With diffraction by crystals, X-ray crystallography is able to provide structural details of the protein at atomic resolution.
1.2 X-RAY SCATTERING
X-rays are electromagnetic waves with a wavelength of 1000-0.1 Å. When electrons with high velocity are decelerated due to collision with metal atoms (conventional X-ray generators) or influenced by a magnetic field, which bends the electron beam (synchrotron radiation source), a part of the kinetic energy is emitted as X-rays.

2
The scattering by an electron is shown in Figure 1-1, where s0 and s1 represent the unit vectors of the incident and scattered waves, respectively.
Figure 1-1 X-ray scattering by one electron e1. The incident and scattered waves are represented using the unit vectors s0 and s1 with a modulus of 1/ . The “reflecting plane” is perpendicular to the scattering vector S.
The scattering vector S, which illustrates the difference between the incident and scattered waves, is given by:
02sin( ),S s s S (1)
The angle between the vectors s0 and s1 (2 ) is the scattering angle. Scattering by an electron can also be regarded as being “reflected” by a plane, which is perpendicular to the scattering vector S (Figure 1-1).
X-ray scattering by different objects, e.g. solutions and crystals of proteins, are based on the same electromagnetic theory; however they differ in their character.
1.2.1 Solution Scattering (Small angle X-ray Scattering)
Small angle X-ray scattering (SAXS), usually observed within a rather small angular range, e.g. 2-5 , occurs in sample solutions containing particles with a colloidal size (tens to several thousand Å). Due to the fact that the particles in solution are randomly orientated, the resulting scattering pattern is centro-symmetric (Figure 1-2a). X-ray scattering of protein solutions is a monotone function of the scattering angle (Figure 1-2b).
Suppose that the solution is considerably diluted, i.e. particles in solution do not interact with one another, it can be assumed that all particles make independent contributions to the intensity, which is given by:
02
sin(2 )( ) 4 ( )2
and ( ) ( )
I p d
p
rSS r rrS
r r r (2)
where ( )p r is a distance distribution function describing the geometrical character of the particles in solution and ( )r gives the probability of finding a volume element

3
Figure 1-2. Small angle scattering experiments (a) a schematic 3D view of solution scattering, colors from blue to white indicate the intensity from low to high. Due to the random orientation of the particles in solution, the pattern of solution scattering is centro-symmetric; (b) The scattering curve (solid) of lumazine synthase from B.subtilis10 overlaid with the calculated scattering function (dashed) for a hollow sphere, where 4 sinh and is the X-ray wavelength; (c) the distance distribution function of the same sample indicates the size of the largest particle in solution.
(e.g. an electron) at a given position represented by the vector r.11, 12 The maximum distance within the particles (Dmax) can be directly obtained from the distance distribution function ( )p r (Figure 1-2c).
In the vicinity of the origin (i.e. small S or small 2 , Figure 1-1), the scattered intensities can be approximated according to the Guinier law13:

4
22 24( ) (0)exp
3 gI I RS S (3)
where the radius of gyration 1
22( ) ( )g V VR dV dVr r r gives a measure of
the distance between the electrons and the center of gravity of the molecule.
In most cases, the application of small angle X-ray scattering can provide structural details such as size, shape and molecular weight indirectly; some of these parameters are calculated by fitting of the experimental data to theoretical scattering curves (Figure 1-2a).
1.2.2 Crystal Scattering
In the case of crystal scattering, an atom can be represented by the electron cloud (r), the total scattering of all electrons of an atom is:
2( )exp[ ]f dr
r r S r (4)
where the integration is over the entire space represented by the vector r. The function f in (4) is called the atomic scattering factor, which represents the scattering power of an atom.
In common diffraction experiments, scattering ( 0f f ) is usually assumed, i.e.
the incident X-rays are mainly scattered by electrons in outer shells, which are considered as free electrons. The scattered beam has no phase change with respect to the incident beam and the atomic scattering factor will have a real value. The diffraction pattern of scattering is centrosymmetric, i.e. ( ) ( )I hkl I hkl . This is the so-called “Friedel’s law”.
However, when the incident X-ray beam has a certain wavelength, close to the absorption edge of the atom (element), the energy of the incident X-rays is strong enough to excite an electron from the inner shell to a higher orbital or just eject it. The resulting scattering will have a phase change with respect to the incident beam. This phenomenon is called anomalous scattering. The atomic scattering factor is then:
0f f f if' ''
A vector cos( ) sin( )A iA in the complex plane can also be written in the exponential
way: exp[ ]A i , where A is the amplitude and is the angle between the vector and the real axis.
Properties of exponential terms:exp[ ] exp[ ] exp[ ]; exp[ ] exp[ ] exp[ ]; exp[ ] {exp[ ]}ka b a b a b a b k a a

5
where f ' and f '' are the real and imaginary components of the anomalous scattering factor. Anomalous scattering varies with change of the wavelength. The presence of anomalous scattering results in the inequivalent changes on phase of the reflections hkl( ) and hkl( ) , which will eventually lead to the breakdown of the Friedel’s law, i.e.
( ) ( )I hkl I hkl . The reflections hkl( ) and hkl( ) are designated as the Bijvoet pairs. Anomalous scattering provides phase information, which can be used for structure determination (See 1.3.3).
Suppose a unit cell has n atoms at positions rj (j = 1, 2, … n) with respect to the origin of the unit cell, the scattering of the unit cell is then the sum of scattering factors of all atoms in it (Figure 1-3):
12( ) exp[ ]
n
j jj
f iF S r S (5)
( )F S is called the structure factor because it depends on the arrangement of atoms (rj, i.e. the structure) in the unit cell.
Figure 1-3. The structure factor is the sum of the scattering by all atoms in the unit cell.
A crystal contains a large number of unit cells arranged in three dimensional space defined by the unit vectors a, b and c. The position of any unit cell in the crystal can be represented by t u va b c , in which t, u and v are whole numbers. The total scattering of the crystal is then obtained by a summation over all unit cells in it:
1 2 3
0 0 02 2 2( ) ( ) exp[ ] exp[ ] exp[ ]
n n n
t u vK F it iu ivS S a S b S c S (6)
where n1, n2 and n3 are the number of unit cells in the directions of a, b and c,respectively.

6
Figure 1-4. Each vector represents the scattering of one unit cell in the crystal. The directions of these vectors are different depending on the phase 2t a S . Because tis a large number, the resulting summation over the total scattering by the unit cells with t = 0 to n1 is almost always equal to zero.
Regarding the term 2exp[ ]ita S in equation (6) as a vector with a phase of 2 ta S (Figure 1-4), the summation over a very large number (e.g. n1) is almost always equal to zero. The same conclusion applies on the other two terms
2exp[ ]iub S and 2exp[ ]ivc S as well. However, when a S , b S and c S are all integers:
hkl
a S =b S =c S =
(7)
namely h, k and l are whole numbers, all vectors have the phases of 2 integer , i.e. the direction of all vectors points to the right, thus the scattering by the crystal can be observed due to the amplification effect. Therefore, a crystal does not scatter X-rays unless equations (7) are fulfilled. They are the well-known “Laue conditions”. The numbers h, k and l in (7) are called the indices of reflections and the vector S can now be written as S(h k l).
Rearranging the Laue conditions (7) as: 1 1 1h k la b cS = ; S = ; S = the
projections of the vectors ha ,
kb and
lc on the scattering vector S all have the same
length of 1 S (Figure 1-5a) .
* Let vectors a and b, with lengths |a| and |b|, be inclined at an angle : The scalar product is the number: cos( )aba b and a b b a . The vector product is a vector c with a length sin( )abc and points to
the direction perpendicular to both a and b. a b b a .

7
Figure 1-5. (a) A 2D unit cell is drawn for simplicity. The endpoints of the vectors a/h, b/k (and c/l) form a lattice plane perpendicular to vector S. d is the distance between these lattice planes. (b) A graphic representation of Bragg’s law. Two lattice planes are separated by a distance d. The condition for constructive interference is that the path difference between the two scattered beams is 2 sind n .
Letting this length 1 dS and from equation (1) where 2sinS , Bragg’s law
emerges14:
2 sin 1d (8)
where d is the distance between two successive lattice planes defined by (h k l).Bragg’s law (Figure 1-5b) describes the condition of diffraction as: the path difference between the beams scattered by two lattice planes is equal to n (n is an integer). It can also be explained as: the phase difference between the beams scattered by two lattice planes is equal to n 2 (n is an integer).
Considering a special vector S(100), it is perpendicular to the plane (100) and has a length 1 100( )d . If we call this vector a*, in the same way S(010) plane (010)
and S(001) plane (001), we can get the vectors b* (with a length of 1 010d( ) ) and
c* (with a length of 1 001d( ) ), respectively. The vectors a*, b* and c* are related to the vectors a, b and c as:
0
***
* * *
hkl
a ab bc c
a b a c b c
(9)
Similar to the vectors a, b and c, which define the real space, the vectors a*, b* and c* define another space called the “reciprocal space”. Applying (9) to the structure factor (5) where
( ) ( * * *)j j j j
j j j
x y z h k l
hx ky lz
r S a b c a b c

8
The structure factor F(S) can then be written as F(hkl) or F(h):
12
n
j j j jj
hkl f i hx ky lz( ) exp[ ( )]F (10)
1.2.3 The Electron Density and the Phase Problem
In equation (10) the summation is over all atoms j in the unit cell. It can also be calculated by the integration of the electron density over the unit cell:
1
2
2
( ) exp[ ( )]
( )exp[ ( )]
n
j j j jj
j j jcell
hkl f i hx ky lz
V xyz i hx ky lz dxdydz
F (11)
The Fourier transformation of the structure factor (11) gives the electron density function ( )xyz (The mathematical principles of the Fourier transformation are given in Drenth, 2002)15:
1 2( ) ( )exp[ ( )]h k l
xyz V hkl i hx ky lzF (12)
Given the structure factors, equation (12) can be used to calculate the electron density of the crystal structure. However in X-ray diffraction experiments, the complete structure factors can not be obtained directly. Instead, only the intensities are accessible, the phase information is lost:
2
2 2
and
( ) ( ) *( )( ) *( ) exp[ ] exp[ ]
( )
( ) ' ( )
I hkl hkl hklhkl hkl i i
c hkl
hkl c I hkl
F FF F
F
F
(13)
where c/c’ are parameters related to absorption and temperature. Shown in (13) only the amplitude of the structure factor ( )hklF is obtained from the intensity; however
the phase is not accessible, because it cancels out in the exponential terms. This is the so-called “phase problem” of X-ray crystallography.
1.3 PROTEIN X-RAY CRYSTALLOGRAPHY 1.3.1 Protein Crystallization and Data Collection
Protein crystallization is a process, which slowly and evenly decreases the protein solubility until the system reaches a supersaturated state. The thermodynamic force can then drive the system to a new equilibrium state with the formation of crystalline or amorphous precipitate.16 The crystallization process may be affected by many factors including the purity and homogeneity of samples, temperature, pressure, pH and the presence of crystallization nuclei. As a consequence, the results of crystallization are to a large degree unpredictable.17

9
In practice, pure and homogenous protein with a concentration higher than 5 mg/ml is normally needed. A number of crystallization conditions may be screened. Crystallization buffers containing one or more precipitating agents such as salts, polyethylene glycol (PEG) and/or other additives like organic compounds, are prepared at different pHs. Protein samples are mixed with buffer solutions and the concentrations of precipitants are slowly increased by vapor diffusion, dialysis or other methods.18 Crystals that are larger than 200 m in all dimensions are usually required in order to obtain diffraction data at high resolution with good quality.
Diffraction data can be collected at either room temperature or cryogenic conditions, i.e. around 100 Kelvin. For data collection at room temperature, the crystal is transferred into a glass capillary. For data collection at low temperatures, a cryoprotectant, typically a water soluble organic material (e.g. 20% glycerol), is added to the crystallization buffer. The cryoprotectant slows down the rate of formation of the crystalline ice, which causes the damage of the crystal.19 Instead, a glass-like solid state is obtained by shock freezing. Collection at low temperature also reduces the radiation damage of the crystals.
Diffraction data are collected by different kinds of detectors, among which the image plate and area detectors are most often used nowadays. The image plate works in a similar way as the conventional film. It “records” the energy of the reflections by an inorganic storage phosphor and the reflections can be read out (scanned) and erased by light with different wavelengths. The original design of the area detector contains a chamber, which is filled with gas e.g. xenon. The gas atoms can be ionized by the diffracted X-rays and the ionization of the gas causes the formation of ions and electrons, which can be amplified and recorded electronically. At synchrotron X-ray beam stations, a new kind of area detector, the charge coupled device (CCD) is widely used to replace the gas chamber of the area detector.
A schematic drawing of the X-ray diffraction by a crystal is shown in Figure 1-6. As the orientation of the crystal is fixed, still exposure of a stationary crystal only contains a small number of reflections (Figure 1-6a). However when the crystal is rotated by a small angle (the oscillation angle ) around an axis, which is perpendicular to the beam, more reflections are collected on one image (Figure 1-6b). In practice, many images, all collected with a small oscillation angle of the crystal, result in a full data set. This procedure is called “rotation method”. Parameters for data collection including the incident wavelength, starting angle, oscillation angle and total rotation range and the distance from crystal to the detector can be optimized according to the absorption, space group, cell dimensions, shape and orientation of the crystal as well as the quality of the crystal (e.g. the mosaicity). A diffraction image from the data collection of lumazine synthase from Aquifexaeolicus is shown in Figure 1-6c.

10
Figure 1-6. (a) A still exposure with a stationary crystal results in only a small number of reflections arranged in a set of narrow ellipses;20 (b) When the crystal is rotated, reflections from the same reciprocal lattice plane form a lune, limited by two ellipses corresponding to the start and end positions;20 (c) X-ray diffraction pattern of lumazine synthase from Aquifex aeolicus. The data were collected at the synchrotron beam line X11 at HASYLAB (EMBL Outstation, DESY, Hamburg). The wavelength of the incident X-ray beam was 0.909 Å and the highest resolution of this image is 2.0 Å.
After a full dataset is collected, the images need to be processed. This procedure is started by finding the spots on one (or several) chosen images. It is usually done by comparing the intensity of the spots and the local background on the image. Once the locations of these spots are determined, the coordinates of the spots (i.e. their positions on the image) are converted and mapped onto the surface of an imaginary Ewald sphere, from where the indices (hkl) of these spots are retrieved. Using the indices, the approximate unit cell parameters (a, b, c, , and ) and the relative orientation of the crystal with respect to the detector can be calculated. These parameters are then used to predict reflections on the image. The positions of the predicted reflections and the observed reflections are compared and the differences between the positions are minimized resulting in a set of refined parameters. The refined parameters, including the unit cell parameters, crystal orientation, beam center, crystal to detector distance, are then used to process the whole dataset. After all reflections are indexed, the data are processed by integrating intensities merging partial reflections and reducing the reflections to a unique set (applying the

11
crystallographic symmetries). Data processing can be done using automated computer programs like XDS and HKL.21, 22
1.3.2 Symmetry and Symmetry Operations
As a consequence of energy optimization, molecular assembly and regular packing, molecules in crystal are usually arranged in a symmetric way.15 A symmetry operation is an action that can be performed on an object without changing the appearance of it. The symmetry operations include: rotation, mirror planes, translation, inversion and certain combinations of them. Mathematically, there are 230 different ways to combine these operations in the crystals leading to 230 possible space groups.23 However not all of them are allowed in protein crystals. The reason is, for instance, a mirror plane would change the chirality of an L-amino acid to a D-amino acid, which does not naturally occur in any protein.
The asymmetric unit of the crystal is the smallest unit required to generate a whole unit cell by the crystallographic symmetry operations (space group symmetry operations). However, the asymmetric unit can also contain more than one molecule, which are often related by symmetries. The symmetry relations between the molecules within an asymmetric unit are called the “non-crystallographic symmetries” (NCS) or the “local symmetries”. The NCS are sometimes not strictly followed due to the flexibility of the molecules. A good example for explaining both the crystallographic symmetries and the non-crystallographic symmetries is the icosahedral capsid in space group I23 (Figure 1-7). The crystallographic symmetries of space group I23 include the 2-fold and 3-fold rotation axes. However, the 5-fold rotation axes are local symmetries.
Figure 1-7. The symmetries of an icosahedral capsid in space group I23: the crystallographic symmetries of the space group I23 including the 2-folds and 3-folds are shown with filled-symbols. The non-crystallographic 5-folds are shown with hollow-symbols.
A translation operation can be mathematically described as:
A x y z A xyz T( ' ' ') ( ) ( )t (14)

12
where (x,y,z) and (x’,y’,z’) are the coordinates before and after the operation and t is the translation vector. In the same way a rotation operation can be described as:
( ' ' ') ( ) ( )A x y z A xyz R C (15)
where R(C) is a rotation operator and C is a 3x3 rotation matrix defining the rotation operation. A rotation operation can be graphically represented in 3D space in different ways, e.g. in the Eulerian system, the three angles 1, 2 and 3 define a rotation (Figure 1-8). The Eulerian angle system can be converted to the polar angle system for the application by different programs, e.g. for construction of 2D stereogram plots.
Figure 1-8. A rotation operation can be represented using Eulerian angles. The rotation can be regarded as rotating the Cartesian system (x, y, z) to (x’, y’, z’), the rotation is defined by the three angles , and .
1.3.3 Phasing Methods
During the past century, a number of methods have been developed for solving the phase problem in X-ray crystallography:
(1) Direct methods, based on statistical and probability analysis, derive phase values ab initio from the observed structure amplitudes. Direct methods are normally used to determine structures of small molecules. It is also, very often in combination with other methods, used to determine heavy atom positions in macromolecules.24-28
(2) Single/multiple isomorphous replacement (SIR/MIR) are used to calculate initial phases by studying the differences in diffraction intensities between the native protein crystals and isomorphous crystals containing heavy atoms like Hg, Ag, Au, Pt … as reference scatterers . Isomorphous replacement was one of the fundamental methods to determine structures of new proteins (new folds). 29-32

13
(3) Single-/Multiple-wavelength anomalous dispersion (SAD/MAD) In the presence of anomalous scattering, Friedel’s law breaks down, i.e.I hkl I hkl( ) ( ) . SAD and MAD extract the information from the differences between the Bijvoet pairs to determine the positions of the anomalous scatters, from which the phase values can be derived. Therefore both methods require the presence of anomalous scatterers, which are either naturally occurring (Fe, Mn, Cu) or specially introduced into the molecule by recombinant techniques (seleno-methionine) or by quick-soaking (bromine). SAD may also use weak anomalous scatters such as sulfur, which exists in almost all proteins. SAD and MAD are regarded as the “high-throughput” phasing methods and are today widely applied for structure determination of new proteins.33-37
(4) Homologous proteins often have similar structures. Molecular replacement(MR) is a method, by which the initial phases are obtained using the structure of a homologous protein as the template. With the increasing number of determined protein structures, MR has become the most commonly used technique in the practice of solving the phase problem.38-40
Molecular replacement, as was used to determine all the structures in this work, will be described briefly in the following sections. For detailed discussions of the phasing methods mentioned above, the reader is referred to relevant literature of crystallography.14, 15
1.3.4 Patterson Function and Patterson Map
The Patterson function plays an essential role for solving the phase problem. It is combined with almost all other phasing methods in the process of phase determination. The Patterson function can be obtained by Fourier transformation ( )of the intensities ( )I h as the Fourier coefficients: 41
I( ) [ ( )] [ ( ) ( )][ ( )] [ ( )]( ) ( )
P u h F h F * hF h F * hr r
(14)
By the convolution integral ( ) ( ) ( )C f gr
u r r f(r) g(u - r)dr we obtain:
( )r
P u (r) (r + u)dr (15)
By insertion of ( )r and ( )r + u , it can be shown, that
21 2( ) ( ) exp[ ]h k l
P V F iu h h u (16)
Mathematically, the Patterson function is the convolution of the electron densities at positions ),,( zyx and ( , , )x u y v z w . The coordinate system (u)

14
defines a space known as the Patterson vector space or the Patterson space. In the Patterson space each peak indicates a vector between two atoms in a crystal unit cell. When positions ),,( zyx and ( , , )x u y v z w are both occupied by atoms, the Patterson Function ( )P u will have a positive value, which is shown as a peak on the so-called “Patterson map”.
The Patterson map is centrosymmetric and the height of each peak is roughly proportional to the product of the atomic numbers of the two atoms that contribute to the vector. As a result, heavy atoms produce much higher peaks in the Patterson map with respect to those of the other peptide atoms. Peaks in a Patterson map are grouped into two categories: (1) those arising from vectors between atoms within the same molecule (self-vectors); (2) those arising from vectors of atoms from different symmetry-equivalent molecules (cross-vectors). In principle, a Patterson map can be directly used to calculate atom positions in a crystal of simple molecules.42 However the Patterson maps of macromolecules, such as proteins, contain a huge number of peaks (theoretically, the number of peaks is NN 2 , where N is the number of atoms in protein) and many of them may overlap making the interpretation of the map very complex. Therefore the analysis of the Patterson map is often combined with correlation methods in the procedure of phase determination.43
1.3.5 Rotation, Translation and Molecular Replacement
Proteins belonging to a same family usually have identical or similar fold. In molecular replacement (MR), a homologous structural template is used to replace the unknown structure in the crystal and provides the information needed for the calculation of initial phases. The phases calculated from the structural template and the diffraction intensities of unknown protein can then be used for Fourier synthesis to compute the electron density map. In order to locate the exact position of the template in the crystal, rotation- and translation-searches are performed in Patterson space.
The rotation search is performed by calculating the rotation function ( )R C(17), in which CrystP is the Patterson function of the crystal calculated using the
observed intensities and ModP is the self-Patterson function of a molecule of the
structural model (containing only the self-vectors). The integration in (17) is over the volume of the Patterson map (u), where the self-Patterson peaks are located. Applying the rotation operation C on ModP , ( )R C will give a maximum when the two
functions, CrystP and ModP , superimpose.38, 44
( ) ( ) ( )Cryst ModR P P dU
C u Cu u (17)
The resulting rotation matrix C (derived from the Eulerian angle triplet, 1, 2,3) defines the relative orientation of template molecule with respect to the unknown

15
structure in its unit cell. After the rotation operation C is applied to the model, translation searches are performed in order to find the absolute position of the model in the crystal:44, 45
( ) ( ) ( , )Cryst ModT P P dU
t u u t u (18)
The translation search is performed in a similar way as the rotation search. However, only cross-vectors are used for the calculations, as self-vectors are not sensitive to translational operations. When cross-vectors of the two Patterson functions superimpose, ( )T t has a maximum value with a corresponding translational vector t.Thus the absolute position of the template structure in crystal of the unknown protein is obtained.39, 40, 46
1.3.6 Density Modification
Initial phases obtained by the phasing methods discussed above contain errors, which may often result in a rather poor agreement between the density map and the model structure. The calculated phases need to be improved so that the quality of the density map can also be improved. This procedure is called “density modification” (DM).47, 48 Density modification combines different methods to improve the quality of both the phase and the electron density map.
Solvent flattening: Protein crystals contain about 30% to 70% solvent,49 most of which is disordered. These solvent molecules often confound the real density with background noise at the initial stage of model building and refinement. Therefore improvement of the electron densities can be obtained by flattening the residual density. The solvent flattening is done by estimating the solvent content of the crystal and defining the protein region using an “envelope” that covers the protein molecule.50-52
Averaging: Most proteins form oligomers. In many cases the asymmetric unit of the protein crystal contains multiple copies of the subunits, which are related by the so called “local symmetries” or non-crystallographic symmetries (NCS). The information on NCS may be used to average the density map.50, 53 In this thesis, the quality of the electron density maps was improved dramatically after averaging against the non-crystallographic 5-fold (icosahedral 5-fold).
Other techniques, such as histogram matching, solvent flipping/ correctionare also used in DM, for detailed discussions, the readers are referred to the relevant literature. 54-58
Solvent flattening is normally performed before crystallographic refinement and model building (1.3.7), however density averaging may be applied throughout the whole refinement procedure.

16
1.3.7 Model Building and Crystallographic Refinement
Model building is a computer aided procedure, in which the model structure is fitted into the density map.59 However, errors introduced by model building may result in deviations between the calculated structure factors ( calcF ) and the observed
structure factors ( obsF ). Crystallographic refinement is then performed in order to
minimize the deviations between obsF and calcF .
2( ( ) ( )) minh o ch
Q W F h F h (19)
where hW is a weight factor.
Least squares refinement: It was shown that structure refinement of macromolecules could be performed in reciprocal space using a least squares method on the individual atomic parameters. As the high resolution 3D structures of all the 20 amino acids are available, the structural information including stereo chemistry, bond length and bond angle can be parameterized and used in the target function for minimization60, 61:
F D TQ w DF w DD w DT (20)
where DF represents the differences between the observed and calculated structure factors; DD restrains the stereochemistry and DT is the deviation of coordinates between the target molecule and the model atoms.61 Least squares refinement requires a high ratio of observations ( obsF ) / the number of parameters to refine (NPR) for
convergence the refinement, therefore it is usually applied for the refinement of small molecules, which can often diffract to very high resolution.
Maximum likelihood refinement: assuming that the best structure model would be most consistent to observations (data), maximum likelihood (ML) can be used for crystallography refinement.62-64
2
2h S
( ( , ) ( ) ( ))
( ( , ) ( ))1 exp max2 ( )( ) 2
calc j obs
calc j obs
P F F
F F
h r h h
h r hhh
(21)
The assumption of using (ML) for crystallographic refinement is that the errors ( ( )h ) in the magnitudes ( ( )obsF h ) are random variables distributed in accordance to
a Gaussian law and the ultimate goal of ML is to find the coordinates ( jr ), which
maximise the probability P, provided the experimental errors are ( )h .
The basic concepts of using ML in crystallographic refinement are (a) given the current model, consistency is measured statistically by the probability that the

17
reflections would be observed; (b) if changes of the model make the observations more probable, the model gets better and the likelihood increases; (c) the probabilities include the effects of all sources of error. As the model gets better, the errors get smaller and the probabilities get sharper, which also would increase the likelihood.
Given a target function, many different mathematical methods can be used for the minimization. The most popular algorithms including the conjugate gradientmethod or simulated-annealing have been integrated in many crystallographic refinement software packages such as REFMAC (CCP4), XPLOR/CNS etc.65-67
The whole refinement procedure is monitored by the so-called crystallographic R-factor:
obs calchkl
obshkl
F k FR
F (22)
where k is a scale factor. It has been noticed that the crystallographic R-factor can be refined to a very low value, which, however does not necessarily stand for a good result. This is due to the bias introduced by model building and refinement. In order to overcome the disadvantage of the crystallographic R-factor, a cross-validation scheme was developed by Brünger:68, 69
obs calchkl Test
freeobs
hkl Test
F k FR
F (23)
The free R-factor or Rfree is calculated using a test set of the reflections (usually 5-10% of the observed reflections), which are set aside from refinement and are therefore unbiased.
The quality of the refined structures can be verified using Ramachandran plot,70, 71 which examines the stereochemistry of the main-chain dihedral angles. The temperature factors 2 28B x ( 2x is the mean square displacement of the atom from its equilibrium position), as one of the refined parameters, can also be used to check the quality of the refined structure. Because the information on mobility is included in the temperature factor, residues that are flexible or poorly defined normally have higher B values. A properly refined structure would have an averaged B factor closed to the statistical value directly obtained from the reflection data from the Wilson plot.72

18
1.4 HYPERTHERMOPHILES AND PROTEINS FROM HYPERTHERMOPHILES
1.4.1 Hyperthermophiles
Hyperthermophiles are microorganisms, which grow optimally at temperatures above 80 C and are usually unable to reproduce at temperatures below 60 C.73-75
Hyperthermophilic microorganisms live and thrive at geothermal and volcanic environments like solfataras, hot springs, geysers and deep-sea vents.76, 77
To adapt to the extreme environment of their biotopes, hyperthermophiles have developed some unique strategies. Most hyperthermophiles are chemolithoautotrophic, i.e. producing energy via inorganic redox reactions (chemolithotrophic) and utilizing only inorganic carbon sources, like CO2, to build up the cell material (autotrophic).78 A novel type of glycerol ether membrane lipids, which dramatically increases the resistance against hydrolysis at high temperature and low pH, has been found in the hyperthermophilic bacterium T. maritima.79-81 Recent studies have revealed that reverse gyrase is the only hyperthermophile-specific protein indicating the important contribution of the enzyme to the stability of DNA molecules in hot environment.82, 83
Figure 1-9. Phylogenetic tree based on 16S rRNA sequences.84 The branches of the hyperthermophiles are highlighted with bold lines.
From a phylogenetic point of view, hyperthermophiles appear in the deepest branches of the genealogy tree (Figure 1-9), studies on these organisms have a great impact on our understanding of the evolution of life on earth.85, 86

19
1.4.2 Aquifex aeolicus
Most hyperthermophiles belong to the domain of archaea, whereas only two families, Thermotogales and Aquificales belong to the domain of bacteria. Aquifexaeolicus, originally found in Sicily, is one of the most thermophilic organisms known to date. Successful isolation from hot springs at 95 C in the Yellowstone National Park has also been reported.87 A. aeolicus is chemolithoautotrophic and can only grow in a medium without organic source such as sugars, amino acid. A. aeolicus was cultured at 85 C in a H2/CO2/O2 (79.5/19.5/1.0) atmosphere. The complete genome of A. aeolicus has been sequenced. Among 1,512 classified open-reading frames (ORFs), 1,105 have been identified for coding of proteins.88, 89
1.4.3 Protein Stability and Folding
Proteins isolated from hyperthermophiles usually display abnormal tolerance against heat, extreme pHs and high salinity. Enzymes from hyperthermophilic organisms often function optimally at temperatures above 80 C.75 Knowledge on the structural basis of protein stability is essential for the understanding of some fundamental problems in biology. The functional and structural features as well as the catalytic mechanisms of thermostable enzymes, once understood, may be used for modification and engineering of enzymes for industrial applications.74, 90, 91
Protein stability and folding are strictly related. The stability of a protein is determined by the difference in the free energy between the Native (folded) state and the Unfolded (denatured) state:92
U U NNG G G (24)
Suppose that the native (N) and the unfolded states (U) are in equilibrium described by the equilibrium constant:
[ ][ ]UKN (25)
a reversible unfolding reaction KNative Unfolded can be represented in terms of thermodynamics. The difference in free energy between the unfolded and native states is then:
lnU U NNG G G RT K (24’)
where R is the universal gas constant and T is the absolute temperature in Kelvin.
There are two important forms of enthalpy as far as protein thermal denaturation is concerned: the Van't Hoff enthalpy ( U vH
N H ), determined from the
temperature dependence of the equilibrium constant and the enthalpy measured experimentally by a calorimeter ( U Cal
N H , the area under the peak, see below). If
20
these enthalpies are equal, i.e. U vH U CalN NH H , the system is considered as a two-
state system. Taking into account both the enthalpic and the entropic contributions to the free energy, a two-state reversible unfolding reaction can be described using the thermodynamic model:
lnU U N U UN N NG G G RT K H T S (24’’)
Equation (24’’) allows the calculation of the change of the free energy upon unfolding ( U
NG ) as a function of the temperature, when the temperature dependence
of UN H and U
N S are both known.
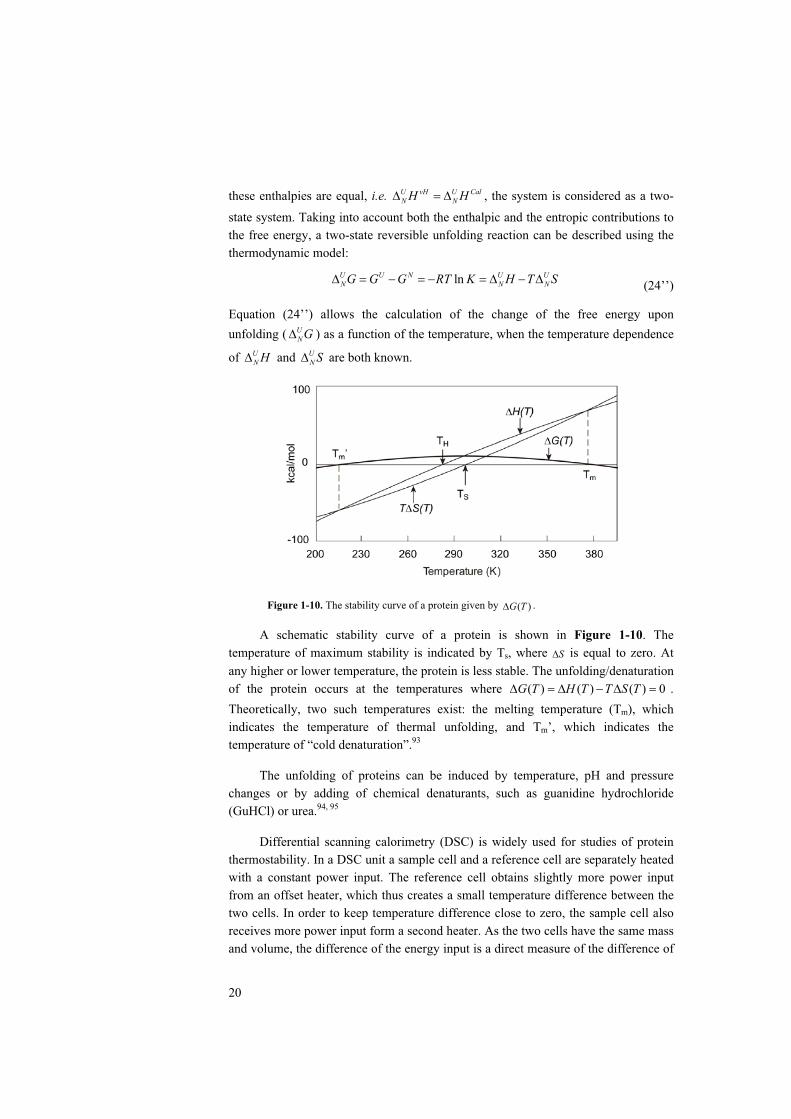
Figure 1-10. The stability curve of a protein given by ( )G T .
A schematic stability curve of a protein is shown in Figure 1-10. The temperature of maximum stability is indicated by Ts, where S is equal to zero. At any higher or lower temperature, the protein is less stable. The unfolding/denaturation of the protein occurs at the temperatures where ( ) ( ) ( ) 0G T H T T S T .Theoretically, two such temperatures exist: the melting temperature (Tm), which indicates the temperature of thermal unfolding, and Tm’, which indicates the temperature of “cold denaturation”.93
The unfolding of proteins can be induced by temperature, pH and pressure changes or by adding of chemical denaturants, such as guanidine hydrochloride (GuHCl) or urea.94, 95
Differential scanning calorimetry (DSC) is widely used for studies of protein thermostability. In a DSC unit a sample cell and a reference cell are separately heated with a constant power input. The reference cell obtains slightly more power input from an offset heater, which thus creates a small temperature difference between the two cells. In order to keep temperature difference close to zero, the sample cell also receives more power input form a second heater. As the two cells have the same mass and volume, the difference of the energy input is a direct measure of the difference of

21
heat capacity. A number of thermodynamic data can be obtained by DSC experiments. The thermal unfolding profile of a DNA binding protein Sso7d measured by differential scanning calorimetry is shown in Figure 1-11 as an example.96 The melting temperature mT is indicated by the peak, the change of
enthalpy ( mH ) is calculated as the area under the curve and change of heat capacity
( PC ) is measured by the difference between the pre- and post- transition baselines
as shown in the picture. The change of free energy ( )G T upon thermal unfolding can be calculated.
Figure 1-11. Differential scanning calorimetric measurement of the thermal unfolding of the DNA binding protein Sso7d from Sulfolobus solfataricus.96
In this thesis, the stability of lumazine synthase is studied by DSC. However the unfolding of this enzyme is irreversible, therefore only apparent melting temperatures were obtained and used for an approximate comparison of thermostability.
1.4.4 Dominant Forces of Protein Stability
It has been shown that proteins isolated from hyperthermophiles display dramatic stability against heat. Considerable effort has been made during the past decades to investigate the dominant forces for protein stability and folding. Structural comparisons, mutagenic studies as well as theoretical calculations have revealed a number of the most important forces responsible for the stability of proteins: the hydrophobic effect, hydrogen bonding, electrostatic interactions, the formation of disulfide-bridges, aromatic interactions, helix dipoles, binding of small molecules or ions, packing and oligomerization. The environmental factors, such as temperature, pH and salts, affect the stability by changing the contributions of the hydrophobic effect, hydrogen bonding and configurational entropy or other forces listed above.97, 98

22
Spassov et al pointed out that the adaptation of proteins from hyperthermophiles to their extreme environment requires optimization of these factors.99
The hydrophobic effect generally refers to the low solubility of hydrophobic (i.e., apolar) compounds in water. The hydrophobic effect in protein can also be defined as the energy associated with the transfer of hydrophobic surface from the protein interior to water.100, 101 In proteins, side chains of the apolar amino acid residues have a tendency to evade water. Applying the hydrophobic concept by Frank & Evans to proteins, Kauzmann suggested that hydrophobic effect is the dominating force for protein folding and thermostability.102-104 This theory has been supported by accumulating experimental evidences.105 Theoretical studies by Privalov, et. al.suggested that the energetically most favorable structure would correspond to those with minimized hydration of apolar atoms. 106, 107
Hydrogen bonding refers to partial sharing of a hydrogen atom between a donor atom, to which it is covalently bound, and an acceptor atom which has a lone pair of electrons. In protein solutions, hydrogen bonds and hydrogen bond networks are formed between the main-chain atoms and side-chain atoms of the amino acids. Water molecules or other compounds from the solvent are also involved in formation of hydrogen bonds. Studies of model compounds and protein mutants suggested that the formation of hydrogen bonds contribute to stability of proteins.108
Earlier work by Perutz and Raidt109, 110 showed that in thermostable proteins the number of surface ion-pairs is increased. This observation was further confirmed by statistical analysis99 and a large number of studies based on structure comparisons.111-
116 With increasing melting temperature, ion-pairs in protein show a tendency of being organized into large networks. A largest increase of the ion-pair content and the largest ion-pair network in hyperthermostable proteins reported to date have been observed in a comparison of glutamate dehydrogenase from Pyrococcus furiosus117
and the enzyme from the mesophilic bacterium Clostridium symbiosum.118 In this thesis, structural studies showed that the number of ion pairs and ionic networks is more than doubled in lumazine synthases form the hyperthermophilic bacterium A.aeolicus with respect to that of the enzyme from the mesophilic B. subtilis (Paper I).
The thermodynamic contribution to stability resulting from an increased number of surface ion pairs, albeit being found in many of the existing structures of hyperthermostable proteins, is still a matter of lively debate. However, in a study of salt-bridge stability at high temperatures, model calculations have suggested that a considerable energy barrier exists for the solvation (breaking) of a salt-bridge and that the height of this barrier increases with temperature.119 A similar barrier is not seen with isosteric hydrophobic groups. The presence of this energy barrier suggests an apparent role of salt-bridges in increasing the kinetic barrier towards thermal inactivation or unfolding. It has also been shown that the desolvation penalty is reduced because of a lower solv at high temperature. 120

23
A number of approaches by site-directed mutagenesis of ion-pair interactions and ionic networks, performed in several laboratories, has still not given the clear answers that were expected. However, Vetriani et al. recently observed that the melting temperature and the half-life of glutamate dehydrogenase from Thermococcus literalis are increased over the values of the native enzyme after introduction of a double mutant.121 The construction of a 16-residue ion-pair network in the less thermostable Thermotoga maritima glutamate dehydrogenase resulted in an enzyme with a half-life of 240 minutes at 85 C, compared to the wild-type protein with a half-life of 210 minutes, suggesting increased kinetic stability of the mutant protein.122
Other important forces and their contributions to protein stability are discussed in detail in Merphy (2001).101
1.5 RIBOFLAVIN AND ENZYMES INVOLVED IN RIBOFLAVIN BIOSYNTHESIS
1.5.1 Riboflavin
Riboflavin (also known as vitamin B2, Figure 1-12a) is the precursor of two coenzymes, namely riboflavin mononucleotide (FMN, Figure 1-12b) and flavin-adenine dinucleotide (FAD, Figure 1-12c), which are fundamental for the metabolism of carbohydrates, fats, and proteins into energy. FAD and FMN serve as electron carriers in oxidation-reduction reactions (Figure 1-13) catalyzed by flavoenzymes (Table 1-1).123, 124
Figure 1-12. The chemical formulae of: (a) riboflavin; (b) riboflavin mononucleotide (FMN) and (c) flavin adenine dinucleotide (FAD).

24
Figure 1-13. FAD and FMN transfer electrons in oxidation-reduction reactions. R represents the ribityl side chain of the compounds.
Table 1-1. Some flavoenzymes
Enzymes Flavin nucleotide
Fatty acyl-CoA dehydrogenase
Dihydrolipoyl dehydrogenase
Succinate dehydrogenase
-Glycerophosphate dehydrogenase
NADH dehydrogenase
Glycolate dehydrogenase
FAD
FAD
FAD
FAD
FMN
FMN
It has been shown that flavoproteins play important roles in many biological processes, such as fetus development in birds and mammal,125, 126 signal transduction in apoptosis,127 regulation of biological clocks128. They are also involved in the biodegradation of aromatic pollutants,129 in photosynthesis as well as in the light dependent DNA reparation.130
Bacteria, archaea, certain yeasts and plants produce riboflavin by endogenous synthesis. However mammals must take up riboflavin through their diet. The fact that
riboflavin is indispensable for living cells makes the enzymes involved in riboflavin
biosynthesis ideal targets for the development of new antimicrobial drugs,131-133 as
highly specific inhibitors are unlikely to interfere with human metabolism.134, 135
1.5.2 Enzymes involved in Riboflavin Biosynthesis
Enzymes involved in the last two steps of riboflavin biosynthesis were first isolated and purified in the form of a large complex from the cell contents of Bacillussubtilis in 1980 at the Technical University of Munich.136 The complex, initially designated as “heavy riboflavin synthase”, has a total molecular weight of more than 1 MDa. Further studies characterized two types of functional subunits in the complex, namely the subunits (MW = 23500 Da) and the subunits (MW = 16000 Da).10, 137
The subunit of heavy riboflavin synthase catalyzes the dismutation of 6,7-dimethyl-8-ribityllumazine (Figure 1-14, 3) resulting in the formation of riboflavin (Figure 1-14, 4) and was therefore named “riboflavin synthase”.138 The mechanism of the dismutation of 6,7-dimethyl-8-ribityllumazine (Figure 1-14, 3) has been investigated in a number of studies.139-142 Recent structural studies of the riboflavin synthase revealed more details of the catalytic mechanism,143-148 further studies on structure and function of riboflavin synthase are underway.149-152

25
Figure 1-14. The biosynthesis of riboflavin.
Earlier ligand binding experiments indicated that the subunit plays a role in catalyzing the biosynthesis of 6,7-dimethyl-8-ribityllumazine (Figure 1-14, 3), the precursor of riboflavin.153, 154 However, the exact function of the subunit remained obscure for almost 40 years until the 4-carbon substrate was identified as (3S)-3,4-dihydroxy-2-butanone-4-phosphate (Figure 1-14, 2).155, 156 It was then clear that the subunit catalyze the formation of 6,7-dimethyl-8-ribityllumazine (Figure 1-14, 3)from 5-amino-6-ribitylamino-2,4-(1H,3H)pyrimidine-dione (Figure 1-14, 1) and (3S)-3,4-dihydroxy-2-butanone-4-phosphate (Figure 1-14, 2). The subunit of heavy riboflavin synthase was thus named as lumazine synthase (LS).136, 157-159
1.5.3 Crystal Structures of Lumazine Synthase
Structural studies of lumazine synthase were pioneered by R. Ladenstein and A. Bacher et al.10, 160-163 The first crystal structure of the enzyme from Bacillus subtilis(LSBS) was determined in the late 80’s of last century.164, 165 It was shown that the enzyme forms a virus-like icosahedral capsid with a molecular weight of about 1MDa. Sixty subunits are arranged in twelve pentameric building blocks on the basis of icosahedral symmetries giving rise to a hollow capsid with inner and outer diameters of 80 Å and 156 Å, respectively.
In the subsequent 15 years, structures of lumazine synthase from a number of other organisms, including Schizosaccharomyces pombe,166 Saccharomycescerevisiae,167 Magnaporthe grisea,168, 169 Mycobacterium tuberculosis,170 Escherichiacoli,171 Spinacia oleracea168, 169, Brucella abortus172, 173 and Aquifex aeolicus174 (this work) have been investigated. It has been shown that the enzyme exists in three association forms in crystals: icosahedral capsids in B. subtilis, E. coli and S.oleracea; pentamers in S. pombe, and S. cerevisiae as well as stacking pentamers in Brucella sp and M. tuberculosis. (Earlier studies on the multiple assembly states of the enzyme in solution are summarized in section 1.5.5.)
26
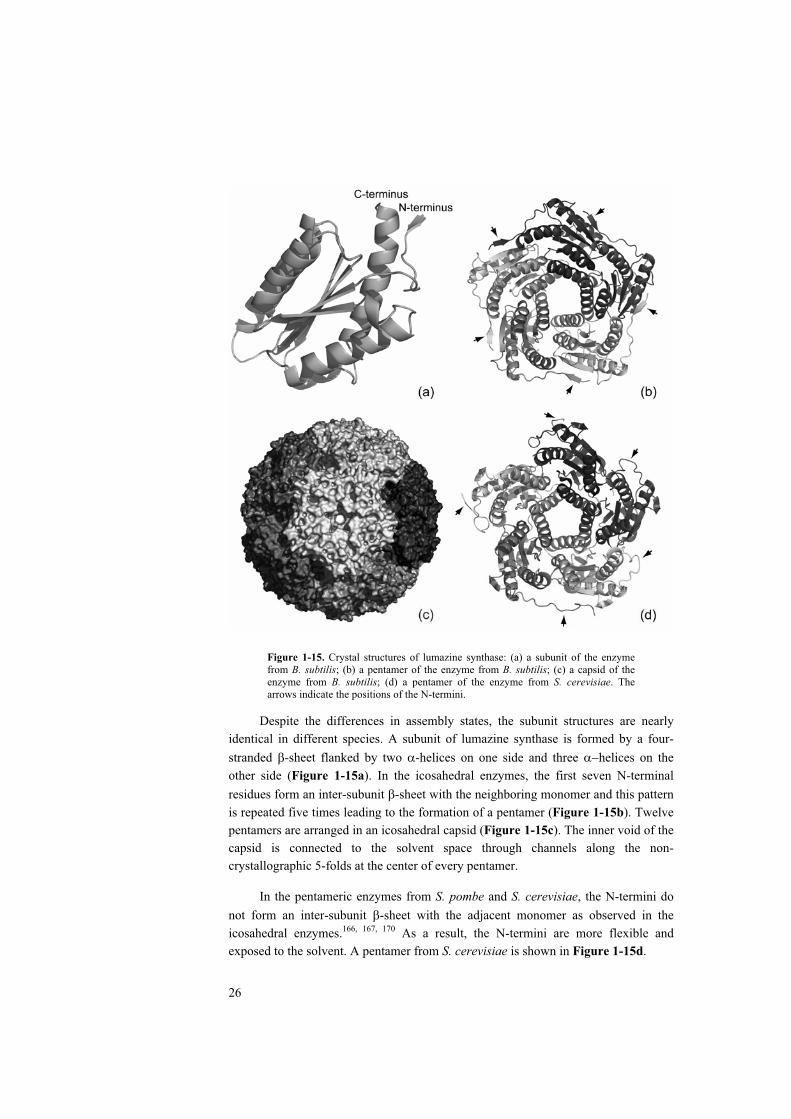
Figure 1-15. Crystal structures of lumazine synthase: (a) a subunit of the enzyme from B. subtilis; (b) a pentamer of the enzyme from B. subtilis; (c) a capsid of the enzyme from B. subtilis; (d) a pentamer of the enzyme from S. cerevisiae. The arrows indicate the positions of the N-termini.
Despite the differences in assembly states, the subunit structures are nearly identical in different species. A subunit of lumazine synthase is formed by a four-stranded -sheet flanked by two -helices on one side and three helices on the other side (Figure 1-15a). In the icosahedral enzymes, the first seven N-terminal residues form an inter-subunit -sheet with the neighboring monomer and this pattern is repeated five times leading to the formation of a pentamer (Figure 1-15b). Twelve pentamers are arranged in an icosahedral capsid (Figure 1-15c). The inner void of the capsid is connected to the solvent space through channels along the non-crystallographic 5-folds at the center of every pentamer.
In the pentameric enzymes from S. pombe and S. cerevisiae, the N-termini do not form an inter-subunit -sheet with the adjacent monomer as observed in the icosahedral enzymes.166, 167, 170 As a result, the N-termini are more flexible and exposed to the solvent. A pentamer from S. cerevisiae is shown in Figure 1-15d.

27
Figure 1-16. Amino acid sequence alignment of lumazine synthase from 8 species. Icosahedral species are highlighted. Fully conserved residues are highlighted. Residues forming the active site pocket are marked with “+”. Position of the N-terminal proline, which is unique for pentameric LS, is marked with “ ”. Positions of the single-site mutations, which lead to the formation of large capsids, are marked with “ ”. The position of the 2-4-residue insertion, which is unique for the pentameric enzymes, is marked with “ ”. Secondary structure elements are labeled according to the crystal structure of lumazine synthase from A. aeolicus.
The substrate binding sites are located at the interface between two adjacent subunits within a pentamer. The secondary structural segments involved in ligand binding are -turns (21-24, 54-58, 81-92) from one subunit and -helices (127’-142’) as well as a -strand (113’-116’) from the neighboring subunit (Figure 1-16). The structures of the active site in enzymes from different species are highly identical. In the structural studies of LS in complex with a number of designed inhibitors, the

28
detailed substrate binding schemes were revealed. The results are briefly summarized in the next section.
1.5.4 Catalytic Mechanism of Lumazine Synthase
The mechanism of this enzymatic reaction was investigated in a number structural and kinetic studies after the function of the -subunit of heavy riboflavin synthase was identified as catalyzing the formation of 6,7-dimethyl-8-ribityllumazine.155, 156
Figure 1-17. Architecture of the active site in LS from B. subtilis. (a) Ribbon and stick representation of the substrate binding site; (b) the inhibitor 6-ribitylamino-5-nitro-2,4(1H,3H)-pyrimidine-dione (RNO2P) forms contacts with the main chain (black) and side chain (red) atoms of the surrounding protein residues.164, 165
Substrate binding and the conformation of the active sites were revealed by structural analysis of the enzymes from Bacillus subtilis, Saccharomyces cerevisiae,Spinacea oleracea and Magnaporthe grisea in complex with different inhibitor compounds. The substrate binding pocket of LS from B. subtilis is shown in Figure 1-17a.164, 165 Arg127 forms a salt bridge with the phosphate ion. It was thus proposed that Arg127 is mainly responsible for the binding of substrate 2 via an ionic contact.165 His88 is in close proximity to the substrate analogue indicating the possibility of serving as a proton donor / acceptor during catalysis.165 The pyrimidine ring of the inhibitor was found in a parallel conformation with respect to the aromatic ring of residue 22 (Phe22/Trp22). Theoretical studies suggested that offset stacking aromatic interactions ( - / - ) in protein energetically stabilize the structure.175-180
Thus it was suggested that Phe22 facilitates the binding of substrate 1 and the reaction intermediate in the active site. The structure of the empty active site of LS has been studied and described in LS from Brucella abortus173 and Aquifex aeolicus(paper I). The observed conformational changes, revealed by comparisons with the occupied active sites of the enzymes in complex with inhibitors, have suggested an induced-fit mechanism of substrate binding in LS. The aromatic ring of Phe22 adapts to the pyrimidine system by a swing movement, which results in a stacking aromatic interaction and proper orientation of the pyrimidine.

29
Hydrogen bond networks are formed between the inhibitor and protein. As shown in the schematic drawing of the inhibitor binding interactions (Figure 1-17b),the hydroxyl groups of the ribityl moiety of the inhibitor compound form hydrogen bonds with the residues Gly55, Ser56, Phe57, Glu58, Phe113’ and Lys135’. The keto groups of the pyrimidine ring form hydrogen bonds with Asn23, Ala56, Thr80 and Ile82. The active site of the pentameric enzymes is similar to that of the LSBS, detailed comparison and analysis have been presented in earlier work.166-170
On the basis of crystal structures of LS, mutagenic studies on LSBS have confirmed a list of residues that are essential for enzyme activity (Table 1-2).181 In addition, in vitro experiments on the wild-type and the mutants of LS have revealed considerable insights into the catalytic mechanism of the enzyme.149, 182-189
Table 1-2. Approximate reaction rates of lumazine synthase mutants (% of wild-type enzyme activity). Gly Val Ala Leu Phe Trp Tyr Ser Thr Cys Asn Gln Asp Glu Lys Arg His Phe22 22 91 40 12 6 12 Ala56 69 Phe57 36 Phe61 8 Val81 3 Ile82 8 Val92 23 Phe113 3 25 5 2 3 Asn23 < 1 20 10 Thr80 47 69 54 3 Ser142 17 92 53 57 55 Glu58 3 62 100 Glu126 49 12 Asp138 78 < 1 < 1 His88 10 6 7 3 12 Arg127 2 2 2 5 2 9 62 Lys131 < 1 8 3 25 Lys135 14 18 5 27
It was shown that the Km value for substrate 2 (130 M) exceeds that of the pyrimidine substrate 1 (5 M) by more than 20 times.184 The kinetic data have also revealed that in absence of the pyrimidine substrate 1, lumazine synthase does not catalyze the proton exchange on the chiral C-3 atom of 2 with solvent water, nor does the enzyme function as a racemase. The spontaneous proton exchange at the methyl group of 2 is not accelerated by the enzyme. It was also shown that the enzyme does not catalyze the formation of diacetyl from 2. It was therefore suggested that the initial step of the enzyme-catalyzed reaction requires the presence of the second substrate (the pyrimidine 1) at the active site. These results indicate an ordered bi-bi mechanism of the reaction, however no conclusive evidence supporting this hypothesis has been obtained.184
Earlier in vitro experiments suggested that the regio-specificity of the reaction is about 90%.190-192 This result was later confirmed by experiment using the 13Clabeled substrate 2, which was prepared in situ. The 13C atoms were found in lumazine and riboflavin at positions shown in Figure 1-18. Taking into account the

30
experimental sensitivity, Kis et al concluded that the regio-specificity of the enzymatic reaction is at least 98%.
Figure 1-18. The regio-specificity of the lumazine synthase catalyzed reaction. The carbon 1 atom of substrate 2 is labeled with an asterisk.
It has also been shown that both enantiomers of the four carbon compound 3,4-dihydroxy-2-butanone-4-phosphate, namely the 3S-2 and 3R-2 enantiomers (Figure1-19), can serve as substrates for lumazine synthase, nonetheless stereo-specificity of the enzyme was detected. The reaction rate of the natural S-enantiomer is about 5-6 times higher than that of the R-enantiomer.
Figure 1-19. The 3S (left) and 3R (right) enantiomers of the substrate 3,4-dihydroxy-2-butanone-4-phosphate. The chiral carbon atom is labeled with an asterisk.
Based on the results of both structural and kinetic studies, Kis et al postulated a reaction mechanism (Figure 1-20) initiated by the formation of a Schiff base (6) upon the reaction of the 5-amino group of the pyrimidine 1 with the carbonyl group of 2.This step could be followed by proton abstraction and phosphate elimination. The resulting intermediate (7) contains a double bond, which is in favorable conjugation with the pyrimidine system. This enolate intermediate could tautomerize under formation of a carbonyl group, which would then be attacked by the 6-amino group, leading to the closure of the second ring. In the final step of the reaction, a water molecule would be released and the heterocyclic double ring system of the product 3would thus be in an energetically favorable conjugation.

31
Figure 1-20. Hypothetical reaction pathway of lumazine biosynthesis.193
1.5.5 Molecular Assembly of Lumazine Synthase
The function of protein oligomers is often related to their assembly state. A paradigm for studying molecular assembly represent the viral proteins, which very often form icosahedral capsids enclosing DNA or RNA molecules.
A simple icosahedral capsid consists of 60 copies of the same subunit, which
is the largest possible number of subunits that can be assembled with each of them in
an identical (equivalent) environment. However, most viral proteins display
icosahedral symmetry but contain more than 60 subunits. In the light of the
pioneering studies of virus structures including the tobacco mosaic virus (TMV),194,
195 tomato bushy stunt virus (TBSV)196 and turnip yellow mosaic virus (TYMV)197, 198,
Caspar and Klug developed the theory of quasi-equivalence of viral protein
assembly.199 Caspar and Klug demonstrated that an icosahedral capsid can be
constructed by inserting pentamers in a hexagonal lattice at certain positions
described by the triangulation number 2 2T h hk k (Figure 1-21).
With T > 1 the resulting capsid consists of more than 60 subunits arranged in hexamers and pentamers, which would show similar conformations and inter-subunit contact environments. Thus the pentameric and hexameric building blocks are regarded as “quasi-equivalent”. Pentamers and hexamers are generally considered as the basic building blocks of a quasi-equivalent icosahedral capsid. (For a detailed

32
discussion of the principles of quasi-equivalence, please refer to Johnson et al. 1997.)200
Figure 1-21. The construction of icosahedral capsids: (a) inserting pentamers into the hexagonal lattices at positions defined by the triangulation number
2 2T h hk k results in a closed icosahedral capsid. Replacing hexamers with pentamers at positions (0, 1) and (1, 0), indicated by the red triangle, results in a closed T = 1 capsid (b); similarly, a T = 3 capsid (c) can be constructed by replacing the hexamers at positions indicated by the blue triangle in (a). Inserting pentamers at positions indicated by the green and yellow triangles in (a) results in icosahedral capsids with T = 7 and T = 13, respectively.
A large body of structural evidence supports the quasi-equivalence theory, however, examples which may disobey part of these rules were also found. It was shown that the same polypeptide chains could assume different conformations in a virus shell. 201-205 The in vitro assembly experiments of the bacterial virus P22 indicated that the formation of pentamers and hexamers did not seem to be obligatory for the association of the icosahedral capsid. It was shown by this experiment that although the matured capsid contains quasi-equivalent pentamers and hexamers, the capsid could be assembled from a solution containing only the monomers but without the presence of pentamers and/or hexamers, i.e. the monomers are the basic building blocks of the capsid .206-209
A theory based on the so-called “local rules”, which illustrates virus shell assembly from a geometrical point of view, was introduced by Berger et al.210 It was proposed that the self-assembly of icosahedral capsids may be regarded as only relying on the “lower-level” interactions of each polypeptide chain with its neighbors, rather than the interactions of large structural building blocks, e.g. pentamers or hexamers. According to this theory, the “lower-level” interactions simply represent all non-covalent inter-molecular interactions. Computer based simulations followed

33
by energy minimization have successfully modeled the assembly of several viral shell proteins.211-213
Together with the pyruvate dehydrogenase (PDH) complex,214-217 the tricorn protease218-221 and the light-harvesting complex of the photosystem in pea chloroplast,222, 223 lumazine synthase is among the four enzymes that are able to form icosahedral capsids. Lumazine synthase was in fact the first known enzyme complex with icosahedral symmetry. As mentioned above, besides the existence of LS pentamers and T = 1 icosahedral capsids, earlier pH-induced dissociation experiments have revealed another association form. LSBS was observed to reassemble to large hollow capsids in Tris hydrochloride buffer at pH > 7.5. Small angle X-ray scattering and negative staining EM analysis showed that the large capsids were about 300 Å in diameter. Ultracentrifugation showed that these aggregates had a molecular mass of about 3 MDa indicating the existence of a T = 3 icosahedral assembly. It was further observed that the substrate analogue, 5-nitroso-6-ribitylamino-2,4(1H,3H)-pyrimidine-dione, could trigger the reassociation of large particles to empty T = 1 capsids.10, 163, 224
Structural determinants that may lead to the difference in assembly forms of lumazine synthases have been discussed extensively. Braden et al. proposed that the enzyme with a five residue-kink, i.e. GxKxG, in the C-terminal helix would form an icosahedral capsid.173 Structure comparisons showed that the N-termini of all the pentameric enzymes contain a proline residue (Figure 1-16). The unique backbone torsion angle of proline may prevent the formation of the inter-subunit -sheet, which is important for capsid assembly and stability. It was therefore proposed that the enzyme with a proline in the N-terminus would favor the pentameric assembly.168 In icosahedral LSs the loop linking helix 4 and helix 5 is positioned at the interface between two pentamers. Sequence alignment showed that an insertion of one to four residues after Gly129 (Figure 1-16) is unique for all pentameric LS. Computer modeling indicated that these extra residues, if inserted in an icosahedral capsid, would lead to clashes at the subunit interface between adjacent pentamers of icosahedral capsid (Figure 1-22).167 It was then proposed that lumazine synthases with the four-residue insertion would rather assemble to pentamers.167 Recent sequence analysis and quaternary structure comparisons of lumazine synthase from different species have identified in total eight regions that appeared to be important for the assembly state.225
As the sixty active sites of icosahedral LS are located close to the inner surface of the capsid, substrates and products need to be transported to and from the active site through a rather densely packed capsid wall. The largest openings in the icosahedral shell are channels with a diameter of 5-12 Å located along the local 5-fold axes. The attempt to block the channels of “heavy riboflavin synthase” (complex formed of 60 LS subunits and 3 RS subunits inside of the LS capsid) from B. subtiliswith a five-fold-symmetric tungsten-compound ([NaP5W30O110]14-) resulted in

34
unperturbed enzymatic activity.161 The results implied that the transport of substrates and products as well as the catalysis seem to be related to the assembly state of LS or to local perturbations causing widening of the icosahedral capsid (see section 5).
Figure 1-22. The subunit interface between adjacent pentamers of the icosahedral LS from B. subtilis (gray); a subunit of the pentameric S. cerevisiae LS (blue) is superimposed on one of the LSBS subunits. Note: the clash of the loop is indicated by an arrow.167

35
2 CRYSTAL STRUCTURE OF LUMAZINE SYNTHASE FROM AQUIFEX AEOLICUS – DETERMINANTS OF THERMOSTABILITY (PAPER I)
To obtain sufficient protein material for structure analysis, an open reading frame (ORF) optimized for the expression of 6,7-dimethyl-8-ribityllumazine synthase from the hyperthermophilic bacterium Aquifex aeolicus (LSAQ) was synthesized and expressed in an Escherichia coli strain to a level of around 15%. The protein was purified by heat-treatment and gel-filtration. The protein was crystallized in the cubic space group I23 with cell dimensions a = b = c = 180.8 Å and diffraction data were collected to 1.6 Å resolution at the synchrotron beam line at DESY (Hamburg, Germany). The structure was solved by molecular replacement using lumazine synthase from Bacillus subtilis as the search model. The structure of the A. aeolicusenzyme was refined to a resolution of 1.6 Å. The empty protein capsid consists of 60 identical subunits with strict icosahedral symmetries. The apparent melting temperature measured by a differential scanning calorimetry (DSC) is 120 C. The structure was compared with other icosahedral and pentameric lumazine synthases and the possible determinants of thermostability were discussed.
2.1 RESULTS AND DISCUSSION
The recombinant lumazine synthase from A. aeolicus was crystallized in a solution containing 12% (W/V) PEG 4000, 0.1 M lithium sulfate and 0.1 M sodium citrate at pH = 5.6 using the sitting drop vapor diffusion method. Cubic crystals were obtained within two weeks and the diffraction data were collected at the synchrotron beam line X11 at HASYLAB (EMBL Outstation, DESY, Hamburg). The parameters for data collection and the results of data evaluation are shown in Table 2-1.
Table 2-1. Data collection and evaluation of LSAQ.
Wavelength (Å) 0.909
Space group I 2 3
Cell dimensions (Å) 180.82
Resolution (Å) 29.3 - 1.6
Observations/Unique (I > 0) 439,597 / 125,175
Completeness (%) 97.4
Rmerge (%) 5.4
The structure of LSAQ was determined by molecular replacement using the structure of lumazine synthase from B. subtilis as the search model. The structure was refined to a resolution of 1.6 Å using the programs Refmac and CNS.64, 67 During refinement, electron density maps were averaged against the non-crystallographic 5-fold axis.71 The stereochemistry of the refined structure was examined using the

36
Ramachandran plot.70 Results of the crystallographic refinement are shown in Table2-2.
Table 2-2. Results of crystallographic refinement
Number of atoms a
Protein 5,885
Ligand 25
Solvent 800
The refinement
Resolution range for refinement (Å) 29.33-1.60
R-factor (%) 21.6
Free R-factor (%) b 23.6
Ramachandran plot
Most favored regions (%)
Allowed regions (%)
Disallowed regions (%)
95.5
4.5
0
B-factors (Å2)
Wilson plot
All atoms
Protein atoms
Solvent atoms
22.44
22.23
19.93
39.15 a Numbers were counted per asymmetric unit.b 5 % of the unique reflections were set aside for the calculation of the free R-factor.
The overall structure of lumazine synthase from A. aeolicus is similar to that of the enzyme from B. subtilis (Figure 2-1). Each asymmetric unit of the crystal cell contains a pentamer. Twelve pentamers are arranged in a T = 1 icosahedral capsid. Apart from the N-terminal and C-terminal ends, the largest difference of main chain C atoms of LSAQ and LSBS are in the loop region between helix 2 and the strand
3 with a maximum deviation of 1.8 Å at residue Lys69. The region is situated at the outer surface of the capsid and is fully exposed to the solvent. The averaged r.m.s deviation of the atom positions between the LSAQ subunit and LSBS subunit is 0.8 Å.
Thermal unfolding experiments of lumazine synthase from B. subtilis, A.aeolicus and S. cerevisiae were performed using differential scanning calorimetry (DSC). Due to the irreversibility of the thermal unfolding process, only the apparent melting temperatures (Tm) could be obtained. The apparent Tm of LSAQ reached 120 C, which is one of the highest melting temperatures for a protein reported to date. Nevertheless, the apparent Tm of LSBS is 93 C, which is unusually high among those of mesophilic proteins. The Tm of the pentameric lumazine synthase from S.cerevisiae was 74 C (Figure 2-2).
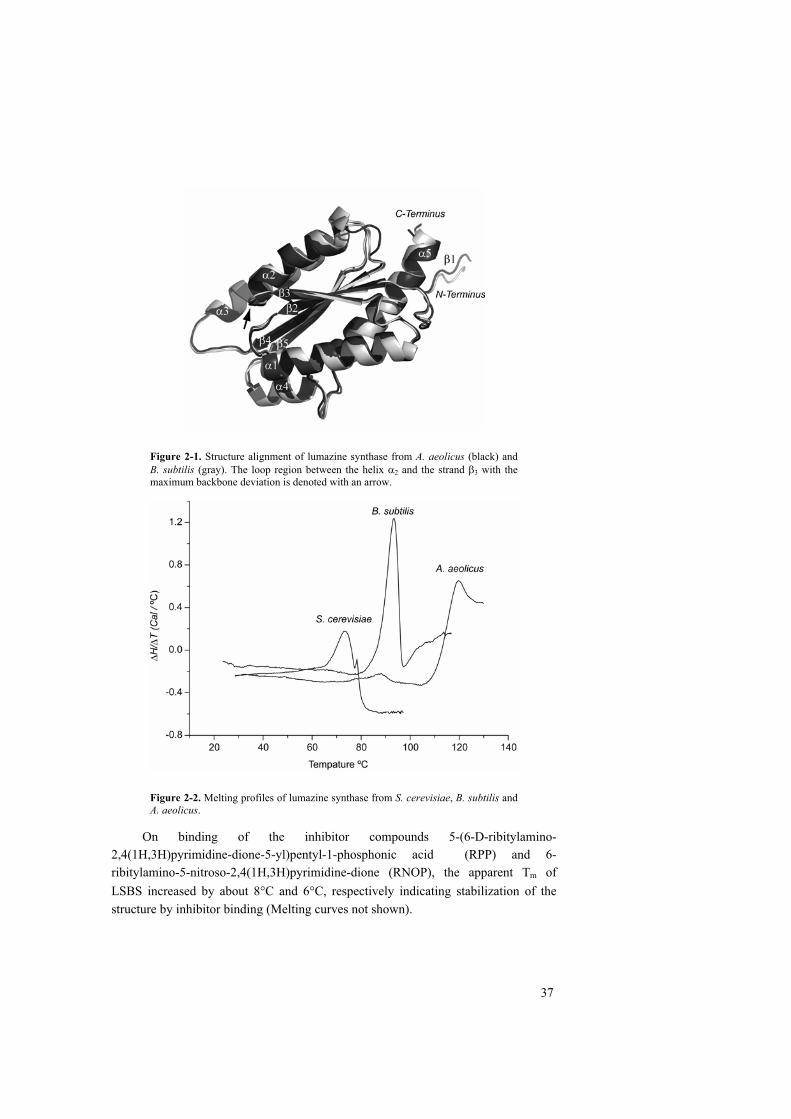
37
Figure 2-1. Structure alignment of lumazine synthase from A. aeolicus (black) and B. subtilis (gray). The loop region between the helix 2 and the strand 3 with the maximum backbone deviation is denoted with an arrow.
Figure 2-2. Melting profiles of lumazine synthase from S. cerevisiae, B. subtilis and A. aeolicus.
On binding of the inhibitor compounds 5-(6-D-ribitylamino-2,4(1H,3H)pyrimidine-dione-5-yl)pentyl-1-phosphonic acid (RPP) and 6-ribitylamino-5-nitroso-2,4(1H,3H)pyrimidine-dione (RNOP), the apparent Tm of LSBS increased by about 8 C and 6 C, respectively indicating stabilization of the structure by inhibitor binding (Melting curves not shown).
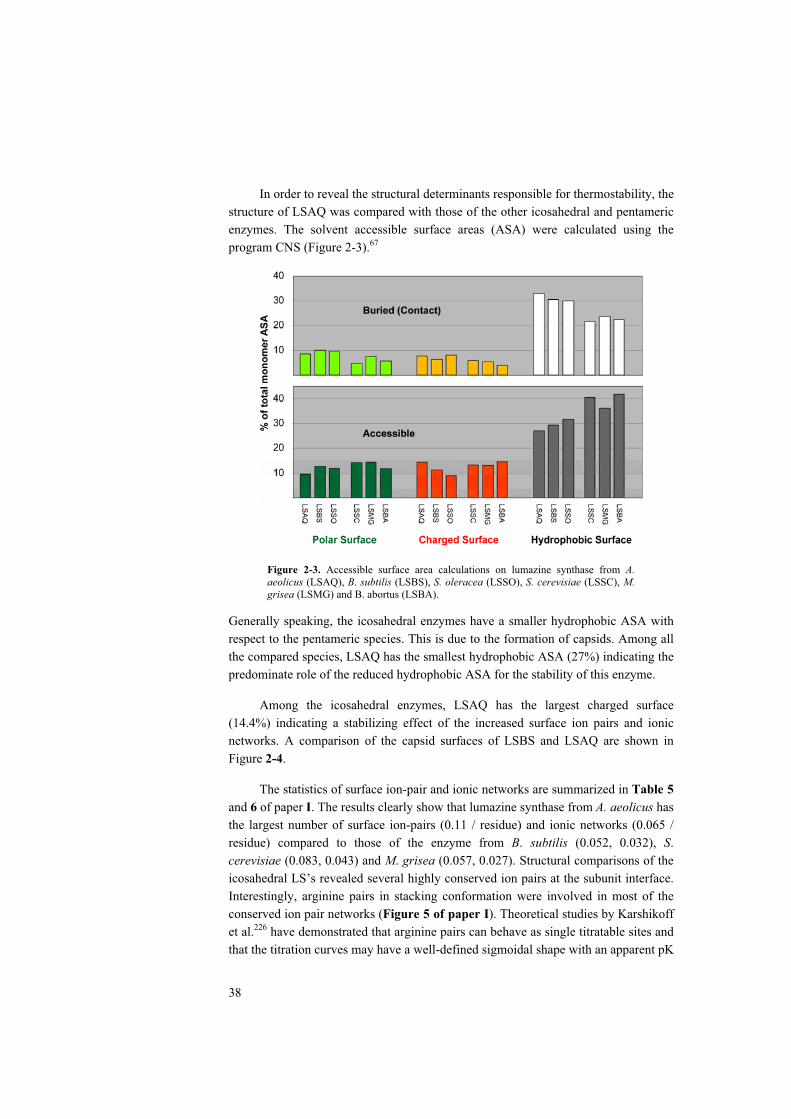
38
In order to reveal the structural determinants responsible for thermostability, the structure of LSAQ was compared with those of the other icosahedral and pentameric enzymes. The solvent accessible surface areas (ASA) were calculated using the program CNS (Figure 2-3).67
Figure 2-3. Accessible surface area calculations on lumazine synthase from A.aeolicus (LSAQ), B. subtilis (LSBS), S. oleracea (LSSO), S. cerevisiae (LSSC), M.grisea (LSMG) and B. abortus (LSBA).
Generally speaking, the icosahedral enzymes have a smaller hydrophobic ASA with respect to the pentameric species. This is due to the formation of capsids. Among all the compared species, LSAQ has the smallest hydrophobic ASA (27%) indicating the predominate role of the reduced hydrophobic ASA for the stability of this enzyme.
Among the icosahedral enzymes, LSAQ has the largest charged surface (14.4%) indicating a stabilizing effect of the increased surface ion pairs and ionic networks. A comparison of the capsid surfaces of LSBS and LSAQ are shown in Figure 2-4.
The statistics of surface ion-pair and ionic networks are summarized in Table 5and 6 of paper I. The results clearly show that lumazine synthase from A. aeolicus has the largest number of surface ion-pairs (0.11 / residue) and ionic networks (0.065 / residue) compared to those of the enzyme from B. subtilis (0.052, 0.032), S.cerevisiae (0.083, 0.043) and M. grisea (0.057, 0.027). Structural comparisons of the icosahedral LS’s revealed several highly conserved ion pairs at the subunit interface. Interestingly, arginine pairs in stacking conformation were involved in most of the conserved ion pair networks (Figure 5 of paper I). Theoretical studies by Karshikoff et al.226 have demonstrated that arginine pairs can behave as single titratable sites and that the titration curves may have a well-defined sigmoidal shape with an apparent pK

39
value of about 7. It was therefore suggested that the earlier reported instability of B.subtilis LS in 0.1 M Tris-HCl at pH > 7 might be due to the disruption of the ionic interactions at the interface between two pentamer blocks, initiated by decharging of the central arginine pair. 163
Figure 2-4. Accessible surface areas of lumazine synthase from (a) A. aeolicus and (b) B. subtilis. Atom-wise color codes are: red for negatively charged surface; blue for positively charged surface; green for polar surface and white for hydrophobic surface.
2.2 CONCLUSIONS
As the apparent Tm of the pentameric LS from S. cerevisiae is much lower than that of the icosahedral LSBS and LSAQ, it is suggested that capsid assembly has a great impact on thermostability of the icosahedral enzymes. Moreover, binding of the inhibitor compounds stabilizes the enzyme.
Compared to that of the pentameric enzymes, the hydrophobic ASA of the icosahedral species are generally smaller, indicating the contribution of the hydrophobic effect to thermostability.
Among all the studied structures, LSAQ has the largest number of surface ion-pairs and networks. Thus the increased ion pairs and ionic networks at the subunit interface are suggested to play a role for capsid assembly and contribute to the overall stability. It is further proposed that the arginine pairs at the subunit interface may be responsible for the pH dependence of the capsid assembly.

40
3 STRUCTURES OF AQUIFEX AEOLICUS LUMAZINE SYNTHASE IN COMPLEX WITH INHIBITORS – A MODEL OF THE REACTION PROCESS (PAPER II)
The enzymes involved in lumazine biosynthesis have been studied in considerable detail. However the mechanism of the enzymatic reaction has remained obscure. In this study, four crystal structures of the enzyme from the hyperthermophilic bacterium Aquifex aeolicus in complex with different inhibitor compounds, which were designed to mimic the substrate, the putative reaction intermediates and the final product, were solved. The structures were refined to resolutions of 1.72 Å, 1.85 Å, 2.05 Å and 2.2 Å, respectively. Earlier kinetic studies of the enzyme from B. subtilis and structural comparisons of the native enzyme from A. aeolicus and the complexes (this work) showed that several highly conserved residues at the active site, namely Phe22, His88, Arg127, Lys135 and Glu138 are most likely involved in catalysis. A structural model of the catalytic process, which illustrates binding of substrates, enantiomer specificity, proton abstraction/donation, inorganic phosphate elimination, formation of the Schiff base and double ring cyclization is proposed.
3.1 RESULTS AND DISCUSSION
The crystal structures of lumazine synthase in complex with four inhibitor compounds140, 227, 228 (Figure 3-1) were determined in this work.
Figure 3-1. The structure formulae of the inhibitors: (a) 6,7-dioxo-5H-8-ribitylaminolumazine (RDL); (b) 3-(7-hydroxy-8-ribityllumazine-6-yl)propionic acid (RPL); (c) 6-ribitylamino-5-nitroso-2,4(1H,3H)pyrimidine-dione (RNOP); (d) 5-(6-D-ribitylamino-2,4(1H,3H)pyrimidine-dione-5-yl)pentyl-1-phosphonic acid (RPP).
Crystals of the complexes were prepared using the sitting drop vapor diffusion method. Crystals of the native protein, used for soaking of the RPL and RPP complexes, were obtained in a solution containing 4% PEG 400 (W/V), 0.3 M lithium sulphate and 0.1 M MOPS (pH 6.5). Protein in complex with RDL and RNOP were co-crystallized by adding 1 l of saturated inhibitor-water solution (ca. 1 M) to 2 lof protein solution (ca. 15 mg/ml) and 3 l of a solution, which contained 0.9 M sodium-potassium tartrate and 0.1M HEPES (pH 7.5). Large crystals were obtained within two weeks.

41
Data collection was performed at HASYLAB, Beam Line X11 (DESY, EMBL Hamburg Outstation). The reflection data were evaluated, merged and scaled using the program package HKL.22 The results of data collection and evaluation are shown in Table 3-1.
Table 3-1. X-ray diffraction data evaluation
LS-RDL LS-RPL LS-RNOP LS-RPP
Wavelength (Å) 0.8482
Space group I 2 3
Cell dimensions (Å) 180.57 180.11 180.12 180.10
Resolution range (Å) 48.22-1.75 48.14-1.82 48.14-2.05 48.13-2.20
Observations/Unique (I > 0) 735750 / 97916 619465 / 86400 480544 / 60538 377239 / 49238
Completeness (%) 99.9 97.6 99.4 100
Overall Rmerge (%) 6.2 5.2 8.7 9.0
All the structures were solved by molecular replacement using the structure of native LSAQ as the search model.174 Results of the crystallographic refinement are summarized in Table 3-2.
Table 3-2. Refinement of A. aeolicus lumazine synthase in complex with inhibitors
RDL RPL RNOP RPP
Number of atoms a
Protein (atoms with 0 occupancy) 5885 (15) 5885 (15) 5885 (25) 5885 (45)
Ligand 115 135 100 135
Solvent 730 585 515 410
Refinement
Resolution range (Å) 48.22-1.75 48.14-1.82 48.14-2.05 48.13-2.2
R-factor (%) 14.2 17.4 15.5 16.1
Free R-factor (%) b 15.7 18.9 18.1 17.7
Ramachandran diagram
Most favored regions (%) 96.2 96.1 95.9 95.9
Allowed regions (%) 3.8 3.2 3.6 4.1
Additionally allowed (%) 0 0.8 0.5 0
B factors (Å2)
Wilson Plot 13.8 21.1 17.7 19.7
All atoms 11.7 17.8 16.7 16.7
Protein 12.5 19.0 17.9 18.0
Ligand 10.8 22.6 13.7 17.0
Solvent 27.0 32.5 29.2 26.8 a Numbers were counted per asymmetric unit.b 5 % of the unique reflections were set aside for calculations of the free R-factor.
The active sites of the enzyme are located at the interface between every two neighboring subunits within a pentamer. The secondary structural elements

42
constructing the active sites are -turns (residues 21-24, 54-58 and 81-92) from one subunit and a -strand (residue 127’-142’) as well as an -helix (residue 113’-116’) from the adjacent subunit. The substrate binding pocket of the enzyme-RPL complex is shown as an example in Figure 3-2a. The superposition of substrate binding sites in the native protein and the four complexes are shown in Figure 3-2b.
Figure 3-2. (a) The substrate binding pocket is located at the interface of two adjacent subunits; (b) Comparison of the active site structures of lumazine synthase in complex with 6,7-dioxo-5H-8-ribitylaminolumazine (RDL, wheat), 3-(7-hydroxy-8-ribityllumazine-6-yl)propionic acid (RPL, green), 6-ribitylamino-5-nitroso-2,4 (1H,3H) pyrimidine-dione (RNOP, blue), and 5-(6-D-ribitylamino-2,4(1H,3H)pyrimidine-dione-5-yl)pentyl-1-phosphonic acid (RPP, yellow) as well as the native enzyme (red) with an empty active site. Note the alternate side-chain locations of Lys135’ and Glu138’ (labels are colored blue) in the complex structures, the adaptation movements of His88 and also the tilted phenyl ring of Phe22 in the structure of the native enzyme.
Shown in Figure 3-2b, the phenyl ring of Phe22 of the complex structures is in an offset parallel conformation with respect to the heteroaromatic ring system of the inhibitor compounds, whereas in the native enzyme it is rotated away by more than 30 . Theoretical studies have shown that the offset-stacking aromatic interactions may stabilize the protein structure.175, 178, 179 It is proposed that the phenyl ring of Phe22 acts like a “gate” controlling the pathway between the active site and the solvent environment. The aromatic interaction stabilizes the bound substrate and the reaction intermediates. After conclusion of the reaction, the phenyl ring rotates back to release the product.
Kinetic studies indicated that the reaction does not commence without binding of substrate 2. Shown in earlier studies and structures in this work, the phosphate group of substrate 2 is in ionic contact with Arg127’. And it also forms hydrogen bonds with Gly64, Ala85 and Thr86. Thus the orientation of substrate 2 is determined mainly by the interactions shown in the schematic drawing (Figure 3-3). It is therefore suggested that Arg127’ and other residues that bind to the phosphate group of substrate 2 are responsible for the regio-specificity of the reaction. Sequence alignments (Figure 1-9) showed that Arg127’ is highly conserved. Replacing Arg127’

43
with a hydrophobic, polar or negatively charged residue led to more than 90% loss of the enzyme activity (Table 1-2). However, replacing Arg127’ with a histidine resulted in a reduction of the activity to only 62% with respective to the wild-type enzyme. It indicates that Arg127’ (or a positively charged residue), which may form a salt-bridge with the phosphate group, is crucial for catalysis.
Figure 3-3. A schematic drawing of the interactions between the phosphate ion and the enzyme residues (contacts via side-chain atoms are marked by frames).
Arg127’, Glu126’ Lys131’ and His132’ form a charged tetrad at the subunit interface. This tetrad constructs a pocket with its counterpart from the neighboring pentamer, which is related by the crystallographic 2-fold symmetry (Figure 3-4). The substrate binding site opens towards the 2-fold pocket, which may serve as an alternate channel for substrate entry. As the charged tetrad is located at the subunit interface, it is proposed that binding of a phosphate ion or an inhibitor / substrate molecule that contains a phosphate group is strictly related to the stability and assembly of the capsid.
Figure 3-4. Solvent-accessible surface representation of the opening at the crystallographic 2-fold. The product analogue 6,7-dioxo-5H-8-ribitylaminolumazine (RDL) is shown by space-filling models. The color codes are: wheat and blue for subunits from one pentamer, salmon and green for subunits from the neighboring pentamer.
According to the reaction pathway proposed by Kis et al (1995),193 the N=C double bond of the Schiff base intermediate must be formed in cis- configuration in order to close the second ring of lumazine (Figure 3-5).

44
Figure 3-5. The cis- configuration of the hypothetical Schiff base intermediate (left) allows nucleophilic attack, which leads to the ring closure; the trans- configuration of the intermediate is shown on the right.
Lys135’ and Glu138’ form an ion pair in the active site. Both residues were observed to adopt either one of the two alternative configurations in all the structures (Figure 3-2b). Lys135’ is able to form a salt bridge with the phosphate group of substrate 2 in one of the conformations. It is therefore suggested that the movement of the side chains of Lys135’ and Glu138’ may facilitate the reorientation of the phosphate moiety of the reaction intermediate leading to a cis- configuration. Following this assumption, the phosphate group of the intermediate may leave the original binding site before the cleavage. Whether the resulting phosphate ion is removed from the active site or bound back to the original site is still unclear. However, it unlikely that the active site would be completely “empty” without either a phosphate ion or a substrate molecule bound to it, because phosphate binding is needed for capsid stabilization. It is therefore proposed that the incoming phosphate-containing substrate (2) would replace the phosphate ion at the active site.
The imidazole group of His 88 is flexible. It is twisted away from the inhibitor by about 0.7 Å in all the complex structures except LSAQ–RNOP, in which the atom N 1on the imidazole ring forms a hydrogen bond with the oxygen atom of the nitroso group of the inhibitor. Earlier studies suggested that the reaction involves several proton transfer steps. Seen from the structures, His88 is in an appropriate position and is therefore a strong candidate for the involvement in the proton transfer steps (Figure 3-6). In the ring closure step of the reaction, the imidazole ring is directly accessible to the oxygen atom of the intermediate, if the S-2 enantiomer is used as the substrate. On the other hand, with the R-2 enantiomer, the distance between His88 and the oxygen atom would not allow a direct proton transfer. It is therefore suggested that His88 is related to the substrate stereo-specificity (note, as mentioned earlier, that the reaction rate of the natural S-2 substrate 2 is about sixfold higher than that of the R-2 enantiomer).

45
Figure 3-6. Proton donation from His88: the His88-N atom serves as a proton donor, which protonates the 15-hydroxyl oxygen of the intermediate (indicated by the arrow). The chiral C15 atom (originally from the S-2 enantiomer of substrate 2)is labeled with an asterisk.
3.2 CONCLUSIONS
Based on kinetic studies and structural comparisons, a reaction mechanism which illustrates key steps of the reaction including substrate binding, nucleophilic attack, formation of the Schiff base, proton transfer, elimination of the phosphate ion, ring closure and the release of the product, is proposed. In this mechanism, the structural features and residues, which may be responsible for the regio-specificity and stereo-specificity of the reaction, were revealed. A hypothetical dual function of the phosphate binding site at Arg127’ is proposed, which is on one hand responsible for capsid stabilization and on the other hand for binding of the phosphate-containing substrate (2).

46
4 MULTIPLE ASSEMBLY STATES OF LUMAZINE SYNTHASES – A MODEL RELATING CATALYTIC FUNCTION AND MOLECULAR ASSEMBLY (MANUSCRIPT III)
Lumazine synthases appear in nature in form of pentamers, dimers of pentamers, icosahedral capsids consisting of 60 subunits and larger capsids with unknown molecular structure. In this paper we describe the analysis of the assembly of wild type and mutant forms of lumazine synthases from Bacillus subtilis and Aquifex aeolicus at various pH values and in presence of different buffers using small angle X-ray scattering and electron microscopy. The data show that multiple assembly states are a general feature of lumazine synthases and there are indications that catalysis is correlated with quaternary structure.
4.1 RESULTS AND DISCUSSION
The pH/buffer dependence of the assembly states of LS from B. subtilis and A.aeolicus as well as a number of mutants was studied using SAXS.
The scattering profiles of LSBS in phosphate (pH = 6.0 ~ 8.0) and Tris hydrochloride (pH = 7.0 ~ 9.0) buffers are shown in Figure 4-1A and 4-1B, respectively. The distance distribution function, p(r), indicates that the assembly of LSBS in phosphate buffers is largely invariant to pH change (Figure 4-1F, curve 1). Capsids with an outer diameter of about 160 Å (which is in line with the size of the icosahedral T = 1 capsid observed in crystal structures) and a small amount of larger particles (yielding a small contribution of distances from 160 to 330 Å, Figure 4-1F curve 1) are formed. In contrast, the distance distribution curves of LSBS in Tris hydrochloride (Figure 4-1F, curves 2-5) point out to large capsids with the outer diameter of about 320 Å. Interestingly, the curves at pH 7.0 and 9.0 (Figure 4-1F, curve 2, 5) display pronounced maxima suggesting rather isometric particle shapes whereas those at pH 7.6 and 8.4 the maxima appear smeared (Figure 4-1F, curve 3, 4), indicating possible polydispersity or deformations of the particles. One can conclude that a structural transition is taking place in Tris at around pH 8.0. In borate buffers (pH = 7.0 ~ 10.0, Figure 4-1C) LSBS displays a similar but even more pronounced dependence of the capsid formation on pH variation than in Tris hydrochloride buffers. At pH 10.0, mostly large particles are formed (Figure 4-1G, curve 4), whereas at other pH 7.0, 8.0 and 9.0 mainly small particles are present (Figure 4-1G, curves 1-3).

47
Figure 4-1. Experimental scattering curves of wild type LSBS and R127 mutant (circles) and fits obtained from the MIXTURE-M program (solid lines). (A) curves (1-5): wild type LSBS in phosphate buffer at pH = 6.0, 6.5, 7.0, 7.5, 8.0, respectively; (B) curves (1-4): wild type LSBS in Tris.HCl buffer at pH = 7.0, 7.6, 8.6 and 9.0, respectively; (C) curves (1-4): wild type LSBS in borate buffer at pH = 7.0, 8.0, 9.0 and 10.0 respectively; (D) curves (1-5): the R127T mutant in phosphate buffer at pH = 6.0, 6.5, 7.0, 7.5 and 8.0, respectively; (E) curves (1-5): the R127T mutant in Tris.HCl buffer at pH = 7.0, 7.5, 8.0, 8.5 and 9.0, respectively. (F):Distance distribution functions p(r), curve (1): wild type LSBS in phosphate buffer pH 6.0; curves (2-5): wild type LSBS in Tris.HCl buffer at pH 7.0, 7.6, 8.4 and 9.0, respectively; (G) curves (1-4): LSBS in borate at pH 7.0, 8.0, 9.0, and 10.0, respectively; (H) curve (1): the R127T mutant in phosphate buffer pH = 6.0; curves (2-6): the mutant in Tris.HCl buffer at pH 7.0, 7.5, 8.0, 8.5 and 9.0, respectively.

48
The scattering curves of the LSBS R127T mutant in phosphate and Tris buffersare shown in Figure 4-1D and 4-1E, respectively. The distance distribution function, p(r), of the mutant in phosphate buffers (Fig. 4-1H, curve 1) indicates the presence of capsids with an outer diameter of about 300 Å. However, in Tris hydrochloride buffers, the distance distribution function (Figure 4-1H, curves 2-6) varies considerably with change of pH. At pH 6.0, the distance distribution function is similar to that of the mutant in phosphate buffer (corresponding to large particles with a diameter of 300 Å), but at pH 7.5 and 8.0 yet larger particles with a diameter of about 320-330 Å appear.
Figure 4-2. Experimental scattering curves of wild type LSAQ and the LSAQ-IDEA mutant (points) and the fits (solid lines) obtained from the program MIXTURE-M. (A) curves (1-5): wild type LSAQ in phosphate buffer at pH = 6.0, 6.5, 7.0, 7.5 and 8.0, respectively; (B) curves (1-5): wild type LSAQ in Tris.HCl buffer at pH = 7.0, 7.5, 8.0, 8.5 and 9.0, respectively; (C) curves 1-3: LSAQ-IDEA mutant in phosphate buffer at pH 6.0, 7.0 and 8.0 respectively; (D) curves (1-5): LSAQ-IDEA mutant in Tris.HCl buffer at pH = 7.0, 7.5, 8.0, 8.5 and 9.0, respectively. (E) the distance distribution function of (1): wild type LSAQ in phosphate buffer at pH 6.0; curve (2), wild type enzyme in Tris.HCl buffer at pH 7.0; curve (3), LSAQ-IDEA mutant in phosphate buffer at pH 6.0; curve(4), LSAQ-IDEA mutant in Tris.HCl buffer at pH 7.0).
The scattering curves (Figure 4-2A, B) and the distance distribution functions of LSAQ in phosphate and Tris hydrochloride buffers (Figure 4-2E, curve 1, 2) are very similar to each other and they display practically no pH dependence. Here, the

49
profile of p(r) corresponds to the small particles (160Å in diameter) with the presence of a minor amount of larger particles (diameter > 300 Å). The LSAQ mutant with an IDEA insertion in phosphate and Tris buffers are similar and show very little pH dependence (Figure 4-2C, D). The p(r) function (Figure 4-2E, curve 3, 4) indicates hollow capsids with an outer diameter of about 275Å. Structural studies of the LSAQ-IDEA mutant are presented in section V (manuscript IV). Detailed results of the SAXS experiments and particle fraction size analyses are summarized in Tables 2 and 3 in manuscript III. The results of the particle size and population analyses obtained using CryoEM are comparable to those of the SAXS experiments (Table 1 in manuscript III).
Mutagenic studies on LSBS have shown that a number of residues at the substrate binding site or the subunit interface are important for both assembly and enzyme activity.
The replacement of Arg127, which is essential for substrate binding, by a polar, hydrophobic or acidic residue results in more than 95% reduction of catalytic activity with respect to the wild type enzyme. The Arg127Thr mutant is not catalytically active, whereas the activity of the Arg127His mutant is reduced to 62%. Both mutants Arg127His (Figure 4-3b) and Arg127Thr (Figure 4-3c) form capsids of at least two different sizes, one resembling the known wild-type T = 1 capsid (Figure 4-3a) and the others with a diameter of at least 280 Å.
Figure 4-3. Negative staining electron micrographs of recombinant lumazine synthase from B. subtilis (A) and the mutants R127T (B) and R127H (C). The scale bar corresponds to 100 nm.
The replacement of either Phe57 or Phe113, involved in substrate binding and inter-subunit interactions, by a serine causes reduction of the reaction rate to 36% and 5%, respectively. These two mutants assemble to both T = 1 icosahedral capsids and large capsids.
Arg21 is involved in a highly conserved ionic network connecting two neighboring pentamer subunits in the capsid. Earlier structural studies suggested that

50
this ionic network is important for capsid assembly. The mutant Arg21Ala has 43% residual catalytic activity with respect to the wild type LSBS. It assembles to both T = 1 capsids and large capsids.
Based on observations presented earlier and in this work, a model relating the capsid assembly and the catalytic function is postulated.
The active site of icosahedral lumazine synthases is located close to the inner wall of the capsids, which implies that the free passage to the solvent is obstructed by the capsid wall. From a structural point of view, the 5-fold channels of the T = 1 capsids could potentially serve as the substrate / product diffusion pathway. However, the available high-resolution structures indicate that the 5-fold channels do not seem wide enough to allow riboflavin, the product of the reaction catalyzed by heavy riboflavin synthase from B. subtilis (the complex [(LS)60(RS)3] described earlier), to pass through. Furthermore, incubation of the enzyme with a tungsten-compound [NaP5W30O110]14-, which was shown to block the 5-fold channels of heavy riboflavin synthase, did not result in a decreased enzymatic activity. Alternative channels close to the 2-fold and 3-fold axis might allow the diffusion of LS substrates and products, but might also be too narrow for riboflavin. This indicates that the transfer of substrates and product could follow another mechanism. An icosahedral LS with a larger number of subunits will most likely be less densely packed and allow for easier access to the binding sites.
As shown in an earlier comparative study, the reaction is unlikely to proceed with rearrangement of the binding site. There is no experimental evidence whether the eliminated phosphate ion would keep being attached to Arg127 during the reaction or at least transiently be removed from the phosphate-binding site. However it was proposed in paper II, that the substrate 2 is moved away from the phosphate-binding site, whereupon phosphate is eliminated from substrate 2. In the subsequent step it might either bind back to the phosphate-binding site or leave the active site. The results and considerations presented in this paper indicate that the formation of proper subunit contacts critically depend on the presence of phosphate or a ligand in the active site and the correct alignment of non-covalent contacts spanning the interface. The absence of these stabilizing contacts, for instance the release of the inorganic phosphate ion from the binding site, would thus destabilize the T = 1 capsids.
Upon conclusion of the reaction, the substrate-binding site could open up involving a widened state of the pentamers (see structural evidence in paper IV). The formation of a large capsid requires a substantial extent of subunit rearrangements, which probably do not proceed on the same time scale as the reaction. It is therefore unlikely that the enzyme would form larger capsids during the catalytic cycle. However, the opening of the active site might be driven by similar forces as the formation of large capsids and eventually lead to a similar local conformation of the

51
pentamers. When the binding site is opened and the interactions in the active site are partially disrupted, the enzyme is not capable of catalysis anymore. This new state might also provide a local widening of the pentamer channel and therefore allow for easier passage of substrates and products through the capsid wall.
As observed by Bacher et al., large capsids are readily converted to T = 1 capsids after incubation with a substrate analogue, 5-nitroso-6-ribitylamino-2,4(1H,3H)-pyrimidine-dione. It is therefore concluded that in the process of substrate binding, the opened and unfunctional binding site rearranges back to the functional state and is thus available for a new catalytic cycle. The rearrangement of LS during the reaction cycle might have a biological and physiological meaning: it has evolved to slow down and control a chemical reaction rather than to accelerate it, in this way keeping the concentration levels of lumazine and riboflavin low in the cell.
4.2 CONCLUSIONS
It has been shown that the wild type lumazine synthase from both B. subtilisand A. aeolicus assembles in multiple states in different buffers. Different assembly forms are in equilibrium, which can be affected by changing of pH. It is suggested that the multiple assembly states are a general feature of icosahedral lumazine synthases.
Studies on the single site mutants of LS from B. subtilis indicate that the capsid assembly is correlated with binding of the substrates (Arg127, Phe57, Phe113) and particular contacts between subunits (e.g. ionic interaction: Arg21, aromatic interaction: Phe57, Phe113). A detailed discussion of structure and assembly of the LSAQ-IDEA mutant is presented in the next section (paper IV).
It is postulated that the substrate binding site in large capsids may have a different conformation with respect to that in the wild type T = 1 capsid. This conformation would allow easier substrate access to and product release from the active site. The local conformational change of the active site may be triggered by binding of the substrate and releasing of the product.

52
5 A 180 SUBUNIT COMPLEX OF A LUMAZINE SYNTHASE MUTANT VIOLATES QUASI-EQUIVALENCE IN CAPSID ASSEMBLY (MANUSCRIPT IV)
As shown above, multiple assembly states are a general feature of lumazine synthases. The sequence alignment indicates that an insertion of one to four residues after Gly138 (S. cerevisiae numbering) in helix 4 is unique for pentameric lumazine synthases. In order to test the hypothesis that introducing this insertion into an icosahedral enzyme could lead to the formation of pentamers, a mutant of Aquifexaeolicus lumazine synthase, in which four amino acids (Ile-Asp-Glu-Ala, IDEA) were inserted after Gly129, was prepared. The structure was determined using cryo-electron microscopy and image reconstruction. Surprisingly, the mutant does not form pentamers, nor does it assemble to the T = 1 capsid like the wild type crystal structure. Instead, it forms large capsids with an outer diameter of 292 Å. The capsid follows icosahedral symmetry with three subunits within each icosahedral asymmetric unit resulting in a complex of 180 subunits. However, the analysis of subunit interactions revealed the lack of quasi-equivalent contacts between subunits indicating that the icosahedral assembly of the LSAQ-IDEA mutant violates the principles of quasi-equivalence.199 Pentameric building blocks in this mutant are widened with respect to that of the native structure. The expanded pentameric structure provides a model for an alternative conformation of the pentamer in the wild-type enzyme as it could be formed during catalysis.
5.1 RESULTS AND DISCUSSION
The LSAQ-IDEA mutant particles were observed on low-dose micrographs as spherical projections with less density in the center (Figure 2 in paper IV).
The refined particle has a diameter of 292 Å, which is 138 Å larger than that of the wild type enzyme, however the thickness of the capsid shell (38 Å) is similar (Figure 5-1 A). The particle shows well-defined 532 symmetry and each icosahedral asymmetric unit contains 3 subunits, i.e. A, B and C (Figure 5-1B), resulting in a capsid with 180 subunits.

53
Figure 5-1. 3D cryo-EM reconstruction of the mutant of lumazine synthase from A.aeolicus with the IDEA insertion. (a) The LSAQ-IDEA mutant follows icosahedral symmetry, the outer diameter is 292 Å. (b) Three unique subunits can be identified in one icosahedral asymmetric unit (the yellow triangle). They are named A, B and C. Filled ovals, triangles and pentagons (green) indicate the location of the icosahedral 2-, 3- and 5-folds, respectively. Open ovals, triangles and hexagons (red) indicate the locations where the quasi 2-, 3- and 6-folds are expected according to the theory of quasi-equivalent assembly. 199
Figure 5-2. The movement needed to transform wildtype LSAQ (T = 1) subunit from its original location (blue) into the position of the LSAQ-IDEA subunit (yellow). The T = 1 subunit must undergo a small translation by 3 Å (along the green line) away from the fivefold axis (grey line) and a rotation by 36° (about the red line).
Earlier modeling studies suggested that introducing the IDEA insertion into an icosahedral enzyme, e.g. LSBS or LSAQ, would lead to clashes at the interface between neighboring pentamers. However, the EM structure shows that pentamers in the mutant are expanded. The diameter of the 5-fold channel is increased is by 8 Å compared to that of the wild-type enzyme. To adopt the opened conformation, each subunit within the pentamer, is to be rotated (as a rigid body) by 36º and translated by 3 Å with respect to the position of the subunits in the native T = 1 capsid (Figure 5-2).
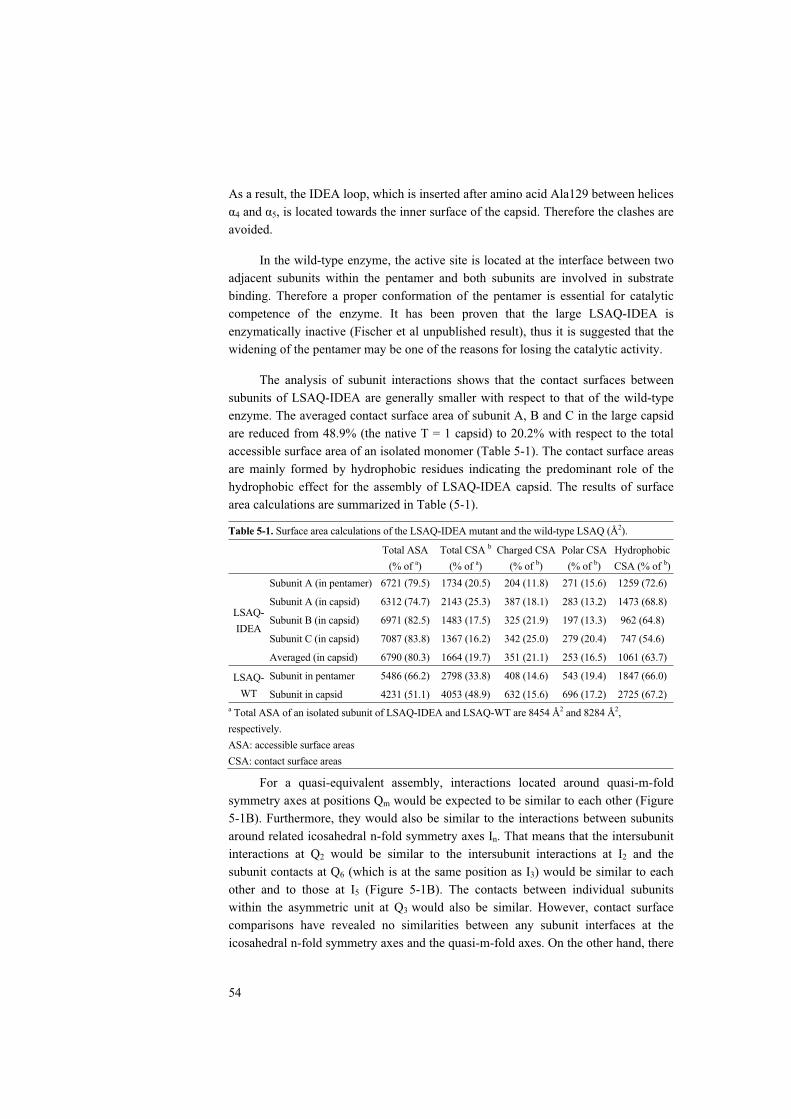
54
As a result, the IDEA loop, which is inserted after amino acid Ala129 between helices 4 and 5, is located towards the inner surface of the capsid. Therefore the clashes are
avoided.
In the wild-type enzyme, the active site is located at the interface between two adjacent subunits within the pentamer and both subunits are involved in substrate binding. Therefore a proper conformation of the pentamer is essential for catalytic competence of the enzyme. It has been proven that the large LSAQ-IDEA is enzymatically inactive (Fischer et al unpublished result), thus it is suggested that the widening of the pentamer may be one of the reasons for losing the catalytic activity.
The analysis of subunit interactions shows that the contact surfaces between subunits of LSAQ-IDEA are generally smaller with respect to that of the wild-type enzyme. The averaged contact surface area of subunit A, B and C in the large capsid are reduced from 48.9% (the native T = 1 capsid) to 20.2% with respect to the total accessible surface area of an isolated monomer (Table 5-1). The contact surface areas are mainly formed by hydrophobic residues indicating the predominant role of the hydrophobic effect for the assembly of LSAQ-IDEA capsid. The results of surface area calculations are summarized in Table (5-1).
Table 5-1. Surface area calculations of the LSAQ-IDEA mutant and the wild-type LSAQ (Å2).
Total ASA (% of a)
Total CSA b
(% of a)Charged CSA
(% of b)Polar CSA
(% of b)HydrophobicCSA (% of b)
Subunit A (in pentamer) 6721 (79.5) 1734 (20.5) 204 (11.8) 271 (15.6) 1259 (72.6)
Subunit A (in capsid) 6312 (74.7) 2143 (25.3) 387 (18.1) 283 (13.2) 1473 (68.8)
Subunit B (in capsid) 6971 (82.5) 1483 (17.5) 325 (21.9) 197 (13.3) 962 (64.8)
Subunit C (in capsid) 7087 (83.8) 1367 (16.2) 342 (25.0) 279 (20.4) 747 (54.6)
LSAQ-IDEA
Averaged (in capsid) 6790 (80.3) 1664 (19.7) 351 (21.1) 253 (16.5) 1061 (63.7)
Subunit in pentamer 5486 (66.2) 2798 (33.8) 408 (14.6) 543 (19.4) 1847 (66.0) LSAQ-WT Subunit in capsid 4231 (51.1) 4053 (48.9) 632 (15.6) 696 (17.2) 2725 (67.2)
a Total ASA of an isolated subunit of LSAQ-IDEA and LSAQ-WT are 8454 Å2 and 8284 Å2,respectively.ASA: accessible surface areas CSA: contact surface areas
For a quasi-equivalent assembly, interactions located around quasi-m-fold symmetry axes at positions Qm would be expected to be similar to each other (Figure 5-1B). Furthermore, they would also be similar to the interactions between subunits around related icosahedral n-fold symmetry axes In. That means that the intersubunit interactions at Q2 would be similar to the intersubunit interactions at I2 and the subunit contacts at Q6 (which is at the same position as I3) would be similar to each other and to those at I5 (Figure 5-1B). The contacts between individual subunits within the asymmetric unit at Q3 would also be similar. However, contact surface comparisons have revealed no similarities between any subunit interfaces at the icosahedral n-fold symmetry axes and the quasi-m-fold axes. On the other hand, there

55
are no similarities between the subunit interfaces among the 3 subunits within the icosahedral asymmetric unit (Figure 8 in paper V).
Modeling studies showed that by a slight expansion of the capsid, the widened LSAQ-IDEA pentamers can be fitted into a wild type T = 1 icosahedral capsid without serious clashes. Recent EM studies have confirmed that the wild-type enzyme from B. subtilis forms an enlarged T = 1 icosahedral capsid with an outer diameter of 186 Å instead of 156 Å (L. Xing, unpublished results). Pentamers in this enlarged T = 1 capsid are also widened and their conformation is similar to that of the pentamers in the LSAQ-IDEA mutant. It is therefore suggested, that the expanded pentameric structure, observed in this study, may serve as a model for an alternative conformation of the wild type LS pentamer as it could also be formed during the catalytic process in the form of local conformational fluctuations (Figure 5-3). Adopting the opened conformation, the product could be released. Furthermore, there would be no steric hindrance for a substrate or product analog, e.g. 5-nitroso-6-ribitylamino-2,4(1H,3H)-pyrimidine-dione, to bind to the more exposed pyrimidine binding site. By this process, sufficient binding energy could be gained, which could trigger the conformational rearrangement leading in a ligand-driven reaction to the reconstitution of the closed pentamer structure, as it is seen in the native T = 1 capsids. 10, 163
Figure 5-3. A hypothetical model of the catalytic cycle, in which the widening of the pentamer may play a role in binding of the substrates (S1 and S2) and in releasing of the products (lumazine and / or the phosphate ion). Note that the phosphate ion could either bind back to the original binding site or be released (paper II).
5.2 CONCLUSIONS
The LSAQ-IDEA mutant does not form either T = 1 capsids or pentamers, it forms large capsids with 180 subunits instead.

56
In the large capsid, the pentameric building blocks are widened compared to that of the wild-type enzyme. The widened pentamers may serve as a model for an alternative conformation of the wild-type enzyme playing a role in catalysis. It is shown that the opened form of the pentamer would exert no steric hindrance to substrate binding or product release. The energy gained by binding of the substrate could trigger the conformational rearrangement from the open enzymatically inactive form of the pentamer structure to the closed enzymatically active form, in accordance with ligand-driven reconstitution of T = 1 capsids from 180 subunit capsids. 10, 163
Although 180 LSAQ-IDEA subunits are assembled into a large icosahedral capsid and the number of subunits fits to the theory of quasi-equivalence assembly with T = 3, contact surface analysis has revealed no similar interactions at the interface between subunits within the icosahedral asymmetric unit or subunits around the icosahedral 2-fold, 3-fold, 5-fold and the quasi 2-fold, 3-fold and 6-fold. It is therefore concluded that the LSAQ-IDEA mutant doesn’t follow the principles of quasi-equivalence in capsid assembly.

57
6 ACKNOWLEDGEMENTS The entire work and every single piece of it are largely due to the expert help of my supervisors. I would like to thank:
Prof. Rudolf Ladenstein, for offering me the opportunity to work in the X-ray group, for guiding me through the entire project, for his endless support, constant encouragement and kindest understanding.
Dr. Winfried Meining, for those valuable advices and fruitful discussions on both theoretical and practical issues, for sharing great ideas on the project and many other scientific topics also for the great company during the past years.
Thanks to all my collaborators:
Prof. Adelbert Bacher, Dr. Markus Fischer, Dr. Ilka Haase, Prof. Mark Cushman, Dr. Dmitri I. Svergun, Dr. Petr V. Konarev, Prof. Holland Cheng, Dr. Li Xing, Josefina Nilsson and Wit Suphamungmee for their contributions, which were indispensable for the project.
Thanks to:
Dr. Assen Koumanov and Mikael Karlström for their enormous help on studying, working and living in Sweden and for their great friendship, which I have always been enjoying very much.
Current and former colleagues in X-ray group: Dr. Andrey Karshikoff, Dr. Bin Ren, Dr. Jordi Benach, Dr. Wei Liu, Dr. Ekaterina Morgunova, Gudrun Tibbelin, Dr. Petras Kundrotas, Linda Arnfors, Dr. Heiko Bönisch, Dr. Joyce Lebbink, Niklas Hellgren, Dr. Stefan Knapp, Dr. Thijs Kaper and Dr. Ida Helene Steen for their help and pleasant company.
Colleagues in CSB: Prof. Kurt Berndt, Prof. Lenart Nilsson, Prof. Hans Hebert, Ana Caballero-Herrera, Dr. Jan Norberg, Dr. Jianxin Duan, Cesar Santiago, Tobias Elgán, Johan Sagemark, Dr. Philip Koeck, Dr. Yijing Xian, Lars Haag, Leif Bergman and other colleagues, whose help and company have been very important for me during that time.
I greatly appreciate:
The IT and service group for their great support, especially Erik Lundgren, for his expert help with computing at CSB.
The administration board, especially Kristina Bergholm and Ingwar Lennerfors for their help throughout these years.
My dearest friends in Sweden:
Jiang Wu, Kejun Li, Shujing Zhang, Li Lan, Roger Eriksson, Xin Ma, Yuli Cao, Yu Shi, Zhong He, Yaofeng Zhao, Jinjing Pei, Wei Jia, Yintong Xue, Yongtao Xue and Hui Zhang for friendship and supports.

58
I shall also thank Prof. Yonggeng Hou and Prof. Lipu Li who introduced me to the field of X-ray crystallography and have been encouraging me all the time.
I am deeply grateful to my wife, Hailin Wang, for her understanding, encouragement and love.
I appreciate my parents and brother, who have given me so much.
This work was supported by Karolinska Institutet, Södertörns Högskola and the Swedish Natural Science Research Council.

59
7 REFERENCES
1. Watson, J. D. & Crick, F. H. (1953). The structure of DNA. Cold Spring Harb Symp Quant Biol 18, 123-131.
2. Kendrew, J. C. (1958). Architecture of a protein molecule. Nature 182, 764-767.
3. Kendrew, J. C., Bodo, G., Dintzis, H. M., Parrish, R. G., Wyckoff, H. & Phillips, D. C. (1958). A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 181, 662-666.
4. Dubochet, J., Adrian, M., Chang, J. J., Homo, J. C., Lepault, J., McDowall, A. W. & Schultz, P. (1988). Cryo-electron microscopy of vitrified specimens. QRev Biophys 21, 129-228.
5. Sakai, H. & Tsukihara, T. (1998). Structures of membrane proteins determined at atomic resolution. J Biochem (Tokyo) 124, 1051-1059.
6. Auer, M. (2000). Three-dimensional electron cryo-microscopy as a powerful structural tool in molecular medicine. J Mol Med 78, 191-202.
7. Campbell, I. D. & Downing, A. K. (1998). NMR of modular proteins. NatStruct Biol 5 Suppl, 496-499.
8. Wüthrich, K. (1986). NMR of Proteins and Nucleic Acids, John Wiley & Sons. 9. Güntert, P. (1998). Structure calculation of biological macromolecules from
NMR data. Q Rev Biophys 31, 145-237. 10. Ladenstein, R., Meyer, B., Huber, R., Labischinski, H., Bartels, K., Bartunik, H.
D., Bachmann, L., Ludwig, H. C. & Bacher, A. (1986). Heavy riboflavin synthase from Bacillus subtilis: Particle dimensions, crystal packing and molecular symmetry. J. Mol. Biol. 187, 87-100.
11. Glatter, O. & Kratky, O. (1982). Small-Angle X-ray Scattering, Academic Press., London.
12. Feigin, L. A. & Svergun, D. I. (1987). Structure Analysis by Small Angle X-ray and Neutron Scattering, Plenum Press., New York.
13. Guinier, A. & Fournet, A. (1955). Small Angle Scattering of X-rays, Wiley, New York.
14. Giacovazzo, C. (2002). Fundamentals of Crystallography. 2nd edit, Oxford University Press, Bari.
15. Drenth, J. (2002). Principles of Protein X-ray Crystallography. 2nd edit, Springer, Heidelberg.
16. McPherson, A. (1994). Crystallization of biological macromolecules, Cold spring harbor laboratory press, New York.
17. McPherson, A. (1991). Useful principles for the crystallization of proteins. In Crystallization of membrane proteins (Michel, H., ed.), pp. 2-51. CRC, Boca Rotan.
18. Unge, T. (1999). Crystallization methods. In Protein Crystallization (Bergfors, T. M., ed.), pp. 7-18. International University Line, California.
19. Garman, E. F. & Doublié, S. (2003). Cryocooling of Macromolecular Crystals: Optimization Methods. Methods in Enzymology 368, 188-216.
20. Dauter, Z. (1999). Data-collection strategies. Acta Crystallogr D Biol Crystallogr 55 ( Pt 10), 1703-1717.
21. Kabsch, W. (1993). Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26, 795-800.
22. Otwinowski, Z. & Minor, W. (1997). Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 276, 307-326.
23. Hahn, T., Ed. (1992). International tables for crystallography. Third edit. Vol. A: Kluwer Academic Publishers.
24. Harker, D. & Kasper, J. S. (1948). Phases of Fourier coefficients directly from crystal diffraction data. Acta Cryst., 70-75.
25. Sayre, D. (1952). The Squaring Method: a New Method for Phase Determination. Acta Cryst., 60-65.

60
26. Karle, J. & Hauptman, H. (1950). The Phases and Magnitudes of the Structure Factors. Acta Cryst., 181-187.
27. Woolfson, M. M. (1987). Direct Methods from Birth to Maturity. Acta Cryst. Section A, 593-612.
28. Giacovazzo, C. (1998). Direct Phasing in Crystallography: Fundamentals and Applications, Oxford University Press, Bari.
29. Blow, D. M. & Rossmann, M. G. (1961). The single isomorphous replacement method. Acta Cryst. 14, 1195-1202.
30. North, A. C. T. (1965). The combination of isomorphous replacement and anomalous scattering data in phase determination of non-centrosymmetric reflexions. Acta Cryst. 18, 212-216.
31. Mathews, B. W. (1966). The determination of the position of anomalously scattering heavy atom groups in protein crystals. Acta Cryst. 20, 230-239.
32. Fourme, R., Shepard, W. & Kahn, R. (1995). Application of the anomalous dispersion of X-rays to macromolecular crystallography. Prog Biophys Mol Biol 64, 167-199.
33. Phillips, J. C. & Hodgson, K. O. (1980). The use of anomalous scattering effects to phase diffraction patterns from macromolecules. Acta Cryst. Section A 36, 856-864.
34. Hendrickson, W. A. (1991). Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science 254, 51-58.
35. Karle, J. (1980). Some developments in anomalous dispersion for the structural investigation of macromolecular systems in biology. International Journal of Quantum Chemistry: Quantum Biology Symposium 7, 357-367.
36. Dauter, Z., Dauter, M. & Dodson, E. (2002). Jolly SAD. Acta Cryst. Section D58, 494-506.
37. Dodson, E. (2003). Is it jolly SAD? Acta Cryst. Section D 59, 1958-1965. 38. Rossmann, M. & Blow, D. (1962). The detection of subunits within the
crystallographic asymmetric unit. Acta Cryst. 15, 24-31. 39. Rossmann, M. G. (1990). The Molecular Replacement Method. Acta Cryst.
Section A 46, 73-82. 40. Rossmann, M. G. (1972). The molecular replacement method, Gordon and
Breach Science Publishers, Inc., New York. 41. Sayre, D. (1953). The double Patterson function. Acta Cryst., 430-431. 42. Hauptman, H. & Karle, J. (1962). The Calculation of Phases from the Patterson
Function. Acta Cryst., 547-550. 43. Terwilliger, T. C. & Kim, S. H. (1987). Generalized method of determining
heavy-atom positions using the difference Patterson function. Acta Cryst. Section A 43, 1-5.
44. Crowther, R. A. & Blow, D. M. (1967). A method of positioning a known molecule in an unknown crystal structure. Acta Cryst., 544-548.
45. Read, R. J. & Schierbeek, A. J. (1988). A phased translation function. J. Appl. Cryst. 21, 490-495.
46. Lattman, E. (1985). Use of the rotation and translation functions. Methods in Enzymology 115, 55-77.
47. Tulinsky, A. (1985). Phase Refinement/Extension by Density Modification. Methods in Enzymology 115, 77-89.
48. Kleywegt, G. J. & Jones, T. A. (1997). Model building and refinement practice. Methods in Enzymology 277, 208-230.
49. Matthews, B. W. (1968). Solvent content of protein crystals. J. Mol. Biol. 33, 491-497.
50. Wang, B. C. (1985). Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol 115, 90-112.
51. Leslie, A. G. W. (1988). CCP4 Study Weekend.52. Terwilliger, T. C. (1999). Reciprocal-space solvent flattening. Acta Crystallogr
D Biol Crystallogr 55, 1863-1871. 53. Vellieux, F. M. D. & Read, R. J. (1997). Noncrystallographic symmetry
averaging in phase refinement and extension. Methods in Enzymology 277, 18-53.

61
54. Zhang, K. Y. J. & Main, P. (1990). Histogram matching as a new density modification technique for phase refinement and extension of protein molecules. Acta Cryst. Section A, 41-46.
55. Abrahams, J. P. & Leslie, A. G. (1996). Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D Biol Crystallogr 52, 30-42.
56. Abrahams, J. P. (1997). Bias reduction in phase refinement by modified interference functions: introducing the gamma correction. Acta Crystallogr D Biol Crystallogr 53, 371-376.
57. Cowtan, K. D. & Zhang, K. Y. (1999). Density modification for macromolecular phase improvement. Prog Biophys Mol Biol 72, 245-270.
58. Zhang, K. Y. J. (2003). Multidimensional Histograms for Density Modification. Methods in Enzymology 374, 188-203.
59. Jones, A. T., Zou, J. Y., Cowtan, J. Y. & Kjeldgaard, M. (1991). Improved methods for building protein models in electron density maps and the location of errors in the model. Acta Crystallogr A 47, 110-119.
60. Engh, R. A. & Huber, R. (1991). Accurate bond and angle parameters for X-ray protein structure refinement. Acta Cryst. Section A, 392-400.
61. Sparks, R. A. (1985). Least-squares refinement. Methods in Enzymology 115, 23-41.
62. Brünger, A. T. (1992). XPLOR manual Version, 3.865 edit. Yale University, New Haven, CT.
63. Adams, P. D., Pannu, N. S., Read, R. J. & Brünger, A. T. (1997). Cross-validated maximum likelihood enhances crystallographic simulated annealing refinement. Proc Natl Acad Sci U S A 94, 5018-5023.
64. Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Cryst.D53, 240-255.
65. Winn, M. D., Murshudov, G. N. & Papiz, M. Z. (2003). Macromolecular TLS Refinement in REFMAC at Moderate Resolutions. Methods in Enzymology374, 300-321.
66. Brünger, A. T. & Rice, L. M. (1997). Crystallographic refinement by simulated annealing: Methods and applications. Methods in Enzymology 277, 243-269.
67. Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Crystallography & NMR system: A new software suite for macromolecular structure determination. ActaCrystallogr D 54, 905-921.
68. Brünger, A. T. (1992). Free R value : a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472-475.
69. Brünger, A. T. (1997). Free R value: Cross-validation in crystallography. Methods Enzymol 277, 366-396.
70. Ramachandran, G. N., Ramakrishnan, C. & Sasisekharan, V. (1963). Stereochemistry of polypeptide chain configurations. J Mol Biol 7, 95-99.
71. Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283-291.
72. Wilson, A. J. C. (1949). The Probability Distribution of X-ray Intensities. ActaCryst. 2, 318-321.
73. Rees, D. C. & Adams, M. W. (1995). Hyperthermophiles: taking the heat and loving it. Structure 3, 251-254.
74. Niehaus, F., Bertoldo, C., Kahler, M. & Antranikian, G. (1999). Extremophiles as a source of novel enzymes for industrial application. Appl Microbiol Biotechnol 51, 711-729.
75. Stetter, K. O. (1999). Extremophiles and their adaptation to hot environments. FEBS Lett 452, 22-25.
76. Madigan, M. T. & Marrs, B. L. (1997). Extremophiles. Sci Am 276, 82-87. 77. Rothschild, L. J. & Mancinelli, R. L. (2001). Life in extreme environments.
Nature 409, 1092-1101.

62
78. Stetter, K. O. (2001). Hyperthermophilic Microorganisms. In Astrobiology: The Quest for the Conditions of Life (Horneck, G. & Baumstark-Khan, C., eds.), pp. 169-184. Springer Verlag, Berlin.
79. Guipaud, O., Marguet, E., Noll, K. M., de la Tour, C. B. & Forterre, P. (1997). Both DNA gyrase and reverse gyrase are present in the hyperthermophilic bacterium Thermotoga maritima. Proc Natl Acad Sci U S A 94, 10606-10611.
80. Guipaud, O. & Forterre, P. (2001). DNA gyrase from Thermotoga maritima.Methods Enzymol 334, 162-171.
81. Bouthier de la Tour, C., Portemer, C., Kaltoum, H. & Duguet, M. (1998). Reverse gyrase from the hyperthermophilic bacterium Thermotoga maritima:properties and gene structure. J Bacteriol 180, 274-281.
82. Forterre, P., Bouthier De La Tour, C., Philippe, H. & Duguet, M. (2000). Reverse gyrase from hyperthermophiles: probable transfer of a thermoadaptation trait from archaea to bacteria. Trends Genet 16, 152-154.
83. Forterre, P. (2002). A hot story from comparative genomics: reverse gyrase is the only hyperthermophile-specific protein. Trends in Genetics 18, 236-237.
84. Hough, D. W. & Danson, M. J. (1999). Extremozymes. Curr Opin Chem Biol3, 39-46.
85. Forterre, P. (1996). A hot topic: the origin of hyperthermophiles. Cell 85, 789-792.
86. Stetter, K. O. (1996). Hyperthermophiles in the history of life. Ciba Found Symp 202, 1-10; discussion 11-18.
87. Huber, R. & Stetter, K. O. (2001). Discovery of hyperthermophilic microorganisms. Methods Enzymol 330, 11-24.
88. Swanson, R. V. (2001). Genome of Aquifex aeolicus. Methods Enzymol 330, 158-169.
89. Deckert, G., Warren, P. V., Gaasterland, T., Young, W. G., Lenox, A. L., Graham, D. E., Overbeek, R., Snead, M. A., Keller, M., Aujay, M., Huber, R., Feldman, R. A., Short, J. M., Olsen, G. J. & Swanson, R. V. (1998). The complete genome of the hyperthermophilic bacterium Aquifex aeolicus. Nature392, 353-358.
90. Gomes, J. & Steiner, W. (2004). The Biocatalytic Potential of Extremophiles and Extremozymes. Food Technol. Biotechnol. 42, 223–235.
91. Burg, B. v. d. (2003). Extremophiles as a source for novel enzymes. CurrentOpinion in Microbiology 6, 213-218.
92. Schellman, J. A. (1987). The thermodynamic stability of proteins. Annu Rev Biophys Biophys Chem 16, 115-137.
93. Privalov, P. L. (1990). Cold denaturation of proteins. Crit Rev Biochem Mol Biol 25, 281-305.
94. Tanford, C. (1968). Protein denaturation. Adv Protein Chem 23, 121-282. 95. Tanford, C. (1970). Protein denaturation. C. Theoretical models for the
mechanism of denaturation. Adv Protein Chem 24, 1-95. 96. Knapp, S., Karshikoff, A., Berndt, K. D., Christova, P., Atanasov, B. &
Ladenstein, R. (1996). Thermal unfolding of the DNA-binding protein Sso7d from the hyperthermophile Sulfolobus solfataricus. Journal of Molecular Biology 264, 1132-1144.
97. Vieille, C. & Zeikus, G. J. (2001). Hyperthermophilic enzymes: sources, uses, and molecular mechanisms for thermostability. Microbiol Mol Biol Rev 65, 1-43.
98. Rose, G. D. & Wolfenden, R. (1993). Hydrogen bonding, hydrophobicity, packing, and protein folding. Annu Rev Biophys Biomol Struct 22, 381-415.
99. Spassov, V. Z., Karshikoff, A. D. & Ladenstein, R. (1995). The optimization of protein-solvent interactions: thermostability and the role of hydrophobic and electrostatic interactions. Protein Science 4, 1516-1527.
100. Dill, K. A. (1990). The meaning of hydrophobicity. Science 250, 297-298. 101. Murphy, K. P. (2001). Stabilization of Protein Structure. In Protein Structure,
Stability, and Folding (Murphy, K. P., ed.), pp. 1-16. Humana press, Iowa City. 102. Stellwagen, E. & Wilgus, H. (1978). Relationship of protein thermostability to
accessible surface area. Nature 275, 342-343.

63
103. Frank, H. S. & Evans, M. W. (1945). Free Volume and Entropy in Condensed Systems III. Entropy in Binary Liquid Mixtures; Partial Molal Entropy in Dilute Solutions; Structure and Thermodynamics in Aqueous Electrolytes. J. Chem. Phy. 13, 507-532.
104. Kauzmann, W. (1959). Some factors in the interpretation of protein denaturation. Adv Protein Chem 14, 1-63.
105. Dill, K. A. (1990). Dominant forces in protein folding. Biochemistry 29, 7133-7155.
106. Makhatadze, G. I. & Privalov, P. L. (1993). Contribution of hydration to protein folding thermodynamics. I. The enthalpy of hydration. J Mol Biol 232, 639-659.
107. Privalov, P. L. & Makhatadze, G. I. (1993). Contribution of hydration to protein folding thermodynamics. II. The entropy and Gibbs energy of hydration. J Mol Biol 232, 660-679.
108. Myers, J. K. & Pace, C. N. (1996). Hydrogen bonding stabilizes globular proteins. Biophys J 71, 2033-2039.
109. Perutz, M. F. (1978). Electrostatic effects in proteins. Science 201, 1187-1191. 110. Perutz, F. M. & Raidt, H. (1975). Stereochemical basis of heat stability in
bacterial ferredoxins and in haemoglobin. Nature 255, 256-259. 111. Russell, R. J. M., Hough, D. W., Danson, M. J. & Taylor, L. T. (1994). The
crystal structure of citrate synthase from the thermophilic archaeon Thermoplasma acidophilum. Structure 2, 1157-1167.
112. Korolev, S., Nayal, M., Barnes, W. M., Di Cera, E. & Waksman, G. (1995). Crystal structure of the large fragment of Thermus aquaticus DNA polymerase I at 2.5 Å resolution: structural basis for thermostability. Proc. Natl. Acad. Sci. USA 92, 9264-9268.
113. Knapp, S., de Vos, W. M., Rice, D. & Ladenstein, R. (1997). Crystal structure of glutamate dehydrogenase from the hyperthermophilic eubacterium Thermotoga maritima at 3.0 Å resolution. J. Mol. Biol. 267, 916-932.
114. Korndörfer, I., Steipe, B., Huber, R., Tomschy, A. & Jaenicke, R. (1995). The crystal structure of holo-glyceraldehyde-3-phosphate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima at 2.5 Å resolution. J. Mol. Biol. 246, 511-521.
115. Kelly, C. A., M., N., Onishi, Y., Beppu, T. & Birktoft, J. J. (1993). Determinants of protein stability in the 1.9 Å crystal structure of malate dehydrogenase from the thermophilic bacterium Thermus flavus. Biochemistry32, 3913-3922.
116. Ren, B., Tibbelin, G., de Pascale, D., Rossi, M., Bartolucci, S. & Ladenstein, R. (1998). A protein disulfide oxidoreductase from the archaeon Pyrococcusfuriosus contains two thioredoxin fold units. Nature Struct. Biol. 7, 602-611.
117. Yip, K. S. P., Stillman, T. J., Britton, K. L., Artymiuk, P. J., Baker, P. J., Sedelnikova, S. E., Engel, P. C., Pasquo, A., Chiaraluce, R., Consalvi, V., Scandurra, R. & Rice, D. (1995). The structure of Pyrococcus furiosusglutamate dehydrogenase reveals a key role for ionpair networks in maintaining enzyme stability at extreme temperatures. Structure 3, 1147-1158.
118. Baker, P. J., Britton, K. L., Engel, P. C., Farrants, G. W., Lilley, K. S., Rice, D. W. & Stillman, T. J. (1992). Subunit assembly and active site location in the structure of glutamate dehydrogenase. Proteins 12, 75-86.
119. Elcock, A. H. (1998). The stability of salt bridges at high temperatures: implications for hyperthermophilic proteins. J. Mol. Biol. 284, 489-502.
120. Karshikoff, A. & Ladenstein, R. (2001). Ion pairs and the thermotolerance of proteins from hyperthermophiles: a "traffic rule" for hot roads. Trends Biochem Sci 26, 550-556.
121. Vetriani, C., Maeder, D. L., Tolliday, N., Yip, K. S., Stillman, T. J., Britton, K. L., Rice, D. W., Klump, H. H. & Robb, F. T. (1998). Protein thermostability above 100 ºC: a key role for ionic interactions. Proceedings of the National Academy of Sciences of the United States of America 95, 12300-12305.
122. Lebbink, J. H., Knapp, S., van der Oost, J., Rice, D., Ladenstein, R. & de Vos, W. M. (1999). Engineering activity and stability of Thermotoga maritimaglutamate dehydrogenase. II: construction of a 16-residue ion-pair network at the subunit interface. Journal of Molecular Biology 289, 357-369.

64
123. Lehninger, A. L., Nelson, D. L. & Cox, M. M. (1993). Principles of biochemistry. 2nd edit, Worth Publishers, Inc., New York.
124. Massey, V. (2000). The chemical and biological versatility of riboflavin. Biochem Soc Trans 28, 283-296.
125. Murty, C. V. & Adiga, P. R. (1982). Pregnancy suppression by active immunization against gestation-specific riboflavin carrier protein. Science 216, 191-193.
126. White, H. B., 3rd & Merrill, A. H., Jr. (1988). Riboflavin-binding proteins. Annu Rev Nutr 8, 279-299.
127. Susin, S. A., Lorenzo, H. K., Zamzami, N., Marzo, I., Snow, B. E., Brothers, G. M., Mangion, J., Jacotot, E., Costantini, P., Loeffler, M., Larochette, N., Goodlett, D. R., Aebersold, R., Siderovski, D. P., Penninger, J. M. & Kroemer, G. (1999). Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397, 441-446.
128. Yagita, K., Tamanini, F., van Der Horst, G. & Okamura, H. (2001). Molecular mechanisms of the biological clock in cultured fibroblasts. Science 292, 278-281.
129. Dagley, S. (1987). Lessons from biodegradation. Annu Rev Microbiol 41, 1-23. 130. Jorns, M. S., Wang, B. & Jordan, S. P. (1987). DNA repair catalyzed by
Escherichia coli DNA photolyase containing only reduced flavin: elimination of the enzyme's second chromophore by reduction with sodium borohydride. Biochemistry 26, 6810-6816.
131. Sassetti, C. M., Boyd, D. H. & Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48, 77-84.
132. Cole, S. T., Eiglmeier, K., Parkhill, J., James, K. D., Thomson, N. R., Wheeler, P. R., Honore, N., Garnier, T., Churcher, C., Harris, D., Mungall, K., Basham, D., Brown, D., Chillingworth, T., Connor, R., Davies, R. M., Devlin, K., Duthoy, S., Feltwell, T., Fraser, A., Hamlin, N., Holroyd, S., Hornsby, T., Jagels, K., Lacroix, C., Maclean, J., Moule, S., Murphy, L., Oliver, K., Quail, M. A., Rajandream, M. A., Rutherford, K. M., Rutter, S., Seeger, K., Simon, S., Simmonds, M., Skelton, J., Squares, R., Squares, S., Stevens, K., Taylor, K., Whitehead, S., Woodward, J. R. & Barrell, B. G. (2001). Massive gene decay in the leprosy bacillus. Nature 409, 1007-1011.
133. Sassetti, C. M. & Rubin, E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100, 12989-12994.
134. Coates, A., Hu, Y., Bax, R. & Page, C. (2002). The future challenges facing the development of new antimicrobial drugs. Nat Rev Drug Discov 1, 895-910.
135. Knowles, J. & Gromo, G. (2003). A guide to drug discovery: Target selection in drug discovery. Nat Rev Drug Discov 2, 63-69.
136. Bacher, A., Baur, R., Eggers, U., Harders, H. D., Otto, M. K. & Schnepple, H. (1980). Riboflavin synthases of Bacillus subtilis. Purification and properties. Journal of Biological Chemistry 255, 632-637.
137. Bacher, A. (1986). Heavy riboflavin synthase from Bacillus subtilis. Methods in Enzymology 122, 192-199.
138. Bacher, A., Schnepple, H., Mailänder, B., Otto, M. K. & Ben-Shaul, Y. (1980). Flavins and Flavoproteins (Yagi, K. Y., T., Ed.), Japan Scientific Societies Press, Tokyo.
139. Plaut, G. W. (1963). Studies on the nature of the enzymic conversion of 6,7-dimethyl-8-ribityllumazine to riboflavin. J Biol Chem 238, 2225-2243.
140. Plaut, G. W. E. (1971). Metabolism of water soluble vitamins. The biosynthesis of riboflavin. In Comprehensive Biochemistry (Florkin, M. & Stotz, E. H., eds.), pp. 11-45. Elsevier, Amsterdam.
141. Wacker, H., Harvey, R. A., Winestock, C. H. & Plaut, G. W. E. (1964). 4-(1'-D-Ribitylamino)-5-amino-2,6-dihydroxypyrimidine, the second product of the riboflavin synthetase reaction. J. Biol. Chem. 239, 3493-3497.
142. Plaut, G. W., Smith, C. M. & Alworth, W. L. (1974). Biosynthesis of water-soluble vitamins. Annual Review of Biochemistry 43, 899-922.

65
143. Liao, D. I., Calabrese, J. C., Wawrzak, Z., Viitanen, P. V. & Jordan, D. B. (2001). Crystal structure of 3,4-dihydroxy-2-butanone 4-phosphate synthase of riboflavin biosynthesis. Structure (Camb) 9, 11-18.
144. Liao, D. I., Wawrzak, Z., Calabrese, J. C., Viitanen, P. V. & Jordan, D. B. (2001). Crystal structure of riboflavin synthase. Structure (Camb) 9, 399-408.
145. Gerhardt, S., Schott, A. K., Kairies, N., Cushman, M., Illarionov, B., Eisenreich, W., Bacher, A., Huber, R., Steinbacher, S. & Fischer, M. (2002). Studies on the reaction mechanism of riboflavin synthase: X-ray crystal structure of a complex with 6-carboxyethyl-7-oxo-8-ribityllumazine. Structure(Camb) 10, 1371-1381.
146. Truffault, V., Coles, M., Diercks, T., Abelmann, K., Eberhardt, S., Luttgen, H., Bacher, A. & Kessler, H. (2001). The solution structure of the N-terminal domain of riboflavin synthase. J Mol Biol 309, 949-960.
147. Eberhardt, S., Zingler, N., Kemter, K., Richter, G., Cushman, M. & Bacher, A. (2001). Domain structure of riboflavin synthase. Eur J Biochem 268, 4315-4323.
148. Meining, W., Eberhardt, S., Bacher, A. & Ladenstein, R. (2001). Crystallization and preliminary crystallographic analysis of the recombinant N-terminal domain of riboflavin synthase. Acta Cryst. D 57, 1296-1299.
149. Haase, I., Mortl, S., Kohler, P., Bacher, A. & Fischer, M. (2003). Biosynthesis of riboflavin in archaea. 6,7-dimethyl-8-ribityllumazine synthase of Methanococcus jannaschii. Eur J Biochem 270, 1025-1032.
150. Zheng, Y. J., Jordan, D. B. & Liao, D. I. (2003). Examination of a reaction intermediate in the active site of riboflavin synthase. Bioorg Chem 31, 278-287.
151. Meining, W., Eberhardt, S., Bacher, A. & Ladenstein, R. (2003). The structure of the N-terminal domain of riboflavin synthase in complex with riboflavin at 2.6A resolution. J Mol Biol 331, 1053-1063.
152. Illarionov, B., Haase, I., Bacher, A., Fischer, M. & Schramek, N. (2003). Presteady state kinetic analysis of riboflavin synthase. J Biol Chem 278, 47700-47706.
153. Bacher, A. & Mailander, B. (1978). Biosynthesis of riboflavin in Bacillussubtilis: function and genetic control of the riboflavin synthase complex. Journal of Bacteriology 134, 476-482.
154. Bacher, A. & Ludwig, H. C. (1982). Ligand-binding studies on heavy riboflavin synthase of Bacillus subtilis. European Journal of Biochemistry 127, 539-545.
155. Volk, R. & Bacher, A. (1990). Studies on the 4-carbon precursor in the biosynthesis of riboflavin. Purification and properties of L-3,4-dihydroxy-2-butanone-4-phosphate synthase. Journal of Biological Chemistry 265, 19479-19485.
156. Volk, R. & Bacher, A. (1991). Biosynthesis of riboflavin. Studies on the mechanism of L-3,4-dihydroxy-2-butanone 4-phosphate synthase. Journal of Biological Chemistry 266, 20610-20618.
157. Bacher, A. & Ladenstein, R. (1990). The Lumazine Synthase/Riboflavin Synthase Complex of Bacillus subtilis. In Chemistry and Biochemistry of Flavoenzymes (Müller, F., ed.), Vol. I., pp. 215-259. Chemical Rubber & Co., Boca Raton, Florida.
158. Schott, K., Ladenstein, R., Konig, A. & Bacher, A. (1990). The lumazine synthase-riboflavin synthase complex of Bacillus subtilis: Crystallization of reconstituted icosahedral -subunit capsids [published erratum appears in J.Biol. Chem., 1990 Oct 15;265(29):18041]. Journal of Biological Chemistry265, 12686-12689.
159. Bacher, A., Fischer, M., Kis, K., Kugelbrey, K., Mortl, S., Scheuring, J., Weinkauf, S., Eberhardt, S., Schmidt-Base, K., Huber, R., Ritsert, K., Cushman, M. & Ladenstein, R. (1996). Biosynthesis of riboflavin: structure and mechanism of lumazine synthase. Biochemical Society Transactions 24, 89-94.
160. Ladenstein, R., Ludwig, H. C. & Bacher, A. (1983). Crystallization and preliminary X-ray diffraction study of heavy riboflavin synthase from Bacillussubtilis. Journal of Biological Chemistry 258, 11981-11983.

66
161. Ladenstein, R., Bacher, A. & Huber, R. (1987). Some observations of a correlation between the symmetry of large heavy-atom complexes and their binding sites on proteins. J. Mol. Biol. 195, 751-753.
162. Ludwig, H. C., Lottspeich, F., Henschen, A., Ladenstein, R. & Bacher, A. (1987). Heavy riboflavin synthase of Bacillus subtilis. Primary structure of the
subunit. Journal of Biological Chemistry 262, 1016-1021. 163. Bacher, A., Ludwig, H. C., Schnepple, H. & Ben-Shaul, Y. (1986). Heavy
riboflavin synthase from Bacillus subtilis. Quaternary structure and reaggregation. J Mol Biol 187, 75-86.
164. Ladenstein, R., Schneider, M., Huber, R., Bartunik, H. D., Wilson, K., Schott, K. & Bacher, A. (1988). Heavy riboflavin synthase from Bacillus subtilis.Crystal structure analysis of the icosahedral beta 60 capsid at 3.3 Å resolution. JMol Biol 203, 1045-1070.
165. Ladenstein, R., Ritsert, K., Huber, R., Richter, G. & Bacher, A. (1994). The lumazine synthase/riboflavin synthase complex of Bacillus subtilis: X-ray structure analysis of hollow reconstituted -subunit capsids. Eur. J. Biochem.223, 1007-1017.
166. Gerhardt, S., Haase, I., Steinbacher, S., Kaiser, J. T., Cushman, M., Bacher, A., Huber, R. & Fischer, M. (2002). The structural basis of riboflavin binding to Schizosaccharomyces pombe 6,7-dimethyl-8-ribityllumazine synthase. J Mol Biol 318, 1317-1329.
167. Meining, W., Mörtl, S., Fischer, M., Cushman, M., Bacher, A. & Ladenstein, R. (2000). The atomic structure of pentameric lumazine synthase from Saccharomyces cerevisiae at 1.85 Å resolution reveals the binding mode of a phosphonate intermediate analogue. J Mol Biol 299, 181-197.
168. Persson, K., Schneider, G., Jordan, D. B., Viitanen, P. V. & Sandalova, T. (1999). Crystal structure analysis of a pentameric fungal and an icosahedral plant lumazine synthase reveals the structural basis for differences in assembly. Protein Sci 8, 2355-2365.
169. Persson, K. (1999). Structural studies of protein assemblies: MHC Class I and Lumazine Synthase, Swedish University of Agricultural Sciences.
170. Morgunova, E., Meining, W., Illarionov, B., Haase, I., Jin, G., Bacher, A., Cushman, M., Fischer, M. & Ladenstein, R. (2005). Crystal structure of lumazine synthase from Mycobacterium tuberculosis as a target for rational drug design: binding mode of a new class of purinetrione inhibitors. Biochemistry 44, 2746-2758.
171. Mörtl, S., Fischer, M., Richter, G., Tack, J., Weinkauf, S. & Bacher, A. (1996). Biosynthesis of riboflavin. Lumazine synthase of Escherichia coli. J. Biol. Chem. 271, 33201-33207.
172. Goldbaum, F. A., Polikarpov, I., Cauerhff, A. A., Velikovsky, C. A., Braden, B. C. & Poljak, R. J. (1998). Crystallization and preliminary x-ray diffraction analysis of the lumazine synthase from Brucella abortus. J Struct Biol 123, 175-178.
173. Braden, B. C., Velikovsky, C. A., Cauerhff, A. A., Polikarpov, I. & Goldbaum, F. A. (2000). Divergence in macromolecular assembly: X-ray crystallographic structure analysis of lumazine synthase from Brucella abortus. J Mol Biol 297, 1031-1036.
174. Zhang, X., Meining, W., Fischer, M., Bacher, A. & Ladenstein, R. (2001). X-ray structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile Aquifex aeolicus at 1.6 Å resolution: determinants of thermostability revealed from structural comparisons. J Mol Biol 306, 1099-1114.
175. Burley, S. K. & Petsko, G. A. (1985). Aromatic-aromatic interaction: a mechanism of protein structure stabilization. Science 229, 23-28.
176. Dosztanyi, Z., Fiser, A. & Simon, I. (1997). Stabilization centers in proteins: identification, characterization and predictions. J Mol Biol 272, 597-612.
177. Meurisse, R., Brasseur, R. & Thomas, A. (2004). Aromatic side-chain interactions in proteins: near- and far-sequence Tyr-X pairs. Proteins 54, 478-490.

67
178. Thomas, A., Meurisse, R. & Brasseur, R. (2002). Aromatic side-chain interactions in proteins. II. Near- and far-sequence Phe-X pairs. Proteins 48, 635-644.
179. Thomas, A., Meurisse, R., Charloteaux, B. & Brasseur, R. (2002). Aromatic side-chain interactions in proteins. I. Main structural features. Proteins 48, 628-634.
180. Toth, G., R, F. M. & Lovas, S. (2001). Stabilization of local structures by pi-CH and aromatic-backbone amide interactions involving prolyl and aromatic residues. Protein Eng 14, 543-547.
181. Fischer, M., Haase, I., Kis, K., Meining, W., Ladenstein, R., Cushman, M., Schramek, N., Huber, R. & Bacher, A. (2003). Enzyme catalysis via control of activation entropy: site-directed mutagenesis of 6,7-dimethyl-8-ribityllumazine synthase. J Mol Biol 326, 783-793.
182. Otto, M. K. & Bacher, A. (1981). Ligand-binding studies on light riboflavin synthase from Bacillus subtilis. European Journal of Biochemistry 115, 511-517.
183. Lee, J., Gibson, B. G., O'Kane, D. J., Kohnle, A. & Bacher, A. (1992). Fluorescence study of the ligand stereospecificity for binding to lumazine protein. European Journal of Biochemistry 210, 711-719.
184. Kis, K., Volk, R. & Bacher, A. (1995). Biosynthesis of riboflavin. Studies on the reaction mechanism of 6,7-dimethyl-8-ribityllumazine synthase. Biochemistry 34, 2883-2892.
185. Kis, K., Kugelbrey, K. & Bacher, A. (2001). Biosynthesis of riboflavin. The reaction catalyzed by 6,7-dimethyl-8-ribityllumazine synthase can proceed without enzymatic catalysis under physiological conditions. J Org Chem 66, 2555-2559.
186. Fischer, M., Haase, I., Feicht, R., Richter, G., Gerhardt, S., Changeux, J. P., Huber, R. & Bacher, A. (2002). Biosynthesis of riboflavin: 6,7-dimethyl-8-ribityllumazine synthase of Schizosaccharomyces pombe. Eur. J. Biochem. 269, 519-526.
187. Haase, I., Fischer, M., Bacher, A. & Schramek, N. (2003). Temperature-dependent presteady state kinetics of lumazine synthase from the hyperthermophilic eubacterium Aquifex aeolicus. J Biol Chem 278, 37909-37915.
188. Schramek, N., Haase, I., Fischer, M. & Bacher, A. (2003). Biosynthesis of riboflavin. Single turnover kinetic analysis of 6,7-dimethyl-8-ribityllumazine synthase. J Am Chem Soc 125, 4460-4466.
189. Scheuring, J., Kugelbrey, K., Weinkauf, S., Cushman, M., Bacher, A. & Fischer, M. (2001). 19F NMR ligand perturbation studies on 6,7-bis(trifluoromethyl)-8-ribityllumazine-7-hydrates and the lumazine synthase complex of Bacillus subtilis. Site-directed mutagenesis changes the mechanism and the stereoselectivity of the catalyzed haloform-type reaction. J Org Chem66, 3811-3819.
190. Bacher, A., LeVan, Q., Buehler, M., Keller, P. J., Eimicke, V. & Floss, H. G. (1982). Biosynthesis of riboflavin. Incorporation of D-[1-13C]ribose. J Am Chem Soc 104, 3754-3755.
191. Bacher, A., LeVan, Q., Keller, P. J. & Floss, H. G. (1985). Biosynthesis of riboflavin. Incorporation of multiply 13C-labeled precursors into the xylene ring. J Am Chem Soc 107, 6380-6385.
192. LeVan, Q., Keller, P. J., Bown, D. H., Floss, H. G. & Bacher, A. (1985). Biosynthesis of riboflavin in Bacillus subtilis: origin of the four-carbon moiety. Journal of Bacteriology 162, 1280-1284.
193. Kis, K. & Bacher, A. (1995). Substrate channeling in the lumazine synthase/riboflavin synthase complex of Bacillus subtilis. Journal of Biological Chemistry 270, 16788-16795.
194. Caspar, D. L. (1960). The structural stability of tocacco mosaic virus. Trans N Y Acad Sci 22, 519-521.
195. Caspar, D. L. (1963). Assembly and Stability of the Tobacco Mosaic Virus Particle. Adv Protein Chem 18, 37-121.
196. Caspar, D. L. (1956). Structure of bushy stunt virus. Nature 177, 475-476.

68
197. Klug, A., Finch, J. T. & Franklin, R. E. (1957). Structure of turnip yellow mosaic virus. Nature 179, 683-684.
198. Klug, A., Finch, J. T. & Franklin, R. E. (1957). The structure of turnip yellow mosaic virus; x-ray diffraction studies. Biochim Biophys Acta 25, 242-252.
199. Caspar, D. L. D. & Klug, A. (1962). Physical principles in the construction of regular viruses. Cold Spring Harbor Symposium on Quantitative Biology 27, 1-24.
200. Johnson, J. E. & Speir, J. A. (1997). Quasi-equivalent viruses: a paradigm for protein assemblies. J. Mol. Biol. 269, 665-675.
201. Caspar, D. L. (1980). Movement and self-control in protein assemblies. Quasi-equivalence revisited. Biophys J 32, 103-138.
202. Rossmann, M. G. (1984). Constraints on the assembly of spherical virus particles. Virology 134, 1-11.
203. Harrison, S. C., Olson, A. J., Schutt, C. E., Winkler, F. K. & Bricogne, G. (1978). Tomato bushy stunt virus at 2,9 Å resolution. Nature 276, 368-373.
204. Liddington, R. C., Yan, Y., Moulai, J., Sahli, R., Benjamin, T. L. & Harrison, S. C. (1991). Structure of simian virus 40 at 3.8-A resolution. Nature 354, 278-284.
205. Prasad, B. V., Prevelige, P. E., Marietta, E., Chen, R. O., Thomas, D., King, J. & Chiu, W. (1993). Three-dimensional transformation of capsids associated with genome packaging in a bacterial virus. J Mol Biol 231, 65-74.
206. Fuller, M. T. & King, J. (1982). Assembly in vitro of bacteriophage P22 procapsids from purified coat and scaffolding subunits. J Mol Biol 156, 633-665.
207. Prevelige, P. E., Jr., Thomas, D., Aubrey, K. L., Towse, S. A. & Thomas, G. J., Jr. (1993). Subunit conformational changes accompanying bacteriophage P22 capsid maturation. Biochemistry 32, 537-543.
208. Prevelige, P. E., Jr., Thomas, D. & King, J. (1988). Scaffolding protein regulates the polymerization of P22 coat subunits into icosahedral shells in vitro. J Mol Biol 202, 743-757.
209. Prevelige, P. E., Jr., Thomas, D. & King, J. (1993). Nucleation and growth phases in the polymerization of coat and scaffolding subunits into icosahedral procapsid shells. Biophys J 64, 824-835.
210. Berger, B., Shor, P. W., Tucker-Kellogg, L. & King, J. (1994). Local rule-based theory of virus shell assembly. Proc Natl Acad Sci U S A 91, 7732-7736.
211. Berger, B., King, J., Schwartz, R. & Shor, P. W. (2000). Local rul mechanism for selecting icosahedral shell geometry. Discrete Applied Mathematics 104, 97-111.
212. Schwartz, R., Garcea, R. L. & Berger, B. (2000). "Local rules" theory applied to polyomavirus polymorphic capsid assemblies. Virology 268, 461-470.
213. Schwartz, R., Shor, P. W., Prevelige, P. E., Jr. & Berger, B. (1998). Local rules simulation of the kinetics of virus capsid self-assembly. Biophys J 75, 2626-2636.
214. Oliver, R. M. & Reed, L. J. (1982). Multienzyme complexes. In Electronmicroscopy of proteins (Harris, J. R., ed.), Vol. 2, pp. 1-48. Academic press, London.
215. Reed, L. J. & Hackert, M. L. (1990). Structure-function relationships in dihydrolipoamide acyltransferases. J. Biol. Chem. 265, 8971-8974.
216. Perham, R. N. (2000). Swing arms and swinging domains in multifunctional enzymes: Catalytic machines for multistep reactions. Annual Review of Biochemistry 69, 961-1004.
217. Izard, T., Aevarsson, A., Allen, M. D., Westphal, A. H., Perham, R. N., de Kok, A. & Hol, W. G. (1999). Principles of quasi-equivalence and Euclidean geometry govern the assembly of cubic and dodecahedral cores of pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. U S A 96, 1240-1245.
218. Tamura, T., Tamura, N., Cejka, Z., Hegerl, R., Lottspeich, F. & Baumeister, W. (1996). Tricorn protease: the core of a modular proteolytic system. Science 274, 1385-1389.

69
219. Walz, J., Tamura, T., Tamura, N., Grimm, R., Baumeister, W. & Koster, A. J. (1997). Tricorn protease exists as an icosahedral supermolecule in vivo. MolCell 1, 59-65.
220. Walz, J., Koster, A. J., Tamura, T. & Baumeister, W. (1999). Capsids of tricorn protease studied by electron cryomicroscopy. J Struct Biol 128, 65-68.
221. Brandstetter, H., Kim, J. S., Groll, M. & Huber, R. (2001). Crystal structure of the tricorn protease reveals a protein disassembly line. Nature 414, 466-470.
222. Hino, T., Kanamori, E., Shen, J. R. & Kouyama, T. (2004). An icosahedral assembly of the light-harvesting chlorophyll a/b protein complex from pea chloroplast thylakoid membranes. Acta Crystallogr D Biol Crystallogr 60, 803-809.
223. Liu, Z., Yan, H., Wang, K., Kuang, T., Zhang, J., Gui, L., An, X. & Chang, W. (2004). Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature 428, 287-292.
224. Ladenstein, R., Meining, W., Zhang, X., Fischer, M. & Bacher, A. (2004). Metamorphosis of an Enzyme. In Conformational Proteomics of Macromolecular Architecture: Approaching the Structure of Large Molecular Assemblies and Their Mechanisms of Action (Cheng, R. H. & Hammar, L., eds.), pp. 198-221. World Scientific Pub. Co., Stockholm.
225. Fornasari, M. S., Laplagne, D. A., Frankel, N., Cauerhff, A. A., Goldbaum, F. A. & Echave, J. (2004). Sequence determinants of quaternary structure in lumazine synthase. Mol Biol Evol 21, 97-107.
226. Karshikoff, A., Spassov, V., Cowan, S. W., Ladenstein, R. & Schirmer, T. (1994). Electrostatic properties of two porin channels from Escherichia coli. J.Mol. Biol. 240, 372-384.
227. Cushman, M., Mavandadi, F., Yang, D., Kugelbrey, K., Kis, K. & Bacher, A. (1999). Synthesis and biochemical evaluation of bis(6,7-dimethyl-8-D-ribityllumazines) as potential bisubstrate analogue inhibitors of riboflavin synthase. J. Org. Chem. 64, 4635-4642.
228. Al-Hassan, S. S., Kulick, R. J., Livingstone, D. B., Suckling, C. J. & Wood, H. C. S. (1980). Specific enzyme inhibitors in vitamin biosynthesis. Part 3. The synthesis and inhibitory properties of some substrates and transition state analogs of riboflavin synthase. J. Chem. Soc. Perkin 1, 2645-2656.