site selective nucleation and size control of gold
TRANSCRIPT

doi.org/10.26434/chemrxiv.7040318.v2
Site Selective Nucleation and Size Control of Gold NanoparticlePhotothermal Antennae on the Pore Structures of a Virus and ConfinedDrug DeliveryCandace Benjamin, Zhuo Chen, Peiyuan Kang, Blake A. Wilson, Na Li, Steven O. Nielsen, Zhenpeng Qin,Jeremiah J. Gassensmith
Submitted date: 04/09/2018 • Posted date: 04/09/2018Licence: CC BY-NC-ND 4.0Citation information: Benjamin, Candace; Chen, Zhuo; Kang, Peiyuan; Wilson, Blake A.; Li, Na; Nielsen,Steven O.; et al. (2018): Site Selective Nucleation and Size Control of Gold Nanoparticle PhotothermalAntennae on the Pore Structures of a Virus and Confined Drug Delivery. ChemRxiv. Preprint.
In this article, we show that the surface of the bacteriophage Qβ is equipped with natural ligands for thesynthesis of small gold nanoparticles. By exploiting disulfides in the protein secondary structure and thegeometry formed from the capsid quaternary structure, we find we can produce regularly arrayed patterns of~6 nm gold nanoparticles across the surface of the virus-like particle. Experimental and computationalanalysis provide insight into the formation and stability of this composite. We further show that the entrappedgenetic material can hold upwards of 500 molecules of the anti-cancer drug Doxorubicin without leaking andwithout interfering with the synthesis of the gold nanoparticles. This direct nucleation of nanoparticles on thecapsid allows for exceptional conduction of photothermal energy upon nanosecond laser irradiation. As aproof of principle, we demonstrate that this energy is capable of rapidly releasing the drug from the capsidwithout heating the bulk solution, allowing for highly targeted cell killing in vitro.
File list (2)
download fileview on ChemRxivSite Selective Nucleation and Size Control of Gold Nano... (693.02 KiB)
download fileview on ChemRxivSite Selective Nucleation and Size Control of Gold Nanopa... (1.70 MiB)

Site Selective Nucleation and Size Control of Gold Nanoparticle Photo-thermal Antennae on the Pore Structures of a Virus and Confined Drug Delivery
Candace E. Benjamin1, Zhuo Chen1, Peiyuan Kang3, Blake A. Wilson1, Na Li1, Steven O. Nielsen1, Zhenpeng Qin,2,3* and Jeremiah J. Gassensmith1,2*
1Department of Chemistry and Biochemistry, 2Department of Biomedical Engineering, 3Department of Mechanical Engi-neering, The University of Texas at Dallas 800 West Campbell Road, Richardson, TX 75080-3021, United States.
KEYWORDS : Virus-Like Particles, Gold Nanoparticles, Photothermal Release, Drug Delivery
ABSTRACT: In this article, we show that the surface of the bacteriophage Qβ is equipped with natural ligands for the synthesis of small gold nanoparticles. By exploiting disulfides in the protein secondary structure and the geometry formed from the capsid quaternary structure, we find we can produce regularly arrayed patterns of ~6 nm gold nanoparticles across the sur-face of the virus-like particle. Experimental and computational analysis provide insight into the formation and stability of this composite. We further show that the entrapped genetic material can hold upwards of 500 molecules of the anti-cancer drug Doxorubicin without leaking and without interfering with the synthesis of the gold nanoparticles. This direct nucleation of nanoparticles on the capsid allows for exceptional conduction of photothermal energy upon nanosecond laser irradiation. As a proof of principle, we demonstrate that this energy is capable of rapidly releasing the drug from the capsid without heating the bulk solution, allowing for highly targeted cell killing in vitro.
INTRODUCTION
The explosive growth in the synthesis of inorganic materi-als, specifically plasmonic gold nanoparticles (AuNPs) with controlled nanostructures, has led to a number of divergent applications in the biomedical and materials fields.1-7 Amongst the possible strategies to create homogenously sized AuNPs, biomolecular templates such as proteins,8 nu-cleic acids,9-10 biological fibers,11 and lipids12-13 have emerged owing to the ordered and well-defined structures these macromolecules form. In addition to these structural advantages, the utilization of bio templates can impart ad-ditional characteristics to these nanomaterials, including solution stability, three-dimensional architectures, molecu-lar and cellular recognition, and tunability for optimized cell uptake.14-17 These emergent characteristics permit ad-vanced applications like concomitant drug delivery and in vivo imaging.
Virus-Like Particles (VLPs),18 the non-infectious19-20 pro-teinaceous nanoparticle analogues of viruses, are proven templates21-23 for the synthesis of metal nanoparticles as they are innately monodisperse in size and can be genet-ically modified24 25 or chemically functionalized26-28 to at-tach stabilizing ligands or residues onto their surface with atomistic precision. While there have been several instruc-tive reports on coating29, absorbing30-39 or growing large AuNPs5, 21-22, 39-43 onto or within viral capsids using surface exposed residues, reports on restricting the diameter of the nanoparticles by constraining their growth to a
homogenous sub-7 nm size have not been forthcoming. This is an important issue because nanoparticle size is critically linked to clearance from blood and tissues.44 While the exact maximum size of a nanoparticle that can be cleared from the body effectively is hard to pin down, it is generally accepted that particles with a diameter greater than 8 nm are too large to pass through the glomerulus of the kidney.45 Conse-quently, with no way to leave the body, these materials be-come trapped in highly vascular tissues and organs of the reticuloendothelial system.46 This is unfortunate, as nano-particles over 20 nm in diameter appear to have the innate capability of localizing in certain tumors47 through the en-hanced permeability and retention (EPR) effect.48 Research-ers have thus found themselves stuck between two irrecon-cilable size constraints—inorganic nanoparticles large enough to exploit the EPR effect are too large to be filtered through the kidney and thus accumulate in the body. To ad-dress this problem, we seek to utilize a multivalent strategy to decorate a proteinaceous VLP that degrades in the liver49 with multiple small plasmonic AuNPs such that the individ-ual nanoparticles are below the kidney’s theoretical cutoff yet, as an ensemble, are large enough to exploit the EPR ef-fect. From here, these small clustered AuNPs could serve as X-ray or PET contrast agents50 as well as photothermal an-tennae for an external stimulus to activate drug release or sensing. Indeed, the growth of AuNPs directly onto the pro-tein surface should increase the efficiency of thermal con-duction following laser induced heating. This improved transfer of thermal energy into the protein by direct tem-plated-growth should cause thermally induced protein

denaturation more efficiently compared to traditional methods to attach gold onto protein surfaces via bioconju-gated spacers or synthetic ligands.51 In principle, this would mean the amount of gold necessary to induce any changes in protein structure could be reduced, decreasing the likeli-hood of detectable bioaccumulation even further. In this pa-per, we report the synthesis and characterization of such a system using VLP Qβ as outlined in Scheme 1. The Qβ capsid itself is an icosahedral virus with 180 identical coat proteins and contains 32 disulfide lined pores.52 We reasoned that these proteins, linked like a daisy chain by either five or six disulfides to form a fixed pore, would act as a natural lig-and53-54 and template for the formation of spherical gold na-noparticles upon in situ reduction of gold salts. We then show that this new composite material works as a proof-of-principle photolytically activated drug delivery vehicle. We can load the capsid with the anticancer drug Doxorubicin (Dox) by non-covalently binding it to the random RNA lo-cated within the capsid, grow gold nanoparticles over the loaded VLP and, finally, initiate release of the RNA-bound Dox from the center of the virus using nanosecond laser ir-radiation to disrupt the proteinaceous shell and release the drug molecules from the interior. Furthermore, we are able to demonstrate highly selective drug release and cell killing in vitro exclusively within the laser path while cells outside the path—even though they are in the same culture—sur-vive.
Scheme 1: Qβ possesses 32 pores, each either 1.5 or 3.0 nanometers in diameter, which are large enough to per-mit A) the diffusion of Doxorubicin into and out of the VLP; after loading, the pores are then B) capped by ~6 nm AuNPs grown in situ.
RESULTS AND DISCUSSION
2.1. Synthesis and Characterization of AuNP@Qβ
The Qβ VLP, structurally depicted in Figure 1, is expressed as a non-infectious nanoparticle that self-assembles around random genomic material within E. coli. Protein crystallo-graphic analysis reveals 20 pores at the three-fold axis and 12 smaller pores at the five-fold axis of symmetry. Key to our strategy is that these pores are lined with solvent ex-posed disulfides and we hoped to benefit from the affinity of disulfides toward gold species. To establish an optimized protocol for site-specific growth of AuNPs, parameters in-cluding the concentrations of Qβ and tetrachloroauric acid, reaction temperature, and incubation time were individu-ally varied until we had minimized the formation of unsta-ble naked gold particles. After significant optimization, the required conditions for AuNP formation turned out to be quite straightforward—specific absorption of gold by disul-fide bonds was achieved by incubating Qβ and tetrachloro-auric acid in water followed by addition of the reducing agent sodium borohydride. Because the water was
unbuffered, the final solution was lowered to a pH of 2 by the gold acid as an alternative pH 7 0.1 M Potassium Phos-phate buffer was used. Once all the reagents were added to the vial, the solution was allowed to sit undisturbed for at least 2 hours, during which time it quickly went from light to dark red (Figure S1). Based upon UV-Vis kinetic analysis following the formation of the surface plasmon, the reaction is 50% complete after 5 min and >99% complete after 20 min (Figure S2). After the reaction, the small amount of pre-cipitated and unbound gold nanoparticles were removed from the crude mixture by passing the reaction solution through a size exclusion column. We noted that the nano-particles in the precipitate increases as we increased the concentration of the acidic gold starting material, which we attribute to a decrease in pH and concomitant protein ag-gregate formation, which can no longer function as a tem-plate.
FIGURE 1: The Qβ VLP is depicted as ribbon model. The VLP has a diameter of 28 nm and contains a total of 32 pores. Depicted are one of the 20 large pores, each of which contains six disul-fide bonds, and one of the 12 smaller pores, each of which con-tains five disulfide bonds.
The selective growth of AuNPs on the pore structures was verified by transmission electron microscopy (TEM) (Fig-ures S3 and S4), which indicated that nearly every VLP con-tained some population of AuNPs, though it appeared that there was a fairly wide distribution of the number of AuNPs per Qβ. The nanoparticle population dispersity prompted an investigation into the average number of nanoparticles per capsid, which was determined to be approximately 6 ± 3 per Qβ (Figure S5) as determined by ICP-MS. DLS found an increase in capsid size from 23 nm to 64.83 ± 0.219 nm, a significant increase in the size of the capsid (Figure S19). Negative stain was used to show the position of Qβ relative to the AuNPs. A crystallographic model of Qβ was used to map the AuNPs over the surface. The micrographs shown in Figure 2 exhibit excellent correlation to the mapped pore patterns on the right. This clear association of the AuNPs to the pore locations strongly suggests that the pores them-selves both selectively nucleate and control the final diam-eter of the AuNPs. The AuNPs appear fairly monodisperse in size by TEM with a diameter of 3.6 ± 1 nm, smaller than the theoretical cutoff limit of the glomerulus of the kidney. Powder X-ray diffraction confirmed the crystallinity of the

as-synthesized AuNPs in the AuNP@Qβ composite and Rietveld refinement of the data was used to confirm that the bulk as-synthesized diameters of the AuNPs is 6.2 ± 0.2 nm. Regardless, both values are within published cutoff limits for glomerular filtration. In addition, X-ray powder diffrac-tion showed the structure type of the AuNP is face centered cubic with the space group Fm3m (Figure S6). The AuNP@Qβ solution is a deep red color and UV-vis spectra shows an absorption centered at 526 nm, which corre-sponds to the surface plasmon (Figure 2B). To elucidate the role of the virus in the nucleation and growth of the AuNPs, we conducted several control experiments. When the same procedure was performed in the absence of any Qβ, a dark precipitate formed in solution. This precipitate was col-lected and was found to be spherical gold nanoparticles as determined by X-Ray diffraction and confirmed by TEM, with a diameter of 48 ± 10 nm as (Figure S6).
FIGURE 2: A) (Left) Illustrated are the relative locations of each nanoparticle on Qβ. Orange dots represent AuNPs on pores fac-ing the reader while grey dots represent AuNPs on pores be-hind the VLP. (Right) TEM micrograph of AuNP@Qβ synthe-sized over the pores of Qβ. B) UV-Vis of Qβ only (black dashed line) and AuNP@Qβ (red line).
To confirm that the disulfides themselves were responsible, we prepared a solution of Qβ and first reduced the disul-fides to the free thiols and then alkylated them with 2-iodo-acetimide. We obtained the same results as when no Qβ was present in the solution—huge particles in a black precipi-tate —confirming the role of the disulfides in the controlled nucleation (Figure S1). In our initial strategy, we assumed that reduction of the disulfides was occurring with sodium borohydride, but we were able to rule this out as we de-tected no such reduction by gel electrophoresis (Figure S8) nor was there any appreciable difference in the Ellman’s as-say before and after addition of borohydride (Figure S10). It is important to point out that, during the synthesis, even spectroscopically pure virus particles are not fully oxi-dized—that is to say, it appears from non-reducing SDS-PAGE that monomeric proteins exist when there should be none55-56 (Figure S9). Curiously, reducing the disulfides to free sulfurs also fails to yield AuNP formation on the surface of the virus. Nevertheless, based on these experiments, it is quite evident that nucleation and size control is occurring as a result of the disulfide and pore geometry as we get un-controlled particle growth in their absence.
AuNP@Qβ are stable at room temperature for more than a month when left undisturbed on the bench. TEM micro-graphs (Figure S11) of a month-old sample that had been left under ambient laboratory conditions show the AuNPs still associated with Qβ, and neither free AuNPs nor aggre-gation was visible. This stability is surprising considering that (1) the nanoparticles are considerably larger than the pores and (2) there are only 10 or 12 sulfurs bound to the nanoparticle. Based on the TEM data, which show varying levels of contrast on the edges of the nanoparticles, we an-ticipated that there were interactions between the virus and the nanoparticles beyond simply Au–S bonding. We thus conducted computational analysis to determine the likely changes in the local environment around the nanoparticle. As seen in Figure 3, we modeled a 6.4 nm nanoparticle bound to the twelve sulfurs of the hexameric pore structure. Molecular Dynamics (MD) simulations provide a reasonable picture of the actual nanoparticle-protein interaction. From these simulations several intriguing results could be in-ferred (See the expanded discussion of the theory in SI and Figures S12-S15). Interestingly, there was no significant change in the size of the hexameric pore.).
However, the pore sulfurs shifted radially inward, which created a deeper cavity for the nanoparticle to sit in and, as shown in Figure 3, allowed more of the loop structures pre-sent near the pore to cover the surface of the nanoparticle. Roughly 23% of the nanoparticle's surface area was pro-tected by surrounding proteins, which does not significantly change the secondary structure of the VLP as shown by cir-cular dichroism (CD) measurements (Figure 4D). The stabil-ity provided by the surrounding proteins makes sense, as 6 nm naked gold nanoparticles are unstable and subject to

rapid Ostwald ripening resulting in precipitation.41, 42 Being embedded in the virus coat protein not only stabilizes but also protects these particles—see supplemental infor-mation for an explanation based on classical nucleation the-ory and an analysis of the smaller five membered pores.
2.2. Laser Activated Drug Delivery
FIGURE 3: MD simulation snapshots of Qβ with the AuNP at the hexameric pore. A) Side-view of the AuNP and surrounding proteins. The protein segments directly attached to the pore sulfurs are shown in a licorice representation, while the rest of the proteins are shown as purple ribbons. B) A snapshot that shows the pore from below. The pore sulfurs are highlighted in yellow. Only the protein segments directly attached to the pore sulfurs are shown.
Gold nanoparticles are well known photothermal agents and are among the best materials for converting incident optical energy into heat.44 This heat can be dissipated dif-fusely into the environment by continuously irradiating the sample, which results in a heating of the bulk solution. Al-ternatively, heat can be generated very locally by pulsed ir-radiation causing the surface of the nanoparticle to heat to several hundred degrees without significantly heating the bulk solution.45 More than a decade ago, this latter form of pulsed irradiation was found to selectively denature pro-teins46 though, recently, the controlled application of photo-thermal irradiation to control protein or nucleotide func-tion without damaging the cell has emerged as a potent method of manipulating specific cells51, 57. Because the growth of the AuNP occurs directly onto the surface of the protein, we reasoned that—even without 100% gold cover-age on each VLP—we would be able to induce a photother-mal response using modest laser power due to close prox-imity of AuNP and protein shell. We thus sought to deter-mine if this mechanism could induce the disruption of the protein shell, triggering release of entrapped small mole-cule drugs. This would enable very localized drug release within a tumor environment without damaging nearby healthy cells.
To do this, we exploited the fact that the self-assembly of recombinant Qβ occurs around random genomic material within E. coli. We hypothesized that this genetic material could be used as a supramolecular host capable of non-co-valently trapping the strong nucleotide intercalator Doxo-rubicin (Dox)—a fluorescent chemotherapeutic used in many different cancer therapies. Binding of Dox to VLP RNA is not without precedent as researchers have shown47 that the RNA inside the red clover necrotic mosaic virus capsid tightly holds upwards of 4300 molecules of Dox. Our tests show that the genetic material inside Qβ does the same—
when Dox is bound inside the VLP the fluorescence in quenched (Figures 4 A-C and S16) and when it is released it fluoresces. The association for Dox to the nucleic acids in-side the Qβ is sufficiently high that after loading, we observe no leakage, even after 24 hours (Figure S17). Thus, we can monitor release by following any fluorescence enhance-ment after laser irradiation.
To create the Dox loaded, gold bespeckled, viral nanoparti-cles, we followed a procedure outlined in Scheme 1. Initially, the Qβ is loaded by incubating the VLP in a 1 mg/mL solu-tion of Dox for 10 min at room temperature. This mixture is then filtered through a GE Sephadex G-25 size exclusion col-umn to remove unbound Dox to create Qβ(Dox). (Figure S18) The filtered solution of Qβ(Dox) was then subjected to the same procedure to grow AuNPs over the surface. This solution was again passed through a GE Sephadex G-25 size exclusion column to remove unattached nanoparticles and excess salts to yield pure AuNP@Qβ(Dox). By TEM, we saw no discernable difference in gold loading over the pores nor any changes in nanoparticle sizes. We were pleased to see that we could load upwards of 500 molecules of Dox—as de-termined by UV-Vis spectroscopy—without interfering with the AuNP formation on the shell.
Figure 4: A) Photographs of Qβ(Dox) prior to irradiation and free
Dox under 365 nm UV-light. The fluorescence is quenched in the
left vial as a result of its interaction with the genetic material en-
trapped in the VLP. B) Photographs of the same vials under white
light. C) AuNP@Qβ(Dox) before (black dashed line) and after (red
solid line) irradiation. The fluorescence intensity increased by
100% following irradiation, indicating the Dox had been released
from the VLP. D) CD Spectra of native Qβ and AuNPs@Qβ before
and after laser radiation showing a change in the protein structure
after laser irradiation. There is no apparent change in the protein
capsid without the presence of AuNPs. The addition of the AuNPs
cause a clear denaturation of the protein structure.
A 70 μL solution at a concentration of 0.04 mg/mL AuNP@Qβ(Dox) was then irradiated with a single six nano-second laser pulse at an energy density of 500 mJ/cm2 and centered at a wavelength of 532 nm. The solution tempera-ture was monitored using a thermocouple and no bulk solu-tion temperature change was observed. Based upon fluores-cence analysis of the irradiated sample, a 100% enhance-ment in fluorescence was seen, indicating that Dox was re-leased from the capsid. Under control conditions, with no laser irradiation, no fluorescence change was observed be-cause the Dox is tightly bound to the RNA (Figure 4 A-C). In a second control, which uses convective heat, we found we could replicate these results by boiling AuNP@Qβ(Dox) in water for five minutes, which completely denatured and de-stroyed the viral capsule. At room temperature, however, the AuNP@Qβ(Dox) was stable for several hours and showed little variation in fluorescence. We conducted CD and DLS spectroscopic studies (Figures 4D and S19) to

determine if the photothermal energy was transferred to the protein. Specifically, we looked at samples of Qβ VLP with and without AuNPs to determine if any obvious change in the spectra of theses samples could be ascertained. When AuNP@Qβ was irradiated, a decrease and slight shift in mo-lar ellipticity could be seen, indicating
FIGURE 5 : A) MTT assay of cells treated with Qβ composites or free Dox and incubated to monitor cell viability after 4 hours. ** and *** denote P values of ≤ 0.01 and 0.001 respectively. Live cell images depicting Dox release after laser irradiation through a pinhole; white line indicates the perimeter of laser irradiation. B) Brightfield. C) Hoechst 34442 D) Dox. E) Merged images acquired with a 10× objective focused near the center of the plate immediately following laser exposure. Cells located close to the aperture experience the release of Dox into the cel-lular space while those further away do not exhibit the same release.
changes in secondary structure and in nanoparticle absorb-ance (Figure 4D) indicating some precipitation of aggre-gates. On the other hand, in a control experiment Qβ VLP, which used laser irradiation at 1064 nm, no change was ev-ident by DLS or agarose band shift (Figures S19 and S21). We thus attribute this heat to localized absorption of the la-ser light to generate thermal energy via localized surface plasmon resonance (LSPR), which in turn denatures the capsid proteins and providing sufficient thermal energy to release the Dox. This localized heat denaturation and drug causes a slight shift in the nanoparticle absorbance (Figure S20), denaturation of the protein capsid and releasing the drug as shown by bandshift assays (Figure S21) in agarose gel electrophoresis. To our knowledge, this is the first ex-perimental proof of using nanoscale-confined protein dena-turation to achieve highly localized and selective drug re-lease.
In order to move toward studies in cellular systems, we first had
to determine the toxicity of our particles via MTT assay using
each of the Qβ constructs without the use of laser activation
(Figure 5A). The assay showed no significant difference be-
tween Qβ(Dox), AuNP@Qβ and AuNP@Qβ(Dox) all of which
have a p value of <0.001 in comparison to nQβ. A key ad-
vantage of this extreme confinement of thermal energy is that it
should enable highly targeted release of therapeutics exclu-
sively within the path of a focused beam of light. In other words,
we should be able to pinpoint the cells we wish to kill in a single
culture without affecting the surrounding cells owing to con-
vective heat loss. To demonstrate the efficacy of our approach
in vitro, cell studies were performed using RAW 264.7 macro-
phages. Three-centimeter glass bottom plates were seeded with
~1 × 106 cells 1-2 days prior to the experiment producing cells
that reached ~80% confluency. The cells were then incubated
with 240 µL of 0.2 mg/mL AuNP@Qβ(Dox) as well as appro-
priate controls for 4 h. The cells were then washed three times
with PBS, stained with 200 nm Hoechst 34442 and washed
again with PBS. The plates were covered with a cardboard mask
pierced with an 18-gauge needle to confine the laser path to a
diameter of 1.27 mm and demonstrate the specificity of release
(Figure 5B-E). The cells were then subject to a single pulse of
500 mJ/cm2 and immediately imaged by live-cell fluorescence
microscopy. Immediately following laser irradiation, the re-
lease of Dox in the targeted “kill-zone” was obvious in the red-
channel of our fluorescence microscope. The morphology of the
cells, as shown in Figure 5E and in higher magnification (inset),
did not immediately change following irradiation, indicating that the initial laser itself did not affect the cells (also shown in Figure S22). Fluorescence imaging, however, revealed ex-tensive Dox release in the kill-zone with the correlation be-tween cells showing release and those within the targeted area being very high.; outside of this area, no red fluores-cence could be discerned. The highly targeted nature of la-ser irradiation makes bulk cell viability assays like MTT in-effective, thus we monitored cell viability via live-cell fluo-rescence imaging for 12 h. Cells outside the kill-zone re-mained intact, adherent, and their morphology was gener-ally unchanged. Within the kill-zone, however, cell mor-phology became clearly different, and the resulting cells largely detached from the plate after 12 h (Figure S23) indi-cating they were killed. When the experiment was repeated

using 1064 nm laser light, which is incapable of exciting the LSPR, the results of live cell imaging show clearly no DOX release and no toxicity over that same time period (Figure S24) demonstrating again, the release is attributed to highly localized optical excitation of the proteinaceous gold com-plex within the living cell.
CONCLUSION
Using the strong affinity between disulfides and gold spe-cies, we have shown that AuNPs can be site selectively syn-thesized in a controlled manner on the pore structures of Qβ through a simple incubation and reducing process. TEM mi-crographs show a well-ordered topology of AuNP@Qβ in ac-cordance with the pattern of pores on Qβ. The growth of the nanoparticles is clearly dependent upon the existence of di-sulfides as reduction to free sulfurs or acylation of these sul-furs fails to yield the composite material. The resulting AuNP@Qβ is stable, when left exposed on the benchtop at RT for more than one month, even though the AuNPs them-selves are prone to self-aggregation. Computational model-ing indicates this stability arises from surface passivation by local protein physisorption. We found that the growth of AuNPs on the VLP is unaffected by loading the interior with the anti-cancer drug Doxorubicin. We successfully demon-strated that this new AuNP@Qβ(Dox) composite can re-lease the Dox upon nanosecond pulsed irradiation without heating the bulk solution and thus offers a pathway to highly localized drug delivery. This proof-of-concept shows great promise in using laser irradiation to trigger the re-lease of materials confined within proteinaceous capsids.
ASSOCIATED CONTENT
Supporting Information. Detailed experimental and synthetic procedures as well as the characterization of the materials pre-sented in this work are available free of charge via the Internet at http://pubs.acs.org.
AUTHOR INFORMATION
Corresponding Author
*Dr. Jeremiah Gassensmith; [email protected] *Dr. Zhengpeng Qin; [email protected] and
Author Contributions
The manuscript was written through contributions of all au-thors. All authors have given approval to the final version of the manuscript.
Funding Sources
C.E.B thanks the National Science Foundation Graduate Re-search Fellows Program (1746053) for their support. J.J.G acknowledges the National Science Foundation (DMR-1654405) and the Cancer Prevention and Research Institute of Texas (CPRIT) (RP170752). Z.Q. acknowledges support from the National Science Foundation (1631910) and CPRIT (RP160770).
ACKNOWLEDGMENT
We thank Professor M.G. Finn and his student Jenny for their helpful advice, guidance, and generous donation of the Qβ plas-mid.
ABBREVIATIONS
VLP, Virus-Like Particle; Dox, Doxorubicin; AuNP, Gold Nano-particle; CD, Circular Dichroism; MD, Molecular Dynamics; TEM, Transmission Electron Microscopy; PXRD, Powder X-Ray Diffraction; EPR, Enhanced Permeability and Retention.
REFERENCES
1. Mao, C.; Solis, D. J.; Reiss, B. D.; Kottmann, S. T.; Sweeney, R. Y.; Hayhurst, A.; Georgiou, G.; Iverson, B.; Belcher, A. M., Virus-Based Toolkit for the Directed Synthesis of Magnetic and Semiconducting Nanowires. Science 2004, 303, 213-217. 2. Bromley, K. M.; Patil, A. J.; Perriman, A. W.; Stubbs, G.; Mann, S., Preparation of high quality nanowires by tobacco mosaic virus templating of gold nanoparticles. J. Am. Chem. Soc. 2008, 18, 4796-4801 3. Fernandes, R.; Li, M.; Dujardin, E.; Mann, S.; Kanaras, A. G., Lig-and-mediated self-assembly of polymer-enveloped gold nanopar-ticle chains and networks. Chem. Comm 2010, 46, 7602-7604. 4. Mann, S., Self-assembly and transformation of hybrid nano-ob-jects and nanostructures under equilibrium and non-equilibrium conditions. Nat. Mater. 2009, 8, 781-792. 5. Zhou, K.; Zhang, J.; Wang, Q., Site-Selective Nucleation and Con-trolled Growth of Gold Nanostructures in Tobacco Mosaic Virus Nanotubulars. Small 2015, 11, 2505-2509. 6. Shenton, W.; Davis, S. A.; Mann, S., Directed Self-Assembly of Na-noparticles into Macroscopic Materials Using Antibody–Antigen Recognition. Adv. Mater. 1999, 11, 449-452. 7. Li, F.; Wang, Q., Fabrication of Nanoarchitectures Templated by Virus-Based Nanoparticles: Strategies and Applications. Small 2014, 10, 230-245. 8. Xie, J.; Zheng, Y.; Ying, J. Y., Protein-Directed Synthesis of Highly Fluorescent Gold Nanoclusters. J. Am. Chem. Soc. 2009, 131, 888-889. 9. Lim, D.-K.; Jeon, K.-S.; Hwang, J.-H.; Kim, H.; Kwon, S.; Suh, Y. D.; Nam, J.-M., Highly uniform and reproducible surface-enhanced Ra-man scattering from DNA-tailorable nanoparticles with 1-nm inte-rior gap. Nat. Nano. 2011, 6, 452-460. 10. Samson, J.; Piscopo, I.; Yampolski, A.; Nahirney, P.; Parpas, A.; Aggarwal, A.; Saleh, R.; Drain, C. M., Fabrication of Size-Tunable Me-tallic Nanoparticles Using Plasmid DNA as a Biomolecular Reactor. Nanomaterials 2011, 1. 11. Scheibel, T.; Parthasarathy, R.; Sawicki, G.; Lin, X.-M.; Jaeger, H.; Lindquist, S. L., Conducting nanowires built by controlled self-as-sembly of amyloid fibers and selective metal deposition. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 4527-4532. 12. Patil, A. J.; Muthusamy, E.; Seddon, A. M.; Mann, S., Higher-Order Synthesis of Organoclay Pipes Using Self-Assembled Lipid Tem-plates. Adv. Mater. 2003, 15, 1816-1819. 13. Burkett, S. L.; Mann, S., Spatial organization and patterning of gold nanoparticles on self-assembled biolipid tubular templates. Chem. Comm. 1996, 321-322. 14. Crookes-Goodson, W. J.; Slocik, J. M.; Naik, R. R., Bio-directed synthesis and assembly of nanomaterials. Chem. Soc. Rev. 2008, 37, 2403-2412. 15. Li, L.; Jianbo, L.; Xiaohai, Y.; Jin, H.; Dinggeng, H.; Xi, G.; Lan, W.; Xiaoxiao, H.; Kemin, W., Biomimetic synthesis of highly biocompat-ible gold nanoparticles with amino acid-dithiocarbamate as a pre-cursor for SERS imaging. Nanotechnology 2016, 27, 105603. 16. Gazit, E., Use of biomolecular templates for the fabrication of metal nanowires. FEBS Journal 2007, 274, 317-322. 17. Huang, J.; Lin, L.; Sun, D.; Chen, H.; Yang, D.; Li, Q., Bio-inspired synthesis of metal nanomaterials and applications. Chem. Soc. Rev.2015, 44, 6330-6374. 18. Chen, Z.; Li, N.; Li, S.; Dharmarwardana, M.; Schlimme, A.; Gas-sensmith, J. J., Viral chemistry: the chemical functionalization of vi-ral architectures to create new technology. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 512-534.

19. Akahata, W.; Yang, Z.-Y.; Andersen, H.; Sun, S.; Holdaway, H. A.; Kong, W.-P.; Lewis, M. G.; Higgs, S.; Rossmann, M. G.; Rao, S.; Nabel, G. J., A virus-like particle vaccine for epidemic Chikungunya virus protects nonhuman primates against infection. Nat. Med. 2010, 16, 334-338. 20. Naskalska, A.; Pyrc, K., Virus like particles as immunogens and universal nanocarriers. Pol. J. Microbiol. 2015, 64, 3-13. 21. Slocik, J. M.; Naik, R. R.; Stone, M. O.; Wright, D. W., Viral tem-plates for gold nanoparticle synthesis. J. Mater. Chem. 2005, 15, 749-753. 22. Zhou, Z.; Bedwell, G. J.; Li, R.; Bao, N.; Prevelige, P. E.; Gupta, A., P22 virus-like particles constructed Au/CdS plasmonic photocata-lytic nanostructures for enhanced photoactivity. Chem. Comm. 2015, 51, 1062-1065. 23. Aljabali, A. A. A.; Barclay, J. E.; Lomonossoff, G. P.; Evans, D. J., Virus templated metallic nanoparticles. Nanoscale 2010, 2, 2596-2600. 24. Fiedler, J. D.; Higginson, C.; Hovlid, M. L.; Kislukhin, A. A.; Cas-tillejos, A.; Manzenrieder, F.; Campbell, M. G.; Voss, N. R.; Potter, C. S.; Carragher, B.; Finn, M. G., Engineered Mutations Change the Structure and Stability of a Virus-Like Particle. Biomacromolecules 2012, 13, 2339-2348. 25. Brown, S. D.; Fiedler, J. D.; Finn, M. G., Assembly of Hybrid Bac-teriophage Qβ Virus-like Particles. Biochemistry 2009, 48, 11155-11157. 26. Yin, Z.; Comellas-Aragones, M.; Chowdhury, S.; Bentley, P.; Ka-czanowska, K.; BenMohamed, L.; Gildersleeve, J. C.; Finn, M. G.; Huang, X., Boosting Immunity to Small Tumor-Associated Carbohy-drates with Bacteriophage Qβ Capsids. ACS Chem. Biol. 2013, 8, 1253-1262. 27. Pokorski, J. K.; Breitenkamp, K.; Liepold, L. O.; Qazi, S.; Finn, M. G., Functional Virus-Based Polymer–Protein Nanoparticles by Atom Transfer Radical Polymerization. J. Am. Chem. Soc. 2011, 133, 9242-9245. 28. Strable, E.; Finn, M. G., Chemical modification of viruses and vi-rus-like particles. Curr. Top. Microbiol. Immunol. 2009, 327, 1-21. 29. Aljabali, A. A. A.; Lomonossoff, G. P.; Evans, D. J., CPMV-Polyelectrolyte-Templated Gold Nanoparticles. Biomacromolecules 2011, 12, 2723-2728. 30. Huang, Y.; Chiang, C.-Y.; Lee, S. K.; Gao, Y.; Hu, E. L.; Yoreo, J. D.; Belcher, A. M., Programmable Assembly of Nanoarchitectures Us-ing Genetically Engineered Viruses. Nano Lett. 2005, 5, 1429-1434. 31. Nagakawa, K.; Niikura, K.; Suzuki, T.; Matsuo, Y.; Igarashi, M.; Sawa, H.; Ijiro, K., Virus Capsid Coating of Gold Nanoparticles via Cysteine-Au Interactions and Their Effective Cellular Uptakes. Chem. Lett. 2012, 41, 113-115. 32. Blaik, R. A.; Lan, E.; Huang, Y.; Dunn, B., Gold-Coated M13 Bac-teriophage as a Template for Glucose Oxidase Biofuel Cells with Di-rect Electron Transfer. ACS Nano 2016, 10, 324-332. 33. Li, F.; Chen, H.; Zhang, Y.; Chen, Z.; Zhang, Z.-P.; Zhang, X.-E.; Wang, Q., Three-Dimensional Gold Nanoparticle Clusters with Tun-able Cores Templated by a Viral Protein Scaffold. Small 2012, 8, 3832-3838. 34. Blum, A. S.; Soto, C. M.; Wilson, C. D.; Cole, J. D.; Kim, M.; Gnade, B.; Chatterji, A.; Ochoa, W. F.; Lin, T.; Johnson, J. E.; Ratna, B. R., Cow-pea Mosaic Virus as a Scaffold for 3-D Patterning of Gold Nanopar-ticles. Nano Lett. 2004, 4, 867-870. 35. Li, F.; Chen, H.; Ma, L.; Zhou, K.; Zhang, Z.-P.; Meng, C.; Zhang, X.-E.; Wang, Q., Insights into Stabilization of a Viral Protein Cage in Templating Complex Nanoarchitectures: Roles of Disulfide Bonds. Small 2014, 10, 536-543. 36. Lee, H.-E.; Lee, H. K.; Chang, H.; Ahn, H.-Y.; Erdene, N.; Lee, H.-Y.; Lee, Y.-S.; Jeong, D. H.; Chung, J.; Nam, K. T., Virus Templated Gold Nanocube Chain for SERS Nanoprobe. Small 2014, 10, 3007-3011. 37. Zahr, O. K.; Blum, A. S., Solution Phase Gold Nanorings on a Viral Protein Template. Nano Lett. 2012, 12, 629-633. 38. Blum, A. S.; Soto, C. M.; Sapsford, K. E.; Wilson, C. D.; Moore, M. H.; Ratna, B. R., Molecular electronics based nanosensors on a viral scaffold. Biosens. Bioelectron. 2011, 26, 2852-2857.
39. Wang, Q.; Lin, T.; Johnson, J. E.; Finn, M. G., Natural Supramolec-ular Building Blocks. Chem. Biol. 2002, 9, 813-819. 40. Vera-Robles, L. I.; González-Gracida, J.; Hernández-Gordillo, A.; Campero, A., Using the M13 Phage as a Biotemplate to Create Mes-oporous Structures Decorated with Gold and Platinum Nanoparti-cles. Langmuir 2015, 31, 9188-9197. 41. Qian, W.; Tianwei, L.; Liang, T.; E., J. J.; G., F. M., Icosahedral Virus Particles as Addressable Nanoscale Building Blocks. Angew. Chem. Int. Ed. 2002, 114, 477-480. 42. Sun, J.; DuFort, C.; Daniel, M.-C.; Murali, A.; Chen, C.; Gopinath, K.; Stein, B.; De, M.; Rotello, V. M.; Holzenburg, A.; Kao, C. C.; Drag-nea, B., Core-controlled polymorphism in virus-like particles. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 1354-1359. 43. Tsvetkova, I. B.; Dragnea, B. G., Encapsulation of Nanoparticles in Virus Protein Shells. In Protein Cages: Methods and Protocols, Or-ner, B. P., Ed. Springer New York: New York, NY, 2015; pp 1-15. 44. De Jong, W. H.; Hagens, W. I.; Krystek, P.; Burger, M. C.; Sips, A. J. A. M.; Geertsma, R. E., Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomater. 2008, 29, 1912-1919. 45. Longmire, M.; Choyke, P. L.; Kobayashi, H., Clearance properties of nano-sized particles and molecules as imaging agents: consider-ations and caveats. Nanomedicine 2008, 3, 703-717. 46. Blanco, E.; Shen, H.; Ferrari, M., Principles of nanoparticle de-sign for overcoming biological barriers to drug delivery. Nat. Bio-tech. 2015, 33, 941-951. 47. Perrault, S. D.; Walkey, C.; Jennings, T.; Fischer, H. C.; Chan, W. C. W., Mediating Tumor Targeting Efficiency of Nanoparticles Through Design. Nano Lett. 2009, 9, 1909-1915. 48. Fang, J.; Nakamura, H.; Maeda, H., The EPR effect: Unique fea-tures of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136-151. 49. Singh, P.; Prasuhn, D.; Yeh, R. M.; Destito, G.; Rae, C. S.; Os-born, K.; Finn, M. G.; Manchester, M., Bio-distribution, toxicity and pathology of cowpea mosaic virus nanoparticles in vivo. J. Control Release. 2007, 120, 41-50. 50. Zhou, C.; Hao, G.; Thomas, P.; Liu, J.; Yu, M.; Sun, S.; Öz, O. K.; Sun, X.; Zheng, J., Near-Infrared Emitting Radioactive Gold Nanoparti-cles with Molecular Pharmacokinetics. Angew. Chem. Int. Ed. 2012, 124, 10265-10269. 51. Kang, P.; Chen, Z.; Nielsen Steven, O.; Hoyt, K.; D'Arcy, S.; Gas-sensmith Jeremiah, J.; Qin, Z., Molecular Hyperthermia: Spatiotem-poral Protein Unfolding and Inactivation by Nanosecond Plas-monic Heating. Small 2017, 13, 1700841. 52. Golmohammadi, R.; Fridborg, K.; Bundule, M.; Valegård, K.; Liljas, L., The crystal structure of bacteriophage Qβ at 3.5 å resolu-tion. Structure 1996, 4, 543-554. 53. Letsinger, R. L.; Elghanian, R.; Viswanadham, G.; Mirkin, C. A., Use of a Steroid Cyclic Disulfide Anchor in Constructing Gold Nanoparticle−Oligonucleotide Conjugates. Bioconjugate Chem. 2000, 11, 289-291. 54. Hakkinen, H., The gold-sulfur interface at the nanoscale. Nat. Chem. 2012, 4, 443-455. 55. Chen, Z.; Boyd, S. D.; Calvo, J. S.; Murray, K. W.; Mejia, G. L.; Ben-jamin, C. E.; Welch, R. P.; Winkler, D. D.; Meloni, G.; D’Arcy, S.; Gas-sensmith, J. J., Fluorescent Functionalization across Quaternary Structure in a Virus-like Particle. Bioconjugate Chem. 2017, 28, 2277-2283. 56. Chen, Z.; Li, N.; Chen, L.; Lee, J.; Gassensmith Jeremiah, J., Dual Functionalized Bacteriophage Qβ as a Photocaged Drug Carrier. Small 2016, 12, 4563-4571. 57. Li, X.; Kang, P.; Chen, Z.; Lal, S.; Zhang, L.; Gassensmith, J. J.; Qin, Z., Rock the nucleus: significantly enhanced nuclear membrane permeability and gene transfection by plasmonic nanobubble in-duced nanomechanical transduction. Chem. Comm. 2018, 54, 2479-2482.

SYNOPSIS TOC (Word Style “SN_Synopsis_TOC”). If you are submitting your paper to a journal that requires a synopsis graphic and/or synopsis paragraph, see the Instructions for Authors on the journal’s homepage for a description of what needs to be provided and for the size requirements of the artwork.
Insert Table of Contents artwork here

download fileview on ChemRxivSite Selective Nucleation and Size Control of Gold Nano... (693.02 KiB)

1
Site Selective Nucleation and Size Control of Gold Nanoparticle
Photothermal Antennae on the Pore Structures of a Virus and
Confined Drug Delivery
Candace E. Benjamin1, Zhuo Chen1, Peiyuan Kang3, Blake A. Wilson1, Na Li1,
Steven O. Nielsen1, Zhenpeng Qin,2,3* and Jeremiah J. Gassensmith1,2*
1Department of Chemistry and Biochemistry, 2Department of Biomedical
Engineering, 3Department of Mechanical Engineering, The University of Texas at
Dallas 800 West Campbell Road, Richardson, TX 75080-3021;
[email protected] and [email protected]
SUPPORTING INFORMATION
Table of Contents
Experimental section: .................................................................................................. 2
Materials ............................................................................................................................... 2
Instruments: ......................................................................................................................... 2
Synthesis of Gold Nanoparticles on Qβ ............................................................................. 2
Alkylation of disulfides of Qβ ............................................................................................. 2
Synthesis of Qβ(Dox)-Au NPs:............................................................................................ 3
Laser irradiation: ................................................................................................................ 3
ICP-MS and Quantitation: ................................................................................................. 3
Ellman’s Assay: .................................................................................................................... 3
Dox Release in Agarose Gel ................................................................................................ 4
Cell Experiments: ................................................................................................................ 4
Ancillary Figures: ........................................................................................................ 5
Molecular Dynamics Simulations ............................................................................. 18
Modeling the Gold Nanoparticle ...................................................................................... 19
Simulating the cg-AuNP at the pore ................................................................................. 20
Measuring the pore diameter ........................................................................................... 21
Measuring the Percent protected Surface Area of the cg-AuNP ................................... 22
cg-AuNP at the pentameric pore ...................................................................................... 23

2
Effect on the critical size of partially protecting AuNPs ................................................ 24
Experimental section:
Materials. Tetrachloroauric (III) trihydrate (HAuCl4·3H2O: 99.9%) and sodium
borohydride (NaBH4: 99%) were purchased from Sigma-Aldrich, Doxorubicin
hydrochloride (C27H29NO11 • HCl) was purchased from Cayman Chemical Company.
All reagents were used without further purification. All aqueous solutions were
prepared using water from Millipore Milli-Q purification system (18.2 MΩ/cm). Qβ
was prepared according to a previously reported method.1 Sephadex G-25 column was
purchased from GE Healthcare.
Instruments: All the TEM (transmission electron microscopy) imaging was performed
on a Tecnai™ G2 Spirit optical system. TEM samples were prepared by evaporating a
drop of the sample solution on a carbon coated grid. Uranyl acetate solution (1%) was
used as a negative stain. UV-vis spectra were recorded on a Shimadzu UV-1601PC UV-
visible spectrophotometer. DLS data was obtained using a Malvern Zetasizer.
Fluorescence spectra were recorded on LS50B Luminescence Spectrometer
(PerkinElmer, Inc.). XRD data were collected using a Bruker D8 Advance powder X-
ray diffractometer operating at 40 kV and 30 mA with CuKa (1.54060 Å) radiation and
a LYNXEYE XE detector. Laser experiments were performed using a Nd:YAG laser
system (Quantel Q-smart 450) with a thermocouple manufactured by HH506A, Omega,
USA. ICP-MS measurements we performed using an Agilent 7800 ICP triple
Quadrupole MS. Fluorescence images were acquired using either an FV3000RS
Confocal Microscope or a Life Technologies EVOS FL Auto Epifluorescence
Microscope with 40 or 60 × objective. Gels were imaged using a Bio-Rad ChemiDoc
MP Imaging System. Circular Dichroism data collected on an AVIV Circular Dichroism
Spectrometer.
Synthesis of Gold Nanoparticles on Qβ. In a solution of Qβ (0.2 mg/mL, 1 mL), 20
μL of HAuCl4 solution (50 mM) was added and mixed well. The mixture was incubated
for 3 h at room temperature. Then 160 μL of NaBH4 (10 mM, freshly prepared and ice
cold) solution was added to the Qβ and HAuCl4 solution with vigorous stirring for 5
min. The solution turned red in color immediately and was incubated without
disturbance for 12 h at RT. The small amount of precipitated and free gold nanoparticles
were removed by running the crude solution through a Sephadex G-25 column.
Alkylation of disulfides of Qβ. A mixture containing 400 μL Qβ solution (1 mg/mL)
and 100 μL of DTT solution (1 M) was incubated at 37 ℃ for 1 h. Next, 1 mL of 100

3
mg/mL iodoacetamide was added and the solution was again incubated at 37 ℃ for 1 h.
The final reaction solution was desalted using a Sephadex G-25 column.
Synthesis of Qβ(Dox)-Au NPs: 300 μL of a Qβ solution (3.3 mg/mL) was mixed with
a 200 μL Dox solution (1 mg/mL) and this mixture was allowed to sit at RT for 10 min.
Free Dox was removed from the system by a Sephadex G-25 column. The collected
Qβ(Dox) protein concentration was confirmed by Bradford assay. This solution of
Qβ(Dox) at a concentration of 1 mg/mL was used in the synthesis of AuNPs on Qβ
using the aforementioned method.
Laser irradiation: The AuNP@Qβ(Dox) solution made above was diluted five-fold
and 70 μL of this solution was used for each experiment. A sample was irradiated with
a Nd:YAG laser system (Quantel Q-smart 450) operated at 532 nm and pulse duration
of six nanoseconds (full width at half maximum, or FWHM). The sample was irradiated
by single laser pulse at 500 mJ/cm2. This was repeated on five samples and the reported
fluorescence and UV-vis spectra are reported as the average solution spectra from the
experiment after five-fold dilution.
ICP-MS and Quantitation: The AuNPs were diluted to a concentration of 0.0002
mg/ml in a 3% nitric and 2% hydrochloric acid aqueous solution. Each sample was
analyzed for Au using both no Gas and Helium mode and compared against prepared
gold standards (Inorganic Ventures) ranging from 12.5 ppb to 1000 ppb. 10 samples
were measured in triplicate for each sample and averaged.
To calculate the number of gold nanoparticles per capsid, we first calculate the
volume of a typical nanoparticle based on the TEM size and the equation for the volume
of a sphere. This value is then divided why the known literature value for the volume
of the gold unit cell, 0.0679 nm3, and subsequently multiplied by four as there are four
gold atoms per unit cell. The result can be converted to molecules using Avogadro’s
number. ICP-MS data can be converted from ppb to molecules of gold and the number
of Qβ capsids was calculated based on concentration used for analysis. The quotient of
the number of available pores on all Qβ capsids to the Au measured yields the
approximate number of particles assuming even distribution.
Circular Dichroism: Each sample was diluted to 0.4 mg/mL and subject to five pulses
of laser irradiation (500 mJ/cm2) each of which lasting six nanoseconds. The samples
were mixed between each pulse. Three samples of each were measure in triplicate using
an AVIV Circular Dichroism Spectrometer (Dynode Cutoff: 650 V, 180-260 nm)
Ellman’s Assay: To prepare Ellman’s reagent solution, 4 mg of Ellman’s reagent was
added into 1 mL of Ellman’s reagent buffer (0.1 M sodium phosphate buffer, 1mM
EDTA, pH 8.00) 125 μL of Ellman’s reagent was added into 6.25 mL of reagent buffer
to make the working solution. 25 μL of nQβ and reoxidized Qβ (2 mg/mL) or cysteine
standards (0 mM, 0.25 mM, 0.5 mM, 0.75 mM, 1.0 mM, 1.25 mM, 1.5 mM) was added
into 255 μL of Ellman’s working solution, followed by incubation at RT for 15 min

4
before reading the absorbance at 412 nm. A standard curve was plotted (Figure S5). The
unfunctionalized cysteine concentrations of Qβ DB conjugates were obtained from the
standard curve. The number of free thiols was calculated using the extinction coefficient
of 2-nitro-5-thiobenzoic acid (TNB) – 14,150M-1cm-1.
Dox Release in Agarose Gel: Utilizing the same samples prepared for circular
dichroism, samples were run in a 1% agarose gel using 1× TBE buffer for 1 h.
Cell Experiments: RAW 264.7 macrophages cultured in Dulbecco’s Modified Eagle
Medium (DMEM) supplemented with 10% Fetal Bovine Serum and 1%
Penicillin/Streptomycin were seeded on three-centimeter glass bottom plates with ~1 ×
106 cells per mL 1–2 days prior to the experiment and grown to 80% confluency. The
cells are incubated with 34 pM Qβ-GFP, AuNP@Qβ, DoxAuNP@Qβ and 4 nM Dox.
The cells are then washed 3× with 1× PBS and stained with an 8 µM solution of Hoechst
33342 nuclear dye. The cells were again washed 3× with 1× PBS and the cells were left
in 1 mL of 1× PBS. The plates were sealed and covered with a piece of 4 × 4 cm
cardstock. The cells were subjected to a single pulse of a 532 nm laser and imaged live
using either a confocal or epifluorescence microscope.
For the time studies in Figure S20, the cells were irradiated and placed in a live cell
imaging setup and incubated at 37 °C and a 5% CO2 environment for 12 hours and
imaged in one-hour increments.

5
Ancillary Figures:
Figure S1. Photographs of the Qβ@AuNP reaction procedure in the presence of Qβ
(left) without Qβ (middle), and on alkylated Qβ (right).
Figure S2. Kinetic study performed using a 0.2 mg/mL of Qβ pre-incubated for 3 h
with 50 mM HAuCl4. An absorbance scan was obtained every 5 min, the first point is

6
representative of the VLPs incubated with 50 mM salts and the second point is
immediately after the addition and mixing of NaBH4.
Figure S3. TOP: Wide field TEM micrograph of AuNPs@Qβ(Dox). Small AuNPs are
shown as gray or black dots on each VLPs. Small nanoparticles can be difficult to see

7
in wide field. Bottom: Sample AuNPs@Qβ with AuNP sizes measured using ImageJ
software based on 20 nm scale bar.
Figure S4. Wide-field TEM micrograph of the as-synthesized (unfiltered) AuNPs@Qβ
prior to running through a size exclusion column. Note the large free black
nanoparticles. Smaller black AuNPs can be seen on all VLPs in the field of vision at
higher magnification.

8
197
Au (No Gas) 197
Au (He)
Sample Concentration
(ppb)
RSD
(%)
Number of
Nanoparticles
per capsid
Concentration
(ppb)
RSD
(%)
Number of
Nanoparticles
per capsid
Qβ-AuNP 559.847 3.37 5.748 555.66 2.79 5.7
Qβ-Dox-
AuNP 596.5548 4.82 6.045 584.1903 4.37 6.0
Figure S5. ICP-MS data of AuNPs@Qβ and AuNPs@Qβ(Dox) used to determine the
number of nanoparticles per capsid in solution and the corresponding calibration curves.
Figure S6. PXRD of AuNPs synthesized with Qβ (AuNPs@Qβ) (top) or without Qβ
(bottom). Red is the calculated pattern, the blue is the experimental, and grey is the
difference between the two. The peaks align indicating the presence of the AuNPs but
the addition of the Qβ capsid broadens the peaks slightly.

9
Figure S7. TEM of AuNPs synthesized without Qβ. The reaction solution formed black
precipitate. TEM shows average size about 50 nm. Scale bars are 2 μm, 0.2 μm and 20
nm respectively.
Figure S8. Non-reducing SDS-PAGE. Lane 1: Unreduced Qβ, 2: Disulfide bonds on
Qβ reduced by TCEP (5 eq), 3: Qβ in 2 mM NaBH4 solution. 4: Qβ in 20 mM NaBH4
solution. This shows that the disulfides on Qβ can be reduced by TCEP, which results
in monomer formation in SDS-PAGE, but they cannot be reduced by NaBH4 (less than
20 mM) to yield the same result.
10
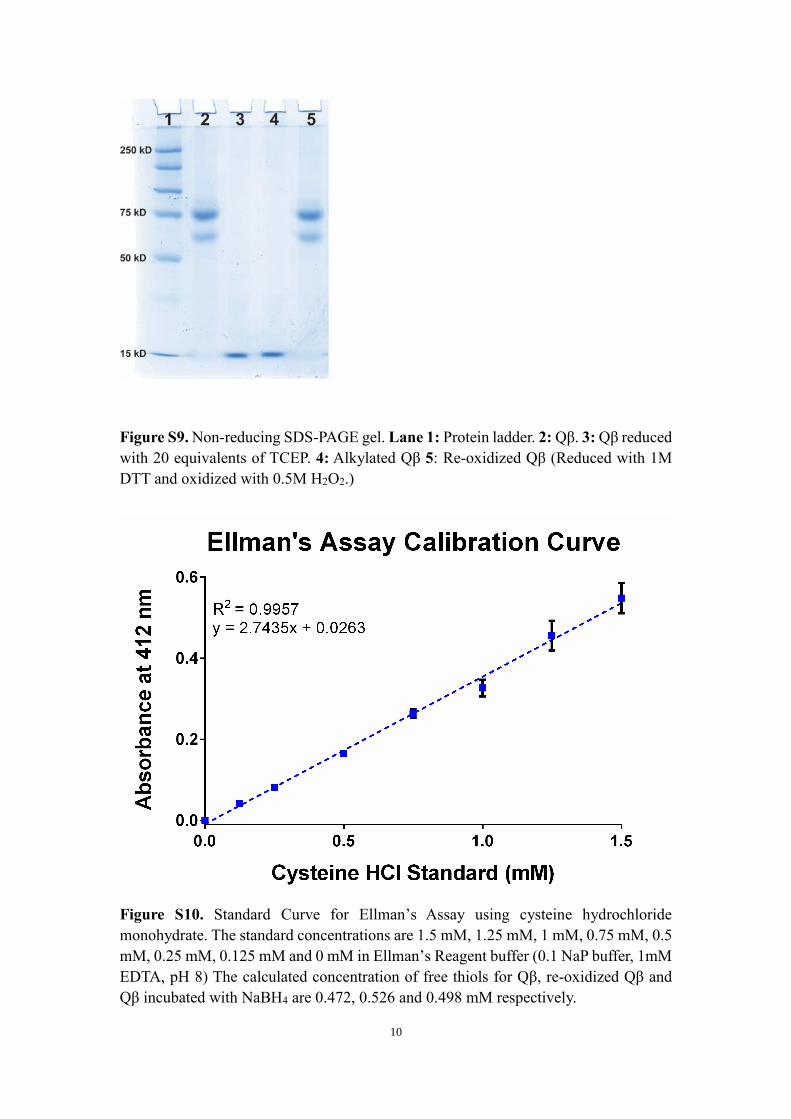
Figure S9. Non-reducing SDS-PAGE gel. Lane 1: Protein ladder. 2: Qβ. 3: Qβ reduced
with 20 equivalents of TCEP. 4: Alkylated Qβ 5: Re-oxidized Qβ (Reduced with 1M
DTT and oxidized with 0.5M H2O2.)
Figure S10. Standard Curve for Ellman’s Assay using cysteine hydrochloride
monohydrate. The standard concentrations are 1.5 mM, 1.25 mM, 1 mM, 0.75 mM, 0.5
mM, 0.25 mM, 0.125 mM and 0 mM in Ellman’s Reagent buffer (0.1 NaP buffer, 1mM
EDTA, pH 8) The calculated concentration of free thiols for Qβ, re-oxidized Qβ and
Qβ incubated with NaBH4 are 0.472, 0.526 and 0.498 mM respectively.

11
Figure S11. A TEM micrograph of AuNPs@Qβ that had been kept at RT for one month.
The Qβ are still studded with gold nanoparticles and the morphology of the VLP has
not changed.
Figures S12-S15 can be found in the Molecular Dynamic Simulation section.

12
Figure S16. Fluorescence spectra (λex = 480 nm λem = 580) of AuNP@ Qβ(Dox) before
(purple line) and after laser radiation (green line) to induce the release of Dox from the
Qβ capsid. Most of the Dox is released after a single pulse form the laser, however total
dox release can be achieved by 5 consecutive pulses (red line) akin to boiling the sample
(blue line).
Figure S17. Time dependent fluorescence spectra of AuNPs@Qβ(Dox) over a 24 h
period in various media (λex = 480 nm λem = 580). A fluorescence increase would be
indicative of Dox being released from the inside of the capsid. The flame denotes the
sample after being placed in boiling water for 10 min to denature the Qβ protein capsid.
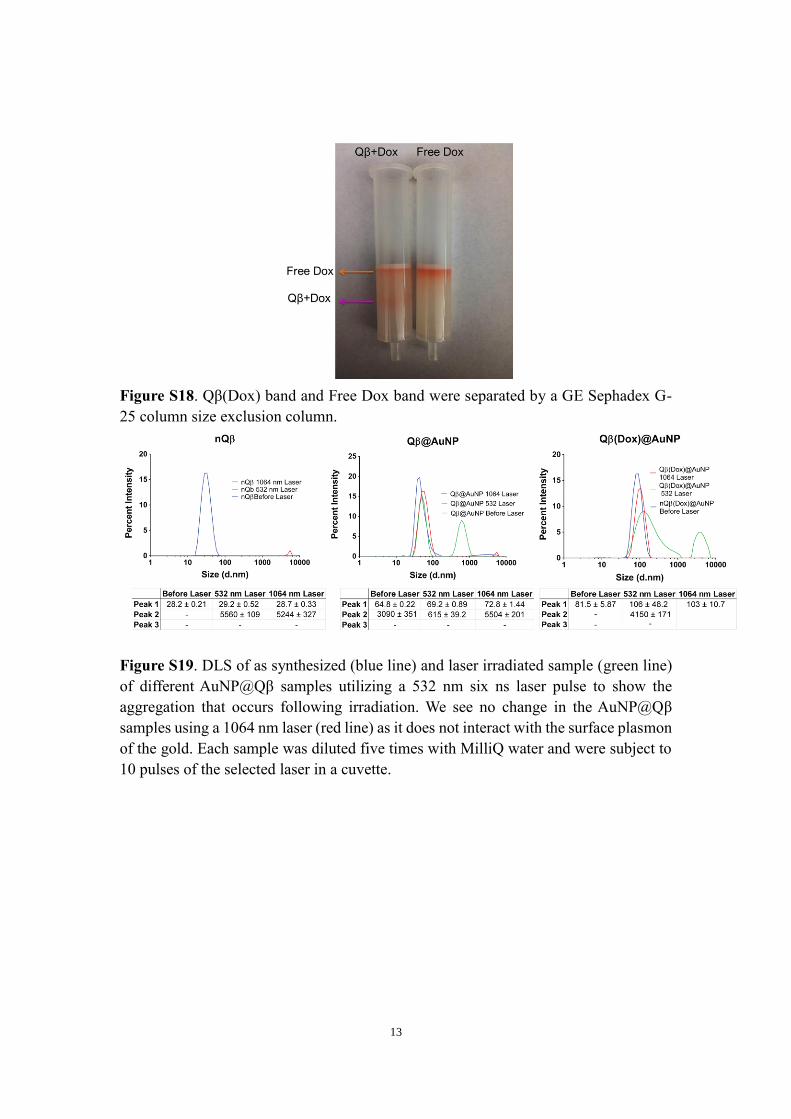
13
Figure S18. Qβ(Dox) band and Free Dox band were separated by a GE Sephadex G-
25 column size exclusion column.
Figure S19. DLS of as synthesized (blue line) and laser irradiated sample (green line)
of different AuNP@Qβ samples utilizing a 532 nm six ns laser pulse to show the
aggregation that occurs following irradiation. We see no change in the AuNP@Qβ
samples using a 1064 nm laser (red line) as it does not interact with the surface plasmon
of the gold. Each sample was diluted five times with MilliQ water and were subject to
10 pulses of the selected laser in a cuvette.

14
Figure S20. UV-vis of AuNPs@Qβ before (red line) and after laser radiation (black
line).

15
Figure S21. Agarose Gel images of Qβ solutions before and after laser irradiation. The
samples were either unirradiated or subject to five pulses of either a 532-nm laser or a
1064-nm laser Lane 1: Qβ 2: Qβ-1064 3: Qβ-532 4: Qβ-boiled 5: Qβ(Dox) 6:
Qβ(Dox)-1064 7: Qβ(Dox)-532 8: Qβ(Dox)-boiled 9: AuNP@Qβ 10: AuNP@Qβ-
1064 11: AuNP@Qβ-532 12: AuNP@Qβ-boiled 13: AuNP@Qβ(Dox) 14:
AuNP@Qβ(Dox)-1064 15: AuNP@Qβ(Dox)-532 16: AuNP@Qβ(Dox)-boiled 18:
Free Dox. Lanes denoted with (-) are empty. The bright field images show the red from
the samples containing gold nanoparticles. As can be seen in lanes 8, 15 and 16 of the
UV image, Doxorubicin is released from the interior of the capsid upon either boiling
or 532 nm laser activation. No release is observed with irradiation by the 1064 nm laser.
The Coomassie staining shows the presence of protein, in lanes in which these are
absent, the capsid shows aggregation either from boiling (lanes 4, 8, 12, and 16) or laser
irradiation (11 and 15)

16
Figure S22. All Fluorescence images were captured after 4 h incubation. Dox samples
were imaged 1 h after incubation. The blank samples show no major change in
morphology after laser exposure indicating no damage to cells. Dox acts quickly and
after 1 h the Dox has entered the cells and has begun to induce cell death. The samples
containing Qβ(Dox) show red fluorescence but no indication of cell death as the Dox
is trapped within the capsid (see MTT assay). Cells containing AuNP@ Qβ(Dox) only
show Dox release after laser irradiation.

17
Figure S23: Epifluorescence images of RAW Macrophages incubated with
AuNP@Qβ(Dox) for 4 h followed by irradiated with a single pulse of the 532 nm laser.
Cell that have been subjected to laser light show Dox release and over the course of 12
h following irradiation and the irradiated cells lose density and de-adhere due to the
effects of the anticancer drug.

18
Figure S24. Fluorescence images of cells after 4-h incubation. Dox samples were
imaged immediately after irradiation. The use of the 1064 nm laser is to depict the
effects of off-LSPR targeting, which, as expected, does not induce the release of Dox.
Molecular Dynamics Simulations
Molecular dynamics simulations were carried out with the NAMD 2.10 2
(http://www.ks.uiuc.edu/Research/namd/) package using the CHARMM 27 all atom
force field modified to include the nanoparticle. All simulations were initially energy
minimized for 1000 steps. The simulations were then run in vacuo at 300 K with a 2-fs
time step for a total time of 4 ns. A cutoff of 4.00 nm with a switching distance of 3.80
nm, and a pair list distance of 4.50 nm were used for the pair-wise (non-bonded)
interactions (a large cutoff was necessary due to scale of the nanoparticle). No periodic
boundary conditions were used.

19
Modeling the Gold Nanoparticle
Although the experimentally synthesized gold nanoparticles (AuNPs) are not truly
spherical, we assumed they could be reasonably approximated as spheres. Therefore,
the gold nanoparticle was modeled as a single site spherical “coarse grain” particle,
which was added to the CHARMM force field using tabulated potentials. The non-
bonded parameters were derived by minima matching of a non-standard Lennard-Jones
(LJ) 80-40 potential, which is given by Eq. 1.
Uij = 4εij [(σij
rij)
80
− (σij
rij)
40
] Eq (1)
for particles i and j separated by a distance rij , to the potential energy between an all
atom gold nanoparticle (aa-AuNP) and a carbon (CHARMM type C) atom. To generate
an aa-AuNP, a slab of gold atoms was generated using the Inorganic Builder extension
in VMD 3 (http://www.ks.uiuc.edu/Research/vmd). A diameter of 6.4 nm (the
experimentally determined diameter plus one standard deviation) was used for the
model nanoparticle, so a spherical selection of radius 3.2 nm was then cut from the
center of the gold slab and used as the aa-AuNP. The potential energy between the aa-
AuNP and the single carbon atom was computed as a function of their center of mass
separation (aa-AuNP rotational degrees of freedom were not included). In this
calculation, the gold atom parameters (Lennard-Jones 12-6) determined by Verde et al.
4 were used with no cutoff; standard Berthelot mixing was used between gold atoms
and the carbon atom. The LJ 80-40 potential was then fit to the potential energy curve
with Rminaa-AuNP-C= 3.03555 nm and єaa-AuNP-C = 1.38759 kcal mol-1. The fit is shown
in Fig. 1. The coarse-grained gold nanoparticle (cg-AuNP) parameters were then
derived by assuming Berthelot mixing between the cg-AuNP and carbon atom LJ
parameters: the values were determined to be σcg-AuNP = 57.8862 Å and εcg-AuNP =
17.5038 kcal mol-1. The cg-AuNP LJ parameters were then mixed with each atom type
in the system and the LJ 80-40 potential with mixed parameters was tabulated with
10000 points up to a maximum distance of 4.5 nm; linear interpolation was used for
intermediate distances.

20
Figure S12 The van der Waals (vdW) energy between aa-AuNP and the CHARMM
type C carbon atom as a function of their center of mass separation.
Simulating the cg-AuNP at the pore
A single hexameric and a single pentameric pore were chosen from the full virus
structure. For each pore of interest, the pore and the surrounding region within ~12.0
nm of the pore center were cut out from the full viral capsid. One starting configuration
of the cg-AuNP was then generated for each pore (two starting configurations in total):
the nanoparticle was placed by hand near the pore in a position outside the capsid. A
harmonic bond was added between the center of the cg-AuNP and each sulfur of the
active pore, modeling a fully chemisorbed nanoparticle. The parameters used for the
harmonic bond between the gold particle and the pore sulfurs were Kb = 100 kcal mol-
1 Å-2 and b0 = 34.0 Å. The choice of force constant was somewhat arbitrary, but the
bond length was chosen to be the cg-AuNP radius plus an additional 2 angstroms (which
is roughly the length of a gold atom to sulfur bond). In order to reduce the computation
time, only the positions of the gold particle and viral segments in the immediate region
of the pore were integrated, while the other protein atoms were fixed. The selections
are shown in S13.

21
Figure S13 Hexameric (Left) and pentameric (Right) pore selections used for
simulations. The pore sulfurs (in yellow) and the surrounding segments (in green) were
allowed to move during the simulations, while the outer segments (in red) were fixed.
Measuring the pore diameter
A relatively simple minimum cone angle measurement was used to approximate the
pore size (without including atom size effects). In this scheme the maximum cross-
sectional diameter of a cone that just overlaps an atom center is computed. A vector is
determined between the geometric center of the pore sulfurs (assumed to be the pore
center) and a radially central reference point. The reference point was taken as the
geometric center of the fixed atoms with distance greater than 10 nm from the cg-AuNP
center in the initial configuration. Then a selection of atoms (not including the cg-AuNP)
within 0.20 nm of the sulfur center was selected. For each of the selected atoms, a vector
was computed from the reference point to the atom center, and the angle between that
vector and the pore center vector was computed. The minimum angle was determined
and was subsequently used to compute the corresponding circular cross section
diameter of the cone. The measurement is graphically depicted in S14.

22
Figure S14. Depiction of the minimum cone angle measurement used to approximate
the pore size.
Measuring the Percent protected Surface Area of the cg-AuNP
We used a Monte Carlo routine to measure the relative amount of the cg-AuNP surface
area that is protected by the surrounding proteins. First the coordinates of protein atoms
within 3.5 nm of the cg-AuNP center were projected onto the surface of the cg-AuNP
(with radius 3.2 nm). An example selection is shown in S15

23
Figure S15. cg-AuNP and the selection of atoms within a distance of 3.5 nm from its
center.
The Monte Carlo trial moves consisted of randomly placing a point on the surface of
the cg-AuNP. These points were flagged as accepted if they did not overlap any atoms.
All atoms were assigned a uniform radius of 0.25 nm (roughly the size of a carbon
atom). A total number of trial moves nt = 20000 was used and the number of
accepted trial moves na was recorded. The Percent protected Surface Area (PSA) was
computed from the acceptance ratio fa =na nt⁄ using Eq. 2.
PSA=(1 − fa)4πrcg-AuNP2 ∗ 100 Eq (2)
cg-AuNP at the pentameric pore
As seen in Figure S13, a 6.4 nm cg-AuNP was modeled at the pentameric pore. The cg-
AuNP was bound to the ten sulfurs of the pentameric pore structure. The cg-AuNP
seemed to have a larger effect on the local structure of the pentameric pore than was
observed in the hexameric pore. By the end of the simulation the diameter of the
pentameric pore had nearly doubled. However, the cg-AuNP did not embed very deep
into the pore structure. Only about 9% of the nanoparticle's surface area was protected
by the pore proteins. That is less than half of the protected area measured for the
hexameric pore (~23%), which suggests that AuNPs at pentameric pores would likely

24
not be stabilized as well as those at hexameric pores.
Effect on the critical size of partially protecting AuNPs
As noted in the main text, 6 nm naked gold nanoparticles (AuNPs) are unstable and
subject to rapid Ostwald ripening, which causes precipitation [main text citations 41,
42]. Being partially embedded in the virus coat protein seems to offer additional
stability and protection for these AuNPs. Since the AuNPs were modeled as spheres
in the MD simulations, we compare both the simulation and experimental results to
classical nucleation theory of spherical particles.
From classical nucleation theory, the free energy associated with a spherical
nanoparticle of radius r is given by Eq. 3,
ΔG(r) = −4
3πr3ΔGV + 4πr2γs Eq. 3
in which ΔGV is the free energy per unit volume of the substance of which the
nanoparticle is composed, and γs is the interfacial tension between the nanoparticle
and the surrounding solvent. The critical radius (corresponding toΔG(r) = 0) is given
by Eq. 4.
rs =2γs
ΔGV Eq. 4
However, if some fraction fp of the nanoparticle's surface area is protected from the
solvent by a protectant with nanoparticle-protectant interfacial tension γp, Eq. 3 can be
re-written as Eq. 5.
ΔG(r) = −4
3πr3ΔGV + (1 − fp)4πr2γs+fp4πr2γp Eq. 5
In Eq. 5 the interfacial free energy cost due to the surface area of the nanoparticle is
split into two components: one for the nanoparticle-solvent contribution, and one for
the nanoparticle-protectant contribution. Likewise, the new critical radius is given by
Eq. 6.
rsp =2[(1−fp)γs+fpγp]
ΔGV Eq. 6
Now we can take the difference between the two critical radii (Eqs. 4 and 6), which is
given in Eq. 7.

25
Δr=rsp–rs =2fp(γp−γs)
ΔGV Eq. 7
We can define the protectant to have a favorable interaction with the nanoparticle, such
that the nanoparticle has a lower interfacial tension with the protectant than with the
solvent (i.e. (γp − γs) < 0). In this case, the difference in critical radii will be less than
zero (i.e. Δr<0), which would imply that the nanoparticle with protectant is stable at a
smaller radius than the bare nanoparticle in solvent.
The simulation results suggest that AuNPs at Qβ pores would be afforded a fair
amount of protection by the surrounding pore proteins; ~23% of the surface area was
protected in the model system for the 6.4 nm cg-AuNP at the hexameric pore and ~9%
for the same particle at the pentameric pore. From experiments, the bare AuNPs that
precipitated from solution had a diameter of 48 ± 10 nm, while those at the Qβ pores
had a diameter of 6.2 ± 0.2 nm. Assuming those diameters correspond to the critical
radii, we can compute the difference in the critical radius to be Δr ≈ −20 nm. This is
a rather large reduction in the size of nanoparticles, which is consistent with the pore
proteins acting as a strong protectant for those AuNPs bound to them.
References
(1) Manzenrieder, F.; Luxenhofer, R.; Retzlaff, M.; Jordan, R.; Finn, M. G.
Angew. Chem. Int. Ed. 2011, 50, 2601 –2605.
(2) Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.;
Chipot C.; Skeel R. D.; Kale L.; Schulten K.; J. Comput. Chem. 2005, 26,
1781-1802.
(3) Humphrey W; Dalke A.; Schulten K.; J. Mol. Graphics.1996, 14, 33-38.
(4) Verde A.V.; Acres J.M.; Maranas J.K.; Biomacromolecules, 2009, 8, 2118-2128.

download fileview on ChemRxivSite Selective Nucleation and Size Control of Gold Nanopa... (1.70 MiB)