synthases table of contents
TRANSCRIPT

Supporting Information for
Mechanistic Insight with HBCH2CoA as A Probe to Polyhydroxybutyrate (PHB) Synthases
Wei Zhang+, Ruben Shrestha+, Rachael M. Buckley≠, Jamie Jewell⊥, Stefan H. Bossmann+, JoAnne Stubbe≠§*, and Ping Li+*
+ Department of Chemistry, Kansas State University, Manhattan, KS 66506
⊥Department of Chemistry, Ohio Dominican University, Columbus, OH 43219
Department of ≠Chemistry and §Biology, Massachusetts Institute of Technology, Cambridge, MA 02139
E-mail: [email protected]; [email protected]
Table of Contents
1. General information………………………………………………………. S2
2. Chemoenzymatic synthesis of HBCH2CoA 1……………………………. S3
3. Inhibition study………………………………............................................ S9
4. Kinetic parameters of PhaCRe in the presence of 0.05% detergent ………. S11
5. Reactions of *sT-PhaCRe and HBCH2CoA……………………………….. S12
6. NMR spectra of synthesized compounds…………………………………. S15
7. HRMS of HBCH2CoA 1………………………………………………….. S36
S-1

1. General information
All chemicals were purchased at the highest purity grade. All solvents were anhydrous. All reactions were performed under argon atmosphere unless otherwise specified. Thin layer chromatography (TLC) was performed using 60 mesh silica gel plates and visualization was performed using short wavelength UV light (254 nm) and basic KMnO4 staining. NMR spectra were recorded on a Varian 400 MHz spectrometer. Chemical shifts of proton (1H NMR) and carbon (13C NMR) were reported in ppm relative to the residual protiated solvent peaks except that methanol was employed as the external reference when D2O was used. Chemical shifts of phosphorus (31P NMR) were reported in ppm relative to the external reference of 85% H3PO4. High resolution mass spectrometry (HRMS) was performed on a Bruker Daltonics Apexiv 4.7 tesla fourier transform ion cyclotron resonance mass spectrometer (FT-ICR-MS). Electrospray ionization mass spectrometry (ESI-MS) was performed on an API 2000 LC/MS/MS system manufactured by AB SCIEX or a Mariner biospectrometry workstation manufactured by Perspective Biosystem. MALDI-TOF MS was performed on an Applied Biosystem Model Voyager DE-STR instrument. HPLC was performed with a Waters Breeze 2 HPLC system consisting of a 1525 HPLC pump and a 2998 photodiode array detector. Absorbance was measured using an Agilent Cary 100 UV-Vis spectrophotometer.
2. Chemoenzymatic synthesis of HBCH2CoA 1
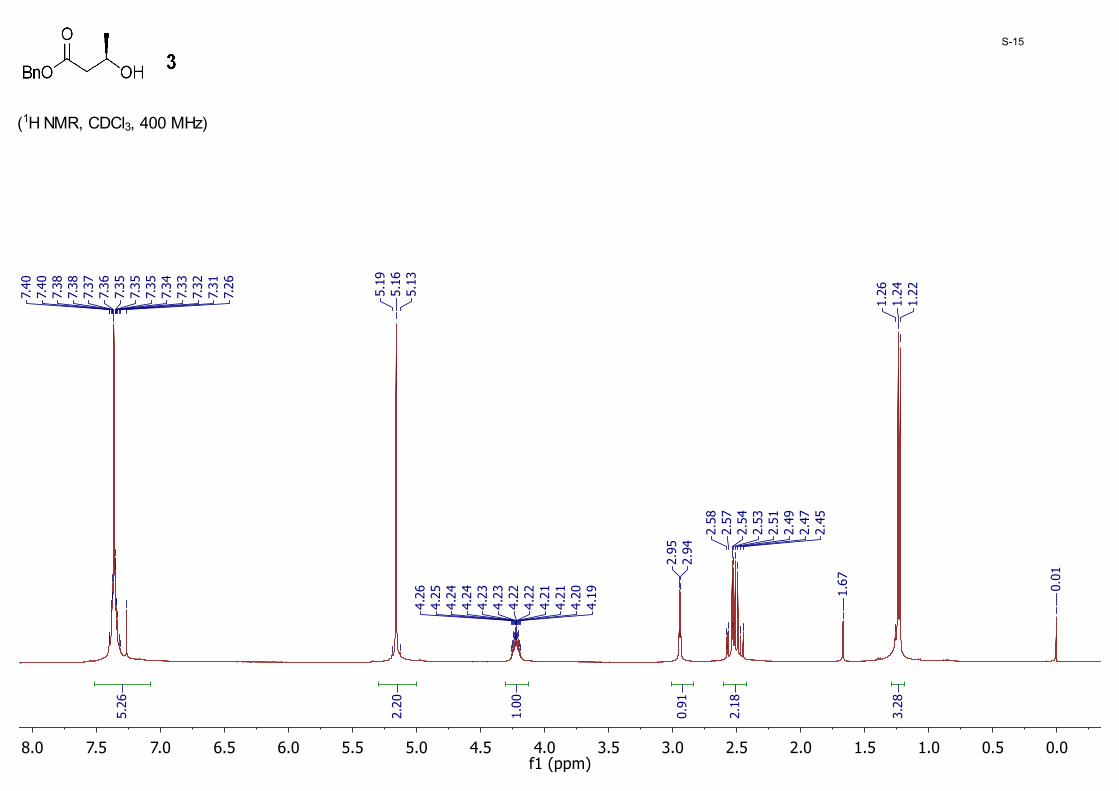
Benzyl (R)-3-hydroxybutanoate 3: To a solution of sodium (R)-3-hydroxybutanoate 2 (5.00 g, 39.7 mmol) in DMF (100 mL) at 0 °C was added drop-wise BnBr (9.40 mL, 79.3 mmol). The resulting mixture was stirred for 12 hours at room temperature. The reaction mixture was poured into water (250 mL) and extracted with ether (100 mL × 3). The ether layer was washed with brine (100 mL), dried with anhydrous MgSO4 and evaporated to dryness. The crude product was purified by silica gel column chromatography eluting with hexane/EtOAc (5/1 to 3/1) to give 3 (6.50 g, 84%) as a colorless oil; 1H NMR (CDCl3, 400 MHz) δ 7.40 – 7.26 (m, 5H), 5.16 (s, 2H), 4.26 – 4.19 (m, 1H), 2.94 (d, J = 3.8 Hz, 1H), 2.58 – 2.45 (m, 2H), 1.24 (t, J = 7.4 Hz, 3H).
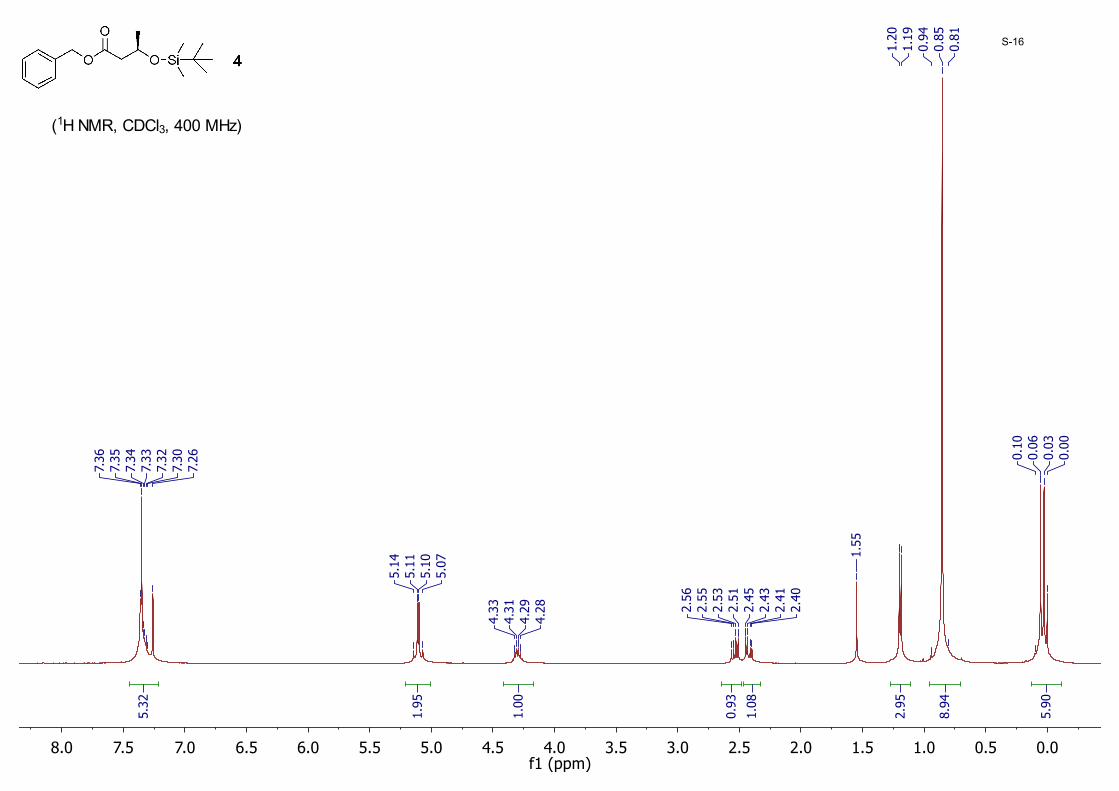
Benzyl (R)-3-((tert-butyldimethylsilyl)oxy)butanoate 4: To a mixture of 3 (7.70 g, 39.6 mmol) and imidazole (5.40 g, 79.3 mmol) in dry DMF (100 mL) was added tert-butylchlorodimethylsilane (TBDMSCl, 7.17 g, 47.6 mmol) at 0 °C. The mixture was then warmed to room temperature and stirred for another 4 hours. On completion, it was poured into 200 mL ice-water and extracted with ether (300 mL). The ether layer was washed with brine, dried with anhydrous MgSO4, and concentrated in vacuo. The crude product was purified by silica gel column chromatography eluting with hexane/EtOAc (40/1) to yield 4 as a colorless oil (10.8 g, 89%); 1H NMR (CDCl3 , 400 MHz) δ 7.36 – 7.26 (m, 5H), 5.11 (dd, J = 12, 5.2 Hz, 2H), 4.33 – 4.28 (m, 1H), 2.54 (dd, J = 14.6, 7.5 Hz, 1H), 2.42 (dd, J = 14.6, 5.3 Hz, 1H), 1.19 (d, J = 6.1 Hz, 3H), 0.85 (s, 9H), 0.06 (s, 3H), 0.03 (s, 3H).
S-2

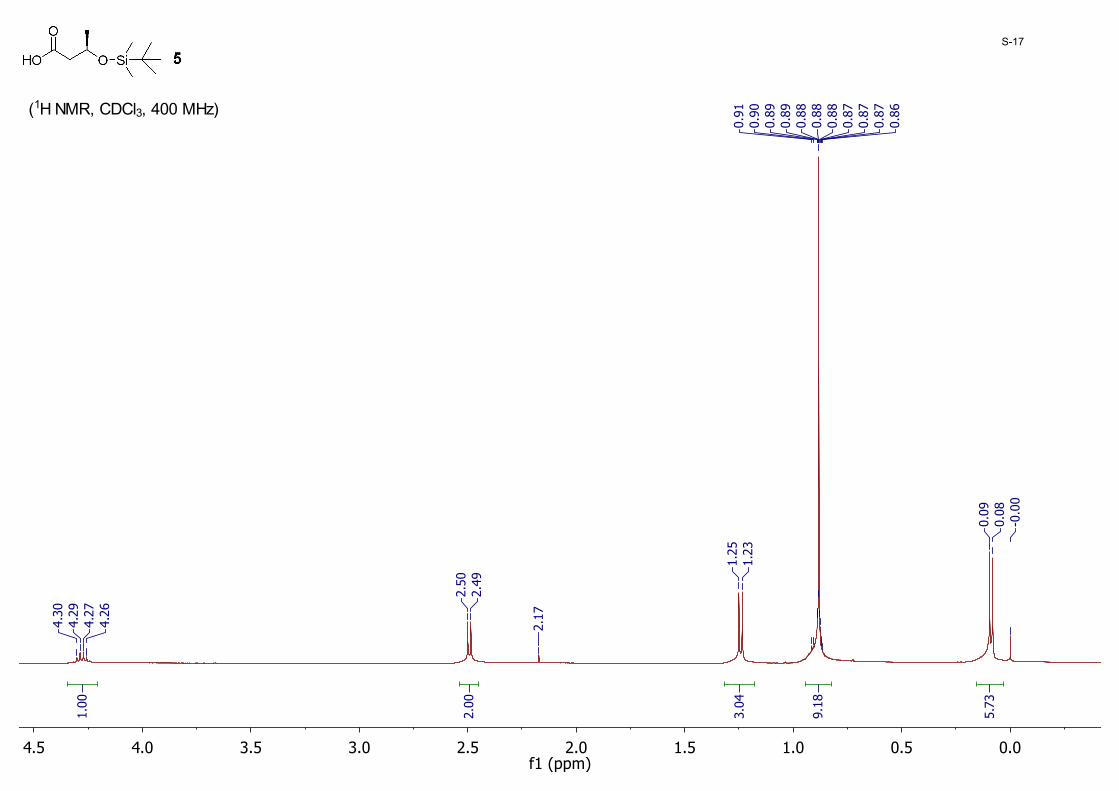
(R)-3-((tert-Butyldimethylsilyl)oxy)butanoic acid 5: A methanol solution of 4 (7.70 g, 24.9 mmol) was added 10% Pd/C powder (0.77 g). The reaction was hydrogenated at atmospheric pressure and stirred for 3 hours at room temperature. The powder was then removed by filtration and washed with MeOH (10 mL × 3). The filtrate was concentrated under high vacuum to give 5 as a colorless oil (5.30 g, 97%); 1H NMR (CDCl3, 400 MHz) δ 4.30 – 4.26 (m, 1H), 2.49 (d, J = 5.8 Hz, 2H), 1.24 (d, J = 6.2 Hz, 3H), 0.88 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H).
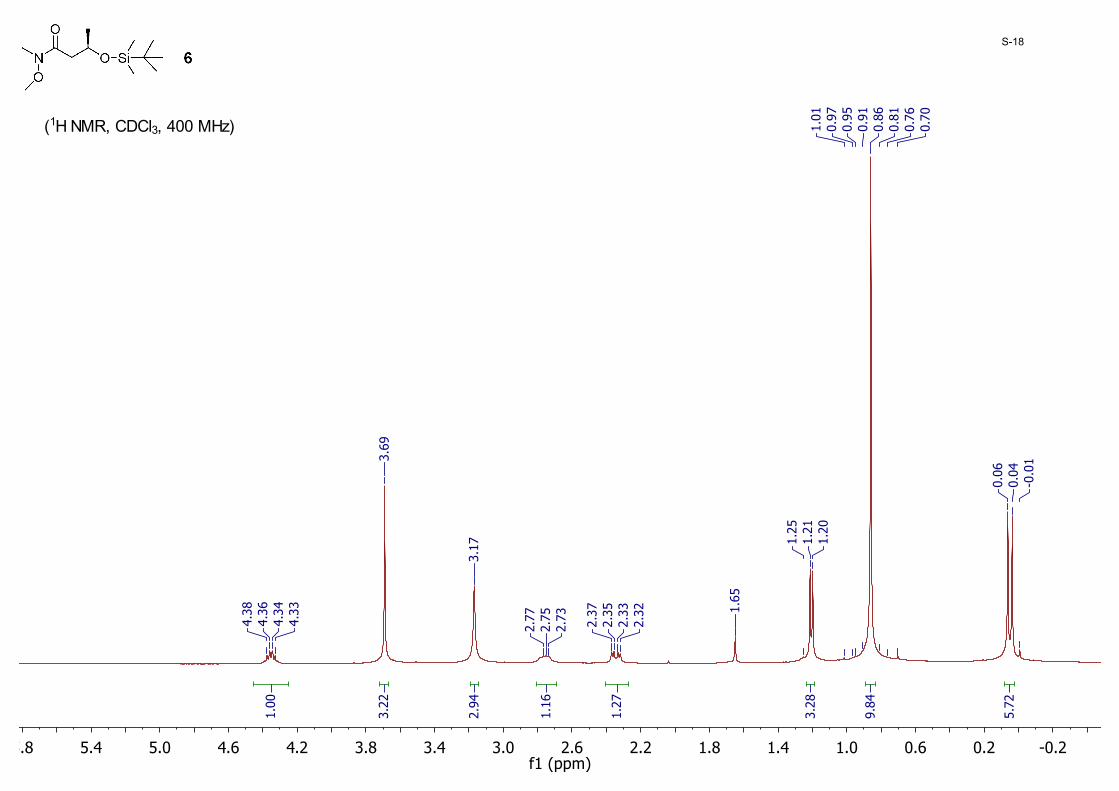
(R)-3-((tert-Butyldimethylsilyl)oxy)-N-methoxy-N-methylbutanamide 6: A mixture of 5 (7.90 g, 36.2 mmol), N,O-dimethylhydroxylamine hydrochloride (3.88 g, 39.8 mmol), 1-ethyl-3-(3-dimethylaminopropyl)carbodimmide (EDCI, 7.63 g, 39.8 mmol), 1-hydroxybenzotriazole (HOBT, 5.38 g, 39.8 mmol), and triethylamine (15.1 mL, 108.5 mmol) in CH2Cl2 (180 mL) was stirred for 3 hours at room temperature. The solution was diluted with 200 mL CH2Cl2 and washed sequentially with 1N HCl (100 mL), an aqueous solution of NaHCO3 (100 mL) and brine (100 mL). The organic layer was dried with anhydrous MgSO4 and concentrated to dryness. The residue was purified by silica gel column chromatography eluting with hexane/EtOAc (20/1 to 10/1) to give 6 (7.85 g, 83%) as a colorless oil; 1H NMR (CDCl3, 400 MHz) δ 4.38 – 4.33 (m, 1H), 3.69 (s, 3H), 3.17 (s, 3H), 2.77 – 2.73 (m, 1H), 2.34 (dd, J = 14.6, 5.5 Hz, 1H), 1.21 (d, J = 6.1 Hz, 3H), 0.86 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H).
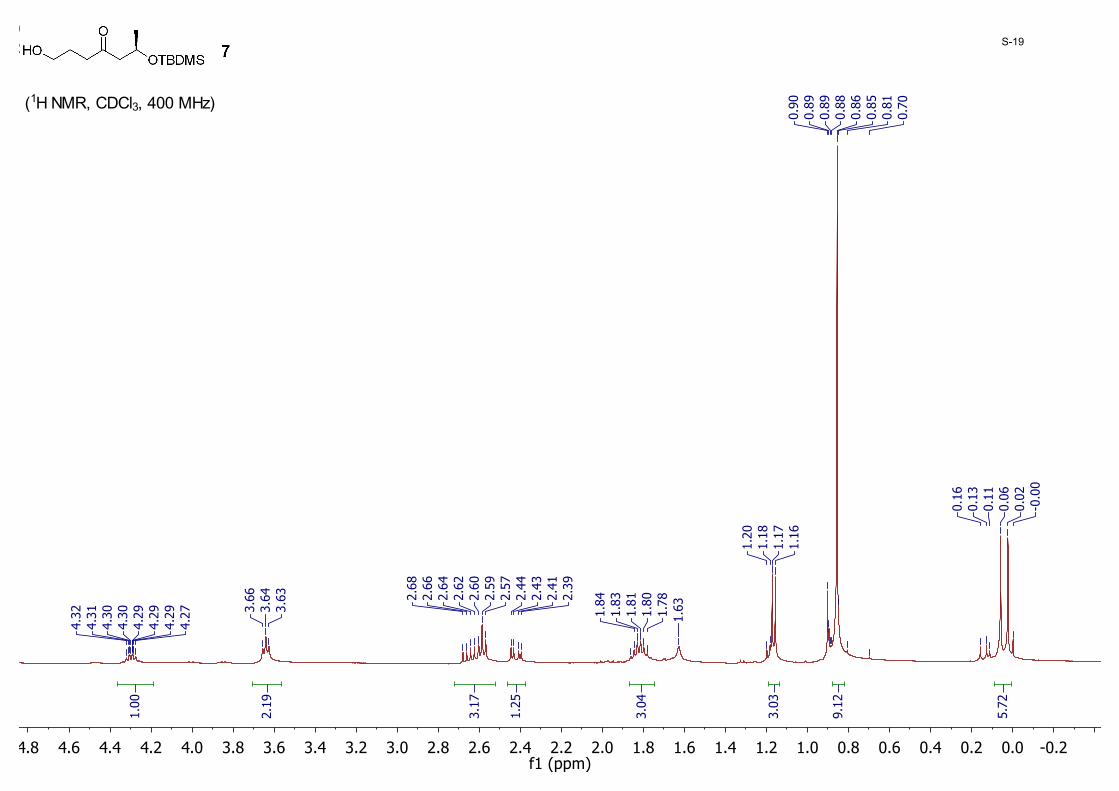
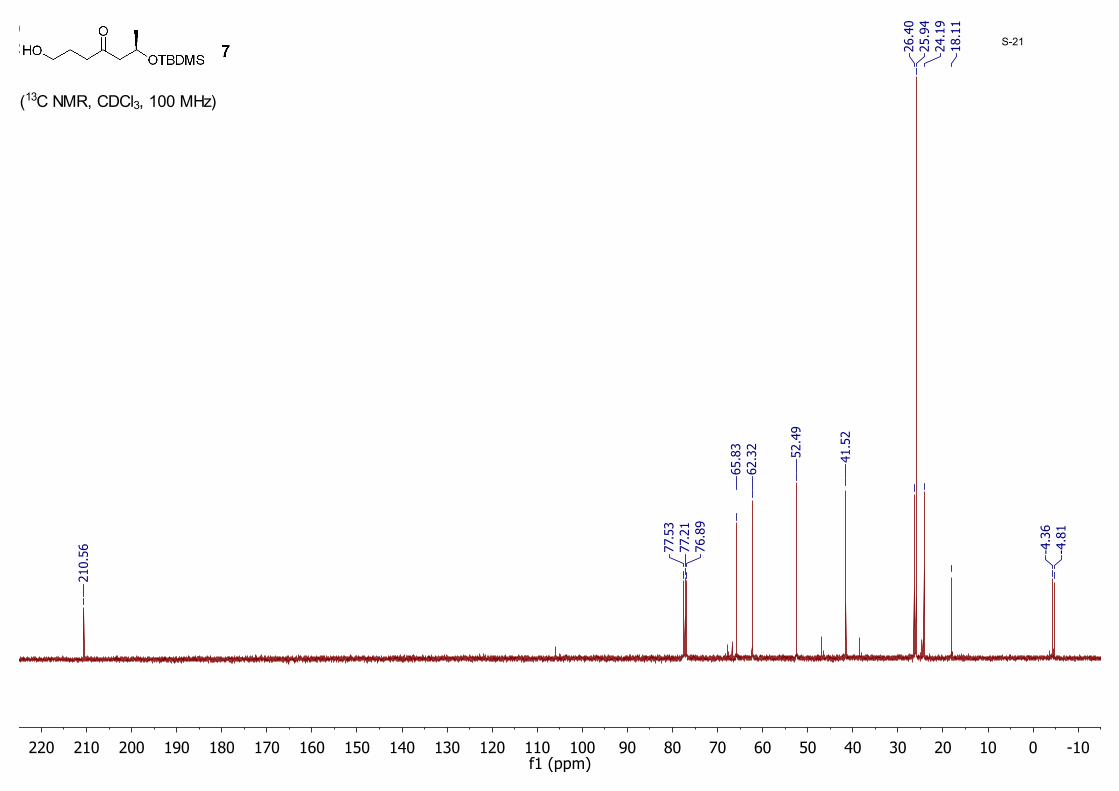
(R)-6-((tert-Butyldimethylsilyl)oxy)-1-hydroxyheptan-4-one (7): MeMgBr (4.90 mL, 14.7 mmol, 3 M in ether) was added drop-wise to a solution of 3-chloropropan-1-ol (1.39 g, 14.7 mmol) in THF (40 mL) cooled to −20 °C. The mixture was warmed to room temperature and then cannulated to another flask containing Mg turnings (0.35 g, 22.0 mmol) followed by addition of catalytic 1, 2-dibromoethane (15.0 μL). The solution was refluxed at 78 °C for 1.5 hours before it was again transferred through cannulation to another flask containing 6 (3.50 g, 13.4 mmol) in THF (60.0 mL) over 20 min at 0 °C. The resulting mixture was stirred for additional 30 min and finally quenched with an aqueous solution of NH4Cl at 0 °C. The organic/water mixture was extracted with EtOAc (40 mL × 3). The organic layer was washed with brine (60 mL), dried with anhydrous MgSO4, and concentrated to dryness. The residue was purified by silica gel column chromatography eluting with hexane/EtOAc (10/1) to yield 7 (2.74 g, 78%) as a colorless oil; 1H NMR (CDCl3, 400 MHz) δ 4.32 – 4.27 (m, 1H), 3.64 (t, J = 6.0 Hz, 2H), 2.65 (dd, J = 14.8, 7.2 Hz, 1H), 2.59 (t, 2H, J = 6.8 Hz), 2.42 (dd, J = 14.9, 5.0 Hz, 1H), 1.82 (quintet, J = 6.0 Hz, 3H), 1.17 (d, J = 6.4 Hz, 3H), 0.86 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13CNMR (CDCl3,10MHz) δ 210.6, 65.8, 62.3, 52.5, 41.5, 26.4, 25.9, 24.2, 18.1, -4.4, -4.8.
(R)-6-((tert-Butyldimethylsilyl)oxy)-4-oxoheptyl methanesulfonate 8: To a solution of 7 (2.74 g, 10.5 mmol) and triethylamine (2.20 mL, 15.8 mmol) in CH2Cl2 (60 mL) was added MeSO2Cl (0.97 mL, 12.6 mmol) at 0 °C. The resulting mixture was stirred for 30 min at 0 °C. The solution was washed sequentially with 1 N HCl, a saturated solution of NaHCO3, and brine.
S-3

The organic layer was dried with anhydrous MgSO4 and concentrated under high vacuum to give crude product 8 (3.50 g) that was directly used for next step.
(R)-1-Azido-6-((tert-butyldimethylsilyl)oxy)heptan-4-one 9:A mixture of 8 (3.50 g, 10.5 mmol) and NaN3 (2.05 g, 31.6 mmol) in dry DMF (60.0 mL) was stirred at 40 °C. When TLC indicated the reaction was complete, water (150 mL) was added and the mixture was extracted with ether (70 mL × 3). The organic layer was dried with anhydrous MgSO4 and then concentrated to dryness. The residue was purified by silica gel column chromatography eluting with hexane/EtOAc (40/1 to 20/1) to give 9 (2.40 g, 81%) as a colorless oil; 1H NMR (CDCl3, 400 MHz) δ 4.32 – 4.27 (m, 1H), 3.31 (t, J = 6.7 Hz, 2H), 2.63 (dd, J = 14.8, 7.5 Hz, 1H), 2.55 (t, J = 7.0 Hz, 2H), 2.40 (dd, J = 14.8, 4.9 Hz, 1H), 1.86 – 1.82 (m, 2H), 1.17 (d, J = 6.1 Hz, 3H), 0.87 (s, 9H), 0.06 (s, 3H), 0.02 (s, 3H); 13CNMR (CDCl3, 100MHz): δ 208.7, 65.8, 52.5, 50.9, 41.3, 25.9, 24.2, 22.8, 18.1, −4.4, −4.8.
(R)-1-Amino-6-((tert-butyldimethylsilyl)oxy)heptan-4-one 10: To a solution of 9 (2.00 g, 7.00 mmol) in CH2Cl2 was added 10% Pd/C powder (0.20 g). The reaction was hydrogenated at atmospheric pressure and vigorously stirred for 12 hours at room temperature. The powder was removed under argon protection by filtration. The liquid filtrate was used directly for next step without concentration or purification.
(R)-3-(2,2,5,5-Tetramethyl-1,3-dioxane-4-carboxamido)propanoic acid 11: To a suspension of D-calcium pantothenate (5.00 g, 21.0 mmol) in DMF (50 mL) was slowly added concentrated H2SO4 (1.11 mL, 21.0 mmol) at room temperature. After stirring for 30 min, 2,2-dimethoxypropane (15.0 mL, 126 mmol) and camphorsulfonic acid (CSA, 0.24 g, 1.10 mmol) was added in one portion. The resulting mixture was stirred for 12 hours at room temperature. DMF was removed under high vacuum and the residue was diluted with water, then extracted with EtOAc. The organic layer was washed with brine and dried with anhydrous MgSO4. The solvent was removed by rotavap and the crude product was recrystallized from hexane/EtOAc (20/1) to give 11 (2.96 g, 55%) as a white powder; 1H NMR (CDCl3, 400 MHz) δ 7.04 (m, 1H), 4.10 (s, 1H), 3.68 (d, J = 11.5 Hz, 1H), 3.64 – 3.56 (m, 1H), 3.53 – 3.46 (m, 1H), 3.28 (d, J = 11.7 Hz, 1H), 2.62 (t, J = 6.2 Hz, 2H), 1.45 (s, 3H), 1.42 (s, 3H), 1.03 (s, 3H), 0.97 (s, 3H).
(R)-N-(3-((R)-6-(tert-butyldimethylsilyloxy)-4-oxoheptylamino)-3-oxopropyl)-2,2,5,5-tetramethyl-1,3-dioxane-4-carboxamide 12:To a solution of amine 10 (1.56 g, 6.00 mmol) in CH2Cl2 (50.0 mL) was added N-ethyl-N isopropylpropan-2-amine (DIPEA, 2.73 mL, 15.8 mmol) and acid 11 (2.36 g, 9.10 mmol) at −20 °C. After stirring for 15 min, (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP, 5.36 g, 10.3 mmol) was added. The resulting mixture was kept stirring until TLC showed the reaction was complete. The mixture was washed with water (100 mL) and the organic layer was dried with anhydrous MgSO4. After removing the solvent by rotavap, the residue was applied to silica gel column eluting with hexane/EtOAc (5/1 to 3/1) to give amide 12 (1.57 g, 52%) as a colorless oil; 1H
S-4

NMR (CDCl3, 400 MHz) δ 7.21 (m, 1H), 4.95 (s, 1H), 4.08 (m, 1H), 4.06 (s, 1H), 3.78 (t, J = 8.7 Hz, 2H), 3.68 (d, J = 14.0 Hz, 1H), 3.57 (m, 2H), 3.27 (d, J = 11.7 Hz, 1H), 2.92 (d, J = 13.5 Hz, 1H), 2.55 – 2.45 (m, 7H), 1.45 (s, 3H), 1.41 (s, 3H), 1.17 (d, J = 6.0Hz, 3H), 1.04 (s, 3H), 0.97 (s, 3H), 0.86 (s, 9H), 0.00 (s, 3H), −0.04 (s, 3H); 13CNMR (CDCl3, 100MHz): δ 170.1, 168.4, 143.2, 112.3, 99.2, 77.7, 71.8, 67.3, 48.5, 40.2, 35.9, 34.4, 33.3, 29.8, 27.7, 26.2, 24.4, 22.5, 19.2, 19.0, 18.4, −4.3, −4.6.
(R)-2,4-dihydroxy-N-(3-((R)-6-hydroxy-4-oxoheptylamino)-3-oxopropyl)-3,3-dimethyl-butanamide 13: A solution of 8% HF in CH3CN (13 mL) was added to amide 12 (0.63 g, 1.30 mmol) dissolved in CH3CN (13.0 mL) at 0 °C. The mixture was stirred for 15 min and then neutralized with basic resin Amberlite IRN-78 until the pH reached 7. The resin was removed by filtration and washed with methanol. After the solvent was removed under vacuum, the crude product was purified by silica gel chromatography eluting with CH2Cl2/MeOH (20/1) to give enzymatic precursor 13 (0.34 g, 79%) as a pale yellow oil; 1H NMR (D2O, 400 MHz) δ 4.28 (m, 1H), 3.99 (s, 1H), 3.58 – 3.46 (m, 3H), 3.40 (d, J = 11.2 Hz, 1H), 3.17 (t, 2H, J = 6.8 Hz), 2.75 – 2.67 (m, 2H), 2.61 (td, J = 7.2, 3.0 Hz, 2H), 2.49 (t, J = 6.5 Hz, 2H), 1.75 (quintet, 2H, J = 7.2Hz), 1.22 (d, J = 8.0 Hz, 3H), 1.20 (s, 3H), 0.93 (s, 3H), 0.89 (s, 3H); 13C NMR (D2O, 100 MHz) δ 214.8, 175.3, 173.96 (s), 75.8, 68.5, 64.2, 51.0, 40.4, 38.8, 35.6, 22.6, 22.2, 20.7, 19.2 (s). MS: calc. for C16H31N2O6
+ [M+H]+: 347.2, found: 347.3.
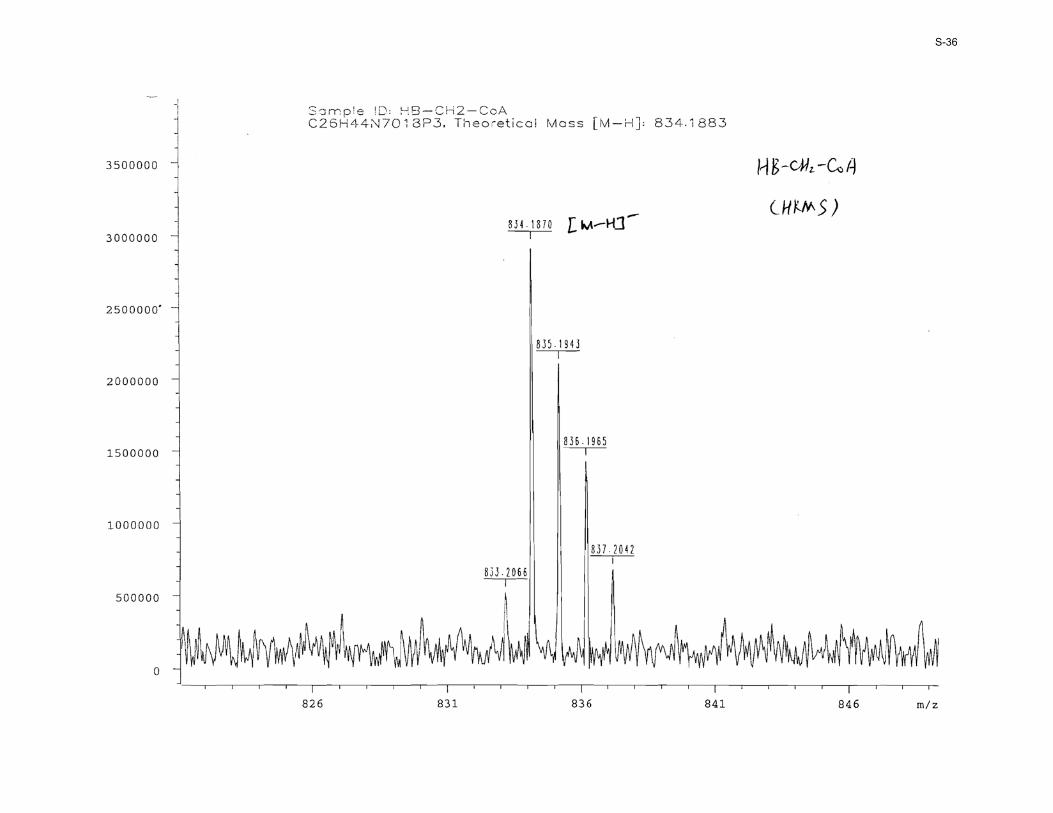
Enzymatic synthesis of HBCH2CoA 1. A 2-mL reaction mixture contained 13 (20.0 mM), ATP (50.0 mM), MgCl2 (10.0 mM), SaCoaA (80.0 µg), EcCoaD (80.0 µg) and EcCoaE (80.0 µg) in 100 mM Tris-HCl (pH 7.60). The reaction was initiated by addition of the enzymes and incubated at 37 °C for 3 h. The reaction was stopped by heating the reaction mixture in a 95 °C water bath for 5 min, and the precipitated protein was removed by centrifugation (14,000 rpm × 5 min). The supernatant was loaded onto a semi-preparative HPLC column (Luna C18-2, 5 µm, 10 mm × 250 mm) that was eluted at 3.00 mL/min using a linear gradient from 0 to 30% methanol in 10.0 mM ammonium acetate (pH 5.00) over 60 min. The fractions containing the product were pooled, concentrated in vacuo, and lyophilized to give a white powder (15.0 mg, 50% yield, HPLC: t = 33 min). The final product was desalted by HPLC using methanol and water as the eluents; 1H NMR (D2O, 400 MHz) δ 8.57 (s, 1H), 8.28 (s, 1H), 6.19 (d, J = 6.0 Hz, 1H), 4.87 (m, 1H), 4.61 (m, 1H), 4.26 (m, 3H), 4.03 (s, 1H), 3.84 (d, J = 6.6 Hz, 1H), 3.58 (d, J = 6.8 Hz, 1H), 3.47 (t, J = 6.3 Hz, 2H), 3.12 (t, J = 6.7 Hz, 2H), 2.66 (m, 2H), 2.56 (t, J = 6.8 Hz, 2H), 2.45 (t, J = 6.2 Hz, 2H), 1.70 (quintet, J = 6.8 Hz, 2H), 1.19 (d, J = 7.2 Hz, 3H), 0.91 (s, 3H), 0.78 (s, 3H); 13C NMR (D2O, 100 MHz) δ 214.7, 174.9, 173.9, 155.4, 152.5, 149.4, 140.2, 86.5, 83.7, 74.3, 73.9, 72.0, 65.4, 64.1, 50.9, 40.3, 38.7, 38.5, 35.6, 22.6, 22.2, 21.0, 18.4; 31P NMR (D2O, 162 MHz) δ 1.16 (s, 1P), −9.77 (d, J = 23.0 Hz, 1P), −10.17 (d, J = 23.0 Hz, 1P); HRMS: calc. for C26H43N7O18P3
− [M−H]−: 834.1883, found: 834.1870.
S-5
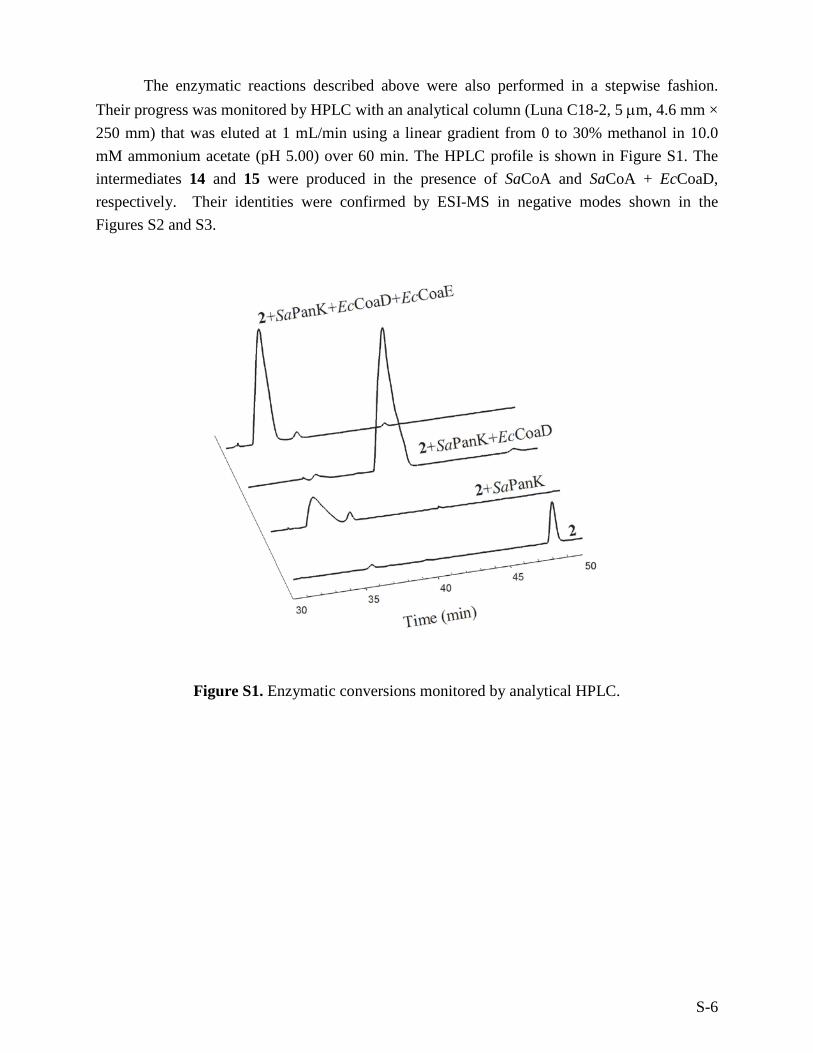
The enzymatic reactions described above were also performed in a stepwise fashion. Their progress was monitored by HPLC with an analytical column (Luna C18-2, 5 µm, 4.6 mm × 250 mm) that was eluted at 1 mL/min using a linear gradient from 0 to 30% methanol in 10.0 mM ammonium acetate (pH 5.00) over 60 min. The HPLC profile is shown in Figure S1. The intermediates 14 and 15 were produced in the presence of SaCoA and SaCoA + EcCoaD, respectively. Their identities were confirmed by ESI-MS in negative modes shown in the Figures S2 and S3.
Figure S1. Enzymatic conversions monitored by analytical HPLC.
S-6
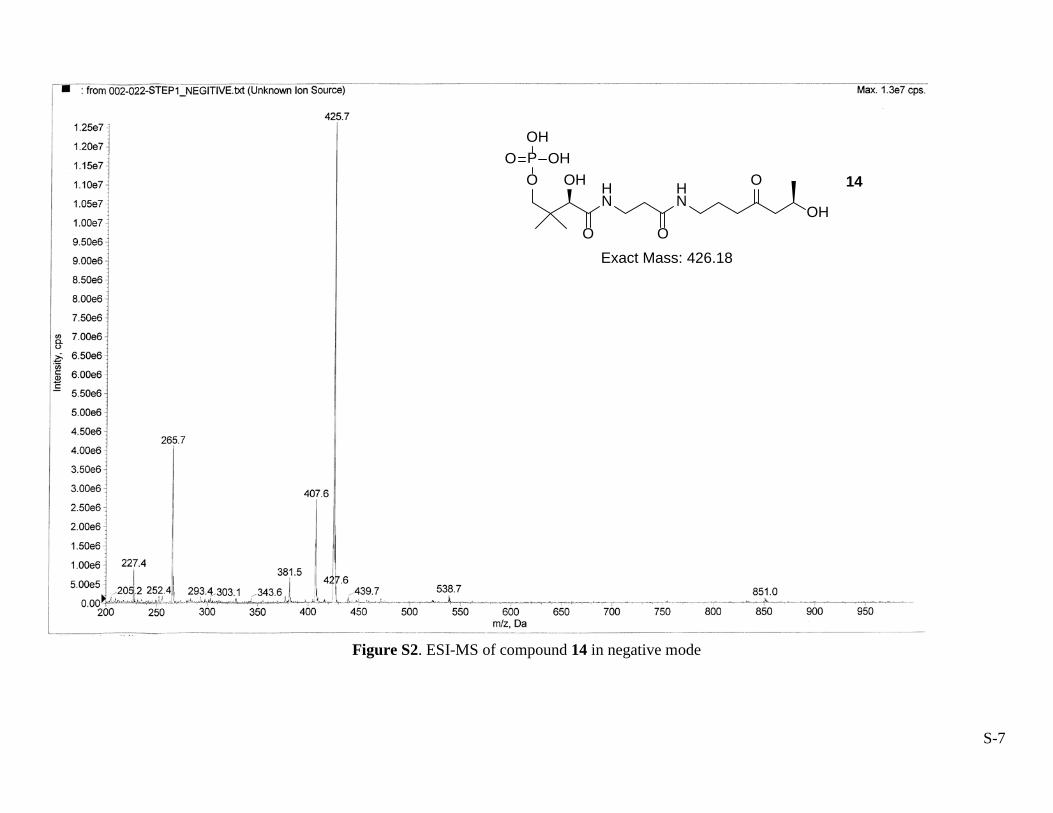
Figure S2. ESI-MS of compound 14 in negative mode
HN
HN
O OH
O O
PO OHOH
OH
O 14
Exact Mass: 426.18
S-7
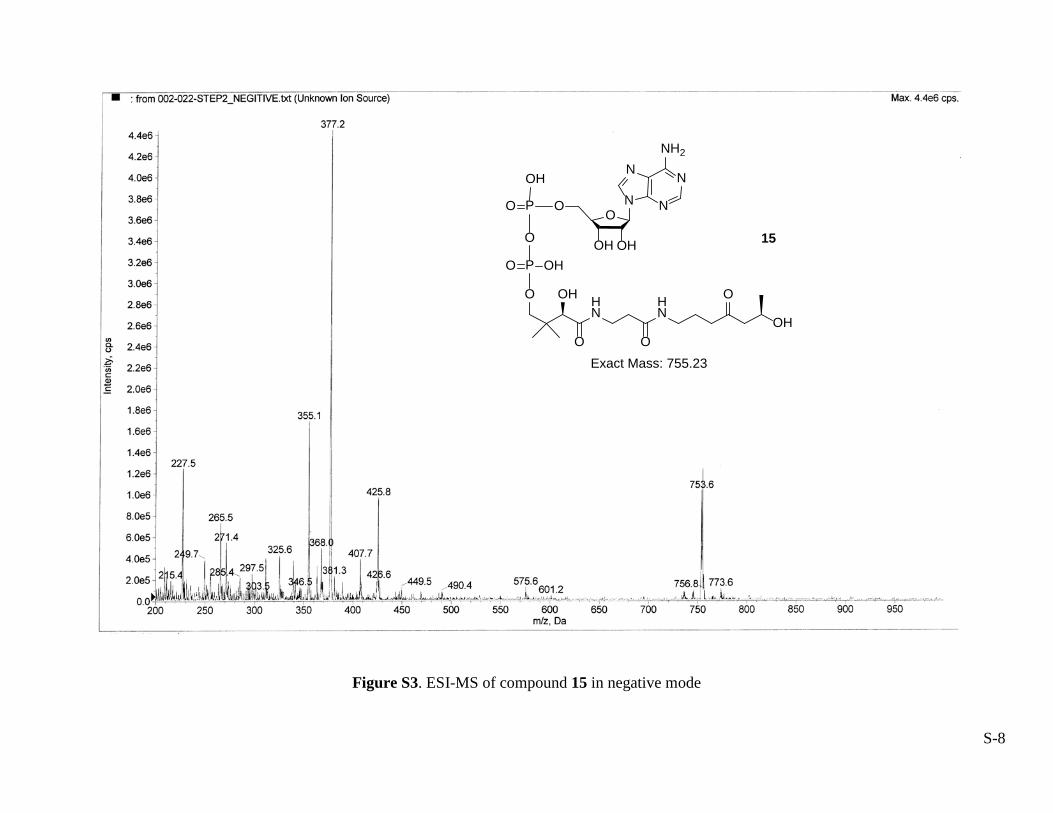
Figure S3. ESI-MS of compound 15 in negative mode
HN
HN
O OH
O O
PO OH
O
P
OH
O OO
N
OH OH
N
N
N
NH2
OH
O
15
Exact Mass: 755.23
S-8
3. Inhibition study
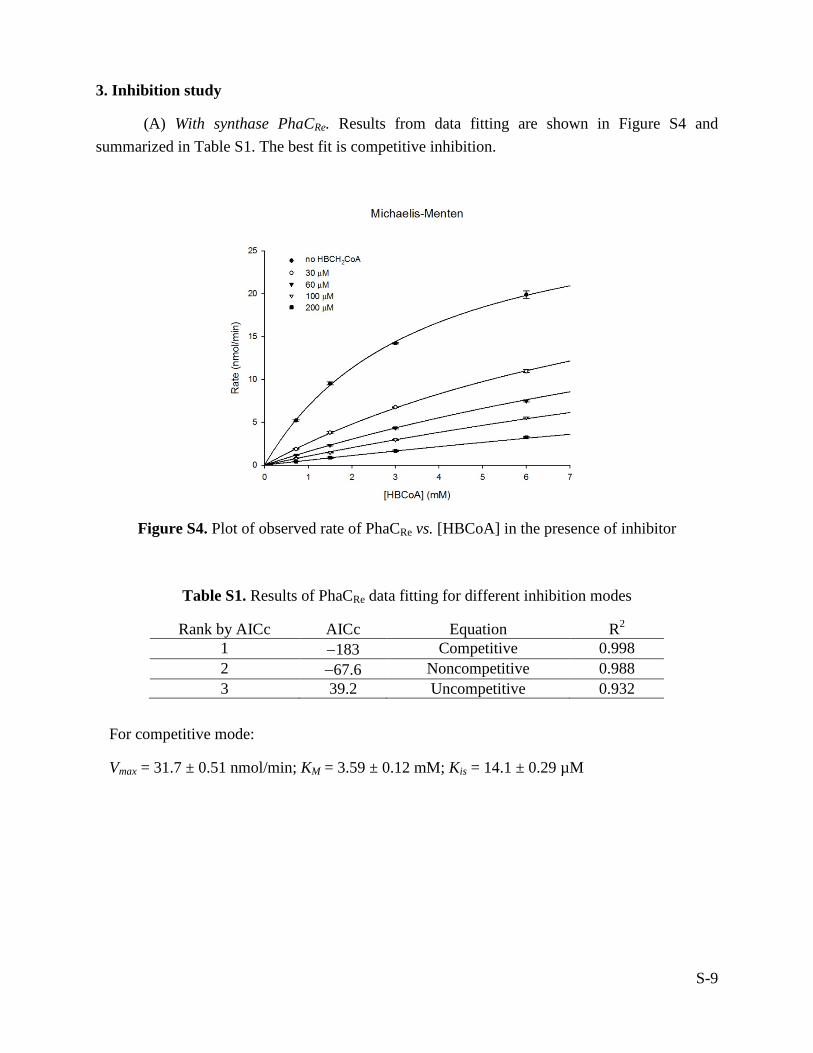
(A) With synthase PhaCRe. Results from data fitting are shown in Figure S4 and summarized in Table S1. The best fit is competitive inhibition.
Figure S4. Plot of observed rate of PhaCRe vs. [HBCoA] in the presence of inhibitor
Table S1. Results of PhaCRe data fitting for different inhibition modes
Rank by AICc AICc Equation R2 1 −183 Competitive 0.998 2 −67.6 Noncompetitive 0.988 3 39.2 Uncompetitive 0.932
For competitive mode:
Vmax = 31.7 ± 0.51 nmol/min; KM = 3.59 ± 0.12 mM; Kis = 14.1 ± 0.29 µM
S-9
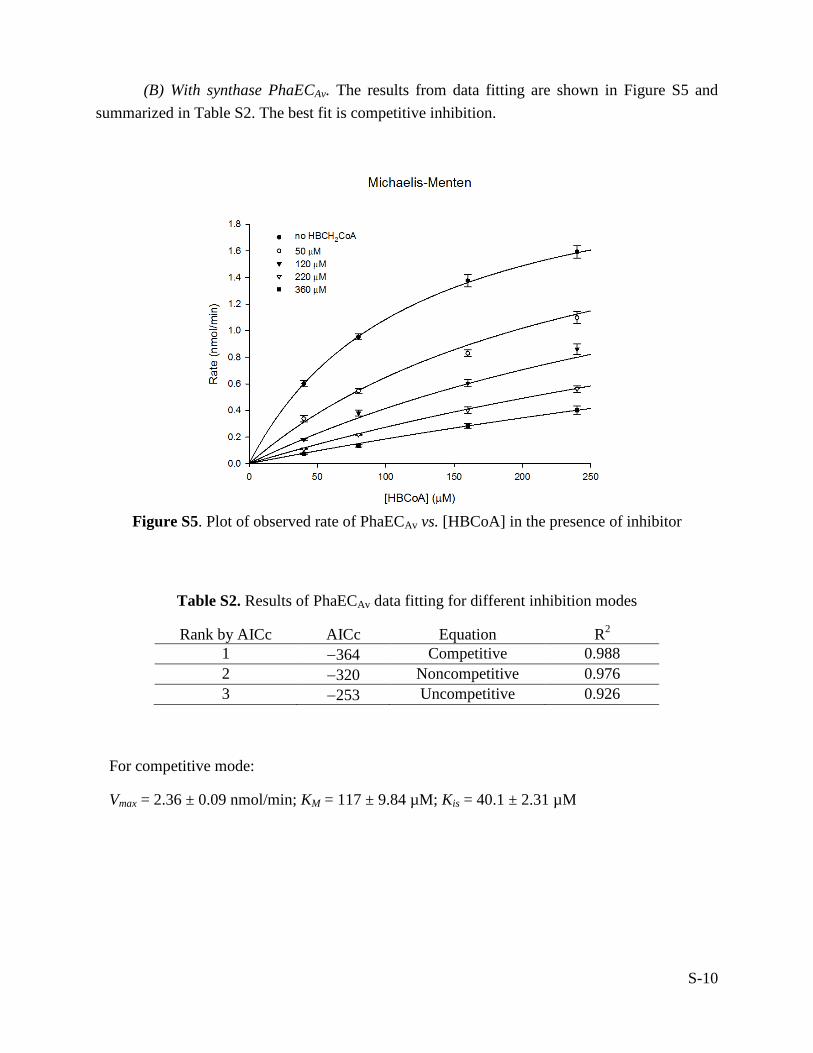
(B) With synthase PhaECAv. The results from data fitting are shown in Figure S5 and summarized in Table S2. The best fit is competitive inhibition.
Figure S5. Plot of observed rate of PhaECAv vs. [HBCoA] in the presence of inhibitor
Table S2. Results of PhaECAv data fitting for different inhibition modes
Rank by AICc AICc Equation R2 1 −364 Competitive 0.988 2 −320 Noncompetitive 0.976 3 −253 Uncompetitive 0.926
For competitive mode:
Vmax = 2.36 ± 0.09 nmol/min; KM = 117 ± 9.84 µM; Kis = 40.1 ± 2.31 µM
S-10
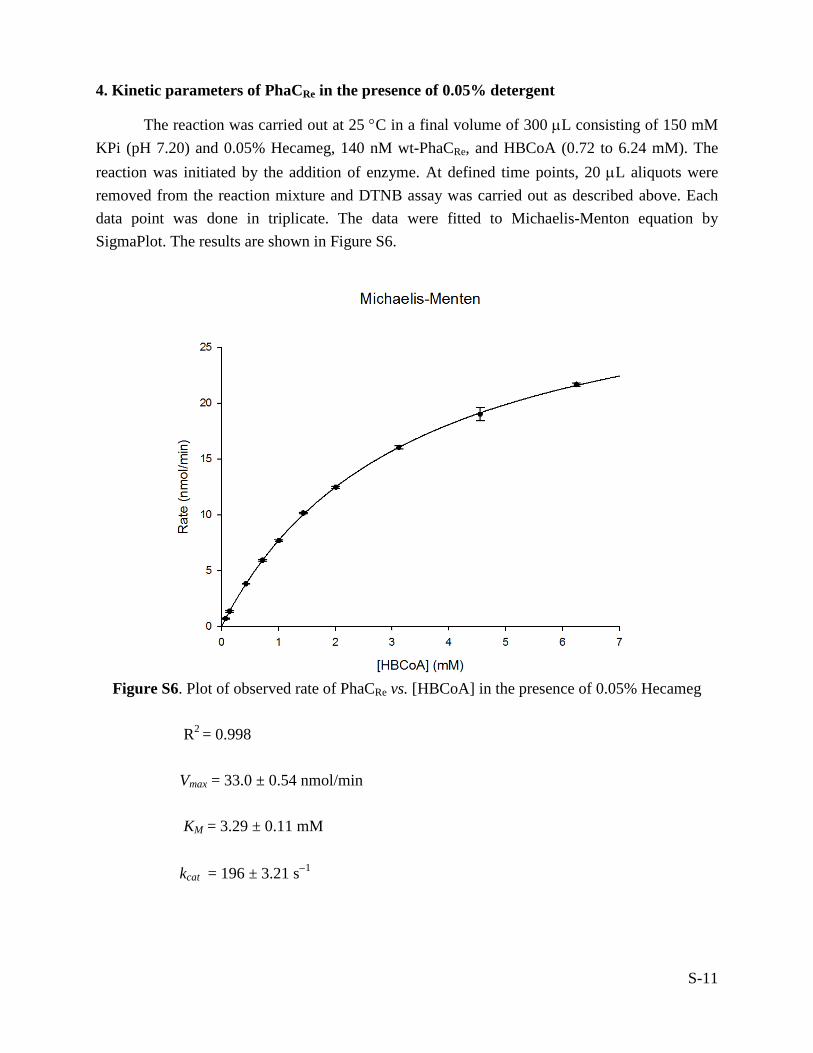
4. Kinetic parameters of PhaCRe in the presence of 0.05% detergent
The reaction was carried out at 25 °C in a final volume of 300 µL consisting of 150 mM KPi (pH 7.20) and 0.05% Hecameg, 140 nM wt-PhaCRe, and HBCoA (0.72 to 6.24 mM). The reaction was initiated by the addition of enzyme. At defined time points, 20 µL aliquots were removed from the reaction mixture and DTNB assay was carried out as described above. Each data point was done in triplicate. The data were fitted to Michaelis-Menton equation by SigmaPlot. The results are shown in Figure S6.
Figure S6. Plot of observed rate of PhaCRe vs. [HBCoA] in the presence of 0.05% Hecameg
R2 = 0.998
Vmax = 33.0 ± 0.54 nmol/min KM = 3.29 ± 0.11 mM kcat = 196 ± 3.21 s−1
S-11
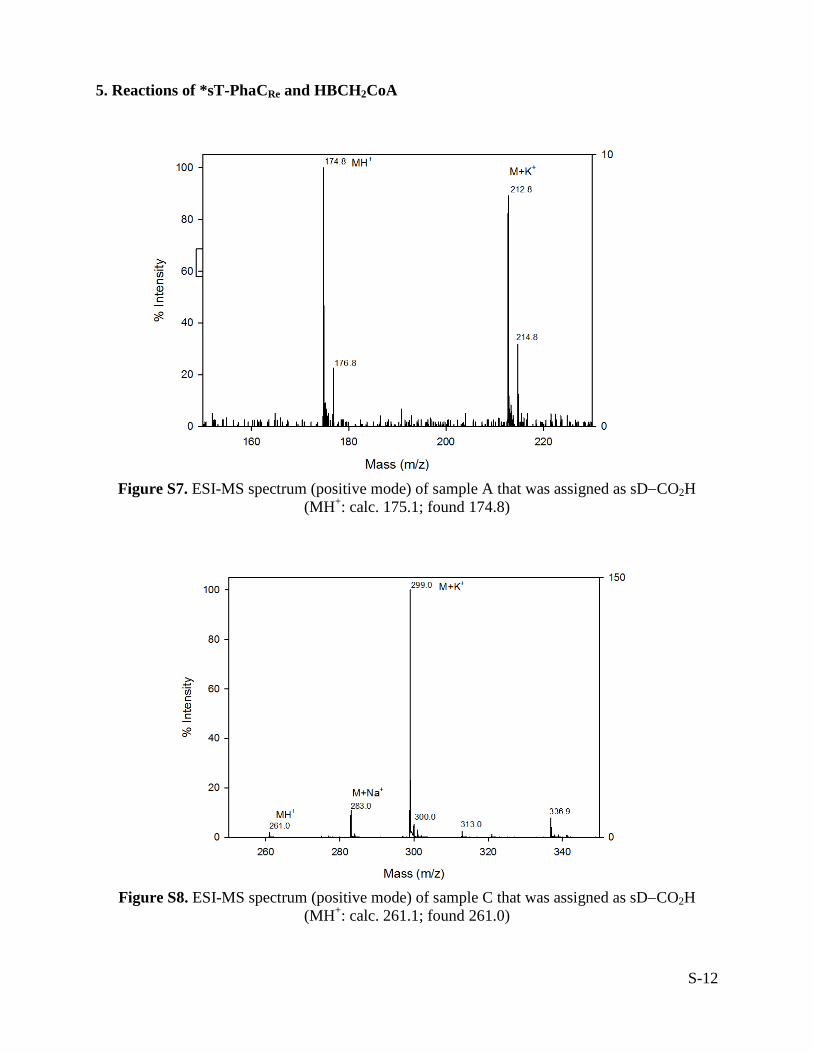
5. Reactions of *sT-PhaCRe and HBCH2CoA
Figure S7. ESI-MS spectrum (positive mode) of sample A that was assigned as sD−CO2H
(MH+: calc. 175.1; found 174.8)
Figure S8. ESI-MS spectrum (positive mode) of sample C that was assigned as sD−CO2H
(MH+: calc. 261.1; found 261.0)
S-12
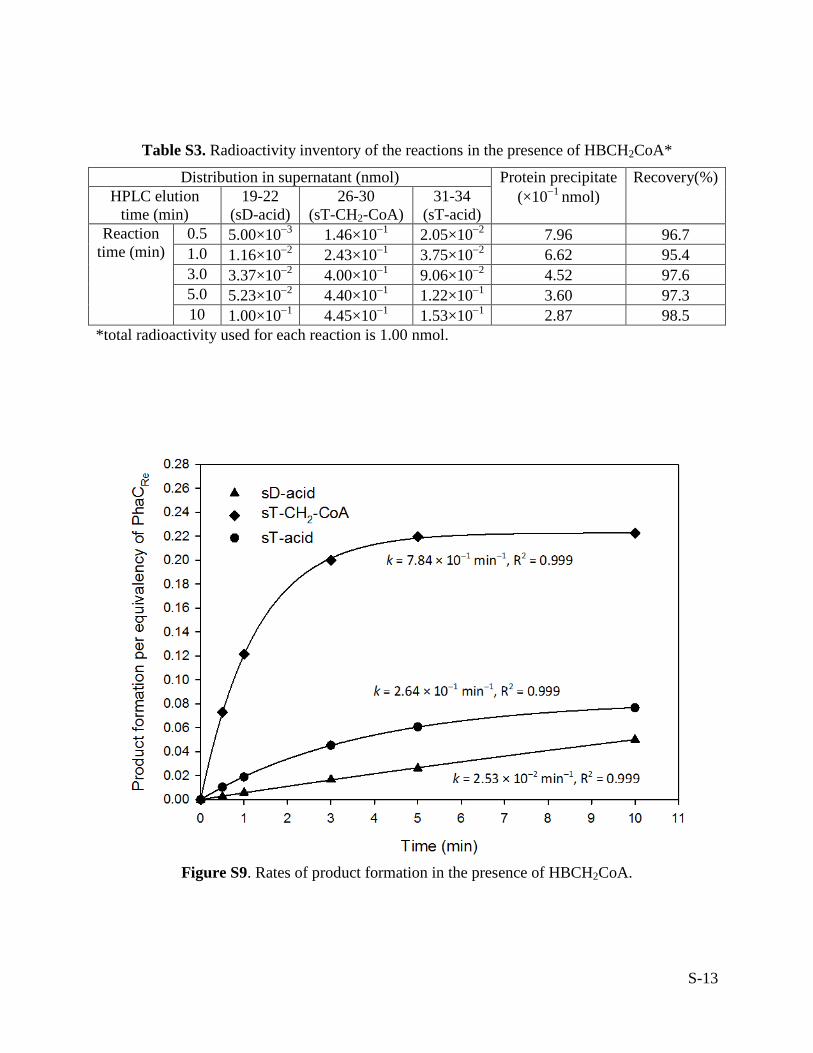
Table S3. Radioactivity inventory of the reactions in the presence of HBCH2CoA*
Distribution in supernatant (nmol) Protein precipitate (×10−1 nmol)
Recovery(%) HPLC elution
time (min) 19-22
(sD-acid) 26-30
(sT-CH2-CoA) 31-34
(sT-acid) Reaction
time (min) 0.5 5.00×10−3 1.46×10−1 2.05×10−2 7.96 96.7 1.0 1.16×10−2 2.43×10−1 3.75×10−2 6.62 95.4 3.0 3.37×10−2 4.00×10−1 9.06×10−2 4.52 97.6 5.0 5.23×10−2 4.40×10−1 1.22×10−1 3.60 97.3 10 1.00×10−1 4.45×10−1 1.53×10−1 2.87 98.5
*total radioactivity used for each reaction is 1.00 nmol.
Figure S9. Rates of product formation in the presence of HBCH2CoA.
S-13
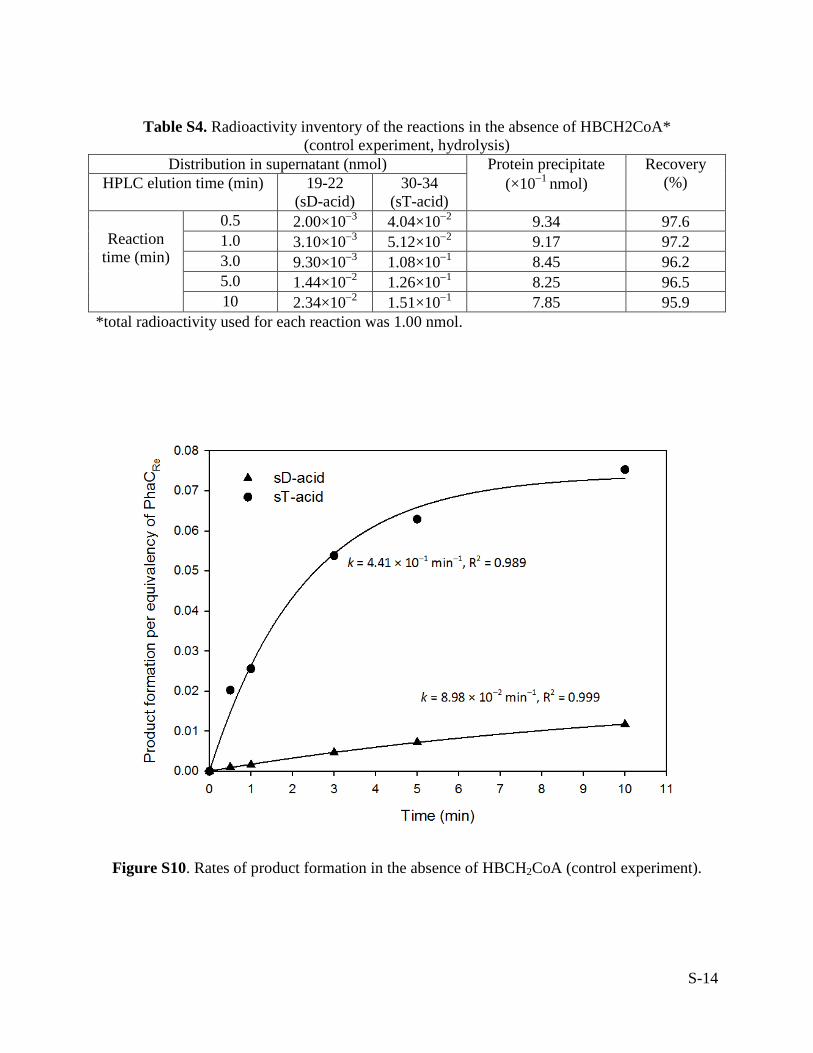
Table S4. Radioactivity inventory of the reactions in the absence of HBCH2CoA* (control experiment, hydrolysis)
Distribution in supernatant (nmol) Protein precipitate (×10−1 nmol)
Recovery (%) HPLC elution time (min) 19-22
(sD-acid) 30-34
(sT-acid)
Reaction time (min)
0.5 2.00×10−3 4.04×10−2 9.34 97.6 1.0 3.10×10−3 5.12×10−2 9.17 97.2 3.0 9.30×10−3 1.08×10−1 8.45 96.2 5.0 1.44×10−2 1.26×10−1 8.25 96.5 10 2.34×10−2 1.51×10−1 7.85 95.9
*total radioactivity used for each reaction was 1.00 nmol.
Figure S10. Rates of product formation in the absence of HBCH2CoA (control experiment).
S-14
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
NMR-compd_3Std Proton parameters
3.28
2.18
0.91
1.00
2.20
5.26
0.01
1.22
1.24
1.26
1.67
2.45
2.47
2.49
2.51
2.53
2.54
2.57
2.58
2.94
2.95
4.19
4.20
4.21
4.21
4.22
4.22
4.23
4.23
4.24
4.24
4.25
4.26
5.13
5.16
5.19
7.26
7.31
7.32
7.33
7.34
7.35
7.35
7.35
7.36
7.37
7.38
7.38
7.40
7.40
(1H NMR, CDCl3, 400 MHz)
S-15
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.0f1 (ppm)
NMR_compd_3Std Proton parameters
5.90
8.94
2.95
1.08
0.93
1.00
1.95
5.32
0.00
0.03
0.06
0.10
0.81
0.85
0.94
1.19
1.20
1.55
2.40
2.41
2.43
2.45
2.51
2.53
2.55
2.56
4.28
4.29
4.31
4.33
5.07
5.10
5.11
5.14
7.26
7.30
7.32
7.33
7.34
7.35
7.36
(1H NMR, CDCl3, 400 MHz)
S-16
0.00.51.01.52.02.53.03.54.04.5f1 (ppm)
compd _5Std Proton parameters
5.73
9.18
3.04
2.00
1.00
-0.0
00.
080.
09
0.86
0.87
0.87
0.87
0.88
0.88
0.88
0.89
0.89
0.90
0.91
1.23
1.25
2.17
2.49
2.50
4.26
4.27
4.29
4.30
(1H NMR, CDCl3, 400 MHz)
S-17
-0.20.20.61.01.41.82.22.63.03.43.84.24.65.05.45.8f1 (ppm)
compd 6Std Proton parameters
5.72
9.84
3.28
1.27
1.16
2.94
3.22
1.00
-0.0
10.
040.
06
0.70
0.76
0.81
0.86
0.91
0.95
0.97
1.01
1.20
1.21
1.25
1.65
2.32
2.33
2.35
2.37
2.73
2.75
2.77
3.17
3.69
4.33
4.34
4.36
4.38
(1H NMR, CDCl3, 400 MHz)
S-18
-0.20.00.20.40.60.81.01.21.41.61.82.02.22.42.62.83.03.23.43.63.84.04.24.44.64.8f1 (ppm)
002-002-11Std Proton parameters
5.72
9.12
3.03
3.04
1.25
3.17
2.19
1.00
-0.0
00.
020.
060.
110.
130.
16
0.70
0.81
0.85
0.86
0.88
0.89
0.89
0.90
1.16
1.17
1.18
1.20
1.631.78
1.80
1.81
1.83
1.842.
392.
412.
432.
442.
572.
592.
602.
622.
642.
662.
68
3.63
3.64
3.66
4.27
4.29
4.29
4.29
4.30
4.30
4.31
4.32
(1H NMR, CDCl3, 400 MHz)
S-19
0.00.40.81.21.62.02.42.83.23.64.04.4f2 (ppm)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
f1 (
ppm
)
{4.29,1.15}
{4.28,2.42}
{4.28,2.64}
{3.61,1.80} {2.56,1.82}
{2.66,2.43}
gCOSY, CDCl3, 400 MHz
S-20
-100102030405060708090100110120130140150160170180190200210220f1 (ppm)
002-007-13CStd Carbon experiment
-4.8
1-4
.36
18.1
124
.19
25.9
426
.40
41.5
2
52.4
9
62.3
265
.83
76.8
977
.21
77.5
3
210.
56(13C NMR, CDCl3, 100 MHz)
S-21
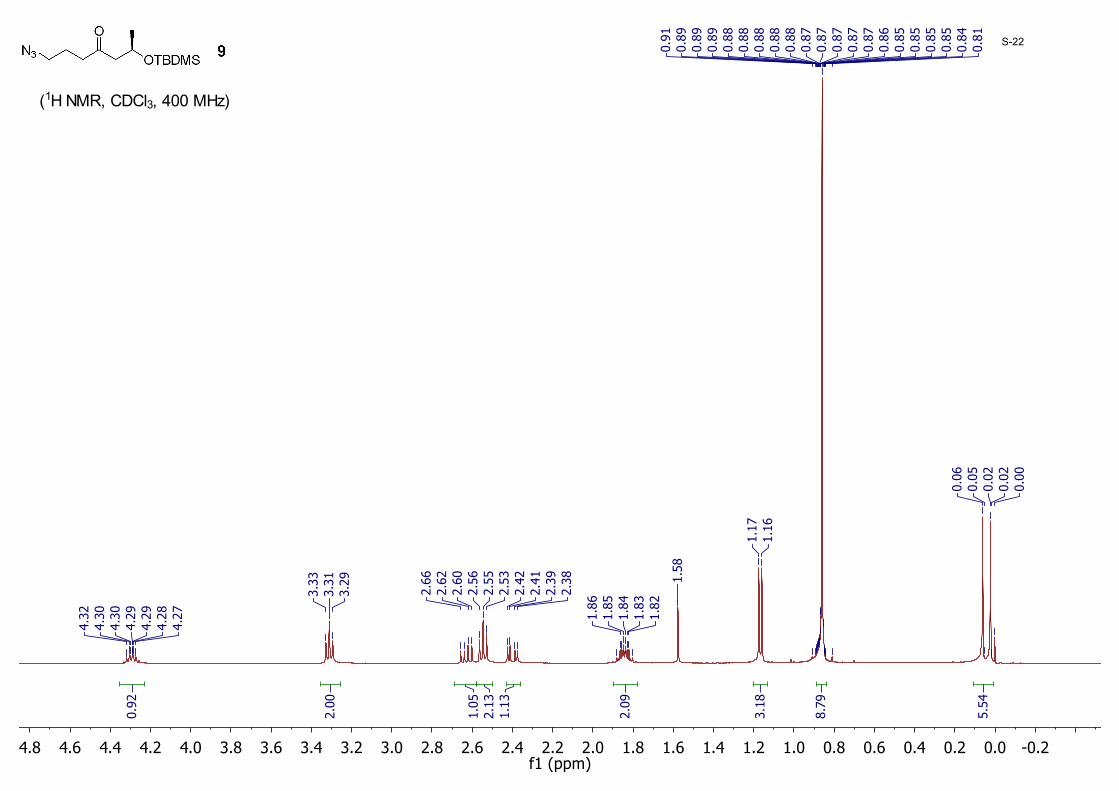
-0.20.00.20.40.60.81.01.21.41.61.82.02.22.42.62.83.03.23.43.63.84.04.24.44.64.8f1 (ppm)
001-163-pureStd Proton parameters
5.54
8.79
3.18
2.09
1.13
2.13
1.05
2.00
0.92
0.00
0.02
0.02
0.05
0.06
0.81
0.84
0.85
0.85
0.85
0.85
0.86
0.87
0.87
0.87
0.87
0.87
0.88
0.88
0.88
0.88
0.88
0.89
0.89
0.89
0.91
1.16
1.17
1.58
1.82
1.83
1.84
1.85
1.86
2.38
2.39
2.41
2.42
2.53
2.55
2.56
2.60
2.62
2.66
3.29
3.31
3.33
4.27
4.28
4.29
4.29
4.30
4.30
4.32
(1H NMR, CDCl3, 400 MHz)
S-22
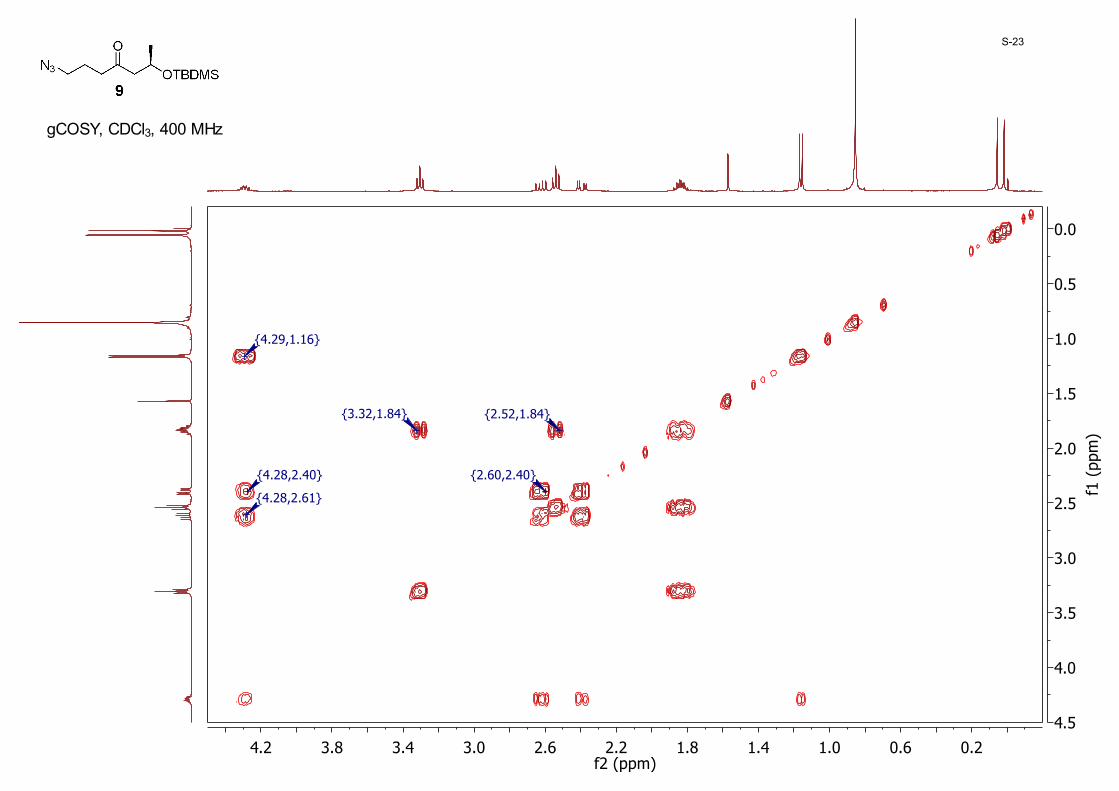
0.20.61.01.41.82.22.63.03.43.84.2f2 (ppm)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
f1 (
ppm
)
{4.28,2.61}
{4.28,2.40}
{4.29,1.16}
{3.32,1.84} {2.52,1.84}
{2.60,2.40}
gCOSY, CDCl3, 400 MHz
S-23
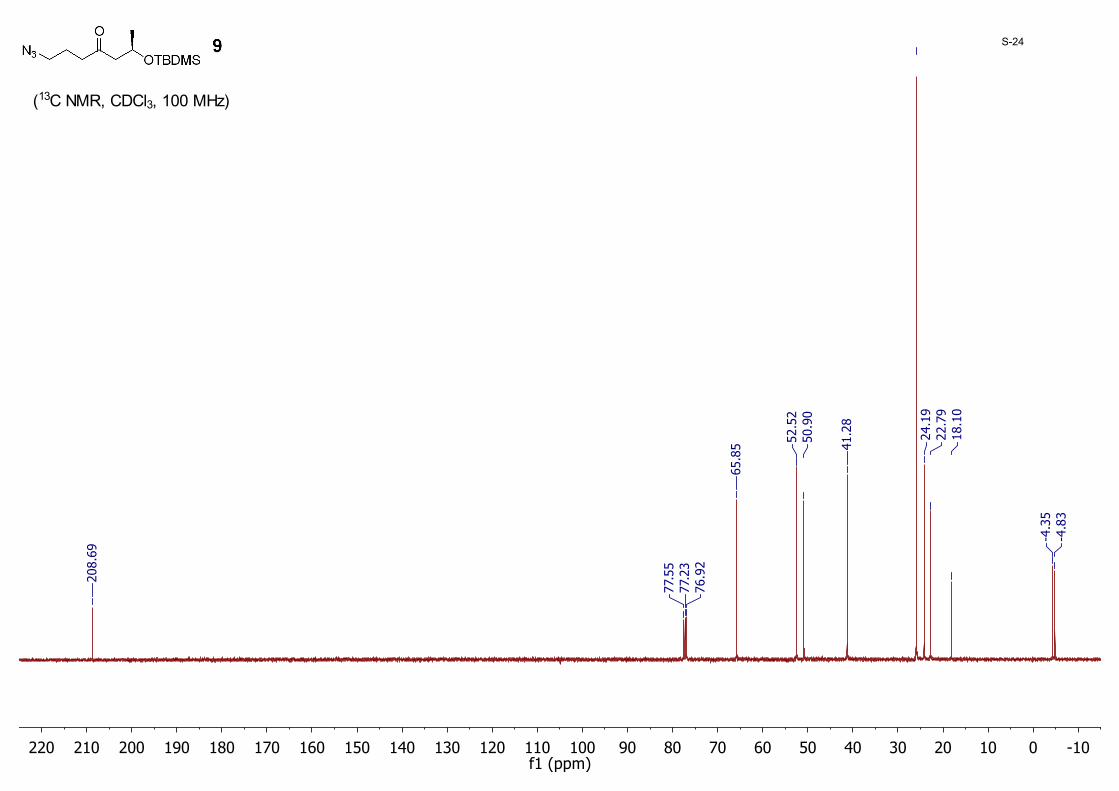
-100102030405060708090100110120130140150160170180190200210220f1 (ppm)
001-163-13CStd Carbon experiment
-4.8
3-4
.35
18.1
022
.79
24.1
9
41.2
8
50.9
052
.52
65.8
5
76.9
277
.23
77.5
5
208.
69(13C NMR, CDCl3, 100 MHz)
S-24
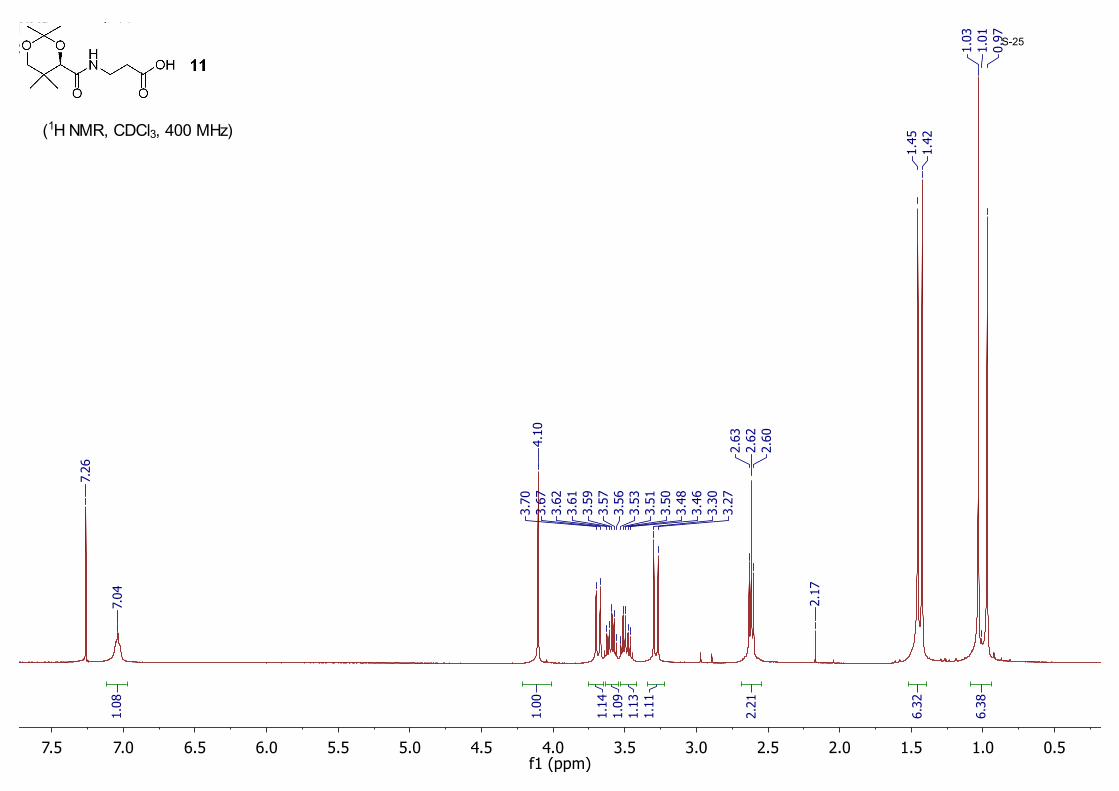
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.5f1 (ppm)
NMR_compd_11Std Proton parameters
6.38
6.32
2.21
1.11
1.13
1.09
1.14
1.00
1.08
0.97
1.01
1.03
1.42
1.45
2.17
2.60
2.62
2.63
3.27
3.30
3.46
3.48
3.50
3.51
3.53
3.56
3.57
3.59
3.61
3.62
3.67
3.70
4.10
7.04
7.26
(1H NMR, CDCl3, 400 MHz)
S-25
0.00.51.01.52.02.53.03.54.04.55.05.56.06.57.0f1 (ppm)
001-171-pureStd Proton parameters
5.87
9.36
2.94
3.12
3.02
2.87
3.20
6.57
1.00
1.08
2.28
1.25
2.15
2.21
1.16
0.73
-0.0
4-0
.00
0.03
0.05
0.06
0.70
0.81
0.86
0.91
0.97
1.01
1.04
1.12
1.16
1.17
1.25
1.36
1.41
1.45
1.62
1.63
2.45
2.47
2.49
2.51
2.53
2.55
2.91
2.94
3.26
3.29
3.66
3.69
3.70
3.78
4.06
4.07
4.09
4.95
7.21
7.26
H2O
S-26
0.00.40.81.21.62.02.42.83.23.64.04.4f2 (ppm)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
f1 (
ppm
)
{4.11,1.17}
{3.69,3.28}
{3.77,2.49} {3.57,2.48} {2.91,2.49}
{2.57,2.46}
{3.66,0.97}
gCOSY, CDCl3, 400 MHz
S-27
-20-100102030405060708090110130150170190210230f1 (ppm)
-4.5
7-4
.28
18.3
819
.03
19.2
122
.50
24.4
326
.15
26.2
027
.66
29.7
833
.29
34.4
435
.91
40.2
148
.53
67.2
671
.82
77.0
577
.37
77.5
077
.69
99.2
4
112.
31
143.
16168.
4117
0.08
(13CNMR, CDCl3, 100 MHz)
S-28
0.81.11.41.72.02.32.62.93.23.53.84.14.4f1 (ppm)
002-015Std Proton parameters
5.99
2.98
2.04
2.08
2.11
1.90
1.96
1.13
3.14
1.00
0.97
0.89
0.93
1.20
1.22
1.71
1.73
1.75
1.77
1.79
2.48
2.49
2.51
2.59
2.60
2.61
2.61
2.62
2.63
2.69
2.70
2.71
2.72
3.14
3.16
3.18
3.20
3.39
3.41
3.46
3.47
3.49
3.51
3.52
3.53
3.53
3.55
3.563.
99
4.26
4.27
4.29
4.31
(1H NMR, D2O, 400 MHz)
S-29
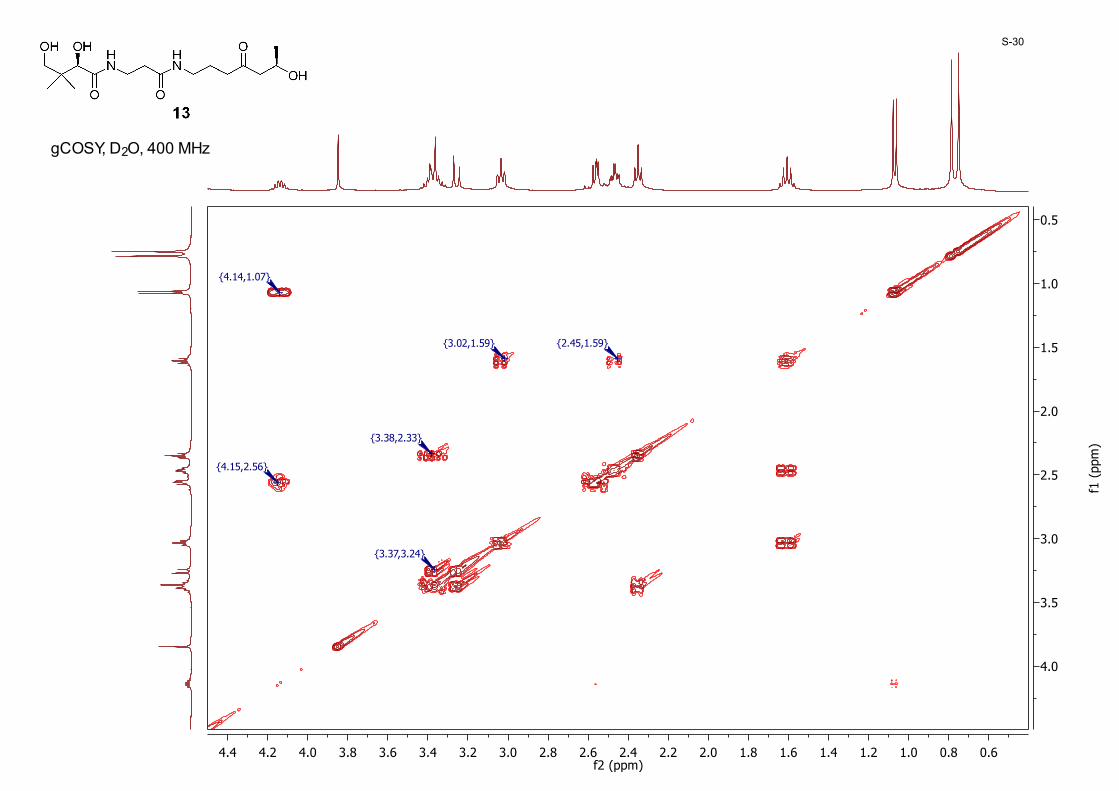
0.60.81.01.21.41.61.82.02.22.42.62.83.03.23.43.63.84.04.24.4f2 (ppm)
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
f1 (
ppm
)
{4.14,1.07}
{4.15,2.56}
{3.02,1.59} {2.45,1.59}
{3.38,2.33}
{3.37,3.24}
gCOSY, D2O, 400 MHz
S-30
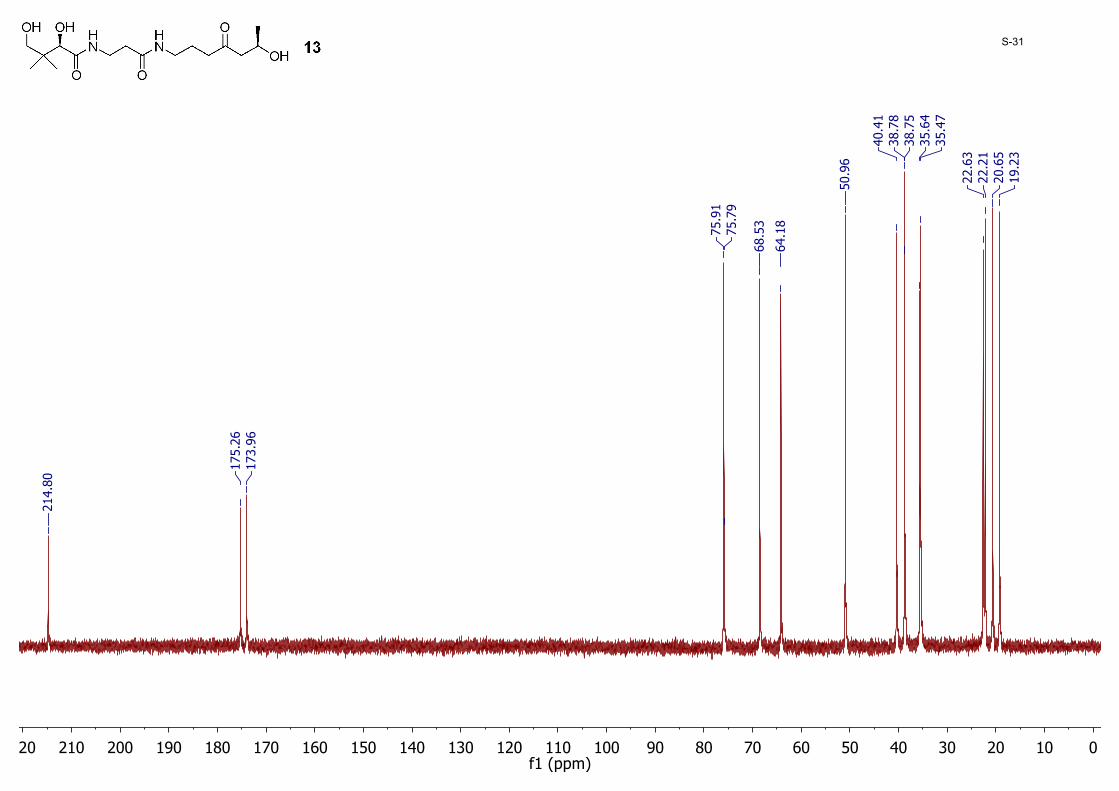
0102030405060708090100110120130140150160170180190200210220f1 (ppm)
002-015-13CStd Carbon experiment
19.2
320
.65
22.2
122
.63
35.4
735
.64
38.7
538
.78
40.4
1
50.9
6
64.1
8
68.5
3
75.7
975
.91
173.
9617
5.26
214.
80S-31
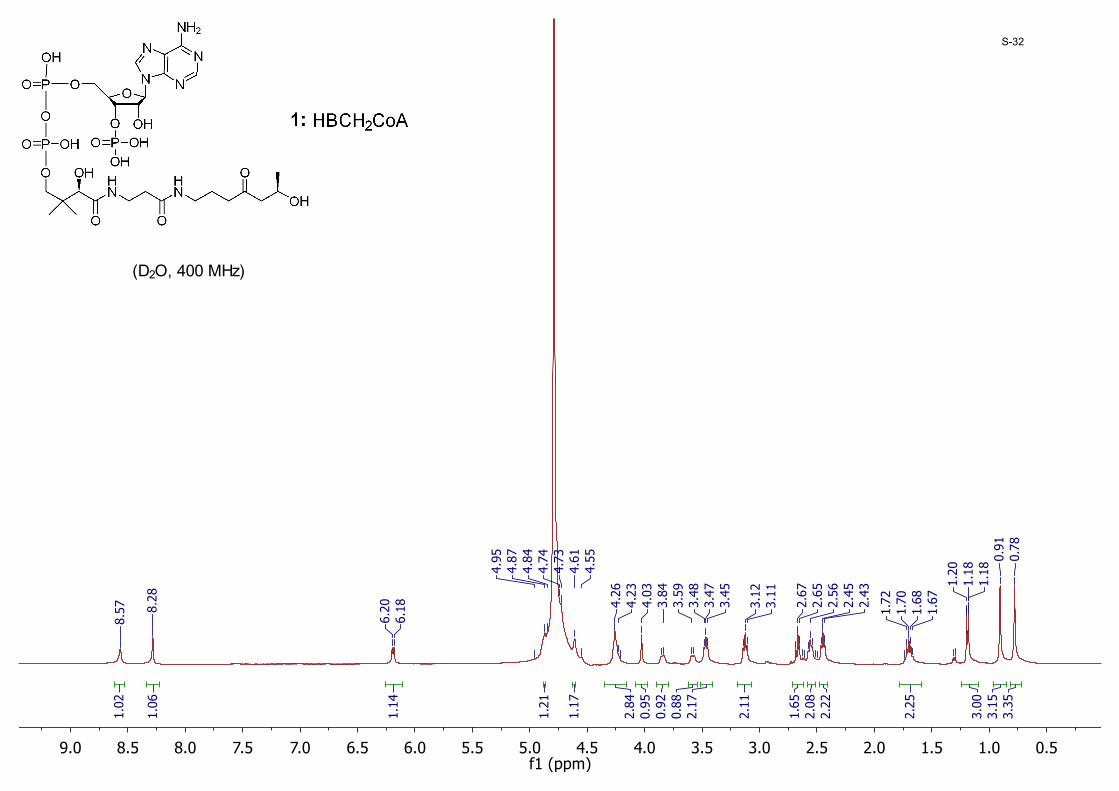
0.51.01.52.02.53.03.54.04.55.05.56.06.57.07.58.08.59.0f1 (ppm)
002-022-1D-D2O
3.35
3.15
3.00
2.25
2.22
2.08
1.65
2.11
2.17
0.88
0.92
0.95
2.84
1.17
1.21
1.14
1.06
1.02
0.78
0.91
1.18
1.18
1.20
1.67
1.68
1.70
1.722.43
2.45
2.56
2.65
2.67
3.11
3.12
3.45
3.47
3.48
3.59
3.84
4.03
4.23
4.26
4.55
4.61
4.73
4.74
4.84
4.87
4.95
6.18
6.208.
28
8.57
(D2O, 400 MHz)
S-32
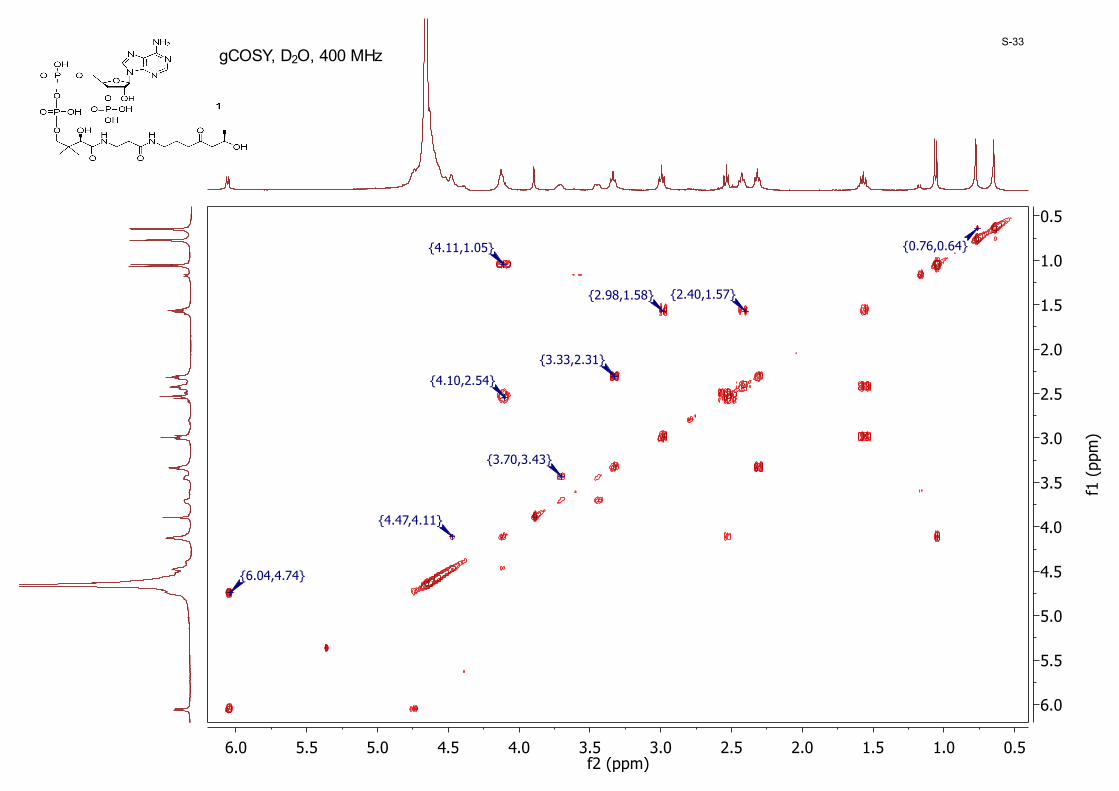
0.51.01.52.02.53.03.54.04.55.05.56.0f2 (ppm)
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
4.5
5.0
5.5
6.0
f1 (
ppm
)
{6.04,4.74}
{4.47,4.11}
{4.10,2.54}
{3.70,3.43}
{4.11,1.05}
{3.33,2.31}
{2.98,1.58} {2.40,1.57}
{0.76,0.64}
gCOSY, D2O, 400 MHzS-33
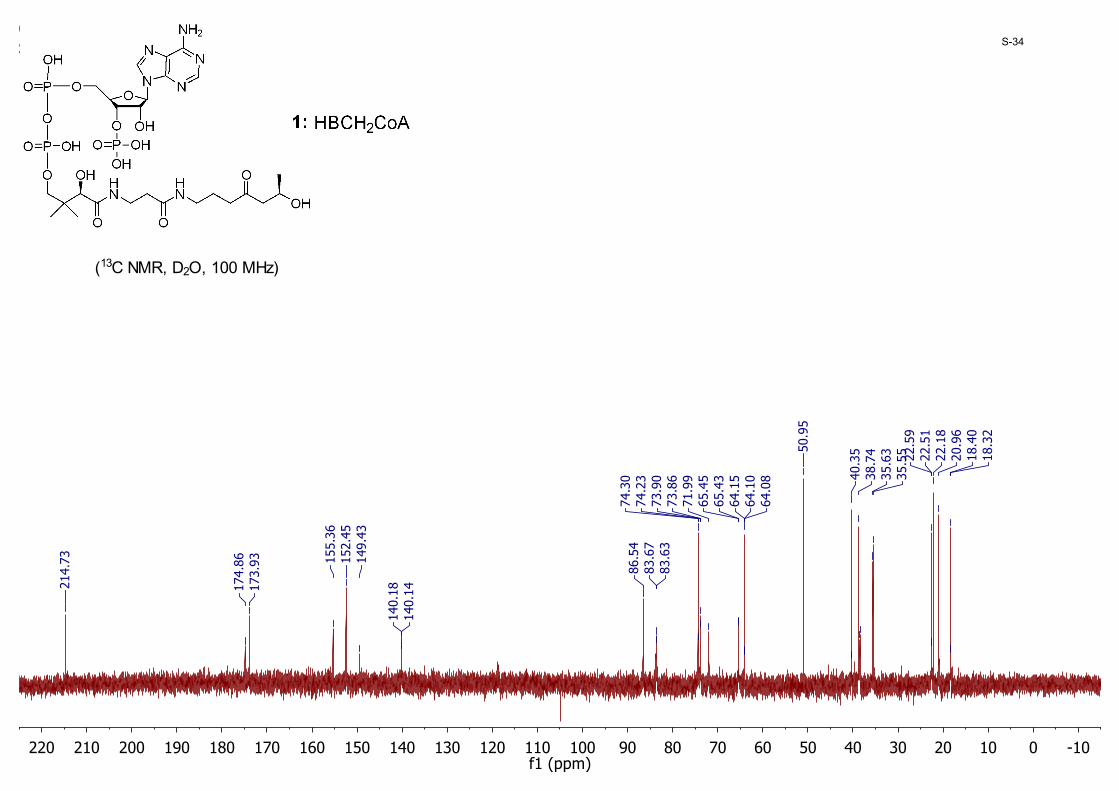
-100102030405060708090100110120130140150160170180190200210220f1 (ppm)
002-022-13CStd Carbon experiment
18.3
218
.40
20.9
622
.18
22.5
122
.59
35.5
535
.63
38.7
440
.3550
.95
64.0
864
.10
64.1
565
.43
65.4
571
.99
73.8
673
.90
74.2
374
.30
83.6
383
.67
86.5
4
140.
1414
0.18
149.
4315
2.45
155.
36
173.
9317
4.86
214.
73
(13C NMR, D2O, 100 MHz)
S-34
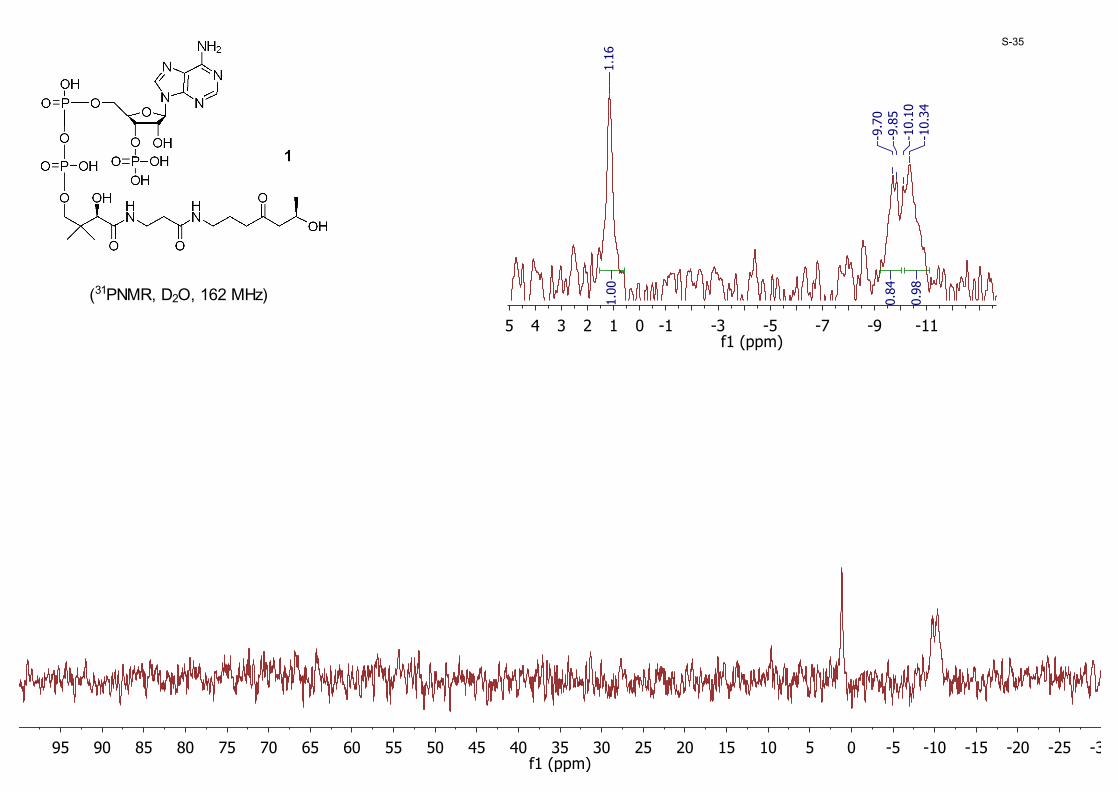
-30-25-20-15-10-505101520253035404550556065707580859095f1 (ppm)
-11-9-7-5-3-1012345f1 (ppm)
0.98
0.84
1.00
-10.
34-1
0.10
-9.8
5-9
.70
1.16
(31PNMR, D2O, 162 MHz)
S-35
S-36